Back to Journals » International Journal of Women's Health » Volume 18
Development and Validation of a Prognostic Model for Cervical Cancer Based on Chlamydia trachomatis-Associated Transcriptional Signatures
Authors Wang M ![]() , Xiong S
, Xiong S ![]() , Qiu X, Hu Z, Xie L
, Qiu X, Hu Z, Xie L ![]() , Zhao H, You C
, Zhao H, You C ![]()
Received 24 March 2026
Accepted for publication 16 June 2026
Published 9 July 2026 Volume 2026:18 611743
DOI https://doi.org/10.2147/IJWH.S611743
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Vinay Kumar
Mei Wang,1,* Shiyin Xiong,2,* Xiufeng Qiu,2 Zhengrui Hu,3 Lang Xie,4 Hao Zhao,2 Cong You2
1Department of Dermatology and Venereology, Tianjin First Central Hospital, Tianjin, 300192, People’s Republic of China; 2Department of Dermatology and Venereology, The First Affiliated Hospital of Gannan Medical University, Ganzhou, 341000, People’s Republic of China; 3College of Pharmacy, Gannan Medical University, Ganzhou, 341000, People’s Republic of China; 4First Clinical Medical College of Gannan Medical University, Ganzhou, 341000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Cong You, Department of Dermatology and Venereology, The First Affiliated Hospital of Gannan Medical University, Ganzhou, 341000, People’s Republic of China, Email [email protected]
Background: Chlamydia trachomatis (CT) infection has been identified as an independent predictor of risk of cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC); however, its prognostic relevance and molecular mechanisms remain poorly understood. Therefore, we aimed to construct a CESC prognostic model based on genes associated with CT infection.
Methods: Transcriptomic data were obtained from GEO and TCGA databases. Differentially expressed genes (DEGs) were identified from GSE63514 (24 normal controls, 28 CESE samples) and GSE158814 (2 negative controls, 2 CT-infection HeLa cells). Overlapping genes were defined as CT-related genes. TCGA-CESC cohort (n = 252) was split into training and validation sets. Univariate Cox regression was used to screen candidate genes, and then multivariate Cox regression were performed to construct a prognostic signature and calculate RiskScore. LASSO-Cox regression and bootstrap analysis evaluated model stability. Patients were stratified by median RiskScore. A nomogram integrating clinical factors and RiskScore was established, followed by evaluation using calibration curves, receiver operating characteristic, and decision curve analyses. Gene set enrichment analysis, immune infiltration, and drug sensitivity analyses were performed to explore the biological and therapeutic implications of the high- and low- RiskScore group.
Results: A total of 31 CT-related CESC genes were identified, among which CSF2RB, LAMA4 and HAL were risk factors, and CSF2RB was a protective factor. A three-gene prognostic model (LAMA4, CSF2RB, HAL) was developed, showing robust predictive performance in both training and validation cohorts. Nomogram analysis confirmed that RiskScore, age, and stage were independent prognostic factors. Functional enrichment revealed that DEGs between the high- and low-risk groups were mainly enriched in immune-related pathways. Immune infiltration analysis indicated that low-risk patients exhibited higher infiltration of effector immune cells (e.g. CD4+, B cells, dendritic cells, M1 macrophages), suggesting a more active antitumor immune microenvironment. Drug sensitivity analysis identified BI-2536, SB505124, and Sepantronium bromide as potential therapeutic agents for high-risk patients.
Conclusion: We established a novel prognostic model for CESC based on CT-related genes in an HPV-positive CESC background, which provides new insights into CT infection-associated immune regulation in CESC and individualized immunotherapy and targeted treatment strategies.
Keywords: cervical cancer, Chlamydia trachomatis, prognostic model, nomogram, immune infiltration
Introduction
Cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC) is the fourth leading cause of cancer-related mortality among women worldwide.1 According to the GLOBOCAN 2022 statistics, there are an estimated 660,000 new cases and more than 340,000 related deaths annually.2 Although the implementation of CESC screening programs and human papillomavirus (HPV) vaccination has markedly reduced incidence in certain regions, the incidence and mortality of CESC remain high in developing countries and resource-limited areas.3,4
Persistent HPV infection has been considered the most important factor in the development of CESC.5 However, accumulating evidence suggests that CESC is a multi-factor, multi-stage disease, in which other microbial infections may also synergistically contribute to tumor occurrence and development.6,7 Chlamydia trachomatis (CT) is a Gram-negative, obligate intracellular bacterium and one of the most common sexually transmitted pathogens worldwide.8 Previous meta-analysis has demonstrated that CT infection is significantly associated with the pathogenesis of CESC,9 and women co-infected with CT and HPV exhibit a higher incidence of CESC compared with HPV-negative patients.10 CT infection can cause epithelial damage and disrupt local immune homeostasis. It impairs antigen presentation through the downregulation of MHC class I and II molecules, thereby suppressing CD4+ T-cell activation. In parallel, CT can evade neutrophil extracellular traps and inhibit macrophage activation, enabling resistance to host immune clearance.11 Moreover, CT-stimulated macrophages produce TNF-α, which can induce apoptosis of neighboring T cells.12 Collectively, these immune evasion and inflammatory mechanisms contribute to chronic cervical inflammation and facilitate the persistence of HPV.13,14 At the genomic level, CT infection has been shown to induce DNA damage in host cells and inhibit the repair of double-strand breaks, thereby promoting malignant transformation.15,16 These biological effects are further reflected at the transcriptomic level, as CT infection can significantly alter host gene expression profiles, including genes involved in inflammation, DNA repair, and cell cycle regulation.17,18 These molecular alterations provide a deeper understanding of how CT infection facilitates the persistence of HPV and its progression to CESC.
Recent advances in high-throughput sequencing technologies and bioinformatics approaches have enabled the prognostic evaluation of tumors using large-cohort, multi-gene models. In CESC, a number of genetic biomarkers have been identified, and several gene-based prognostic models have been developed. For instance, Ding et al19 identified that ZNF695 as a potential prognostic biomarker and immunotherapeutic target in patients with CESC. Zhang et al20 reported CEP78, DOCK7, DPY19L4, and POM121 as key genes associated with CESC based on computational analyses. Zeng et al21 demonstrated that hsa_circ_0000021 promotes cervical cancer (CC) progression by regulating the miR-3940-3p/KPNA2 axis. Furthermore, Du et al established a ferroptosis-related genes signature for prognostic model in CC,22 while Chen et al constructed a prognostic model based on immune-related long noncoding RNA in CC patient.23 Collectively, these studies have shown promising performance in predicting overall survival (OS) and guiding therapeutic strategies. However, most existing models are based on general tumor-related or pathway-specific genes, and no studies have specifically focused on constructing a prognostic model based on CT infection-related genes in CESC, leaving a significant gap in both research and clinical application.
In this study, we aimed to address this gap by integrating transcriptomic datasets from the Gene Expression Omnibus (GEO) and the Cancer Genome Atlas (TCGA) databases to identify differentially expressed genes (DEGs) between CESC and normal tissues. These DEGs were then intersected with those derived from HeLa cell models before and after CT infection to identify candidate genes associated with CT infection. Subsequently, Cox regression analysis was performed to construct a prognostic risk model in the TCGA-CESC cohort.
Materials and Methods
Data Source
The R package GEOquery was used to download transcriptomic data from the GEO database. Briefly, the GEO database was systematically searched using the keywords “Chlamydia trachomatis” and “Cervical cancer”. Specifically, the GSE63514 database, which includes 24 normal samples and 28 CESC samples, was used to screen DEGs between human CESC and normal cervical tissues. Meanwhile, GSE158814, containing two HeLa cell samples without CT infection (negative controls) and two HeLa cell samples collected 20 hours post-infection with CT, was applied to obtain transcriptomic profiles altered by CT infection in HeLa cells. Transcriptomic and clinical data for CESC were retrieved from the TCGA database, containing 306 tumor samples, which were used for model construction and validation. Exclusion criteria: samples with incomplete clinical information, missing survival data, or invalid OS time (≤ 0). Finally, a total of 252 CESC samples with complete transcriptomic and clinical data were retained for subsequent analyses (Table 1). A flowchart of the study design is shown in Figure 1.
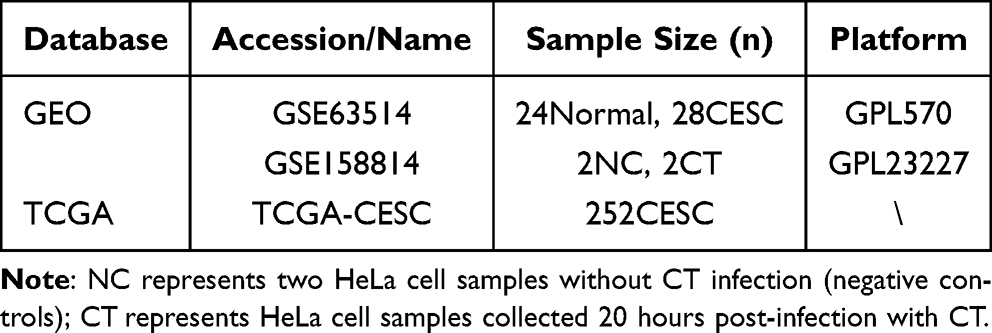 |
Table 1 Data Source |
 |
Figure 1 The flow diagram of study design. |
Identification and Analysis of DEGs
For GSE63514, probe annotation was performed using the GPL570 platform, and the expression values of duplicate genes were averaged. DEGs were identified using the limma package with the thresholds of p < 0.05 and |log2 fold change| > 2. For GSE158814, gene annotation was conducted using the Human.GRCh38.p13.annot.tsv.gz file, and DEGs were identified with the DESeq2 package under the same thresholds (p < 0.05 and |log2 fold change| > 2). Finally, the intersection of DEGs from GSE63514 and GSE158814 was taken to obtain the overlapping gene set. The two datasets were jointly analyzed to intersect CT-associated genes and CC-related genes, ensuring that the selected genes were closely associated with both CT infection and cervical malignancy.
CT-Related Genes RiskScore Model Construction and Validation
The 252 cases in TCGA-CESC cohort were randomly divided into a training set (n = 126) and a validation set (n = 126) at a 1:1 ratio. In the training set, univariate and multivariate Cox regression analyses were performed to screen the overlapping genes and identify CT infection-related prognostic genes. The risk score was calculated using the formula:
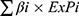
where ExPi represents the mRNA expression level of each overlapping gene, and βi denotes the regression coefficient derived from multivariate Cox analysis. To evaluate the stability of the selected prognostic genes, LASSO-Cox regression and bootstrap analysis were further performed in the test cohort. Based on the median risk score, CESC patients were stratified into low- and high-risk groups, and Kaplan-Meier survival analysis was conducted to compare OS between the two groups. Receiver operating characteristic (ROC) curves were further generated to assess the predictive performance of the model for 1-, 3-, and 5-year outcomes.
Nomogram Construction
Clinical characteristics of CESC patients, including age, stage, and grade, were integrated with the RiskScore. Univariate and multivariate Cox regression analyses were performed to identify independent prognostic factors of CC. A nomogram was then constructed based on the RiskScore and other clinicopathological variables using the R package regplot. The calibration, discrimination, and clinical utility of the nomogram were evaluated using calibration curves, ROC curves, and decision curve analysis (DCA), respectively.
Gene Set Enrichment Analysis (GSEA)
To investigate the biological pathways associated with differential expression between the high- and low-risk groups defined by the prognostic RiskScore, GSEA24–26 was performed using the C5 gene sets “c5.all.v2025.1.Hs.entrez.gmt” and C2 gene set “c2.all.v2025.1.Hs.entrez.gmt” downloaded from the MSigDB database (https://www.gsea-msigdb.org/gsea/msigdb/). DEGs were identified using thresholds of p < 0.05 and |log2 fold change| > 1. GSEA was then conducted with the following significance criteria: |Normalized Enrichment Score (NES)| > 1, nominal p-value < 0.05, and false discovery rate (FDR) q-value < 0.25. For the gene set “c5.all.v2025.1.Hs.entrez.gmt”, the Gene Ontology (GO) results were categorized into three main functional domains: molecular function (MF), biological process (BP), and cellular component. The enrichment results were visualized using ridge plots.
Immune Infiltration Analysis
xCell, an ssGSEA-based method, estimates the abundance of 64 cell types in the tumor microenvironment, including immune, stromal, and epithelial cells, based on gene expression data.27 To assess the differences in immune cell infiltration between high- and low-risk groups of TCGA-CESC patients, we performed immune infiltration analysis using xCell (v1.1.0). The results were visualized using box plots.
Gene-Gene Interaction (GGI) Analysis
GeneMANIA investigates the functions of genes and gene sets. It utilizes a large amount of functional association data to find other genes related to the input gene set. The data include protein and genetic interactions, pathways, co-expression, co-localization and protein domain similarity.28 GeneMANIA was used to explore the interaction relationship of prognostic genes.
Drug Sensitivity Analysis
OncoPredict integrates publicly available cell line drug sensitivity data with corresponding gene expression profiles (eg., Genomics of Drug Sensitivity in Cancer, Cancer Therapeutic Response Portal) to construct linear predictive models and generate drug response predictions. In this study, the GDSC2 dataset within OncoPredict was applied to estimate the drug IC50 values for CESC patients.29 Box plots of drug sensitivity between groups were generated using ggplot2 and other R packages, and significance was assessed by the Wilcoxon rank-sum test.
Statistical Analysis
All analyses were conducted in R software (version 4.4.2). Kaplan-Meier analysis was used to plot survival curves, and the Wilcoxon rank-sum test was applied to assess statistical significance. A two-sided p < 0.05 was considered statistically significant.
Results
Identification of Overlapping DEGs Between CESC and CT Infection in an HPV-Positive Context
The data from GSE63514 and GSE158814 were well normalized, and a total of 673 DEGs were identified between the CESC and normal groups, including 343 downregulated and 330 upregulated genes (Figure 2A–C). In the GSE158814 dataset, which was generated from HPV-positive HeLa cells, 1,202 DEGs were identified between the CT-infection and negative control groups, comprising 214 downregulated and 988 upregulated genes (Figure 2D). These genes represent CT infection-associated transcriptional alterations under an HPV-positive CC cell background. The intersection of these two datasets yielded 31 overlapping genes associated with both CT-related transcriptional changes infection and CESC (Figure 2E).
 |
Figure 2 Screening of intersecting genes. (A) Boxplots showing the data quality of GSE63514. Green boxes represent the control group, and purple boxes represent the experimental group. (B) Volcano plots of GSE63514. Green dots indicate downregulated genes, and purple dots indicate upregulated genes. The top five genes ranked by |log2 fold change| are labeled. (C) Boxplots showing the data quality of GSE158814. (D) Volcano plots showing significantly altered genes in GSE158814. Green dots indicate downregulated genes, and purple dots indicate upregulated genes. The top five genes ranked by |log2 fold change| are labeled. (E) Venn diagram showing the intersecting DEGs between GSE63514 and GSE158814. |
Establishing a Prognostic Model Based on LAMA4, CSF2RB, and HAL
Among the 31 overlapping genes derived from CT infection-associated transcriptional alterations under an HPV-positive CESC cell background, univariate and multivariate Cox regression analyses (Figure 3A and B) identified three genes with p < 0.05 in multivariate analysis: LAMA4, CSF2RB, and HAL. Of these, LAMA4 and HAL were identified as risk factors (HR > 1), whereas CSF2RB acted as a protective factor (HR < 1). A prognostic model was constructed using these three genes in the training set and validated in the testing set. The risk score calculated as follows: RiskScore = (LAMA4 × 0.06023) + (CSF2RB × –0.09137) + (HAL × 0.14401). LASSO, Cox regression and bootstrap analysis further confirmed the robustness of prognostic genes (Table S1 and 2). Kaplan–Meier survival analysis demonstrated significant differences in both the training and testing sets (p < 0.05), with patients in the high-risk group exhibiting worse survival compared with the low-risk group (Figure 3C and D). ROC curves showed that the model had good predictive performance. In the training set, the area under the curve (AUC) values at 1-, 3-, and 5-years were 0.801, 0.741, and 0.766, respectively, indicating good predictive accuracy (Figure 3E). In the test set, the corresponding AUC values were 0.897, 0.636, and 0.622, suggesting an acceptable decline in predictive ability for mid- to long-term survival (Figure 3F).
 |
Figure 3 Construction and validation of the prognostic model. Results of univariate (A) and multivariate (B) Cox regression analyses. Green boxes indicate HR > 1, and purple boxes indicate HR < 1. Kaplan–Meier survival curves of high- and low-risk groups in the training (C) and test (D) cohorts. ROC curves evaluating the predictive performance and accuracy of the prognostic model in the training (E) and test (F) cohorts. |
Construction and Validation of the Nomogram
Univariate and multivariate Cox regression analyses integrating RiskScore with age, stage, and grade were performed (Table S3). The results showed that RiskScore, age, and stage were independent prognostic factors. Based on these findings, a nomogram was constructed incorporating RiskScore, age, and stage. The nomogram indicated higher scores to patients in the high-risk group and those with stage III–IV disease (Figure 4A). Calibration plots in both the training and testing sets showed good agreement between predicted and observed 1-, 3-, and 5-year OS (Figure 4B and D). The AUC values of nomogram for predicting 1-, 3-, and 5-year OS were 0.838, 0.720, and 0.793 in the training set, and 0.827, 0.684, and 0.747 in the testing set, respectively (Figure 4C and E). DCA further revealed that the nomogram yielded higher net benefit at 5 years compared with 1- and 3-year predictions (Figure S1).
 |
Figure 4 Construction and validation of the nomogram. (A) Nomogram constructed based on RiskScore, age, and stage to predict patient survival probability. (B) Calibration plots in the training cohorts. (C) ROC curves in the training cohorts. (D) Calibration plots in the test cohorts. (E) ROC curves in the test cohorts. |
Functional Enrichment and GGI Analysis Reveals Key Immune- and Tumor-Related Pathways
Differential expression analysis between the high- and low-risk groups identified 225 DEGs under the defined screening criteria. GSEA was performed, and the top five enriched pathways were visualized (Figure 5A and B). For the C5 gene sets, the top five enriched terms in BP included adaptive immune response, lymphocyte-mediated immunity, collagen fiber organization, leukocyte-mediated immunity, and immune response-regulating cell surface receptor signaling pathway (Figure 5A). For the C2 gene sets, the top five enriched pathways were Anastassiou multicancer invasiveness signature, Mclachlan dental caries up, Wieland up by HBV infection, Zhang FH deficient RCC C1 VS other up, Wallace prostate cancer race up (Figure 5B). Detailed enrichment results are provided in Tables S4 and S5.
 |
Figure 5 Functional enrichment and immune infiltration analyses. Gene set enrichment analysis (GSEA) results for the C5 (A) and C2 (B) gene sets. (C) GeneMANIA network analysis illustrating potential functional interactions among the model-related genes. (D) Immune infiltration analysis between the high- and low-risk groups. Green and purple boxes represent the high- and low-risk groups, respectively. Cell types with significant intergroup differences (p < 0.05) are showed on the x-axis. (E) Potential therapeutic agents predicted for the high-risk group. Note: * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001, ns p ≥ 0.05. |
GeneMANIA network analysis (Figure 5C) indicated that the three prognostic genes (HAL, CSF2RB, and LAMA4) are functionally interconnected with a shared interaction network associated with immune response, extracellular matrix organization, and metabolic regulation. This suggests that these genes may participate in coordinated biological processes rather than acting independently.
Immune Infiltration Differences Reveal Potential Immune Regulatory Mechanisms
Immune infiltration analysis demonstrated significant differences in multiple immune cell populations between the high- and low-risk groups (Figure 5D). Immune-activating cells including T cell subsets (CD4⁺ T cells, CD8⁺ central memory T cells, Tregs), B cells (B cells, memory B cells, naive B cells), dendritic cells (DCs, pDCs, aDCs), and M1 macrophages were significantly enriched in the low-risk group, suggesting a potential role in controlling tumor progression. In contrast, stromal-derived cells such as mesenchymal stem cells (MSCs) and smooth muscle cells were enriched in the high-risk group, indicating a more immunosuppressive tumor microenvironment. As shown in Figure S2, the ImmuneScore and MicroenvironmentScore were significantly higher in the low-risk group, reflecting enhanced immune infiltration and overall microenvironmental activity, which is consistent with better prognosis. No significant difference in StromaScore was observed between the groups, suggesting comparable stromal content.
Potential Therapeutic Agents for the High-Risk Group
Drug sensitivity analysis revealed that the median IC50 values of BI-2536_1086, SB505124_1194, and Sepantronium bromide_1941 were significantly lower in the high-risk group than in the low-risk group (Figure 5E, p < 0.05). These findings suggest that high-risk patients with CESC may exert increased sensitivity to these agents. Detailed drug sensitivity data are provided in Table S6.
Discussion
CT infection may synergistically promote the cervical carcinogenesis and enhance HPV E6/E7-mediated immunosuppression.30 Moreover, CT infection has been identified as an independent predictor of CC risk.31 Nevertheless, no studies have developed a prognostic model for CESC based on CT infection-related genes in an HPV-positive CESC background. In this study, we systematically investigated CT infection-related genes in CESC by integrating transcriptomic data from GEO and TCGA databases, and constructed a novel prognostic model comprising three genes (LAMA4, CSF2RB, and HAL) associated with CT infection. This model demonstrated stable prognostic performance in both the training and validation cohorts, and its predictive accuracy was further improved by a nomogram incorporating age and stage. Furthermore, our findings support the hypothesis that CT infection may contribute to cervical carcinogenesis and potentially facilitate HPV persistence and malignant transformation through multiple mechanisms, including chronic inflammation, immune suppression, and genomic instability.
We identified 31 overlapping genes between CT-infected HeLa cells and CESC tissues. Among them, three genes (LAMA4, CSF2RB, and HAL) were screened through univariate and multivariate Cox regression analysis, and subsequently incorporated into a prognostic risk model. Notably, LAMA4 and HAL acted as risk factors, whereas CSF2RB served as a protective factor. Laminin subunit α4 (LAMA4), a member of the laminin family, is an extracellular glycoprotein involved in regulating cell differentiation, migration, and adhesion.32 Previous studies have demonstrated that LAMA4 promotes the migration, proliferation, and survival of endothelial, blood, and cancer cells in multiple cancers,33,34 which is consistent with our findings that LAMA4 is associated with poor prognosis in CESC. Histidine ammonia-lyase (HAL) is the rate-limiting enzyme in histidine catabolism and catalyzes the conversion of L-histidine into urocanate and ammonia in the liver and skin.35 Emerging evidence suggests that HAL is involved in tumor metabolic reprogramming and may contribute to tumor progression.36 Colony stimulating factor 2 receptor subunit beta (CSF2RB) is the common subunit of the type I cytokine receptors for granulocyte-macrophage colony-stimulating factor (GM-CSF), interleukin (IL)-3 and IL-5.37 CSF2RB has been implicated in immune regulation and inflammation responses, and is highly expressed in Treg cells, where it may participate in antitumor immune responses.38 Zhu et al further reported that CSF2RB serves as an important biomarker in lung adenocarcinoma and may function as a tumor suppressor gene by inhibiting the growth, proliferation, and differentiation of lung adenocarcinoma cells.39 Collectively, the differential roles of these genes are consistent with their contributions to tumor progression, thereby supporting their biological relevance and prognostic value in CESC.
Functional enrichment analysis provided mechanistic insights into the molecular underlying prognostic model. GSEA revealed that DEGs between high- and low-risk groups were significantly associated with immune responses and tumor-related signaling pathways. Notably, the enrichment of adaptive immune processes and lymphocyte-mediated immunity in the low-risk group underscores the importance of effective anti-tumor immunity in improving survival.40 Consistent with the GSEA findings, significant differences in immune cell infiltration were observed between the two risk groups. Low-risk groups exhibited increased infiltration of multiple immune cell populations, including T cell subsets (CD4+ T cells, CD8⁺ central memory T cells, Tregs), B cells (B cells, memory B cells, naïve B cells), dendritic cells (DCs, pDCs, aDCs), and M1 macrophages. These immune cells are well known to play pivotal roles in antitumor immunity. Previous studies have demonstrated that activated CD4+ and CD8+ T cells exert potent antitumor effects.41 Cytotoxic CD8+ T cells act as primary effectors in eliminating pathogens and tumor cells, while CD4+ T cells are essential for sustaining CD8+ T-cell responses and preventing their functional exhaustion.42,43 Treg cells are considered a subset of T cells with immunosuppressive function.44 However, the role of Tregs in cancer procession remains complex and highly context-dependent. A large meta-analyses suggests that the prognostic significance of FoxP3⁺ Tregs varies across tumor types and may even correlate with improved survival outcome in certain malignancies.45 Furthermore, increased Tregs infiltration has paradoxically been associated with improved prognosis in several cancers, highlighting the functional heterogeneity of these cells.46 Tregs may suppress tumor-promoting inflammation, thereby contributing to improved prognosis under specific tumor microenvironment conditions.47 These findings underscore that the prognostic and functional significance of Tregs should be interpreted with broader immune microenvironment rather than evaluated as an isolated cell population. In addition, M1-polarized macrophages exhibit antitumor activity through both intrinsic phagocytic capacity and the promotion of pro-inflammatory immune responses.48 The enrichment of these immune cells in the low-risk group suggests a more active antitumor immune response, which may contribute to the favorable prognosis observed in these patients. In contrast, high-risk patients displayed enrichment of stromal-derived cells, including mesenchymal stem cells and smooth muscle cells, indicative of a more immunosuppressive tumor microenvironment.49 This immune landscape may partially explain the poorer clinical outcomes observed in high-risk patients.
Importantly, drug sensitivity analysis revealed potential therapeutic implications. The high-risk group showed lower predicted IC50 values for BI-2536, SB505124, and Sepantronium bromide, suggesting that increased sensitivity to these agents in patients with poor prognosis. BI-2536, a Polo-like kinase 1 inhibitor, has demonstrated anti-proliferative activity in cervical and other cancers.50 SB505124 targets transforming growth factor-beta signaling, which is frequently dysregulated in tumor immunosuppression.51 Sepantronium bromide is a survivin inhibitor that can induce apoptosis in cancer cells.52 Collectively, these findings highlight the potential value of integrating molecular risk stratification with precision therapeutic strategies in CESC.
Despite these promising findings, several limitations should be acknowledged. First, the CT-related genes were derived from an in vitro infection model based on HeLa cells. Because HeLa cells are HPV-positive, the transcriptional response to CT infection may be influenced by an underlying HPV-driven oncogenic background. Therefore, the identified CT-related genes may reflect host responses in the context of HPV-positive CESC rather than CT-specific effects alone. Second, the CT-infected cell dataset (GSE158814) included only two infected samples and two control samples and was limited to a single HeLa cell model, which may limit ability to capture the heterogeneity of CESC subtypes. Third, HPV infection is a key driver of CESC, and adjusting for HPV status could help mitigate potential confounding effects. However, complete and reliable HPV status (eg., HPV16/18 positivity or viral load) is not available in the TCGA-CESC cohort, which is a common limitation of public CESC omics datasets. In addition, immune cell infiltration was inferred using the xCell algorithm rather than validated through experimental assays. While xCell is a validated and widely computational approach, in silico estimations cannot fully substitute for experimental validation. Therefore, further studies incorporating clinical specimens and functional experiments are required to validate these findings. Finally, the biological functions of LAMA4, CSF2RB, and HAL in CESC remain to be experimentally validated, particularly regarding their mechanistic roles in CT-HPV co-infection and immune regulation.
Conclusions
In this study, we constructed and validated a three-gene risk model (LAMA4, CSF2RB, HAL) based on CT infection-associated transcriptional alterations in CESC. The model demonstrated stable predictive performance in independent cohorts. A nomogram combining RiskScore with clinical factors further enhanced prognostic accuracy and clinical applicability. Immune infiltration analyses revealed that differences in tumor immune microenvironment underlie the prognostic stratification, while drug sensitivity analyses suggested potential therapeutic strategies for high-risk patients. In summary, our findings provide a novel prognostic tool for CESC and offer additional insights into CT infection-associated molecular and immune-related alterations in CESC, which may facilitate future studies on individualized prognosis assessment and therapeutic strategies.
Abbreviations
BP, biological process; CC, cervical cancer; CESC, Cervical squamous cell carcinoma and endocervical adenocarcinoma; CSF2RB, Colony stimulating factor 2 receptor subunit beta; CT, Chlamydia trachomatis; DCA: decision curve analysis; DEGs, differentially expressed genes; FDR, false discovery rate; GEO, Gene Expression Omnibus; GGI, Gene-gene interaction; GO, Gene Ontology; GSEA, Gene set enrichment analysis; HAL, Histidine ammonia-lyase; HPV, human papillomavirus; LAMA4, Laminin subunit α4; MF, molecular function; NES, Normalized Enrichment Score; OS, overall survival; ROC, Receiver operating characteristic; TCGA, the Cancer Genome Atlas.
Data Sharing Statement
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Ethics Approval and Informed Consent
This study conforms to the principles outlined in the Declaration of Helsinki. All data used in this study are publicly available, and the studies have obtained approval from their respective institutional review boards. No additional ethical approval was required, as this study was exempt from approval based on national legislation guidelines, such as item 1 and 2 of Article 32 of the Measures for Ethical Review of life Science and Medical Research involving Human subjects dated February 18, 2023, China.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the Science and Technology Project of Jiangxi Health Commission (Grant No.: 202510462), the Science and Technology research project of Jiangxi Education Department (Grant No.: GJJ2401311), and the Young Scientists Fund of the Natural Science Foundation of Jiangxi Province (Grant No.: 20242BAB20427).
Disclosure
Mei Wang and Shiyin Xiong are co-first authors for this study. The authors report no conflicts of interest in this work.
References
1. Arbyn M, Weiderpass E, Bruni L, et al. Estimates of incidence and mortality of cervical cancer in 2018: a worldwide analysis. Lancet Glob Health. 2020;8(2):e191–14. doi:10.1016/S2214-109X(19)30482-6
2. Bray F, Laversanne M, Sung H, et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2024;74(3):229–263. doi:10.3322/caac.21834
3. Bedell SL, Goldstein LS, Goldstein AR, Goldstein AT. Cervical cancer screening: past, present, and future. Sexual Med Rev. 2020;8(1):28–37. doi:10.1016/j.sxmr.2019.09.005
4. Wu J, Jin Q, Zhang Y, et al. Global burden of cervical cancer: current estimates, temporal trend and future projections based on the GLOBOCAN 2022. J. Natl. Cancer Cent. 2025;5(3):322–329. doi:10.1016/j.jncc.2024.11.006
5. Abu-Rustum NR, Yashar CM, Bean S, et al. NCCN Guidelines Insights: cervical Cancer, Version 1.2020: featured Updates to the NCCN Guidelines. J Natl Compr Cancer Netw. 2020;18(6):660–666. doi:10.6004/jnccn.2020.0027
6. Xie L, Li Q, Dong X, et al. Investigation of the association between ten pathogens causing sexually transmitted diseases and high-risk human papilloma virus infection in Shanghai. Mol Clin Oncol. 2021;15(1):132. doi:10.3892/mco.2021.2294
7. Fong Amaris WM, PPd A, Valadares LJ, Moreira FC. Microbiota changes: the unseen players in cervical cancer progression. Review. Front Microbiol. 2024;15. doi:10.3389/fmicb.2024.1352778.
8. Rodrigues R, Sousa C, Vale N. Chlamydia trachomatis as a current health problem: challenges and opportunities. Diagnostics. 2022;12(8):1795. doi:10.3390/diagnostics12081795
9. Bhuvanendran Pillai A, Mun Wong C, N DIZA, et al. Chlamydia infection as a risk factor for cervical cancer: a systematic review and meta-analysis. Iran J Public Health. 2022;51(3):508–517. doi:10.18502/ijph.v51i3.8926
10. Mosmann JP, Zayas S, Kiguen AX, Venezuela RF, Rosato O, Cuffini CG. Human papillomavirus and Chlamydia trachomatis in oral and genital mucosa of women with normal and abnormal cervical cytology. BMC Infect Dis. 2021;21(1):422. doi:10.1186/s12879-021-06118-3
11. Bugalhão JN, Mota LJ. The multiple functions of the numerous Chlamydia trachomatis secreted proteins: the tip of the iceberg. Microb Cell. 2019;6(9):414–449. doi:10.15698/mic2019.09.691
12. Rother M, Teixeira da Costa Ana R, Zietlow R, Meyer Thomas F, Rudel T. Modulation of host cell metabolism by Chlamydia trachomatis. Microbiol. Spectr. 2019;7(3). doi:10.1128/microbiolspec.bai-0012-2019
13. Arcia Franchini AP, Iskander B, Anwer F, et al. The role of Chlamydia trachomatis in the pathogenesis of cervical cancer. Cureus. 2022;14(1):e21331. doi:10.7759/cureus.21331
14. Wang X, Wu H, Fang C, Li Z. Insights into innate immune cell evasion by Chlamydia trachomatis. Front Immunol. 2024;15:1289644. doi:10.3389/fimmu.2024.1289644
15. Chumduri C, Gurumurthy RK, Zadora PK, Mi Y, Meyer TF. Chlamydia infection promotes host DNA damage and proliferation but impairs the DNA damage response. Cell Host Microbe. 2013;13(6):746–758. doi:10.1016/j.chom.2013.05.010
16. Yang X, Siddique A, Khan AA, et al. Chlamydia trachomatis infection: their potential implication in the etiology of cervical cancer. J Cancer. 2021;12(16):4891–4900. doi:10.7150/jca.58582
17. Dzakah EE, Huang L, Xue Y, et al. Host cell response and distinct gene expression profiles at different stages of Chlamydia trachomatis infection reveals stage-specific biomarkers of infection. BMC Microbiol. 2021;21(1):3. doi:10.1186/s12866-020-02061-6
18. Olagoke O, Chittaranjan S, Dean D. Transcriptional profiling of Chlamydia trachomatis and its host in an ex vivo endocervical primary cell culture system using dual RNA sequencing. Original Research. Front Cell Infect Microbiol. 2025;15. doi:10.3389/fcimb.2025.1613922.
19. Ding X, Wan A, Qi X, Jiang K, Liu Z, Chen B. ZNF695, A potential prognostic biomarker, correlates with immune infiltrates in cervical squamous cell carcinoma and endoce rvical adenocarcinoma: bioinformatic analysis and experimental verification. Curr Gene Ther. 2024;24(5):441–452. doi:10.2174/0115665232285216240228071244
20. Zhang H, Li J, Xu H, Zhang S, Lu Y, Feng S. Identifying key epigenetic modification-related genes for cervical squamous cell carcinoma and endocervical adenocarcinoma and cellular validation. Curr Gene Ther. 2025. doi:10.2174/0115665232432474251103064112
21. Zeng Q, Feng K, Yu Y, Lv Y. Hsa_Circ_0000021 Sponges miR-3940-3p/KPNA2 Expression to Promote Cervical Cancer Progression. Curr Mol Pharmacol. 2024;17:e170223213775. doi:10.2174/1874467216666230217151946
22. Du H, Tang Y, Ren X, et al. A prognostic model for cervical cancer based on ferroptosis-related genes. Original Research. Front Endocrinol. 2022;Volume 13. doi:10.3389/fendo.2022.991178
23. Chen P, Gao Y, Ouyang S, et al. A prognostic model based on immune-related long non-coding RNAs for patients with cervical cancer. original research. Front Pharmacol. 2020;11. doi:10.3389/fphar.2020.585255
24. Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1(6):417–425. doi:10.1016/j.cels.2015.12.004
25. Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 27(12):1739–1740. doi:10.1093/bioinformatics/btr260
26. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. doi:10.1073/pnas.0506580102
27. Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18(1):220. doi:10.1186/s13059-017-1349-1
28. Warde-Farley D, Donaldson SL, Comes O, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38(suppl_2):W214–W220. doi:10.1093/nar/gkq537
29. Maeser D, Gruener RF, Huang RS. oncoPredict: an R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Brief Bioinform.;226:10.1093/bib/bbab260
30. Challagundla N, Chrisophe-Bourdon J, Agrawal-Rajput R. Chlamydia trachomatis infection co-operatively enhances HPVE6-E7oncogenes mediated tumorigenesis and immunosuppression. Microb Pathogenesis. 2023;175:105929. doi:10.1016/j.micpath.2022.105929
31. Koskela P, Anttila T, Bjørge T, et al. Chlamydia trachomatis infection as a risk factor for invasive cervical cancer. Int, J, Cancer. 2000;85(1):35–39. doi:10.1002/(sici)1097-0215(20000101)85:1
32. Zheng B, Qu J, Ohuchida K, et al. LAMA4 upregulation is associated with high liver metastasis potential and poor survival outcome of Pancreatic Cancer. Theranostics. 2020;10(22):10274–10289. doi:10.7150/thno.47001
33. Wang M, Li C, Liu Y, Wang Z. Effect of LAMA4 on prognosis and its correlation with immune infiltration in gastric cancer. Biomed Res Int. 2021;2021:6428873. doi:10.1155/2021/6428873
34. Yang ZX, Zhang B, Wei J, et al. MiR-539 inhibits proliferation and migration of triple-negative breast cancer cells by down-regulating LAMA4 expression. Cancer Cell Int. 2018;18:16. doi:10.1186/s12935-018-0512-4
35. Nakagawa S, Yamaguchi K, Takane K, Tabata S, Ikenoue T, Furukawa Y. Wnt/β-catenin signaling regulates amino acid metabolism through the suppression of CEBPA and FOXA1 in liver cancer cells. Commun. Biol. 2024;7(1):510. doi:10.1038/s42003-024-06202-9
36. Nakagawa S, Yamaguchi K, Takane K, Tabata S, Ikenoue T, Furukawa Y. Abstract 7079: the Wnt/β-catenin signaling pathway regulates cellular metabolism through the suppression of HAL and ARG1 in liver cancer cells. Cancer Res. 2024;84(6_Supplement):7079. doi:10.1158/1538-7445.AM2024-7079
37. Hercus TR, Dhagat U, Kan WL, et al. Signalling by the βc family of cytokines. Cytokine Growth Factor Rev. 2013;24(3):189–201. doi:10.1016/j.cytogfr.2013.03.002
38. Côrte-Real BF, Arroyo Hornero R, Dyczko A, Hamad I, Kleinewietfeld M. Dissecting the role of CSF2RB expression in human regulatory T cells. Original Research. Front Immunol. 2022;13. doi:10.3389/fimmu.2022.1005965.
39. Zhu N, Yang Y, Wang H, et al. CSF2RB Is a Unique Biomarker and Correlated With Immune Infiltrates in Lung Adenocarcinoma. Front Oncol. 2022;12:822849. doi:10.3389/fonc.2022.822849
40. Li K, Zhang C, Zhou R, et al. Single cell analysis unveils B cell-dominated immune subtypes in HNSCC for enhanced prognostic and therapeutic stratification. Int J Oral Sci. 2024;16(1):29. doi:10.1038/s41368-024-00292-1
41. Speiser DE, Chijioke O, Schaeuble K, Münz C. CD4+ T cells in cancer. Nat Cancer. 2023;4(3):317–329. doi:10.1038/s43018-023-00521-2
42. Raskov H, Orhan A, Christensen JP, Gögenur I. Cytotoxic CD8+ T cells in cancer and cancer immunotherapy. Br. J. Cancer. 2021;124(2):359–367. doi:10.1038/s41416-020-01048-4
43. Zander R, Schauder D, Xin G, et al. CD4+ T Cell Help Is Required for the Formation of a Cytolytic CD8+ T Cell Subset that Protects against Chronic Infection and Cancer. Immunity. 2019;51(6):1028–1042.e4. doi:10.1016/j.immuni.2019.10.009
44. Zhang J, Zhan J, Guan Z, et al. The prognostic value of Th17/Treg cell in cervical cancer: a systematic review and meta-analysis. Front Oncol. 2024;14:1442103. doi:10.3389/fonc.2024.1442103
45. Shang B, Liu Y, Jiang SJ, Liu Y. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: a systematic review and meta-analysis. Sci Rep. 2015;5:15179. doi:10.1038/srep15179
46. Pan Y, Zhou H, Sun Z, et al. Regulatory T cells in solid tumor immunotherapy: effect, mechanism and clinical application. Cell Death Dis. 2025;16(1):277. doi:10.1038/s41419-025-07544-w
47. Li Y, Zhang C, Jiang A, et al. Potential anti-tumor effects of regulatory T cells in the tumor microenvironment: a review. J Transl Med. 2024;22(1):293. doi:10.1186/s12967-024-05104-y
48. Liu J, Geng X, Hou J, Wu G. New insights into M1/M2 macrophages: key modulators in cancer progression. Can Cell Inter. 2021;21(1):389. doi:10.1186/s12935-021-02089-2
49. Ridge SM, Sullivan FJ, Glynn SA. Mesenchymal stem cells: key players in cancer progression. Mol Cancer. 2017;16(1):31. doi:10.1186/s12943-017-0597-8
50. Li Z, Yang C, Li X, et al. The dual role of BI 2536, a small-molecule inhibitor that targets PLK1, in induction of apoptosis and attenuation of autophagy in neuroblastoma cells. J Cancer. 2020;11(11):3274–3287. doi:10.7150/jca.33110
51. Almuraikhi N. Inhibition of TGF-β type I receptor by SB505124 down-regulates osteoblast differentiation and mineralization of human mesenchymal stem cells. Cell Biochem Funct. 2023;41(5):564–572. doi:10.1002/cbf.3812
52. Ganbat D, Jugder B-E, Ganbat L, et al. Use of the Naphthoquinone YM155 (Sepantronium Bromide) in the Treatment of Cancer: a Systematic Review and Meta-Synthesis. Oncologie. 2022;24(2):195–225. doi:10.32604/oncologie.2022.022299
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Nomogram to Predict Radiation Enteritis in Cervical Squamous Cell Carcinoma
Wang J, Hu G
Cancer Management and Research 2022, 14:3303-3311
Published Date: 25 November 2022
A Novel Gene Signature Associated with Inflammatory Responses and Immune Status Assists in Prognosis and Intervention for Patients with HCC
Lu G, Du R, Feng B, Wang J, Zhang F, Pei J, Wang Y, Shang Y
Journal of Inflammation Research 2022, 15:6729-6743
Published Date: 13 December 2022
Exploring Prognosis, Tumor Microenvironment and Tumor Immune Infiltration in Hepatocellular Carcinoma Based on ATF/CREB Transcription Factor Family Gene-Related Model
Shen H, Gu X, Li H, Tang M, Li X, Zhang Y, Su F, Wang Z
Journal of Hepatocellular Carcinoma 2023, 10:327-345
Published Date: 27 February 2023
Construction and Assessment of a Prognostic Risk Model for Cervical Cancer Based on Lactate Metabolism-Related lncRNAs
Gao Y, Liu H, Wan J, Chang F, Zhang L, Wang W, Zhang Q, Feng Q
International Journal of General Medicine 2023, 16:2943-2960
Published Date: 11 July 2023
Prognostic Value of Body Composition and Systemic Inflammatory Markers in Patients with Locally Advanced Cervical Cancer Following Chemoradiotherapy
Guo H, Feng S, Li Z, Yin Y, Lin X, Yuan L, Sheng X, Li D
Journal of Inflammation Research 2023, 16:5145-5156
Published Date: 10 November 2023