Back to Journals » International Journal of Nanomedicine » Volume 21
Bioengineered Exosome-Magnetic Nanoplatform for Precision Therapy of Hepatocellular Carcinoma via Dual-Targeted Drug Delivery
Authors Han Y, Yan S, Wan R, Li X, Wu G
Received 27 October 2025
Accepted for publication 14 April 2026
Published 8 July 2026 Volume 2026:21 576489
DOI https://doi.org/10.2147/IJN.S576489
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Krishna Nune
Yongsheng Han1, Shixian Yan2, Ruohan Wan3, Xingjing Li1, Guanlin Wu4
1Department of Hepatobiliary Pancreatic Surgery, Henan Provincial People’s Hospital & People Hospital of Zhengzhou University, Zhengzhou, 450003, People’s Republic of China; 2Department of General Surgery, The First Affiliated Hospital of Dalian Medical University, Dalian, 116011, People’s Republic of China; 3Department of Cardiac Surgery, Heart Centre of Henan Provincial People’s Hospital, Central China Fuwai Hospital of Zhengzhou University, Zhengzhou, 451464, People’s Republic of China; 4School of Clinical Medicine, Shanghai University of Medicine & Health Sciences, Shanghai, 201318, People’s Republic of China
Correspondence: Guanlin Wu, Email [email protected]; [email protected]
Introduction: Hepatocellular carcinoma (HCC) continues to pose a significant threat to global health, contributing substantially to worldwide cancer-related mortality, particularly in high-incidence regions such as Asia, where current treatment strategies are often limited by poor drug delivery efficiency, systemic toxicity, and drug resistance.
Methods: To address these critical challenges, we developed an innovative dual-targeted nanoplatform (Exo-SPIONs-SRF/CGA) that synergistically combines the natural tumour-homing capability of HCC-derived exosomes with the magnetic guidance of superparamagnetic iron oxide nanoparticles (SPIONs) for precision drug targeting. This nanoplatform co-encapsulates SRF and CGA to improve the therapeutic index by enhancing desired responses and minimizing undesired side effects.
Results: The exosome component provides inherent biological targeting to HCC cells. At the same time, the incorporated SPIONs enable external magnetic field-guided spatial control, collectively ensuring superior tumour accumulation compared to conventional delivery systems. Furthermore, the platform’s tumour microenvironment-responsive release characteristics ensure localized drug activation, maximizing the therapeutic index through spatial and temporal control of drug availability. In vitro and in vivo evaluations demonstrated that this nanoplatform significantly enhances tumour suppression and drug retention while reducing systemic side effects compared to monotherapies or single-modality nanocarriers.
Discussion: The Exo-SPIONs-SRF/CGA platform represents a promising strategy in HCC treatment, addressing fundamental limitations of current therapies by simultaneously overcoming biological barriers to drug delivery, enhancing therapeutic efficacy through synergistic drug combinations, and minimizing collateral damage to healthy tissues, thereby advancing the frontier of precision oncology toward more effective and safer HCC management.
Keywords: hepatocellular carcinoma, exosome, drug delivery, dual targeting, precise treatment
Introduction
Hepatocellular carcinoma (HCC) is the primary pathological type of liver cancer, accounting for 85–90% of cases.1,2 According to the 2022 Global Cancer Statistics, HCC has 865,000 new cases and 758,000 deaths annually.3 It is the sixth most commonly diagnosed malignancy and the third leading cause of cancer death worldwide.4 The disease burden shows significant regional differences, with the highest incidence rates in East Asia and Africa.5 China accounts for over 40% of the global HCC cases and deaths, which are 42.5% and 41.8%, respectively.6 The epidemiological prediction model showed that the global annual incidence of HCC would increase by 55% between 2020 and 2040, and there would be 1.4 million new cases and 1.3 million deaths worldwide in 2040.7 HCC is characterized by high heterogeneity, difficulty in early diagnosis, and a significant clinical challenge is that the majority of cases (around 70%) are not detected until the mid or advanced stages, and prone to recurrence and metastasis after treatment and a 5-year recurrence rate >70%. These characteristics collectively lead to an inferior prognosis, with a dismal five-year overall survival rate of merely 20%.8 Of note, the onset of HCC has distinct gender and age characteristics.9 The incidence rate is the highest among men aged 60–70, with a male-to-female ratio of approximately 3:1.10 Moreover, the vast majority of cases (about 90%) occur in the context of liver cirrhosis caused by chronic liver diseases. The long-term inflammatory microenvironment of the liver promotes malignant transformation by inducing genomic instability and epigenetic changes.11 Consequently, the complex pathogenesis and severe clinical course of HCC underscore the urgent need for more effective therapeutic strategies.
Current clinical management of HCC primarily encompasses surgical resection, liver transplantation, local ablation therapy, transarterial chemoembolization (TACE), targeted drug therapy and immunotherapy,12,13 etc. However, these treatment methods are subject to various limitations. For instance, surgical resection is only applicable to approximately 30% of early-stage patients, and many patients are unable to undergo surgical treatment due to impaired liver function or complex tumour locations.14 Although liver transplantation can provide radical therapy for some patients, its application scope is relatively limited due to the scarcity of liver sources and the high cost.15 Local ablation therapy can provide effective treatment for early-stage liver cancer, but its indications are relatively narrow, and the size and location of the tumour greatly influence its efficacy.16 TACE controls tumour growth by blocking the blood supply to tumours and injecting chemotherapy drugs, but its efficacy is limited in some patients and may be accompanied by severe side effects.17 Targeted drug therapy has achieved specific therapeutic effects in some patients, yet both its monotherapy effective rate and objective response rate remain relatively low. Immunotherapy has also failed to bring significant improvement to all liver cancer patients, especially monotherapy with immune checkpoint inhibitors.18 Although it has shown specific effects in some clinical trials, its effective rate is only 15% to 30%, which has not reached the ideal treatment level.
Leveraging the rapid progress in nanotechnology, drug delivery systems utilizing nanomedicine have become a major focus in oncology research.19,20 Nanocarriers such as liposomes, polymer micelles, and inorganic nanoparticles have emerged as a highly promising strategy due to their advantages in improving drug solubility, prolonging drug circulation time, and enhancing targeting.21 However, traditional nanocarriers still face challenges such as poor biocompatibility, high immunogenicity, and limited tumour penetration ability.22,23 Especially for tumours with special blood supply characteristics like liver cancer, existing nanomedicine delivery systems’ passive targeting (EPR effect) efficiency often fails to achieve the desired results.24 These issues have prompted scientists to constantly explore new drug delivery systems to enhance the targeting and efficacy of treatments.
Exosomes, as endogenous nano-delivery carriers, have demonstrated unique biological advantages in tumour-targeted therapy.25 These 30–150nm membranous vesicles naturally secreted by cells have a natural bilayer lipid membrane structure, which endows them with favorable biocompatibility and stability.26 The surface of exosomes is rich in members of the four-transmembrane protein family, such as CD9, CD63, and CD81.27 These molecules serve as characteristic markers and achieve precise drug delivery by mediating cell-specific recognition. Exosomes have significantly reduced immunogenicity compared to synthetic nanocapsules and can effectively evade immune clearance. Their natural membrane penetration capability enables them to cross multiple physiological barriers, including the blood-brain barrier. As a natural medium for intercellular communication, exosomes can efficiently deliver a variety of therapeutic molecules through membrane fusion mechanisms and protect their contents from enzymatic degradation.28–30 This property renders exosomes suitable as drug delivery platform. In liver cancer treatment, exosomes derived from liver cancer cells have special biological significance as delivery carriers. Studies have shown that tumour-derived exosomes retain the specific membrane proteins and integrin profiles of the parent cells on their surfaces and can be preferentially taken up by primary and metastatic liver cancer cells through the enhanced cellular affinity.31,32 This specific targeting ability effectively promotes drug accumulation within tumors and addresses the key limitation of target failure caused by tumour heterogeneity in traditional nanocarriers. In addition, liver cancer exosomes have unique response characteristics to the tumour microenvironment (TME). They can achieve controllable drug release by stimulating pH value, enzyme activity, etc., significantly reducing systemic toxicity.33 It is worth noting that these exosomes can also improve their accumulation in tumour tissues by leveraging the enhanced permeability and retention effect (EPR). The matrix metalloproteinases (MMPs) they carry can degrade the extracellular matrix and promote the diffusion of drugs in the dense tumour stroma.34 These characteristics make the exosomes of liver cancer origin ideal carriers with efficient, targeted delivery and immune escape capabilities.
Sorafenib (SRF) is the first molecular targeted drug approved for treating advanced HCC.35,36 It exerts dual effects of anti-tumour proliferation and anti-angiogenesis by inhibiting the RAF/MEK/ERK signalling pathway and targeting multiple signalling pathways such as VEGFR and PDGFR. Therefore, it has become the cornerstone of clinical treatment.37 The clinical application of SRF, however, is hampered by its poor water solubility, low bioavailability, high systemic toxicity, and propensity to induce drug resistance.38 Chlorogenic Acid (CGA), a natural polyphenol compound, possesses multiple biological activities such as antioxidation (by activating the Nrf2 pathway), anti-inflammation and anti-tumour.39 Studies have shown that CGA can inhibit tumour growth by regulating ROS-mediated signalling pathways such as NF-κB and MAPK. In addition, CGA may enhance the anti-tumor effect of SRF while reducing its toxicity,40,41 and thus, it is regarded as an ideal candidate for combination therapy.
To overcome the limitations of traditional treatment methods, this study proposes a “dual targeting” strategy, combines the innate homing targeting of exosomes with external magnetic targeting of superparamagnetic iron oxide nanoparticles (SPIONs) to construct a multifunctional composite nanoplatform Exo-SPIONs-SRF/CGA, to achieve highly efficient targeted anti-tumour effects (Figure 1). Exosomes derived from liver cancer cells serve as the core carriers, retaining the natural targeting ability for liver cancer. Meanwhile, the loaded SPIONs can further increase the accumulation concentration of drugs at the tumour site guided by an external magnetic field. This system is jointly loaded with SRF and CGA enabling synergistic multiple anti-tumour effects: SRF inhibits tumour proliferation and angiogenesis, while CGA enhances antioxidant effects and reduces toxicity and side effects. The system’s TME responsive release characteristic ensures the controllable release of drugs at the targeted site, thereby minimizing systemic toxicity. This design not only resolves the bottleneck problems of traditional nanocarriers in terms of insufficient targeting and poor permeability but also represents a novel therapeutic approach to precision therapy for liver cancer. The main advantages of this strategy include: (1) Taking liver cancer-derived exosomes as the core carrier and leveraging their natural targeting property and excellent biocompatibility; (2) Combining with SPIONs to achieve magnetic navigation, precisely controlling the enrichment of drugs at the tumour site through an external magnetic field; (3) Co-loading SRF and CGA to synergistically exert anti-tumour proliferation, anti-angiogenesis, and antioxidant/anti-inflammatory effects; (4) Achieving TME-responsive drug release, which minimizes systemic toxicity. The Exo-SPIONs-SRF/CGA composite platform constructed in this study not only significantly enhanced the anti-tumour effect but also effectively reduced toxic and side effects, providing a new solution for overcoming multidrug resistance and metastasis and recurrence of liver cancer and promoting the realization of the treatment goal of high efficiency, low toxicity and precision.
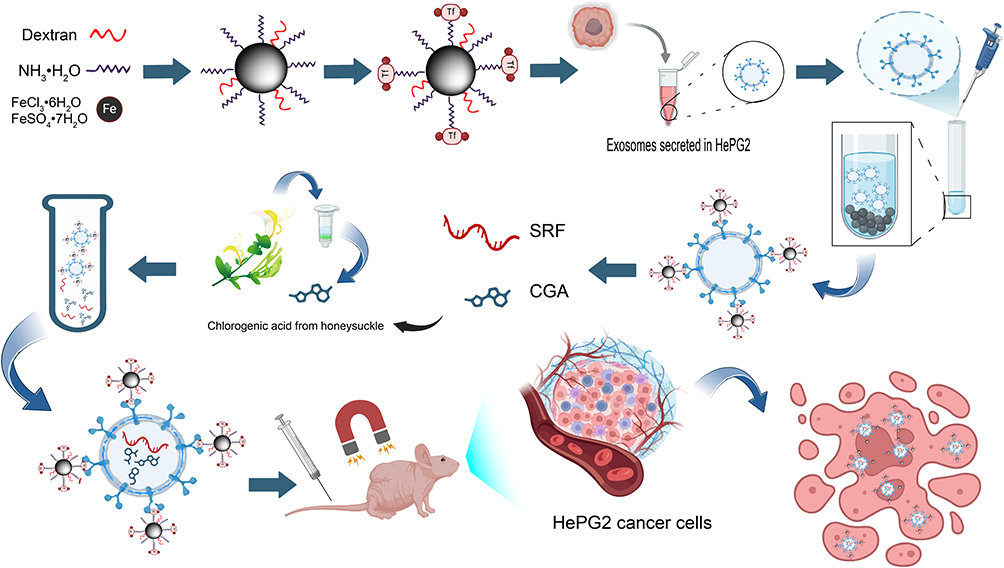 |
Figure 1 Schematic design of Exo-SPIONs-mediated drug delivery and tumor treatment. |
Experiment Section
Materials and Reagents
FeSO4·7H2O and FeCl3·6H2O were purchased from aladdin scientific Co., Ltd. (Shanghai, China). Sorafenib (SRF), chlorogenic acid, and the CCK-8 assay kit were obtained from MedChemExpress Co., Ltd. (Monmouth Junction, NJ, USA). Transferrin (Tf) was sourced from Merck Life Science Co., Ltd. (Shanghai, China). HepG2 cells were obtained from the Cell Bank of the Chinese Academy of Sciences. Cell culture reagents, including DMEM, fetal bovine serum (FBS), and penicillin-streptomycin solution, were acquired from Thermo Fisher Scientific Co., Ltd. (Shanghai, China). Anhydrous ethanol, toluene, and acetone were supplied by Sinopharm Co., Ltd. (Shanghai, China). 4% paraformaldehyde fixative solution, and DAPI staining solution were obtained from Beyotime Biotechnology Co., Ltd. (Shanghai, China).
Synthesis and Modification of SPIONs
A mixture of 3 g FeSO4·7H2O and 5.4 g FeCl3·6H2O was dissolved in deionized water, followed by the addition of NH3·H2O as a precipitating agent until the pH>10. The reaction was carried out under stirring in a water bath at constant temperature for 30 min. The resulting precipitate was repeatedly washed with distilled water until a neutral pH was achieved, after which the supernatant was removed. Then dried at 60 °C, ground into fine powder, and collected as SPIONs. The purified SPIONs were then functionalized by incubation with carboxylated dextran under stirring at 60 °C overnight, followed by magnetic separation and purification to obtain dextran-coated SPIONs.
Preparation of Tf-SPIONs
Tf-conjugated SPIONs were synthesized through a two-step process. First, dextran-coated SPIONs were conjugated with transferrin using EDC and NHS to activate the carboxyl groups on the surface of SPIONs. The mixture was incubated overnight under gentle stirring. Finally, the Tf-SPIONs were isolated by magnetic separation and stored at 4 °C.
Preparation of Exo-SPIONs
HepG2 cell cultures were grown in RPMI 1640 medium with a 10% FBS supplement at 37 °C under 5% CO2. Upon reaching 80% confluence at passage 3, cells were washed with PBS and serum-starved overnight. The conditioned medium was collected and sequentially centrifuged at 300 × g (10 min) and 2000 × g (10 min) to remove cellular debris. The clarified supernatant was then incubated with Tf-SPIONs for 30 min at 4 °C with gentle agitation. Exo-SPIONs were harvested via magnetic separation and followed by resuspension in PBS.
Drug Loading into Exo-SPIONs
Drug loading was achieved by incubating Exo-SPIONs with SRF/CGA in PBS at 4 °C for 4 h, resulting in the formation of the Exo-SPIONs-SRF/CGA nanocomplex, followed by 37 °C incubation for 30 min. To isolate the drug-loaded nanocomplex, unincorporated drugs were removed magnetically, and the prepared Exo-SPIONs-SRF/CGA was resuspended in PBS. The drug loading efficiency was determined by measuring the supernatant absorbance at 480 nm against a standard curve (0.5–10 μg/mL). The encapsulation efficiency (EE) was then calculated using the formula:
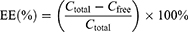
Where Ctotal and Cfree represent the initial and unencapsulated drug concentrations, respectively.
Characterization
The morphology and size distribution of Exo-SPIONs-SRF/CGA were characterized by transmission electron microscopy (JEOL-1010, JEOL Ltd., Japan) and dynamic light scattering (ZS90, Malvern Panalytical, UK). The hydrodynamic diameter and zeta potential were measured in PBS at 4 °C over 14 days using the same DLS system. The optical properties of the synthesized samples were analyzed using a UV-Vis spectrophotometer (UH-5300, Hitachi, Japan) and fluorescence spectrophotometer (Hitachi, Japan). The phase composition and crystalline structure of the sample were characterized by X-ray diffraction (X-TRA, Rigaku, Japan), while chemical composition and functional groups were analyzed using Fourier transform infrared spectroscopy (NEXUS870, Nicolet, USA). Cellular imaging was conducted using a laser scanning confocal microscope (ZEISS LSM900, Germany), and absorbance measurements were performed with a microplate reader (Infinite M200 PRO, Tecan, Switzerland).
In vitro Drug Release
The drug release of SRF/CGA from Exo-SPIONs was evaluated in PBS (pH 6.0 and 7.0) using dialysis bags. Samples were dialyzed against 25 mL PBS at 37 °C with gentle agitation. At predetermined time intervals (1, 2, 6, 12, 24, and 48 h), 3 mL aliquots were withdrawn and replaced with fresh PBS. The released SRF/CGA concentrations were quantified spectrophotometrically at 480 nm.
Cellular Uptake Study
HepG2 cells were seeded at a density of 4×103 cells per well in confocal dishes and cultured in complete RPMI-1640 medium until reaching 80% confluence. The cells were then treated with either free SRF/CGA (10 μg/mL) or an equivalent dose of Exo-SPIONs-SRF/CGA for 6, 12, and 24 h, with an equal volume of PBS serving as the control. After treatment, the cells were washed three times with PBS, fixed with 4% paraformaldehyde for 30 min, and rinsed 2–3 times with PBS. Subsequently, the nuclei were stained with DAPI for 15 min in the dark, followed by three additional PBS washes. Cellular uptake was visualized using a confocal microscope, and quantitative analysis was performed using ImageJ software.
Hemolysis Assay
Fresh rat red blood cells (RBCs) were washed with phosphate-buffered saline (PBS) and centrifuged. The supernatant was discarded, and the pelleted RBCs were resuspended in PBS to obtain a 4% (v/v) suspension. This suspension was then mixed with different concentrations of Exo, Exo-SPIONs, SRF/CGA, and Exo-SPIONs-SRF/CGA, followed by incubation at 37 °C for 1 h. After incubation, the samples were centrifuged, and the absorbance of the supernatant was measured at 570 nm to assess hemolysis. Triton X-100 (0.1%, v/v) and PBS were used as positive and negative controls, respectively. The hemolysis rate was calculated according to the following formula:
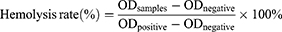
Where ODsample, ODnegative, and ODpositive represent the absorbance values of the test sample, negative control (PBS), and positive control (0.1% Triton X-100), respectively.
In vitro Targeting Ability Study
HepG2 cells (4×103 cells/well) were cultured in RPMI-1640 medium until reaching 80% confluence in confocal dishes. Cells were then treated with Exo-SPIONs-SRF/CGA. During the 2 h incubation period, a static magnetic field was applied by positioning a magnet beneath the confocal dish to provide magnetic targeting of Exo-SPIONs-SRF/CGA. Following incubation, uninternalized compounds were removed by PBS washing, and cells were fixed with 4% paraformaldehyde (30 min, RT). After additional PBS rinsing, nuclei were stained with DAPI (15 min, dark), and cellular uptake was visualized using confocal microscopy.
In vitro Anti-Cancer Cell Effect Study
Cell Counting Kit-8 was used to examine the anticancer ability of free SRF/CGA and Exo-SPIONs-SRF/CGA. HepG2 cells were seeded at a density of 4×103 cells/well and incubated for 24 h at 37 °C in a 5% CO2 atmosphere. Subsequently, the cells were treated with Exo-SPIONs, free SRF/CGA, or Exo-SPIONs-SRF/CGA and further incubated for 12, 24, and 48 h. Following incubation, we quantified cell viability using the CCK-8 assay according to the manufacturer’s protocol.42
In vivo Antitumor Efficacy and Biosafety Evaluation
Tumor Model Establishment
Female BALB/c nude mice were obtained from the Animal Center in the School of Clinical Medicine, Shanghai University of Medicine & Health Sciences. The mice were six weeks old and weighed between 18 and 22 g at the start of the experiment. All animal procedures were conducted in accordance with guidelines approved by the Institutional Animal Care and Use Committee (2024-HXXM-01-340823199208070073) and complied with international animal welfare guidelines. HepG2 cells were cultured, passaged to the third generation, trypsinized, and resuspended in PBS. After disinfecting the dorsal skin, each mouse received a subcutaneous injection of 1×107 cells. Tumor growth was subsequently monitored, and the volume (V) was calculated with the formula:
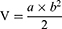
Where *a* and *b* represent the longest and shortest tumor diameters, respectively.
Animal Grouping and Treatment
When tumor volumes reached approximately 50 mm3, the nude mice were randomly divided into five groups. Each group contained five mice (n=5) for intravenous administration of different formulations: PBS served as the control group, while experimental groups received Exo-SPIONs, free SRF/CGA, Exo-SPIONs-SRF/CGA, or Exo-SPIONs-SRF/CGA + Magnetic. For the Exo-SPIONs-SRF/CGA + Magnetic group, following tail vein injection in mice, A permanent magnet (NdFeB disk-shaped magnet, 25 mm diameter, 10 mm thick, 0.3 T) was fixed above the tumor site in mice to promote drug enrichment in tumors. All treatments were administered every 48 h, body weight and tumor size were monitored regularly throughout the study to assess treatment efficacy and potential systemic toxicity.
Histological Analysis
At the end of the treatment process, the tumor and major organs were collected, fixed, and processed for histological examination. Tissue sections were stained with hematoxylin for 3–5 min, rinsed with distilled water, and subsequently counterstained with eosin for 5 min. Dehydration was performed using graded ethanol (85% and 95%), followed by clearing and mounting. Finally, histopathological evaluation of the stained tissue sections was performed by light microscopy.
Blood Biochemical Analysis
At the endpoint, blood samples were collected and processed by centrifugation at 5000 rpm for 5 min to obtain serum for subsequent biochemical analysis. Blood routine analysis was performed using the detection kit, and the following nine indicators were measured. The levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), blood urea nitrogen (BUN), creatinine (CR), uric acid (UA), total bilirubin (TBIL), γ-glutamyl transferase (γ-GT), and albumin (ALB) were used to evaluate the function of liver and kidney.
Prussian Blue Staining for Iron Detection
On day 18 after intravenous injection of samples, nude mice were euthanized, dissected, and the tumors were removed, sectioned, placed in staining solution for 1 h, and washed twice with ultrapure water. The nuclei were stained for 3 min before rinsing with running water. The tissue sections were examined under a microscope for iron deposition after dehydration and sealing, to assess Exo-SPIONs distribution.
ELISA for Inflammatory Cytokines
TNF-α and IL-6 levels in serum were quantified using ELISA kits.43 Samples were incubated in antibody-coated 96-well plates, followed by enzyme-conjugated secondary antibodies and substrate addition. Following measurement of the absorbance at 450 nm, cytokine concentrations were calculated based on their respective standard curves.
Statistical Analysis
All results were obtained from at least three independent replicates and expressed as mean ± standard deviation (SD). Data were analyzed using two-way ANOVA (or repeated-measures ANOVA for longitudinal data). Differences were considered significant at p < 0.05.
Results and Discussion
Preparation and characterization of Tf-SPIONs
TEM morphologically characterized the prepared Tf (transferrin)-SPIONs. Figure 2A–C shows that the prepared Tf-SPIONs present a monodisperse nanospherical structure with uniform particle size distribution, and no evident agglomeration phenomenon was observed. This monodispersity may stem from the steric hindrance effect of Tf-SPIONs during the thermal decomposition preparation process. Its long-chain structure of dextran and coupled transferrin effectively prevented the aggregation of nanoparticles during high-temperature synthesis. Elemental mapping analysis (Figure 2E) further confirmed the homogeneous distribution of C, N, O, and Fe elements in the nanoparticles. The uniform dispersion of the Fe element indicated that the Fe3O4 core was encapsulated entirely by ligands. The elemental composition was quantitatively analyzed by EDS spectroscopy (Figure 2D). The results showed that the weight percentage of carbon (C) was 86.35%, that of nitrogen (N) was 0.62%, that of oxygen (O) was 11.53%, and that of iron (Fe) was 1.50%. The higher ratio of C and present N indicated that dextran coupled with Tf successfully coated the prepared SPIONs.
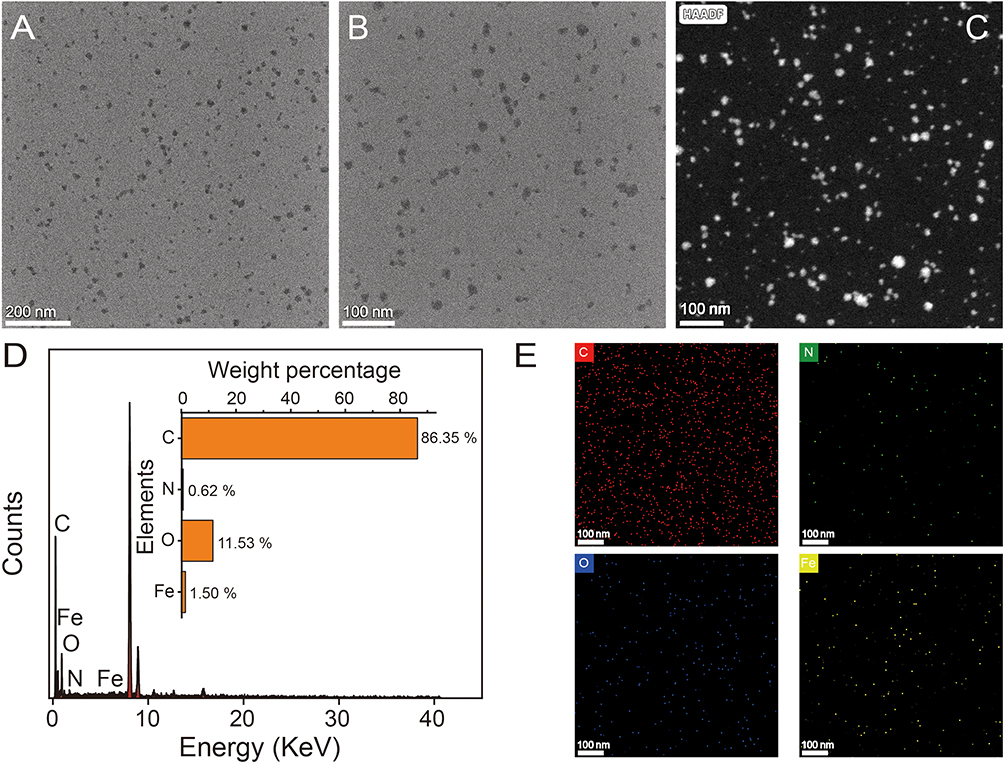 |
Figure 2 Morphological analysis of Tf-SPIONs. TEM images of Tf-SPIONs with low magnification (A) and high magnification (B and C), and the corresponding (D) EDS spectra and (E) mapping (carbon, red; oxygen, blue; nitrogen, green; iron, yellow). |
Transferrin (Tf) serves as the principal transporter for iron in plasma, delivering both dietary iron and iron derived from red blood cell degradation. Tf and transferrin receptors can bind specifically. The transferrin receptor is a type II transmembrane glycoprotein that is relatively abundant on the surface of cell membranes. In this study, Tf was bound to SPIONs, and exosome-like vesicles were magnetically enriched by utilizing the binding mode of transferrin and transferrin receptors. Transferrin was modified on dextran-SPIONs by the EDC/NHS chemistry, and the Tf-SPIONs were used to separate the exosomes from HepG2 cells. The properties of Tf-SPIONs were studied systematically, as shown in Figure 3A, The diffraction peaks of X-ray diffraction pattern at 2θ angles of 18.30°, 30.10°, 35.45°, 37.09°, 43.09°, 47.18°, 56.98°, 62.57°, 65.79°, 70.99° and 81.93° respectively corresponded to (111), (220), (311), (222), (400), (331), (511), (440), (531), (620) and (711) of the cubic phase Fe3O4 (PDF#65-3107),44 confirming that it had a cubic phase Fe3O4 structure.
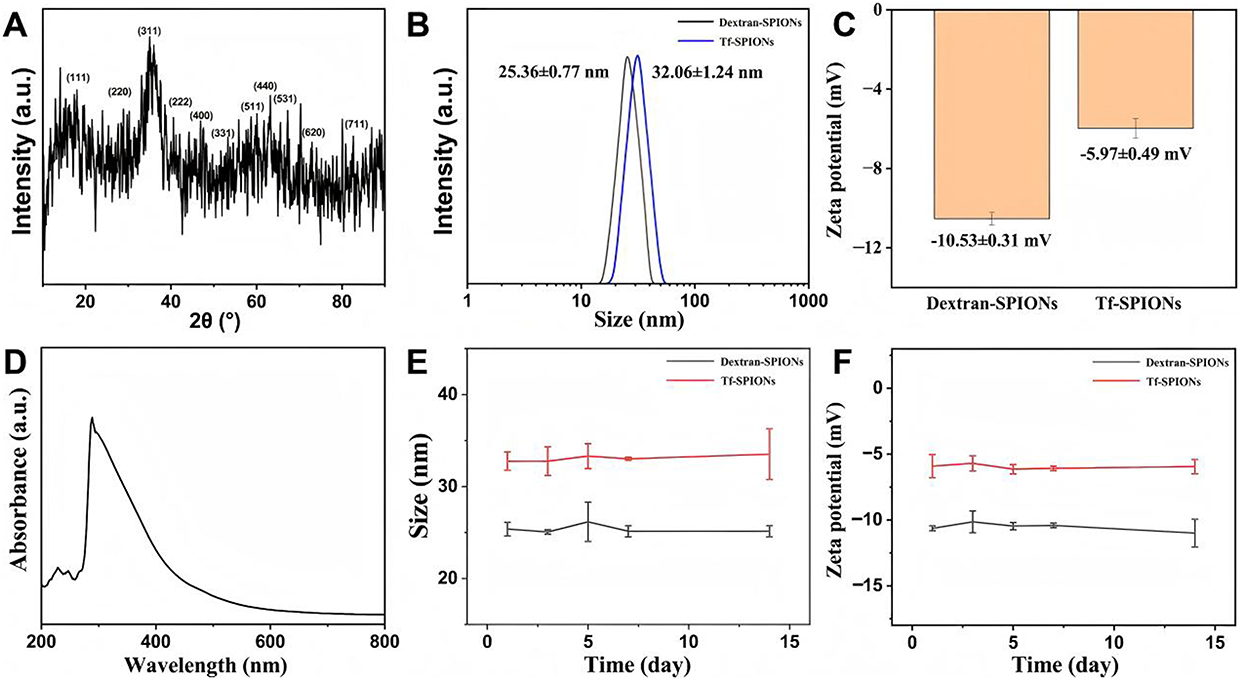 |
Figure 3 Characterization of physicochemical properties of Tf-SPIONs. (A) XRD patterns, (B) hydrodynamic size and (C) zeta potential, (D) UV absorption peak, (E) hydrodynamic size changes, and (F) zeta potential changes within the 14 days of Tf-SPIONs. |
The dynamic light scattering (DLS) test (Figure 3B) showed that the hydrodynamic size of dextran-SPIONs was 25.36 ± 0.77 nm, and the size changed to 32.06±1.24 nm after coupling Tf. Both the size of dextran-SPIONs and Tf-SPIONs showed the normal distribution, indicating that the nanoparticles had good monodispersity. The surface potential test showed that its zeta potential of dextran-SPIONs and Tf-SPIONs was −10.53 ± 0.31 mV and −5.97 ± 0.49 mV, respectively (Figure 3C). This moderate negative charge is conducive to the stable dispersion of nanoparticles in aqueous solution. The change of size and zeta potential revealed that the Tf was coupled to the SPIONs successfully. The UV-Vis spectrum of Tf-SPIONs showed a broad absorption characteristic of iron oxide (Figure 3D) further validating the formation of Fe3O4 nanoparticles. Furthermore, a 14-day stability monitoring experiment (Figure 3E and F) showed that neither the hydration diameter nor the zeta potential of the nanoparticles changed significantly, confirming that dextran modification endowed the nanoparticles with excellent colloidal stability, which provides an important guarantee for their application in the biomedical field.
HepG2 cells were used as the exosome donor, exosomes in the cell culture supernatant were collected by differential centrifugation and incubated with Tf-SPIONs. Subsequently, Exo-SPIONs were harvested by magnetic separation and then loaded with the SRF and CGA via incubation to prepare Exo-SPIONs-SRF/CGA. FTIR and XPS spectroscopy were used to investigate the chemical composition and surface functional groups of Exo-SPIONs-SRF/CGA. As shown in Figure 4A, within the range of 4000–500 cm−1, the absorption band observed at 3355.26 cm−1 corresponded to stretching vibrations of O-H and N-H groups. This likely originated from the hydrophilic headgroups (P-O-H) of exosomal membrane phospholipids, the amide N-H groups of transferrin (Tf), as well as the phenolic hydroxyl groups of the loaded drug CGA. The band broadening suggested the presence of a multi-hydrogen-bonding network, confirming the interactions between biomolecules and the inorganic core within the Exo-SPIONs complex. The intense peak at 1642.61 cm−1 was assigned to C=O stretching vibrations, potentially arising from the peptide backbone of Tf, exosomal surface transmembrane proteins, and the amide carbonyl group of the drug SRF. The peak at 1509.78 cm−1 corresponded to N-H in-plane bending and C-N stretching vibrations. Together with the C=O stretching vibration at 1643 cm−1, it constituted the fingerprint region for protein secondary structures, confirming that Tf and exosomal proteins maintain their conformational integrity within the composite. The peak at 1382.76 cm−1 was assigned to the symmetric bending vibration of -CH3, originating from the alkyl chains of exosomal membrane phospholipids. The peak at 1046.32 cm−1 was attributed to the overlapping vibrations of the phosphate ester bond (P=O) and the sugar ring C-O-C, confirming the successfully synthesis of Tf-SPIONs. The peak at 888.49 cm−1 was ascribed to the anomeric vibration of the sugar ring, reflecting the glycan structures within the exosomal surface glycocalyx. The peak at 588.45 cm−1 arise from the Fe-O lattice vibration, matching the cubic spinel structure of the SPIONs core, confirming the stable crystalline phase of the magnetic nanoclusters after complex formation.
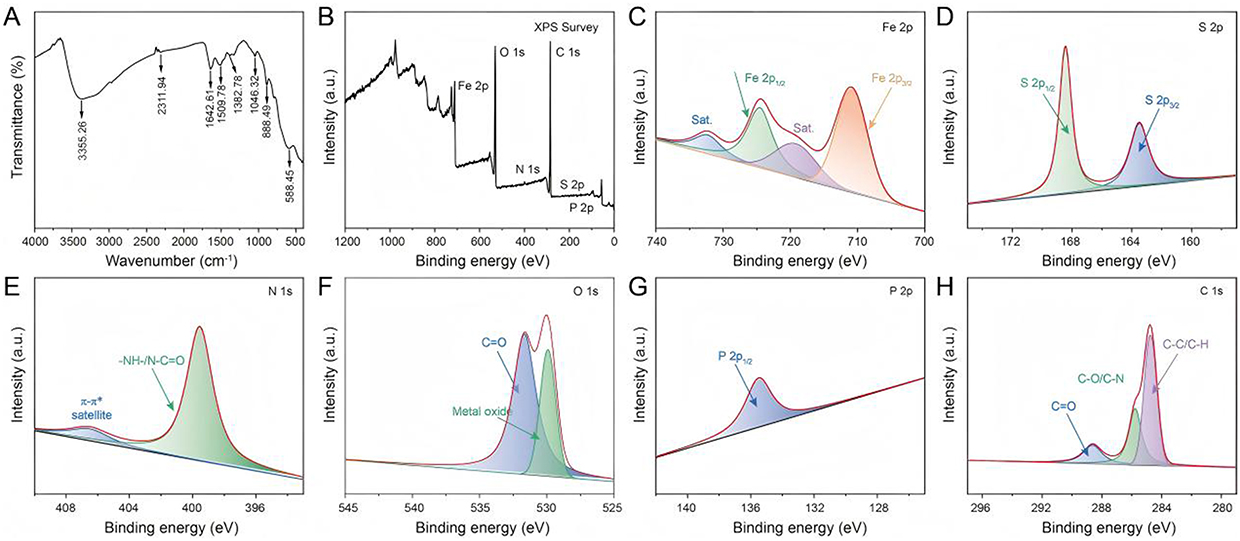 |
Figure 4 FTIR and XPS spectra of Exo-SPIONs-SRF/CGA (A) FTIR spectra, (B) XPS Survey scan, (C) Fe 2p, (D) S 2p, (E) N 1s, (F) O 1s, (G) P 2p, and (H) C 1s spectra. |
Subsequently, XPS survey scan (Figure 4B) confirmed the presence of six elements in the sample: Fe, O, C, N, S, and P. The characteristic signals of SPIONs were identified as Fe 2p (711.01 eV) and O 1s (530.6 eV). The N 1s (399.65 eV) and S 2p (168.36 eV) signals primarily originated from transferrin and exosomal protein components. The P 2p signal was attributed to phospholipid groups in the exosomal membrane, while the C 1s peak (284.8 eV) encompasses contributions from the organic shell (dextran, Tf). This elemental composition demonstrated the successful synthesis of exosome encapsulated SPIONs loaded with SRF/CGA . High-resolution XPS spectra were then deconvoluted (Figure 4C–H). Figure 4C shows the Fe 2p spectrum, exhibiting a doublet at binding energies of 710.8 eV (Fe 2p3/2) and 724.3 eV (Fe 2p1/2), accompanied by a satellite peak (Sat)., which is characteristic of the Fe3+ oxidation state, confirming the stability of the SPIONs core. The S 2p spectrum (Figure 4D) displays a doublet at 163.8 eV (S 2p3/2) and 168.4 eV (S 2p1/2), assigned to thioether bonds (-C-S-) derived from proteins, and sulfonic acid/sulfate groups (-OSO3−), reflecting the exosomal surface glycocalyx structure. Critically, the signal at 168.4 eV serves as key evidence for intact exosomal encapsulation. The N 1s spectrum (Figure 4E) shows a peak at 399.5 eV, reflecting peptide bonds (-NH-/N-C=O) in proteins. The O 1s spectrum (Figure 4F) shows a peak at 529.9 eV corresponding to metal oxide lattice oxygen (Fe-O-Fe), while the peak at 531.6 eV is attributed to organic carbonyl/carboxyl groups (C=O) from proteins and the drug CGA. The P 2p spectrum (Figure 4G) exhibits a single peak at 135.5 eV, assigned to the phosphodiester bond (-O-P(O)2-O-) in phospholipids, matching the exosomal membrane structure. The C 1s spectrum (Figure 4H) displays three components: 284.8 eV (C-C/C-H), 286.3 eV (C-O/C-N), and 288.2 eV (C=O), corresponding to alkyl chains, sugar rings/amino acid side chains, and carbonyl groups, respectively. The above results demonstrate that the Exo-SPIONs-SRF/CGA was successfully constructed.
HepG2 cells were used as the exosome donor, cell debris was removed by differential centrifugation, followed by magnetic enrichment of extracellular vesicles using Tf-SPIONs. The morphology of Exo-SPIONs-SRF/CGA was depicted in Figure 5A, B. The Tf-SPIONs were bound to the surface of exosomes, which displayed a typical oval-shaped morphology. DLS analysis indicated that the hydrodynamic diameters of Exo-SPIONs-SRF/CGA presented an unimodal normal distribution, with an average particle size of 167.53 ± 0.75 nm (Figure 5C). Surface potential analysis revealed that the Zeta potential of the complex was −15.22 ± 0.90 mV. This negative charge characteristic mainly originated from the carboxyl and sulfate groups in the phospholipid bilayer on the surface of the exosome membrane (Figure 5D). These results demonstrate that the Exo-SPIONs-SRF/CGA delivery system has been successfully constructed. Its excellent monodispersity and moderate surface potential are conducive to maintaining the colloidal stability of the system,45 laying a foundation for subsequent biomedical applications.
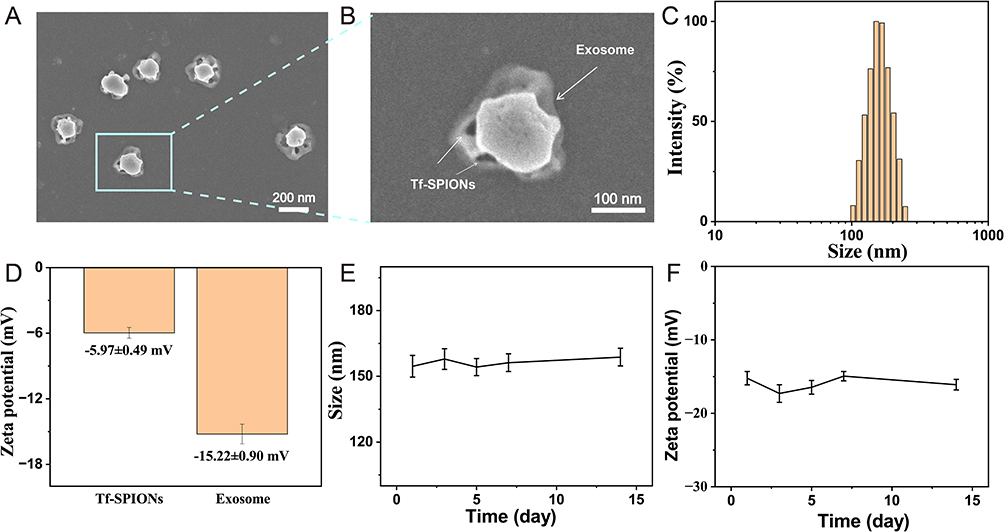 |
Figure 5 Characterization of Exo-SPIONs-SRF/CGA. (A) TEM images of Exo-SPIONs-SRF/CGA with the morphology of the magnified area (B). (C) Hydrodynamic particle size distribution of Exo-SPIONs-SRF/CGA. (D) Zeta potential of Tf-SPIONs and Exo-SPIONs-SRF/CGA. (E) hydrodynamic size and zeta potential changing (F) over time of Exo-SPIONs-SRF/CGA. |
Subsequently, the biocompatibility of Exo-SPIONs-SRF/CGA was evaluated by the RBC hemolysis assay. As shown in the supernatant images (Figure 6A), the positive control well exhibited a deep orange-red color, indicating extensive RBC rupture and hemoglobin release, while the negative control showed only faint coloration, suggesting minimal hemolysis. Only the free SRF/CGA displayed slight color, the supernatants of the other experimental groups at different concentrations displayed color intensity similar to that of the negative control, implying low levels of RBCs disruption. Quantitative analysis of relative hemolysis rates (Figure 6B) further demonstrated that the positive control reached nearly 100% hemolysis, whereas the negative control remained close to 0%.
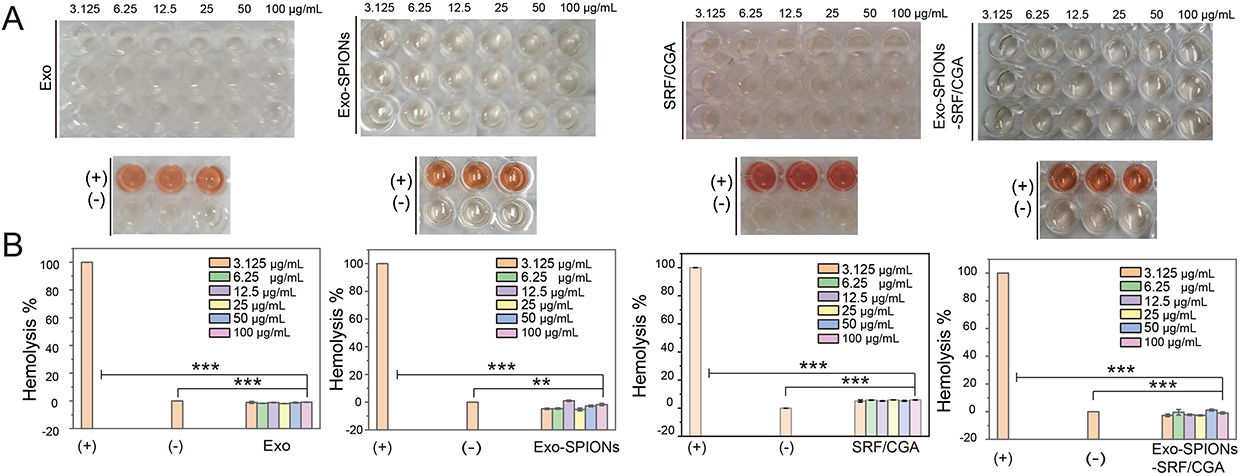 |
Figure 6 Cytotoxicity assessment of Exo-SPIONs-SRF/CGA. Images of the supernatant (A) and relative hemolysis rates (B) of RBC samples treated with Exo, Exo-SPIONs, SRF/CGA, and Exo-SPIONs-SRF/CGA. **p<0.01, ***p<0.001. |
The groups treated with Exo, Exo-SPIONs, and Exo-SPIONs-SRF/CGA exhibited significantly lower hemolysis rates compared to the positive control and showed results comparable to those of the negative control across all tested concentrations. The free SRF/CGA showed a higher hemolysis rate compared to other groups, this might be caused by the direct contact of anti-cancer drugs with cells. However, there was no hemolysis phenomenon after the drugs were encapsulated by Exo-SPIONs, which also indicates that Exo-SPIONs can reduce nonspecific exposure to non-target cells before being delivered to the targeted area. Therefore, we can infer that Exo-SPIONs can effectively protect tissue cells and reduce the toxicity and side effects of drugs. The above results indicate that none of the samples induced significant hemolysis within a broad concentration range (3.125–100 μg/mL), demonstrating low cytotoxicity and favorable biocompatibility, which supports their potential applicability in biomedical fields.
Exosomes have become highly promising drug delivery carriers due to their natural biocompatibility, excellent biological barrier penetration ability and targeted homing characteristics.46 Based on this, an Exo-SPIONs-SRF/CGA nanocomposite delivery system was constructed by loading SRF/CGA on exosomes derived from HepG2 cells. The drug encapsulation efficiency and drug loading capacity were 50.73±1.56% and 19.87±1.37%, respectively. Its drug release characteristics and in vitro anti-tumour cells effects were systematically evaluated (Figure 7). In addition, we systematically investigated the in vitro release of SRF and CGA from Exo-SPIONs-SRF/CGA. The release kinetics were evaluated using PBS at two different pH values (6.0 and 7.0). As illustrated in Figure 7A, the acidic condition (pH 6.0) promoted the release of SRF and CGA compared to the neutral condition (pH 7.0). This accelerated release can be attributed to the protonation of SRF and CGA under acidic conditions, which increases their solubility and diffusion rates. This pH-responsive release behavior is particularly promising for targeted drug delivery within the tumor microenvironment, which is typically characterized by acidic conditions. This characteristic enables more efficient drug accumulation and release at the tumor site. In the HepG2 cells cytotoxicity experiment (Figure 7B–D), the anti-cancer effects of Exo-SPIONs, free SRF/CGA and Exo-SPIONs-SRF/CGA at different concentrations and time points (12, 24 and 48 h) were systematically evaluated. Importantly, the Exo-SPIONs vector demonstrates good biocompatibility, and no significant cytotoxicity was observed even at the highest test concentration (cell survival rate approaches 100%), which provides an essential basis for its safety as a drug delivery system. Notably, Exo-SPIONs-SRF/CGA demonstrated a significantly enhanced anti-cancer effect. After 24 h of culture at the equivalent concentration of 1.25 µg/mL SRF/CGA, the survival rate of HepG2 cells in the free drug group was 80.05 ± 1.41%. The Exo-SPIONs-SRF/CGA decreased to 55.83 ± 0.99%. This enhancing effect might stem from exosomes’ natural targeting and penetrating ability.
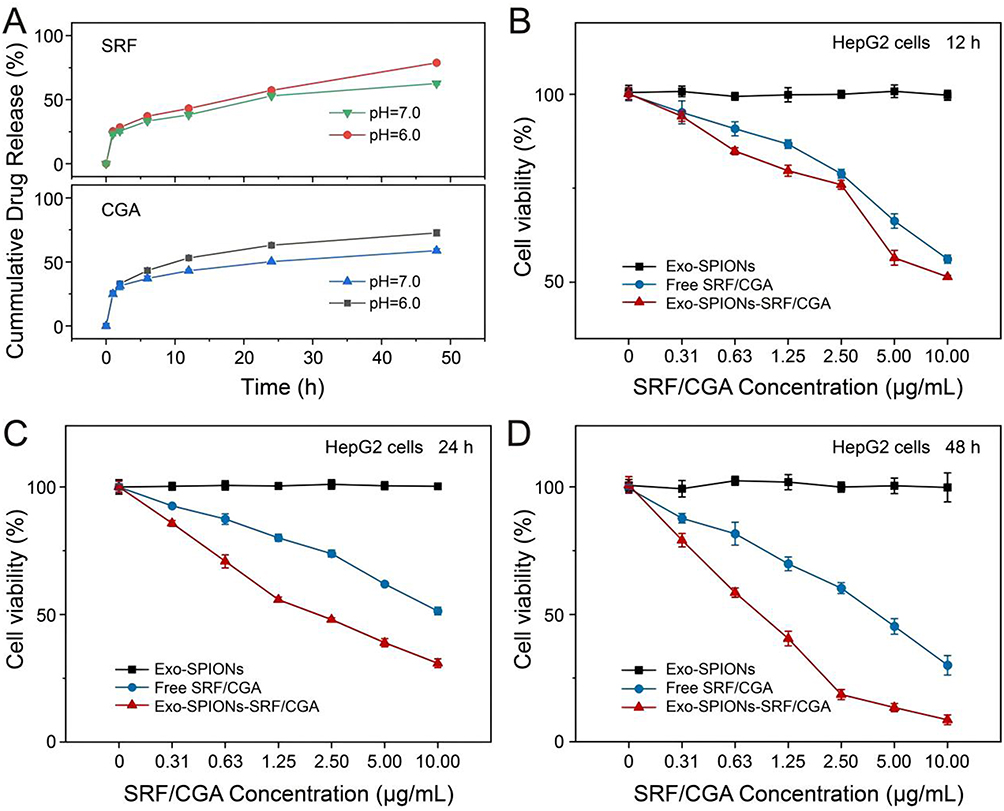 |
Figure 7 Drug release and anticancer activity of Exo-SPIONs-SRF/CGA. (A) Drug release profile in vitro at pH 6.0 and 7.0. (B-D) Cytotoxicity of Exo-SPIONs-SRF/CGA against HepG2 cells at 12, 24, and 48 h. |
Furthermore, as the incubation time was extended to 48 h, a pronounced enhancement in the anti-cancer efficacy of Exo-SPIONs-SRF/CGA was observed. Moreover, the cell survival rate of the Exo-SPIONs-SRF/CGA group further decreased to 18.54 ± 1.97% at the drug concentration of 2.50 µg/mL. It was significantly lower than 60.29 ± 2.15% of the free drug group, confirming that this delivery system can effectively maintain drug activity and achieve continuous release. To assess the potential cellular toxicity of Exo-SPIONs under magnetic field, we examined the effect of Exo-SPIONs on HepG2 cell viability under the condition of applying a magnetic field. As can be seen in Figure S1, Exo-SPIONs did not produce toxic effects on HepG2 under magnetic field. The feasibility of Exo-SPIONs for drug delivery under magnetic field was demonstrated. These results collectively indicate that the delivery system based on HepG2-derived exosomes has excellent biological safety and significantly enhances the bioavailability and targeted delivery efficiency of SRF/CGA drugs, providing a new strategy for treating liver cancers.
Subsequently, the cellular delivery capability and magnetic targeting potential of prepared Exo-SPIONs were investigated. As shown in Figure 8A, in the study of cellular delivery capacity, HepG2 cells were co-cultured with SRF/CGA-loaded Exo-SPIONs (Exo-SPIONs-SRF/CGA), and the distribution of the carrier was tracked using fluorescent labeling. Following 6 hours of incubation, red fluorescence was observed within the cells. With prolonged incubation time (12 h, 24 h), the intensity and distribution range of the red fluorescence signal significantly increased, visually demonstrating the progressive accumulation of the carrier within the cells. Quantitative fluorescence analysis (Figure 8B) further corroborated these findings, showing a continuous increase in the mean fluorescence intensity over time. This further verified that Exo-SPIONs-SRF/CGA could effectively deliver drugs into cells, with delivery efficiency exhibiting time-dependence.
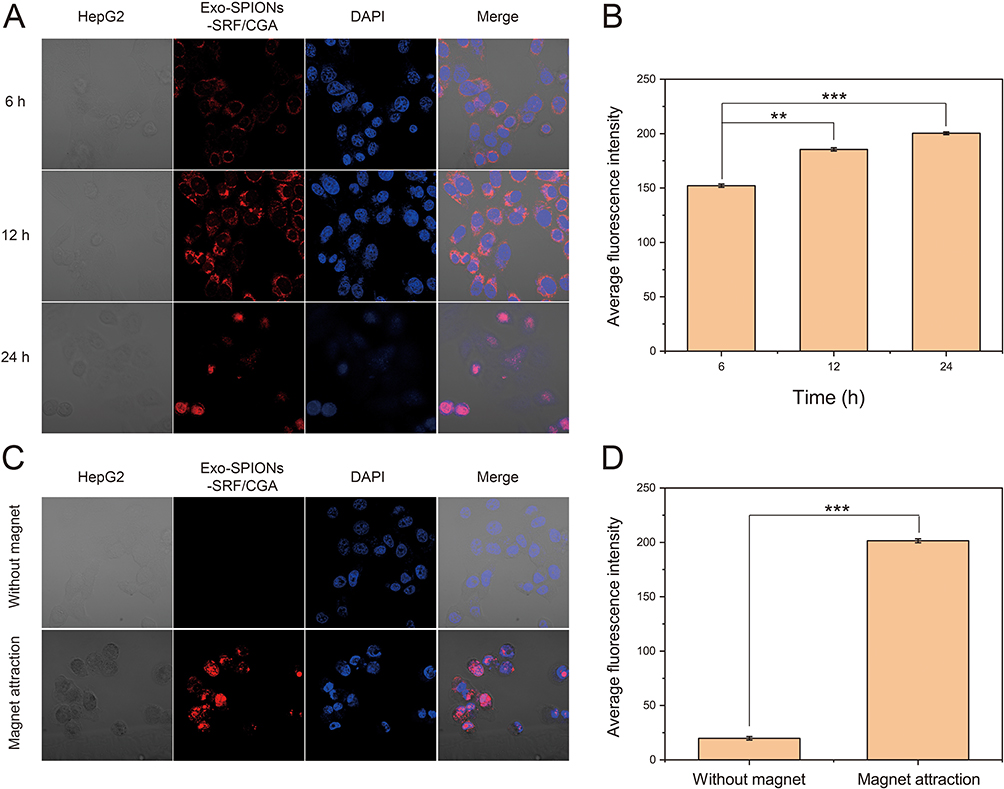 |
Figure 8 Intracellular delivery and magnetic targeting of Exo-SPIONs-SRF/CGA in HepG2 cells. (A) Fluorescence microscopy images showing intracellular localization of Exo-SPIONs-SRF/CGA in HepG2 cells incubation with different time. Nuclei were stained with DAPI (blue). (B) Average intracellular fluorescence intensity in HepG2 cells, quantitatively analyzed by ImageJ. (C) Magnetic targeted delivery and enrichment at the cellular level in vitro facilitated by Exo-SPIONs in combination with magnet attraction. (D) Average intracellular fluorescence intensity in cells, quantitatively analyzed by ImageJ. **p<0.01, ***p<0.001. |
To elucidate the magnetic targeting properties of prepared Exo-SPIONs, a magnetic intervention experiment was designed. Exo-SPIONs-SRF/CGA were co-cultured with HepG2 cells, and a magnet was placed under the confocal dish to induce targeted carrier accumulation. Confocal imaging results (Figure 8C) revealed that without magnetic intervention, only minimal fluorescence distribution was observed within the cells. Following the application of a magnetic field, the intracellular red fluorescence intensity significantly increased, indicating the guiding effect of the magnetic field on carrier delivery. Quantitative fluorescence statistics (Figure 8D) showed that the mean fluorescence intensity after magnetic field application was substantially higher than that without the field, confirming that the magnetic field could significantly promote the targeted accumulation of Exo-SPIONs-SRF/CGA at the cellular level. In summary, these results demonstrate that Exo-SPIONs possess efficient cellular delivery capability and can achieve targeted regulation via magnetic responsiveness. This provides experimental evidence at the cellular level for their application in precision drug delivery, highlighting the potential value of this carrier system in scenarios such as tumor-targeted therapy.
Exo-SPIONs-SRF/CGA achieves dual targeting and synergistic therapy for tumour tissues by integrating exosome homing targeting property and the magnetic targeting property of Tf-SPIONs. To evaluate the in vivo therapeutic effect of the system, the HepG2 tumour-bearing mouse model was established, and the PBS, Free SRF/CGA, Exo-SPIONs-SRF/CGA and Exo-SPIONs-SRF/CGA +Magnet were set up for comparative studies. As shown in Figure 9B, the tumour tissue was dissected after 18 days of treatment. Compared with the PBS control group, it can be seen that the tumour tissue was minimal after 18 days of Exo-SPIONs-SRF/CGA+Magnet treatment. Furthermore, as shown in the experimental results of Figure 9C, the tumours in the PBS control group presented a rapid proliferation trend. The Free SRF/CGA only showed a limited tumour suppressive effect, and the tumour volume change on the 18th day was (V/V0=80 ± 23%). In contrast, the Exo-SPIONs-SRF/CGA group demonstrated significantly enhanced anti-tumour activity on the 18th day (V/V0=19 ± 2%), which was mainly attributed to the exosome-mediated enhanced cellular affinity effect promoting the specific accumulation of the drug in tumour tissues. Importantly, under the guidance of an external magnetic field, the Exo-SPIONs-SRF/CGA+Magnet group demonstrated the most outstanding therapeutic effect on the 18th day (V/V0=6 ± 2%), and the tumour volume was significantly reduced compared with other groups. Notably, during the 18-day treatment period, the body weight of all mice in the treatment groups maintained a stable growth trend (Figure 9D), indicating that the delivery system had good biocompatibility and did not cause significant systemic toxicity.
 |
Figure 9 In vivo antitumor efficacy of Exo-SPIONs-SRF/CGA. (A) Schematic of tumor model establishment and treatment protocol. (B) Representative tumor images from HepG2-xenografted nude mice after 18 days. (C) Tumor volume progression. (D) Body weight changes. (E) TUNEL and H&E staining of the tumor. |
TUNEL and H&E staining were performed on the tumour tissues after 18 days of treatment for histopathological analysis (Figure 9E). It was found that H&E staining showed that the tumour tissue structure of the magnetic field-guided group (Exo-SPIONs-SRF/CGA+Magnet) was significantly loose, and the cell arrangement was disordered. The evaluation of TUNEL staining in tumour tissues of different groups reflects the situation of cell apoptosis. It can be seen that the proportion of apoptotic cells in the Exo-SPIONs-SRF/CGA+Magnet group is significantly higher than that in other treatment groups. This synergistic enhancement effect stems from the organic combination of dual targeting mechanisms: SPIONs achieve efficient enrichment at tumour sites under magnetic field. At the same time, exosomes promote intracellular drug internalization through membrane fusion. Coupled with the drug release triggered by the pH of the tumour microenvironment, they jointly form an efficient and precise drug delivery system. These findings provide crucial experimental evidence for developing combined targeted therapeutic strategies based on exosomes and magnetic nanoparticles.
SRF has been discussed due to its low bioavailability, high systemic toxicity and toxic effect on normal liver cells.47 Exo-SPIONs-SRF/CGA+Magnet effectively delivers SRF to the lesion site, and at the same time, CGA reduces the toxicity of SRF to normal organs and tissues, as shown in Figure S2. The biosafety of the drug was evaluated in vivo through routine blood analysis. The treatment in the free SRF/CGA and Exo-SPIONs-SRF/CGA groups resulted in a slight increase in blood routine in the liver and kidney groups of mice’s. The effects caused by Exo-SPIONs-SRF/CGA+Magnet were almost the same as those of the control group, suggesting that the dual targeting system with added magnets could produce effects at the lesion site more quickly and accurately, reduce the damage of organs and tissues, and reduce the pressure on organs.
The integration of exosomes with magnetic nanomaterials enables a “dual targeting” effect, which enhances drug delivery efficiency while minimizing the toxic effects of SRF/CGA on healthy organs and tissues. Intravenous SRF/CGA administration can trigger an inflammatory response. Elevated levels of inflammatory factors are established key markers for cardiotoxicity and potentially heart failure. Serum levels of specific inflammatory factors (interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-α), and monocyte chemoattractant protein-1 (MCP-1) were quantified using ELISA kits following injection of different SRF/CGA formulations. As shown in Figure S3, prolonged administration of SRF/CGA markedly elevated IL-6, TNF-α, and MCP-1 levels. Conversely, exosomal encapsulation of SRF/CGA substantially suppressed this inflammatory response. The results indicate that, relative to free SRF/CGA, Exo-SPIONs-SRF/CGA significantly attenuated the inflammatory response without inducing detectable systemic immunotoxicity in vivo. This effect is likely attributed to SRF/CGA encapsulation within exosomes preventing direct exposure to serum components post-injection. By leveraging the inherent homing capability of exosomes in conjunction with the magnetic guidance of synthesized Tf-SPIONs. Exo-SPIONs-SRF/CGA, under magnetic guidance, exhibited notably enhanced targeting and potent anti-tumor effects. Consequently, this dual-targeting strategy minimized SRF/CGA accumulation in non-target tissues, thereby mitigating both the inflammatory response and its consequential cytotoxicity.
Discussion
In this study, the Exo-SPIONs-SRF/CGA dual-targeting nanoplatform was successfully constructed, which integrates the biological homing ability of HCC-derived exosomes with the magnetic guidance property of SPIONs to achieve precise drug delivery. The platform exhibited excellent pH-responsive release characteristics and good biocompatibility. Both in vitro and in vivo experiments demonstrated that the dual-targeting strategy could significantly enhance drug accumulation and retention at the tumor site compared with free drug or single targeting vehicle. Moreover, the synergistic effect of SRF and CGA can effectively inhibit tumor growth and reduce systemic toxicity.
At the same time, there are still several limitations of this study that need to be explored in depth in future studies. First, we effectively removed residual cells and large debris by differential centrifugation (300×g, 2000×g) to reduce the contamination of active carcinogenic components at the source. Simultaneously, transferrin-modified SPIONs (Tf-SPIONs) were used for magnetic separation, and the specific binding of transferrin to the receptor was used to selectively capture mature exosomes, further excluding abnormal vesicles, to purify potentially harmful components. Moreover, through the efficient loading and targeted delivery of SRF/CGA, it is difficult to drive tumor progression under the action of therapeutic doses, even if the exosomes carry trace amounts of cancer-promoting signals. Although the risk can be mitigated by a purification step, testing for carcinogenic components in Exo-SPIONs is still necessary to verify their safety. In the future, omics methods such as small RNA sequencing and protein profiling should be used to comprehensively characterize its molecular cargo and verify whether it has completely eliminated known oncogenic factors such as miR-21 and TGF-β, which is the key premise to ensure the safety of its clinical application. Second, although the subcutaneously transplanted tumor model used in this study is easy to manipulate, it cannot fully mimic the orthotopic HCC, such as the tumor microenvironment in the background of liver fibrosis. In the future, it is necessary to further verify the targeting and therapeutic effects of this platform in more clinically relevant orthotopic models, especially liver cirrhosis models.
In summary, the Exo-SPIONs-SRF/CGA nanoplatform was successfully constructed, which provides a novel strategy for precise, efficient and low toxicity treatment of HCC through the exquisite integration of biological and physical targeting strategies. Future work should focus on comprehensive safety evaluation in large animal models, clinical translation optimization of magnetic guidance technology, and exploration of the synergistic effect of combined application with immunotherapy, so as to ultimately benefit the majority of liver cancer patients.
Conclusions
This work developed a dual-targeted nanoplatform, Exo-SPIONs-SRF/CGA, to overcome the limitations of current HCC therapies, including poor drug delivery efficiency, systemic toxicity, and drug resistance. By combining the natural tumour-homing capability of HCC-derived exosomes with the magnetic guidance of SPIONs, this platform achieves exact and efficient drug delivery to tumour sites. The co-encapsulation of SRF and CGA enables synergistic anti-tumour effect: SRF inhibiting tumour proliferation and angiogenesis, while CGA enhances antioxidant activity and reduces toxicity. Furthermore, the TME-responsive drug release ensures localized activation, thereby enhancing therapeutic outcomes while reducing systemic side effects. Experimental validation demonstrated that Exo-SPIONs-SRF/CGA significantly enhances tumour suppression and drug retention compared to conventional monotherapies or single-modality nanocarriers. The dual-targeting strategy, integrating biological and magnetic navigation, overcomes traditional nanocarriers’ insufficient targeting and poor permeability. Additionally, the system’s ability to controllably release drugs in response to the tumour microenvironment reduces systemic toxicity, addressing a critical challenge in HCC treatment. This work presents a promising strategy in precision oncology, offering a high-efficiency, low-toxicity solution for HCC therapy. By leveraging the natural properties of exosomes and the external controllability of SPIONs, the platform improves drug delivery and may provide a potential strategy for overcoming multidrug resistance and metastasis. At the same time, the modular design can be easily adapted to different tumor types by simply changing the source of exosomes, providing flexible strategies for precision oncology. In clinical practice, SPIONs can be used as integrated treatment agents for magnetic resonance imaging-guided therapy. In summary, this platform could provide a clinically feasible, highly effective and low-toxicity solution for HCC patients, and extend to other malignancies where precision drug delivery is difficult.
Author Contributions
All authors made a significant, contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
Dr Shixian Yan reports a patent “MACC1-Inhibitor for Targeted Therapy of MACC1-Induced Metastasis” licensed to Espacenet. The authors report no other conflicts of interest in this work.
References
1. Shin D, Kim Y, Park J, Kim Y. High-throughput proteomics-guided biomarker discovery of hepatocellular carcinoma. Biomed J. 2025;48(1):100752. doi:10.1016/j.bj.2024.100752
2. Hamed M, Fawzy Montasser IM, Salah-Eldin MM, Hassan RS. Characteristics and outcomes of hepatocellular carcinoma in patients with c-viral chronic liver disease and non-viral chronic liver disease. QJM. 2024;117. doi:10.1093/qjmed/hcae175.993
3. Li H, Liu R, Qiu H, et al. Tumor burden score stratifies prognosis of patients with intrahepatic cholangiocarcinoma after hepatic resection: a retrospective, multi-institutional study. Front Oncol. 2022;12:829407. doi:10.3389/fonc.2022.829407
4. Calvisi DF. Aberrant ubiquitination causing liver cancer: the ADRM1-ΔEx9- FBXW7 connection. J Hepatol. 2025;83(1):15–20. doi:10.1016/j.jhep.2025.03.032
5. Singh SP, Madke T, Chand P. Global epidemiology of hepatocellular carcinoma. J Clin Exp Hepatol. 2025;15(2):102446. doi:10.1016/j.jceh.2024.102446
6. Li Y, Zhu Y, Fang J, et al. Apoptin regulates apoptosis and autophagy by modulating reactive oxygen species (ROS) levels in human liver cancer cells. Front Oncol. 2020;10:1026. doi:10.3389/fonc.2020.01026
7. Rumgay H, Arnold M, Ferlay J, et al. Global burden of primary liver cancer in 2020 and predictions to 2040. J Hepatol. 2022;77(6):1598–1606. doi:10.1016/j.jhep.2022.08.021
8. Harimoto N, Tsukagoshi M, Seki T, et al. Predictors for early recurrence beyond up-to-7 or distant metastasis after hepatocellular carcinoma resection: proposal for borderline resectable HCC. Int J Clin Oncol. 2024;29(2):195–204. doi:10.1007/s10147-023-02434-7
9. Liu Z, Zhang Z, Mei H, Mao J, Zhou X. Distribution and clinical relevance of phospholipids in hepatocellular carcinoma. Hepatol Int. 2020;14(4):544–555. doi:10.1007/s12072-020-10056-8
10. Lasagni S, Leonardi F, Pivetti A, et al. Endothelial angiopoietin-2 overexpression in explanted livers identifies subjects at higher risk of recurrence of hepatocellular carcinoma after liver transplantation. Front Oncol. 2022;12:960808. doi:10.3389/fonc.2022.960808
11. Luo M-Y, Han Z, Wang J, Zhong C, Chen J. TARDBP is a candidate diagnostic biomarker promoting tumor progression via impacting tumor immunity and tumor microenvironment. J Cancer. 2024;15(13):4113–4127. doi:10.7150/jca.96800
12. Agudile EP, Vega EA, Salirrosas O, et al. Temporal trends of health disparity in the utilization of curative-intent treatments for hepatocellular carcinoma: are we making progress? J Gastrointest Surg. 2024;28(9):1392–1399. doi:10.1016/j.gassur.2024.05.015
13. Dadrass F, Sher A, Kim E. Update on locoregional therapies for liver cancer: radiation segmentectomy. Curr Oncol. 2023;30(12):10075–10084. doi:10.3390/curroncol30120732
14. Sarno G, Montalti R, Giglio MC, et al. Hepatocellular carcinoma in patients with chronic renal disease: challenges of interventional treatment. Surg Oncol. 2021;36:42–50. doi:10.1016/j.suronc.2020.11.007
15. Ding ZWH, Wen Z. Overexpression of C‑sis inhibits H2O2‑induced Buffalo rat liver cell apoptosis in vitro and alleviates liver injury in a rat model of fulminant hepatic failure. IntJ Mol Med. 2018;42:873–882. doi:10.3892/ijmm.2018.3684
16. Li Y, Xu C, Sun B, Zhong F, Cao M, Yang L. Sema3d restrained hepatocellular carcinoma progression through inactivating Pi3k/Akt signaling via interaction with FLNA. Front Oncol. 2022;12:913498. doi:10.3389/fonc.2022.913498
17. Chen H, Cheng H, Wu W, et al. The blooming intersection of transcatheter hepatic artery chemoembolization and nanomedicine. Chin Chem Lett. 2020;31(6):1375–1381. doi:10.1016/j.cclet.2020.03.024
18. Liu Y, Yang H, Li T, Zhang N. Immunotherapy in liver cancer: overcoming the tolerogenic liver microenvironment. Front Immunol. 2024;15:1460282. doi:10.3389/fimmu.2024.1460282
19. Liu G, Yang L, Chen G, et al. A review on drug delivery system for tumor therapy. Front Pharmacol. 2021;12:735446. doi:10.3389/fphar.2021.735446
20. Shao X, Zhao X, Wang B, Fan J, Wang J, An H. Tumor microenvironment targeted nano-drug delivery systems for multidrug resistant tumor therapy. Theranostics. 2025;15(5):1689–1714. doi:10.7150/thno.103636
21. Joy R, George J, John F. Brief outlook on polymeric nanoparticles, micelles, niosomes, hydrogels and liposomes: preparative methods and action. ChemistrySelect. 2022;7. doi:10.1002/slct.202104045
22. Wang R, Yan H, Yu A, Ye L, Zhai G. Cancer targeted biomimetic drug delivery system. J Drug Delivery Sci Technol. 2021;63:102530. doi:10.1016/j.jddst.2021.102530
23. Han X, Gong C, Yang Q, Zheng K, Wang Z, Zhang W. Biomimetic nano-drug delivery system: an emerging platform for promoting tumor treatment. Int J Nanomed. 2024;19:571–608. doi:10.2147/IJN.S442877
24. Guo C, Zhang J, Cai X, et al. Emerging nanomedicine strategies for hepatocellular carcinoma therapy. iMetaOmics. 2024;1(1):e12. doi:10.1002/imo2.12
25. Li T, Li X, Han G, et al. The therapeutic potential and clinical significance of exosomes as carriers of drug delivery system. Pharmaceutics. 2022;15:21. doi:10.3390/pharmaceutics15010021
26. Yin W, Ma H, Qu Y, et al. Targeted exosome-based nanoplatform for new-generation therapeutic strategies. Biomed Mater. 2024;19:032002. doi:10.1088/1748-605X/ad3310
27. Malla RR, Pandrangi S, Kumari S, Gavara MM, Badana AK. Exosomal tetraspanins as regulators of cancer progression and metastasis and novel diagnostic markers. Asia Pac J Clin Oncol. 2018;14(6):383–391. doi:10.1111/ajco.12869
28. Saka OM, Dora DD, Kibar G, Tevlek A. Expanding the role of exosomes in drug, biomolecule, and nanoparticle delivery. Life Sci. 2025;368:123499. doi:10.1016/j.lfs.2025.123499
29. Tanziela T, Shaikh S, Jiang H, Lu Z, Wang X. Efficient encapsulation of biocompatible nanoparticles in exosomes for cancer theranostics. Nano Today. 2020;35:100964. doi:10.1016/j.nantod.2020.100964
30. Zhang Y, Zhang C, Wu N, et al. The role of exosomes in liver cancer: comprehensive insights from biological function to therapeutic applications. Front Immunol. 2024;15:1473030. doi:10.3389/fimmu.2024.1473030
31. Wang Q, Wang G, Niu L, et al. Exosomal MiR-1290 promotes angiogenesis of hepatocellular carcinoma via targeting SMEK1. J Oncol. 2021;2021:6617700. doi:10.1155/2021/6617700
32. Wang G, Li J, Bojmar L, et al. Tumour extracellular vesicles and particles induce liver metabolic dysfunction. Nature. 2023;618(7964):374–382. doi:10.1038/s41586-023-06114-4
33. Pan J-H, Zhou H, Zhao -X-X, et al. Role of exosomes and exosomal microRNAs in hepatocellular carcinoma: potential in diagnosis and antitumour treatments. Int J Mol Med. 2018;41(4):1809–1816. doi:10.3892/ijmm.2018.3383
34. Liu JJJ, Liu D, To SKY, Wong AST. Exosomes in cancer nanomedicine: biotechnological advancements and innovations. Mol Cancer. 2025;24(1):166. doi:10.1186/s12943-025-02372-0
35. Hu Y, Luo Z, Cai S, Xie Q, Zheng S. Glycyrrhizic acid attenuates sorafenib resistance by inducing ferroptosis via targeting mTOR signaling in hepatocellular carcinoma. Scand J Gastroenterol. 2024;59(6):730–736. doi:10.1080/00365521.2024.2315317
36. Luo A-L, Zheng W-Y, Zhang Q, et al. COPS5 triggers ferroptosis defense by stabilizing MK2 in hepatocellular carcinoma. Adv Sci. 2025;12(22):e2416360. doi:10.1002/advs.202416360
37. Wang X-Y, Liao Y, Wang R-Q, et al. Tribbles pseudokinase 3 converts sorafenib therapy to neutrophil-mediated lung metastasis in hepatocellular carcinoma. Adv Sci. 2025;12(13):e2413682. doi:10.1002/advs.202413682
38. Ye S, Chen J, Zheng Y, et al. Targeting USP18 overcomes acquired resistance in hepatocellular carcinoma by regulating NCOA4 deISGylation and ferroptosis. Cell Death Dis. 2025;16(1):448. doi:10.1038/s41419-025-07772-0
39. Huang Q, Shan Q, Ma F, Li S, Sun P. Chlorogenic acid mitigates heat stress-induced oxidative damage in bovine mammary epithelial cells by inhibiting NF-κB-mediated NLRP3 inflammasome activation via upregulating the Nrf2 signaling pathway. Int J Biol Macromol. 2025;301:140133. doi:10.1016/j.ijbiomac.2025.140133
40. Tian L, Su C-P, Wang Q, et al. Chlorogenic acid: a potent molecule that protects cardiomyocytes from TNF-α–induced injury via inhibiting NF-κB and JNK signals. J Cell Mol Med. 2019;23(7):4666–4678. doi:10.1111/jcmm.14351
41. Zhang H, Shan G, Liu M, et al. Harnessing ROS amplification and GSH depletion using a carrier-free nanodrug to enhance ferroptosis-based cancer therapy. Small. 2025;21(6):e2409250. doi:10.1002/smll.202409250
42. Li J, Li J, Yao Y, et al. Biodegradable electrospun nanofibrous platform integrating antiplatelet therapy-chemotherapy for preventing postoperative tumor recurrence and metastasis. Theranostics. 2022;12(7):3503–3517. doi:10.7150/thno.69795
43. da Silva KLC, Camacho AP, Mittestainer FC, et al. Atorvastatin and diacerein reduce insulin resistance and increase disease tolerance in rats with sepsis. J Inflamm. 2018;15(1):8. doi:10.1186/s12950-018-0184-9
44. Eslami P, Albino M, Scavone F, et al. Smart magnetic nanocarriers for multi-stimuli on-demand drug delivery. Nanomaterials. 2022;12:303. doi:10.3390/nano12030303
45. Bi H, Han X. Magnetic field triggered drug release from lipid microcapsule containing lipid-coated magnetic nanoparticles. Chem Phys Lett. 2018;706:455–460. doi:10.1016/j.cplett.2018.06.051
46. Liang Y, Iqbal Z, Lu J, et al. Cell-derived nanovesicle-mediated drug delivery to the brain: principles and strategies for vesicle engineering. Mol Ther. 2023;31(5):1207–1224. doi:10.1016/j.ymthe.2022.10.008
47. Zhang W, Hong X, Xiao Y, Wang H, Zeng X. Sorafenib resistance and therapeutic strategies in hepatocellular carcinoma. Biochim Biophys Acta Rev Cancer. 2025;1880:189310. doi:10.1016/j.bbcan.2025.189310
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Impaired Autophagy Response in Hepatocellular Carcinomas Enriches Glypican-3 in Exosomes, Not in the Microvesicles
Koksal AR, Thevenot P, Aydin Y, Nunez K, Sandow T, Widmer K, Nayak L, Scott J, Delk M, Moehlen MW, Cohen AJ, Dash S
Journal of Hepatocellular Carcinoma 2022, 9:959-972
Published Date: 7 September 2022
Identification of the Level of Exosomal Protein by Parallel Reaction Monitoring Technology in HCC Patients
Huang H, Zhang Q, Zhang Y, Sun X, Liu C, Wang Q, Huang Y, Li Q, Wu Z, Pu C, Sun A
International Journal of General Medicine 2022, 15:7831-7842
Published Date: 14 October 2022
Surface Engineering of HEK293 Cell-Derived Extracellular Vesicles for Improved Pharmacokinetic Profile and Targeted Delivery of IL-12 for the Treatment of Hepatocellular Carcinoma
Zhang J, Song H, Dong Y, Li G, Li J, Cai Q, Yuan S, Wang Y, Song H
International Journal of Nanomedicine 2023, 18:209-223
Published Date: 13 January 2023
A Single-Step Immunocapture Assay to Quantify HCC Exosomes Using the Highly Sensitive Fluorescence Nanoparticle-Tracking Analysis
Koksal AR, Ekmen N, Aydin Y, Nunez K, Sandow T, Delk M, Moehlen M, Thevenot P, Cohen A, Dash S
Journal of Hepatocellular Carcinoma 2023, 10:1935-1954
Published Date: 2 November 2023
Nanocomposites to Overcoming Sorafenib Resistance in Hepatocellular Carcinoma Therapy
Liu J, Zhou S, Wei Y, Du J, Jia F, Dong W, Cao Y, Shi Z, Mu H, Chen L, Yu S
International Journal of Nanomedicine 2025, 20:15723-15767
Published Date: 26 December 2025