Back to Journals » Journal of Inflammation Research » Volume 16
The Chains of Ferroptosis Interact in the Whole Progression of Atherosclerosis
Authors Wan X, Zhang H, Tian J ![]() , Hao P, Liu L, Zhou Y, Zhang J, Song X, Ge C
, Hao P, Liu L, Zhou Y, Zhang J, Song X, Ge C
Received 17 July 2023
Accepted for publication 3 October 2023
Published 16 October 2023 Volume 2023:16 Pages 4575—4592
DOI https://doi.org/10.2147/JIR.S430885
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Xueqi Wan,* Huan Zhang,* Jinfan Tian,* Peng Hao, Libo Liu, Yuquan Zhou, Jing Zhang, Xiantao Song, Changjiang Ge
Department of Cardiology, Beijing Anzhen Hospital, Capital Medical University, Beijing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Changjiang Ge; Xiantao Song, Email [email protected]; [email protected]
Abstract: Atherosclerosis (AS), a category of cardiovascular disease (CVD) that can cause other more severe disabilities, increasingly jeopardizes human health. Owing to its imperceptible and chronic symptoms, it is hard to determine the pathogenesis and precise therapeutics for AS. A novel type of programmed cell death called ferroptosis was discovered in recent years that is distinctively different from other traditional cell death pathways in morphological and biochemical aspects. Characterized by iron overload, redox disequilibrium, and accumulation of lipid hydroperoxides (L-OOH), ferroptosis influences endothelial cells, vascular smooth muscle cells (VSMCs), and macrophages, as well as inflammation, partaking in the pathology of many cardiovascular diseases such as atherosclerosis, stroke, ischemia-reperfusion injury, and heart failure. The mechanisms behind ferroptosis are so sophisticated and interwoven that many molecules involved in this procedure are unknown. This review systematically depicts the initiation and modulation of ferroptosis and summarizes the contribution of ferroptosis to AS, which may open a feasible approach for target treatment in the alleviation of AS progression.
Keywords: ferroptosis, iron overload, oxidation, lipid peroxidation, atherosclerosis
Introduction
Due to lifestyle changes in recent decades, the number of patients suffering from cardiovascular diseases (CVDs), including atherosclerosis, myocardial infarction, heart attack, stroke, heart failure, arrhythmia, and heart valve disorders, has been mounting. Although numerous treatments have been applied to improve the prognosis, CVDs-associated morbidity and mortality increase yearly. CVDs have become a leading problem worldwide.1 Among them, atherosclerosis (AS) has become a tough issue affecting a multitude of patients, even at an early age. Although improved treatments and interventions, such as those to lower lipids, considerably elevate patients’ life quality, culminating complications of AS are lethal.2 According to reports, ischemic heart disease triggered by atherosclerotic plaque rupture accounts for 42.5% of mortality caused by CVDs.3 Hence, novel targets and strategies are urgently needed to give those patients suffering from AS optimal management for a better prognosis.
Regarding factors shaping the initiation and progression of AS, a mountain of literature attests that macrophage, VSMC, endothelium, and inflammatory responses are the hallmarks that traditionally lead to necrotic core formation and plaque destabilization that worsen AS.4–11 Cell death penetrates these key processes, which lays a fundamental mechanism of AS progression.12 The traditional methods dictating cell death are autophagy, apoptosis, and necrosis. Distinct from these forms of cell death, ferroptosis is an iron-dependent form of regulated cell death characterized by iron burden, ROS release, and excessive lipid peroxidation.13,14 Based on the crosstalk of oxidation and lipid accumulation, research into the involvement of ferroptosis in AS pathogenesis and progression has received much attention. Interestingly, ferroptosis was recently found to be partially associated with certain key processes in AS, such as the dysfunction of endothelial cells (ECs), foam cell build-up, and irritation of inflammation.15–17
As for specific mechanisms, there is a wealth of modulators behind ferroptosis, including glutathione peroxidase 4 (GPX4), nuclear factor erythroid 2-related factor2 (Nrf2), and heme oxygenase-1 (HMOX-1). Additionally, coenzyme Q10 (CoQ10), nicotinamide adenine dinucleotide phosphate (NADPH), fibroblast specific protein (FSP), and heat shock proteins (HSP) are implicated in ferroptosis via diverse signaling. Although controversies and uncertainties exist concerning the mechanisms behind ferroptosis, the potential risk of iron and ferroptosis in atherosclerosis has been described in direct and indirect experiments.18 For example, research has shown the vital role of gut microbiota in AS,19,20 while the exact mechanism is seldom elucidated. Interestingly, certain research has proven the correlation between ferroptosis and gut microbiota; hence, ferroptosis may be an internal mechanism for the impact of gut microbiota on AS.21 Zou et al conducted a bioinformatic analysis to study the iron metabolism-related genes (IMRG); interestingly, most differentially expressed IMRGs are involved in the ferroptosis and atherosclerosis pathway displayed by KEGG enrichment.22 Further, another GO and KEGG analysis revealed that ten candidate ferroptosis-related biomarkers are among the most abundant in reaction to lipid oxidation, ferroptosis, and AS.23 The above data demonstrates the potentially close correlation between ferroptosis and atherogenesis.
Given the essential role of ferroptosis in AS, certain inhibitors targeting iron overload and lipid peroxidation have been tested in cell and animal models but still need to be examined in clinical practice.24–26 Therefore, we speculate that ferroptosis is a promising and valuable target for improving AS. To test this, we screened PubMed, Web of Science, and Medline databases using a combination of keywords, such as ferroptosis, oxidation stress, iron metabolism, CVD, cardiovascular diseases, AS, and atherosclerosis, and included relevant reviews and original articles. Our review comprehensively summarizes the complex connections incorporated in ferroptosis and investigates how ferroptosis is involved in AS. Additionally, we categorize the related inhibitors and their targeting molecules to offer a better understanding of the relationship between ferroptosis and AS. The following information may pave an avenue for the in-depth exploration of optimized strategies for inhibiting ferroptosis and managing AS.
The Chain Reaction of Ferroptosis
Ferroptosis is a form of programmed cell death. Its main feature is the iron-dependent accumulation of lipid hydroperoxides to lethal levels, as depicted in Figure 1. To date, many experiments have been conducted to comprehensively understand ferroptosis. Although some other molecules and their interactions remain unclear, a few sophisticated and subtle mechanisms have been distinguished in the literature.
 |
Figure 1 The characteristics of ferroptosis. Iron overload, lipid peroxidation, and redox imbalance are the hallmark conditions orchestrating ferroptosis. Extracellular iron is introduced into the cell in two different ways. First, Fe3+ carried by transferrin binds TfR anchored in the cytoplasm, where it can be stored in the form of ferritin that releases free iron via a process called ferritinophagy. NCOA4 is a cargo receptor for the disintegration of ferritin to liberate the free iron. Upon entry into cells, it converts to ferrous iron in the endosome and moves to the cytoplasm through several transport systems, such as DMT1. In the second process, Fe2+ diffuses into the cell through binding with low-molecular-weight complexes, including ATP, citrate, ascorbate, and the like. Fe2+ is mainly stored in the endosome, while the redundant iron is exported by ferroportin. Intracellular ferrous iron can be converted into Fe3+ along with the generation of ROS via the Fenton reaction, such as superoxide anion, hydrogen peroxide, and even hydroxyl radicals, with the outcome of H2O2 and Fe-S being released from mitochondria. The ROS produced by the Fenton reaction participates in PL-O formation from PUFA. LOX and ROS serve as catalysts for the peroxidation of PUFA. First, ACSL4 drives the binding of free AA/AdA with CoA to form AA/AdA-CoA derivatives, followed by the biosynthesis of AA/AdA-CoA and membrane PE mediated by LPCAT3 to build up AA/AdA-PE. Finally, LOX directs the peroxidation of AA/AdA-PE to yield AA/AdA-PE-OOH, accelerating ferroptosis. Abbreviations: TfR, transferrin receptor; NCOA4, nuclear receptor coactivator 4; DMT1, divalent metal transporter 1; ROS, reactive oxygen species; H2O2, hydrogen peroxide; Fe-S, iron-sulfur protein; PL-O, phospholipids oxidation; PUFA, polyunsaturated fatty acids; LOX, lipoxygenase; ACSL4, acyl-CoA synthetase long-chain family member 4; AA/AdA-PE-OOH, arachidonic acid/adrenic acid-phosphatidylethanolamine-hydroperoxides; PE, phosphatidylethanolamine; LPCAT3, lysophosphatidylcholine acyltransferase 3. |
The Overall Characterization and Mechanism of Ferroptosis
Iron overload, lipid peroxidation, and redox imbalance are the typical hallmarks orchestrating ferroptosis.27 Extracellular iron is introduced into the cell in two different processes. First, Fe3+ carried by transferrin binds to the transferrin receptor (TfR) anchored in the cytoplasm.28 The Fe3+ can be stored in the form of ferritin and releases free iron via a process called ferritinophagy. In the second process, Fe2+ diffuses into the cell by binding low-molecular-weight complexes, including ATP, citrate, ascorbate, and the like. Fe2+ is mainly stored in the endosome, while the redundant iron is exported by ferroportin.28,29 Intracellular ferrous iron can be converted into Fe3+ along with the generation of reactive oxygen species (ROS) via the Fenton reaction, with hydrogen peroxide (H2O2) and iron-sulfur protein (Fe-S) released from mitochondria.28 Nuclear receptor coactivator 4 (NCOA4) is a cargo receptor for the disintegration of ferritin to liberate free iron, leading to ferroptosis.28
Heat shock protein beta-1 (HSPB1) is an antagonist for iron uptake.30 Iron chelators such as deferoxamine (DFO) and ciclopirox (CPX) can decrease the iron pool.31,32 The •OH produced by the Fenton reaction participates in the formation of phospholipids oxidation (PL-O) from polyunsaturated fatty acids (PUFA), accelerating the process of ferroptosis.33,34 Ferroptosis antagonists ferrostatin-1 (Fer-1), Liproxstatin, and vitamin E (Vit E) block PUFA oxidation.26 GXP4 is also a key modulator for ferroptosis that can govern the Xc-system, composed of transporter solute carrier family 7 member 11 (SLC7A11) and SLC3A2, enabling the exchange of intracellular glutamate and extracellular cystine to promote the generation of glutathione (GSH), a potent antioxidant.3 GSH can transfer the potentially poisonous, multi-unsaturated L-OOH into the relatively less harmful and active lipid alcohols (L-OH).26 RAS-selective lethal 3 (RSL3), ML162, and FIN56 promote the deletion of GPX4.35,36 Ferroptosis agonists erastin, sorafenib, and BAY117085 target GXP4.29,37 In contrast, heat shock protein 5 (HSPA5) can enhance GXP4 expression by upregulating activating transcription factor 4 (ATF4).28 Meanwhile, NADPH and CoQ10 are endogenous inhibitors of ferroptosis due to their anti-oxidative exertion, which can be regulated by GXP4. FSP can be recruited in the plasma membrane and reduces the CoQ10 dependent on NADPH to impede the formation of lipid peroxidation (LPO).38 Nrf2 is the prominent negative regulator of ferroptosis and is in charge of the heme oxygenase-1 (HO-1).39 Although proper levels of HO-1 can exert a protective effect against ROS invasion, excessive HO-1 promotes ferrous iron accumulation, increasing its susceptibility in response to ferroptosis.40,41 Nrf2 also increases the expression of quinone oxidoreductase (NQO1), ferritin heavy chain 1 (FTH1), TXNRD1, and SRXN1 to alleviate ferroptosis.22,41 Additionally, Nrf2 upregulates the expression of SLC7A11. The mevalonate pathway and the voltage-dependent anion channel (VDAC) molecule are also implicated in ferroptosis.29,42
The Rudimentary Features of Ferroptosis
Iron Metabolism Dysfunction
Iron homeostasis is essential for maintaining a broad spectrum of biological processes that ensure cell viability.43 Iron deficiency or supplementation can lead to a pathological reaction in the cell. Although the exact link between iron and ferroptosis remains unclear, there is no doubt that iron plays a pivotal part in this type of cell death regulation. Early in 1981, iron was discovered to be a potential risk factor for CVD. Over the past few decades, literature has revealed the crucial role of iron balance in a constellation of pathophysiological processes of common human diseases like AS.25,44
Iron absorption, transportation, release, and storage must be balanced to sustain normal organelle function. Food contains two types of iron, known as heme and nonheme iron.45 Due to deficiencies in transferrin (Tf) and transferrin receptor 1 (TfR1), intestinal cells take up nutrition-derived Fe2+ through the divalent metal transporter (DMT1) and heme carrier protein 1 (HCP1).46,47 Fe3+ can be transformed into Fe2+ by duodenal cytochrome B reductase (DCYTB), promoted by gastric acid and vitamin C.45,46,48 The iron absorbed by the intestine is transported to essential tissues and cells once demanded. Iron absorbed by enterocytes exits via the transportation of ferroportin.49,50 There are two processes by which iron reaches the non-intestinal cell. In the first, Fe3+ enters the cell with Tf and is transported via TfR1.14 Tf acts as a storage pool for retaining iron circulating in the plasma until the iron is required, at which point Tf directs it towards target tissues to be introduced into the cell.46 Fe3+ is then packaged into ferritin. NCOA4 is a cargo receptor for the disintegration of ferritin to liberate free iron, leading to ferroptosis.13,51 Upon entry into cells, Fe3+ converts to ferrous iron in the endosome and is then stored in the labile iron pool (LIP), while redundant iron is exported by ferroportin.26,27,35 In the second process, Fe2+ diffuses into cells through binding low-molecular-weight complexes, including adenosine triphosphate, citrate, ascorbate, peptides, or phosphatases.52,53 Mechanistically, ferrous iron enters the cytoplasm mostly through several transport systems, such as DMT1 and L-type voltage-dependent Ca2+ channels (LTCC). While investigating exosomes stemming from human umbilical cord blood (HUCB-MSC), Song et al ascertained that the mitigation of ferroptosis and myocardial injury in this exosome was attributed to the inactivation of DMT1, which is brought about by delivering miR-23a-3p in mouse models of acute myocardial infarction.54 A liver peptide was recently shown to regulate ferroptosis by controlling serum iron via the degradation of ferroportin in enterocytes and macrophages.55 Other regulatory proteins like HSPB1, expectedly, can govern the sensitivity of ferroptosis via the impact of the iron swing.30 This may grant novel insight into targeting iron as a therapeutic aimed at ferroptosis. Under physiological conditions, the quantity of iron in LIP is low to prevent ROS production. By contrast, excessive iron can generate ROS via the Fenton and Haber-Weiss reactions,56 such as superoxide anion, hydrogen peroxide, and even hydroxyl radicals, which yield chemical reactions with lipids and damage cells, entailing the abnormal procedure involved in several diseases.25,29,56
Redox Imbalance and Lipid Peroxidation
The well-known homeostasis between oxidation and reduction plays a vital role in various physiological activities, such as immune defense and inflammatory response. Although ROS caused by specific stimulation contributes to the clearance of pathogens and cancer cells, its abundance can lead to DNA and protein damage and even cell death.27 The classic mechanism for ferroptosis is the deficiency of GSH and inactivation of GPX4 caused by the absence of cystine, which results in the accumulation of lipid ROS and ultimately leads to cell death.32,57
By contrast, GSH, like other potent and common reductants, exerts anti-oxidative effects such as superoxide dismutase (SOD) and coenzyme CoQ10, the decrease of which is a crucial hallmark in ferroptosis.58 A heap of oxidative and antioxidant reagents, acting together on autophagy machinery, promote the process of lipid peroxidation during ferroptosis.58 Multiple ROS generated by mitochondria-mediated electron transport chain or other related enzymes like transmembrane NADPH oxidases (NOXs) and VDAC induce lipid peroxidation. Accumulated oxysterols are detrimental to atheroma lesions.12 Oxysterols, particularly 7β-hydroxycholesterol and 7-ketocholesterol (7keto), increase intracellular levels of ROS, nudging the macrophage to switch towards the pro-inflammatory type.59 Cluster of differentiation (CD) 74 can reportedly enhance inflammation, and its deficiency can mitigate the severity of atheroma lesions.60 It is noteworthy that CD74 expression becomes elevated with worsening AS lesions. Furthermore, being exposed to 7-ketocholesterol, the macrophage is loaded with observably higher CD74, inextricably linked with higher ferritin and cell death.60
Aberrant Lipid Peroxidation
It is broadly acknowledged that free radical oxidation of PUFAs in lipoproteins or cell membranes, allegedly called lipid peroxidation (LPO), is one of the fairly crucial shaping factors in atherosclerosis. The accumulation of products of lipid peroxide, including phosphatidylcholine, cardiolipin, and phosphatidylethanolamine, are hallmarks of ferroptosis in response to the oxidative degradation of lipids followed by lipid peroxyl radical and hydroperoxide release.26,59 Over the decades, peroxidation of phospholipids (PLs) with polyunsaturated fatty acyl tails was discovered as the primary drive for ferroptosis.26 As a PUFA, it is more susceptible to oxidative attack.33 In general, the formation of lipid hydroperoxides is intimately associated with catalyzed activities of lipoxygenase (LOX), an enzyme that can actuate the di-oxygenation of free and esterified PUFAs, including arachidonic acid (AA) and adrenic acid (AdA), to catalyze lipid peroxidation immediately.35 First, Acyl-CoA synthetase long-chain family member 4 (ACSL4) drives the binding of free AA/AdA with CoA to form AA/AdA-CoA derivatives, followed by the biosynthesis of AA/AdA-CoA and membrane phosphatidylethanolamine (PE) mediated by lysophosphatidylcholine acyltransferase 3 (LPCAT3) to build up AA/AdA-PE. Lastly, LOX directs the peroxidation of AA/AdA-PE to yield AA/AdA-PE-OOH, accelerating the process of ferroptosis.34,61 Another essential enzyme largely implicated in lipid oxidation is acetaldehyde dehydrogenase 2 (ALDH2), which is considered beneficial in CVD protection, such as in AS and stroke.49,62
Lipid-modified oxidative stress is involved in various anomalies of diseases such as AS. Doxorubicin (Dox), a widely used drug, is associated with heart injury and failure through ferroptosis in several patients. After DOX administration, excess free iron is released in the heart via heme degradation mediated by the Nrf2/HMOX-1 pathway, which deposits in mitochondria and results in lipid peroxidation on its membrane.63 Studies have shown that cardiac toxicity generated through oxidative phospholipids is implicated in cell death.64
Prominent Molecules Governing Ferroptosis
GXP4
GPX4 plays an essential role in ferroptosis, and its inactivation can lead to overwhelming lipid peroxidation. The cystine/glutamate antiporter (Xc-system)/GSH/GPX4 axis is the main pathway involved in ferroptosis, which intoxicates lipid hydroperoxides to alcohol and converts potentially toxic L-OOH to non-toxic L-OH, thus maintaining the normal physiological function of the cell.14 Alleviating oxidative stress protects endothelial cells from disruption.65 Several ferroptosis triggers like glutamate and erastin downregulate GSH and abrogate the enzymatic activity of GPX4 through retarding the import of cystine mediated by this axis.32,42,66 Still, erastin binds with and inhibits VDAC to fuel ferroptosis in cardiomyocytes.29 In a myocardial ischemia-reperfusion (MIR) model, Lu et al found that by administering britanin (Bri), a drug exerting excellent anti-inflammatory, anti-oxidative, and anti-tumor activities, the infarction area was observably mitigated through ferroptosis inhibition. They further investigated the mechanism regulating ferroptosis and eventually demonstrated that upregulation of GXP4 via the AMPK/GSK3b/Nrf2 pathway alleviated ferroptosis-associated MIR injury.67 SLC7A11, which is targeted by Nrf2,40 is a crucial member of the Xc-system, accounting for the uptake of cystine so as to synthesize GSH.68 Solute carrier family 3 member 2 (SLC3A2) is another component of this system.28 It was confirmed by string data analysis to be a hub gene in iron-metabolism-related genes.22 In addition, RSL3, ML162, and FIN56 inhibit GPX4, thus resulting in L-OOH overload in cells.35 In contrast, heat shock protein 5 (HSPA5) can enhance GXP4 expression via ATF4 upregulation (Figure 2).28
 |
Figure 2 The GXP4 mechanism is involved in ferroptosis. GXP4 is a crucial modulator for ferroptosis that can govern the Xc-system. It is composed of SLC7A11 and SLC3A2, enabling the exchange of intracellular glutamate and extracellular cystine to promote the generation of GSH, a potent anti-oxidant that facilitates the production of NADPH. GSH can transform the potentially poisonous, multi-unsaturated L-OOH into the relatively less harmful and active L-OH. RSL3, ML162, and FIN56 promote the deletion of Gpx4. Erastin, sorafenib, BA Y117089, and RSL3 are agonists of ferroptosis via GXP4 targeting. In contrast, heat shock protein 5 (HSPA5) can enhance GXP4 expression via ATF4 upregulation. NADPH and CoQ10 are endogenous ferroptosis inhibitors due to their anti-oxidative exertion, which can regulate GXP4. Abbreviations: GXP4, glutathione peroxidase 4; SLC7A11, solute carrier family 7 member 11; GSH, glutathione; NADPH, nicotinamide adenine dinucleotide phosphate; CoQ10, coenzyme Q10; L-OOH, lipid hydroperoxides; RSL3, RAS-selective lethal 3. |
Nrf2
Early data uncovered the importance of Nrf2, which showed that it protects cells against deleterious contexts like oxidative stress. Under the normal state, Nrf2 is steadily located in the cytoplasm with the help of its combination with Keap1. Once cellular homeostasis is disrupted by certain stimulations, such as ROS, it exits the complex and moves to the nucleus to be activated in order to exert its protective function, accompanied by the initiation of an array of cytoprotective genes.40
In a study by Fang et al.63 DOX administration in mice induced cardiomyopathy with high iron accumulation mediated by HMOX-1, which is upregulated by Nrf2. Conversely, Nrf2 is a well-established key transcription factor in defense of excessive oxidative stress to preserve cellular redox homeostasis and prohibit ferroptosis.39,41,69 This discrepancy in whether Nrf2 brings about beneficial or detrimental effects may depend on experimental variations in conditions and cell types. For instance, britanin treatment restrained ROS and iron levels in H9C2 cells and prevented MIR injury. However, these alterations were weakened after the siRNA knockdown of Nrf2.67 Still, other experiments have engaged in the verification of Nrf2 for regulating ferroptosis using several associated inhibitors and agonists.70,71 Lower expression of Nrf2 was discovered in patients with AS, while cells treated with ox-LDL showed a remarkable decrease in Nrf2 profiling.38,72 Prenyl diphosphate synthase subunit 2 (PDSS2), a master regulator in AS, suppressed ferroptosis and degeneration of vascular endothelial cells by activating Nrf2. To make the evidence more convincing, atherosclerotic plaque lesions significantly increased in mice with Nrf2 deficiency in an AS animal model.38
Tanshinone IIA (TSA) protects endothelial tissues from being damaged. A study aimed to investigate the underlying mechanism for TSA concluded that it participated in the process of ferroptosis by abrogating the decline of the potent reductant GSH and attenuating its release in human coronary artery ECs. By applying this natural compound, the cell death initiated by either erastin or RSL3 was significantly inhibited. The study then discovered the elevation of Nrf2 associated with TSA. Of note, rescue experiments indicated that the knockdown of Nrf2 subverted the beneficial action on cells.39 Additionally, Nrf2 upregulated the expression of 1NQO1, HMOX-1, and FTH1, the elimination of which could promote ferroptosis by erastin and sorafenib41 (Figure 3).
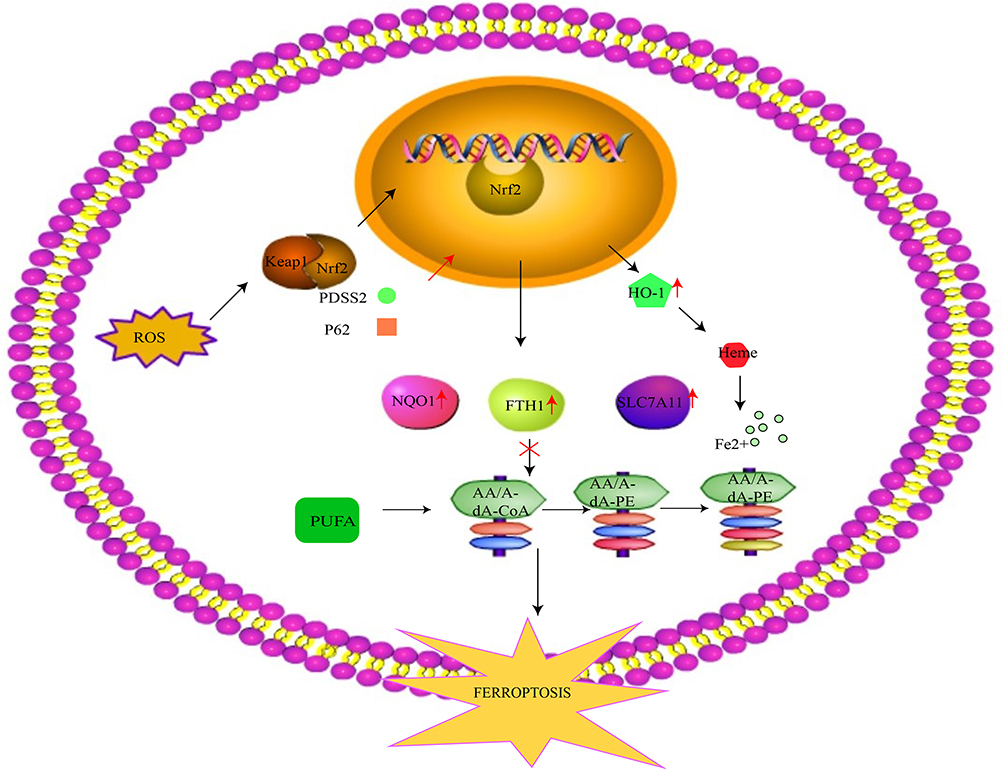 |
Figure 3 The Nrf2 mechanism involved in ferroptosis. Under the normal state, Nrf2 is steadily located in the cytoplasm with the help of Keap1 binding. Upon stimulation from oxidative stress, it disengages from Keap1 and translocates to the nucleus, where it combines with AREs such as ferritin and HO-1. Although proper levels of HO-1 can exert a protective effect against ROS invasion, excessive HO-1 promotes ferrous iron accumulation, increasing susceptibility to ferroptosis. Nrf2 also increases the expression of NQO1 and FTH1 to alleviate ferroptosis. Additionally, it upregulates the expression of SLC7A11, thus suppressing ferroptosis through the reversal of lipid peroxidation. PDSS2 suppresses ferroptosis by activating Nrf2, while P62 downregulates Nrf2. Abbreviations: Nrf2, nuclear factor erythroid 2-related factor 2; ARE, promoter antioxidant response element; HO-1, heme oxygenase-1; ROS, reactive oxygen species; NQO1, quinone oxidoreductase 1; FTH1, ferritin heavy chain 1; SLC7A11, solute carrier family 7 member 11; PDSS2, prenyl diphosphate synthase subunit 2. |
ACSL4
Acyl-CoA synthetase long-chain family member 4 (ACSL4) was significantly upregulated in the advanced stage of AS within coronary artery samples.4 The positive correlation between the grade and stage of AS and the level of ACSL4 was analyzed by the Spearman correlation coefficient. In breast cancer cells, eliminating the suppressor of fused homolog (SUFU) augmented the susceptibility to RSL3-induced ferroptosis via ACSL4 upregulation.73 According to prior data, ACSL4 participates in a positive feedback loop model for lipid peroxidation and ferroptosis initiation. Park et al found that bromelain strongly depressed mutant colorectal cancer in its blockage of cell ferroptosis by directing the decline of ACSL4.74 To be more concise, PKCβII senses lipid peroxidation and activates, followed by ACSL4 dimerization and phosphorylation at the Thr328 site to accelerate polyunsaturated‐fatty‐acid‐containing phospholipids (PUFA‐PL) formation and enlarge lipid peroxide deposits, leading to ferroptosis.75
HMOX1
Regulated by the Nrf2, HMOX-1 fuels the transformation of heme to biliverdin, carbon monoxide (CO), and iron in cooperation with NADPH and cytochrome P450 upon activation by stimuli like chemicals or oxidative stress. It was previously determined to protect against detrimental conditions, including cell apoptosis, autophagy, and stress, which is advantageous for cardiovascular diseases such as AS.76,77 Like a double-edged sword, however, it can also damage cells due to its force on ferroptosis, which concurs with its dichotomous activity in ROS regulation.53,78 As it is reportedly to be involved in the process of ferroptosis in cardiomyopathy,63 HOMX1 currently attracts a lot of attention for its role in comprehending the mechanisms behind ferroptosis. Observation of mice with sickle cell disease (SCD) exhibited clues that HMOX-1 augmentation could induce ferroptosis and cardiac assault. Strikingly, abolishing or enhancing HMOX-1 alleviated or amplified the ferroptosis associated with cardiomyopathy in SCD mice, respectively.79 Endothelial cells isolated from high-fat diet (HFD)-fed ApoE-/- diabetic mice harbored elevated expression of HMOX1. Of note, the inhibition of ferroptosis enabled mouse aortic endothelial cells and human umbilical vein endothelial cells exposed to high glucose or lipid levels to maintain a lower level of HMOX1. Furthermore, the knockdown of HMOX1 moderately restored GXP4 and GSH levels and decreased ROS generation, abrogating ferroptosis in vascular insult.24 On the other hand, ferroptotic cell death triggered by HMOX1 may have its upside. As shown by Chang’s investigation, potent anti-tumor BAY 11–7085 killed cancer cells via ferroptosis directed by the Nrf2-SLC7A11-HOMX-1 pathway, in which HOMX-1 was a pivotal mediator in response to the state of oxidation.37 Intriguingly, evidence also exists connecting the negative manipulation of HMOX-1 to ferroptosis. For example, renal proximal tubule cells (PTCs) depleted of HMOX-1 became more susceptible to cell death in reaction to two ferroptosis inducers, erastin or RSL3, compared with the cell enrichment in cells with HMOX-1.80 This information arouses many questions concerning the vital role of HMOX1 in controlling ferroptosis. The causes of the discrepancy in its impact on ferroptosis are poorly defined, although previous scholars attempted to explain it by the amount of ROS or different conditions.53,81 Moreover, studies on ferroptosis dependent on HMOX1 in AS are insufficient, demanding more sophisticated and delicate analysis and trials.
Other Pathways Involved in Ferroptosis
Endogenous antioxidants, such as NADPH and CoQ10, also play a vital part in ferroptosis.32 Apoptosis-inducing factor mitochondrion-associated 2 (AIFM2, also named FSP1) is another electron transporter and lipid-soluble molecule in defense of oxidation, which nudges CoQ10 reduction via NADPH to decrease LPO generation and blocks ferroptosis.42 PDSS2 is the cardinal enzyme for CoQ10 production, along with the positive regulation of Nrf2.38 Alternatively, current studies have revealed that FSP1 manipulates LPO through an endosomal sorting complex required for the transport (ESCRT)-III dependent cell repair mechanism by activating charged multivesicular body protein 5 (CHMP5) and CHMP6.82 In addition, the mevalonate pathway is also involved in ferroptosis via the generation of CoQ10, isopentenyl-pyrophosphate (IPP), and farnesyl-pyrophosphate (FPP) to negatively regulate ferroptosis.42
Ferroptosis: A Crucial Role in Atherosclerosis
Pathogenesis of atherosclerosis. AS is a chronic inflammatory disease characterized by endothelial damage.1 During the initiation and development of AS, deviant VSMCs and macrophage proliferation and apoptosis, excessive lipid deposit, oxidative stress and inflammation can exacerbate the insult to the endothelium.83,84 In turn, EC-induced injury can augment the release of inflammatory mediators, facilitating VSMC migration, macrophage infiltration, formation of foam cells, and so on. Oxidized LDL, the initial atherogenic signal, induces oxidative stress on VSMCs and ECs, which promotes cell transition to the atherosclerotic phenotype.83 Macrophage cells engulf ox-LDL and other lipids, transforming them into foam cells through scavenger receptors.85 The foam cells cluster in the intima and form a core region with extracellular lipids, which are surrounded by a cap enriched in VSMCs and collagen materials. Other inflammatory cells infiltrate the shoulder area, where atheroma develops.10,86 Therefore, the increased apoptosis of VSMCs results in the decreased thickness of the fibrous cap due to the lack of collagen, which destabilizes atherosclerotic plaque.83,86
It is well-established that lipid peroxidation is associated with the formation and worsening of AS. Of note, the pathogenesis of AS is also tightly connected to dysregulated iron metabolism, increased levels of ROS and rampant ferroptosis in pertinent types of cells like macrophages, VSMCs, and endothelial cells31 (Table 1, Figure 4).
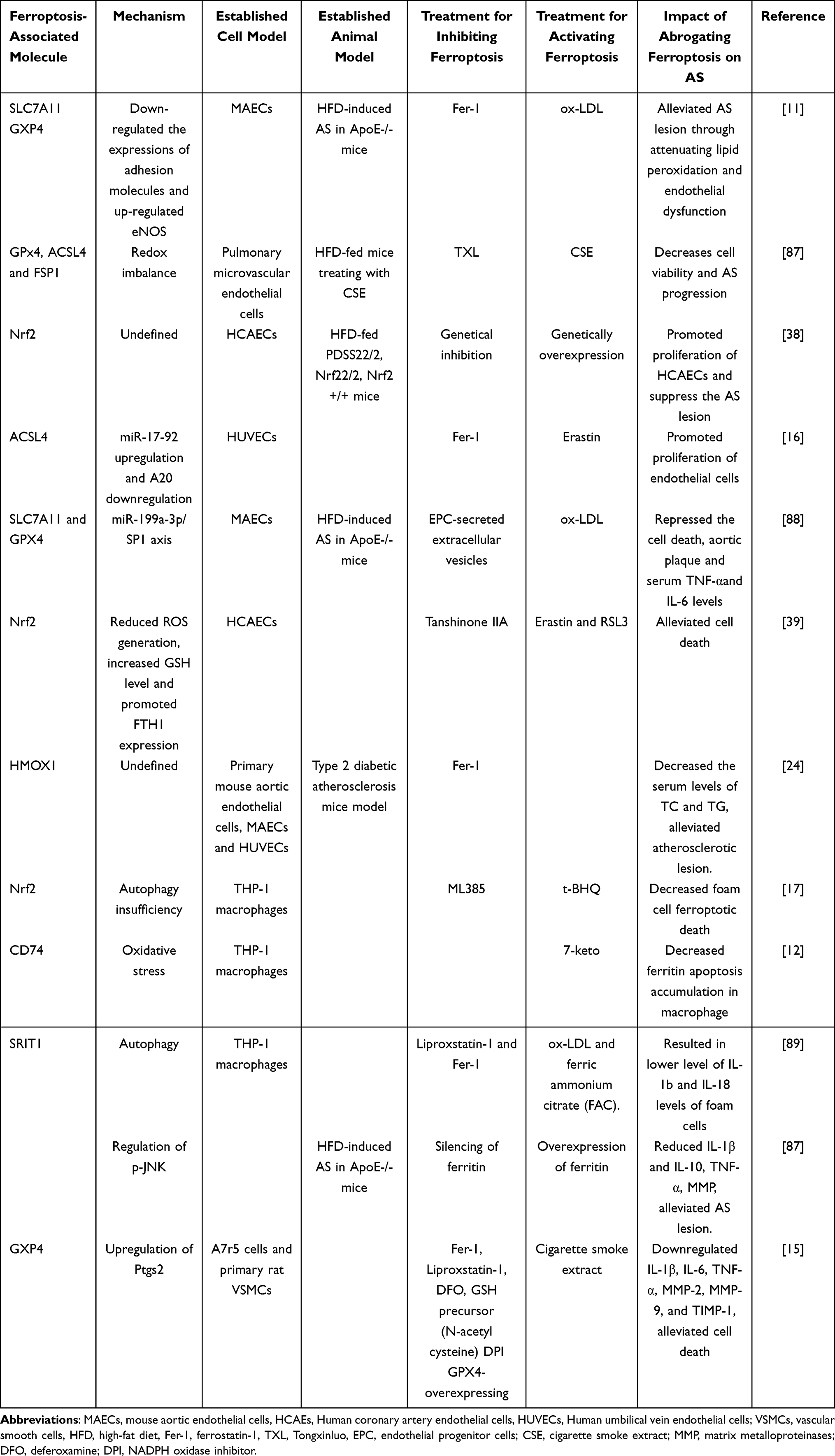 |
Table 1 The Related Researches About Ferroptosis for AS |
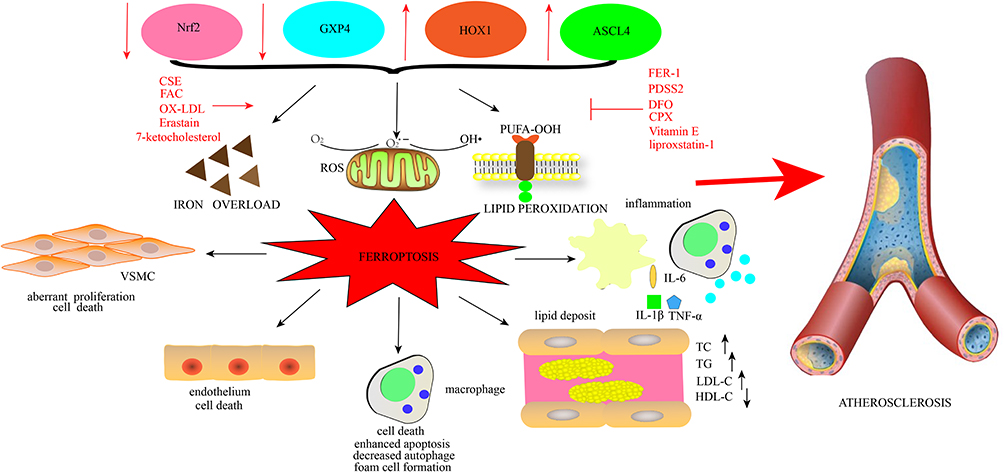 |
Figure 4 The interplay between ferroptosis and AS. Iron overload, lipid peroxidation, and oxidative stress are the typical hallmarks orchestrating ferroptosis. Ferroptosis is governed by regulators such as Nrf2, GXP4, ACSL44, and HOMX1 and is implicated in many pathological cell activities. It can decrease cell viability in VSMCs, facilitate the death of macrophages, and enhance apoptosis while suppressing autophagy. Additionally, it promotes foam cell formation. It also facilitates the death of endothelial cells. Further, ferroptosis is involved in the generation of inflammation via the activation of inflammatory signaling and the release of related inflammatory factors, such as IL-6, IL-1β, and TNF-α. Lipid accumulation is also led by ferroptosis, with increases in LDL, TG, and TC and decreases in HDL. Concerning the five crucial factors correlated with the generation and development of AS, ferroptosis ultimately prompts AS. Ox-LDL, FAC, CSE, erastin, and 7-ketocholesterol activate ferroptosis, while Fer-1, PDSS2, DFO, CPX, liproxstatin-1, and Vit E inhibit ferroptosis. Abbreviations: Nrf2, nuclear factor erythroid 2-related factor 2; GXP4, glutathione peroxidase 4; ACSL44, Acyl-CoA synthetase long-chain family member 4; HOMX1, heme oxygenase-1; VSMCs, vascular smooth muscle cells; IL-6, interleukin-6; IL-1β, interleukin-1β; TNF-α, tumor necrosis factor; LDL, low-density lipoprotein; TG, triglyceride; TC, total cholesterol; HDL, high-density lipoprotein; AS, atherosclerosis; ox-LDL, oxidized LDL; FAC, ferric ammonium citrate; CSE, cigarette smoke extract; Fer-1, ferrostatin-1; PDSS2, prenyl diphosphate synthase subunit 2; DFO, deferoxamine; CPX, ciclopirox; Vit E, vitamin E. |
Oxidative Stress
Mitochondria are the major organelle generating ROS. Well-documented data show that mtDNA damage results in the mitochondrial membrane potential change, impairing mitochondrial iron homeostasis and culminating in higher levels of iron. As previously discussed, Fe2+ can accelerate the process of H2O2 conversion into the highly reactive hydroxyl radical (•OH) and other oxidants.90 Excessive ROS can disrupt the oxidative equilibrium.27 According to early studies, redox imbalance is intimately related to the course of AS. First and foremost, the ox-LDL, an oxidized lipid that can promote the formation of foam cells and damage endothelial cells, worsens AS.11,91 Moreover, iron-induced oxidative stress has been implicated in various pathological conditions of AS.29 It was reported to be the perpetrator of the elevated activation of the scavenger receptor (SR) of SMC, the formation of foam cells, and the disintegration of the fibrous wall, leading to vulnerable plaques.92
Endothelial Cells
Endothelial cells play an indispensable role in the pathology and development of AS and the accompanying sequelae.93 As a matter of fact, the cell death of endothelium is closely correlated with the pathological mechanism of AS.16 Ferroptosis, as a novel cell death pathway, aggravates the decline in the viability of human umbilical vein endothelial cells accompanied by the increased release of ROS. This phenomenon could be rescued by Fer-1, a kind of ferroptosis antagonist. Additionally, researchers have figured out a specific miR-17-92/A20/ACSL4 network to manipulate this procedure by transducing cells with lentivirus vectors carrying associated genes.16 The experiment may indicate the modality of how the detriment of ferroptosis can be controlled. Yang et al38 illustrated that PDSS2 increased the proliferation and decreased the ferroptosis of human coronary vascular endothelial cells exposed to ox-LDL via upregulating Nrf2.
Likewise, the intracellular release of ROS was decreased. Further, endothelial cells treated with ox-LDL were analyzed to verify how ferroptosis performed in endothelial cell dysfunction, specifically whether erastin or the iron level heightened by HTF could expedite cell death. Conversely, these consequences could be restored by Fer-1. Moreover, Fer-1 administration also protected ECs against injury induced by ox-LDL. In HFD-fed ApoE-/- mice, administration of the ferroptosis inhibitor Fer-1 substantially alleviated the degree of plaque lesion compared with control groups. The treatment efficacy is equivalent to that of simvastatin (SIM), a ubiquitous lipid-lowering drug.11 Endothelial progenitor cells (EPCs) were reported to defend endothelial despair and reduce AS severity. A study focused on the relationship between EPC-derived extracellular vesicles (EPC-Evs) and AS unveiled that these vesicles extracted from EPCs mitigated the ROS release, lipid accumulation, and cell ferroptosis through the miR-199a-3p/SP1 axis in vitro in cultured endothelial cells. In HFD-fed ApoE-/- mice injected with Evs exhibited lower proinflammatory factors such as tumor necrosis factor (TNF)-α and interleukin-6 (IL-6).
Regarding the ferroptosis-associated index, there were decreased levels of GSH, SLC7A11, and GPX4 expression. Furthermore, the plaque area was ameliorated after transferring Evs. The study suggested that Evs delivered miR-199a-3p, which silenced SP1 and depressed ferroptosis in ECs, postponing endothelial injury and conferring a cardioprotective effect.88
The alveolar-capillary barrier mainly consists of the alveolar epithelium and capillary endothelium, which can maintain cell permeability in response to inflammatory mediators.89 The disruption of the pulmonary microvascular endothelial barrier can affect AS accompanying COPD. COPD mice complicated with AS showed more pronounced endothelial barrier dysfunction compared with control groups, accompanied by a heightened ferroptosis, as reflected by the increase of ACSL4 and decrease of FSP1 and GXP4. Accordantly, in vivo experiments determined that impaired endothelial cells displayed elevated ferroptosis and an oxidation/antioxidation imbalance. This result suggested that ferroptosis may be involved in AS progression due to COPD pathology.87
Apart from respiratory system diseases, AS is pervasively accompanied by diabetes and persists as the primary cause of disability among patients suffering from type 2 diabetes. According to a comprehensive bioinformatic assay conducted by Meng et al, ferroptosis and HMOX1 are upregulated in atherosclerotic vascular disease.24 Importantly, in vivo experiments exhibited that the ferroptosis inhibitor Fer-1 markedly reduced serum lipid profiles and plaque lesions in HFD-fed ApoE-/- mice. Furthermore, cellular experiments verified that HMOX1 knockdown reduced iron content and ROS, reducing ferroptosis in diabetic human endothelial cells. This discovery indicates that HMOX1 is responsible for ferroptosis activation orchestrating diabetic AS.
Macrophages
Despite the poor understanding of the underlying mechanisms, several seminal findings have indirectly elucidated the interplay between ferroptosis and macrophages in AS. Acetaldehyde dehydrogenase 2 (ALDH2), an enzyme critical for lipid metabolism, detoxifies the lipid aldehydes obtained from lipid peroxidation under the oxidant context. The protective effect of ALDH2 in CVD has been demonstrated in certain publications. Zhong et al found that ALDH2/LDL receptor-KO mice had decelerated atherosclerosis compared with solely LDL receptor-KO mice.94 When ALDH2 was genetically manipulated in ApoE-/- mice, however, AS incidence increased. These results show the complicated interaction of ALDH2 with the LDL receptor in AS. The researchers also discovered that ALDH2 modulated the foam cell formation coordinated by LDL receptor, a crucial factor for absorbing LDL in macrophages and promoting plaque formation. Consequently, the link between ferroptosis and AS may partly be attributed to lipid peroxidation. Refreshingly, Peng’s team concluded that the Nrf2 pathway inversely dictates the demise of foam cells.17 Additionally, the negative Nrf2 effect, harboring incongruity with its broadly acknowledged antioxidant execution, was substantiated by inadequate autophagy.
The accumulation of 7-ketocholesterol (7ket) is involved in the atherosclerotic inflammatory response and foam cell formation in macrophages. In one study, 7ket induced the higher expression of CD74, contributing to macrophage apoptosis accompanied by elevated ferritin.60 Using ferric ammonium citrate (FAC) to coax ferroptosis, Su et al95 observed that treating with FAC can decrease foam cell viability, increase lipid ROS levels, decrease GPX4 expression, inhibit expression of SIRT1, which is a novel target for treating AS, and increase IL-1β and IL-18 levels. These pathological changes were restored by Liproxstatin-1 and Fer-1. Interestingly, a similar result was observed via activating autophagy, while further inhibiting autophagy can reverse this change. Ferritin is essential in maintaining iron balance, and a ferritin disorder can lead to ferroptosis.
Currently, more attention has been shifted to how iron disruption mediates the biogenesis of ferroptosis; thus, ferritin has become a target for study. Zheng et al96 designed ferritin-overexpressing and silencing constructs in the HFD-fed mouse model. They ultimately discovered that the overexpression of ferritin resulted in higher profiles of interleukin (IL) and matrix metalloproteinases (MMPs), two types of key biomarkers for AS plaque lesions. Furthermore, in a recent clinical study from sixty-one carotid samples, the surface molecule CD74 was surprisingly heightened in THP-1 apoptotic macrophages induced by 7-ketocholesterol. Further, severe plaque lesions involved higher ferritin levels and apoptotic cell death. This may indicate that CD74 expression is associated with necrotic core formation, plaque rupture, macrophage apoptosis, and ferritin. In an array of tests, the study demonstrated that aberrant ferritin production induced by oxidized lipids might be closely related to macrophage apoptosis and plaque instability in atherosclerosis.12 Likewise, in samples collected from human atherosclerotic plaques, macrophages challenged with ferryl Hb displayed a more proinflammatory phenotype, as reflected by a greater release of IL-1β and TNF-α, the upregulation of HMOX-1 and H-ferritin and the accumulation of iron.97 These consequences may suggest that ferroptosis is likely to become a target for AS amelioration.
Vascular Smooth Muscle Cells
Iron-dependent free radicals cause ox-LDL accumulation in smooth muscle cells, and iron chelators can prevent vascular smooth muscle proliferation.42,98 A study showed that the anti-oxidant reagent Vit E prevents atherosclerosis in vivo, the function of which might be attributed to abolishing ferroptosis by lowering the oxidative modification of LDL.42,99 Additionally, cigarette smoke extract (CSE) caused the death of VSMCs characterized by lipid peroxidation and intracellular GSH consumption. These data suggest that ferroptosis is probably the shaping factor for losing VSMCs, as evidenced by the protective effects of several ferroptosis inhibitors.15
Lipid Accumulation
Dyslipidemia is intimately associated with AS formation and advancement. A previous study illustrated that ApoE-/- mice fed an HFD displayed more serious plaque lesions and serum lipid levels concomitant with a higher level of ferroptosis.87 Bai et al11 found that inhibiting ferroptosis slightly reduced total cholesterol (TC) levels and enhanced the HDL-C content in AS mice. In addition, the serum LDL-C and triglyceride (TG) remarkably declined. The effect of Fer-1 on serum lipids is concordant with the clinical drug for hyperlipidemia. Apart from these changes, it alleviated lipid peroxidation in mice. The above evidence demonstrates that ferroptosis may be a precursor for lipid disturbance.
Inflammation
Atherosclerosis is viewed as a ubiquitous chronic inflammatory disease. Undoubtedly, inflammation plays a crucial part in the pathogenesis and development of AS.100 Refreshingly, literature concerning the link between inflammation and ferroptosis is available. Research has revealed the pro-inflammatory role of ferroptosis and the eradication of ferroptosis in mitigating inflammation.101 Damage-associated molecular pattern molecules (DAMPs) are obvious prerequisites for innate and inflammatory responses. In Wen’s102 study, DAMPs were conspicuously released by classic activators of ferroptosis, like erastin, in an autophagy-dependent manner. Mechanistically, the autophagy-directed histone deacetylase (HDAC) abolishment aggravated high-mobility group box-1 (HMGB1), a DAMP acetylation, and gave rise to HMGB release during ferroptosis. In light of the vital role of ROS in ferroptosis, the crosstalk between ROS and inflammation indicates that the major ROS product lipoxygenases promoted the recruitment of inflammatory cells such as macrophages and the release of pro-inflammatory molecules, which was pivotal for atherogenesis.103,104 Additionally, ferroptosis-oxidized lipids were illustrated to drive pro-inflammatory responses and were of great importance in advancing AS.105 Liu et al103 found that cadmium telluride quantum dots (CdTe QDs), yielded by the waste from our environment, were able to trigger ferroptosis in macrophages and mice via the negative modulation of Nrf2 and positive regulation of extracellular signal-regulated kinase (ERK), which profoundly contributed to both systemic and local inflammatory responses. More strikingly, ferroptosis was implicated in the pro-inflammatory response, as evidenced by the increased release of IL-1β and IL-18 within foam cells in AS.95 In contrast, the levels of IL-1β, IL-6 and TNF-α were subdued by the administration of Fer-1 in VSMCs exposed to CSE.15
Promising Therapeutics for AS with Desirable Clinical Value
The explosion of interest in ferroptosis in recent decades has enabled extensive investigations concerning its inhibition in the improvement and alleviation of AS in both in vivo and in vitro experiments. Due to the sophisticated mechanisms and mystery behind this form of cell death, mounting numbers of antagonists have been revealed. At the broadest level, inhibitors targeting the phenomenon are roughly divided into five categories. First, the canonical inhibitors of crucial molecules mediating ferroptosis, known as Fer-1, administered by HFD to ApoE-/- mice increased the expression of SLC7A11 and GXP4 and ultimately undermined ferroptosis.11 The role of foam cells in the development of AS is self-evident, and Fer-1 alleviated the inflammation and ferroptosis-decreased viability in foam cells.95 Second, iron chelators such as deferoxamine DFO and CPX can clear the iron overload to control ferroptosis.11,32 Third, antioxidative reagents such as Vit E (alpha-tocopherol) decreased lipid accumulation and ferroptosis in a striatal cell model by suppressing the activity of 15-lipoxygenase.99 Fer-1 serves as a sort of antioxidant for overwhelming lipid peroxidation.14 Mitochondria-oriented antioxidant MitoTEMPO also subverted lipid peroxidation and ferroptosis in heart tissue, ameliorating the DOX-triggered cardiomyopathy.63 Last, the lipid peroxidation antagonist liproxstatin-1 impedes the generation of lipid peroxyl radicals, efficiently reversing ferroptosis and even IL-1β and IL-18 liberation.95,106 Further, the genetic upregulation or downregulation of targeted genes is also plausible and accessible.35,107 Although the promotion of these anti-ferroptosis drugs is small, the application of these drugs in targeting various points in cell or animal models offers much light for the clinical treatment of AS.
Conclusion and Outlook
Our review has described the essential role of ferroptosis in the generation and progression of AS and certain possible mechanisms, which provides excellent support for the exploration of ferroptosis-based therapeutics. At the same time, some problems remain to be solved. First, the specific underlying mechanisms directing the players in this process and exerting the deleterious repercussions for the heart and vasculature are unclear. We have no idea in which segment it induces the abnormality of the circulation system, which makes precision in novel drug discovery difficult. Second, ferroptosis inhibitors are more applied in research performed in mice instead of clinical trials, bringing the uncertainty of the benefits of ferroptosis inhibition in people. Third, several studies on the regulation of some key molecules for this type of cell death, called the dual effect, are controversial. For this reason, we cannot predict whether the positive outcomes override negative ones in all AS conditions. In addition, other outstanding regulators for ferroptosis remain unclear. Last, despite the crucial role of miRNA in governing cell death, few regulating networks targeting it have been explored. Concerning the unsolved questions and meaningful value of ferroptosis for ameliorating AS, a more in-depth exploration of the interplay of ferroptosis and oxidation imbalance on the pathogenesis and advancement of AS is required. From what we discussed above, it is reasonable to deduce that the amelioration of ferroptosis can be an effective target for treating AS patients. In short, given the intricate execution in a myriad of CVDs, although several preclinical studies suggest ferroptosis to be a potential drug target, the underlying molecular signaling pathways and networks in cells associated with the circulation system remain to be well understood. Prior to clinical promotion, it is wise to use ferroptosis-targeting treatments in specific disease circumstances to comprehensively harness their potential benefits to patients.
Data Sharing Statement
Data sharing does not apply to this article, as no datasets were generated or analyzed during the study.
Acknowledgments
Xueqi Wan, Huan Zhang, and Jinfan Tian are co-first authors for this study.
Author Contributions
All authors made a significant contribution to the work reported. XW, HZ, JT drafted and written the article, PH, LL, YZ, and JZ substantially revised the article and XS, CG critically reviewed the article. All authors gave final approval of the version to be published; agreed on the journal to which the article has been submitted; and agreed to be accountable for the contents of the article.
Funding
This work was supported by the Beijing Natural Science Foundation of China (NO. 7202039 and 7232043), Beijing Municipal Science and Technology Project (Z161100000516139), National Nature Science Foundation (82270341 and 82100486), “qingmiao” plan (QML20210603), and “Deng Feng” Talent Training Program (DFL20220603).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Chen Z, Yan Y, Qi C, Liu J, Li L, Wang J. The role of ferroptosis in cardiovascular disease and its therapeutic significance. Front Cardiovasc Med. 2021;8:733229. doi:10.3389/fcvm.2021.733229
2. Fang M, Li Y, Wu Y, Ning Z, Wang X, Li X. miR-185 silencing promotes the progression of atherosclerosis via targeting stromal interaction molecule 1. Cell Cycle. 2019;18(6–7):682–695. doi:10.1080/15384101.2019.1580493
3. Chen X, Li X, Xu X, et al. Ferroptosis and cardiovascular disease: role of free radical-induced lipid peroxidation. Free Radic Res. 2021;55(4):405–415. doi:10.1080/10715762.2021.1876856
4. Zhou Y, Zhou H, Hua L, et al. Verification of ferroptosis and pyroptosis and identification of PTGS2 as the hub gene in human coronary artery atherosclerosis. Free Radic Biol Med. 2021;171:55–68. doi:10.1016/j.freeradbiomed.2021.05.009
5. Durham AL, Speer MY, Scatena M, Giachelli CM, Shanahan CM. Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc Res. 2018;114(4):590–600. doi:10.1093/cvr/cvy010
6. Xiao Q, Che X, Cai B, et al. Macrophage autophagy regulates mitochondria-mediated apoptosis and inhibits necrotic core formation in vulnerable plaques. J Cell Mol Med. 2020;24(1):260–275. doi:10.1111/jcmm.14715
7. Wang F, Zhang Z, Fang A, et al. Macrophage foam cell-targeting immunization attenuates atherosclerosis. Front Immunol. 2018;9:3127. doi:10.3389/fimmu.2018.03127
8. Bi X, Du C, Wang X, et al. Mitochondrial damage-induced innate immune activation in vascular smooth muscle cells promotes chronic kidney disease-associated plaque vulnerability. Adv Sci. 2021;8(5):2002738. doi:10.1002/advs.202002738
9. Wang J, Uryga AK, Reinhold J, et al. Vascular smooth muscle cell senescence promotes atherosclerosis and features of plaque vulnerability. Circulation. 2015;132(20):1909–1919. doi:10.1161/circulationaha.115.016457
10. Liang SJ, Zeng DY, Mai XY, et al. Inhibition of orai1 store-operated calcium channel prevents foam cell formation and atherosclerosis. Arterioscler Thromb Vasc Biol. 2016;36(4):618–628. doi:10.1161/atvbaha.116.307344
11. Bai T, Li M, Liu Y, Qiao Z, Wang Z. Inhibition of ferroptosis alleviates atherosclerosis through attenuating lipid peroxidation and endothelial dysfunction in mouse aortic endothelial cell. Free Radic Biol Med. 2020;160:92–102. doi:10.1016/j.freeradbiomed.2020.07.026
12. Li W, Sultana N, Yuan L, Forssell C, Yuan XM. CD74 in apoptotic macrophages is associated with inflammation, plaque progression and clinical manifestations in human atherosclerotic lesions. Metabolites. 2022;12(1). doi:10.3390/metabo12010054
13. Hu H, Chen Y, Jing L, Zhai C, Shen L. The link between ferroptosis and cardiovascular diseases: a novel target for treatment. Front Cardiovasc Med. 2021;8:710963. doi:10.3389/fcvm.2021.710963
14. Wu X, Li Y, Zhang S, Zhou X. Ferroptosis as a novel therapeutic target for cardiovascular disease. Theranostics. 2021;11(7):3052–3059. doi:10.7150/thno.54113
15. Sampilvanjil A, Karasawa T, Yamada N, et al. Cigarette smoke extract induces ferroptosis in vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2020;318(3):H508–H518. doi:10.1152/ajpheart.00559.2019
16. Xiao FJ, Zhang D, Wu Y, et al. miRNA-17-92 protects endothelial cells from erastin-induced ferroptosis through targeting the A20-ACSL4 axis. Biochem Biophys Res Commun. 2019;515(3):448–454. doi:10.1016/j.bbrc.2019.05.147
17. Peng Q, Liu H, Luo Z, Zhao H, Wang X, Guan X. Effect of autophagy on ferroptosis in foam cells via Nrf2. Mol Cell Biochem. 2022;477(5):1597–1606. doi:10.1007/s11010-021-04347-3
18. Le Y, Zhang Z, Wang C, Lu D. Ferroptotic cell death: new regulatory mechanisms for metabolic diseases. Endocr Metab Immune Disord Drug Targets. 2021;21(5):785–800. doi:10.2174/1871530320666200731175328
19. Koren O, Spor A, Felin J, et al. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc Natl Acad Sci U S A. 2011;108 Suppl 1(Suppl 1):4592–4598. doi:10.1073/pnas.1011383107
20. Jie Z, Xia H, Zhong SL, et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat Commun. 2017;8(1):845. doi:10.1038/s41467-017-00900-1
21. Zhou W, Cheng Y, Zhu P, Nasser MI, Zhang X, Zhao M. Implication of gut microbiota in cardiovascular diseases. Oxid Med Cell Longev. 2020;2020:5394096. doi:10.1155/2020/5394096
22. Zou HX, Qiu BQ, Lai SQ, et al. Iron metabolism and idiopathic pulmonary arterial hypertension: new insights from bioinformatic analysis. Biomed Res Int. 2021;2021:5669412. doi:10.1155/2021/5669412
23. Chen G, Li L, Tao H. Bioinformatics identification of ferroptosis-related biomarkers and therapeutic compounds in ischemic stroke. Front Neurol. 2021;12:745240. doi:10.3389/fneur.2021.745240
24. Meng Z, Liang H, Zhao J, et al. HMOX1 upregulation promotes ferroptosis in diabetic atherosclerosis. Life Sci. 2021;284:119935. doi:10.1016/j.lfs.2021.119935
25. Vinchi F, Porto G, Simmelbauer A, et al. Atherosclerosis is aggravated by iron overload and ameliorated by dietary and pharmacological iron restriction. Eur Heart J. 2020;41(28):2681–2695. doi:10.1093/eurheartj/ehz112
26. Li N, Jiang W, Wang W, Xiong R, Wu X, Geng Q. Ferroptosis and its emerging roles in cardiovascular diseases. Pharmacol Res. 2021;166:105466. doi:10.1016/j.phrs.2021.105466
27. Mao H, Zhao Y, Li H, Lei L. Ferroptosis as an emerging target in inflammatory diseases. Prog Biophys Mol Biol. 2020;155:20–28. doi:10.1016/j.pbiomolbio.2020.04.001
28. Li JY, Liu SQ, Yao RQ, Tian YP, Yao YM. A novel insight into the fate of cardiomyocytes in ischemia-reperfusion injury: from iron metabolism to ferroptosis. Front Cell Dev Biol. 2021;9:799499. doi:10.3389/fcell.2021.799499
29. Kobayashi M, Suhara T, Baba Y, Kawasaki NK, Higa JK, Matsui T. Pathological roles of iron in cardiovascular disease. Curr Drug Targets. 2018;19(9):1068–1076. doi:10.2174/1389450119666180605112235
30. Sun X, Ou Z, Xie M, et al. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene. 2015;34(45):5617–5625. doi:10.1038/onc.2015.32
31. Ouyang S, You J, Zhi C, et al. Ferroptosis: the potential value target in atherosclerosis. Cell Death Dis. 2021;12(8):782. doi:10.1038/s41419-021-04054-3
32. Stockwell BR, Friedmann Angeli JP, Bayir H, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273–285. doi:10.1016/j.cell.2017.09.021
33. Bochkov VN, Oskolkova OV, Birukov KG, Levonen AL, Binder CJ, Stöckl J. Generation and biological activities of oxidized phospholipids. Antioxid Redox Signal. 2010;12(8):1009–1059. doi:10.1089/ars.2009.2597
34. Kagan VE, Mao G, Qu F, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13(1):81–90. doi:10.1038/nchembio.2238
35. Huang F, Yang R, Xiao Z, et al. Targeting ferroptosis to treat cardiovascular diseases: a new continent to be explored. Front Cell Dev Biol. 2021;9:737971. doi:10.3389/fcell.2021.737971
36. Shimada K, Skouta R, Kaplan A, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol. 2016;12(7):497–503. doi:10.1038/nchembio.2079
37. Chang LC, Chiang SK, Chen SE, Yu YL, Chou RH, Chang WC. Heme oxygenase-1 mediates BAY 11-7085 induced ferroptosis. Cancer Lett. 2018;416:124–137. doi:10.1016/j.canlet.2017.12.025
38. Yang K, Song H, Yin D. PDSS2 Inhibits the ferroptosis of vascular endothelial cells in atherosclerosis by activating Nrf2. J Cardiovasc Pharmacol. 2021;77(6):767–776. doi:10.1097/fjc.0000000000001030
39. He L, Liu YY, Wang K, et al. Tanshinone IIA protects human coronary artery endothelial cells from ferroptosis by activating the NRF2 pathway. Biochem Biophys Res Commun. 2021;575:1–7. doi:10.1016/j.bbrc.2021.08.067
40. Shaw P, Chattopadhyay A. Nrf2-ARE signaling in cellular protection: mechanism of action and the regulatory mechanisms. J Cell Physiol. 2020;235(4):3119–3130. doi:10.1002/jcp.29219
41. Sun X, Ou Z, Chen R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63(1):173–184. doi:10.1002/hep.28251
42. Yu Y, Yan Y, Niu F, et al. Ferroptosis: a cell death connecting oxidative stress, inflammation and cardiovascular diseases. Cell Death Discov. 2021;7(1):193. doi:10.1038/s41420-021-00579-w
43. Bogdan AR, Miyazawa M, Hashimoto K, Tsuji Y. Regulators of iron homeostasis: new players in metabolism, cell death, and disease. Trends Biochem Sci. 2016;41(3):274–286. doi:10.1016/j.tibs.2015.11.012
44. Ravingerová T, Kindernay L, Barteková M, et al. The molecular mechanisms of iron metabolism and its role in cardiac dysfunction and cardioprotection. Int J Mol Sci. 2020;21(21):7889. doi:10.3390/ijms21217889
45. Piskin E, Cianciosi D, Gulec S, Tomas M, Capanoglu E. Iron absorption: factors, limitations, and improvement methods. ACS omega. 2022;7(24):20441–20456. doi:10.1021/acsomega.2c01833
46. MacKenzie EL, Iwasaki K, Tsuji Y. Intracellular iron transport and storage: from molecular mechanisms to health implications. Antioxid Redox Signal. 2008;10(6):997–1030. doi:10.1089/ars.2007.1893
47. Woloshun RR, Yu Y, Xu X, et al. Four AAs increase DMT1 abundance in duodenal brush-border membrane vesicles and enhance iron absorption in iron-deprived mice. Blood Adv. 2022;6(10):3011–3021. doi:10.1182/bloodadvances.2021005111
48. Zhang Y, Xin L, Xiang M, et al. The molecular mechanisms of ferroptosis and its role in cardiovascular disease. Biomed Pharmacother. 2022;145:112423. doi:10.1016/j.biopha.2021.112423
49. Guo JM, Liu AJ, Zang P, et al. ALDH2 protects against stroke by clearing 4-HNE. Cell Res. 2013;23(7):915–930. doi:10.1038/cr.2013.69
50. Balusikova K, Dostalikova-Cimburova M, Tacheci I, Kovar J. Expression profiles of iron transport molecules along the duodenum. J Cell Mol Med. 2022;26(10):2995–3004. doi:10.1111/jcmm.17313
51. Hou W, Xie Y, Song X, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12(8):1425–1428. doi:10.1080/15548627.2016.1187366
52. Paffetti P, Perrone S, Longini M, et al. Non-protein-bound iron detection in small samples of biological fluids and tissues. Biol Trace Elem Res. 2006;112(3):221–232. doi:10.1385/bter:112:3:221
53. Chiang SK, Chen SE, Chang LC. A dual role of heme oxygenase-1 in cancer cells. Int J Mol Sci. 2018;20(1):39. doi:10.3390/ijms20010039
54. Song Y, Wang B, Zhu X, et al. Human umbilical cord blood-derived MSCs exosome attenuate myocardial injury by inhibiting ferroptosis in acute myocardial infarction mice. Cell Biol Toxicol. 2021;37(1):51–64. doi:10.1007/s10565-020-09530-8
55. Camaschella C, Nai A, Silvestri L. Iron metabolism and iron disorders revisited in the hepcidin era. Haematologica. 2020;105(2):260–272. doi:10.3324/haematol.2019.232124
56. Valko M, Jomova K, Rhodes CJ, Kuča K, Musílek K. Redox- and non-redox-metal-induced formation of free radicals and their role in human disease. Arch Toxicol. 2016;90(1):1–37. doi:10.1007/s00204-015-1579-5
57. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59(2):298–308. doi:10.1016/j.molcel.2015.06.011
58. Kuang F, Liu J, Tang D, Kang R. Oxidative damage and antioxidant defense in ferroptosis. Front Cell Dev Biol. 2020;8:586578. doi:10.3389/fcell.2020.586578
59. Saha S, Profumo E, Togna AR, Riganò R, Saso L, Buttari B. Lupeol counteracts the proinflammatory signalling triggered in macrophages by 7-keto-cholesterol: new perspectives in the therapy of atherosclerosis. Oxid Med Cell Longev. 2020;2020:1232816. doi:10.1155/2020/1232816
60. Sun J, Hartvigsen K, Chou MY, et al. Deficiency of antigen-presenting cell invariant chain reduces atherosclerosis in mice. Circulation. 2010;122(8):808–820. doi:10.1161/circulationaha.109.891887
61. Yuan H, Li X, Zhang X, Kang R, Tang D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun. 2016;478(3):1338–1343. doi:10.1016/j.bbrc.2016.08.124
62. Pan C, Xing JH, Zhang C, et al. Aldehyde dehydrogenase 2 inhibits inflammatory response and regulates atherosclerotic plaque. Oncotarget. 2016;7(24):35562–35576. doi:10.18632/oncotarget.9384
63. Fang X, Wang H, Han D, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A. 2019;116(7):2672–2680. doi:10.1073/pnas.1821022116
64. Koleini N, Nickel BE, Edel AL, Fandrich RR, Ravandi A, Kardami E. Oxidized phospholipids in Doxorubicin-induced cardiotoxicity. Chem Biol Interact. 2019;303:35–39. doi:10.1016/j.cbi.2019.01.032
65. Singh A, Rangasamy T, Thimmulappa RK, et al. Glutathione peroxidase 2, the major cigarette smoke-inducible isoform of GPX in lungs, is regulated by Nrf2. Am J Respir Cell Mol Biol. 2006;35(6):639–650. doi:10.1165/rcmb.2005-0325OC
66. Qiu Y, Cao Y, Cao W, Jia Y, Lu N. The application of ferroptosis in diseases. Pharmacol Res. 2020;159:104919. doi:10.1016/j.phrs.2020.104919
67. Lu H, Xiao H, Dai M, Xue Y, Zhao R. Britanin relieves ferroptosis-mediated myocardial ischaemia/reperfusion damage by upregulating GPX4 through activation of AMPK/GSK3β/Nrf2 signalling. Pharm Biol. 2022;60(1):38–45. doi:10.1080/13880209.2021.2007269
68. Konstorum A, Tesfay L, Paul BT, Torti FM, Laubenbacher RC, Torti SV. Systems biology of ferroptosis: a modeling approach. J Theor Biol. 2020;493:110222. doi:10.1016/j.jtbi.2020.110222
69. Kovac S, Angelova PR, Holmström KM, Zhang Y, Dinkova-Kostova AT, Abramov AY. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim Biophys Acta. 2015;1850(4):794–801. doi:10.1016/j.bbagen.2014.11.021
70. Ma S, He L, Zuo Q, Zhang G, Guo Y. Canagliflozin regulates ferroptosis, potentially via activating AMPK/PGC-1α/Nrf2 signaling in HFpEF Rats. Cardiovasc Innov Appl. 2023;7(1). doi:10.15212/cvia.2022.0024
71. Wang X, Chen X, Zhou W, et al. Ferroptosis is essential for diabetic cardiomyopathy and is prevented by sulforaphane via AMPK/NRF2 pathways. Acta Pharm Sin B. 2022;12(2):708–722. doi:10.1016/j.apsb.2021.10.005
72. Yu J, Zhou L, Song H, et al. (−)-Epicatechin gallate blocked cellular foam formation in atherosclerosis by modulating CD36 expression in vitro and in vivo. Food Funct. 2023;14(5):2444–2458. doi:10.1039/d2fo03218j
73. Fang K, Du S, Shen D, et al. SUFU suppresses ferroptosis sensitivity in breast cancer cells via Hippo/YAP pathway. iScience. 2022;25(7):104618. doi:10.1016/j.isci.2022.104618
74. Park S, Oh J, Kim M, Jin EJ. Bromelain effectively suppresses Kras-mutant colorectal cancer by stimulating ferroptosis. Anim Cells Syst. 2018;22(5):334–340. doi:10.1080/19768354.2018.1512521
75. Lei G, Horbath A, Li Z, Gan B. PKCβII-ACSL4 pathway mediating ferroptosis execution and anti-tumor immunity. Cancer Commun. 2022;42(7):583–586. doi:10.1002/cac2.12319
76. Loboda A, Damulewicz M, Pyza E, Jozkowicz A, Dulak J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell Mol Life Sci. 2016;73(17):3221–3247. doi:10.1007/s00018-016-2223-0
77. Zhang X, Yu Y, Lei H, et al. The Nrf-2/HO-1 signaling axis: a ray of hope in cardiovascular diseases. Cardiol Res Pract. 2020;2020:5695723. doi:10.1155/2020/5695723
78. Vítek L, Schwertner HA. The heme catabolic pathway and its protective effects on oxidative stress-mediated diseases. Adv Clin Chem. 2007;43:1–57. doi:10.1016/s0065-2423(06)43001-8
79. Menon AV, Liu J, Tsai HP, et al. Excess heme upregulates heme oxygenase 1 and promotes cardiac ferroptosis in mice with sickle cell disease. Blood. 2022;139(6):936–941. doi:10.1182/blood.2020008455
80. Adedoyin O, Boddu R, Traylor A, et al. Heme oxygenase-1 mitigates ferroptosis in renal proximal tubule cells. Am J Physiol Renal Physiol. 2018;314(5):F702–F714. doi:10.1152/ajprenal.00044.2017
81. Ryter SW. Heme oxygenase-1, a cardinal modulator of regulated cell death and inflammation. Cells. 2021;10(3):515. doi:10.3390/cells10030515
82. Dai E, Zhang W, Cong D, Kang R, Wang J, Tang D. AIFM2 blocks ferroptosis independent of ubiquinol metabolism. Biochem Biophys Res Commun. 2020;523(4):966–971. doi:10.1016/j.bbrc.2020.01.066
83. Tulenko TN, Laury-Kleintop L, Walter MF, Mason RP. Cholesterol, calcium and atherosclerosis: is there a role for calcium channel blockers in atheroprotection? Int J Cardiol. 1997;62 Suppl 2:S55–S66. doi:10.1016/s0167-5273(97)00242-8
84. Mury P, Chirico EN, Mura M, Millon A, Canet-Soulas E, Pialoux V. Oxidative stress and inflammation, key targets of atherosclerotic plaque progression and vulnerability: potential impact of physical activity. Sports Med. 2018;48(12):2725–2741. doi:10.1007/s40279-018-0996-z
85. Hong L, Xie ZZ, Du YH, et al. Alteration of volume-regulated chloride channel during macrophage-derived foam cell formation in atherosclerosis. Atherosclerosis. 2011;216(1):59–66. doi:10.1016/j.atherosclerosis.2011.01.035
86. Rudijanto A. The role of vascular smooth muscle cells on the pathogenesis of atherosclerosis. Acta Med Indones. 2007;39(2):86–93.
87. Wang Y, Kuang X, Yin Y, et al. Tongxinluo prevents chronic obstructive pulmonary disease complicated with atherosclerosis by inhibiting ferroptosis and protecting against pulmonary microvascular barrier dysfunction. Biomed Pharmacother. 2022;145:112367. doi:10.1016/j.biopha.2021.112367
88. Li L, Wang H, Zhang J, Chen X, Zhang Z, Li Q. Effect of endothelial progenitor cell-derived extracellular vesicles on endothelial cell ferroptosis and atherosclerotic vascular endothelial injury. Cell Death Discov. 2021;7(1):235. doi:10.1038/s41420-021-00610-0
89. Komarova YA, Kruse K, Mehta D, Malik AB. Protein interactions at endothelial junctions and signaling mechanisms regulating endothelial permeability. Circ Res. 2017;120(1):179–206. doi:10.1161/circresaha.116.306534
90. Quan Y, Xin Y, Tian G, Zhou J, Liu X. Mitochondrial ROS-modulated mtDNA: a potential target for cardiac aging. Oxid Med Cell Longev. 2020;2020:9423593. doi:10.1155/2020/9423593
91. Hua Z, Ma K, Liu S, Yue Y, Cao H, Li Z. LncRNA ZEB1-AS1 facilitates ox-LDL-induced damage of HCtAEC cells and the oxidative stress and inflammatory events of THP-1 cells via miR-942/HMGB1 signaling. Life Sci. 2020;247:117334. doi:10.1016/j.lfs.2020.117334
92. Kattoor AJ, Pothineni NVK, Palagiri D, Mehta JL. Oxidative stress in atherosclerosis. Curr Atheroscler Rep. 2017;19(11):42. doi:10.1007/s11883-017-0678-6
93. Gimbrone MA Jr, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. 2016;118(4):620–636. doi:10.1161/circresaha.115.306301
94. Zhong S, Li L, Zhang YL, et al. Acetaldehyde dehydrogenase 2 interactions with LDLR and AMPK regulate foam cell formation. J Clin Invest. 2019;129(1):252–267. doi:10.1172/jci122064
95. Su G, Yang W, Wang S, Geng C, Guan X. SIRT1-autophagy axis inhibits excess iron-induced ferroptosis of foam cells and subsequently increases IL-1Β and IL-18. Biochem Biophys Res Commun. 2021;561:33–39. doi:10.1016/j.bbrc.2021.05.011
96. Zheng M, Li L, Liu Y, Liang Y, Qi X. Silencing ferritin alleviates atherosclerosis in mice via regulating the expression levels of matrix metalloproteinases and interleukins. Acta Biochim Pol. 2021;68(4):705–710. doi:10.18388/abp.2020_5605
97. Potor L, Hendrik Z, Patsalos A, et al. Oxidation of hemoglobin drives a proatherogenic polarization of macrophages in human atherosclerosis. Antioxid Redox Signal. 2021;35(12):917–950. doi:10.1089/ars.2020.8234
98. Shah SV, Alam MG. Role of iron in atherosclerosis. Am J Kidney Dis. 2003;41(3 Suppl 1):S80–S83. doi:10.1053/ajkd.2003.50091
99. Hinman A, Holst CR, Latham JC, et al. Vitamin E hydroquinone is an endogenous regulator of ferroptosis via redox control of 15-lipoxygenase. PLoS One. 2018;13(8):e0201369. doi:10.1371/journal.pone.0201369
100. Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12(3):204–212. doi:10.1038/ni.2001
101. Sun Y, Chen P, Zhai B, et al. The emerging role of ferroptosis in inflammation. Biomed Pharmacother. 2020;127:110108. doi:10.1016/j.biopha.2020.110108
102. Wen Q, Liu J, Kang R, Zhou B, Tang D. The release and activity of HMGB1 in ferroptosis. Biochem Biophys Res Commun. 2019;510(2):278–283. doi:10.1016/j.bbrc.2019.01.090
103. Liu N, Liang Y, Wei T, et al. The role of ferroptosis mediated by NRF2/ERK-regulated ferritinophagy in CdTe QDs-induced inflammation in macrophage. J Hazard Mater. 2022;436:129043. doi:10.1016/j.jhazmat.2022.129043
104. Bäck M, Hansson GK. Leukotriene receptors in atherosclerosis. Ann Med. 2006;38(7):493–502. doi:10.1080/07853890600982737
105. Hammad SM, Twal WO, Barth JL, et al. Oxidized LDL immune complexes and oxidized LDL differentially affect the expression of genes involved with inflammation and survival in human U937 monocytic cells. Atherosclerosis. 2009;202(2):394–404. doi:10.1016/j.atherosclerosis.2008.05.032
106. Zilka O, Shah R, Li B, et al. On the mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent Sci. 2017;3(3):232–243. doi:10.1021/acscentsci.7b00028
107. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi:10.1016/j.cell.2012.03.042
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Shikonin Could Be Used to Treat Tubal Pregnancy via Enhancing Ferroptosis Sensitivity
Lai Y, Zeng F, Chen Z, Feng M, Huang Y, Qiu P, Zeng L, Ke Y, Deng G, Gao J
Drug Design, Development and Therapy 2022, 16:2083-2099
Published Date: 1 July 2022
Research Progress on Relationship Between Iron Overload and Lower Limb Arterial Disease in Type 2 Diabetes Mellitus
Wang Z, Fang S, Ding S, Tan Q, Zhang X
Diabetes, Metabolic Syndrome and Obesity 2022, 15:2259-2264
Published Date: 30 July 2022
The Emerging Role of Ferroptosis in Sepsis, Opportunity or Challenge?
Huang Q, Ding Y, Fang C, Wang H, Kong L
Infection and Drug Resistance 2023, 16:5551-5562
Published Date: 23 August 2023
Targeting Ferroptosis in Bone-Related Diseases: Facts and Perspectives
Chen H, Han Z, Wang Y, Su J, Lin Y, Cheng X, Liu W, He J, Fan Y, Chen L, Zuo H
Journal of Inflammation Research 2023, 16:4661-4677
Published Date: 18 October 2023
The Regulatory Landscape of Ferroptosis and Iron Homeostasis: Pathophysiological Mechanisms and Therapeutic Horizons in Cardiovascular Disease
Zhang T, Xiao S, Xia Y, Chen K, Liu H, Zhang P, Luo L
Drug Design, Development and Therapy 2026, 20:581236
Published Date: 1 May 2026