Back to Journals » Journal of Inflammation Research » Volume 16
The Emerging Roles of Ferroptosis in Neonatal Diseases
Authors Chen W ![]() , Zheng D
, Zheng D ![]() , Yang C
, Yang C ![]()
Received 26 March 2023
Accepted for publication 13 June 2023
Published 26 June 2023 Volume 2023:16 Pages 2661—2674
DOI https://doi.org/10.2147/JIR.S414316
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tara Strutt
Wenqian Chen,1 Dali Zheng,2 Changyi Yang1
1Department of Neonatology, Fujian Maternity and Child Health Hospital, Affiliated Hospital of Fujian Medical University, Fuzhou, People’s Republic of China; 2Key Laboratory of Stomatology of Fujian Province, School and Hospital of Stomatology, Fujian Medical University, Fuzhou, People’s Republic of China
Correspondence: Changyi Yang, Department of Neonatology, Fujian Maternity and Child Health Hospital, Affiliated Hospital of Fujian Medical University, Fuzhou, 350001, People’s Republic of China, Tel +86 15959000599, Fax +86 87279676, Email [email protected]
Abstract: Ferroptosis is a novel type of programmed cell death involved in many diseases’ pathological processes. Ferroptosis is characterized by lipid peroxidation, reactive oxygen species accumulation, and iron metabolism disorder. Newborns are susceptible to ferroptosis due to their special physiological state, which is prone to abnormal iron metabolism and the accumulation of reactive oxygen species. Recent studies have linked ferroptosis to a variety of diseases in the neonatal period (including hypoxic-ischemic encephalopathy, bronchopulmonary dysplasia, and necrotizing enterocolitis). Ferroptosis may become an effective target for the treatment of neonatal-related diseases. In this review, the ferroptosis molecular mechanism, metabolism characteristics of iron and reactive oxygen species in infants, the relationship between ferroptosis and common infant disorders, and the treatment of infant diseases targeted for ferroptosis are systematically summarized.
Keywords: ferroptosis, neonatal disease, reactive oxygen species, iron metabolism, lipid peroxidation
Introduction
The concept of ferroptosis was first proposed by Dr. Brent R Stockwell in 2012, which is a programmed cell death mode caused by iron-dependent lipid peroxidation and reactive oxygen species accumulation.1 Unlike other forms of cell death discovered so far, it has distinct characteristics regarding cytogenetics, biochemistry, metabolism, and morphology. A wave-like spreading pattern can be seen in its spread through cellular populations.2 During ferroptosis, lipid peroxides (eg, malondialdehyde (MDA) and 4-hydroxynonoleic acid (4-HNE)) increase, ferroptosis-related gene changes (eg, SLC7A11, ACSL4, TFR1 up-regulated, RGS4 down-regulated) and specific changes in cell morphology (decrease in mitochondrial volume, increase in mitochondrial membrane density, decrease or disappearance of mitochondrial cristae, rupture of mitochondrial outer membrane, despite normal nucleus size, without nuclear condensation or chromatin marginalization), which is reversed by ferroptosis inhibitors like ferrostatin-1.3 In various neonatal diseases, ferroptosis plays a significant role in the pathological process and may serve as a potential therapeutic target.4–7 In this article, we will review ferroptosis’ molecular mechanisms and its potential role in neonatal diseases. The relationship between ferroptosis and neonatal hypoxic-ischemic encephalopathy, neonatal bronchopulmonary dysplasia, and neonatal necrotizing enterocolitis was emphatically introduced.
Molecular Mechanism of Ferroptosis
Ferroptosis is a form of programmed cell death caused by iron-dependent lipid peroxidation. Lipid peroxidation, reactive oxygen species (ROS) accumulation, and iron metabolism imbalance are the basic and central link in the molecular mechanism of ferroptosis3. (Figure 1, which modified from Zhang, X.2023)8.
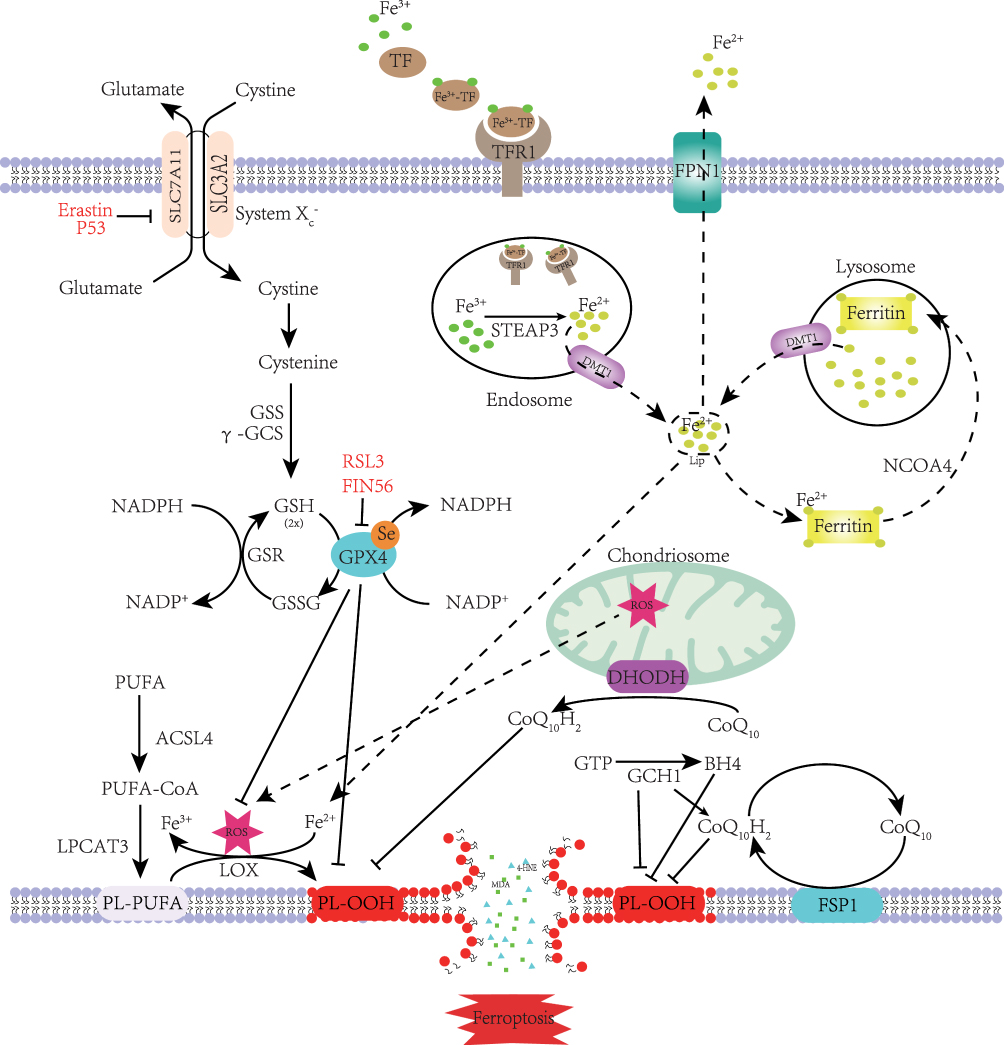 |
Figure 1 Mechanism of ferroptosis. PUFA became a part of the cell membrane under the action of ACSL4 and LPCAT3.Through the Fenton reaction and enzymatic reaction, PL-PUFA on the cell membrane undergoes a lipid peroxidation reaction to form PL-OOH. Due to the consumption of PL-PUFA and the toxic effect of peroxidation products, the cell membrane ruptures, eventually causing ferroptosis. Cystine enters the cytoplasm through System Xc- and rapidly transforms into cysteine to participate in GSH synthesis. GPX4 inhibits lipid peroxidation while reducing GSH to GSSG. Lipid peroxidation is also inhibited by FSP1 and DHODH on the cell membrane, which reduce CoQ10 to CoQ10H2. GCH1 can inhibit lipid peroxidation directly or indirectly. When the TF binds to the TFR on the cell membrane, it is endocytosed into the cell. Under the action of STEAP3 and DMT1, Fe2+ enters the cytoplasm and forms labile iron pools. Most Fe2+ is kept in ferritin. When intracellular iron is deficient, ferritin is transported to the lysosomes by NCOA4 to release Fe2+ through iron autophagy. Excess intracellular iron is excreted out of the cell through FPN1. When intracellular iron metabolism is imbalanced, the Fenton reaction and increased iron-containing enzyme activity cause ferroptosis. Note: Adapted from Zhang X, Ma Y, Lv G, Wang H. Ferroptosis as a therapeutic target for inflammation-related intestinal diseases. Front Pharmacol. 2023;14:1095366.8 |
Iron Metabolism and Ferroptosis
Iron is an essential trace element in the human body, playing a crucial role in hematopoiesis, ATP production, and DNA synthesis.9 Maintaining iron homeostasis is vital, as disruptions in this balance are closely associated with ferroptosis.10 Excessive iron leads to lipid peroxidation and ferroptosis through the Fenton reaction or increased activity of iron-containing enzymes.
In newborns, breast milk and formula are the sole sources of iron.11 Lactoferrin (LF) in breast milk binds to the lactoferrin receptor (LFR) and is absorbed by intestinal epithelial cells through clathrin-mediated endocytosis.12–14 Iron is stored by ferritin (FTN) in the cytoplasm or exported by ferroportin-1 (FPN1).15 Transferrin (TF) transports circulating iron in complex form to various tissues. Moreover, in addition to iron absorption from the digestive tract, macrophages can phagocytize aging red blood cells and degrade Fe2+ in hemoglobin through heme oxygenase-1 (HO-1).16 Overexpression of HO-1, leading to increased iron release, can promote ferroptosis.17
Hepcidin regulates the balance of circulating iron. It is produced by the liver in response to high iron levels in the body. In enterocytes and macrophages, hepcidin binds to FPN1, leading to its degradation.18 This negative regulation of iron levels reduces the uptake of dietary iron and the release of iron from macrophages. Enterocytes, with a short lifespan of 3–4 days, shed the iron that is not transported into circulation.19 The synthesis of hepcidin is influenced by factors such as erythropoietin, inflammation, and iron levels in the body. High iron levels or inflammation increase the release of hepatic hepcidin while decreasing FPN1 expression. Conversely, anemia, hypoxia, and low iron levels result in decreased hepcidin expression, increasing FPN1 activity and iron mobilization.20 Increased hepcidin expression can alleviate ferroptosis in hypoxia-reperfused rat cardiomyocytes.21
Extracellular transferrin binds to the transferrin receptor (TFR) on the cell membrane, undergoes endocytosis, and forms endosomes.22 Within the endosomes, a protein called six-transmembrane epithelial antigen of the prostate 3 (STEAP3) reduces Fe3+ to Fe2+, which is then transported to the cytoplasm by the divalent metal ion transporter 1 (DMT1).23 The TFRs are recycled back to the cell membrane. Intracellular Fe2+ is utilized in enzymatic reactions and heme synthesis. Cellular iron homeostasis is maintained by either pumping excess iron out of the cell membrane through FPN1 or storing it in ferritin. When intracellular Fe2+ levels decrease, ferritin is transported to lysosomes via nuclear receptor coactivator 4 (NCOA4). In the lysosomes, ferritin undergoes degradation through iron autophagy, releasing iron ions that are exported to the cytoplasm via DMT1 on the lysosome.24 This process helps maintain intracellular iron balance. TF and TFR promote ferroptosis by importing iron into cells.25 Conversely, iron export mechanisms can render cells resistant to ferroptosis.26 Iron autophagy also regulates ferroptosis.27,28 For example, specific knockout of NCOA4 can inhibit ferroptosis induced by Erastin, controlled by autophagy-related genes ATG5 and ATG7.29
Iron regulatory proteins 1 and 2 (IRP1 and 2) are crucial for regulating intracellular iron levels.9 They interact with iron-responsive elements (IREs) in genes related to iron metabolism, controlling processes such as absorption, storage, utilization, and output of iron.30,31 When intracellular iron is deficient, IRP1 is detached from the iron-sulfur cluster and binds to IRE located at the 3 “end of the gene, increasing gene transcription of DMT1 and TFR and promoting iron uptake. In addition, IRP1 can bind to the ferritin gene”s IRE at its 5’end and negatively regulate FTN, thereby reducing iron storage. Upon intracellular iron excess, IRP1 rebinds to iron-sulfur clusters, resulting in increased FTN and decreased TFR and DMT1, thereby reducing iron levels. Similar to IRP1 in structure, IRP2 is degraded by ubiquitin upon iron accumulation and modulates gene expression to regulate iron metabolism.16 During ferroptosis, IRP plays a key role. The expression of IRP1 and IRP2 is increased during ferroptosis of melanoma cells, and inhibition of IRP1 can significantly inhibit ferroptosis.32 Overexpression of IRP2 in hepatic stellate cells reduces liver fibrosis through ferroptosis.33
Lipid Peroxidation and Ferroptosis
Lipid peroxidation is a key step in ferroptosis.34 It occurs when lipids have one or more carbon-carbon double bonds. Active hydrogen atoms are located between the methylene bridges (-CH2-) on both sides of the double bond. Free radicals acquire hydrogen atoms from cell membranes through these bridges, leading to cell damage through free radical chain reactions.35 Lipid oxidation results in the accumulation of lipid peroxidation products in cells, altering cell membrane fluidity and permeability, ultimately causing structural and functional damage.36 Phospholipids containing polyunsaturated fatty acids (PUFAs) are particularly susceptible to lipid peroxidation and ferroptosis due to their multiple double bonds. Deuterium isotope of hydrogen, found at the bi-allylic position of PUFAs, inhibits PUFA peroxidation and blocks ferroptosis.37 Increasing the synthesis of monounsaturated fatty acids (MUFAs) or adding palmitoleic acid or oleic acid can also inhibit ferroptosis.38 MUFAs may displace PUFAs in cell membranes, inhibiting lipid peroxidation.39 However, not all PUFAs cause lipid peroxidation.3 ROS attack phosphatidylethanolamines with specific fatty acid groups, making them major targets for lipid peroxidation.8
The lipid peroxidation process that causes ferroptosis can be divided into two steps: phospholipid synthesis with PUFAs as substrates and peroxidation reaction. First, Lysophosphatidylcholine acyltransferase 3 (LPCAT3) integrates PUFA-CoA into cellular membranes after synthesis by acyl-CoA synthetase 4 (ACSL4).40 Phospholipids-polyunsaturated fatty acids (PL-PUFA) will participate in subsequent peroxidation reactions as peroxidation substrates. In the peroxidation step, either a Fenton reaction or enzymatic reaction induces lipid peroxidation.41 The Fenton reaction converts Fe2+ to Fe3+, generating ROS that attack PL-PUFA and cause lipid peroxidation.42 In the lipoxygenase-promoted lipid peroxidation pathway,43 PL-PUFA is oxidized by 15-lipoxygenase (15-LOX), which binds to phosphatidylethanolamine-binding protein 1 (PEP1). PUFA-PE-OOH is then produced, resulting in ferroptosis in the cells.44,45 In the endoplasmic reticulum, NADH-cytochrome B5 reductase (CB5R) converts electrons from nicotinamide adenine dinucleotide phosphate (NADPH) into superoxide (O2-) which is then used to initiate lipid peroxidation directly or indirectly by acquiring methylene hydrogen from PL-PUFA or by reducing ferric iron.46 Since the cells are in an oxidative imbalance, Lipid peroxidation will continue until PL-PUFA is exhausted. As PL-PUFA is an integral part of cell structure, its excessive consumption will eventually result in cell ruptures and perforations. Furthermore, major lipid peroxidation products (PL-OOH, MDA, 4-HNE, etc.) produced during lipid peroxidation do further damage to the cell membrane, resulting in membrane pores and ferroptosis.47
ROS and Ferroptosis
Lipid Peroxidation Induced by ROS
ROS, including superoxides (O2-), peroxides (H2O2), and free radicals (HO- and RO-), are products of O2 gaining electrons.48 These reactive oxygen species play a central role in promoting lipid peroxidation. Increased accumulation of ROS in cells makes them more vulnerable to ferroptosis.
Regulatory Mechanism of ROS
The body clears lipid peroxides caused by ROS through the GSH/GPX4 system and GPX4-independent regulatory mechanisms. Once these regulatory mechanisms are abnormal, intracellular ROS accumulation and lipid peroxidation will occur, leading to ferroptosis.
GSH/GPX4 Inhibits ROS
GPX4, a selenium protein, is the primary enzyme in mammalian cells responsible for reducing PL-OOH, a lipid peroxide.49 It catalyzes the reduced glutathione (GSH) to the conversion of oxidized glutathione (GSSG) and reduces cytotoxic PL-OOH to non-toxic phospholipid alcohol (PL-OH),50 thus inhibiting ferroptosis. However, compounds like RSL3 and FIN56 can directly inhibit or degrade GPX4, promoting ferroptosis.3
Glutathione (GSH), a tripeptide, acts as an intracellular antioxidant and inhibits ferroptosis. It is synthesized from glutamic acid, glycine, and cysteine, with cystine being crucial for GSH biosynthesis.51 Cystine enters cells through the cystine/glutamate antiporter (system Xc-), facilitated by the proteins SLC7A11 and SLC3A2L. Inside the cell, cystine is rapidly reduced and then converted into GSH through enzymatic reactions.52 GSH, under the influence of GPX4, helps to neutralize ROS, reducing lipid peroxidation and preventing ferroptosis.53 Inhibitors like erastin and P53 target SLC7A11, hindering cystine uptake and suppressing GSH synthesis, ultimately leading to lipid peroxidation and ferroptosis.1,54 N-acetylcysteine can counteract ferroptosis by promoting GSH synthesis.55
Regulation of Lipid ROS Independent of GPX4
In addition to GSH/GPX4, three GSH/GPX4-independent signaling pathways have been identified to regulate ferroptosis: the FSP1-CoQ10-NADPH pathway, the DHODH-CoQ10 pathway, and the GCH1-BH4 pathway.3
By using NADPH, ferroptosis suppressor protein 1 (FSP1), which is translocated to the cell membrane, converts ubiquinone (also known as CoQ10) into ubiquinol (CoQ10H2). Antioxidant ubiquinol traps lipid-ROS to form ubiquinone, inhibiting lipid peroxidation in this process and attenuating ferroptosis.56,57 Researchers have found that inhibiting FSP1 can induce ferroptosis in tumor cells.34
A flavin-dependent enzyme, dihydroorotate dehydrogenase (DHODH) is present in the mitochondrial membrane. DHODH can oxidize dihydro whey to whey acid and reduce CoQ10 to CoQ10H2 in the mitochondrial membrane, thus inhibiting lipid peroxidation and ferroptosis.58 Through mTOR signaling, DHODH promotes ferroptosis in cervical cancer cells.59
Tetrahydrobiopterin (BH4), synthesized from GTP catalyzed by GTP cyclohydrolase 1 (GCH1), is a lipophilic antioxidant with a function like CoQ10H2 to prevent lipid peroxidation.60 In addition to catalyzing the synthesis of BH4, GCH1 can also lead to the remodeling of the lipid membrane environment, increase CoQ10H2, and consume PL-PUFA.3 Thus, by overexpressing GCH1 or supplementing with BH4, ferroptosis can be reduced.61
Neonates are Susceptible to Ferroptosis
Newborns’ susceptibility to ferroptosis is due to imbalanced iron metabolism and increased ROS levels. Their delicate physiology and unique metabolism make them prone to this process. As a result, various neonatal diseases are closely linked to ferroptosis. (Figure 2, which modified from Du, X. 2022).62
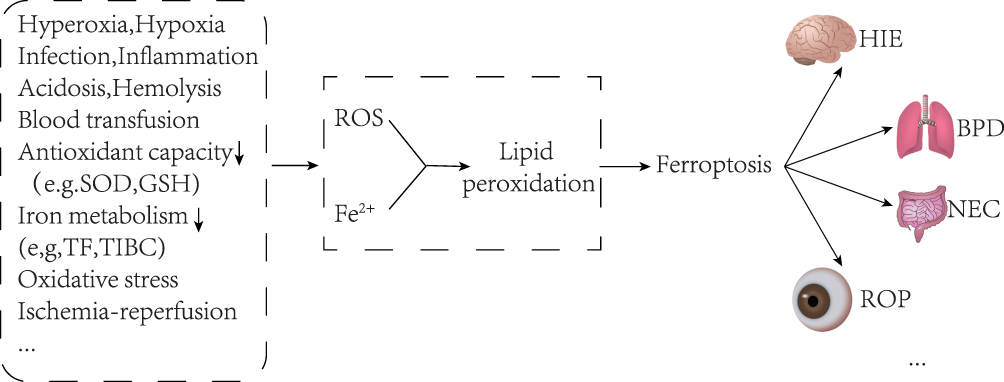 |
Figure 2 Neonatal disease and ferroptosis. Newborns are easily exposed to oxidative stress environments such as hypoxia, hyperoxia, infection, inflammation, acidosis, and blood transfusion after birth, and their antioxidant capacity and iron metabolism capacity are poor, which will lead to increased ROS and free iron, and then cause ferroptosis through lipid peroxidation. A variety of neonatal diseases may be caused by ferroptosis, such as hypoxic-ischemic encephalopathy, bronchopulmonary dysplasia, necrotizing enterocolitis, and retinopathy of prematurity. Note: Adapted from Du X, Dong R, Wu Y, Ni B. Physiological effects of ferroptosis on organ fibrosis. Oxid Med Cell Longev. 2022;2022:5295434.62 |
Newborns are Prone to Increase Free Iron
Newborns are prone to the accumulation of non-transferrin-bound iron (NTBI) due to factors such as perinatal acidosis, recurrent hypoxic ischemia, and immature antioxidant system.63 Red blood cell transfusions in newborns also contribute to NTBI accumulation.64 In addition, newborns are at risk for NTBI through red blood cell transfusions.65 The increased extracellular NTBI can elevate intracellular iron levels,66,67 leading to oxidative stress and lipid peroxidation, ultimately resulting in ferroptosis.
Newborns are Prone to ROS Accumulation
The accumulation of ROS in newborns is due to an imbalance between oxidative stress and antioxidants. High oxidative stress during pregnancy, delivery, and the postnatal period exposes neonates to increased ROS production. Maternal conditions like preeclampsia, chronic hypertension, obesity, and infections contribute to elevated oxidative stress.68 Maternal elevated oxidative stress also impact newborn ROS levels.69 Neonates experience a transition from intrauterine hypoxia to postnatal hyperoxia, leading to ROS generation.70 The body degrades ROS through enzymatic (SOD, CAT, GPX) and non-enzymatic antioxidants (glutathione, tocopherol, etc.).68 Newborns, especially preterm infants, have immature antioxidant systems and imbalanced oxidation.71 Neonatal diseases like BPD, ROP, NEC, and HIE are associated with increased ROS levels.72 Excess ROS triggers ferroptosis via lipid peroxidation.73 Inhibiting the ferroptosis signaling pathway can attenuate tissue damage caused by these diseases.7,74–76
The Role of Ferroptosis in Common Neonatal Diseases
The Role of Ferroptosis in Neonatal HIE
Neonatal hypoxic-ischemic encephalopathy (HIE) is a severe neurological injury caused by hypoxia-ischemia of brain tissue.77 Despite mild hypothermia, half of infants with moderate to severe HIE still die or suffer neurological sequelae.78
Oxidative stress is a key factor in neonatal hypoxic-ischemic encephalopathy (HIE) due to decreased activity of antioxidant enzymes in the immature brain.79 Hypoxia-ischemia and hyperoxia in newborns lead to the production of excessive ROS in the brain, causing lipid peroxidation. Infants with perinatal asphyxia show significantly elevated levels of malondialdehyde (MDA) in cord blood and blood, indicating increased oxidative stress.80 Elevated serum lipid peroxides in neonates can serve as a sensitive diagnostic marker for HIE.81 High concentrations of MDA in the cerebrospinal fluid of neonates with HIE suggest lipid peroxidation in nerve cells.82 Mild hypothermia is the main therapeutic approach for HIE and works by inhibiting ROS production in the nervous system.83 Hypothermia treatment has been shown to reduce MDA levels compared to normothermic controls.84
Due to the Fenton reaction, non-transferrin-bound iron (NTBI) generates free radicals, leading to lipid peroxidation and triggering ferroptosis. NTBI has been detected in significant amounts in the cerebrospinal fluid and serum of neonates after hypoxic-ischemic encephalopathy (HIE).82,85 The concentration of NTBI is directly related to the severity of brain injury and can predict long-term neurodevelopmental outcomes in infants.85 During cerebral ischemia-reperfusion, increased levels of ROS cause the release of iron from binding proteins and red blood cells, exacerbating brain damage.20,86 Iron-induced free radicals disrupt the blood-brain barrier and contribute to endothelial necrosis.87 Intracellular iron accumulation in microglia following hypoxia contributes to the death of oligodendrocytes and axonal swelling.88 Iron released from heme disruption in hypoxic-ischemia-induced intracerebral hemorrhage leads to lipid peroxidation and ferroptosis.89 Treatment options such as deferoxamine, an iron chelator, and erythropoietin, which promotes iron sequestration, have shown beneficial effects in reducing brain injury after HIE.90,91 Heat shock protein B (HSPB1) plays a role in reducing cerebral infarction and brain ferritin levels in HIE by regulating iron metabolism, promoting antioxidant enzyme expression, and alleviating ferroptosis.92
The expressions of SLC7A11, GSH, and GPX4 were decreased in HIE neonatal rat brain tissues, indicating the activation of ferroptosis-related signaling pathways.93 Inhibition of ferroptosis through intraventricular injection of a ferroptosis inhibitor increased GPx4 expression and reduced MDA and iron levels in the hippocampi of HIE rats. The SIRT1/NRF2/GPx4 signaling pathway plays a crucial role in this process.94 TLR4 also contributes to ferroptosis in HIE, and the use of a TLR4 inhibitor called TAK-242 elevated SLC7A11 and GPX4 levels in hippocampal tissue, both in vivo and in vitro. It also reduced the expression of genes associated with ferroptosis.95 Lipocalin 2 (LCN2) was identified as a critical gene associated with HIE, promoting ferroptosis through the NF-κB/STAT3 pathway. LCN2 can serve as a biomarker for the diagnosis of HIE.96
The Role of Ferroptosis in Neonatal BPD
Bronchopulmonary dysplasia (BPD) is a common lung disorder in premature infants. Oxidative stress plays a significant role in its development.68,97 Premature infants have immature type II alveolar cells, leading to the accumulation of intracellular ROS.98 Excessive ROS damages the lung epithelium and vascular endothelium, impairing lung development and contributing to BPD.99 The pathogenesis of BPD involves lipid peroxidation, supporting the association with ferroptosis. Lipid hydrogen peroxide (LOOH) is generated during lipid peroxidation, causing damage to biological macromolecules. Infants with BPD exhibit higher levels of LOOH in their alveolar lavage fluid, likely due to ROS overload.100
Hyperoxia is a risk factor for bronchopulmonary dysplasia (BPD) and is commonly used to create an animal model of BPD.101 Ferroptosis has been observed in neonatal rats experiencing hyperoxia-induced lung injury, indicating its involvement in lung development impairment.102 The increased transcription and activity of P53, along with enhanced acetylated P53 expression, are observed as a result of hyperoxia exposure.103 P53 inhibits SLC7A11, leading to decreased glutathione (GSH) synthesis and GPX4 activity, resulting in ROS accumulation and triggering ferroptosis.104
Excessive iron supplementation in infants with very low birth weight increases the risk of developing BPD.105 Experiments on a mouse model of lung injury demonstrated the accumulation of iron ions in the lung, which can induce ferroptosis.106 Inflammation also plays a role in BPD, and the activation of the KEAP1-NRF2/HO-1 pathway by panaxydol inhibits ferroptosis, thereby reducing LPS-induced pneumonitis.107,108
In summary, hyperoxia, oxidative stress, inflammation, and excessive iron are key factors contributing to the development of BPD, and their association with ferroptosis suggests a significant role of ferroptosis in BPD pathology. However, more research is required to elucidate the specific regulatory mechanisms underlying ferroptosis in BPD.
The Role of Ferroptosis in Neonatal NEC
Neonatal necrotizing enterocolitis (NEC) is a severe gastrointestinal disorder. Factors such as oxidative stress, ischemia-reperfusion, and inflammation play a role in its development. Ferroptosis is closely associated with the pathogenesis of NEC and may contribute to its progression.
First, NEC is characterized by intestinal ischemia.109 Ischemia-reperfusion generates ROS and free iron, worsening intestinal injury.110 Elevated free iron levels enhance the Fenton response, leading to the production of highly toxic free radicals.111 Free iron and ROS cause ferroptosis through lipid peroxidation, contributing to tissue damage in NEC.
Second, NEC development is linked to lipid peroxidation damage. NEC-affected infants had higher total oxidative state and oxidative stress index, which correlated with NEC severity.112 NEC infants’ cord blood had higher levels of NTBI and ROS compared to non-NEC infants.113 High levels of NTBI and ROS lead to tissue damage through lipid peroxidation. Lipid peroxidation products in cord blood strongly correlate with NEC.113 Preterm infants with NEC had increased intestinal MDA.114,115 Animals with NEC exhibited decreased levels of GPX, SOD, and GSH in intestinal tissues, along with significantly elevated MDA levels.116 Recent research discovered that Lactobacillus can alleviate NEC by reducing lipid peroxidation in enterocytes.117
Third, Ferroptosis is linked to TLR4, which is important in NEC. The TLR4 signaling pathway plays a role in embryonic crypt development and intestinal epithelial cell differentiation.118 Persistent TLR4 pathway hyperactivation in intestinal epithelial cells after birth is associated with gut damage in NEC. Inhibiting TLR4 signaling effectively reduces the occurrence and severity of NEC.119 LTR4 is also closely related to ferroptosis. Inhibition of TLR4 attenuates ferroptosis in hippocampal neurons during hypoxic-ischemic injury.95 TLR4 promotes ferroptosis in heart failure cardiomyocytes.120 During renal ischemia-reperfusion injury, TLR4/NOX4 induce oxidative stress and ferroptosis.121
Fourth, excessive inflammation is a major pathological change in NEC. In NEC intestinal tissue, immune cells release inflammatory factors like IL-6 and TNF-α. IL-6 promotes lipid peroxidation and ferroptosis in epithelial cells by increasing ferrous ions.122 TNF-α promotes ferroptosis by increasing the production of acyl-CoA synthetase long-chain family member 1 (ACSL1), which enhances the production of polyunsaturated lipids and subsequent lipid peroxidation in human endothelial cells.123 IFN-γ downregulates SLC3A2 and SLC7A11, causing lipid peroxidation and ferroptosis.124 Ferroptosis of intestinal epithelial cells further triggers an inflammatory response by releasing cellular contents. For instance, the lipid peroxidation product 4-HNE activates the NF-κB pathway, promoting inflammation.125 Release of HMGB1, a protein found in ferroptosis cells, also contributes to NEC-related inflammation by activating the TLR4-mediated NF-κB signaling pathway.126,127 The evidence suggests a close association between inflammation and ferroptosis in NEC.8
In addition, In an NEC model created by anemia-blood transfusion, elevated levels of free iron and MDA were observed in the intestines, along with mitochondrial dysfunction. Ferroptosis inhibitors were found to alleviate intestinal injury in NEC.6 The analysis of genetic data in premature infants with NEC revealed that specific genes related to NEC were significantly enriched in the ferroptosis signaling pathway.128 While there is evidence linking NEC to ferroptosis, the precise mechanisms by which ferroptosis contributes to NEC and how it can be regulated to impact NEC are not fully understood.
The Role of Ferroptosis in Other Common Neonatal Diseases
Premature infants are susceptible to retinal diseases such as retinopathy of prematurity (ROP), which can result in blindness. The mouse model of oxygen-induced retinopathy showed lipid peroxidation and ferroptosis markers in the mouse retina were found to increase significantly, suggesting that ferroptosis is critical in the pathological process of ROP, and ferroptosis inhibitors can alleviate the process of ROP middle retinal vascular occlusion and pathological neovascularization.5,129 Intracranial hemorrhage is a common complication in neonates, especially premature infants. In the intracranial hemorrhage model of neonatal rats, ferroptosis-related genes were detected higher 72 hours after hemorrhage, implicating ferroptosis in hemorrhage-induced brain injury pathology.130 Lipocalin 2 (LCN2) inhibition attenuated inflammation and ferroptosis in neonate acute respiratory distress syndrome (ARDS) through MAPK/ERK signaling.131 In addition, ferroptosis may play a critical role in the neuronal damage caused by hemolytic hyperbilirubinemia in the brain.132
New Therapeutic Targets for Neonatal Diseases Based on Ferroptosis
In neonatal diseases, ferroptosis plays an important role, so targeting ferroptosis may be a promising treatment strategy (Table 1). For example, Vitamin D is an important antioxidant, which can increase the production of NRF2 and HO-1, lead to the increase of GPX4, SOD and GSH, reduce MDA and ROS, and reduce ferroptosis in brain injury.133 Intraventricular injection of ferroptosis inhibitor (Ferrostatin-1 or Resveratrol) can reduce neonatal cerebral ischemia-reperfusion injury.94 Glycyrrhizin attenuated RSL3-induced ferroptosis and inflammatory response in neonatal rat brain tissue by inhibiting HMGB1.134 Melatonin can inhibit the process of ferroptosis in hypoxic-ischemic encephalopathy by activating GPX4 activity and playing a protective role in the brain.135 Hyperoxia-induced lung damage can be prevented by cathelicidins, which have both antioxidant and ferroptosis inhibition properties.136 Deferoxamine would be able to attenuate intestinal damage in neonatal necrotizing enterocolitis by binding excess free iron in neonates.137 Flavonoid glycosides are potent antioxidants that attenuate endothelial ROS production and free iron accumulation by specifically binding to HO-1 and MAO-B, thereby inhibiting ferroptosis in the ischemia-reperfused intestine in a dose-dependent manner.138 All-trans retinoic acid treatment decreased intestinal MDA, increased SOD and GPX activity, and attenuated NEC by inhibiting lipid peroxidation.114 Boric acid and 2-aminoethoxydiphenyl borate can reduce NEC by increasing GSH expression and inhibiting lipid peroxidation.139 Further clinical studies are required to confirm the safety and effectiveness of novel medications targeting ferroptosis for the treatment of newborn illnesses.
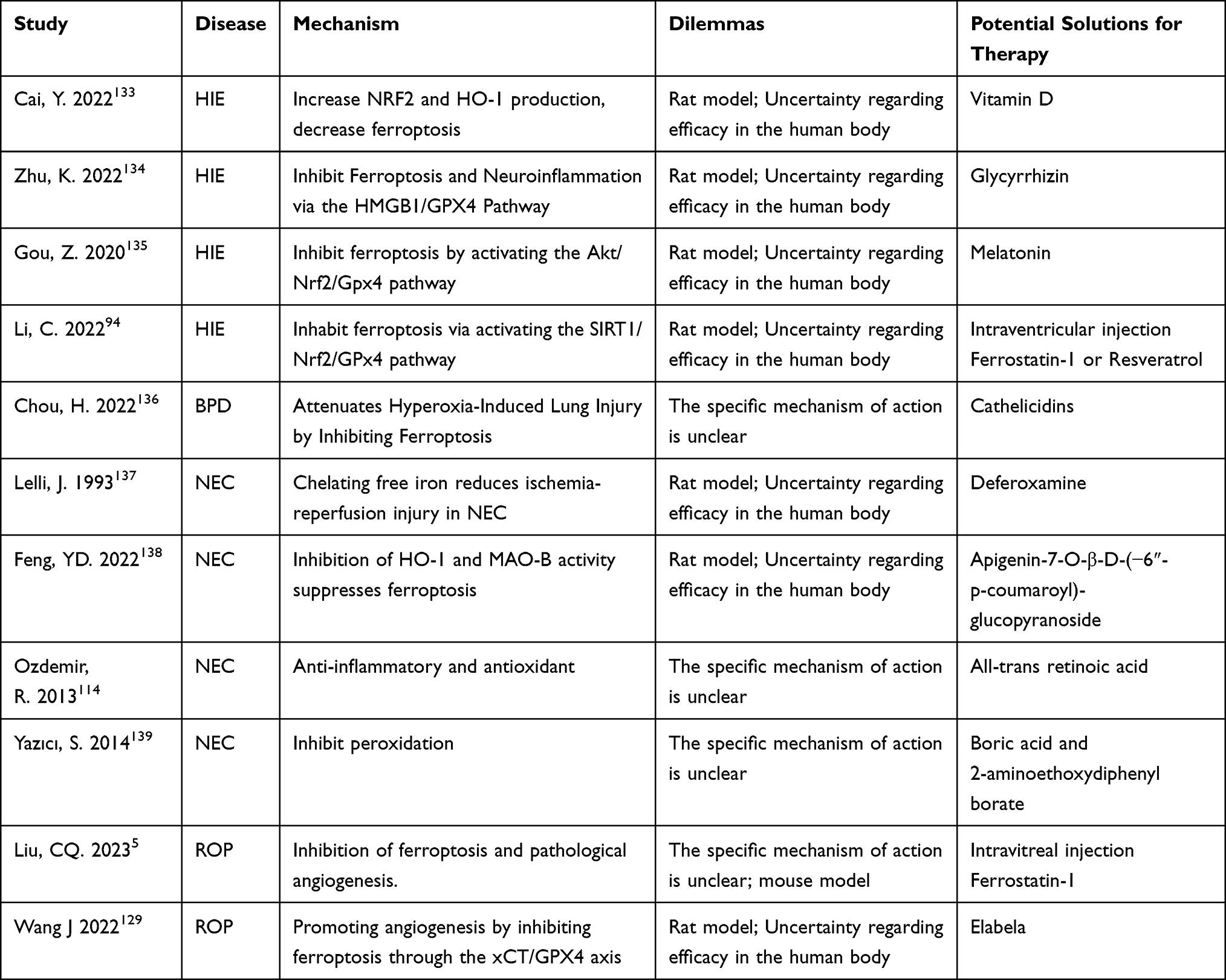 |
Table 1 Targeted Drugs for Ferroptosis and Their Mechanisms in Common Diseases of Newborns |
Conclusion
Ferroptosis is a newly discovered form of programmed cell death triggered by free iron and ROS, leading to tissue damage through lipid peroxidation. Newborns, due to their unique physiological state and metabolic characteristics, are prone to accumulate free iron and ROS, resulting in organ damage via ferroptosis. Studies have shown that ferroptosis is involved in the development of neonatal diseases such as hypoxic-ischemic encephalopathy, bronchopulmonary dysplasia, and necrotizing enterocolitis, and inhibiting ferroptosis can alleviate these conditions. Consequently, ferroptosis has emerged as a potential therapeutic target for newborn disorders. However, most research conducted so far has been limited to animal and cell studies, necessitating further clinical investigations to validate these findings.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi:10.1016/j.cell.2012.03.042
2. Riegman M, Sagie L, Galed C, et al. Ferroptosis occurs through an osmotic mechanism and propagates independently of cell rupture. Nat Cell Biol. 2020;22(9):1042–1048. doi:10.1038/s41556-020-0565-1
3. Stockwell BR. Ferroptosis turns 10: emerging mechanisms, physiological functions, and therapeutic applications. Cell. 2022;185(14):2401–2421. doi:10.1016/j.cell.2022.06.003
4. Li LY, Wang Q, Deng L, et al. Chlorogenic acid alleviates hypoxic-ischemic brain injury in neonatal mice. Neural Regen Res. 2023;18(3):568–576. doi:10.4103/1673-5374.350203
5. Liu CQ, Liu XY, Ouyang PW, et al. Ferrostatin-1 attenuates pathological angiogenesis in oxygen-induced retinopathy via inhibition of ferroptosis. Exp Eye Res. 2023;226:109347. doi:10.1016/j.exer.2022.109347
6. Dang D, Meng Z, Zhang C, Li Z, Wei J, Wu H. Heme induces intestinal epithelial cell ferroptosis via mitochondrial dysfunction in transfusion-associated necrotizing enterocolitis. FASEB J. 2022;36(12):e22649. doi:10.1096/fj.202200853RRR
7. Deng X, Bao Z, Yang X, et al. Molecular mechanisms of cell death in bronchopulmonary dysplasia. Apoptosis. 2022;28(1–2):39–54. doi:10.1007/s10495-022-01791-4
8. Zhang X, Ma Y, Lv G, Wang H. Ferroptosis as a therapeutic target for inflammation-related intestinal diseases. Front Pharmacol. 2023;14:1095366. doi:10.3389/fphar.2023.1095366
9. Silva B, Faustino P. An overview of molecular basis of iron metabolism regulation and the associated pathologies. Biochim Biophys Acta. 2015;1852(7):1347–1359. doi:10.1016/j.bbadis.2015.03.011
10. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22(4):266–282. doi:10.1038/s41580-020-00324-8
11. Cerami C. Iron Nutriture of the Fetus, Neonate, Infant, and Child. Ann Nutr Metab. 2017;71(Suppl 3):8–14. doi:10.1159/000481447
12. Jiang R, Lopez V, Kelleher SL, Lönnerdal B. Apo- and holo-lactoferrin are both internalized by lactoferrin receptor via clathrin-mediated endocytosis but differentially affect ERK-signaling and cell proliferation in Caco-2 cells. J Cell Physiol. 2011;226(11):3022–3031. doi:10.1002/jcp.22650
13. Matsuzaki T, Nakamura M, Nogita T, Sato A. Cellular uptake and release of intact lactoferrin and its derivatives in an intestinal enterocyte model of caco-2 cells. Biol Pharm Bull. 2019;42(6):989–995. doi:10.1248/bpb.b19-00011
14. Akiyama Y, Oshima K, Kuhara T, et al. A lactoferrin-receptor, intelectin 1, affects uptake, sub-cellular localization and release of immunochemically detectable lactoferrin by intestinal epithelial Caco-2 cells. J Biochem. 2013;154(5):437–448. doi:10.1093/jb/mvt073
15. Tian H, Xiong Y, Zhang Y, et al. Activation of NRF2/FPN1 pathway attenuates myocardial ischemia-reperfusion injury in diabetic rats by regulating iron homeostasis and ferroptosis. Cell Stress Chaperones. 2021;27(2):149–164. doi:10.1007/s12192-022-01257-1
16. Eid R, Arab NT, Greenwood MT. Iron mediated toxicity and programmed cell death: a review and a re-examination of existing paradigms. Biochim Biophys Acta Mol Cell Res. 2017;1864(2):399–430. doi:10.1016/j.bbamcr.2016.12.002
17. Kwon MY, Park E, Lee SJ, Chung SW. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget. 2015;6(27):24393–24403. doi:10.18632/oncotarget.5162
18. Ganz T, Nemeth E, Rivella S, et al. TMPRSS6 as a therapeutic target for disorders of erythropoiesis and iron homeostasis. Adv Ther. 2023;40:1317–1333. doi:10.1007/s12325-022-02421-w
19. Sharp P, Srai SK. Molecular mechanisms involved in intestinal iron absorption. World J Gastroenterol. 2007;13(35):4716–4724. doi:10.3748/wjg.v13.i35.4716
20. Wang Y, Wu Y, Li T, Wang X, Zhu C. Iron metabolism and brain development in premature infants. Front Physiol. 2019;10:463. doi:10.3389/fphys.2019.00463
21. Peng Y, Liao B, Zhou Y, Zeng W, Zeng ZY. Atorvastatin inhibits ferroptosis of H9C2 cells by regulatingSMAD7/Hepcidin expression to improve ischemia-reperfusion injury. Cardiol Res Pract. 2022;2022:3972829. doi:10.1155/2022/3972829
22. Kawabata T. Iron-induced oxidative stress in human diseases. Cells. 2022;11(14):2152. doi:10.3390/cells11142152
23. Graham RM, Chua AC, Herbison CE, Olynyk JK, Trinder D. Liver iron transport. World J Gastroenterol. 2007;13(35):4725–4736. doi:10.3748/wjg.v13.i35.4725
24. Rochette L, Dogon G, Rigal E, Zeller M, Cottin Y, Vergely C. Lipid peroxidation and iron metabolism: two corner stones in the homeostasis control of ferroptosis. Int J Mol Sci. 2022;24(1):449. doi:10.3390/ijms24010449
25. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59(2):298–308. doi:10.1016/j.molcel.2015.06.011
26. Brown CW, Amante JJ, Chhoy P, et al. Prominin2 drives ferroptosis resistance by stimulating iron export. Dev Cell. 2019;51(5):575–586.e574. doi:10.1016/j.devcel.2019.10.007
27. Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26(9):1021–1032. doi:10.1038/cr.2016.95
28. Zhou B, Liu J, Kang R, Klionsky DJ, Kroemer G, Tang D. Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol. 2020;66:89–100. doi:10.1016/j.semcancer.2019.03.002
29. Hou W, Xie Y, Song X, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12(8):1425–1428. doi:10.1080/15548627.2016.1187366
30. Gozzelino R, Arosio P. Iron homeostasis in health and disease. Int J Mol Sci. 2016;17(1):130. doi:10.3390/ijms17010130
31. Recalcati S, Minotti G, Cairo G. Iron regulatory proteins: from molecular mechanisms to drug development. Antioxid Redox Signal. 2010;13(10):1593–1616. doi:10.1089/ars.2009.2983
32. Yao F, Cui X, Zhang Y, et al. Iron regulatory protein 1 promotes ferroptosis by sustaining cellular iron homeostasis in melanoma. Oncol Lett. 2021;22(3):657. doi:10.3892/ol.2021.12918
33. Li Y, Jin C, Shen M, et al. Iron regulatory protein 2 is required for artemether -mediated anti-hepatic fibrosis through ferroptosis pathway. Free Radic Biol Med. 2020;160:845–859. doi:10.1016/j.freeradbiomed.2020.09.008
34. Zhao X, Zhang J, Zhang W, et al. A chiral fluorescent Ir(iii) complex that targets the GPX4 and ErbB pathways to induce cellular ferroptosis. Chem Sci. 2023;14(5):1114–1122. doi:10.1039/D2SC06171F
35. Yin H, Xu L, Porter NA. Free radical lipid peroxidation: mechanisms and analysis. Chem Rev. 2011;111(10):5944–5972. doi:10.1021/cr200084z
36. Yin X, Yang Q, Li H, Kang Y, Li Z. Vancomycin induced ferroptosis in renal injury through the inactivation of recombinant glutathione peroxidase 4 and the accumulation of peroxides. Drug Des Devel Ther. 2023;17:283–295. doi:10.2147/DDDT.S392813
37. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A. 2016;113(34):E4966–4975. doi:10.1073/pnas.1603244113
38. Tesfay L, Paul BT, Konstorum A, et al. Stearoyl-CoA desaturase 1 protects ovarian cancer cells from ferroptotic cell death. Cancer Res. 2019;79(20):5355–5366. doi:10.1158/0008-5472.CAN-19-0369
39. Zhang X, Li W, Ma Y, et al. High-fat diet aggravates colitis-associated carcinogenesis by evading ferroptosis in the ER stress-mediated pathway. Free Radic Biol Med. 2021;177:156–166. doi:10.1016/j.freeradbiomed.2021.10.022
40. Doll S, Proneth B, Tyurina YY, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91–98. doi:10.1038/nchembio.2239
41. Xing L, Liu XY, Zhou TJ, Wan X, Wang Y, Jiang HL. Photothermal nanozyme-ignited Fenton reaction-independent ferroptosis for breast cancer therapy. J Control Release. 2021;339:14–26. doi:10.1016/j.jconrel.2021.09.019
42. Zhang Z, Pan Y, Cun JE, et al. A reactive oxygen species-replenishing coordination polymer nanomedicine disrupts redox homeostasis and induces concurrent apoptosis-ferroptosis for combinational cancer therapy. Acta Biomater. 2022;151:480–490. doi:10.1016/j.actbio.2022.07.055
43. Tomita K, Takashi Y, Ouchi Y, et al. Lipid peroxidation increases hydrogen peroxide permeability leading to cell death in cancer cell lines that lack mtDNA. Cancer Sci. 2019;110(9):2856–2866. doi:10.1111/cas.14132
44. Kagan VE, Tyurina YY, Vlasova II, et al. Redox epiphospholipidome in programmed cell death signaling: catalytic mechanisms and regulation. Front Endocrinol. 2020;11:628079. doi:10.3389/fendo.2020.628079
45. Anthonymuthu TS, Tyurina YY, Sun WY, et al. Resolving the paradox of ferroptotic cell death: ferrostatin-1 binds to 15LOX/PEBP1 complex, suppresses generation of peroxidized ETE-PE, and protects against ferroptosis. Redox Biol. 2021;38:101744. doi:10.1016/j.redox.2020.101744
46. Zou Y, Li H, Graham ET, et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol. 2020;16(3):302–309. doi:10.1038/s41589-020-0472-6
47. Pedrera L, Espiritu RA, Ros U, et al. Ferroptotic pores induce Ca(2+) fluxes and ESCRT-III activation to modulate cell death kinetics. Cell Death Differ. 2021;28(5):1644–1657. doi:10.1038/s41418-020-00691-x
48. Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2014;10(1):9–17. doi:10.1038/nchembio.1416
49. Ursini F, Maiorino M, Gregolin C. The selenoenzyme phospholipid hydroperoxide glutathione peroxidase. Biochim Biophys Acta. 1985;839(1):62–70. doi:10.1016/0304-4165(85)90182-5
50. Maiorino M, Conrad M, Ursini F. GPx4, lipid peroxidation, and cell death: discoveries, rediscoveries, and open issues. Antioxid Redox Signal. 2018;29(1):61–74. doi:10.1089/ars.2017.7115
51. Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting ferroptosis to iron out cancer. Cancer Cell. 2019;35(6):830–849. doi:10.1016/j.ccell.2019.04.002
52. Wang L, Liu Y, Du T, et al. ATF3 promotes erastin-induced ferroptosis by suppressing system Xc(). Cell Death Differ. 2020;27(2):662–675. doi:10.1038/s41418-019-0380-z
53. Forman HJ, Zhang H, Rinna A. Glutathione: overview of its protective roles, measurement, and biosynthesis. Mol Aspects Med. 2009;30(1–2):1–12. doi:10.1016/j.mam.2008.08.006
54. Shao S, Liu Y, Hong W, et al. Influence of FOSL1 inhibition on vascular calcification and ROS generation through ferroptosis via P53-SLC7A11 axis. Biomedicines. 2023;11(2):635. doi:10.3390/biomedicines11020635
55. Hu M, Zhang Y, Ma S, et al. Suppression of uterine and placental ferroptosis by N-acetylcysteine in a rat model of polycystic ovary syndrome. Mol Hum Reprod. 2021;27(12). doi:10.1093/molehr/gaab067
56. Doll S, Freitas FP, Shah R, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575(7784):693–698. doi:10.1038/s41586-019-1707-0
57. Bersuker K, Hendricks JM, Li Z, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575(7784):688–692. doi:10.1038/s41586-019-1705-2
58. Mao C, Liu X, Zhang Y, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593(7860):586–590. doi:10.1038/s41586-021-03539-7
59. Jiang M, Song Y, Liu H, Jin Y, Li R, Zhu X. DHODH inhibition exerts synergistic therapeutic effect with cisplatin to induce ferroptosis in cervical cancer through regulating mTOR pathway. Cancers. 2023;15(2):546. doi:10.3390/cancers15020546
60. Soula M, Weber RA, Zilka O, et al. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat Chem Biol. 2020;16(12):1351–1360. doi:10.1038/s41589-020-0613-y
61. Kraft VAN, Bezjian CT, Pfeiffer S, et al. GTP Cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci. 2020;6(1):41–53. doi:10.1021/acscentsci.9b01063
62. Du X, Dong R, Wu Y, Ni B. Physiological effects of ferroptosis on organ fibrosis. Oxid Med Cell Longev. 2022;2022:5295434. doi:10.1155/2022/5295434
63. Moreno-Fernandez J, Ochoa JJ, Latunde-Dada GO, Diaz-Castro J. Iron deficiency and iron homeostasis in low birth weight preterm infants: a systematic review. Nutrients. 2019;11(5):1090. doi:10.3390/nu11051090
64. Buonocore G, Perrone S, Longini M, et al. Non protein bound iron as early predictive marker of neonatal brain damage. Brain. 2003;126(Pt 5):1224–1230. doi:10.1093/brain/awg116
65. Baker RD, Greer FR. Diagnosis and prevention of iron deficiency and iron-deficiency anemia in infants and young children (0–3 years of age). Pediatrics. 2010;126(5):1040–1050. doi:10.1542/peds.2010-2576
66. Park J, Lee DG, Kim B, et al. Iron overload triggers mitochondrial fragmentation via calcineurin-sensitive signals in HT-22 hippocampal neuron cells. Toxicology. 2015;337:39–46. doi:10.1016/j.tox.2015.08.009
67. Salvador GA, Oteiza PI. Iron overload triggers redox-sensitive signals in human IMR-32 neuroblastoma cells. Neurotoxicology. 2011;32(1):75–82. doi:10.1016/j.neuro.2010.11.006
68. Cannavò L, Perrone S, Viola V, Marseglia L, Di Rosa G, Gitto E. Oxidative stress and respiratory diseases in preterm newborns. Int J Mol Sci. 2021;22(22):12504. doi:10.3390/ijms222212504
69. Argüelles S, Machado MJ, Ayala A, Machado A, Hervías B. Correlation between circulating biomarkers of oxidative stress of maternal and umbilical cord blood at birth. Free Radic Res. 2006;40(6):565–570. doi:10.1080/10715760500519834
70. D’Angelo G, Chimenz R, Reiter RJ, Gitto E. Use of melatonin in oxidative stress related neonatal diseases. Antioxidants. 2020;9(6). doi:10.3390/antiox9060477
71. Matyas M, Hasmasanu MG, Zaharie G. Antioxidant capacity of preterm neonates assessed by hydrogen donor value. Medicina. 2019;55(11). doi:10.3390/medicina55110720
72. de Almeida VO, Pereira RA, Amantéa SL, Rhoden CR, Colvero MO. Neonatal diseases and oxidative stress in premature infants: an integrative review. J Pediatr. 2022;98(5):455–462. doi:10.1016/j.jped.2021.11.008
73. Lembo C, Buonocore G, Perrone S. Oxidative Stress in Preterm Newborns. Antioxidants. 2021;10(11). doi:10.3390/antiox10111672
74. Zheng D, Liu J, Piao H, Zhu Z, Wei R, Liu K. ROS-triggered endothelial cell death mechanisms: focus on pyroptosis, parthanatos, and ferroptosis. Front Immunol. 2022;13:1039241. doi:10.3389/fimmu.2022.1039241
75. ArulJothi KN, Kumaran K, Senthil S, et al. Implications of reactive oxygen species in lung cancer and exploiting it for therapeutic interventions. Med Oncol. 2022;40(1):43. doi:10.1007/s12032-022-01900-y
76. Sun H, Li X, Guo Q, Liu S. Research progress on oxidative stress regulating different types of neuronal death caused by epileptic seizures. Neurol Sci. 2022;43(11):6279–6298. doi:10.1007/s10072-022-06302-6
77. Peeples ES, Genaro-Mattos TC. Ferroptosis: a promising therapeutic target for neonatal hypoxic-ischemic brain injury. Int J Mol Sci. 2022;23(13):7420. doi:10.3390/ijms23137420
78. Lee AC, Kozuki N, Blencowe H, et al. Intrapartum-related neonatal encephalopathy incidence and impairment at regional and global levels for 2010 with trends from 1990. Pediatr Res. 2013;74(Suppl 1):50–72. doi:10.1038/pr.2013.206
79. Panfoli I, Candiano G, Malova M, et al. Oxidative stress as a primary risk factor for brain damage in preterm newborns. Front Pediatr. 2018;6:369. doi:10.3389/fped.2018.00369
80. Mondal N, Bhat BV, Banupriya C, Koner BC. Oxidative stress in perinatal asphyxia in relation to outcome. Indian J Pediatr. 2010;77(5):515–517. doi:10.1007/s12098-010-0059-4
81. Barrera-de León JC, Cervantes-Munguía R, Vásquez C, Higareda-Almaraz MA, Bravo-Cuellar A, González-López L. Usefulness of serum lipid peroxide as a diagnostic test for hypoxic ischemic encephalopathy in the full-term neonate. J Perinatol. 2013;33(1):15–20. doi:10.1038/jp.2012.38
82. Shouman BO, Mesbah A, Aly H. Iron metabolism and lipid peroxidation products in infants with hypoxic ischemic encephalopathy. J Perinatol. 2008;28(7):487–491. doi:10.1038/jp.2008.22
83. McManus T, Sadgrove M, Pringle AK, Chad JE, Sundstrom LE. Intraischaemic hypothermia reduces free radical production and protects against ischaemic insults in cultured hippocampal slices. J Neurochem. 2004;91(2):327–336. doi:10.1111/j.1471-4159.2004.02711.x
84. Yang T, Li S. Efficacy of different treatment times of mild cerebral hypothermia on oxidative factors and neuroprotective effects in neonatal patients with moderate/severe hypoxic-ischemic encephalopathy. J Int Med Res. 2020;48(9):300060520943770. doi:10.1177/0300060520943770
85. Dorrepaal CA, Berger HM, Benders MJ, van Zoeren-Grobben D, Van de Bor M, Van Bel F. Nonprotein-bound iron in postasphyxial reperfusion injury of the newborn. Pediatrics. 1996;98(5):883–889. doi:10.1542/peds.98.5.883
86. Ciccoli L, Rossi V, Leoncini S, et al. Iron release, superoxide production and binding of autologous IgG to band 3 dimers in newborn and adult erythrocytes exposed to hypoxia and hypoxia-reoxygenation. Biochim Biophys Acta. 2004;1672(3):203–213. doi:10.1016/j.bbagen.2004.04.003
87. Perrone S, Tataranno LM, Stazzoni G, Ramenghi L, Buonocore G. Brain susceptibility to oxidative stress in the perinatal period. J Matern Fetal Neonatal Med. 2015;28(1):2291–2295. doi:10.3109/14767058.2013.796170
88. Rathnasamy G, Ling EA, Kaur C. Iron and iron regulatory proteins in amoeboid microglial cells are linked to oligodendrocyte death in hypoxic neonatal rat periventricular white matter through production of proinflammatory cytokines and reactive oxygen/nitrogen species. J Neurosci. 2011;31(49):17982–17995. doi:10.1523/JNEUROSCI.2250-11.2011
89. Wu Y, Song J, Wang Y, Wang X, Culmsee C, Zhu C. The potential role of ferroptosis in neonatal brain injury. Front Neurosci. 2019;13:115. doi:10.3389/fnins.2019.00115
90. Peeters-Scholte C, Braun K, Koster J, et al. Effects of allopurinol and deferoxamine on reperfusion injury of the brain in newborn piglets after neonatal hypoxia-ischemia. Pediatr Res. 2003;54(4):516–522. doi:10.1203/01.PDR.0000081297.53793.C6
91. Bailey DM, Lundby C, Berg RM, et al. On the antioxidant properties of erythropoietin and its association with the oxidative-nitrosative stress response to hypoxia in humans. Acta Physiol. 2014;212(2):175–187. doi:10.1111/apha.12313
92. Dai Y, Hu L. HSPB1 overexpression improves hypoxic-ischemic brain damage by attenuating ferroptosis in rats through promoting G6PD expression. J Neurophysiol. 2022;128(6):1507–1517. doi:10.1152/jn.00306.2022
93. Lin W, Zhang T, Zheng J, Zhou Y, Lin Z, Fu X. Ferroptosis is involved in hypoxic-ischemic brain damage in neonatal rats. Neuroscience. 2022;487:131–142. doi:10.1016/j.neuroscience.2022.02.013
94. Li C, Wu Z, Xue H, et al. Ferroptosis contributes to hypoxic-ischemic brain injury in neonatal rats: role of the SIRT1/Nrf2/GPx4 signaling pathway. CNS Neurosci Ther. 2022;28(12):2268–2280. doi:10.1111/cns.13973
95. Zhu K, Zhu X, Sun S, et al. Inhibition of TLR4 prevents hippocampal hypoxic-ischemic injury by regulating ferroptosis in neonatal rats. Exp Neurol. 2021;345:113828. doi:10.1016/j.expneurol.2021.113828
96. Luo L, Deng L, Chen Y, Ding R, Li X. Identification of Lipocalin 2 as a ferroptosis-related key gene associated with hypoxic-ischemic brain damage via STAT3/NF-κB signaling pathway. Antioxidants. 2023;12:1.
97. Perez M, Robbins ME, Revhaug C, Saugstad OD. Oxygen radical disease in the newborn, revisited: oxidative stress and disease in the newborn period. Free Radic Biol Med. 2019;142:61–72. doi:10.1016/j.freeradbiomed.2019.03.035
98. Frank L, Groseclose EE. Preparation for birth into an O2-rich environment: the antioxidant enzymes in the developing rabbit lung. Pediatr Res. 1984;18(3):240–244. doi:10.1203/00006450-198403000-00004
99. Wang J, Dong W. Oxidative stress and bronchopulmonary dysplasia. Gene. 2018;678:177–183. doi:10.1016/j.gene.2018.08.031
100. Fabiano A, Gavilanes AW, Zimmermann LJ, et al. The development of lung biochemical monitoring can play a key role in the early prediction of bronchopulmonary dysplasia. Acta Paediatr. 2016;105(5):535–541. doi:10.1111/apa.13233
101. Xia S, Vila Ellis L, Winkley K, et al. Neonatal hyperoxia induces activated pulmonary cellular states and sex-dependent transcriptomic changes in a model of experimental bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol. 2022;324:L123–L140. doi:10.1152/ajplung.00252.2022
102. Chou HC, Chen CM. Hyperoxia induces ferroptosis and impairs lung development in neonatal mice. Antioxidants. 2022;11:4.
103. Hori YS, Kuno A, Hosoda R, Horio Y. Regulation of FOXOs and p53 by SIRT1 modulators under oxidative stress. PLoS One. 2013;8(9):e73875. doi:10.1371/journal.pone.0073875
104. Guan Z, Chen J, Li X, Dong N. Tanshinone IIA induces ferroptosis in gastric cancer cells through p53-mediated SLC7A11 down-regulation. Biosci Rep. 2020;40(8). doi:10.1042/BSR20201807
105. Patel RM, Knezevic A, Yang J, et al. Enteral iron supplementation, red blood cell transfusion, and risk of bronchopulmonary dysplasia in very-low-birth-weight infants. Transfusion. 2019;59(5):1675–1682. doi:10.1111/trf.15216
106. Zhou H, Li F, Niu JY, et al. Ferroptosis was involved in the oleic acid-induced acute lung injury in mice. Sheng Li Xue Bao. 2019;71(5):689–697.
107. Holzfurtner L, Shahzad T, Dong Y, et al. When inflammation meets lung development-an update on the pathogenesis of bronchopulmonary dysplasia. Mol Cell Pediatr. 2022;9(1):7. doi:10.1186/s40348-022-00137-z
108. Li J, Lu K, Sun F, et al. Panaxydol attenuates ferroptosis against LPS-induced acute lung injury in mice by Keap1-Nrf2/HO-1 pathway. J Transl Med. 2021;19(1):96. doi:10.1186/s12967-021-02745-1
109. Yazji I, Sodhi CP, Lee EK, et al. Endothelial TLR4 activation impairs intestinal microcirculatory perfusion in necrotizing enterocolitis via eNOS-NO-nitrite signaling. Proc Natl Acad Sci U S A. 2013;110(23):9451–9456. doi:10.1073/pnas.1219997110
110. Zhou L, Han S, Guo J, Qiu T, Zhou J, Shen L. Ferroptosis-A new dawn in the treatment of organ ischemia-reperfusion injury. Cells. 2022;11(22):3653. doi:10.3390/cells11223653
111. Marseglia L, Gitto E, Laschi E, et al. Antioxidant effect of melatonin in preterm newborns. Oxid Med Cell Longev. 2021;2021:6308255. doi:10.1155/2021/6308255
112. Aydemir C, Dilli D, Uras N, et al. Total oxidant status and oxidative stress are increased in infants with necrotizing enterocolitis. J Pediatr Surg. 2011;46(11):2096–2100. doi:10.1016/j.jpedsurg.2011.06.032
113. Perrone S, Tataranno ML, Negro S, et al. May oxidative stress biomarkers in cord blood predict the occurrence of necrotizing enterocolitis in preterm infants? J Matern Fetal Neonatal Med. 2012;25(1):128–131. doi:10.3109/14767058.2012.663197
114. Ozdemir R, Yurttutan S, Sari FN, et al. All-trans-retinoic acid attenuates intestinal injury in a neonatal rat model of necrotizing enterocolitis. Neonatology. 2013;104(1):22–27. doi:10.1159/000350510
115. Perrone S, Laschi E, Buonocore G. Biomarkers of oxidative stress in the fetus and in the newborn. Free Radic Biol Med. 2019;142:23–31. doi:10.1016/j.freeradbiomed.2019.03.034
116. Aceti A, Beghetti I, Martini S, Faldella G, Corvaglia L. Oxidative stress and necrotizing enterocolitis: pathogenetic mechanisms, opportunities for intervention, and role of human milk. Oxid Med Cell Longev. 2018;2018:7397659. doi:10.1155/2018/7397659
117. Lai Z, Gong F. Protective effects of lactobacillus reuteri on intestinal barrier function in a mouse model of neonatal necrotizing enterocolitis. Am J Perinatol. 2022. doi:10.1055/s-0042-1755554
118. Sodhi CP, Neal MD, Siggers R, et al. Intestinal epithelial Toll-like receptor 4 regulates goblet cell development and is required for necrotizing enterocolitis in mice. Gastroenterology. 2012;143(3):708–718.e705. doi:10.1053/j.gastro.2012.05.053
119. Huang D, Wang P, Chen J, et al. Selective targeting of MD2 attenuates intestinal inflammation and prevents neonatal necrotizing enterocolitis by suppressing TLR4 signaling. Front Immunol. 2022;13:995791. doi:10.3389/fimmu.2022.995791
120. Chen X, Xu S, Zhao C, Liu B. Role of TLR4/NADPH oxidase 4 pathway in promoting cell death through autophagy and ferroptosis during heart failure. Biochem Biophys Res Commun. 2019;516(1):37–43. doi:10.1016/j.bbrc.2019.06.015
121. Feng R, Xiong Y, Lei Y, et al. Lysine-specific demethylase 1 aggravated oxidative stress and ferroptosis induced by renal ischemia and reperfusion injury through activation of TLR4/NOX4 pathway in mice. J Cell Mol Med. 2022;26(15):4254–4267. doi:10.1111/jcmm.17444
122. Han F, Li S, Yang Y, Bai Z. Interleukin-6 promotes ferroptosis in bronchial epithelial cells by inducing reactive oxygen species-dependent lipid peroxidation and disrupting iron homeostasis. Bioengineered. 2021;12(1):5279–5288. doi:10.1080/21655979.2021.1964158
123. Jung HS, Shimizu-Albergine M, Shen X, et al. TNF-α induces acyl-CoA synthetase 3 to promote lipid droplet formation in human endothelial cells. J Lipid Res. 2020;61(1):33–44. doi:10.1194/jlr.RA119000256
124. Wang W, Green M, Choi JE, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569(7755):270–274. doi:10.1038/s41586-019-1170-y
125. Jang EJ, Kim DH, Lee B, et al. Activation of proinflammatory signaling by 4-hydroxynonenal-Src adducts in aged kidneys. Oncotarget. 2016;7(32):50864–50874. doi:10.18632/oncotarget.10854
126. Wiernicki B, Maschalidi S, Pinney J, et al. Cancer cells dying from ferroptosis impede dendritic cell-mediated anti-tumor immunity. Nat Commun. 2022;13(1):3676. doi:10.1038/s41467-022-31218-2
127. Yu R, Jiang S, Tao Y, Li P, Yin J, Zhou Q. Inhibition of HMGB1 improves necrotizing enterocolitis by inhibiting NLRP3 via TLR4 and NF-κB signaling pathways. J Cell Physiol. 2019;234(8):13431–13438. doi:10.1002/jcp.28022
128. Dang D, Zhang C, Meng Z, et al. Integrative analysis links ferroptosis to necrotizing enterocolitis and reveals the role of ACSL4 in immune disorders. iScience. 2022;25(11):105406. doi:10.1016/j.isci.2022.105406
129. Wang J, Zhang Q, Chen E, Zhao P, Xu Y. Elabela promotes the retinal angiogenesis by inhibiting ferroptosis during the vaso-obliteration phase in mouse oxygen-induced retinopathy model. FASEB J. 2022;36(5):e22257. doi:10.1096/fj.202101785RRR
130. Song J, Nilsson G, Xu Y, et al. Temporal brain transcriptome analysis reveals key pathological events after germinal matrix hemorrhage in neonatal rats. J Cereb Blood Flow Metab. 2022;42(9):1632–1649. doi:10.1177/0271678X221098811
131. Wang X, Zhang C, Zou N, et al. Lipocalin-2 silencing suppresses inflammation and oxidative stress of acute respiratory distress syndrome by ferroptosis via inhibition of MAPK/ERK pathway in neonatal mice. Bioengineered. 2022;13(1):508–520. doi:10.1080/21655979.2021.2009970
132. Viktorinova A. Iron-mediated oxidative cell death is a potential contributor to neuronal dysfunction induced by neonatal hemolytic hyperbilirubinemia. Arch Biochem Biophys. 2018;654:185–193. doi:10.1016/j.abb.2018.07.022
133. Cai Y, Li X, Tan X, et al. Vitamin D suppresses ferroptosis and protects against neonatal hypoxic-ischemic encephalopathy by activating the Nrf2/HO-1 pathway. Transl Pediatr. 2022;11(10):1633–1644. doi:10.21037/tp-22-397
134. Zhu K, Zhu X, Liu S, Yu J, Wu S, Hei M. Glycyrrhizin attenuates hypoxic-ischemic brain damage by inhibiting ferroptosis and neuroinflammation in neonatal rats via the HMGB1/GPX4 pathway. Oxid Med Cell Longev. 2022;2022:8438528. doi:10.1155/2022/8438528
135. Gou Z, Su X, Hu X, et al. Melatonin improves hypoxic-ischemic brain damage through the Akt/Nrf2/Gpx4 signaling pathway. Brain Res Bull. 2020;163:40–48. doi:10.1016/j.brainresbull.2020.07.011
136. Chou HC, Chen CM. Cathelicidin attenuates hyperoxia-induced lung injury by inhibiting ferroptosis in newborn rats. Antioxidants. 2022;11:12.
137. Lelli JL Jr, Pradhan S, Cobb LM. Prevention of postischemic injury in immature intestine by deferoxamine. J Surg Res. 1993;54(1):34–38. doi:10.1006/jsre.1993.1006
138. Feng YD, Ye W, Tian W, et al. Old targets, new strategy: apigenin-7-O-β-d-(−6″-p-coumaroyl)-glucopyranoside prevents endothelial ferroptosis and alleviates intestinal ischemia-reperfusion injury through HO-1 and MAO-B inhibition. Free Radic Biol Med. 2022;184:74–88. doi:10.1016/j.freeradbiomed.2022.03.033
139. Yazıcı S, Akşit H, Korkut O, Sunay B, Çelik T. Effects of boric acid and 2-aminoethoxydiphenyl borate on necrotizing enterocolitis. J Pediatr Gastroenterol Nutr. 2014;58(1):61–67. doi:10.1097/MPG.0b013e3182a7e02b
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Shikonin Could Be Used to Treat Tubal Pregnancy via Enhancing Ferroptosis Sensitivity
Lai Y, Zeng F, Chen Z, Feng M, Huang Y, Qiu P, Zeng L, Ke Y, Deng G, Gao J
Drug Design, Development and Therapy 2022, 16:2083-2099
Published Date: 1 July 2022
The Relationship Between Ferroptosis and Diseases
Lv J, Hou B, Song J, Xu Y, Xie S
Journal of Multidisciplinary Healthcare 2022, 15:2261-2275
Published Date: 6 October 2022
Targeting Ferroptosis in Bone-Related Diseases: Facts and Perspectives
Chen H, Han Z, Wang Y, Su J, Lin Y, Cheng X, Liu W, He J, Fan Y, Chen L, Zuo H
Journal of Inflammation Research 2023, 16:4661-4677
Published Date: 18 October 2023
Ferroptosis in Osteoarthritis: Current Understanding
Liu Y, Zhang Z, Fang Y, Liu C, Zhang H
Journal of Inflammation Research 2024, 17:8471-8486
Published Date: 7 November 2024
Post-Translational Modification Networks in Ferroptosis: Orchestrating Defense, Drug Resistance, and Therapeutic Opportunities in Hepatocellular Carcinoma
Chen J, Li L, Fang S, Zhou H
Journal of Hepatocellular Carcinoma 2026, 13:607656
Published Date: 21 May 2026