Back to Journals » Journal of Hepatocellular Carcinoma » Volume 13
Post-Translational Modification Networks in Ferroptosis: Orchestrating Defense, Drug Resistance, and Therapeutic Opportunities in Hepatocellular Carcinoma
Authors Chen J, Li L, Fang S, Zhou H
Received 7 March 2026
Accepted for publication 14 May 2026
Published 21 May 2026 Volume 2026:13 607656
DOI https://doi.org/10.2147/JHC.S607656
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Mohamed Shaker
Jie Chen,* Lingling Li,* Sufang Fang, Haitao Zhou
Department of Hepatobiliary & Pancreatic Surgery, The Affiliated Yangming Hospital of Ningbo University (Yuyao People’s Hospital), Ningbo, Zhejiang, People’s Republic of China
*Contributed equally to this work
Correspondence: Haitao Zhou, Department of Hepatobiliary & Pancreatic Surgery, The Affiliated Yangming Hospital of Ningbo University (Yuyao People’s Hospital), Ningbo, Zhejiang, People’s Republic of China, Email [email protected]
Abstract: Hepatocellular carcinoma (HCC) remains a leading cause of cancer-related mortality, with therapeutic efficacy severely limited by drug resistance. Ferroptosis, an iron-dependent form of cell death driven by lipid peroxidation, has emerged as a critical tumor-suppressive mechanism with particular relevance to HCC given the liver’s central role in iron homeostasis. Despite growing interest, a systematic examination of how post-translational modifications (PTMs) coordinately regulate ferroptotic susceptibility across all major defense systems and how this regulatory logic is exploited during drug resistance has been lacking. This review addresses this gap across four interconnected themes. We first dissect the three major ferroptosis defense systems in HCC—the system Xc−-GSH-GPX4 axis, the ACSL4-LPCAT3-PUFA peroxidation cascade, and the FSP1/CoQ10-DHODH parallel pathways—revealing that each node is governed by a combinatorial “PTMs code” involving ubiquitination, phosphorylation, acetylation, and lipid modifications that collectively determine the ferroptotic threshold. We then characterize iron metabolism reprogramming, demonstrating how the TFRC↑/FTH1↑/FPN1↓ triad creates an iron-rich milieu that primes tumor cells for ferroptotic execution while engaging pro-survival pathways through PTM-dependent mechanisms. A central focus is ferroptosis evasion as a unifying mechanism underlying resistance to sorafenib, lenvatinib, immunotherapy, and radiotherapy. We reveal that despite remarkable molecular diversity, resistance mechanisms converge on reinforcement of ferroptosis defense through multi-layered PTMs inputs—from NRF2-driven transcription and SLC7A11/GPX4 stabilization to recently discovered metabolite-sensitive modifications including lactylation, ISGylation, and O-GlcNAcylation. Emerging therapeutic strategies, including natural products, nanotechnology-based platforms, and rational drug combinations targeting the ubiquitin-proteasome system, are critically evaluated. Our synthesis highlights three key insights: (i) ferroptotic susceptibility is determined by the combinatorial state of multiple concurrent PTMs rather than any single modification; (ii) metabolite-sensitive PTMs serve as a molecular code linking the altered metabolic state of resistant cells directly to ferroptosis evasion; and (iii) the temporal hierarchy of PTMs deployment during resistance evolution defines a narrowing therapeutic window informing optimal intervention timing. Finally, we identify critical knowledge gaps and propose future directions for translating the PTMs-ferroptosis nexus into clinical benefit for HCC patients.
Keywords: hepatocellular carcinoma, ferroptosis, post-translational modifications, drug resistance, iron metabolism, lipid peroxidation
Introduction
Epidemiology and Therapeutic Challenges of HCC
Hepatocellular carcinoma (HCC) constitutes the predominant histological subtype of primary liver cancer, accounting for approximately 75–85% of all cases, and represents one of the most lethal malignancies worldwide.1,2 According to recent global cancer statistics, HCC ranks as the sixth most commonly diagnosed cancer and the third leading cause of cancer-related mortality, with an estimated 906,000 new cases and 830,000 deaths annually.1,2 The geographic distribution of HCC is markedly heterogeneous: in the Asia-Pacific region, particularly in China, chronic hepatitis B virus (HBV) infection remains the dominant etiological driver, with China alone contributing approximately 367,700 new cases per year.3 In Western countries, the epidemiological landscape has undergone a notable shift, with hepatitis C virus (HCV) infection, alcohol-associated liver disease (ALD), and most recently metabolic dysfunction-associated steatotic liver disease (MASLD, formerly NAFLD/NASH) emerging as the fastest-growing risk factors for HCC development.2,4 In the United States, MASLD has already become the leading indication for liver transplantation among women and patients with HCC.5 Additional established risk factors include chronic aflatoxin B1 exposure, hereditary hemochromatosis, and alpha-1 antitrypsin deficiency, all of which converge on a common pathological trajectory of chronic hepatic inflammation, progressive fibrosis, and cirrhosis—the principal substrate upon which HCC arises.1,4
The therapeutic armamentarium for HCC has expanded considerably over the past two decades but remains constrained by formidable clinical challenges. For patients diagnosed at early stages (Barcelona Clinic Liver Cancer [BCLC] stage 0-A), potentially curative interventions include surgical resection, radiofrequency ablation (RFA), and liver transplantation, with the latter offering 5-year survival rates exceeding 70% under optimized selection criteria.6,7 However, the insidious nature of HCC, coupled with the lack of sensitive and specific early-detection biomarkers, means that a substantial proportion of patients present with intermediate or advanced disease, at which point curative options are no longer applicable. For intermediate-stage HCC (BCLC-B), transarterial chemoembolization (TACE) has served as the standard of care, while systemic therapy dominates the treatment of advanced-stage disease (BCLC-C).8 The introduction of sorafenib in 2007 and lenvatinib in 2018 as first-line multikinase inhibitors marked a paradigm shift in systemic HCC therapy; however, the survival benefits conferred by these agents remain modest, with median overall survival (OS) rarely exceeding 13–15 months.8,9 The landmark IMbrave150 trial, which demonstrated the superiority of the immune checkpoint inhibitor (ICI) combination atezolizumab plus bevacizumab over sorafenib as first-line therapy for unresectable HCC, ushered in the era of immunotherapy and has since become the preferred standard of care.10,11 Subsequent studies have further validated ICI-based combination regimens, including durvalumab plus tremelimumab (HIMALAYA trial), cementing immunotherapy as a cornerstone of advanced HCC management.8,9
Despite these therapeutic advances, several critical challenges persist. First, the 5-year recurrence rate following curative resection remains disturbingly high at approximately 60–70%, driven by intrahepatic metastasis and multicentric de novo carcinogenesis on the background of a “field cancerization” effect in the cirrhotic liver.6 Second, intrinsic and acquired resistance to both targeted therapies and immunotherapy severely limits durable responses in a substantial proportion of patients, with objective response rates to atezolizumab–bevacizumab reaching only approximately 30%.10,11 Third, the efficacy of ICI-based therapy exhibits striking heterogeneity across HCC etiologies, representing an area of active disagreement and conflicting evidence in the field. Accumulating evidence suggests that patients with non-viral (MASLD/ALD-related) HCC may derive less benefit from immunotherapy compared with those harboring viral hepatitis-associated tumors, although conflicting reports exist—some studies have found no significant etiological difference or even opposite trends—and this remains a subject of intense debate. The mechanistic basis for this potential discrepancy is not fully understood but may involve differences in the tumor immune microenvironment (eg, MASLD-associated HCC often exhibits exhausted CD8+ T cell profiles and altered antigen presentation), adding another layer of complexity to patient stratification for ferroptosis-based combinations.12 Fourth, the immunosuppressive tumor microenvironment (TME) in HCC—characterized by enrichment of regulatory T cells, exhausted CD8+ T cells, M2-polarized macrophages, and myeloid-derived suppressor cells—poses a formidable barrier to the efficacy of immune-based strategies. Collectively, these limitations underscore the urgent need to explore novel biological mechanisms of tumor cell elimination and drug resistance, among which ferroptosis—an iron-dependent form of regulated cell death (RCD)—has attracted rapidly growing interest as a promising therapeutic avenue.
Ferroptosis: An Iron-Dependent Death Pathway with Unique Relevance to HCC
For much of the late 20th century, RCD research was dominated by an apoptosis-centric paradigm, wherein caspase-dependent, morphologically stereotyped cell dismantling was considered the principal genetically encoded mechanism of cell elimination in physiology and pathology.13 Over the past two decades, this framework has been fundamentally reshaped by the identification of multiple non-apoptotic RCD modalities, each governed by distinct molecular machineries. The Nomenclature Committee on Cell Death (NCCD) now recognizes over a dozen mechanistically defined RCD subroutines, including necroptosis (RIPK1/RIPK3/MLKL-dependent), pyroptosis (gasdermin-mediated inflammatory death), ferroptosis (iron-dependent lipid peroxidation), autophagy-dependent cell death, and parthanatos.13 This expanded taxonomy has profound implications for cancer biology: tumour cells frequently evade one or more death pathways, while therapies may exploit alternative RCD routes to overcome apoptosis resistance.13,14
Among the recently delineated RCD modalities, ferroptosis—first defined in 2012 by Dixon et al—has emerged as the most extensively investigated non-apoptotic death pathway in HCC biology.15 Ferroptosis is driven by the uncontrolled peroxidation of polyunsaturated fatty acid (PUFA)-containing phospholipids, a process that requires catalytic iron to generate reactive oxygen species (ROS) via Fenton chemistry and that overwhelms the cell’s intrinsic antioxidant defenses.15,16 Three major intracellular defense systems counteract ferroptotic lipid peroxidation: (i) the system Xc−-GSH-GPX4 axis, which constitutes the canonical ferroptosis-suppressive pathway; (ii) the ACSL4–LPCAT3 lipid remodeling axis, which determines the availability of oxidizable PUFA-phosphatidylethanolamines in cellular membranes; and (iii) the FSP1/CoQ10 and DHODH parallel pathways, which provide GPX4-independent, compartment-specific radical-trapping defense.16 Disruption of any of these defense systems—particularly when combined with elevated intracellular iron—triggers the ferroptotic cascade of lipid peroxidation, membrane damage, and cell death.15,16 The liver, as the central organ of systemic iron homeostasis and the exclusive production site of hepcidin, occupies a uniquely privileged metabolic position that renders hepatocytes—and by extension HCC cells—particularly susceptible to ferroptotic perturbation, making HCC an ideal model to dissect the contributions of ferroptosis to tumor biology and therapeutic vulnerability.14,17,18
Beyond its intrinsic molecular machinery, the TME and metabolic reprogramming in HCC exert decisive influences on sensitivity to ferroptosis. HCC cells undergo extensive metabolic rewiring—including enhanced glycolysis (the Warburg effect), altered fatty acid synthesis, reprogrammed amino acid metabolism, and dysregulated mitochondrial oxidative phosphorylation—all of which directly affect the availability of substrates (PUFAs, GSH, iron) and cofactors (CoQ10, selenium) that govern ferroptotic susceptibility.19,20 The immunological landscape of the TME further modulates ferroptosis outcomes: ferroptotic tumor cells release immunostimulatory damage-associated molecular patterns (DAMPs) that promote anti-tumor immunity and dendritic cell maturation, while metabolite-mediated crosstalk within the TME can paradoxically suppress immune cell function and drive ferroptosis of anti-tumor effector cells—illustrating the complex, context-dependent interplay between cell death, metabolism, and immunity that characterizes HCC.14,17,21
Post-Translational Modifications as Master Regulators
Post-translational modifications (PTMs) constitute a diverse repertoire of covalent chemical modifications that occur after ribosomal synthesis, vastly expanding proteome complexity and enabling rapid, reversible fine-tuning of cellular signaling networks in response to environmental cues.22,23 In ferroptosis regulation, PTMs have emerged as indispensable regulatory layers that orchestrate the balance between pro-survival and pro-death signaling, often with exquisite selectivity for individual nodes of the ferroptosis machinery.24
A growing body of literature has delineated the critical roles of multiple PTMs categories in ferroptosis regulation. Ubiquitination governs the stability and turnover of key regulators—including SLC7A11, GPX4, ACSL4, and NCOA4—through E3 ligase‑mediated proteasomal degradation and deubiquitinase (DUB)-mediated stabilization.22,24,25 Phosphorylation, catalyzed by kinase cascades such as AMPK, MAPK, and PI3K/AKT/mTOR, modulates the activity of BECN1, NRF2, and other ferroptosis-relevant substrates at the intersection of autophagy and oxidative stress signaling.22,23 Acetylation—most notably p53 acetylation at specific lysine residues—regulates SLC7A11 transcriptional repression, while histone acetylation broadly shapes the epigenetic landscape governing ferroptosis gene expression in HCC.24,26 Arginine methylation, SUMOylation, and other PTMs have likewise been implicated in the control of RNA-binding proteins, STAT1 phosphorylation interplay, and ferroptosis-related mRNA stability.22–24
Importantly, several recently discovered or re-contextualized PTMs have opened new frontiers in cell death biology. Protein lactylation, a metabolite-derived lysine modification coupling the Warburg effect to gene regulation, has been shown to modulate ferroptosis resistance through both histone and non-histone substrates—exemplified by the ZNF207-PRDX1-K67la-NRF2 axis in drug-resistant HCC.27 O-GlcNAcylation, a sugar-based PTM responsive to cellular metabolic state, participates in a ROS-mediated oxidation-O-GlcNAcylation cascade that governs ferroptosis sensitivity.22 ISGylation, an interferon-stimulated ubiquitin-like modification, regulates NCOA4-mediated ferritinophagy and thereby controls the iron supply for ferroptotic execution.28 Furthermore, epitranscriptomic modifications—particularly N6-methyladenosine (m6A) RNA methylation mediated by writers (METTL3/METTL14), erasers (ALKBH5/FTO), and readers (YTHDF, IGF2BP families)—have emerged as potent post-transcriptional regulators of ferroptosis gene expression in HCC, blurring the traditional boundary between transcriptional and PTMs-based control.24,26,29
Despite this rapidly expanding knowledge, existing reviews on ferroptosis in HCC have typically focused on individual ferroptosis pathways (eg, the GPX4 axis or iron metabolism alone) or on specific PTM types (eg, ubiquitination or m6A modification) in isolation, without systematically examining how the full spectrum of PTMs coordinately regulates all three major ferroptosis defense systems within the specific biological context of HCC. Moreover, no prior review has linked PTMs-mediated ferroptosis regulation to multi-drug resistance mechanisms—spanning sorafenib, lenvatinib, immunotherapy, and radiotherapy—in a unified analytical framework. This gap is particularly significant given that: (i) HCC occupies a unique metabolic niche as the primary organ of iron homeostasis, rendering it intrinsically primed for ferroptotic perturbation; (ii) the three major ferroptosis defense systems—system Xc−-GSH-GPX4, ACSL4-LPCAT3-PUFA, and FSP1/CoQ10-DHODH—are each subject to multi-layered PTMs-mediated regulation that dynamically calibrates the ferroptotic threshold; and (iii) PTMs-dependent mechanisms, including recently discovered metabolite-sensitive modifications such as lactylation, ISGylation, and O-GlcNAcylation, increasingly underpin drug resistance to first-line HCC therapies. An integrated discussion of the PTMs-ferroptosis nexus that bridges molecular mechanism, drug resistance, and therapeutic strategy is therefore both timely and essential.
To address this gap, we organize our review around four interconnected themes: (1) we delineate the core molecular pathways of the three major ferroptosis defense systems and their disease-specific regulatory modules in HCC, highlighting PTMs-mediated control at each node throughout the relevant discussion (Three Major Ferroptosis Defense Systems and Their Perturbation in HCC); (2) we characterize the unique landscape of iron metabolism reprogramming in HCC and its convergence with lipid peroxidation to create synergistic ferroptotic vulnerability (Iron Metabolism Reprogramming in HCC); (3) we systematically dissect the ferroptosis evasion mechanisms underlying drug resistance across therapeutic modalities—sorafenib, lenvatinib, immunotherapy, and emerging agents—highlighting PTMs as nodal resistance regulators (Drug Resistance and Metabolic Adaptation: Ferroptosis Evasion as a Central Resistance Mechanism in HCC); and (4) we evaluate emerging therapeutic strategies that exploit PTMs-regulated ferroptosis to overcome drug resistance and improve clinical outcomes in HCC (Section 4.7). Finally, we synthesize the evidence into an integrative framework, identify critical knowledge gaps, and propose a translational roadmap for future research (Conclusion and Future Perspectives).
Three Major Ferroptosis Defense Systems and Their Perturbation in HCC
The System Xc−-GSH-GPX4 Axis
The system Xc−-glutathione (GSH)-glutathione peroxidase 4 (GPX4) axis constitutes the most extensively characterized intracellular defense against ferroptosis and serves as the canonical suppressive pathway whose disruption initiates lethal lipid peroxidation.16,30 System Xc− is a heterodimeric antiporter comprising the light-chain subunit SLC7A11 (xCT), which confers cystine/glutamate exchange specificity, and the heavy-chain subunit SLC3A2 (4F2hc/CD98), required for membrane localization and stability.15,16 By importing cystine in exchange for glutamate, system Xc− provides the rate-limiting substrate for cysteine generation, which fuels two-step GSH synthesis: first, γ-glutamylcysteine ligase (GCL, comprising GCLC and GCLM) forms γ-glutamylcysteine; second, glutathione synthetase (GSS) conjugates glycine to produce GSH.16,30,31 GSH then serves as the essential cofactor for GPX4, the only glutathione peroxidase capable of directly reducing phospholipid hydroperoxides (PL-OOH) to non-toxic phospholipid alcohols (PL-OH), thereby terminating lipid peroxidation and preventing ferroptotic membrane rupture.16,30,32 GPX4 is a selenoprotein requiring selenocysteine (Sec) at its catalytic active site (U46); its biosynthesis depends on mevalonate pathway-derived isopentenylation of selenocysteine-tRNA, linking GPX4 expression to broader metabolic circuits.30,32 The obligate dependence of this pathway on extracellular cystine import, GSH biosynthesis, and GPX4 catalytic integrity creates a three‑node vulnerability extensively exploited for ferroptosis induction in HCC (Figure 1).
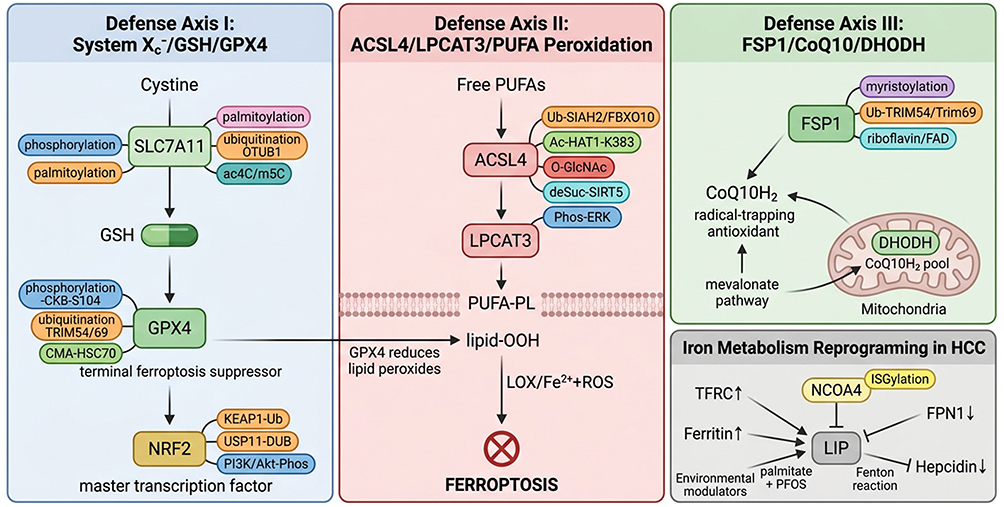 |
Figure 1 Three interconnected ferroptosis defense systems and their PTMs regulatory network in HCC. Three biochemically distinct defense axes are depicted with PTMs annotations on each key regulatory node. Left: The system Xc−-GSH-GPX4 axis. SLC7A11 imports cystine for GSH biosynthesis; GPX4 reduces phospholipid hydroperoxides to prevent membrane damage. SLC7A11 is regulated by palmitoylation,33 ubiquitination (auraptene,34 rottlerin35), and epitranscriptomic modifications (ac4C,36 m5C37); GPX4 by phosphorylation (CKB-S104),38 ubiquitination (TRIM54,39 Trim6940), and chaperone-mediated autophagy (CMA). The NRF2-KEAP1 hub transcriptionally controls both nodes and is itself regulated by ubiquitination,41,42 deubiquitination (USP11),43 and phosphorylation (PI3K/Akt).44 Center: The ACSL4-LPCAT3-PUFA peroxidation axis. ACSL4 activates PUFAs for membrane incorporation; subsequent LOX/Fe2+-driven peroxidation triggers ferroptosis. ACSL4 is subject to five PTMs types: ubiquitination (SIAH2,45 FBXO1046), acetylation (HAT1-K383),47 O-GlcNAcylation,48 desuccinylation (SIRT5),49 and phosphorylation (ERK).50 Right: The FSP1/CoQ10 and DHODH pathways provide GPX4-independent radical-trapping defense. FSP1 is regulated by myristoylation,51 ubiquitination (TRIM54,39 RNF852), and riboflavin/FAD availability.52 Bottom: Iron metabolism reprogramming (TFRC↑/FPN1↓/hepcidin↓) expands the labile iron pool;16,17,53 NCOA4-mediated ferritinophagy is controlled by ISGylation.28 Arrows (→) indicate pathway direction or regulatory consequence. Upward arrows (↑) denote upregulation and downward arrows (↓) denote downregulation in HCC. The crossed circle symbol represents ferroptotic cell death execution. Bold text indicates key regulatory proteins. The dashed line represents the phospholipid membrane bilayer. Colored labels denote distinct PTM types: palmitoylation (purple), ubiquitination (Orange), acetylation (teal), O-GlcNAcylation (red), desuccinylation (blue), phosphorylation (dark blue), and CMA (green). |
In HCC, SLC7A11 has emerged as a central node of ferroptosis resistance, with its expression subject to multi-layered transcriptional and post‑transcriptional regulation that collectively determines the ferroptotic threshold.54 The NRF2-KEAP1 axis serves as the master transcriptional regulator of SLC7A11. Under oxidative stress, NRF2 dissociates from KEAP1, translocates to the nucleus, and activates SLC7A11 transcription.41,42 In HCC, this pathway is constitutively activated by p62/SQSTM1-mediated sequestration of KEAP1, creating an autophagic-antioxidant feed-forward loop that shields tumor cells from ferroptosis.41,42 Conversely, the tumor suppressor p53 transcriptionally represses SLC7A11 by directly binding its promoter, linking p53 activity to ferroptosis induction.55 At the post‑transcriptional level, SLC7A11 and its associated GSH synthesis machinery are regulated by a network of epitranscriptomic modifiers and non-coding RNAs (Table 1). These regulators act through diverse mechanisms—including m6A‑dependent mRNA stabilization (KIAA1429, METTL14), direct mRNA stabilization (lncRNA CASC11), and ceRNA-mediated derepression (lncRNA HCG18)—to collectively tune the ferroptotic threshold in HCC cells.
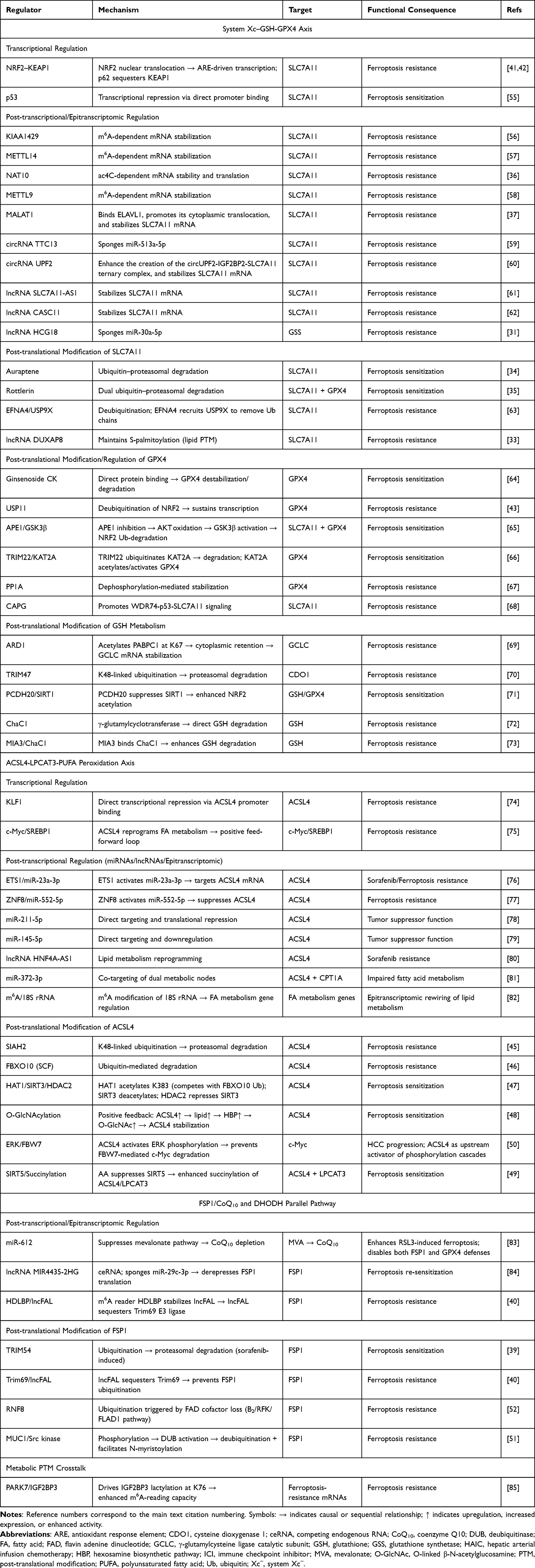 |
Table 1 Multi-Layered Regulatory Mechanisms of the Three Major Ferroptosis Defense Systems in HCC |
Beyond transcriptional and post-transcriptional control, SLC7A11 itself is subject to direct PTMs that governs its membrane stability, trafficking, and functional activity in HCC—establishing PTMs as decisive regulators of the ferroptotic threshold at the transporter level. At the level of protein stability, the natural product auraptene targets SLC7A11 for proteasomal degradation, inducing ferroptosis in HCC cells,34 while rottlerin orchestrates simultaneous ubiquitin-proteasomal degradation of both SLC7A11 and GPX4, functioning as a dual degrader of central ferroptosis defense nodes.35 In a distinct pathway, EFNA4 recruits the deubiquitinase USP9X to SLC7A11, removing ubiquitin chains and stabilizing the transporter; this EFNA4-USP9X-SLC7A11 axis inhibits ferroptosis and promotes HCC proliferation and metastasis, identifying a novel deubiquitination-mediated evasion mechanism.63 Perhaps most strikingly, Shi et al uncovered that the lncRNA DUXAP8 maintains SLC7A11 stability through S-palmitoylation, a lipid modification essential for membrane anchoring and transporter function; loss of DUXAP8 triggers de-palmitoylation, protein destabilization, and synergistic enhancement of sorafenib‑induced ferroptosis.33
Collectively, these findings reveal that SLC7A11 is regulated by at least four distinct PTM types—ubiquitination (auraptene and rottlerin), deubiquitination (USP9X), and palmitoylation (DUXAP8)—which can function either independently or in concert to calibrate transporter stability. Notably, the relative contribution of each PTMs under different therapeutic contexts remains poorly defined, and whether these modifications compete for overlapping lysine residues or operate on distinct protein domains has not been systematically investigated. Addressing these questions will be essential for the rational design of combinatorial strategies targeting SLC7A11 stability.
GPX4, the terminal effector of the system Xc−-GSH-GPX4 axis, is subject to intricate PTMs-mediated regulation in HCC that determines the overall capacity of ferroptosis defense. The natural product ginsenoside compound K (CK) has been identified as a novel GPX4 degrader, directly binding to and destabilizing GPX4 protein to promote ferroptosis—representing the first reported phytochemical-based GPX4 degradation inducer in HCC.64 The transcriptional control of GPX4 is itself modulated through the NRF2-KEAP1 ubiquitin-proteasome axis: Kong et al demonstrated that the deubiquitinase USP11 stabilizes NRF2 by removing ubiquitin chains, sustaining SLC7A11/GPX4 expression; USP11 depletion accelerates NRF2 degradation, suppresses GPX4, and sensitizes HCC cells to ferroptosis.43 Complementarily, Du et al revealed that inhibition of the dual‑function enzyme APE1 in HCC increases AKT oxidation, impairs AKT phosphorylation, activates GSK3β, and facilitates ubiquitin-proteasome-dependent NRF2 degradation, resulting in coordinated downregulation of both SLC7A11 and GPX4 and robust ferroptosis induction.65 Furthermore, an acetylation-ubiquitination crosstalk wherein the E3 ligase TRIM22 promotes degradation of the acetyltransferase KAT2A; since KAT2A acetylates and transcriptionally activates GPX4, TRIM22-mediated KAT2A destruction indirectly suppresses GPX4 and triggers ferroptosis.66 Zhou et al further demonstrated that the protein phosphatase PP1A modulates lenvatinib plus ICI efficacy by inhibiting ferroptosis through dephosphorylation‑mediated stabilization of GPX4, establishing phosphorylation as another PTM layer governing GPX4 turnover.67
The intermediate node of the axis, GSH itself, is similarly regulated at the metabolic and PTMs levels in HCC. Using genome-wide CRISPR screening and targeted metabolomics, Liu et al identified the acetyltransferase ARD1 as a pivotal facilitator of de novo GSH synthesis.69 Mechanistically, ARD1 acetylates the RNA‑binding protein PABPC1 at lysine 67, enhancing its cytoplasmic retention and enabling it to stabilize GCLC mRNA, thereby increasing GSH biosynthesis and conferring ferroptosis resistance.69 Notably, this axis is feedback-regulated by oxidative stress, which suppresses ARD1 ubiquitination and degradation.69 At the level of precursor diversion, the E3 ligase TRIM47 promotes K48-linked ubiquitination and degradation of cysteine dioxygenase 1 (CDO1) in HCC; since CDO1 diverts cysteine away from GSH synthesis toward taurine, TRIM47-mediated CDO1 destruction expands the cysteine pool available for GSH production, suppressing ferroptosis and promoting HCC progression.70 Furthermore, protocadherin 20 (PCDH20) promotes ferroptosis by suppressing SIRT1, leading to enhanced NRF2 acetylation, reduced NRF2 transcriptional activity, and consequently diminished GSH synthesis and GPX4 expression.71 The GSH-degrading enzyme ChaC1 has also been implicated: Yu et al demonstrated that ChaC1‑based drug screening identified a synergistic lethal interaction between auranofin and proteasome inhibitors through enhanced GSH consumption,72 while MIA3 promotes GSH degradation by directly binding to ChaC1, further accelerating HCC progression.73
An emerging theme from these studies is that GSH metabolism in HCC is bidirectionally regulated at the PTMs level: while ARD1-mediated acetylation and TRIM47-dependent ubiquitination expand the GSH pool by stabilizing biosynthetic enzymes or removing competitive metabolic shunts, ChaC1-mediated degradation acts as a counterbalancing force. However, how these opposing mechanisms are coordinated within the same tumor cell—and whether their relative dominance shifts during drug resistance acquisition—remains an open question that warrants further investigation.
The mechanistic and PTMs-level understanding of the system Xc−-GSH-GPX4 axis has catalyzed diverse therapeutic strategies targeting this pathway in HCC. Sorafenib, the first systemic agent approved for advanced HCC, functions in part as a ferroptosis inducer by inhibiting system Xc−-mediated cystine import;86,87 its ferroptotic activity can be potentiated by NRF2 inhibitors such as brusatol via nanoplatform-mediated co-delivery that dismantles both transcriptional and metabolic defenses.88 Lenvatinib similarly engages ferroptotic pathways, with efficacy modulated by PP1A-mediated dephosphorylation of GPX4 representing a druggable resistance mechanism.67 Multiple additional agents induce ferroptosis through this axis, including echinacoside (via TP53/SLC7A11/GPX4),55 picropodophyllin (via AKT/NRF2/SLC7A11),89 erianin (via JAK2/STAT3/SLC7A11),90 nelfinavir (via ER stress-mediated GPX4/GSH downregulation),91 and Pien-Tze-Huang (via direct SLC7A11-GSH-GPX4 suppression).92 Nanotechnology-based approaches further expand the arsenal: targeted xCT-mediated ferroptosis using functionalized nanoparticles eliminates HCC cells and repolarizes protumoral macrophages, enhancing anti-PD-1/PD-L1 efficacy,93 while dual GSH-depleting strategies show remarkable synergistic potential.94 Combining ferroptosis induction with MDSC blockade sensitizes both primary tumors and liver metastases to immune checkpoint inhibition.95 These diverse therapeutic strategies—ranging from repurposing first‑line agents like sorafenib as ferroptosis inducers, to nanotechnology‑enabled combination delivery, and to synergistic integration with immunotherapy—collectively underscore the vast potential of targeting the system Xc−-GSH-GPX4 axis in HCC. Looking forward, a key challenge lies in deciphering the compensatory crosstalk among the three nodes of this axis; developing multi-pronged strategies that simultaneously dismantle transcriptional, post-translational, and metabolic layers of defense will be essential to fully unleash the therapeutic power of ferroptosis induction and overcome intrinsic or acquired resistance.
It should be noted, however, that the majority of these therapeutic studies rely on two-dimensional cell culture and subcutaneous xenograft models, which may not faithfully recapitulate the iron-rich, immunologically complex microenvironment of human HCC. Validation in patient-derived organoids and orthotopic syngeneic models with intact immune systems will be critical to confirm the translational relevance of these findings.
The ACSL4-LPCAT3-PUFA Peroxidation Axis
The execution of ferroptosis begins with the incorporation of oxidation-susceptible PUFAs into membrane phospholipids. Specifically, ACSL4 catalyzes the thioesterification of long‑chain PUFAs—notably arachidonic acid (AA) and adrenic acid (AdA)—to form PUFA‑CoA thioesters. These activated PUFAs are then esterified into membrane phospholipids by LPCAT3, which preferentially generates peroxidation‑prone phosphatidylethanolamines (PUFA-PEs).15,16,30 Once incorporated, PUFA‑PEs undergo oxidation via both enzymatic (lipoxygenase-mediated) and non‑enzymatic (Fenton chemistry) mechanisms, culminating in the generation of toxic PL-OOH that execute ferroptotic membrane damage.15,16,30 In this context, Xue et al recently demonstrated that exogenous AA drives ferroptosis in HCC cells through the SIRT5-ACSL4/LPCAT3/ALOX15 axis: AA suppresses the mitochondrial desuccinylase SIRT5, enhancing succinylation-dependent activation of ACSL4 and LPCAT3, which amplifies PUFA-PE incorporation and subsequent ALOX15‑mediated lipid peroxidation, culminating in mitochondrial dysfunction and ferroptosis.49 This finding illustrates that the ACSL4-LPCAT3-LOX cascade operates not merely as a constitutive lipid supply pathway but as a dynamically regulated pro-ferroptotic module whose output is fine-tuned by protein succinylation—a PTM that directly controls the catalytic efficiency of rate-limiting enzymes.
In HCC, ACSL4 exhibits considerable biological complexity, playing context‑dependent dual roles that both promote tumor progression and sensitize cells to ferroptosis—a paradox that has stimulated extensive investigation. Cui et al comprehensively reviewed its diagnostic and therapeutic significance, delineating its involvement in tumor initiation, metastasis, immune modulation, and resistance, and proposing ACSL4-mediated ferroptosis as a targetable vulnerability.96 From a prognostic perspective, multiple clinical studies have established ACSL4 as a valuable biomarker: Sun and Xu first reported that ACSL4 overexpression correlates with advanced tumor stage and poor survival,97 a finding reinforced by Toshida et al, who demonstrated its association with cancer-associated fibroblast (CFA) abundance and immune microenvironment composition.98 Notably, another study established ACSL4 as a predictive biomarker of sorafenib sensitivity in HCC patients receiving postoperative adjuvant TACE, identifying those more likely to benefit from ferroptosis-inducing therapies.99 The clinical relevance extends beyond viral hepatitis-driven HCC: Classon et al recently provided the first systematic characterization of long-chain acyl-CoA synthetases in MASLD-driven HCC and ferroptosis, demonstrating that altered ACSL4 expression contributes to the ferroptotic landscape in this increasingly prevalent metabolic etiology.100 However, the relationship between ACSL4 and HCC outcome is not unidirectional: Grube et al reported that in certain mouse models, ACSL4-dependent ferroptosis does not suppress tumor growth; rather, ACSL4 promotes HCC progression through ferroptosis-independent mechanisms involving lipid metabolic reprogramming—a paradox underscoring the need for nuanced therapeutic targeting.101 This constitutes a clear area of conflicting evidence in the ferroptosis field: while the majority of studies position ACSL4 as a pro-ferroptotic sensitizer and favorable prognostic marker for ferroptosis-inducing therapies, Grube et al provide contradictory evidence that ACSL4 can drive HCC progression independent of its ferroptotic function. Such context-dependent duality—where ACSL4 acts as either a tumor suppressor (via ferroptosis) or a tumor promoter (via metabolic reprogramming)—likely reflects differences in experimental models, genetic backgrounds, or the presence of additional co-factors, and remains an unresolved disagreement requiring systematic comparative studies.
The expression of ACSL4 in HCC is governed by multi-layered transcriptional and post-transcriptional regulation, which dynamically shapes the ferroptotic potential of tumor cells. At the transcriptional level, the transcription factor KLF1 is upregulated in HCC and directly represses ACSL4 promoter activity, thereby inhibiting the ACSL4/LPCAT3 execution axis and promoting tumor growth.74 Conversely, ACSL4 itself reprograms fatty acid metabolism via the c-Myc/SREBP1 pathway, creating a positive feed-forward loop between lipid remodeling and oncogenic transcription.75 At the post-transcriptional level, ACSL4 is negatively regulated by a diverse set of microRNAs (Table 1). These miRNAs fine-tune the ferroptotic threshold in HCC: some, like miR-23a-3p and miR-552-5p, are specifically linked to therapy resistance by suppressing ACSL4,76,77 while others, such as miR-211-5p and miR-145-5p, modulate tumor progression and immune evasion.78,79 Beyond direct miRNA targeting, lncRNAs add another layer of control; for instance, lncRNA HNF4A-AS1 reprograms lipid metabolism to facilitate sorafenib resistance through ACSL4-related pathways.80 The convergence of transcriptional (KLF1, ETS1), post-transcriptional (miR-23a-3p, miR-211-5p, miR-145-5p, miR-552-5p), and epitranscriptomic (m6A-mediated regulation via 18S rRNA modification82) regulatory layers collectively demonstrates that ACSL4 expression in HCC is not a fixed parameter but a dynamically controlled output determining the threshold for ferroptotic cell death.
Beyond transcriptional and post‑transcriptional control, ACSL4 itself is subject to extensive PTMs that governs its stability, enzymatic activity, and subcellular function in HCC—establishing PTMs as decisive determinants of the ferroptotic threshold. At the level of protein turnover, ACSL4 is regulated by ubiquitin-proteasome-mediated degradation through multiple E3 ligase systems. For instance, SIAH2 directly mediates K48-linked ubiquitination and degradation of ACSL4 in HCC, suppressing ferroptosis susceptibility and impairing CD8+ T cell-mediated anti-tumor immunity; pharmacological SIAH2 inhibition stabilizes ACSL4, restores ferroptotic sensitivity, and enhances immune checkpoint blockade efficacy.45 Besides, FBXO10, a component of the SCF E3 ligase complex, as another ubiquitin-mediated regulator that promotes ACSL4 degradation, with FBXO10 silencing stabilizing ACSL4 and potentiating ferroptosis.46 More recently, Zhou et al elucidated a competitive PTM switch that calibrates ACSL4 abundance: acetylation at lysine 383 (K383) by HAT1 directly blocks FBXO10-mediated K48-linked ubiquitination, thereby stabilizing ACSL4 and enhancing ferroptotic sensitivity.47 Conversely, this acetyl group can be removed by the mitochondrial deacetylase SIRT3, which itself is epigenetically repressed by HDAC2, adding a layer of transcriptional control to this PTM circuit.47 These findings establish a paradigm in which the pro-ferroptotic activity of ACSL4 is quantitatively determined by the dynamic equilibrium between competing PTMs—acetylation (stabilizing, HAT1-dependent) versus ubiquitination (destabilizing, SIAH2/FBXO10-dependent)—with deacetylases (SIRT3) and epigenetic modulators (HDAC2) serving as upstream tuning elements.
In addition to the ubiquitination–acetylation axis, ACSL4 in HCC is subject to at least two other PTMs-mediated regulatory mechanisms that modulate ferroptotic susceptibility and tumor progression. Wang et al revealed a positive feedback loop between ACSL4 expression and O-GlcNAcylation: elevated ACSL4 promotes lipid accumulation, enhancing the hexosamine biosynthetic pathway and global O-GlcNAcylation, which in turn stabilizes ACSL4 protein—a self-reinforcing metabolic–PTM circuit that sustains both lipids reprogramming and the ferroptosis-prone membrane landscape.48 Furthermore, Chen et al identified a phosphorylation‑dependent cascade wherein ACSL4-driven lipid changes activate ERK-mediated phosphorylation, preventing FBW7-mediated degradation of c-Myc and thereby sustaining oncogenic transcriptional programs—demonstrating that ACSL4 not only serves as a PTMs substrate but also acts as an upstream activator of phosphorylation signaling.50 Additionally, the SIRT5-dependent desuccinylation axis represents another PTM layer: arachidonic acid suppresses SIRT5, enhancing succinylation and catalytic activity of ACSL4 and LPCAT3 to amplify PUFA-PE peroxidation via ALOX15.49 Collectively, ACSL4 in HCC exists at a nexus of at least five distinct PTM types—ubiquitination (SIAH2, FBXO10), acetylation (HAT1/K383), O-GlcNAcylation, succinylation (SIRT5-regulated), and phosphorylation (ERK signaling)—each modulating different aspects of ACSL4 biology, including protein abundance, enzymatic activity, and downstream signaling output.
The convergence of mechanistic, prognostic, and PTMs-regulatory data has catalyzed growing interest in pharmacologically targeting the ACSL4-LPCAT3-PUFA axis in HCC. Curcumin promotes ferroptosis by upregulating ACSL4 and amplifying lipid peroxidation,102 while the herb pair Astragali Radix-Curcumae Rhizoma enhances sorafenib efficacy by inducing ACSL4‑dependent ferroptosis and activating Th1 anti-tumor immunity.103 Additional strategies include dipeptidyl peptidase 9 (DPP9) inhibition, which improves sorafenib sensitivity through ACSL4-related lipid metabolic pathways;104 RRM2-targeted nanocarriers that amplify ferroptosis following radiofrequency ablation via enhanced ACSL4 expression and immune remodeling;105 and sorafenib-loaded metal-organic framework nanoparticles that synergistically potentiate ferroptosis and reverse immunosuppression.106 These diverse therapeutic strategies—ranging from natural compounds and nanomedicines to combination immunotherapies—highlight the broad potential of targeting the ACSL4 axis. Notably, ACSL4 also holds translational potential as an intraoperative imaging biomarker; fluorescent probes targeting ACSL4 enable real‑time visualization of HCC for ferroptosis‑guided surgical margin assessment.107 Collectively, these advances position ACSL4 as a promising theranostic target in HCC. However, the context-dependent dual role of ACSL4—both promoting tumor progression and sensitizing cells to ferroptosis—underscores the critical need for future research to decipher the molecular switches that govern these opposing functions, thereby enabling precise therapeutic intervention that maximizes ferroptotic tumor suppression while minimizing potential pro-tumorigenic effects.
The FSP1/CoQ10 and DHODH Parallel Pathways
Beyond the canonical System Xc−-GSH-GPX4 axis, mammalian cells employ at least three parallel, compartment-specific anti-ferroptotic surveillance mechanisms. Two landmark 2019 studies identified ferroptosis suppressor protein 1 (FSP1) as a critical extra-mitochondrial defender.108,109 Localized to the plasma membrane via N-myristoylation, FSP1 acts as an NAD(P)H-dependent oxidoreductase that reduces coenzyme Q10 (CoQ10) to its radical-trapping antioxidant form, ubiquinol (CoQ10H2), thereby directly quenching lipid peroxyl radicals at the membrane surface. In stark contrast to the plasma membrane-localized defense of FSP1, an independent mitochondrial fortress was concurrently unveiled. Concurrently, Mao et al discovered that dihydroorotate dehydrogenase (DHODH), a mitochondrial inner membrane enzyme essential for de novo pyrimidine biosynthesis, constitutes an independent mitochondrial defense system, reducing CoQ to CoQH2 within the mitochondrial compartment to suppress lipid peroxidation in a manner spatially and genetically separable from both cytosolic GPX4 and plasma membrane FSP1.110 More recently, mitochondrial respiratory chain complex I (MCI) has been identified as a third defense layer that maintains CoQH2 levels within mitochondria, operating in parallel with DHODH and GPX4; pharmacologic MCI inhibition by IACS-010759 induces ferroptosis and suppresses tumor growth in vivo.111 Collectively, these pathways—FSP1/CoQ10 at the plasma membrane, DHODH/CoQH2 in the mitochondrial inner membrane, and MCI/CoQH2 in the mitochondrial matrix, forming an integrated, multi-compartmental anti-ferroptotic network that provides layered protection against lipid peroxidation across distinct subcellular membranes.
The efficacy of the FSP1/CoQ10 axis is fundamentally dependent on intracellular CoQ10 availability, which is endogenously synthesized via the mevalonate (MVA) pathway. In HCC, the MVA pathway critically sustains ferroptosis resistance through a dual mechanism: it not only supplies CoQ10 to fuel FSP1-mediated radical trapping, but also supports selenocysteine-tRNA modification—essential for GPX4 translation—thereby reinforcing both GPX4-dependent and -independent anti-ferroptotic defenses.112 Conversely, disruption of CoQ10 biosynthesis sensitizes hepatocytes to ferroptosis. PDSS2, a key enzyme in CoQ10 synthesis, acts as a tumor suppressor in HCC; its loss promotes hepatocarcinogenesis by impairing mitochondrial respiration and reprogramming glucose metabolism,113 while reduced PDSS2 expression stratifies patients at elevated risk of post-resection recurrence.114 Consistent with this framework, miR‑612 enhances RSL3-induced ferroptosis in HCC cells by suppressing the MVA pathway, thereby depleting CoQ10 and simultaneously disabling both FSP1- and GPX4-mediated defenses.83
Accumulating evidence validates FSP1 as a high-priority therapeutic target in HCC. Cheu et al provided the first systematic characterization, demonstrating that genetic or pharmacological FSP1 inhibition triggers robust ferroptosis in HCC cells while remodeling the tumor immune microenvironment, promoting dendritic cell maturation and CD8+ T cell infiltration, thereby simultaneously eliminating tumor cells and reversing immunosuppression.115 Building on this concept, Tang et al developed a co-delivery nanosystem releasing sorafenib and an FSP1 inhibitor (iFSP1) within the tumor, achieving dual ferroptosis induction in both HCC cells and immunosuppressive M2-like tumor-associated macrophages (TAMs), converting immunologically “cold” HCC into an immunostimulatory state and enhancing anti-PD-1 efficacy.116 Pharmacologically, multiple natural products have been identified as FSP1-targeting ferroptosis inducers in HCC: ginsenoside (20)S-APPT and ginsenoside RK1 directly bind FSP1 to inhibit its CoQ10 reductase activity,117,118 while a novel oridonin derivative overcomes FSP1/DHODH-mediated defense by disrupting NADPH supply via G6PD/PGD modulation, impairing the reductive capacity of both enzymes.119
The DHODH-mediated mitochondrial ferroptosis defense pathway has emerged as a targetable vulnerability in HCC, particularly through its intersection with pyrimidine metabolism. Mao et al established that in cancer cells with low GPX4 expression, DHODH inhibition alone suffices to trigger mitochondrial ferroptosis, whereas GPX4‑high cells require combined DHODH and GPX4 blockade for potent synthetic lethality.110 This metabolic vulnerability is especially pronounced in HCC given the high pyrimidine demand of proliferating hepatoma cells. DHODH catalyzes dihydroorotate oxidation, coupling this reaction to mitochondrial CoQ reduction; inhibiting DHODH thus simultaneously depletes pyrimidine nucleotides and removes the mitochondrial CoQH2 radical ferroptosis suppressor protein 1trapping shield, creating a dual metabolic and oxidative stress crisis.110,111 Supporting this concept, exogenous uridine induces ferroptosis in HCC cells by modulating nucleotide pools and redox balance,120 while computational drug repurposing analyses have identified DHODH-targeting strategies as promising HCC treatment avenues via pyrimidine starvation.121 Furthermore, an oridonin derivative achieves anti‑HCC activity by concurrently disabling FSP1 and DHODH through G6PD/PGD-mediated NADPH depletion, demonstrating that blockade of both GPX4‑independent defense arms can overcome compensatory redundancy.119
Critically, the activity, stability, and subcellular localization of FSP1 are governed by multi‑layered PTMs, establishing PTMs as decisive modulators of the FSP1/CoQ10 ferroptosis defense axis in HCC. At the level of protein turnover, FSP1 stability is controlled by competing ubiquitination and deubiquitination circuits. Liu et al demonstrated that sorafenib promotes ferroptosis in HCC cells partly by inducing TRIM54-mediated ubiquitination and proteasomal degradation of FSP1, directly coupling this first‑line therapeutic to dismantlement of GPX4‑independent ferroptosis defense.39 Conversely, the long non‑coding RNA lncFAL stabilizes FSP1 by sequestering the E3 ligase Trim69, preventing its ubiquitination; the nuclear m6A reader HDLBP stabilizes lncFAL itself, creating an epitranscriptomic-PTMs cascade (HDLBP-lncFAL-Trim69-FSP1) that confers ferroptosis resistance.40 A recent CRISPR screen further identified RNF8 as an E3 ligase that triggers FSP1 ubiquitination when FSP1 fails to bind its FAD cofactor, establishing a direct link between vitamin B2 metabolism, cofactor binding, and PTM‑dependent FSP1 quality control.52 In intrahepatic cholangiocarcinoma, MUC1 recruits Src kinase to phosphorylate and activate a deubiquitinase that stabilizes FSP1, while simultaneously facilitating FSP1 N-myristoylation, a lipid modification essential for membrane targeting and CoQ10 reductase activity, illustrating how phosphorylation, deubiquitination, and lipidation converge to reinforce a single ferroptosis‑resistance node.51
A key unresolved question is the relative contribution of each GPX4-independent defense arm—FSP1, DHODH, and MCI—to ferroptosis resistance in different HCC subtypes. While Mao et al demonstrated that GPX4-low cells are preferentially dependent on DHODH, the extent to which FSP1 and DHODH provide redundant versus non-overlapping protection in GPX4-high HCC remains unclear. Resolving this hierarchy is clinically important, as it will determine whether single-target or multi-target inhibition of parallel pathways is required for effective ferroptosis induction.
In addition to direct protein‑level PTMs, the FSP1/CoQ10 defense axis is subject to extensive post‑transcriptional and epitranscriptomic regulation in HCC that indirectly shapes FSP1 protein abundance and ferroptosis susceptibility. Zhang et al demonstrated that the long non-coding RNA MIR4435-2HG functions as a competing endogenous RNA (ceRNA) to sponge miR-29c-3p, relieving miR-29c-3p-mediated translational repression of FSP1 and conferring lenvatinib resistance; silencing MIR4435‑2HG restores miR-29c-3p activity, downregulates FSP1, and re-sensitizes resistant HCC cells to ferroptosis.84 The intersection of metabolic PTMs with ferroptosis regulation is further exemplified by protein lactylation. Moreover, Zhu et al recently revealed that PARK7 drives IGF2BP3 lactylation at lysine 76 (K76la) in HCC, enhancing its m6A-reading capacity to stabilize mRNAs of ferroptosis‑resistance genes, including those governing the FSP1/CoQ10 axis, thereby conferring resistance to hepatic arterial infusion chemotherapy.85 Collectively, the multi-layered PTMs landscape governing these five core ferroptosis regulators—SLC7A11, GPX4, NRF2, ACSL4, and FSP1—is summarized as a combinatorial “PTMs code” map in Figure 2.
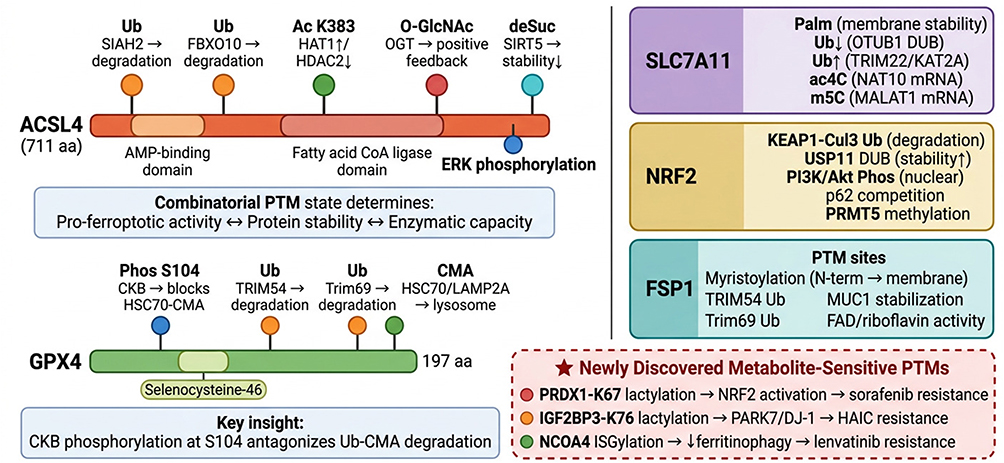 |
Figure 2 The “PTMs code” on key ferroptosis regulators: multi-site combinatorial modification maps in HCC. Detailed protein-level PTMs maps illustrate how the combinatorial modification state—not individual PTMs—determines ferroptotic susceptibility. Left, top: ACSL4 (711 aa) with five PTMs sites shown as lollipop markers on the protein bar: Ub (SIAH2,45 FBXO1046), Ac-K383 (HAT1/HDAC2),47 O-GlcNAc (OGT),48 desuccinylation (SIRT5),49 and phosphorylation (ERK).50 Left, bottom: GPX4 (197 aa) with CKB-mediated phosphorylation at S10438 antagonizing ubiquitin/CMA-dependent degradation,39,40 establishing a phosphorylation–degradation switch.67 Right: PTMs convergence maps for SLC7A11 (Palm33/Ub34/ac4C36/m5C37), NRF2 (KEAP1-Ub41,42/USP11-DUB43/Akt-Phos44/p6241,42), and FSP1 (Myr51/Ub39/MUC151/FAD52). Bottom: Three newly discovered metabolite-sensitive PTMs: PRDX1-K67 lactylation (sorafenib resistance),27 IGF2BP3-K76 lactylation (HAIC resistance),85 and NCOA4 ISGylation (lenvatinib resistance)28—each linking metabolic rewiring directly to ferroptosis evasion. Symbols: Colored lollipop markers on protein bars indicate distinct PTM sites; each color corresponds to a specific modification type. Arrows (→) indicate regulatory consequences; upward arrows (↑) indicate enhanced expression or activity, and downward arrows (↓) indicate reduced expression or activity. Double-headed arrows (↔) denote bidirectional functional relationships. The star symbol (★) highlights newly discovered metabolite-sensitive PTMs. Bold text indicates protein names and key functional concepts. The dashed red border box highlights three recently identified metabolite-sensitive PTMs linked to drug resistance. Colored protein boxes represent distinct ferroptosis regulators: purple (SLC7A11), yellow (NRF2), and teal (FSP1). |
Iron Metabolism Reprogramming in HCC
The liver, as the principal organ of systemic iron homeostasis and the exclusive production site of hepcidin, presents a unique metabolic landscape that renders hepatocytes—and by extension HCC cells—particularly sensitive to ferroptotic perturbation.16,18 Under physiological conditions, hepatocytes maintain iron equilibrium through coordinated activities of transferrin receptor 1 (TFRC) for iron uptake, ferritin (FTH1/FTL) for safe storage, and ferroportin (FPN1) for regulated export, with hepcidin serving as the master endocrine switch controlling FPN1 degradation.18,19 In HCC, this homeostatic circuit is profoundly reprogrammed to establish intracellular iron overload: tumor cells upregulate TFRC to enhance iron acquisition, increase FTH1 to buffer the labile iron pool, and downregulate FPN1 to restrict efflux.16,17,19 This TFRC↑/FTH1↑/FPN1↓ triad creates an iron‑rich intracellular milieu that, while essential for sustaining tumor proliferation, simultaneously primes HCC cells for ferroptotic death when antioxidant defenses are compromised.16,17,53 Liu et al recently provided a comprehensive overview of multi-target ferroptosis regulation in HCC, emphasizing that iron metabolism reprogramming constitutes a defining vulnerability of this cancer type.53 Complementarily, Elmetwalli systematically reviewed how iron‑dependent ferroptosis intersects with the cGAS-STING innate immune pathway in HCC, proposing that precision nano-immuno-theranostic platforms can exploit iron overload to trigger ferroptosis and activate STING-mediated immunogenic responses, thereby reversing drug resistance through the convergence of metabolic and immunological reprogramming.122
Critically, the functional consequences of iron reprogramming in HCC extend far beyond providing catalytic iron for Fenton chemistry‑driven lipid peroxidation. In a landmark study, Qian et al demonstrated that transferrin—the principal circulating iron carrier and TFRC ligand—promotes fatty acid oxidation (FAO) and liver tumor growth through an iron-dependent, non-ferroptotic mechanism.123 Mechanistically, transferrin-mediated iron delivery activates prolyl hydroxylase domain 2 (PHD2) in an iron‑dependent manner, which catalyzes hydroxylation of peroxisome proliferator-activated receptor alpha (PPARα), a master transcriptional regulator of hepatic FAO.123 PHD2-mediated PPARα hydroxylation enhances its transcriptional activity, driving expression of FAO enzymes (including CPT1A and ACADL) to fuel tumor cell bioenergetics and proliferation.123 This finding fundamentally redefines transferrin/TFRC as a dual-function node in HCC: TFRC-mediated iron import expands the labile iron pool and sensitizes cells to ferroptosis, yet the same iron pool activates PHD2-PPARα-driven FAO to support tumor growth. This duality represents a critical conflicting evidence in the field: the same iron metabolism pathway that primes HCC cells for ferroptotic death also activates a pro-survival metabolic program. Such conflicting roles pose a therapeutic dilemma—simple iron-loading strategies intended to boost ferroptosis may inadvertently promote FAO-dependent tumor progression. This paradox has profound therapeutic implications, as strategies increasing intracellular iron to promote ferroptosis may simultaneously stimulate pro-survival FAO programs. The identification of the transferrin-PHD2-PPARα hydroxylation axis also adds an important post-translational dimension to iron metabolism reprogramming, positioning prolyl hydroxylation—an oxygen- and iron-dependent PTMs—as a critical regulatory node coupling iron availability to transcriptional control of lipid metabolism in HCC.
The hepcidin-ferroportin regulatory axis, the central endocrine circuit governing systemic iron homeostasis, is pathologically dysregulated in HCC in ways that directly shape ferroptotic susceptibility. Under normal conditions, hepatocyte-derived hepcidin binds ferroportin (FPN1) to trigger its internalization and degradation, thereby restricting iron efflux.18,19 In a study linking lipid metabolism to iron homeostasis, HCC cells chronically exposed to palmitate—recapitulating the lipotoxic microenvironment of metabolic dysfunction-associated steatotic liver disease (MASLD)—acquire ferroptosis resistance through downregulation of glutamine‑driven hepcidin expression.124 Mechanistically, chronic palmitate reprograms glutamine metabolism, suppressing hepcidin transcription; reduced hepcidin sustains FPN1 surface expression, enhancing iron export and paradoxically depleting the labile iron pool (LIP) that fuels Fenton chemistry, thereby conferring resistance to ferroptosis‑inducing agents.124 This finding is clinically significant given the rising incidence of MASLD-driven HCC, suggesting that such tumors harbor an intrinsic metabolic adaptation—palmitate‑mediated hepcidin suppression—that limits their amenability to ferroptosis‑based therapies.100,124 The study identifies glutamine metabolism as an upstream regulator of hepcidin, establishing a novel metabolic–endocrine axis (palmitate → glutamine reprogramming → hepcidin↓ → FPN1↑ → LIP↓ → ferroptosis resistance) interconnecting lipotoxicity, amino acid metabolism, and iron homeostasis.124 Therapeutically, these data argue that combinatorial strategies in MASLD-associated HCC should consider incorporating glutamine metabolism modulators or direct iron supplementation to restore ferroptotic vulnerability.
A critical mechanism linking iron storage to ferroptosis execution is NCOA4-mediated ferritinophagy, a selective autophagic process in which nuclear receptor coactivator 4 (NCOA4) binds FTH1, delivers ferritin to lysosomes for degradation, and liberates catalytic Fe2+ into the labile iron pool (LIP), thereby providing the iron necessary for lipid peroxidation.16,30 The balance between ferritin‑mediated iron sequestration and NCOA4-driven ferritinophagy thus represents a pivotal toggle determining whether intracellular iron is safely buffered or mobilized for ferroptotic execution. This toggle is subject to transcriptional control: Yang et al demonstrated that excessive expression of the transcription factor SOX8 reprograms both energy and iron metabolism by coordinately upregulating iron uptake genes (including TFRC) while suppressing iron storage (FTH1) and antioxidant programs, thereby tilting the metabolic equilibrium toward an iron-rich, peroxidation-prone state.125 SOX8 thus functions as a master transcriptional switch integrating iron metabolism and ferroptotic threshold in HCC, with potential utility as a predictive biomarker.125 Additionally, emerging evidence implicates novel molecular actors such as C12ORF49 in this regulatory network, suggesting that the machinery governing iron-dependent cell death extends beyond canonical players.126
Beyond cell-autonomous genetic and metabolic reprogramming, emerging evidence demonstrates that environmental factors can reshape ferroptotic vulnerability in HCC by modulating iron metabolism and its downstream consequences. Hong et al reported that exposure to environmental pollutants—specifically perfluorooctane sulfonate (PFOS) and 6:2 Cl-PFESA—reshapes ferroptosis vulnerability in liver cancer, likely through perturbation of iron-dependent metabolic pathways and redox homeostasis.127 Given that PFOS and related compounds are ubiquitous environmental contaminants with established hepatotoxicity and have been epidemiologically linked to elevated HCC risk, this finding suggests that chronic environmental exposures may pre-condition the hepatic iron-redox landscape, potentially contributing to geographic and demographic disparities in HCC treatment responses.127 Furthermore, the bidirectional crosstalk between iron metabolism and the lipid peroxidation machinery underscores the interconnectedness of the iron and PUFA axes in HCC. Iron-catalyzed Fenton chemistry generates hydroxyl radicals that initiate non‑enzymatic peroxidation of ACSL4/LPCAT3-esterified PUFA-PEs, while enzymatic peroxidation via lipoxygenases (particularly ALOX15) requires iron at the catalytic center.15,49 Besides, exogenous arachidonic acid drives ferroptosis in HCC through the SIRT5-ACSL4/LPCAT3/ALOX15 axis, with succinylation-dependent activation of ACSL4 and LPCAT3 amplifying PUFA-PE incorporation and subsequent iron-dependent peroxidation.49 Furthermore, chlorogenic acid induces ferroptosis in HCC via the PTGS2/AKR1C3/GPX4 axis, demonstrating that pharmacological reprogramming of arachidonic acid metabolism can synergize with iron-catalyzed peroxidation to overwhelm antioxidant defenses.128 These findings collectively illustrate that iron metabolism reprogramming converges with the ACSL4-LPCAT3-PUFA peroxidation axis to create a synergistic pro-ferroptotic environment: iron overload provides the catalytic engine, while PUFA-PE-enriched membranes supply the oxidizable substrate.
The mechanistic understanding of iron metabolism reprogramming in HCC has catalyzed diverse therapeutic strategies exploiting the iron-ferroptosis nexus. Sorafenib triggers ferroptosis in part by inhibiting the HBXIP/SCD axis, shifting membrane phospholipids toward peroxidation-susceptible PUFAs—a mechanism inherently dependent on elevated intracellular iron in HCC cells.87 Beyond conventional therapies, innovative nanotechnology-based platforms simultaneously manipulate iron metabolism and immune responses. Tang et al developed a tumor-targeted FABP5/STING cascade nanosystem amplifying radiofrequency ablation‑induced ferroptosis while remodeling the intratumoral immune landscape.129 Wang et al engineered an immunogenic magnetothermodynamic platform using iron‑containing nanoparticles to synergize magnetothermal ablation with ferroptosis induction and reverse immunosuppression.130 Luo et al further demonstrated that targeting iron and lipid metabolism can enhance TACE efficacy in preclinical models.131 Looking forward, clinical translation of iron-targeted ferroptosis strategies requires nuanced consideration of the dual role of transferrin/TFRC in both ferroptosis sensitization and FAO-driven tumor promotion,123 etiology‑dependent variation in hepcidin regulation (particularly in MASLD-HCC),100,124 and environmental modifiers that may precondition ferroptotic vulnerability.127
Developing combinatorial regimens that simultaneously amplify iron-dependent Fenton chemistry, block compensatory antioxidant pathways (GPX4 and FSP1), and harness the immunogenic consequences of ferroptotic cell death represents the most promising avenue for maximizing therapeutic potential. Critically, the success of such strategies hinges on a deeper understanding of the PTMs codes governing iron metabolism nodes: targeting TFRC palmitoylation to disrupt its membrane localization, modulating FPN1 phosphorylation to enhance iron retention, or manipulating NCOA4 ubiquitination to fine‑tune ferritinophagy—each represents an underexplored opportunity to precisely calibrate intracellular iron availability. By integrating the iron axis with the previously discussed PUFA and antioxidant defense networks, future ferroptosis‑based therapies can move beyond single-node inhibition toward multi-layered, systems-level disruption of the metabolic vulnerabilities that sustain HCC.
It is worth highlighting a key paradox in targeting iron metabolism for ferroptosis induction: as demonstrated by Qian et al, the same transferrin/TFRC-mediated iron import that sensitizes cells to ferroptosis also activates the PHD2-PPARα-FAO pro-survival axis. This duality cautions against simplistic iron-loading strategies and argues for approaches that simultaneously amplify Fenton chemistry while blocking compensatory FAO activation—a combinatorial logic that has yet to be tested in preclinical models.
Drug Resistance and Metabolic Adaptation: Ferroptosis Evasion as a Central Resistance Mechanism in HCC
Resistance to first-line systemic agents remains a formidable clinical challenge in advanced HCC. A rapidly expanding body of evidence has converged on ferroptosis evasion as a central mechanism underpinning both intrinsic and acquired drug resistance.132,133 Sorafenib, originally developed as a multi-kinase inhibitor targeting RAF/MEK/ERK and VEGFR/PDGFR signaling. It was subsequently recognized as a potent inducer of ferroptosis through its inhibition of system Xc−-mediated cystine import and suppression of the HBXIP/SCD lipid desaturation axis.87,134 This dual identity—kinase inhibitor and ferroptosis inducer—means that resistance mechanisms frequently involve the reactivation or compensatory upregulation of anti-ferroptotic defenses.132,133,135 Lenvatinib, another first-line multi-kinase inhibitor, similarly engages ferroptotic pathways, yet its resistance profile encompasses both shared and mechanistically distinct molecular circuits7,8. Additionally, emerging therapeutic modalities—including regorafenib, hepatic arterial infusion chemotherapy (HAIC), immunotherapy, and radiotherapy—are each subject to ferroptosis-mediated resistance involving novel PTMs and TME remodeling.24,122 In this section, we systematically dissect the ferroptosis evasion mechanisms underlying resistance to each major therapeutic modality in HCC, organized by drug class (Figure 3).
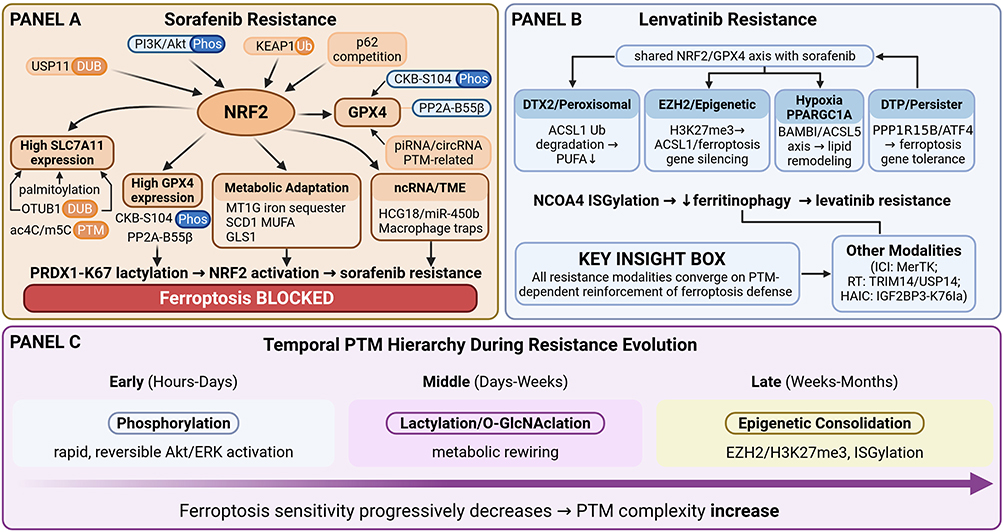 |
Figure 3 PTMs-dependent ferroptosis evasion as a convergent mechanism driving multi-drug resistance in HCC. (A) Sorafenib resistance architecture. NRF2 serves as the central hub (stabilized by USP11 DUB,43 PI3K/Akt Phos,44 p6241,42), driving SLC7A11 upregulation (Palm33/Ub34/ac4C36/m5C37) and GPX4 stabilization (CKB-S104 Phos,38 piRNA136/circRNA137). Additional modules include metabolic adaptation (MT1G,138 SCD1,87 GLS1139) and TME remodeling. Star box: PRDX1-K67 lactylation activates NRF2 to suppress ferroptosis.27 (B) Lenvatinib resistance. Beyond shared NRF2/GPX4 mechanisms, four distinct circuits are highlighted: DTX2-mediated ACSL1 ubiquitination,140 EZH2/H3K27me3 epigenetic silencing,141 hypoxia-driven PPARGC1A/BAMBI/ACSL5 rewiring,142 and ATF4-dependent drug-tolerant persister emergence.143 Star box: NCOA4 ISGylation suppresses ferritinophagy.28 Additional modalities (ICI/radiotherapy/HAIC) are summarized. (C) Proposed conceptual model based on the synthesis of current evidence; this temporal hierarchy has not been experimentally validated in longitudinal studies. Proposed “temporal PTMs hierarchy” model: early phosphorylation (hours–days) → intermediate lactylation/O-GlcNAcylation (days–weeks) → late epigenetic consolidation (weeks–months), with progressive narrowing of the therapeutic window. Symbols: Arrows (→) indicate regulatory or causal relationships. Downward arrows (↓) denote decreased activity or expression. Bold text denotes core regulatory proteins. In Panel D, the gradient arrow represents the progressive decrease in ferroptosis sensitivity during resistance evolution, with increasing PTM complexity over time. Panel D depicts the proposed “temporal PTMs hierarchy” model, a conceptual framework that has not been experimentally validated in longitudinal studies. |
Sorafenib Resistance: A Multi-Layered Ferroptosis Defense Architecture
NRF2-KEAP1 as the Master Transcriptional Hub
At the epicenter of sorafenib-induced ferroptosis resistance stands the NRF2-KEAP1 antioxidant response pathway. Upon sorafenib exposure, HCC cells activate NRF2 nuclear translocation, leading to transcriptional upregulation of a coordinated suite of ferroptosis-protective genes including SLC7A11, FTH1, heme oxygenase-1 (HMOX1), and metallothionein-1G (MT1G), thereby restoring GSH-GPX4 defense capacity and conferring robust ferroptosis resistance.133,138,144 Michilli et al recently performed comprehensive transcriptomic profiling of sorafenib-resistant HCC cells, revealing an NRF2-mediated redox and metabolic reprogramming signature characterized by coordinated upregulation of antioxidant, detoxification, and metabolic genes—confirming NRF2 activation as a systemic rather than single-gene resistance mechanism.145 Multiple upstream regulators converge on NRF2 stabilization in resistant cells: PIP5K1A suppresses ferroptosis and drives sorafenib resistance by directly stabilizing NRF2 protein;146 MCM4 potentiates ferroptosis evasion through NRF2 signaling;147 and FNDC5 activates the PI3K/AKT/NRF2 cascade to shield HCC cells from sorafenib-triggered lipid peroxidation.44 Conversely, pharmacological NRF2 suppression—by brusatol, curcumin (via p62-KEAP1-NRF2 axis), camptothecin, withaferin A, or silencing approaches—restores ferroptotic sensitivity and overcomes sorafenib resistance,148–150 while arsenic trioxide (ATO) induces ferroptosis and augments immunogenic cell death via cGAS-STING-IFN pathway activation in HCC, with NRF2 silencing further potentiating its efficacy.151,152 These findings collectively establish NRF2 as the master transcriptional hub of sorafenib-induced ferroptosis resistance and a high-priority therapeutic target for combination strategies.
SLC7A11: A Convergence Node of Epitranscriptomic, Post-Transcriptional, and PTMs Regulation
SLC7A11, the functional subunit of system Xc− and a direct NRF2 transcriptional target, has emerged as the single most frequently implicated molecular node in sorafenib-associated ferroptosis resistance.54 Beyond NRF2-driven transcriptional upregulation, SLC7A11 expression and function in resistant HCC cells are regulated by an extraordinarily diverse array of mechanisms spanning epitranscriptomic, post-transcriptional, and post-translational layers (Table 1). At the epitranscriptomic level, NAT10-mediated N4-acetylcytidine (ac4C) modification of SLC7A11 mRNA enhances its stability and translation, promoting ferroptosis resistance and metastasis.36 Moreover, SIX2 sensitizes HCC to sorafenib by modulating the METTL9-SLC7A11 axis, establishing a methyltransferase-dependent control point for ferroptosis resistance,58 while 5-methylcytosine (m5C) modification of the lncRNA MALAT1 promotes sorafenib resistance through ELAVL1/SLC7A11-mediated ferroptosis suppression, directly linking RNA methylation to drug resistance.37 At the non-coding RNA level, circTTC13 confers sorafenib resistance by sponging miR-513a-5p to upregulate SLC7A11;59 exosome-derived circUPF2 redeployes ferroptosis sensitivity through SLC7A11-centered exosomal communication between resistant and sensitive cell populations;60 and the lncRNA SLC7A11-AS1 directly stabilizes SLC7A11 mRNA as a cis-acting ferroptosis resistance factor.61 At the protein level, CAPG inhibits ferroptosis to drive sorafenib resistance through the WDR74-p53-SLC7A11 signaling pathway, mechanistically coupling cytoskeletal remodeling to ferroptosis evasion.68 This multi-layered regulatory architecture underscores the remarkable plasticity of ferroptosis resistance, wherein tumor cells deploy transcriptional, epitranscriptomic, post-transcriptional, and protein-level mechanisms in a coordinated fashion to maintain cystine import capacity under sorafenib pressure.
GPX4 Stabilization Through Diverse Molecular Mechanisms
GPX4, the terminal effector of the GSH-dependent ferroptosis defense, represents another critical resistance node stabilized through diverse mechanisms under sorafenib-selective pressure. Song et al identified that SSR2 (signal sequence receptor subunit 2) directly interacts with GPX4 protein to inhibit its degradation, establishing a novel protein–protein interaction that confers sorafenib resistance.153 In a mechanistically distinct pathway, Ji et al uncovered a circRNA-SORE/UBQLN1/GPX4 axis in which circRNA-SORE recruits the ubiquitin-like protein UBQLN1 to stabilize GPX4, preventing its proteasomal degradation.137 The RNA splicing factor DDX39B further contributes to GPX4-dependent resistance by facilitating the splicing and cytoplasmic export of GPX4 pre-mRNA, ensuring efficient GPX4 translation under sorafenib stress.154 Furthermore, PLAG1 interacts with GPX4 to conquer sorafenib-induced ferroptosis vulnerability through a PVT1/miR-195-5p axis-dependent mechanism.155 At the PTMs level, the protein phosphatase PP2A-B55β mediates mitochondrial GPX4 dephosphorylation through p53 retrograde signaling, and this dephosphorylation event is required for sorafenib-induced ferroptosis—suggesting that phospho-GPX4 represents a functionally active, pro-ferroptotic form whose dephosphorylation constitutes a resistance mechanism.38 Targeting GPX4 directly overcomes sorafenib resistance by inducing ferroptosis in human HCC cells,156 while the piRNA hsa_piR_016975 abates Maspin/GPX4-mediated ferroptosis to boost resistance through a novel piRNA-dependent mechanism.136 Collectively, GPX4 serves as a convergence point where post-transcriptional (circRNAs, piRNAs, splicing factors), protein–protein interaction (SSR2, PLAG1, UBQLN1), and phosphorylation-dependent mechanisms synergistically maintain ferroptosis defense.
Metabolic Adaptation: Iron Restriction, Lipid Desaturation, and Alternative Amino Acid Acquisition
Beyond the GSH–GPX4 axis, sorafenib-resistant HCC cells exhibit profound metabolic adaptation involving iron homeostasis reprogramming, lipid metabolic rewiring, and amino acid metabolism modulation. In the iron axis, SCARA5 (scavenger receptor class A member 5) deficiency inhibits ferroptosis by regulating iron homeostasis and results in sorafenib resistance, positioning SCARA5-mediated ferritin receptor function as a gatekeeper of ferroptotic iron supply.157 The CCT3/ACTN4/TFRC axis cooperatively inhibits TFRC-mediated iron endocytosis, reducing the labile iron pool and conferring ferroptosis resistance,158 while mitochondrial GCN5L1 acts as a positive regulator of iron homeostasis whose loss disrupts mitochondrial iron metabolism and contributes to resistance.159 In the lipid metabolism dimension, URI (unconventional prefoldin RPB5 interactor) alleviates TKI-induced ferroptosis by reprogramming lipid metabolism in p53 wild-type liver cancers, demonstrating that metabolic adaptation is genotype-dependent.160 Decreased lncRNA HNF4A-AS1 facilitates resistance to sorafenib-induced ferroptosis through comprehensive lipid metabolism reprogramming,80 while SLC27A4-mediated selective uptake of monounsaturated fatty acids (MUFAs) competitively displaces PUFAs from membrane phospholipids, reducing the oxidizable substrate pool.161 Targeting fatty acid synthase (FASN) modulates sorafenib sensitivity through ferroptosis, establishing de novo lipogenesis as a druggable resistance node.162 In amino acid metabolism, macropinocytosis serves as an alternative pathway of cysteine acquisition that mitigates sorafenib-induced ferroptosis, effectively bypassing system Xc− inhibition through bulk extracellular fluid uptake,163 while GLS1 inhibition by CB-839 overcomes 5-FU resistance by disrupting glutamine metabolism,139 and canagliflozin reduces chemoresistance through PKM2–c-Myc complex-mediated glutamine starvation.164 These diverse metabolic adaptation strategies highlight the remarkable metabolic plasticity of resistant HCC cells under sustained therapeutic pressure.
Non-Coding RNA Networks and Genome-Wide Screening Discoveries
The non-coding RNA (ncRNA) landscape of sorafenib resistance has expanded dramatically, revealing regulatory networks encompassing lncRNAs, circRNAs, miRNAs, and piRNAs that modulate virtually every node of the ferroptosis machinery.165,166 The lncRNA URB1-AS1 suppresses sorafenib-induced ferroptosis through a particularly elegant mechanism: driving ferritin phase separation via liquid-liquid phase separation (LLPS), thereby sequestering iron within biomolecular condensates and preventing its release into the labile iron pool.167 Complementing candidate-gene approaches, genome-wide CRISPR screens have emerged as powerful unbiased tools for discovering novel ferroptosis regulators. Tian et al performed an in vivo CRISPR screen identifying POU3F3 as a novel ferroptosis resistance regulator via retinoic acid signaling;168 Chen et al identified PSTK (phosphoseryl-tRNA kinase) as a protective regulator of chemotherapy-induced ferroptosis;169 Yao et al revealed that targeting TRIM34 enhances ferroptosis sensitivity and augments immunotherapy efficacy;170 and Li et al identified CRTC3 as a regulator of IFN-γ-induced ferroptosis.171 At the RNA processing level, the splicing factor SF3B4 was identified through genome-wide CRISPR screening as a driver of HCC,172 while the epigenetic regulator SETDB1 has been characterized as a key component of cancer stem cells and drug resistance.173 These unbiased screening approaches have uncovered previously unsuspected connections between retinoic acid signaling, tRNA metabolism, interferon response, and RNA splicing with ferroptosis regulation.
Tumor Microenvironment Remodeling and Immune Evasion
Ferroptosis evasion in sorafenib-resistant HCC does not occur in isolation but fundamentally reshapes the TME and immune landscape. Chao et al demonstrated that FAD synthase confers ferroptosis resistance and simultaneously restrains CD8+ T cell recruitment, mechanistically linking flavin metabolism, ferroptosis defense, and immune evasion through a single molecular node.174 The MVP-LCN2 axis triggers ferroptosis evasion to drive hepatocarcinogenesis and sorafenib resistance, with lipocalin-2 serving dual functions as an iron-sequestering protein and a TME-modulating secreted factor.175 Mu et al demonstrated that targeting ferroptosis-elicited inflammation suppresses HCC metastasis and enhances sorafenib efficacy, revealing that ferroptotic cell death can paradoxically promote metastasis through the release of pro-inflammatory DAMPs when occurring at sub-lethal levels.176 Sorafenib-induced macrophage extracellular traps via the ARHGDIG/IL4/PADI4 axis confer drug resistance through inhibiting ferroptosis, revealing that innate immune cells within the TME actively participate in resistance.177 These findings underscore that overcoming sorafenib resistance requires therapeutic strategies that simultaneously address tumor cell-intrinsic metabolic defenses and TME-mediated immunosuppressive barriers.
Lenvatinib Resistance: Shared Foundations and Distinct Ferroptosis Escape Circuits
NRF2-GPX4 Axis Activation and Lenvatinib-Tolerant Persister Cells
Lenvatinib resistance in HCC shares certain mechanistic features with sorafenib resistance—particularly NRF2 activation and GPX4 stabilization—but also involves distinct molecular circuits that reflect its unique pharmacological profile.178,179 At the NRF2 level, ponicidin, a natural diterpenoid, enhances ferroptosis and overcomes lenvatinib resistance by promoting KEAP1-mediated NRF2 degradation,180 while PP1A modulates lenvatinib plus ICI efficacy by inhibiting ferroptosis through dephosphorylation-mediated GPX4 stabilization, establishing phosphorylation as a critical PTMs governing combined immunotherapy response.67 A particularly important recent discovery is the identification of lenvatinib-tolerant persister (LTP) cells—a drug-tolerant subpopulation that survives initial treatment and serves as a reservoir for the emergence of stably resistant clones. LTP cells are characterized by PPP1R15B-ATF4 axis activation; targeting this axis induces GPX4-mediated ferroptosis and eliminates the persister population, providing a strategy for preventing resistance emergence rather than merely overcoming established resistance.143 This concept of ferroptosis-targeted persister cell elimination represents a paradigm shift in lenvatinib resistance management, moving from reactive treatment of resistance to proactive eradication of resistance reservoirs.
Peroxisomal Lipid Metabolism, Epigenetic Silencing, and Hypoxia-Driven Rewiring
In a mechanistically distinct pathway that sets lenvatinib resistance apart from sorafenib resistance, Zhang et al uncovered DTX2 as a lenvatinib resistance factor that attenuates ferroptosis by suppressing docosahexaenoic acid (DHA) biosynthesis through HSD17B4-dependent peroxisomal β-oxidation—a finding that connects peroxisomal lipid metabolism to ferroptosis resistance for the first time in HCC.140 Since DHA is a highly oxidation-susceptible omega-3 PUFA, its depletion via peroxisomal degradation effectively removes a key substrate for lipid peroxidation, thereby shielding lenvatinib-resistant cells from ferroptotic death. This peroxisomal axis is mechanistically orthogonal to the SCD1-mediated MUFA desaturation pathway described in sorafenib resistance, suggesting that different TKIs may select for distinct lipid metabolic adaptations. At the epigenetic level, EZH2, the catalytic subunit of the polycomb repressive complex 2, confers lenvatinib resistance by epigenetically suppressing ACSL1-mediated ferroptosis through H3K27me3-dependent transcriptional silencing.141 Notably, EZH2 also reduces sorafenib sensitivity through epigenetic regulation of TFR2,181 positioning EZH2 as a shared epigenetic resistance node across multiple TKIs with distinct downstream targets. The hypoxia-responsive PPARGC1A/BAMBI/ACSL5 axis has been identified as a driver of lenvatinib resistance through metabolic reprogramming under the chronically hypoxic conditions characteristic of HCC,142 while the lncRNA HAND2-AS1 promotes ferroptosis to reverse lenvatinib resistance via the TLR4/NOX2/DUOX2 axis, connecting innate immune signaling to ferroptosis regulation.182 Collectively, these findings demonstrate that lenvatinib resistance involves a multi-layered defense architecture encompassing GPX4 phospho-stabilization, peroxisomal DHA catabolism, H3K27me3-mediated gene silencing, and hypoxia-driven metabolic rewiring—providing multiple targetable vulnerabilities for combinatorial strategies.
Ferroptosis Resistance Across Emerging Therapeutic Modalities
Beyond the first-line agents sorafenib and lenvatinib, ferroptosis evasion has been increasingly implicated in resistance to a broader range of HCC therapeutic modalities, with recently discovered PTMs emerging as nodal regulators. In regorafenib-resistant HCC, Yang et al uncovered the ZNF207-PRDX1-NRF2 axis, in which ZNF207 facilitates lactylation of the peroxiredoxin PRDX1 at lysine 67, promoting its nuclear translocation and hyperactivation of NRF2 to drive ferroptosis escape—a finding that mechanistically links protein lactylation, a Warburg effect-derived PTMs, directly to multi-kinase inhibitor resistance.27 In the context of HAIC resistance, Zhu et al demonstrated that PARK7 drives IGF2BP3 lactylation at K76, enhancing its m6A-reading capacity to stabilize mRNAs of ferroptosis-resistance genes, establishing a lactylation-epitranscriptomic cascade governing treatment response.85 A bioinformatic cross-talk analysis further identified STMN1 and PRDX1 as therapeutic targets at the intersection of lactylation and ferroptosis in HCC prognosis.183 Beyond lactylation, a ROS-mediated oxidation–O-GlcNAcylation cascade governing ferroptosis, in which oxidative stress triggers sequential protein oxidation followed by O-GlcNAcylation that modulates ferroptosis sensitivity.184 In an equally novel finding, targeting USP18 overcomes acquired resistance by regulating NCOA4 deISGylation and ferroptosis; ISGylation of NCOA4—the ferritinophagy receptor—blocks its autophagic function, preventing iron release from ferritin, while USP18-mediated deISGylation restores NCOA4 activity and ferroptotic sensitivity.28
The intersection of ferroptosis with immunotherapy resistance represents a rapidly expanding frontier. Disruption of MerTK (a TAM receptor kinase) increases checkpoint inhibitor efficacy by enhancing ferroptosis and immune response,185 while matrix stiffness-dependent PD-L2 deficiency improves SMYD3/xCT-mediated ferroptosis and anti-PD-1 efficacy, connecting the physical TME to ferroptosis sensitivity and immunotherapy response.186 Wang et al demonstrated that cabozantinib combined with sulfasalazine targets ferroptosis to overcome resistant immunotherapy in advanced HCC, providing a clinically actionable combination strategy.187 Intriguingly, lenvatinib-activated NDUFA4L2/IL33/PADI4 pathway induces neutrophil extracellular traps that inhibit cuproptosis rather than ferroptosis in HCC, suggesting that different TKIs may engage distinct metal-dependent death pathways through TME-mediated resistance.188 In the radiotherapy context, Yue et al demonstrated that targeting the TRIM14/USP14 axis enhances radiotherapy efficacy by inducing GPX4 degradation and disrupting ferroptotic defense, establishing deubiquitinase targeting as a strategy for radiosensitization.189 These findings collectively underscore that ferroptosis resistance operates across the full therapeutic spectrum in HCC—from TKIs and immunotherapy to radiotherapy and locoregional modalities—necessitating modality-specific resistance biomarkers and tailored combinatorial approaches.
Notably, these resistance-associated PTMs—PRDX1 lactylation at K67, IGF2BP3 lactylation at K76, and NCOA4 ISGylation—are not isolated phenomena. They collectively reveal a deeper principle: under therapeutic pressure, tumor cells dynamically remodel their PTM landscapes to rapidly tune the activity and stability of the ferroptosis defense network. These findings serve as concrete mechanistic examples of how PTMs function as molecular switches governing ferroptosis sensitivity—a theme that will be systematically explored in the following section. The convergence of diverse therapeutic modalities on PTMs-mediated resistance mechanisms underscores the centrality of post-translational regulation as a nodal control point in HCC ferroptosis biology.
Therapeutic Strategies for Overcoming Ferroptosis Resistance
Natural Products as Ferroptosis-Restoring Agents
Natural products represent a rich and expanding source of ferroptosis-restoring agents in drug-resistant HCC.190 Decursin induces ferroptosis via the NRF2/GPX4/SLC11A2 axis and suppresses migration;191 curcumin modulates the p62-KEAP1-NRF2 pathway to sensitize resistant cells18; dihydroartemisinin (DHA) targets the ATF4-xCT pathway to induce ferroptosis across the hepatitis-to-HCC cascade;192,193 ponicidin overcomes lenvatinib resistance through KEAP1/NRF2 regulation;180 triptolide disrupts CARMA3 signaling to reverse sorafenib resistance;194 zingiberensis new saponin targets the lncRNA TCONS-00026762/AKR1C1 axis by modulating autophagy and ferroptosis;195 tiliroside targets TBK1 to sensitize sorafenib treatment;196 glycyrrhizic acid attenuates sorafenib resistance by targeting mTOR signaling;197 and dicoumarol sensitizes HCC cells to erastin-induced ferroptosis.198 The natural exosome-like nanovesicles from Smilax China rhizome induce mitophagy-dependent ferroptosis via GPX4/ACSL4 axis, representing a biomimetic delivery approach for ferroptosis-inducing phytochemicals.199 These natural products engage distinct resistance nodes—NRF2, GPX4, system Xc−, mTOR, autophagy, and lipid metabolism—highlighting the pharmacological diversity available for combinatorial resistance-overcoming strategies (Figure 4).
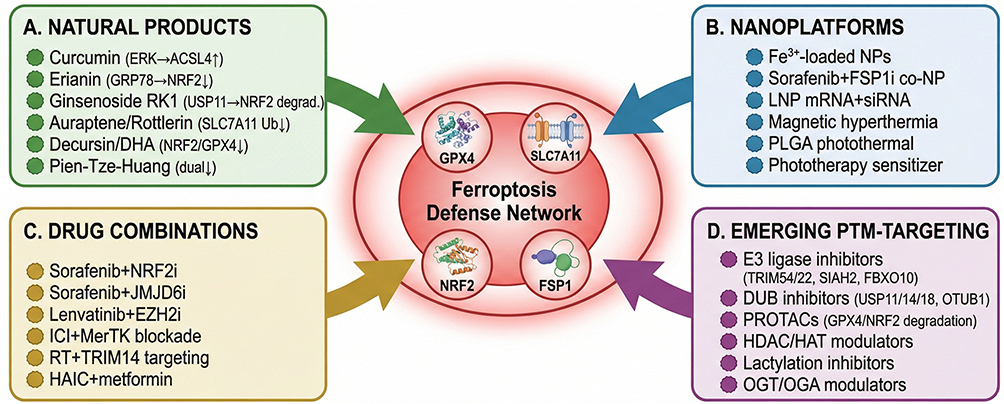 |
Figure 4 Therapeutic strategies targeting the PTMs-ferroptosis axis to overcome drug resistance in HCC. Hub-and-spoke layout with the ferroptosis defense network (GPX4/SLC7A11/NRF2/FSP1) at center. Four converging strategies: (A) Natural products restoring ferroptosis sensitivity (curcumin,102 erianin,90 ginsenoside RK1,118 auraptene,34 rottlerin,35 decursin,191 DHA,192,193 Pien-Tze-Huang92) targeting NRF2/SLC7A11/GPX4/ACSL4 nodes. (B) Nanoplatforms enabling multi-axis targeting: Fe3+-loaded NPs,130 sorafenib/FSP1i co-NPs,116 LNP mRNA/siRNA co-delivery,200 magnetic hyperthermia,201 and photothermal systems.202 (C) Rational drug combinations: sorafenib + NRF2i,88 lenvatinib + EZH2i,141 ICI + MerTK blockade,185 RT + TRIM14 targeting,189 HAIC + metformin.203 (D) Proposed therapeutic strategies; these modalities have not yet been tested in HCC ferroptosis models. Emerging PTM-targeting modalities: E3 ligase inhibitors, DUB inhibitors, PROTACs, HDAC/HAT modulators, lactylation inhibitors, and OGT/OGA modulators. Symbols: Arrows (→) indicate pathway connections or translational workflow direction. Upward arrows (↑) denote upregulation and downward arrows (↓) denote downregulation or inhibition. Colored bullets distinguish four therapeutic strategy categories: green (natural products), blue (nanoplatforms), gold (rational drug combinations), and purple (emerging PTM-targeting modalities). Bold text indicates specific therapeutic agents and target proteins. Curved arrows indicate convergence of each strategy category on the central ferroptosis defense network. The strategies listed in (D) (E3 ligase inhibitors, PROTACs, lactylation inhibitors, and OGT/OGA modulators for ferroptosis targeting) represent proposed therapeutic concepts that have not yet been validated in HCC ferroptosis models. |
Nanotechnology-Based Multi-Target Platforms
Nanotechnology-based platforms offer particularly promising approaches for overcoming ferroptosis resistance through simultaneous multi-target engagement. Liao et al developed optimized lipid nanoparticles for co-delivery of mRNA and siRNA therapeutics in refractory liver cancer, enabling concurrent gene silencing and protein expression within resistant tumor cells.200 Chen et al engineered a multi-functional metal-coordinated nanoplatform for synergistic ferroptosis enhancement through controlled iron release and antioxidant depletion.204 Tang et al created intracellular magnetic hyperthermia nanoparticles that sensitize sorafenib to orthotopic HCC via amplified ferroptosis through magnetically-induced Fenton chemistry enhancement.201 Dong et al designed tumor-targeted PLGA nanospheres combining lenvatinib with photothermal/photodynamic therapy, achieving multi-modal ferroptosis induction,202 while Qin et al developed a phototherapy nanosensitizer-unlocked multi-target strategy to inhibit tumor stemness and potentiate sorafenib response.205 These nanotechnology approaches share a common design principle: co-delivering ferroptosis inducers with agents that simultaneously dismantle compensatory resistance mechanisms, thereby converting the “whack-a-mole” challenge of single-target therapy into a coordinated multi-hit assault on the ferroptosis defense network.
Rational Drug Combinations and Ubiquitin-Proteasome Targeting
Rational drug combinations represent a particularly promising strategy for overcoming ferroptosis resistance in HCC. Cabozantinib combined with sulfasalazine targets ferroptosis to overcome resistant immunotherapy;187 triptolide co-treatment with RSL3 induces both apoptosis and ferroptosis, engaging dual death pathways;206 metformin promotes ferroptosis and sorafenib sensitivity via ATF4/STAT3;203 and the JMJD6/PPARγ/GPX4 axis has been identified as a targetable vulnerability for enhancing ferroptosis and therapeutic efficacy.207 A particularly innovative therapeutic frontier is the targeting of ubiquitin–proteasome circuits to dismantle GPX4 stability. The TRIM14/USP14 axis enhances radiotherapy efficacy by inducing GPX4 degradation and disrupting ferroptotic defense,189 and COPS5 triggers ferroptosis defense by stabilizing MK2 in HCC208—both establishing deubiquitinase targeting as a viable strategy for dismantling ferroptosis resistance. The concept of targeting drug-tolerant persister cells through PPP1R15B/ATF4-mediated GPX4 ferroptosis induction represents a proactive paradigm for preventing, rather than merely treating, resistance emergence.143 Looking forward, the integration of predictive biomarkers—such as NRF2 activation status, SLC7A11 expression levels, GPX4 phosphorylation status, and ACSL4/ACSL1 epigenetic silencing—with rational combinatorial regimens tailored to the specific therapeutic modality and resistance mechanism will be essential for realizing the clinical potential of ferroptosis-targeted strategies in HCC.
In summary, ferroptosis-mediated drug resistance in HCC, despite its extraordinary molecular complexity across therapeutic modalities, reveals a unifying principle: resistance mechanisms converge on the reinforcement of the three core ferroptosis defense axes—System Xc−-GSH-GPX4, FSP1-CoQ10, and ACSL4-PUFA peroxidation—through multi-layered regulatory inputs. Sorafenib resistance exemplifies this through NRF2-driven transcriptional reprogramming of SLC7A11/GPX4, multi-layered SLC7A11 stabilization, and metabolic plasticity encompassing iron restriction and lipid desaturation. Lenvatinib resistance, while sharing NRF2/GPX4-dependent features, distinguishes itself through peroxisomal DHA catabolism (DTX2/HSD17B4) and EZH2-mediated epigenetic silencing of ACSL1—demonstrating that different TKIs select for distinct metabolic adaptations that nonetheless serve the same defensive end. Resistance to emerging modalities—regorafenib, HAIC, immunotherapy, and radiotherapy—introduces novel PTMs including lactylation (PRDX1-K67la, IGF2BP3-K76la), O-GlcNAcylation, and ISGylation (NCOA4) as rapid-response mechanisms for ferroptosis evasion. Critically, these resistance-associated molecular events—NRF2-driven transcription, m6A/m5C/ac4C RNA modifications, protein lactylation, O-GlcNAcylation cascades, and ISGylation—represent quintessential examples of post-translational and epitranscriptomic modifications serving as nodal control points of ferroptosis sensitivity.
Methodological Caveats and Future Directions
A critical caveat, however, is the heterogeneity in experimental models across the studies summarized above: resistance mechanisms identified in established resistant cell lines may not fully recapitulate the gradual, multi-step resistance evolution observed clinically, and findings from immunodeficient xenograft models cannot capture the immunological dimensions of ferroptosis resistance—particularly the complex crosstalk between ferroptotic tumor cells and CD8+ T cells, macrophages, and neutrophils within the tumor immune microenvironment. Notably, conflicting observations persist in the literature regarding the relative contribution of individual resistance nodes across different model systems; for instance, while some studies report NRF2 activation as essential for sorafenib resistance, others demonstrate NRF2-independent mechanisms under identical treatment conditions in distinct HCC cell lines, suggesting context-dependent hierarchies that remain mechanistically unresolved.
The field would benefit from standardized resistance-induction protocols and greater adoption of immunocompetent preclinical models to validate these mechanisms. Addressing these caveats will not only strengthen the mechanistic bridge to the subsequent discussion of PTMs-regulated ferroptosis in HCC—which offers a unifying framework for understanding how tumor cells rapidly tune ferroptosis sensitivity through post-translational mechanisms—but also provide a compelling rationale for developing combinatorial strategies that simultaneously dismantle multiple layers of the ferroptosis defense network. Such strategies should target not just individual nodes in isolation, but the regulatory logic that coordinates their reinforcement under therapeutic pressure.
Conclusion and Future Perspectives
Integrative Summary: The PTMs-Ferroptosis Nexus in HCC
The collective evidence reviewed herein establishes that ferroptosis in HCC is not governed by a single linear pathway but rather by three interconnected yet biochemically distinct defense systems—each subject to multi-layered post-translational regulation that dynamically calibrates the ferroptotic threshold. The canonical system Xc−-GSH-GPX4 axis, the most extensively characterized defense module, is regulated at virtually every node by PTMs: SLC7A11 stability is controlled by palmitoylation and ubiquitination,43,64 GPX4 is stabilized through phosphorylation at S104 by CKB and protected from ubiquitin-mediated degradation by multiple mechanisms,38,136,156 and GSH biosynthesis itself is subject to metabolic and PTMs-level modulation via CHAC1-dependent degradation.86,87 The ACSL4-LPCAT3 pro-ferroptotic axis exhibits perhaps the most striking example of combinatorial PTM control: ACSL4 is simultaneously regulated by SIAH2- and FBXO10-mediated ubiquitination, HAT1/HDAC2-dependent acetylation at K383, O-GlcNAcylation, SIRT5-mediated desuccinylation, and ERK-driven phosphorylation47,48,50,102—with the combinatorial state of these modifications determining whether ACSL4 functions as a pro-tumorigenic or pro-ferroptotic effector. The FSP1/CoQ10 and DHODH parallel pathways, which provide GPX4-independent radical-trapping defense, are likewise governed by TRIM54-mediated ubiquitination, Trim69-dependent degradation, myristoylation-dependent membrane anchoring, and riboflavin metabolism-linked stabilization.51,52,84,85 We propose that the collective PTMs acting on a given ferroptosis-regulatory protein—such as the simultaneous ubiquitination, acetylation, O‑GlcNAcylation, succinylation, and phosphorylation of ACSL4—constitute a combinatorial PTMs code. This code, rather than any single modification in isolation, determines the net functional output (pro‑ferroptotic vs. anti‑ferroptotic) and dynamically calibrates the ferroptotic threshold in HCC cells.
Superimposed upon these cell-autonomous defense systems, the unique iron metabolism landscape of the liver—characterized by TFRC upregulation, ferritin accumulation, ferroportin downregulation, and pathological suppression of the hepcidin-ferroportin axis—creates a constitutively elevated labile iron pool that primes HCC cells for ferroptotic execution.123,125,126 This iron-rich milieu intersects with PTMs-regulated ferritinophagy through NCOA4, whose ISGylation status determines the rate of iron release from ferritin stores, establishing a direct PTMs-iron-ferroptosis regulatory circuit.28 Environmental factors, including dietary palmitate exposure and persistent organic pollutants such as PFOS, further reshape the ferroptosis vulnerability landscape through metabolic reprogramming of hepcidin expression and lipid peroxidation substrates,124,127 underscoring that the PTMs-ferroptosis nexus in HCC operates within a broader metabolic and environmental context.
The drug resistance landscape further reinforces the centrality of PTMs-regulated ferroptosis in HCC biology. Across all therapeutic modalities examined—sorafenib, lenvatinib, ICIs, radiotherapy, and locoregional therapies—resistance mechanisms converge on the reinforcement of one or more ferroptosis defense axes through PTMs-dependent mechanisms. In sorafenib resistance, NRF2 stabilization via USP11-mediated deubiquitination and PI3K/Akt-driven phosphorylation constitutes the master transcriptional hub,43,44,146 while SLC7A11 is stabilized through an extraordinary diversity of epitranscriptomic and post-translational inputs including ac4C modification, m5C methylation, palmitoylation, and deubiquitination.36,37,43,64 In lenvatinib resistance, EZH2-mediated H3K27me3 silencing of ACSL1 and ferroptosis-related genes provides an epigenetic dimension to PTMs-regulated ferroptosis evasion,142,182 while hypoxia-driven PPARGC1A/BAMBI/ACSL5 axis activation and PPP1R15B/ATF4-dependent persister cell emergence represent mechanistically distinct escape circuits.141,183 Critically, the recently discovered resistance-associated PTMs—PRDX1 lactylation at K67, IGF2BP3 lactylation at K76, and NCOA4 ISGylation—share a common property: they are metabolite-sensitive modifications that directly link the altered metabolic state of resistant tumor cells to ferroptosis evasion, thereby constituting a metabolic PTMs code for drug resistance.27,28,85
Knowledge Gaps and Technical Challenges
Despite the substantial progress documented in this review, several critical knowledge gaps and technical challenges must be addressed before the PTMs–ferroptosis paradigm can be fully translated into clinical benefit. First and foremost, the crosstalk and hierarchical relationships among different PTMs on the same ferroptosis-regulatory protein remain largely unexplored. As illustrated by the case of ACSL4—which is simultaneously subject to at least five distinct PTMs types—it remains unknown whether these modifications operate independently, compete for the same or adjacent lysine residues, or form obligate sequential cascades in which one PTMs event primes or prevents another. Similarly, the observation that GPX4 stability is governed by both CKB-mediated phosphorylation at S104 and multiple ubiquitin-dependent degradation pathways raises the unresolved question of whether phosphorylation directly antagonizes ubiquitination or whether these modifications are regulated by independent upstream signals.38,136 Resolving these questions will require systematic application of quantitative PTMs proteomics—including parallel enrichment of phospho-, ubiquitin-, and acetyl-peptides from the same biological samples—combined with site-directed mutagenesis of individual modification sites to dissect combinatorial logic.
A second major gap—and one that we propose to frame as the “temporal PTMs hierarchy” model—concerns the temporal dynamics of PTMs-mediated ferroptosis regulation during the acquisition of drug resistance. The current literature provides largely static snapshots—comparing sensitive versus resistant cell lines or treatment-naïve versus post-treatment tumors—without capturing the progressive evolution of the PTMs landscape through the drug-tolerant persister (DTP) state to stably resistant populations. The identification of lenvatinib-tolerant persister cells with ATF4-dependent ferroptosis evasion141,183 and the emergence of PARK7-driven IGF2BP3 lactylation in HAIC-resistant cells85 suggest that different PTMs classes may dominate at distinct stages of resistance evolution—rapid, reversible phosphorylation events during early drug exposure, metabolite-driven lactylation and O-GlcNAcylation during the persister state, and epigenetic consolidation through histone methylation (EZH2/H3K27me3) in stably resistant clones.142,181,182,187 This temporal PTMs hierarchy, if validated, would provide a conceptual framework for understanding how resistance evolves stepwise and for designing stage‑specific intervention strategies. Testing this “temporal PTMs hierarchy” model will require longitudinal single-cell multi-omics profiling of HCC cells at defined timepoints during drug exposure.
Third, the overwhelming reliance on two-dimensional cell culture models and xenograft systems in the ferroptosis field raises concerns about the physiological relevance of many reported mechanisms. Standard cell culture conditions impose non-physiological oxygen tensions (approximately 21% versus 3–8% in the liver parenchyma), supraphysiological glucose concentrations, and absence of stromal interactions—all of which profoundly influence redox balance, iron homeostasis, and lipid metabolism, the very parameters that govern ferroptotic susceptibility.209,210 The TME dimensions of ferroptosis regulation—including macrophage-mediated iron recycling, T cell-derived IFNγ-induced ferroptosis sensitization, and CFA remodeling of the lipid microenvironment45,98,177—are difficult to recapitulate in conventional models. The adoption of patient-derived organoids, syngeneic mouse models with intact immune systems, and emerging spatial multi-omics technologies (spatial transcriptomics, imaging mass cytometry) will be essential to validate key PTMs–ferroptosis regulatory nodes identified in cell-based studies and to map the spatial heterogeneity of ferroptosis vulnerability within the complex HCC ecosystem.
Fourth, the etiological heterogeneity of HCC represents a critical but underexplored variable in the PTMs–ferroptosis equation. The rapidly increasing incidence of metabolic dysfunction-associated steatotic liver disease (MASLD)-driven HCC—now representing the fastest-growing etiological category—introduces a fundamentally different metabolic backdrop characterized by chronic lipotoxicity, insulin resistance, and altered iron-handling capacity. Kim et al demonstrated that chronic palmitate exposure downregulates glutamine-driven hepcidin expression, conferring ferroptosis resistance through a mechanism entirely distinct from the NRF2-dependent pathways that dominate in viral hepatitis-associated HCC.124 Meanwhile, Classon et al revealed through single-cell RNA sequencing and spatial transcriptomics that ACSL4 is heterogeneously enriched in MASLD-HCC tumors relative to non-cancerous MASLD tissue, suggesting etiology-specific ferroptosis vulnerability profiles.100 Whether the PTMs landscape—particularly metabolite-sensitive modifications such as lactylation, O-GlcNAcylation, and succinylation—differs systematically between viral, alcohol-related, and MASLD-driven HCC remains virtually unexplored, yet answering this question is essential for the rational design of etiology-stratified ferroptosis-based therapies.
Finally, the field lacks validated ferroptosis-specific biomarkers suitable for clinical application. Whereas lipid peroxidation end-products (malondialdehyde, 4-hydroxynonenal), PTGS2/COX-2 induction, and transferrin receptor expression have been employed as surrogate markers of ferroptosis in preclinical studies,13,14 none has been prospectively validated in clinical cohorts for the purpose of patient stratification or treatment monitoring. The PTMs-centric perspective of this review suggests that site-specific PTMs signatures on ferroptosis-regulatory proteins—such as the phosphorylation status of GPX4-S104, the ubiquitination pattern of SLC7A11, or the lactylation level of PRDX1-K67—could serve as more mechanistically informative biomarkers than bulk metabolite measurements. However, the development of clinically deployable PTMs-based assays will require advances in targeted mass spectrometry workflows, antibody development against specific modification sites, and standardized sample processing protocols compatible with formalin-fixed, paraffin-embedded clinical specimens (Figure 5).
 |
Figure 5 From bench to bedside: a clinical translation roadmap for PTMs-ferroptosis-targeted therapy in HCC. Proposed translational roadmap representing the authors’ conceptual synthesis; projected timelines are estimates based on current preclinical progress. A four-column matrix maps translational barriers from Knowledge Gaps through Required Research and Enabling Technologies to Clinical Goals across four rows: Row 1: PTMs crosstalk hierarchy (eg, five concurrent PTMs on ACSL4) → multi-PTM proteomics and mutagenesis → SEPTM/AI prediction → PTM-informed target selection.45–50 Row 2: Temporal PTMs dynamics during resistance evolution → longitudinal single-cell multi-omics → scRNA-seq/spatial metabolomics/live-cell reporters → optimal intervention timing.85,143 Row 3: Model limitations (2D culture artifacts) → patient-derived organoids and syngeneic models → spatial transcriptomics/imaging mass cytometry → in vivo validation.209,210 Row 4: Etiology-specific PTM landscapes and biomarker gap → stratified cohort studies → targeted mass spectrometry/anti-PTM antibodies/liquid biopsy → etiology-guided patient selection (GPX4-pS104,38 PRDX1-K67la27). Bottom: translational vision (descriptive cataloging → systems-level PTM code → precision ferroptosis medicine) and projected timeline (mechanistic discovery → Phase I/II within 4–6 years). Symbols: Arrows (→) indicate the translational progression from knowledge gaps through required research and enabling technologies to clinical goals. Color-coded column headers denote four translational stages: red (knowledge gaps), Orange (required research), blue (enabling technologies), and green (clinical goals). Bold text indicates row category labels and column headers. The question mark in “Approval?” indicates uncertainty in the projected timeline. The bottom timeline bar illustrates the estimated progression from current discovery to potential clinical approval. This figure represents a proposed translational roadmap based on the authors’ conceptual synthesis; projected timelines are estimates based on current preclinical progress. |
Clinical Translation Prospects
Despite these challenges, several features of the PTMs-ferroptosis regulatory network in HCC offer tangible opportunities for near-term clinical translation. The convergence of multiple resistance mechanisms on the NRF2-KEAP1 hub across therapeutic modalities identifies NRF2 as a particularly attractive pharmacological target, and several NRF2 pathway inhibitors (brusatol, ML385, trigonelline) have demonstrated synergistic activity with sorafenib in preclinical HCC models.44,88,147 The ubiquitin-proteasome system, which regulates the stability of SLC7A11, GPX4, ACSL4, FSP1, and NRF2, offers a particularly rich landscape of druggable targets. E3 ligases (TRIM54, TRIM22, SIAH2, FBXO10) and deubiquitinases (USP11, USP14, USP18) represent emerging targets for which small-molecule inhibitors or proteolysis-targeting chimeras (PROTACs) could be developed to selectively destabilize ferroptosis-suppressive proteins or stabilize pro-ferroptotic effectors (Table 1). The advent of PROTAC technology in oncology provides a particularly promising framework for degrading previously undruggable ferroptosis regulators with high selectivity.
The nanoplatform-based strategies reviewed in Therapeutic Strategies for Overcoming Ferroptosis Resistance represent the most clinically advanced ferroptosis-exploiting approaches in HCC. Multi-functional nanocarriers capable of simultaneous drug delivery (sorafenib, FSP1 inhibitors), iron supply (Fe3+-loaded nanoparticles), GSH depletion, and immune microenvironment remodeling have achieved impressive preclinical efficacy by simultaneously targeting multiple ferroptosis defense axes. The integration of photothermal therapy and magnetic hyperthermia with ferroptosis induction further amplifies therapeutic potency through synergistic ROS generation and membrane destabilization.200,201,204 For these platforms to advance toward clinical trials, standardized manufacturing protocols, pharmacokinetic characterization in large-animal models, and biomarker-guided patient selection criteria will be essential. The combination of ferroptosis inducers with ICIs holds particular promise, given the immunostimulatory properties of ferroptotic cell death through DAMPs release and dendritic cell maturation,95,182 and the emerging evidence that ferroptosis evasion underlies resistance to anti-PD-1/PD-L1 therapy in HCC.186,187
Looking further ahead, the integration of artificial intelligence (AI) and multi-omics profiling offers transformative potential for the PTMs-ferroptosis field. Machine learning algorithms trained on integrated datasets encompassing PTMs proteomics, metabolomics, single-cell transcriptomics, and spatial profiling could generate predictive models of ferroptosis vulnerability for individual patient tumors, enabling truly personalized therapeutic selection. The genome-wide CRISPR screens that have already identified novel ferroptosis regulators in HCC—including PSTK, TRIM34, CRTC3, and SF3B4169–172—could be extended to systematically map the PTMs enzymes (kinases, E3 ligases, acetyltransferases, glycosyltransferases) whose loss or gain of function most potently sensitizes HCC cells to ferroptosis. Such functional genomics approaches, when integrated with phosphoproteomic and ubiquitinomic profiling, would provide an unbiased catalog of actionable PTMs targets and accelerate the transition from descriptive cataloging of individual PTMs events to a systems-level understanding of the ferroptosis regulatory network.
In conclusion, the PTMs-ferroptosis nexus in HCC represents a rapidly maturing field that has transitioned from the initial recognition of individual PTMs events to an increasingly integrated understanding of how multi-layered post-translational regulation orchestrates ferroptotic susceptibility, metabolic reprogramming, and drug resistance. The central insight that emerges from this review is that ferroptosis defense in HCC is not a fixed property but a dynamically tunable state, continuously recalibrated by the combinatorial PTMs landscape on key regulatory nodes. This dynamic nature, while increasing biological complexity, simultaneously creates therapeutic opportunity: by targeting the PTMs enzymes that maintain the ferroptosis-resistant state—rather than individual ferroptosis effectors alone—it may be possible to collapse multiple defense layers simultaneously and restore ferroptotic vulnerability across diverse resistance contexts. Realizing this potential will require concerted efforts spanning mechanistic biochemistry, multi-omics technology development, etiology-stratified preclinical modeling, and biomarker-guided clinical trial design. As the ferroptosis field moves toward its second decade, the convergence of PTMs biology, cancer metabolism, and translational oncology holds promise for transforming ferroptosis from a laboratory curiosity into a clinically actionable therapeutic modality for patients with HCC.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
There is no funding to report.
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. Hwang SY, Danpanichkul P, Agopian V, et al. Hepatocellular carcinoma: updates on epidemiology, surveillance, diagnosis and treatment. Clin Mol Hepatol. 2025;31(Suppl):S228–33. doi:10.3350/cmh.2024.0824
2. Kale SR, Karande G, Gudur A, Garud A, Patil MS, Patil S. Recent trends in liver cancer: epidemiology, risk factors, and diagnostic techniques. Cureus. 2024;16(10):e72239. doi:10.7759/cureus.72239
3. Song P, Tang W, Kokudo N. Expert consensus on sequential surgery after immune-targeted conversion therapy for advanced hepatocellular carcinoma in China. Biosci Trends. 2024;18(6):495–496.
4. Zeng RW, Ong CEY, Ong EYH, et al. Global prevalence, clinical characteristics, surveillance, treatment allocation, and outcomes of alcohol-associated hepatocellular carcinoma. Clin Gastroenterol Hepatol. 2024;22(12):2394–2402.e2315. doi:10.1016/j.cgh.2024.06.026
5. Younossi ZM, Kalligeros M, Henry L. Epidemiology of metabolic dysfunction-associated steatotic liver disease. Clin Mol Hepatol. 2025;31(Suppl):S32–s50. doi:10.3350/cmh.2024.0431
6. Magyar CTJ, O’Kane GM, Aceituno L, et al. Liver transplantation for hepatocellular carcinoma: an expanding cornerstone of care in the era of immunotherapy. J Clin Oncol. 2025;43(5):589–604. doi:10.1200/JCO.24.00857
7. Ma YN, Jiang X, Song P, Tang W. Neoadjuvant therapies in resectable hepatocellular carcinoma: exploring strategies to improve prognosis. Biosci Trends. 2024;18(1):21–41. doi:10.5582/bst.2023.01436
8. Yoo HJ, Yoo JJ, Kim SG, Kim YS. Current perspectives on the pharmacological treatment of advanced hepatocellular carcinoma: a narrative review. Ewha Med J. 2024;47(4):e53. doi:10.12771/emj.2024.e53
9. Hwang SY, Lee SL, Liu H, Lee SS. Second-line treatment after failure of immune checkpoint inhibitors in hepatocellular carcinoma: tyrosine kinase inhibitor, retrial of immunotherapy, or locoregional therapy? Liver Cancer. 2024;13(3):246–255. doi:10.1159/000534303
10. Piscaglia F, Masi G, Martinelli E, et al. Atezolizumab plus bevacizumab as first-line treatment of unresectable hepatocellular carcinoma: interim analysis results from the phase IIIb AMETHISTA trial. ESMO Open. 2025;10(2):104110. doi:10.1016/j.esmoop.2024.104110
11. Allaire M, Thiam EM, Amaddeo G, et al. Real-world outcomes of atezolizumab-bevacizumab in hepatocellular carcinoma: the prospective French CHIEF cohort. Liver Int. 2025;45(10):e70337. doi:10.1111/liv.70337
12. Rossari F, Tada T, Suda G, et al. Disease etiology impact on outcomes of hepatocellular carcinoma patients treated with atezolizumab plus bevacizumab: a real-world, multicenter study. Liver Cancer. 2024;13(5):522–536. doi:10.1159/000537915
13. Galluzzi L, Vitale I, Aaronson SA, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25(3):486–541. doi:10.1038/s41418-017-0012-4
14. Liu RJ, Yu XD, Yan SS, Guo ZW, Zao XB, Zhang YS. Ferroptosis, pyroptosis and necroptosis in hepatocellular carcinoma immunotherapy: mechanisms and immunologic landscape (Review). Int J Oncol. 2024;64(6):63. doi:10.3892/ijo.2024.5651
15. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi:10.1016/j.cell.2012.03.042
16. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22(4):266–282. doi:10.1038/s41580-020-00324-8
17. Chen Q, Wang J, Li N, Cai H, Lu Y. Crosstalk between ROS and ferroptosis, cuproptosis, and PANoptosis in liver cancer: mechanisms and therapeutic strategies. Crit Rev Oncol/Hematol. 2025;215:104924. doi:10.1016/j.critrevonc.2025.104924
18. Liang J, Wang R, Wu H, Huang Z, Zhang R, Jiang F. Copper metabolism and cuproptosis: broad perspectives in the treatment of hepatocellular carcinoma. Front Oncol. 2025;15:1555858. doi:10.3389/fonc.2025.1555858
19. Zhu K, Cai Y, Lan L, Luo N. Tumor metabolic reprogramming and ferroptosis: the impact of glucose, protein, and lipid metabolism. Int J Mol Sci. 2024;25(24):13413. doi:10.3390/ijms252413413
20. Sun J, Liu J, Chen F, Wang X, Wu G. Exploring the significance of fatty acid metabolism reprogramming in the pathogenesis of cancer and anticancer therapy. Annals Med. 2025;57(1):2445774. doi:10.1080/07853890.2024.2445774
21. Cui K, Wang K, Huang Z. Ferroptosis and the tumor microenvironment. J Exp Clin Cancer Res. 2024;43(1):315. doi:10.1186/s13046-024-03235-0
22. Xie X, Pang Q, Luo L. Post-translational modifications in ferroptosis: mechanisms and therapeutic potential. Int J Biol Sci. 2026;22(4):1868–1905. doi:10.7150/ijbs.120624
23. Wang Y, Yan D, Liu J, Tang D, Chen X. Protein modification and degradation in ferroptosis. Redox Biol. 2024;75:103259. doi:10.1016/j.redox.2024.103259
24. Bolandi S, Dodge S, Zahed Z, et al. Epigenetic and post-translational modifications in ferroptosis regulation and hepatocellular carcinoma: new frontiers in therapeutic targeting. Pathol Res Pract. 2025;270:155991. doi:10.1016/j.prp.2025.155991
25. Wang Y, Hu J, Wu S, et al. Targeting epigenetic and posttranslational modifications regulating ferroptosis for the treatment of diseases. Signal Transduct Target Ther. 2023;8(1):449. doi:10.1038/s41392-023-01720-0
26. Chunlian Z, Qi W, Rui Z. The role of pyruvate kinase M2 posttranslational modification in the occurrence and development of hepatocellular carcinoma. Cell Biochem Funct. 2024;42(7):e4125. doi:10.1002/cbf.4125
27. Yang T, Zhang S, Nie K, et al. ZNF207-driven PRDX1 lactylation and NRF2 activation in regorafenib resistance and ferroptosis evasion. Drug Resistance Updates. 2025;82:101274.
28. Ye S, Chen J, Zheng Y, et al. Targeting USP18 overcomes acquired resistance in hepatocellular carcinoma by regulating NCOA4 deISGylation and ferroptosis. Cell Death Dis. 2025;16(1):448.
29. Jiang Q, Peng C, Mao W, et al. METTL3-mediated m6A modification of FDX1 confers resistance to cuproptosis and promotes hepatocellular carcinoma progression. Commun Biol. 2026;9(1):222.
30. Stockwell BR. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell. 2022;185(14):2401–2421.
31. Zhan T, Liu Y, Duan S, et al. Targeting HCG18 counteracts ferroptosis resistance via blocking the miR-30a-5p/RRM2/GSS pathway in hepatocellular carcinoma. Int J Biol Sci. 2025;21(6):2550–2567. doi:10.7150/ijbs.104127
32. Yang WS, SriRamaratnam R, Welsch ME, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1–2):317–331. doi:10.1016/j.cell.2013.12.010
33. Shi Z, Li Z, Jin B, et al. Loss of LncRNA DUXAP8 synergistically enhanced sorafenib induced ferroptosis in hepatocellular carcinoma via SLC7A11 de-palmitoylation. Clin Transl Med. 2023;13(6):e1300. doi:10.1002/ctm2.1300
34. Li D, Li Y, Chen L, et al. Natural product auraptene targets SLC7A11 for degradation and induces hepatocellular carcinoma ferroptosis. Antioxidants. 2024;13(8):1015.
35. Luo H, Jin X, Gao C, et al. Rottlerin triggers dual degradation of SLC7A11 and GPX4 to drive ferroptosis and chemosensitization in hepatocellular carcinoma. Cell Death Discov. 2026;12(1):89. doi:10.1038/s41420-026-02942-1
36. Wang C, Liu J, Chang H, et al. NAT10-mediated N4-acetylation of SLC7A11 mRNA promotes hepatocellular carcinoma progression and metastasis by repressing ferroptosis. J Biochem Mol Toxicol. 2025;39(9):e70496. doi:10.1002/jbt.70496
37. Shi CJ, Pang FX, Lei YH, et al. 5-methylcytosine methylation of MALAT1 promotes resistance to sorafenib in hepatocellular carcinoma through ELAVL1/SLC7A11-mediated ferroptosis. Drug Resistance Updates. 2025;78:101181. doi:10.1016/j.drup.2024.101181
38. Qian B, Che L, Du ZB, et al. Protein phosphatase 2A-B55β mediated mitochondrial p-GPX4 dephosphorylation promoted sorafenib-induced ferroptosis in hepatocellular carcinoma via regulating p53 retrograde signaling. Theranostics. 2023;13(12):4288–4302. doi:10.7150/thno.82132
39. Liu MR, Shi C, Song QY, et al. Sorafenib induces ferroptosis by promoting TRIM54-mediated FSP1 ubiquitination and degradation in hepatocellular carcinoma. Hepatol Commun. 2023;7(10):e0246. doi:10.1097/HC9.0000000000000246
40. Yuan J, Lv T, Yang J, et al. HDLBP-stabilized lncFAL inhibits ferroptosis vulnerability by diminishing Trim69-dependent FSP1 degradation in hepatocellular carcinoma. Redox Biol. 2022;58:102546. doi:10.1016/j.redox.2022.102546
41. Su B, Wang S, Xiong Z, et al. Hua Zheng San Ji Fang promotes ferroptosis through Keap1/Nrf2 pathway in hepatocellular carcinoma cells. J Ethnopharmacol. 2025;352:120165. doi:10.1016/j.jep.2025.120165
42. Gupta N, Dubey V, Koiri RK. KEAP1/NRF2 mediated activation of oxidative stress in aflatoxin B1 induced early and advanced stage of hepatocellular carcinoma. J Biochem Mol Toxicol. 2025;39(12):e70624. doi:10.1002/jbt.70624
43. Kong S, Zhao C, Li J, Pan X, Li Y. USP11 is involved in the sensitivity of liver cancer cells to ferroptosis and taxanes through the regulation of NRF2 ubiquitin-mediated degradation. Transl Oncol. 2025;62:102553. doi:10.1016/j.tranon.2025.102553
44. Liu H, Zhao L, Wang M, et al. FNDC5 causes resistance to sorafenib by activating the PI3K/Akt/Nrf2 pathway in hepatocellular carcinoma cells. Front Oncol. 2022;12:852095. doi:10.3389/fonc.2022.852095
45. Shu F, Shi Y, Shan X, Zha W, Fan R, Xue W. SIAH2-mediated degradation of ACSL4 inhibits the anti-tumor activity of CD8+ T cells in hepatocellular carcinoma. Crit Rev Eukaryotic Gene Exp. 2024;34(5):1–13. doi:10.1615/CritRevEukaryotGeneExpr.2024051981
46. Feng S, Jia J, Wang K, Zhao H, Lv G. FBXO10 inhibits ferroptosis and promotes the progression of esophageal squamous cell carcinoma by post-translational mediation of ACSL4 degradation. J Mol Histol. 2025;56(4):214. doi:10.1007/s10735-025-10497-1
47. Zhou P, Peng X, Zhang K, et al. HAT1/HDAC2 mediated ACSL4 acetylation confers radiosensitivity by inducing ferroptosis in nasopharyngeal carcinoma. Cell Death Dis. 2025;16(1):160. doi:10.1038/s41419-025-07477-4
48. Wang J, Wang Z, Yuan J, Wang J, Shen X. The positive feedback between ACSL4 expression and O-GlcNAcylation contributes to the growth and survival of hepatocellular carcinoma. Aging. 2020;12(9):7786–7800. doi:10.18632/aging.103092
49. Xue P, Zhang J, Wang Z, et al. Arachidonic acid induces ferroptosis in hepatocellular carcinoma via the SIRT5-ACSL4/LPCAT3/ALOX15 axis, leading to lipid peroxidation and mitochondrial dysfunction. Phytomedicine. 2025;148:157450. doi:10.1016/j.phymed.2025.157450
50. Chen J, Ding C, Chen Y, et al. ACSL4 promotes hepatocellular carcinoma progression via c-Myc stability mediated by ERK/FBW7/c-Myc axis. Oncogenesis. 2020;9(4):42. doi:10.1038/s41389-020-0226-z
51. Zhao Y, Yang S, Huang L, et al. MUC1 drives ferroptosis resistance in ICC via Src-mediated FSP1 deubiquitination and myristoylation. Clin Transl Med. 2025;15(10):e70495. doi:10.1002/ctm2.70495
52. Deol KK, Harris CA, Tomlinson SJ, et al. Vitamin B2 metabolism promotes FSP1 stability to prevent ferroptosis. Nat Struct Mol Biol. 2026;33(3):525–536. doi:10.1038/s41594-026-01759-x
53. Liu Y, Fu J, Shi L, Chen C, Bao T. Progress in targeted ferroptosis regulation and mechanism of action in hepatocellular carcinoma. Cell Signal. 2026;138:112283. doi:10.1016/j.cellsig.2025.112283
54. Li T, Yi J, Wu H, Wang K, Zhou B. SLC7A11 in hepatocellular carcinoma: potential mechanisms, regulation, and clinical significance. Am J Cancer Res. 2024;14(5):2326–2342.
55. Wang P, Lin J, Su D. Echinacoside as a novel ferroptosis inducer in hepatocellular carcinoma: mechanistic insights from TP53/SLC7A11/GPX4 pathway modulation. Int J Mol Sci. 2025;27(1):411. doi:10.3390/ijms27010411
56. Wang H, Chen W, Cui Y, Gong H, Li H. KIAA1429 protects hepatocellular carcinoma cells from ferroptotic cell death with a m(6) A-dependent posttranscriptional modification of SLC7A11. J Cell Mol Med. 2023;27(24):4118–4132. doi:10.1111/jcmm.17997
57. He X, Gu Y, Lu G, et al. Reprogramming cysteine metabolism via METTL14-SLC7A11 axis promotes the progression of NAFLD and hepatocellular carcinoma. Life Sci. 2025;380:123965. doi:10.1016/j.lfs.2025.123965
58. Lu J, Cai D, Qian L, et al. Targeting SIX2 as a novel sensitization strategy of sorafenib treatment on advanced hepatocellular carcinoma through modulating METTL9-SLC7A11 axis. NPJ Precision Oncol. 2025;9(1):186. doi:10.1038/s41698-025-01004-6
59. Zhang Y, Yao R, Li M, et al. CircTTC13 promotes sorafenib resistance in hepatocellular carcinoma through the inhibition of ferroptosis by targeting the miR-513a-5p/SLC7A11 axis. Mol Cancer. 2025;24(1):32. doi:10.1186/s12943-024-02224-3
60. Dong FL, Xu ZZ, Wang YQ, Li T, Wang X, Li J. Exosome-derived circUPF2 enhances resistance to targeted therapy by redeploying ferroptosis sensitivity in hepatocellular carcinoma. J Nanobiotechnol. 2024;22(1):298. doi:10.1186/s12951-024-02582-6
61. Yuan X, Wang Y, Jiao S, et al. Identification of SLC7A11-AS1/SLC7A11 pair as a ferroptosis-related therapeutic target for hepatocellular carcinoma. J Cell Mol Med. 2024;28(13):e18496. doi:10.1111/jcmm.18496
62. Chen F, Wang L. Long noncoding RNA CASC11 suppresses sorafenib-triggered ferroptosis via stabilizing SLC7A11 mRNA in hepatocellular carcinoma cells. Discover Oncol. 2023;14(1):145. doi:10.1007/s12672-023-00761-9
63. Zhong X, Zhu Z, Du Y, et al. EFNA4-enhanced deubiquitination of SLC7A11 inhibits ferroptosis in hepatocellular carcinoma. Apoptosis. 2025;30(1–2):349–363. doi:10.1007/s10495-024-02042-4
64. Jiang Y, Ma P, Yang Y, Shi X, Liu X, Sun M. The Achilles’ heel of hepatocellular carcinoma: ginsenoside compound K as a novel GPX4 degrader promotes ferroptosis in hepatocellular carcinoma. J Transl Med. 2026;24(1):277. doi:10.1186/s12967-025-07587-9
65. Du Y, Zhou Y, Yan X, et al. APE1 inhibition enhances ferroptotic cell death and contributes to hepatocellular carcinoma therapy. Cell Death Differ. 2024;31(4):431–446. doi:10.1038/s41418-024-01270-0
66. Wang W, Chen X, Wei W. TRIM22 mechanism promoting KAT2A ubiquitination degradation to regulate ferroptosis in hepatocellular carcinoma cell invasion and metastasis. Histol Histopathol. 2025;40(8):1295–1307. doi:10.14670/HH-18-856
67. Zhou J, Gao M, Zhang S, et al. PP1A modulates the efficacy of lenvatinib plus ICIs therapy by inhibiting ferroptosis in hepatocellular carcinoma. Adv Sci. 2025;12(27):e2501730. doi:10.1002/advs.202501730
68. Quan B, Yao F, Liu W, et al. Increased CAPG inhibits ferroptosis to drive tumor proliferation and sorafenib resistance in hepatocellular carcinoma via the WDR74-p53-SLC7A11 pathway. Int J Biol Sci. 2025;21(12):5476–5495. doi:10.7150/ijbs.111419
69. Liu Y, Liu F, Liu J, et al. The acetyltransferase ARD1 induces glutathione synthesis to facilitate ferroptosis evasion in hepatocellular carcinoma. Cancer Res. 2025;85(21):4212–4232. doi:10.1158/0008-5472.CAN-24-4015
70. Zhang J, Yimamu M, Cheng Z, et al. TRIM47-CDO1 axis dictates hepatocellular carcinoma progression by modulating ferroptotic cell death through the ubiquitin‒proteasome system. Free Radic Biol Med. 2024;219:31–48. doi:10.1016/j.freeradbiomed.2024.04.222
71. Jun L, Chen W, Han L, Yanmin L, Qinglei Z, Pengfei Z. Protocadherin 20 promotes ferroptosis by suppressing the expression of Sirtuin 1 and promoting the acetylation of nuclear factor erythroid 2-related factor 2 in hepatocellular carcinoma. Int J Biochem Cell Biol. 2023;156:106363. doi:10.1016/j.biocel.2023.106363
72. Yu C, Liu J, Jian H, et al. ChaC1-based drug screenings identify a synergistic lethal effect of auranofin and proteasome inhibitors in hepatocellular carcinoma cells. Cell Death Discov. 2025;11(1):532. doi:10.1038/s41420-025-02838-6
73. Wanbiao Z, Jing M, Shi Z, Tengxiang C, Xueke Z, Haiyang L. MIA3 promotes the degradation of GSH (glutathione) by binding to CHAC1, thereby promoting the progression of hepatocellular carcinoma. Mol Cell Biochem. 2024;479(10):2769–2784. doi:10.1007/s11010-023-04850-9
74. Chen Z, Zhang C, Yang J, Peng Y. KLF1 exerts pro-tumour role in liver cancer via inhibiting ACSL4/LPCAT3-regulated ferroptosis. J Cell Mol Med. 2026;30(3):e71033. doi:10.1111/jcmm.71033
75. Chen J, Ding C, Chen Y, et al. ACSL4 reprograms fatty acid metabolism in hepatocellular carcinoma via c-Myc/SREBP1 pathway. Cancer Lett. 2021;502:154–165. doi:10.1016/j.canlet.2020.12.019
76. Lu Y, Chan YT, Tan HY, et al. Epigenetic regulation of ferroptosis via ETS1/miR-23a-3p/ACSL4 axis mediates sorafenib resistance in human hepatocellular carcinoma. J Exp Clin Cancer Res. 2022;41(1):3. doi:10.1186/s13046-021-02208-x
77. Yang H, Sun W, Bi T, et al. ZNF8-miR-552-5p axis modulates ACSL4-mediated ferroptosis in hepatocellular carcinoma. DNA Cell Biol. 2023;42(6):336–347. doi:10.1089/dna.2022.0582
78. Qin X, Zhang J, Lin Y, Sun XM, Zhang JN, Cheng ZQ. Identification of MiR-211-5p as a tumor suppressor by targeting ACSL4 in Hepatocellular Carcinoma. J Transl Med. 2020;18(1):326. doi:10.1186/s12967-020-02494-7
79. Wang D, Huang W, Li G. miR-145-5p regulates hepatocellular carcinoma malignant advancement and immune escape via down-regulation of AcylCoA synthase ACSL4. Biomol Biomed. 2025;25(5):1184–1196. doi:10.17305/bb.2024.11209
80. Zhao Y, Han S, Zeng Z, et al. Decreased lncRNA HNF4A-AS1 facilitates resistance to sorafenib-induced ferroptosis of hepatocellular carcinoma by reprogramming lipid metabolism. Theranostics. 2024;14(18):7088–7110. doi:10.7150/thno.99197
81. Phetkong C, Boonto T, Thamjamrassri P, Ariyachet C, Tangkijvanich P. MicroRNA-372-3p impairs fatty acid metabolism in hepatocellular carcinoma cells by targeting CPT1A and ACSL4. BioImpacts. 2025;15:31075. doi:10.34172/bi.31075
82. Peng H, Chen B, Wei W, et al. N(6)-methyladenosine (m(6)A) in 18S rRNA promotes fatty acid metabolism and oncogenic transformation. Nat Metab. 2022;4(8):1041–1054. doi:10.1038/s42255-022-00622-9
83. Xing K, Bian X, Shi D, et al. miR-612 enhances RSL3-induced ferroptosis of hepatocellular carcinoma cells via mevalonate pathway. J Hepatocell Carcinoma. 2023;10:2173–2185. doi:10.2147/JHC.S433332
84. Zhang C, Sun M, Shi Y, et al. Targeting the MIR4435-2HG/miR-29c-3p/FSP1 axis overcomes lenvatinib resistance by inducing ferroptosis in hepatocellular carcinoma. Cancer Cell Int. 2025;26(1):46. doi:10.1186/s12935-025-04147-5
85. Zhu Z, Xia X, Lu Y, et al. PARK7-driven IGF2BP3-K76 lactylation mediates ferroptosis and HAIC resistance in hepatocellular carcinoma. Redox Biol. 2025;87:103869. doi:10.1016/j.redox.2025.103869
86. Huang CY, Chen LJ, Chen G, Chao TI, Wang CY. SHP-1/STAT3-signaling-axis-regulated coupling between BECN1 and SLC7A11 contributes to sorafenib-induced ferroptosis in hepatocellular carcinoma. Int J Mol Sci. 2022;23(19):11092. doi:10.3390/ijms231911092
87. Zhang L, Li XM, Shi XH, et al. Sorafenib triggers ferroptosis via inhibition of HBXIP/SCD axis in hepatocellular carcinoma. Acta Pharmacol Sin. 2023;44(3):622–634. doi:10.1038/s41401-022-00981-9
88. Liu F, Li S, Huang C, et al. Self-assembled nanoplatform-mediated co-delivery of brusatol to sensitize sorafenib for hepatocellular carcinoma treatment. RSC Adv. 2025;15(15):11675–11687. doi:10.1039/D5RA00108K
89. Zheng J, Liu Y, Zhu F, et al. Picropodophyllin induces ferroptosis via blockage of AKT/NRF2/SLC7A11 and AKT/NRF2/SLC40A1 axes in hepatocellular carcinoma as a natural IGF1R inhibitor. Phytomedicine. 2025;143:156840. doi:10.1016/j.phymed.2025.156840
90. Chen L, Sun R, Fang K. Erianin inhibits tumor growth by promoting ferroptosis and inhibiting invasion in hepatocellular carcinoma through the JAK2/STAT3/SLC7A11 pathway. Pathol Int. 2024;74(3):119–128. doi:10.1111/pin.13403
91. Zhang L, Wang X. Nelfinavir triggers ferroptosis by inducing ER stress mediated downregulation of GPX4/GSH system, upregulation of NRF2/HO-1 axis, and mitochondrial impairment in hepatocellular carcinoma cells. Cell Death Discov. 2025;11(1):444. doi:10.1038/s41420-025-02761-w
92. Yan X, Liu Y, Li C, et al. Pien-Tze-Huang prevents hepatocellular carcinoma by inducing ferroptosis via inhibiting SLC7A11-GSH-GPX4 axis. Cancer Cell Int. 2023;23(1):109. doi:10.1186/s12935-023-02946-2
93. Tang B, Zhu J, Wang Y, et al. Targeted xCT-mediated ferroptosis and protumoral polarization of macrophages is effective against HCC and enhances the efficacy of the Anti-PD-1/L1 response. Adv Sci. 2023;10(2):e2203973. doi:10.1002/advs.202203973
94. Yang H, Chen X, Huang S, et al. A novel GSH depletor for simultaneous ferroptosis and cuproptosis Activation in hepatocellular carcinoma. Biochem Pharmacol. 2026;243(Pt 1):117488. doi:10.1016/j.bcp.2025.117488
95. Conche C, Finkelmeier F, Pešić M, et al. Combining ferroptosis induction with MDSC blockade renders primary tumours and metastases in liver sensitive to immune checkpoint blockade. Gut. 2023;72(9):1774–1782. doi:10.1136/gutjnl-2022-327909
96. Cui Y, Sun M, Wu J, et al. ACSL4 as a potential ferroptosis target in hepatocellular carcinoma: from mechanisms to implications. Eur J Med Res. 2026;31(1). doi:10.1186/s40001-026-03993-x
97. Sun XJ, Xu GL. Overexpression of Acyl-CoA Ligase 4 (ACSL4) in patients with hepatocellular carcinoma and its prognosis. Med Sci Monit. 2017;23:4343–4350. doi:10.12659/MSM.906639
98. Toshida K, Itoh S, Iseda N, et al. Impact of ACSL4 on the prognosis of hepatocellular carcinoma: association with cancer-associated fibroblasts and the tumour immune microenvironment. Liver Int. 2024;44(4):1011–1023. doi:10.1111/liv.15839
99. Feng J, Bin JL, Liao XW, et al. The prognostic role of ACSL4 in postoperative adjuvant TACE-treated HCC: implications for therapeutic response and mechanistic insights. J Exp Clin Cancer Res. 2024;43(1):306. doi:10.1186/s13046-024-03222-5
100. Classon P, Wixom AQ, Calixto Mancipe N, et al. Role of long-chain acyl-CoA synthetases in MASH-driven hepatocellular carcinoma and ferroptosis. Am J Physiol Gastrointest Liver Physiol. 2025;329(5):G571–g584. doi:10.1152/ajpgi.00096.2025
101. Grube J, Woitok MM, Mohs A, et al. ACSL4-dependent ferroptosis does not represent a tumor-suppressive mechanism but ACSL4 rather promotes liver cancer progression. Cell Death Dis. 2022;13(8):704. doi:10.1038/s41419-022-05137-5
102. Jiang Y, Hui D, Pan Z, et al. Curcumin promotes ferroptosis in hepatocellular carcinoma via upregulation of ACSL4. J Cancer Res Clin Oncol. 2024;150(9):429. doi:10.1007/s00432-024-05878-0
103. Wang C, Hu H, Gao H, Zhu Z, Zhao H. Astragali radix - Curcumae rhizoma herb pair enhances Sorafenib’s efficacy by inducing ferroptosis and activates Th1 cell immune response synergistically against hepatocellular carcinoma. Phytomedicine. 2025;148:157326. doi:10.1016/j.phymed.2025.157326
104. Li Q, Wang Y, Zou J. Inhibition of dipeptidyl peptidase 9 improves sorafenib sensitivity by inducing ferroptosis in hepatocellular carcinoma. J Cancer Res Clin Oncol. 2025;151(9):243. doi:10.1007/s00432-025-06300-z
105. Hou W, Hong W, Cai S, et al. RRM2-targeted nanocarrier enhances radiofrequency ablation efficacy in hepatocellular carcinoma through ferroptosis amplification and immune remodeling. iMeta. 2025;4(5):e70067. doi:10.1002/imt2.70067
106. Yan Y, Hu J, Han N, et al. Sorafenib-loaded metal-organic framework nanoparticles for anti-hepatocellular carcinoma effects through synergistically potentiating ferroptosis and remodeling tumor immune microenvironment. Mater Today Bio. 2025;32:101848. doi:10.1016/j.mtbio.2025.101848
107. Tan J, Feng S, Dang W, et al. Fluorescent probes targeting Acyl-CoA synthetase long-chain family member 4 for intraoperative imaging of hepatocellular carcinoma. J Control Release. 2025;387:114161. doi:10.1016/j.jconrel.2025.114161
108. Bersuker K, Hendricks JM, Li Z, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575(7784):688–692. doi:10.1038/s41586-019-1705-2
109. Doll S, Freitas FP, Shah R, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575(7784):693–698. doi:10.1038/s41586-019-1707-0
110. Mao C, Liu X, Zhang Y, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593(7860):586–590. doi:10.1038/s41586-021-03539-7
111. Deng R, Fu L, Liang H, et al. Inhibition of mitochondrial complex I induces mitochondrial ferroptosis by regulating CoQH2 levels in cancer. Cell Death Dis. 2025;16(1):254. doi:10.1038/s41419-025-07510-6
112. Chen Y, Lee D, Kwan KK, et al. Mevalonate pathway promotes liver cancer by suppressing ferroptosis through CoQ10 production and selenocysteine-tRNA modification. J Hepatol. 2025;83(6):1338–1352. doi:10.1016/j.jhep.2025.06.034
113. Li Y, Lin S, Li L, et al. PDSS2 deficiency induces hepatocarcinogenesis by decreasing mitochondrial respiration and reprogramming glucose metabolism. Cancer Res. 2018;78(16):4471–4481. doi:10.1158/0008-5472.CAN-17-2172
114. Kanda M, Sugimoto H, Nomoto S, et al. Clinical utility of PDSS2 expression to stratify patients at risk for recurrence of hepatocellular carcinoma. Int J Oncol. 2014;45(5):2005–2012. doi:10.3892/ijo.2014.2637
115. Cheu JW, Lee D, Li Q, et al. Ferroptosis suppressor protein 1 inhibition promotes tumor ferroptosis and anti-tumor immune responses in liver cancer. Cell Mol Gastroenterol Hepatol. 2023;16(1):133–159. doi:10.1016/j.jcmgh.2023.03.001
116. Tang C, He C, Wang D, et al. Co-delivery of sorafenib and an FSP1 inhibitor triggers dual ferroptosis in tumor cells and immunosuppressive macrophages for enhanced immunotherapy in mouse models of hepatocellular carcinoma. Nat Commun. 2025;16(1):10096. doi:10.1038/s41467-025-65056-9
117. Liu FY, Yang YJ, Wang XL, et al. Ginsenoside (20)S-APPT induces ferroptosis in hepatocellular carcinoma and cholangiocarcinoma by targeting FSP1. Acta Pharmacol Sin. 2025;46(12):3273–3290. doi:10.1038/s41401-025-01589-5
118. Jiang Y, Yu Y, Pan Z, Wang Z, Sun M. Ginsenoside RK1 induces ferroptosis in hepatocellular carcinoma cells through an FSP1-Dependent pathway. Pharmaceuticals. 2024;17(7):871. doi:10.3390/ph17070871
119. Ma C, Han L, Yao H, et al. Modulating G6PD/PGD to overcome FSP1/DHODH-mediated ferroptosis defence: a novel oridonin derivative suppresses liver cancer. Br J Pharmacol. 2026;183(2):249–267. doi:10.1111/bph.70160
120. Zi L, Ma W, Zhang L, et al. Uridine inhibits hepatocellular carcinoma cell development by inducing ferroptosis. J Clin Med. 2023;12(10):3552. doi:10.3390/jcm12103552
121. Talubo NDD, Dela Cruz EWB, Fowler P, Tsai PW, Tayo LL. QSAR-based drug repurposing and RNA-seq metabolic networks highlight treatment opportunities for hepatocellular carcinoma through pyrimidine starvation. Cancers. 2025;17(5):903. doi:10.3390/cancers17050903
122. Elmetwalli A. Ferroptosis and the cGAS-STING pathway into precision nano-immuno-theranostics: a mechanistic paradigm for reversing drug resistance in hepatocellular carcinoma. Drug Resistance Updates. 2026;84:101326. doi:10.1016/j.drup.2025.101326
123. Qian X, Zhou Q, Ouyang Y, et al. Transferrin promotes fatty acid oxidation and liver tumor growth through PHD2-mediated PPARα hydroxylation in an iron-dependent manner. Proc Natl Acad Sci USA. 2025;122(5):e2412473122. doi:10.1073/pnas.2412473122
124. Kim DH, Kim MK, Kim D, et al. Liver cancer chronically exposed to palmitate acquires ferroptosis resistance via the downregulation of glutamine-driven hepcidin expression. Metabolism. 2026;176:156469. doi:10.1016/j.metabol.2025.156469
125. Yang X, Gu C, Cai J, et al. Excessive SOX8 reprograms energy and iron metabolism to prime hepatocellular carcinoma for ferroptosis. Redox Biol. 2024;69:103002. doi:10.1016/j.redox.2023.103002
126. Liu Y, Jia L, Yang L, Ning Z, Li Y. Targeting C12ORF49-mediated ferroptosis in hepatocellular carcinoma. JGH Open. 2026;10(2):e70353. doi:10.1002/jgh3.70353
127. Hong J, Du K, Wu T, et al. Environmental PFOS and 6:2 Cl-PFESA reshape ferroptosis vulnerability in liver cancer. Environ Sci Technol. 2026;60(8):6096–6110. doi:10.1021/acs.est.5c16490
128. Wu L, Chen HY, Zhang JT, et al. Chlorogenic acid induces hepatocellular carcinoma cell ferroptosis via PTGS2/AKR1C3/GPX4 axis-mediated reprogramming of arachidonic acid metabolism. World J Gastrointest Oncol. 2025;17(3):98844. doi:10.4251/wjgo.v17.i3.98844
129. Tang B, Zhang X, Sun Y, et al. Tumor-targeted FABP5/STING cascade promote radiofrequency ablation induced ferroptosis and intratumoral immune rewiring in hepatocellular carcinoma. Adv Sci. 2025;12(45):e07864. doi:10.1002/advs.202507864
130. Wang X, Yan B, Li H, et al. Reprogrammed IDO-induced immunosuppressive microenvironment synergizes with immunogenic magnetothermodynamics for improved cancer therapy. ACS Appl Mater Interfaces. 2024;16(24):30671–30684. doi:10.1021/acsami.4c02740
131. Luo Y, Yang Y, Ye M, Zuo J. Targeting metabolic reprogramming promotes the efficacy of transarterial chemoembolization in the rabbit VX2 liver tumor model. Oncol Lett. 2024;27(3):111. doi:10.3892/ol.2024.14244
132. Wang Z, Zhou C, Zhang Y, et al. From synergy to resistance: navigating the complex relationship between sorafenib and ferroptosis in hepatocellular carcinoma. Biomed Pharmacother. 2024;170:116074. doi:10.1016/j.biopha.2023.116074
133. Liu R, Cui H, Li D, et al. Roles and mechanisms of ferroptosis in sorafenib resistance for hepatocellular carcinoma. J Hepatocell Carcinoma. 2024;11:2493–2504. doi:10.2147/JHC.S500084
134. Galmiche A, Chauffert B, Barbare JC. New biological perspectives for the improvement of the efficacy of sorafenib in hepatocellular carcinoma. Cancer Lett. 2014;346(2):159–162. doi:10.1016/j.canlet.2013.12.028
135. Yousef EH, El Gayar AM, El-Magd NFA. Insights into Sorafenib resistance in hepatocellular carcinoma: mechanisms and therapeutic aspects. Crit Rev Oncol/Hematol. 2025;212:104765. doi:10.1016/j.critrevonc.2025.104765
136. Feng W, Xu J, Chen B, et al. Hsa_piR_016975 is a novel target of nanotherapy that boosts hepatoma progression and sorafenib resistance by abating Maspin/GPX4-mediated ferroptosis. Biomater Res. 2025;29:0225. doi:10.34133/bmr.0225
137. Ji L, Ruan Y, Tong M, et al. circRNA-SORE/UBQLN1/GPX4 mediates the acquisition of sorafenib resistance in hepatocellular carcinoma through inhibition of ferroptosis. MedComm. 2025;6(12):e70488. doi:10.1002/mco2.70488
138. Guo L, Hu C, Yao M, Han G. Mechanism of sorafenib resistance associated with ferroptosis in HCC. Front Pharmacol. 2023;14:1207496. doi:10.3389/fphar.2023.1207496
139. Wang H, Wang XY, Ji JB, Zheng ZX, Shang PF, Guo XL. GLS1 inhibitor CB-839 inhibits the malignant progression of 5-FU resistant hepatoma cells by regulating glutamine metabolism. Chem Biol Interact. 2026;423:111812. doi:10.1016/j.cbi.2025.111812
140. Zhang Z, Zhou Q, Li Z, et al. DTX2 attenuates Lenvatinib-induced ferroptosis by suppressing docosahexaenoic acid biosynthesis through HSD17B4-dependent peroxisomal β-oxidation in hepatocellular carcinoma. Drug Resistance Updates. 2025;81:101224. doi:10.1016/j.drup.2025.101224
141. Zhang Y, Lin Y, Cai H, Zhou T. EZH2 confers lenvatinib resistance in hepatocellular carcinoma by suppressing ACSL1-Mediated ferroptosis. BMC Cancer. 2025;25(1):1638. doi:10.1186/s12885-025-15086-9
142. Zhang Q, Xiong L, Wei T, et al. Hypoxia-responsive PPARGC1A/BAMBI/ACSL5 axis promotes progression and resistance to lenvatinib in hepatocellular carcinoma. Oncogene. 2023;42(19):1509–1523. doi:10.1038/s41388-023-02665-y
143. Chen MY, Lai SW, Cheng YC, et al. Targeting PPP1R15B and ATF4 axis in hepatocellular carcinoma: a novel strategy for overcoming lenvatinib-tolerant persister cells through GPX4-mediated ferroptosis induction. Eur J Pharmaceut Sci. 2026;218:107434. doi:10.1016/j.ejps.2026.107434
144. Sun X, Niu X, Chen R, et al. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology. 2016;64(2):488–500. doi:10.1002/hep.28574
145. Michilli A, Bassi C, Moshiri F, et al. Transcriptomic analysis reveals an NRF2-mediated redox and metabolic reprogramming in sorafenib-resistant hepatocellular carcinoma cells. Biotech. 2026;15(1):18.
146. Guo M, Chen S, Sun J, et al. PIP5K1A suppresses ferroptosis and induces sorafenib resistance by stabilizing NRF2 in hepatocellular carcinoma. Adv Sci. 2025;12(30):e04372. doi:10.1002/advs.202504372
147. Liu X, Zhang F, Fan Y, Qiu C, Wang K. MCM4 potentiates evasion of hepatocellular carcinoma from sorafenib-induced ferroptosis through Nrf2 signaling pathway. Int Immunopharmacol. 2024;142(Pt A):113107. doi:10.1016/j.intimp.2024.113107
148. Deng J, Wu Z, Ning S, et al. Curcumin induces ferroptosis in hepatocellular carcinoma by regulating the P62-KEAP1-NRF2-signaling pathway. BMC Cancer. 2025;26(1):90. doi:10.1186/s12885-025-15307-1
149. Elkateb AS, Nofal S, Ali SA, Atya HB. Camptothecin sensitizes hepatocellular carcinoma cells to sorafenib- induced ferroptosis via suppression of Nrf2. Inflammation. 2023;46(4):1493–1511. doi:10.1007/s10753-023-01823-4
150. Zhang Y, Tan Y, Liu S, et al. Implications of Withaferin A for the metastatic potential and drug resistance in hepatocellular carcinoma cells via Nrf2-mediated EMT and ferroptosis. Toxicol Mech Meth. 2023;33(1):47–55. doi:10.1080/15376516.2022.2075297
151. Li X, Pan YF, Chen YB, et al. Arsenic trioxide augments immunogenic cell death and induces cGAS-STING-IFN pathway activation in hepatocellular carcinoma. Cell Death Dis. 2024;15(4):300. doi:10.1038/s41419-024-06685-8
152. Huang M, Li D, Xia Z, et al. Silencing NRF2 enhances arsenic trioxide-induced ferroptosis in hepatocellular carcinoma cells. PLoS One. 2025;20(5):e0322746. doi:10.1371/journal.pone.0322746
153. Song Z, Zhou M, Song X, Jia H, Yang X. SSR2 promotes sorafenib resistance via interacting with GPX4 to inhibit ferroptosis. Curr Mol Med. 2026. doi:10.2174/0115665240405981251201125139
154. Li Q, Yuan H, Zhao G, et al. DDX39B protects against sorafenib-induced ferroptosis by facilitating the splicing and cytoplasmic export of GPX4 pre-mRNA in hepatocellular carcinoma. Biochem Pharmacol. 2024;225:116251. doi:10.1016/j.bcp.2024.116251
155. Li J, Li Y, Wang D, Liao R, Wu Z. PLAG1 interacts with GPX4 to conquer vulnerability to sorafenib induced ferroptosis through a PVT1/miR-195-5p axis-dependent manner in hepatocellular carcinoma. J Exp Clin Cancer Res. 2024;43(1):143. doi:10.1186/s13046-024-03061-4
156. Tang HH, Hsu CP, Su PY, et al. Targeting GPX4 to overcome sorafenib resistance of human hepatocellular carcinoma by inducing ferroptosis. J Cell Physiol. 2025;240(8):e70078. doi:10.1002/jcp.70078
157. Chen C, Wang L, Cui XF, et al. SCARA5 deficiency inhibits ferroptosis via regulating iron homeostasis and results in sorafenib resistance in hepatocellular carcinoma. Cell Signal. 2025;129:111656. doi:10.1016/j.cellsig.2025.111656
158. Zhu H, Liu Q, Meng Q, et al. CCT3/ACTN4/TFRC axis protects hepatocellular carcinoma cells from ferroptosis by inhibiting iron endocytosis. J Exp Clin Cancer Res. 2024;43(1):245. doi:10.1186/s13046-024-03169-7
159. Hu X, Zhang P, Li S, et al. Mitochondrial GCN5L1 acts as a novel regulator for iron homeostasis to promote sorafenib sensitivity in hepatocellular carcinoma. J Transl Med. 2024;22(1):593. doi:10.1186/s12967-024-05404-3
160. Ding Z, Pan Y, Shang T, et al. URI alleviates tyrosine kinase inhibitors-induced ferroptosis by reprogramming lipid metabolism in p53 wild-type liver cancers. Nat Commun. 2023;14(1):6269. doi:10.1038/s41467-023-41852-z
161. Li Z, Liao X, Hu Y, et al. SLC27A4-mediated selective uptake of mono-unsaturated fatty acids promotes ferroptosis defense in hepatocellular carcinoma. Free Radic Biol Med. 2023;201:41–54. doi:10.1016/j.freeradbiomed.2023.03.013
162. Li Y, Yang W, Zheng Y, et al. Targeting fatty acid synthase modulates sensitivity of hepatocellular carcinoma to sorafenib via ferroptosis. J Exp Clin Cancer Res. 2023;42(1):6. doi:10.1186/s13046-022-02567-z
163. Byun JK, Lee S, Kang GW, et al. Macropinocytosis is an alternative pathway of cysteine acquisition and mitigates sorafenib-induced ferroptosis in hepatocellular carcinoma. J Exp Clin Cancer Res. 2022;41(1):98. doi:10.1186/s13046-022-02296-3
164. Zeng Y, Jiang H, Zhang X, et al. Canagliflozin reduces chemoresistance in hepatocellular carcinoma through PKM2-c-Myc complex-mediated glutamine starvation. Free Radic Biol Med. 2023;208:571–586. doi:10.1016/j.freeradbiomed.2023.09.006
165. Tian Y, Ouyang K, Wu H, et al. Adaptive drug resistance mechanisms driven by non-coding RNA-protein interaction networks in hepatocellular carcinoma. Crit Rev Oncol/Hematol. 2026;218:105060. doi:10.1016/j.critrevonc.2025.105060
166. Mohammadi Y, Mahdiabadi MA, Ourang Z, Bahreini E, Abaszade-Cheragheali A. Targeting ferroptosis by noncoding RNAs as a novel therapeutic strategy for hepatocellular carcinoma. Discover Oncol. 2025;16(1):2303. doi:10.1007/s12672-025-04062-1
167. Gao Y, Tong M, Wong TL, et al. Long noncoding RNA URB1-Antisense RNA 1 (AS1) suppresses sorafenib-induced ferroptosis in hepatocellular carcinoma by driving ferritin phase separation. ACS Nano. 2023;17(22):22240–22258. doi:10.1021/acsnano.3c01199
168. Tian Y, Bao X, Lei S, et al. In vivo CRISPR screening identifies POU3F3 as a novel regulator of ferroptosis resistance in hepatocellular carcinoma via retinoic acid signaling. Cell Commun Signal. 2025;23(1):329. doi:10.1186/s12964-025-02285-x
169. Chen Y, Li L, Lan J, et al. CRISPR screens uncover protective effect of PSTK as a regulator of chemotherapy-induced ferroptosis in hepatocellular carcinoma. Mol Cancer. 2022;21(1):11. doi:10.1186/s12943-021-01466-9
170. Yao F, Zhou S, Zhang R, et al. CRISPR/Cas9 screen reveals that targeting TRIM34 enhances ferroptosis sensitivity and augments immunotherapy efficacy in hepatocellular carcinoma. Cancer Lett. 2024;593:216935. doi:10.1016/j.canlet.2024.216935
171. Li L, Xing T, Chen Y, et al. In vitro CRISPR screening uncovers CRTC3 as a regulator of IFN-γ-induced ferroptosis of hepatocellular carcinoma. Cell Death Discov. 2023;9(1):331. doi:10.1038/s41420-023-01630-8
172. Guo Y, Xu M, Xue H, et al. Genome-wide CRISPR screen identifies splicing factor SF3B4 in driving hepatocellular carcinoma. Sci Adv. 2025;11(41):eadw7181. doi:10.1126/sciadv.adw7181
173. Padelli M, Desterke C, Devocelle A, Uzan G, Lemoine A, Giron-Michel J. The epigenetic regulator SETDB1 as a key component of cancer stem cells and drug resistance in primary liver cancer. Cell Oncol Dordr. 2026;49(1):18. doi:10.1007/s13402-025-01157-3
174. Chao J, Liang Y, Wang H, et al. FAD synthase confers ferroptosis resistance and restrains CD8(+) T cell recruitment in hepatocellular carcinoma. Nat Commun. 2025;16(1):9547. doi:10.1038/s41467-025-64572-y
175. Xu J, Wang B, Liu Q, et al. MVP-LCN2 axis triggers evasion of ferroptosis to drive hepatocarcinogenesis and sorafenib resistance. Drug Resistance Updates. 2025;81:101246. doi:10.1016/j.drup.2025.101246
176. Mu M, Huang CX, Qu C, et al. Targeting ferroptosis-elicited inflammation suppresses hepatocellular carcinoma metastasis and enhances sorafenib efficacy. Cancer Res. 2024;84(6):841–854. doi:10.1158/0008-5472.CAN-23-1796
177. Huang X, Yi N, Zhu P, Gao J, Lv J. Sorafenib-induced macrophage extracellular traps via ARHGDIG/IL4/PADI4 axis confer drug resistance through inhibiting ferroptosis in hepatocellular carcinoma. Biol Direct. 2024;19(1):110. doi:10.1186/s13062-024-00560-4
178. Chen R, Hu X, Huang Y, et al. Regulated cell death in lenvatinib resistance of hepatocellular carcinoma: from molecular mechanisms to therapeutic strategies. Int J Biol Sci. 2025;21(5):2012–2026. doi:10.7150/ijbs.107195
179. Lv X, Lan G, Zhu L, Guo Q. Breaking the barriers of therapy resistance: harnessing ferroptosis for effective hepatocellular carcinoma therapy. J Hepatocell Carcinoma. 2024;11:1265–1278. doi:10.2147/JHC.S469449
180. Zhang L, Wang H, Liang B, et al. Ponicidin promotes ferroptosis to enhance treatment sensitivity in Lenvatinib-resistant hepatocellular carcinoma cells through regulation of KEAP1/NRF2. Phytomedicine. 2025;143:156824. doi:10.1016/j.phymed.2025.156824
181. Lai Y, Han X, Xie B, et al. EZH2 suppresses ferroptosis in hepatocellular carcinoma and reduces sorafenib sensitivity through epigenetic regulation of TFR2. Cancer Sci. 2024;115(7):2220–2234. doi:10.1111/cas.16186
182. Song Z, Zhang Y, Luo W, et al. HAND2-AS1 promotes ferroptosis to reverse lenvatinib resistance in hepatocellular carcinoma by TLR4/NOX2/DUOX2 axis. Curr Cancer Drug Targets. 2025;25(2):144–158. doi:10.2174/0115680096279597240219055135
183. Meng J, Liang C, Li L, et al. A lactylation-ferroptosis cross-talk gene signature predicts hepatocellular carcinoma prognosis and reveals STMN1/PRDX1 as therapeutic targets. Front Immunol. 2025;16:1677089. doi:10.3389/fimmu.2025.1677089
184. Zhang H, Ma J, Hou C, et al. A ROS-mediated oxidation-O-GlcNAcylation cascade governs ferroptosis. Nat Cell Biol. 2025;27(8):1288–1300. doi:10.1038/s41556-025-01722-w
185. Wang S, Zhu L, Li T, et al. Disruption of MerTK increases the efficacy of checkpoint inhibitor by enhancing ferroptosis and immune response in hepatocellular carcinoma. Cell Rep Med. 2024;5(2):101415. doi:10.1016/j.xcrm.2024.101415
186. Wang S, Yuan X, Yang Z, et al. Matrix stiffness-dependent PD-L2 deficiency improves SMYD3/xCT-mediated ferroptosis and the efficacy of anti-PD-1 in HCC. J Adv Res. 2025;73:265–282. doi:10.1016/j.jare.2024.08.021
187. Wang B, Kong G, Meng C, Nie C, Wan D. A novel cabozantinib-sulfasalazine combination targeting ferroptosis to overcome resistant immunotherapy in advanced hepatocellular carcinoma. Pak J Pharm Sci. 2025;38(5):1906–1912. doi:10.36721/PJPS.2025.38.5.REG.15087.1
188. Yi N, Zhang L, Huang X, Ma J, Gao J. Lenvatinib-activated NDUFA4L2/IL33/PADI4 pathway induces neutrophil extracellular traps that inhibit cuproptosis in hepatocellular carcinoma. Cell Oncol Dordr. 2025;48(2):487–504. doi:10.1007/s13402-024-01013-w
189. Yue X, Xiang Z, Yi Y, et al. Targeting the TRIM14/USP14 axis enhances radiotherapy efficacy by inducing GPX4 degradation and disrupting ferroptotic defense in HCC. Cell Death Dis. 2025;16(1):481. doi:10.1038/s41419-025-07807-6
190. Tang D, Kroemer G, Kang R. Ferroptosis in hepatocellular carcinoma: from bench to bedside. Hepatology. 2024;80(3):721–739. doi:10.1097/HEP.0000000000000390
191. Li J, Wang Z, Wei K, Liu Q, Liang L, Lin J. Decursin induces ferroptosis via the NRF2/GPX4/SLC11A2 axis and suppresses migration in hepatocellular carcinoma. Biomed Pharmacother. 2026;195:118941. doi:10.1016/j.biopha.2025.118941
192. Wang T, Jiang W, Li J, et al. Dihydroartemisinin: a promising therapeutic agent against the hepatitis-to-hepatocellular carcinoma cascade. Drug Des Devel Ther. 2025;19:11145–11162. doi:10.2147/DDDT.S554484
193. Ji J, Cheng Z, Zhang J, et al. Dihydroartemisinin induces ferroptosis of hepatocellular carcinoma via inhibiting ATF4-xCT pathway. J Cell Mol Med. 2024;28(8):e18335. doi:10.1111/jcmm.18335
194. Wang WY, Hsu TW, Su YH, et al. Disrupting CARMA3 signaling with triptolide reverses sorafenib resistance in hepatocellular carcinoma. Int J Med Sci. 2025;22(16):4353–4364. doi:10.7150/ijms.121880
195. Luo L, Zhou P, He K. Zingiberensis new saponin reverses sorafenib resistance by targeting lncRNA TCONS-00026762/AKR1C1 and modulating autophagy and ferroptosis in hepatocellular carcinoma. Toxicol Appl Pharmacol. 2026;507:117664. doi:10.1016/j.taap.2025.117664
196. Yang C, Lu T, Liu M, et al. Tiliroside targets TBK1 to induce ferroptosis and sensitize hepatocellular carcinoma to sorafenib. Phytomedicine. 2023;111:154668. doi:10.1016/j.phymed.2023.154668
197. Hu Y, Luo Z, Cai S, Xie Q, Zheng S. Glycyrrhizic acid attenuates sorafenib resistance by inducing ferroptosis via targeting mTOR signaling in hepatocellular carcinoma. Scand J Gastroenterol. 2024;59(6):730–736. doi:10.1080/00365521.2024.2315317
198. Yang Z, Han T, Yang R, et al. Dicoumarol sensitizes hepatocellular carcinoma cells to ferroptosis induced by imidazole ketone erastin. Front Immunol. 2025;16:1531874.
199. Fang X, Liang Z, Zhang Z, et al. Natural exosome-like nanovesicles from Smilax China rhizome induce mitophagy-dependent ferroptosis in hepatocellular carcinoma via GPX4/ACSL4 axis. Biochem Biophys Res Commun. 2026;800:153270. doi:10.1016/j.bbrc.2026.153270
200. Liao Y, Zeng X, Zhang X, et al. Optimized lipid nanoparticles for co-delivery of mRNA and siRNA therapeutics in refractory liver cancer. Adv Mat. 2026;38(11):e19473. doi:10.1002/adma.202519473
201. Tang Q, Wang Y, Yan B, et al. Intracellular magnetic hyperthermia sensitizes sorafenib to orthotopic hepatocellular carcinoma via amplified ferroptosis. ACS Nano. 2024;18(43):29804–29819.
202. Dong M, Liu Y, Xiao Y, et al. Tumor-Targeted PLGA nanospheres enhance therapeutic effect of lenvatinib in hepatocellular carcinoma via photothermal and photodynamic therapy. ACS Appl Mater Interfaces. 2025;17(31):44922–44938.
203. Hu Z, Zhao Y, Li L, et al. Metformin promotes ferroptosis and sensitivity to sorafenib in hepatocellular carcinoma cells via ATF4/STAT3. Mol Biol Rep. 2023;50(8):6399–6413. doi:10.1007/s11033-023-08492-4
204. Chen X, Ji J, Li S, et al. Synergistic enhancement of ferroptosis via a multi-functional metal-coordinated nanoplatform for cancer therapy. Colloids Surf B. 2026;258:115205. doi:10.1016/j.colsurfb.2025.115205
205. Qin T, Lu H, Wang Q, et al. A phototherapy nanosensitizer-unlocked multihit strategy to inhibit tumor stemness and potentiate sorafenib response in hepatocellular carcinoma. ACS Appl Mater Interfaces. 2025;17(39):54537–54552. doi:10.1021/acsami.5c13211
206. Liu W, Wu G, Wang J, Wu S, Chen Z. Co‑treatment with triptolide and RSL3 induces hepatocellular carcinoma cell apoptosis and ferroptosis. Mol Med Rep. 2025;32(1):1–10. doi:10.3892/mmr.2025.13567
207. Li J, Mao S, Yang S, et al. Targeting JMJD6/PPARγ/GPX4 axis overcomes ferroptosis resistance and enhances therapeutic efficacy in hepatocellular carcinoma. Oncogene. 2025;44(45):4377–4390. doi:10.1038/s41388-025-03581-z
208. Luo AL, Zheng WY, Zhang Q, et al. COPS5 triggers ferroptosis defense by stabilizing MK2 in hepatocellular carcinoma. Adv Sci. 2025;12(22):e2416360. doi:10.1002/advs.202416360
209. Kang R, Liu J, Wang J, Kroemer G, Tang D. Translating ferroptosis into oncology: challenges, opportunities and future directions. Nat Rev Clin Oncol. 2026. doi:10.1038/s41571-026-01128-z
210. Wahida A, Conrad M. Decoding ferroptosis for cancer therapy. Nat Rev Cancer. 2025;25(12):910–924. doi:10.1038/s41568-025-00864-1
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
The Relationship Between Ferroptosis and Diseases
Lv J, Hou B, Song J, Xu Y, Xie S
Journal of Multidisciplinary Healthcare 2022, 15:2261-2275
Published Date: 6 October 2022
The Emerging Roles of Ferroptosis in Neonatal Diseases
Chen W, Zheng D, Yang C
Journal of Inflammation Research 2023, 16:2661-2674
Published Date: 26 June 2023
Application and Resistance Mechanisms of Lenvatinib in Patients with Advanced Hepatocellular Carcinoma
Tao M, Han J, Shi J, Liao H, Wen K, Wang W, Mui S, Li H, Yan Y, Xiao Z
Journal of Hepatocellular Carcinoma 2023, 10:1069-1083
Published Date: 10 July 2023
Breaking the Barriers of Therapy Resistance: Harnessing Ferroptosis for Effective Hepatocellular Carcinoma Therapy

Lv X, Lan G, Zhu L, Guo Q
Journal of Hepatocellular Carcinoma 2024, 11:1265-1278
Published Date: 2 July 2024
Ferroptosis in Osteoarthritis: Current Understanding
Liu Y, Zhang Z, Fang Y, Liu C, Zhang H
Journal of Inflammation Research 2024, 17:8471-8486
Published Date: 7 November 2024