Back to Journals » Journal of Multidisciplinary Healthcare » Volume 15
The Relationship Between Ferroptosis and Diseases
Authors Lv J, Hou B, Song J, Xu Y, Xie S ![]()
Received 16 July 2022
Accepted for publication 22 September 2022
Published 6 October 2022 Volume 2022:15 Pages 2261—2275
DOI https://doi.org/10.2147/JMDH.S382643
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Jinchang Lv, Biao Hou, Jiangang Song, Yunhua Xu, Songlin Xie
Department of Hand and Foot Microsurgery, The affiliated Nanhua Hospital of University of South China, Hengyang, People’s Republic of China
Correspondence: Songlin Xie, Department of Hand and Foot Microsurgery, The affiliated Nanhua Hospital of the University of South China, Hengyang, People’s Republic of China, Tel +86 13975404959, Email [email protected]
Abstract: Ferroptosis is an iron-dependent mode of cell death. It can occur through two major pathways, exogenous (or transporter-dependent) and endogenous (or enzyme-regulated) pathways are activated by biological or chemical inducers, and glutathione peroxidase activity is inhibited, which causes intracellular iron accumulation and lipid Peroxidation. Ferroptosis is closely related to the pathological process of many diseases. How to intervene in the occurrence and development of related diseases by regulating ferroptosis has become a hot research topic. At present, studies have shown that ferroptosis is found in common diseases such as tumors, inflammatory diseases, bacterial infections, pulmonary fibrosis, hepatitis, inflammatory bowel disease, neurodegenerative diseases, kidney injury, ischemia-reperfusion injury and skeletal muscle injury. This article reviews the characteristics and mechanism of ferroptosis, and summarizes how ferroptosis participates in the pathophysiological process in various systemic diseases of the body, which may provide new references for the treatment of clinical diseases in the future.
Keywords: ferroptosis, mechanisms of ferroptosis, iron metabolism, cell death, systemic diseases
Introduction
The cell is the basic unit in the body. Cell death is the end of cell life, apoptosis and necrosis are recognized as the main forms of cell death. Ferroptosis is a new mode of cell death discovered in recent years, which emerged after the discovery that a small molecule compound, erastin and RSL-3, could induce a unique form of cell death in cells.1,2 In 2012, this method of death was officially named “Ferroptosis” by Dixon.3 The research in the past decade has exponentially due to the wide impact of ferroptosis on human health and disease.4,5 Ferroptosis has expanded from mammalian systems to plants, protozoans, and fungi.6–9 We discussed the characteristics and mechanism of ferroptosis, and reviewed the relationship between ferroptosis and various systemic diseases. We expect that ferroptosis can provide new ideas in clinical treatment.
An Overview of Ferroptosis
The Discovery of Ferroptosis
The term ferroptosis arose after the discovery that a small-molecule compound, erastin, could induce a unique form of cell death in cells.1,2 Although ferroptosis has only been proposed in recent years, similar forms of death have been discovered before, but no one officially proposed a name at the time. In the mid-20th century, Eagle found in experiments that the lack of cysteine can lead to cell death, while increasing the endogenous synthesis of cysteine can avoid the cell death caused by the loss of cysteine.10 In 2003, Dolma discovered that erastin-induced cell death was different from camptothecin (CPT)-induced cell death, and proposed that erastin-induced cell death was a novel form of cell death.2 Subsequently, Yagoda elaborated the mechanism of erastin inducing death in cells,11 and Yang found another new compound that can cause this form of cell death—Ras-selective-lethal compound 3 (RSL3).1 According to its characteristics, this form of cell death was officially named ferroptosis by Dixon in 2012.3
The Characteristics of Ferroptosis
Common cell death methods include apoptosis, autophagy, pyroptosis, and necrosis (Table 1). Apoptosis is a type of programmed cell death that depends on genes. Studies have shown that ferroptosis can increase the sensitivity of cells to apoptosis. As a tumor suppressor gene, p53 can not only induce ferroptosis in cells, but also hinder the cell cycle and promote cell apoptosis,12 which confirms the possibility that ferroptosis and apoptosis can synergistically promote cell death. Autophagy is mediated by a lysosome-dependent degradation pathway. Ma confirmed that the activation of the autophagy pathway can degrade ferritin in cells and promote ferroptosis.13 Cell necrosis is a passive death caused by pathological factors and injury, and is not regulated by a program. Pyroptosis is a signaling pathway stimulated by the inflammasome, which activates Caspase-1 and ultimately activates inflammatory factors leading to cell death.14 Pyroptosis is mainly manifested by cell membrane rupture, DNA fragmentation, chromatin condensation, cytoskeleton destruction and other phenomena, which are not present in ferroptosis.15 However, iron can not only induce ferroptosis but also induce pyroptosis through the Tom20-Bax-caspase-GSDME pathway,16 suggesting that ferroptosis may coexist with pyroptosis.
 |
Table 1 The Features of Ferroptosis, Necroptosis, Apoptosis, Autophagy, and Pyroptosis |
Morphologically, the main features are small and deformed mitochondria, reduction or disappearance of mitochondrial cristae, shrunken mitochondrial membrane, and rupture of outer membrane,1,3 but the cytoplasmic membrane remains intact and Normal nuclear volume. Biochemically, It is manifested as a decrease in intracellular glutathione (GSH), a decrease in the activity of Glutathione peroxidase 4 (GPX4), resulting in the accumulation of lipid peroxides and the accumulation of Fe2+ causes the Fenton reaction to occur, which produces excess Reactive oxygen species (ROS) promote oxidative stress in organelles such as mitochondria,1,17 endoplasmic reticulum, and Golgi apparatus.18–20 Meanwhile, ROS reacts with polyunsaturated fatty acids (PUFAs) on the cell membrane to promote ferroptosis.3 It is worth noting that mitochondrial features are by no means unique and that the morphology of mitochondria can vary considerably within a single cell. Therefore, the change of mitochondrial morphology is not enough to judge the occurrence of ferroptosis in cells, and a comprehensive judgment needs to be combined with biochemical indicators.
Mechanisms of Ferroptosis
With the deepening of research, we have a deeper understanding of the molecular mechanism of ferroptosis. The activation of ferroptosis requires two key signals, the inhibition of the SLC7A11-GSH-GPX4 antioxidant axis and the accumulation of iron, and the production of this process requires a series of regulations (Figure 1). The genetic, transcriptional and translational of this process were systematically described by Chen.21
 |
Figure 1 The regulatory pathways of ferroptosis. The figure shows the regulatory pathways of ferroptosis, which can be roughly divided into three categories. The first is regulated by the GSH/GPX4 pathway, such as the cystine/glutamate anti-transporter SystemXC−, the sulfur transport pathway, and arachidonic acid and other related pathways. The second is the regulatory mechanisms of iron metabolism, such as the regulation of NCOA4 and IREB2 related to ferritin metabolism, and the regulatory pathway of ferroportin-related STEAP3, all have an effect on free iron content, ultimately causing the Fenton reaction. The third category is related pathways of lipid metabolism, such as ACSL4, LPCAT3, ALOXs, etc., which have the role of lipid regulation and ferroptosis. Abbreviations: PUFA, Polyunsaturated fatty acid; PE, Phosphatidylethanolamine; ACSL4, Acyl-CoA synthetase long-chain family member 4; LPCAT3, Lysophosphatidylcholine Acyltransferase 3; ALoxs, Lipoxygenase; AA, Arachidonic acid; AdA, Adrenaline acid; CoA, Coenzyme A; L-OOH, Lipid hydroperoxide; L-OH, Lipid alcohol; SLC7A11, Solute carrier family, member11; SLC3A2, Solute carrier family 3; member 2; GSSG, Oxidized glutathione; IREB2, Iron response element binding protein 2; DMT-1, Divalentmetal- iontransporter-1; ZIP, Zinc-Iron regulatory protein; STEAP3, Six transmembrane epithelial antigen of prostate3; VDACs, Voltage-dependent anion channels; CoQ10, Coenzyme Q10; NCOA4, nuclear receptor coactivator 4; FSP1, Ferroptosis-suppressor-protein 1. |
Amino Acid Metabolic Pathway
GSH is a tripeptide that participates in various metabolic activities of cells and plays a crucial role in cellular oxidative stress. Murphy found that the levels of GSH can be reduced when cells were exposed to glutamate or low concentrations of cystine, and the level of intracellular peroxides is increased, leading to oxidative stress and cell death.22 With further research, Dixon deduced that glutamate-induced cell death is similar to ferroptosis,3 and that cystine uptake depends on the cystine(Cys)/glutamate(Glu) antiporter (systemXC−) on the cell membrane.23 SystemXC− is an amino acid antiporter widely distributed on the cell membrane, and it is a heterodimer composed of the light chain subunit SLC7A11 and the heavy chain subunit SLC3A2.24 It can mediate the import of extracellular Cys and the export of intracellular Glu.25 Cys is the rate-limiting substrate for the synthesis of GSH.26 Cysteine entering cells is rapidly reduced to two cysteines, and then cysteine combines with glutamate and glycine to generate glutathione. Glutathione plays an anti-oxidative stress role.27 Another study confirmed that the lack of Cys not only inhibits the synthesis of GSH, but also induces the accumulation of Glu and promotes ferroptosis.28 Therefore, inhibiting the activity of systemXC− can reduce the concentration of intracellular cystine and GSH synthesis, leading to the occurrence of ferroptosis.
GPX4 is a key enzyme in reducing peroxides. As a phospholipid hydroperoxidase, the expression and activity of GPX4 are affected by GSH. Under the action of GSH, the degradation of intracellular hydrogen peroxide and hydroperoxide can be accelerated, and the generation of lipid reactive oxygen species can be inhibited.29 Yang found that the reduction of GSH would lead to a decrease in the activity of GPXs.1 And Li discovered in their experiments that the use of a GPX4 activator can reduce the level of intracellular ROS and inhibit ferroptosis.30 Thus, GSH can inhibit lipid peroxidation and ferroptosis in cells by increasing the activity of GPX4.
Lipid Metabolism
The cell membrane is a phospholipid bilayer structure, which can maintain a variety of biological functions and homeostasis in the cell. Intracellular lipids are mainly stored in lipid droplets, which are eventually hydrolyzed into lipids in the cytoplasm for cellular use. PUFAs are separated from membrane phospholipids and form lipid droplets in response to cellular oxidative damage. Although lipid droplets do not directly affect ferroptosis, PUFAs are sensitive to lipid peroxidation, which is an essential feature of ferroptosis.31 Although there are three types of fatty acids (saturated fatty acids, monounsaturated fatty acids, and polyunsaturated fatty acids), studies have confirmed that lipoxygenases (LOXs) are more important for the PUFAs.32
Free PUFAs are necessary for lipid synthesis, and the existence of a diallyl matrix makes PUFAs susceptible to free radicals and LOXs, which make PUFAs more prone to ferroptosis.33 The PUFAs chain produces ROS in cells after a series of reactions.34 PUFAs serve as substrates for the synthesis of lipid signaling mediators, which must be esterified into membrane phospholipids and further oxidized to participate in ferroptosis.35 The activation of ferroptosis requires the participation of two enzymes, namely acyl-CoA synthetase long-chain family member 4 (ACSL4) and Lysophosphatidylcholine Acyltransferase 3 (LPCAT3), ACSL4 binds coenzyme A to long-chain polyunsaturated fatty acids, Lysophospholipids are esterified by LPCAT3 via long-chain polyunsaturated fatty acids and then induce ferroptosis in cells.35
Iron Metabolism
Iron is an important trace element in the body. Decreased iron content will lead to iron-deficiency disease.36 Contrarily, overload of iron content will generate free radicals through the Fenton reaction, resulting in oxidative stress in cells, which eventually leads to cell death.37 Iron comes primarily from dietary iron and can also be recovered from the body’s liver and aging red blood cells (RBCs). Dietary iron is mainly ferric ions (Fe3+), which are absorbed by intestinal epithelial cells in the duodenum and upper jejunum, binds to transferrin (TF) to form a complex, It enters cells under the mediation of transferrin receptor (TFR) and is subsequently reduced to ferrous ions (Fe2+) for a series of biochemical reactions (Figure 2). Iron plays a crucial role in maintaining ATP energy reserves during oxidative phosphorylation of the inner mitochondrial membrane.38
 |
Figure 2 (A) Mechanism of iron absorption in intestinal mucosa. (B) Mechanism of cellular iron uptake. Abbreviations: Hcp-1, Heme carrier protein 1; HO-1, Heme oxygenase 1; Dcytb, Duodenal cytochrome b; DMT-1, Divalentmetal- iontransporter-1; Fpn-1, Ferroportin 1; Hepe, Hephaestin; Tf, transferrin; TfR, Transferrin receptor; NTBI, Non—transferrin-bound iron; ZIP, Zinc-Iron regulatory protein; STEAP3, Six transmembrane epithelial antigen of prostate3. |
When the iron-binding complexes in the body approach saturation, excess iron begins to deposit inside the cells. Dixon found that the sensitivity of cells to ferroptosis can be changed by regulating the content of intracellular iron, increased levels of transferrin (TF) and transferrin receptor-1 (TFR-1) can promote ferroptosis in cells.3 Cellular iron homeostasis is mainly regulated by the iron regulatory proteins IRP1 and IRP2, which are involved in the regulation of intracellular iron uptake and distribution.39 The accumulation of Fe2+ will participate in the oxygen reaction in cells, thereby initiating the Fenton reaction to generate ROS such as hydroxyl radicals and hydrogen peroxide. The accumulation of ROS is closely related to lipid peroxidation and tissue damage.40
In vivo, Fe2+ is mainly stored in ferritin, which consists of ferritin light chain (FTL) and ferritin heavy chain 1 (FTH1), but also in iron pools. Iron-responsive element-binding protein 2 (IREB2) is a major transcription factor that inhibits iron metabolism, and IREB2 can inhibit erastin-induced ferroptosis by increasing the expression of FTL and FTH1.41 TFR-1 is also an important factor in promoting ferroptosis. Studies have found that erastin-induced ferroptosis can be inhibited by silencing the gene TFRC encoding TFR1.42 Heat shock protein β-1 (HSPB1) can reduce intracellular iron content by inhibiting the expression of TRF1, so inhibiting the expression of HSPB1 can promote ferroptosis.43 Likewise, iron export was blocked by inhibiting the expression of Solute Carrier family 11 member A3 (SLC11A3) promoting erastin-induced ferroptosis in neuroblastomas cells.44 Lipophilic iron chelators can chelate intracellular free iron through the cell membrane,45 and inhibit the lipid peroxidation of PUFAs and the generation of ROS by preventing Fe2+ from donating electrons.46 Gao has found that the overexpression of NCOA4 in cells can activate ferroautophagy and increase the free iron content in cells, which is an important regulator involved in the occurrence of ferroptosis.47
p53 and Ferroptosis
P53 is the most important and common tumor suppressor gene, and it is also one of the tumor suppressor genes related to ferroptosis early discovered, and plays a dual role in ferroptosis (Figure 3). Jiang demonstrated that the activity of H1299 cells remained unchanged when treated with ROS after silencing the P53 gene.48 However, cell death increases after p53 activation. In their experiments, they confirmed that when the expression of p53 gene was up-regulated, the messenger RNA and protein expression of SLC7A11 was significantly reduced, indicating that SLC7A11 is a target of p53 gene and the activation of p53 can mediate the down-regulation of SLC7A11 to promote ferroptosis.49 The suppressor of Cytokine Signaling-1(SOCS1) can control the phosphorylation of p53 by regulating the expression of p53 target genes. In addition, several studies have demonstrated that both SLC7A11 and SAT1 are SOCS1-dependent p53 targets, suggesting that the SOCS1-p53 axis is involved in the ferroptosis pathway.50 SAT1 is an important rate-limiting enzyme in polyamine catabolism and upregulation of SAT1 expression can accelerate ROS-induced ferroptosis. This suggests that the P53-SAT1 axis is also involved in the regulation of ferroptosis.51 Further studies confirmed that P53 and dipeptidyl peptidase-4(DPP4) were also involved in the ferroptosis in Golgi.19
 |
Figure 3 Dual mechanism of p53 in ferroptosis. Abbreviations: Cys, Cysteine; GSH, Glutathione; GPX4, Glutathione Peroxidase 4; SLC7A11, Solute carrier family7member11; GLS2, Glutaminase 2; PTGS2, prostaglandin-endoperoxide synthase 2; SAT1, Spermidine/spermine N(1)-acetyltransferase 1; PUFA, Polyunsaturated fatty acid; L-OOH, Lipid hydroperoxide; L-OH, Lipid alcohol; NOX1, Nicotinamide adenine dinucleotide phosphate oxidase 1; ROS, Reactive oxygen species; DPP4, Dipeptidyl peptidase-4; ALox, Lipoxygenase; CDKN1A, Cyclin-dependent kinase inhibitor 1A. |
Conversely, p53 also inhibits ferroptosis under certain conditions. Tarangelo’s research has indicated that stable wild-type P53 can reduce the activity of systemXC− and the susceptibility to ferroptosis.52 Deletion of p53 prevents the accumulation of DPP4 in the nucleus, promotes the localization of DPP4 on the plasma membrane, and further enables DPP4 to form a complex with NOX1 (NADPH oxidase 1) for lipid peroxidation.53 While P53 can inhibit DPP4 and limit ferroptosis in colorectal cancer cells.53 In human fibrosarcoma cells, p53-mediated transcription of CDKN1A/p21 (cyclin-dependent kinase inhibitor 1A) can slow down GSH depletion and delay ferroptosis induced by cystine deprivation.52 P53 has shown the ability of dual regulation of ferroptosis, but the specific regulatory mechanism of promoting or inhibiting ferroptosis still needs to be further studied.
Other Regulatory Pathways
Other researchers have found that ferroptosis may also be regulated by other pathways. As a voltage-dependent anion channel (VDACs) on the mitochondrial outer membrane, VDACs play an important role in iron metabolism.54 In 2007, Yagoda found that erastin can act on VDACs on mitochondria,11 and change the shape and structure of mitochondria, leading to mitochondrial dysfunction.2 The final activation of the RAS-RAF-MEK pathway leads to the disturbance of intracellular iron metabolism and ferroptosis.3 Under oxidative stress, methionine converts itself into cystine through the transsulfuration pathway, and finally synthesizes GSH to assist GPX4 in anti-oxidation.55 Thus, the transsulfuration pathway can inhibit ferroptosis in cells. Cysteine can also be synthesized through the transsulfuration pathway in other cell types.55 Cysteine desulfurase (NFS1) is an enzyme that can synthesize iron-sulfur clusters (ISCs) and ISCs are capable of transporting electrons in mitochondria.56 The reduction of NFS1 activity can inhibit the synthesis of ISCs and lead to disturbance of mitochondrial electron transport, leading to the occurrence of ferroptosis. Doll indicated that ferroptosis suppressor protein 1 (FSP1) is an independent pathway to resist ferroptosis, exerting ferroptosis resistance by catalyzing coenzyme Q10.57 Meanwhile, another study found that FSP1 traps free radicals at the plasma membrane, preventing the diffusion of lipid peroxidation. Bersuker et al found that the ability of cancer cells to resist ferroptosis is related to the level of FSP1 expression in hundreds of cancer cell lines.58 Recent new findings confirm that FSP1 acts as a reductase that reduces vitamin K to vitamin K hydroquinone (VKH2), which protects cells through the non-canonical vitamin K cycle for vitamin K to produce antioxidant effects protected from ferroptosis.59
In summary, the regulation of ferroptosis is complex. There are multiple pathways involved in the regulation of ferroptosis, which together lead to lipid peroxidation and ferroptosis.
The Detection of Ferroptosis
With the deepening of ferroptosis research, there are more and more detection methods. In terms of methods, iron metabolism and lipid metabolism are mainly detected. The levels of intracellular Fe3+ and Fe2+ were analyzed using fluorescent probe kits, and the FRET Iron Probe 1 (FIP-1) probe could also be used to detect unstable iron pools.60 Using antibodies to detect TFRC is also a feasible method.61 The imbalance of lipid metabolism is an essential part of ferroptosis, and detection of lipid ROS, metabolic end products (such as MDA or 4HNE) or antioxidant components (such as GSH, GPX4, etc.) is the most commonly used method. MitoPerOx, MitoPeDPP and MitoCLox can detect lipid peroxidation in mitochondria. MitoSOX can detect superoxide in cells. These are the most commonly used probes to detect ferroptosis. However, all these methods, including antibody-based methods, are still not specific. Therefore, there are only two reliable methods for detecting ferroptosis currently: a) reversal of cell death by free radical scavengers and iron chelators, and b) (oxy-)lipidomics by mass spectrometry. Exploring specific detection methods for ferroptosis is still one of the important directions for future research.
The Role of Ferroptosis in Various Systemic Diseases
The in-depth study of ferroptosis has gradually become the focus and hotspot for the treatment and improvement of the prognosis of related systemic diseases (Figure 4).
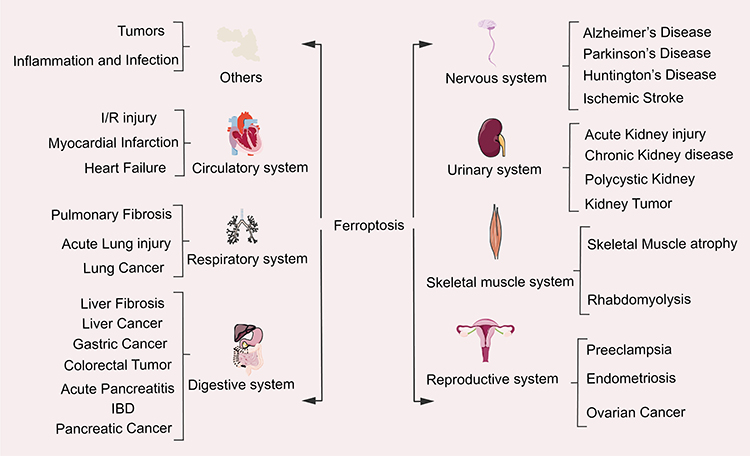 |
Figure 4 The relationship between ferroptosis and system diseases. |
Tumors
Ferroptosis is closely related to tumors, and the progression and spread of tumors require iron to participate, so the iron requirement of cancer cells is much greater than that of normal cells.62 However, the high iron environment in cells is the reason why cancer cells are prone to trigger ferroptosis. Eling confirmed that in pancreatic cancer cells, artesunate can induce ferroptosis and inhibit cancer cell growth.63 Subsequently, Lin also found that ferroptosis could be detected in head and neck squamous cell carcinoma after the use of dihydroartemisinin.64 In another study, Louandre et al found that hepatoma cells were exposed to sorafenib to increase cell death, which may be caused by sorafenib-induced ferroptosis in hepatoma cells.65 Ma et al found that siramesine and lapatinib can induce ferroptosis in breast cancer cells, and finally achieve the purpose of treatment.66 Belavgeni found that the human ACC NCI-H295R cell line was highly sensitive to the induction of ferroptosis.67 Ferroptosis was completely inhibited in H295R cells after ferrostatin-1 treatment, suggesting that induction of ferroptosis may be a novel approach for the treatment of ACCs. Cisplatin is a traditional oncology drug and an inducer of ferroptosis. Recent studies have shown that the occurrence of ferroptosis may enhance the anticancer effect of cisplatin on cancer cells.68 The above studies suggest that inhibiting the growth of cancer cells through targeted induction of ferroptosis may be a new strategy for cancer treatment in the future.
Inflammation and Infection
Inflammation is the body’s protective response to tissue damage and is an important physiological process of the body, mainly manifested as redness, swelling, heat, pain and dysfunction. The inflammatory response has an appropriate range, beyond which it will cause damage to the body. Studies have found that PUFAs and their metabolic enzymes are important regulators involved in the process of the body’s inflammatory response.69 In inflammation, GPX4 can inhibit the activation of arachidonic acid (AA) and nuclear factor kappa B (NF-κB) pathways and reduce the level of ROS generated by lipid peroxidation, which indicates that inflammation is closely related to ferroptosis.30 Jiao et al found that curcumin can inhibit the synthesis of hepcidin (a peptide hormone mainly produced in the liver), affect the body’s iron metabolism, and reduce inflammation caused by lipopolysaccharide (LPS).70 Dexamethasone is mainly used for autoimmune inflammatory diseases. Recent studies by Mässenhausen have demonstrated that dexamethasone reduces GSH levels by upregulating GSH metabolic regulatory protein dipeptidase-1 (DPEP1) in a glucocorticoid receptor (GR)-dependent manner, depleting intracellular GSH and increasing cellular response to Sensitivity to ferroptosis.71 This is an unprecedented discovery that makes a major contribution to the study of complications arising from dexamethasone treatment of the disease.
Intracellular bacterial survival is a major factor leading to infection. This often leads to treatment failure, and studies have found that tissue infections may be associated with ferroptosis. Macrophages are the predominant cells in the early stages of bacterial infection. A recent study demonstrated that ferroptotic stress can assist macrophages to induce bacterial death, as demonstrated in a mouse infection model.72 In addition, Dar et al found that the activity of pLoxA may determine the susceptibility of P. aeruginosa isolates to ferroptosis.73 This enables ferroptosis in airway epithelial cells under the action of Pseudomonas aeruginosa. We speculate that targeted induction of ferroptosis may be a novel approach to achieving therapeutic targets for intracellular bacterial infections.
Circulatory System
It has been found that ferroptosis is closely related to cardiovascular system damage. Especially in ischemic heart disease, ferroptosis has been extensively studied. In a mouse model of cardiac ischemia/reperfusion (I/R) injury, the application of ferroptosis inhibitors can significantly reduce cardiac injury and improve myocardial function. Fang demonstrated that ferroptosis is a mechanism of cardiac injury caused by doxorubicin (DOX) and I/R, and confirmed that the free iron released during Heme degradation is the cause of cardiac injury.74 Ferrostatin-1 and iron chelators can maintain mitochondrial function by inhibiting cellular lipid peroxidation and avoid ferroptosis-induced cardiac damage. In systemic inflammation after heart transplantation, Li et al found that ferroptosis promotes the adhesion of neutrophils and coronary endothelial cells through the signaling pathway of TLR4/TRIF/IFN-1.75 The administration of ferrostatin-1 prevents neutrophil recruitment after cardiac transplantation, inhibits inflammation and reduces cardiomyocyte death. These findings provide a new platform for the treatment of systemic inflammation after heart transplantation. Liproxstatin-1 (Lip-1) is a ferroptosis inhibitor, Feng illustrated that the level of GPX4 was increased, the level of ROS and the size of myocardial infarct(MI) were decreased, and cardiomyocyte ferroptosis was inhibited in IRI mice after administration of Lip-1.76 Park demonstrated that the finding of ferroptosis during myocardial infarction.77 Using a rat MI model, they performed quantitative proteomics analysis and found that GPX4 is down-regulated in early and mid-MI, increasing the sensitivity of primary neonatal rat ventricular myocytes to ferroptosis. Bulluck confirmed that the residual myocardial iron in patients with intramyocardial hemorrhage (IMH) myocardial infarction may be a new therapeutic target for patients with poor left ventricular remodeling through follow-up of patients with ST-segment elevation myocardial infarction after interventional therapy.78 Cardiomyocyte injury plays an important role in heart failure (HF). Chen discovered knockdown of TLR4 and NOX4 (NADPH oxidase 4) could inhibit autophagy and ferroptosis and delay heart failure.79 In addition, Li et al established a septic cardiomyopathy model by injecting LPS to study its relationship with ferroptosis.80 They found that LPS could increase the expression of NCOA4 and the level of Fe2+, resulting in elevated levels of ROS in mitochondria. This suggests that ferroptosis may be associated with sepsis-induced cardiac damage. Patients in the intensive care unit (ICU) frequently die from multiple organ dysfunction syndromes (MODS). Van et al speculated that the severity of multiple organ dysfunction may be related to ferroptosis by analyzing Fe and MDA in the plasma of critically ill patients, and confirmed this speculation in mouse experiments.81 Interestingly, this phenomenon was not found in a mouse model of sepsis-induced MODs. This is different from the findings of Li et al and the reason for this difference needs to be proved by further experiments.80 Reducing cardiomyocyte death and improving myocardial remodeling by inhibiting ferroptosis may be a new strategy for preventing cardiac disease in the future.
Respiratory System
Among respiratory diseases, pulmonary fibrosis (PF) is a deadly disease. In bleomycin (BLM)-induced rat PF lung tissue, In bleomycin (BLM)-induced rat PF lung tissue, Yang demonstrated that inhibition of lncRNA ZFAS1 could down-regulate SLC38A1 attenuating ferroptosis progression and attenuating BLM-induced inflammation, lipid peroxidation, and the development of PF.82 In the tissues of acute lung injury (ALI), Li discovered that panaxoxynol (PX) may inhibit ferroptosis and alleviate LPS-induced inflammatory response, which may be related to upregulating the pathways of Keap1-Nrf2/HO-1 by PX.83 Levels of ALOX15 are elevated in bronchial epithelial cells following infection with Pseudomonas aeruginosa, leading to ferroptosis.73 Amaral considered that induction of ferroptosis may be one of the mechanisms by which Mycobacterium tuberculosis kills host cells, but further research is needed to verify this.84 The researchers found that P53 can induce ferroptosis in lung cancer A549 cells. When erastin acts on lung cancer A549 cells, P53 is up-regulated and activated, inhibiting the activity of SLC7A11, inducing the accumulation of ROS, and finally leading to ferroptosis.56 In recent years, it has been found in coronavirus studies that coronaviruses can degrade serum iron levels in cells, reduce GSH activity and increase the amount of ROS.85 However, in patients with high levels of GSH activity, symptoms of COVID-19 recovered more quickly,86 suggesting that coronavirus may increase clinical symptoms in patients by mediating ferroptosis.
Digestive System
As ferroptosis is gradually recognized, its relationship in the digestive system is becoming clearer. Wang proved that ferrostatin-1 (Fer-1) can slow down the anti-fibrotic effect of artemether (ART) on liver by inhibiting ferroptosis and found that P53 is an upstream molecule that induces ferroptosis in hepatic stellate cells (HSC).87 Yu used the hepatocyte-specific Trf knockout mouse (Trf LKO) model and found that after treatment with ferrostatin-1, the liver fibrosis of the Trf LKO mice was alleviated, and hepatic transferrin played a role in maintaining liver function.88 This suggests that ferroptosis may be helpful in the treatment of certain liver diseases. It was also mentioned before that Sorafenib can induce ferroptosis in retinoblastoma (Rb), and Rb dysfunction is an important factor in the progression of liver cancer.65 In nonalcoholic steatohepatitis, RSL-3 can significantly increase the levels of inflammatory factors and hepatitis-related markers, indicating that inhibiting ferroptosis may be a new idea for the treatment of this disease.89,90 Hao found that erastin can induce ferroptosis in gastric cancer (GC) cells, while cysteine dioxygenase1 (CDO1) competitively absorbs cysteine and restricts the synthesis of GSH.91 Therefore, silencing CDO1 can restore the level of GSH in GC cells, maintain mitochondrial morphological stability, and inhibit ferroptosis. Another study showed that the ability of GSH synthesis was increased in gastrointestinal tumor cells with high CD44 expression, which made GC cells have a certain degree of ferroptosis resistance.92 P53 has a bidirectional effect on ferroptosis. P53 can induce ferroptosis dependent on lipid peroxidation. Conversely, P53 can also induce ferroptosis resistance in colorectal tumor cells by inhibiting the activity of DPP4.53 The combination of cisplatin and erastin can enhance the anti-tumor effect.93 Several studies have confirmed that ferroptosis inhibitors can upregulate the activity of GPX4 and alleviate IBD.94 Ferroptosis is also associated with pancreatic disease. The research of Eling demonstrated that artesunate can inhibit the growth of pancreatic cancer cells by inducing ferroptosis.63 Knockout of Arntl, a core component of the circadian clock, increased the incidence of acute pancreatitis, while administration of liproxstatin-1 ameliorated the incidence of acute pancreatitis. This provides a new idea for the treatment of pancreatic diseases.
Nervous System
The accumulation of lipid peroxidation and iron is closely related to the normal physiological function of the brain and the occurrence of neurological diseases. Several researches found that multiple regions of the brain (the globus pallidus, putamen, substantia nigra, caudate nucleus, etc.) showed the accumulation of iron. Chen found that knockout of GPX4 in mice can cause paralysis symptoms and accelerate the death of mice, and also observed that the motor neurons in the spinal cord of mice also degenerate.95 But after the use of vitamin E, this phenomenon was delayed, confirming that GPX4 has a key role in the motor neuron system. Disturbances of iron metabolism are found in various neurological diseases such as Alzheimer’s disease (AD),96 Parkinson’s disease and others.97 AD is the most common neurodegenerative disease, and Raven found elevated levels of iron in the hippocampus of AD patients by MRI instrumentation.98 Do confirmed in another study that ferropstatin-1 could block the toxic effects of 1-Methy1-4-Pheny1-1,2,3,6-etrahydropyridine (MPTP) on dopaminergic neurons, and also demonstrated that DFO (iron chelator) increases dopamine activity and improves motor symptoms.97 This indicates that the progression of Parkinson’s disease (PD) is likely to be related to ferroptosis. It should be known that the main pathological feature of PD is the degeneration of dopaminergic neurons. In their research, Agrawal demonstrated that iron could impair mitochondrial function as a mediator in a mouse model of Huntington’s disease (HD) and suggested that targeting the iron-mitochondrial pathway may have a protective effect.99 Skoutal also confirmed that Ferrostatin-1 can inhibit nerve cell death and restore the number of healthy neurons in the HD model.100 Recent studies have shown that the serine protease thrombin can promote ferroptosis by promoting arachidonic acid mobilization and esterification of acyl-CoA synthase long-chain family member 4 (ACSL4), and inhibition of thrombin-ACSL4 axis conduction can improve ischemic Neuronal damage during stroke.101 This provides a new treatment idea for improving the sequela caused by ischemic stroke.
Urinary System
The kidneys are the organs in the body that metabolize and reabsorb and retain water and other useful substances. Acute kidney injury (AKI) is a clinical syndrome with a series of complications caused by a sharp decline in renal function caused by different etiologies, with a high mortality rate. Ferroptosis has been shown to play an important role in the progression of AKI. Friedmann found an important role of the GSH/Gpx4 axis in the Gpx4 knockout mouse model of acute renal failure, and GPX4 is a negative regulator of AKI.17 A more important study provides genetic evidence for the association of AKI with ferroptosis. Tonnus demonstrated that loss of FSP1 or GPX4 dysfunction can increase the sensitivity of renal tubules to ferroptosis.102 In addition, they developed a new ferroptosis inhibitor: Nec-1f. Notably, no differences were detected in GPX4 in the cisplatin-induced AKI model. This phenomenon suggests that ferroptosis due to GPX4 dysfunction requires certain conditions, which require further research. In addition, Muller also found that the increased expression of ACSL4, a key fatty acid metabolism enzyme that regulates ferroptosis, may aggravate the degree of AKI.103 In a mouse model of AKI induced by folic acid (a nephrotoxic drug), ferroptosis was the main mechanism for cell death. On the contrary, inhibition of ferroptosis by ferropstatin-1 resulted in a significant improvement in renal function.104 In another study, Deng found tubular damage and disturbances of renal physiology in cisplatin-treated CD1 mice.105 MIOX is a proximal tubular-specific enzyme and was discovered in recent years. Upregulation of MIOX expression can promote the generation of ROS and aggravate renal tubular damage. However, evidence of ferroptosis was reduced after cisplatin treatment, suggesting that ferroptosis is regulated by MIOX expression in the pathogenesis of cisplatin-induced AKI. In chronic kidney disease (CKD), inflammation and fibrosis are important pathways for its progression. Luo et al found that Fer-1 could alleviate HFD-induced pathological changes and functional impairments (such as fibrosis, inflammatory factor expression, and inflammatory cell infiltration) in Fer-1-pretreated high-fat diet (HFD) mice, confirming that ferroptosis can reduce the progression of renal injury in CKD, and provides a new treatment idea for fibrotic diseases.106 In polycystic kidney disease, cells secrete chloride ions through the chloride channels cystic fibrosis transmembrane conductance regulator (CFTR) and TMEM16A, which promote cyst enlargement. Schreiber studied tissue samples from patients with polycystic kidney disease, embryonic kidney culture and MDCK in vitro cyst model and found that inhibition of ferroptosis or ROS generation would lead to decreased TMEM16A activity and decreased cyst proliferation.107 Among kidney tumors, clear cell carcinoma is the most common. Yang screened cancer cell lines and found through sensitivity analysis that diffuse large B-cell lymphoma and renal cell carcinoma were particularly sensitive to GPX4-mediated ferroptosis.108 In addition, Miess also found that inhibition of GSH synthesis can prevent the growth of kidney tumors, which is through the inhibition of fatty acid metabolism and the GSH/GPX pathway.109 These findings provide a new idea for tumor treatment. For some tumors that are not sensitive to conventional radiation and chemotherapy, the ferroptosis mechanism may be an executable new therapy.
Skeletal Muscle System
Skeletal muscle is an important part of the motor system, and the body completes various actions through muscle contraction. Due to trauma or other reasons, the content of the fascial compartment increases, the volume of the fascial compartment decreases, and the internal pressure increases, resulting in acute compartment syndrome (ACS), which can further aggravate skeletal muscle injury. Van and other researchers have found that iron metabolism or mitochondrial dysfunction are associated with skeletal muscle atrophy.110 Wang confirmed that down-regulation of cystathionine γ-lyase/hydrogen sulfide (CSE/H2S) signaling promotes ferroptosis and enhances acetylation of related proteins in skeletal muscle of mice, which is associated with Skeletal muscle injury and aging through MuRF-1-dependent pathways.111 Other research has shown that oxidative stress can hinder the self-healing of skeletal muscle after ischemic injury. Yuan found that dexmedetomidine (DEX) combined with α2 receptor can up-regulate the level of Nrf2 in skeletal muscle, increase the expression of HO-1 downstream of Nrf2, and enhance the antioxidant effect.112 Rhabdomyolysis (RM) is a relatively common syndrome caused by severe damage to skeletal muscle. A recent study confirmed that ferroptosis can aggravate the development of RM. He increased the expression level of GPX4 in muscle cells by inhibiting ACSL4, decreased lipid peroxidation products, and improved the progress of RM, which to a certain extent inhibited the effect of exertional heat stroke (EHS) in the Muscle cells.113 The above studies suggest that intervening in ferroptosis-related regulators (such as Nrf2/HO-1, ACSL4) may be a new therapeutic strategy for improving skeletal muscle-related diseases.
Reproductive System
Disturbance of iron metabolism determines the functional stability of the reproductive system in some aspects. Compared with other systems, there is less research on the relationship between the reproductive system and ferroptosis and needs to be further improved. In the male reproductive system, testicular damage can promote the death of germ cells and sertoli cells, so protection of cells from death can restore testicular function to some extent. Bromfield found the existence of ferroptosis in male mouse germ cells, sperm cells may be mediated to trigger the occurrence of ferroptosis by ACSL4, and the ACSL4-ALOX15 pathway may be involved in this phenomenon.114 In addition, Ghoochani demonstrated that intervention with erastin and RSL3 can delay the growth of prostate cancer cells, and combined use with second-generation anti-androgens for advanced prostate cancer can better prevent tumor progression, which may be related to High expression of SLC7A11, SLC3A2 and GPX4 in prostate cancer.115 Likewise, in the female reproductive system, disturbances in iron metabolism have been implicated in reproductive disorders such as pregnancy, preeclampsia, and endometriosis. In pregnant women, iron requirements are increased compared to normal, and the associated response to the sharp increase in oxygen and iron requirements is amplified during pregnancy. The consequent membrane lipid peroxidation and ferroptosis at the maternal-fetal interface (mainly trophoblasts) can remodel maternal spiral arteries and superficial intravascular infiltration of extravillous cytotrophoblasts (EVCTs), which are responsible for the formation of preeclampsia (PE) pathological features.116 Endometriosis is a common disease in gynecology. At present, it is believed that the occurrence of the disease is mainly due to the reverse flow of menstrual blood, the reverse shedding of endometrial tissue through the fallopian tube to the pelvis and abdominal cavity, and the high levels of iron can promote inflammation, induce lipid peroxidation and inhibit the growth of endometriotic cells. Conversely, in women with elevated cholesterol, the activity of the mevalonate cholesterol biosynthesis pathway is increased, protecting endometriotic cells from ferroptosis pathway regulation.117 This is related to the enhancement of cellular ferroptosis resistance by mevalonate-driven coenzyme Q10 as an endogenous lipophilic antioxidant.118 Artesunate (ART) is a good antimalarial drug, and it also has antitumor activity, which can trigger ferroptosis by the production of ROS in tumor cells and inhibit the progression of ovarian cancer.119 Overall, there is a lot of room for exploration in the study of ferroptosis in the reproductive system, which may be a new therapeutic strategy in the control and treatment of related diseases in the future.
Conclusion and Outlook
Over the past decade, we have witnessed a boom in ferroptosis research. As a new way of cell death, ferroptosis involves a variety of regulatory pathways, and ultimately leads to cell death due to ROS-induced accumulation of lipid peroxides. Whether ferroptosis has a more profound relationship with several other different cell death methods remains to be explored. Although a large number of studies have confirmed the existence of ferroptosis in various disease models, so far no specific marker has been found to prove the occurrence of ferroptosis. In order to better apply ferroptosis to clinical disease treatment, transcriptomics or metabolomics should be used to explore specific markers of ferroptosis in the future. There are still huge challenges in how to translate basic research into clinical applications. The discovery of ferroptosis provides a new idea for the treatment of the disease.
Acknowledgments
We thank those who contributed to this review for their cooperation and support.
Funding
This work was financially supported by the 2020 Hunan Provincial Health and Family Planning Commission Major Project (No. 20201906).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. 2008;15(3):234–245. doi:10.1016/j.chembiol.2008.02.010
2. Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3(3):285–296. doi:10.1016/s1535-6108(03)00050-3
3. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi:10.1016/j.cell.2012.03.042
4. Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res. 2019;29(5):347–364. doi:10.1038/s41422-019-0164-5
5. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22(4):266–282. doi:10.1038/s41580-020-00324-8
6. Stockwell BR, Jiang X, Gu W. Emerging mechanisms and disease relevance of ferroptosis. Trends Cell Biol. 2020;30(6):478–490. doi:10.1016/j.tcb.2020.02.009
7. Ingold I, Berndt C, Schmitt S, et al. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell. 2018;172(3):409–422.e21. doi:10.1016/j.cell.2017.11.048
8. Bogacz M, Krauth-Siegel RL. Tryparedoxin peroxidase-deficiency commits trypanosomes to ferroptosis-type cell death. Elife. 2018;7. doi:10.7554/eLife.37503
9. Distéfano AM, Martin MV, Córdoba JP, et al. Heat stress induces ferroptosis-like cell death in plants. J Cell Biol. 2017;216(2):463–476. doi:10.1083/jcb.201605110
10. Eagle H, Piez KA, Oyama VI. The biosynthesis of cystine in human cell cultures. J Biol Chem. 1961;236(5):1425–1428. doi:10.1016/S0021-9258(18)64190-0
11. Yagoda N, von Rechenberg M, Zaganjor E, et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 2007;447(7146):864–868. doi:10.1038/nature05859
12. Zheng DW, Lei Q, Zhu JY, et al. Switching apoptosis to ferroptosis: metal-organic network for high-efficiency anticancer therapy. Nano Lett. 2017;17(1):284–291. doi:10.1021/acs.nanolett.6b04060
13. Ma S, Dielschneider RF, Henson ES, et al. Ferroptosis and autophagy induced cell death occur independently after siramesine and lapatinib treatment in breast cancer cells. PLoS One. 2017;12(8):e0182921. doi:10.1371/journal.pone.0182921
14. Liu X, Xia S, Zhang Z, Wu H, Lieberman J. Channelling inflammation: gasdermins in physiology and disease. Nat Rev Drug Discov. 2021;20(5):384–405. doi:10.1038/s41573-021-00154-z
15. Yu P, Zhang X, Liu N, Tang L, Peng C, Chen X. Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther. 2021;6(1):128. doi:10.1038/s41392-021-00507-5
16. Zhou B, Zhang JY, Liu XS, et al. Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis. Cell Res. 2018;28(12):1171–1185. doi:10.1038/s41422-018-0090-y
17. Friedmann Angeli JP, Schneider M, Proneth B, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16(12):1180–1191. doi:10.1038/ncb3064
18. Gaschler MM, Hu F, Feng H, Linkermann A, Min W, Stockwell BR. Determination of the subcellular localization and mechanism of action of ferrostatins in suppressing ferroptosis. ACS Chem Biol. 2018;13(4):1013–1020. doi:10.1021/acschembio.8b00199
19. Alborzinia H, Ignashkova TI, Dejure FR, et al. Golgi stress mediates redox imbalance and ferroptosis in human cells. Commun Biol. 2018;1:210. doi:10.1038/s42003-018-0212-6
20. Wang H, Liu C, Zhao Y, Gao G. Mitochondria regulation in ferroptosis. Eur J Cell Biol. 2020;99(1):151058. doi:10.1016/j.ejcb.2019.151058
21. Chen X, Li J, Kang R, Klionsky DJ, Tang D. Ferroptosis: machinery and regulation. Autophagy. 2021;17(9):2054–2081. doi:10.1080/15548627.2020.1810918
22. Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron. 1989;2(6):1547–1558. doi:10.1016/0896-6273(89)90043-3
23. Hayano M, Yang WS, Corn CK, Pagano NC, Stockwell BR. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 2016;23(2):270–278. doi:10.1038/cdd.2015.93
24. Lin CH, Lin PP, Lin CY, et al. Decreased mRNA expression for the two subunits of system xc(-), SLC3A2 and SLC7A11, in WBC in patients with schizophrenia: evidence in support of the hypo-glutamatergic hypothesis of schizophrenia. J Psychiatr Res. 2016;72:58–63. doi:10.1016/j.jpsychires.2015.10.007
25. Bannai S. Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J Biol Chem. 1986;261(5):2256–2263.
26. Seelig GF, Simondsen RP, Meister A. Reversible dissociation of gamma-glutamylcysteine synthetase into two subunits. J Biol Chem. 1984;259(15):9345–9347.
27. Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem. 1999;274(17):11455–11458. doi:10.1074/jbc.274.17.11455
28. Kang YP, Mockabee-Macias A, Jiang C, et al. Non-canonical glutamate-cysteine ligase activity protects against ferroptosis. Cell Metab. 2021;33(1):174–189.e7. doi:10.1016/j.cmet.2020.12.007
29. Ursini F, Maiorino M, Hochstein P, Ernster L. Microsomal lipid peroxidation: mechanisms of initiation. The role of iron and iron chelators. Free Radic Biol Med. 1989;6(1):31–36. doi:10.1016/0891-5849(89)90156-1
30. Li C, Deng X, Xie X, Liu Y, Friedmann Angeli JP, Lai L. Activation of glutathione peroxidase 4 as a novel anti-inflammatory strategy. Front Pharmacol. 2018;9:1120. doi:10.3389/fphar.2018.01120
31. Pratt DA, Tallman KA, Porter NA. Free radical oxidation of polyunsaturated lipids: new mechanistic insights and the development of peroxyl radical clocks. Acc Chem Res. 2011;44(6):458–467. doi:10.1021/ar200024c
32. Wenzel SE, Tyurina YY, Zhao J, et al. PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell. 2017;171(3):628–641.e26. doi:10.1016/j.cell.2017.09.044
33. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A. 2016;113(34):E4966–E4975. doi:10.1073/pnas.1603244113
34. Cheng Z, Li Y. What is responsible for the initiating chemistry of iron-mediated lipid peroxidation: an update. Chem Rev. 2007;107(3):748–766. doi:10.1021/cr040077w
35. Kagan VE, Mao G, Qu F, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13(1):81–90. doi:10.1038/nchembio.2238
36. Sinha S, Pereira-Reis J, Guerra A, Rivella S, Duarte D. The role of iron in benign and malignant hematopoiesis. Antioxid Redox Signal. 2021;35(6):415–432. doi:10.1089/ars.2020.8155
37. Hou W, Xie Y, Song X, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12(8):1425–1428. doi:10.1080/15548627.2016.1187366
38. Oexle H, Gnaiger E, Weiss G. Iron-dependent changes in cellular energy metabolism: influence on citric acid cycle and oxidative phosphorylation. Biochim Biophys Acta. 1999;1413(3):99–107. doi:10.1016/s0005-2728(99)00088-2
39. Rouault TA, Stout CD, Kaptain S, Harford JB, Klausner RD. Structural relationship between an iron-regulated RNA-binding protein (IRE-BP) and aconitase: functional implications. Cell. 1991;64(5):881–883. doi:10.1016/0092-8674(91)90312-m
40. Iwai K, Drake SK, Wehr NB, et al. Iron-dependent oxidation, ubiquitination, and degradation of iron regulatory protein 2: implications for degradation of oxidized proteins. Proc Natl Acad Sci U S A. 1998;95(9):4924–4928. doi:10.1073/pnas.95.9.4924
41. Gammella E, Recalcati S, Rybinska I, Buratti P, Cairo G. Iron-induced damage in cardiomyopathy: oxidative-dependent and independent mechanisms. Oxid Med Cell Longev. 2015;2015:230182. doi:10.1155/2015/230182
42. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59(2):298–308. doi:10.1016/j.molcel.2015.06.011
43. Sun X, Ou Z, Xie M, et al. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene. 2015;34(45):5617–5625. doi:10.1038/onc.2015.32
44. Geng N, Shi BJ, Li SL, et al. Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells. Eur Rev Med Pharmacol Sci. 2018;22(12):3826–3836. doi:10.26355/eurrev_201806_15267
45. Petrat F, de Groot H, Rauen U. Subcellular distribution of chelatable iron: a laser scanning microscopic study in isolated hepatocytes and liver endothelial cells. Biochem J. 2001;356(Pt 1):61–69. doi:10.1042/0264-6021:
46. Schaich KM, Borg DC. Fenton reactions in lipid phases. Lipids. 1988;23(6):570–579. doi:10.1007/bf02535600
47. Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26(9):1021–1032. doi:10.1038/cr.2016.95
48. Jiang L, Hickman JH, Wang SJ, Gu W. Dynamic roles of p53-mediated metabolic activities in ROS-induced stress responses. Cell Cycle. 2015;14(18):2881–2885. doi:10.1080/15384101.2015.1068479
49. Jiang L, Kon N, Li T, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520(7545):57–62. doi:10.1038/nature14344
50. Saint-Germain E, Mignacca L, Vernier M, Bobbala D, Ilangumaran S, Ferbeyre G. SOCS1 regulates senescence and ferroptosis by modulating the expression of p53 target genes. Aging. 2017;9(10):2137–2162. doi:10.18632/aging.101306
51. Ou Y, Wang SJ, Li D, Chu B, Gu W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci U S A. 2016;113(44):E6806–E6812. doi:10.1073/pnas.1607152113
52. Tarangelo A, Magtanong L, Bieging-Rolett KT, et al. p53 suppresses metabolic stress-induced ferroptosis in cancer cells. Cell Rep. 2018;22(3):569–575. doi:10.1016/j.celrep.2017.12.077
53. Xie Y, Zhu S, Song X, et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Rep. 2017;20(7):1692–1704. doi:10.1016/j.celrep.2017.07.055
54. Skonieczna M, Cieslar-Pobuda A, Saenko Y, et al. The impact of DIDS-induced inhibition of Voltage-Dependent Anion Channels (VDAC) on cellular response of lymphoblastoid cells to ionizing radiation. Med Chem. 2017;13(5):477–483. doi:10.2174/1573406413666170421102353
55. McBean GJ. The transsulfuration pathway: a source of cysteine for glutathione in astrocytes. Amino Acids. 2012;42(1):199–205. doi:10.1007/s00726-011-0864-8
56. Alvarez SW, Sviderskiy VO, Terzi EM, et al. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature. 2017;551(7682):639–643. doi:10.1038/nature24637
57. Doll S, Freitas FP, Shah R, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575(7784):693–698. doi:10.1038/s41586-019-1707-0
58. Bersuker K, Hendricks JM, Li Z, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575(7784):688–692. doi:10.1038/s41586-019-1705-2
59. Mishima E, Ito J, Wu Z, et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature. 2022. doi:10.1038/s41586-022-05022-3
60. Aron AT, Loehr MO, Bogena J, Chang CJ. An endoperoxide reactivity-based FRET probe for ratiometric fluorescence imaging of labile iron pools in living cells. J Am Chem Soc. 2016;138(43):14338–14346. doi:10.1021/jacs.6b08016
61. Feng H, Schorpp K, Jin J, et al. Transferrin receptor is a specific ferroptosis marker. Cell Rep. 2020;30(10):3411–3423.e7. doi:10.1016/j.celrep.2020.02.049
62. Torti SV, Torti FM. Iron and cancer: more ore to be mined. Nat Rev Cancer. 2013;13(5):342–355. doi:10.1038/nrc3495
63. Eling N, Reuter L, Hazin J, Hamacher-Brady A, Brady NR. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience. 2015;2(5):517–532. doi:10.18632/oncoscience.160
64. Lin R, Zhang Z, Chen L, et al. Dihydroartemisinin (DHA) induces ferroptosis and causes cell cycle arrest in head and neck carcinoma cells. Cancer Lett. 2016;381(1):165–175. doi:10.1016/j.canlet.2016.07.033
65. Louandre C, Marcq I, Bouhlal H, et al. The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepatocellular carcinoma cells. Cancer Lett. 2015;356(2Pt B):971–977. doi:10.1016/j.canlet.2014.11.014
66. Ma S, Henson ES, Chen Y, Gibson SB. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016;7(7):e2307. doi:10.1038/cddis.2016.208
67. Belavgeni A, Bornstein SR, von Mässenhausen A, et al. Exquisite sensitivity of adrenocortical carcinomas to induction of ferroptosis. Proc Natl Acad Sci U S A. 2019;116(44):22269–22274. doi:10.1073/pnas.1912700116
68. Roh JL, Kim EH, Jang H, Shin D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol. 2017;11:254–262. doi:10.1016/j.redox.2016.12.010
69. Çolakoğlu M, Tunçer S, Banerjee S. Emerging cellular functions of the lipid metabolizing enzyme 15-Lipoxygenase-1. Cell Prolif. 2018;51(5):e12472. doi:10.1111/cpr.12472
70. Jiao Y, Wilkinson J, Di X, et al. Curcumin, a cancer chemopreventive and chemotherapeutic agent, is a biologically active iron chelator. Blood. 2009;113(2):462–469. doi:10.1182/blood-2008-05-155952
71. von Mässenhausen A, Zamora Gonzalez N, Maremonti F, et al. Dexamethasone sensitizes to ferroptosis by glucocorticoid receptor-induced dipeptidase-1 expression and glutathione depletion. Sci Adv. 2022;8(5):eabl8920. doi:10.1126/sciadv.abl8920
72. Ma R, Fang L, Chen L, Wang X, Jiang J, Gao L. Ferroptotic stress promotes macrophages against intracellular bacteria. Theranostics. 2022;12(5):2266–2289. doi:10.7150/thno.66663
73. Dar HH, Tyurina YY, Mikulska-Ruminska K, et al. Pseudomonas aeruginosa utilizes host polyunsaturated phosphatidylethanolamines to trigger theft-ferroptosis in bronchial epithelium. J Clin Invest. 2018;128(10):4639–4653. doi:10.1172/jci99490
74. Fang X, Wang H, Han D, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A. 2019;116(7):2672–2680. doi:10.1073/pnas.1821022116
75. Li W, Feng G, Gauthier JM, et al. Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J Clin Invest. 2019;129(6):2293–2304. doi:10.1172/jci126428
76. Feng Y, Madungwe NB, Imam Aliagan AD, Tombo N, Bopassa JC. Liproxstatin-1 protects the mouse myocardium against ischemia/reperfusion injury by decreasing VDAC1 levels and restoring GPX4 levels. Biochem Biophys Res Commun. 2019;520(3):606–611. doi:10.1016/j.bbrc.2019.10.006
77. Park TJ, Park JH, Lee GS, et al. Quantitative proteomic analyses reveal that GPX4 downregulation during myocardial infarction contributes to ferroptosis in cardiomyocytes. Cell Death Dis. 2019;10(11):835. doi:10.1038/s41419-019-2061-8
78. Bulluck H, Rosmini S, Abdel-Gadir A, et al. Residual myocardial iron following intramyocardial hemorrhage during the convalescent phase of reperfused ST-segment-elevation myocardial infarction and adverse left ventricular remodeling. Circ Cardiovasc Imaging. 2016;9(10):e004940. doi:10.1161/circimaging.116.004940
79. Chen X, Xu S, Zhao C, Liu B. Role of TLR4/NADPH oxidase 4 pathway in promoting cell death through autophagy and ferroptosis during heart failure. Biochem Biophys Res Commun. 2019;516(1):37–43. doi:10.1016/j.bbrc.2019.06.015
80. Li N, Wang W, Zhou H, et al. Ferritinophagy-mediated ferroptosis is involved in sepsis-induced cardiac injury. Free Radic Biol Med. 2020;160:303–318. doi:10.1016/j.freeradbiomed.2020.08.009
81. Van Coillie S, Van San E, Goetschalckx I, et al. Targeting ferroptosis protects against experimental (multi)organ dysfunction and death. Nat Commun. 2022;13(1):1046. doi:10.1038/s41467-022-28718-6
82. Yang Y, Tai W, Lu N, et al. lncRNA ZFAS1 promotes lung fibroblast-to-myofibroblast transition and ferroptosis via functioning as a ceRNA through miR-150-5p/SLC38A1 axis. Aging. 2020;12(10):9085–9102. doi:10.18632/aging.103176
83. Li J, Lu K, Sun F, et al. Panaxydol attenuates ferroptosis against LPS-induced acute lung injury in mice by Keap1-Nrf2/HO-1 pathway. J Transl Med. 2021;19(1):96. doi:10.1186/s12967-021-02745-1
84. Amaral EP, Costa DL, Namasivayam S, et al. A major role for ferroptosis in Mycobacterium tuberculosis-induced cell death and tissue necrosis. J Exp Med. 2019;216(3):556–570. doi:10.1084/jem.20181776
85. Zhao K, Huang J, Dai D, Feng Y, Liu L, Nie S. Serum iron level as a potential predictor of coronavirus disease 2019 severity and mortality: a retrospective study. Open Forum Infect Dis. 2020;7(7):ofaa250. doi:10.1093/ofid/ofaa250
86. Horowitz RI, Freeman PR, Bruzzese J. Efficacy of glutathione therapy in relieving dyspnea associated with COVID-19 pneumonia: a report of 2 cases. Respir Med Case Rep. 2020;30:101063. doi:10.1016/j.rmcr.2020.101063
87. Wang L, Zhang Z, Li M, et al. P53-dependent induction of ferroptosis is required for artemether to alleviate carbon tetrachloride-induced liver fibrosis and hepatic stellate cell activation. IUBMB Life. 2019;71(1):45–56. doi:10.1002/iub.1895
88. Yu Y, Jiang L, Wang H, et al. Hepatic transferrin plays a role in systemic iron homeostasis and liver ferroptosis. Blood. 2020;136(6):726–739. doi:10.1182/blood.2019002907
89. Qi J, Kim JW, Zhou Z, Lim CW, Kim B. Ferroptosis affects the progression of nonalcoholic steatohepatitis via the modulation of lipid peroxidation-mediated cell death in mice. Am J Pathol. 2020;190(1):68–81. doi:10.1016/j.ajpath.2019.09.011
90. Tsurusaki S, Tsuchiya Y, Koumura T, et al. Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis. 2019;10(6):449. doi:10.1038/s41419-019-1678-y
91. Hao S, Yu J, He W, et al. Cysteine dioxygenase 1 mediates erastin-induced ferroptosis in human gastric cancer cells. Neoplasia. 2017;19(12):1022–1032. doi:10.1016/j.neo.2017.10.005
92. Ishimoto T, Nagano O, Yae T, et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell. 2011;19(3):387–400. doi:10.1016/j.ccr.2011.01.038
93. Guo J, Xu B, Han Q, et al. Ferroptosis: a novel anti-tumor action for cisplatin. Cancer Res Treat. 2018;50(2):445–460. doi:10.4143/crt.2016.572
94. Mayr L, Grabherr F, Schwärzler J, et al. Dietary lipids fuel GPX4-restricted enteritis resembling Crohn’s disease. Nat Commun. 2020;11(1):1775. doi:10.1038/s41467-020-15646-6
95. Chen L, Hambright WS, Na R, Ran Q. Ablation of the ferroptosis inhibitor glutathione peroxidase 4 in neurons results in rapid motor neuron degeneration and paralysis. J Biol Chem. 2015;290(47):28097–28106. doi:10.1074/jbc.M115.680090
96. Ashraf A, Jeandriens J, Parkes HG, So PW. Iron dyshomeostasis, lipid peroxidation and perturbed expression of cystine/glutamate antiporter in Alzheimer’s disease: evidence of ferroptosis. Redox Biol. 2020;32:101494. doi:10.1016/j.redox.2020.101494
97. Do Van B, Gouel F, Jonneaux A, et al. Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol Dis. 2016;94:169–178. doi:10.1016/j.nbd.2016.05.011
98. Raven EP, Lu PH, Tishler TA, Heydari P, Bartzokis G. Increased iron levels and decreased tissue integrity in hippocampus of Alzheimer’s disease detected in vivo with magnetic resonance imaging. J Alzheimers Dis. 2013;37(1):127–136. doi:10.3233/jad-130209
99. Agrawal S, Fox J, Thyagarajan B, Fox JH. Brain mitochondrial iron accumulates in Huntington’s disease, mediates mitochondrial dysfunction, and can be removed pharmacologically. Free Radic Biol Med. 2018;120:317–329. doi:10.1016/j.freeradbiomed.2018.04.002
100. Skouta R, Dixon SJ, Wang J, et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc. 2014;136(12):4551–4556. doi:10.1021/ja411006a
101. Tuo QZ, Liu Y, Xiang Z, et al. Thrombin induces ACSL4-dependent ferroptosis during cerebral ischemia/reperfusion. Signal Transduct Target Ther. 2022;7(1):59. doi:10.1038/s41392-022-00917-z
102. Tonnus W, Meyer C, Steinebach C, et al. Dysfunction of the key ferroptosis-surveilling systems hypersensitizes mice to tubular necrosis during acute kidney injury. Nat Commun. 2021;12(1):4402. doi:10.1038/s41467-021-24712-6
103. Müller T, Dewitz C, Schmitz J, et al. Necroptosis and ferroptosis are alternative cell death pathways that operate in acute kidney failure. Cell Mol Life Sci. 2017;74(19):3631–3645. doi:10.1007/s00018-017-2547-4
104. Martin-Sanchez D, Ruiz-Andres O, Poveda J, et al. Ferroptosis, but not necroptosis, is important in nephrotoxic folic acid-induced AKI. J Am Soc Nephrol. 2017;28(1):218–229. doi:10.1681/asn.2015121376
105. Deng F, Sharma I, Dai Y, Yang M, Kanwar YS. Myo-inositol oxygenase expression profile modulates pathogenic ferroptosis in the renal proximal tubule. J Clin Invest. 2019;129(11):5033–5049. doi:10.1172/jci129903
106. Luo Y, Chen H, Liu H, et al. Protective effects of ferroptosis inhibition on high fat diet-induced liver and renal injury in mice. Int J Clin Exp Pathol. 2020;13(8):2041–2049.
107. Schreiber R, Buchholz B, Kraus A, et al. Lipid peroxidation drives renal cyst growth in vitro through activation of TMEM16A. J Am Soc Nephrol. 2019;30(2):228–242. doi:10.1681/asn.2018010039
108. Yang WS, SriRamaratnam R, Welsch ME, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1–2):317–331. doi:10.1016/j.cell.2013.12.010
109. Miess H, Dankworth B, Gouw AM, et al. The glutathione redox system is essential to prevent ferroptosis caused by impaired lipid metabolism in clear cell renal cell carcinoma. Oncogene. 2018;37(40):5435–5450. doi:10.1038/s41388-018-0315-z
110. van der Meer P, van der Wal HH, Melenovsky V. Mitochondrial function, skeletal muscle metabolism, and iron deficiency in heart failure. Circulation. 2019;139(21):2399–2402. doi:10.1161/circulationaha.119.040134
111. Wang Y, Yu R, Wu L, Yang G. Hydrogen sulfide guards myoblasts from ferroptosis by inhibiting ALOX12 acetylation. Cell Signal. 2021;78:109870. doi:10.1016/j.cellsig.2020.109870
112. Yuan PG, Xue BB, Lin B, et al. Nrf2/ARE通路介导右美托咪定减轻肢体缺血/再灌注损伤中的作用 [Nrf2/ARE pathway mediates the reducing effect of Dexmedeto-midine on ischemia/reperfusion injury in skeletal muscle]. Zhongguo Ying Yong Sheng Li Xue Za Zhi. 2016;32(3):250–254. Chinese. doi:10.13459/j.cnki.cjap.2016.03.016
113. He S, Li R, Peng Y, et al. ACSL4 contributes to ferroptosis-mediated rhabdomyolysis in exertional heat stroke. J Cachexia Sarcopenia Muscle. 2022. doi:10.1002/jcsm.12953
114. Bromfield EG, Walters JLH, Cafe SL, et al. Differential cell death decisions in the testis: evidence for an exclusive window of ferroptosis in round spermatids. Mol Hum Reprod. 2019;25(5):241–256. doi:10.1093/molehr/gaz015
115. Ghoochani A, Hsu EC, Aslan M, et al. Ferroptosis inducers are a novel therapeutic approach for advanced prostate cancer. Cancer Res. 2021;81(6):1583–1594. doi:10.1158/0008-5472.Can-20-3477
116. Ng SW, Norwitz SG, Norwitz ER. The impact of iron overload and ferroptosis on reproductive disorders in humans: implications for preeclampsia. Int J Mol Sci. 2019;20(13). doi:10.3390/ijms20133283
117. Ng SW, Norwitz SG, Taylor HS, Norwitz ER. Endometriosis: the role of iron overload and ferroptosis. Reprod Sci. 2020;27(7):1383–1390. doi:10.1007/s43032-020-00164-z
118. Shimada K, Skouta R, Kaplan A, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol. 2016;12(7):497–503. doi:10.1038/nchembio.2079
119. Greenshields AL, Shepherd TG, Hoskin DW. Contribution of reactive oxygen species to ovarian cancer cell growth arrest and killing by the anti-malarial drug artesunate. Mol Carcinog. 2017;56(1):75–93. doi:10.1002/mc.22474
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
The Emerging Role of Ferroptosis in Various Chronic Liver Diseases: Opportunity or Challenge
Zhu L, Luo S, Zhu Y, Tang S, Li C, Jin X, Wu F, Jiang H, Wu L, Xu Y
Journal of Inflammation Research 2023, 16:381-389
Published Date: 31 January 2023
The Emerging Roles of Ferroptosis in Neonatal Diseases
Chen W, Zheng D, Yang C
Journal of Inflammation Research 2023, 16:2661-2674
Published Date: 26 June 2023
Targeting Ferroptosis in Bone-Related Diseases: Facts and Perspectives
Chen H, Han Z, Wang Y, Su J, Lin Y, Cheng X, Liu W, He J, Fan Y, Chen L, Zuo H
Journal of Inflammation Research 2023, 16:4661-4677
Published Date: 18 October 2023
Unraveling the Molecular Regulation of Ferroptosis in Respiratory Diseases
Zhu L, Zhou J, Yu C, Gu L, Wang Q, Xu H, Zhu Y, Guo M, Hu M, Peng W, Fang H, Wang H
Journal of Inflammation Research 2024, 17:2531-2546
Published Date: 24 April 2024
Ferroptosis in Osteoarthritis: Current Understanding
Liu Y, Zhang Z, Fang Y, Liu C, Zhang H
Journal of Inflammation Research 2024, 17:8471-8486
Published Date: 7 November 2024