Back to Journals » International Journal of Nanomedicine » Volume 20
Nanomaterial-Enhanced Immunotherapy: Advancing T-Cell-Based Treatments for Bladder Cancer
Authors Chen J ![]() , Fu Y, Zhang Z
, Fu Y, Zhang Z ![]() , Zhao J
, Zhao J ![]() , Zuo J, Ye X, Xiong Q, Nie Z, Dong H, Shi H, Tan Z
, Zuo J, Ye X, Xiong Q, Nie Z, Dong H, Shi H, Tan Z ![]() , Wang C, Chen B, Wang Z, Li X, Chen P
, Wang C, Chen B, Wang Z, Li X, Chen P ![]() , Wang H, Fu S
, Wang H, Fu S
Received 3 August 2025
Accepted for publication 1 December 2025
Published 18 December 2025 Volume 2025:20 Pages 15235—15275
DOI https://doi.org/10.2147/IJN.S557690
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Junhao Chen,1,* Yuanzhi Fu,2,* Zhongsong Zhang,3,* Junxian Zhao,4,* Jieming Zuo,1,* Xinni Ye,1 Qiao Xiong,1 Zuqing Nie,5 Haonan Dong,1 Hongjin Shi,1 Zhiyong Tan,1 Chengjie Wang,6 Bo Chen,7 Zhengyan Wang,8 Xiangyun Li,1 Peng Chen,1 Haifeng Wang,1 Shi Fu1
1Department of Urology, The Second Affiliated Hospital of Kunming Medical University, Kunming, Yunnan, People’s Republic of China; 2Department of Clinical Medicine, Kunming University of Science and Technology, Kunming, People’s Republic of China; 3School of Clinical Medicine, Chengdu Medical College, Chengdu, 610550, People’s Republic of China; 4Department of Urology, 920th Hospital of Joint Logistics Support Force of Chinese People’s Liberation Army, Kunming, Yunnan, People’s Republic of China; 5Department of Central Lab, The Second Affiliated Hospital of Kunming Medical University, Kunming, Yunnan, People’s Republic of China; 6School of Stomatology, Xinjiang Second Medical College, Karamay, People’s Republic of China; 7Department of Urology, Qujing Second People’s Hospital, Qujing, Yunnan, People’s Republic of China; 8Department of Urology, Honghe Hospital Affiliated to Kunming Medical University/South Yunnan Central Hospital of Yunnan Province (The First People’s Hospital of Honghe Hani and Yi Autonomous Prefecture), Honghe, China; Kunming Medical University, Kunming, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Haifeng Wang, Email [email protected] Shi Fu, Email [email protected]
Abstract: Bladder cancer (BC) is a prevalent urinary malignancy characterized by high recurrence rates and suboptimal long-term outcomes from traditional treatments such as surgery, chemotherapy, and radiotherapy. T-cell-based immunotherapy has emerged as a promising approach, harnessing T cells’ capacity to target and destroy tumor cells, yet it faces challenges from the immunosuppressive tumor microenvironment (TME), immune evasion, and T-cell exhaustion. Nanomaterials offer innovative solutions by enabling targeted delivery of antigens, checkpoint inhibitors, and immunomodulators; remodeling the TME through metabolic interventions (eg, hypoxia alleviation and adenosine reduction); and enhancing T-cell infiltration and persistence with stimulus-responsive systems like pH-sensitive nanoparticles and biomimetic vesicles. This review systematically examines nanomaterial integration to amplify T-cell-mediated immunity in BC, covering T-cell origins, differentiation (eg, CD8+ cytotoxic and CD4+ helper subsets), roles in the TME, and exhaustion mechanisms driven by factors like PD-1 and TOX. We discuss key strategies including direct immune enhancement via immunogenic cell death induction, metabolic reprogramming to optimize T-cell function, and sustained activation for improved persistence. In conclusion, these nanomaterial-enhanced therapies address critical barriers, promoting precise and synergistic immune responses. Future prospects highlight AI-driven designs, personalized medicine, and clinical translation to tackle heterogeneity, biosafety, and resistance for durable BC remission.
Keywords: bladder cancer, T cell, immunotherapy, nanotechnology, nanomaterials, precision medicine
Introduction
BC is a common malignancy of the urinary system that primarily affects the epithelial lining of the bladder. The most prevalent subtype is bladder urothelial carcinoma (BUC), accounting for over 90% of all BC cases.1,2 BC typically manifests as a mass within the bladder, disrupting its normal function. Due to the subtlety of early symptoms, it is often diagnosed at an advanced stage.3 Non-muscle-invasive bladder cancer (NMIBC) exhibits a particularly high recurrence rate, with approximately 70–80% of patients experiencing tumor recurrence after initial treatment. In a subset of patients, NMIBC may progress over time, infiltrating the bladder muscle layer and developing into muscle-invasive bladder cancer (MIBC). MIBC is associated with increased invasiveness, a higher risk of metastasis, and significantly worsened prognosis.4 If left untreated, MIBC can spread to distant organs such as lymph nodes, lungs, and bones.5 Standard treatment options for BC include surgery, chemotherapy, and radiotherapy, all of which can exert adverse effects on patients. Although cystectomy can eradicate the tumor, it profoundly impacts the patient’s quality of life due to the need for urinary diversion or neobladder reconstruction. Chemotherapy and radiotherapy are also associated with side effects such as immunosuppression, alopecia, and nausea, which further affect overall health and life quality.6
In recent years, immunotherapy has emerged as a promising strategy in cancer treatment due to its unique mechanism of action and reduced systemic toxicity. BC cells often exhibit robust immune evasion capabilities, making immunotherapy an attractive treatment modality. Bacillus Calmette-Guérin (BCG) immunotherapy remains the gold standard for high-risk NMIBC, especially in patients with high-grade or recurrent tumors. BCG induces a strong localized immune response and effectively prevents tumor recurrence and progression. Approximately 70% of high-risk NMIBC patients benefit from BCG therapy.7,8 The advent of immune checkpoint inhibitors has further expanded the role of immunotherapy in BC. Clinical trials have demonstrated that PD-1/PD-L1 inhibitors significantly improve overall survival in patients with metastatic BC.9 For example, pembrolizumab has been approved for patients ineligible for surgery or chemotherapy, particularly those with high PD-L1 expression.10 In patients with advanced or metastatic disease, PD-1/PD-L1 blockade has been approved as a treatment option, especially when BCG therapy fails or resistance develops. From the perspective of T cells, BC—being a prevalent malignancy of the urinary system—exhibits a high degree of immune evasion, making T cell function and activation central to effective immunotherapeutic strategies. BC cells evade immune surveillance through multiple mechanisms, among which suppression of T cell activity is one of the most critical.11 Tumor cells frequently suppress T cell function by expressing immune checkpoint molecules, such as PD-L1, thereby facilitating immune evasion. The central objective of immunotherapy is to activate or restore T cell function in order to potentiate the immune system’s capacity to eliminate BC cells. Current immunotherapeutic modalities include immune checkpoint inhibitors, BCG immunotherapy, and chimeric antigen receptor T (CAR-T) cell therapy, among others. T cells play a central role in the antitumor immune response against BC, primarily through the recognition and elimination of tumor cells, regulation of immune signaling pathways, and maintenance of immunological memory. Immunotherapy enhances the antitumor activity of T cells by promoting their activation, alleviating immunosuppressive mechanisms, and modulating the tumor microenvironment. With continuous advancements in immunotherapeutic strategies, T cells are anticipated to play an increasingly critical role in the treatment of BC, particularly in patients with BCG-resistant, advanced-stage, or metastatic disease.
With the rapid development of nanotechnology, nanomaterials have demonstrated tremendous potential in the field of cancer therapy. Beyond serving as carriers for targeted drug delivery to improve intratumoral accumulation of therapeutic agents, nanomaterials can also synergize with immunotherapy to enhance antitumor immune responses. For instance, rationally designed nanocarriers can precisely deliver tumor antigens, immune adjuvants, or gene-editing tools, thereby promoting robust and sustained activation of T cells. Furthermore, nanomaterials have the capacity to reshape the tumor immune microenvironment, facilitating T cell infiltration and enhancing cytotoxic activity. In the context of BC, the integration of nanotechnology with T cell-based immunotherapy offers a promising strategy to overcome the limitations of conventional treatments. This combinatorial approach holds the potential to provide more efficient and safer therapeutic outcomes, paving the way for personalized and precision immunotherapy.12 In this review, we focus on recent advances in the application of nanomaterials for enhancing T cell-mediated immunotherapy in BC. We further discuss current research gaps and future directions in this emerging field, with the aim of accelerating the development of effective T cell-based immunotherapeutic strategies against BC.
T Cells as the Biological Foundation of Immunotherapy in BC
Origin and Functional Differentiation of T Cells
T lymphocytes (T cells) are a vital subset of leukocytes within the human immune system, primarily responsible for recognizing and eliminating pathogens, tumor cells, and other abnormal cells. As integral components of the adaptive immune system, T cells play a pivotal role in mounting antigen-specific immune responses against infections and malignancies. T cells originate from hematopoietic stem cells in the bone marrow; however, their maturation and functional differentiation predominantly occur in the thymus. Upon completion of thymic development, mature T cells enter the peripheral circulation and migrate to various tissues, where they participate in immune surveillance and responses. Based on their functional roles and surface receptor expression, T cells can be classified into several subtypes. Among these, the two most common and well-characterized subsets are cytotoxic T cells (CD8⁺ T cells) and helper T cells (CD4⁺ T cells).13,14 Among T cell subsets, cytotoxic T lymphocytes (CD8⁺ T cells) are the primary effector cells responsible for antiviral and antitumor immunity. These cells eliminate infected or malignant cells by recognizing and attacking those that present specific antigens on their surface. Recognition is mediated through T cell receptors (TCRs), which bind to antigenic peptides displayed by infected or tumor cells, initiating a cytolytic immune response. Activated cytotoxic T cells release effector molecules such as perforin and granzymes, leading to apoptosis of the target cells.15 TCRs recognize foreign antigens presented by major histocompatibility complex (MHC) molecules. Specifically, CD8⁺ T cells interact with antigens presented by MHC class I, while CD4⁺ T cells recognize antigens presented by MHC class II molecules.16 Helper T cells (CD4⁺ T cells) play a crucial immunoregulatory role by assisting other immune cells. They promote B cell differentiation and antibody production, activate CD8⁺ T cells, and enhance macrophage-mediated phagocytosis. These functions are mediated via the secretion of cytokines such as interleukin-2 (IL-2) and interferon-gamma (IFN-γ). Based on their cytokine expression profiles, CD4⁺ T cells are further categorized into subsets including Th1, Th2, Th17, and regulatory T cells (Tregs).17 Tregs are a specialized CD4⁺ T cell subset that function to suppress immune responses and prevent autoimmunity. They maintain immune tolerance by inhibiting overactive immune cells through the secretion of immunosuppressive cytokines such as transforming growth factor-beta (TGF-β) and interleukin-10 (IL-10)18 (Figure 1).
 |
Figure 1 Phenotypic Comparison Between Functional and Exhausted Effector CD8⁺ T Cells. (A) Functional effector CD8⁺ T cells exhibit high proliferative capacity and cytotoxicity. They maintain functional mitochondria with increased mitochondrial mass and proper polarization, leading to upregulation of effector genes (eg, IFN-γ and TNF-α) and robust cytokine production. Expression of inhibitory receptors is low. (B) Exhausted effector CD8⁺ T cells display impaired proliferation and cytotoxicity. They have dysfunctional mitochondria with depolarization and increased ROS (reactive oxygen species) accumulation. These cells upregulate exhaustion-associated genes such as PDCD1 and TOX, produce fewer cytokines, and express high levels of inhibitory receptors (PD-1, LAG-3, TIGIT, CTLA-4). Adapted from Zhong, Tao et al. The mechanisms and clinical significance of CD8+ T cell exhaustion in anti-tumor immunity. Cancer biology and medicine vol. 22,5 (2025), under the Creative Commons Attribution-NonCommercial 4.0 International License.19 |
T cells originate from the bone marrow, which serves as the primary site for hematopoiesis and the production of all immune cells, including stem cells and hematopoietic progenitors. Hematopoietic stem cells (HSCs) within the bone marrow give rise to various blood cell lineages, including red blood cells, platelets, and leukocytes such as T cells and B cells.20 T cell precursor cells, referred to as thymic progenitor T cells or pro-T cells, originate in the bone marrow and, following partial differentiation, migrate via the bloodstream to the thymus, where they undergo further maturation and selection. Upon antigen recognition, the T cell receptor (TCR) engages with antigen–MHC complexes presented on the surface of antigen-presenting cells. This interaction triggers the phosphorylation of immunoreceptor tyrosine-based activation motifs (ITAMs) located within the CD3 complex, which is physically associated with the TCR. This phosphorylation event is mediated by Src family kinases, primarily Lck, and leads to the activation of several downstream signaling cascades, including the phosphoinositide 3-kinase (PI3K)/Akt, mitogen-activated protein kinase (MAPK), and nuclear factor-kappa B (NF-κB) pathways. These signaling networks coordinate the transcriptional and functional reprogramming of T cells, ultimately resulting in their proliferation, differentiation, and cytotoxic activity against target cells.21,22 In the thymus, T cells undergo a process known as TCR rearrangement, during which the genes encoding the T cell receptor are somatically recombined to generate a diverse repertoire of antigen specificities. As a result of this process, each mature T cell expresses a unique TCR, enabling it to recognize a specific antigen. The TCR thus serves as the physiological antigen recognition structure common to all T cells. In contrast, chimeric antigen receptor (CAR) T cells are genetically engineered T cells that express synthetic transmembrane receptors. A CAR typically consists of an extracellular antigen-binding domain derived from a monoclonal antibody (usually in the form of a single-chain variable fragment, scFv), a hinge region, and one or more intracellular signaling domains derived from native T cell receptor components. Similar to monoclonal antibody-based therapies, CAR-expressing T cells exhibit high antigen specificity. However, unlike natural TCRs—which require peptide antigen presentation by MHC molecules—CARs directly bind to tumor-associated antigens (TAAs) on the cell surface independently of MHC presentation. This MHC-unrestricted recognition enables CAR-T cells to target antigens that may evade traditional TCR-mediated detection, thus providing a complementary and potent approach to conventional T cell-based immunotherapy.23,24 Through this mechanism, CAR-T cells bypass the restriction imposed by MHC molecules, enabling them to recognize antigens that conventional TCRs are unable to detect. The single-chain variable fragment (scFv) region of the CAR mediates direct recognition and binding to tumor-associated antigens expressed on the surface of cancer cells, regardless of whether these antigens are presented by MHC molecules. This MHC-independent recognition endows CAR-T cells with the capacity to target tumor-specific or tumor-associated markers that may otherwise evade immune surveillance. Upon antigen engagement, the intracellular signaling domain—typically composed of the CD3ζ chain—triggers downstream activation cascades. These include the MAPK, PI3K/Akt, and NF-κB pathways, which collectively enhance T cell proliferation, cytokine production, and cytotoxic activity.25 Through CAR-mediated recognition, T cells can rapidly identify and eliminate tumor cells. In addition to direct cytotoxicity, CAR-T cells also secrete pro-inflammatory cytokines, such as interferon-gamma (IFN-γ), which contribute to remodeling the TME and amplifying local immune responses, thereby potentiating antitumor immunity.
Mechanisms of T Cells and Immunotherapy in BC
CD8+ T
CD8⁺ T cells are key effector cells in antitumor immune responses and are referred to as cytotoxic T lymphocytes (CTLs) due to their ability to directly kill tumor cells. As part of the αβ T cell lineage, they express T cell receptors (TCRs) that recognize tumor-associated antigens presented by MHC class I molecules on the surface of tumor cells, mediating antigen-specific cytotoxicity.26 Studies have shown that the spatial distribution of CD8⁺ T cells in the TME is closely associated with clinical outcomes in BC. High intratumoral CD8⁺ T cell density correlates with prolonged overall survival, whereas high CD8⁺ T cell density in the tumor stroma is often associated with poorly differentiated tumors and worse prognosis.27 In BC, CD8⁺ T cells can differentiate into various functional subsets, including effector CD8⁺ T cells and exhausted CD8⁺ T cells (Table 1).
 |
Table 1 The Role of Different T Cell Subsets in the Immunity of BC and Its Immunotherapeutic Mechanism |
Effector CD8⁺ T
Effector CD8⁺ T cells are the principal cytotoxic effectors in adaptive immunity. Upon antigen recognition and activation, naive CD8⁺ T cells differentiate into this subset, which is characterized by potent cytolytic activity and robust secretion of inflammatory cytokines. Structurally, effector CD8⁺ T cells express both TCRs and the CD8 co-receptor. The TCR binds to antigen–MHC I complexes, while CD8 facilitates this interaction and recruits Lck kinase to initiate intracellular signaling. Functionally, they highly express cytotoxic mediators such as perforin, granzyme B, IFN-γ, and TNF-α, enabling them to directly kill target cells.26 Phenotypically, these cells are typically CD44⁺, CD69⁺, CD25⁺, and CCR7−, indicating their activation and migration from lymphoid organs. Effector CD8⁺ T cells kill tumor cells by releasing perforin, which forms pores in the target cell membrane, allowing granzyme B to enter and activate caspase-dependent apoptosis. Simultaneously, secretion of IFN-γ and TNF-α enhances antigen presentation, inhibits tumor angiogenesis, and strengthens local immune responses.28 This mechanism enables effector CD8⁺ T cells not only to directly kill tumor cells but also to serve as immunological coordinators, amplifying the antitumor immune response. In BC, the presence and activation status of effector CD8⁺ T cells are critical determinants of effective tumor clearance. Studies have demonstrated that a higher intratumoral density of granzyme B⁺ and IFN-γ⁺ CD8⁺ T cells correlates with improved patient outcomes, indicating that these cells have entered an active effector state with cytotoxic capability. However, T cell infiltration alone is insufficient; what matters is whether these T cells express perforin and granzyme B and are functionally active. Several studies have revealed that the bladder TME often suppresses perforin expression in CD8⁺ T cells via the TGF-β2 and ICAM-1 signaling pathways, leading to a state of phenotypic activation but functional silencing, rendering the T cells ineffective. For instance, immunosuppressive mediators such as TGF-β and indoleamine 2,3-dioxygenase (IDO) have been shown to markedly reduce perforin expression and impair cytotoxic activity.29 Moreover, chronic antigen exposure and inhibitory signaling—notably through the PD-L1/PD-1 axis—can drive effector CD8⁺ T cells into functional exhaustion, transitioning them into a non-cytolytic state. Modern immunotherapies, particularly immune checkpoint inhibitors (ICIs), are designed to restore the function of effector CD8⁺ T cells by relieving such suppressive signals. As such, both the presence and functional activation of effector CD8⁺ T cells have become key predictive markers of response to immunotherapy.
While earlier studies did not yet distinguish functional effector CD8⁺ T cells from other subsets, they laid the foundation by establishing a relationship between CD8⁺ T cell infiltration and tumor prognosis. Ferris et al30 identified immunogenic peptides derived from p53 (8–11 amino acids in length) and tested their binding affinity and stability with common HLA class I molecules (eg, HLA-A2, A24, B44, B51). CD8⁺ T cells from 16 BC patients were stimulated with these peptides, and IFN-γ secretion was measured using ELISPOT assays. Their findings indicated that tumor-infiltrating CD8⁺ T cells could recognize and respond to p53-derived tumor-associated antigens, demonstrating antigen-specific reactivity and establishing a basis for subsequent functional analyses. Later research shifted focus toward the cytotoxic function of these cells. Tajima et al31 reported that upregulation of granzyme B and IFN-γ was a defining feature of functional effector CD8⁺ T cells. In murine models, initially non-cytotoxic Tc17 cells (IL-17⁺CD8⁺ T cells) could be reprogrammed by IL-12 to acquire an effector phenotype characterized by expression of IFN-γ and granzyme B (Tc17/IFN-γ), gaining cytotoxic potential comparable to conventional Tc1 cells. This study highlighted that the expression of cytotoxic molecules, rather than the cell’s original phenotype, is the true determinant of effector function. This functional reprogramming was confirmed to be IL-12–induced and associated with the upregulation of granzyme B and perforin, rather than being a superficial phenotypic change. Nonetheless, subsequent findings revealed that even CD8⁺ T cells with appropriate phenotypic markers often lose cytotoxic function due to complex immunosuppressive signals within the TME. Subsequent research efforts have increasingly focused on elucidating how tumors selectively silence cytotoxic T cells that are otherwise equipped with effector potential. In the context of BC, immunological investigations have begun to uncover that tumor cells exploit immunosuppressive signaling pathways—most notably those involving transforming growth factor-beta (TGF-β) and intercellular adhesion molecule-1 (ICAM-1)—to suppress the expression of perforin and granzyme B in CD8⁺ T cells, thereby establishing a mechanism of immune evasion. Critically, studies conducted during this period not only confirmed the existence of such effector function suppression, but also demonstrated that this dysfunction is potentially reversible. These findings point to actionable molecular targets and underscore the therapeutic potential of interventions aimed at reinvigorating T cell cytotoxicity within the TME. Hartana et al29 described an immune evasion mechanism in BC mediated by ICAM-1 and TGF-β2, which suppresses perforin expression in CD8⁺ T cells. In their study, CD8⁺ T cells were isolated from the peripheral blood, sentinel lymph nodes, and tumor tissues of patients with urothelial bladder cancer (UBC). Notably, perforin expression was significantly reduced in CD8⁺ T cells derived from the sentinel lymph nodes. Mass spectrometry analysis revealed that UBC cells secrete ICAM-1 and TGF-β2. CD8⁺ T cells were treated with tumor cell-conditioned medium or ICAM-1/TGF-β2, and it was confirmed that these treatments suppressed perforin expression. Under Tc1-promoting conditions (such as with IL-12), perforin expression could be partially restored. The tumor induces CD8⁺ T cell exhaustion and functional inactivation through ICAM-1 and TGF-β2 signaling, which represents a mechanism of immune escape, suggesting that dual targeting of these two molecules may improve the efficacy of immunotherapy. This key study found that ICAM-1 and TGF-β2 secreted by BC cells can suppress perforin expression in CD8⁺ T cells within tumor-draining sentinel lymph nodes (SNs), while granzyme B levels remain unchanged. Most of these perforin-deficient CD8⁺ T cells exhibited an exhausted effector memory phenotype (TEM/PD-1⁺/GATA3⁺), indicating that the tumor had induced a population of “functionally hollow” T cells. This reveals an important immune escape pathway: even when T cells appear phenotypically activated, they have in fact lost their cytotoxic function.
T Cell Exhaustion
Under conditions of chronic antigen stimulation, such as persistent viral infections or tumors, CD8⁺ T cells remain continuously activated but eventually enter a functionally impaired state known as T cell exhaustion.32 This phenomenon was first described in chronic viral infections and has since been recognized as a major mechanism underlying the failure of antitumor immunity in solid tumors, including BC. Exhausted CD8⁺ T cells arise under prolonged antigen exposure in the tumor or chronic infection environment. Although they are not completely inactive, their cytotoxic function is significantly diminished. Specifically, they exhibit reduced expression of key cytolytic molecules such as granzyme B, perforin, IFN-γ, and TNF-α, leading to impaired antitumor activity. Additionally, they show weakened proliferative capacity, limiting their clonal expansion. These cells are also characterized by persistent high expression of multiple co-inhibitory receptors, including PD-1, TIM-3, LAG-3, CTLA-4, and TIGIT, which restrain their activation. At the transcriptional level, they undergo reprogramming, with upregulation of transcription factors such as TOX, NFAT, and Eomes, and downregulation of T-bet. Moreover, exhausted T cells display metabolic dysregulation, including mitochondrial dysfunction and reduced glucose metabolism, further contributing to their functional impairment.33 Together, these alterations constitute the molecular basis of CD8⁺ T cell exhaustion. T cell exhaustion is not a binary state but a progressive continuum, generally categorized into progenitor exhausted and terminally exhausted stages. Early exhausted cells are characterized by a PD-1⁺TCF1⁺SLAMF6⁺TOX− phenotype, retain self-renewal capacity, and are responsive to PD-1 blockade therapy, making them the primary targets of immune checkpoint inhibitors (ICIs). In contrast, terminally exhausted cells exhibit a PD-1⁺TOX⁺TCF1−TIM-3⁺ profile, are functionally impaired, possess weak cytotoxicity, and respond poorly to ICIs. In BC, sustained tumor antigen exposure and an immunosuppressive microenvironment (eg, TGF-β, PD-L1) promote the progression of CD8⁺ T cells toward exhaustion. Studies have shown a high abundance of PD-1⁺TOX⁺CD8⁺ T cells within bladder tumors. Although these cells can infiltrate tumors, their expression of multiple co-inhibitory receptors—such as PD-1 and TIGIT—severely limits their effector function.34 Further single-cell transcriptomic analyses have revealed that exhausted CD8⁺ T cells in tumors commonly express high levels of TOX, LAG-3, and CTLA-4, which are closely associated with immunotherapy resistance and poor prognosis.35 Nevertheless, these cells are still considered potentially reactivatable therapeutic targets. For example, Han et al34 discovered that PD-1⁺TOX⁺CD8⁺ T cells harbor tumor antigen–specific TCR clones, and following combined PD-1 and TIGIT blockade, these cells could re-express effector cytokines such as IFN-γ and TNF-α, suggesting that a subset belongs to the progenitor exhausted population and may respond to checkpoint-based immunotherapy.
CD4+ T
CD4⁺ T cells are key regulators within the adaptive immune system, orchestrating and guiding immune responses through diverse mechanisms. They play central roles in antiviral and antitumor immunity, maintenance of immune homeostasis, and tissue repair. These cells become activated upon recognizing peptide antigens presented by major histocompatibility complex class II (MHC II) molecules on antigen-presenting cells (APCs) such as dendritic cells, in the presence of costimulatory signals. Following activation, CD4⁺ T cells differentiate into distinct functional subsets depending on the local cytokine milieu, including Th1, Th2, and regulatory T cells (Tregs).36 In terms of cellular immunity, Th1 cells promote antitumor and antiviral defense primarily by secreting interferon-gamma (IFN-γ) and interleukin-2 (IL-2), which activate macrophages and CD8⁺ cytotoxic T lymphocytes, enhancing the clearance of intracellular pathogens. In contrast, Th2 cells are involved in humoral immunity by producing IL-4, IL-5, and IL-13, which facilitate B cell activation, class-switch recombination, and responses against extracellular parasites.36 BC tissues are often enriched with regulatory T cells (Tregs), a subset of CD4⁺ T cells characterized by FOXP3 expression. Tregs exert immunosuppressive functions by producing cytokines such as IL-10 and TGF-β, which inhibit the activity of effector T cells and dendritic cells, thereby dampening antitumor immune responses. High levels of Treg infiltration are generally associated with poor prognosis, suggesting that tumors may exploit Tregs to sustain an immunosuppressive microenvironment conducive to tumor progression. In addition, BC cells can directly impair CD4⁺ T cell function to promote immune evasion. Studies have shown that BC cell lines such as T24 can significantly suppress T cell activity and even induce apoptosis in vitro, indicating that tumor cells themselves possess the capacity to actively inhibit immune cell function.37
Th1
The differentiation of Th1 cells is regulated by specific cytokine signals. Naïve CD4⁺ T cells, upon encountering antigens presented by APCs such as dendritic cells and being stimulated by IL-12 and IFN-γ, initiate Th1 lineage commitment through activation of the key transcription factor T-bet.38 This process is also regulated by the STAT4 signaling pathway. In the context of tumor immune surveillance, Th1 cells are considered a central force in initiating and maintaining antitumor immune responses. Enhanced Th1 responses have been associated with improved treatment efficacy and prognosis in cancer patients. Conversely, a shift in the Th1/Th2 balance toward Th2 dominance often correlates with immune suppression, tumor immune escape, and disease progression. In BC, the correlation between Th1 immune activity and therapeutic response has also been demonstrated.39 For instance, the efficacy of BCG immunotherapy depends on a strong Th1 response that induces local expression of IFN-γ and IL-12, which in turn activates tumor-associated immune responses. The absence of an effective Th1 response is often associated with BCG treatment failure or higher recurrence risk.40
Research on Th1 cells in BC was initially driven by efforts to understand the mechanism of BCG immunotherapy. Since the clinical application of BCG for NMIBC in the 1980s, it has become evident that its efficacy is closely related to host immune system activation.41 In the early 2000s, studies showed that BCG treatment induces a Th1-type immune response, characterized by upregulation of inflammatory cytokines such as IFN-γ and IL-2, establishing the central role of Th1 cells in antitumor immunity.42 Subsequent work by Luo et al42 confirmed that BCG can significantly elevate Th1-type cytokines, including IFN-γ and TNF-α, which are critical for promoting local immune activation within the TME. These findings laid the foundation for the positive correlation between Th1 responses and BCG efficacy. Attention then shifted to the regulatory role of Th1/Th2 balance in BC immunity. For example, Satyam et al43 observed a Th2-skewed immune profile in BC patients, marked by elevated Th2 cytokines (eg, IL-4, IL-10) and reduced Th1 cytokines (eg, IFN-γ), a shift associated with immune evasion and therapeutic resistance. Concurrently, researchers began exploring ways to enhance Th1 responses via recombinant genetic engineering. Luo and colleagues44 developed recombinant BCG strains that secrete Th1-promoting cytokines, which led to enhanced antitumor immunity and improved therapeutic outcomes. With the clinical introduction of immune checkpoint inhibitors (ICIs) such as PD-1/PD-L1 antibodies, interest has grown in Th1 cells as predictive biomarkers of immunotherapy response. In 2014, Ingels et al45 reported that bladder tumors with high levels of Th1-associated markers (eg, IFN-γ expression, CD8⁺ T cell infiltration) were associated with better prognosis and stronger therapeutic responses. Subsequent studies linked the Th1 gene expression signature within tumors to both treatment efficacy and overall survival. Several research groups have analyzed BC samples from the TCGA database and found that patients with upregulated expression of Th1-related genes, particularly IFNG, tend to exhibit higher immune activity, better response rates to immunotherapy, and improved overall survival.46 This finding has promoted interest in developing “Th1 gene signatures” as potential tools for personalized treatment stratification in BC. In recent studies, Th1-based vaccines have emerged as promising strategies to actively induce antitumor immune responses in BC therapy. For example, Dang et al.47 A multi-epitope Th1-selective vaccine platform was developed specifically targeting TAAs that are highly expressed in BC patients, including CDC20, TOPO2A, PBK, and MELK. The researchers screened TCGA BC datasets to identify a panel of TAAs that are highly expressed in tumors, strongly immunogenic, and minimally expressed in normal tissues. They then applied MHC class I and class II binding prediction tools to precisely select Th1-biased epitope peptides capable of inducing both CD8⁺ cytotoxic T cell and CD4⁺ helper T cell responses. Their findings suggested that such tumor epitope–based Th1-inducing vaccines can effectively activate antigen-specific immune responses, bypass tumor immune escape mechanisms, and hold potential as an alternative to BCG therapy or as a component of combination immunotherapy strategies for BC.
Th2
T helper type 2 (Th2) cells, a major subset of CD4⁺ T cells, play a key role in regulating humoral immunity, promoting B cell differentiation, antibody production, and immune tolerance. Th2 cells differentiate under the influence of interleukin-4 (IL-4) and characteristically secrete cytokines such as IL-4, IL-5, IL-6, IL-10, and IL-13.48,49 Under physiological conditions, Th2 cells are involved in anti-parasitic responses, allergic reactions, and tissue repair. However, in the TME, overactivation of Th2 cells may contribute to the formation of an immunosuppressive niche by suppressing Th1-type responses and promoting the recruitment of tumor-associated macrophages (TAMs) and regulatory T cells (Tregs), thereby facilitating immune evasion. In BC, increased expression of Th2-associated cytokines, especially IL-4 and IL-10, has been observed. This Th2-skewed immune imbalance is strongly associated with tumor progression, immune escape, and failure of BCG immunotherapy.50 Furthermore, some studies suggest that a tumor-driven Th2-dominant environment can suppress effective T cell responses and limit the efficacy of immune checkpoint inhibitors (ICIs).51 In recent years, restoring Th1/Th2 balance and reversing Th2 polarization has emerged as a therapeutic strategy in cancer immunotherapy. Inhibiting Th2 cytokines or enhancing Th1 responses may improve clinical responses to immunotherapy in BC patients.
Early studies revealed a Th2-biased immune phenotype in the BC immune microenvironment. This skewing not only contributes to immune escape but may also impact immunotherapeutic efficacy. For instance, Abhigyan et al43 elevated expression of Th2 cytokines such as IL-4 and IL-10, along with a marked decrease in Th1 cytokines (eg, IFN-γ and IL-2), has been observed in BC patients. This immunological skewing is thought to contribute to tumor immune evasion, as Th2 cytokines often suppress Th1 responses, thereby weakening cell-mediated antitumor immunity. Researchers have found that such an imbalance may foster immune tolerance within the TME, suppressing effective antitumor immune responses. With the advent of BC immunotherapies such as BCG treatment, increasing attention has been paid to the role of the Th2 response in modulating therapeutic efficacy. BCG acts primarily by promoting a Th1-dominant immune profile to exert antitumor effects. However, in some patients, an enhanced Th2 response may counteract BCG’s efficacy. Pichler et al52 observed that BCG responders exhibited a higher GATA-3/T-bet ratio, suggesting that interactions between Th2 cells (marked by GATA-3) and Th1 cells (marked by T-bet) within the TME may enhance overall immune activation and improve clinical outcomes. Therefore, Th2 cells may exert dual roles in immunotherapy response: both supporting Th1 responses and modulating the immune milieu to enhance efficacy. Immune evasion in BC is not solely due to suppressed Th1 activity. Th2 cells may directly regulate immune escape via the secretion of immunosuppressive cytokines. It has been shown that Th2-polarized immune responses are frequently hyperactivated in bladder tumors and closely linked to mechanisms of tumor immune escape. For instance, Th2 cytokines like IL-4 and IL-10 inhibit effective Th1 responses, thereby attenuating antitumor immunity and promoting tumor progression. Activation of eosinophils, a hallmark of Th2 responses, may further support immune escape by secreting cytokines and participating in immunomodulation. In 2023, Villoldo et al53 introduced the concept of a “Th2-score”, a quantitative metric based on the expression ratio of GATA3 to T-bet, to classify the immune status of BC patients. Given that GATA3 is a canonical Th2 transcription factor and T-bet a key Th1 lineage marker, this ratio reflects the Th2/Th1 immune inclination. Analyzing samples from NMIBC patients undergoing BCG therapy, they found that patients with a high Th2-score had greater response rates to BCG and longer recurrence-free survival, suggesting that a Th2-dominant state may not simply indicate immune suppression, but rather a plastic and responsive immune system, offering a new prognostic biomarker for therapeutic efficacy. Meanwhile, the application of single-cell RNA sequencing (scRNA-seq) and spatial transcriptomics has significantly advanced the in-depth identification of intratumoral Th2 cells. Traditional immunohistochemistry can only measure bulk expression levels, whereas these emerging technologies allow researchers to precisely localize T cells within tumor tissues, as well as to determine their quantity and functional status. Specifically, studies utilizing spatial transcriptomic platforms have observed that Th2 cells may cooperate with regulatory T cells (Tregs) and M2-type tumor-associated macrophages (TAMs) to form a coordinated suppressive network, collectively shaping an immunosuppressive microenvironmentTME.54–57 This “spatial clustering effect” suggests that Th2 cells are not merely bystanders but rather critical nodes within the immune evasion axis. Through the IL-4/IL-13 signaling axis, Th2 cells can induce macrophage polarization, promote Treg stabilization, and suppress CD8⁺ T cell activity. In addition, some studies have identified a population of Th2-like CD4⁺ T cells within tumor tissues that exhibit a functionally exhausted phenotype, characterized by high expression of inhibitory receptors such as PD-1 and CD200. This indicates that prolonged antigen stimulation may drive these Th2 cells into a loss-of-function state.58 This finding links Th2 cells to resistance against immune checkpoint blockade, highlighting the possibility that successful combination immunotherapy may require simultaneously targeting Th2-mediated immune escape mechanisms to restore global T cell functionality. In summary, the development of Th2 scoring systems and advances in spatial transcriptomics mark a shift from merely qualitative descriptions of Th2 cells to a new era of quantitative, spatially resolved, and functionally defined immunological profiling. These innovations lay a robust theoretical and technical foundation for future personalized immunotherapeutic strategies.
Regulatory T Cells
Regulatory T cells (Tregs) are a critical immunosuppressive subset responsible for maintaining immune homeostasis, preventing excessive immune activation, and limiting autoimmune responses. Most Tregs belong to the CD4⁺CD25⁺Foxp3⁺ lineage, with the transcription factor Foxp3 (forkhead box P3) serving as the master regulator of Treg development and function.59 Tregs can be categorized into two groups based on their origin: naturally occurring thymic Tregs (nTregs) and peripherally induced Tregs (iTregs). The former develop tolerance to self-antigens within the thymus, while the latter arise in peripheral tissues under the influence of TGF-β and IL-2, particularly in inflammatory or tumor environments. In BC, elevated proportions of Tregs have been observed in both the peripheral blood and tumor tissues of patients, especially those with high-grade or MIBC. Tregs exert immunosuppressive effects through multiple mechanisms: high CD25 expression allows them to consume IL-2 competitively, limiting effector T cell expansion; CTLA-4 on Tregs binds to CD80/CD86 on APCs, inhibiting costimulatory signaling; and they can induce indoleamine 2,3-dioxygenase (IDO) expression in dendritic cells, further suppressing immune activity via metabolic reprogramming.60,61 Together, these mechanisms establish a prototypical “immune-suppressive niche” within the bladder TME. Although Tregs are predominantly viewed as promoters of tumor immune evasion, their role is not exclusively detrimental. Some studies have shown that Tregs may limit tumor invasiveness by suppressing matrix metalloproteinase-2 (MMP2) expression, thereby exerting protective, anti-invasive effects under specific contexts.62 Recent studies have begun to explore the potential of regulatory T cells (Tregs) as predictive biomarkers for immunotherapy responsiveness. By establishing an immune scoring model based on the infiltration levels of Tregs and natural killer (NK) cells, researchers have demonstrated its effectiveness in predicting the sensitivity of BC patients to immune checkpoint inhibitors (ICIs), thereby offering promise for personalized treatment strategies.63 In BC, Tregs contribute to tumor progression and therapeutic resistance through multiple immunosuppressive mechanisms. However, their functions appear to exhibit a degree of context dependency, varying with the TME. As such, Treg-targeted immunotherapeutic approaches—including combinatorial checkpoint blockade, chemokine axis inhibition, and selective depletion techniques—are emerging as important avenues to enhance treatment response rates in BC.
The immunological importance of regulatory T cells (Tregs) in tumors was initially recognized through the discovery of CD4⁺Foxp3⁺ cells exerting immunosuppressive functions across multiple cancer types. In a foundational study in 2007, Brandau et al64 first reported a marked accumulation of Tregs in both tumor tissues and peripheral blood of BC patients. These Tregs expressed Foxp3, and their presence was associated with high levels of IL-10 and TGF-β, suggesting that Tregs play a central role in both local and systemic immunosuppression, contributing to the formation of a so-called “immune-cold” TME in BC. Subsequent research shifted from simply quantifying Tregs to dissecting their suppressive mechanisms and regulatory pathways. It became widely accepted that Tregs not only increase in number but also reshape the TME via multiple immunosuppressive mechanisms. Early studies demonstrated that BC-associated Tregs suppress the cytotoxic activity of CD8⁺ T cells and NK cells by secreting IL-10 and TGF-β. Additionally, they inhibit antigen presentation by dendritic cells through CTLA-4–mediated competition for CD80/CD86, collectively forming the core of the tumor immunosuppressive network.65 Meanwhile, researchers have begun to recognize that the function of Tregs is not entirely detrimental. Winerdal66 reported in a 2015 study that FOXP3⁺ cells in BC tissues exhibit considerable heterogeneity. Notably, a subset of these cells appeared to suppress tumor invasiveness by downregulating pro-invasive factors such as MMP2, suggesting that Tregs may exert a “restrictive regulatory” role in specific TME. This bidirectional modulatory capacity has led to a paradigm shift in the understanding of Tregs, transforming them from being regarded solely as “immunosuppressive agents” to “complex regulators.” With the widespread application of immune checkpoint inhibitors (ICIs) and BCG immunotherapy in BC, Tregs have increasingly been identified as a critical mechanism contributing to immunotherapy resistance. While BCG can robustly activate T cell-mediated immunity, it also induces the expansion of PD-L1⁺Foxp3⁺ Treg populations, potentially undermining therapeutic efficacy. Fenner et al67 demonstrated that suppressive PD-L1-expressing Treg subsets were significantly elevated in tumor tissues following BCG treatment, indicating that these cells may counteract BCG-induced antitumor responses. The authors proposed that combining BCG with immune checkpoint blockade could enhance therapeutic outcomes. Simultaneously, Treg trafficking mechanisms have emerged as a focus of research. Maeda et al68 found in a canine BC model that Tregs are highly dependent on the CCR4 chemotactic axis. Treatment with a CCR4-blocking antibody, such as mogamulizumab, significantly depleted intratumoral Tregs and prolonged the survival of the animals. This study proposed a Treg migration blockade strategy, providing a basis for clinical translational application. With the advancement of technologies such as single-cell sequencing and spatial transcriptomics, researchers are now able to precisely localize Treg cells within the tissue microenvironment and identify their subpopulation states. For example, studies have demonstrated the presence of functionally heterogeneous Treg subsets in BC tissues, including populations expressing PD-1⁺, ICOS⁺, and LAG-3⁺, some of which exhibit metabolically active or exhausted phenotypes. Yang et al63 developed an immune risk prediction model by integrating the infiltration scores of Tregs and NK cells, which can effectively predict the responsiveness of BC patients to immunotherapy and provide a foundation for individualized treatment stratification.
T Cells in the Tumor Microenvironment of BC
The tumor microenvironment (TME) plays a pivotal role in tumor initiation, progression, metastasis, and invasion. T cells not only act as direct effectors of anti-tumor immunity, but also serve as key indicators of therapeutic efficacy and prognosis due to their abundance, spatial distribution, and functional state within the TME (Figure 2). Investigating the interplay between T cells and the BC microenvironment has thus become a crucial avenue for developing targeted therapies. TheTME of BC comprises tumor cells, cancer-associated fibroblasts (CAFs), tumor-associated macrophages (TAMs), vascular and lymphatic endothelial cells, as well as a variety of immune cell subsets. These components interact through chemokines, metabolic intermediates, and extracellular matrix (ECM) components to determine whether T cells can effectively infiltrate the tumor and maintain their effector functions. Recent studies have highlighted that immunosuppressive cytokines (such as TGF-β), metabolic competition (eg, lactic acid accumulation), and inhibitory surface molecules (eg, PD-L1) collectively impair the function of CD8⁺ T cells and T helper 1 (Th1) responses, thereby facilitating immune evasion by the tumor.69,70 Within this context, T cells play multifaceted and dual roles in the BC microenvironment—not only as central mediators of anti-tumor immunity but also as key contributors to immune escape and therapeutic failure. Emerging evidence suggests that in many bladder tumors, T cells fail to effectively eliminate malignant cells, and may even exacerbate local inflammation or autoimmune-like tissue injury. The functional activity of T cells within the bladder is subject to modulation by local mucosal immune factors, commensal microbiota, and tumor-derived signals. In in vitro studies, BC cell lines such as EJ and T24 have been shown to directly suppress T cell activity and induce apoptosis.71 This indicates that BC cells possess intrinsic immunosuppressive properties, enabling them to evade recognition and destruction by the immune system.
 |
Figure 2 T cell subsets in the BC tumor microenvironment. Naïve CD8⁺ T cells are activated by antigen-presenting cells (APCs) through MHC–TCR and CD28–CD80/86 interactions, with cytokine support from CD4⁺ T cells. Activated effector CD8⁺ T cells release granzyme B and perforin to kill cancer cells. During chronic tumor antigen exposure, CD8⁺ T cells progressively lose effector functions and express inhibitory receptors (PD-1, CTLA-4, TIM3, LAG3), leading to T cell exhaustion. Immunotherapeutic agents such as anti–PD-1 and anti–CTLA-4 antibodies can restore T cell function and promote tumor clearance. Adapted from Ahmed, Hossain et al. Role of T cells in cancer immunotherapy: Opportunities and challenges. Cancer pathogenesis and therapy vol. 1,2 116–126. 20.72 |
In addition, Lv et al73 utilized human BC specimens to demonstrate that downregulation of SIRT4 enhances glycolytic activity and promotes an immunosuppressive phenotype in tumor cells, leading to a significant reduction in CD8⁺ T cell cytotoxicity. Another study involving deep sequencing of T cell receptors (TCRs) from 119 patients with MIBC revealed that low TCR diversity and a low frequency of circulating T cells were closely associated with poor overall survival.74 Furthermore, TCR sequencing of tumor-infiltrating T cells in BC tissues showed that a high clonal expansion of TCRs correlated with favorable responses to immunotherapy. These findings suggest that in BC, T cell-mediated immunity is not solely dependent on cell quantity, but is also critically influenced by antigen specificity and clonal expansion dynamics.75,76 To date, single-cell RNA sequencing (scRNA-seq) and spatial transcriptomics have demonstrated that T cells within the bladder TME exhibit high heterogeneity among BC patients (Table 1). Adoptive T-cell therapy, particularly tumor-infiltrating lymphocyte (TIL) therapy, has been extensively studied in the context of BC. This therapy involves extracting T-cells from the patient’s tumor, expanding them ex vivo, and reintroducing them back into the patient. When combined with interleukin-2 (IL-2), a cytokine that promotes T-cell proliferation, TIL therapy has shown promise in improving anti-tumor responses and prolonging survival in patients with advanced BC. Although clinical studies involving TIL and IL-2 combination therapies for metastatic BC are being pursued, similar approaches using TIL therapy have been tested in other cancers, and promising results are still emerging.77,78 Immune checkpoint inhibitors, such as PD-1/PD-L1 inhibitors like atezolizumab, nivolumab, and pembrolizumab, have also become critical components of T-cell-based treatments for BC. These inhibitors work by blocking the immune checkpoints that suppress T-cell activation, thereby enabling T-cells to better recognize and attack BC cells. Clinical trials have demonstrated that these drugs significantly improve progression-free survival and overall response rates, especially in patients with locally advanced or metastatic BC. Notable trials include the IMvigor210 study, which found that atezolizumab provided durable responses in patients with advanced BC who were unresponsive to standard therapies.79 A variety of combination strategies are currently being tested in clinical trials. One promising direction is combining T-cell therapies, such as CAR-T cell therapy, with immune checkpoint inhibitors. Early-phase studies have shown that this combination may help overcome the tumor’s resistance mechanisms and improve the efficacy of T-cell therapies.80 Additionally, combination treatments with chemotherapy or anti-angiogenic therapies are being explored to enhance T-cell infiltration into the tumor and to counteract tumor-induced immune suppression.81 These trials collectively highlight the growing potential of T-cell-based immunotherapies for BC. While more research is needed to confirm long-term efficacy, these therapies offer new hope for more targeted and effective treatment options, particularly for patients with advanced or metastatic BC.
T cell exhaustion (TEX) within the TME of BC has become a focal point of current research, offering new insights for clinical immunotherapy strategies. For instance, Xue et al82 employed single-cell RNA sequencing (scRNA-seq) and bulk transcriptomic analysis to investigate TEX-associated immunosuppressive signatures in the BC TME. In their study, a 28-gene TEX signature score was constructed to stratify patients into TEX^high and TEX^low subgroups. It was found that patients in the TEX^high group had worse prognosis and exhibited reduced responsiveness to immune checkpoint inhibitors (ICIs). This study provided a meaningful link between molecular profiling and clinical decision-making, suggesting potential applications in personalized immunotherapy. Moreover, Liu et al83 used single-cell transcriptomic mapping to reveal that cancer-associated fibroblast (CAF)-derived CXCL12/CXCL14 interact with CXCR4 on exhausted T cells, forming a CAF–CXCL axis that spatially sequesters exhausted T cells within the TME, restricting their access to tumor target cells. These findings underscore the dual role of T cells in BC—as both guardians of immune surveillance and facilitators of immune evasion when functionally impaired. Although T cells have the capacity to recognize and eliminate tumor cells, the cancer microenvironment may induce exhaustion, suppress immune function, and remodel cellular context, allowing tumors to escape immune control. Therefore, restoring T cell quantity, diversity, and functionality remains a key therapeutic objective in enhancing immunotherapy efficacy for BC (Table 2).
 |
Table 2 High Heterogeneity of T Cells in BC Tumor Microenvironment of BC |
Nanomaterials Directly Enhance T Cell–Mediated Immunotherapy in BC
Immunotherapy for BC, particularly T cell–based strategies, has made remarkable progress in recent years. However, multiple challenges remain that hinder its overall efficacy and clinical applicability.84–86 Against this backdrop, the integration of nanomaterials into immunotherapeutic regimens has emerged as a novel approach to overcoming these immunological barriers.87,88 Nanomaterials offer a versatile platform for precise delivery of immunomodulators, tumor antigens, or siRNA, and have been shown to promote immunogenic cell death (ICD) within the TME. Additionally, they can help alleviate metabolic stress, remodel the physical barriers of the tumor stroma, and enhance T cell infiltration and cytotoxic function12,89 (Figure 3). In the following sections, we highlight the recent advances and underlying mechanisms by which nanomaterials are being applied to potentiate T cell–mediated immunotherapy in BC, with a particular focus on distinct T cell subsets.
 |
Figure 3 Application of Nanomaterials in BC Therapy. Nanoparticles have emerged as a promising strategy for targeted BC treatment. By incorporating surface ligands that recognize specific biomarkers on BC cells, these nanocarriers can precisely deliver therapeutic agents—such as small interfering RNA (siRNA), circular RNA (circRNA), chemotherapeutic drugs (eg, vincristine), and PD-L1 inhibitors—into tumor cells via linker-mediated conjugation. The morphology of nanoparticles significantly influences their biodistribution and cellular uptake efficiency. A variety of nanodelivery platforms have been explored, including nanodiamonds, gold nanorods, exosomes, chitosan, polymeric nanoparticles, porous materials, and liposomes. Each of these systems offers distinct advantages in enhancing drug stability, targeting specificity, and bioavailability, thereby contributing to improved therapeutic efficacy in BC treatment. Adapted from Zhao, Xinming et al. A Novel Approach for BC Treatment: Nanoparticles as a Drug Delivery System. International journal of nanomedicine, Copyright © 2025 by the authors.86 |
CD8⁺ T
In recent years, CD8⁺ T cells have emerged as central effectors of antitumor immunity, particularly in the context of cancer immunotherapy. However, within the immunosuppressive TME of BC, CD8⁺ T cells often exhibit functional exhaustion or suppression, leading to diminished cytotoxic activity. To overcome these limitations, nanomaterial-based strategies have been explored to reactivate and sustain CD8⁺ T cell functions. Due to their high surface-area-to-volume ratio, programmability, and excellent biocompatibility, nanomaterials can serve as precision delivery systems for tumor antigens, adjuvants, immune checkpoint inhibitors, and regulatory agents. These platforms enhance cross-presentation by dendritic cells (DCs), improve T cell priming, and promote robust antigen-specific CD8⁺ T cell activation. For example, nanovaccines co-delivering TLR7/8 agonists and peptide antigens have demonstrated superior immunostimulatory capacity compared to conventional adjuvants, significantly increasing the population of antigen-specific CD8⁺ T cells.90 Moreover, smart nanocarriers have been employed to transport small-molecule drugs, checkpoint inhibitors, or redox modulators directly into tumor sites, facilitating local immune activation, CD8⁺ T cell infiltration, and sustained cytotoxic responses.91 Some multifunctional platforms integrate photothermal therapy (PTT), chemotherapy, and immune stimulation. These systems induce immunogenic cell death (ICD) and release tumor-associated antigens, further boosting CD8⁺ T cell priming and memory formation, resulting in durable antitumor immunity.92
In the BC context, conventional therapies such as BCG or PD-1/PD-L1 blockade may partially activate T cell responses, yet often fall short due to T cell exhaustion or suboptimal antigen delivery. Nanomaterial-enabled immunomodulation has thus emerged as a promising strategy to overcome these limitations. Early designs focused on PLGA-based nanovaccines, co-loading tumor antigens with TLR agonists for efficient delivery to lymphoid tissues. Kim et al90 developed a PLGA nanoparticle containing a TLR7/8 agonist and tumor peptide, which significantly increased CD44⁺CD8⁺ T cells in murine BC and outperformed BCG in durability of immune response. With the continuous advancement of nanoparticle delivery strategies, researchers have developed multifunctional biomimetic nanomaterials that enable synergistic targeting of therapeutic agents and antibodies. For instance, Zhou et al93 engineered exosome-like nanovesicles cloaked with macrophage membranes, co-loaded with PD-L1 monoclonal antibodies and CD73 inhibitors. These vesicles mimic innate immune cells’ tumor-infiltrating behavior, facilitating deep penetration across the tumor barrier and significantly enhancing CD8⁺ T cell recruitment and cytotoxic activation within the TME, ultimately suppressing tumor progression. Recent breakthroughs have also incorporated nanomotor propulsion systems and tissue-penetrating mechanisms to further improve T cell access. Choi et al94 designed a urease-driven nanomotor, whose surface was functionalized with a STING pathway agonist. This self-propelling system enabled non-catheter-based mucosal delivery, penetrating the bladder epithelium autonomously and delivering immune-stimulatory signals directly to the tumor site, thereby robustly enhancing CD8⁺ T cell activation and accumulation—an approach significantly superior to conventional instillation methods (Figure 4). Moreover, researchers have explored the integration of magneto-responsive nanomaterials with localized hyperthermia, inducing immunogenic tumor cell death (ICD), releasing tumor-associated antigens, and promoting the activation of local dendritic cells (DCs). This cascade enhances subsequent CD8⁺ T cell proliferation and memory formation, presenting a unified “therapy-plus-vaccine” immunoengineering strategy.92 Collectively, these studies underscore the evolution of the “nanomaterials + CD8⁺ T cell modulation” paradigm in BC immunotherapy—from simple adjuvant delivery to targeted, penetrating, and multi-modal smart systems. This transition marks a pivotal shift in nanomedicine’s role, from an auxiliary component to a central element in immune-centric therapeutic designs, unlocking new avenues for precision and efficacy in BC treatment.
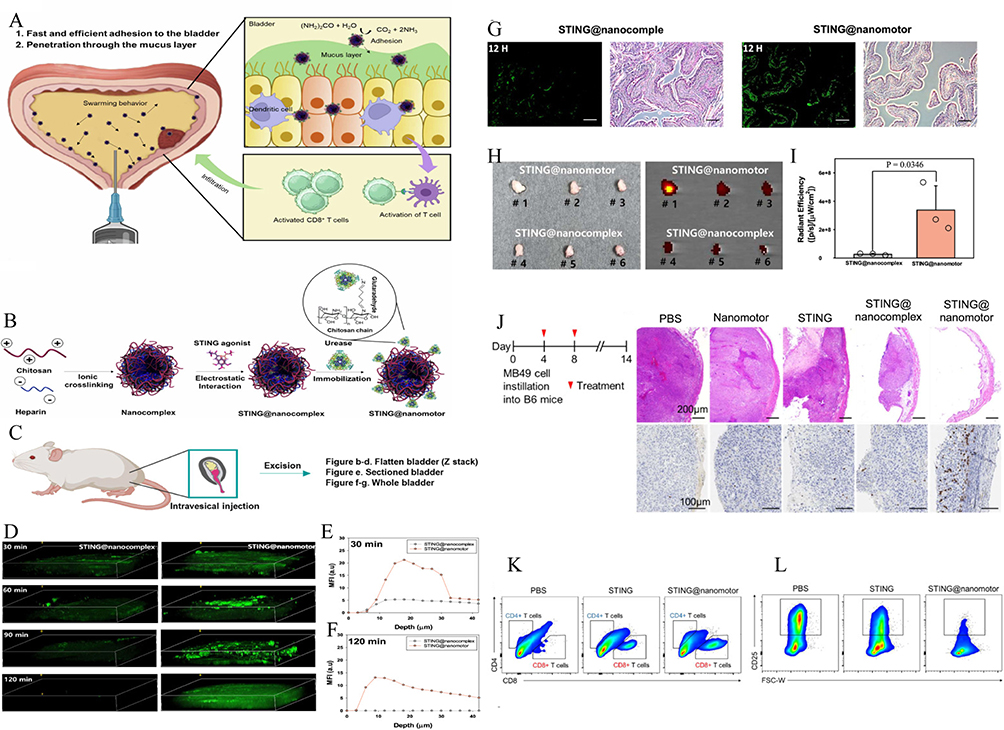 |
Figure 4 Intravesical Delivery and Immune Activation by STING@Nanomotors. (A and B) Schematic illustration of urease-powered nanomotor. (C–I) In vivo penetration and retention of STING@nanomotor after intravesical instillation. (C) Illustration of three administration methods for assessing nanomotor penetration and retention post-intravesical instillation. (D) 3D fluorescence imaging and corresponding mean fluorescence intensity (MFI) at (E) 30 min and (F) 120 min show enhanced bladder accumulation of STING@nanomotors compared to STING@nanocomplex. (G) Bladder sections at 12 h post-instillation reveal deeper tissue penetration by STING@nanomotors (scale bar = 100 μm). (H) Ex vivo bladder images and IVIS imaging, and (I) quantification of total radiant efficiency confirm superior retention. Data represent mean ± S.D. (n = 3); statistical significance determined by two-sided t-test. (J): STING@nanomotor to inhibit BC growth by inducing antitumor immunity. (K and L): Immune response and pro-tumor response of STING@nanomotor. The representative flow cytometry plots of (K) CD4+ and CD8+ T cells, and (L) regulatory T cells. Adapted from Choi, Hyunsik et al. Urease-powered nanomotor containing STING agonist for BC immunotherapy. Nature communications vol. 15,1 9934. 15 Nov, under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License. Copyright © 2024 by the authors.94 |
CD4⁺ T
Th1
Recent studies have increasingly explored how nanomaterials can potentiate Th1-mediated immune responses to enhance the efficacy of BC immunotherapy, particularly in the context of BCG-based immunotherapy and nanovaccine strategies. One of the key mechanisms involves the use of nanomaterials as adjuvant-antigen delivery systems, which activate dendritic cells (DCs) and promote the secretion of Th1-polarizing cytokines such as IL-12 and IFN-γ, thereby directing naive T cell differentiation toward a Th1 phenotype. For example, MgAl-layered double hydroxide (LDH) nanoparticles conjugated with CpG oligodeoxynucleotides significantly increased the IgG2a/IgG1 ratio in murine models, indicating a shift from Th2 to Th1 immunity. This strategy not only enhanced antigen-specific humoral responses but also improved antitumor efficacy in melanoma models through reinforced Th1 polarization, a mechanism with potential translatability to BC therapy.95 In the field of BC, Nano-BCG represents one of the most promising advancements to date. Conventional Bacillus Calmette–Guérin (BCG) therapy exerts its anti-tumor effect primarily through the induction of a Th1-type immune response. However, the application of nanodelivery systems significantly enhances the local stability and cellular uptake of BCG, thereby boosting the expression of Th1-associated cytokines such as interferon-gamma (IFN-γ) and improving the overall efficacy of immunotherapy.12 In addition to delivering conventional agents, certain functional nanomaterials themselves exhibit intrinsic immunomodulatory properties. For instance, Gd@C82(OH)22, a fullerene-based nanoparticle, can promote Th1 immune polarization and induce the release of cytokines such as tumor necrosis factor-alpha (TNF-α) and IFN-γ. This enhanced Th1 bias may help activate cytotoxic CD8⁺ T cells and macrophages, leading to more effective clearance of BC cells.96 Recent studies have begun to explore how to reconstruct a Th1-dominant TME through genetic engineering, gut microbiota modulation, or localized immune delivery systems. For example, some research teams are using targeted nanoparticles to deliver IL-12 or IFN-γ in order to enhance local Th1 responses; others are attempting to systemically improve anti-tumor Th1 immunity by regulating the gut microbiota (such as increasing Th1-inducing bacteria). Recent studies have begun to explore how to reconstruct a Th1-dominant tTME through genetic engineering, gut microbiota modulation, or localized immune delivery systems. Some teams are using targeted nanoparticles to deliver IL-12 or IFN-γ to enhance local Th1 responses; others are attempting to systemically boost anti-tumor Th1 immunity by modulating the gut microbiota (such as enhancing Th1-inducing bacteria). Although current studies on Th1-oriented nanomaterials in BC are mostly confined to animal models, accumulating evidence supports their potential in immune polarization and tumor suppression. Future investigations may focus on integrating Th1-associated transcription factors (eg, T-bet) or chemokines (eg, CXCL10) into nano-platforms to enable more precise activation of Th1 immunity and improved therapeutic outcomes in BC.
Th2
Although direct studies specifically targeting “nanomaterial-mediated regulation of Th2 immunity in BC immunotherapy” are currently lacking, existing research offers relevant clues and mechanistic insights suggesting that nanomaterials may influence Th2-type responses and thereby affect therapeutic efficacy. On one hand, nanomaterials have been extensively utilized to improve the delivery efficiency, antigen presentation, and local immune activation of BCG immunotherapy. In particular, while enhancing Th1 responses, they may concurrently suppress Th2 polarization, thereby disrupting the immunosuppressive TME. For example, Zeng et al12 reviewed that Nano-BCG, via stabilized delivery, can promote dendritic cell maturation and cross-presentation of antigens, inducing stronger IL-12 and IFN-γ expression, which in turn may suppress Th2-biased responses. On the other hand, certain intelligent nano-platforms (such as metallic nanoparticles and polymer-based nanocarriers) have been developed to construct “nanovaccines” or to deliver Toll-like receptor (TLR) agonists and immunomodulatory cytokines. Although most of these studies focus on Th1 activation or CD8⁺ T cell stimulation, they may also indirectly modulate the function or frequency of Th2 cells by remodeling the local immune microenvironment. For instance, Tian et al reported that some nano-systems can induce tumor necrosis and release pro-inflammatory factors, potentially transforming “cold tumors” into “hot tumors”—a process that might involve Th1/Th2 immune reprogramming.97 Taken together, although current research on direct targeting or modulation of Th2 cells by nanomaterials in BC remains in the exploratory stage, emerging mechanisms suggest that nanotechnology could alleviate Th2-dominant immunosuppression by promoting Th1-skewed polarization, thus improving immunotherapeutic outcomes. Future studies are warranted to identify specific Th2-related targets and regulatory strategies for nanoplatform design.
Regulatory T Cells (Treg)
Nanomaterials have shown great promise in remodeling the TME in BC immunotherapy, with regulatory T cells (Tregs) emerging as a critical target. Tregs facilitate immune evasion in BC and are often associated with therapeutic resistance. Therefore, nanomaterial-mediated targeting of Tregs to attenuate their immunosuppressive activity has become an emerging strategy. A cutting-edge study introduced a “Reconstructed Synthetic Nanopathogen” (RSnP) system, which combines an inactivated Mycobacterium cell wall skeleton with a TLR7/8 agonist to elicit potent anti-tumor immune responses without pathogen-associated toxicity. This system significantly enhances the activation of CD8⁺ T cells, natural killer (NK) cells, and Th17 cells while reducing the proportion of immunosuppressive cells such as myeloid-derived suppressor cells (MDSCs) and M2-type macrophages. However, the robust immune activation also leads to a compensatory expansion of CCR8⁺FoxP3⁺ Tregs within the TME. Co-administration of anti-CCR8 monoclonal antibodies can effectively deplete tumor-specific Tregs, further improving therapeutic efficacy and reducing the risk of autoimmune side effects.98 Moreover, Zeng et al highlighted that the Nano-BCG platform enhances T cell activation, improves antigen presentation, and boosts local immune responses, potentially reprogramming immune lineages within the TME and indirectly suppressing Treg expansion to enhance treatment response.12 In terms of delivery strategies, intelligent nanocarriers have been engineered to deliver TLR agonists, antibodies, or siRNAs that target Treg differentiation or recruitment. For instance, Li et al developed multifunctional PICO nanoparticles (PICO NPs) that co-deliver a TLR7 agonist and OKT3 antibody. This platform not only inhibits Treg function but also boosts the activity of APCs, leading to robust antigen-specific immune responses.99 In summary, nanomaterials have become a pivotal strategy in modulating Treg cell function and abundance, beyond merely improving drug delivery. Treg-targeted nanoplatforms hold potential to overcome resistance barriers in current immunotherapy and offer more precise and effective clinical interventions.
Nanomaterial-Mediated T Cell Delivery and Activation
In recent years, the development of nanomaterials has introduced new strategies for BC immunotherapy. Through rational design, nanocarriers can efficiently deliver drugs or biological agents to tumors and immune organs, enhancing anti-tumor immune responses while minimizing systemic side effects.100,101 Although numerous preclinical studies have demonstrated the promise of nanomaterials in enhancing T cell–mediated immunotherapy for BC, the translation into clinical trials remains limited and primarily focused on drug delivery or tumor ablation, rather than immune modulation. For instance, a Phase I trial investigated nab-paclitaxel, a nanoparticle-bound formulation of paclitaxel, for non-muscle-invasive BC. This approach improved local drug retention and reduced systemic toxicity.102 Another example is VAX014, a genetically engineered oncolytic bacterial nanoparticle designed for integrin-targeted delivery. It has completed early-phase trials showing preliminary safety and potential to stimulate anti-tumor immune responses.103 Recent studies also highlight developments in Nano-BCG immunotherapy, where nanotechnology enhances the delivery and immune activation of Bacillus Calmette-Guérin. These platforms aim to improve treatment durability while reducing toxicity.12 Despite these advances, most clinical applications are still in early stages and have not yet addressed the more complex goals of T cell activation, metabolic reprogramming, or precise immunosuppression reversal that are being demonstrated in preclinical nanomedicine platforms.91,104 Various types of nanomaterials—with distinct structures and functionalities—have been developed to improve the therapeutic efficacy of BC immunotherapy. These materials can be broadly categorized into organic nanomaterials, inorganic nanomaterials, biomimetic membrane-coated systems, and stimuli-responsive nanocarriers. In the following section, we provide an in-depth review of the design principles, targeting performance, and research progress of nanomaterials for enhancing T cell–mediated immunotherapy against BC.
Organic Nanomaterials
Organic nanomaterials are typically composed of biocompatible constituents such as polymers, lipids, or proteins—examples include poly(lactic-co-glycolic acid) (PLGA) nanoparticles, chitosan nanoparticles, liposomes, and dendrimers. These carriers are commonly prepared using methods like self-assembly or emulsion–solvent evaporation and can be surface-modified to introduce functional moieties.105 Their advantages include excellent biocompatibility, biodegradability, high drug loading capacity, and chemical modifiability, making them suitable for co-delivery of multiple drugs or nucleic acid molecules.106–108 For example, PLGA-based nanoparticles have been used to encapsulate gene therapeutics or hydrophobic drugs, enabling controlled release.109 The typical size range of organic nanoparticles (tens to hundreds of nanometers) allows for passive accumulation in tumor tissues via the enhanced permeability and retention (EPR) effect.110 For example, we have observed that such nanomaterials are commonly employed to deliver tumor neoantigens or adjuvants in a targeted manner to dendritic cells (DCs) and lymph nodes, thereby activating APCs and inducing CD8⁺ T cell responses.86,111 For instance, PLGA nanovaccines carrying TLR7/8 agonists (eg, IMDQ, R848) have been shown to upregulate co-stimulatory molecules on DCs, promote CD8⁺ T cell expansion, and inhibit tumor growth in BC models.112 Additionally, surface functionalization enables the conjugation of peptides, antibodies, or ligands for active targeting. In the context of BC, considering the unique characteristics of intravesical drug administration, organic nanocarriers can be engineered to enhance mucosal adhesion and tissue penetration. For instance, cationic chitosan nanomicelles can electrostatically adsorb to urothelial mucins, thereby improving intravesical retention and tumor tissue penetration.113 Moreover, chitosan can transiently disrupt tight junctions, improving trans-epithelial drug permeability, making it an ideal high-molecular-weight carrier for intravesical applications.114 These properties allow organic nanomaterials to deliver immunotherapeutics more effectively to the bladder tumor site, improving treatment specificity.
In one study, attenuated BCG was formulated into chitosan nanoparticles for intravesical immunotherapy. The resulting nano-BCG formulation demonstrated good biocompatibility and reduced toxicity; bladder instillation of nano-BCG significantly enhanced anti-tumor immune responses and prolonged survival in mice, while minimizing systemic side effects.115,116 In another study, researchers constructed a liposome-encapsulated formulation of BCG cell wall skeleton (CWS) as an alternative to live BCG. This nano-CWS system overcame the poor solubility and cellular uptake limitations of free CWS; approximately 95% of BC (MBT-2) cells internalized the CWS nanoparticles, resulting in marked tumor growth inhibition.117 In animal studies, intravesical instillation of nano-CWS led to a dose-dependent reduction in tumor volume and induced a Th1-type immune response, as evidenced by increased IFN-γ–producing cells and decreased IL-4 expression. These findings indicate that nano-CWS elicits superior immune activation compared to conventional BCG. Collectively, the results demonstrate that organic nanomaterials possess favorable in vivo adaptability and targeted drug delivery capabilities for BC immunotherapy and gene therapy applications.
Inorganic Nanomaterials
Inorganic nanomaterials include metallic or non-metallic nanoparticles such as gold nanoparticles (AuNPs), magnetic iron oxide nanoparticles (Fe3O4), silica nanoparticles (SiNPs), and manganese dioxide nanozymes (MnO2). These materials typically possess a stable inorganic framework, offer precise control over size and morphology, and feature a large surface area that facilitates conjugation with a variety of functional groups or biomolecules.118,119 Moreover, inorganic nanomaterials often exhibit unique physicochemical properties: for instance, gold nanorods (AuNRs) efficiently convert near-infrared (NIR) light into thermal energy (photothermal effect); magnetic iron oxide nanoparticles respond to external magnetic fields, enabling tumor targeting or magnetic hyperthermia; and manganese dioxide nanoparticles can generate oxygen to relieve tumor hypoxia.120–122 These characteristics position inorganic nanomaterials as promising platforms for theranostics in cancer immunotherapy. For example, gold nanoparticle–Listeriolysin O (LLO) peptide-based nanovaccines can mimic pathogenic patterns to enhance immunogenicity, leading to significantly increased intratumoral infiltration of CD8⁺ T cells and dendritic cells (DCs), while concurrently suppressing immunosuppressive Tregs and myeloid-derived suppressor cells (MDSCs).123 Additionally, Paolo Armanetti et al demonstrated that AuNR-assisted photoacoustic imaging combined with photothermal therapy enabled visualization and ablation of residual tumor foci smaller than 1 mm in a mouse BC model. This approach offers a potential strategy for monitoring and treating residual disease post-surgery, providing valuable prognostic insights for BC patients.124 Manganese dioxide or iron oxide nanoparticles can be employed for magnetic hyperthermia and co-delivery of STING agonists such as cyclic GMP–AMP (cGAMP), thereby activating the stimulator of interferon genes (STING) pathway and promoting dendritic cell (DC) maturation and T cell activation.125
Despite their tunable size and surface characteristics, inorganic nanoparticles are typically rigid and poorly degradable, leading to partial clearance by the mononuclear phagocyte system during circulation.126 To enhance tumor targeting, surface modification with polymers or targeting ligands can prolong circulation time and improve active accumulation. For instance, mesoporous silica nanoparticles functionalized with RGD peptides targeting tumor vasculature showed preferential accumulation in bladder tumors and significant tumor growth inhibition in nude mice.104 Moreover, inorganic nanoparticles can deposit within the bladder cavity. For example, glucose aldehyde-functionalized AuNR/Fe3O4 hydrogels can selectively adhere to collagen in bladder tumor tissue, enabling localized intravesical drug delivery.126
Héctor Terán-Navarro et al127 designed a gold nanoparticle–bacterial toxin peptide composite nanovaccine (GNP–LLO_91–99) for BC immunotherapy. In this system, the immunostimulatory peptide (LLO_91–99) from Listeriolysin O was conjugated to the surface of AuNPs to enhance anti-tumor immunity. In a subcutaneous BC mouse model, a single injection of the nanovaccine reduced tumor burden by approximately 4.7-fold and elicited a potent Th1-biased immune response.127 Mechanistically, GNP–LLO was internalized by DCs, which subsequently secreted pro-inflammatory cytokines and induced immunogenic tumor cell apoptosis. This led to increased intratumoral infiltration of cytotoxic T cells and DCs, along with a reduction in immunosuppressive cell populations. In animal models, the nanovaccine alone significantly inhibited tumor growth and exhibited synergistic efficacy when combined with PD-1/CTLA-4 immune checkpoint blockade. Another study utilized a composite hydrogel containing AuNRs and iron oxide nanoparticles (IONs) to construct a “tri-modal” therapy platform that achieved near-complete tumor eradication in a mouse BC (MB49) model.126 Under NIR irradiation, AuNRs generated localized heat for photothermal ablation, while high concentrations of iron ions induced ferroptosis and reprogrammed tumor-associated macrophages (TAMs) from an M2 to an anti-tumor M1 phenotype. Immunofluorescence analysis revealed a marked reduction in CD11b⁺ M2 macrophages and an increase in CD80⁺ M1 macrophages. Following AuNR&ION hydrogel treatment and NIR exposure, tumors became nearly undetectable within 20 days, and the survival rate significantly improved. Manganese dioxide (MnO2) nanozymes have also demonstrated promising applications in BC photodynamic therapy (PDT). Lin et al developed a HSA–MnO2–Ce6 nanoplatform capable of generating oxygen from endogenous H2O2 within tumors to relieve hypoxia, thereby enhancing the production of reactive oxygen species (ROS) by the photosensitizer Ce6.128 In an orthotopic mouse BC PDT model, this system increased intratumoral oxygenation and significantly prolonged survival, improving therapeutic outcomes. Collectively, these examples highlight the diverse applications of inorganic nanomaterials in multi-modal BC immunotherapy. Through rational design, these platforms not only mediate direct tumor destruction but also remodel the local immune microenvironment to enhance T cell–mediated anti-tumor responses (Figure 5).
 |
Figure 5 O2-generating MnO2 nanoparticles for enhanced photodynamic therapy of BC by ameliorating hypoxia. (A) Schematic representation of the synthesis of HSA-MnO2-Ce6 NPs. (B and C) HSA-MnO 2-Ce6 NPs’ application in enhanced PDT therapy for orthotopic BC by ameliorating hypoxia. (D–F) The physicochemical properties of HSA-MnO2, HSA-Ce6, and HSA-MnO2-Ce6 nanoparticles were characterized by UV–vis spectroscopy, DLS, and TEM, confirming their distinct optical features, size distributions, and morphologies. (G–J) The biodistribution and bladder-targeting ability of HSA-MnO2-Ce6 nanoparticles were validated by in vivo and ex vivo fluorescence imaging, showing enhanced bladder accumulation and reduced off-target distribution compared to HSA-Ce6. (K–M) HSA-MnO2-Ce6 nanoparticles combined with photodynamic therapy significantly reduced bladder tumor volume, as evidenced by imaging, CEUS/US monitoring, and histological analysis, demonstrating effective therapeutic response within 12 days. Adapted from Lin, Tingsheng et al. O2-generating MnO2 nanoparticles for enhanced photodynamic therapy of BC by ameliorating hypoxia. Theranostics vol. 8,4 990–1004. (https://creativecommons.org/licenses/by-nc/4.0/). Copyright © 2020 by the authors.128 |
Membrane Biomimetic Nanomaterials
Cell membrane– or biomimetic membrane–coated nanoplatforms achieve immune-targeted delivery by cloaking nanoparticles with T cell membranes, tumor cell membranes, or other biological membranes.100 For instance, nanoparticles coated with T cell membranes can utilize native surface receptors to target tumor cells while evading immune clearance.129 In addition, stimuli-responsive and composite nanoplatforms can release payloads under specific conditions of the TME—such as pH, enzymatic activity, or redox potential—thus enabling precise drug delivery. Peng et al125 developed a “nanomotor” system that forms in situ within the bladder via self-polymerization, generating nanoparticles (DMCU) loaded with a STING agonist. The system is propelled by urease-driven motion, which enhances mucosal retention and intravesical delivery efficiency, resulting in robust immune activation (Figure 6). Similarly, Choi et al94 designed a urease-powered nanomotor functionalized with STING agonists. These autonomous particles penetrate the urothelium and deliver immunostimulatory signals without requiring catheterization, thereby inducing potent activation and recruitment of CD8⁺ T cells—markedly outperforming traditional infusion methods. Due to the presence of source cell-derived surface receptors and ligands, biomimetic membrane-coated nanoparticles can target specific microenvironments. For example, tumor cell membrane–camouflaged nanovaccines can carry tumor-specific antigens and home to their parental tumor site, enabling personalized immune activation. Exosome-like vesicles (EMVs) derived from macrophage membranes possess intrinsic inflammation-homing properties and can transport payloads to inflamed tumor regions.130,131 In BC, recent reports have described the use of macrophage-derived EMVs to deliver agents that modulate the immunosuppressive microenvironment.93 These vesicles inherit membrane proteins from macrophages, allowing them to evade immune clearance and selectively target immunosuppressive components in tumor tissue, thereby improving drug delivery efficiency132 For instance, Zhou et al93 constructed macrophage membrane–derived biomimetic nanovesicles (EMVs) capable of co-delivering the CD73 inhibitor AB680 and anti–PD-L1 monoclonal antibodies (αPD-L1) for BC treatment. This EMV-based nanosystem exhibited excellent biocompatibility and stability both in vitro and in vivo, along with strong bladder tumor–targeting capability. Mechanistically, AB680 carried by the macrophage membrane vesicles inhibits CD73 within the tumor, reducing the production of immunosuppressive adenosine. Meanwhile, αPD-L1 antibodies released from the vesicles bind to tumor cells, reversing PD-L1–mediated immune escape. In tumor-bearing mice, this dual-delivery biomimetic nano-therapy significantly enhanced the activation and proliferation of tumor-infiltrating T lymphocytes. A robust increase in cytotoxic T cell infiltration was observed within the tumor, leading to inhibited tumor growth and prolonged survival.133 In another study, Hu et al designed a platelet membrane–camouflaged hydrogel delivery system incorporating CSPG4-targeted CAR-T cells, IL-15–loaded PLGA nanoparticles, and platelets conjugated with anti–PD-L1 antibodies.134 Upon postoperative implantation into the tumor bed, local inflammation triggered the platelets to release PD-L1 antibodies, neutralizing tumor-derived PD-L1, while the hydrogel slowly released IL-15 to sustain CAR-T cell activity. In a melanoma model, this biomimetic platform exhibited superior tumor suppression compared to conventional CAR-T cell infusion. Notably, due to prolonged CAR-T cell retention within the hydrogel, the system also suppressed contralateral untreated tumors in a bilateral tumor model—suggesting a durable systemic immune response.134 These findings imply that locally implanted biomimetic carriers may prolong the in vivo persistence and function of T cells, thereby sustaining anti-tumor immune pressure. In summary, membrane-biomimetic nanomaterials, by mimicking the natural attributes of immune or tumor cells, enable precise targeting of the TME and co-delivery of multiple immunotherapeutic agents. They represent a promising emerging strategy to enhance the efficacy of innate and adaptive cancer immunotherapy.
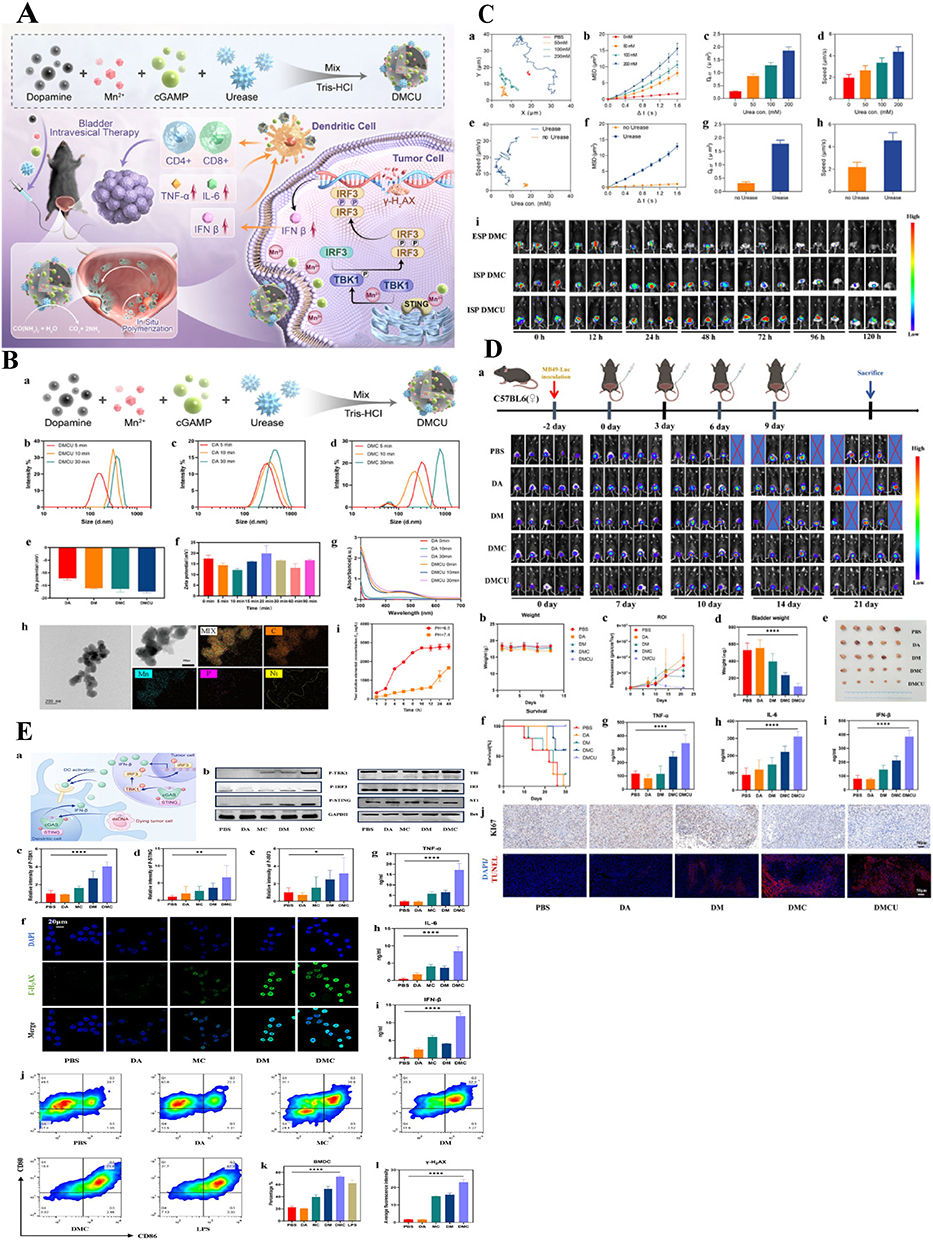 |
Figure 6 Design, characteristics and effects of Mn-cGAMP@PDA-urease (DMCU) nanoparticles. (A) Schematic diagram of the preparation of in-situ polymerized nanomedicine and the mechanism of nanoparticle formation. (B) The construction and characterization of self-propelled in situ polymerized nanodrugs were systematically illustrated, including a schematic of the polymerization process, variations in particle size and zeta potential, UV–vis absorption changes, TEM imaging with elemental mapping, and drug release profiles under different conditions. (C) The bladder retention time of nanomedicine formulations in vivo was evaluated to assess the exercise performance of DMCU. (D) DMCU inhibits the growth of BC in vivo (E) In vitro investigation of DNA damage mechanisms and STING pathway activation with DMC. *p < 0.05, **p < 0.01, ***p < 0.001. Adapted from Peng, Lei et al. Self-Propelled In Situ Polymerized Nanoparticles Activating the STING Pathway for Enhanced BC Immunotherapy. Advanced science (Weinheim, Baden-Wurttemberg, Germany) vol. 12,25 (2025): e2502750. Copyright © 2025 by the authors.125 |
Stimuli-Responsive Nanomaterials
Stimuli-responsive nanomaterials are engineered to incorporate functional units that undergo physicochemical transformations upon exposure to specific endogenous or exogenous stimuli, thereby enabling on-demand drug release or therapeutic activation. Stimuli can be broadly categorized into tumor-internal microenvironmental triggers (eg, acidic pH, elevated levels of reducing glutathione [GSH], overexpressed enzymes, or hypoxia and high hydrogen peroxide) and exogenous physical triggers (eg, light, ultrasound, magnetic fields, or temperature changes).135,136 Common design strategies include: acid-labile linkers that release drugs under acidic conditions, redox-sensitive polymers that disassemble in response to high GSH levels, enzyme-cleavable peptides sensitive to tumor-associated proteases (eg, MMPs), and photothermal/magnetothermal conversion nanoparticles.136,137 These designs allow nanomaterials to remain inert and stable under physiological conditions but become activated upon reaching the tumor site, significantly increasing local drug concentration while minimizing toxicity to normal tissues.
Moreover, stimuli-responsive behavior itself can serve as a targeting mechanism by exploiting the aberrant features of the TME. Most solid tumors exhibit mildly acidic pH (~6.5–6.8), elevated reductive potential (high GSH), increased H2O2 levels, and hypoxia.138 Accordingly, various smart nanocarriers have been developed, such as pH-sensitive release systems, GSH-triggered drug-releasing micelles, and H2O2-driven oxygen-generating nanozymes. Once these carriers reach the TME, they are “unlocked” by specific stimuli to release payloads or exert therapeutic effects precisely at the tumor site.139,140 For instance, Hunter et al141 utilized an HSA–MnO2 nanozyme that responded to high intratumoral H2O2 levels to release oxygen, enhancing photodynamic therapy (PDT) in bladder tumors. As the bladder is a surface-accessible organ, it is also suitable for exogenous physical triggers such as light. NIR light can penetrate the bladder wall to activate photosensitizers or photothermal agents, enabling localized energy release and tumor ablation.142 This facilitates novel opportunities for multimodal BC therapy.
Chen et al143 developed a multifunctional nanoparticle system (GOx@MBSA–PPy–MnO2) responsive to both glucose and GSH for combination BC therapy. In this system, polypyrrole (PPy) components generate heat under NIR light for photothermal therapy (PTT); meanwhile, the encapsulated glucose oxidase (GOx) catalyzes glucose into H2O2 and gluconic acid, lowering pH and triggering MnO2 decomposition to release Mn2⁺. Mn2⁺ contributes in two ways: (1) it facilitates Fenton-like reactions under high H2O2 levels, generating cytotoxic hydroxyl radicals (·OH) for chemodynamic therapy (CDT); and (2) it enhances oxygen generation by decomposing H2O2, thereby alleviating hypoxia.144 Similarly, Hao et al designed an RGD-modified platinum nanozyme system (PtNPs) co-loaded with a GSH-responsive prodrug (PTX–SS–HPPH/Pt@RGD–NP) for targeted BC therapy. Their in vitro and in vivo studies demonstrated that the synergistic effects of PTT, CDT, and metabolic modulation achieved significantly better tumor ablation than monotherapies.145 Guo et al’s AuNR&ION hydrogel system is another classic example of stimuli-responsive design: the gold nanorod (AuNR) component requires NIR irradiation to exert photothermal effects, while the ferrite nanoparticles (IONs) are activated by H2O2 and iron ions in the TME to trigger lipid peroxidation chain reactions, leading to ferroptosis and reactive oxygen species (ROS) production.126 In another study, urease-driven nanomotors were developed for BC therapy. These nanocarriers had surface-anchored urease that catalyzed urea in bladder urine to generate gas bubbles, propelling the particles deep into tumor tissues.146 When labeled with radioactive iodine-131 (^131I), these nanorobots enabled intravesical radionuclide therapy, reducing tumor volume by ~90% and increasing nanoparticle accumulation within the tumor eightfold compared to passive diffusion.146 In summary, stimuli-responsive nanomaterials provide a powerful strategy for spatiotemporal control of therapeutic activation. By sensitively responding to endogenous or exogenous cues, they enable precise delivery of potent therapies to tumor sites, enhancing efficacy and minimizing adverse effects in BC immunotherapy (Figure 7).
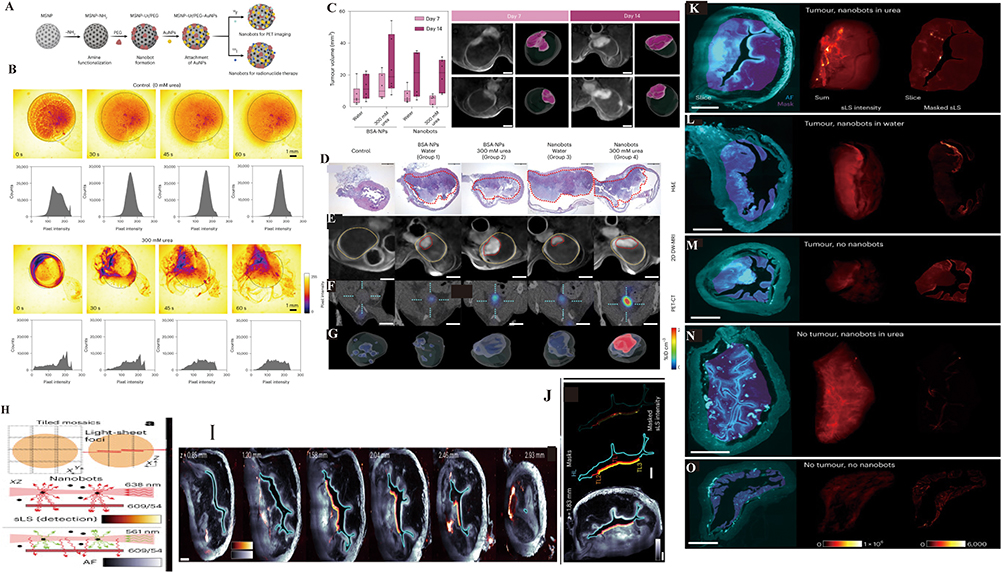 |
Figure 7 Urea-Driven Nanobot Design, Motion, and Multimodal Imaging Reveal Enhanced Bladder Tumor Targeting and Retention. (A) Schematic representation of the nanobot fabrication process and radiolabelling. (B) Snapshots depicting the nanobot motion dynamics in the absence and presence (300 mM) of urea as fuel, and corresponding pixel intensity histograms for the ROI marked by a circle. (C) Tumor volumes on days 7 and 14 after cell implantation for the different study groups. (D–G) Multimodal imaging including H&E staining, DW-MRI, PET/CT, and 3D reconstruction demonstrated tumor presence, bladder localization, and enhanced radioactive accumulation in representative animals across treatment groups. (H) The principle of laser being scattered by particles (such as nanorobots) and detected through a filter. (I) Optical sections of bladder tissue at different depths with tissue layer annotations for quantification. (J) tumour and healthy layers of tissues along bladder cavity, detected and annotated (masks) in three dimensions. (K–O) Volumetric light-sheet imaging and voxel intensity analysis revealed that nanobots administered with urea achieved markedly higher retention and signal intensity in both tumor-bearing and healthy bladders, whereas nanobots without urea or control groups showed minimal signal, confirming urea-driven active accumulation. Adapted from Simó, Cristina et al. Urease-powered nanobots for radionuclide BC therapy. Nature nanotechnology vol. 19,4 (2024): 554–564. https://creativecommons.org/licenses/by-nc/4.0/. Copyright © 2024 by the authors.146 |
Functional Nanomaterials for Enhancing T Cell Immunity in BC
BC is characterized by a high recurrence rate and notable resistance to therapy, largely attributable to its highly immunosuppressive TME and the unique anatomical features of the urinary tract, which together impede T cell infiltration and effector function.147 Although current T cell–based therapies—such as PD-1/PD-L1 immune checkpoint inhibitors and adoptive cell transfer approaches (eg, CAR-T, TCR-T, TIL)—have demonstrated durable responses in certain advanced cases, their overall response rates remain limited.113 To overcome these challenges, nanotechnology has emerged as a novel platform for drug delivery and immune modulation owing to its unique biological advantages. It is rapidly becoming a central strategy in optimizing T cell–based immunotherapy.106,135,148 The use of engineered nanomaterials is now reshaping the therapeutic landscape for BC T cell immunotherapy.149 In general, both organic (eg, liposomes, polymeric micelles) and inorganic (eg, mesoporous silica, gold nanoparticles) nanomaterials ranging from 1 to 100 nm in size offer innovative solutions to address the limitations of conventional approaches. Their high surface area, programmable surface chemistry, and intelligent, stimuli-responsive drug release capabilities are particularly advantageous.150 Its core advantages include the following: First, through surface functionalization with targeting ligands—such as tumor-homing peptides or anti-CD3 antibodies—nanocarriers can achieve precise accumulation of therapeutic molecules (eg, antigens, adjuvants, cytokines, or immune checkpoint inhibitors) at tumor sites or in the vicinity of T cells, thereby significantly reducing systemic toxicity.151 Second, their capacity for multi-component co-delivery enables synergistic T cell activation and reversal of microenvironmental immunosuppression. For example, co-delivery of tumor antigens and PD-L1 inhibitors, in combination with pH-, enzyme-, or ROS-responsive release mechanisms, enhances localized immune stimulation within tumors.152 Furthermore, the nanoscale characteristics of these materials facilitate penetration through the dense stromal barriers of bladder tumors, thereby addressing the issue of insufficient T cell infiltration. Meanwhile, biomimetic modifications—such as T cell membrane coating—can enhance the ability of the nanocarriers to evade immune clearance. What is particularly important is that the anatomical accessibility of the urinary system makes nanomedicine especially suitable for intravesical administration, greatly improving bioavailability. For instance, liposome-encapsulated BCG antigens can enhance dendritic cell cross-presentation, while pH-responsive hydrogel-based sustained release of IL-15 helps maintain the activity of tissue-resident memory T cells in the bladder mucosa.153 Recent progress has further confirmed these advantages. Self-propelling, in situ–polymerized nanoparticles significantly boosted the immunotherapeutic efficacy against BC by activating the STING pathway.125
Nanomaterials for the Delivery of siRNA and Antibodies
In BC immunotherapy, a critical strategy involves regulating the expression of immune-related molecules in tumor cells and the TME, such as silencing immunosuppressive genes (eg, PD-L1, IDO) or delivering exogenous antibodies/receptors (eg, anti–PD-1 antibodies or immune-stimulatory cytokines).154,155 However, small interfering RNA (siRNA) and plasmid DNA are highly susceptible to degradation in vivo and have poor membrane permeability, while systemic delivery of macromolecular antibodies is often limited by poor tumor targeting and adverse side effects.97,156 Nanocarriers serve as ideal platforms for delivering siRNA and antibodies, as they protect biomacromolecules, enhance targeting, and facilitate cellular uptake.
In general, nanomaterials designed for gene delivery are expected to possess high loading capacity, efficient cellular uptake, and produce no toxic byproducts upon degradation. Commonly used carriers include cationic polymeric nanoparticles, which form complexes with nucleic acids to facilitate cellular internalization, and mesoporous silica nanoparticles (MSNs), which load nucleic acids within their porous structures and protect them via pore sealing.157,158 These platforms improve the targeting specificity and therapeutic efficacy of gene therapies and are regarded as promising strategies for nucleic acid delivery in BC. For protein-based therapeutics such as antibodies, nanomaterials can be employed via surface conjugation or encapsulation to enhance their accumulation in tumor tissues.159 A representative example is the delivery of PD-L1 antibodies using macrophage-derived biomimetic extracellular vesicles (EMVs). Researchers successfully utilized these vesicles to enrich antibodies at bladder tumor sites while facilitating rapid clearance from the circulatory system, thereby reducing systemic exposure. In vivo studies demonstrated that compared with free antibodies, EMV-delivered PD-L1 antibodies elicited comparable immune activation—such as enhanced T cell activity following PD-L1 blockade in the TME—while significantly reducing systemic toxicity.97 In addition, several stimuli-responsive nanocarriers have been developed to further improve the performance of antibody-based therapies. For example, near-infrared photoimmunotherapy (NIR-PIT) uses antibodies conjugated with photosensitive moieties that, upon binding to tumor cells and subsequent light exposure, release localized cytotoxic effects.160 Applications of NIR-PIT in BC have been reported, targeting molecules such as EGFR and PD-L1, resulting in enhanced local immune-mediated tumor killing with minimal side effects.161,162
Broadly speaking, the delivery of siRNA/genes and antibodies via nanocarriers can relieve immunosuppression at the molecular level, thereby indirectly enhancing T cell–mediated anti-tumor responses.163 For instance, one study demonstrated that PD-L1 downregulation via RGD-functionalized mesoporous silica nanoparticles (RGD-MSNs) increased tumor susceptibility to cytotoxic T lymphocyte (CTL)–mediated killing. At the same time, delivery of miR-34a was shown to reduce cancer stem cell–like properties, helping to eliminate antigen-negative, drug-resistant clones. Furthermore, a combination therapy involving EMV-based co-delivery of PD-L1 antibodies and a CD73 inhibitor successfully reversed two key immunosuppressive mechanisms—PD-L1 expression and adenosine accumulation—in the TME. This led to a substantial increase in the number of intratumoral CD8⁺ cytotoxic T cells and significantly inhibited tumor growth.164 Currently, multiple nanomedicine-based strategies for the delivery of immune-related genes and antibodies have entered preclinical or clinical stages. Lipid nanoparticle (LNP)–based mRNA technology, for example, has already been utilized in personalized cancer vaccines for other malignancies and holds promise for translation to BC.165 In parallel, an innovative approach has been proposed using amphiphilic polymeric micelle carriers to deliver plasmids encoding chimeric antigen receptors (CARs) directly into T cells.113,166 This strategy aims to “nano-transfect” patient-derived T cells in vivo, converting them into CAR-T cells capable of targeting BC and other solid tumors—representing a frontier in the convergence of gene-engineered immunotherapy and nanotechnology. Looking ahead, continued translation of these nanocarrier platforms from bench to bedside is anticipated to broaden therapeutic options and improve the efficacy of T cell–based immunotherapy for BC.
Nanomaterials for Inducing Immunogenic Cell Death (ICD)
Immunogenic cell death (ICD) refers to a form of tumor cell death triggered by specific therapeutic interventions, during which highly immunostimulatory damage-associated molecular patterns (DAMPs)—such as ATP, HMGB1, and heat shock proteins—are released along with tumor-associated antigens. This process initiates and enhances anti-tumor immune responses by the host.167 ICD plays a pivotal role in the concept of “in situ tumor vaccination,” as it promotes antigen uptake, maturation of dendritic cells (DCs), and subsequent activation of T cell–mediated immunity168 However, the efficacy of conventional radiotherapy or chemotherapy in inducing ICD is often suboptimal, and tumor cells may escape immune surveillance by upregulating tolerance mechanisms.140 To overcome these limitations, nanomaterials have been employed to amplify the immunogenicity of ICD through multimodal interventions—including physical, chemical, and biological strategies. These materials not only directly kill tumor cells but also deliver abundant danger signals and antigenic stimuli to support robust T cell activation, thereby significantly enhancing the effectiveness of T cell–based immunotherapy in BC.169,170
Physically, nanomaterial-induced ICD primarily relies on photothermal therapy (PTT) and photodynamic therapy (PDT), both of which exploit the photoresponsive properties of nanoparticles to generate localized heat or reactive oxygen species (ROS), inducing immunogenic damage to BC cells.171 For example, gold nanorods (AuNRs) under near-infrared (NIR) irradiation can efficiently convert light into heat (photothermal conversion efficiency >90%), causing disruption of the tumor cell membrane and subsequent release of DAMPs172 Moreover, when AuNRs are combined with iron oxide nanoparticles (IONPs), they can further induce ferroptosis—an iron-dependent, lipid peroxidation–driven form of non-apoptotic cell death. This enhances the release of ROS and oxidized phospholipids, amplifying DAMP exposure and polarizing tumor-associated macrophages (TAMs) toward a pro-inflammatory M1 phenotype. This immune remodeling creates a more favorable environment for T cell infiltration and activation.173 In BC models, such composite systems have not only achieved direct tumor ablation but also enhanced cytotoxic T lymphocyte (CTL) infiltration and immune memory formation, providing resistance against tumor rechallenge.124 In addition, Deng et al recently reported a T cell membrane–camouflaged nanoparticle system (Tim3@PHSM@IC), which combines PTT-induced ICD with blockade of the Tim-3 checkpoint pathway. This dual-functional nanoplatform overcomes the immunosuppressive TME and significantly improves the response rate to T cell–based therapies (Figure 8).129 These physical mechanisms offer the advantage of precise spatial and temporal control. However, they often require further surface modification (eg, PEGylation) to enhance biocompatibility and tumor targeting efficiency.
 |
Figure 8 Therapeutic Design, Characterization, and Antitumor Efficacy of Tim3\@PHSM\@IC Nanoparticles for BC Treatment Schematic illustration showing the preparation (A) of Tim3@PHSM@IC and the proposed mechanism (B) of Tim3@PHSM@IC to treat BCa. (C) Tim3@PHSM@IC nanoparticles were successfully constructed and characterized by TEM, size and zeta-potential measurements, drug release profiling, protein staining, Western blot, and flow cytometry, confirming their structural integrity, protein incorporation, and Tim3 expression. (D) Tim3@PHSM@IC nanoparticles showed enhanced cellular uptake and tumor targeting compared to controls, as demonstrated by fluorescence imaging, flow cytometry, and biodistribution analyses in cells and mice, with significantly higher accumulation in tumors. (E) Tim3@PHSM@IC nanoparticles significantly inhibited orthotopic bladder tumor growth in mice, as evidenced by reduced tumor fluorescence, smaller tumor size and weight, and statistical analysis confirming superior therapeutic efficacy over control treatments. (F) In a subcutaneous BC model, Tim3@PHSM@IC nanoparticles markedly suppressed tumor growth, reduced tumor weight, improved survival, and showed minimal toxicity, with histological analysis confirming decreased proliferation and effective therapeutic impact. *p < 0.05, **p < 0.01, ***p < 0.001. Adapted from Deng, Wen et al. Genetically engineered T cell membrane-camouflaged nanoparticles triggered cuproptosis for synergistic BC photothermal-immunotherapy. Journal of nanobiotechnology vol. 23,1 425. 7. https://creativecommons.org/licenses/by-nc/4.0/. Copyright © 2025 by the authors.129 |
Chemically, ICD can be enhanced through the delivery of pro-oxidative or pro-apoptotic agents via nanocarriers, which directly modulate cell death pathways to increase the immunogenicity of dying tumor cells.174 For example, polymeric nanoparticles such as PLGA loaded with anthracycline-based chemotherapeutics (eg, doxorubicin) or oxidized phosphonates can be used to locally release these agents within the tumor, inducing surface exposure of calreticulin (CRT) and ATP efflux. These DAMP signals promote dendritic cell (DC) maturation and activate downstream T cell responses175 Additionally, Nectin-4–targeted PLGA nanoparticles delivering gene therapies such as MT50 have demonstrated dual functionality—suppressing tumor proliferation (with >70% inhibition) and chemically inducing ICD to release tumor-associated antigens, thereby stimulating antigen-specific T cell responses176 Another innovative platform is the hybrid membrane–coated nanoparticle (HM-MPS NP), which co-delivers oxidative agents alongside anti–PD-1 antibodies. This system amplifies ICD, reverses immune suppression in the TME, and enhances T cell–mediated tumor clearance.177 These chemically functionalized nanomaterials are typically synthesized via emulsion–solvent evaporation techniques, with particle sizes controlled between 50–200 nm to optimize the enhanced permeability and retention (EPR) effect. However, parameters such as drug loading efficiency (>80%) and potential toxicity must be carefully evaluated to ensure safe delivery.178
From a biological perspective, nanomaterials can mimic infection signals or deliver microbial components to further strengthen ICD-associated immune stimulation. For instance, gold nanoparticles loaded with Listeriolysin O (LLO) peptides simulate bacterial membrane perforation, triggering stress-induced necrosis and DAMP release. This facilitates DC recruitment and debris clearance, followed by activation of secondary immunogenic apoptosis. Experiments have shown that 35–55% of tumor cells undergo DC-mediated ICD, while direct cytotoxic effects remain minimal.125 Moreover, self-propelling, in situ–polymerized nanoparticles (eg, DMCU) can activate the STING pathway by delivering microbial mimics, promoting both DC maturation and T cell recruitment, thereby enhancing BC immunotherapy outcomes.179 These biologically inspired nanomaterials are often biomimetically modified (eg, with membrane coatings) to improve targeting accuracy and evade immune clearance, enabling efficient ICD induction within the bladder’s mucosal environment. Through a combination of physical, chemical, and biological strategies, nanomaterials can synergistically induce ICD, effectively transforming tumor cells into “vaccine donors” that recruit and activate T cells. Such strategies have been validated in BC models for their capacity to enhance anti-tumor T cell responses.126 For instance, the AuNR&ION nanoplatform has been shown to induce systemic immune memory capable of suppressing distant tumors.127 GNP–LLO nanovaccines increase infiltration of CTLs and DCs while reducing immunosuppressive cells,180 and HM-MPS NPs in combination with anti–PD-1 therapy further amplify therapeutic outcomes.177 Therefore, nanomaterial-induced ICD represents a promising approach to substantially improve the responsiveness of BC to T cell–based immunotherapy, and to facilitate the transition from an immunologically “cold” to a “hot” tumor phenotype.
Nanomaterials for Dendritic Cell Activation
Dendritic cells (DCs) are the key antigen-presenting cells responsible for initiating tumor antigen–specific T cell responses. Effective BC immunotherapy requires the uptake of tumor antigens by a large number of mature DCs, followed by their migration to lymph nodes to activate T cells.181 However, DCs in the bladder TME are often poorly activated and sparsely infiltrated. Moreover, conventional vaccines—such as tumor lysates and peptide vaccines—typically lack sufficient adjuvant stimulation, resulting in suboptimal DC maturation.182,183 Nanotechnology offers powerful tools to construct novel vaccine and adjuvant delivery systems that markedly enhance antigen uptake and maturation of DCs in BC.
One widely studied strategy involves nanoparticle-based cancer vaccines that co-deliver tumor antigens and immune adjuvants directly to DCs. For example, Nie et al designed a hybrid membrane nanoparticle vaccine (HM@NP) composed of fused tumor cell membrane and bacterial (E. coli) membrane components, thereby combining autologous tumor antigens with microbial adjuvants on a single nanoplatform.182 This vaccine elicited robust anti-tumor immunity and long-term immune memory in murine models of colon and breast cancers.182 For BC, patient-derived tumor cell membranes could be similarly fused with bacterial components (eg, Acinetobacter, Mycobacterium) to formulate autologous nanovaccines aimed at DC activation.184 Another strategy is the use of nanoparticle-based adjuvant delivery, in which potent immunostimulatory molecules are encapsulated in nanocarriers for targeted delivery to DCs.181 For instance, CpG oligodeoxynucleotides (TLR9 agonists) or STING agonists encapsulated in nanoparticles can be injected intratumorally or intravesically to locally trigger DC maturation and type I interferon production, converting “cold tumors” into “hot tumors” that are more readily recognized by T cells.185 A representative example is hafnium-based metal–organic frameworks (Hf-MOFs) loaded with CpG; combined with radiotherapy, this nano-adjuvant eliminated primary tumors, inhibited metastasis, and established strong anti-tumor immune memory in murine models.186
BCG, a classical BC immunotherapy, functions partly by stimulating DC uptake of BCG and tumor antigens in the bladder epithelium, thereby initiating both innate and adaptive immunity.113 Based on this mechanism, nanotechnology has been applied to optimize BCG delivery and safety. As reviewed by Yu et al, chitosan-based nano-BCG significantly increased recruitment and activation of immune cells such as DCs. In mice, intravesical instillation of nano-BCG induced stronger local inflammatory responses and lower systemic toxicity, suggesting more efficient DC activation within the bladder mucosa and reduced risk of systemic BCG infection.187 In another study, Ren et al constructed chitosan/graphene oxide nanoparticles (CS/GO-NPs) loaded with GM-CSF for combined photothermal therapy (PTT) and immunotherapy in an orthotopic mouse model of BC. These nanoparticles were anchored to the tumor surface via biotinylation, enabling sustained local release of GM-CSF, effective recruitment and activation of DCs, and enhanced anti-tumor immunity. The thermal effects of PTT further increased DC and T cell infiltration, reinforcing the immune response.188 Additionally, using nanoparticles as artificial antigen-presenting cells (aAPCs) is an emerging concept. For instance, PLGA nanoparticles functionalized with MHC-I/peptide complexes, co-stimulatory molecules, and cytokines have been developed to directly interact with and activate T cells in vitro, mimicking the role of natural APCs.189 Although not yet applied to BC specifically, this proof-of-concept highlights the potential for nano-platforms to perform antigen presentation without relying on host cells, paving the way for future antigen-specific vaccine design.
The ultimate outcome of DC activation via nanotechnology is the efficient cross-priming of naïve T cells and enhancement of both effector and memory T cell quantity and quality.190 In the case of nano-delivered BCG, improved DC activation correlated with stronger anti-tumor responses and superior long-term tumor-free survival compared to conventional BCG.191 Similarly, Th1 polarization induced by nano-formulated CWS indicates that the resulting CD8⁺ cytotoxic and Th1 helper T cells exhibit heightened effector functions. For example, in the study by Li et al, the hybrid membrane nanoparticle vaccine HM@NP induced long-lasting immune memory in various tumor models, significantly extending postoperative tumor-free survival. Upon tumor rechallenge, these mice rapidly cleared cancer cells, demonstrating durable immune protection.192 In conclusion, effective DC activation through nanotechnology can be seen as the establishment of an “immune arsenal” within the host, continuously generating high-quality tumor-specific T cells and significantly amplifying the depth and breadth of BC immunotherapy.189
Nanomaterials for Enhancing the Formation of Memory T Cells
The generation and maintenance of memory T cells are critical for the long-term success of cancer immunotherapy, as these cells provide sustained tumor surveillance and rapid immune responses that help prevent tumor recurrence.193 However, inducing durable memory T cells in solid tumors remains challenging. Persistent tumor antigen exposure can lead to T cell exhaustion,194 while the absence of post-treatment immune stimulation may cause gradual apoptosis or functional decline of memory T cells.195 Thus, therapeutic strategies aimed at promoting memory T cell differentiation and survival have become a key focus of current research. Nanomaterials contribute to this objective primarily through two mechanisms: (1) serving as sustained-release depots to provide prolonged antigen/adjuvant exposure, mimicking chronic infection to drive memory differentiation;196 and (2) delivering survival factors such as cytokines (eg, IL-7, IL-15) or co-stimulatory signals to T cells, preventing apoptosis and reinforcing the memory phenotype.197
Currently, hydrogel-based nanocomposites are frequently utilized as post-surgical implantable vaccine depots. One notable example is the CAR-T hydrogel system developed by Gu et al, which incorporates sustained release of IL-15 to support the long-term proliferation of CAR-T cells, effectively mimicking the function of a memory T cell–nourishing cytokine.134 Results demonstrated that CAR-T cells retained within the hydrogel remained viable and functional over extended periods in vivo. Remarkably, tumor suppression was also observed in contralateral, untreated tumor sites, indicating that the persistent effector T cells exerted systemic protective effects.134 Similarly, controlled release of nano-vaccines can avoid transient immune stimulation caused by single-dose immunization, instead enabling continuous antigen exposure that promotes memory T cell differentiation.198 In one study, tumor-derived neoantigen peptides were co-encapsulated with CpG adjuvant in PLGA nanoparticles and injected intramuscularly. Compared with free vaccines, the slow-release formulation significantly increased the proportion of CD8⁺ memory T cells in mice and improved tumor-free survival following rechallenge.199 Moreover, certain immunoregulatory cytokines—such as IL-2, IL-7, and IL-15—are known to favor the development of memory T cell responses.200 Nanocarriers offer a means to deliver these potent cytokines locally, thereby minimizing systemic toxicity. For example, in a murine BC study, IL-12 and the chemotherapeutic agent pirarubicin (THP) were co-encapsulated in fluorinated chitosan nanoparticles for intravesical instillation. The chitosan derivative enhanced drug penetration into tumor tissues, while IL-12 activated local lymphocytes. The combination effectively eradicated primary tumors and significantly reduced recurrence rates.93 These findings suggest that IL-12 promoted T cell differentiation and IFN-γ secretion within the bladder TME, making residual cancer cells less likely to survive, and thus exerting a “relapse-preventing” effect. Similarly, local delivery of cytokines such as IL-15 and IL-21 using nanoparticle platforms may further enhance the memory phenotype of effector T cells. Notably, nanoparticle-mediated delivery of the IL-15 superagonist ALT-803 has been shown to dramatically expand the pool of tumor-specific memory T cells, which persisted in vivo and exhibited rapid recall responses upon re-exposure to tumor antigens.201
To evaluate the impact of nanomaterial-based strategies on memory T cell formation, tumor rechallenge experiments or long-term survival assessments are typically conducted.202 To date, multiple murine studies have demonstrated that following nanoparticle-based immunotherapeutic interventions, animals often exhibit delayed or completely inhibited tumor growth upon rechallenge with tumor cells.100 For example, in a 4T1 breast cancer model, mice treated with a nanocomposite therapy co-delivering PD-L1–targeted siRNA and oncolytic viral proteins showed complete clearance of the primary tumor on the left flank. When rechallenged with 4T1 cells on the contralateral side, 25% of the mice developed no secondary tumors, while the remainder showed significantly delayed tumor growth—providing strong evidence of robust immune memory.199 Similarly, in the aforementioned hybrid vaccine model composed of E. coli membrane and tumor membrane components, mice that received postoperative vaccination remained tumor-free for up to 90 days, whereas most control animals experienced recurrence within several weeks.182 Collectively, these findings suggest that nanomaterials, by providing sustained antigenic stimulation and targeted immune modulation, can effectively promote the formation and maintenance of memory T cells. This offers a powerful immunological foundation for achieving long-term immune surveillance and relapse prevention in BC therapy.
Nanomaterials for Reshaping the Tumor Metabolic Environment
In the TME of BC, metabolic dysregulation is often intricately linked with immune suppression. Tumor cells and immunosuppressive myeloid-derived cells competitively consume essential nutrients such as glucose, tryptophan, and arginine, while producing high levels of lactic acid and adenosine. This leads to a nutrient-deprived, acidic, and hypoxic environment that impairs T cell survival and function, thereby posing a significant challenge to effective immunotherapy.203,204 In addition, several metabolic enzymes—such as indoleamine 2,3-dioxygenase (IDO), arginase, and CD73—are overexpressed by tumor cells or suppressive immune cells. These enzymes deplete amino acid pools critical for T cell proliferation or generate immunosuppressive metabolites, rendering T cells dysfunctional.205 Encouragingly, recent research has shown that remodeling the metabolic landscape of the TME—essentially reshaping its “soil”—can significantly enhance the efficacy of immunotherapy.84,206 Based on this, nanomaterials—with their inherent targeting capabilities—are increasingly being applied to precisely modulate intratumoral metabolic parameters and alleviate T cell–suppressive conditions.
One approach involves the delivery of small-molecule inhibitors or genetic regulators via nanocarriers to directly block immunometabolic suppressive pathways within the (TME.207 For instance, Zhou et al utilized extracellular vesicle–mimetic (EMV) nanovesicles to deliver the CD73 inhibitor AB680. CD73 catalyzes the degradation of ATP into adenosine, a potent suppressor of T cell activity. Nano-enabled delivery of AB680 effectively reduced adenosine accumulation in the TME and restored T cell functionality.93 Similarly, loading IDO inhibitors (eg, NLG-919) or siRNA targeting IDO into nanocarriers can suppress local tryptophan catabolism, thereby relieving restrictions on T cell proliferation.168 Although not yet demonstrated in BC, such approaches have been validated in melanoma and other tumor models, highlighting their translational potential.168,208 A second strategy focuses on the clearance or reprogramming of metabolic byproducts. For example, MnO2 nanoparticles act as nanozymes that decompose H2O2 to produce oxygen, thereby relieving tumor hypoxia and acidity and enhancing oxygen availability for T cells and NK cells.128 Glucose oxidase (GOx)-loaded nanoparticles simultaneously deplete glucose—limiting energy supplies to tumor and regulatory cells—and generate H2O2, which can synergistically induce tumor cell death and immune activation.209 In one system, GOx was co-encapsulated with catalase (CAT) to convert H2O2 into O2 immediately upon generation, minimizing oxidative damage to T cells while maintaining local oxygen concentration to support T cell effector function.143 Compared to the aforementioned strategies, a third approach focuses on reprogramming metabolism-associated immune cells to counteract the immunosuppressive conditions of the TME.210 Tumor-associated macrophages (TAMs), which are abundant in the TME, play a pivotal role in immunometabolic regulation by secreting arginase—which depletes arginine—and vascular endothelial growth factor (VEGF)—which promotes abnormal angiogenesis and hypoxia.211 To target TAMs, Feng et al developed upconversion nanoparticles camouflaged with TAM membranes (NPR@TAMM) to deliver a colony-stimulating factor 1 receptor (CSF1R) inhibitor into tumors. This strategy exerts a dual function: on one hand, it depletes the TAM survival factor CSF-1, disrupting TAM–tumor cell interactions; on the other, it induces immunogenic cell death (ICD) via photodynamic therapy, thereby directly eliminating a subset of TAMs.212 As a result, TAMs were repolarized from a tumor-promoting M2 phenotype to an antitumor M1 phenotype, with concurrent blockade of the CSF-1/CSF1R axis, leading to significant attenuation of immunosuppression within the TME.212 This dual modulation of metabolism and immunity markedly enhanced T cell infiltration and activation. Similarly, neutrophil membrane–mimetic nanoparticles have been designed to adsorb and deplete myeloid-derived suppressor cells (MDSCs), another key suppressive population in the TME. Wang et al reported that granulocyte membrane–coated nanoparticles (pCS) significantly reduced MDSC accumulation in both tumor tissues and lymphoid organs in murine tumor models. This intervention restored T lymphocyte function and, when combined with anti–PD-1 therapy, led to a substantial extension in overall survival.213 Collectively, these findings demonstrate that targeting metabolism-regulatory immune cells via nanomaterials provides an effective means to reshape the immunometabolic landscape of tumors, thereby indirectly optimizing the metabolic fitness of T cells and enhancing their antitumor activity.
Remodeling the tumor metabolic environment confers multifaceted benefits to T cells. For instance, in a murine model treated with nano-delivered AB680 (a CD73 inhibitor) in combination with αPD-L1 antibody, the reduction of adenosine and PD-L1 blockade synergistically led to a marked increase in tumor-infiltrating cytotoxic T lymphocytes (CTLs), eliciting potent antitumor activity.93 Similarly, oxygen-releasing MnO2 nanozymes alleviated tumor hypoxia, thereby enhancing the efficacy of photodynamic therapy (PDT) in bladder tumors. This not only resulted in direct tumor ablation but also downregulated hypoxia-inducible immunosuppressive factors such as HIF-1α and VEGF, thereby improving the responsiveness of subsequent immunotherapeutic interventions.128 In the context of TAM/MDSC depletion or repolarization, murine models frequently exhibit simultaneous inhibition of both primary and metastatic tumor growth. These outcomes are largely attributed to global immunological reprogramming, characterized by reduced regulatory T cell (Treg) populations and elevated levels of Th1-polarizing cytokines such as IL-12, ultimately enhancing T cell–mediated immune surveillance.214 In conclusion, precise metabolic intervention using nanomaterials offers a robust strategy to eliminate physiological barriers impeding T cell functionality. By targeting both tumor metabolism and immunosuppressive cellular components, these platforms provide the necessary conditions for unleashing effective antitumor immunity—thus representing a powerful paradigm in the integration of immunotherapy and metabolic modulation.
Improve T-Cell Tumor Infiltration and Persistence
Effective tumor infiltration and sustained functionality of T cells are pivotal for the long-term success of immunotherapy in BC. However, the tumor immune microenvironment is typically enriched with immunosuppressive cells, inhibitory cytokines, and physical barriers such as the extracellular matrix (ECM), which collectively restrict T cell infiltration and induce functional exhaustion.202 To address these challenges, nanomaterials have emerged as multifunctional regulatory platforms that enhance T cell infiltration and persistence by precisely delivering immunomodulators and remodeling the TME.215 For instance, as reviewed by Suphiya Parveen et al, organic nanomaterials such as poly(lactic-co-glycolic acid) (PLGA) nanoparticles can be used to encapsulate interleukin-12 (IL-12), promoting T cell proliferation and interferon-γ (IFN-γ) secretion, thereby reversing the immunosuppressive TME and significantly increasing CD8⁺ T cell infiltration into tumors. The biodegradability and controlled release properties of PLGA (particle size ~100–200 nm; drug loading efficiency >80%) ensure sustained IL-12 exposure at the tumor site while minimizing systemic toxicity and enhancing tumor targeting via the enhanced permeability and retention (EPR) effect.216 However, organic nanomaterials alone may not suffice to overcome physical barriers within tumors. Inorganic nanomaterials such as mesoporous silica nanoparticles (MSNs) are commonly employed to deliver ECM-degrading enzymes (eg, hyaluronidase) to break down stromal components and facilitate T cell penetration.135 MSNs, with a high surface area (>800 m2/g) and pH-responsive release profiles, allow for precise enzyme delivery under acidic TME conditions, markedly improving T cell infiltration into tumor cores and enhancing antitumor efficacy in BC models.217 Moreover, membrane-biomimetic nanomaterials enhance tumor targeting via cell membrane camouflage (eg, macrophage membrane coating). These materials retain critical surface markers such as CD47, which transmits a “don’t phagocytose me” signal to immune cells, thereby prolonging systemic circulation (>48 h) and enabling the targeted depletion of myeloid-derived suppressor cells (MDSCs), ultimately promoting sustained T cell infiltration and functional persistence.218
In terms of persistence, nanomaterials can mitigate T cell exhaustion by modulating cellular metabolism. For instance, inorganic gold nanoparticles (AuNPs) loaded with rapamycin have been shown to inhibit the mTOR pathway, thereby promoting the differentiation of T cells into memory phenotypes and providing antioxidant protection to preserve mitochondrial function—ultimately extending T cell survival within the TME. In addition, stimulus-responsive nanomaterials—such as pH- or ROS-sensitive systems—can locally release metabolic supplements (eg, glucose) within tumors, enhancing glycolytic metabolism in T cells and downregulating the expression of exhaustion markers such as PD-1 and LAG-3.219 Oxidative stress is another major contributor to T cell dysfunction. Nanomaterials with intrinsic antioxidant properties, such as cerium oxide nanoparticles (CeNPs), are capable of scavenging reactive oxygen species (ROS), thereby preserving mitochondrial integrity and sustaining T cell effector functions.220 One study demonstrated that CeNPs significantly reduced intracellular ROS levels in T cells and maintained their cytotoxic capacity within tumors.221 This approach is particularly beneficial in oxidative-stress-prone tumors such as BC.86 Furthermore, nanomaterials can be engineered into scaffolds or hydrogels to mimic lymphoid niches, supporting T cell metabolism and functional maintenance.222 For example, nanofiber-based hydrogel scaffolds have been shown to enhance the metabolic activity of T cells in vitro while downregulating exhaustion markers like PD-1 and LAG-3.223 In the context of BC therapy, such technologies may be integrated with adoptive T cell transfer strategies to further improve clinical efficacy.91 Collectively, these multifunctional nanoplatforms not only alleviate physical and biological barriers to T cell infiltration but also enhance metabolic adaptability and long-term functionality, offering promising avenues for improving both the response rate and durability of immunotherapy in BC models.
The Risks and Limitations of Nanomaterials
While nanomaterials hold great potential in enhancing the efficacy of cancer therapies, it is essential to consider their potential risks and limitations. The size, shape, and surface characteristics of nanomaterials play a crucial role in their biocompatibility and toxicity.224 Long-term exposure to nanomaterials may lead to their accumulation in the body, causing immune responses, cytotoxicity, or tissue damage.225 For instance, certain nanoparticles, such as metallic oxide and metal nanoparticles, have been shown to induce oxidative stress or damage cellular structures, resulting in inflammation or tissue injury. In addition to their direct impact on tissues, the long-term accumulation of nanomaterials may pose chronic toxicity risks, especially with repeated usage in cancer therapies. Incomplete clearance or retention of nanomaterials in the body can lead to adverse effects on various organs. Therefore, the safety profile of nanomaterials must be thoroughly evaluated in preclinical and clinical trials to ensure that their benefits outweigh any potential harm. Moreover, the environmental impact of nanomaterials must not be overlooked. The production, use, and disposal of nanomaterials can lead to their release into the environment, where they may affect ecosystems, particularly aquatic organisms. This environmental concern necessitates careful monitoring and regulation of nanomaterial use to prevent ecological disruptions. Efforts to mitigate these risks include designing nanomaterials with improved biodegradability, lower toxicity, and enhanced biocompatibility.226 Personalized treatment approaches, such as precision medicine, may also help reduce adverse effects by tailoring therapies to individual patients, thus optimizing safety. Additionally, more research is needed to develop methods for the safe disposal or recycling of nanomaterials, ensuring their environmental sustainability. These considerations highlight the need for continued research on the safety and environmental impact of nanomaterials in cancer treatment. By improving their design and developing better safety protocols, nanomaterials can be utilized more effectively while minimizing risks.
Conclusions and Future Research Directions
Nanotechnology is rapidly reshaping the paradigm of T cell–based immunotherapy for BC. Innovations span from urease-powered nanomotors capable of self-propelling deep into bladder mucosa to deliver STING agonists—achieving over a 10-fold increase in CD8⁺ T cell infiltration and a tumor inhibition rate of 94.2% in murine models94 —to macrophage-mimicking membrane vesicles that co-deliver CD73 inhibitors and αPD-L1 antibodies, effectively disrupting the adenosine–PD-L1 dual immunosuppressive axis.93 Other advances include immune–photothermal hybrid nanoplatforms that encapsulate cuproptosis inducers and photothermal agents within Tim-3⁺ T cell membranes, simultaneously overcoming thermotolerance and T cell exhaustion.129 These frontier approaches highlight the potential of multifunctional, programmable, and biomimetic nanocarriers to penetrate bladder barriers, modulate metabolic and checkpoint axes, and catalyze a shift toward the “fourth-generation” of logic-gated, multimodal immunotherapeutics.86,227,228
Despite these promising advancements, several critical challenges continue to hinder the clinical translation of nanomaterial-assisted T cell immunotherapy for BC.86 One of the foremost obstacles lies in the intratumoral heterogeneity across BC subtypes, particularly between NMIBC and MIBC. These subtypes exhibit distinct TME compositions and immune evasion mechanisms, which may lead to inconsistent therapeutic responses.135,229,230 From the perspective of nanomaterials, biosafety concerns remain paramount, especially regarding their long-term biocompatibility and immunogenicity.231,232 For instance, although metallic nanoparticles such as gold nanorods (AuNRs) have demonstrated robust photothermal activation of immune responses, they may also induce off-target inflammatory reactions or accumulate in non-tumor tissues, thereby exacerbating adverse effects. Moreover, the enhanced permeability and retention (EPR) effect, a foundational principle for passive tumor targeting by nanoparticles, has shown limited reliability in human tumors compared to xenograft models. This underscores an urgent need for more physiologically relevant, patient-derived tumor models to more accurately predict clinical outcomes and nanoparticle performance.219 The promising potential of nanomaterial-based therapies for BC, but several barriers to their clinical application remain. The scalability of nanoparticle production remains an obstacle. Achieving consistent quality and reproducibility at a large scale is a significant hurdle, as even small variations in particle size, surface properties, or formulation can affect their therapeutic efficacy and biodistribution. Additionally, tumor targeting and penetration are still major challenges. The dense extracellular matrix and high interstitial pressure of the TME hinder effective delivery and penetration of nanoparticles. Lastly, regulatory considerations pose a significant challenge, as nanomedicines do not fully fit within existing drug approval frameworks.104,233 Regulatory agencies, such as the FDA and EMA, are still developing appropriate guidelines for nanomedicines, requiring tailored testing protocols for long-term toxicity, immune responses, biodegradability, and manufacturing standards. In conclusion, while nanomaterial-based therapies for BC show great promise, addressing these barriers—ranging from safety and production to regulatory frameworks—will be essential for their clinical translation. Overcoming these challenges will enable the development of safer and more effective therapeutic options for BC patients.
We propose that future research in nanomaterial-enhanced T cell immunotherapy for BC should focus on four key directions: First, integrating multi-dimensional single-cell and spatial transcriptomics to decode the complex crosstalk among nanomaterials, T cells, metabolism, and the microbiome. This will guide personalized nanotherapeutic design, offering tailored treatment and prognosis strategies for BC patients and advancing precision medicine;234,235 Second, the development of cascade-responsive or logic-gated nanoplatforms capable of on-demand release of metabolic modulators, immune checkpoint inhibitors, and memory-supporting cytokines, thereby enabling multifunctional theranostics and achieving a one-stop transition from “cold” to “hot” to “memory” tumors;146,236 Third, employing 3D organoid–microfluidic systems combined with real-time imaging to dynamically assess pharmacokinetics, pharmacodynamics, and immune responses, facilitating a seamless translation from bench to bedside.237,238 In parallel, clinical challenges in BC treatment should actively inform basic science, uncovering new questions and inspiring novel mechanistic insights.166 In recent years, artificial intelligence (AI) and machine learning (ML) have rapidly emerged as a “second engine” in nanomedicine research. Their potential is especially prominent in BC T cell immunotherapy, where multi-parametric optimization—encompassing nanoparticle size, surface ligands, delivery kinetics, and immunomodulatory profiles—is essential. Emerging studies suggest that AI-driven nanomaterial design is shifting from “predict-and-optimize” to “generate-and-self-evolve”, with the prospect of creating ultra-personalized delivery systems adapted to the high-salinity, flushing-prone bladder environment and patient-specific antigenic landscapes.239–242 However, current models remain constrained by limited datasets, measurement noise, and batch-to-batch variability across laboratories. Most datasets lack negative results and long-term toxicity annotations, limiting model generalizability and interpretability, and failing to meet GMP-grade safety and quality standards for regulatory approval.149,243 Looking forward, we believe that only under a triadic framework combining precision delivery, systemic immune modulation, and industrial-grade quality control, can nanotechnology truly break through the ceiling of recurrence and therapeutic resistance. This would ultimately lead to a new era of reproducible, affordable, and sustainable precision immunotherapy for BC.219,244 We look forward to future breakthroughs and the emergence of diverse, effective strategies to combat BC.
Data Sharing Statement
No datasets were generated or analysed during the current study.
Consent for Publication
All the authors have read and approved the manuscript.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research is supported by the National Natural Science Foundation of China (grant Nos. 82260609, 82160583). This research was supported by the Graduate Innovation Fund project of Kunming Medical University: 2025S249.
Disclosure
The authors declare no competing interests.
References
1. Tanaka MF, Sonpavde G. Diagnosis and management of urothelial carcinoma of the bladder. Postgraduate Med. 2011;123(3):43–55. doi:10.3810/pgm.2011.05.2283
2. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
3. Avinash BM, Raman JD, Kaag MG. Bladder cancer: overview, epidemiology, initial presentation and diagnosis. In: Trabulsi EJ, Lallas CD, Lizardi-Calvaresi AE, editors. Chemotherapy and Immunotherapy in Urologic Oncology: A Guide for the Advanced Practice Provider. Cham: Springer International Publishing; 2021:141–157.
4. Xylinas E, Kluth LA, Lotan Y, et al. Blood- and tissue-based biomarkers for prediction of outcomes in urothelial carcinoma of the bladder. Urol Oncol. 2014;32(3):230–242. doi:10.1016/j.urolonc.2013.06.009
5. Green DA, Rink M, Xylinas E, et al. Urothelial carcinoma of the bladder and the upper tract: disparate twins. J Urol. 2013;189(4):1214–1221.
6. Sverrisson EF, Espiritu PN, Spiess PE. New therapeutic targets in the management of urothelial carcinoma of the bladder. Res Rep Urol. 2013;5:53–65. doi:10.2147/RRU.S29131
7. Wołącewicz M, Hrynkiewicz R, Grywalska E, et al. Immunotherapy in bladder cancer: current methods and future perspectives. Cancers. 2020;12(5):1181. doi:10.3390/cancers12051181
8. Hashizume A, Umemoto S, Yokose T, et al. Enhanced expression of PD-L1 in non-muscle-invasive bladder cancer after treatment with Bacillus Calmette-Guerin. Oncotarget. 2018;9(75):34066–34078. doi:10.18632/oncotarget.26122
9. Heo JH, Jin HA, Kang YH, Kim KH, Hong SJ, Han KS. Abstract A16: expression of PD-L1 and BCG immunotherapy in non-muscle invasive bladder cancer. Cancer Res. 2016;76(15_Supplement):A16. doi:10.1158/1538-7445.TME16-A16
10. Aurilio G, Cimadamore A, Lopez-Beltran A, et al. Urology: narrative review: update on immunotherapy and pathological features in patients with bladder cancer. Transl Androlo Urol. 2021;10(3):1521–1529. doi:10.21037/tau-20-1436
11. Lim CJ, Nguyen PHD, Wasser M, et al. Immunological Hallmarks for clinical response to BCG in bladder cancer. Front Immunol. 2020;11:615091. doi:10.3389/fimmu.2020.615091
12. Zeng S, Xing S, Zhang Y, Wang H, Liu Q. Nano-Bacillus Calmette-Guerin immunotherapies for improved bladder cancer treatment. J Zhejiang Univ Sci B. 2024;25(7):557–567. doi:10.1631/jzus.B2300392
13. Moro-García MA, Mayo JC, Sainz RM, Alonso-Arias R. Influence of inflammation in the process of T lymphocyte differentiation: proliferative, metabolic, and oxidative changes. Front Immunol. 2018;9:339. doi:10.3389/fimmu.2018.00339
14. Arsenio J, Metz PJ, Chang JT. Asymmetric cell division in T lymphocyte fate diversification. Trends Immunol. 2015;36(11):670–683. doi:10.1016/j.it.2015.09.004
15. Luan J, Liu Y, Cao M, Guo X, Guo N. The pathogenic response of cytotoxic T‑lymphocytes, a common therapeutic target for cancer, has a direct impact on treatment outcomes (Review). Oncol Rep. 2024;52(1):1–9. doi:10.3892/or.2024.8757
16. Deo AS, Shrijana SUS, Karun S, Bisaria K, Sarkar K. Participation of T cells in generating immune protection against cancers. Pathol Res Pract. 2024;262:155534. doi:10.1016/j.prp.2024.155534
17. Zimmerman CM, Robino RA, Cochrane RW, Dominguez MD, Ferreira LMR. Redirecting human conventional and regulatory T cells using chimeric antigen receptors. Methods Mol Biol. 2024;2748:201–241.
18. Shokeen N, Saini C, Sapra L, et al. Role of Regulatory T Lymphocytes in Health and Disease. In: Singh S, editor. Systems and Synthetic Immunology. Singapore: Springer Singapore; 2020:201–243.
19. Zhong T, Sun S, Zhao M, Zhang B, Xiong H. The mechanisms and clinical significance of CD8(+) T cell exhaustion in anti-tumor immunity. Cancer Biol Med. 2025;22(5):460–480.
20. Kharkwal SS, Porcelli SA. T Cell Immunity. In: Segal BH, editor. Management of Infections in the Immunocompromised Host. Cham: Springer International Publishing; 2018:27–41.
21. Shah K, Al-Haidari A, Sun J, Kazi JU. T cell receptor (TCR) signaling in health and disease. Signal Transduct Target Ther. 2021;6(1):412. doi:10.1038/s41392-021-00823-w
22. Vico-Barranco I, Arbulo-Echevarria MM, Serrano-García I, et al. A novel, LAT/Lck double deficient T cell Subline J.CaM1.7 for combined analysis of early TCR signaling. Cells. 2021;10(2):343. doi:10.3390/cells10020343
23. Li R, Ma C, Cai H, Chen W. The CAR T-Cell Mechanoimmunology at a Glance. Adv Sci. 2020;7(24):2002628. doi:10.1002/advs.202002628
24. Wu L, Brzostek J, Sankaran S, et al. Targeting CAR to the peptide-MHC complex reveals distinct signaling compared to that of TCR in a Jurkat T cell model. Cancers. 2021;13(4):867. doi:10.3390/cancers13040867
25. Shahid S, Cai W, Burns ER, et al. Enhancing CAR T cell efficacy by modulating PI3K signaling via a synthetic CD28 rheostat. Blood. 2024;144:4802. doi:10.1182/blood-2024-209964
26. Mørch AM, Bálint Š, Santos AM, Davis SJ, Dustin ML. Coreceptors and TCR signaling – the strong and the weak of it. Front Cell Develop Biol. 2020;8:597627. doi:10.3389/fcell.2020.597627
27. Debatin NF, Bady E, Mandelkow T, et al. Intraepithelial CD8+ cytotoxic T-cells are the main component of the immune tumor microenvironment that drive prognosis in muscle-invasive bladder cancer. Am J Clin Pathol. 2023;160(Supplement_1):S45–S46. doi:10.1093/ajcp/aqad150.102
28. Kelso A, Costelloe EO, Johnson BJ, Groves P, Buttigieg K, Fitzpatrick DR. The genes for perforin, granzymes A–C and IFN‐γ are differentially expressed in single CD8+ T cells during primary activation. Int Immunol. 2002;14(6):605–613. doi:10.1093/intimm/dxf028
29. Hartana CA, Ahlén Bergman E, Zirakzadeh AA, et al. Urothelial bladder cancer may suppress perforin expression in CD8+ T cells by an ICAM-1/TGFβ2 mediated pathway. PLoS One. 2018;13(7):e0200079. doi:10.1371/journal.pone.0200079
30. Ferriès E, Connan F, Pagès F, et al. Identification of p53 peptides recognized by CD8+ T lymphocytes from patients with bladder cancer. Hum Immunol. 2001;62(8):791–798. doi:10.1016/S0198-8859(01)00266-X
31. Tajima M, Wakita D, Satoh T, Kitamura H, Nishimura T. IL-17/IFN-γ double producing CD8+ T (Tc17/IFN-γ) cells: a novel cytotoxic T-cell subset converted from Tc17 cells by IL-12. Int Immunol. 2011;23(12):751–759. doi:10.1093/intimm/dxr086
32. Cheng H, Ma K, Zhang L, Li G. The tumor microenvironment shapes the molecular characteristics of exhausted CD8+ T cells. Cancer Lett. 2021;506:55–66. doi:10.1016/j.canlet.2021.02.013
33. Zhang B, Liu J, Mo Y, Zhang K, Huang B, Shang D. CD8(+) T cell exhaustion and its regulatory mechanisms in the tumor microenvironment: key to the success of immunotherapy. Front Immunol. 2024;15:1476904. doi:10.3389/fimmu.2024.1476904
34. Han HS, Jeong S, Kim H, et al. TOX-expressing terminally exhausted tumor-infiltrating CD8+ T cells are reinvigorated by co-blockade of PD-1 and TIGIT in bladder cancer. Cancer Lett. 2021;499:137–147. doi:10.1016/j.canlet.2020.11.035
35. Kim K, Park S, Park SY, et al. Single-cell transcriptome analysis reveals TOX as a promoting factor for T cell exhaustion and a predictor for anti-PD-1 responses in human cancer. Genome Med. 2020;12(1):22. doi:10.1186/s13073-020-00722-9
36. Tavakolpour S, Darvishi M. The roles of CD4+ T-cells in tumor immunity. In: Rezaei N, editor. Cancer Immunology: A Translational Medicine Context. Cham: Springer International Publishing; 2020:63–90.
37. Swatler J, De Luca M, Rotella I, Lise V, Mazza EMC, Lugli E. CD4+ regulatory T cells in human cancer: subsets, origin, and molecular regulation. Cancer Immunol Res. 2024;12(4):393–399. doi:10.1158/2326-6066.CIR-23-0517
38. Zhang Y, Zhang Y, Gu W, Sun B. TH1/TH2 cell differentiation and molecular signals. Adv Exp Med Biol. 2014;841:15–44.
39. Martínez R, Tapia G, De Muga S, et al. Combined assessment of peritumoral Th1/Th2 polarization and peripheral immunity as a new biomarker in the prediction of BCG response in patients with high-risk NMIBC. OncoImmunology. 2019;8(8):1602460. doi:10.1080/2162402X.2019.1602460
40. Singh AK, Praharaj M, Lombardo KA, et al. Recombinant BCG overexpressing STING agonist elicits trained immunity and improved antitumor efficacy in non-muscle invasive bladder cancer. Urol Oncol. 2020;38(12):899. doi:10.1016/j.urolonc.2020.10.030
41. Brosman SA. Experience with bacillus Calmette-Guerin in patients with superficial bladder carcinoma. J Urol. 1982;128(1):27–30. doi:10.1016/S0022-5347(17)52736-6
42. Luo Y, Chen X, O’Donnell MA. Role of Th1 and Th2 cytokines in BCG-induced IFN-γ production: cytokine promotion and simulation of BCG effect. Cytokine. 2003;21(1):17–26. doi:10.1016/S1043-4666(02)00490-8
43. Satyam A, Singh P, Badjatia N, Seth A, Sharma A. A disproportion of TH1/TH2 cytokines with predominance of TH2, in urothelial carcinoma of bladder. Urol Oncol. 2011;29(1):58–65. doi:10.1016/j.urolonc.2009.06.002
44. Luo Y, Henning J, O′Donnell MA. Th1 cytokine-secreting recombinant mycobacterium bovis bacillus calmette-guérin and prospective use in immunotherapy of bladder cancer. J Immunol Res. 2011;2011(1):728930.
45. Ingels A, Sanchez Salas RE, Ravery V, et al. T-helper 1 immunoreaction influences survival in muscle-invasive bladder cancer: proof of concept. Ecancermedicalscience. 2014;8:486. doi:10.3332/ecancer.2014.486
46. Liu L, Liu Z, Fan L, et al. An evaluation of the tumor microenvironment through CALR, IL1R1, IFNB1, and IFNG to assess prognosis and immunotherapy response in bladder cancer patients. eLife. 2024;13:RP95326.
47. Yushe D, Lauren C, Erin R, et al. 1177 Identification of and vaccination with antigen specific Th1 selective epitopes in bladder cancer: a step towards a multi-antigen vaccine for bladder cancer prevention. J Immunother Cancer. 2022;10(Suppl 2):1.
48. León B, Ballesteros-Tato A. Modulating Th2 cell immunity for the treatment of asthma. Front Immunol. 2021;12:637948. doi:10.3389/fimmu.2021.637948
49. Zhu J, Paul WE. CD4 T cells: fates, functions, and faults. Blood. 2008;112(5):1557–1569. doi:10.1182/blood-2008-05-078154
50. Tan C, Li C, Ge R, et al. Mcl-1 downregulation enhances BCG treatment efficacy in bladder cancer by promoting macrophage polarization. Can Cell Inter. 2025;25(1):48. doi:10.1186/s12935-025-03676-3
51. Pan S, Li S, Zhan Y, et al. Immune status for monitoring and treatment of bladder cancer. Front Immunol. 2022;13:963877.
52. Pichler R, Gruenbacher G, Culig Z, et al. Intratumoral Th2 predisposition combines with an increased Th1 functional phenotype in clinical response to intravesical BCG in bladder cancer. Cancer Immunol Immunother. 2017;66(4):427–440. doi:10.1007/s00262-016-1945-z
53. Villoldo GM, Pombo MT, Aris M, et al. A Th2-score in the tumor microenvironment as a predictive biomarker of response to Bacillus Calmette Guérin in patients with non-muscle invasive bladder carcinoma: a retrospective study. Oncol Res. 2023;31(2):207–220. doi:10.32604/or.2023.028163
54. Arora R, Cao C, Kumar M, et al. Spatial transcriptomics reveals distinct and conserved tumor core and edge architectures that predict survival and targeted therapy response. Nat Commun. 2023;14(1):5029. doi:10.1038/s41467-023-40271-4
55. Zhu J, Yamane H, Cote-Sierra J, Guo L, Paul WE. GATA-3 promotes Th2 responses through three different mechanisms: induction of Th2 cytokine production, selective growth of Th2 cells and inhibition of Th1 cell-specific factors. Cell Res. 2006;16(1):3–10. doi:10.1038/sj.cr.7310002
56. Fujimura T, Kambayashi Y, Furudate S, Kakizaki A, Aiba S. A possible mechanism in the recruitment of eosinophils and Th2 cells through CD163+ M2 macrophages in the lesional skin of eosinophilic cellulitis. Eur J Dermatol. 2014;24:180–185. doi:10.1684/ejd.2014.2283
57. Roider T, Baertsch MA, Fitzgerald D, et al. Multimodal and spatially resolved profiling identifies distinct patterns of T cell infiltration in nodal B cell lymphoma entities. Nat Cell Biol. 2024;26(3):478–489. doi:10.1038/s41556-024-01358-2
58. Jancewicz I, Szarkowska J, Konopiński R, et al. PD-L1 overexpression, SWI/SNF complex deregulation, and profound transcriptomic changes characterize cancer-dependent exhaustion of persistently activated CD4+ T cells. Cancers. 2021;13:4148. doi:10.3390/cancers13164148
59. Xu Z, Jiang X, Dai X, Li B. The dynamic role of FOXP3(+) tregs and their potential therapeutic applications during SARS-CoV-2 infection. Front Immunol. 2022;13:916411. doi:10.3389/fimmu.2022.916411
60. Togashi Y, Shitara K, Nishikawa H. Regulatory T cells in cancer immunosuppression — implications for anticancer therapy. Nat Rev Clin Oncol. 2019;16(6):356–371. doi:10.1038/s41571-019-0175-7
61. Tekguc M, Wing JB, Osaki M, Long J, Sakaguchi S. Treg-expressed CTLA-4 depletes CD80/CD86 by trogocytosis, releasing free PD-L1 on antigen-presenting cells. Proc Natl Acad Sci USA. 2021;118(30):e2023739118. doi:10.1073/pnas.2023739118
62. Winerdal ME, Krantz D, Hartana CA, et al. Urinary bladder cancer tregs suppress MMP2 and potentially regulate invasiveness. Cancer Immunol Res. 2018;6(5):528–538. doi:10.1158/2326-6066.CIR-17-0466
63. Yang YJ, Xu XQ, Zhang YC, Hu PC, Yang WX. Establishment of a prognostic model related to tregs and natural killer cells infiltration in bladder cancer. World J Clin Cases. 2023;11(15):3444–3456. doi:10.12998/wjcc.v11.i15.3444
64. Brandau S. Local and systemic immune suppression in bladder cancer. J Urol. 2007;177(1):12–13. doi:10.1016/j.juro.2006.10.013
65. Chen T, Wang H, Zhang Z, et al. A novel cellular senescence gene, SENEX, is involved in peripheral regulatory T cells accumulation in aged urinary bladder cancer. PLoS One. 2014;9(2):e87774. doi:10.1371/journal.pone.0087774
66. Winerdal ME. Regulatory T Cells – Molecular and Clinical Aspects [dissertation]. Sweden: Karolinska Institutet; 2015.
67. Fenner A. BCG enriches Treg cells. Nat Rev Urol. 2018;15(10):591. doi:10.1038/s41585-018-0075-0
68. Maeda S, Murakami K, Inoue A, Yonezawa T, Matsuki N. CCR4 blockade depletes regulatory T cells and prolongs survival in a canine model of bladder cancer. Cancer Immunol Res. 2019;7(7):1175–1187. doi:10.1158/2326-6066.CIR-18-0751
69. Ye F, Cai Z, Wang B, et al. TGFβ antagonizes IFNγ-mediated adaptive immune evasion via activation of the AKT–Smad3–SHP1 axis in lung adenocarcinoma. Cancer Res. 2023;83(13):2262–2277. doi:10.1158/0008-5472.CAN-22-3009
70. Chatterjee S, Chatterjee A, Jana S, et al. Transforming growth factor beta orchestrates PD-L1 enrichment in tumor-derived exosomes and mediates CD8 T-cell dysfunction regulating early phosphorylation of TCR signalome in breast cancer. Carcinogenesis. 2021;42(1):38–47. doi:10.1093/carcin/bgaa092
71. Jun L, Feng-shuo JI, Qian-shen LI, et al. The role of bladder cancer cell in T cell functional inhibition in vitro. Cancer Res Prev Treat. 2004;31(03):148–150.
72. Ahmed H, Mahmud AR, Siddiquee MF, et al. Role of T cells in cancer immunotherapy: opportunities and challenges. Cancer Pathog Ther. 2023;1(2):116–126. doi:10.1016/j.cpt.2022.12.002
73. Lv J, Zhang Y, Wu Q, Jiang P, Lin Y. Inhibition of SIRT4 promotes bladder cancer progression and immune escape via attenuating CD8(+) T cells function. Int Immunopharmacol. 2025;147:113906. doi:10.1016/j.intimp.2024.113906
74. Kristjánsdóttir N, Kjær A, Nordentoft I, et al. Abstract PR003: t cell receptor repertoire and diversity are prognostic markers in bladder cancer. Clin Cancer Res. 2024;30(10_Supplement):PR003. doi:10.1158/1557-3265.BLADDER24-PR003
75. Sankin A, Chand D, Schoenberg M, Zang X. Human urothelial bladder cancer generates a clonal immune response: the results of T-cell receptor sequencing. Urol Oncol. 2019;37(11):810.e811–810.e815. doi:10.1016/j.urolonc.2019.04.011
76. Choudhury NJ, Kiyotani K, Yap KL, et al. Low T-cell receptor diversity, high somatic mutation burden, and high neoantigen load as predictors of clinical outcome in muscle-invasive bladder cancer. Eur Urol Focus. 2016;2(4):445–452. doi:10.1016/j.euf.2015.09.007
77. Geukes Foppen MH, Donia M, Svane IM, Haanen JB. Tumor-infiltrating lymphocytes for the treatment of metastatic cancer. Mol Oncol. 2015;9(10):1918–1935. doi:10.1016/j.molonc.2015.10.018
78. Matsueda S, Chen L, Li H, Yao H, Yu F. Recent clinical researches and technological development in TIL therapy. Cancer Immunol Immunother. 2024;73(11):232. doi:10.1007/s00262-024-03793-4
79. Inman BA, Longo TA, Ramalingam S, Harrison MR. Atezolizumab: a PD-L1–blocking antibody for bladder cancer. Clin Cancer Res. 2017;23(8):1886–1890. doi:10.1158/1078-0432.CCR-16-1417
80. Khalil DN, Smith EL, Brentjens RJ, Wolchok JD. The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol. 2016;13(5):273–290. doi:10.1038/nrclinonc.2016.25
81. Bocca P, Di Carlo E, Caruana I, et al. Bevacizumab-mediated tumor vasculature remodelling improves tumor infiltration and antitumor efficacy of GD2-CAR T cells in a human neuroblastoma preclinical model. Oncoimmunology. 2017;7(1):e1378843. doi:10.1080/2162402X.2017.1378843
82. Xue Y, Zhao G, Pu X, Jiao F. Construction of T cell exhaustion model for predicting survival and immunotherapy effect of bladder cancer based on WGCNA. Front Oncol. 2023;13:1196802. doi:10.3389/fonc.2023.1196802
83. Liu Z, Mao X, Xie Y, et al. Single-cell RNA sequencing reveals a fibroblast gene signature that promotes T-cell infiltration in muscle-invasive bladder cancer. Commun Biol. 2025;8(1):696. doi:10.1038/s42003-025-08094-9
84. Shin J, Park JW, Kim SY, Lee JH, Choi WS, Kim HS. Strategies for Overcoming Immune Evasion in Bladder Cancer. Int J Mol Sci. 2024;25(6):3105. doi:10.3390/ijms25063105
85. Davarpanah NN, Yuno A, Trepel JB, Apolo AB. Immunotherapy: a new treatment paradigm in bladder cancer. Curr Opin Oncol. 2017;29(3):184–195. doi:10.1097/CCO.0000000000000366
86. Zhao X, Qi X, Liu D, Che X, Wu G. A novel approach for bladder cancer treatment: nanoparticles as a drug delivery system. Int J Nanomed. 2024;19:13461–13483. doi:10.2147/IJN.S498729
87. Wang ZQ, Qu TR, Zhang ZS, et al. A transformable specific-responsive peptide for one-step synergistic therapy of bladder cancer. Small. 2024;20(35):e2310416. doi:10.1002/smll.202310416
88. Liu K, Wang L, Peng J, et al. Drug-loaded bacillus calmette-guérin bacteria for immuno-chemo combo therapy in bladder cancer. Adv Mater. 2024;36(19):e2310735. doi:10.1002/adma.202310735
89. Liao S-Y, Wu L-T, Chou J, Lee JK. Abstract B008: development of intravesical adoptive NECTIN4 chimeric antigen receptor (CAR) T cell therapy for non-muscle invasive bladder cancer. Cancer Immunol Res. 2025;13(2_Supplement):B008. doi:10.1158/2326-6074.IO2025-B008
90. Kim H, Niu L, Larson P, et al. Abstract 2348: nanovaccine: a novel immunotherapeutic strategy to treat bladder cancer. Cancer Res. 2016;76(14_Supplement):2348. doi:10.1158/1538-7445.AM2016-2348
91. Li G, Wu S, Chen W, et al. Designing intelligent nanomaterials to achieve highly sensitive diagnoses and multimodality therapy of bladder cancer. Small Methods. 2023;7(2):2201313. doi:10.1002/smtd.202201313
92. Tao K, Liu S, Wang L, et al. Targeted multifunctional nanomaterials with MRI, chemotherapy and photothermal therapy for the diagnosis and treatment of bladder cancer. Biomater Sci. 2020;8(1):342–352. doi:10.1039/C9BM01377F
93. Zhou Q, Ding W, Qian Z, et al. Immunotherapy strategy targeting programmed cell death ligand 1 and CD73 with macrophage-derived mimetic nanovesicles to treat bladder cancer. Mol Pharm. 2021;18(11):4015–4028. doi:10.1021/acs.molpharmaceut.1c00448
94. Choi H, Jeong S-H, Simó C, et al. Urease-powered nanomotor containing STING agonist for bladder cancer immunotherapy. Nat Commun. 2024;15(1):9934. doi:10.1038/s41467-024-54293-z
95. Yan S, Rolfe BE, Zhang B, Mohammed YH, Gu W, Xu ZP. Polarized immune responses modulated by layered double hydroxides nanoparticle conjugated with CpG. Biomaterials. 2014;35(35):9508–9516. doi:10.1016/j.biomaterials.2014.07.055
96. Liu Y, Jiao F, Qiu Y, et al. The effect of Gd@C82(OH)22 nanoparticles on the release of Th1/Th2 cytokines and induction of TNF-α mediated cellular immunity. Biomaterials. 2009;30(23):3934–3945. doi:10.1016/j.biomaterials.2009.04.001
97. Tian Y, Liu Z, Wang J, et al. Nanomedicine for Combination Urologic Cancer Immunotherapy. Pharmaceutics. 2023;15(2):546. doi:10.3390/pharmaceutics15020546
98. Lee SN, Park SH, Choi J-H, et al. Virulence-free reconstituted synthetic nanopathogen empowered by timely-activating TLR agonist promotes heterologous cancer immunotherapy with depletion of tumor-specific treg cells. Adv Funct Mater. 2025;35(1):2411747.
99. Li R, Huang X, Li X, Liu H, Zhou J, Shen J. A multi-functional drug delivery nanosystem release of TLR-7 immunostimulant and OKT3 induced efficient cancer immunotherapy. Photodiagn Photodyn Ther. 2023;44:103834. doi:10.1016/j.pdpdt.2023.103834
100. Chen Z, Yue Z, Wang R, Yang K, Li S. Nanomaterials: a powerful tool for tumor immunotherapy. Front Immunol. 2022;13:979469. doi:10.3389/fimmu.2022.979469
101. Guo X, Zhang Y, Liu Q, et al. Progress on liposome delivery systems in the treatment of bladder cancer. RSC Adv. 2025;15(18):14315–14336. doi:10.1039/D5RA00746A
102. McKiernan JM, Barlow LJ, Laudano MA, Mann MJ, Petrylak DP, Benson MC. A phase I trial of intravesical nanoparticle albumin-bound paclitaxel in the treatment of bacillus Calmette-Guérin refractory nonmuscle invasive bladder cancer. J Urol. 2011;186(2):448–451. doi:10.1016/j.juro.2011.03.129
103. Lerner SP, Karsh LI, O’Donnell MA, et al. VAX014 for instillation in subjects with nonmuscle-invasive bladder cancer. J Clin Oncol. 2022;40(6_suppl):488. doi:10.1200/JCO.2022.40.6_suppl.488
104. Xu Y, Luo C, Wang J, et al. Application of nanotechnology in the diagnosis and treatment of bladder cancer. J Nanobiotechnol. 2021;19(1):393. doi:10.1186/s12951-021-01104-y
105. Kim H, Khanna V, Kucaba TA, et al. TLR7/8 agonist-loaded nanoparticles augment NK cell-mediated antibody-based cancer immunotherapy. Mol Pharm. 2020;17(6):2109–2124. doi:10.1021/acs.molpharmaceut.0c00271
106. Irvine DJ, Dane EL. Enhancing cancer immunotherapy with nanomedicine. Nat Rev Immunol. 2020;20(5):321–334. doi:10.1038/s41577-019-0269-6
107. Ni Q, Xu F, Wang Y, et al. Nanomaterials with changeable physicochemical property for boosting cancer immunotherapy. J Control Release. 2022;342:210–227. doi:10.1016/j.jconrel.2022.01.003
108. Han X, Alu A, Liu H, et al. Biomaterial-assisted biotherapy: a brief review of biomaterials used in drug delivery, vaccine development, gene therapy, and stem cell therapy. Bioact Mater. 2022;17:29–48. doi:10.1016/j.bioactmat.2022.01.011
109. Shahidi M, Abazari O, Dayati P, et al. Using chitosan-stabilized, hyaluronic acid-modified selenium nanoparticles to deliver CD44-targeted PLK1 siRNAs for treating bladder cancer. Nanomedicine. 2023;18(3):259–277. doi:10.2217/nnm-2022-0198
110. Pereira Santos Carvalho I, Bueno silva L, Luis Ferraz Do Amaral R, et al. Evaluation of in vivo and in vitro efficacy of solasonine/solamargine-loaded lipid-polymer hybrid nanoparticles against bladder cancer. Int J Pharm. 2024;661:124411. doi:10.1016/j.ijpharm.2024.124411
111. Parayath NN, Stephan SB, Koehne AL, Nelson PS, Stephan MT. In vitro-transcribed antigen receptor mRNA nanocarriers for transient expression in circulating T cells in vivo. Nat Commun. 2020;11(1):6080. doi:10.1038/s41467-020-19486-2
112. Kim H, Niu L, Larson P, et al. Polymeric nanoparticles encapsulating novel TLR7/8 agonists as immunostimulatory adjuvants for enhanced cancer immunotherapy. Biomaterials. 2018;164:38–53. doi:10.1016/j.biomaterials.2018.02.034
113. Winnicka A, Brzeszczyńska J, Saluk J, Wigner-Jeziorska P. Nanomedicine in bladder cancer therapy. Int J Mol Sci. 2024;25(19):10388. doi:10.3390/ijms251910388
114. Al-Shadidi J, Al-Shammari S, Al-Mutairi D, Alkhudhair D, Thu HE, Hussain Z. Chitosan nanoparticles for targeted cancer therapy: a review of stimuli-responsive, passive, and active targeting strategies. Int J Nanomed. 2024;19:8373–8400. doi:10.2147/IJN.S472433
115. Larsen ES, Joensen UN, Poulsen AM, Goletti D, Johansen IS. Bacillus Calmette-Guérin immunotherapy for bladder cancer: a review of immunological aspects, clinical effects and BCG infections. APMIS. 2020;128(2):92–103. doi:10.1111/apm.13011
116. Erdoğar N, Iskit AB, Eroğlu H, Sargon MF, Mungan NA, Bilensoy E. Antitumor efficacy of bacillus calmette-guerin loaded cationic nanoparticles for intravesical immunotherapy of bladder tumor induced rat model. J Nanosci Nanotechnol. 2015;15(12):10156–10164. doi:10.1166/jnn.2015.11690
117. Nakamura T, Fukiage M, Higuchi M, et al. Nanoparticulation of BCG-CWS for application to bladder cancer therapy. J Control Release. 2014;176:44–53. doi:10.1016/j.jconrel.2013.12.027
118. Brokesh AM, Gaharwar AK. Inorganic biomaterials for regenerative medicine. ACS Appl Mater Interfaces. 2020;12(5):5319–5344. doi:10.1021/acsami.9b17801
119. Jiang S, Lin J, Huang P. Nanomaterials for NIR-II photoacoustic imaging. Adv Healthcare Mater. 2023;12(16):e2202208. doi:10.1002/adhm.202202208
120. Xu C, Dobson HE, Yu M, et al. STING agonist-loaded mesoporous manganese-silica nanoparticles for vaccine applications. J Control Release. 2023;357:84–93. doi:10.1016/j.jconrel.2023.03.036
121. Li Q, Yang M, Sun X, et al. NIR responsive nanoenzymes via photothermal ablation and hypoxia reversal to potentiate the STING-dependent innate antitumor immunity. Mater Today Bio. 2023;19:100566. doi:10.1016/j.mtbio.2023.100566
122. Cao X, Yang C, Zhu X, et al. Synergistic enhancement of chemotherapy for bladder cancer by photothermal dual-sensitive nanosystem with gold nanoparticles and PNIPAM. Chin Chem Lett. 2024;35(8):109199. doi:10.1016/j.cclet.2023.109199
123. Wu T, Duan X, Hu C, et al. Synthesis and characterization of gold nanoparticles from Abies spectabilis extract and its anticancer activity on bladder cancer T24 cells. Artif Cells Nanomed Biotechnol. 2019;47(1):512–523. doi:10.1080/21691401.2018.1560305
124. Armanetti P, Locatelli I, Venegoni C, et al. Gold nanorod-assisted theranostic solution for nonvisible residual disease in bladder cancer. Proc Natl Acad Sci USA. 2024;121(37):e2411583121. doi:10.1073/pnas.2411583121
125. Peng L, Zhao A, Li R, et al. Self-propelled in situ polymerized nanoparticles activating the STING pathway for enhanced bladder cancer immunotherapy. Adv Sci. 2025;12(25):e2502750. doi:10.1002/advs.202502750
126. Guo P, Wang L, Shang W, et al. Intravesical In Situ Immunostimulatory Gel for Triple Therapy of Bladder Cancer. ACS Appl Mater Interfaces. 2020;12(49):54367–54377. doi:10.1021/acsami.0c15176
127. Terán-Navarro H, Zeoli A, Salines-Cuevas D, et al. Gold glyconanoparticles combined with 91-99 peptide of the bacterial toxin, Listeriolysin O, are efficient immunotherapies in experimental bladder tumors. Cancers. 2022;14(10):2413. doi:10.3390/cancers14102413
128. Lin T, Zhao X, Zhao S, et al. O(2)-generating MnO(2) nanoparticles for enhanced photodynamic therapy of bladder cancer by ameliorating hypoxia. Theranostics. 2018;8(4):990–1004. doi:10.7150/thno.22465
129. Deng W, Chen Y, Bai Y, et al. Genetically engineered T cell membrane-camouflaged nanoparticles triggered cuproptosis for synergistic bladder cancer photothermal-immunotherapy. J Nanobiotechnol. 2025;23(1):425. doi:10.1186/s12951-025-03511-x
130. Zeng YY, Gu Q, Li D, et al. Immunocyte membrane-derived biomimetic nano-drug delivery system: a pioneering platform for tumour immunotherapy. Acta Pharmacol Sin. 2024;45(12):2455–2473. doi:10.1038/s41401-024-01355-z
131. Zhang S, Chen W, Zhou Y, et al. Intelligent nanoplatform integrating macrophage and cancer cell membrane for synergistic chemodynamic/immunotherapy/photothermal therapy of breast cancer. ACS Appl Mater Interfaces. 2023;15(51):59117–59133. doi:10.1021/acsami.3c12560
132. Baek S, Jeon M, Jung HN, et al. M1 macrophage-derived exosome-mimetic nanovesicles with an enhanced cancer targeting ability. ACS Appl Bio Mater. 2022;5(6):2862–2869. doi:10.1021/acsabm.2c00246
133. Guo H, Li F, Qiu H, et al. Synergistically enhanced mucoadhesive and penetrable polypeptide nanogel for efficient drug delivery to orthotopic bladder cancer. Research. 2020;2020:8970135. doi:10.34133/2020/8970135
134. Hu Q, Li H, Archibong E, et al. Inhibition of post-surgery tumour recurrence via a hydrogel releasing CAR-T cells and anti-PDL1-conjugated platelets. Nat Biomed Eng. 2021;5(9):1038–1047. doi:10.1038/s41551-021-00712-1
135. Ashrafizadeh M, Zarrabi A, Karimi-Maleh H, et al. (Nano)platforms in bladder cancer therapy: challenges and opportunities. Bioeng Transl Med. 2023;8(1):e10353. doi:10.1002/btm2.10353
136. Zhang J, Zhou J, Tang L, et al. Custom-design of multi-stimuli-responsive degradable silica nanoparticles for advanced cancer-specific chemotherapy. Small. 2024;20(35):e2400353. doi:10.1002/smll.202400353
137. El-Tanani M, Satyam SM, Rabbani SA, et al. Revolutionizing drug delivery: the impact of advanced materials science and technology on precision medicine. Pharmaceutics. 2025;17(3):375. doi:10.3390/pharmaceutics17030375
138. Nejabat M, Samie A, Khojastehnezhad A, et al. Stimuli-responsive covalent organic frameworks for cancer therapy. ACS Appl Mater Interfaces. 2024;16(39):51837–51859. doi:10.1021/acsami.4c07040
139. Zhu Y, Bai Y, He J, Qiu X. Advances in the stimuli-responsive mesoporous silica nanoparticles as drug delivery system nanotechnology for controlled release and cancer therapy. 3 Biotech. 2023;13(8):274. doi:10.1007/s13205-023-03651-7
140. Miao Z, Li J, Zeng S, et al. Endoplasmic reticulum-targeting AIE photosensitizers to boost immunogenic cell death for immunotherapy of bladder carcinoma. ACS Appl Mater Interfaces. 2024;16(1):245–260. doi:10.1021/acsami.3c14068
141. Hunter BA, Eustace A, Irlam JJ, et al. Expression of hypoxia-inducible factor-1α predicts benefit from hypoxia modification in invasive bladder cancer. Br J Cancer. 2014;111(3):437–443. doi:10.1038/bjc.2014.315
142. Liang G, Han J, Xing D. Precise tumor photothermal therapy guided and monitored by magnetic resonance/photoacoustic imaging using A safe and pH-Responsive Fe(III) Complex. Complex. 2021;10(3):2001300.
143. Chen WH, Yu KJ, Jhou JW, et al. Glucose/glutathione co-triggered tumor hypoxia relief and chemodynamic therapy to enhance photothermal therapy in bladder cancer. ACS Appl Bio Mater. 2021;4(10):7485–7496. doi:10.1021/acsabm.1c00741
144. Zhang L, Wan -S-S, Li C-X, Xu L, Cheng H, Zhang X-Z. An adenosine triphosphate-responsive autocatalytic fenton nanoparticle for tumor ablation with self-supplied H2O2 and acceleration of Fe(III)/Fe(II) conversion. Nano Lett. 2018;18(12):7609–7618. doi:10.1021/acs.nanolett.8b03178
145. Hao Y, Chen Y, He X, et al. RGD peptide modified platinum nanozyme Co-loaded glutathione-responsive prodrug nanoparticles for enhanced chemo-photodynamic bladder cancer therapy. Biomaterials. 2023;293:121975. doi:10.1016/j.biomaterials.2022.121975
146. Simó C, Serra-Casablancas M, Hortelao AC, et al. Urease-powered nanobots for radionuclide bladder cancer therapy. Nat Nanotechnol. 2024;19(4):554–564. doi:10.1038/s41565-023-01577-y
147. Sun B, Hyun H, Li LT, Wang AZ. Harnessing nanomedicine to overcome the immunosuppressive tumor microenvironment. Acta Pharmacol Sin. 2020;41(7):970–985. doi:10.1038/s41401-020-0424-4
148. Aljabali AAA, Obeid MA, Gammoh O, et al. Nanomaterial-driven precision immunomodulation: a new paradigm in therapeutic interventions. Cancers. 2024;16(11):2030. doi:10.3390/cancers16112030
149. Zhang C, Zhao J, Wang W, Geng H, Wang Y, Gao B. Current advances in the application of nanomedicine in bladder cancer. Biomed Pharmacothe. 2023;157:114062. doi:10.1016/j.biopha.2022.114062
150. Mainini F, Eccles MR. Lipid and polymer-based nanoparticle siRNA delivery systems for cancer therapy. Molecules. 2020;25(11):2692. doi:10.3390/molecules25112692
151. Hao Y, Ji Z, Zhou H, et al. Lipid-based nanoparticles as drug delivery systems for cancer immunotherapy. MedComm. 2023;4(4):e339. doi:10.1002/mco2.339
152. Zhang T, Tian H, Qin S, et al. Nanoengineering Calcium Peroxide-Based Site-Specific Delivery Platform to Efficiently Activate the cGAS-STING Pathway for Cancer Immunotherapy by Amplified Endoplasmic Reticulum Stress. Adv Funct Mater. 2024;34(18):2313384.
153. Lv M, Shang S, Liu K, et al. Revitalizing bacillus calmette-guérin immunotherapy for bladder cancer: nanotechnology and bioengineering approaches. Pharmaceutics. 2024;16(8):1067. doi:10.3390/pharmaceutics16081067
154. Wang YS, Zheng AH, Zhao JW, et al. Anti-PD-L1 antibody retains antitumour effects while mitigating immunotherapy-related colitis in bladder cancer-bearing mice after CT-mediated intratumoral delivery. Int Immunopharmacol. 2024;137:112417. doi:10.1016/j.intimp.2024.112417
155. Wang W, Yang F, Zhang L, et al. Targeting DNA damage and repair machinery via delivering WEE1 inhibitor and platinum (IV) prodrugs to stimulate STING pathway for maximizing chemo-immunotherapy in bladder cancer. Adv Mater. 2024;36(1):e2308762. doi:10.1002/adma.202308762
156. Zhao Z, Ukidve A, Kim J, Mitragotri S. Targeting strategies for tissue-specific drug delivery. Cell. 2020;181(1):151–167. doi:10.1016/j.cell.2020.02.001
157. Yan G, Xiao Q, Zhao J, et al. Brucea javanica derived exosome-like nanovesicles deliver miRNAs for cancer therapy. J Control Release. 2024;367:425–440. doi:10.1016/j.jconrel.2024.01.060
158. Xie H, Zhan H, Gao Q, et al. Synthetic artificial “long non-coding RNAs” targeting oncogenic microRNAs and transcriptional factors inhibit malignant phenotypes of bladder cancer cells. Cancer Lett. 2018;422:94–106. doi:10.1016/j.canlet.2018.02.038
159. Crumrine N, Reda M, Kumar P, et al. Abstract A052: development of PD-L1 and STING-targeted nanoparticles for lung cancer treatment. Cancer Immunol Res. 2025;13(2_Supplement):A052. doi:10.1158/2326-6074.IO2025-A052
160. Fukushima H, Turkbey B, Pinto PA, Furusawa A, Choyke PL, Kobayashi H. Near-Infrared Photoimmunotherapy (NIR-PIT) in urologic cancers. Cancers. 2022;14(12):2996. doi:10.3390/cancers14122996
161. Huber F, Wolf I, Storz J, et al. Antibody-dye conjugates targeting EGFR and HER2 for the photoimmunotherapy of bladder cancer. Anticancer Res. 2024;44(5):1837–1844. doi:10.21873/anticanres.16985
162. Fukushima H, Takao S, Furusawa A, et al. Near-infrared photoimmunotherapy targeting Nectin-4 in a preclinical model of bladder cancer. Cancer Lett. 2024;585:216606. doi:10.1016/j.canlet.2023.216606
163. Won JE, Byeon Y, Wi TI, et al. Immune checkpoint silencing using RNAi-incorporated nanoparticles enhances antitumor immunity and therapeutic efficacy compared with antibody-based approaches. J Immunother Cancer. 2022;10(2):e003928. doi:10.1136/jitc-2021-003928
164. Su Z, Dong S, Chen Y, et al. Microfluidics-enabled nanovesicle delivers CD47/PD-L1 antibodies to enhance antitumor immunity and reduce immunotoxicity in lung adenocarcinoma. Adv Sci. 2023;10(20):e2206213. doi:10.1002/advs.202206213
165. Sun J. Nanoparticle therapies: targeted treatment for bladder cancer with reduced side effects [Letter]. Int J Nanomed. 2025;20:989–990. doi:10.2147/IJN.S513952
166. Wang Y-F, Shen Z-F, Xiang F-Y, Wang H, Zhang Q. Application of targeted drug delivery based on nano platform in diagnosis and treatment of bladder cancer. J Drug Delivery Sci Technol. 2023;87:104873. doi:10.1016/j.jddst.2023.104873
167. Roussot N, Ghiringhelli F, Rébé C. Tumor immunogenic cell death as a mediator of intratumor CD8 T-cell recruitment. Cells. 2022;11(22):3672. doi:10.3390/cells11223672
168. Jin M, Hou Y, Quan X, Chen L, Gao Z, Huang W. Smart polymeric nanoparticles with pH-Responsive and PEG-Detachable Properties (II): co-delivery of paclitaxel and VEGF siRNA for synergistic breast cancer therapy in mice. Int J Nanomed. 2021;16:5479–5494. doi:10.2147/IJN.S313339
169. Guo Y, Ma R, Zhang M, Cao Y, Zhang Z, Yang W. Nanotechnology-assisted immunogenic cell death for effective cancer immunotherapy. Vaccines. 2023;11(9):1440. doi:10.3390/vaccines11091440
170. Zeng X, Luo D, Zhang S, et al. High-dose radiation-induced immunogenic cell death of bladder cancer cells leads to dendritic cell activation. PLoS One. 2024;19(9):e0307024. doi:10.1371/journal.pone.0307024
171. Zhang W, Cai K, Sun Z, et al. Elevating second near-infrared photothermal conversion efficiency of hollow gold nanorod for a precise theranostic of orthotopic bladder cancer. ACS Nano. 2023;17(19):18932–18941. doi:10.1021/acsnano.3c04175
172. Wang J, Jin N, Xie Z, et al. Gold nanorods coated with self-assembled silk fibroin for improving their biocompatibility and facilitating targeted photothermal-photodynamic cancer therapy. Nanoscale. 2025;17(8):4624–4635. doi:10.1039/D4NR03641G
173. Lin Z, Nie F, Hou J, et al. Development of pH-responsive porphyran-coated gold nanorods for tumor photothermal and immunotherapy. Int J Biol Macromol. 2024;275:133460. doi:10.1016/j.ijbiomac.2024.133460
174. Duan X, Chan C, Lin W. Nanoparticle-mediated immunogenic cell death enables and potentiates cancer immunotherapy. Angew Chem. 2019;58(3):670–680. doi:10.1002/anie.201804882
175. Kim J, Choi Y, Yang S, et al. Sustained and long-term release of doxorubicin from PLGA nanoparticles for eliciting anti-tumor immune responses. Pharmaceutics. 2022;14(3):474.
176. Wang Z, Miao F, Gu L, et al. Stimulator of interferon genes-activated biomimetic dendritic cell nanovaccine as a chemotherapeutic booster to enhance systemic fibrosarcoma treatment. ACS Nano. 2024;18(35):24219–24235. doi:10.1021/acsnano.4c05657
177. Wu J, Chen Z, Shang H, et al. Genetically engineered hybrid membrane-coated nanoparticles enable triple activation of dendritic cells for synergistic bladder cancer immunotherapy. Chem Eng J. 2025;510:161652. doi:10.1016/j.cej.2025.161652
178. Choi Y, Yoon HY, Kim J, et al. Doxorubicin-loaded PLGA nanoparticles for cancer therapy: molecular weight effect of PLGA in doxorubicin release for controlling immunogenic cell death. Pharmaceutics. 2020;12(12):1165. doi:10.3390/pharmaceutics12121165
179. Ma C, Cheng Z, Tan H, et al. Nanomaterials: leading immunogenic cell death-based cancer therapies. Front Immunol. 2024;15:1447817. doi:10.3389/fimmu.2024.1447817
180. Zhu Z, Ma AH, Zhang H, et al. Phototherapy with cancer-specific nanoporphyrin potentiates immunotherapy in bladder cancer. Clin Cancer Res. 2022;28(21):4820–4831. doi:10.1158/1078-0432.CCR-22-1362
181. Liu J, Zhang Y, Yang B, et al. Synergistic glutathione depletion and STING activation to potentiate dendritic cell maturation and cancer vaccine efficacy. Angew Chem Int Ed. 2024;63(10):e202318530.
182. Chen L, Qin H, Zhao R, et al. Bacterial cytoplasmic membranes synergistically enhance the antitumor activity of autologous cancer vaccines. Sci trans med. 2021;13(601):eabc2816. doi:10.1126/scitranslmed.abc2816
183. Chen J, Qiao Y, Chen G, et al. Salmonella flagella confer anti-tumor immunological effect via activating Flagellin/TLR5 signalling within tumor microenvironment. Acta pharmaceutica Sinica B. 2021;11(10):3165–3177. doi:10.1016/j.apsb.2021.04.019
184. Patel RB, Ye M, Carlson PM, et al. Development of an in situ cancer vaccine via combinational radiation and bacterial-membrane-coated nanoparticles. Adv Mater. 2019;31(43):e1902626. doi:10.1002/adma.201902626
185. Deng G, Sun Z, Li S, et al. Cell-membrane immunotherapy based on natural killer cell membrane coated nanoparticles for the effective inhibition of primary and abscopal tumor growth. ACS Nano. 2018;12(12):12096–12108. doi:10.1021/acsnano.8b05292
186. Liu N, Zhu J, Zhu W, et al. X-ray-induced release of nitric oxide from hafnium-based nanoradiosensitizers for enhanced radio-immunotherapy. Adv Mater. 2023;35(29):e2302220. doi:10.1002/adma.202302220
187. Yu C, Wang S, Lai WF, Zhang D. The progress of chitosan-based nanoparticles for intravesical bladder cancer treatment. Pharmaceutics. 2023;15(1):211. doi:10.3390/pharmaceutics15010211
188. Ren W, Cai X, Chen J, et al. GM-CSF-loaded nanoparticles for photothermal-assisted immunotherapy against orthotopic bladder cancer. Oncologie. 2021;23(3):359–371. doi:10.32604/Oncologie.2021.018605
189. Zhou S, Huang Y, Chen Y, et al. Engineering ApoE3-incorporated biomimetic nanoparticle for efficient vaccine delivery to dendritic cells via macropinocytosis to enhance cancer immunotherapy. Biomaterials. 2020;235:119795. doi:10.1016/j.biomaterials.2020.119795
190. Hong E, Dobrovolskaia MA. Antigen-specific stimulation of CD8(+) T cells by murine bone marrow-derived dendritic cells after treatment with nanoparticles. Methods Mol Biol. 2024;2789:171–184.
191. Garris CS, Wong JL, Ravetch JV, Knorr DA. Dendritic cell targeting with Fc-enhanced CD40 antibody agonists induces durable antitumor immunity in humanized mouse models of bladder cancer. Sci trans med. 2021;13(594):eabd1346. doi:10.1126/scitranslmed.abd1346
192. Li C, Clauson R, Bugada LF, et al. Antigen-clustered nanovaccine achieves long-term tumor remission by promoting B/CD 4 T cell crosstalk. ACS Nano. 2024;18(13):9584–9604. doi:10.1021/acsnano.3c13038
193. Han J, Zhao Y, Shirai K, et al. Resident and circulating memory T cells persist for years in melanoma patients with durable responses to immunotherapy. Nat Cancer. 2021;2(3):300–311. doi:10.1038/s43018-021-00180-1
194. Snell LM, MacLeod BL, Law JC, et al. CD8(+) T cell priming in established chronic viral infection preferentially directs differentiation of memory-like cells for sustained immunity. Immunity. 2018;49(4):678–694.e675. doi:10.1016/j.immuni.2018.08.002
195. Choi H, Kim Y, Jung YW. The function of memory CD8+ T cells in immunotherapy for human diseases. Immun net. 2023;23(1):e10. doi:10.4110/in.2023.23.e10
196. Fujiki F, Morimoto S, Katsuhara A, et al. T cell-intrinsic vitamin A metabolism and its signaling are targets for memory T cell-based cancer immunotherapy. Front Immunol. 2022;13:935465. doi:10.3389/fimmu.2022.935465
197. Reina-Campos M, Heeg M, Kennewick K, et al. Metabolic programs of T cell tissue residency empower tumour immunity. Nature. 2023;621(7977):179–187. doi:10.1038/s41586-023-06483-w
198. Wang F, Su H, Wang Z, et al. Supramolecular filament hydrogel as a universal immunomodulator carrier for immunotherapy combinations. ACS Nano. 2023;17(11):10651–10664. doi:10.1021/acsnano.3c01748
199. Guo Z, Hu Y, Zhao M, et al. Prodrug-based versatile nanomedicine with simultaneous physical and physiological tumor penetration for enhanced cancer chemo-immunotherapy. Nano Lett. 2021;21(9):3721–3730. doi:10.1021/acs.nanolett.0c04772
200. Martomo SA, Lu D, Polonskaya Z, et al. Single-dose anti-PD-L1/IL-15 fusion protein KD033 generates synergistic antitumor immunity with robust tumor-immune gene signatures and memory responses. Mol Cancer Ther. 2021;20(2):347–356. doi:10.1158/1535-7163.MCT-20-0457
201. Batra SA, Rathi P, Guo L, et al. Glypican-3-specific CAR T cells coexpressing IL15 and IL21 have superior expansion and antitumor activity against hepatocellular carcinoma. Cancer Immunol Res. 2020;8(3):309–320. doi:10.1158/2326-6066.CIR-19-0293
202. Zhang Z, Zheng Y, Liu Y. Nanomaterials for regulating cancer immune responses. Sci Bull. 2021;66(18):2288–2302. doi:10.1016/j.scib.2021.06.020
203. Li G, Tao T, Deng D, et al. Collagen-targeted tumor-specific transepithelial penetration enhancer mediated intravesical chemoimmunotherapy for non-muscle-invasive bladder cancer. Biomaterials. 2022;283:121422. doi:10.1016/j.biomaterials.2022.121422
204. Liu F, Li K, Zhu Q. Targeting metabolic reprogramming in bladder cancer immunotherapy: a precision medicine approach. Biomedicines. 2025;13(5):1145.
205. Izawa M, Tanaka N, Murakami T, et al. Single-cell phenotyping of CD73 expression reveals the diversity of the tumor immune microenvironment and reflects the prognosis of bladder cancer. Lab Invest. 2023;103(4):100040. doi:10.1016/j.labinv.2022.100040
206. Zhang H, Lu Y, Chen X, et al. A new strategy to authenticate prognosis and tumor immunity model based on cancer-associated fibroblasts lncRNA in bladder cancer. J Biomed Nanotechnol. 2024;20:1169–1186. doi:10.1166/jbn.2024.3848
207. Wang Y, Chen Q, Luo Y, et al. Metabolic nanoregulators induce ferroptosis and change metabolite flow to reverse immunosuppressive tumor microenvironment. ACS Nano. 2024;18(51):34996–35012. doi:10.1021/acsnano.4c13425
208. Liu Q, Wang C, Zheng Y, et al. Virus-like nanoparticle as a co-delivery system to enhance efficacy of CRISPR/Cas9-based cancer immunotherapy. Biomaterials. 2020;258:120275. doi:10.1016/j.biomaterials.2020.120275
209. Dang Y, Guan J. Nanoparticle-based drug delivery systems for cancer therapy. Smart Mater Med. 2020;1:10–19. doi:10.1016/j.smaim.2020.04.001
210. Wang J, He Y, Hu F, et al. Metabolic reprogramming of immune cells in the tumor microenvironment. Int J Mol Sci. 2024;25(22):12223.
211. Li L, Tian Y. The role of metabolic reprogramming of tumor-associated macrophages in shaping the immunosuppressive tumor microenvironment. Biomed Pharmacother. 2023;161:114504.
212. Chen C, Song M, Du Y, et al. Tumor-associated-macrophage-membrane-coated nanoparticles for improved photodynamic immunotherapy. Nano Lett. 2021;21(13):5522–5531. doi:10.1021/acs.nanolett.1c00818
213. Li S, Wang Q, Shen Y, et al. Pseudoneutrophil cytokine sponges disrupt myeloid expansion and tumor trafficking to improve cancer immunotherapy. Nano Lett. 2020;20(1):242–251. doi:10.1021/acs.nanolett.9b03753
214. Lu Q, Kou D, Lou S, et al. Nanoparticles in tumor microenvironment remodeling and cancer immunotherapy. J hematol oncol. 2024;17(1):16.
215. Ma J, Yuan H, Zhang J, et al. An ultrasound-activated nanoplatform remodels tumor microenvironment through diverse cell death induction for improved immunotherapy. J Control Release. 2024;370:501–515. doi:10.1016/j.jconrel.2024.05.001
216. Parveen S, Konde DV, Paikray SK, et al. Nanoimmunotherapy: the smart trooper for cancer therapy. Explor Target Anti Tumor Ther. 2025;6:1002308. doi:10.37349/etat.2025.1002308
217. Dong X, Wu Z, Li X, et al. The size-dependent cytotoxicity of amorphous silica nanoparticles: a systematic review of in vitro studies. Int J Nanomed. 2020;15:9089–9113. doi:10.2147/IJN.S276105
218. Liu N, Wang X, Wang Z, et al. Nanomaterials-driven in situ vaccination: a novel frontier in tumor immunotherapy. J hematol oncol. 2025;18(1):45.
219. Ramos B, Rogers D. Immunotherapies to nano-immunotherapies: advances in immune targeting in bladder cancer. Ann Urol Oncol. 2025;8(1):1–11. doi:10.32948/auo.2025.02.01
220. Li F, Wang Y, Chen D, Du Y. Nanoparticle-based immunotherapy for reversing T-cell exhaustion. Int J Mol Sci. 2024;25(3):1396. doi:10.3390/ijms25031396
221. Hsiao CH, Lin YW, Liu CH, Nguyen HT, Chuang AE. Light-driven green-fabricated artificial intelligence-enabled micro/nanorobots for multimodal phototherapeutic management of bladder cancer. Adv Healthcare Mater. 2024;13(32):e2402864. doi:10.1002/adhm.202402864
222. Livingston NK, Hickey JW, Sim H, et al. In vivo stimulation of therapeutic antigen-specific T cells in an artificial lymph node matrix. Adv Mater. 2024;36(23):e2310043. doi:10.1002/adma.202310043
223. Angelopoulou A. Nanostructured biomaterials in 3D tumor tissue engineering scaffolds: regenerative medicine and immunotherapies. Int J Mol Sci. 2024;25(10):5414. doi:10.3390/ijms25105414
224. Singh R, Sharma A, Saji J, Umapathi A, Kumar S, Daima HK. Smart nanomaterials for cancer diagnosis and treatment. Nano Converg. 2022;9(1):21. doi:10.1186/s40580-022-00313-x
225. Zhu R, Zhang F, Peng Y, Xie T, Wang Y, Lan Y. Current progress in cancer treatment using nanomaterials. Front Oncol. 2022;12:930125. doi:10.3389/fonc.2022.930125
226. Cheng Z, Li M, Dey R, Chen Y. Nanomaterials for cancer therapy: current progress and perspectives. J hematol oncol. 2021;14(1):85.
227. Shang H, Xia D, Geng R, et al. Thermosensitive resiquimod-loaded lipid nanoparticles promote the polarization of tumor-associated macrophages to enhance bladder cancer immunotherapy. ACS Nano. 2025;19(21):19599–19621. doi:10.1021/acsnano.4c17444
228. Fu Y, Zhang Y, Zhang Y, et al. Nanoreactors with cascade catalytic activity reprogram the tumor microenvironment for enhanced immunotherapy by synchronously regulating treg and macrophage cells. ACS Appl Mater Interfaces. 2024;16(37):49053–49068. doi:10.1021/acsami.4c09830
229. Lopez-Beltran A, Cookson MS, Guercio BJ, Cheng L. Advances in diagnosis and treatment of bladder cancer. BMJ. 2024;384:e076743. doi:10.1136/bmj-2023-076743
230. Alonso JCC, Souza BRD, Leme G, et al. Crosstalk among T-cell CX3CR1, immune checkpoints and toll-like receptor 4 signaling pathway in BCG-unresponsive nonmuscle invasive bladder cancer: mechanism of action of OncoTherad Nano-Immunotherapy. J Clin Oncol. 2022;40(16_suppl):e16551. doi:10.1200/JCO.2022.40.16_suppl.e165
231. Catalán J, Norppa H. Safety aspects of bio-based nanomaterials. Bioengineering. 2017;4(4):94. doi:10.3390/bioengineering4040094
232. Ge Y, Wu L, Mei S, Wu J. Nanomaterials: promising tools for the diagnosis and treatment of myocardial infarction. Int J Nanomed. 2025;20:1747–1768. doi:10.2147/IJN.S500146
233. Tomlinson B, Lin TY, Dall’Era M, Pan CX. Nanotechnology in bladder cancer: current state of development and clinical practice. Nanomedicine. 2015;10(7):1189–1201. doi:10.2217/nnm.14.212
234. Grausenburger R, Herek P, Shariat SF, Englinger B. Recent contributions of single-cell and spatial profiling to the understanding of bladder cancer. Curr Opin Urol. 2024;34(4):236–243. doi:10.1097/MOU.0000000000001183
235. Wu J, Gao F, Meng R, et al. Single-cell and multi-omics analyses highlight cancer-associated fibroblasts-induced immune evasion and epithelial mesenchymal transition for smoking bladder cancer. Toxicology. 2024;504:153782. doi:10.1016/j.tox.2024.153782
236. Ye F, Ye C, Zhao Y, et al. Engineered Cu-VT composite nanoparticles induce copper-dependent cell death in bladder cancer: insights from single-cell spatial transcriptomics. Adv Compos Hybrid Mater. 2024;8(1):81. doi:10.1007/s42114-024-01153-5
237. Chen Z, Deng K, Sun L, et al. 3D laparoscopic treatment of bladder cancer with pelvic multi-organ invasion: a case report and literature review. Front Oncol. 2023;13:1249389. doi:10.3389/fonc.2023.1249389
238. Mazonakis M, Lyraraki E, Tolia M, Damilakis J. Risk for second bladder and rectal malignancies from cervical cancer irradiation. J Appl Clin Med Phys. 2021;22(7):103–109. doi:10.1002/acm2.13274
239. Wei Z, Zhuo S, Zhang Y, et al. Machine learning reshapes the paradigm of nanomedicine research. Acta Pharmaceutica Sinica B. 2025. doi:10.1016/j.apsb.2025.05.014
240. Gong D, Ben-Akiva E, Singh A, et al. Machine learning guided structure function predictions enable in silico nanoparticle screening for polymeric gene delivery. Acta Biomater. 2022;154:349–358. doi:10.1016/j.actbio.2022.09.072
241. Ortiz-Perez A, Van Tilborg D, van der Meel R, Grisoni F, Albertazzi L. Machine learning-guided high throughput nanoparticle design. Digit Discov. 2024;3(7):1280–1291. doi:10.1039/d4dd00104d
242. Rahman T, Tahmid A, Arman SE, et al. Leveraging generative neural networks for accurate, diverse, and robust nanoparticle design. Nanoscale Adv. 2025;7(2):634–642. doi:10.1039/D4NA00859F
243. Kuppe C, Klümper N, Goni O, et al. Abstract 768: decode the antibody-drug conjugate surfaceome in metastatic urothelial cancer (DECODE-ADC). Cancer Res. 2025;85(8_Supplement_1):768. doi:10.1158/1538-7445.AM2025-768
244. Ren C, Li Y, Cong Z, Li Z, Xie L, Wu S. Bioengineered bacterial outer membrane vesicles encapsulated Polybia-mastoparan I fusion peptide as a promising nanoplatform for bladder cancer immune-modulatory chemotherapy. Front Immunol. 2023;14:1129771. doi:10.3389/fimmu.2023.1129771
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Immunotherapy for Urothelial Carcinoma: Focus on Clinical Utility of Nivolumab
Chiang RS, Glover MJ, Khaki AR, Srinivas S
OncoTargets and Therapy 2022, 15:1259-1269
Published Date: 20 October 2022
Targeted Treatment of Advanced Endometrial Cancer: Focus on Pembrolizumab
El-ghazzi N, Durando X, Giro A, Herrmann T
OncoTargets and Therapy 2023, 16:359-369
Published Date: 2 June 2023
Systemic Treatment-Decision Algorithms in Muscle-Invasive Bladder Cancer: Clinical Complexities and Navigating for Improved Outcomes
Giles M, Crabb SJ
Research and Reports in Urology 2023, 15:321-331
Published Date: 7 July 2023
Integrating Bulk and Single-Cell RNA Sequencing Reveals Heterogeneity, Tumor Microenvironment, and Immunotherapeutic Efficacy Based on Sialylation-Related Genes in Bladder Cancer
Tan Z, Chen X, Zuo J, Fu S, Wang J, Wang H
Journal of Inflammation Research 2023, 16:3399-3417
Published Date: 14 August 2023
CD69 is a Promising Immunotherapy and Prognosis Prediction Target in Cancer
Li Y, Gu Y, Yang P, Wang Y, Yu X, Li Y, Jin Z, Xu L
ImmunoTargets and Therapy 2024, 13:1-14
Published Date: 9 January 2024