Back to Journals » Hepatic Medicine: Evidence and Research » Volume 15
Disulfidptosis-Associated lncRNAs are Potential Biomarkers for Predicting Immune Response and Prognosis Within Individuals Diagnosed with Hepatocellular Carcinoma
Authors Wei Q ![]() , Hou YC, Mao FF
, Hou YC, Mao FF ![]() , Feng JK, Wang X, Cheng SQ
, Feng JK, Wang X, Cheng SQ
Received 16 August 2023
Accepted for publication 12 December 2023
Published 27 December 2023 Volume 2023:15 Pages 249—264
DOI https://doi.org/10.2147/HMER.S435726
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Gerry Lake-Bakaar
Qian Wei,1,2,* Yu-Chao Hou,3,* Fei-Fei Mao,4,* Jin-Kai Feng,2 Xu Wang,3 Shu-Qun Cheng1,2,*
1The First Clinical Medicine School, Guangdong Pharmaceutical University, Guangzhou, People’s Republic of China; 2Department of Hepatic Surgery VI, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, Shanghai, People’s Republic of China; 3Cancer Center, Yueyang Hospital of Integrated Traditional Chinese and Western Medicine, Shanghai University of Traditional Chinese Medicine, Shanghai, People’s Republic of China; 4Tongji University Cancer Center, Shanghai 10th People’s Hospital, School of Medicine, Tongji University, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Shu-Qun Cheng, The First Clinical Medicine School, Guangdong Pharmaceutical University, 19 Nonglin Xia Road, Guangzhou, 510080, People’s Republic of China, Email [email protected]
Purpose: Hepatocellular carcinoma (HCC) is a prevalent form of cancer that is distributed globally. Disulfidptosis, characterized by the fragility of the actin cytoskeleton, represents a distinct type of cell death and holds promise for novel cancer therapies. Nevertheless, the connection among disulfidptosis-associated long non-coding RNAs (lncRNAs) and HCC is still unexplored. This study uses an in silico approach to provide the novel biomarkers of disulfidptosis-associated lncRNAs for predicting the immune response and prognosis with HCC.
Methods: In order to address this gap, we integrated transcriptomic data of HCC from The Cancer Genome Atlas (TCGA) and identified genes that exhibit differential expression with disulfidptosis and lncRNAs. Through co-expression analysis, we identified disulfidptosis-related lncRNAs. Afterwards, by employing univariate Cox regression analysis and the least absolute shrinkage and selection operator (LASSO), a model for disulfidptosis-associated lncRNA was constructed. The risk model underwent assessment through the utilization of diverse analytical methodologies, including functional enrichment annotation, Kaplan-Meier analysis, principal component analysis (PCA), immune infiltration and immune status analysis, as well as tumor mutation analysis. Furthermore, we discussed the implications of the model in predicting drug sensitivity.
Results: Our study culminated in the construction of a disulfidptosis-related lncRNA model comprising four prognostic disulfidptosis-related lncRNAs (ACYTOR, NRAV, AL080248.1, and AC069307.1). This model demonstrates exceptional diagnostic value for HCC patients and holds practical implications for guiding clinicians in personalizing immunotherapy and drug selection based on individual variations.
Conclusion: In summary, our research introduces a novel predictive tool utilizing disulfidptosis-related lncRNAs, offering potential guidance for the therapeutic management of HCC.
Keywords: disulfidptosis, hepatocellular carcinoma, long noncoding RNA, prognosis, immune response
Introduction
Liver malignancy is recognized as the sixth most commonly occurring malignant tumor globally and holds the position of the 4th primary reason for cancer-associated mortality.1,2 Liver cancer comprises largely of two primary histologic types: hepatocellular carcinoma (HCC) and cholangiocarcinoma (CCA), with HCC constituting nearly 80% of cases worldwide.3 The majority of cases of primary liver cancer are attributed to HCC and can be attributed to various factors such as chronic viral hepatitis, alcoholism, non-alcoholic fatty liver disorders, and food toxins exposure like aflatoxins.4 In addition to surgery, immunotherapy, immune checkpoint blockers (ICBs), and transcatheter arterial chemoembolization (TACE) are used in the clinical management of HCC patients with different stages.5 Despite advancements in the diagnosis and management of HCC, the prognosis of HCC patients, especially those with advanced HCC, is still poor due to high rates of recurrence and drug resistance. Hence, it is crucial to investigate the underlying processes of tumorigenesis and proliferation, enabling the development of prognostic models and targeted therapies.
Numerous studies have linked different forms of cell death to cancer prevention. For instance, in genetically modified mice, increased vulnerability to tumor-selective ferroptosis has been shown to impede the development and spread of pancreatic cancer.6,7 Recently, a study published in Nature Cell Biology introduced the concept of “disulfidptosis”, a type of cell apoptosis induced via disulfide stress.8 Disulfide stress occurs due to an abnormal accumulation of intracellular disulfides, particularly cystine, which can be highly toxic to cells.9,10 To counter disulfide stress and maintain cell viability, reduced nicotinamide adenine dinucleotide phosphate (NADPH) offers essential redox capabilityT.8 The cytoplasmic NADPH pool is produced through the pentose phosphate pathway, primarily fueled by glucose.8 The overexpression of solute carrier family 7 member 11 (SLC7A11; frequently denoted as xCT), is commonly observed in cancer cells, a cysteine transporter that enhances the uptake of cystine and diminishes its conversion into cysteine. When combined with glucose deprivation, this leads to NADPH depletion, resulting in an intracellular disulfide compound excessive buildup and prompt cell apoptosis.9,10 Disulfidoptosis refers to a type of cellular death that was previously unrecognized, is a result of abnormal intracellular disulfides accumulation within cells with elevated levels of SLC7A11, when subjected to conditions of glucose deprivation.8 The recognition and classification of cellular death pathways not only improve our comprehension of intracellular homeostasis but also provide pivotal perspectives into the management of many different diseases, including cancer.11 However, the connection among disulfidptosis and HCC is still demonstrating poor comprehension.
LncRNAs are a class of RNA molecules that possess a length exceeding 200 nucleotides and are known to exert significant influence on the occurrence of numerous diseases.12–14 In cancer, lncRNAs participate in a wide range of biological pathways, including DNA damage, epigenetic regulation, metabolic disorders, chemoresistance, immune evasion, epithelial-mesenchymal transition (EMT), and cell stemness, as evidenced by numerous studies.15 These findings emphasize the prospective utility of lncRNAs as novel indicators and targets for therapeutic intervention in the management of cancer. Although the abundance of research conducted on the function of lncRNAs in HCC progression, recurrence, immunotherapy response, and various biological processes including m6A methylation, hypoxia, ferroptosis, autophagy, and energy metabolism,16–20 fewer investigations have explored the involvement of lncRNAs in disulfidptosis. Thus, investigating the properties of disulfidptosis-related lncRNAs and their implications for HCC diagnosis and treatment would be valuable.
In this study, our objective was to develop a disulfidptosis-related lncRNA model with the capability to predict prognosis, Kaplan-Meier analysis, PCA, tumor immune infiltration, and tumor mutation burden (TMB) in HCC. In order to accomplish this, the expression profiles of 16,876 lncRNAs and 10 disulfidptosis-related genes were provided by the TCGA dataset. Through Pearson correlation analysis, we successfully identified lncRNAs related to disulfidptosis. Subsequently, a novel prognostic model has been established for disulfidptosis to anticipate OS in HCC individuals. Lastly, by leveraging publicly accessible drug sensitivity databases, we identified potential drug candidates that specifically target the lncRNA signature associated with disulfidptosis.
Materials and Methods
Information Gathering About HCC Patients
On 17 March 2023, we obtained clinical data and RNA sequencing information related to HCC patients using the TCGA database, containing 50 normal tissues and 374 HCC tissues (https://portal.gdc.cancer.gov/). We then screened 10 genes associated with disulfidptosis from previous literature, including NDUFS1, GYS1, LRPPRC, OXSM, NDUFA11, NUBPL, NCKAP1, RPN1, SLC3A2, and SLC7A11. Clinical data on patients with HCC was also extracted as well, which included variables such as gender, age, stage, tumor-node-metastasis (TNM) stage, tumor differentiation grade, survival status, and survival time. To ensure statistical integrity, HCC patients without available overall OS data were not included in the investigation. We summarized the clinical characteristics of 371 individuals (Table 1). Consequently, we processed the dataset using the Strawberry Perl software and ultimately extracted 19,938 mRNAs and 16,876 lncRNAs.
 |
Table 1 The Clinical Characteristics of HCC Patients in the TCGA Dataset |
Selection of Genes and lncRNAs Associated with Disulfidptosis
Based on a comprehensive review of relevant literature, we identified 10 genes known to be associated with disulfidptosis, namely NDUFS1, GYS1, LRPPRC, OXSM, NDUFA11, NUBPL, NCKAP1, RPN1, SLC3A2, and SLC7A11. To identify disulfidptosis-associated lncRNAs, we performed Pearson correlation analysis with specific parameters: correlation coefficient = 0.4 and p-value< 0.001. Visualization of the relationship between disulfidptosis-associated genes and lncRNAs was accomplished utilizing the “dplyr”, “ggalluvial”, and “ggplot2” packages, which allowed us to generate Sankey diagrams for clear representation.
The Risk Signature Development and Verification
We randomly divided the complete TCGA dataset into a training set and a test set and used both sets of data to develop and validate a model for disulfidptosis-associated lncRNAs. There were no statistically significant variations detected in the clinical features among the training and testing groups (p > 0.05). We used Cox regression analyses, including univariate Cox regression analyses for the assessment of prognostic significance, and the R software packages “survival”, “caret”, “survminer”, and “timeROC” for analyses utilizing On the TCGA dataset, we used univariate Cox regression to analyze the predictive relevance of 317 disulfidptosis-associated lncRNAs (p < 0.05). We also used the “glmnet” package in R to do LASSO Cox regression analysis and found that the four lncRNAs related to disulfide were strongly linked to OS in people who had been diagnosed with HCC (calculated using penalty parameters from 10-fold cross-validation). By multifactorial Cox regression analysis, we analysed the relationship between four disulfidptosis-associated lncRNAs and OS. We calculate the risk score using the following formula:
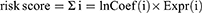
Coef (i) and Expr (i) demonstrate the regression coefficient and a specific level of lncRNA expression, respectively. Based on the median risk score derived from the lncRNA model, we categorised the study participants into two groups: low and high risk.
PCA and Kaplan-Meier Survival Analysis
We reduced the dimensionality of the gene expression profiles using the PCA method, which included the full set of gene expression data, 10 disulfide genes, and 317 disulfidptosis-associated lncRNAs, and the risk model relied on the 317 lncRNAs expression patterns. We compared the OS of patients in the high-risk group with those in the low-risk group using Kaplan-Meier analysis and performed it using the R software packages “survMiner” and “survival”.
Functional Analysis
For greater understanding, the study conducted pathway analysis and functional annotation on a set of differentially expressed genes (DEGs) utilizing various tools such as Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), and gene set enrichment analysis (GSEA). GO analysis was performed to recognize DEGs in the high- and low-risk groups utilizing packages as “clusterProfiler”, “org.Hs.eg.db”, “enrichplot”, “ggplot2”, “circlize”, “RColorBrewer”, “dplyr”, “ComplexHeatmap”, and “ggpubr”. The study conducted KEGG analysis to determine the pathways linked to the high- and low-risk groups utilizing packages such as “clusterProfiler”, “org.Hs.eg.db”, “enrichplot”, “ggplot2”, “circlize”, “RColorBrewer”, and “dplyr”. Using programs such as “limma”, “org.Hs.eg.db”, “clusterProfiler”, and “enrichplot”, We performed GSEA analyses to identify the top 5 pathways in different gene sets. P-value: statistically significant level of enrichment analysis; in general, p-value < 0.05 change function to p-value corrected for enrichment term p.adjust. Q-value: q-value for a statistical test of p-value.
Analysing the Model in Immunotherapeutic Therapy
We used the R package “maftools” to analyse and summarise the mutation data and calculate the TMB by analysing the genes specifically altered in tumor cells. We used the Tumor Immune Dysfunction and Exclusion (TIDE) approach to assess the likelihood of response to immunotherapeutic drugs and visualised the results using software packages such as “ggpubr” and “limma”.
Independence of the lncRNA Model Connected to Disulfidptosis
The study conducted both univariate and multivariate Cox regression analyses to assess the autonomy of the lncRNA model in the context of diverse clinical features, including age, gender, grade, and stage, in individuals suffering from HCC. Through the establishment of nomograms and calibration, we assessed the accuracy of the models in predicting prognosis using the software packages “survival”, “regplot”, “rms”, and “survcomp”. To identify potential drugs for the treatment of HCC patients using the disulfidptosis-related lncRNAs model, we determined the half-maximal inhibitory concentration (IC50) of compounds downloaded from the GDSC website using the TCGA HCC dataset. We used R packages such as “limma”, “ggplot2”, and “ggpubr” to predict IC50 data in patients diagnosed with HCC downloaded from the GDSC website.
Statistical Analysis
For statistical analyses, we used the R programming language version 4.2.2 and Strawberry Perl version 5.30.0. A p-value of less than 0.05 is considered statistically significant, unless we state otherwise.
Results
Determination of HCC-Associated IncRNAs with Variable Disulfidptosis Expression
Flow chart illustrating the process of establishing the risk model and conducting further analysis (Figure 1). To identify HCC-related lncRNAs with variable expression related to disulfidptosis, a Pearson correlation analysis was conducted, utilizing a correlation coefficient threshold of 0.4 and a p-value threshold of 0.001. This examination led to the discovery of 317 disulfidptosis-related lncRNAs. The Sankey diagram visualizes the co-expression patterns between the genes connected to disulfidptosis and the disulfidptosis-associated lncRNAs (Figure 2A).
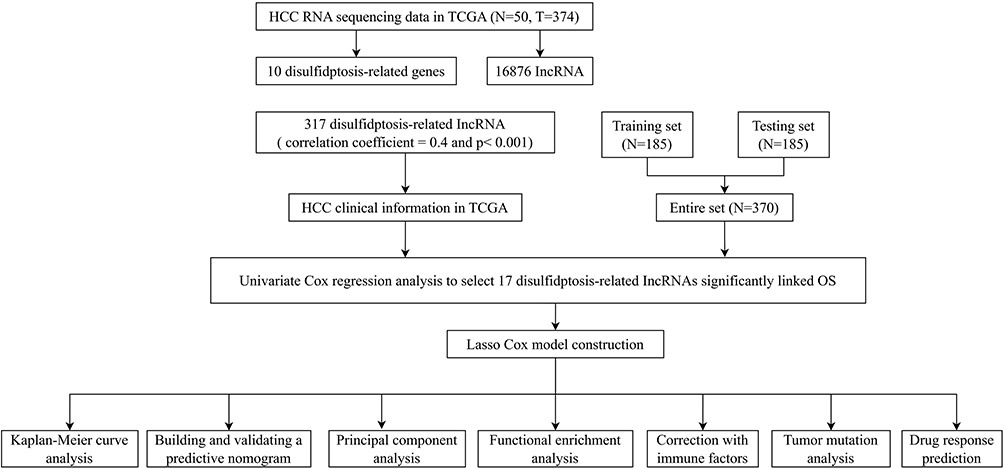 |
Figure 1 Flow chart of this study. |
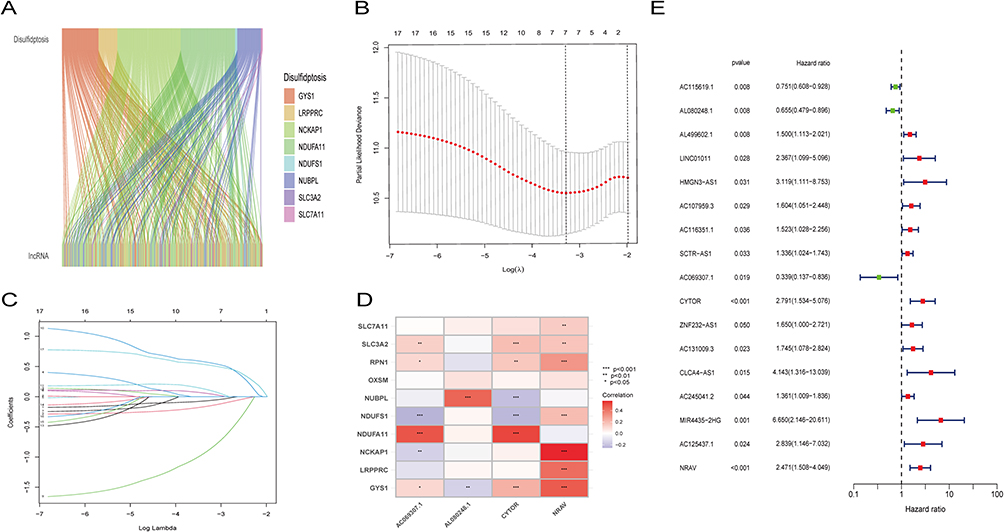 |
Figure 2 Creation of a lncRNA predictive signature associated with disulfidptosis. (A)Sankey diagram displaying the co-expression of genes and lncRNAs associated with disulfidptosis. (B) LASSO coefficient profiles of the disulfidptosis-related abnormally expressed lncRNAs in HCC samples. (C) Selection of the ideal parameter (λ) in the LASSO model. (D) Heatmap showing the co-expression of disulfide death-related genes and bisulfide death survival-related lncRNAs (*p < 0.05, **p<0.01,***p<0.001).(E) Forest plots representing the results of the univariate Cox regression analysis of the abnormally expressed lncRNAs linked to disulfidptosis. |
Creation of a lncRNA Predictive Signature Associated to Disulfidptosis
To develop a prognostic signature for disulfidptosis, the TCGA training set was employed to perform a univariate Cox regression analysis on 317 disulfidptosis-associated lncRNAs. Among them, 17 lncRNAs showed significant association towards OS. LASSO-penalized Cox analysis was then employed in order to refine the model, considering regularization and variable selection (Figures 2B and C). Based on this analysis, a risk model was constructed using four key disulfidptosis-related lncRNAs: CYTOR, NRAV, AL080248.1, and AC069307.1. Figure 2D illustrates the correlation between these four lncRNAs and the ten genes connected with disulfidptosis. The forest plot displays the confidence intervals and hazard ratios for the association between the 17 disulfidptosis-related lncRNAs and OS within the TCGA dataset (Figure 2E).
Evaluation of the HCC lncRNA Signature Associated with Disulfidptosis
A chi-square test was utilized in order to make a comparison of the clinical features within the training and test groups. The outcomes indicated that there were no significant variations identified among the two groups (p > 0.05). The outcomes suggest that individuals categorized as low-risk group exhibited a notably superior OS outcome in comparison to their high-risk counterparts within the high-risk group. This outcome was consistent across those of the training, testing, and combined sets (p<0.001, p=0.005, p<0.001; Figure 3J–L). The dot plots displayed the risk scores of individuals with HCC within the training, test, and combined sets, along with their corresponding survival status (Figure 3A–F). The investigation revealed that the results of the high-risk group exhibited a significantly elevated incidence of mortality. The four key disulfidptosis-associated lncRNAs expression levels (CYTOR, NRAV, AL080248.1, and AC069307.1) were examined within the high- and low-risk groups. The study demonstrated that the outcomes of the high-risk group indicated increased levels of CYTOR and NRAV expression, indicating that these lncRNAs could potentially function as unfavorable prognostic indicators. Conversely, the high-risk group findings exhibited decreased AL080248.1 and AC069307.1 expression levels, indicating their potential as protective markers (Figure 3G–I).
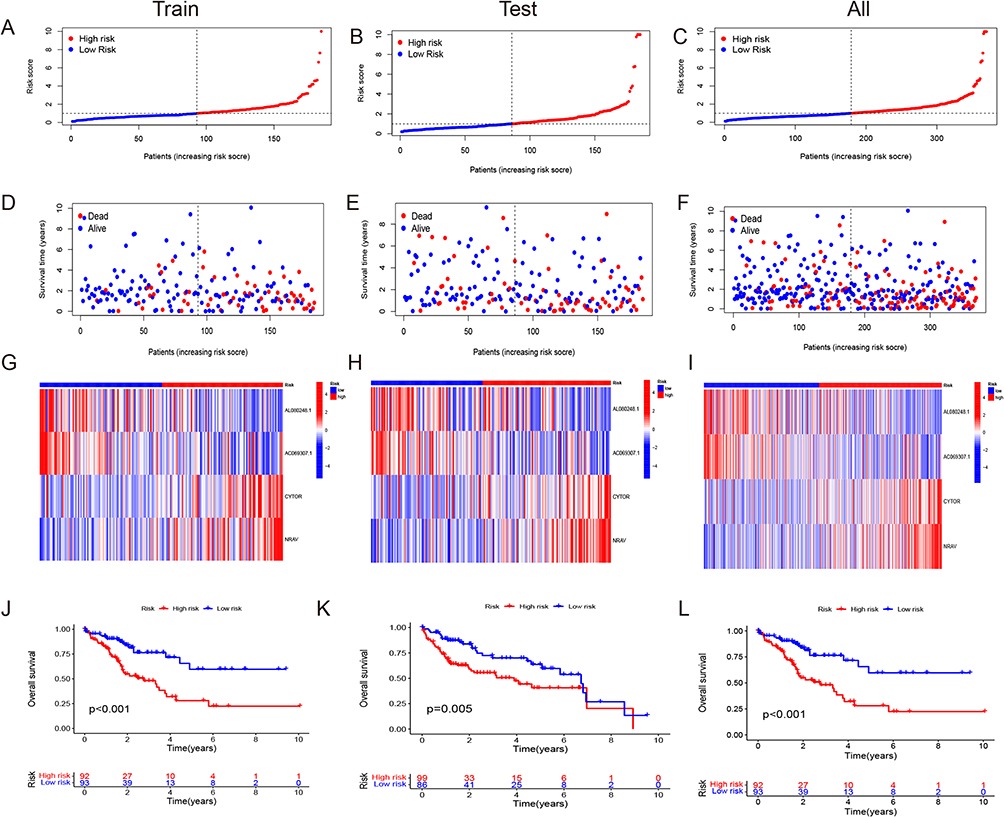 |
Figure 3 The predictive value of HCC is being evaluated and validated in testing, training, and all sets. (A–C) Increasing risk scores are shown in the patient distribution. (D–F) Patient survival rates and risk levels. (G–I) The heat map shows the expression of four important lncRNAs with rising risk scores. (J–L) OS of HCC patients were compared between high- and low-risk sets using the Kaplan-Meier method. |
Receiver operating characteristic (ROC) curves were established to compare the prognostic performance of the risk score with clinicopathological features. The area under the curve (AUC) for the risk score at 1-, 3-, and 5-year survival times were 0.669, 0.660, and 0.711, independently, demonstrating the prognostic performance of the lncRNA signature (Figure 4A). The risk and stage scores showed larger AUC values compared to other clinicopathological features (Figure 4B). The risk score’s concordance index exhibited consistent superiority over other clinical indicators, demonstrating its better prognostic accuracy (Figure 4C). Furthermore, individuals who exhibited greater risk scores demonstrated poorer progression-free survival (PFS) in comparison to individuals with lower risk scores (p < 0.001; Figure 4D). Furthermore, survival curves were plotted for different clinical stages (stages I–II and stages III–IV), showing a statistically significant variance among the high- and low-risk groups in both stage categories (p < 0.001 for stages I–II; p = 0.029 for stages III–IV). Individuals who were identified as high-risk patients exhibited a greater probability of early mortality in contrast with individuals who were categorized as low-risk (Figure 4E and F). Overall, the evaluation of the lncRNA signature associated with disulfidptosis demonstrated its reliable and favorable prognostic assessment capacity in HCC, even in the earliest and most advanced phases of the disease.
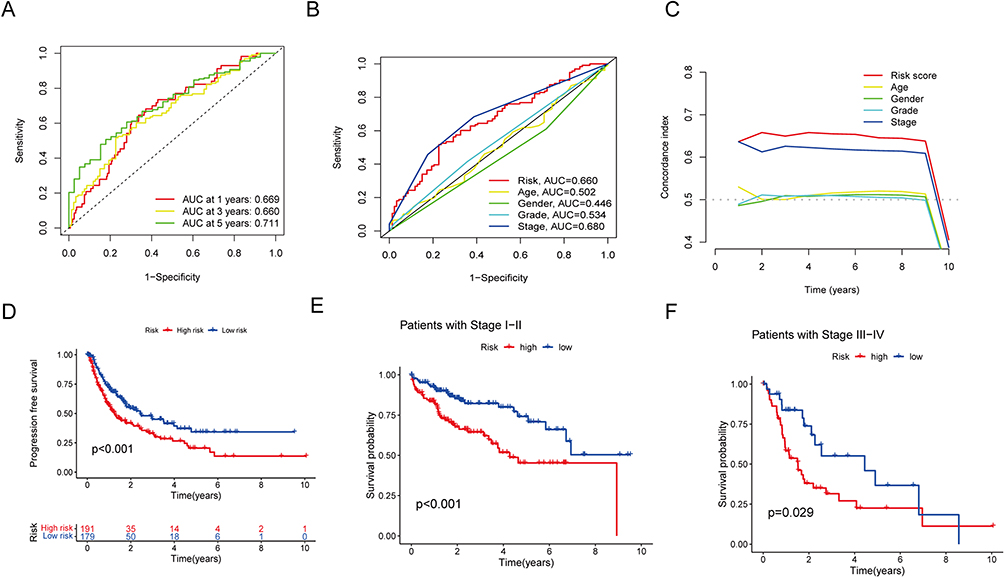 |
Figure 4 Evaluation of the disulfidptosis-related lncRNAs’ predictive risk model and the clinical characteristics of HCC in the full TCGA dataset. (A) This prognostic model’s sensitivity and specificity are evaluated using a time-dependent ROC analysis. (B) Risk score and clinical characteristics ROC curves. (C) C-index for risk assessment and clinical features (D) The progression-free survival for the entire HCC patient population. (E and F) The Kaplan–Meier contours for low- and high-risk HCC patients in stages I–II (E) and stages III–IV (F). |
The Independence of the Disulfidptosis-Associated lncRNAs Signature in Terms of Prognosis
The investigation conducted both univariate and multivariate Cox regression analyses to evaluate the predicted value of the lncRNA signature associated with disulfidptosis in terms of its independence, considering other clinical factors. The results confirmed the predictive independence of the lncRNA signature with respect to the OS in the entire dataset. The univariate Cox analysis revealed a hazard ratio (HR) of 1.116 (95% CI: 1.060–1.1176), and the multivariate Cox analysis revealed that HR results were 1.099 (95% CI: 1.031–1.171), demonstrating a significant correlation between the lncRNA signature and OS (Figure 5A and B). Similarly, stage has been recognized as an independent predictive indicator of other variables, with an HR of 1.680 (95% CI: 1.369–2.062) in the univariate analysis and an HR of 1.676 (95% CI: 1.359–2.066) within the multivariate analysis (Figure 5A and B). A prognostic nomogram has been developed utilizing the signature risk score and clinical characteristics of the entire dataset to enable OS quantitative analysis in individuals suffering from HCC (Figure 5C). This nomogram provides a visual instrument to anticipate the likelihood of 1-, 3-, and 5-year OS for each individual (*p<0.05; Figure 5D).
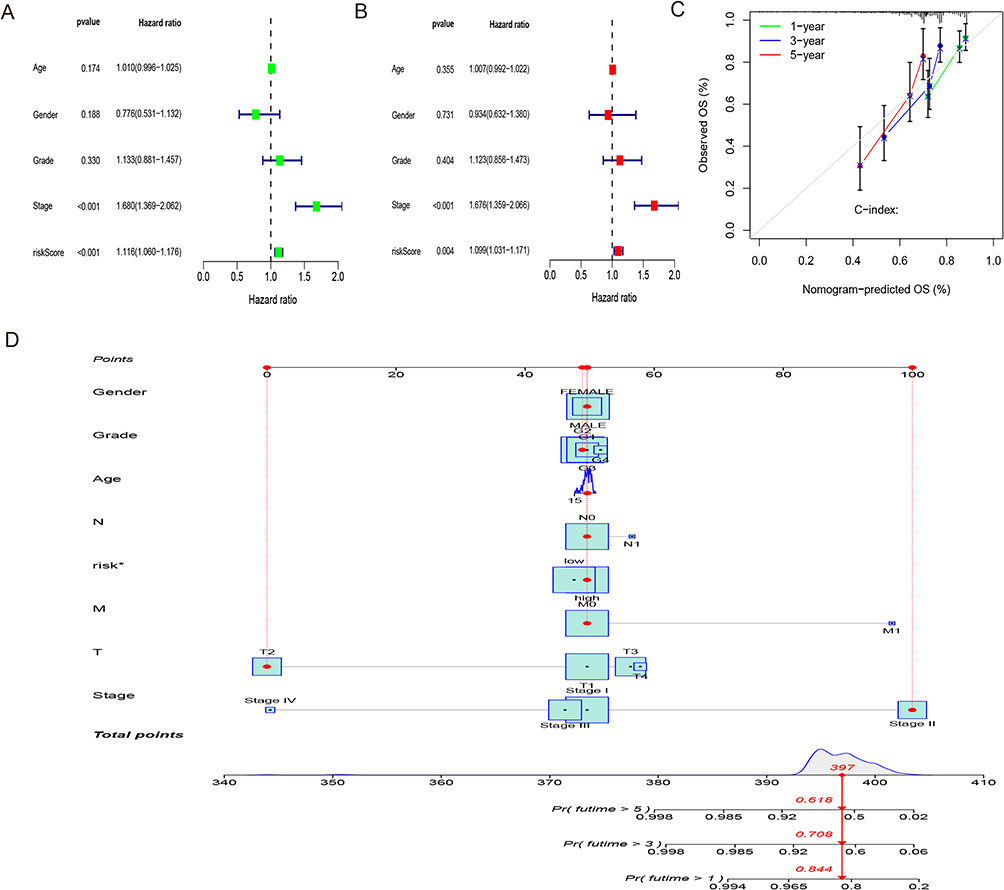 |
Figure 5 Nomogram and independent prognostic analysis. (A) Forest plot of independent univariate prognostic analysis. (B) Forest plot of multivariate independent prognostic analysis. (C) Nomogram for estimating the OS of HCC patients at different time points. (D) Calibration curves for the nomogram predictions (*p < 0.05). |
PCA is an unsupervised pattern recognition method for statistical analysis of multivariate data that converts a set of potentially correlated variables into a set of linearly uncorrelated variables by orthogonal transformation, and this converted variable is called the principal component. The original data is compressed into three principal components to characterize the original dataset: PC1 means the most significant feature that can describe the multidimensional data matrix; PC2 means the most significant feature that can describe the data matrix other than PC1, PC3, and so on. The utilization of PCA was implemented to investigate the variances in distribution among the low- and high-risk groups, considering the whole profiles of gene expression, 10 disulfidptosis-associated genes, 317 disulfidptosis-related lncRNAs, and the risk model. The PCA findings demonstrated that the risk model in the low- and high-risk groups was well separated, demonstrating the strong prognostic significance of the risk model (Figures 6A–D).
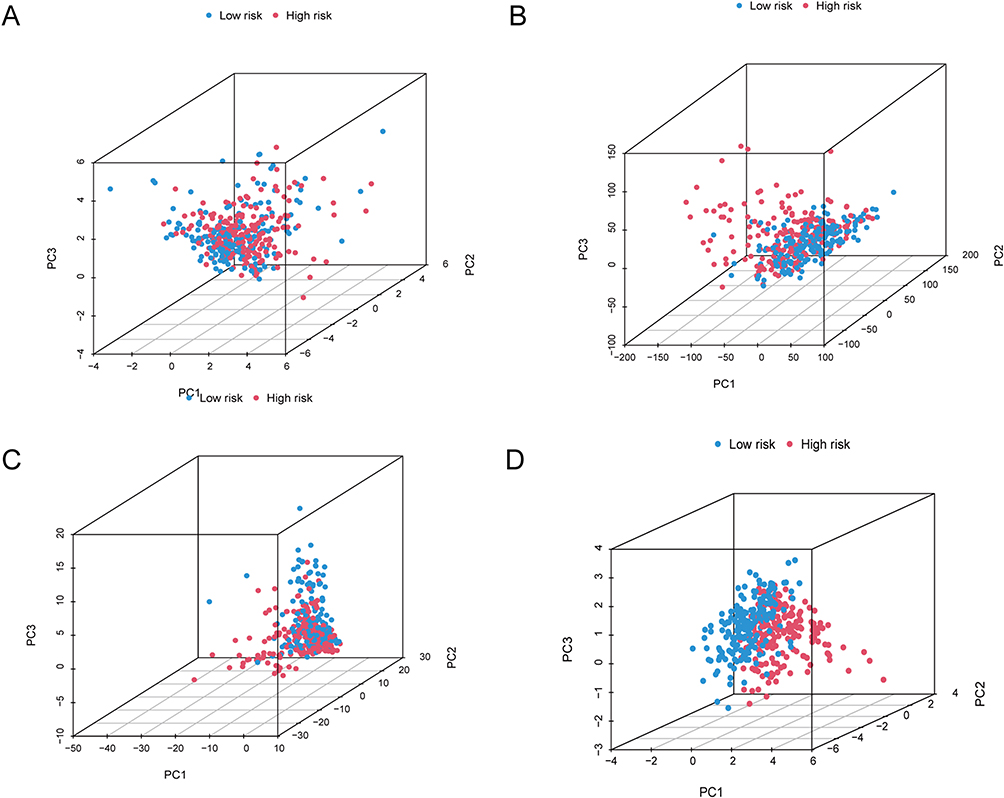 |
Figure 6 Using principal component analysis, compare the risk groupings at high and low levels depending on. (A–D) all gene expression profiles (A), 10 disulfidptosis-related genes (B), 317 disulfidptosis-related lncRNAs (C), and risk model based on the representation profiles of the 317 disulfidptosis-related lncRNAs (D) in the TCGA entire set. |
Functional Enrichment Analysis
In order to do functional enrichment analysis and understand the biological processes underlying the risk scores, DEGs were utilized. GO analysis revealed that biological processes (BP) correlated with responding to xenobiotic stimuli and positively regulating cell adhesion, cellular components (CC) associated with the extracellular matrix, including collagen, and molecular functions (MF) primarily involved in peptidase regulator activity and structural components of the extracellular matrix (Figure 7A and B). KEGG pathway analysis findings indicated that the DEGs have been primarily linked to the human papillomavirus infection, proteoglycans in cancer, the PI3K-Akt signaling pathway, and focal adhesion, particularly the NF-kappaB signaling pathway (Figure 7C and D). Analysis of KEGG pathways revealed that these pathways are mainly associated with tumorigenesis and tumor progression. Additionally, the results obtained from the high-risk group demonstrated activity in “cell adhesion molecules” and “cytokine-cytokine receptor interaction”, whereas those acquired from the low-risk group demonstrated significant enrichment in “fatty acid metabolism” and “drug metabolism cytochrome P450” (Figure 7E and F). The functional enrichment analysis outcomes demonstrated a strong association among the immune pathways and metabolic processes. Overall, these outcomes suggest that the lncRNA signature linked to disulfidptosis serves as an independent predictive biomarker for HCC individuals and can offer significant perspectives into the fundamental biological pathways and mechanisms implicated in the incidence of HCC.
 |
Figure 7 Functional enrichment analysis of DEGs associated with disulfidptosis. (A–D) The TCGA cohort’s most significant GO enrichment (A and B) and KEGG pathways (C and D). (E and F) Based on the entire TCGA high-risk (E) and low-risk (F) groups, GSEA revealed the top five pathways with the most significant enrichment. |
Immune Status and Immune Infiltration of Individuals Belonging to Distinct Risk Categories
The disulfidptosis-related lncRNA model was also investigated with regard to immune status and immune infiltration in individuals diagnosed with HCC. The immune score, representing the immune microenvironment, was found to exhibit a statistically significant variation among the low- and high-risk groups (*p<0.05; Figure 8A). Analysis of 22 immune cell types revealed notable distinctions in B cells naive, T cells CD8, monocytes, and macrophages M0 among the two risk groups (*p < 0.05,***p<0.001; Figure 8B and C). Furthermore, 19 immune function scores demonstrated statistically significant variations among the low- and high-risk groups, with aDCs, CCR, macrophages, MHC class I, Tregs, and type II IFN responses being particularly distinct (*p< 0.05, **p<0.01,***p<0.001; Figure 8D).
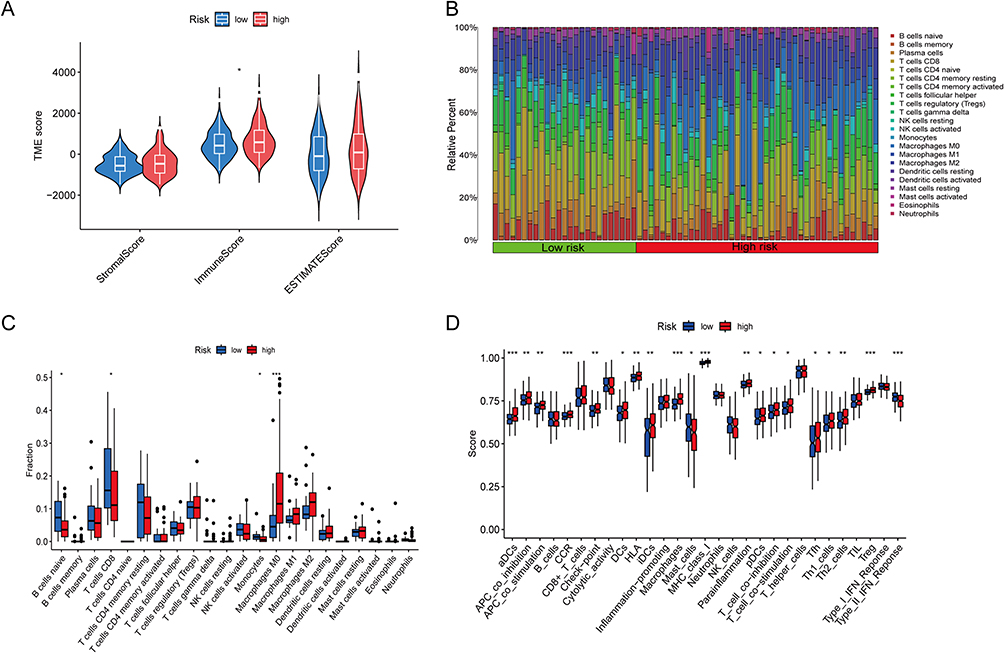 |
Figure 8 Estimation of the tumor immune microenvironment and cancer immunotherapy response utilizing the disulfidptosis-related lncRNA model in the TCGA complete data set. (A) TME score in the patients at high- and low-risk (*p < 0.05). (B and C) Boxplots are used to depict the relative percent and percentage of 22 immune cells (*p < 0.05, ***p<0.001). (D) Boxplots show the results of 29 immune-related activities (*p < 0.05, **p<0.01, ***p<0.001). |
Tumor Mutation Analysis
Utilization of tumor mutation analysis was employed to assess the number of genetic mutations in HCC specimens. The percentage of samples with genetic alterations was similar between the low and high-risk groups, with approximately 79.89% and 82.49% of samples affected, respectively (Figure 9A and B). The TMB did not significantly differ among the low- and high-risk groups (Figure 9C). However, the outcomes of the Kaplan-Meier analysis indicate that the group with high TMB exhibited significantly reduced survival rates in comparison with the group of low TMB (p<0.001; Figure 9D). Further analysis revealed significant differences in survival rates between the high-TMB + high-risk group, high-TMB + low-risk group, low-TMB + high-risk group, and low-TMB + low-risk group (p<0.001; Figure 9E). The current investigation outcomes indicate that the integration of the risk model with TMB can enhance predictive accuracy. Additionally, the outcomes regarding the high-risk group revealed a greater propensity to benefit from immunotherapy, indicating that the disulfidptosis-associated lncRNA model could serve as a predictive factor for TIDE (*** p<0.001; Figure 9F).
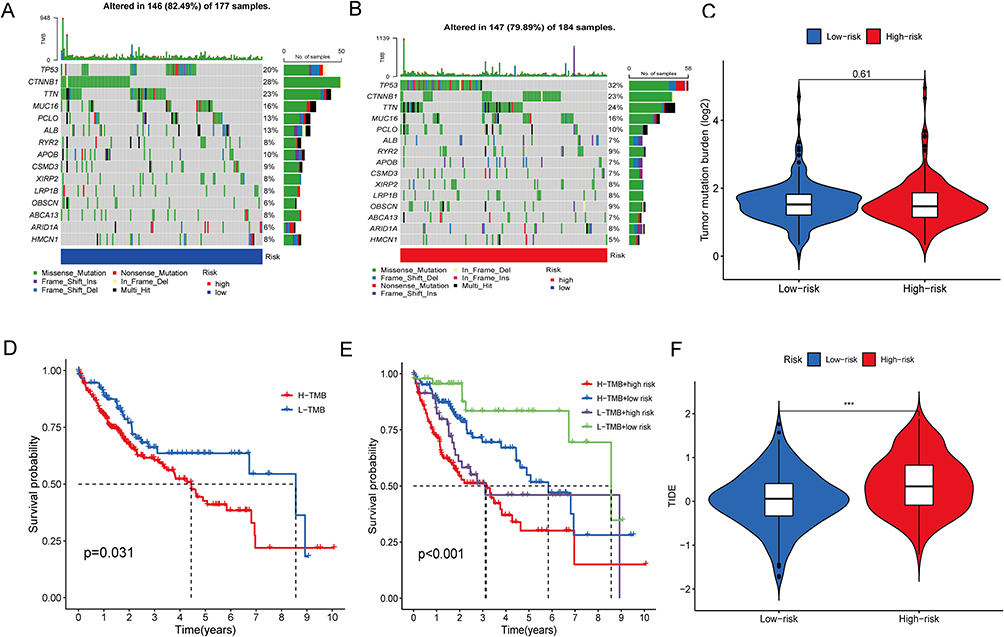 |
Figure 9 Tumor mutation analysis. (A and B) The distribution of the top 15 commonly mutated genes in patients with HCC in the high-risk (A) and low-risk (B) groups. (C) The burden of tumor mutation in the high-risk and low-risk groups. (D) OS of the high- and low-TMB groups. (E) OS of the high-TMB + high-risk group, high-TMB + low-risk group, low-TMB + high-risk group, and low-TMB + low-risk group. (F) The difference in TIDE score between the high- and low-risk groups. TIDE, tumor immune dysfunction and exclusion (***p<0.001). |
The Discovery of New Potential Drugs That Target the Disulfidptosis-Related lncRNAs Model
The pRRophetic method was utilized for discovering potential drugs that target the disulfidptosis-associated lncRNA model for the intervention and management of HCC. The findings of the IC50 for 95 compounds available within the GDSC database has been calculated by the aforementioned approach.There are four specific drugs that could be further explored for potential treatments in individuals with HCC. The IC50 values of Sorafenib, Oxaliplatin, and Gemcitabine in the high-risk group are higher than those in the low-risk group, indicating that the anti-tumor efficacy of the low-risk group is better than that of the high-risk group, whereas the IC50 values of 5-Fluorouracil in the low-risk group are higher than those in the high-risk group, indicating that the anti-tumor efficacy of the high-risk group is better than that of the low-risk group (Supplementary Figure 1). Among them, Gemcitabine has the best anti-tumor activity among the four drugs because its IC50 value is the lowest. Predicting sensitivity to chemotherapy is important, and adjuvant chemotherapy is an alternative treatment for HCC that can help clinicians choose the best chemotherapy regimen.
Discussion
The global impact of HCC is significant and exhibits a growing pattern.21,22 Despite the available treatment options such as surgery, radiation, and targeted therapies, the 5-year OS rate and quality of life for individuals with HCC have not improved significantly. Therefore, there exists a necessity to investigate innovative prognostic biomarkers and indicators of therapeutic response, particularly in the context of immunotherapy. The induction of disulfide stress has been found to trigger a regulated cell death process known as disulfidptosis, which has been identified as a prospective therapeutic option for the management of metabolic cancer.23 The discovery of disulfidptosis opens the door to novel anticancer therapies that target the pathophysiological function of disulfide stress.24 However, the possible impact of disulfidptosis-related lncRNA on the prediction of HCC has yet to be explored.
The four disulfidptosis-related lncRNAs involved in the prognostic model are CYTOR, NRAV, AL080248.1, and AC069307.1. The research has revealed that CYTOR is capable of providing selective targets to the microRNA-125a-5p/LASP1 axis, thereby facilitating the proliferation of HCC. CYTOR has been found to target the microRNA-125a-5p/LASP1 axis, promoting the growth of HCC. It also controls the miR-125b-5p/KIAA1522 axis, influencing growth, cell cycle, and HCC cells death.25 NRAV, another lncRNA, promotes the progression and invasion of HCC cells through altering the Wnt/β-catenin signaling pathway.26 On the other hand, AL080248.1 and AC069307.1 require further investigation as their roles in HCC have not been extensively studied yet. The aforementioned findings enhance our comprehension of the mechanisms that underlie the progression of cancer and provide potential targets for future therapeutic interventions.
To gain insights into the biological mechanisms, our findings indicate that the lncRNA signature associated with regulatory disulfidptosis could potentially enhance metabolic interactions in HCC. In the age of precision oncology, the strategy of selectively eliminating cancer cells by targeting cancer metabolism has gained significant popularity.27 Several pathways, including fatty acid metabolism, the PI3K-Akt signaling pathway, drug metabolism cytochrome P450, and the NF-kappaB signaling pathway, may be contributed to this process. The proper modulation of the PI3K-Akt signaling pathway is crucial for fundamental biological activities including cell development, death, survival, and metabolism.28 The onset and progression of various malignancies have been linked to the dysregulation of this pathway. Genetic polymorphisms of drug-metabolizing enzymes, such as cytochrome P450, may also play a role in hepatocarcinogenesis related to tobacco use.29 CD147, via the P38/PPAR and Akt/mTOR/SREBP1c pathways, modulates the metabolism of fatty acids within HCC cells.30 Liver cell IKK-dependent NF-kappaB signaling prevents the growth of liver cancer by enhancing hepatocyte survival.31
This study also measured the enrichment scores of correlations between immune cells and their corresponding activities, revealing significant variations between the low and high-risk groups concerning B cells naïve, T cells CD8, monocytes, and particularly macrophages M0. The scores for aDCs, CCR, macrophages, MHC class I, Tregs, and type II IFN response also showed the greatest statistical variance among the two risk score groups. The high-risk group revealed elevated counts of Treg cells and macrophages. Previous research has shown that the enrollment of macrophages and T-regulatory cells by tumor-related neutrophils contributes to sorafenib resistance and HCC progression.32 Increased tumor-associated macrophages,33 or Treg cells,34 have been connected with a negative prognostic value within HCC as a result of their function in immunological evasion. Increased tumor-related macrophages, or Treg cells have been connected with a negative prognostic value within HCC due to their role in immunological evasion. Thus, individuals at high risk may have compromised antitumor immunity, which could explain their poor prognosis. The utilization of TMB as an indicator has gained prominence due to its high sensitivity in predicting the response to immunotherapy across various malignancies, including lung cancer,35,36 Advanced Melanoma37 and gastric cancer.38 Throughout the current investigation, somatic alterations underwent detection in 79.89% of high-risk individuals and 82.49% of low-risk individuals. Similar to various previous research, the presence of genes exhibiting the highest frequency of mutations were TP53, CTNNB1, and TTN.39–41 Although there was no clear connection among TMB and the disulfidptosis-associated lncRNA signature, an elevated TMB was discovered to be correlated with lower survival rates. Targeting elevated TMB in combination with the high-risk disulfidptosis-related lncRNA signature improved patient prognosis. Additionally, individuals included in high-risk scores for HCC had a significant score in their TIDE algorithm that was statistically greater contrasted with those of the low-risk group, implying that immunotherapy could reveal a viable alternative for such individuals.Four drugs identified according to the lncRNAs model targeting disulfidptosis.Sorafenib, oxaliplatin, gemcitabine, 5-fluorouracil can be further explored as potential drugs for the treatment of HCC patients.Anticipating the responsiveness to chemotherapy is crucial, and adjuvant chemotherapy serves as a viable therapeutic option for HCC, aiding doctors in selecting the optimal chemotherapy regimen.
In our study, this unique model was verified in a variety of ways, and we can now choose the most effective model and apply it fairly. Without external data validation, we thought that the prediction model was adequate. We are also aware that this research has several flaws and restrictions. The complete understanding of the molecular mechanism of disulfidptosis-related lncRNAs remains unclear, so external validation by further clinical information would confer benefits. Consequently, we will collect a greater number of clinical specimens and increase the sample size. Throughout our further study, we will investigate the function of lncRNAs and their interaction with the disulfidptosis genes, as well as try to evaluate the correctness of our model through additional external tests.
Conclusion
In conclusion, our study culminated in the construction of a disulfidptosis-related lncRNA model comprising four prognostic disulfidptosis-related lncRNAs (ACYTOR, NRAV, AL080248.1, and AC069307.1). We created a prognosis-associated lncRNA signature that was strongly prognostic of HCC results and correlated it with functional enrichment pathways, immunotherapeutic response, targeted drug sensitivity, and TMB, as well as offering novel perspectives into disulfidptosis-associated lncRNAs function. This adds to our understanding of immunotherapy and targeted treatment for HCC.
Ethics Approval and Consent to Participate
All procedures in this study were conducted following the World Medical Association’s Declaration of Helsinki tenets. The current research follows the TCGA data access policies and publication guidelines.
Acknowledgments
We thank the TCGA contributor (https://cancergenome.nih.gov/) for making the LIHC dataset freely accessible. We are grateful to all of the volunteers who helped make this research possible.
Funding
This work was supported by the Clinical Research Plan of Shanghai Hospital Development Center (SHDC2020CR1004A), the Key Project of the National Natural Science Foundation of China(81730097), the National Natural Science Foundation of China(82072618), the National Key Research and Development Program of China (2022YFC2503700), and the National Natural Science Foundation of China (82002480).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Jemal A, Ward EM, Johnson CJ, et al. Annual report to the nation on the status of cancer, 1975–2014, featuring survival. J Natl Cancer Inst. 2017;109(9). doi:10.1093/jnci/djx030
2. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
3. Rumgay H, Ferlay J, de Martel C, et al. Global, regional and national burden of primary liver cancer by subtype. Eur J Cancer. 2022;161:108–118. doi:10.1016/j.ejca.2021.11.023
4. Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol. 2019;16(10):589–604. doi:10.1038/s41575-019-0186-y
5. Cucarull B, Tutusaus A, Rider P, et al. Hepatocellular carcinoma: molecular pathogenesis and therapeutic advances. Cancers. 2022;14(3):621. doi:10.3390/cancers14030621
6. Tang R, Xu J, Zhang B, et al. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J Hematol Oncol. 2020;13(1):110. doi:10.1186/s13045-020-00946-7
7. Badgley MA, Kremer DM, Maurer HC, et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science. 2020;368:
8. Liu X, Nie L, Zhang Y, et al. Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis. Nat Cell Biol. 2023;25(3):404–414. doi:10.1038/s41556-023-01091-2
9. Liu X, Olszewski K, Zhang Y, et al. Cystine transporter regulation of pentose phosphate pathway dependency and disulfide stress exposes a targetable metabolic vulnerability in cancer. Nat Cell Biol. 2020;22(4):476–486. doi:10.1038/s41556-020-0496-x
10. Joly JH, Delfarah A, Phung PS, Parrish S, Graham NA. A synthetic lethal drug combination mimics glucose deprivation-induced cancer cell death in the presence of glucose. J Biol Chem. 2020;295(5):1350–1365. doi:10.1016/S0021-9258(17)49891-7
11. Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res. 2019;29(5):347–364. doi:10.1038/s41422-019-0164-5
12. Shi X, Sun M, Liu H, Yao Y, Song Y. Long non-coding RNAs: a new frontier in the study of human diseases. Cancer Lett. 2013;339(2):159–166. doi:10.1016/j.canlet.2013.06.013
13. Fan Y, Li J, Yang Q, et al. Dysregulated long non-coding RNAs in Parkinson’s disease contribute to the apoptosis of human neuroblastoma cells. Front Neurosci. 2019;13:1320. doi:10.3389/fnins.2019.01320
14. Ruan X, Li P, Ma Y, et al. Identification of human long noncoding RNAs associated with nonalcoholic fatty liver disease and metabolic homeostasis. J Clin Invest. 2021;131(1). doi:10.1172/JCI136336
15. Jiang MC, Ni JJ, Cui WY, Wang BY, Zhuo W. Emerging roles of lncRNA in cancer and therapeutic opportunities. Am J Cancer Res. 2019;9(7):1354–1366.
16. Zhou C, Zhang H, Lu L. Identification and validation of hypoxia-related lncRNA signature as a prognostic model for hepatocellular carcinoma. Front Genet. 2021;12:744113. doi:10.3389/fgene.2021.744113
17. Li L, Xie R, Lu G. Identification of m6A methyltransferase-related lncRNA signature for predicting immunotherapy and prognosis in patients with hepatocellular carcinoma. Biosci Rep. 2021;41:6.
18. Xu Z, Peng B, Liang Q, et al. Construction of a ferroptosis-related nine-lncRNA signature for predicting prognosis and immune response in hepatocellular carcinoma. Front Immunol. 2021;12:719175. doi:10.3389/fimmu.2021.719175
19. Yang S, Zhou Y, Zhang X, et al. The prognostic value of an autophagy-related lncRNA signature in hepatocellular carcinoma. BMC Bioinf. 2021;22(1):217. doi:10.1186/s12859-021-04123-6
20. Bai Y, Lin H, Chen J, Wu Y, Yu S. Identification of prognostic glycolysis-related lncRNA signature in tumor immune microenvironment of hepatocellular carcinoma. Front Mol Biosci. 2021;8:645084. doi:10.3389/fmolb.2021.645084
21. Llovet JM, Zucman-Rossi J, Pikarsky E, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2016;2:16018. doi:10.1038/nrdp.2016.18
22. Villanueva A. Hepatocellular carcinoma. N Engl J Med. 2019;380(15):1450–1462. doi:10.1056/NEJMra1713263
23. Zheng P, Zhou C, Ding Y, Duan S. Disulfidptosis: a new target for metabolic cancer therapy. J Exp Clin Cancer Res. 2023;42(1):103. doi:10.1186/s13046-023-02675-4
24. Zheng T, Liu Q, Xing F, Zeng C, Wang W. Disulfidptosis: a new form of programmed cell death. J Exp Clin Cancer Res. 2023;42(1):137. doi:10.1186/s13046-023-02712-2
25. Hu B, Yang XB, Yang X, Sang XT. LncRNA CYTOR affects the proliferation, cell cycle and apoptosis of hepatocellular carcinoma cells by regulating the miR-125b-5p/KIAA1522 axis. Aging. 2020;13(2):2626–2639. doi:10.18632/aging.202306
26. Wang Q, Tang Y, Ge Y, Zhang S, Zheng M. Long non-coding RNA NRAV enhances proliferation and invasion of hepatocellular carcinoma cells by modulating the Wnt/β-catenin signaling pathway. Bioengineered. 2022;13(4):10026–10037. doi:10.1080/21655979.2022.2062977
27. Stine ZE, Schug ZT, Salvino JM, Dang CV. Targeting cancer metabolism in the era of precision oncology. Nat Rev Drug Discov. 2022;21(2):141–162. doi:10.1038/s41573-021-00339-6
28. Haddadi N, Lin Y, Travis G, Simpson AM, Nassif NT, McGowan EM. PTEN/PTENP1: ‘Regulating the regulator of RTK-dependent PI3K/Akt signalling’, new targets for cancer therapy. Mol Cancer. 2018;17(1):37. doi:10.1186/s12943-018-0803-3
29. Imaizumi T, Higaki Y, Hara M, et al. Interaction between cytochrome P450 1A2 genetic polymorphism and cigarette smoking on the risk of hepatocellular carcinoma in a Japanese population. Carcinogenesis. 2009;30(10):1729–1734. doi:10.1093/carcin/bgp191
30. Li J, Huang Q, Long X, et al. CD147 reprograms fatty acid metabolism in hepatocellular carcinoma cells through Akt/mTOR/SREBP1c and P38/PPARα pathways. J Hepatol. 2015;63(6):1378–1389. doi:10.1016/j.jhep.2015.07.039
31. He G, Karin M. NF-κB and STAT3 - key players in liver inflammation and cancer. Cell Res. 2011;21(1):159–168. doi:10.1038/cr.2010.183
32. Zhou SL, Zhou ZJ, Hu ZQ, et al. Tumor-associated neutrophils recruit macrophages and T-regulatory cells to promote progression of hepatocellular carcinoma and resistance to sorafenib. Gastroenterology. 2016;150(7):1646–1658.e1617. doi:10.1053/j.gastro.2016.02.040
33. Zhang Q, He Y, Luo N, et al. Landscape and dynamics of single immune cells in hepatocellular carcinoma. Cell. 2019;179(4):829–845.e820. doi:10.1016/j.cell.2019.10.003
34. Fu J, Xu D, Liu Z, et al. Increased regulatory T cells correlate with CD8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology. 2007;132(7):2328–2339. doi:10.1053/j.gastro.2007.03.102
35. Sakai K, Tsuboi M, Kenmotsu H, et al. Tumor mutation burden as a biomarker for lung cancer patients treated with pemetrexed and cisplatin (the JIPANG-TR). Cancer Sci. 2021;112(1):388–396. doi:10.1111/cas.14730
36. Sholl LM, Hirsch FR, Hwang D, et al. The promises and challenges of tumor mutation burden as an immunotherapy biomarker: a perspective from the international association for the study of lung cancer pathology committee. J Thorac Oncol. 2020;15(9):1409–1424. doi:10.1016/j.jtho.2020.05.019
37. Hodi FS, Wolchok JD, Schadendorf D, et al. TMB and inflammatory gene expression associated with clinical outcomes following Immunotherapy in advanced melanoma. Cancer Immunol Res. 2021;9(10):1202–1213. doi:10.1158/2326-6066.CIR-20-0983
38. Cheng Y, Bu D, Zhang Q, et al. Genomic and transcriptomic profiling indicates the prognosis significance of mutational signature for TMB-high subtype in Chinese patients with gastric cancer. J Adv Res. 2022;2022:1.
39. Khemlina G, Ikeda S, Kurzrock R. The biology of hepatocellular carcinoma: implications for genomic and immune therapies. Mol Cancer. 2017;16(1):149. doi:10.1186/s12943-017-0712-x
40. Zhu X, Jiang S, Wu Z, et al. Long non-coding RNA TTN antisense RNA 1 facilitates hepatocellular carcinoma progression via regulating miR-139-5p/SPOCK1 axis. Bioengineered. 2021;12(1):578–588. doi:10.1080/21655979.2021.1882133
41. Wen J, Min X, Shen M, et al. ACLY facilitates colon cancer cell metastasis by CTNNB1. J Exp Clin Cancer Res. 2019;38(1):401. doi:10.1186/s13046-019-1391-9
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Decaprenyl Diphosphate Synthase Subunit 1 (PDSS1): A Potential Prognostic Biomarker and Immunotherapy-Target for Hepatocellular Carcinoma
Yang Y, Li J, Tang M, Nie B, Huang W
Cancer Management and Research 2022, 14:1627-1639
Published Date: 3 May 2022
Identification of KRBA1 as a Potential Prognostic Biomarker Associated with Immune Infiltration and m6A Modification in Hepatocellular Carcinoma
Liu Y, Fu B, Yu Z, Song G, Zeng H, Gong Y, Ding Y, Huang D
Journal of Hepatocellular Carcinoma 2022, 9:497-516
Published Date: 31 May 2022
Low MARCO Expression is Associated with Poor Survival in Patients with Hepatocellular Carcinoma Following Liver Transplantation
Zhang Q, Wei Y, Li Y, Jiao X
Cancer Management and Research 2022, 14:1935-1944
Published Date: 11 June 2022
Histological Severity of Cirrhosis Influences Surgical Outcomes of Hepatocellular Carcinoma After Curative Hepatectomy
Liang BY, Gu J, Xiong M, Zhang EL, Zhang ZY, Lau WY, Wang SF, Guan Y, Chen XP, Huang ZY
Journal of Hepatocellular Carcinoma 2022, 9:633-647
Published Date: 23 July 2022
Nomogram for the Preoperative Prediction of the Macrotrabecular-Massive Subtype of Hepatocellular Carcinoma
Shan Y, Yu X, Yang Y, Sun J, Wu S, Mao S, Lu C
Journal of Hepatocellular Carcinoma 2022, 9:717-728
Published Date: 10 August 2022