Back to Journals » Infection and Drug Resistance » Volume 19
Ventilation-Associated Differences in Lower Airway Microbial Signatures and Peripheral Blood Transcriptome Among Critically Ill COVID-19 Patients
Authors Li Y, Huang M, Mao Z, Liu C, Qu W, Ni J, Xu W, Li A, Bao J, Han D, Yu F, Shen Y, Wang Y ![]() , Chen W, Zheng S
, Chen W, Zheng S
Received 18 December 2025
Accepted for publication 6 April 2026
Published 8 June 2026 Volume 2026:19 586598
DOI https://doi.org/10.2147/IDR.S586598
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Oliver Planz
Yuxia Li,1,* Mingzhu Huang,1,* Zheying Mao,1,* Chang Liu,1,* Wenxin Qu,1 Jili Ni,1 Wang Xu,2 Aijing Li,2 Jiaqi Bao,1,3,4 Dongsheng Han,1,3,4 Fei Yu,1,3,4 Yifei Shen,1,3,4 Yuefeng Wang,5 Weizhen Chen,1,3,4 Shufa Zheng1,3,4
1Department of Laboratory Medicine, the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, People’s Republic of China; 2Jinan Microecological Biomedicine Shandong Laboratory, Jinan, People’s Republic of China; 3Key Laboratory of Clinical in Vitro Diagnostic Techniques of Zhejiang Province, Hangzhou, People’s Republic of China; 4Institute of Laboratory Medicine, Zhejiang University, Hangzhou, People’s Republic of China; 5Department of Blood Transfusion, Shaoxing Maternity and Child Health Care Hospital, Shaoxing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Weizhen Chen, Email [email protected] Shufa Zheng, Email [email protected]
Background: Invasive mechanical ventilation (IMV) is a critical intervention for severe respiratory failure and has been widely used in the clinical management of COVID-19 patients. The relationship between such microbiota changes and the host transcriptome in IMV patients remains poorly documented.
Methods: We prospectively enrolled 69 critically ill COVID-19 patients, among whom 41 received IMV. Correlation analyses were conducted to investigate the relationship between the lung microbiome and host immune status in IMV COVID-19 patients.
Results: Compared to the Non-mechanical ventilation (NMV) group, patients in the IMV group exhibited significantly reduced alpha diversity, while no significant difference was observed in beta diversity. The abundance of Streptococcus genus was significantly higher in the NMV group, primarily dominated by Streptococcus oralis and Streptococcus mitis. Transcriptomic enrichment analysis revealed significant upregulation of inflammation-related pathways in the IMV group, including “positive regulation of inflammatory response” and “cellular response to interleukin-1”. The decreased relative abundance of Streptococcus genus in the IMV group showed significant correlations with upregulation of genes including CXCL8, PLAU, SELENOK, SDC4, RPL17, RPS23, TOMM7, and PLK3. These upregulated genes promoted the recruitment of immune cells to inflammatory sites through activation of pathways including chemotaxis, leukocyte migration, and leukocyte cell-cell adhesion, ultimately triggering a robust innate immune response.
Conclusion: IMV COVID-19 ARDS patients demonstrated significantly reduced pulmonary microbial diversity, with a marked decrease in the abundance of Streptococcus—primarily Streptococcus oralis and Streptococcus mitis. Transcriptomic profiling further revealed substantial upregulation of inflammatory pathways in IMV patients, including “positive regulation of inflammatory response”.
Keywords: COVID-19, critically ill, mechanical ventilation, lower respiratory tract microbiota, blood transcriptome
Introduction
Since the outbreak of COVID-19 in early 2020, more than 7.1 million deaths have been reported globally.1 Although existing studies indicate a declining trend in the severe case rate of SARS-CoV-2 infection, recent evidence indicates that emerging viral variants exhibit enhanced affinity for human upper respiratory tract tissues, which significantly increases their transmission efficiency.2 Consequently, the virus continues to pose an ongoing threat to global public health. In terms of treatment, significant progress has been achieved in antiviral drug development, with novel therapeutics effectively reducing the incidence of severe cases.3 However, it is noteworthy that elderly individuals and immunocompromised patients remain at high risk of progressing to serious complications such as acute respiratory distress syndrome (ARDS) post-infection. Therefore, the development and refinement of clinical management strategies for severe cases are of critical importance.
Currently, treatment options for severe patients remain limited. Invasive mechanical ventilation (IMV), which utilizes ventilators to maintain airway patency, improve ventilation and oxygenation, and prevent hypoxia and carbon dioxide retention, serves as a critical therapeutic intervention.4,5 It enables the body to potentially overcome respiratory failure caused by underlying conditions while creating favorable conditions for treating the primary disease.4,6 IMV plays an essential role in managing respiratory failure, particularly when respiratory muscles fail to maintain adequate pulmonary ventilation due to acute or chronic respiratory dysfunction caused by pulmonary or systemic injuries. By assisting gas exchange and acid-base balance, it provides vital life-sustaining support and serves as a crucial transitional measure for lung recovery.7 However, studies have reported complications associated with IMV, including barotrauma, hemodynamic instability, and infections, which may further harm the host.4,8,9 More recently, studies have demonstrated that IMV can alter the lower respiratory tract microbiota, potentially contributing to adverse clinical outcomes.8,9
The critical role of the lower respiratory tract microbiome has gained increasing recognition in both infectious and noninfectious respiratory diseases. Recent findings indicate a reciprocal interplay between pulmonary microbial dynamics and alveolar pathology: alterations in the lung microbiome may trigger inflammation, tissue damage, and subsequent edema, while conversely, the onset of edema may modify nutrient availability in the alveolar space, thereby reshaping microbial composition.10 In our preliminary research, we were the first to reveal the characteristics of the respiratory tract microbiota in COVID-19 patients and to investigate its prognostic relevance.11 Emerging evidence has found that IMV can alter the composition of patients’ respiratory tract microbiota, thereby affecting clinical outcomes.4,12 However, few studies have investigated how such microbiota alterations contribute to severe disease progression by influencing the host transcriptome.
In this investigation, we conducted a comparative assessment of the lower respiratory tract microbiota between IMV and Non-mechanical ventilation (NMV) COVID-19 patients, combined with host transcriptomic data, aiming to elucidate the MV-associated microbiome characteristics and their relationship with host transcriptome in COVID-19 patients. This research is expected to provide scientific evidence to guide the optimization MV therapy outcomes.
Materials and Methods
Study Design and Participants
This study included sixty-nine critically ill COVID-19 Patients (41 IMV patients and 28 NMV patients) from the First Affiliated Hospital of Zhejiang University. The critically ill COVID-19 Patients met any of the following criteria: (1) respiratory failure and required MV, (2) shock, and (3) complicating nonfunction of other organs.13 This study included a total of 60 samples from the lower respiratory tract (35 IMV patients and 25 NMV patients). Nine patients were excluded based on predefined quality control criteria, primarily for inadequate sample quality or lack of essential paired clinical data. Additionally, PBMC samples were obtained from 43 patients (27 IMV patients and and 16 NMV patients) on the same period as respiratory sample collection (2 days before and after). We extracted epidemiological, clinical, and laboratory data, along with treatment details and outcomes, from hospital electronic medical records. All data were validated by a trained team of doctors. The overview of the experimental design is shown in Figure S1. The study protocol was approved by the Ethics Committee of the First Affiliated Hospital, Zhejiang University School of Medicine, China (IIT20250125B). The ethics committee granted a waiver of patient informed consent, as the data used contain no personally identifiable information.
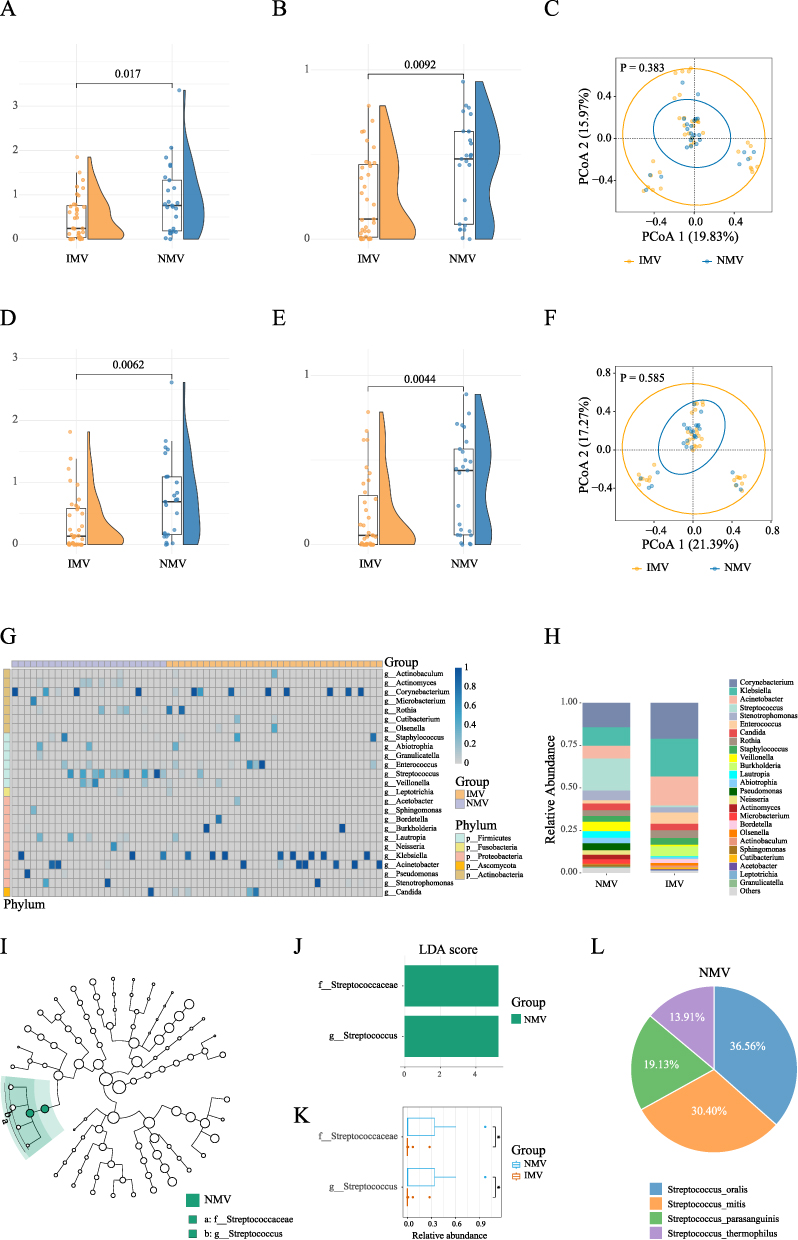 |
Figure 1 Metagenomic Analysis of the Sputum Microbiome in Invasive mechanical ventilation versus Non-mechanical ventilation COVID-19 Patients (A–C): Comparisons of α-diversity (A) Shannon index; (B) Simpson index) and β-diversity (C) between the Invasive mechanical ventilation (IMV) and Non-mechanical ventilation (NMV) group at the species level. (D–F): Comparisons of α-diversity (D Shannon index; (E) Simpson index) and β-diversity (F) between the IMV group and NMV group at the genus level. (G): Heatmap of the microbial community composition between the IMV group and NMV group. (H): Relative abundances of bacteria at the genus level in IMV group and NMV group. (I and J): LEfSe Analysis and LDA Scores for differential microbial taxa between the IMV and NMV groups. (K): Comparative Analysis of the Relative Abundance of Differentially Abundant Species Between the IMV and NMV Groups. (L): Species composition of the Streptococcus genus in the NMV group. Note: * P < 0.05. |
Sample Collection, PBMC Transcriptome Sequencing and Metagenome Sequencing
This study obtained lower respiratory specimens, including sputum from conscious patients and bronchial aspirates from those unconscious. For bronchial aspirates, sterile and single-use closed-suction systems were employed. All procedures were performed by trained personnel using aseptic techniques. Blood samples were collected in dedicated collection tubes. PBMCs were separated by standard density gradient centrifugation and subjected to RNA-seq. To monitor and control for potential contamination, extraction blanks and negative controls were included in every batch of nucleic acid extraction and subsequent library preparation. Consistent with the previous study,11 in simple terms, after processing the samples according to the protocol, we performed PBMC transcriptome sequencing on the Illumina NovaSeq 6000 platform and metagenome sequencing on the Illumina HiSeq 2500 platform.
Metagenomic Analysis Pipeline
We utilized the R packages vegan (v 2.6–6.1) and microeco (v 1.10.0) to analyze microbial community composition and diversity.14 Alpha diversity was evaluated using the Shannon and Simpson indices, reflecting the microbial diversity within individual samples. Beta diversity was calculated using Bray-Curtis distance, which measures the relative differences in microbial composition between two samples. The resulting distance matrix subsequently underwent Principal Coordinate Analysis (PCoA), with the first and second axes visualized to illustrate differences in microbial community structure among sample groups. To identify microbial features with significant differences between groups, the linear discriminant analysis effect size (LEfSe) algorithm from the microeco package was employed.15 Prior to analysis, low-abundance taxa with a relative abundance below 0.1% across all samples were removed to reduce noise and enhance statistical power. Adjusted P < 0.05 were considered statistically significant for screening potential biomarker taxa.
Transcriptome Analysis Pipeline
Differentially expressed genes (DEGs) were identified via the DESeq2 package (v 1.44.0).16 To screen for candidate genes with strong functional relevance, the STRING database (v 12.0) was further used to build a protein-protein interaction (PPI) network, resulting in 392 differentially expressed genes as the core candidate set. Pathway enrichment analysis (including GO and KEGG) was conducted on these 392 genes using the clusterProfiler package (v 4.12.0).17 Combined with the differentially abundant microbial taxa identified in the metagenomic analysis, Spearman correlation analysis was performed to integrate host transcriptome and microbiome data. Genes were screened based on a stringent criterion: they must be significantly correlated with differential bacteria and simultaneously enriched in at least three biological pathways, thereby identifying high-confidence candidate genes associated with the microbiome. To further screen for high-confidence genes, centrality analysis (based on metrics such as degree, closeness, and betweenness) of the PPI network was performed using the Cytoscape plugin cytoHubba (v 0.1) to identify key regulatory hub genes in the network. The intersection of the pathway enrichment results and the network centrality analysis results was used as the initial feature gene set for the machine learning stage. We employed three mainstream machine learning algorithms to independently screen feature genes: LASSO regression (glmnet, v 4.1–8), random forest (randomForest, v 4.7–1.1), and support vector machine based on recursive feature elimination (SVM-RFE, e1071, v 1.7–14). The final key genes were defined as the intersection of the gene sets identified by all three algorithms. Their diagnostic efficacy was evaluated by calculating the area under the receiver operating characteristic curve (AUC) via the pROC package (v 1.18.5). In the above process, we first reduced the number of candidate genes for machine learning by applying two filtering strategies: (1) selection based on significant correlation with differentially abundant microbial taxa, and (2) centrality-based filtering from the PPI network. This initial step helped mitigate overfitting. Subsequently, we further prevented overfitting through LASSO regularization, hyperparameter optimization in random forest and support vector machine models, and evaluated model performance using k-fold cross-validation to detect potential overfitting. Differential expression patterns were displayed via a heatmap created using the ComplexHeatmap package (v 2.22.0), with the key genes identified via machine learning highlighted. Subsequently, we performed immune infiltration analysis using xCell (v 1.1.0) and further computed the correlations between the machine learning-identified key genes and various differential immune cell types to explore the possible functions of these genes in regulating the pulmonary immune microenvironment.
Multi-Omics Data Integration Analysis
To elucidate the intricate association of the lung microbiome with host immune status in IMV COVID-19 patients, we performed an integrated multi-omics correlation analysis. First, a Sankey diagram was generated using the ggsankey package (v 0.0.99999) to visualize the connections between key differentially expressed genes and major biological pathways (eg., GO and KEGG). Subsequently, information on differentially abundant microbial taxa was incorporated to construct a triple-interaction network encompassing “microbes–differential genes–pathways,” using Spearman correlation coefficients to quantify associations across omics layers. Furthermore, based on immune infiltration profiles, an association network integrating differential immune cells, differentially expressed genes, and pathway enrichment results was established using Spearman correlation analysis. This approach provides an exploratory framework to systematically deciphers the pathological mechanism through which pulmonary microbial disturbances drive alterations in host immune responses in critically ill IMV COVID-19 patients.
Statistical Analysis
Descriptive statistics for clinical characteristics are expressed as median (interquartile range, IQR) for continuous variables (skewed distribution) or percentage (%) for categorical variables. Group comparisons were assessed using non-parametric tests: the Mann–Whitney U-test for continuous variables, and the chi-square test or Fisher’s exact test (adjusted based on expected frequencies) for categorical variables. We performed all statistical analyses using the R package compareGroups (v 4.8.0), based on non-missing values. The significance threshold was defined as a two-tailed P < 0.05, with multiple testing correction performed using the Benjamini–Hochberg procedure to control the false discovery rate (FDR).
Results
Patient Characteristics
Table 1 displays the demographic and clinical characteristics of COVID-19 patients in the IMV and NMV groups. Patients in two groups did not have significant differences in age or gender. The majority of patients (84.1%) had underlying medical conditions, with hypertension (53.6%), diabetes (34.8%), and chronic renal insufficiency (24.6%) being the most common. No statistically significant differences were found regarding underlying diseases between the two groups. Fever (100%), cough (65.2%), sputum production (55.1%), and chest tightness (53.6%) were the most frequently reported clinical manifestations, but no significant differences were found between the IMV and NMV groups. Most patients received corticosteroid therapy (88.4%), with fewer undergoing continuous renal replacement therapy (24.6%) or receiving immunosuppressants (10.1%), though no significant differences were observed between the two groups. Regarding clinical outcomes, the mortality rate was significantly higher in the IMV group than in the NMV group (46.3% vs. 17.9%, P = 0.029). In initial laboratory tests upon admission, Lymphocyte count (P = 0.019), Lactate (P = 0.027), and CK-MB (P = 0.001) were significantly higher in the IMV group compared to the NMV group, while PH (P = 0.014) was significantly lower (Table S1).
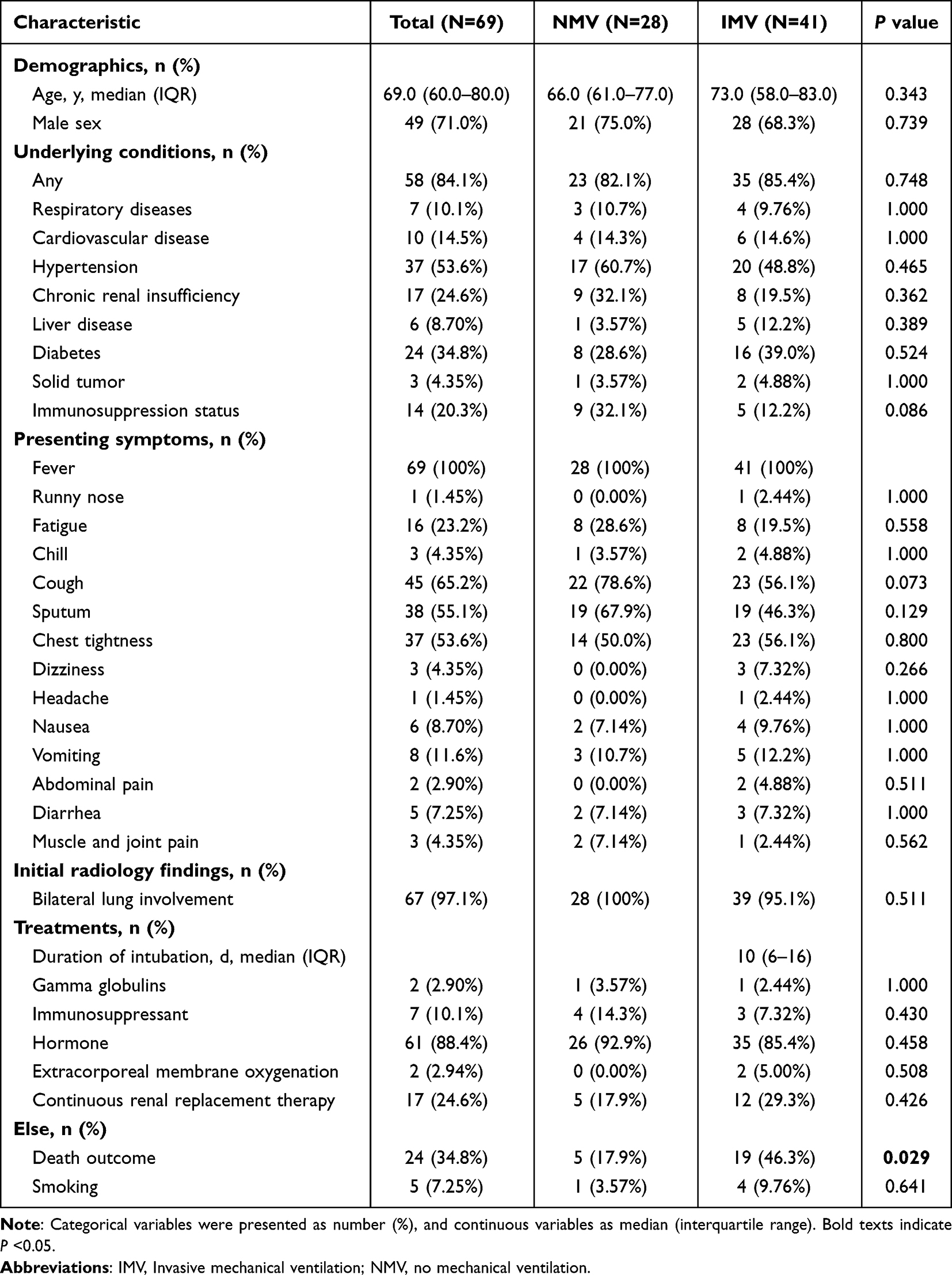 |
Table 1 Clinical Characteristics and Treatments of Mechanically Ventilated versus Non-Ventilated COVID-19 Patients |
Differences in Lung Microbiota Between IMV and NMV Patients Were Observed in Terms of Species Diversity and Taxonomic Composition
At the species level, α-diversity was significantly lower in the IMV group compared to the NMV group, as indicated by the Shannon index (P = 0.017) and Simpson index (P = 0.0092). However, no significant difference was found in β-diversity (P = 0.383) (Figure 1A–1C). Similarly, at the genus level, the biodiversity in the IMV group showed consistent trends with those at the species level: decreased Shannon index (P = 0.0062), decreased Simpson index (P = 0.0044), and no statistically significant difference in β-diversity (P = 0.585) (Figure 1D–1F). In the heatmap of genus-level taxa, Corynebacterium, Burkholderia, Klebsiella, and Acinetobacter exhibited dominant infection trends. After infection, these genera often showed near-monopolistic relative abundance (Figure 1G). The comparison revealed that Corynebacterium, Burkholderia, Klebsiella, and Acinetobacter had relatively higher abundances in the IMV group, whereas Streptococcus showed a relatively higher abundance in the NMV group (Figure 1H). We further conducted differential analysis of species between the two groups. LEfSe results indicated that both the Streptococcaceae and the Streptococcus were significantly reduced in the IMV group, primarily driven by decreases in Streptococcus oralis and Streptococcus mitis (Figure 1I–1L).
Host Transcriptome Analysis of IMV and NMV Patients
Differential gene analysis revealed that compared to the NMV group, the IMV group had a total of 3,282 differentially expressed genes, among which 1,446 genes were up-regulated and 1,836 genes were down-regulated (Figure S2A). Based on protein-protein interaction (PPI) network analysis, 392 core differentially expressed genes were screened. Enrichment analysis showed that the IMV group was significantly enriched in inflammation-related pathways such as positive regulation of inflammatory response and cellular response to Interleukin-1. Meanwhile, oxidative phosphorylation, response to oxidative stress, ribosome function, and intrinsic apoptotic signaling pathway were also significantly up-regulated (Figure S2B).
We further ranked the 392 differentially expressed genes in the PPI network using six centrality metrics via the CytoHubba algorithm, and obtained 10 hub genes by taking the intersection of the top 50 genes (Table S2). Simultaneously, from the 37 candidate genes obtained from differential microbiota correlation analysis, bidirectional screening was performed based on the frequency of their associated pathways, ultimately identifying 10 genes and their corresponding 19 pathways. After removing duplicate genes, a total of 19 candidate genes were included in the machine learning workflow. The expression profiles of these 19 differentially expressed genes in the two groups are shown in Figure 2A. Using three algorithms—LASSO regression, support vector machine (SVM), and random forest—core gene sets were identified, resulting in 12, 15, and 10 genes, respectively (Figure 2B–2F). The intersection of these three sets yielded seven stable hub genes: FAU (AUC = 0.861), PDGFRB (AUC = 0.794), PSMA6 (AUC = 0.877), RPL17 (AUC = 0.898), RPL30 (AUC = 0.905), SELENOK (AUC = 0.935), and SPN (AUC = 0.889) (Figure 3A and 3B). Box plots further validated the differential expression levels of the seven hub genes: in the IMV group, SELENOK, RPL30, RPL17, FAU, and PSMA6 were significantly up-regulated, while SPN and PDGFRB were significantly down-regulated (Figure 3C).
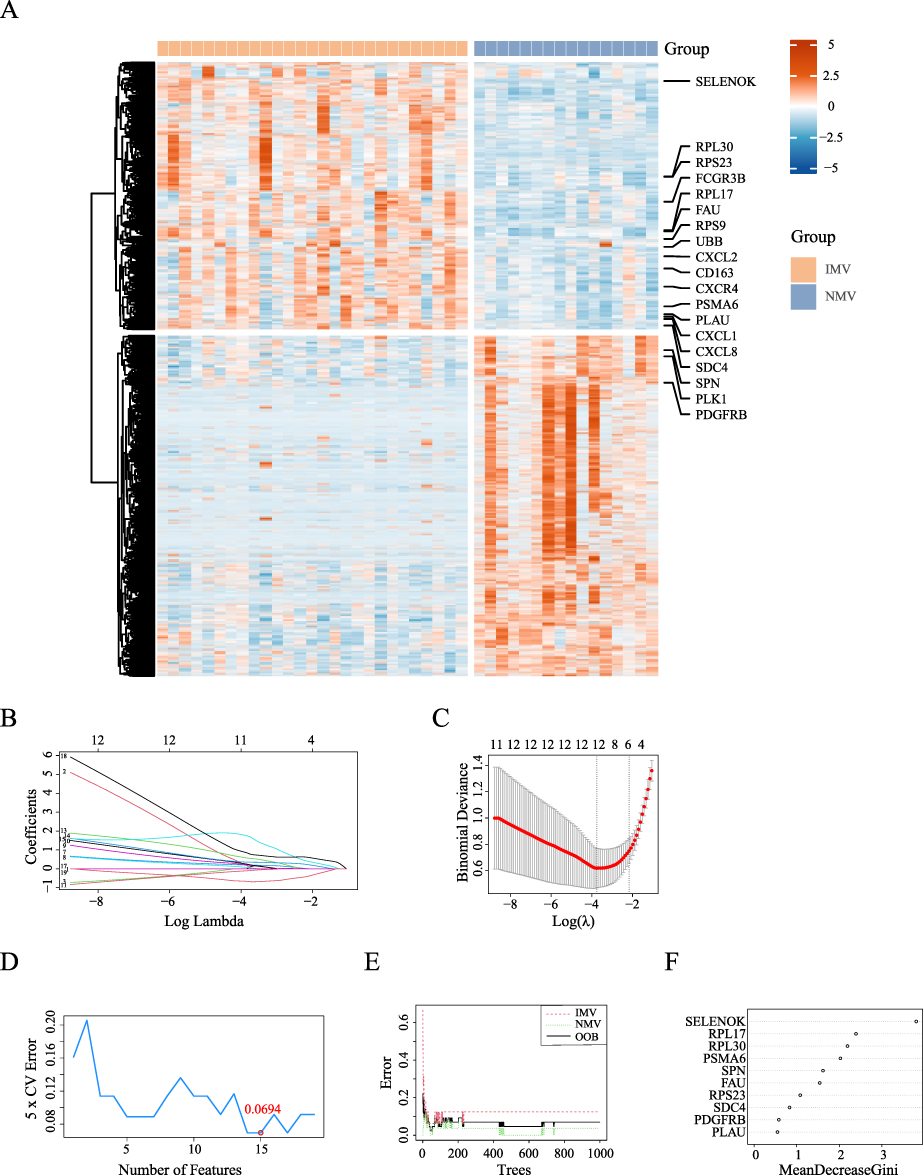 |
Figure 2 Expression Profiles and Machine Learning-Based Screening of Differentially Expressed Genes in Invasive mechanical ventilation versus Non-mechanical ventilation COVID-19 Patients. (A): The heatmap of differentially expressed genes between the Invasive mechanical ventilation (IMV) and the Non-mechanical ventilation (NMV) groups, with the 19 feature genes selected for machine learning highlighted. (B and C): LASSO coefficient profiles of the candidate genes were plotted, and the optimal lambda was determined when the binomial deviance reached its minimum value. In Figure (B) each coefficient curve corresponds to a single gene. In Figure (C) the solid vertical lines represent the binomial deviance, and the number of genes (n = 12) at the lowest point of the curve is considered the most suitable for LASSO. (D): The effect of the number of features on the 5-fold cross-validation error. The line plot shows how the 5-fold cross-validation error changes as the number of features increases. (E and F): The OOB error of the random forest model varies with the number of trees. Figure (E) shows OOB error for the intubation group (dashed red line) and the non-intubation group (dashed green line), with the overall OOB error represented by the solid black line. In Figure (F) the relative importance of the candidate genes was calculated using the random forest model. Importance scores are plotted on the x-axis, and the genetic variables are shown on the y-axis. |
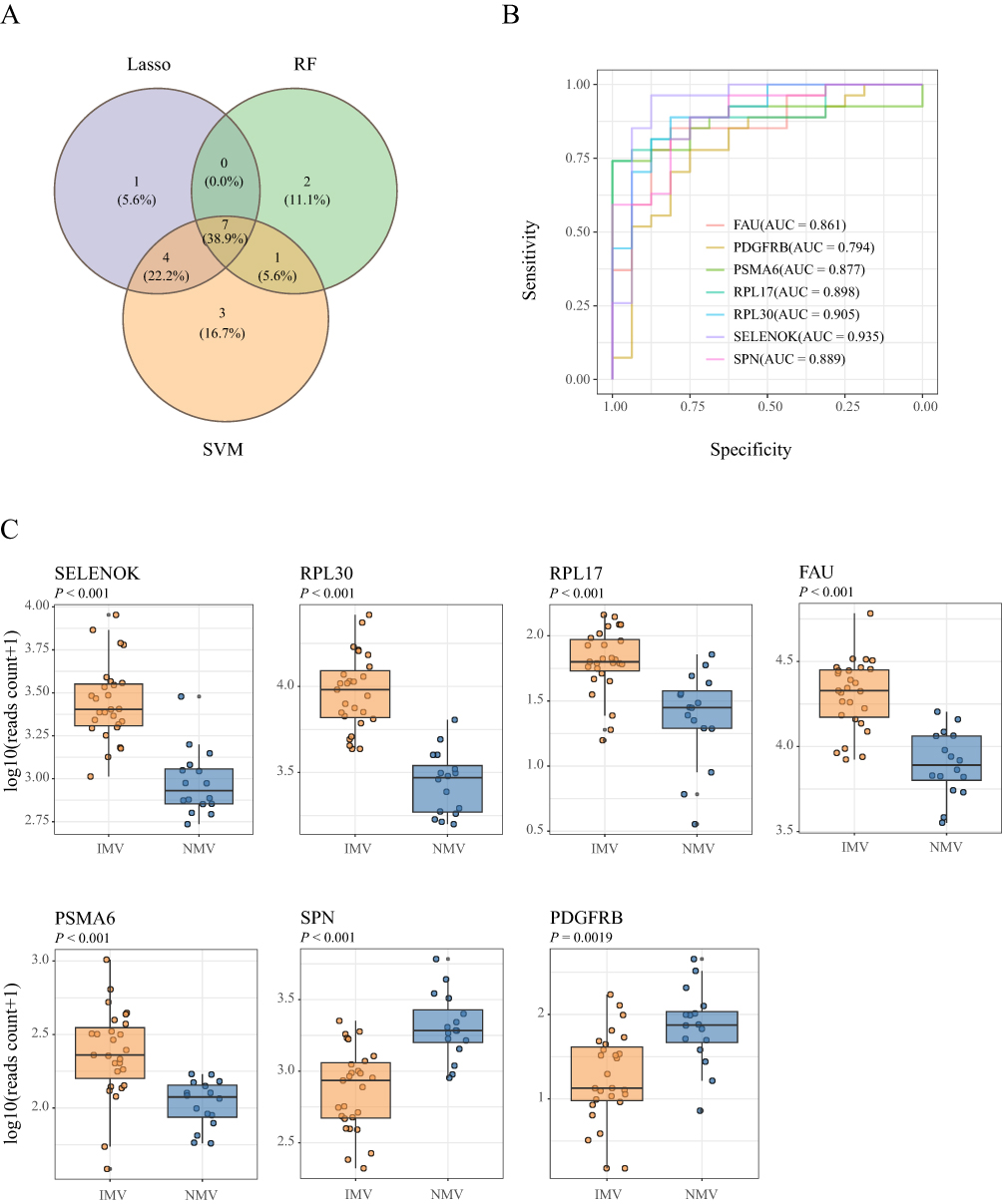 |
Figure 3 ROC curves and expression box plots of the key genes identified by machine learning (A): The intersection of genes selected by three machine learning algorithms was visualized using a Venn diagram, identifying seven common genes. (B): ROC curves and AUC values of the seven intersection genes identified by three machine learning algorithms. (C): Comparison of expression levels of the 7 intersecting genes identified by machine learning between the Invasive mechanical ventilation (IMV) and the Non-mechanical ventilation (NMV) groups. |
Correlation Analysis of Metagenomic, Transcriptomic, and Clinical Indicators
We performed pairwise Spearman correlation analysis among differential clinical indicators, differential bacteria, and the differentially expressed genes selected for pathway enrichment to explore the relationships between multi-omics data and clinical indicators in IMV and NMV patients. We observed that Streptococcaceae family and Streptococcus genus were negatively correlated with inflammation- and oxidative stress-related genes such as CXCL8, SELENOK, and PLK3, as well as with ribosomal protein-related genes including RPL30, RPL17, RPS23, and MRPL33, while showing positive correlations with genes such as PDGFRB and CIT (Figure 4A). Among the differential clinical indicators, Streptococcaceae family and Streptococcus genus were only negatively correlated with CK-MB (Figure 4B). In addition, correlation analysis between clinical indicators and DEGs revealed that CK-MB was positively correlated with signal transduction and cell communication-related genes such as IL1R2, IL18RAP, and EFNA1, whereas pH was positively correlated with platelet coagulation function-related genes including GP1BA and GP6 (Figure 4C). Finally, internal correlation analysis among the five differential clinical indicators revealed that lactate was positively correlated with CK-MB, while pH was negatively correlated with lactate (Figure 4D).
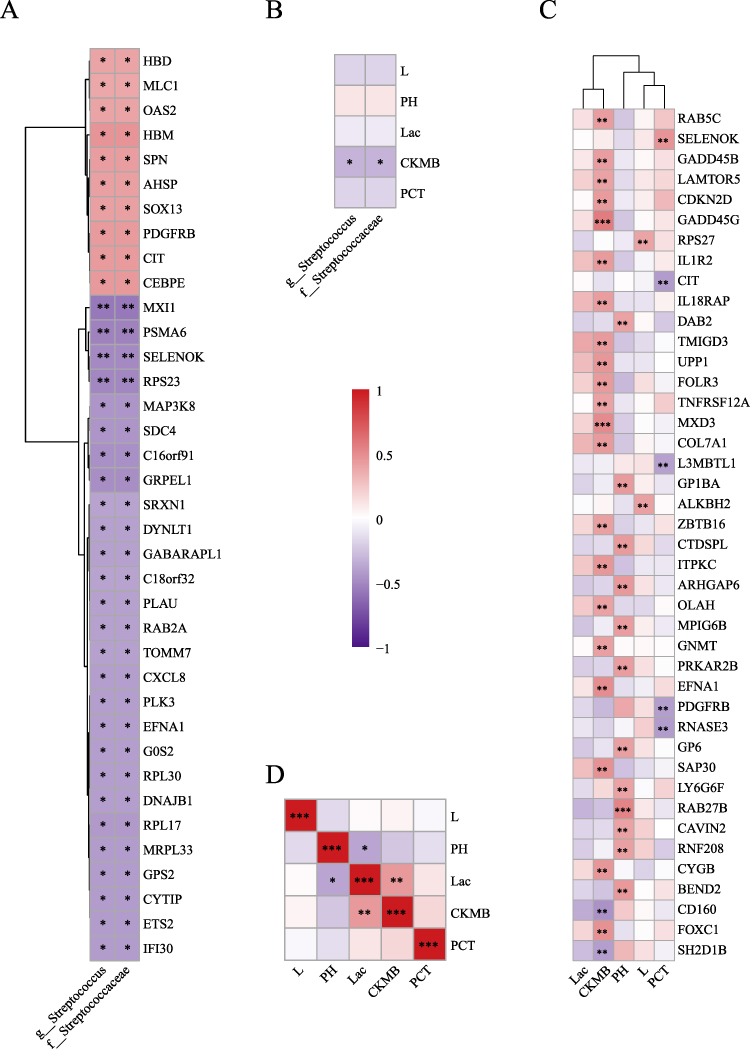 |
Figure 4 Correlation analysis among differential clinical indicators, differential microbiota, and differentially expressed genes in Invasive mechanical ventilation versus Non-mechanical ventilation COVID-19 patients (A): Heatmap of correlations between differential microbiota and the 392 differentially expressed genes, showing only statistically significant results. (B): Heatmap of correlations between differential microbiota and differential clinical indicators. (C): Heatmap of correlations between differential clinical indicators and the 392 differentially expressed genes, showing only statistically significant results with P < 0.01. (D): Heatmap of correlations among differential clinical indicators. Note: * P < 0.05, ** P < 0.01, *** P < 0.001. |
Immune Cell Infiltration Analysis
To investigate the activation or suppression of immune cells in IMV versus NMV patients, we performed immune infiltration analysis. The results revealed that compared to the NMV group, the IMV group exhibited significant upregulation of CD4+ Effector Memory T cells (CD4+ Tem), Neutrophils, Natural Killer T cells (NKT), Plasma cells, and Regulatory T cells (Tregs), while Common Myeloid Progenitor (CMP), Mast cells, Megakaryocytes, Platelets, Preadipocytes, and Th2 cells were significantly downregulated (Figure 5A and 5B). Subsequently, correlation analysis of these dysregulated immune cells indicated positive relationships between Platelets and Megakaryocytes (r = 0.8) as well as Preadipocytes (r = 0.7) (Figure 5C). We further analyzed the relationship between 18 union feature genes identified from three machine learning approaches and these dysregulated immune cells, and observed extensive associations, particularly with Neutrophils, NKT, Tregs, and the feature genes (Figure 5D). Clearly, the differences in core gene expression between the two groups are directly reflected in immune dysfunction.
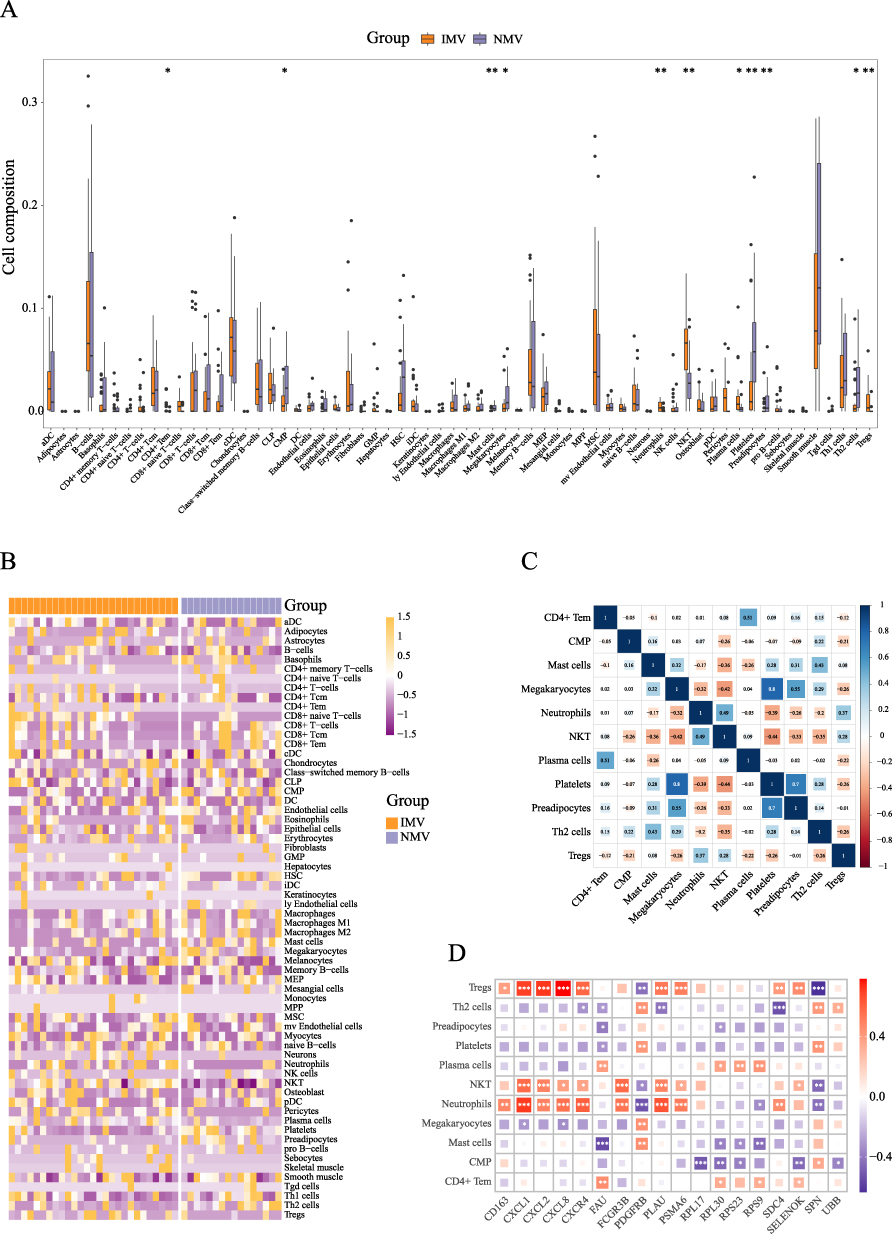 |
Figure 5 Immune Infiltration Analysis in Invasive mechanical ventilation versus Non-mechanical ventilation COVID-19 Patients (A): Compositional differences of 64 immune cell types between the Invasive mechanical ventilation (IMV) and the Non-mechanical ventilation (NMV) in SARS-CoV-2 infected patients. (B): Heatmap displaying expression patterns of immune cell types across different samples. (C): Correlation analysis among 11 immune cell types showing significant differences between IMV and NMV groups. (D): Correlation analysis between the union of machine learning-identified genes and the 11 differentially abundant immune cells. Note: * P < 0.05, ** P < 0.01, *** P < 0.001. |
Bridging Analysis of Differential Bacteria, Differential Genes, Enriched Pathways, and Immune Infiltrating Cells
Compared to the NMV group, the decreased relative abundance of Streptococcus genus in the IMV group showed a significant correlation with the upregulation of genes including CXCL8, PLAU, SELENOK, SDC4, RPL17, RPS23, TOMM7, and PLK3. As shown in Figure 6A, further Spearman correlation analysis suggested a significant association between the reduced relative abundance of Streptococcus in the IMV group and a robust inflammatory response and oxidative stress. On one hand, upregulated genes such as CXCL8, PLAU, SELENOK, and SDC4 in the IMV group promoted the recruitment of immune cells, including neutrophils and NKT cells, to inflammatory sites via the activation of pathways such as chemotaxis, leukocyte migration, and leukocyte cell-cell adhesion, thereby triggering a strong innate immune response (Figure 6A–6C). The production of Tregs was also subsequently upregulated. Meanwhile, the upregulation of CXCL8 and PLAU further exacerbated the immune response by activating the NF-kappa B signaling pathway, which initiated the production of downstream cytokines (Figure 6A). On the other hand, upregulated genes such as RPL17, RPS23, and FAU in the IMV group promoted plasma cell antibody production by protein translation pathways, including cytosolic ribosome and cytoplasmic translation, in response to high viral load infections (Figure 6A–6C). Additionally, the upregulation of SELENOK, TOMM7, and PLK3 genes activated pathways related to oxidative phosphorylation, import into the mitochondrion, response to oxidative stress, and the intrinsic apoptotic signaling pathway, intensifying the oxidative stress response in patients (Figure 6A and S2B).
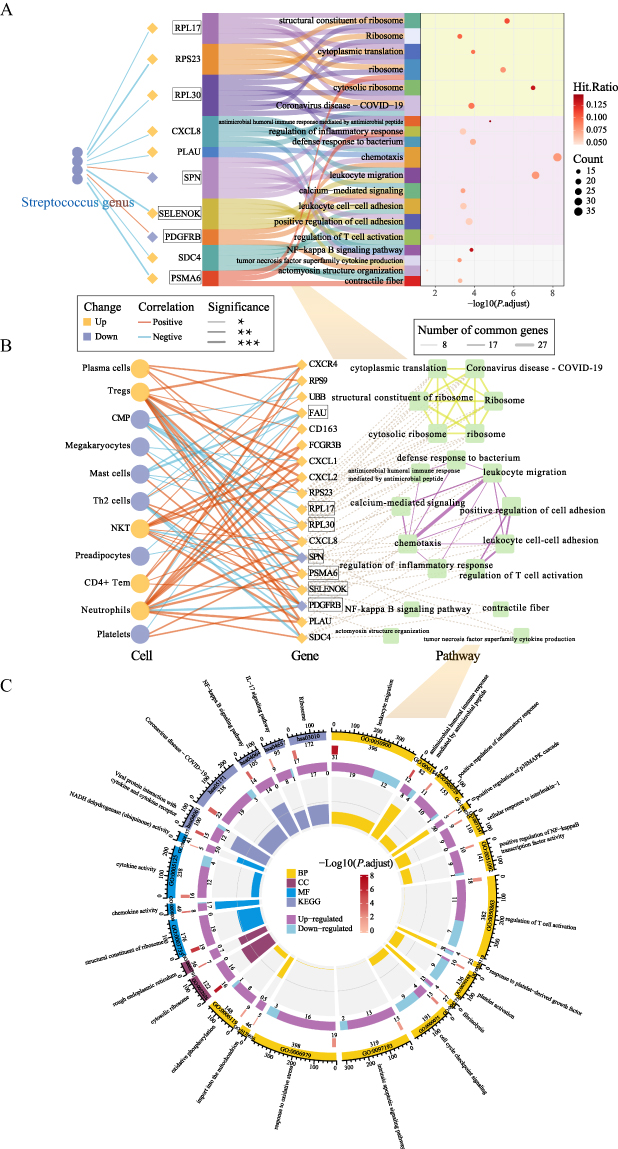 |
Figure 6 Multi-omics Integrative Analysis in Invasive mechanical ventilation versus Non-mechanical ventilation COVID-19 Patients (A): Sankey diagram illustrating relationships between microbiota-associated genes and pathways. Left-side connections show correlations between differential microbiota and differentially expressed genes, while right-side sections display pathway enrichment results. Colors of microbiota and gene nodes represent directional changes (up/down-regulation) in the Invasive mechanical ventilation (IMV) and the Non-mechanical ventilation (NMV). Connection line colors and thickness indicate the direction (positive/negative) and statistical significance of correlations respectively. Genes highlighted with boxes represent the intersecting genes identified by machine learning. (B): Network diagram demonstrating relationships among machine learning-analyzed differentially expressed genes (including microbiota-associated genes), immune infiltrating cells, and pathways. Line thickness between pathways indicates the number of shared genes. Boxed genes represent the machine learning-identified intersecting genes. (C): Circular enrichment plot displaying comprehensive pathway analysis. From outer to inner rings: First ring: Pathway IDs and background gene numbers for each pathway. Second ring: Number of differentially expressed genes enriched in each pathway. Third ring: Breakdown of up-regulated and down-regulated genes from the second ring. Fourth ring: Rich factor values for each pathway. Note: * P < 0.05, ** P < 0.01, *** P < 0.001. |
Discussion
Relevant studies have revealed that the lung microbiome contributes critically to maintaining pulmonary function and correlates with the pathogenesis of diseases including bronchiectasis and COPD.18–20 This is largely attributable to the semi-open nature of the lungs, which is constantly exposed to the external environment through the airways.4 Consequently, the pulmonary microbiota engages in a persistent exchange of microorganisms with the upper airways (including the nasal cavity, oral cavity, pharynx, and trachea) and the external environment, thereby forming a dynamic microbial cycle and a constantly fluctuating pulmonary microbial community. Meanwhile, the lung microbiota relies on absorbing necessary nutrients from the airways, alveoli, and their cellular components to sustain its dynamic equilibrium.4,18 Therefore, compared with microbiomes in other sites such as the gut, the composition of the lung microbiome is more profoundly shaped by environmental factors, resulting in a distinct local microbial ecosystem with unique characteristics.
IMV is a widely used life-support method in patients with respiratory failure. Relevant studies monitoring the dynamic changes of the respiratory microbiota in MV patients have suggested that invasive MV may alter the lung microbiome diversity. Research by Kelly et al found a significant reduction in microbial diversity within 24 hours after post-intubation compared with the initial 24-hour period.21 Similarly, Woo S et al, in a study monitoring the respiratory microbiota of severe pneumonia patients in successful extubation versus failed extubation groups, also observed reduced bacterial diversity in tracheal aspirates from day 1 and day 7 of invasive MV.12 In our study, we likewise found that alpha diversity was significantly lower in the IMV group compared with the NMV group. However, no significant difference in beta diversity was observed between the groups, which may be attributed to secondary infections in some patients leading to the dominance of specific bacterial taxa, thereby masking intergroup differences. This finding is consistent with the study by Sulaiman I et al, which also reported no significant difference in beta diversity between IMV and NMV COVID-19 patients.22
In this study, we observed higher relative abundances of common nosocomial pathogens such as Corynebacterium, Burkholderia, Klebsiella, and Acinetobacter in the IMV group, suggesting a heightened susceptibility to secondary infections among mechanically ventilated patients. Relevant studies indicate that MV increases the risk of secondary infections by 6 to 21 times and represents a major risk factor for severe outcomes and mortality.4,6 Multiple factors contribute to secondary bacterial infections in IMV patients. First, endotracheal intubation compromises the protective laryngeal barrier, leading to drying of the airway mucosa, impaired ciliary function, and reduced clearance of secretions, thereby facilitating bacterial colonization.4 Second, ventilation-induced lung injury increases alveolar-capillary permeability, which may promote the translocation of mediators, lipopolysaccharides, and bacteria into the systemic circulation.23 Third, studies have shown that the inserted tube, as a foreign body, becomes rapidly coated with host proteins—such as fibrin and fibronectin—forming a “conditioning film” that provides an ideal anchor for initial bacterial adhesion.24 Finally, frequent medical interventions (eg., suctioning and tube adjustments) significantly increase the risk of cross-infection.
The normal lung microbiome in healthy individuals is generally characterized by low microbial biomass (approximately 103–105 bacteria per gram of tissue) and is primarily composed of genera such as Streptococcus, Prevotella, Veillonella, Fusobacterium, and Haemophilus.18 A study by Kitsios GD et al found that high abundances of Prevotella, Streptococcus, and Haemophilus genera in IMV patients had a protective effect and were associated with reduced mortality.25 In our study, we also observed that NMV COVID-19 patients exhibited a higher relative abundance of Streptococcus in the lung microbiome compared to IMV patients, while no statistically significant differences were found in other typical respiratory microbial taxa. Previous research has shown that probiotic interventions using Streptococcus thermophilus in ventilator-assisted patients significantly reduced the incidence of ventilator-associated pneumonia and lowered mortality rates.26 Additionally, studies have demonstrated that Streptococcus oralis in the respiratory tract plays a protective role against severe pneumonia caused by respiratory viral infections such as influenza.27
Multiple studies have confirmed that CXCL8 expression is significantly elevated in patients infected with SARS-CoV, MERS-CoV, and SARS-CoV-2, and its levels have been independently identified as a predictor of mortality risk in severe cases, highlighting its critical role in virus-induced immunopathological processes.28–30 Our study also found that CXCL8 expression was significantly upregulated in critically ill COVID-19 patients requiring MV and closely associated with their immune infiltration profile. Specifically, the mechanically ventilated group showed markedly increased proportions of neutrophils, NKT cells, and regulatory T cells (Tregs), and these cell subsets were positively correlated with CXCL8 levels. Further functional enrichment analysis revealed significant activation of CXCL8-involved pathways, including chemotaxis, leukocyte migration, and the NF-κB signaling pathway. The above results reveal that CXCL8 not only acts as a chemokine to recruit effector cells to sites of pulmonary inflammation but may also promote the production of additional pro-inflammatory factors by activating downstream NF-κB transcriptional programs, thereby forming a positive feedback loop that accelerates the development of a cytokine storm. The mutual regulatory relationship between CXCL8 and the NF-κB signaling pathway has been reported in multiple studies, indicating a tight coupling in amplifying inflammatory responses and driving immunopathology.31–33
Although Tregs are conventionally recognized as core immunoregulatory units responsible for anti-inflammatory functions and maintaining immune homeostasis, our data demonstrate that despite a significant increase in Treg proportion in mechanically ventilated severe COVID-19 patients, they fail to effectively suppress excessive inflammatory responses. This suggests that under severe viral infection-induced hyperinflammatory conditions, Tregs may undergo functional transformation-shifting from a typical immunosuppressive phenotype toward a pro-inflammatory or even tissue-destructive subtype. Previous studies have confirmed that Tregs in severe COVID-19 patients are not only increased but also exhibit distinct subtypes, characterized by destructive properties and loss of conventional suppressive function.34,35 Thus, the dual role of Tregs in severe viral infections highlights their high context-dependency and functional plasticity in inflammatory regulation. These findings provide new insights into the mechanisms of immune dysregulation in critically ill patients and suggest that targeting Treg reprogramming may represent a promising future strategy for intervening in cytokine storms.
In this study, we found that mechanically ventilated COVID-19 patients exhibited significantly enhanced oxidative stress responses and specific immune activation profiles. Pathway enrichment analysis revealed significant upregulation of multiple biological processes closely related to oxidative stress and apoptosis, including import into the mitochondrion, NADH dehydrogenase (ubiquinone) activity, oxidative phosphorylation, response to oxidative stress, and intrinsic apoptotic signaling pathway. Previous studies have also indicated that severe COVID-19 patients often experience persistent oxidative stress, accompanied by accumulated DNA damage and programmed cell death.36–38 Further investigation revealed that NF-κB functions as a critical molecular redox switch capable of either activating or suppressing oxidative stress, while itself being modulated by post-translational modifications induced by excessive ROS. This bidirectional regulation forms a self-modulating inflammatory-oxidative homeostatic circuit.39,40 Under conditions of severe viral infection, this self-regulatory homeostasis may shift toward positive feedback amplification, driving the organism into a state of intense oxidative stress. The resulting oxidative DNA damage subsequently activates the intrinsic apoptotic pathway.41
Furthermore, patients in the mechanically ventilated group exhibited significantly elevated expression levels of ribosomal pathway-related genes, which showed a positive correlation with plasma cell proportions. This indicates an enhanced adaptive immune response in these individuals. The phenomenon may reflect higher viral loads and stronger antigenic stimulation in mechanically ventilated patients, thereby driving B cell differentiation into plasma cells and promoting the secretion of high-titer antibodies. Consistently our previous study also confirmed that severe COVID-19 patients experience prolonged viral persistence and higher viral loads, which is highly consistent with the observed immune activation pattern.42
In conclusion, we conducted the first integrated analysis of the respiratory microbiome and transcriptome in MV COVID-19 patients. Comparative analysis between mechanically ventilated and non-ventilated COVID-19 patients revealed that differentially expressed genes in multiple key immune-related pathways showed a significant negative association with the abundance of Streptococcus in the lower respiratory tract. Further analysis revealed that these differentially expressed genes were not only enriched in innate immune response, inflammatory response, and oxidative stress pathways, but also closely associated with the proportions of immune cell subsets (such as neutrophils, NKT cells, and Tregs). This suggests that Streptococcus may mitigate the progression of systemic inflammatory storms and oxidative damage by modulating the host innate immune barrier. Previous studies have reported that enrichment of Streptococcus enhances innate immunity and suppresses influenza virus infection,27 indicating a potential similar protective role in severe COVID-19 progression. However, whether Streptococcus attenuates disease progression by regulating host-microbial interactions requires further validation through functional intervention experiments in prospective cohorts.
Several limitations are present in this study. First, as a single-center study with a relatively small cohort size, this study has the potential for selection bias. Second, we were unable to collect specimens before and after MV for comparative analysis, limiting our ability to fully evaluate the impact of MV on the respiratory microbiome. Finally, although we identified characteristic microbial communities and their associated immune pathways, we did not perform mechanistic studies to further investigate the role and underlying mechanisms of the respiratory microbiota in this context.
Conclusions
This study found that the IMV group exhibited a significant decrease in the α-diversity of the pulmonary microbiota compared to the NMV group, with a notable reduction in the genus Streptococcus, primarily driven by decreases in Streptococcus oralis and Streptococcus mitis. Through transcriptomic analysis, pathway enrichment, immune infiltration, and Spearman correlation analysis, it was discovered that the genus Streptococcus may influence inflammation and oxidative stress-related pathways by regulating the expression of host differential genes and modulating immune cell infiltration patterns. These findings suggest that Streptococcus may inhibit excessive inflammatory responses by reshaping host immune homeostasis, thereby improving prognosis.
Data Sharing Statement
The datasets used and/or analysed during the current study are available from the corresponding author ([email protected]) on reasonable request.
Ethics Approval and Consent to Participate
The study was approved by the Ethics Committee of the First Affiliated Hospital, Zhejiang University School of Medicine, China (IIT20250125B). The study complies with the Declaration of Helsinki.
Acknowledgments
We thank Prof. Yu Chen, Renyong Guo and Bin Lou from Department of Laboratory Medicine, the First Affiliated Hospital, Zhejiang University School of Medicine for advice regarding the clinical study, data analysis, and preparation of the manuscript.
Author Contributions
Yuxia Li, Mingzhu Huang, Chang Liu and Zheying Mao are listed as co-first authors. All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysisand interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Zhejiang Provincial Natural Science Foundation for Distinguished Young Scholar (grant number LR23H200002).
Disclosure
The authors declare that they have no competing interests.
References
1. World health organization. Coronavirus disease (COVID-19) outbreak situation. Available from: https://www.who.int/emergencies/diseases/novelcoronavirus-2019.
2. Liu Y, Li J, Li P, et al. ARNLE model identifies prevalence potential of SARS-CoV-2 variants. Nat Mach Intell. 2025;7:18–17. doi:10.1038/s42256-024-00919-2
3. Ibrahim S, Siemieniuk RAC, Oliveros MJ, et al. Drug treatments for mild or moderate covid-19: systematic review and network meta-analysis. BMJ. 2025;389:e081165. doi:10.1136/bmj-2024-081165
4. Liu C, Wu K, Sun T, et al. Effect of invasive mechanical ventilation on the diversity of the pulmonary microbiota. Crit Care. 2022;26(1):252. doi:10.1186/s13054-022-04126-6
5. Kalil AC, Metersky ML, Klompas M, et al. Management of adults with hospital-acquired and ventilator-associated pneumonia: 2016 clinical practice guidelines by the infectious diseases society of America and the American Thoracic Society. Clin Infect Dis. 2016;63(5):e61–e111. doi:10.1093/cid/ciw353
6. American Thoracic Society; Infectious Diseases Society of America. Guidelines for the management of adults with hospital-acquired, ventilator-associated, and healthcare-associated pneumonia. Am J Respir Crit Care Med. 2005;171(4):388–416. doi:10.1164/rccm.200405-644ST.
7. Goligher EC, Ferguson ND, Brochard LJ. Clinical challenges in mechanical ventilation. Lancet. 2016;387(10030):1856–1866. doi:10.1016/S0140-6736(16)30176-3
8. Brodsky MB, Levy MJ, Jedlanek E, et al. Laryngeal injury and upper airway symptoms after oral endotracheal intubation with mechanical ventilation during critical care: a systematic review. Crit Care Med. 2018;46(12):2010–2017. doi:10.1097/CCM.0000000000003368
9. Papazian L, Klompas M, Luyt CE. Ventilator-associated pneumonia in adults: a narrative review. Intensive Care Med. 2020;46(5):888–906. doi:10.1007/s00134-020-05980-0
10. Ruff WE, Greiling TM, Kriegel MA. Host-microbiota interactions in immune-mediated diseases. Nat Rev Microbiol. 2020;18(9):521–538. doi:10.1038/s41579-020-0367-2
11. Shen Y, Yu F, Zhang D, et al. Dynamic alterations in the respiratory tract microbiota of patients with covid-19 and its association with microbiota in the gut. Adv Sci. 2022;9(27):e2200956. doi:10.1002/advs.202200956
12. Woo S, Park SY, Kim Y, Jeon JP, Lee JJ, Hong JY. The dynamics of respiratory microbiota during mechanical ventilation in patients with pneumonia. J Clin Med. 2020;9(3):638. doi:10.3390/jcm9030638
13. National health and family planning commission of the people’s republic of china. guideline for diagnosis and treatment of SARS-CoV-2. Available from: https://www.gov.cn/zhengce/zhengceku/2023-01/06/content5735343.htm.
14. Liu C, Cui Y, Cui Y, et al. Microeco an R package for data mining in microbial community ecology. FEMS Microbiol. Ecol. 2021;97(2):fiaa255. doi:10.1093/femsec/fiaa255
15. Segata N, Izard J, Waldron L, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12(6):R60. doi:10.1186/gb-2011-12-6-r60
16. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi:10.1186/s13059-014-0550-8
17. Wu T, Hu E, Xu S, et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation. 2021;2(3):100141. doi:10.1016/j.xinn.2021.100141
18. Wypych TP, Wickramasinghe LC, Marsland BJ. The influence of the microbiome on respiratory health. Nat Immunol. 2019;20(10):1279–1290. doi:10.1038/s41590-019-0451-9
19. Dicker AJ, Lonergan M, Keir HR, et al. The sputum microbiome and clinical outcomes in patients with bronchiectasis: a prospective observational study. Lancet Respir Med. 2021;9(8):885–896. doi:10.1016/S2213-2600(20)30557-9
20. Yan Z, Chen B, Yang Y, et al. Multi-omics analyses of airway host-microbe interactions in chronic obstructive pulmonary disease identify potential therapeutic interventions. Nat Microbiol. 2022;7(9):1361–1375. doi:10.1038/s41564-022-01196-8
21. Kelly BJ, Imai I, Bittinger K, et al. Composition and dynamics of the respiratory tract microbiome in intubated patients. Microbiome. 2016;4:7. doi:10.1186/s40168-016-0151-8
22. Sulaiman I, Chung M, Angel L, et al. Microbial signatures in the lower airways of mechanically ventilated COVID-19 patients associated with poor clinical outcome. Nat Microbiol. 2021;6(10):1245–1258. doi:10.1038/s41564-021-00961-5
23. Slutsky AS, Ranieri VM. Ventilator-induced lung injury. N Engl J Med. 2013;369(22):2126–2136. doi:10.1056/NEJMra1208707
24. Donlan RM. Biofilms and device-associated infections. Emerg Infect Dis. 2001;7(2):277–281. doi:10.3201/eid0702.010226
25. Kitsios GD, Yang H, Yang L, et al. Respiratory tract dysbiosis is associated with worse outcomes in mechanically ventilated patients. Am J Respir Crit Care Med. 2020;202(12):1666–1677. doi:10.1164/rccm.201912-2441OC
26. Bo L, Li J, Tao T, et al. Probiotics for preventing ventilator-associated pneumonia. Cochrane Database Syst Rev. 2014;2014(10):CD009066. doi:10.1002/14651858.CD009066.pub2
27. Zou X, Cao H, Hong L, et al. Enrichment of Streptococcus oralis in respiratory microbiome enhance innate immunity and protects against influenza infection. Signal Transduct Target Ther. 2025;10(1):272. doi:10.1038/s41392-025-02365-x
28. Chen J, Subbarao K. The Immunobiology of SARS. Annu Rev Immunol. 2007;25(1):443–472. doi:10.1146/annurev.immunol.25.022106.141706
29. Del Valle DM, Kim-Schulze S, Huang HH, et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat Med. 2020;26(10):1636–1643. doi:10.1038/s41591-020-1051-9
30. Hamed ME, Naeem A, Alkadi H, et al. Elevated expression levels of lung complement anaphylatoxin, neutrophil chemoattractant chemokine il-8, and rantes in mers-cov-infected patients: predictive biomarkers for disease severity and mortality. J Clin Immunol. 2021;41(7):1607–1620. doi:10.1007/s10875-021-01061-z
31. Li Q, Verma IM. NF-κB regulation in the immune system. Nat Rev Immunol. 2(10):725–734. doi:10.1038/nri910
32. Hariharan A, Hakeem AR, Radhakrishnan S, Reddy MS, Rela M. The role and therapeutic potential of nf-kappa-b pathway in severe covid-19 patients. Inflammopharmacol. 2021;29(1):91–100. doi:10.1007/s10787-020-00773-9
33. Lin HY, Wang WK, Lin CH, et al. The IL-8/NF-κB feedback loop confers a paclitaxel-sensitive/doxorubicin-resistant phenotype in triple-negative breast cancer. Free Radic Biol Med. 2025;238:316–328. doi:10.1016/j.freeradbiomed.2025.06.049
34. Galván-Peña S, Leon J, Chowdhary K, et al. Profound Treg perturbations correlate with COVID-19 severity. Proc Natl Acad Sci USA. 2021;118(37):e2111315118. doi:10.1073/pnas.2111315118
35. Vick SC, Frutoso M, Mair F, et al. A regulatory T cell signature distinguishes the immune landscape of COVID-19 patients from those with other respiratory infections. Sci Adv. 2021;7(46):eabj0274. doi:10.1126/sciadv.abj0274
36. Laforge M, Elbim C, Frère C, et al. Tissue damage from neutrophil-induced oxidative stress in COVID-19. Nat Rev Immunol. 2020;20(9):515–516. doi:10.1038/s41577-020-0407-1
37. Kundura L, Gimenez S, Cezar R, et al. Angiotensin II induces reactive oxygen species, DNA damage, and T-cell apoptosis in severe COVID-19. J Allergy Clin Immunol. 2022;150(3):594–603.e2. doi:10.1016/j.jaci.2022.06.020
38. LE V-VE, Bulthuis MLC, van der Gun BTF, et al. Systemic oxidative stress associates with the development of post-COVID-19 syndrome in non-hospitalized individuals. Redox Biol. 2024;76:103310. doi:10.1016/j.redox.2024.103310
39. Morgan MJ, Gang LZ. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011;21(1):103–115. doi:10.1038/cr.2010.178
40. Sies H, Berndt C, Jones DP. Oxidative Stress. Annu Rev Biochem. 2017;86(1):715–748. doi:10.1146/annurev-biochem-061516-045037
41. Yuan J, Ofengeim D. A guide to cell death pathways. Nat Rev Mol Cell Biol. 2024;25(5):379–395. doi:10.1038/s41580-023-00689-6
42. Zheng S, Fan J, Yu F, et al. Viral load dynamics and disease severity in patients infected with SARS-CoV-2 in Zhejiang province, China, January-March 2020: retrospective cohort study. BMJ. 2020:1443. doi:10.1136/bmj.m1443.
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Determinants of Pneumothorax Among Mechanically Ventilated COVID-19 Intensive Care Unit Patients, a Single Centre Study
Hundie TG, Alemu ZA, Getachew LZ, Abera LA, Seyoum AB, Mogus LS, Admasu NM, Regassa GB, Tilahun YB, Bareamichael PI, Tessema AG, Derese TN
Journal of Multidisciplinary Healthcare 2023, 16:3977-3989
Published Date: 11 December 2023
A Multicenter Study Assessing the Optimal Anticoagulation Strategies in COVID-19 Critically Ill Patients with New-Onset Atrial Fibrillation: Balancing Effectiveness and Safety
Al Sulaiman K, Aljuhani O, Korayem GB, Altebainawi AF, Alharbi A, Alalawi M, Joharji H, Almohsen RA, Faden RM, Alotaibi N, Alshalawi BS, Alkhushaym N, Alanazi FF, Alharbi A, Alqarni A, Samkari S, Alharbi B, Alshehab N, Alshehri RA, Vishwakarma R
International Journal of General Medicine 2024, 17:5611-5622
Published Date: 28 November 2024
Immune Cell-Based versus Albumin-Based Ratios as Outcome Predictors in Critically Ill COVID-19 Patients
Zeba S, Surbatovic M, Udovicic I, Stanojevic I, Vojvodic D, Rondovic G, Mladenovic K, Abazovic T, Hasanovic A, Ilic AN, Abazovic D, Khan W, Djordjevic D
Journal of Inflammation Research 2025, 18:73-90
Published Date: 3 January 2025
The Impact of Early Protein Advancement in Critically Ill Patients with COVID-19: A Multicenter Cardinality Matching Study
Alissa A, Korayem GB, Aljuhani O, AlFaifi M, Alnajjar LI, Souaan NAL, Albassam M, Alrayes A, Albishi S, Alqahtani RA, Alharthi AF, Alkhushaym N, Alhammad MA, Vishwakarma R, Alharbi A, Alsohimi S, Ababtain A, Al-Dorzi HM, Alqahtani R, Almuaither G, Alarifi LA, Almutairi A, Alharbi MB, Alghamdi AA, Alhmoud MF, Al Sulaiman K
Therapeutics and Clinical Risk Management 2025, 21:177-189
Published Date: 21 February 2025
Clinical Characteristics and Prognostic Factors of Severe COVID-19
Yang Y, Huang Z, Pei Y, Huang Y, Chen C, Zhou T
Infection and Drug Resistance 2025, 18:3117-3125
Published Date: 25 June 2025