Back to Journals » OncoTargets and Therapy » Volume 19
The Role of DNA Methylation in Osteosarcoma Pathogenesis and Therapy
Received 15 November 2025
Accepted for publication 19 February 2026
Published 23 February 2026 Volume 2026:19 581744
DOI https://doi.org/10.2147/OTT.S581744
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr John Maher
Xiaolan Zhou,1 Bingbing Li,1 Peigan Tao2
1Department of Pediatrics, Shaoxing Central Hospital, Affiliated Central Hospital of Shaoxing University, Shaoxing City, Zhejiang Province, 312030, People’s Republic of China; 2Department of General Practice, Shaoxing Central Hospital, Affiliated Central Hospital of Shaoxing University, Shaoxing City, Zhejiang Province, 312030, People’s Republic of China
Correspondence: Peigan Tao, Email [email protected]
Abstract: Osteosarcoma (OS) is a highly malignant bone tumor primarily affecting children and adolescents. Clinical treatment has consistently encountered challenges, including chemotherapy resistance, high recurrence rates, and metastasis. Research has demonstrated that epigenetic regulation, particularly DNA methylation, can stably modify the DNA sequence without altering it, playing a key role in the development and progression of OS. Compared with normal tissue, OS exhibits distinctive alterations in DNA methylation, characterized by genome-wide hypomethylation and hypermethylation of specific gene promoter regions. This “dual pattern” not only promotes tumor proliferation, invasion, and metastasis but also maintains cancer stem cell characteristics and modulates the tumor immune microenvironment (TIME). Molecular classification based on DNA methylation profiles offers a new tool for the diagnosis and prognosis of OS. Drugs targeting DNA methylation, such as decitabine, have shown promising results for reversing gene silencing and suppressing tumor progression. This article systematically reviews the core mechanisms by which DNA methylation contributes to OS development, progression, and metastasis, and examines its potential for clinical translation.
Keywords: biomarkers, DNA methylation, epigenetics, osteosarcoma, targeted therapy, tumor microenvironment
Introduction
OS is an ordinary malignant bone tumor that mainly occurs among children and adolescents.1 Owing to its fast growth rate, strong metastatic potential, and intricate tumor microenvironment (TME), the tumor is highly malignant.2 According to an epidemiological survey conducted in the year 2009, the incidence rates observed among males younger than 24 years old were 4.3 among each million and slightly lower among females.3 Currently, the mainstay of treatment involves neoadjuvant chemotherapy followed by surgical resection and adjuvant chemotherapy. Surgery and several chemotherapy regimens are the standard clinical management strategies. For far-distant metastatic patients, the survival rate is around 5 years about 20%.4,5 Despite early diagnosis, the five-year survival rate is merely around 60%, and the patients tend to encounter invasive therapies, including amputation.6,7 Though the therapies increased the survival rates through these therapies, the complications like chemotherapy resistance and elevated recurrences remain and signify the necessity to boost the effectiveness and quality of life among the patients.8,9 Various studies are now working on the immunotherapies treating the OS, including the cGAS-STING signaling pathway. However, some underlying mechanisms remain undefined and the translation to clinic still poses difficulties. Early, accurate, and effective therapeutic remedies against the OS are therefore required on an emergency basis.10
With the explosion of epigenetics, interest has intensified on interactions between epigenetic mechanisms and variations of the DNA nucleotide sequence and also on consequences thereof on biology, disease and evolution. Epigenetics is alterations to the molecules of the DNA to control the activity of the genes without modifying the nucleotide sequence.11
Epigenetic changes have become an overriding mechanism leading to the development of numerous diseases.12 They control gene expression by varying the local and global accessibility of the epigenetic codes within the chromatin and therefore affect various physiological and pathological processes.11,13,14 In the progression of cancer, particularly the earlier phases, aberrant metabolic processes and environmental exposures commonly cause epigenetic changes. Indeed, modification such as DNA methylation, histone modification, microRNA expression, and nucleosome remodeling are specifically critical. They cause the disruption of fine regulation within the genome within cells and result in carcinogenic processes involving the unassociated division of cells, unassociated differentiation, and evasion from apoptosis.15 Therefore, epigenetic changes represent the hallmarks of cancer.16–19
DNA methylation is an extremelystable epigenetic mark that greatly affects gene expression and controls the phenotype and working of cells by quelling genes without the altering of the DNA nucleotide sequences.20 DNA methylation comprises various sorts, such as 5mC, 6mA, and 4mC,21,22 where among them 5mC is predominant incellular organisms.23 This biochemical process involves the covalent attachment of a methyl group from S-adenosylmethionine (SAM) to the fifth carbon atom of cytosine, forming 5-methylcytosine (5mC), catalyzed by enzymes known as DNA methyltransferases (DNMTs). This reaction predominantly occurs at cytosines within CpG dinucleotides, where cytosine is paired with guanine.24,25 The DNMT enzyme family includes Dnmt1, Dnmt3a, and Dnmt3b. Another member, Dnmt3L, lacks a catalytic domain and thus has no enzymatic activity.26,27 DNMT3a and DNMT3b are de novo methyltransferases, which establish new methylation patterns at previously unmethylated CpG sites during embryonic development and cell differentiation, thereby setting initial gene expression states.28 Although Dnmt3L has no catalytic function, it can interact with DNMT3a and DNMT3b to enhance their methylation activity.29 DNMT1 is a maintenance methyltransferase that specifically recognizes and binds to hemimethylated DNA after replication, methylating corresponding cytosines in daughter strands.30 This ensures faithful inheritance of epigenetic information during cell division and preserves cellular identity24,31 (Figure 1).
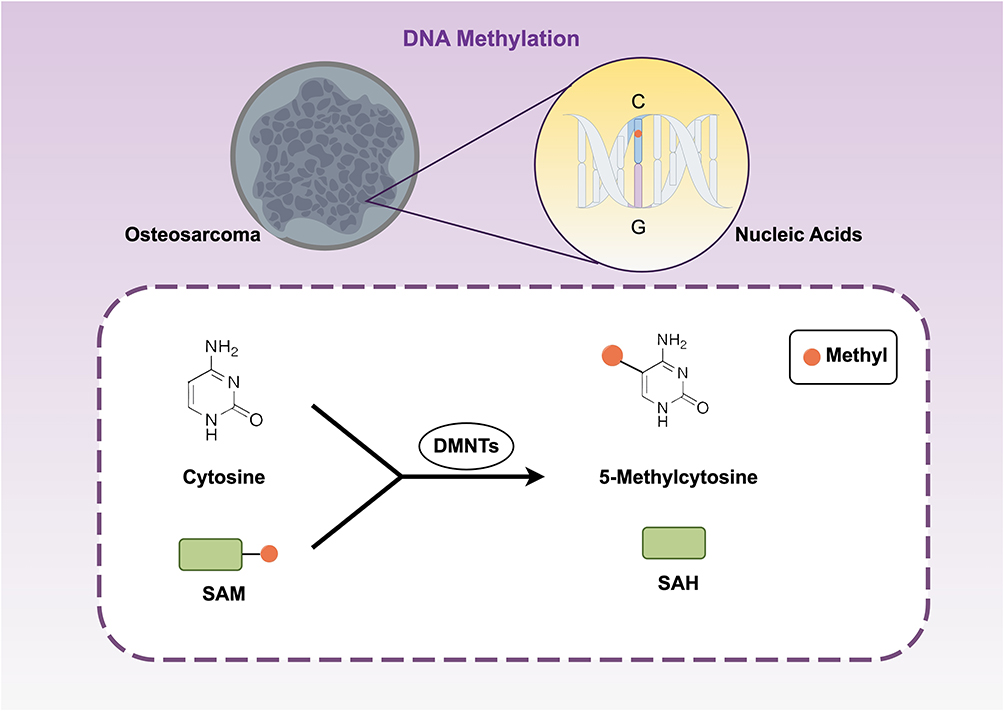 |
Figure 1 Under DNMT catalysis, cytosine bases are converted into 5mC. A methyl group from the donor SAM is covalently attached to the fifth carbon atom of cytosine, primarily at CpG dinucleotide sites, generating SAH as a byproduct. |
DNA methylation distribution across the genome is not uniform, exhibiting significant regional specificity associated with various biological functions. CpG dinucleotide distribution is highly uneven: while most mammalian DNA lacks CpG dinucleotides,32 specific genomic regions, known as CpG islands (CGIs), have high G+C content and near-expected CpG frequencies.33 Bioinformatically, CGIs are typically defined as DNA segments longer than 200 bp, with G+C content exceeding 50%, and an observed-to-expected CpG ratio of at least 0.6.33 Most CpG sites outside CGIs are methylated, whereas gene promoters typically contain unmethylated CGIs, enabling transcription. Approximately 50,200 CGIs exist within the human genome.34 Increased CpG sites are often linked to hypomethylation, as these sites are protected from methyltransferases by DNA-binding proteins like Sp1.35,36 Promoter hypermethylation is a major dysregulation mechanism in cancer, silencing about 70% of genes with CpG islands.37,38 Hypermethylation physically prevents transcription factor binding and recruits methyl-CpG-binding proteins (MBDs) and histone-modifying enzymes (eg, HDACs), leading to chromatin condensation and transcriptional silencing.39 In contrast to promoters, gene bodies often exhibit moderate methylation, positively correlated with active gene expression. Although it does not impede transcriptional elongation, gene body methylation may influence RNA splicing.40,41 Furthermore, methylation of repetitive sequences and transposable elements is essential for transcriptional repression and genomic stability.42
Epigenetic variation is common in cancer.43,44 Dysregulated expression of oncogenes and tumor suppressor genes plays a central role in cancer, driven significantly by epigenetic abnormalities.19 DNA methylation changes are particularly prominent and occur more frequently than genetic mutations. Therefore, epigenetic changes are promising therapeutic targets and consistent clinical diagnosis markers. Epigenetic changes can be involved in the progression of OS and can serve potential indicators of prediction and therapy.45 This review demystifies the distinct roles played by DNA methylation within OS, highlighting key signaling pathways and corresponding genes and discusses its translational clinical potential and future research direction.
DNA Methylation in OS Pathogenesis
Global Methylation Patterns
DNA methylation, an important epigenetic modification, has a distinct “dual pattern” expression during the development and progression of OS: genome-wide hypomethylation and localized hypermethylation. Compared to normal tissue, OS has more differentially methylated sites.46 Another study detected 2845 differentially methylated sites within the OS specimens, of which 1379 were hypermethylated and 169 hypomethylated promoter sites. Such two apparently opposite processes work together to promote tumor progression.47–49 Genome-wide hypomethylation primarily happens within non-coding areas. This ubiquitous hypomethylation directly destroys genome stability because the hypermethylation of repeat sequences usually keeps repeat sequences and chromosomes transcriptionally silenced. Removing the methylation activates the genomic “dark matter” to cause chromosomal translocations, gene fusions and aneuploidy. Besides, hypomethylation can indirectly activate proto-oncogenes. Demethylation within the promoters of the oncogenes eliminates the actedness of transcription repression and thus stimulates the expression of the genes to cause uncontrolled proliferation of the cells.
In contrast, hypermethylation predominantly occurs in CpG islands, especially in gene promoter regions.47 CpG islands are normally unmethylated and allow gene transcription. However, aberrant hypermethylation attracts the binding of methylation-binding proteins and histone deacetylases and results in the formation of bulky heterochromatin complexes. This physically impedes the binding of transcription factors, leading to permanent gene silencing. Large-scale epigenomic data confirm the observation. For example, about 3.8% and 2.2% of the CpG sites are respectively hypermethylated and hypomethylated within OS tissues. This aberrant pattern is more intensified within recurrent or metastatic specimens and is highly correlated with the degree of disease aggressiveness and adverse outcome.50 Promoter CpG island hypermethylation is the prevailing mechanism responsible for tumor suppressor gene silencing commonly illustrated within the malignancies of human beings, including OS.51 Model studies involving the OS cell lines U2OS and MG63 experimentally confirmed the pattern of this methylation. Deep high-throughput sequencing identified hundreds of gene promoters with aberrant hypermethylation and hypomethylation. They included genes related to cell cycle regulation, DNA repair, apoptosis, signal transduction pathways, differentiation, and invasion. For example, cyclin-dependent kinase inhibitor gene (CDKN2A) promoter hypermethylation abolishes cell cycle check-points, and MGMT gene silencing impairs chemosensitivity against chemotherapy agents.48
Importantly, these “dual patterns” mutually interact extensively. Genome-wide hypomethylation-induced relaxed chromatin structure is postulated to facilitate an favored “epigenetic landscape” for localized hypermethylation to induce malignant transformation. Consequently, the simultaneous existence of worldwide hypomethylation and localized hypermethylation is the defining epigenetic characteristic of OS. It accounts for primary tumor behaviors, such as unlimited proliferation, escape from growth inhibition, and anti-apoptosis. This opens to clinicians new avenues: methylation profiles can be used as biomarkers to diagnose and categorize diseases and predict progression. Furthermore, because DNA methylation is reversible, demethylating agents are promising therapeutic interventions to restore tumor suppressor gene expression, break through chemotherapy resistance, and suppress metastasis.
Methylation of Genes in Key Signaling Pathways
In OS, DNA methylation precisely controls key signaling pathways responsible for its malignant phenotype. Hypermethylation of gene promoters can inactivate crucial tumor suppressor pathways, supporting tumor proliferation, survival, invasion, and metastasis. It is important to note that several well-characterized mechanisms of methylation-mediated silencing, identified primarily in other cancers, provide valuable references for understanding epigenetic dysregulation in OS. For instance, studies across various solid tumors have shown that hypermethylation of the tissue inhibitor of metalloproteinases 3 (TIMP3) promoter silences its expression, leading to impaired natural inhibition of matrix metalloproteinases (MMPs) and disruption of angiogenic balance. This mechanism may similarly contribute to promoting angiogenesis, local invasion, and distant metastasis in OS. Likewise, research in other cancer models indicates that hypermethylation of the DNA repair protein O6-methylguanine-DNA methyltransferase (MGMT) reduces cellular capacity to repair DNA damage, thereby accelerating genomic mutation accumulation and tumor evolution. The potential role of this pathway in OS warrants investigation. Furthermore, studies have demonstrated that expression of death-associated protein kinase (DAPK) can be suppressed by hypermethylation, resulting in apoptosis evasion and chemotherapy resistance. This offers clues for exploring therapeutic resistance mechanisms in OS. These findings from other tumor models illustrate the general role of methylation-mediated inactivation of tumor suppressor genes in driving malignant transformation, providing significant insights for osteosarcoma research.52–58
Besides classical tumor suppressors, OS progression also depends on abnormal activation of major signaling pathways. The Wnt/β-catenin pathway regulates tumor immunity, bone remodeling, angiogenesis, hypoxia response, and epithelial-mesenchymal transition (EMT).59 Aberrant activation involves precise DNA methylation regulation. For example, the Wnt pathway inhibitor gene APCDD1 undergoes hypermethylation mediated by DNMT3a, leading to pathway activation, increased invasion, and EMT.60
Research has expanded from protein-coding genes to non-coding RNAs (ncRNAs), whose dysregulation is closely linked to tumor progression.61 Their regulatory mechanisms often involve epigenetic modifications. Some long non-coding RNAs (lncRNAs) recruit DNMTs to specific gene promoters, modifying methylation levels and downstream gene expression.62 In OS, lncRNA THAP9-AS1 recruits DNMTs to silence suppressor of cytokine signaling 3 (SOCS3), a negative regulator of the JAK2/STAT3 pathway, enhancing tumorigenesis, metastasis, and oxidative stress resistance.63 Similarly, DNA methylation regulates microRNAs (miRNAs), which modulate signaling pathways and invasive behaviors, representing therapeutic targets64–66 (Figure 2).
 |
Figure 2 Cancer genomes exhibit a “dual pattern” of DNA methylation, characterized by genome-wide hypomethylation coexisting with hypermethylation at specific gene promoters. This pattern drives tumor proliferation, invasion, and metastasis by regulating key genes and reshaping the TIME. Genes such as APCDD1, and TIMP3 are silenced by promoter hypermethylation, affecting signaling pathways including Wnt/β-catenin, JAK/STAT, and PI3K/Akt, thereby promoting tumor progression. Conversely, genes like IRX1 are activated by promoter hypomethylation, further contributing to tumor progression. These mechanisms collectively form the core epigenetic regulatory network in cancer. |
Methylation and OS Stem Cells (OSCs)
Progresses in the epigenetic evaluation technology have further elaborated the function of DNA methylation involvement in the maintenance and differentiation of stem cells.67 Epigenetic processes manage the expression of genes during the differentiation and self-renewal of stem cells by adjusting the structure of chromatin. Among these processes, the study on DNA methylation is highly intensive.68 Clinical challenges in OS, such as chemotherapy resistance, relapse, and heterogeneity of tumors, are all related to OSCs. They are thought to be responsible for tumor initiation, maintenance, progression, and chemotherapy failure. OSCs’ self-renewal, tumorigenicity, and chemotherapy resistance are controlled not just by genetic mutations but also by epigenetics, particularly by DNA methylation. DNA methylation downregulates tumor suppressor genes and key pathways involving differentiation control and cell cycle check points and apoptosis and thus widens the stemness of OSCs. This sheds new evidence on OS progression and chemotherapy resistance.
Recent research has elucidated the roles of distinct molecular pathways. For example, the aberrant DNMT1 activation sustains OSC stemness by facilitating the hypermethylation of the miR-34a promoter and thereby silencing its expression. miR-34a is an established tumor suppressor protein inhibiting self-renewal and proliferation through the targets stemness-associated proto-oncogenes Notch and Bcl-2. Consequently, the silencing of miR-34a is therefore augmented OSC stemness by freeing its inhibition on downstream signaling pathways. This finding explains the key involvement of the DNMT1/miR-34a axis in OSC biology and lends themselves to the exploitation in the generation of new therapeutic interventions aimed at OSCs.69
Exogenous molecules also affect OSC characteristics by altering DNA methylation profiles. For instance, the cardiac glycoside ouabain suppresses Na+/K+-ATPase and activates intracellular signaling pathways in OS cells. Ouabain increases intracellular sodium levels, subsequently raising intracellular calcium (Ca2+) by the sodium-calcium exchanger (NCX). Calcium signaling thus generated alters calcium-dependent enzymes and causes massive DNA methylation alterations at stemness transcription factor promoter areas including Oct4, Sox2, and Nanog. Epigenetic reprogramming by these changes downregulates these genes and decreases stemness and chemotherapy resistance. Pharmacological intervention therefore on DNA methylation assumes significance as an attractive strategy to suppress OS progression and metastasis.70
Continued research on the involvement of DNA methylation in cell fate determination will clarify the differentiation barrier of OS. DNMT3B controls the fate of mesenchymal stem cells through the hypermethylation of the tumor suppressor gene PTEN’s promoter. PTEN silence stimulates the PI3K/Akt pro-survival signaling pathway and suppresses osteoblastic differentiation to preserve cells within an undifferentiated and proliferative state. Such an epigenetic mechanism is potential bait to be pursued in bone regeneration studies and clarifies PTEN silencing commonly seen within OS. PTEN promoter hypermethylation is the leading cause of malignant proliferation, dysfunctional differentiation, and stem-like characteristics’ acquirement within OS cells.71
In summary, DNA methylation controls the non-coding RNAs, key stemness transcription factors, and essential signaling pathways. Such an intricate control system accurately regulates OSC stemness, self-renewal, differentiation inhibition, and therapeutically induced resistance.
DNA Methylation and OS Progression and Metastasis
Methylation of Genes Related to Invasion and Metastasis
Though chemotherapy and surgery have improved, the five-year survival rate is low for metastatic OS. So there is an imperative to identify biomarkers to allow early diagnosis and to introduce new therapeutic strategies. IRX1 (Iroquois homeobox 1) is an OS pro-metastatic gene induced by DNA hypomethylation. Hypomethylation results in the increased expression of IRX1 and highly elevated the migration and invasion characteristics of tumor cells and anti-apoptosis by the activation of the CXCL14/NF-κB signaling pathway and thereby induced lung metastasis. Hypomethylation of IRX1 can be used as an indication marker to predict metastasis and by restoring its methylation status new anti-metastasis therapies can be introduced.72,73
It is important to note that the aforementioned methylation alterations in these genes do not occur in isolation; environmental factors play a significant driving role. Exposure to environmental factors such as carcinogens, radiation, or specific nutritional factors can induce aberrant DNA methylation patterns either genome-wide or at specific gene loci.74–76 This occurs by interfering with DNA methyltransferase activity or affecting the availability of methyl donors. Environmental carcinogens may directly cause DNA damage and trigger abnormal localized methylation reprogramming, while radiation could indirectly affect the epigenetic regulatory network through pathways like oxidative stress. These environmentally induced methylation changes may act as early events, synergizing with genetic variations to collectively shape the epigenomic landscape of osteosarcoma, thereby promoting tumor initiation, invasion, and metastasis. Therefore, incorporating consideration of environmental factors is essential for understanding the etiology behind methylation changes in osteosarcoma, providing a broader perspective for disease prevention and risk intervention.
Methylation and TME and Extracellular Matrix (ECM)
DNA methylation has an important determinant role to play in the development and progression of cancers. Its involvement is not limited to tumor cells but is also fundamentally important to the shape the TME takes, specifically by shaping localized immune responses.77–79 DNA methylation controls the expression of genes related to immunity precisely. Modulations within the methylation state at gene promoters/regulator elements can cause altered gene expression to produce specific DNA methylation patterns (IMPs). According to these patterns, OS can be stratified by immune subtypes: an immunosuppressive TME and an immune-activated TME.
In the immune-activated TME, specific DNA methylation patterns promote the upregulation of genes beneficial to anti-tumor immunity. For example, the promoters of chemokines (CXCL9, CXCL10) and antigen-presenting molecules (HLA family genes) exhibit hypomethylation. This increases their transcription, enhancing recruitment, infiltration, and activation of CD8+ T cells and NK cells at the tumor site, thus establishing an effective anti-tumor immune response. Clinical studies show that this immune-activated TME significantly correlates with improved treatment response and prognosis in OS. In contrast, the immunosuppressive TME features abnormal DNA methylation, causing dysregulated expression of immunosuppressive genes. Specifically, DNA methylation promotes tumor immune escape through the regulation of multiple key mechanisms: Promoters of immune checkpoint molecules and anti-inflammatory cytokines may become demethylated and upregulated, while pro-inflammatory genes may be silenced by hypermethylation. This epigenetic imbalance, particularly the abnormal demethylation and upregulated expression of immune checkpoint molecules such as PD-L1 and CTLA-4, directly weakens the immune system’s ability to attack tumor cells. Simultaneously, it recruits and activates immunosuppressive cells, including γδ T cells, naïve CD4+ T cells, regulatory T cells (Tregs), and myeloid-derived suppressor cells (MDSCs). These cells inhibit cytotoxic T and NK cell functions through cytokine secretion or direct cell interactions, creating a highly immunosuppressive microenvironment. This TME protects tumor cells, promotes tumor growth, angiogenesis, invasion, and metastasis, and closely associates with poor patient prognosis.80 Therefore, DNA methylation plays a pivotal role in shaping the TME by precisely regulating the expression of immune checkpoints, immunosuppressive factors, and related cellular functions. It determines the intensity of the immune response and mediates interactions between tumor cells and stromal immune cells. Together, these mechanisms constitute critical pathways of immune escape in osteosarcoma.
Beyond the immune compartment, DNA methylation also critically regulates the extracellular matrix, a key non-cellular component of the TME that provides structural support and biochemical cues essential for tumor progression.81 Aberrant DNA methylation can directly influence the expression of genes encoding ECM components and ECM-remodeling enzymes, such as matrix metalloproteinases (MMPs) and lysyl oxidases (LOXs).82 Promoter hypomethylation of specific genes can lead to their overexpression, enhancing ECM degradation and facilitating local invasion and metastasis of osteosarcoma cells.83 This epigenetic reprogramming of the ECM creates a physical path for tumor spread and releases growth factors and alters mechano-signaling, further driving proliferation and survival. Therefore, the interplay between DNA methylation and ECM dynamics is a crucial layer of regulation in osteosarcoma invasion and metastasis, warranting further investigation as a potential therapeutic target.
Clinicopathological Correlations
For many years, tissue morphology has served as the primary tool in anatomical pathology for cancer diagnosis, particularly in bone tumors.84 However, some tumors share similar morphologies and are difficult to distinguish. DNA methylation patterns simultaneously reflect the cell of origin and tumorigenesis-related changes, enabling precise identification of histological subtypes.85,86 Genome-wide methylation profiling combined with machine learning methods has facilitated the creation of various clinicopathological classifiers, increasingly employed as diagnostic tools for diverse tumors.87 Researchers developed a sarcoma classifier based on methylation profiles, including OS and normal tissues, detecting CpG methylation levels for accurate pathological classification and prognosis prediction.88–90 This classifier demonstrated high accuracy in pediatric sarcomas.91 For example, methylation profiles distinguish undifferentiated round cell sarcomas (URCS) harboring the EWSR1–NFATc2 fusion from Ewing sarcoma (EwS) with the classic TET–ETS fusion.92 Integration of methylation profiles with WHO classification identified four correspondence types: complete matches with WHO entities, intra-entity heterogeneity, merging similar biological entities, and identifying new disease types. This molecular classification has strong correlations to clinical behaviors in OS, individual subtypes of methylation predict various tumor grades and malignancy. High-risk patterns of methylation are also often observed in advanced and metastatic examples. Also, categories of methylation can be an independent category used to predict prognosis. For example, TLR4 promoter status of the methylation highly correlates to the five-year event-free survival and is therefore an important marker to predict recurrence50. Despite the addition of data on the methylation along with CNV analysis to better predict chemotherapy resistance and metastatic potential and overall survival is also improved by adding CNV analysis to the data on the methylation.88 Consequently, DNA methylation Profiling is an effective instrument that not only aide accurate diagnosis and classification of the OS but also is an important differentiator to be used to make clinical decisions on risk stratification and individualized therapy.
Clinical Translational Applications
Prognostic and Predictive Markers
DNA methylation is an important epigenetic mechanism with immense clinical translational potential in OS, acting as an epigenetic biomarker to predict prognosis and monitor therapeutic efficacy. One study involving database integration identified three critical methylation-driven genes: COL13A1, MXI1, and TBRG1.93 Type XIII collagen is encoded by COL13A1 and is a hypermembrane protein whose reduced expression is caused by its hypermethylation to facilitate tumor advancement through the enhancement of cell invasion. MXI1 is a tumor suppressor that regulates growth and differentiation through competitive inhibition to form the MYC-MAX complex and has its hypermethylation related to adverse prognosis.94–96 TBRG1 is a growth inhibitor and has tumor-suppressive action through the induction of the p53 action and chromosomal stabilization but has its action inhibited by its hypermethylation.97 According to mRNA expression of these genes, an mRNA expression-derived risk score model with strong predictive power was established. Characterization of these methylation-promoted biomarkers sheds light on OS epigenetic mechanisms and initiates an innovative clinical risk prediction tool. Another study generated an assay to generate a risk prediction model from the basis of the data on the methylation data through nine genes (ARHGAP9, CADM1, CPE, DUSP3, FGFR1, GALNT3, IGF2BP3, KIF26A, and ZFP3) that showed high prognostic ability.98 Likewise, four DNA methylation loci-based prognostic models were generated by Zhang et al.99 Successful predictive capability by these models suggests the potential to monitor the gene-specific methylation status to allow future individualized therapeutic efficacy monitoring and tailoring.93 Looking ahead, the integration of emerging technologies such as single-cell sequencing and spatial transcriptomics holds great promise for advancing DNA methylation research in osteosarcoma. These cutting-edge approaches enable the precise mapping of methylation changes at the single‑cell resolution and within specific tissue architectures, thereby uncovering spatial and cellular heterogeneity in the tumor microenvironment. By correlating methylation profiles with transcriptional activity and cellular phenotypes in situ, these tools may refine prognostic models, identify novel methylation-driven subpopulations, and ultimately guide more personalized therapeutic strategies.
Targeting DNA Methylation
Decitabine, a DNA methylation inhibitor, reverses hypermethylation of gene promoters in OS, thus inhibiting tumor proliferation and metastasis. Decitabine also reverses methylation of the GADD45A gene’s 5′ CpG island, activating its expression and re-establishing the GADD45A-mediated apoptotic pathway, specifically inducing OS cell apoptosis.100 Additionally, decitabine restores expression of the estrogen receptor α (ERα) by reversing promoter hypermethylation, thus activating osteogenic differentiation and reducing proliferation and metastasis markers.101 Furthermore, decitabine upregulates expression of the NKG2D ligand genes MICB and ULBP1, enhancing γδ T-cell-mediated immune recognition and killing of OS cells via the NKG2D–NKG2DL axis.102 Azacitidine, another DNA methylation inhibitor, similarly reverses or blocks aberrant methylation in cancer cells.103 In chemotherapy-resistant OS, azacitidine combined with methionine restriction therapy (o-rMETase) effectively inhibited tumor growth by reducing the methylation capacity essential for cancer cell survival.104 Thus, DNA methylation inhibitors like decitabine and azacitidine provide comprehensive anti-OS effects, ranging from proliferation inhibition and induction of apoptosis and differentiation to immune activation105 (Table 1).
 |
Table 1 Mechanisms of DNA Methylation-Targeting Agents in OS via Gene Regulation |
However, demethylating drugs alone may be insufficient for permanently reversing highly stable epigenetic states. Stable DNA methylation states depend on complex regulatory circuits. Studies indicate that imprinting control regions exhibit “epigenetic bistability”, where methylation states are maintained by positive feedback involving DNA-binding factors and chromatin modification complexes.106–108 Future strategies could target specific components of these stable complexes, enabling more precise and lasting desilencing of tumor suppressor genes, and providing new therapeutic approaches for OS treatment. Although preclinical studies demonstrate the potential of DNA methylation-targeting drugs, their clinical translation in osteosarcoma faces three major challenges. Current agents are broad-spectrum demethylating agents that may non-specifically activate proto-oncogenes and cause off-target effects. These drugs often induce side effects such as myelosuppression and gastrointestinal reactions, limiting dosage and duration, with increased risks when combined with chemotherapy. Tumor cells may develop resistance through upregulation of drug-metabolizing enzymes, altered drug transport, or activation of compensatory epigenetic pathways. Therefore, future research should focus on improving target specificity, optimizing dosing regimens to balance efficacy and toxicity, and exploring combination strategies to overcome resistance, thereby advancing the safe and effective application of such epigenetic therapies in osteosarcoma.
Conclusions
In summary, DNA methylation, as a crucial epigenetic regulatory mechanism, plays a multifaceted and central role in the initiation and progression of osteosarcoma. It directly regulates the expression of numerous key genes, including tumor suppressor genes, DNA repair genes, and metastasis-related genes. This regulation occurs through promoter hypermethylation leading to gene silencing or hypomethylation activating proto-oncogenes, thereby driving tumor proliferation, invasion, metastasis, and therapy resistance. DNA methylation profiling demonstrates significant advantages in OS research, providing powerful tools for pathological classification, subtype identification, prognostic prediction, and treatment response evaluation.109–111 Studies indicate that methylation profiling offers independent and more robust chemotherapy-response predictions than other molecular markers, such as miRNAs. Methylation profiles effectively complement other molecular patterns and can jointly identify clinically relevant tumor subtypes.112 However, clinical translation faces several challenges. The primary issue is that current risk-prediction models based on multi-gene methylation markers mainly rely on small cohorts, lacking validation through large-scale, multicenter, prospective studies, thus limiting their reliability and universality.112,113 Additionally, many studies remain at the bioinformatics analysis stage without subsequent functional validation experiments, making the clinical significance of these findings uncertain.49 Future research must expand sample sizes and conduct multicenter validations to enhance the predictive efficacy of methylation biomarkers. Moreover, integrating functional experiments to clarify the mechanisms by which key methylation events drive OS progression is essential for converting biomarkers into therapeutic targets. Ultimately, combining DNA methylation data with other omics data to establish comprehensive molecular classification systems will facilitate precision diagnosis and treatment of OS.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Simpson E, Brown HL. Understanding osteosarcomas. JAAPA off J Am Acad Physician Assist. 2018;31(8):15–13. doi:10.1097/01.JAA.0000541477.24116.8d
2. Iacobescu GL, Corlatescu AD, Costin HP, Spiridonica R, Popa MIG, Cirstoiu C. Molecular and glycosylation pathways in osteosarcoma: tumor microenvironment and emerging strategies toward personalized oncology. Curr Issues Mol Biol. 2025;47(8):629. doi:10.3390/cimb47080629
3. Mirabello L, Troisi RJ, Savage SA. International osteosarcoma incidence patterns in children and adolescents, middle ages and elderly persons. Int, J, Cancer. 2009;125(1):229–234. doi:10.1002/ijc.24320
4. Guo J, Reddick WE, Glass JO, et al. Dynamic contrast-enhanced magnetic resonance imaging as a prognostic factor in predicting event-free and overall survival in pediatric patients with osteosarcoma. Cancer. 2012;118(15):3776–3785. doi:10.1002/cncr.26701
5. Han G, Wang Y, Bi WZ, et al. Magnetic resonance imaging is appropriate for determining the osteotomy plane for appendicular osteosarcoma after neoadjuvant chemotherapy. Med Oncol. 2012;29(2):1347–1353. doi:10.1007/s12032-011-9861-8
6. Misaghi A, Goldin A, Awad M, Kulidjian AA. Osteosarcoma: a comprehensive review. SICOT-J. 2018;4:12. doi:10.1051/sicotj/2017028
7. Luetke A, Meyers PA, Lewis I, Juergens H. Osteosarcoma treatment - where do we stand? A state of the art review. Cancer Treat Rev. 2014;40(4):523–532. doi:10.1016/j.ctrv.2013.11.006
8. Meltzer PS, Helman LJ. New horizons in the treatment of osteosarcoma. N Engl J Med. 2021;385(22):2066–2076. doi:10.1056/NEJMra2103423
9. Jafari F, Javdansirat S, Sanaie S, et al. Osteosarcoma: a comprehensive review of management and treatment strategies. Ann Diagn Pathol. 2020;49:151654. doi:10.1016/j.anndiagpath.2020.151654
10. Li B, Zhang C, Xu X, Shen Q, Luo S, Hu J. Manipulating the cGAS-STING axis: advancing innovative strategies for osteosarcoma therapeutics. Front Immunol. 2025;16:1539396. doi:10.3389/fimmu.2025.1539396
11. Li Y. Modern epigenetics methods in biological research. Methods. 2021;187:104–113. doi:10.1016/j.ymeth.2020.06.022
12. Miranda Furtado CL, Dos Santos Luciano MC, Silva Santos RD, Furtado GP, Moraes MO, Pessoa C. Epidrugs: targeting epigenetic marks in cancer treatment. Epigenetics. 2019;14(12):1164–1176. doi:10.1080/15592294.2019.1640546
13. Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi:10.1016/j.cell.2007.02.005
14. Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429(6990):457–463. doi:10.1038/nature02625
15. Werner RJ, Kelly AD, Issa JPJ. Epigenetics and precision oncology. Cancer J. 2017;23(5):262–269. doi:10.1097/PPO.0000000000000281
16. Murphy SK, Jirtle RL. Imprinting evolution and the price of silence. BioEssays News Rev Mol Cell Dev Biol. 2003;25(6):577–588. doi:10.1002/bies.10277
17. Das R, Hampton DD, Jirtle RL. Imprinting evolution and human health. Mamm Genome off J Int Mamm Genome Soc. 2009;20(9–10):563–572. doi:10.1007/s00335-009-9229-y
18. Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150(1):12–27. doi:10.1016/j.cell.2012.06.013
19. Ilango S, Paital B, Jayachandran P, Padma PR, Nirmaladevi R. Epigenetic alterations in cancer. Front Biosci Landmark Ed. 2020;25(6):1058–1109. doi:10.2741/4847
20. Nishiyama A, Nakanishi M. Navigating the DNA methylation landscape of cancer. Trends Genet. 2021;37(11):1012–1027. doi:10.1016/j.tig.2021.05.002
21. Chen K, Zhao BS, He C. Nucleic acid modifications in regulation of gene expression. Cell Chem Biol. 2016;23(1):74–85. doi:10.1016/j.chembiol.2015.11.007
22. Zhang G, Huang H, Liu D, et al. N6-methyladenine DNA modification in drosophila. Cell. 2015;161(4):893–906. doi:10.1016/j.cell.2015.04.018
23. Jones PA, Takai D. The role of DNA methylation in mammalian epigenetics. Science. 2001;293(5532):1068–1070. doi:10.1126/science.1063852
24. Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacol off Publ Am Coll Neuropsychopharmacol. 2013;38(1):23–38. doi:10.1038/npp.2012.112
25. Sun T, Liu D, Wu J, et al. Decreased expression of miR-195 mediated by hypermethylation promotes osteosarcoma. Open Med Wars Pol. 2022;17(1):441–452. doi:10.1515/med-2022-0441
26. Aapola U, Kawasaki K, Scott HS, et al. Isolation and initial characterization of a novel zinc finger gene, DNMT3L, on 21q22.3, related to the cytosine-5-methyltransferase 3 gene family. Genomics. 2000;65(3):293–298. doi:10.1006/geno.2000.6168
27. Hata K, Okano M, Lei H, Li E. Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Dev Camb Engl. 2002;129(8):1983–1993. doi:10.1242/dev.129.8.1983
28. Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99(3):247–257. doi:10.1016/s0092-8674(00)81656-6
29. Jurkowska RZ, Anspach N, Urbanke C, et al. Formation of nucleoprotein filaments by mammalian DNA methyltransferase Dnmt3a in complex with regulator Dnmt3L. Nucleic Acids Res. 2008;36(21):6656–6663. doi:10.1093/nar/gkn747
30. Bestor TH, Ingram VM. Two DNA methyltransferases from murine erythroleukemia cells: purification, sequence specificity, and mode of interaction with DNA. Proc Natl Acad Sci U S A. 1983;80(18):5559–5563. doi:10.1073/pnas.80.18.5559
31. Martisova A, Holcakova J, Izadi N, Sebuyoya R, Hrstka R, Bartosik M. DNA methylation in solid tumors: functions and methods of detection. Int J Mol Sci. 2021;22(8):4247. doi:10.3390/ijms22084247
32. Bird AP. DNA methylation and the frequency of CpG in animal DNA. Nucleic Acids Res. 1980;8(7):1499–1504. doi:10.1093/nar/8.7.1499
33. Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol. 1987;196(2):261–282. doi:10.1016/0022-2836(87)90689-9
34. Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. doi:10.1038/35057062
35. Höller M, Westin G, Jiricny J, Schaffner W. Sp1 transcription factor binds DNA and activates transcription even when the binding site is CpG methylated. Genes Dev. 1988;2(9):1127–1135. doi:10.1101/gad.2.9.1127
36. Macleod D, Charlton J, Mullins J, Bird AP. Sp1 sites in the mouse aprt gene promoter are required to prevent methylation of the CpG island. Genes Dev. 1994;8(19):2282–2292. doi:10.1101/gad.8.19.2282
37. Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011;11(10):726–734. doi:10.1038/nrc3130
38. Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005;6(8):597–610. doi:10.1038/nrg1655
39. Li S, Tollefsbol TO. DNA methylation methods: global DNA methylation and methylomic analyses. Methods San Diego Calif. 2021;187:28–43. doi:10.1016/j.ymeth.2020.10.002
40. Ball MP, Li JB, Gao Y, et al. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat Biotechnol. 2009;27(4):361–368. doi:10.1038/nbt.1533
41. Hellman A, Chess A. Gene body-specific methylation on the active X chromosome. Science. 2007;315(5815):1141–1143. doi:10.1126/science.1136352
42. Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13(7):484–492. doi:10.1038/nrg3230
43. Recillas-Targa F. Cancer epigenetics: an overview. Arch Med Res. 2022;53(8):732–740. doi:10.1016/j.arcmed.2022.11.003
44. Nacev BA, Jones KB, Intlekofer AM, et al. The epigenomics of sarcoma. Nat Rev Cancer. 2020;20(10):608–623. doi:10.1038/s41568-020-0288-4
45. Twenhafel L, Moreno D, Punt T, Kinney M, Ryznar R. Epigenetic changes associated with osteosarcoma: a comprehensive review. Cells. 2023;12(12):1595. doi:10.3390/cells12121595
46. Chen XG, Ma L, Xu JX. Abnormal DNA methylation may contribute to the progression of osteosarcoma. Mol Med Rep. 2018;17(1):193–199. doi:10.3892/mmr.2017.7869
47. Pires SF, de Barros JS, da Costa SS, et al. DNA methylation patterns suggest the involvement of DNMT3B and TET1 in osteosarcoma development. Mol Genet Genomics MGG. 2023;298(3):721–733. doi:10.1007/s00438-023-02010-8
48. Sadikovic B, Yoshimoto M, Al-Romaih K, Maire G, Zielenska M, Squire JA. In vitro analysis of integrated global high-resolution DNA methylation profiling with genomic imbalance and gene expression in osteosarcoma. PLoS One. 2008;3(7):e2834. doi:10.1371/journal.pone.0002834
49. Xu J, Li D, Cai Z, et al. An integrative analysis of DNA methylation in osteosarcoma. J Bone Oncol. 2017;9:34–40. doi:10.1016/j.jbo.2017.05.001
50. Rosenblum JM, Wijetunga NA, Fazzari MJ, et al. Predictive properties of DNA methylation patterns in primary tumor samples for osteosarcoma relapse status. Epigenetics. 2015;10(1):31–39. doi:10.4161/15592294.2014.989084
51. Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349(21):2042–2054. doi:10.1056/NEJMra023075
52. Hou P, Ji M, Yang B, et al. Quantitative analysis of promoter hypermethylation in multiple genes in osteosarcoma. Cancer. 2006;106(7):1602–1609. doi:10.1002/cncr.21762
53. Bachman KE, Herman JG, Corn PG, et al. Methylation-associated silencing of the tissue inhibitor of metalloproteinase-3 gene suggest a suppressor role in kidney, brain, and other human cancers. Cancer Res. 1999;59(4):798–802.
54. Pegg AE. Mammalian O6-alkylguanine-DNA alkyltransferase: regulation and importance in response to alkylating carcinogenic and therapeutic agents. Cancer Res. 1990;50(19):6119–6129.
55. Esteller M, Garcia-Foncillas J, Andion E, et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000;343(19):1350–1354. doi:10.1056/NEJM200011093431901
56. Inbal B, Cohen O, Polak-Charcon S, et al. DAP kinase links the control of apoptosis to metastasis. Nature. 1997;390(6656):180–184. doi:10.1038/36599
57. Rosas SL, Koch W, da Costa Carvalho MG, et al. Promoter hypermethylation patterns of p16, O6-methylguanine-DNA-methyltransferase, and death-associated protein kinase in tumors and saliva of head and neck cancer patients. Cancer Res. 2001;61(3):939–942.
58. Tang X, Khuri FR, Lee JJ, et al. Hypermethylation of the death-associated protein (DAP) kinase promoter and aggressiveness in stage I non-small-cell lung cancer. J Natl Cancer Inst. 2000;92(18):1511–1516. doi:10.1093/jnci/92.18.1511
59. McQueen P, Ghaffar S, Guo Y, Rubin EM, Zi X, Hoang BH. The wnt signaling pathway: implications for therapy in osteosarcoma. Expert Rev Anticancer Ther. 2011;11(8):1223–1232. doi:10.1586/era.11.94
60. Han W, Liu J. Epigenetic silencing of the wnt antagonist APCDD1 by promoter DNA hyper-methylation contributes to osteosarcoma cell invasion and metastasis. Biochem Biophys Res Commun. 2017;491(1):91–97. doi:10.1016/j.bbrc.2017.07.049
61. Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013;154(1):26–46. doi:10.1016/j.cell.2013.06.020
62. Wang W, Chen G, Wang B, et al. Long non-coding RNA BZRAP1-AS1 silencing suppresses tumor angiogenesis in hepatocellular carcinoma by mediating THBS1 methylation. J Transl Med. 2019;17(1):421. doi:10.1186/s12967-019-02145-6
63. Yang S, Wang B, Liu C, et al. THAP9-AS1 promotes tumorigenesis and reduces ROS generation through the JAK2/STAT3 signaling pathway by increasing SOCS3 promoter methylation in osteosarcoma. Oxid Med Cell Longev. 2021;2021(1):5620475. doi:10.1155/2021/5620475
64. Romano G, Veneziano D, Acunzo M, Croce CM. Small non-coding RNA and cancer. Carcinogenesis. 2017;38(5):485–491. doi:10.1093/carcin/bgx026
65. Correia de Sousa M, Gjorgjieva M, Dolicka D, Sobolewski C, Foti M. Deciphering miRNAs’ action through miRNA editing. Int J Mol Sci. 2019;20(24):6249. doi:10.3390/ijms20246249
66. Otoukesh B, Abbasi M, Gorgani HOL, et al. MicroRNAs signatures, bioinformatics analysis of miRNAs, miRNA mimics and antagonists, and miRNA therapeutics in osteosarcoma. Cancer Cell Int. 2020;20:254. doi:10.1186/s12935-020-01342-4
67. Berdasco M, Esteller M. DNA methylation in stem cell renewal and multipotency. Stem Cell Res Ther. 2011;2(5):42. doi:10.1186/scrt83
68. Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447(7143):425–432. doi:10.1038/nature05918
69. Liang X, Xu C, Wang W, Li X. The DNMT1/miR-34a axis is involved in the stemness of human osteosarcoma cells and derived stem-like cells. Stem Cells Int. 2019;2019:7028901. doi:10.1155/2019/7028901
70. Guo W, Wei B, Cheng T, Xu X, Ruan F, Xiang M. The na+/K+ ATPase inhibitor ouabain attenuates stemness and chemoresistance of osteosarcoma cells. Med Sci Monit Int Med J Exp Clin Res. 2019;25:9426–9434. doi:10.12659/MSM.919266
71. Shen WC, Lai YC, Li LH, et al. Methylation and PTEN activation in dental pulp mesenchymal stem cells promotes osteogenesis and reduces oncogenesis. Nat Commun. 2019;10(1):2226. doi:10.1038/s41467-019-10197-x
72. Lu J, Wang J. IRX1 hypomethylation in osteosarcoma metastasis. Oncotarget. 2015;6(19):16802–16803. doi:10.18632/oncotarget.4696
73. Lu J, Song G, Tang Q, et al. IRX1 hypomethylation promotes osteosarcoma metastasis via induction of CXCL14/NF-κB signaling. J Clin Invest. 2015;125(5):1839–1856. doi:10.1172/JCI78437
74. Wang H, Liu B, Chen H, Xu P, Xue H, Yuan J. Dynamic changes of DNA methylation induced by benzo(a)pyrene in cancer. Genes Environ off J Jpn Environ Mutagen Soc. 2023;45(1):21. doi:10.1186/s41021-023-00278-1
75. Miousse IR, Ewing LE, Kutanzi KR, Griffin RJ, Koturbash I. DNA methylation in radiation-induced carcinogenesis: experimental evidence and clinical perspectives. Crit Rev Oncog. 2018;23(1–2):1–11. doi:10.1615/CritRevOncog.2018025687
76. Boughanem H, Hernandez-Alonso P, Tinahones A, et al. Association between serum vitamin B12 and global DNA methylation in colorectal cancer patients. Nutrients. 2020;12(11):3567. doi:10.3390/nu12113567
77. Yang J, Xu J, Wang W, Zhang B, Yu X, Shi S. Epigenetic regulation in the tumor microenvironment: molecular mechanisms and therapeutic targets. Signal Transduct Target Ther. 2023;8(1):210. doi:10.1038/s41392-023-01480-x
78. Duan Q, Zhang H, Zheng J, Zhang L. Turning cold into hot: firing up the tumor microenvironment. Trends Cancer. 2020;6(7):605–618. doi:10.1016/j.trecan.2020.02.022
79. Zhou L, Yu CW. Epigenetic modulations in triple-negative breast cancer: therapeutic implications for tumor microenvironment. Pharmacol Res. 2024;204:107205. doi:10.1016/j.phrs.2024.107205
80. Shi D, Mu S, Pu F, et al. Integrative analysis of immune-related multi-omics profiles identifies distinct prognosis and tumor microenvironment patterns in osteosarcoma. Mol Oncol. 2022;16(11):2174–2194. doi:10.1002/1878-0261.13160
81. Duan J, Zhong B, Fan Z, et al. DNA methylation in pulmonary fibrosis and lung cancer. Expert Rev Respir Med. 2022;16(5):519–528. doi:10.1080/17476348.2022.2085091
82. Zuccato JA, Patil V, Mansouri S, et al. DNA methylation-based prognostic subtypes of chordoma tumors in tissue and plasma. Neuro-Oncol. 2022;24(3):442–454. doi:10.1093/neuonc/noab235
83. Odagiri H, Kadomatsu T, Endo M, et al. The secreted protein ANGPTL2 promotes metastasis of osteosarcoma cells through integrin α5β1, p38 MAPK, and matrix metalloproteinases. Sci Signal. 2014;7(309):ra7. doi:10.1126/scisignal.2004612
84. Lam SW, van IJzendoorn DGP, Cleton-Jansen AM, Szuhai K, Bovée JVMG. Molecular pathology of bone tumors. J Mol Diagn JMD. 2019;21(2):171–182. doi:10.1016/j.jmoldx.2018.11.002
85. Capper D, Jones DTW, Sill M, et al. DNA methylation-based classification of central nervous system tumours. Nature. 2018;555(7697):469–474. doi:10.1038/nature26000
86. Capper D, Stichel D, Sahm F, et al. Practical implementation of DNA methylation and copy-number-based CNS tumor diagnostics: the Heidelberg experience. Acta Neuropathol. 2018;136(2):181–210. doi:10.1007/s00401-018-1879-y
87. Papanicolau-Sengos A, Aldape K. DNA methylation profiling: an emerging paradigm for cancer diagnosis. Annu Rev Pathol. 2022;17(1):295–321. doi:10.1146/annurev-pathol-042220-022304
88. Koelsche C, Schrimpf D, Stichel D, et al. Sarcoma classification by DNA methylation profiling. Nat Commun. 2021;12(1):498. doi:10.1038/s41467-020-20603-4
89. Lyskjaer I, De Noon S, Tirabosco R, et al. DNA methylation-based profiling of bone and soft tissue tumours: a validation study of the “DKFZ sarcoma classifier. J Pathol Clin Res. 2021;7(4):350–360. doi:10.1002/cjp2.215
90. Roohani S, Ehret F, Perez E, et al. Sarcoma classification by DNA methylation profiling in clinical everyday life: the charité experience. Clin Clin Epigenet. 2022;14(1):149. doi:10.1186/s13148-022-01365-w
91. Silva FLT, Euzébio MF, Ruas JS, et al. Classification of pediatric soft and bone sarcomas using DNA methylation-based profiling. BMC Cancer. 2024;24(1):1428. doi:10.1186/s12885-024-13159-9
92. Koelsche C, Kriegsmann M, Kommoss FKF, et al. DNA methylation profiling distinguishes ewing-like sarcoma with EWSR1-NFATc2 fusion from ewing sarcoma. J Cancer Res Clin Oncol. 2019;145(5):1273–1281. doi:10.1007/s00432-019-02895-2
93. Kang Y, Li G, Wang G, et al. Development of a risk score model for osteosarcoma based on DNA methylation-driven differentially expressed genes. J Oncol. 2022;2022:7596122. doi:10.1155/2022/7596122
94. Zervos AS, Gyuris J, Brent R. Mxi1, a protein that specifically interacts with max to bind myc-max recognition sites. Cell. 1993;72(2):223–232. doi:10.1016/0092-8674(93)90662-a
95. Hurlin PJ, Huang J. The MAX-interacting transcription factor network. Semin Cancer Biol. 2006;16(4):265–274. doi:10.1016/j.semcancer.2006.07.009
96. Ayer DE, Kretzner L, Eisenman RN. Mad: a heterodimeric partner for max that antagonizes myc transcriptional activity. Cell. 1993;72(2):211–222. doi:10.1016/0092-8674(93)90661-9
97. Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130(2):223–233. doi:10.1016/j.cell.2007.07.003
98. Zhao W, Meng H, Dai Z, et al. Prediction of patients with high-risk osteosarcoma on the basis of XGBoost algorithm using transcriptome and methylation data from SGH-OS cohort. JCO Precis Oncol. 2025;9(9):e2400732. doi:10.1200/PO-24-00732
99. Zhang X, Zheng Y, Li G, Yu C, Ji T, Miao S. Identifying four DNA methylation gene sites signature for predicting prognosis of osteosarcoma. Transl Cancer Res. 2020;9(11):7299–7309. doi:10.21037/tcr-20-3204
100. Al-Romaih K, Sadikovic B, Yoshimoto M, Wang Y, Zielenska M, Squire JA. Decitabine-induced demethylation of 5’ CpG island in GADD45A leads to apoptosis in osteosarcoma cells. Neoplasia N Y N. 2008;10(5):471–480. doi:10.1593/neo.08174
101. Lillo Osuna MA, Garcia-Lopez J, El Ayachi I, et al. Activation of estrogen receptor alpha by decitabine inhibits osteosarcoma growth and metastasis. Cancer Res. 2019;79(6):1054–1068. doi:10.1158/0008-5472.CAN-18-1255
102. Wang Z, Wang Z, Li S, et al. Decitabine enhances Vγ9Vδ2 T cell-mediated cytotoxic effects on osteosarcoma cells via the NKG2DL-NKG2D axis. Front Immunol. 2018;9:1239. doi:10.3389/fimmu.2018.01239
103. Glover AB, Leyland-Jones B. Biochemistry of azacitidine: a review. Cancer Treat Rep. 1987;71(10):959–964.
104. Higuchi T, Sugisawa N, Yamamoto J, et al. The combination of oral-recombinant methioninase and azacitidine arrests a chemotherapy-resistant osteosarcoma patient-derived orthotopic xenograft mouse model. Cancer Chemother Pharmacol. 2020;85(2):285–291. doi:10.1007/s00280-019-03986-0
105. Christman JK. 5-azacytidine and 5-aza-2’-deoxycytidine as inhibitors of DNA methylation: mechanistic studies and their implications for cancer therapy. Oncogene. 2002;21(35):5483–5495. doi:10.1038/sj.onc.1205699
106. Butz S, Schmolka N, Karemaker ID, et al. DNA sequence and chromatin modifiers cooperate to confer epigenetic bistability at imprinting control regions. Nat Genet. 2022;54(11):1702–1710. doi:10.1038/s41588-022-01210-z
107. Acurzio B, Cecere F, Giaccari C, et al. The mismatch-repair proteins MSH2 and MSH6 interact with the imprinting control regions through the ZFP57-KAP1 complex. Epigenet Chromatin. 2022;15(1):27. doi:10.1186/s13072-022-00462-7
108. Anvar Z, Cammisa M, Riso V, et al. ZFP57 recognizes multiple and closely spaced sequence motif variants to maintain repressive epigenetic marks in mouse embryonic stem cells. Nucleic Acids Res. 2016;44(3):1118–1132. doi:10.1093/nar/gkv1059
109. Sonaglio V, de Carvalho AC, Toledo SRC, et al. Aberrant DNA methylation of ESR1 and p14ARF genes could be useful as prognostic indicators in osteosarcoma. Oncol Targets Ther. 2013;6:713–723. doi:10.2147/OTT.S44918
110. Cui Q, Jiang W, Guo J, et al. Relationship between hypermethylated MGMT gene and osteosarcoma necrosis rate after chemotherapy. Pathol Oncol Res POR. 2011;17(3):587–591. doi:10.1007/s12253-010-9354-7
111. Kresse SH, Rydbeck H, Skårn M, et al. Integrative analysis reveals relationships of genetic and epigenetic alterations in osteosarcoma. PLoS One. 2012;7(11):e48262. doi:10.1371/journal.pone.0048262
112. Lietz CE, Newman ET, Kelly AD, et al. Genome-wide DNA methylation patterns reveal clinically relevant predictive and prognostic subtypes in human osteosarcoma. Commun Biol. 2022;5(1):213. doi:10.1038/s42003-022-03117-1
113. Shi J, Huang D, Zhang G, Zhao F, Yang L. A DNA methylation-associated nomogram predicts the overall survival of osteosarcoma. Medicine. 2020;99(51):e23772. doi:10.1097/MD.0000000000023772
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Rett Syndrome and MECP2 Duplication Syndrome: Disorders of MeCP2 Dosage
Collins BE, Neul JL
Neuropsychiatric Disease and Treatment 2022, 18:2813-2835
Published Date: 29 November 2022
Association Between Promoter Methylation of Vitamin D Metabolic Pathway Genes and Tuberculosis and Diabetes Comorbidity in a Chinese Han Population: A Case–Control Study
Chen Y, Peng AZ, Li K, Liu L, Zhang F, Chen J, Zhang H, Li L, Yang H, Xu X, Zhang Q
Journal of Inflammation Research 2022, 15:6831-6842
Published Date: 23 December 2022
Development and Validation of a Propionate Metabolism-Related Gene Signature for Prognostic Prediction of Hepatocellular Carcinoma
Xiao J, Wang J, Zhou C, Luo J
Journal of Hepatocellular Carcinoma 2023, 10:1673-1687
Published Date: 2 October 2023
Advances in the Epigenetic Mechanisms of Diabetic Nephropathy Pathogenesis
Zhang C, Xue S, Ren P, Han S, Zhou Y, Si Y, Han X, Zhang X, Zhang Y, Chen N, He H, Feng R, Shang L
Diabetes, Metabolic Syndrome and Obesity 2025, 18:2629-2639
Published Date: 30 July 2025
Advances of Drug-Loaded Microsphere Technology for Targeted Immunotherapy Against Prostate Cancer
Feng W
International Journal of Nanomedicine 2025, 20:11479-11489
Published Date: 20 September 2025