")
Back to Journals » Neuropsychiatric Disease and Treatment » Volume 18
Rett Syndrome and MECP2 Duplication Syndrome: Disorders of MeCP2 Dosage
Authors Collins BE , Neul JL
Received 8 September 2022
Accepted for publication 14 November 2022
Published 29 November 2022 Volume 2022:18 Pages 2813—2835
DOI https://doi.org/10.2147/NDT.S371483
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Yuping Ning
Bridget E Collins,1 Jeffrey L Neul2
1Medical Scientist Training Program, Vanderbilt University, Nashville, TN, USA; 2Vanderbilt Kennedy Center, Departments of Pediatrics, Pharmacology, and Special Education, Vanderbilt University Medical Center and Vanderbilt University, Nashville, TN, USA
Correspondence: Jeffrey L Neul, PMB 40, 230 Appleton Place, Nashville, TN, 37203, Tel +1 615-322-8242, Fax +1 615-322-8236, Email [email protected]
Abstract: Rett syndrome (RTT) is a neurodevelopmental disorder caused predominantly by loss-of-function mutations in the gene Methyl-CpG-binding protein 2 (MECP2), which encodes the MeCP2 protein. RTT is a MECP2-related disorder, along with MECP2 duplication syndrome (MDS), caused by gain-of-function duplications of MECP2. Nearly two decades of research have advanced our knowledge of MeCP2 function in health and disease. The following review will discuss MeCP2 protein function and its dysregulation in the MECP2-related disorders RTT and MDS. This will include a discussion of the genetic underpinnings of these disorders, specifically how sporadic X-chromosome mutations arise and manifest in specific populations. We will then review current diagnostic guidelines and clinical manifestations of RTT and MDS. Next, we will delve into MeCP2 biology, describing the dual landscapes of methylated DNA and its reader MeCP2 across the neuronal genome as well as the function of MeCP2 as a transcriptional modulator. Following this, we will outline common MECP2 mutations and genotype–phenotype correlations in both diseases, with particular focus on mutations associated with relatively mild disease in RTT. We will also summarize decades of disease modeling and resulting molecular, synaptic, and behavioral phenotypes associated with RTT and MDS. Finally, we list several therapeutics in the development pipeline for RTT and MDS and available evidence of their safety and efficacy.
Keywords: neurodevelopmental disorders, epigenetics, DNA methylation, disease modeling, therapeutics
Review
One afternoon in Vienna in 1954, pediatrician Andreas Rett walked into the waiting room of his clinic for children with disabilities and noticed two girls sitting in their mothers’ laps, both with their arms gently restrained.1 The mothers, by happenstance, released their grip simultaneously and both girls began to make unusual, stereotyped movements. With further research, Rett found that these girls shared similar developmental and clinical histories, and, upon analysis of the clinic’s records, identified an additional six girls that did too. Rett believed he was observing a previously unrecognized syndrome and reported his findings in a little-known Austrian newsletter one year later, in 1966.2 This seminal article, titled “On a remarkable syndrome of cerebral atrophy associated with hyperammonemia in childhood”, would remain virtually unrecognized for 15 years, until a group of European researchers would give a name to what pediatric neurologists now recognize as Rett syndrome.3
Rett Syndrome (RTT)
Andreas Rett’s 1966 report described 22 young girls, each with an uncomplicated birth history and typical development until approximately 1 year of age.2 However, at this age, Rett noted that the girls exhibited a consistent pattern of disruption in typical development, including shared characteristics of absent speech, motor dysfunction, apraxic gait, hand stereotypies, and epilepsy. Rett termed this syndrome “cerebral atrophy associated with hyperammonemia” and published his findings to raise awareness in the pediatric neurology community. Unfortunately, Rett’s work was not widely read in the Austrian newsletter Wiener Medizinische Wochenschrift, and his finding of hyperammonemia was not replicated by others (and subsequently discovered to be a lab error), preventing broad acknowledgement of this childhood syndrome. Broad acknowledgement came only in 1983, when Swedish neurologist Bengt Hagberg and colleagues published a synthesis of 35 cases of “developmental stagnation…followed by rapid deterioration of higher brain functions” in young girls from France, Portugal and Sweden.3 Noting similarities to the cases described in Rett’s 1966 report, Hagberg termed this clinical pattern Rett’s syndrome.
In the years following Hagberg’s report, growing attention to this disorder led to the need for improved clinical characterization. With his broad expertise, Hagberg proposed a four-stage system describing the temporal progression of the disease: (1) early onset, (2) regression, (3) plateau, and (4) late motor deterioration (Figure 1).4 Prior to onset of symptoms, children with Rett syndrome (RTT) undergo apparently typical development. In the first stage, early onset, girls with RTT may show delays in psychomotor skill progression (developmental delay) and growth, specifically, a decrease in the rate of head circumference growth, with some individuals having acquired microcephaly. The second stage of the disease, regression (termed “rapid deterioration” by Hagberg, but subsequent work determined that regression is not always rapid), involves partial or complete loss of previously acquired skills pertaining to spoken language and hand use. This stage also includes the onset of hand stereotypies such as hand wringing/squeezing and gait abnormalities or the inability to ambulate. Children with RTT may also develop respiratory abnormalities (eg, hyperventilation, apneas) at this time, although these features often do not manifest until the plateau phase. The regression stage has a finite duration, after which affected individuals enter the plateau stage around 3–5 years of age. During the plateau stage, further loss of skills does not occur and behavioral and cognitive function may stabilize, while other medical conditions, such as seizures, gastrointestinal and nutritional problems (notably constipation), other movement disorders, autonomic abnormalities, and other medical issues manifest.5,6 Finally, in the late motor deterioration stage, individuals with RTT may experience decreasing mobility, onset of parkinsonian features, and scoliosis, leading to significant motor disability and, in some cases, the loss of the ability to ambulate.7 It is important to note that this staging system is a general characterization of clinical progression and that not all girls with RTT will experience each stage or each clinical feature.
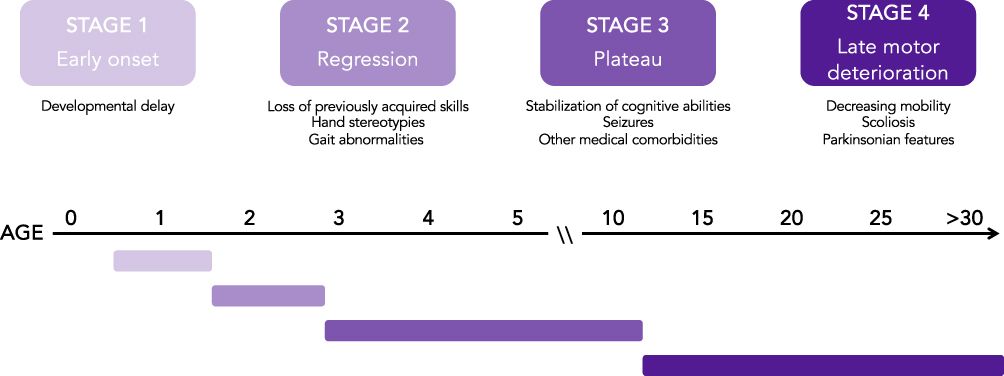 |
Figure 1 Disease stages in Rett syndrome. The progression of Rett syndrome can be described in four stages of disease. In Stage 1 (early onset), children develop mostly typically with possible developmental delay. In Stage 2 (regression), children undergo developmental regression over the course of weeks, months, or years with loss of previously acquired skills including purposeful hand movements and spoken language. Children also begin to develop breathing and gait abnormalities during this period. In Stage 3 (plateau), children usually experience stabilization of cognitive abilities and have onset of other medical conditions such as seizures. In Stage 4 (late motor deterioration) individuals with RTT experience decreasing mobility and may develop parkinsonian features. |
The clinical diagnosis of RTT is made according to consensus guidelines published in 2010 by the RettSearch Consortium.8 Typical, or classic, RTT is defined by the presence of the characteristic disease progression of RTT (a period of regression followed by recovery or stabilization), with the presence of 4 main criteria: 1) loss of acquired purposeful hand skills; 2) loss of acquired spoken language; 3) development of gait abnormalities or the inability to walk; and 4) presence of characteristic hand stereotypies. With the broader recognition of RTT, clinicians have also identified individuals that display some, but not all, of the features of typical RTT. These individuals are described to have atypical, or variant, RTT. Atypical RTT is defined by the presence of a period of regression followed by recovery or stabilization, as well as at least 2 of the main 4 criteria for typical RTT and at least 5 out of the 11 supporting criteria (eg, breathing disturbances when awake, peripheral vasomotor disturbances, intense eye communication – “eye pointing”). Though diagnostic criteria have varied over the almost four decades since RTT was recognized globally, researchers have been able to glean epidemiologic data as to the incidence of RTT. RTT is a rare neurodevelopmental disorder, occurring with a cumulative incidence of 1.09 per 10,000 females by age 12 years.9
The nearly exclusive occurrence of RTT in girls led the research community to predict that the disease is caused by an X-linked dominant mutation with lethality in males, the first part of which turned out to be true and the latter not.3,10–14 Extensive gene mapping of rare familial cases of RTT narrowed down the genetic region of interest to the Xq28 region, and in 1999 Ruthie Amir and colleagues identified a causative gene.13–18 Using a systematic gene screening approach in sporadic cases of RTT, Amir et al identified disease-causing mutations in the MECP2 gene encoding methyl-CpG-binding protein 2 (MeCP2). Since then, it has been found that MECP2 mutations account for over 95% of typical RTT cases and approximately 75% of atypical RTT cases.19,20 MECP2 mutations occur largely in a sporadic manner; however, a small percentage of RTT cases occur in a familial manner through parental MECP2 mutations.21,22
The discovery of a genetic basis to RTT provided insight into the disease’s marked manifestation in girls and women. While the notion of male lethality continues to mistakenly permeate the literature, genotyping of affected individuals and their parents led to the discovery that de novo MECP2 mutations nearly exclusively derive from the paternal X-chromosome.21,23,24 Consistently, a large portion of MECP2 mutations involve C→T transitions at CpG sites, which, when methylated, are prone to spontaneous deamination and conversion to thymine.25 Male germ cells are highly methylated on the X-chromosome, providing more chance for CpG mutation.26 As the paternal X-chromosome is only inherited by female children, RTT occurs nearly exclusively in girls and women. Occasionally, the MECP2 mutation is inherited on the maternal X-chromosome.12,16,27 In this case, mothers typically carry one allele with a MECP2 mutation and have skewed X-chromosome inactivation in preference of the non-mutant allele. These mothers often do not fulfill criteria for RTT and often are completely unaffected, but some may display cognitive impairment or a learning disability.
While most individuals with RTT are female, MECP2 mutations do cause disease in males with an array of clinical phenotypes.12 These mutations manifest disease in males in one of three ways: (1) inheritance of two X-chromosomes (47XXY, Klinefelter syndrome),28,29 (2) development of a postzygotic MECP2 mutation with resulting somatic mosaicism,30 and (3) inheritance of a MECP2 mutation leading to RTT.31 The array of MECP2 mutations that causes disease in males also differs from those causing disease in females. Reported mutations in males include those causing RTT in females but also a subset of likely pathogenic mutations not observed in females with RTT. This subset includes A140V, G118E, N126I, F157L, R167W, P176H, R306P, R309W, and P322S.32 The A140V mutation, for example, is not considered to be associated with RTT in females, but can cause cognitive, motor, and psychiatric manifestations in males.33 MECP2-associated disease in males ranges from cognitive impairment to typical and atypical RTT, progressive encephalopathy, and neonatal encephalopathy. Overall, disease tends to be more severe in males than in females, with males more commonly exhibiting abnormal initial development, ventilator dependency, and early death.32 Males tend to have more severe disease because they express MECP2 from a single X-chromosome, whereas females undergo X-chromosome inactivation and variably express the non-mutant or mutant MECP2 allele in each cell (therefore retaining some expression of full-length MeCP2). To better classify the different presentation and progression of MECP2-associated disease in males, the term “male RTT encephalopathy” has been proposed to describe the subset of males that meet criteria for RTT.32
MECP2 Duplication Syndrome (MDS)
In the mid-2000s came the clinical discovery that overexpression of MeCP2 in people causes a different MECP2-related disorder, called MECP2 duplication syndrome (MDS).34,35 Specifically, Meins et al reported on a boy with severe intellectual disability and features of RTT who was determined to have a submicroscopic ~430 kb duplication within the Xq28 region containing the MECP2 gene.34 Following this, Van Esch et al reported on four families with individuals displaying intellectual disability that had duplications 400–800 kb in size containing several genes, including at least L1CAM (L1 cell adhesion molecule) and MECP2.35 Several lines of evidence pointed specifically to MECP2 as the genetic cause of MDS. First, the duplication of the individual described by Meins et al did not involve L1CAM, suggesting that MECP2 duplication is sufficient to cause MDS. Second, mouse modeling work suggested that MeCP2 overexpression leads to a progressive neurological disease involving seizures, spasticity, and premature death, and that higher MeCP2 level is associated with more severe disease.36
The duplications that cause MDS are thought to occur via several molecular mechanisms.37,38 The described MECP2 duplications are non-recurrent, meaning that duplications have variable breakpoints and do not span the same genomic interval across unrelated individuals. In general, the Xq28 region has higher GC content and higher Alu repeat density compared to random sequences of the genome, possibly increasing regional genomic instability.37 Furthermore, most duplications have a distal breakpoint within a 215 kb genomic interval (47 kb telomeric to MECP2) that contains complex low copy repeat (LCR) sequences, suggesting that these LCRs may have a role in the origin of genomic rearrangement. Analysis of individual duplication breakpoints suggests that multiple mechanisms may be involved. For example, the MECP2 duplication of one individual was proposed to occur via non-homologous end joining (NHEJ) due to the presence of proximal and distal microhomology.37 However, the duplications of other individuals have been proposed to occur via complex rearrangements, which can result in embedded triplicated segments, segments of non-duplicated sequence within the duplicated region, and inversion of the duplicated sequence. These complex rearrangements may occur via Fork Stalling and Template Switching or, more generally, microhomology-mediated break-induced replication (FoSTeS/MMBIR), a replication-based mechanism that involves stalled replication forks switching templates through complementary template microhomology.39 Further complicating this, duplicated segments of the Xq28 region have been found in tandem on the X-chromosome (Xq-Xp rearrangements), on the Y-chromosome (Xq-Yq translocations), as well as on autosomes (X-autosome translocations).40
Despite the wide variation of MECP2 duplication events in mechanism of origin, genetic content, and genomic location, they collectively result in a consistent clinical MDS phenotype.40,41 Individuals with MDS typically have severe developmental delay, early infantile hypotonia with progressive acquired spasticity, feeding difficulties, and recurrent respiratory infections.41,42 While developmental delay and hypotonia are commonly observed in neurodevelopmental disorders, the predisposition to infection distinguishes MDS. Infections range from otitis media to pneumonia, pyelonephritis, and sepsis.41 While incompletely understood, gastroesophageal reflux and immune dysfunction may contribute to infection risk, due to aspiration and immunoglobulin deficiency, respectively.43,44 As individuals with MDS age, over half will meet formal criteria for autism spectrum disorder (ASD) with gaze avoidance, impaired social interactions, and stereotypical movements.45,46 Nearly all individuals have limited or absent spoken language.47 Seizures are also present at least half of individuals, although further research is needed to definitively determine the overall incidence and prevalence of seizures in MDS. This is particularly important because seizure onset and progression in MDS leads to marked decline in affected individuals with associated loss of skills and intellectual regression.42 Overall, MDS is a severely debilitating disorder that requires lifelong specialized care. Often, recurrent infections coupled with refractory epilepsy and neurological deterioration lead to early death.47
While individual MECP2 duplication events are rare, MDS accounts for as much as 1% of unexplained X-linked intellectual disability.48 Most MECP2 duplications are transmitted to male children from a carrier mother.35 Carrier mothers typically exhibit extreme (>90%) skewing with preference for the X-chromosome bearing one copy of MECP2 and are asymptomatic or exhibit neuropsychiatric symptoms. Girls who inherit a MECP2 duplication and do not have extreme skewing can manifest variable degrees of MDS-like symptoms, ranging from mild cognitive disability to severe disease similar to that observed in males.49,50 MECP2 duplication can also occur de novo in males and females. In this case, males and females are affected similarly because the MECP2 duplication is often expressed on an autosome.40,47
Due to the rarity of MDS, clinical characterization and genotype–phenotype correlations in MDS are limited. However, work by Peters et al showed that larger duplication size is correlated with increased severity across total clinical severity and motor behavioral assessment inventory scores.51 Additionally, some evidence suggests that duplication of neighboring genes contributes to additional clinical phenotypes. For example, Filamin A (FLNA) gene duplication has been proposed to contribute to intestinal and bladder dysfunction and development of distinct facial features amongst individuals with MDS.52 Furthermore, other genes that can be duplicated with MECP2 have been associated independently with disease: GDP dissociation inhibitor 1 (GDI1, cognition), Ras-related protein Rab-39B (RAB39B, intellectual disability), and SRSF protein kinase 3 (SRPK3, muscle degeneration).53–55
DNA Methylation and Methyl-CpG-Binding Protein 2 (MeCP2)
DNA methylation is an epigenetic mark defined by the addition of a single methyl (-CH3) functional group onto the 5-position of DNA base cytosine (5-methyl-cytosine, 5mC). DNA methylation is integral for many cellular processes in the developing and developed organism, including cellular differentiation, genomic imprinting, X-chromosome inactivation (XCI), and silencing of repetitive DNA.56,57 Approximately 1% of all DNA bases and 70–80% of all CpG dinucleotides are symmetrically methylated in somatic cells of the vertebrate genome.58 DNA methylation occurs at high levels in constitutive heterochromatin, where MeCP2 is also densely localized.59 Other regions of the genome are methylated as well, including transcription start sites (TSS), gene bodies, enhancer elements, insulator elements, and intergenic regions.57 DNA methylation functions differently at each of these regions, making for a complex system of epigenetic regulation. For example, the TSS of most active genes contain unmethylated CpG islands (CGIs); however, promoters of inactive genes often contain methylated CGIs, and these are associated with long-term stability of a repressed transcriptional state (eg, imprinted genes, genes on the inactive X-chromosome).60 In contrast, gene body methylation is not associated with repression and is instead positively correlated with gene expression.61 These examples illustrate the context-dependency of DNA methylation.
In addition to CpG methylation, non-CG methylation (mCH, where H=adenine, cytosine, or thymine) occurs in an asymmetric manner throughout the genome, particularly in the developed brain of mice and humans.62,63 mCH increases in brain tissue of mice and humans during early postnatal development, most rapidly during the period of synaptogenesis (2–4 weeks of age in mice; 2 years of age in humans).64–66 Intriguingly, MeCP2 levels also increase during this period of development.67 mCH levels reach a maximum of ~1.5% of CH genome positions and is predominantly present in the mCA context.62,64 In another level of complexity, methylated cytosines can also be modified by ten-eleven translocation (TET) dioxygenase enzymes that are crucial for DNA demethylation.68,69 During the demethylation process, intermediate hydroxymethylated cytosine residues (5hmC) are generated and retain the capacity to interact with DNA methylation reader proteins.70,71 Like non-CG methylation, 5hmC residues accumulate in the brain during development and in a cell type-specific manner. 5hmC is enriched in gene bodies of active genes and is thought to “functionally demethylate” these regions (generate lower occupancy of MeCP2 via decreasing high-affinity mCG availability) while retaining capacity to interact with DNA methylation readers to mediate chromatin organization.72,73
One of these DNA methylation readers is MeCP2, discovered and cloned in 1992.74 A chromatin-associated protein, MeCP2 binds symmetrically methylated cytosines in a CpG context via a methyl-binding domain (MBD, amino acids 78–162).75 The MBD spans an 85-amino acid sequence at the N-terminal part of MeCP2 and has a DNA binding footprint of approximately 12 nucleotides (Figure 2). In addition to symmetric CpG methylation, MeCP2 can bind to methylated cytosines in a wider variety of non-CpG contexts when methylated in an asymmetric manner.76–78 MeCP2 has particular affinity for mCA amongst mCH contexts, especially in a CAC sequence.76,78 Structural work of the MBD shows that MeCP2 likely binds as a monomer to methylated DNA, recognizing the hydration in the major groove of methylated DNA.79,80
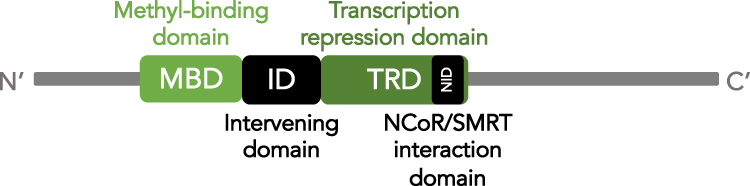 |
Figure 2 Schematic of MeCP2 functional domains. Schematic illustration of MeCP2 protein from the N-terminus (right) to C-terminus (left). The two main protein domains are indicated in shades of green: methyl-binding domain (MBD) and transcription repression domain (TRD). The location of the NCoR/SMRT interaction domain (NID) within the TRD is indicated in black. The intervening domain (ID) between the MBD and TRD is also indicated in black. |
Once bound to DNA, MeCP2 interacts with the transcriptional repressors and activators to modulate transcription. In a well-characterized interaction, MeCP2 binds to the transcriptional co-repressor nuclear receptor co-repressor/silencing mediator of retinoid and thyroid hormone receptors (NCoR/SMRT) through its 29-amino acid NCoR interaction domain (NID, amino acids 285–313) (Figure 2).81–83 However, MeCP2 has also been reported to interact with a variety of other binding partners, including transcriptional co-activators, other transcriptional co-repressors, chromatin remodelers, chromatin modifiers, and splicing factors.82,84–94 While MeCP2 has been shown to act as a transcriptional repressor in an in vitro context,81 transcriptomics of mice with MeCP2 deficiency or MeCP2 overexpression have shown that not all differentially expressed genes are upregulated in MeCP2 deficient mice or downregulated in MeCP2 overexpression mice, as would be expected for a pure transcriptional repressor.77,94,95 Though it is not clear if these transcriptional changes are direct or indirect effects of MeCP2 dosage, further evidence of a complex transcriptional regulatory role for MeCP2 is provided by the finding that MeCP2 loss globally reduces (instead of elevates) transcription and translation in human embryonic stem cell (ESC) derived neurons.96
A large portion, ~60%, of the MeCP2 sequence is disordered in solution, making structural analysis challenging.97,98 However, in addition to the MBD and NID, MeCP2 contains three AT-hook-like domains. AT-hooks are short DNA-binding motifs that interact with the minor groove of AT-rich DNA with the consensus sequence RGRP.99 The three AT-hook-like domains in MeCP2 occur in 1) the interdomain (ID) between the MBD and NID at amino acids 184–195, 2) the transcription repression domain (TRD) at amino acids 264–273, and 3) the C-terminus of the protein at amino acids 341–364 (Figure 2). The second of these AT-hook-like domains has been shown to alter chromatin structure when MeCP2 is bound, suggesting that these regions can stabilize or modulate chromatin structure around MeCP2 binding sites.100,101
MeCP2 is expressed predominantly in the central and peripheral nervous systems (CNS, PNS), particularly in postmitotic neurons and less so in glia.102 MeCP2 is expressed to a lesser extent in the periphery, where is it thought to help maintain muscle function required for exercise and bone integrity.103 As a nuclear protein, MeCP2 is localized to neuronal nuclei at levels approaching that of histone octamers.104 With such high levels of protein, MeCP2 shows widespread binding throughout the genome and tracks the densities of mCG, mCH, and hmC.77,104,105 Predominant binding sites occur in intergenic and intronic regions of the genome, but MeCP2 can also interact with elements such as promoters, enhancers, and gene bodies.106 The expanse of MeCP2’s DNA footprint suggests that it functions in a highly context-dependent manner. As an example of this, MeCP2 decreases the rate of transcriptional initiation of highly methylated long genes via interaction of gene body MeCP2 with the TSS.107 Further, at enhancers containing a high density of mCG and mCA, MeCP2 represses enhancer activity.108 These functions are likely mediated by the repressive action of MeCP2 in a context-dependent manner. Adding to the complexity of MeCP2 function, several phosphorylation sites (S308, S421) are activity-dependent and may provide an additional level of transcriptional control. While MeCP2 S308 phosphorylation reduces interaction with NCoR/SMRT, MeCP2 S421 phosphorylation permits brain-derived neurotrophic factor (Bdnf) transcription, dendritic growth, and spine maturation.109,110 Interestingly, MeCP2 has also been found to alter chromatin structure and promote chromatin compaction, introducing the notion that MeCP2 functions outside of transcriptional repression.111,112
MECP2 and Disease-Causing Mutations
The MECP2 gene contains four exons from which two transcript isoforms are expressed, MeCP2_e1 and MeCP2_e2.113,114 The MeCP2_e1 transcript isoform excludes exon 2 via alternative splicing and is translated from a start codon in exon 1, producing a slightly longer 498 amino acid protein with a unique 21 amino acid N-terminus in humans.113,114 The MeCP2_e2 transcript isoform contains all four exons and has a translation start codon in exon 2, producing a 486 amino acid protein with a unique 9 amino acid N-terminus in humans.115 MeCP2_e1 is more highly expressed in adult brain, while MeCP2_e2 is more highly expressed in peripheral tissues.116
More than 95% of individuals with typical RTT and approximately 75% of individuals with atypical RTT have a mutation in the MECP2 gene.19,20 Over 200 different MECP2 mutations have been documented as causative of RTT (RettBase: http://mecp2.chw.edu.au). However, some mutations are more commonly observed than others. Namely, there are 8 major point mutations that account for over 60% of all typical RTT cases: R106W, R133C, T158M, R168X, R255X, R270X, R294X, and R306C (Figure 3).19 Additionally, there is a subset of C-terminal mutations, predominately small deletions, that accounts for approximately 5–10% of typical RTT cases. Less common MECP2 mutations include those in exon 1, short deletions, large deletions, insertions, mutations at splice sites, and other point mutations.20 Apart from RTT, MECP2 mutations have also rarely been associated with other neurodevelopmental disorders including ASD,117–120 Angelman syndrome,121,122 and X-linked intellectual disability.123
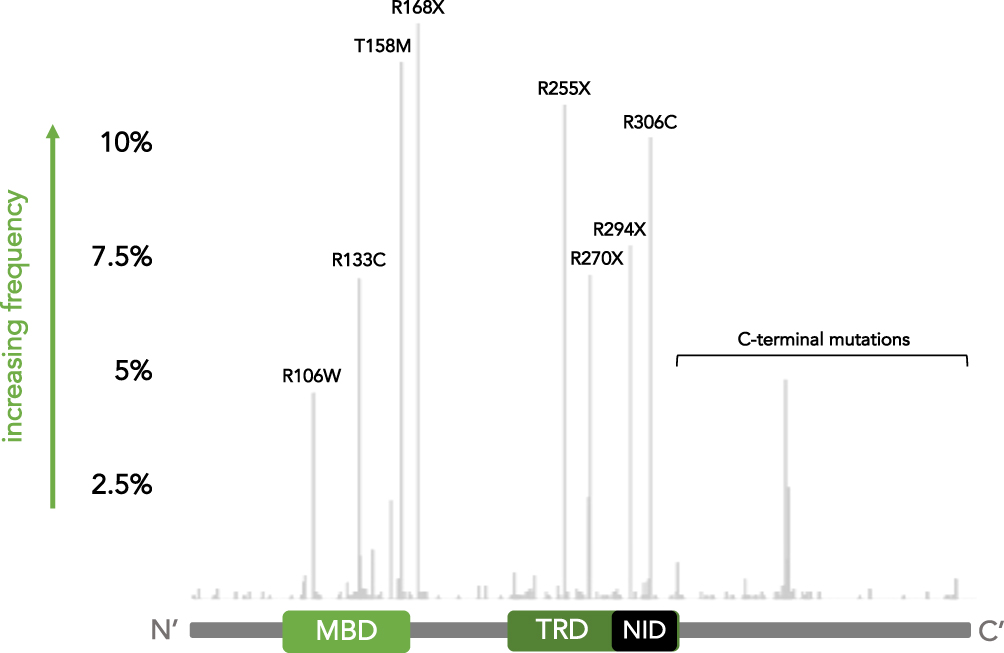 |
Figure 3 Common MECP2 mutations in Rett syndrome. Frequency map of MECP2 mutations identified in girls with RTT from the NIH Natural History Study of Rett Syndrome and Related Disorders. Mutation frequency is depicted on the y-axis while the start of the mutation along the protein sequence is indicated on the x-axis. A schematic of the MeCP2 protein domains is provided for reference. The 8 common point mutations and set of C-terminal mutations are indicated. |
While most MECP2 mutations that cause RTT are in exons 3–4 and affect both transcript isoforms, rare mutations in exon 1 of MECP2 have been reported, as mentioned above.124,125 These are predominantly early frameshift mutations predicted to cause nonsense-mediated decay of the MeCP2_e1 transcript. To date, no mutations that affect only exon 2 (and therefore only MeCP2_e2) have been identified. Some have proposed that the reason for this may be that MeCP2_e1 can functionally compensate for loss of MeCP2_e2, but not vice versa. However, one study in mice showed that transgenic expression of MeCP2_e2 was able to rescue of phenotypes of Mecp2 null mice, demonstrating that work clarifying the functions of MeCP2 isoforms in humans is needed.126
Genotype-Phenotype Correlations in RTT
A high degree of variability in clinical severity is observed within RTT, across clinical features such as language and motor function.19 There are several possible contributors to this variability, including XCI status, MECP2 mutation type, genetic modifiers of MECP2 mutations, and environment. The first, XCI, occurs in a random manner as one X-chromosome is silenced in each cell during early female embryogenesis.127 XCI status dictates relative expression of mutant to non-mutant MeCP2 across cell and tissue types in girls. Higher preference for the non-mutant MECP2 allele is thought to contribute to lower clinical severity.16,22,128 The second, MECP2 mutation type, influences the degree to which MeCP2 function is preserved. Mutations that are partial loss-of-function (eg, R133C, R294X) tend to yield a milder clinical course than mutations that are complete loss-of-function (eg, R255X, large deletions).19 The third, genetic modifiers of MECP2 mutations, may alter clinical phenotypes via direct effects on MECP2 or indirect effects on MeCP2’s function.129 Finally, behavioral interventions such as environmental enrichment have the potential to reduce functional deficits in RTT.130,131
Though it cannot explain the full complement of clinical variability, MECP2 mutation type has emerged as a significant source for both typical and atypical RTT.19,20 Generally, genotype–phenotype correlations map onto both typical and atypical RTT; however, the phenotypes tend to be more highly skewed in atypical RTT.20 For example, complete loss-of-function mutations such as R255X and large deletions are associated with higher clinical severity in both typical and atypical RTT, but the degree of increase in severity is greater in atypical than typical RTT (and vice versa for partial loss-of-function mutations associated with lower clinical severity). Additionally, these correlations appear to affect the entire disease course, as individuals with mild MECP2 mutations tend to have a later diagnosis and less severe disease as children (and vice versa). Despite this difference in baseline severity, clinical severity does increase with age across most mutations. The largest contributors to clinical severity are hand use and onset of stereotypies, with smaller contributions from growth, motor dysfunction, and communication dysfunction. Ultimately, these genotype–phenotype correlations denote a pattern, but individuals with RTT may not have disease that exactly ascribes to this pattern. Other factors, including XCI, genetic modifiers, and environment, can also alter disease course. Intriguingly, individuals have been identified with RTT-causing mutations in MECP2 and random XCI who have neurodevelopmental problems but do not have the characteristic RTT features (ie, regression) and overall clinical severity of people with RTT, pointing to such other genetic factors influencing the ultimate phenotypical manifestation of alteration in MeCP2 function.132
MECP2 Mutations Associated with Mild Disease in RTT
MECP2 mutations associated with mild disease include R133C, R294X, and C-terminal mutations. The R133C mutation has been shown to partially disrupt binding to methylated DNA, particularly to hydroxymethylated cytosines, leading to partial loss-of-function of MeCP2.72,105,133 In contrast, the later R294X and C-terminal mutations do not disrupt the MBD and instead disrupt the NCoR/SMRT interaction domain (NID) sequence (R294X) or the 3’ end of the transcript (C-terminal mutations). Recent work has shed light on the partial loss-of-function mechanism of R294X; however, the pathogenic mechanism of C-terminal mutations remains largely unknown.
The R294X mutation, one of the 8 common point mutations, occurs in approximately 6% of individuals with typical RTT and approximately 4% of individuals with atypical RTT.19,20 R294X is an arginine-to-stop mutation that occurs within the NID sequence. It is predicted to retain methylated DNA binding capacity and interrupt binding to the transcriptional co-repressor complex NCoR/SMRT. Recent mouse modeling work has shown that male hemizygous Mecp2R294X/Y (R294X) mice exhibit RTT-like phenotypes with delayed onset and increased lifespan compared to mouse models of other MECP2 mutations.134 These mild RTT-like phenotypes are associated with production of stable truncated MeCP2, unlike other models of nonsense MECP2 mutations. Collectively, this work suggests that the R294X mutation acts through partial loss-of-function to cause RTT. Molecular work confirmed that the R294X truncation product binds to chromatin; however, interestingly, the truncation product bears higher chromatin binding affinity compared to wild type (WT) MeCP2.134
C-terminal domain (CTD) mutations are a class of MECP2 mutations grouped by genomic location. They occur in approximately 9% of individuals with typical RTT and approximately 17% of individuals with atypical RTT.20 CTD mutations span a region of more than 500 bp and are highly variable, with over 100 different C-terminal MECP2 mutations reported (RettBase: http://mecp2.chw.edu.au). However, a hotspot of small deletion-frameshift mutations starting between c.1157–1164 comprises about 60% of all CTD mutations. Two of these hotspot mutations have been modelled in mice: c.1157_1197del41 and c.1164_1207del44.135 Mice with the c.1157_1197del41 mutation displayed severe RTT-like phenotypes and reduced lifespan; correspondingly, mice expressed very low levels of truncated MeCP2. However, mice with the c.1164_1207del44 mutation did not display severe RTT-like phenotypes and had no difference in lifespan compared to WT mice. Interestingly, when the mouse c.1164_1207del44 Mecp2 allele was “humanized” with several added point mutations around the mutation to more closely match the human c.1164_1207del44 MECP2 allele, truncated protein levels were significantly reduced in a cellular model. This work suggested that human-specific sequences around the CTD mutation hotspot might impact protein stability. Other studies have implicated the C-terminus in facilitating chromatin binding and in directing chromatin architecture, suggesting that CTD mutations might impact function as well as stability.97,136 Still, a clear picture of how specific CTD mutations affect MeCP2 function is needed.
Disease Modeling of RTT
While several animal models of RTT have been developed, including mice, zebrafish, and monkeys, this review will focus on mouse models given their widespread use and utility in modeling mutation-specific aspects of disease.137,138 The first mouse models of RTT were developed in the early 2000s, from the labs of Adrian Bird and Rudolf Jaenisch.139,140 Both models involved deletion of exons shared by the two isoforms of Mecp2 (Bird null: exons 3–4; Jaenisch null: exon 3). Global deficiency of MeCP2 led to a constellation of RTT-like phenotypes that exhibited sex-specific onset. Hemizygous male Mecp2−/Y mice were significantly underweight from 4 weeks of age (on the C57BL/6 background) and developed stiff gait, decreased activity, and tremor between 3 and 8 weeks of age.139,140 Mecp2−/Y mice also developed hindlimb clasping and irregular breathing, with early death at approximately 10 weeks of age. In contrast, heterozygous female Mecp2± mice displayed a similar set of RTT-like phenotypes, including stiff gait, decreased activity, hindlimb clasping, and irregular breathing, beginning much later, at 3–4 months of age.139,140 The phenotypes exhibited by mice with global MeCP2 deficiency recapitulate features of RTT in humans, permitting use of this system for disease modelling.
The predominance of neurological features in RTT suggested that the central nervous system may be the predominant site of MeCP2 dysfunction. To test this, both the Bird and Jaenisch labs crossed mice expressing a loxP-flanked Mecp2 allele with Nestin-Cre driver mice. This cross results in deficiency of MeCP2 in the CNS and PNS, including neuronal and glial precursors. Nestin-Cre conditional mutant mice were indistinguishable from global knockout (KO) mice. While unexpected due to the broad expression of MeCP2 throughout the body, this result indicated that phenotypes of Mecp2−/Y and Mecp2± mice are attributable to loss of MeCP2 in the nervous system.139,140 Furthermore, the Jaenisch lab crossed mice expressing a loxP-flanked Mecp2 allele with CamK-Cre93 driver mice to yield MeCP2 deficiency in postnatal forebrain, hippocampus, and brainstem neurons. These CamK-Cre93 conditional mutant mice displayed typical development until 3 months, at which time they began to show similar RTT-like phenotypes of stiff gait and decreased activity.139 This result suggested that RTT-like phenotypes are attributable to MeCP2 deficiency in neurons rather than glia. Consistently, follow-up work showed that astrocyte-specific reactivation of Mecp2 in globally MeCP2 deficient mice partially rescued behavioral abnormalities and had very minimal effect on lifespan,141 while microglia-specific reactivation of Mecp2 in globally MeCP2 deficient mice did not rescue behavioral abnormalities.142,143
The Jaenisch lab’s work suggested that MeCP2 deficiency in post-mitotic neurons is sufficient to cause disease. This was confirmed and extended by experiments that induced MeCP2 deficiency when mice reached maturity. Specifically, a tamoxifen-inducible Mecp2 knockout strategy was applied to mice aged 60 days or older, eliminating MeCP2 only during adulthood.144 These mice exhibited RTT-like phenotypes similar to germline Mecp2 knockout mice, including hypoactivity, abnormal gait, hindlimb clasping, and premature death with similar median time to death after dosing (~10–12 weeks). This result demonstrated that MeCP2 expression is required continuously in the developed brain and argued that, though the onset of RTT correlates with neurodevelopment, the disease may not be attributable to disruption of the developing brain.
Given these findings, the field raised the reverse question of whether reactivation of Mecp2 after symptom onset could provide therapeutic benefit. This was shown in a tamoxifen-inducible Cre recombination experiment, whereby a loxP-flanked stop sequence is excised upon administration of tamoxifen.145 Reactivation of Mecp2 after symptom onset in hemizygous male Mecp2 mutant mice rescued phenotypic score, improved motor behavior and breathing phenotypes, and preserved survival up to 30 weeks of age.145,146 Reactivation of Mecp2 after symptom onset (~30 weeks of age) in heterozygous female Mecp2 mutant mice similarly significantly rescued phenotypic score, representing observation of activity, gait, hindlimb clasping, tremor, breathing, and coat condition.145 Together, these experiments provided early hope for the potential of disease-modifying therapeutics, through reversal of MeCP2 loss after onset of symptoms and diagnosis in people.
Since then, numerous studies have used complex mouse genetics to achieve cell-type specific loss of MeCP2 in WT mice or reactivation of Mecp2 in global knockout mice, to determine the cell types, brain regions, and circuits involved in RTT pathogenesis. Selective reduction of MeCP2 has been behaviorally characterized in multiple different brain regions, including postnatal forebrain,139,147 basal ganglia,148 hypothalamus,149 and cerebellum,150 as well as in specific cell types, including glutamatergic neurons,151 GABAergic neurons,152 dopaminergic and noradrenergic neurons,153 serotonergic neurons,153 parvalbumin-expressing interneurons,154 and somatostatin-expressing interneurons.154 Though the details of behavioral consequences of region- and cell type-specific MeCP2 loss are beyond the scope of this review, collectively this work demonstrates that MeCP2 function is critical across a variety of brain regions that, together, contribute to the complex behaviors observed in RTT.
Additionally, extensive work has gone into the development of mouse models harboring common MECP2 mutations to understand their pathophysiological mechanism. To date, mouse models of all 8 common MECP2 point mutations – R106W, R133C, T158M, R168X, R255X, R270X, R294X, and R306C – have been generated (Table 1).82,133,134,155–161 These mutations fall into two categories – missense (R106W, R133C, T158M, R306C) and nonsense/truncating (R168X, R255X, R270X, R294X) mutations. Each of the missense mutations yield a protein product in mice at varying levels compared to full-length WT MeCP2. In comparison, amongst the nonsense mutations, R168X and R255X produce no detectable protein product while R270X and R294X mutations yield produce stable truncation products in mice. However, R270X truncation product was not detected in a human brain lysate from a heterozygous female individual with RTT, suggesting that features of the R270X mouse model may not reflect human pathophysiology.160 Despite differences in protein production amongst the 8 common point MECP2 mutations, they each cause RTT via complete or partial loss-of-function. In the latter case, mutations disrupting the MBD are predicted to decrease binding to methylated DNA, while mutations disrupting the NID are predicted to decrease interaction with transcriptional co-repressors. Furthermore, different mutations within the MBD differentially affect binding to methylated DNA.162 For example, the R106W mutation nearly abolishes methylated DNA binding capacity, while R133C retains partial binding capacity. Apart from the 8 common point mutations, an additional 2 C-terminal mutations have been modelled in mice – c.1157_1197del41 (p.L386HfsX5) and c.1164_1207del44 (p.P389X) (Table 1).135 L386HfsX5 mice display RTT-like phenotypes and decreased survival, consistent with other models of MECP2 mutation. However, P389X mice did not display RTT-like behavioral phenotypes and had survival comparable to WT mice up to 1 year. As discussed in the previous section, differences in the MeCP2 C-terminus between mice and humans may explain this discrepancy; however, more work is needed to understand the mechanism underlying C-terminal MECP2 mutation.
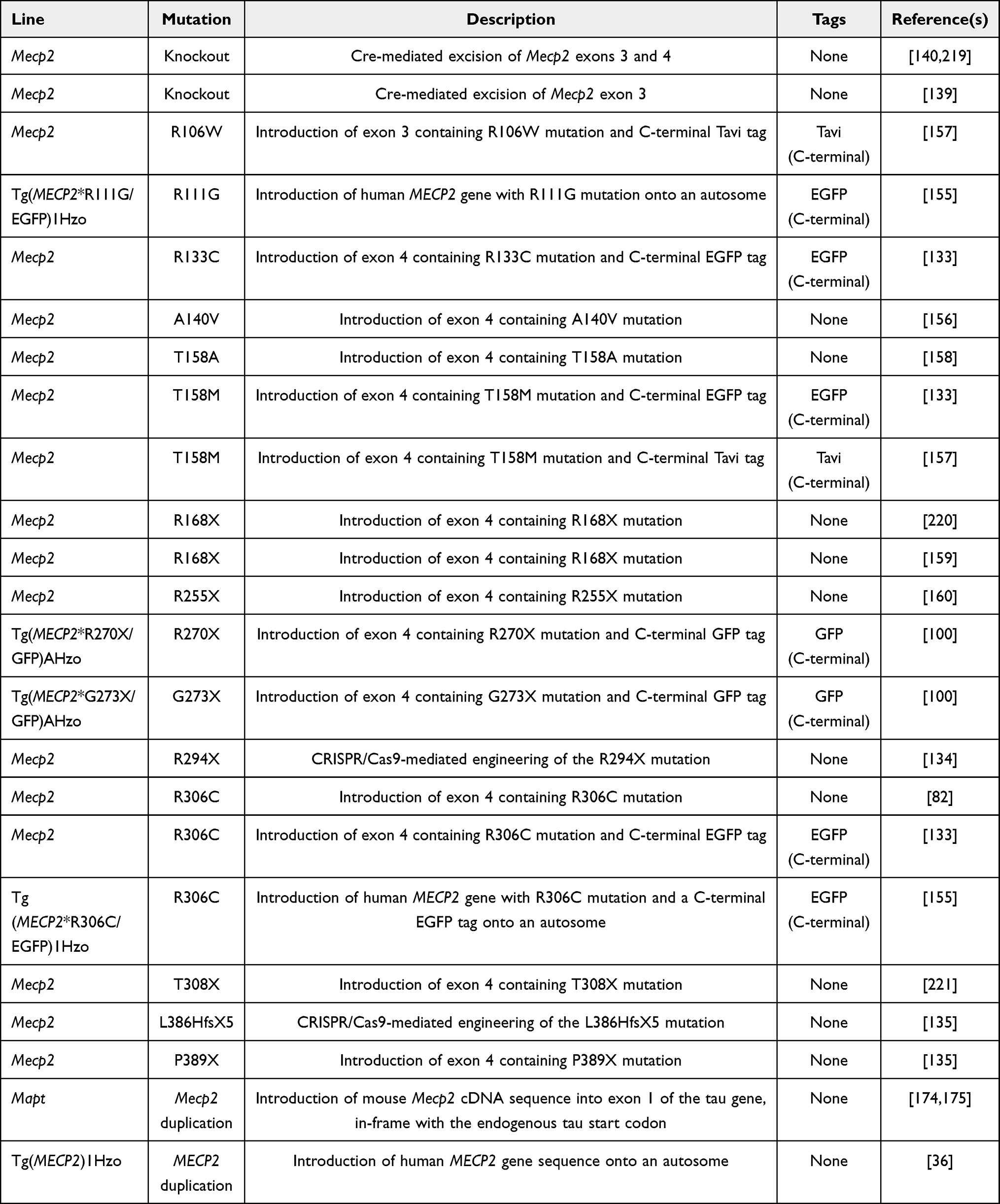 |
Table 1 Mouse Models of MECP2 Mutation and Duplication |
With the knowledge that MECP2 loss-of-function predominantly affects neurons in the brain, the field moved to determine synaptic effects of MeCP2 loss. Basal synaptic transmission was first evaluated by input-output (I/O) curves, which correlate presynaptic volleys as input (representing depolarization of presynaptic terminals) with the slope of evoked field excitatory postsynaptic potentials as output (field EPSPs). At Schaffer collateral-CA1 synapses, male Mecp2−/Y and female Mecp2± mice have I/O ratios unchanged from WT, indicating similar basal synaptic transmission.145,163 MeCP2 loss does, however, affect presynaptic function. One metric of this is paired-pulse facilitation (PPF), a form of short-term plasticity. PPF is measured by delivering two stimuli within a short millisecond interstimulus interval and measuring field EPSP responses. Typically, the second recorded field EPSP is enhanced due to elevated presynaptic calcium concentration following the first stimulus, leading to a greater release of synaptic vesicles following the second stimulus. Pre-symptomatic Mecp2 mutant mice do not display differences in PPF compared to WT littermates; however, symptomatic Mecp2 mutant mice display decreased PPF, suggesting increased release probability.163–165 This PPF phenotype may be caused by direct effects of MeCP2 protein level on calcium concentration in the presynaptic terminal or indirect alterations of presynaptic proteins involved in neurotransmitter release, that take effect only during postnatal development.166
Long-term potentiation (LTP) and long-term depression (LTD) are forms of long-term synaptic plasticity in which different frequency stimulations cause a persistent increase or decrease in synaptic strength. Male hemizygous Mecp2−/Y and female heterozygous Mecp2± mice both show attenuated LTP and LTD in the CA1 region of the hippocampus.145,163,164 Of note, and consistent with the LTP phenotype, Mecp2−/Y mice express lower levels of N-methyl-D-aspartate (NMDA) receptor subunit NR2A and higher levels of subunit NR2B compared to WT.163 The NR2B subunit is expressed predominantly at immature synapses, and the NR2A subunit is more efficient than NR2B in generating NMDA receptor-dependent LTP at adult CA1 synapses.167 A final form of plasticity affected by MeCP2 loss is homeostatic plasticity, the ability of neurons to restore their activity to a setpoint following changes to network activity. One specific form of homeostatic plasticity is synaptic scaling, characterized by cell-wide changes in synaptic strength following neuronal activity. MeCP2 deficiency prevents an activity-dependent decrease in the α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptor subunit GluR2 expression and synaptic scaling in hippocampal cultures,168 as well as synaptic scaling in the neocortex after sensory deprivation.169
Loss of MECP2 expression is also associated with changes to synaptic structure. Mouse and human neurons deficient in MeCP2 have smaller neuronal cell size and increased density of neurons.96,139,170,171 Additionally, post-mortem brain tissue and mouse neurons deficient in MeCP2 show low dendritic spine density and decreased dendritic branching complexity.102,170,172,173 These results argue that MeCP2 is involved in the structural maturation of neurons, including dendritic spine development and dendritic arborization.
Disease Modeling of MDS
With the successful generation of mouse models of RTT, two mouse models of MDS were subsequently developed. The first, Tg(MECP2), was created by the Zoghbi lab via the introduction of the human MECP2 genomic sequence (including the genomic DNA containing the promoter and regulatory sequences) into an autosome, effectively globally duplicating protein levels throughout the life of the animal (Table 1).36 Tg(MECP2) mice demonstrate typical development until approximately 10–12 weeks of age, at which time they show forepaw clasping, enhanced motor learning, and enhanced fear response in a contextual fear conditioning paradigm. As they age, these Tg(MECP2) mice develop seizures, hypoactivity, and spasticity with early death. Importantly, analysis of several transgenic lines with differing MeCP2 protein levels found that increasing levels of MeCP2 protein correlated with increased severity of MDS-like phenotypes, reinforcing the notion that MECP2 gene dosage drives MDS. A second mouse model of MDS, Mapttm1(Mecp2), was created by the Jaenisch lab via the introduction of the mouse Mecp2 coding sequencing in-frame into the first exon of the Tau gene, effectively globally duplicating the gene in post-mitotic neurons (Table 1).174 This tau-MeCP2 fusion protein contains the first 31 amino acids of Tau followed by the MeCP2 E2 protein and, when combined with endogenous mouse MeCP2, yields approximately twice the typical levels of MeCP2 in the brain.174,175 The Mapttm1(Mecp2) mice display motor dysfunction, increased anxiety-like behavior, and enhanced fear response in contextual and cued conditioning paradigms.
Both Tg(MECP2) and Mapttm1(Mecp2) MDS mouse models have behavioral phenotypes redolent of MDS features, including deficits in motor coordination and increased anxiety-like behavior. However, there are differences between the two; for example, the Mapttm1(Mecp2) mice do not display decreased lifespan while the Tg(MECP2) mice do display decreased lifespan. Discrepancies such as this may be attributed to differences in transcriptional control of the transgene (ie, the native MECP2 human promotor and regulatory sequences vs those of mouse Tau) or differences in function of the protein (ie, human MeCP2 E1 and E2 vs tau-MeCP2-E2 fusion). Complicating this picture, the Tg(MECP2) model of MDS has been reported to show mouse strain-specific phenotypes, indicating that background affects phenotypic expression. To circumvent this, several studies have used F1 hybrid mice with success.155,176,177 Due to these complicating issues, the contributions of specific cell types and brain regions to MDS-like phenotypes have not been as thoroughly dissected as in RTT. However, one study showed reversal of social recognition deficits in MDS model mice via normalization of MeCP2 levels in the medial prefrontal cortex.178 Additionally, elevated anxiety-like behavior and decreased social approach in MDS model mice were reversed by genetic reduction of corticotropin-releasing hormone (CRH) or opioid receptor mu (μ) 1 (OPRM1), respectively.177
Apart from MDS-like behavior, mouse models have provided insight into the synaptic underpinnings of the disease. Electrophysiological analysis of MDS mouse model hippocampal brain slices showed impairments in synaptic plasticity. These experiments found normal I/O curves, indicating that Schaffer collateral input onto CA1 neurons is intact.175 However, PPF, a form of short-term synaptic plasticity, was enhanced in hippocampal slices of both Tg(MECP2) and Mapttm1(Mecp2) mice, suggesting a presynaptic effect of MeCP2 overexpression leading to a decrease in release probability.36,175 Consistently, some cognitive and motor behavioral phenotypes and synaptic dysfunction can be normalized by treatment with a non-competitive GABAA receptor antagonist, picrotoxin.179 Additionally, MDS model mice show differences in LTP in the hippocampus.36,175 While hippocampal LTP was enhanced in Tg(MECP2) mice, hippocampal LTP was attenuated in Mapttm1(Mecp2) mice; these differences may be due to pattern of MeCP2 expression and require further study. Spontaneous neurotransmission was evaluated alongside evoked neurotransmission in the Mapttm1(Mecp2) mice. Recordings from cultured hippocampal neurons found increased resting excitatory drive, with enhanced mini-excitatory postsynaptic currents (mEPSCs) and no changes to mini-inhibitory postsynaptic currents (mIPSCs).175 Collectively, these studies of neurotransmission suggest that MeCP2 overexpression causes alterations to excitatory neurotransmission in the hippocampus, with increased resting excitatory drive.
While the molecular mechanism for these electrophysiological alterations is not well understood, MeCP2 overexpression has been associated with alterations to structural features of synapses. For example, in mouse hippocampal CA1, MeCP2 overexpression leads to increased glutamatergic synaptic density during early postnatal development.180 Consistently, cortical neurons derived from several different MECP2 duplication induced pluripotent stem cell (iPSC) lines show increased synaptogenesis and dendritic complexity.181 However, overexpression of WT MECP2 in rat hippocampal pyramidal slices leads to a transient decrease in the density of spines, specifically of mature spines, without affecting dendritic complexity or length.172 These studies suggested that the structural effects of MeCP2 overexpression are model- and timepoint-dependent. Follow-up work used the Tg(MECP2) mouse model to study L5 pyramidal neurons in the barrel cortex (region of somatosensory cortex that receives input from whiskers via the thalamus) in vivo.182 In this model, dendritic spine density was initially increased until 12 weeks of age, after which point MDS model animals showed decreased dendritic spine density compared to WT littermates. Interestingly, spontaneous spine turnover remains increased in aged MDS model animals, with higher spine gain and loss rates biased towards spine loss.
In addition to baseline differences in spine density, the structural plasticity of spines is also affected by MeCP2 overexpression. In WT mice, rotarod motor training induces an increase in bouton elimination rate in the motor cortex.183,184 MeCP2-overexpressing mice do not exhibit this increase in bouton elimination, and instead show excessive formation and stabilization of dendritic spine clusters, consistent with the behavioral phenotype of motor overperformance. These molecular and behavioral changes are reversible by pharmacological inhibition of ERK, suggesting that Ras-ERK signaling hyperactivity contributes to these phenotypes in MDS model animals.185 Collectively, this work supports the notion that excessive MeCP2 contributes to inappropriate consolidation of clustered synaptic connections during motor learning, in addition to having baseline effects on spine density.
Therapeutic Development for RTT and MDS
Prior to the systematic evaluation of animal models of RTT, treatment trials using folate and betaine to increase DNA methylation were undertaken, but this treatment did not result in clinical improvement in people with RTT.186 The reversibility of RTT-like phenotypes in mouse models after symptom onset has provided hope that disease-modifying treatment could be developed for individuals diagnosed with RTT. Rescue experiments have shown that elevation of MeCP2 levels in symptomatic mice significantly improves, but does not fully rescue, overall RTT-like phenotypes, survival, and respiratory and motor function.145,146 The lack of complete rescue may be due to incomplete restoration of MeCP2 levels across the brain, but further work is needed to determine this. Regardless, a cautious but hopeful outlook for RTT therapy has spurred preclinical work and clinical trials in this space.
One of the first targets of RTT therapeutic development was BDNF, an important mediator of neuronal and synaptic maturation. In heterozygous female mouse models of RTT and in humans with RTT, BDNF transcript is downregulated with loss of functional MeCP2.187 Further, overexpression of BDNF in Mecp2 null male mice extends survival, rescues specific motor deficits, and reverses electrophysiological abnormalities.188 Therapeutic efforts were directed towards increasing BDNF levels as an indirect method of restoring MeCP2 function. One of these methods was treatment with ampakines, drugs that enhance glutamatergic AMPA receptor activity to upregulate endogenous BDNF. Chronic treatment of hemizygous male Mecp2−/Y mice with the ampakine CX546 was able to restore normal breathing rate and minute volume/weight.189 Another drug shown to increase endogenous BDNF is fingolimod, a sphingosine-1 phosphate receptor agonist that crosses the blood brain barrier, and administration of fingolimod to hemizygous male Mecp2−/Y mice normalized rotarod motor performance and extended lifespan by approximately 1 month.190 A clinical trial assessing the safety and efficacy of oral fingolimod in children older than 6 years found that the drug is safe but found no evidence of an effect on clinical, laboratory, or imaging measures of disease (FINGORETT; NCT02061137).191
Unfortunately, the blood brain barrier penetrance of BDNF is low, making its application as a therapeutic challenging. Insulin-like growth factor 1 (IGF1), however, confers similar effects on neuronal survival and maturation through overlapping signaling pathways and crosses the blood brain barrier, especially through its metabolite glycine-proline-glutamate (GPE).192–194 Treatment of hemizygous male Mecp2−/Y mice with GPE showed distinct benefits after initiating dosing from a young age (P15-P18).195 Specifically, GPE promoted survival, improved structural brain abnormalities (eg, brain weight, spine density), and improved behavioral and physiological abnormalities (eg, breathing irregularity, activity). Additionally, treatment of adult female heterozygous Mecp2± mice with GPE stabilized cortical plasticity to WT levels.195 This encouraging data provided the impetus for further preclinical studies of IGF1, GPE, and GPE analogues like trofinetide in RTT. Trofinetide, the proline-methylated analogue of GPE, harbors an improved pharmacokinetic profile compared to GPE and a Phase II clinical trial in individuals with RTT showed that oral trofinetide is generally safe and well tolerated (NCT01703533).196 Promisingly, trofinetide at 200 mg/kg twice daily in children/adolescents with RTT improves core features across multiple disease domains as assessed by clinicians and caregivers (NCT02715115).197 A recently completed Phase III trial of trofinetide in children and adolescents with RTT (NCT04181723) reported success in meeting the primary clinical efficacy outcomes (https://acadia.com/media/news-releases/acadia-pharmaceuticals-announces-positive-top-line-results-from-the-pivotal-phase-3-lavender-trial-of-trofinetide-in-rett-syndrome/).
Another modulator of the BDNF signaling pathway is the NMDA receptor antagonist ketamine. Previous work in mouse models has shown that ketamine can act through NMDA receptor antagonism to release a translational block of Bndf.198 Acute systemic treatment with ketamine in female heterozygous Mecp2± mice normalizes pre-pulse inhibition of acoustic startle, and chronic treatment of Mecp2−/Y mice improves locomotor activity, breathing, and survival.199,200 A Phase II clinical trial of oral ketamine in RTT is ongoing (NCT03633058).
A final modulator of BDNF signaling is glatiramer acetate, a collection of artificial polypeptides currently used for treatment of multiple sclerosis in the United States.201 Though its mechanism is incompletely understood, it is thought to activate peripheral T-cells that cross the blood–brain barrier and stimulate secretion of neurotrophic factors such as BDNF in the brain.202,203 A Phase II clinical trial of glatiramer acetate in girls with RTT showed improvement in clinical features such as gait velocity, memory, and breath-holding.204 However, multiple severe post-injection adverse reactions were later observed in an open-label clinical trial in girls with RTT, raising significant safety concerns with glatiramer acetate in this population.205 Apart from modulation of BNDF signaling, therapeutic efforts for RTT have also targeted other downstream mediators of MeCP2 function, such as neurotransmitter systems (eg, GABA, dopamine, serotonin) with variable success in mouse models.206
Direct methods to restore MeCP2 function aimed to elevate functional MeCP2 protein. These include 1) gene therapy, 2) X-chromosome reactivation, 3) genome editing, and 4) RNA editing strategies. First, gene therapy employs viral delivery of the MECP2 gene to permit increased protein expression. Numerous studies have evaluated the safety and efficacy of modelled gene therapy in hemizygous Mecp2−/Y mice and have demonstrated rescue of survival, body weight, and select behavioral phenotypes.207–212 However, only one study thus far has evaluated the safety and efficacy of gene therapy in heterozygous Mecp2+/- mice, where authors demonstrated that systemic viral delivery in adulthood rescued gross phenotypic progression as well as motor and cognitive behavioral phenotypes.207 Collectively these studies have also identified potential barriers to the use of gene therapy in people, most prominently hepatotoxicity occurring with systemic delivery.209,212 Second, X-chromosome reactivation aims to pharmacologically de-repress MECP2 transcription from the inactive X-chromosome in female neurons.213 Third, genome editing aims to correct the mutant MECP2 allele to via approaches such as CRISPR/Cas9 (clustered regularly interspaced short palindromic sequences).214 Finally, RNA editing employs adenosine deaminase enzymes to program site-specific adenine-to-inosine editing of RNAs and correction of G > A mutations in Mecp2 at the transcript level.215,216
In contrast to those for RTT, current efforts to develop therapeutics for MDS have focused on reducing the levels of MeCP2. One approach has been through antisense oligonucleotides (ASOs), which are short, synthetic single strands of chemically modified DNA designed to alter mRNA expression and subsequently protein level.217 A human-specific MECP2-ASO has been shown to reduce MeCP2 levels in brain and rescue behavioral and EEG phenotypes in mice with one human copy of MECP2.176 This work showed that the benefits from ASO therapy were apparent approximately 4 weeks later than benefits from genetic rescue, likely due to gradual reduction of MeCP2 levels. More recent work has shown benefit of a human-specific MECP2-ASO in mice harboring two copies of the human MECP2 allele.218 This experiment importantly introduced the potential to titrate MECP2-ASO and determine the optimal dosage. Promisingly, no toxicity associated with sub-therapeutic MeCP2 levels was observed across the tested ASO doses. This study did find that molecular changes in response to therapy precede behavioral changes by almost 2 months, suggesting that molecular information could be used as an early biomarker for dosing guidance.
Conclusion
Rett syndrome (RTT) and MECP2 duplication syndrome (MDS) are related neurodevelopmental disorders that each result from errant MeCP2 function – loss and gain, respectively. This review has highlighted the genetic origin, clinical characteristics, cellular features, and therapeutic approaches under development for both RTT and MDS, summarized in Table 2. While these disorders stem from opposite effects on MeCP2 function, they share some clinical and cellular features. For example, both individuals with RTT and MDS can have seizures, impaired speech, gait abnormalities, and GI issues. However, there are RTT-specific features (eg, developmental regression, breathing abnormalities) and MDS-specific features (eg, infantile hypotonia, recurrent respiratory infections) as well. Furthermore, MeCP2 loss and gain of function have largely opposite synaptic-level effects, though there is evidence that both RTT and MDS model mice exhibit attenuated LTP in hippocampus, suggesting that MeCP2 loss and elevation differentially affect hippocampal circuitry. This review has also described a set of therapeutic approaches under development for RTT and MDS. Perhaps most currently promising among those for RTT is trofinetide, which recently completed a Phase III clinical trial (LAVENDER; NCT04181723) with reportedly encouraging results. Though therapeutic development for MDS is still in the preclinical stage, MECP2-ASO therapy provides a potentially titratable, disease-modifying approach to reducing MeCP2 protein levels. Collectively, these efforts provide hope for the development of efficacious therapies for MECP2-related neurodevelopmental disorders RTT and MDS in the coming years.
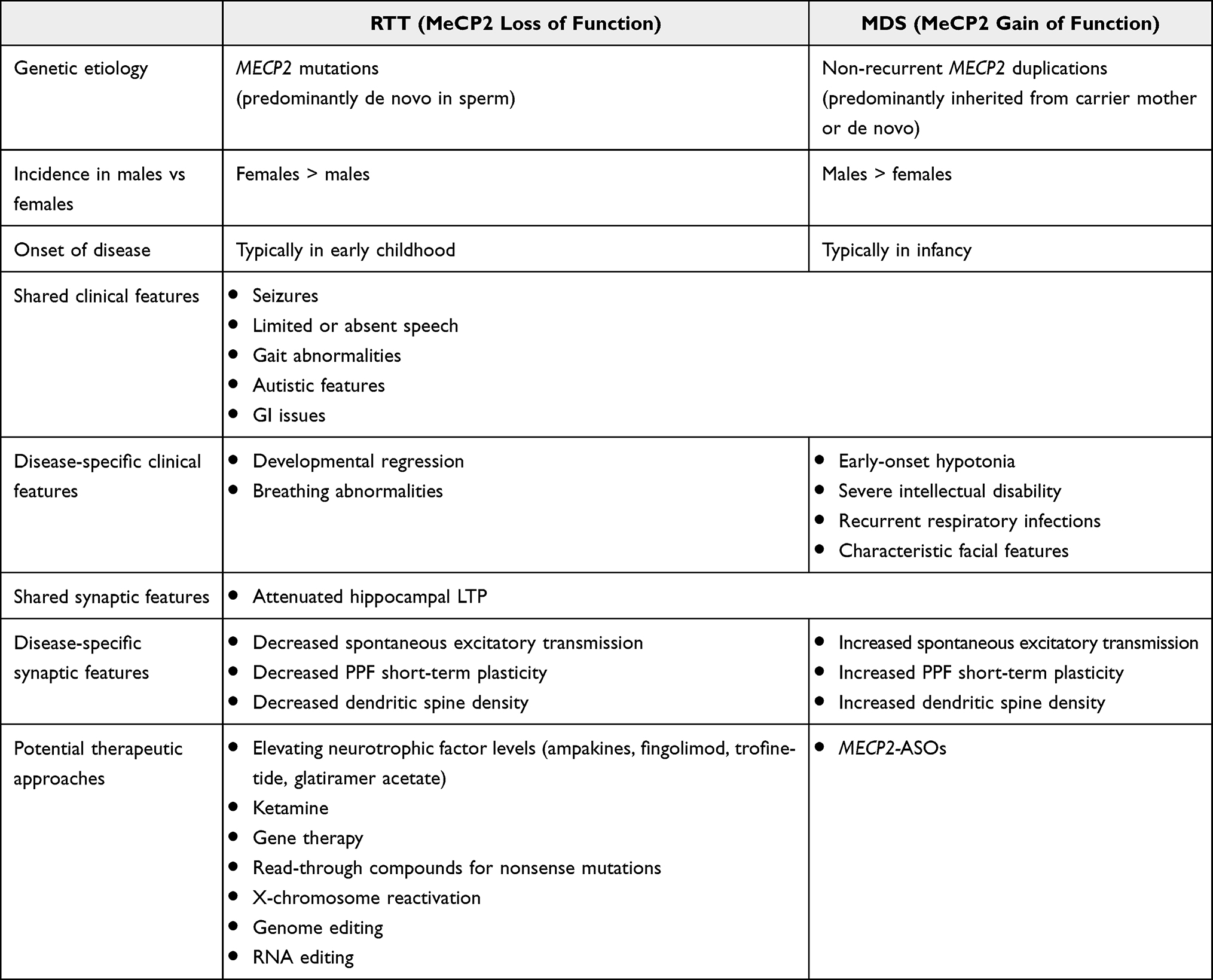 |
Table 2 Similarities and Differences Between RTT and MDS |
Abbreviations
AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid; ASD, autism spectrum disorder; ASO, antisense oligonucleotide; BDNF, brain-derived neurotrophic factor; BRM, BRAHMA; CGI, CpG island; CNS, central nervous system; CRH, corticotropin-releasing hormone; CRISPR, clustered regularly interspaced short palindromic repeats; CTD, C-terminal domain; DNMT, DNA methyltransferase; EPSC, excitatory postsynaptic current; EPSP, excitatory postsynaptic potential; ESC, embryonic stem cell; FLNA, Filamin A; FoSTeS, fork stalling and template switching; GABA, γ-aminobutyric acid; GDI1, GDP dissociation inhibitor 1; GFP, green fluorescent protein; GPE, glycine-proline-glutamate; HP1, heterochromatin protein 1; ID, interdomain; I/O, input-output; IGF1, insulin-like growth factor 1; iPSC, induced pluripotent stem cell; IPSC, inhibitory postsynaptic current; KO, knockout; L1CAM, L1 cell adhesion molecule; LCR, low copy repeat; LOF, loss-of-function; LTD, long-term depression; LTP, long-term potentiation; LUHMES, Lund human mesencephalic; MBD, methyl-binding domain; MDS, MECP2 duplication syndrome; MeCP2, methyl-CpG-binding protein 2; MMBIR, microhomology-mediated break-induced replication; NCoR, nuclear receptor co-repressor; NHEJ, non-homologous end joining; NID, NCoR/SMRT interaction domain; NMDA, N-methyl-D-aspartate; OPRM1, opioid receptor mu (μ) 1; PHF14, PHD finger protein 14; PNS, peripheral nervous system; PPF, paired-pulse facilitation; RAB39B, Ras-related protein Rab-39B; RNA, ribonucleic acid; RTT, Rett syndrome; SMRT, silencing mediator of retinoid and thyroid hormone receptors; SRPK3, SRSF protein kinase 3; TET, ten-eleven translocation; TRD, transcription repression domain; TSS, transcription start site; WT, wild type; YBX1, Y-box binding protein 1; YY1, Yin Yang 1; XCI, X-chromosome inactivation.
Acknowledgment
This work was supported by funding from the National Institutes of Health grants U54HD083211 (JLN), HD083181 (JLN), F30MH122064 (BEC), and the Annette Schaffer Eskind Chair Fund (JLN). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosure
This paper was based on a thesis chapter presented to Vanderbilt University as a PhD thesis with interim findings. The thesis was published in the Vanderbilt University repository (https://ir.vanderbilt.edu/handle/1803/17347). BEC reports grants from NIMH, during the conduct of the study. JLN was a blinded investigator for the Neuren supported Phase 2 trials of trofinetide in RTT and is currently a blinded investigator for the Acadia supported Phase 3 trial of trofinetide in RTT. JLN received funding for consultation from Neuren, Acadia, GW Pharmaceuticals, AveXis, Tasha, Neurogene, GW, Signant Health, and Analysis Group but has no financial stake in any pharmaceutical company; also personal fees from Ovid, personal fees from Roche, personal fees from Alcyone, personal fees from Taysha, personal fees from Myrtelle, personal fees from PeerViewInstitute, personal fees from Acadia, personal fees from Analysis Group, personal fees from Horizon PCORI, other from Lizarbio, personal fees from Medscape, personal fees from NeuroGene, personal fees from Signant, personal fees from SymBiosis, grants from International Rett Syndrome Foundation, grants from Rett Syndrome Research Trust, and grants from National Institutes of Health, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Haas RH. The history and challenge of rett syndrome. J Child Neurol. 1988;3(1_suppl):S3–S5. doi:10.1177/0883073888003001S02
2. Rett A. On a unusual brain atrophy syndrome in hyperammonemia in childhood. Wiener Medizinische Wochenschrift. 1966;116(37):723–726.
3. Hagberg B, Aicardi J, Dias K, Ramos O, Progressive A. Syndrome of Autism, Dementia, Ataxia, and Loss of Purposeful Hand Use in Girls: rett’s Syndrome: report of 35 Cases. Ann Neurol. 1983;14(4):471–479.
4. Hagberg B, Rett Syndrome: W-EI. A suggested staging system for describing impairment profile with increasing age towards adolescence. Am J Med Genet. 1986;1:47–59.
5. Fu C, Armstrong D, Marsh E, et al. Multisystem comorbidities in classic Rett syndrome: a scoping review. BMJ Paediatr Open. 2020;4:e000731.
6. Motil KJ, Barrish JO, Lane J, et al. Vitamin D Deficiency is Prevalent in Females with Rett Syndrome. J Pediatr Gastroenterol Nutr. 2011;53(5):569–574.
7. Killian JT, Lane JB, Lee H-S, et al. Scoliosis in Rett Syndrome: progression, Comorbidities, and Predictors. Pediatr Neurol. 2017;70:20–25.
8. Neul JL, Kaufmann WE, Glaze DG, et al. Rett syndrome: revised Diagnostic Criteria and Nomenclature. Ann Neurol. 2010;68(6):944–950.
9. Laurvick CL, de Klerk N, Bower C, et al. Rett syndrome in Australia: a review of the epidemiology. J Pediatr. 2006;148(3):347–352.
10. Zoghbi ZH. Genetic Aspects of Rett Syndrome. J Child Neurol. 1988;3(1_suppl):S76–S78. doi:10.1177/0883073888003001S15
11. Zoghbi HY, Percy AK, Schultz RJ, Fill C. Patterns of X Chromosome Inactivation in the Rett Syndrome. Brain Dev. 1990;12(1):131–135.
12. Schanen C, Francke U, Severely Affected A. Male Born into Rett Syndrome Kindred Supports X-linked Inheritance and Allows Extension of the Exclusion Map. Am J Hum Genet. 1998;63(1):267–269.
13. Schanen NC, Dahle EJR, Capozzoli F, et al. Syndrome Family Consistent with X-Linked Inheritance Expands the X chromosome Exclusion Map. Am J Hum Genet. 1997;61(3):634–641.
14. Ellison KA, Fill CP, Terwilliger J, et al. Examination of X chromosome Markers in Rett Syndrome: exclusion Mapping with a Novel Variation on Multilocus Linkage Analysis. Am J Hum Genet. 1992;50(2):278–287.
15. Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23(2):185–188.
16. Sirianni N, Naidu S, Pereira J, Pillotto RF, Hoffman EP. Rett Syndrome: confirmation of X-linked Dominant Inheritance, and Localization of the Gene to Xq28. Am J Hum Genet. 1998;63(5):1552–1558.
17. Curtis ARJ, Headland S, Lindsay S, et al. X chromosome linkage studies in familial Rett syndrome. Hum Genet. 1993;90(5):551–555.
18. Archidiacono N, Lerone M, Rocchi M, et al. Rett syndrome: exclusion mapping following the hypothesis of germinal mosaicism for new X-linked mutations. Hum Genet. 1991;86(6):604–606.
19. Neul JL, Fang P, Barrish J, et al. Specific mutations in Methyl-CpG-Binding Protein 2 confer different severity in Rett syndrome. Neurology. 2008;70(16):1313–1321.
20. Cuddapah VA, Pillai RB, Shekar KV, et al. Methyl-CpG-binding protein 2 (MECP2) mutation type is associated with disease severity in Rett syndrome. J Med Genet. 2014;51(3):152–158.
21. Trappe R, Laccone F, Cobilanschi J, et al. MECP2 Mutations in Sporadic Cases of Rett Syndrome Are Almost Exclusively of Paternal Origin. Am J Hum Genet. 2001;68(5):1093–1101.
22. Wan M, Lee SSJ, Zhang X, et al. Rett Syndrome and Beyond: recurrent Spontaneous and Familial MECP2 Mutations at CpG Hotspots. Am J Hum Genet. 1999;65(6):1520–1529.
23. Girard M, Couvert P, Carrié A, et al. Parental origin of de novo MECP2 mutations in Rett syndrome. Eur J Hum Genet. 2001;9(3):231–236.
24. Zhu X, Li M, Pan H, Bao X, Zhang J, Wu X. Analysis of the Parental Origin of De Novo MECP2 Mutations and X chromosome Inactivation in 24 Sporadic Patients With Rett Syndrome in China. J Child Neurol. 2010;25(7):842–848.
25. Duncan BK, Miller JH. Mutagenic deamination of cytosine residues in DNA. Nature. 1980;287(5782):560–561.
26. Oakes CC, La Salle S, Smiraglia DJ, Robaire B, Trasler JM. A unique configuration of genome-wide DNA methylation patterns in the testis. PNAS. 2007;104(1):228–233.
27. Augenstein K, Lane JB, Horton A, Schanen C, Percy AK. Variable phenotypic expression of a MECP2 mutation in a family. J Neurodev Disord. 2009;1(4):313–317.
28. Vorsanova SG, Yurov YB, Ulas VY, et al. Cytogenetic and molecular-cytogenetic studies of Rett syndrome (RTT): a retrospective analysis of a Russian cohort of RTT patients (the investigation of 57 girls and three boys). Brain Dev. 2001;23(Suppl 1):S196–S201.
29. Schwartzman JS, Bernardino A, Nishimura A, Gomes RR, Zatz M. Rett Syndrome in a Boy with a 47, XXY Karyotype Confirmed by a Rare Mutation in the MECP2 Gene. Neuropediatrics. 2001;32(3):162–164.
30. Clayton-Smith J, Watson P, Ramsden S, Black G. Somatic mutation in MECP2 as a non-fatal neurodevelopmental disorder in males. Lancet. 2000;356(9232):830–832.
31. Dayer AG, Bottani A, Bouchardy I, et al. MECP2 mutant allele in a boy with Rett syndrome and his unaffected heterozygous mother. Brain Dev. 2007;29(1):47–50.
32. Neul JL, Benke TA, Marsh ED, et al. The array of clinical phenotypes of males with mutations in Methyl-CpG binding protein 2. Am J Med Genet. 2019;180(1):55–67.
33. Cohen D, Lazar G, Couvert P, et al. MECP2 Mutation in a Boy With Language Disorder and Schizophrenia. Am J Psychiatry. 2002;159(1):148–149.
34. Meins M, Lehmann J, Gerresheim F, et al. Submicroscopic duplication in Xq28 causes increased expression of the MECP2 gene in a boy with severe mental retardation and features of Rett syndrome. J Med Genet. 2005;42(2):e12.
35. Van Esch H, Bauters M, Ignatius J, et al. Duplication of the MECP2 Region is a Frequent Cause of Severe Mental Retardation and Progressive Neurological Symptoms in Males. Am J Hum Genet. 2005;77(3):442–453.
36. Collins AL, Levenson JM, Vilaythong AP, et al. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum Mol Genet. 2004;13(21):2679–2689.
37. Bauters M, Van Esch H, Friez MJ, et al. Nonrecurrent MECP2 duplications mediated by genomic architecture-driven DNA breaks and break-induced replication repair. Genome Res. 2008;18(6):847–858.
38. Carvalho CMB, Zhang F, Liu P, et al. Complex rearrangements in patients with duplications of MECP2 can occur by fork stalling and template switching. Hum Mol Genet. 2009;18(12):2188–2203.
39. Zhang F, Khajavi M, Connolly AM, Towne CF, Batish SD, Lupski JR. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat Genet. 2009;41(7):849–853.
40. Sanlaville D, Prieur M, de Blois MC, et al. Functional disomy of the Xq28 chromosome region. Eur J Hum Genet. 2005;13(5):579–585.
41. Friez MJ, Jones JR, Clarkson K, et al. Recurrent Infections, Hypotonia, and Mental Retardation Caused by Duplication of MECP2 and Adjacent Region in Xq28. Pediatrics. 2006;118(6):e1687–e1695.
42. Ta D, Downs J, Baynam G, Wilson A, Richmond P, Leonard H. A brief history of MECP2 duplication syndrome: 20‑years of clinical understanding. Orphanet J Rare Dis. 2022;17(1):131.
43. Yang T, Ramocki MB, Neul JL, et al. Overexpression of Methyl-CpG Binding Protein 2 Impairs TH1 Responses. Sci Transl Med. 2012;4(163):163ra158.
44. Bauer M, Kölsch U, Krüger R, et al. Infectious and Immunologic Phenotype of MECP2 Duplication Syndrome. J Clin Immunol. 2015;35(2):168–181.
45. Ramocki MB, Peters SU, Tavyev YJ, et al. Autism and Other Neuropsychiatric Symptoms Are Prevalent in Individuals With MECP2 Duplication Syndrome. Ann Neurol. 2009;66(6):771–782.
46. Peters SU, Hundley RJ, Wilson AK, et al. The Behavioral Phenotype in MECP2 Duplication Syndrome: a Comparison with Idiopathic Autism. Autism Res. 2013;6(1):42–50.
47. Van Esch H. MECP2 Duplication Syndrome. Mol Syndromol. 2012;2(3–5):128–136.
48. Lugtenberg D, Kleefstra T, Oudakker AR, et al. Structural variation in Xq28: MECP2 duplications in 1% of patients with unexplained XLMR and in 2% of male patients with severe encephalopathy. Eur J Hum Genet. 2009;17(4):444–453.
49. Reardon W, Donoghue V, Murphy AM, et al. Progressive cerebellar degenerative changes in the severe mental retardation syndrome caused by duplication of MECP2 and adjacent loci on Xq28. Eur J Pediatr. 2010;169(8):941–949.
50. Schwoerer JS, Laffin J, Haun J, Raca G, Friez MJ, Giampietro PF. MECP2 Duplication: possible Cause of Severe Phenotype in Females. Am J Med Genet Part A. 2014;164A(4):1029–1034.
51. Peters S, Fu C, Suter B, et al. Characterizing the Phenotypic Effect of Xq28 Duplication Size in MECP2 Duplication Syndrome. Clin Genet. 2019;95(5):575–581.
52. Clayton-Smith J, Walters S, Hobson E, et al. Xq28 duplication presenting with intestinal and bladder dysfunction and a distinctive facial appearance. Eur J Hum Genet. 2009;17(4):434–443.
53. Nakagawa O, Arnold M, Nakagawa M, et al. Centronuclear myopathy in mice lacking a novel muscle-specific protein kinase transcriptionally regulated by MEF2. Genes Dev. 2005;19(17):2066–2077.
54. Vandewalle J, Van Esch H, Govaerts K, et al. Dosage-Dependent Severity of the Phenotype in Patients with Mental Retardation Due to a Recurrent Copy-Number Gain at Xq28 Mediated by an Unusual Recombination. Am J Hum Genet. 2009;85(6):809–822.
55. Vanmarsenille L, Giannandrea M, Fieremans N, et al. Increased Dosage of RAB39B Affects Neuronal Development and Could Explain the Cognitive Impairment in Male Patients with Distal Xq28 Copy Number Gains. Hum Mutat. 2014;35(3):377–383.
56. Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14(3):204–220.
57. Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13(7):484–492.
58. Ehrlich M, Gama-Sosa MA, Huang L-H, et al. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues or cells. Nucleic Acids Res. 1982;10(8):2709–2721.
59. Meehan RR, Lewis JD, Bird AP. Characterization of MeCP2, a vertebrate DNA binding protein with affinity for methylated DNA. Nucleic Acids Res. 1992;20(19):5085–5092.
60. Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. PNAS. 2006;103(5):1412–1417.
61. Ball MP, Li JB, Gao Y, et al. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat Biotechnol. 2009;27(4):361–368.
62. Xie W, Barr CL, Kim A, et al. Base-Resolution Analyses of Sequence and Parent-of-Origin Dependent DNA Methylation in the Mouse Genome. Cell. 2012;148(4):816–831.
63. Varley KE, Gertz J, Bowling KM, et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 2013;23(3):555–567.
64. Lister R, Mukamel EA, Nery JR, et al. Global Epigenomic Reconfiguration During Mammalian Brain Development. Science. 2013;341(6146):1237905.
65. Huttenlocher PR, Dabholkar AS. Regional Differences in Synaptogenesis in Human Cerebral Cortex. J Comp Neurol. 1997;387(2):167–178.
66. De Felipe J, Marco P, Fairén A, Jones EG. Inhibitory Synaptogenesis in Mouse Somatosensory Cortex. Cereb Cortex. 1997;7(7):619–634.
67. Shahbazian MD, Antalffy BA, Armstrong DL, Zoghbi HY. Insight into Rett syndrome: meCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum Mol Genet. 2002;11(2):115–124.
68. Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-Methylcytosine by TET1 Promotes Active DNA Demethylation in the Adult Brain. Cell. 2011;145(3):423–434.
69. Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL Partner TET1. Science. 2009;324(5929):930–935.
70. Globisch D, Münzel M, Müller M, et al. Tissue Distribution of 5-Hydroxymethylcytosine and Search for Active Demethylation Intermediates. PLoS One. 2010;5(12):e15367.
71. Kriaucionis S, Heintz N. The Nuclear DNA Base 5-Hydroxymethylcytosine Is Present in Purkinje Neurons and the Brain. Science. 2009;324(5929):929–930.
72. Mellén M, Ayata P, Dewell S, Kriaucionis S, MeCP2 HN. Binds to 5hmC Enriched within Active Genes and Accessible Chromatin in the Nervous System. Cell. 2012;151(7):1417–1430.
73. Mellén M, Ayata P, Heintz N. 5-Hydroxymethylcytosine accumulation in postmitotic neurons results in functional demethylation of expressed genes. PNAS. 2017;114(37):E7812–E7821.
74. Lewis JD, Meehan RR, Henzel WJ, et al. Purification, Sequence, and Cellular Localization of a Novel Chromosomal Protein That Binds to Methylated DNA. Cell. 1992;69(6):905–914.
75. Nan X, Meehan RR, Bird A. Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res. 1993;21(21):4886–4892.
76. Guo JU, Su Y, Shin JH, et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat Neurosci. 2014;17(2):215–222.
77. Chen L, Chen K, Lavery LA, Andrew S, Shaw CA, MeCP2 LW. binds to non-CG methylated DNA as neurons mature, influencing transcription and the timing of onset for Rett syndrome. PNAS. 2015;112(17):5509–5514.
78. Gabel HW, Kinde B, Stroud H, et al. Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature. 2015;522(7554):89–93.
79. Ho KL, McNae IW, Schmiedeberg L, Klose RJ, Bird AP, Walkinshaw MD. MeCP2 Binding to DNA Depends upon Hydration at Methyl-CpG. Mol Cell. 2008;29(4):525–531.
80. Wakefield RID, Smith BO, Nan X, et al. The Solution Structure of the Domain from MeCP2 that Binds to Methylated DNA. J Mol Biol. 1999;291(5):1055–1065.
81. Nan X, Campoy FJ, MeCP2 BA. is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell. 1997;88(4):471–481.
82. Lyst MJ, Ekiert R, Ebert DH, et al. Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat Neurosci. 2013;16(7):898–902.
83. Kruusvee V, Lyst MJ, Taylor C, Tarnauskaite Z, Bird AP, Cook AG. Structure of the MeCP2–TBLR1 complex reveals a molecular basis for Rett syndrome and related disorders. Proc Natl Acad Sci. 2017;114(16):E3243–E3250.
84. Jones PL, Veenstra GJC, Wade PA, et al. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19(2):187–191.
85. Nan X, Ng H, Johnson CA, et al. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393(6683):386–389.
86. Kokura K, Kaul SC, Wadhwa R, et al. The Ski Protein Family Is Required for MeCP2-mediated Transcriptional Repression. J Biol Chem. 2001;276(36):34115–34121. doi:10.1074/jbc.M105747200
87. Nan X, Hou J, Maclean A, et al. Interaction between chromatin proteins MECP2 and ATRX is disrupted by mutations that cause inherited mental retardation. PNAS. 2007;104(8):2709–2714.
88. Kernohan KD, Jiang Y, Tremblay DC, et al. ATRX Partners with Cohesin and MeCP2 and Contributes to Developmental Silencing of Imprinted Genes in the Brain. Dev Cell. 2010;18(2):191–202. doi:10.1016/j.devcel.2009.12.017
89. Kernohan KD, Vernimmen D, Gloor GB, Bérubé NG. Analysis of neonatal brain lacking ATRX or MeCP2 reveals changes in nucleosome density, CTCF binding and chromatin looping. Nucleic Acids Res. 2014;42(13):8356–8368. doi:10.1093/nar/gku564
90. Harikrishnan KN, Chow MZ, Baker EK, et al. Brahma links the SWI/SNF chromatin-remodeling complex with MeCP2-dependent transcriptional silencing. Nat Genet. 2005;37(3):254–264. doi:10.1038/ng1516
91. Fuks F, Hurd PJ, Wolf D, Nan X, Bird AP, Kouzarides T. The Methyl-CpG-Binding Protein MeCP2 Links DNA Methylation to Histone Methylation. J Biol Chem. 2003;278(6):4035–4040. doi:10.1074/jbc.M210256200
92. Zhou J, Hamdan H, Yalamanchili HK, et al. Disruption of MeCP2-TCF20 complex underlies distinct neurodevelopmental disorders. PNAS. 2022;119(4):e2119078119.
93. Young JI, Hong EP, Castle JC, et al. Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. PNAS. 2005;102(49):17551–17558.
94. Chahrour M, Jung SY, Shaw C, et al. MeCP2, a Key Contributor to Neurological Disease, Activates and Represses Transcription. Science. 2008;320(5880):1224–1229.
95. Ben-Shachar S, Chahrour M, Thaller C, Shaw CA, Zoghbi HY. Mouse models of MeCP2 disorders share gene expression changes in the cerebellum and hypothalamus. Hum Mol Genet. 2009;18(13):2431–2442.
96. Li Y, Wang H, Muffat J, et al. Global Transcriptional and Translational Repression in Human-Embryonic-Stem-Cell-Derived Rett Syndrome Neurons. Cell Stem Cell. 2013;13(4):446–458.
97. Adams VH, McBryant SJ, Wade PA, Woodcock CL, Hansen JC, Disorder I. Autonomous Domain Function in the Multifunctional Nuclear Protein, MeCP2. J Biol Chem. 2007;282(20):15057–15064.
98. Ghosh RP, Nikitina T, Horowitz-Scherer RA, et al. Unique Physical Properties and Interactions of the Domains of Methylated DNA Binding Protein 2. Biochemistry. 2010;49(20):4395–4410.
99. Aravind L, Landsman D. AT-hook motifs identified in a wide variety of DNA-binding proteins. Nucleic Acids Res. 1998;26(19):4413–4421.
100. Baker SA, Chen L, Wilkins AD, Yu P, Lichtarge O, Zoghbi HY. An AT-hook Domain in MeCP2 Determines the Clinical Course of Rett Syndrome and Related Disorders. Cell. 2013;152(5):984–996. doi:10.1016/j.cell.2013.01.038
101. Lyst MJ, Connelly J, Merusi C, Bird BA. Sequence-specific DNA binding by AT-hook motifs in MeCP2. FEBS Lett. 2016;590(17):2927–2933. doi:10.1002/1873-3468.12328
102. Kishi N, Macklis JD. MECP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Mol Cell Neurosci. 2004;27(3):306–321. doi:10.1016/j.mcn.2004.07.006
103. Ross PD, Guy J, Selfridge J, et al. Exclusive expression of MeCP2 in the nervous system distinguishes between brain and peripheral Rett syndrome-like phenotypes. Hum Mol Genet. 2016;25(20):4389–4404. doi:10.1093/hmg/ddw269
104. Skene PJ, Illingworth RS, Webb S, et al. Neuronal MeCP2 Is Expressed at Near Histone-Octamer Levels and Globally Alters the Chromatin State. Mol Cell. 2010;37(4):457–468. doi:10.1016/j.molcel.2010.01.030
105. Ibrahim A, Papin C, Mohideen-Abdul K, et al. MeCP2 is a microsatellite binding protein that protects CA repeats from nucleosome invasion. Science. 2021;372(6549):eabd5581.
106. Yasui DH, Peddada S, Bieda MC, et al. Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. PNAS. 2007;104(49):19416–19421.
107. Boxer LD, Renthal W, Greben AW, et al. MeCP2 Represses the Rate of Transcriptional Initiation of Highly Methylated Long Genes. Mol Cell. 2020;77(2):294–309.
108. Clemens AW, Wu DY, Moore JR, Christian DL, Zhao G, Gabel HW. MeCP2 Represses Enhancers through Chromosome Topology-Associated DNA Methylation. Mol Cell. 2020;77(2):279–293.
109. Zhou Z, Hong EJ, Cohen S, et al. Brain-specific Phosphorylation of MeCP2 Regulates Activity-Dependent Bdnf Transcription, Dendritic Growth, and Spine Maturation. Neuron. 2006;52(2):255–269.
110. Ebert DH, Gabel HW, Robinson ND, et al. Activity-dependent phosphorylation of MeCP2 threonine 308 regulates interaction with NCoR. Nature. 2013;499(7458):341–345.
111. Georgel PT, Horowitz-Scherer RA, Adkins N, Woodcock CL, Wade PA, Hansen JC. Chromatin Compaction by Human MeCP2. J Biol Chem. 2003;278(34):32181–32188.
112. Ghosh RP, Horowitz-Scherer RA, Nikitina T, Shlyakhtenko LS, Woodcock CL. MeCP2 Binds Cooperatively to Its Substrate and Competes with Histone H1 for Chromatin Binding Sites. Mol Cell Biol. 2010;30(19):4656–4670.
113. Mnatzakanian GN, Lohi H, Munteanu I, et al. A previously unidentified MECP2 open reading frame defines a new protein isoform relevant to Rett syndrome. Nat Genet. 2004;36(4):339–341. doi:10.1038/ng1327
114. Kriaucionis S, Bird A. The major form of MeCP2 has a novel N-terminus generated by alternative splicing. Nucleic Acids Res. 2004;32(5):1818–1823. doi:10.1093/nar/gkh349
115. Coy JF, Sedlacek Z, Bächner D, Delius H, Poustka A. A complex pattern of evolutionary conservation and alternative polyadenylation within the long 3’-untranslated region of the methyl-CpG-binding protein 2 gene (MeCP2) suggests a regulatory role in gene expression. Hum Mol Genet. 1999;8(7):1253–1262. doi:10.1093/hmg/8.7.1253
116. Dragich JM, Kim Y-H, Arnold AP, Schanen NC. Differential Distribution of the Mecp2 Splice Variants in the Postnatal Mouse Brain. J Comp Neurol. 2007;501(4):526–542. doi:10.1002/cne
117. Zappella M, Meloni I, Longo I, et al. Study of MECP2 Gene in Rett Syndrome Variants and Autistic Girls. Am J Med Genet. 2003;119B(1):102–107.
118. Mount RH, Charman T, Hasting RP, Reilly S, Cass H. Features of Autism in Rett Syndrome and Severe Mental Retardation. J Autism Dev Disord. 2003;33(4):435–442.
119. Carney RM, Wolpert CM, Ravan SA, et al. Identification of MeCP2 Mutations in a Series of Females with Autistic Disorder. Pediatr Neurol. 2003;28(3):205–211.
120. Shibayama A, Cook EH, Feng J, et al. MECP2 Structural and 3′-UTR Variants in Schizophrenia, Autism and Other Psychiatric Diseases: a Possible Association with Autism. Am J Med Genet. 2004;128B(1):50–53.
121. Hitchins MP, Rickard S, Dhalla F, et al. Investigation of UBE3A and MECP2 in Angelman syndrome (AS) and Patients With Features of AS. Am J Med Genet. 2004;125A(2):167–172.
122. Watson P, Black G, Ramsden S, et al. Angelman syndrome phenotype associated with mutations in MECP2, a gene encoding a methyl CpG binding protein. J Med Genet. 2001;38(4):224–228.
123. Couvert P, Bienvenu T, Aquaviva C, et al. MECP2 is highly mutated in X-linked mental retardation. Hum Mol Genet. 2001;10(9):941–946.
124. Amir RE, Fang P, Yu Z, et al. Mutations in exon 1 of MECP2 are a rare cause of Rett syndrome. J Med Genet. 2005;42(2):e15. doi:10.1136/jmg.2004.026161
125. Saunders CJ, Minassian BE, Chow EWC, Zhao W, Vincent JB, Exon N. 1 Mutations in MECP2 Implicate Isoform MeCP2_e1 in Classical Rett Syndrome. Am J Med Genet Part A. 2009;149A:1019–1023.
126. Kerr B, Soto CJ, Saez M, Abrams A, Walz K, Young JI. Transgenic complementation of MeCP2 deficiency: phenotypic rescue of Mecp2-null mice by isoform-specific transgenes. Eur J Hum Genet. 2012;20(1):69–76. doi:10.1038/ejhg.2011.145
127. Lyon MF. Gene Action in the X-chromosome of the Mouse (Mus musculus L.). Nature. 1961;190:372–373.
128. Young JI, Zoghbi HY. X-Chromosome Inactivation Patterns Are Unbalanced and Affect the Phenotypic Outcome in a Mouse Model of Rett Syndrome. Am J Hum Genet. 2004;74(3):511–520.
129. Enikanolaiye A, Ruston J, Zeng R, et al. Suppressor mutations in Mecp2-null mice implicate the DNA damage response in Rett syndrome pathology. Genome Res. 2020;30(4):540–552.
130. Achilly NP, Wang W, Zoghbi HY. Presymptomatic training mitigates functional deficits in Rett syndrome mice. Nature. 2021;592(7855):596–600.
131. Downs J, Rodger J, Li C, et al. Environmental enrichment intervention for Rett syndrome: an individually randomised stepped wedge trial. Orphanet J Rare Dis. 2018;13(1):3.
132. Suter B, Treadwell-Deering D, Zoghbi HY, Glaze DG, Neul JL. MECP2 Mutations in People without Rett Syndrome. J Autism Dev Disord. 2014;44(3):703–711.
133. Brown K, Selfridge J, Lagger S, et al. The molecular basis of variable phenotypic severity among common missense mutations causing Rett syndrome. Hum Mol Genet. 2016;25(3):558–570.
134. Merritt JK, Collins BE, Erickson KR, Dong H, Neul JL. Pharmacological read-through of R294X Mecp2 in a novel mouse model of Rett syndrome. Hum Mol Genet. 2020;29(15):2461–2470.
135. Guy J, Alexander-Howden B, Fitzpatrick L, et al. A mutation-led search for novel functional domains in MeCP2. Hum Mol Genet. 2018;27(14):2531–2545.
136. Nikitina T, Shi X, Ghosh RP, Horowitz-Scherer RA, Hansen JC, Woodcock CL. Multiple Modes of Interaction between the Methylated DNA Binding Protein MeCP2 and Chromatin. Mol Cell Biol. 2007;27(3):864–877.
137. Pietri T, Roman AC, Guyon N, et al. The first mecp2-null zebrafish model shows altered motor behaviors. Front Neural Circuits. 2013;7(118):1–10. doi:10.3389/fncir.2013.00118
138. Chen Y, Yu J, Niu Y, et al. Modeling Rett Syndrome Using TALEN-Edited MECP2 Mutant Cynomolgus Monkeys. Cell. 2017;169(5):945–955. doi:10.1016/j.cell.2017.04.035
139. Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27(3):327–331.
140. Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27(3):322–326.
141. Lioy DT, Garg SK, Monaghan CE, et al. A role for glia in the progression of Rett’s syndrome. Nature. 2011;475(7357):497–500.
142. Cronk JC, Derecki NC, Ji E, et al. Methyl-CpG Binding Protein 2 Regulates Microglia and Macrophage Gene Expression in Response to Inflammatory Stimuli. Immunity. 2015;42(4):679–691.
143. Wang J, Wegener E, Huang T-W, et al. Wild-type microglia do not reverse pathology in a mouse models of Rett syndrome. Nature. 2015;521(7552):E1–E4.
144. McGraw CM, Samaco RC, Zoghbi HY. Adult neural function requires MeCP2. Science. 2011;333(6039):186.
145. Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of Neurological Defects in a Mouse Model of Rett Syndrome. Science. 2007;315(5815):1143–1147.
146. Robinson L, Guy J, McKay L, et al. Morphological and functional reversal of phenotypes in a mouse model of Rett syndrome. Brain. 2012;135(9):2699–2710.
147. Gemelli T, Berton O, Nelson ED, Perrotti LI, Jaenisch R, Monteggia LM. Postnatal Loss of Methyl-CpG Binding Protein 2 in the Forebrain is Sufficient to Mediate Behavioral Aspects of Rett Syndrome in Mice. Biol Psychiatry. 2006;59(5):468–476.
148. Su S-H, Kao F-C, Huang Y-B LW. MeCP2 in the Rostral Striatum Maintains Local Dopamine Content Critical for Psychomotor Control. J Neurosci. 2015;35(15):6209–6220.
149. Fyffe SL, Neul JL, Samaco RC, et al. Deletion of Mecp2 in Sim1-Expressing Neurons Reveals a Critical Role for MeCP2 in Feeding Behavior, Aggression, and the Response to Stress. Neuron. 2008;59(6):947–958.
150. Achilly NP, He LJ, Kim OA, et al. Deleting Mecp2 from the cerebellum rather than its neuronal subtypes causes a delay in motor learning in mice. Elife. 2021;10:e64833.
151. Meng X, Wang W, Lu H, et al. Manipulations of MeCP2 in glutamatergic neurons highlight their contributions to Rett and other neurological disorders. Elife. 2016;5:e14199.
152. Chao H-T, Chen H, Samaco RC, et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. 2010;468(7321):263–269.
153. Samaco RC, Mandel-Brehm C, Chao HT, et al. Loss of MeCP2 in aminergic neurons causes cell-autonomous defects in neurotransmitter synthesis and specific behavioral abnormalities. PNAS. 2009;106(51):21966–21971.
154. Ito-Ishida A, Ure K, Chen H, Swann JW, Zoghbi HY. Loss of MeCP2 in Parvalbumin-and Somatostatin-Expressing Neurons in Mice Leads to Distinct Rett Syndrome-like Phenotypes. Neuron. 2015;88(4):651–658.
155. Heckman LD, Chahrour MH, Zoghbi HY. Rett-causing mutations reveal two domains critical for MeCP2 function and for toxicity in MECP2 duplication syndrome mice. Elife. 2014;3:e02676.
156. Rangasamy S, Olfers S, Gerald B, Hilbert A, Svejda S, Narayanan V. Reduced neuronal size and mTOR pathway activity in the Mecp2 A140V Rett syndrome mouse model. F1000Research. 2016;5(2269):1–16.
157. Johnson B, Zhao Y, Fasolino M, et al. Biotin tagging of MeCP2 in mice reveals contextual insights into the Rett syndrome transcriptome. Nat Med. 2017;23(10):1203–1214.
158. Goffin D, Allen M, Zhang L, et al. Rett syndrome mutation MeCP2 T158A disrupts DNA binding, protein stability and ERP responses. Nat Neurosci. 2012;15(2):274–283.
159. Wegener E, Brendel C, Fischer A, Hülsmann S, Gärtner J, Huppke P. Characterization of the MeCP2R168X Knockin Mouse Model for Rett Syndrome. PLoS One. 2014;9(12):e115444.
160. Pitcher MR, Herrera JA, Buffington SA, et al. Rett syndrome like phenotypes in the R255X Mecp2 mutant mouse are rescued by MECP2 transgene. Hum Mol Genet. 2015;24(9):2662–2672.
161. Brendel C, Belakhov V, Werner H, et al. Readthrough of nonsense mutations in Rett syndrome: evaluation of novel aminoglycosides and generation of a new mouse model. J Mol Med. 2011;89(4):389–398.
162. Ghosh RP, Horowitz-Scherer RA, Nikitina T, Gierasch LM, Woodcock CL. Rett Syndrome-causing Mutations in Human MeCP2 Result in Diverse Structural Changes That Impact Folding and DNA Interactions. J Biol Chem. 2008;283(29):20523–20534.
163. Asaka Y, Jugloff DGM, Zhang L, Eubanks JH, Fitzsimonds RM. Hippocampal synaptic plasticity is impaired in the Mecp2-null mouse model of Rett syndrome. Neurobiol Dis. 2006;21(1):217–227.
164. Moretti P, Levenson JM, Battaglia F, et al. Learning and Memory and Synaptic Plasticity are Impaired in a Mouse Model of Rett Syndrome. J Neurosci. 2006;26(1):319–327.
165. Nelson ED, Bal M, Kavalali ET, Monteggia LM. Selective impact of MeCP2 and associated histone deacetylases on the dynamics of evoked excitatory neurotransmission. J Neurophysiol. 2011;106(1):193–201.
166. Na ES, Nelson ED, Kavalali ET, Monteggia LM. The Impact of MeCP2 Loss-or Gain-of-Function on Synaptic Plasticity. Neuropsychopharmacology. 2013;38(1):212–219.
167. Liu L, Wong TP, Pozza MF, et al. Role of NMDA Receptor Subtypes in Governing the Direction of Hippocampal Synaptic Plasticity. Science. 2004;304(5673):1021–1024.
168. Qiu Z, Sylwestrak EL, Lieberman DN, Zhang Y, Liu XY, Ghosh A. The Rett Syndrome Protein MeCP2 Regulates Synaptic Scaling. J Neurosci. 2012;32(3):989–994.
169. Blackman MP, Djukic B, Nelson SB, Turrigiano GG. A critical and cell-autonomous role for MeCP2 in synaptic scaling up. J Neurosci. 2012;32(39):13529–13536.
170. Fukuda T, Itoh M, Ichikawa T, Washiyama K, Goto YI. Delayed Maturation of Neuronal Architecture and Synaptogenesis in Cerebral Cortex of Mecp2-Deficient Mice. J Neuropathol Exp Neurol. 2005;64(6):537–544.
171. Marchetto MCN, Carromeu C, Acab A, et al. A Model for Neural Development and Treatment of Rett Syndrome Using Human Induced Pluripotent Stem Cells. Cell. 2010;143(4):527–539.
172. Chapleau CA, Calfa GD, Lane MC, et al. Dendritic spine pathologies in hippocampal pyramidal neurons from Rett syndrome brain and after expression of Rett-associated MECP2 mutations. Neurobiol Dis. 2010;35(2):219–233.
173. Schüle B, Armstrong DD, Vogel H, Oviedo A, Francke U. Severe congenital encephalopathy caused by MECP2 null mutations in males: central hypoxia and reduced neuronal dendritic structure. Clin Genet. 2008;74(2):116–126.
174. Luikenhuis S, Giacometti E, Beard CF, Jaenisch R. Expression of MeCP2 in postmitotic neurons rescues Rett syndrome in mice. PNAS. 2004;101(16):6033–6038.
175. Na ES, Nelson ED, Adachi M, et al. A Mouse Model for MeCP2 Duplication Syndrome: meCP2 Overexpression Impairs Learning and Memory and Synaptic Transmission. J Neurosci. 2012;32(9):3109–3117.
176. Sztainberg Y, Chen H, Swann JW, et al. Reversal of phenotypes in MECP2 duplication mice using genetic rescue or antisense oligonucleotides. Nature. 2015;528(7580):123–126.
177. Samaco RC, Mandel-Brehm C, McGraw CM, Shaw CA, McGill BE, Zoghbi HY. Crh and Oprm1 mediate anxiety-related behavior and social approach in a mouse model of MECP2 duplication syndrome. Nat Genet. 2012;44(2):206–211.
178. Yu B, Yuan B, Dai J-K, et al. Reversal of Social Recognition Deficit in Adult Mice with MECP2 Duplication via Normalization of MeCP2 in the Medial Prefrontal Cortex. Neurosci Bull. 2020;36(6):570–584.
179. Na ES, Morris MJ, Nelson ED, Monteggia LM. GABAa Receptor Antagonism Ameliorates Behavioral and Synaptic Impairments Associated with MeCP2 Overexpression. Neuropsychopharmacology. 2014;39(8):1946–1954.
180. Chao HT, Zoghbi HY, MeCP2 RC. Controls Excitatory Synaptic Strength by Regulating Glutamatergic Synapse Number. Neuron. 2007;56(1):58–65.
181. Nageshappa S, Carromeu C, Trujillo CA, et al. Altered neuronal network and rescue in a human MECP2 duplication model. Mol Psychiatry. 2016;21(2):178–188.
182. Jiang M, Ash RT, Baker SA, et al. Dendritic Arborization and Spine Dynamics are Abnormal in the Mouse Model of MECP2 Duplication Syndrome. J Neurosci. 2013;33(50):19518–19533.
183. Ash RT, Fahey PG, Park J, Zoghbi HY, Smirnakis SM. Increased axonal bouton stability during learning in the mouse model of MECP2 duplication syndrome. eNeuro. 2018;5(3):ENEURO.0056–17.2018.
184. Ash RT, Park J, Suter B, Smirnakis SM, Zoghbi HY. Excessive formation and stabilization of dendritic spine clusters in the mecp2-duplication syndrome mouse model of autism. eNeuro. 2021;8(1):ENEURO.0282–20.2020.
185. Ash RT, Buffington SA, Park J, et al. Inhibition of Elevated Ras-MAPK Signaling Normalizes Enhanced Motor Learning and Excessive Clustered Dendritic Spine Stabilization in the MECP2-Duplication Syndrome Mouse Model of Autism. eNeuro. 2021;8(4):ENEURO.0056–21.2021.
186. Glaze DG, Percy AK, Motil KJ, et al. A Study of the Treatment of Rett Syndrome With Folate and Betaine. J Child Neurol. 2009;24(5):551–556.
187. Renthal W, Boxer LD, Hrvatin S, et al. Characterization of human mosaic Rett syndrome brain tissue by single-nucleus RNA sequencing. Nat Neurosci. 2018;21(12):1670–1679.
188. Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. The Disease Progression of Mecp2 Mutant Mice Is Affected by the Level of BDNF Expression. Neuron. 2006;49(3):341–348.
189. Ogier M, Wang H, Hong E, Wang Q, Greenberg ME, Katz DM. Brain-Derived Neurotrophic Factor Expression and Respiratory Function Improve After Ampakine Treatment in a Mouse Model of Rett Syndrome. J Neurosci. 2007;27(40):10912–10917.
190. Deogracias R, Yazdani M, Dekkers MPJ, et al. Fingolimod, a sphingosine-1 phosphate receptor modulator, increases BDNF levels and improves symptoms of a mouse model of Rett syndrome. PNAS. 2012;109(35):14230–14235.
191. Naegelin Y, Kuhle J, Schädelin S, et al. Fingolimod in children with Rett syndrome: the FINGORETT study. Orphanet J Rare Dis. 2021;16(1):19.
192. D’Ercole AJ, Ye P, Calikoglu AS, Gutierrez-Ospina G. The role of the insulin-like growth factors in the central nervous system. Mol Neurobiol. 1996;13(3):227–255.
193. Zheng W, Quirion R. Comparative signaling pathways of insulin-like growth factor-1 and brain-derived neurotrophic factor in hippocampal neurons and the role of the PI3 kinase pathway in cell survival. J Neurochem. 2004;89(4):844–852.
194. O’Kusky JR, Ye P, D’Ercole AJ. Insulin-like growth factor-I promotes neurogenesis and synaptogenesis in the hippocampal dentate gyrus during postnatal development. J Neurosci. 2000;20(22):8435–8442.
195. Tropea D, Giacometti E, Wilson NR, et al. Partial reversal of Rett syndrome-like symptoms in MeCP2 mutant mice. Proc Natl Acad Sci. 2009;106(6):2029–2034.
196. Glaze DG, Neul JL, Percy A, et al. A Double-Blind, Randomized, Placebo-Controlled Clinical Study of Trofinetide in the Treatment of Rett Syndrome. Pediatr Neurol. 2017;76:37–46.
197. Glaze DG, Neul JL, Kaufmann WE, et al. Double-blind, randomized, placebo-controlled study of trofinetide in pediatric Rett syndrome. Neurology. 2019;92(16):e1912–e1925.
198. Autry AE, Adachi M, Nosyreva E, et al. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature. 2011;475(7354):91–96.
199. Kron M, Howell CJ, Adams IT, et al. Brain activity mapping in Mecp2 mutant mice reveals functional deficits in forebrain circuits, including key nodes in the default mode network, that are reversed with ketamine treatment. J Neurosci. 2012;32(40):13860–13872.
200. Patrizi A, Picard N, Simon AJ, Gunner G, Andrews NA, Fagiolini M. Chronic administration of the N-methyl-D-aspartate receptor antagonist ketamine improves Rett syndrome phenotype. Biol Psychiatry. 2016;79(9):755–764.
201. Arnon R, Aharoni R. Mechanism of action of glatiramer acetate in multiple sclerosis and its potential for the development of new applications. Proc Natl Acad. 2004;101(Suppl 2):14593–14598. doi:10.1073/pnas.0404887101
202. Ziemssen T, Kümpfel T, Klinkert WEF, Neuhaus O, Hohlfeld R. Glatiramer acetate-specific T-helper 1- and 2-type cell lines produce BDNF: implications for multiple sclerosis therapy. Brain. 2002;125(11):2381–2391. doi:10.1093/brain/awf252
203. Aharoni R, Kayhan B, Eilam R, Sela M, Arnon R. Glatiramer acetate-specific T cells in the brain express T helper 2/3 cytokines and brain-derived neurotrophic factor in situ. Proc Natl Acad. 2003;100(24):14157–14162. doi:10.1073/pnas.2336171100
204. Djukic A, Holtzer R, Shinnar S, et al. Pharmacologic Treatment of Rett Syndrome With Glatiramer Acetate. Pediatr Neurol. 2016;61(2016):51–57. doi:10.1016/j.pediatrneurol.2016.05.010
205. Nissenkorn A, Kidon M, Ben-Zeev B, Potential Life-Threatening A. Reaction to Glatiramer Acetate in Rett Syndrome. Pediatr Neurol. 2017;68(2017):40–43. doi:10.1016/j.pediatrneurol.2016.11.006
206. Lombardi LM, Baker SA, Zoghbi HY. MECP2 disorders: from the clinic to mice and back. J Clin Invest. 2015;125(8):2914–2923.
207. Garg SK, Lioy DT, Cheval H, et al. Systemic Delivery of MeCP2 Rescues Behavioral and Cellular Deficits in Female Mouse Models of Rett Syndrome. J Neurosci. 2013;33(34):13612–13620.
208. Gadalla KK, Bailey ME, Spike RC, et al. Improved Survival and Reduced Phenotypic Severity Following AAV9/MECP2 Gene Transfer to Neonatal and Juvenile Male Mecp2 Knockout Mice. Mol Ther. 2013;21(1):18–30.
209. Gadalla KKE, Vudhironarit T, Hector RD, et al. Development of a Novel AAV Gene Therapy Cassette with Improved Safety Features and Efficacy in a Mouse Model of Rett Syndrome. Mol Ther. 2017;5:180–190.
210. Sinnett SE, Hector RD, Gadalla KKE, et al. Improved MECP2 Gene Therapy Extends the Survival of MeCP2-Null Mice without Apparent Toxicity after Intracisternal Delivery. Mol Ther. 2017;5:106–115.
211. Matagne V, Ehinger Y, Saidi L, et al. A codon-optimized Mecp2 transgene corrects breathing deficits and improves survival in a mouse model of Rett syndrome. Neurobiol Dis. 2017;99:1–11.
212. Matagne V, Borloz E, Ehinger Y, Saidi L, Villard L, Roux J-C. Severe off target effects following intravenous delivery of AAV9-MECP2 in a female mouse model of Rett syndrome. Neurobiol Dis. 2021;149:105235.
213. Przanowski P, Wasko U, Zheng Z, et al. Pharmacological reactivation of inactive X-linked Mecp2 in cerebral cortical neurons of living mice. PNAS. 2018;115(31):7991–7996.
214. Huong Le TT, Tran NT, Lan Dao TM, et al. Efficient and Precise CRISPR/Cas9-Mediated MECP2 Modifications in Human-Induced Pluripotent Stem Cells. Front Genet. 2019;10:625.
215. Sinnamon JR, Kim SY, Corson GM, et al. Site-directed RNA repair of endogenous Mecp2 RNA in neurons. PNAS. 2017;114(44):E9395–E9402.
216. Sinnamon JR, Kim SY, Fisk JR, et al. In Vivo Repair of a Protein Underlying a Neurological Disorder by Programmable RNA Editing. Cell Rep. 2020;32(2):107878.
217. Rinaldi C, Wood MJA. Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat Rev Neurol. 2018;14(1):9–21. doi:10.1038/nrneurol.2017.148
218. Shao Y, Sztainberg Y, Wang Q, et al. Antisense oligonucleotide therapy in a humanized mouse model of MECP2 duplication syndrome. Sci Transl Med. 2021;13(583):eaaz7785. doi:10.1126/scitranslmed.aaz7785
219. Samaco RC, Mcgraw CM, Ward CS, Sun Y, Neul JL, Zoghbi HY. Female Mecp2± mice display robust behavioral deficits on two different genetic backgrounds providing a framework for pre-clinical studies. Hum Mol Genet. 2013;22(1):96–109.
220. Lawson-Yuen A, Liu D, Han L, et al. Ube3a mRNA and protein expression are not decreased in Mecp2R168X mutant mice. Brain Res. 2007;1180(1):1–6. doi:10.1016/j.brainres.2007.08.039
221. Shahbazian MD, Young JI, Yuva-Paylor LA, et al. Mice with Truncated MeCP2 Recapitulate Many Rett Syndrome Features and Display Hyperacetylation of Histone H3. Neuron. 2002;35(2):243–254.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.