Back to Journals » International Journal of Nanomedicine » Volume 21
Nanotherapeutic Interventions for Parkinson’s Disease: Modulating Pathogenic Mechanisms and Overcoming Therapeutic Obstacles
Authors Yin X, Wang C, Lin W, Huang S, Chen T ![]()
Received 26 September 2025
Accepted for publication 22 January 2026
Published 7 February 2026 Volume 2026:21 570452
DOI https://doi.org/10.2147/IJN.S570452
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Eng San Thian
Xuanying Yin,* Chen Wang,* Weisen Lin,* Shuiqing Huang, Tongkai Chen
Science and Technology Innovation Center, Guangzhou University of Chinese Medicine, Guangzhou, 510405, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Tongkai Chen, Science and Technology Innovation Center, Guangzhou University of Chinese Medicine, 12 Jichang Road, Guangzhou, 510405, People’s Republic of China, Email [email protected]
Abstract: Parkinson’s disease (PD) is the second most prevalent neurodegenerative disorder globally. Despite significant research, effective clinical treatments for PD remain limited due to its complex pathogenesis and the selective nature of PD-related lesions. Advances in nanotechnology, however, have opened new avenues for PD therapy. Nanomedicines and nanomaterials, developed through interdisciplinary research, have shown the potential to enable precise and targeted drug delivery while modulating key pathogenic mechanisms involved in PD. This review first outlines the key pathophysiological mechanisms underlying PD before providing an overview of nanomedicine-based strategies designed to target PD lesions by altering these mechanisms. We also summarize clinical trials on emerging PD therapeutics, with particular focus on the development and translational potential of nanomedicines. Although nanotechnology-based treatments for PD are still in their infancy, and a definitive cure remains out of reach, this field shows considerable promise. Finally, we explore future prospects to guide the continued development of PD nanotherapies. Overall, this review highlights the transformative potential of nanomedicine in the treatment of PD and underscores its potential for the development of more effective and targeted PD therapies.
Keywords: Parkinson’s disease, nanomedicines, pyroptosis, ferroptosis, autophagy
Introduction
Parkinson’s disease (PD), the second most prevalent neurodegenerative disorder in the world, is a progressive condition characterized by tremors, akinesia, rigidity, and postural instability, primarily resulting from the degeneration of dopaminergic neurons.1 At present, pharmacological interventions with dopaminergic agents, anticholinergics, monoamine oxidase B inhibitors, and catechol-O-methyltransferase inhibitors remain the cornerstone of PD management.2 While these treatments provide symptomatic relief, they fail to target the pathophysiology underlying PD.3 The lack of disease-modifying therapies, alongside the increase in global population aging, positions PD as an escalating public health concern.
Understanding the pathogenic mechanisms underlying PD is crucial for alleviating its public health burden and developing targeted therapies.4 The pathological hallmark of PD is the progressive degeneration of dopaminergic neurons in the substantia nigra (SN) coupled with the deposition of α-synuclein (α-syn), leading to the formation of insoluble Lewy bodies (LBs).5 Recent studies have revealed several pathogenic mechanisms contributing to PD — including α-syn aggregation,6 apoptosis,7 pyroptosis,8 ferroptosis,9 autophagy,10 neuroinflammation,11 and alterations in the gut microbiota12 — all of which have been implicated in progressive dopaminergic neuron loss. The multifactorial nature of PD highlights the need to address these mechanisms in order to develop advanced therapeutic interventions for PD.
To address these challenges, researchers have proposed innovative therapeutic strategies for PD, including lifestyle modifications and disease-modifying therapies that directly target PD pathology.13 However, clinical efficacy remains limited, largely due to factors such as: (1) the physiological barriers of the central nervous system (CNS), particularly the blood–brain barrier (BBB),14 (2) the complex and multifactorial pathophysiology of PD, compounded by the current lack of targeted approaches,15 and (3) the irreversible degeneration of the nigrostriatal pathway.16 Although there has been some progress in the development of small-molecule drugs and monoclonal antibodies, these innovations alone are insufficient to address the complexities of PD, underscoring the need for more comprehensive treatment strategies. Accordingly, drug delivery systems with high loading capacity and efficient brain-targeting capabilities are critical for improving therapeutic outcomes in PD.
In recent decades, the integration of nanotechnology into biomedicine has significantly advanced drug delivery systems. Nanotechnology has helped in addressing the key limitations of conventional anti-PD drugs, including drug instability, poor solubility, and low bioavailability. Nanomaterials improve brain-targeting capabilities by facilitating BBB penetration and enhancing drug accumulation within the CNS.17 Various strategies have been employed to enhance delivery efficiency to the brain parenchyma, including photothermal modulation,18 ultrasound-mediated BBB disruption,19 intranasal delivery,20 and biomimetics-based approaches.21 Moreover, nanomaterials offer considerable versatility and can be tailored for targeted delivery through techniques such as membrane coating,22 chemical conjugation,23 and other methods that ensure both therapeutic efficacy and biocompatibility. Collectively, owing to these innovations, nanomaterials have been recognized as a promising platform for developing more effective, precise, and personalized PD therapies.
To advance PD treatment, two key challenges must be addressed: (1) achieving targeted drug delivery across the BBB to lesion sites and (2) regulating pathogenic factors. This review first outlines the fundamental pathogenic mechanisms of PD, including α-syn aggregation, apoptosis, pyroptosis, ferroptosis, autophagy dysfunction, neuroinflammation, and gut microbiota alterations, and discusses their pathophysiological impact on PD. It then evaluates current nanotherapeutic strategies targeting these mechanisms, with a focus on design principles and their therapeutic applications. Finally, the review addresses the prevailing challenges in the field, highlighting the clinical relevance and future prospects of nanomaterial-based platforms, and proposes strategies to overcome existing barriers and improve therapeutic outcomes.
Key Pathophysiological Mechanisms in PD
A thorough comprehension of the physiological characteristics and molecular mechanisms underlying PD is essential for the development of targeted therapeutic strategies. This section provides an in-depth examination of the major pathological processes driving PD and its progression, including α-syn aggregation (Figure 1A), apoptosis (Figure 1B), pyroptosis (Figure 1C), ferroptosis (Figure 1D), autophagy dysfunction (Figure 1E), neuroinflammation (Figure 1F), and alterations in the gut–brain axis (Figure 1G).
 |
Figure 1 Pathological role of biological processes in PD. (A) α-syn aggregation: Misfolded α-syn forms Lewy bodies, disrupting mitochondria and dopamine balance. (B) Apoptosis: Programmed neuronal death via DNA fragmentation and apoptotic bodies. (C) Pyroptosis: Inflammatory cell lysis driven by membrane pore formation and cytokine release. (D) Ferroptosis: Excess iron–catalyzed ROS trigger lipid peroxidation, causing membrane damage and cell death. (E) Autophagy: Lysosomal clearance of damaged proteins/organelles; its failure leads to α-syn buildup. (F) Neuroinflammation: Chronic microglial activation releases cytokines that worsen neuron loss. (G) Gut-brain axis dysbiosis: Microbiome shifts alter vagal, immune, endocrine, and circulatory signals, impairing SN function. |
α-Syn Aggregation and Pathological Progression in PD
α-syn is a small, acidic protein predominantly expressed in the SN, where it plays a crucial role in regulating neurotransmitter release and reuptake.24 Under normal physiological conditions, α-syn exists in a dynamic equilibrium among multiple conformational states, maintaining a balance between its unstructured monomeric form and tetrameric assembly.25 However, when this equilibrium is disrupted, α-syn undergoes conformational changes, resulting in the aggregation of α-syn into oligomers, protofibrils, and fibrils.26 These misfolded aggregates eventually form intracellular inclusions known as LBs (Figure 2A), which accumulate in the dopaminergic neurons of the substantia nigra pars compacta (SNpc), a region particularly susceptible to damage due to its extensive axonal branching and high metabolic demands.27 Pathological α-syn aggregation disrupts critical cellular processes, including calcium homeostasis, synaptic vesicle trafficking, and dopamine release, thereby intensifying neuroinflammation.28 Ultimately, the accumulation of α-syn within LBs impairs dopaminergic signaling in the SNpc, resulting in motor dysfunction and accelerating PD progression (Figure 2B).29
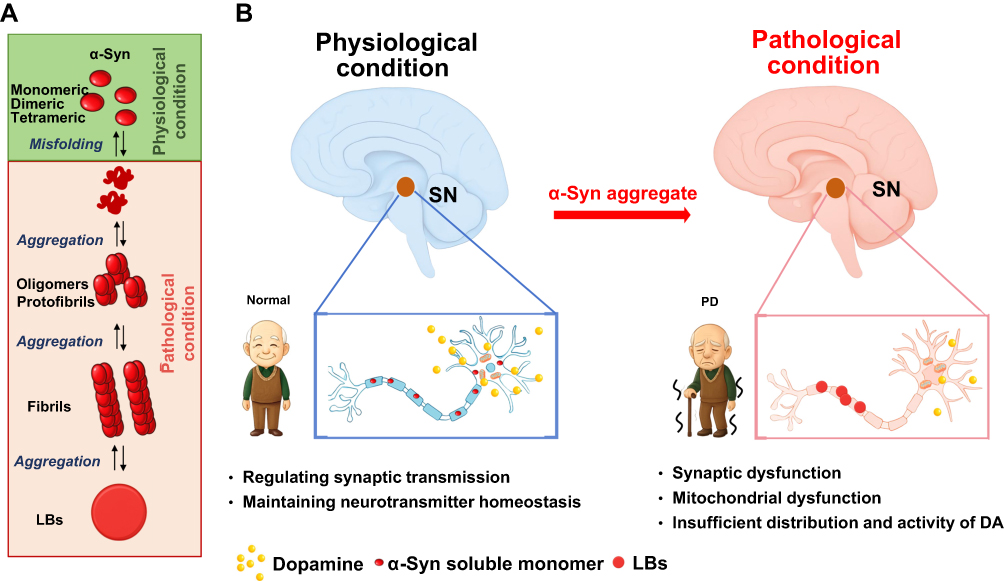 |
Figure 2 The role of α-syn in both physiological and pathological conditions associated with PD. (A) Under normal physiological conditions, α-syn primarily exists as a monomer, playing a pivotal role in synaptic transmission and neurotransmitter release. In contrast, under pathological conditions, the delicate equilibrium between the synthesis and clearance of α-syn is disrupted, leading to the aggregation of α-syn into toxic oligomers, protofibrils, and fibrils, which ultimately form Lewy bodies. (B) These oligomers exert detrimental effects, particularly on dopaminergic neurons, which are inherently vulnerable. This results in mitochondrial dysfunction, impaired synaptic integrity, and widespread cellular disruption, all of which contribute to the onset and progression of PD. |
Given the central role of α-syn aggregation in PD pathogenesis, reducing its accumulation and toxicity is vital for halting disease progression. Various therapeutic strategies have therefore focused on stabilizing α-syn in its native, non-aggregated tetrameric form to prevent the formation of toxic aggregates; modulating intracellular calcium homeostasis to preserve neuronal integrity and maintain synaptic vesicle dynamics; and targeting neuroinflammation, since α-syn aggregation stimulates microglial activation, which exacerbates neurodegeneration. Additional approaches include enhancing the selective clearance of pre-formed LBs via proteasomal or autophagic degradation pathways, improving α-syn-targeting drug delivery across the BBB through surface modifications or ligand-based systems, and employing diagnostic tools for the real-time imaging and monitoring of α-syn aggregation in the brain. These strategies represent a promising avenue for halting α-syn aggregation, thereby offering potentially effective PD therapeutic strategies.
Apoptosis Pathways in PD
Apoptosis is a tightly regulated, evolutionarily conserved form of programmed cell death observed across metazoans and certain eukaryotic organisms.30 It is orchestrated through two primary signaling cascades: the intrinsic (mitochondrial) and extrinsic (death receptor) pathways,31 both of which culminate in the formation of apoptotic bodies that are subsequently cleared by phagocytes to maintain tissue homeostasis. Accumulating evidence shows that apoptosis acts as a “double-edged sword”: it maintains neuronal homeostasis under physiological conditions but causes deleterious effects when dysregulated. In PD, persistent mitochondrial dysfunction and excessive ROS abnormally activate apoptotic signaling, transforming apoptosis from a homeostatic mechanism to a pathological factor driving dopaminergic neuron loss.
Given that dysregulated apoptosis drives dopaminergic loss in PD, targeting this mechanism can slow down PD progression. PD is associated with excessive apoptosis, and the intrinsic apoptotic pathway is initiated by mitochondrial outer membrane permeabilization (MOMP), which facilitates the release of cytochrome c.32 This activates the apoptosome, leading to the activation of caspase-9 and its downstream effector caspase-3, ultimately resulting in cellular apoptosis.33 This process is tightly regulated by BCL-2 family proteins, with pro-apoptotic members such as BAX promoting MOMP, while anti-apoptotic proteins like BCL-2 inhibit it.34 In contrast, the extrinsic pathway is triggered by the binding of ligands such as tumor necrosis factor-α (TNFα) and Fas ligand (FasL) to their corresponding death receptors. These interactions induce receptor oligomerization, leading to the recruitment of adaptor proteins that activate initiator caspases, particularly caspase-8 and caspase-10.35 These caspases mediate cellular fate decisions and modulate inflammation, culminating in apoptosis.36 Both pathways share a common endpoint, namely, the release of signaling molecules like cleaved receptors and phosphatidylserine (PS), which act as “eat me” signals to facilitate phagocytosis.37 Apoptosis plays a critical role in the loss of dopaminergic neurons in PD, as evidenced by DNA fragmentation and caspase-3 activation in dopaminergic neurons of the SNpc.38 Apoptotic mitochondrial dysfunction is exacerbated by reactive oxygen species (ROS) accumulation and sustained cytochrome c release, further accelerating cell death.39 Additionally, mitochondrial complex I inhibition by neurotoxins such as MPTP40 and rotenone amplifies oxidative stress, enhancing dopaminergic neuron apoptosis and contributing to PD pathogenesis (Figure 3).41 These findings underscore the pivotal role of mitochondrial dysfunction and apoptotic signaling in PD.
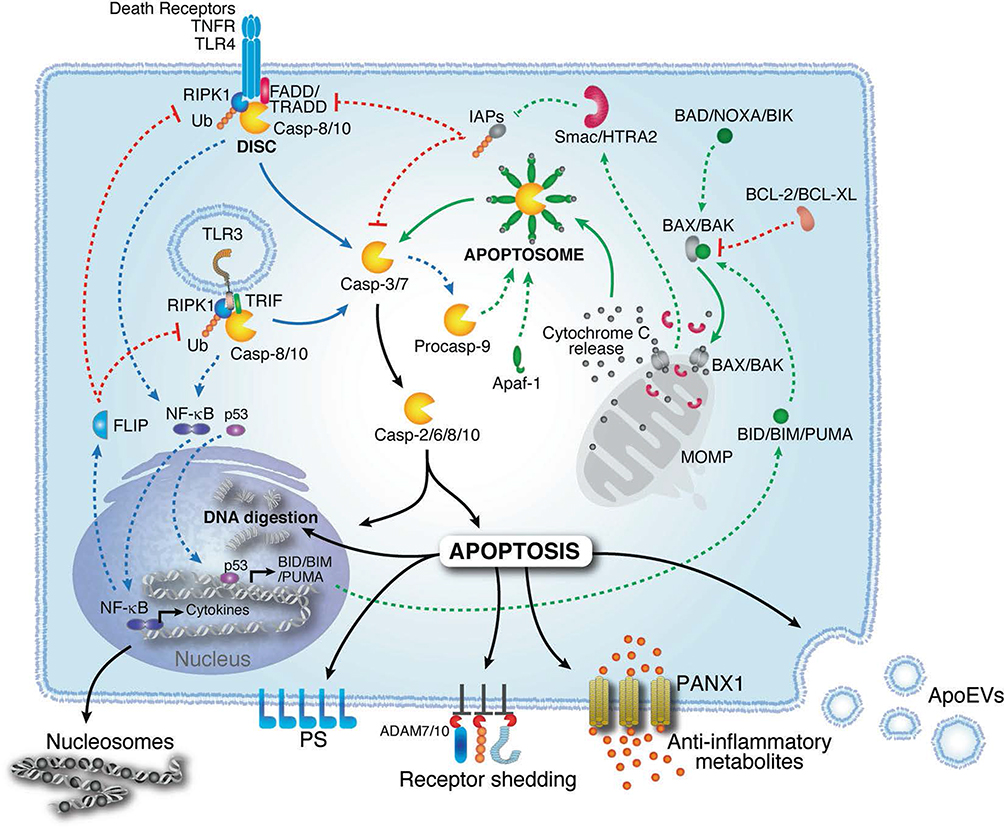 |
Figure 3 Mechanism of apoptosis. Apoptosis is mediated by intrinsic and extrinsic pathways. The intrinsic pathway is initiated by BAX/BAK oligomerization and MOMP, which together induce cytochrome c release and caspase-9 activation. The extrinsic pathway is triggered by receptor activation (eg, TNFR1, TLRs), resulting in caspase-8 activation. Both pathways converge to activate executioner caspases (eg, caspase-2, caspase-6, caspase-8, caspase-10), causing cell death. Apoptotic cells release “eat-me” signals, including nucleosomal structures and phosphatidylserine (PS), to facilitate phagocytosis. Adapted from41 with permission. |
Given the central role of apoptosis in PD, nanomaterial-based interventions must be designed with a multifaceted approach. These include addressing mitochondrial dysfunction as a central driver of apoptosis by stabilizing mitochondrial membranes to prevent MOMP and preserve cellular energy metabolism, modulating the expression and activity of pro- and anti-apoptotic BCL-2 family proteins to rebalance apoptotic signaling, and scavenging ROS to mitigate oxidative damage and support mitochondrial function. Additionally, the strategies must aim to inhibit cytochrome c release and downstream caspase-9 and caspase-3 activation to preserve dopaminergic neuron viability. Moreover, they must promote the efficient phagocytic clearance of apoptotic debris and reduce secondary neuroinflammation to protect surrounding neurons. Finally, they must enable the targeted inhibition of initiator and effector caspases to suppress the apoptotic cascade, thereby offering neuroprotection. Together, these strategies could be used to harness the therapeutic potential of nanomaterial-based platforms in slowing down PD progression by directly targeting apoptosis.
Pyroptosis and Its Role in PD
Pyroptosis is a form of programmed cell death characterized by inflammatory necrosis. The term “pyroptosis” originates from the Greek words pyro (fire) and ptosis (fall), reflecting its inflammatory nature.42
Pyroptosis is triggered by inflammasomes, which are multi-protein complexes that recognize pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs).43 Once activated, key inflammasome proteins, such as AIM2, members of the NOD-like receptor (NLR) family, and Pyrin, recruit the adaptor protein ASC to activate caspase-1.44 Activated caspase-1 cleaves pro-inflammatory cytokines, including interleukin-1β (IL-1β) and IL-18,45 as well as Gasdermin D (GSDMD),46 generating membrane pores that mediate cytokine release.47 In addition to this canonical pathway, cytosolic lipopolysaccharide also directly activates caspase-4/11 (caspase-5 in humans), which cleaves GSDMD and further promotes NLRP3 inflammasome activation, forming the non-canonical pathway. The pyroptotic cascade is further regulated through various mechanisms, including NLRP3 phosphorylation,48 ubiquitination,49 and the Endosomal Sorting Complex Required for Transport (ESCRT) pathway,50 which collectively modulate the amplitude of the inflammatory response. In PD, pyroptosis contributes to disease progression by exacerbating neuroinflammation, compromising the BBB, and impairing cognitive function.49 Under PD-related oxidative stress, Nox4-dependent mitochondrial ROS activates the NLRP3/GSDMD signal axis. Subsequently, cell death promotes glial cell activation and amplifies inflammation,48,51–53 forming a feed-forward inflammatory loop. In addition, pyroptosis also increases BBB permeability, facilitating the infiltration of peripheral immune cells and inflammatory mediators in the brain parenchyma.54 As these inflammatory signals spread within the CNS, they can cause cognitive impairments and depressive behavior (Figure 4).55
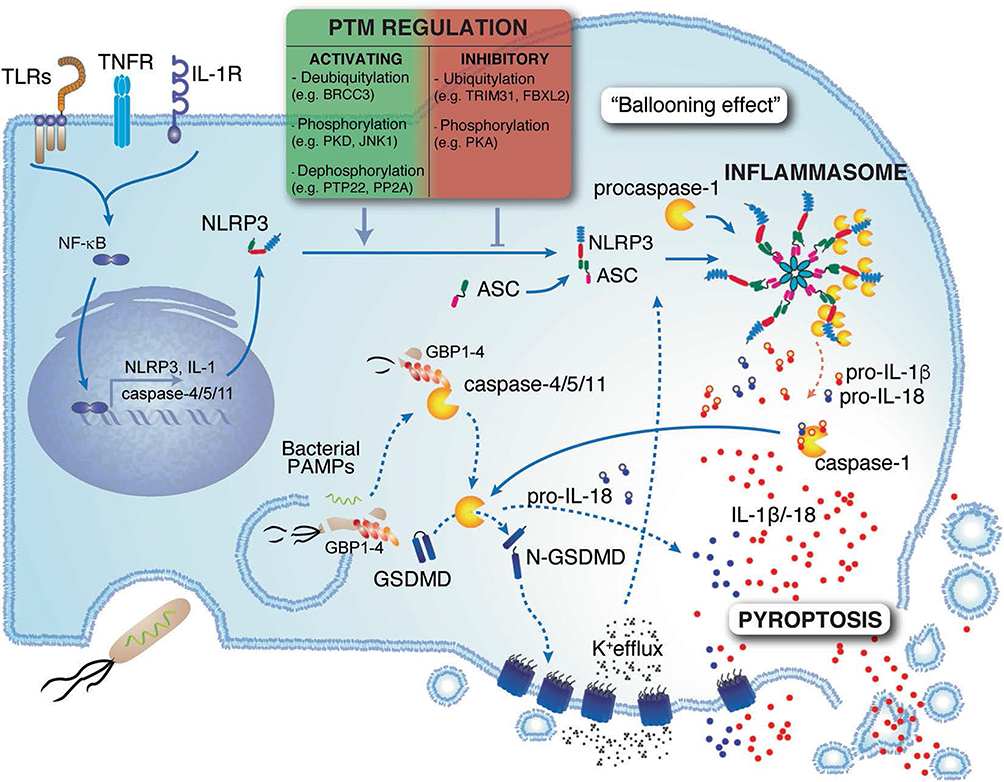 |
Figure 4 Pyroptosis is a form of inflammatory cell death triggered by sensors such as NLRP3. The activation of this protein triggers inflammasome formation, activating caspase-1, which cleaves IL-1β, IL-18, and GSDMD. This induces pore formation, cytokine release, and cell swelling. The recognition of lipopolysaccharide activates caspase-4/11, further promoting pyroptosis. This process is regulated by TLRs, TNFR, IL-1R priming, and various posttranslational modifications, including deubiquitination, phosphorylation, and dephosphorylation. Adapted from41 with permission. |
Nanodrugs that target PD-related pyroptosis aim to have the ability to modulate pyroptotic pathways through the inhibition of inflammasomes, particularly NLRP3, to prevent caspase-1 activation and cytokine release. Further, they can also regulate GSDMD to orchestrate pyroptotic cell death, thereby preventing membrane rupture and cellular lysis, which are intrinsic to the pyroptotic process. Further, these agents may additionally suppress IL-1β and IL-18 production to impede the inflammatory signaling cascade causing pyroptosis, thereby mitigating neuroinflammation. Pyroptosis-modulating approaches for PD management can be developed based on these considerations.
Ferroptosis and Dysregulation of Iron Homeostasis in PD
Ferroptosis is an iron-dependent form of programmed cell death characterized by perturbations in thiol, lipid, and iron metabolism, which lead to the accumulation of lipid peroxides and ROS.56 These molecules overwhelm the cell’s antioxidant defense system, triggering uncontrolled lipid peroxidation,57 membrane damage,58 and ultimately cell death.
The key mechanisms driving ferroptosis — including ferritinophagy,59 the Fenton reaction,60 and the activity of enzymes such as arachidonate lipoxygenases61 and cytochrome P450 oxidoreductase58 — facilitate lipid peroxidation. Polyunsaturated fatty acids (PUFAs), activated by acyl-CoA synthetase long-chain family member 4 (ACSL4)62 and lysophosphatidylcholine acyltransferase 3,63 are incorporated into cellular membranes, making them susceptible to peroxidation and intensifying ferroptotic cell death. Mitochondrial and peroxisomal dysfunction further promote lipid peroxide accumulation, exacerbating the ferroptotic process.64 Therefore, ferroptosis can be mitigated by antioxidants such as glutathione (GSH) and GPX4 or by increasing coenzyme Q10 (CoQ10) activity.65 The inhibitory mechanisms involving ferroptosis suppressor protein 1 (FSP1),66 dihydroorotate dehydrogenase (DHODH),67 and GTP cyclohydrolase 1 (GCH1)68 help modulate antioxidant responses (Figure 5). The PKC/Nrf2 pathway and transsulfuration pathway also mitigate ferroptosis by enhancing antioxidant defenses and cysteine biosynthesis.69,70 In PD, disruptions in iron metabolism, lipid peroxidation, and α-syn aggregation, including abnormal iron accumulation and dopamine oxidation, exacerbate ferroptosis and damage dopaminergic neurons.71 Microglial activation, in combination with changes in iron-related proteins such as P2RY12 and heme oxygenase-1 (HO-1), contributes to inflammation and neurodegeneration.72,73 Thus, ferroptosis, through its association with iron and lipid metabolism, plays a major role in the progression of PD.
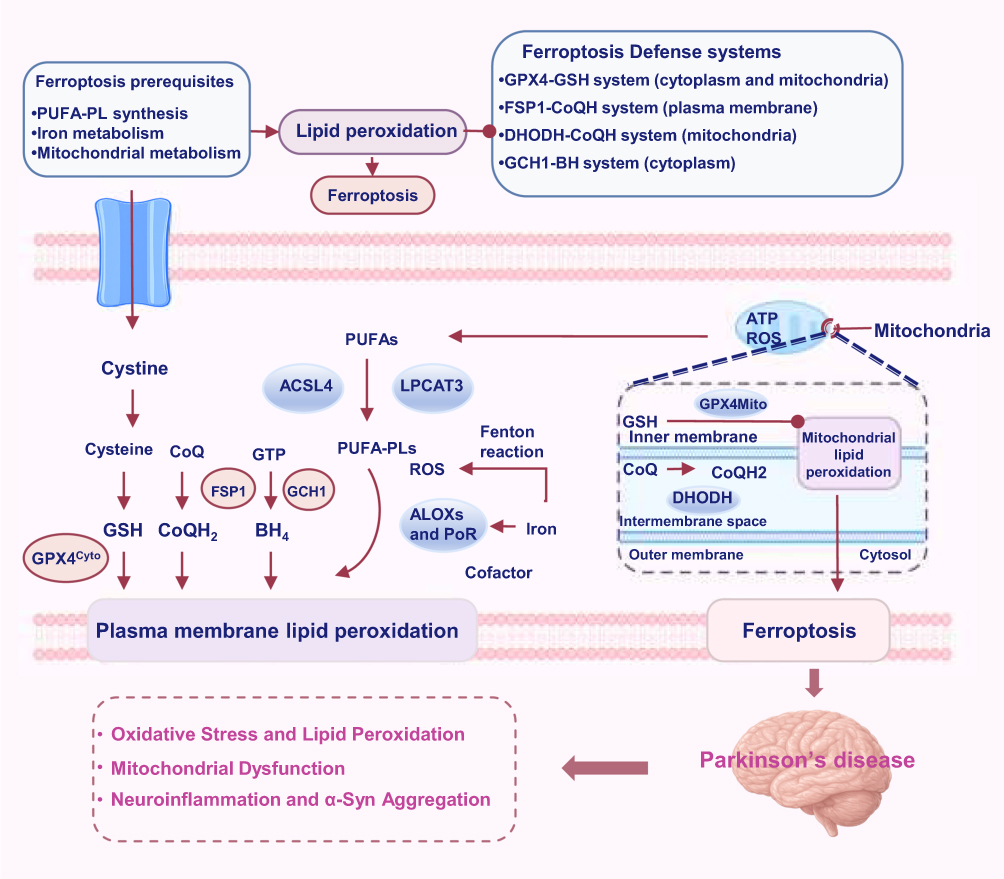 |
Figure 5 Ferroptosis is an iron-dependent programmed cell death process characterized by lipid peroxidation and ROS accumulation. The key mechanisms contributing to ferroptosis include the oxidation of PUFAs, which is facilitated by ACSL4 and lysophosphatidylcholine acyltransferase 3, mitochondrial dysfunction, and the Fenton reaction. Antioxidant systems such as GPX4, FSP1, DHODH, and GCH1 play crucial roles in regulating ferroptosis by controlling lipid peroxidation and maintaining the cellular redox balance. These processes collectively drive membrane damage and cell death. |
To design therapeutic strategies targeting ferroptosis for PD management, some key aspects need to be addressed. Iron metabolism should be modulated through the regulation of iron-related proteins or chelators to reduce intracellular iron overload and inhibit ROS production. Furthermore, lipid peroxidation can be inhibited by targeting enzymes like ACSL4 and lysophosphatidylcholine acyltransferase 3, which lower PUFA levels, prevent lipid peroxides, and minimize membrane damage. Mitochondrial function can be enhanced by incorporating antioxidants like CoQ10 into nanomaterials or targeting DHODH to modulate mitochondrial ferroptosis via the DHODH-CoQ10 axis. Furthermore, microglial inflammation can be modulated by targeting iron-related proteins such as P2RY12 and HO-1 to reduce inflammation and mitigate PD-related neurodegeneration. In summary, targeting ferroptosis through these combined strategies could help in the management of PD.
Autophagic Dysregulation in PD
Autophagy is a process that maintains cellular homeostasis by degrading and recycling proteins and organelles.73 However, dysregulated or excessive autophagic activity can disrupt this balance, potentially causing cell death. Autophagy is tightly regulated through a series of key steps.74 Specifically, AMP-activated protein kinase (AMPK) inhibits mTORC1, which in turn activates the ULK1 complex to initiate autophagic vesicle formation (Figure 6a).75 The Beclin-1/VPS34 complex, activated by the JNK-mediated phosphorylation of BCL-2 and BIM, generates phosphatidylinositol 3-phosphate (PI3P), a lipid that facilitates vesicle nucleation and elongation (Figure 6b).76 The ATG12/ATG5/ATG16L complex completes polymerization and fuses with autophagic vesicles, while microtubule-associated protein 1 light chain 3 (LC3) is processed into LC3-II and incorporated into the autophagosomal membrane (Figure 6c and d).77 In the final stage, the autophagosome fuses with lysosomes via the Soluble N-ethylmaleimide-sensitive factor Attachment protein REceptor (SNARE) complex, leading to the formation of autolysosomes that degrade cellular components (Figure 6e).78
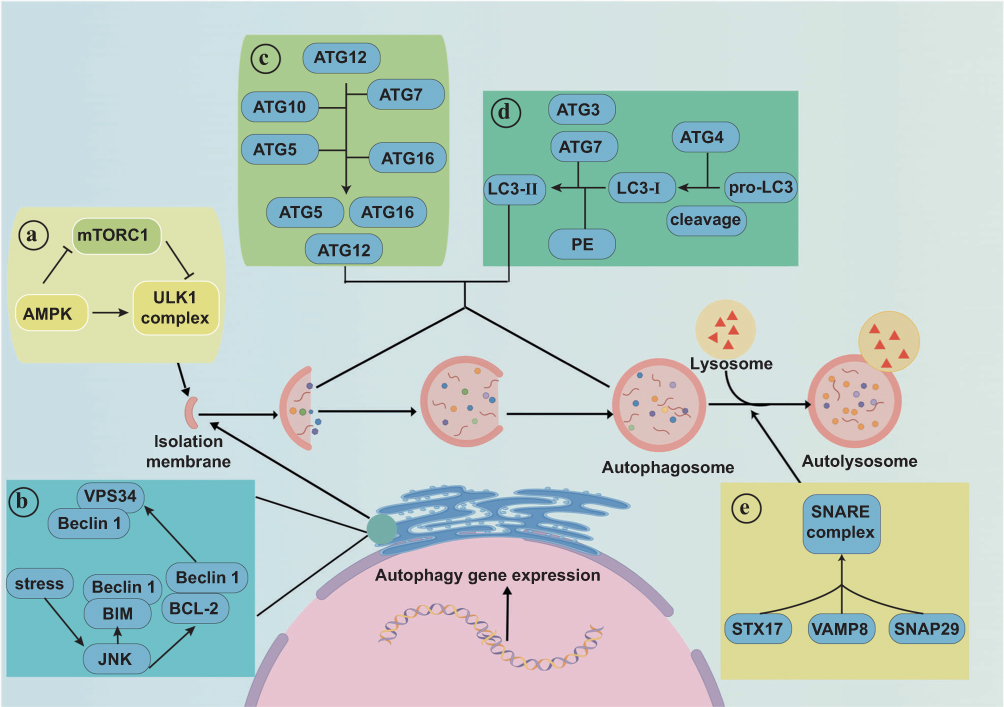 |
Figure 6 Autophagy is a self-degradation process involving the following key steps. (a) AMPK inhibits mTORC1, promoting ULK1 complex formation and autophagic vesicle production. (b) The Beclin-1/VPS34 complex enables the formation of larger vesicles, with JNK activating Beclin-1 by phosphorylating BCL-2 and BIM. (c) The ATG12/ATG5/ATG16L complex forms and fuses with vesicles. (d) LC3 is cleaved by ATG4 and processed by ATG3 and ATG7 to form LC3-II, which is inserted into autophagosomes. (e) Autophagosomes fuse with lysosomes to form autolysosomes, and this process is facilitated by the STX17/SNAP29/VAMP8 SNARE complex. Adapted from74 with permission. |
Impaired autophagy leads to the accumulation of misfolded proteins, such as α-syn, which contribute to PD.79 Dysregulation of the autophagic–lysosomal pathway impairs the removal of damaged proteins and organelles, resulting in oxidative stress and neuronal damage.80 Genetic mutations in LRRK2, SNCA, PINK1, and Parkin disrupt mitophagy and chaperone-mediated autophagy, worsening the pathology of PD.81 For instance, LRRK2 mutations activate mTORC182 and inhibit autophagy, while PINK1 and Parkin mutations impair mitochondrial clearance.83 The aggregation of α-syn further impedes lysosomal function, triggering a cycle of autophagic dysfunction.84 Notably, restoring autophagic homeostasis by targeting the autophagy–lysosome pathway is believed to enhance neuronal survival and inhibit PD progression.85
Therapeutic strategies intended to restore autophagic equilibrium in PD should modulate autophagy pathways by targeting AMPK/mTORC1 or the Beclin-1/VPS34 complex to promote autophagosome formation and expansion. Furthermore, they should enhance mitophagy by targeting PINK1 or Parkin to promote the clearance of damaged mitochondria. Therapies restoring lysosomal function could optimize autophagosome–lysosome fusion via the SNARE complex, enhancing lysosomal activity and promoting α-syn degradation. Finally, gene-specific targeting via CRISPR-Cas9, RNAi, aptamers, or antibodies could correct mutations in LRRK2, SNCA, PINK1, Parkin and enable the selective binding of mutant α-syn. These strategies may restore autophagic equilibrium, enhance protein degradation, and thereby mitigate PD-related neuronal damage.
Mechanisms of Neuroinflammation in PD
Neuroinflammation is a multifaceted response to brain injury and is marked by glial activation, inflammatory mediator release, and ROS and reactive nitrogen species generation.86 Neuroinflammation exacerbates neuronal injury by disrupting cellular homeostasis, compromising BBB integrity, and triggering a chronic, dysregulated inflammatory cycle.87
The inflammatory receptors on the surfaces of immune cells, particularly glial cells, function as critical sensors for cellular damage.88 Upon recognizing DAMPs or PAMPs, these receptors activate signal transducers and transcription factors, promoting the production of pro-inflammatory mediators that amplify the inflammatory response.89 Under certain conditions, this inflammation is not fully resolved, exacerbating neurotoxic effects, protein aggregation, and neurodegeneration through DAMP release90 (Figure 7). Immune dysregulation is especially deleterious in PD, as neuroinflammation is known to drive disease progression. While α-syn aggregation is a hallmark of PD, it is not the primary cause of neuronal death, as neuronal loss occurs even before LBs are formed. Instead, owing to microglial activation, neuroinflammation precedes the loss of dopaminergic neurons.91 The misfolded α-syn released from damaged neurons activates microglia and astrocytes, leading to an amplified cycle of harmful effects.92 Importantly, α-syn–induced NLRP3 inflammasome activation can trigger the pyroptotic release of IL-1β and IL-18, amplifying neuroinflammatory signals and creating a bridge between protein aggregation and inflammatory cell death. Accumulating evidence suggests that neuroinflammation, driven by both innate and adaptive immune responses, actively contributes to neuronal death in PD.93
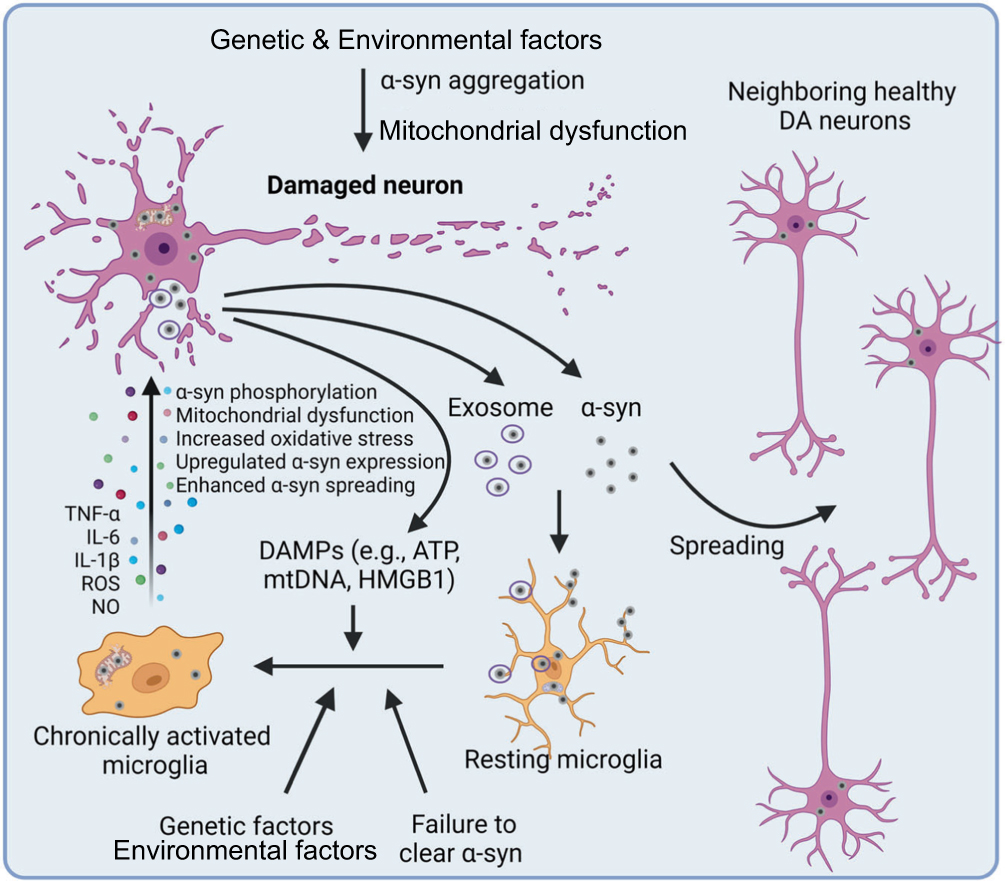 |
Figure 7 In PD, α-syn aggregation, triggered by genetic and environmental factors, causes mitochondrial dysfunction and neurotoxicity. Mutations in proteins such as LRRK2, PINK1, PARK7, and PRKN further exacerbate this dysfunction. Excess α-syn accumulation and impaired α-syn clearance both activate microglia, causing the spread of α-syn to neighboring neurons, which expands neurotoxic effects. The DAMPs released by dying neurons further amplify microglial activation, promoting oxidative stress and mitochondrial dysfunction and thereby accelerating disease progression. Adapted from90 with permission. |
Targeting immune dysregulation in the early stages of PD could help in its therapeutic management. Such treatment strategies could focus on modulating microglial and astrocytic responses to mitigate neuroinflammation through targeted drug delivery in regions of inflammation, especially when BBB integrity is compromised. Such targeted delivery could be achieved through the surface modification of nanomedicines with ligands, antibodies, or small molecules that regulate inflammatory pathways. Incorporating materials with anti-inflammatory properties, such as those that target the NF-κB signaling pathway, NLRP3 inflammasome activation, or other pathways that exacerbate neuroinflammation, could also serve as a valuable strategy. Finally, integrating antioxidant and α-syn clearance effects could reduce immune-mediated neuronal damage. Through such well-designed strategies, nanomaterials could be used to restore autophagic balance, enhance protein degradation, and mitigate neuronal damage in PD.
Microbiota–Gut–Brain Axis in PD
Owing to its vital physiological role, the gut influences the onset and progression of various extraintestinal diseases.94 The gut microbiome represents a complex ecosystem that comprises commensal bacteria, archaea, fungi, and viruses that interact with and modulate host physiology.95
The brain regulates gut function through the hypothalamic–pituitary–adrenal axis and the autonomic nervous system.96 For instance, the brain releases norepinephrine, which can promote the proliferation of certain gut pathogens.97 Conversely, the gut exerts a modulatory effect on CNS function by releasing microbial metabolites,98 neuroactive substances,99 and gut hormones.100 These factors travel through the enteric nervous system (ENS),101 vagus nerve,102 and immune pathways,103 thereby modulating brain activity. This bidirectional communication between the gut and the brain is integral for maintaining physiological homeostasis and mediating disease progression.104 The early stages of PD are associated with alterations in the gut and ENS, before CNS pathology can be detected.105 Studies suggest that PD-related pathological changes induce gastrointestinal dysfunction. Disruptions in gut microbiota via the gut–brain axis contribute to PD through several interconnected mechanisms. First, the altered microbiota and microbial metabolites induce inflammation, disrupt the intestinal barrier, activate immune cells, and promote α-syn aggregation (Figure 8a).106 Second, the increased intestinal permeability facilitates the entry of microbial signaling molecules, immune cells, and metabolites into systemic circulation,107 amplifying systemic inflammation (Figure 8b). Third, misfolded α-syn in the gut can be transmitted to the brain through the vagus nerve,108 likely in a bidirectional manner (Figure 8c). Finally, the compromised BBB and disrupted vagal pathways allow the entry of pathological products into the brain,109 activating resident immune cells such as microglia and astrocytes (Figure 8d). This cascade triggers neuroinflammation, leading to the degeneration of dopaminergic neurons and PD progression.
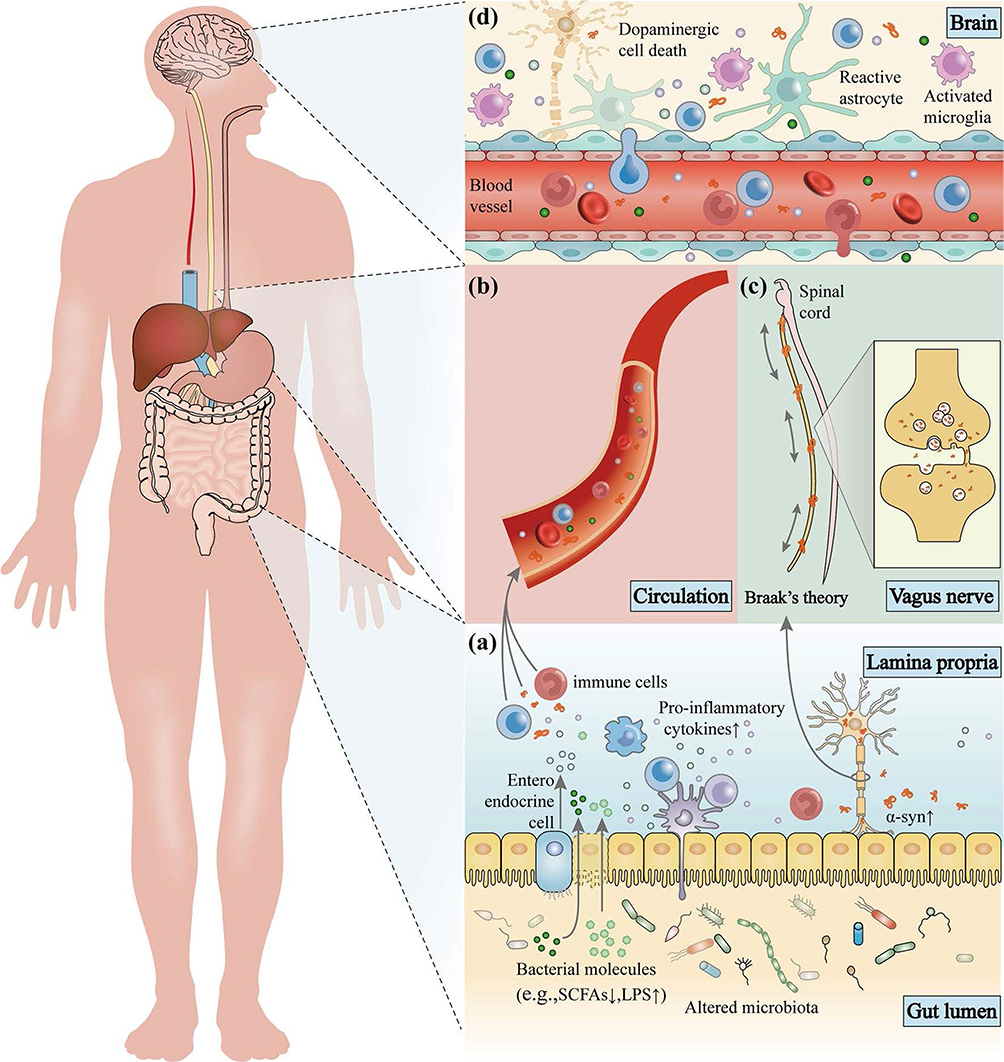 |
Figure 8 Gut microbiota dysbiosis contributes to PD via the immune, endocrine, and nervous systems. (a) Alterations in microbial populations induce gut inflammation, leading to the disruption of the intestinal barrier and promoting α-syn aggregation. (b) This increased intestinal permeability allows microbial and immune signals to cause systemic inflammation. (c) Misfolded α-syn is transported from the gut to the brain via the vagus nerve. (d) Compromised BBB and vagal pathways enable α-syn entry, triggering neuroinflammation and dopaminergic neuron loss in PD. Adapted from106 with permission. |
While designing nanomedicines that target the gut microbiome for PD treatment, several critical factors must be considered. Mainly, there should be a focus on modulating the gut microbiome to restore microbial balance. Further, intestinal barrier integrity can be enhanced by administering anti-inflammatory agents or tight junction modulators to prevent the translocation of pro-inflammatory cytokines and α-syn into the systemic circulation. The vagus nerve can also be targeted to inhibit the transmission of misfolded α-syn via the ENS. Finally, optimizing BBB penetration and implementing controlled release systems could ensure the precise, stimuli-responsive delivery of therapeutic agents to target sites, maximizing therapeutic efficacy. Accordingly, nanomedicines can be designed to modulate the gut–brain axis, alleviating PD symptoms and preventing disease progression.
Strategies for Overcoming Biological Barriers and Multifunctional Design
Strategies That Enable Nanomaterials to Cross the BBB
The BBB remains one of the main pharmacological challenges in the treatment of PD, which prevents most small molecules and almost all macromolecules from entering the CNS.79 To overcome this limitation, endogenous BBB transport systems and alternative access routes have become increasingly utilized in nanomaterial design. Among these, receptor-mediated transcytosis has attracted considerable attention. Angiopep-2 can target LRP1 and continuously enhance the accumulation of polymeric and protein nanocarriers in the brain,110 the rabies virus-derived peptide Rabies Virus Glycoprotein 29 (RVG29) can bind to nicotinic acetylcholine receptors to deliver antioxidants and regulatory RNAs via systemic or intranasal administration.111 Other validated receptor-mediated transcytosis pathways include transferrin receptor–mediated transport of neuroprotective agents.112 Glucose Transporter 1-mediated transport using glucose- or trehalose-modified nanodots carrying α-syn modulators,113 and the ApoE–LRP1 axis, which can promote endothelial internalization.114 Ferritin nanocages have been shown to cross the BBB through synergistic interaction with the transferrin receptor and Scavenger Receptor Class A Member 5.115
In addition to the receptor-specific strategies, adsorptive-mediated transcytosis using cationic peptides such as Trans-Activator of Transcription provides an additional pathway for the transport of neurotrophic factors.116 Biomimetic platforms, including macrophage-mimetic vesicles, Poly (2-methacryloyloxyethyl phosphorylcholine)-coated nanocarriers, and mesenchymal stem cell (MSC)-derived exosomes, use natural membrane components to traverse the BBB and preferentially accumulate in inflamed brain regions.117–119 Some nanomaterials also exhibit intrinsic BBB permeability: for example, the chirality-engineered nanozyme Ptzyme@D-ZIF enters the brain through clathrin- and caveolae-mediated pathways,120 whereas ultrasmall black phosphorus nanosheets can directly enter the central nervous system and remodel α-syn fibrils.121 Other methods, such as focused ultrasound-mediated BBB opening19 and intranasal drug delivery,122 also offer temporary or non-invasive approaches to bypass the barrier. Together, these diverse mechanisms form a practical and flexible framework for constructing nanotherapeutics capable of reaching and modulating the key pathological targets of PD.
Multifunctional Nanomaterials Targeting Multiple Pathological Pathways in PD
Given the multifactorial nature of PD pathology and the convergence of oxidative stress, mitochondrial dysfunction, neuroinflammation, ferroptosis, and protein aggregation, single-target therapeutics are unlikely to achieve meaningful disease modification. Therefore, increasing attention has shifted toward integrating multiple therapeutic mechanisms into a single multifunctional nanomaterial platform. Nanozymes are a particularly effective example: PtCu alloy nanozymes have been shown to scavenge ROS, inhibit the intercellular transmission of pathogenic α-syn, and reduce dopaminergic neurodegeneration in the PD model.123 More recently, Co–Cu diatomic nanozymes have been engineered to couple superoxide dismutase– and catalase-like activity, thus inhibiting mitochondrial oxidative injury and NLRP3 inflammasome activation in the PD-related inflammatory microenvironment.124 Single-atom nanozyme platforms combine catalytic, antioxidant, and cytoprotective functions within a single Fe–N–C framework to regulate ferroptosis- and inflammation-related pathways in neurodegenerative diseases.125 A related concept involves microenvironment-responsive engineering: linkers such as thioether bonds or ROS-responsive polymer shells remain inert in circulation but are selectively activated in the high-ROS or acidic microenvironment of the substantia nigra, triggering on-demand drug release.126
Another complementary strategy focuses on multi-target regulation through biological network interactions. Extracellular vesicle-based nanotherapies naturally integrate lipids, proteins, and nucleic acids, allowing the simultaneous modulation of mitochondrial stress responses, synaptic function, glial activation, and cytokine release, while also providing innate compatibility for delivering therapeutic miRNAs or antioxidant peptides. For example, MSC-derived exosomes promote repair in PD models by inducing autophagy and inhibiting neuroinflammation,127 whereas dental pulp stem cell-derived exosomes protect dopaminergic neurons from 6-OHDA-induced apoptosis.128 Recent studies have also highlighted the importance of systemic factors, especially gut–brain axis dysfunction, in driving neuroinflammation and α-syn pathology, showing that engineered nanomaterials capable of reinforcing intestinal barrier integrity, restructuring microbial communities, and neutralizing pro-inflammatory metabolites can reduce peripheral inflammation and indirectly ameliorate central pathology.129,130 Studies of metal–microbiome interactions further suggest that nanoplatforms modulating microbial metal metabolism may influence the inflammatory and oxidative pathways implicated in PD.131 Multifunctional designs have also been extended to integrated theranostics. For instance, nanozymes containing iron oxide cores can be used for magnetic resonance imaging-guided tracking to achieve synchronous monitoring and therapeutic intervention.132 In conclusion, these advances show that nanotherapy is progressing towards multi-target, system-level strategies capable of targeting both central and peripheral mechanisms that drive PD progression.
Multifaceted Nanomedicine-Based Therapeutic Strategies for PD
The primary goals of current PD treatment strategies are neuroprotection (preventing dopaminergic neuron damage)133 and neuro-restoration (slowing or reversing neuronal degeneration).134 While several therapeutic approaches have demonstrated some potential, their effectiveness is hindered by challenges such as limited BBB penetration,135 suboptimal pharmacokinetics,136 and unwanted pharmacodynamic effects.137 In recent years, advancements in nanomaterial-based therapies for drug delivery,138 gene therapy,139 immune modulation,140 and oxidative stress management141 have created new avenues for PD treatment. This section reviews the latest nanotherapeutic strategies targeting PD-related pathological processes. Additionally, it discusses design considerations for future nanomedicines, offering insights guiding the development of more effective nanotherapeutic approaches (Table 1).
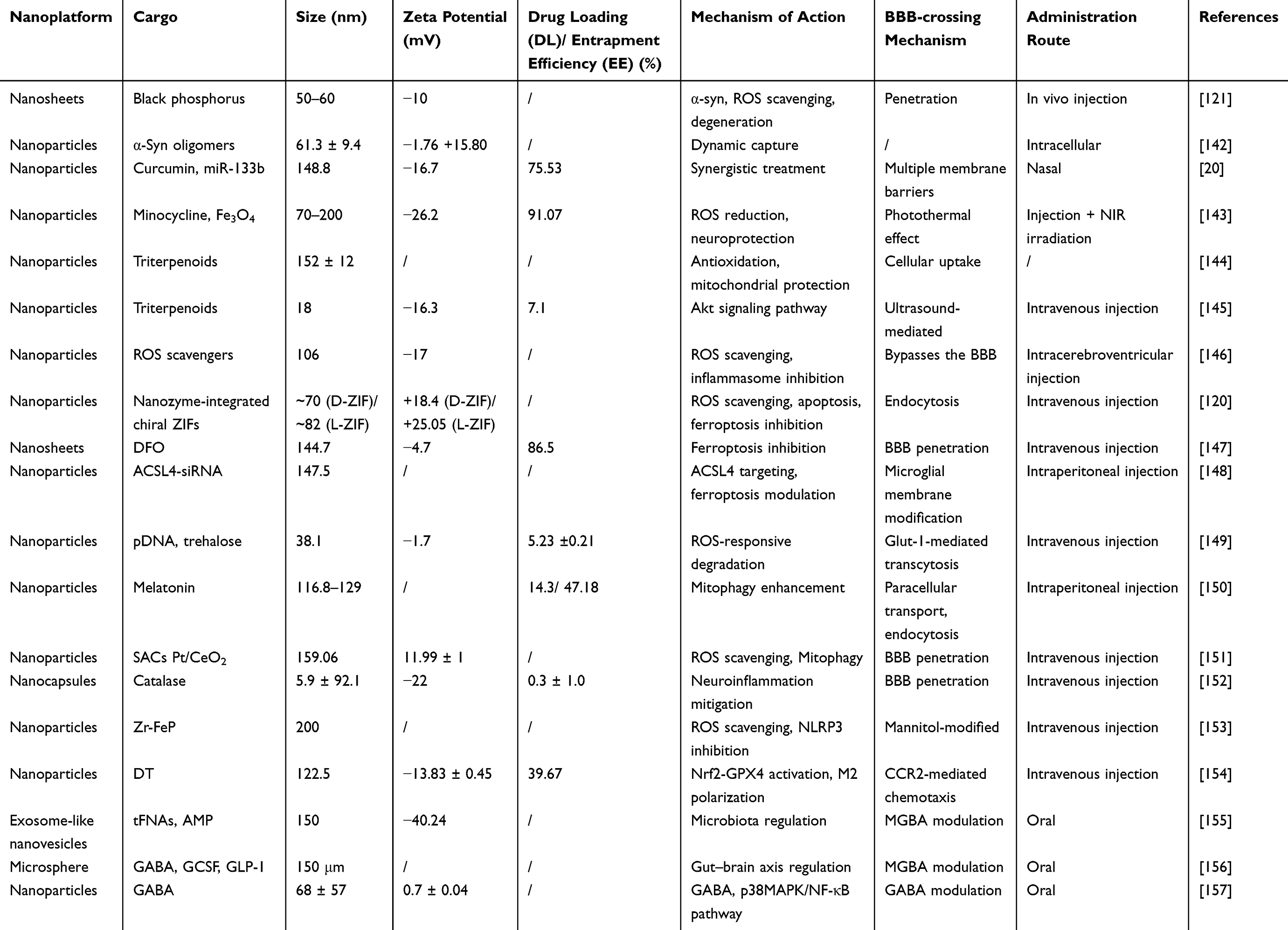 |
Table 1 The Application of Nanomaterial-Based Platforms for Neuroprotection and Neuronal Restoration |
Nanomaterial-Mediated Inhibition of α-Syn Aggregation in PD
The role of α-syn in PD progression has been thoroughly explored in previous sections. This section focuses on the recent advancements in nanomedicines targeting α-syn to mitigate PD.
To maintain α-syn homeostasis, nanoformulations must have the ability to mitigate its abnormal accumulation and neutralize ROS. Black phosphorus nanosheets (BPNSs) have shown promise in this regard due to their biocompatibility,158 BBB permeability,159 and potent antioxidant properties.160 BPNSs demonstrate affinity for amyloid β (Aβ40), acting as multifunctional nanomedicines for eliminating both α-syn and ROS.161 In vitro, BPNS-PEG-Cy5 nanoparticles were found to exhibit a cellular uptake efficiency of 53.60% ± 2.465%, confirming their effective internalization. Thus, they have emerged as promising agents for treating PD (Figure 9A).121 In a C. elegans PD model, BPNSs displayed neuroprotective effects against 6-hydroxydopamine-induced neurotoxicity (Figure 9B), reduced α-syn aggregation, promoted α-syn aggregate degradation, and activated autophagy to enhance α-syn clearance (Figure 9C). Notably, BPNSs induced the dissociation of α-syn fibrils after 24 h of treatment (Figure 9D), reducing ThT fluorescence to 37.38% of baseline, markedly lowering both cellular and mitochondrial ROS, and restoring LC3B puncta from approximately half the levels detected in the control back to baseline levels (Figure 9E and F).
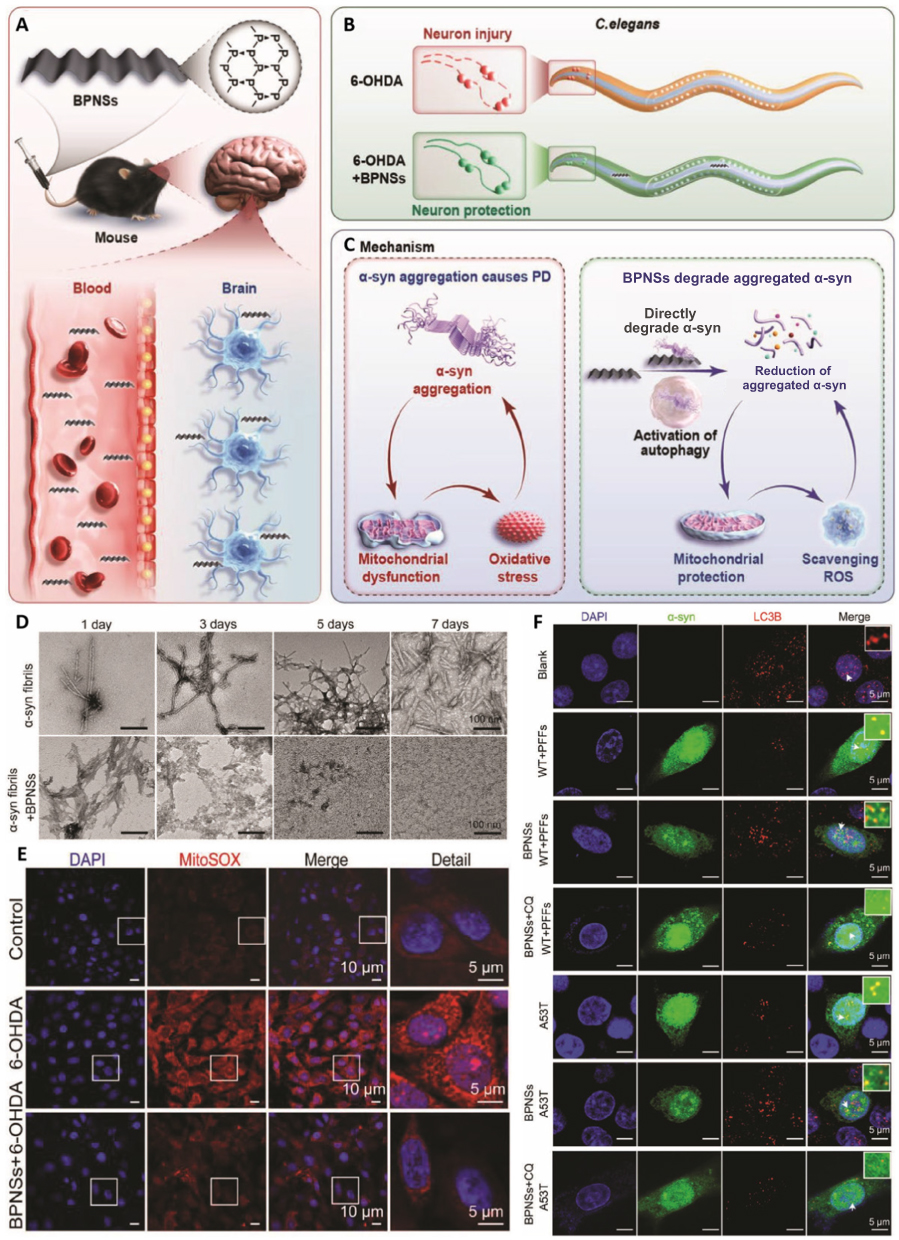 |
Figure 9 (A–C) Schematic illustration showing the neuroprotective effect of BPNSs in C. elegans and mouse models of PD. (D) Transmission electron microscopy (TEM) images of pre-formed α-synuclein fibrils at different time intervals (1, 3, 5, and 7 days) in the absence (top) and presence (bottom) of BPNS treatment; scale bars: 100 nm (E) Mitochondrial ROS scavenging effects of BPNSs in cell models of PD, with white squares indicating magnified regions; white scale bars = 10 µm (main) and 5 µm (detailed). (F) Effects of BPNSs on the expression levels of LC3B in PC12 cells after transfecting α-syn Aggregation Assay Kit and A53T plasmids for 48 h; white arrows indicate co-localization of α-syn and LC3B. Scale bar, 5 μm. Adapted from121 with permission. |
Membrane binding is a critical mechanism in the pathogenesis of α-syn-associated PD.162 The engineered nanochaperone αS-nChap was found to inhibit fibril formation by binding to monomeric α-syn, preventing its oligomerization and reducing ThT fluorescence by 90% at 72 h, and promoting the lysosomal degradation of aggregates, thereby lowering intracellular α-syn deposition from 64.6 ± 10.1% to 16.2 ± 8.3%.142 However, the BBB poses a significant challenge, limiting drug delivery to the brain parenchyma. To address this, a self-targeting nanotherapeutic platform (PR-EXO/PP@Cur) was developed for synergistic PD treatment.20 This system integrated curcumin, which promotes α-syn clearance, with MRI-tracked superparamagnetic iron oxide nanoparticles (SPIONs). These components were encapsulated within a MSC -derived shell modified with the RVG29 peptide for neuron-specific targeting. Upon intranasal administration, the nanocarrier could cross the nasal–brain barrier, target dopaminergic neurons via acetylcholine receptor recognition, and release its payload (miR-133b, which promotes neuroregeneration, and curcumin for α-syn clearance and anti-inflammatory effects) through membrane fusion and ROS-triggered micelle disintegration. This triple-action platform integrated real-time tracking, BBB penetration, and microenvironment-responsive drug release, offering a precise and minimally invasive approach for comprehensive PD management.
To design a nanomedicine targeting α-syn for PD treatment, the following critical factors must be considered: (1) Targeting α-syn aggregation: The nanodrug should inhibit α-syn aggregation and induce the disaggregation of pre-formed toxic fibrils to mitigate neurotoxicity; (2) Antioxidant properties: The nanodrug should efficiently scavenge ROS to protect neurons from oxidative stress; (3) BBB penetration: The nanodrug must effectively cross the BBB to deliver therapeutic agents to brain cells; (4) Nanomaterial selection: Multifunctional nanomaterials, such as BPNSs, should be selected owing to their high affinity for α-syn and their antioxidant properties; (5) Membrane binding and α-syn interaction: The nanodrug should inhibit the binding of α-syn to lipid membranes to prevent aggregation; (6) Synergistic action: The nanomedicine should provide anti-aggregation, anti-inflammatory, and neuroprotective effects to enable comprehensive treatment; (7) Real-time monitoring and targeting: Nanocarriers with real-time tracking abilities should be employed to monitor drug distribution and efficacy. Overall, addressing these factors will facilitate the development of a comprehensive and effective α-syn-targeting nanodrug for PD treatment.
Nanomaterials as Apoptosis Modulators in PD
In the previous section, we explored the associations between excessive oxidative stress, mitochondrial dysfunction, apoptosis, and PD progression. Although ROS dysregulation and neuroinflammation serve as upstream triggers, recent nanomaterial-based approaches increasingly emphasize the direct regulation of apoptotic pathways. This section reviews the latest advancements in nanomaterials designed to inhibit neuro-apoptosis for PD treatment by modulating key apoptotic nodes, including caspase activation, the BCL-2/BAX ratio, and mitochondrial membrane permeabilization.
Emerging research indicates that minocycline, a tetracycline antibiotic, exerts neuroprotective effects in PD by reducing microglial activation163 while inhibiting pro-inflammatory cytokine (eg, IL-1β) production164 and ROS accumulation, thereby attenuating downstream caspase-3–mediated signaling and mitigating apoptosis. Photosensitizer-based nanoplatforms such as iron oxide (Fe3O4) NPs can enhance drug delivery to the brain by mediating photothermal effect-induced BBB traversal upon near-infrared (NIR) light irradiation.143 When minocycline (MIN) is loaded into hollow porous Fe3O4 NPs (Fe3O4-MIN NPs) and activated by NIR, it improves BBB penetration in PD models, promotes cerebral drug accumulation, and scavenges ROS. This leads to the inhibition of caspase-3 activation and protects dopaminergic neurons from apoptotic death.
Extracellular vesicles (EVs) are membrane-bound particles with sizes ranging from the nanometer to the micrometer scale. These vesicles are released by cells in response to various physiological and pathological stimuli.165 Owing to their intrinsic biocompatibility and cell-targeting potential, EVs have emerged as next-generation nanocarriers and are known to overcome several biological barriers. EVs from Salvia sclarea and Salvia dominica have been isolated using plant hairy root bio-factories and meticulously characterized.144 These plant HR-derived EVs have been found to mitigate 6-hydroxydopamine-induced neurotoxicity and apoptosis, highlighting their potential as innovative therapeutics for PD management. Moreover, the EVs reduce cleaved caspase-3 levels while preserving neuronal viability, demonstrating a specific anti-apoptotic effect that extends beyond antioxidant effects.
Neuronal apoptosis in PD is linked to an altered BCL-2/BAX ratio166 and elevated caspase-3 activity,167 underscoring the need for therapies that preserve synaptic integrity. Targeting Akt kinase, which regulates cell survival and neurogenesis, can enhance the BCL-2/BAX ratio and inhibit apoptotic signaling. SC79, a small-molecule Akt phosphorylation agonist, has demonstrated potential in alleviating excitotoxicity and has been encapsulated in Cu2-χSe NPs to improve its BBB penetration. The release of Cu2+ ions from these NPs has been found to promote neuronal proliferation and migration via Akt and ERK pathway activation. Additionally, evidence shows that coating the NPs with RAW 264.7 cell membranes (CSS@CM NPs) enables the precise targeting of neurons via α4β1–VCAM-1 interactions, enhancing neurite outgrowth, reducing synaptic degeneration and apoptosis, and improving therapeutic efficacy in PD models (Figure 10A).145 Notably, it also directly modulates intrinsic apoptotic pathways by restoring the BCL-2/BAX ratio, suppressing cleaved caspase-3, and maintaining mitochondrial membrane potential (Figure 10B). In MPP⁺-induced neurotoxicity models, the co-administration of the Akt inhibitor MK-2206 reverses the effect of CSS@CM NPs on cleaved caspase-3 expression and the BCL-2/BAX ratio (Figure 10C and D). This effect is mediated by the elevation of the p-Akt/Akt ratio (Figure 10E and F). Additionally, CSS@CM NPs mitigate MPP⁺-induced apoptotic cell death, as evidenced by the quantification of the cellular apoptosis ratio (Figure 10G). Furthermore, JC-1 staining shows that the mitochondrial membrane potential is preserved following CSS@CM treatment (Figure 10H). In vivo, CSS@CM-SC79 administration can restore the expression of tyrosine hydroxylase (TH), IBA-1, and GFAP to near-normal levels, reflecting the attenuation of synaptic damage and recovery from apoptosis.
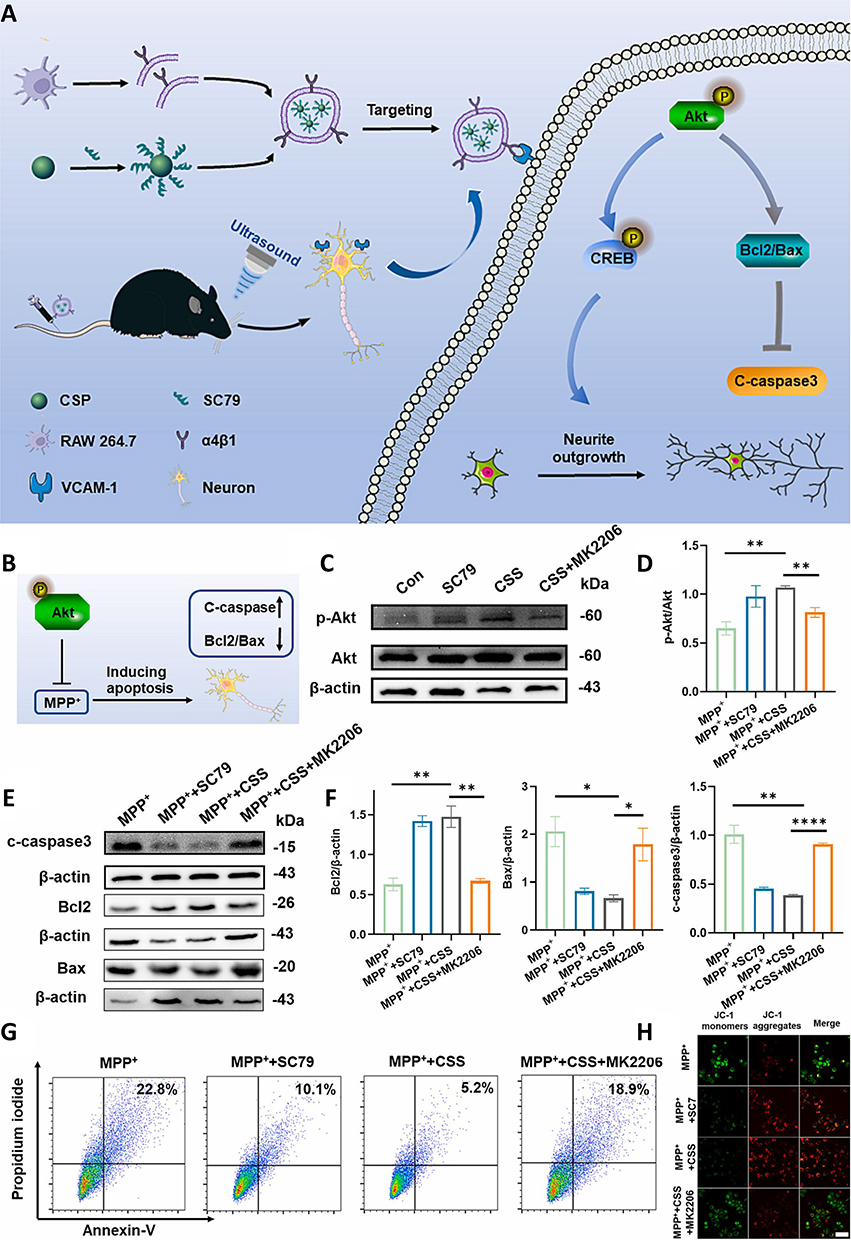 |
Figure 10 (A) CSS@CM NPs promote neurite outgrowth, inhibit apoptosis, and protect neurons from synaptic damage in PD models. (B) CSS NPs enhance BCL-2 expression and inhibit BAX signaling in MPP+-treated cells. (C and D) Western blot analysis shows differences in p-Akt and Akt expression between untreated PC12 cells and PC12 cells treated with SC79, CSS NPs, and CSS NPs + MK2206. (E and F) BCL-2, BAX, and cleaved caspase-3 levels in different groups of cells. (G) Apoptosis rates analyzed by flow cytometry. (H) and mitochondrial depolarization observed via JC-1 staining and CLSM; white scale bars = 10 µm. Statistical significance assessed using the Student’s t-test (mean ± SD, *P < 0.05, **P < 0.01, and ****P < 0.0001). Adapted from145 with permission. |
Building on these characteristics, the design of apoptosis-targeted nanoplatforms for PD should consider the following: (1) Optimizing BBB penetration through photothermal/photoacoustic triggers or receptor-mediated targeting for enhanced brain accumulation; (2) Constructing multifunctional payloads showing anti-inflammatory, antioxidant, and neurotrophic effects with gene/nucleic acid therapeutics and designing stimuli-responsive release mechanisms for precise control; (3) Enhancing biocompatibility and immune evasion using plant- or mammalian cell-derived EVs, biomimetic membranes, PEG modifications, or “don’t eat me” signals while minimizing long-term accumulation; (4) Inducing the targeted inhibition of dopaminergic neuron apoptosis through the suppression of neuroinflammation, ROS scavenging, or the activation of the PI3K/Akt/CREB axis; (5) Beyond anti-apoptotic effects, achieving synergistic anti-inflammatory, antioxidative, and neuroregenerative effects. By focusing on these aspects, a comprehensive and effective nanodrug for PD treatment via apoptosis inhibition could be developed.
Nanomaterials Suppressing Pyroptosis Pathways in PD
As discussed previously, pyroptosis is closely linked to the release of inflammatory factors that cause tissue damage and exacerbate neuroinflammation, contributing to PD progression. This section explores the nanomedicine strategies aimed at targeting pyroptosis for PD treatment, with a particular focus on the molecular execution processes of this pathway, including caspase-1 activation, GSDMD cleavage, and pore formation, which distinguish pyroptosis from general inflammatory signaling.
As discussed earlier, there are several mechanisms through which pyroptosis contributes to PD. The NLRP3 inflammasome, expressed in microglia, plays a central role in PD progression. In the presence of PAMPs or DAMPs, the NLRP3 inflammasome is rapidly assembled in microglia, leading to the activation of caspase-1.168 This, in turn, cleaves the IL-1β precursor into its mature form, stimulating pro-inflammatory responses and cleaving GSDMD to initiate the membrane pore formation that executes pyroptosis. Therefore, blocking NLRP3 is a critical strategy for treating PD.169
Over the past decade, artificial nanozymes have garnered increasing interest due to their adjustable catalytic activities,170 multifunctional enzyme-like activities,171 and high stability.172 Prussian blue, an FDA-approved antidote for poisoning caused by thallium and other radioactive elements,173 has been combined with nanozymes to develop Prussian blue nanozyme (PBzyme). Notably, PBzyme exhibits intrinsic enzyme-mimicking catalytic functions, including ROS scavenging (eg, •OH, •OOH, H2O2).146 Additionally, PBzyme also functions as a direct pyroptosis inhibitor, mitigating PD progression by suppressing the assembly of the NLRP3 inflammasome and subsequent GSDMD-mediated membrane rupture, which is characteristic of pyroptotic cell death. Furthermore, PBzyme can inhibit microglial pyroptosis in both in vivo and in vitro PD models (Figure 11A). The cytoprotective effect of PBzyme, mediated by pyroptosis attenuation, is evidenced by its ability to reduce pore formation in BV2 cells (Figure 11B). Western blot analysis has confirmed that PBzyme significantly suppresses the expression of cleaved caspase-1 and cleaved GSDMD, both of which are markedly upregulated by MPTP/ATP treatment (Figure 11C–E). This indicates that PBzyme can attenuate microglial NLRP3 inflammasome activation and disrupt the execution phase of pyroptosis. Furthermore, PBzyme reduced IL-1β and IL-18 levels in BV2 cells, as evidenced by enzyme-linked immunosorbent assay (ELISA) results (Figure 11F and G). Meanwhile, immunocytochemical staining showed that PBzyme treatment can diminish NLRP3 expression and ASC speck formation in MPTP/ATP-treated microglia (Figure 11H). Furthermore, immunofluorescence staining for TH, a marker of dopaminergic neurons, confirmed that PBzyme treatment significantly preserves TH-positive cells in MPTP/ATP-treated cultures (Figure 11I). Finally, PBzyme was also found to ameliorate the mitochondrial depolarization induced by PD pathology (Figure 11J).
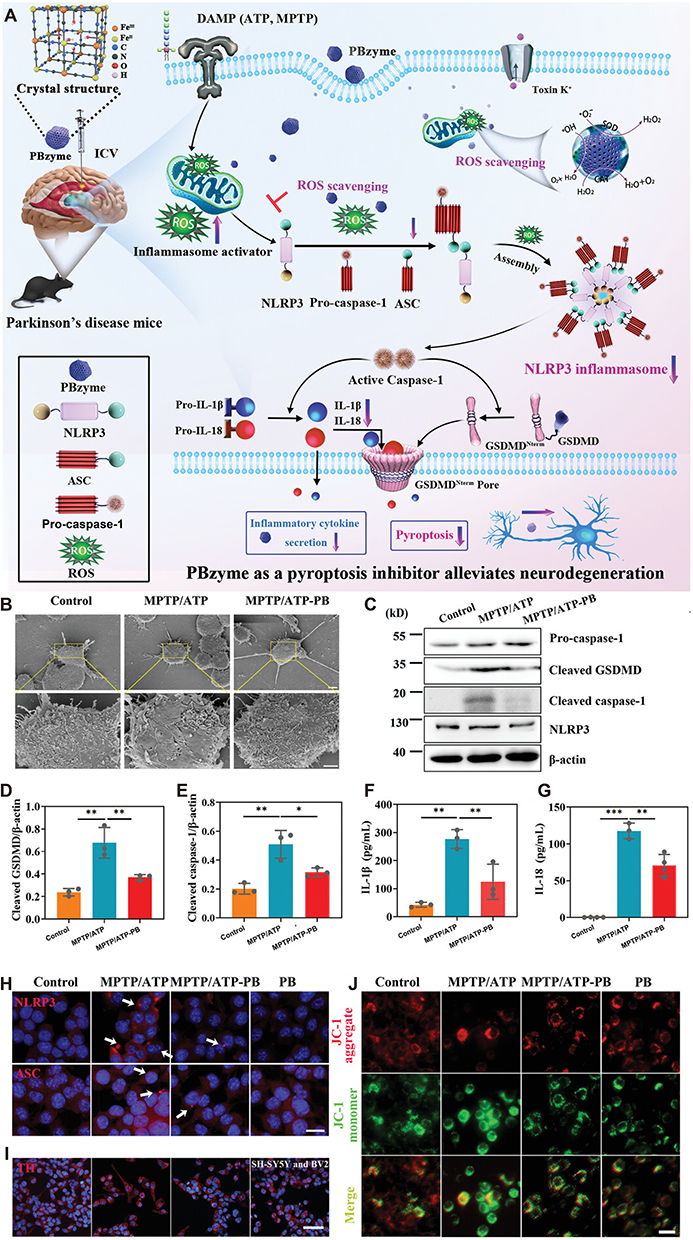 |
Figure 11 (A) Mechanistic action of PBzyme as a pyroptosis inhibitor for PD treatment. (B) SEM images of pores on BV2 cell membranes after MPTP/ATP treatment; scale bars: 5 and 1 µm. (C) Western blot analysis of pro-caspase-1, cleaved GSDMD, cleaved caspase-1, and NLRP3 levels in BV2 cell supernatants and lysates. (D and E) Quantification of cleaved GSDMD (D) and cleaved caspase-1 (E), normalized based on β-actin levels (n = 3). (F and G) ELISA results for IL-1β (F) and IL-18 (G) levels in BV2 cells (n = 3–4). (H) Immunocytochemistry for NLRP3 and ASC in BV2 cells; scale bar: 100 µm; white arrows show ASC puncta. (I) TH immunofluorescence image; scale bar: 50 µm. (J) Mitochondrial membrane potential in BV2 cells treated with MPTP/ATP, with or without PBzyme; scale bar: 25 µm. (mean ± SD, *P < 0.05, **P < 0.01, and ***P < 0.001. Adapted from146 with permission. |
Inspired by the pyroptosis-targeting strategies of PBzyme, some key considerations for designing nanoparticle (NP)-based therapeutics for PD can be identified. These include: (1) Elucidating the role of pyroptosis in PD, particularly with regard to GSDMD cleavage, pore formation, and the accompanying cytokine release and using nanomaterials to modulate this pathway to prevent neuronal loss; (2) Targeting the NLRP3 inflammasome, which is activated by oxidative stress and α-syn aggregation, to halt pyroptosis at both the inflammasome assembly and execution stages; (3) Utilizing nanozymes to scavenge ROS and block the biochemical triggers required for NLRP3 activation and subsequent GSDMD processing, thereby mitigating PD progression; (4) Preserving mitochondrial function, as mitochondrial dysfunction is central to PD pathogenesis. Nanomaterials designed based on such strategies hold significant promise in directly modulating pyroptotic signaling while also alleviating neuroinflammation and inhibiting the progression of PD.
Nanomaterials Restoring Ferroptosis in PD
In the previous section, we discussed the mechanism of ferroptosis and its relationship with PD. As a form of cell death induced by iron overload and lipid peroxides, ferroptosis has been implicated in the early stages of PD, often preceding apoptosis.174 Given the role of ferroptosis in PD progression, this section focuses on emerging nanotherapeutic strategies designed to target ferroptosis and provide neuroprotection.
Traditional therapies utilizing free enzymes often fail to meet clinical requirements due to their susceptibility to inactivation,175 short half-life,176 and limited membrane permeability.177 Therefore, when developing nanomedicines for PD, it is crucial to optimize antioxidant activity, stability, retention time, and transmembrane transport capacity. In recent years, various nanozymes with superoxide dismutase (SOD)-,178 catalase-,179 and uricase-like activities124 have been explored for biomedical applications. To address the neuronal damage caused by PD-related excessive ROS production and inflammatory dysregulation, novel nanozyme platforms, Ptzyme@L-ZIF and Ptzyme@D-ZIF, were designed.120 These systems incorporated chiral metal–organic frameworks (MOFs) that encapsulate Ptzymes within L- and D-type chiral ZIF shells, designed to target both apoptosis and ferroptosis in PD. The protective effects of these agents against oxidative stress in PD mouse brains were evaluated by measuring ROS and malondialdehyde (MDA) levels (Figure 12A). Ptzyme@D-ZIF demonstrated superior ROS scavenging capabilities when compared to Ptzyme@L-ZIF (Figure 12B), with significantly lower MDA levels observed after Ptzyme@D-ZIF treatment in MPTP-induced PD mouse models (Figure 12C). Moreover, the expression of the neuroinflammation markers GFAP and Iba-1 was markedly reduced in MPTP-induced PD mice following Ptzyme@D-ZIF treatment, and the gene expression levels of pro-inflammatory factors like TNF-α, IL-6, and IL-1β were also downregulated (Figures 12D–G). Importantly, the enhanced brain accumulation of Ptzyme@D-ZIF can be attributed to its superior BBB penetration via clathrin- and caveolae-mediated transcytosis, as well as its prolonged plasma residence time relative to the L-enantiomer. In conclusion, Ptzyme@D-ZIF not only exhibited strong cascade SOD- and catalase-like mimetic activity but also demonstrated efficient brain penetration, improved brain accumulation, and plasma stability, offering a promising ferroptosis-targeted therapeutic strategy for PD.
 |
Figure 12 Therapeutic mechanisms of nanozyme-integrated chiral ZIFs in PD mice, addressing oxidative damage and inflammation. (A). Measurement of ROS (B) and malondialdehyde levels (C) in the brains of PD mice after treatment with nanozyme-integrated chiral ZIFs (n = 3). Immunohistochemical analysis of GFAP and Iba-1 expression in brain sections from various groups of PD mice (D); scale bar: 50 µm. Inflammatory markers including (E) TNF-α, (F) IL-6, and (G) IL-1β quantified in brain tissues from different groups (n = 3). Data are presented as the mean ± SD, with comparisons performed using one-way ANOVA followed by Tukey’s post hoc test for multiple comparisons. “ns” indicates not significant. Adapted from120 with permission. |
The labile iron pool (LIP) is a critical driver of ferroptosis within dopaminergic neurons in PD, as it disrupts iron homeostasis and promotes ROS generation.71 The LIP serves as a rapid source of Fe2+ and Fe3+, and excess free Fe2+ reacts with H2O2 to generate Fe3+, causing ion dyshomeostasis, lipid peroxides accumulation, and ferroptosis.180 To halt this cascade, engineered deferoxamine (DFO)-integrated nanosheets (BDPR NSs) that target the LIP were developed in a previous study.147 Functionalized with the RVG29 peptide and nicotinic acetylcholine receptor ligands, these nanosheets showed enhanced BBB penetration. Owing to polydopamine (PDA) loading, the BDPR NSs could effectively deliver DFO to mitochondria in PD models, reducing mitochondrial Fe2⁺ and ROS accumulation. This chelation of free iron and consumption of ROS mitigated LIP dysregulation and prevented LPO, thereby providing neuroprotection.
Beyond targeting iron homeostasis, the modulation of ACSL4 expression has also shown therapeutic potential in PD. ACSL4 inhibition alleviates behavioral deficits, reduces neuroinflammation, and protects against neuronal degeneration in PD.181 Evidence shows that RNA interference (RNAi), via small interfering RNAs (siRNAs), can be employed to target ACSL4. However, due to the inherent instability of siRNAs, NP-based delivery systems are essential for protection and targeted delivery. To enhance BBB penetration, PLGA NPs can be modified with cell membranes. One such integrated delivery system, GM@ACSL4-siRNA/NPs — consisting of LPS-stimulated microglial cell membrane-coated PLGA NPs — was developed to cross the BBB and target the SN in PD mouse models. This system restored iron homeostasis, serving as an effective intervention for PD-related ferroptosis.148 This integrated approach that targeted both the LIP and ACSL4 offered a comprehensive strategy for mitigating ferroptosis and providing neuroprotection in PD.
Designing nanoplatform-based drugs for ferroptosis therapy in PD requires the careful consideration of several key factors to enhance efficacy and specificity: (1) Targeting the mechanism of ferroptosis, specifically iron overload and LPO; (2) Enhancing antioxidant activity through SOD and catalase mimetic functions; (3) Reducing neuroinflammation by downregulating pro-inflammatory cytokines such as TNF-α, IL-6, and IL-1β; (4) Ensuring effective BBB penetration for optimal brain delivery; (5) Incorporating iron-chelating agents, such as deferoxamine, to regulate the LIP and suppress ROS generation; (6) Ensuring the stability, prolonged half-life, and biocompatibility of the nanomaterials. In conclusion, a well-designed nanodrug system targeting ferroptosis holds significant potential for effective PD treatment.
Nanomaterials as Autophagy Modulators in PD
As discussed, autophagy dysfunction exacerbates α-syn aggregation and accelerates PD progression. Thus, targeting autophagy to clear protein aggregates could help in PD treatment. This section discusses some nanomaterials designed to modulate autophagy pathways for effective PD management.
Recent advances in gene therapy show potential in inhibiting α-syn synthesis but fail to eliminate pre-formed aggregates. In this context, trehalose has been explored for its ability to modulate autophagy and promote α-syn degradation.182 However, both gene therapy and trehalose treatment show limited efficacy due to BBB-related brain delivery challenges. Glucose transporter 1, which facilitates glucose uptake into the brain, can be leveraged to enhance nanomaterial delivery across the BBB. Using this approach, researchers developed ultra-small, positively charged particles with surface carboxyl groups called polymer dots (CDs) and functionalized them with glucose and trehalose via PEG and ROS-sensitive thioketal linkers. These NPs were subsequently loaded with plasmid DNA (pDNA) to generate Glu-Tre-PEG-CDs@pDNA NPs.149 This multifunctional system successfully silenced SNCA gene expression, restored autophagy, and alleviated α-syn-induced neurotoxicity, providing a novel therapeutic approach for PD.
Mitophagy, the selective autophagic degradation of damaged mitochondria, also plays a critical role in PD. Mutations in key mitophagy regulators such as PINK1183 and Parkin184 are thus associated with autosomal recessive PD. Impaired mitophagy leads to the accumulation of dysfunctional mitochondria and increased oxidative stress. Melatonin, a well-known antioxidant184 and neurotransmitter,185 promotes mitophagy through receptor-mediated pathways, ubiquitin-independent mechanisms,150 and the activation of the PINK1-Parkin pathway, thereby mitigating the oxidative stress caused by mitochondrial dysfunction.150 Polycomb group proteins, particularly the BMI1 complex, also regulate mitophagy via chromatin modifications. In one study, human serum albumin NPs (HSAnps) loaded with melatonin (M@HSA NPs) were found to modulate BMI1-PTEN interactions, reduce neurotoxicity, and alleviate PD symptoms, highlighting the potential of mitophagy modulation for PD treatment.150 However, despite the potential of inorganic catalytic NPs in inducing mitophagy and suppressing ROS production, challenges such as limited brain-to-blood ratio, complicated synthesis, and large particle size (>10 nm) still remain. However, natural antioxidants like lycopene, a carotenoid known for its high singlet oxygen (1O2) scavenging capacity, offer an alternative approach to modulate autophagy in PD and overcome these challenges.186 However, the therapeutic potential of lycopene is also hindered by its low retention time, poor BBB penetration, and limited bioavailability. To address these limitations, a biocompatible neural enrichment platform was developed using recombinant human H-ferritin (rHuHF) nanocages.187 These nanocages, which supported receptor-mediated transcytosis across the BBB and contained an ideal loading cavity, were modified with lipophilic triphenylphosphonium (TPP). The NPs could easily cross the BBB and subsequently enhance mitochondrial homeostasis, and promote PINK1/Parkin-mediated mitophagy in dopaminergic neurons, offering a viable PD therapeutic strategy.
Cost-effective inorganic catalysts with multi-enzyme activities and high stability are emerging as agents for combating ROS production. Single-atom catalysts (SACs), which demonstrate superior ROS decomposition kinetics, have shown promise in reducing neuroinflammation. However, they primarily eliminate extracellular and cytoplasmic ROS, without addressing the root cause of ROS generation. Moreover, the ROS levels often rebound following catalyst removal. Studies show that Pt/CeO2 not only decomposes ROS but also decreases the cytoplasmic NAD+/NADH ratio, facilitates the dissipation of mitochondrial membrane potential (Δψm), and indirectly induces mitophagy. To improve BBB penetration, Pt/CeO2 NPs can be coated with an activated HL-60 cell membrane (AHM) and functionalized with RVG29, leading to the fabrication of the RVG29@AHM@Pt/CeO2 NP system (Figure 13A).151 In cells with CCCP-induced mitochondrial membrane potential loss, treatment with Pt/CeO2 led to NP accumulation in mitochondria owing to electrostatic attraction (Figure 13B). JC-1 staining confirmed a marked reduction in Δψm after treatment with Pt/CeO2 (Figure 13C). Further, mitochondrial autophagosome and lysosomal co-localization assays demonstrated robust mitophagy induction after Pt/CeO2 treatment (Figures 13D–F). These findings validate the ability of RVG29@AHM@Pt/CeO2 to induce autophagy and enhance mitophagic activity.
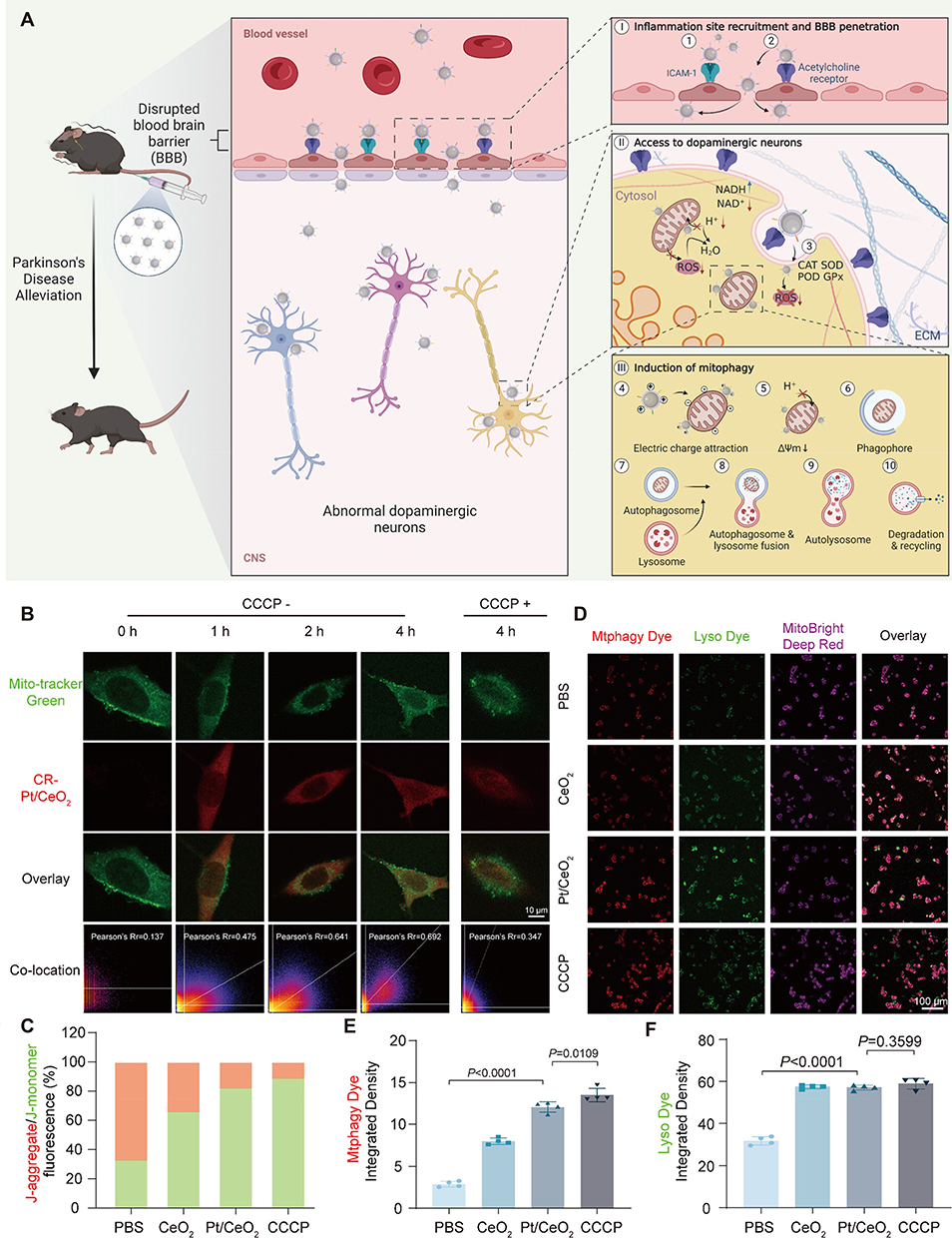 |
Figure 13 (A) In vivo action of core–shell structured single-atom catalysts (SACs). I) SACs bind to ICAM-1 at the BBB inflammation site via LFA-1, Mac-1, and β2 integrin, with RVG29 peptide enabling BBB penetration. II) SACs enter dopaminergic neurons through RVG29-mediated acetylcholine receptor binding and endocytosis, then scavenge intracellular ROS. III) SACs accumulate around abnormal mitochondria via electrostatic attraction, consuming H+ and disrupting mitochondrial H+ supply pathways, leading to mitochondrial depolarization and mitophagy induction. (B) Confocal laser scanning microscopy (CLSM) images of mouse MN9D cells co-cultured with Pt/CeO2 for 0, 1, 2, and 4 h (CCCP− group and CCCP+ group, in which the MN9D cells were treated with CCCP for 4 h and then co-cultured with Pt/CeO2 for 4 h); scale bar: 10 µm. (C) Quantitative analysis of J-aggregates and J-monomers in fluorescence microscopy images. (D) CLSM images showing mitophagy; scale bar: 100 µm. (E) Integrated density of mitochondrial autophagosome dye (Mtphagy dye) from (D) (n =4). (F) Integrated density of lysosomal dye (Lyso dye) from (D) (n=4). Adapted from151 with permission. |
In conclusion, PD pathophysiology involves α-syn aggregation, mitochondrial and lysosomal dysfunction, and mutations in autophagy-related genes. Nanoparticle-based strategies can address these challenges by integrating three essential features: (1) BBB penetration, (2) Nuclear localization, and (3) Cell-specific targeting. This could enhance the translational potential of α-syn-targeted gene therapies. To effectively alleviate mitophagy defects, the focus should be placed on key molecular mechanisms, including (1) Hypoxia-induced mitophagy, (2) Δψm-dependent mitophagy, and (3) Key regulatory proteins associated with mitophagy. By targeting autophagy and mitophagy, integrated nanomaterial-based strategies could offer a comprehensive approach for restoring cellular homeostasis and mitigating cellular dysfunction in PD.
Nanomaterials Mitigating Neuroinflammatory Responses in PD
Although neuroinflammation is initially protective, its persistence can become detrimental, causing oxidative stress, α-syn aggregation, and neuronal damage and impairing cellular degradation systems. Collectively, these alterations contribute to PD progression. This section highlights emerging nanotherapeutic strategies that target oxidative stress and neuroinflammation in PD.
Conventional nanocarriers can accumulate within the neuronal milieu and hinder microglial phagocytosis.188 Thus, there is an urgent need for carrier-free, biocompatible nanomaterials that are capable of delivering multi-modal treatment agents for PD management. Carrier-free nanocapsules, fabricated from DA and 4-formylphenylboronic acid, show superior biocompatibility and do not exhibit the safety issues inherent to exogenous carriers. By incorporating cyclo (CRGDfK) (cRGD)-modified catalase into these nanocapsules, one study exploited the elevated integrin expression on brain endothelial cells in PD to enable targeted delivery at lesion sites and mitigate neuroinflammation.152 Moreover, the peptide ligand Angiopep-2 (Ang), which shows high affinity to low-density lipoprotein receptor-related protein-1 (LRP-1) at the BBB, was also previously used to achieve efficient BBB transcytosis.189 This carrier-free nanoformulation effectively shielded DA from oxidation while concurrently ensuring targeted delivery at lesion sites, providing comprehensive PD therapy. Recent studies have also shown that regulating chemokine communication between neurons and immune cells can block the amplification of inflammatory responses. Based on a similar carrier-free concept, a microgel based on functional monomers was assembled to prevent the rigidity and safety issues of traditional carriers. In this carrier, MPC enabled acetylcholine-like BBB targeting, while PDA degradation supplied dopamine to support neurogenesis. Moreover, Cu–PDA–imidazole networks enabled stable SOD/CAT-like cascade catalysis to reduce oxidative stress. By lowering neuronal ROS levels and limiting microglial activation, the microgel could inhibit CX3CL1 release and suppress CX3CR1-dependent NF-κB–NLRP3 signaling, thereby producing synergistic anti-inflammatory and neuroprotective effects.190
Meanwhile, other studies have shown that targeting neuroinflammation via strategies like modulating the NLRP3 inflammasome can also boost anti-PD therapeutic efficacy. NLRP3 activation leads to the release of pro-inflammatory cytokines like IL-6 and IL-1β, exacerbating inflammation and promoting dopaminergic degeneration.191 To combat this, MOF-derived nanozymes with a high surface area, good porosity, and diverse functional groups have been developed to enhance gas adsorption and storage, catalytic activity, and molecular sieving.192 Specifically, a hollow spherical Zr-FeP MOF nanozyme, coordinated with Fe-TCPP (Fe-5,10,15,20-tetra(4-carboxyphenyl) porphyrin), benzoic acid, and triphenylphosphine, was engineered to target NLRP3-mediated inflammation.153 This nanozyme was functionalized with ligand-targeting moieties and conjugated with mannitol (Man) to enhance delivery at the site of inflammation. Treatment with Zr-FeP MOF@Man liposomes was found to alleviate mitochondrial dysfunction. ROS levels were reduced by approximately 130% and MDA levels by 44%.
While conventional microglia-targeted therapies can transiently reduce neuroinflammation, they fail to address the sustained influx of peripheral immune cells. This perpetuates a hyperactivated cerebral immune environment in PD.193 To restore the neuroimmune balance, an EV-based nanoformulation (EVN) that suppresses microglial inflammation and prevents peripheral immune cell infiltration was engineered.154 C-C motif chemokine ligand 2 (CCL2), a key chemokine secreted by activated microglia and astrocytes, binds to C-C chemokine receptor 2 (CCR2), causing CCR2 overexpression at the lesion site in PD. This interaction further drives inflammatory factor release from microglia, further exacerbating the imbalance in the brain’s immune environment. In this context, EVNs can be engineered to modulate immune responses and restore homeostasis. In a previous study, EVNs conjugated with the MG1 peptide displayed selective affinity for pro-inflammatory M1 microglia, enabling targeted delivery. To inhibit the CCR2–CCL2 axis, mesenchymal stem cell-derived EVs enriched with CCR2 (MSCCCR2 EVs) were engineered as delivery shells. Recent studies have also shown that engineered EVs derived from human umbilical cord MSCs can regulate microglial activation and inhibit NLRP3-associated neuroinflammation in PD models.194
Nrf2 is a transcription factor regulating anti-inflammatory responses via the Keap1–Nrf2 system.195 Dihydrotanshinone I (DT), an active compound from Salvia miltiorrhiza, promotes the nuclear import of Nrf2 and inhibits its export.196 By loading DT into MSCCCR2 EVs and integrating them with diselenide-bridged mesoporous silica NPs (MSeN-DT), targeted delivery to high-ROS environments can be achieved, eliminating over-oxidation via enhanced Nrf2 expression (Figure 14A). In a previous study on this system, immunofluorescence imaging showed reduced Iba-1 expression following EVN treatment (Figure 14B). EVN administration induced a shift from the M1 to the M2 phenotype in SN microglia: CD86⁺ M1 cells were downregulated, such that CD206⁺ M2 cells were markedly upregulated (relative to the PBS group) (Figure 14C). Furthermore, the EVNs significantly reduced GFAP expression in the SN (Figure 14D). Additionally, they downregulated pro-inflammatory markers (TNF-α, IL-6) and upregulated anti-inflammatory factors (IL-10, Arg-1) in the striatum, creating a favorable anti-inflammatory environment (Figure 14E–H). These results confirm that EVNs can promote the DT-dependent activation of the Nrf2–GPX4 pathway, facilitate M2 polarization, restore neuronal function, and provide potent anti-inflammatory effects, collectively offering a synergistic nanotherapeutic strategy for PD.
 |
Figure 14 (A) Schematic illustration showing the preparation of extracellular vesicle-based nanoformulations overexpressing CCR2 and their therapeutic role in PD. (B) Representative immunofluorescence images and corresponding quantitative analysis of Iba-1 expression (red) in the SN of mice (n = 3). (C) Phenotypic analysis of microglial cells in the midbrain of mice via flow cytometry and the corresponding percentage of CD86+ and CD206+ microglial cells under different conditions (n = 3). (D) Representative immunofluorescence images and corresponding quantitative analysis of GFAP (red) in the SN of mice (n = 3). (E) IL-10, (F) Arg-1, (G) TNF-α, and (H) IL-6 expression in the striatum of mice (n = 5). I: sham, II: PBS, III: MSCCCR2 EVs, IV: MSeN-DT, and V: EVN. Adapted from154 with permission. |
When designing nanoplatform-based therapies targeting neuroinflammation in PD, several key aspects should be prioritized: (1) Microglial targeting: Nanomedicines should specifically target M1 microglia and promote their polarization toward the anti-inflammatory M2 phenotype; (2) Antioxidant delivery: The effective delivery of antioxidants is critical for reducing ROS levels and alleviate neuroinflammation in PD; (3) Improved BBB penetration: Ligand-functionalized NPs should be employed to ensure efficient BBB crossing and brain accumulation; (4) Immune modulation: The activation of key regulatory pathways such as the Nrf2–GPX4 pathway can modulate the immune response and support neuronal survival; (5) NLRP3 inflammasome inhibition: Nanoparticles that suppress NLRP3 inflammasome activation can alleviate chronic inflammation and protect dopaminergic neurons. Neuroinflammation-targeting nanotherapeutic strategies developed based on these principles offer significant promise in improving PD management and slowing down disease progression.
Nanomaterials Modulating Gut Microbiota for PD Therapy
The gut–brain axis is a complex bidirectional communication network between the gut microbiota and CNS, affecting behavior, immune responses, and neurochemical signaling. In PD, gastrointestinal symptoms often exacerbate both motor and non-motor impairments. As gut microbiota influence neurotransmitter function and disease progression, the gut–brain axis has garnered growing interest as a target for PD treatment.
Emerging evidence indicates that gut commensals such as Escherichia coli (E. coli),197 Segmentomycetes,198 and Akkermansia muciniphila199 contribute to the initiation and progression of PD by modulating immune responses and influencing neural activity. DNA-based nanomaterials, particularly tetrahedral framework nucleic acids (tFNAs), have gained attention for their programmability,200 stability,201 and biocompatibility.202 These tFNAs can be engineered with antimicrobial peptides and targeting ligands to precisely modulate the gut microbiota for PD therapy. Interestingly, EVs offer a promising platform for the delivery of these agents, enabling the targeted release of tFNAs to modulate macrophages, microglia, and enteroendocrine cells.155 This approach not only alleviates neuroinflammation but also enhances serotonin and dopamine precursor synthesis, mitigating α-syn deposition.
Optogenetic techniques, in which light is used to induce gene expression in engineered gut microbes, also provide a non-invasive and efficient strategy to control microbial activity in the gut.203 To ensure precise delivery to the small intestine, Lactococcus lactis can be encapsulated in pH-responsive sodium alginate microspheres, which ensures payload release in the small intestine. Meanwhile, modification with proton-dependent transporter 1 (PepT1) facilitates targeted delivery. Engineered Lactococcus lactis strains, designed to synthesize therapeutic compounds such as GABA, GCSF, and GLP-1 upon light irradiation, can serve as powerful tools to modulate PD pathology.156 These strains can be manipulated through an optogenetic NIR-light-triggered upconversion nanosystem, enabling precise CNS modulation via the gut–brain axis. Such integrated nanoplatforms provide a promising multi-modal approach for addressing PD through targeted gut microbiota modulation and neural signaling enhancement. In addition to intestinal-guided optogenetic regulation, the nano-optogenetic platform could also realize minimally invasive control of PD-related neural circuits. Up-conversion nanoparticles (UCNPs) convert near-infrared light into visible light, which can activate deep motor nuclei expressing opsins and restore basal ganglia activity in PD models without implanted fibers.204 Wireless transcranial systems combining UCNPs with high-sensitivity opsins can further achieve two-way intracranial regulation of subcortex–striatum projections under near-infrared irradiation, enabling precise circuit control while minimizing tissue damage.205 In addition to intestinal-based optogenetic regulation, these nanotechnology-driven neuroregulation strategies demonstrate how phototransresponsive materials enable multi-level interventions along the intestinal–brain axis, providing a unified and tunable platform for next-generation PD treatment.
Microbial therapies aimed at short-chain fatty acid (SCFA) generation have demonstrated therapeutic efficacy against conditions such as colorectal cancer,206 enteritis,207 and diabetes mellitus.208 Given the vulnerability of microbial therapeutics to environmental stressors during gastrointestinal transit, strategies to enhance microbial resilience are essential. In this regard, an intelligent microbial therapy agent inspired by dormant bodies, which is activated within 6–48 h and is then eliminated, has shown considerable promise.209 A recent study described an efficient strategy for fabricating biomimetic dormant bodies. This strategy involved coating with a pH-responsive anionic copolymer of methacrylic acid and ethyl acrylate (L30D-55) through supramolecular self-assembly at the biological interface. These structures remained dormant even when exposed to the strong acids in simulated gastric fluid. Moreover, they maintained mucosal adhesion, colonization, and proliferation until they reached the intestine, where the pH was different (Figure 15A). The gastrointestinal bioavailability of dormant probiotics (L.p@L30D-55) was found to be over 3.5-fold greater than that of unmodified microorganisms.157
 |
Figure 15 (A) Schematic of probiotics dormant body preparation and BBB penetration. (B) Mechanism of PD symptom amelioration via p38MAPK/NF-κB inhibition in microglia. (C) Immunofluorescence analysis of TH expression in the striatum across various experimental groups. (D) TUNEL staining in the striatum across various experimental groups. (E) Randomized movement trajectories in the open-field test. (F) Path length in the open-field test (mean ± SD; *P < 0.05). (G) Randomized movement trajectories in the forced-swimming test. (H) Immobility time in the forced-swimming test (mean ± SD; *P < 0.05). Adapted from157 with permission. |
PD therapeutic efficacy can also be substantially enhanced through the in situ microbial production of the inhibitory neurotransmitter γ-amino-butyric acid (GABA) following probiotic administration. Evidence shows that GABA antagonizes the p38MAPK/NF-κB signaling axis and ameliorates PD phenotypes in murine models (Figure 15B). In a previous study, immunofluorescence analysis demonstrated that L.p@L30D-55 preserves TH expression and significantly reduces dopaminergic neuron apoptosis in the striatum (Figure 15C and D). Both the open-field and forced-swimming tests—which are standard assays for evaluating spontaneous motor activity in PD models—revealed improvements in PD-related motor symptoms after L.p@L30D-55 treatment. Compared with wild-type mice, the L.p@L30D-55-treated cohort crossed more gridlines and exhibited greater rearing behavior in the open-field test. Similarly, in the forced-swimming test, this group achieved higher swimming scores and demonstrated enhanced spontaneous locomotor activity (Figure 15E–H). In summary, these findings showed that biomimetic dormant bodies can be reactivated under specific conditions to enable the production of bioactive therapeutics in situ. Such oral bionic systems could enable the synthesis of small-molecule drugs such as SCFAs, 5-HT, and DA directly at target sites to treat PD.
The gut microbiota-based interventions for PD developed so far share several key hallmarks: (1) The gut–brain axis is used to deliver engineered microbial strains that are capable of producing neuroactive compounds (eg, GABA, 5-HT, DA precursors, GCSF, GLP-1) that attenuate neuroinflammation, inhibit α-syn aggregation, and preserve dopaminergic neurons; (2) Biocompatible and pH-responsive nanomaterials systems can optimize the retention time, mucosal adhesion, and colonization and enable precise delivery at lesion sites; (3) Temporal control via external stimuli (eg, NIR-triggered upconversion enabling blue-light-activated gene expression) or pH-dependent “dormant body” coatings that allow the programmable activation of bacterial therapeutics can be employed.
Based on these insights, we infer that gut microbiota-based nanotherapeutic systems for PD should prioritize: (1) The selection and safety of probiotic strains that secrete disease-modifying factors; (2) The development of stimuli-responsive, gastroresistant carriers to maximize intestinal bioavailability and minimize off-target release and effects; (3) The integration of spatiotemporal control mechanisms to coordinate microbial activity with the therapeutic windows; (4) Rigorous in vivo assessment of biocompatibility, immunogenicity, and long-term colonization dynamics to ensure safety and sustained efficacy. Overall, restoring gut microbiota balance and expanding anti-inflammatory bacterial populations could improve BBB function and alleviate PD symptoms.
Status of Clinical Trials
Current PD treatments mainly focus on symptom management, and therapies targeting the underlying pathological mechanisms of PD are largely unavailable. Therefore, there is an urgent need to develop novel drugs targeting the root causes of PD in order to improve patients’ overall quality of life. α-Syn, a key marker of PD, is also considered a central target for PD treatment. Some clinical trials, such as the Phase I trial NCT06445465, are exploring the efficacy of α-syn-targeting therapeutics for PD. In addition to α-syn-targeting therapies, other therapeutic strategies based on levodopa-sparing agents, gene therapies, neuroprotective agents, and immunotherapies have also been developed and tested in clinical trials.
Neuroprotective agents can protect dopaminergic neurons and inhibit neurodegeneration. Notably, the NCT06612593 trial evaluated the impact of a novel neuroprotective agent on neuronal survival. Meanwhile, the goal of levodopa-sparing agents is to reduce levodopa dosage, thereby minimizing its long-term side effects. For example, the Phase II clinical trial NCT04778176, which has now been completed, evaluated the safety and efficacy of a novel levodopa-sparing agent. Although the gene therapy trial NCT04287543, designed to correct PD-associated mutations, was withdrawn, it provided a valuable proof-of-concept for PD-related gene therapy.
Recently, the role of the gut–brain axis and gut microbiota in PD has gained significant attention, highlighting the importance of gastrointestinal dysfunction in disease progression. Accordingly, the NCT04730245/NCT04451096 trials are currently exploring the role of gut microbiota in PD, and higher-phase trials are being conducted, despite some delays due to the COVID-19 pandemic. At the same time, therapies targeting metabolic pathways and mitochondrial dysfunction are also emerging as promising avenues for addressing the multifaceted pathological mechanisms of PD.
Personalized and precision medicine are expected to become increasingly important in the future. To ensure the sustainability and safety of PD treatments, long-term follow-up studies will be essential. Despite ongoing challenges, numerous clinical trials based on different drug formulations and mechanisms are currently underway, signaling a shift towards multi-targeted, diversified approaches in PD treatment (Table 2). Beyond conventional drug trials, rapid progress in artificial intelligence (AI) could also revolutionize the field of drug development. By harnessing the capabilities of AI, therapeutics that are more precisely tailored to individual patient profiles can be produced, enhancing therapeutic efficacy and expediting their adoption.
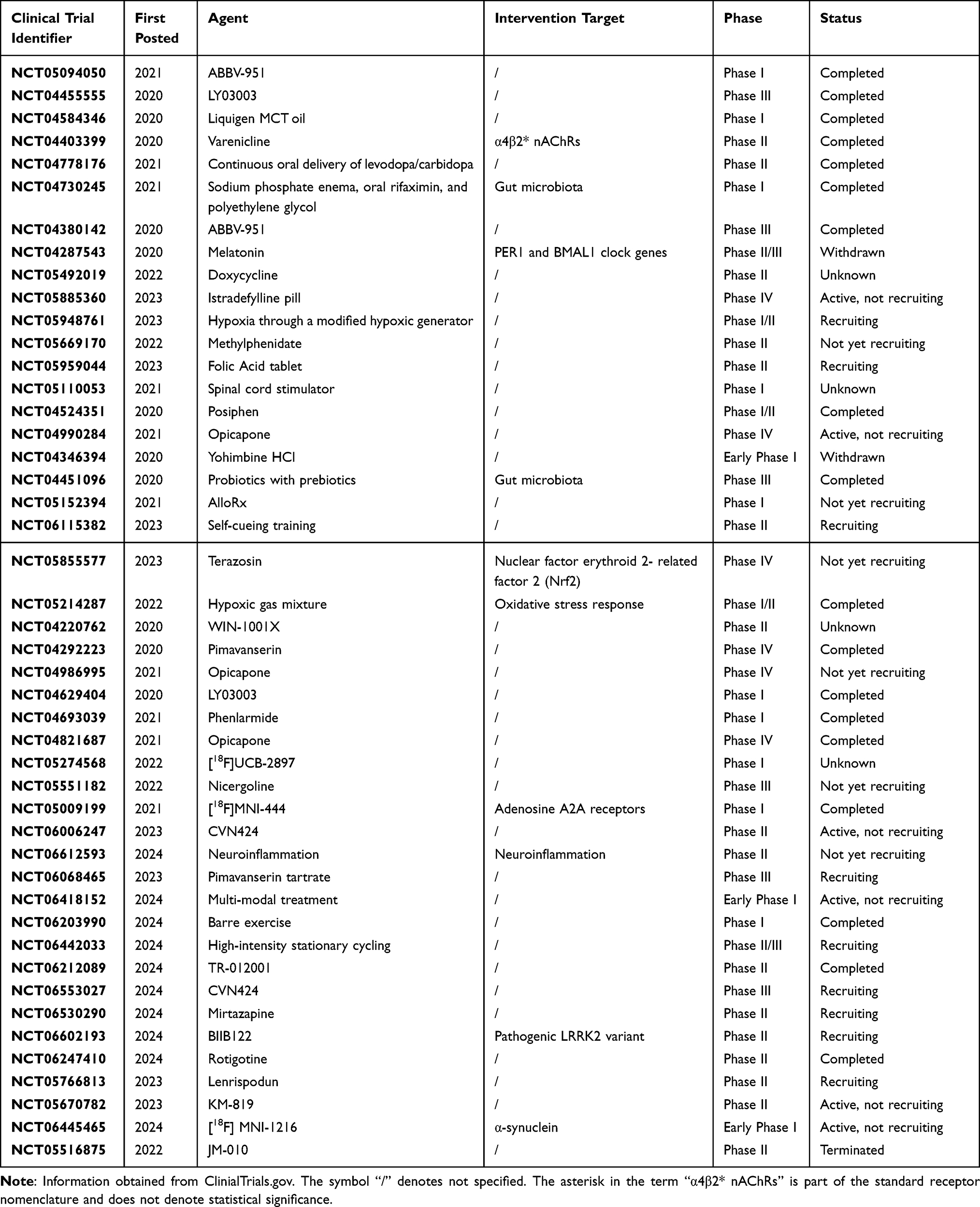 |
Table 2 Clinical Trials for PD Treatment in Recent years |
Conclusion and Future Perspectives
Nanotechnology holds immense promise for revolutionizing the treatment of PD by enabling drugs to cross the BBB and enhancing drug accumulation within PD lesions. Advances in nanomedicine have enabled the targeted distribution and prolonged retention of PD therapeutics in the SNpc. Owing to their versatility, biocompatibility, and precision, nanomedicines have emerged as a powerful tool for PD therapy, guiding the development of more effective, personalized treatments for this neurodegenerative disease.
This review discusses the key pathophysiological mechanisms driving PD and categorizes different nanotherapeutic strategies based on the pathogenic factors they target. Accordingly, we highlight various nanomedicine-based treatment approaches for PD. For instance, nanomedicines can serve as nanochaperones that chelate misfolded proteins, preventing their further aggregation. Alternatively, they can be engineered to develop nanosponges that are capable of adsorbing and promoting the degradation of pathogenic proteins. Moreover, some nanomaterials allow the precise delivery of therapeutic agents at target sites. Diverse nanomedicine platforms — including nanozymes, EVs, nutritional agents, and gene delivery systems — have been developed for PD treatment. Broadening the application of these technologies to not only eliminate disease-driving factors but also support tissue regeneration represents a key direction for future research. The multifunctional nature of nanomedicines underscores their potential to go beyond conventional drug delivery and address complex neurodegenerative pathways.
Although nanotechnology has only been applied in the field of PD therapy over the past few decades, it has significantly improved treatment efficacy when compared to traditional drug formulations. However, a cure for PD is still unavailable. Based on the current developments in this field, we believe that future studies should prioritize the following aspects to achieve enhanced anti-PD efficacy with nanomedicines.
- Safety: While the biocompatibility of anti-PD nanomedicines has been rigorously evaluated in preclinical animal models, intrinsic species differences hinder a comprehensive understanding of their clinical safety in humans. Moreover, some inorganic nanomaterials may produce metabolites that elicit deleterious responses in the human body. Therefore, meticulous studies are required to optimize the therapeutic advantages of anti-PD nanomedicines while mitigating their potential toxicity.
- Preparation: Ensuring reproducibility in the synthesis of anti-PD nanomedicines remains a significant challenge. Even in small-scale laboratory settings, these formulations frequently exhibit heterogeneity, which can compromise their consistency and efficacy.
- Drug availability: In some cases, nanocarriers function exclusively as delivery vesicles, and the therapeutic efficacy of the encapsulated drug determines the final treatment effects. Hence, more potent and disease-specific drugs are required for maximizing the therapeutic potential of nanomedicine-based PD treatments.
- Targeting efficiency: To be effective, nanomedicines must target three critical disease components: lesions, diseased cells, and pathogenic factors. This is even more challenging in PD therapy because of the additional hindrance created by the BBB. Although various agents capable of targeting the BBB, neurons, and microglia have been developed, concerns about specificity remain. Alternative administration routes, such as nasal or intrathecal administration, help in bypassing the BBB but present additional challenges, including undefined brain entry mechanisms and the absence of suitable delivery devices. Therefore, more refined targeting strategies are required to enhance the therapeutic efficacy of nanomedicines while minimizing off-target effects.
- Targeting key pathological processes: In the mechanism-based treatment of PD, diverse nanomedicine-based strategies can be used to modulate key pathological processes: (a) α-Syn aggregation: Nanodrugs should inhibit α-syn aggregation and promote aggregate clearance, and surface modifications should be optimized to enhance BBB penetration and selective targeting. (b) Apoptosis: Nanomedicines should stabilize mitochondria, regulate apoptotic proteins, scavenge ROS, and clear apoptotic debris to reduce inflammation and neuronal loss. (c) Pyroptosis: Nanodrugs should inhibit the activation of inflammasomes, particularly NLRP3, to inhibit pro-inflammatory cytokine release and protect mitochondrial function. Meanwhile, BBB penetration should also be enhanced. (d) Ferroptosis: Nanomedicines should regulate iron metabolism, prevent LPO, and enhance antioxidant activity, restoring mitochondrial function and mitigating oxidative stress in PD brains. (e) Autophagy: Nanodrugs should modulate key autophagic pathways to enhance α-syn degradation and restore mitophagy, supporting neuronal health. (f) Neuroinflammation: Nanomedicines should target microglia, astrocytes, and inflammatory pathways such as the NF-κB axis to reduce neuroinflammation, ensuring effective targeting and BBB penetration. (g) Gut–brain axis: Nanomedicines should restore intestinal barrier function and modulate gut microbiota to prevent the transmission of misfolded α-syn to the brain, optimizing therapeutic efficacy through targeted, stimuli-responsive delivery.
- Effectiveness of pre-clinical models: The use of animal models remains one of the most significant challenges in PD research. Current models are typically established through drug treatments or gene editing and cannot fully replicate the complex pathological features of PD. Thus, it is essential to develop animal models that more accurately reflect the clinical manifestations of PD to improve the clinical translation of PD therapeutics.
In conclusion, nanotechnology presents an innovative therapeutic opportunity for PD treatment, as evidenced by preclinical studies. Although the number of clinical trials remains limited at present, ongoing research targeting multiple disease mechanisms holds considerable potential for the gradual clinical integration of anti-PD nanomedicines. Therefore, researchers must continuously stay informed regarding the latest advancements in PD research. In the future, the clinical advancement of precise, multi-targeted nanomedicines will be instrumental in addressing the complexities of this key neurodegenerative disorder, heralding a new era in PD management.
Acknowledgments
We deeply appreciate the support from all participants.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was financially supported through grants from the National Natural Science Foundation of China (82474607) and the Key Fields of Biomedicine and Health Foundation of Colleges and Universities in Guangdong Province (2022ZDZX2017).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Zhao X, Kang Z, Han R. et al. JWA binding to NCOA4 alleviates degeneration in dopaminergic neurons through suppression of ferritinophagy in Parkinson’s disease. Redox Biol. 2024;73:103190. doi:10.1016/j.redox.2024.103190
2. Liu LL, Han Y, Zhang ZJ, et al. Loss of DJ-1 function contributes to Parkinson’s disease pathogenesis in mice via RACK1-mediated PKC activation and MAO-B upregulation. Acta Pharmacol Sin. 2023;44(10):1948–39. doi:10.1038/s41401-023-01104-8
3. Jankovic J, Tan EK. Parkinson’s disease: etiopathogenesis and treatment. J Neurol Neurosurg Psychiatry. 2020;91(8):795–808. doi:10.1136/jnnp-2019-322338
4. Su D, Cui Y, He C, et al. Projections for prevalence of Parkinson’s disease and its driving factors in 195 countries and territories to 2050: modelling study of Global Burden of Disease Study 2021. BMJ. 2025;388:e080952. doi:10.1136/bmj-2024-080952
5. Koeglsperger T, Rumpf SL, Schließer P, et al. Neuropathology of incidental Lewy body & prodromal Parkinson’s disease. Mol Neurodegener. 2023;18(1):32. doi:10.1186/s13024-023-00622-7
6. Pan L, Li C, Meng L, et al. Tau accelerates α-synuclein aggregation and spreading in Parkinson’s disease. Brain. 2022;145(10):3454–3471. doi:10.1093/brain/awac171
7. Han X, Liu Y, Dai Y, et al. Neuronal SH2B1 attenuates apoptosis in an MPTP mouse model of Parkinson’s disease via promoting PLIN4 degradation. Redox Biol. 2022;52:102308. doi:10.1016/j.redox.2022.102308
8. Moujalled D, Strasser A, Liddell JR. Molecular mechanisms of cell death in neurological diseases. Cell Death Differ. 2021;28(7):2029–2044. doi:10.1038/s41418-021-00814-y
9. Wang ZL, Yuan L, Li W, Li JY. Ferroptosis in Parkinson’s disease: glia-neuron crosstalk. Trends Mol Med. 2022;28(4):258–269. doi:10.1016/j.molmed.2022.02.003
10. Lizama BN, Chu CT. Neuronal autophagy and mitophagy in Parkinson’s disease. Mol Aspects Med. 2021;82:100972. doi:10.1016/j.mam.2021.100972
11. Tansey MG, Wallings RL, Houser MC, Herrick MK, Keating CE, Joers V. Inflammation and immune dysfunction in Parkinson disease. Nat Rev Immunol. 2022;22(11):657–673. doi:10.1038/s41577-022-00684-6
12. Zhao Z, Ning J, Bao XQ, et al. Fecal microbiota transplantation protects rotenone-induced Parkinson’s disease mice via suppressing inflammation mediated by the lipopolysaccharide-TLR4 signaling pathway through the microbiota-gut-brain axis. Microbiome. 2021;9(1):226. doi:10.1186/s40168-021-01107-9
13. Charvin D, Medori R, Hauser RA, Rascol O. Therapeutic strategies for Parkinson disease: beyond dopaminergic drugs. Nat Rev Drug Discov. 2018;17(11):804–822. doi:10.1038/nrd.2018.136
14. Qian K, Jiang X, Liu ZQ, et al. Revisiting the critical roles of reactive astrocytes in neurodegeneration. Mol Psychiatry. 2023;28(7):2697–2706. doi:10.1038/s41380-023-02061-8
15. Vijiaratnam N, Simuni T, Bandmann O, Morris HR, Foltynie T. Progress towards therapies for disease modification in Parkinson’s disease. Lancet Neurol. 2021;20(7):559–572. doi:10.1016/s1474-4422(21)00061-2
16. Chu Y, Hirst WD, Federoff HJ, Harms AS, Stoessl AJ, Kordower JH. Nigrostriatal tau pathology in parkinsonism and Parkinson’s disease. Brain. 2024;147(2):444–457. doi:10.1093/brain/awad388
17. Nayab DE, Din FU, Ali H, et al. Nano biomaterials based strategies for enhanced brain targeting in the treatment of neurodegenerative diseases: an up-to-date perspective. J Nanobiotechnology. 2023;21(1):477. doi:10.1186/s12951-023-02250-1
18. Zhang C, Zheng Q, Li C, et al. A multifunctional electrochemical biosensor based on near-infrared light-responsive hydrogel for in vivo recording and modulation. Anal Chem. 2025. doi:10.1021/acs.analchem.5c00640
19. Wang J, Li Z, Pan M, et al. Ultrasound-mediated blood-brain barrier opening: an effective drug delivery system for theranostics of brain diseases. Adv Drug Deliv Rev. 2022;190:114539. doi:10.1016/j.addr.2022.114539
20. Peng H, Li Y, Ji W, et al. Intranasal administration of self-oriented nanocarriers based on therapeutic exosomes for synergistic treatment of Parkinson’s disease. ACS Nano. 2022;16(1):869–884. doi:10.1021/acsnano.1c08473
21. Zheng Q, Liu H, Zhang H, et al. Ameliorating mitochondrial dysfunction of neurons by biomimetic targeting nanoparticles mediated mitochondrial biogenesis to boost the therapy of Parkinson’s disease. Adv Sci. 2023;10(22):e2300758. doi:10.1002/advs.202300758
22. Lei T, Li C, Liu Y, et al. Microfluidics-enabled mesenchymal stem cell derived Neuron like cell membrane coated nanoparticles inhibit inflammation and apoptosis for Parkinson’s disease. J Nanobiotechnology. 2024;22(1):370. doi:10.1186/s12951-024-02587-1
23. Kampen S, Duy Vo D, Zhang X, et al. Structure-guided design of G-protein-coupled receptor polypharmacology. Angew Chem Int Ed Engl. 2021;60(33):18022–18030. doi:10.1002/anie.202101478
24. Praschberger R, Kuenen S, Schoovaerts N, et al. Neuronal identity defines α-synuclein and tau toxicity. Neuron. 2023;111(10):1577–1590.e11. doi:10.1016/j.neuron.2023.02.033
25. Wang J, Dai L, Chen S, Zhang Z, Fang X, Zhang Z. Protein-protein interactions regulating α-synuclein pathology. Trends Neurosci. 2024;47(3):209–226. doi:10.1016/j.tins.2024.01.002
26. Xu CK, Meisl G, Andrzejewska EA, et al. α-Synuclein oligomers form by secondary nucleation. Nat Commun. 2024;15(1):7083. doi:10.1038/s41467-024-50692-4
27. Martirosyan A, Ansari R, Pestana F, et al. Unravelling cell type-specific responses to Parkinson’s Disease at single cell resolution. Mol Neurodegener. 2024;19(1):7. doi:10.1186/s13024-023-00699-0
28. Sharma M, Burré J. α-Synuclein in synaptic function and dysfunction. Trends Neurosci. 2023;46(2):153–166. doi:10.1016/j.tins.2022.11.007
29. Travagli RA, Browning KN, Camilleri M. Parkinson disease and the gut: new insights into pathogenesis and clinical relevance. Nat Rev Gastroenterol Hepatol. 2020;17(11):673–685. doi:10.1038/s41575-020-0339-z
30. Newton K, Strasser A, Kayagaki N, Dixit VM. Cell death. Cell. 2024;187(2):235–256. doi:10.1016/j.cell.2023.11.044
31. Tian X, Srinivasan PR, Tajiknia V, et al. Targeting apoptotic pathways for cancer therapy. J Clin Invest. 2024;134(14). doi:10.1172/jci179570
32. Du CH, Wu YD, Yang K, et al. Apoptosis-resistant megakaryocytes produce large and hyperreactive platelets in response to radiation injury. Mil Med Res. 2023;10(1):66. doi:10.1186/s40779-023-00499-z
33. Hou Q, Zhu Q, Lu W, Zhang W. Protein S-nitrosylation regulates postmortem beef apoptosis through the intrinsic mitochondrial pathway. J Agric Food Chem. 2022;70(4):1252–1260. doi:10.1021/acs.jafc.1c06516
34. Fowler-Shorten DJ, Hellmich C, Markham M, Bowles KM, Rushworth SA. BCL-2 inhibition in haematological malignancies: clinical application and complications. Blood Rev. 2024;65:101195. doi:10.1016/j.blre.2024.101195
35. Horn S, Hughes MA, Schilling R, et al. Caspase-10 negatively regulates caspase-8-mediated cell death, switching the response to CD95L in favor of NF-κB activation and cell survival. Cell Rep. 2017;19(4):785–797. doi:10.1016/j.celrep.2017.04.010
36. Cho M, Dho SH, Shin S, et al. Caspase-10 affects the pathogenesis of primary biliary cholangitis by regulating inflammatory cell death. J Autoimmun. 2022;133:102940. doi:10.1016/j.jaut.2022.102940
37. Zargarian S, Shlomovitz I, Erlich Z, et al. Phosphatidylserine externalization, “necroptotic bodies” release, and phagocytosis during necroptosis. PLoS Biol. 2017;15(6):e2002711. doi:10.1371/journal.pbio.2002711
38. Amo-Aparicio J, Daly J, Højen JF, Dinarello CA. Pharmacologic inhibition of NLRP3 reduces the levels of α-synuclein and protects dopaminergic neurons in a model of Parkinson’s disease. J Neuroinflammation. 2023;20(1):147. doi:10.1186/s12974-023-02830-w
39. Brokatzky D, Dörflinger B, Haimovici A, et al. A non-death function of the mitochondrial apoptosis apparatus in immunity. EMBO j. 2019;38(11). doi:10.15252/embj.2018100907
40. González-Rodríguez P, Zampese E, Stout KA, et al. Disruption of mitochondrial complex I induces progressive parkinsonism. Nature. 2021;599(7886):650–656. doi:10.1038/s41586-021-04059-0
41. Bertheloot D, Latz E, Franklin BS. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol Immunol. 2021;18(5):1106–1121. doi:10.1038/s41423-020-00630-3
42. Robinson N, Ganesan R, Hegedűs C, Kovács K, Kufer TA, Virág L. Programmed necrotic cell death of macrophages: focus on pyroptosis, necroptosis, and parthanatos. Redox Biol. 2019;26:101239. doi:10.1016/j.redox.2019.101239
43. Hu Y, Wang B, Li S, Yang S. Pyroptosis, and its role in central nervous system disease. J Mol Biol. 2022;434(4):167379. doi:10.1016/j.jmb.2021.167379
44. Franchi L, Muñoz-Planillo R, Núñez G. Sensing and reacting to microbes through the inflammasomes. Nat Immunol. 2012;13(4):325–332. doi:10.1038/ni.2231
45. Ceballos-Olvera I, Sahoo M, Miller MA, Del Barrio L, Re F. Inflammasome-dependent pyroptosis and IL-18 protect against Burkholderia pseudomallei lung infection while IL-1β is deleterious. PLoS Pathog. 2011;7(12):e1002452. doi:10.1371/journal.ppat.1002452
46. Miao R, Jiang C, Chang WY, et al. Gasdermin D permeabilization of mitochondrial inner and outer membranes accelerates and enhances pyroptosis. Immunity. 2023;56(11):2523–2541.e8. doi:10.1016/j.immuni.2023.10.004
47. Schiffelers LDJ, Tesfamariam YM, Jenster LM, et al. Antagonistic nanobodies implicate mechanism of GSDMD pore formation and potential therapeutic application. Nat Commun. 2024;15(1):8266. doi:10.1038/s41467-024-52110-1
48. Coll RC, Schroder K, Pelegrín P. NLRP3 and pyroptosis blockers for treating inflammatory diseases. Trends Pharmacol Sci. 2022;43(8):653–668. doi:10.1016/j.tips.2022.04.003
49. Liao Y, Zhang W, Zhou M, Zhu C, Zou Z. Ubiquitination in pyroptosis pathway: a potential therapeutic target for sepsis. Cytokine Growth Factor Rev. 2024;80:72–86. doi:10.1016/j.cytogfr.2024.09.001
50. Nozaki K, Miao EA. Bucket lists must be completed during cell death. Trends Cell Biol. 2023;33(9):803–815. doi:10.1016/j.tcb.2023.02.008
51. Zeng C, Duan F, Hu J, et al. NLRP3 inflammasome-mediated pyroptosis contributes to the pathogenesis of non-ischemic dilated cardiomyopathy. Redox Biol. 2020;34:101523. doi:10.1016/j.redox.2020.101523
52. Luo T, Zhou X, Qin M, et al. Corilagin restrains NLRP3 inflammasome activation and pyroptosis through the ROS/TXNIP/NLRP3 pathway to prevent inflammation. Oxid Med Cell Longev. 2022;2022:1652244. doi:10.1155/2022/1652244
53. Yan H, Luo B, Wu X, et al. Cisplatin Induces Pyroptosis via Activation of MEG3/NLRP3/caspase-1/GSDMD Pathway in Triple-Negative Breast Cancer. Int J Biol Sci. 2021;17(10):2606–2621. doi:10.7150/ijbs.60292
54. Lawrence JH, Patel A, King MW, Nadarajah CJ, Daneman R, Musiek ES. Microglia drive diurnal variation in susceptibility to inflammatory blood-brain barrier breakdown. JCI Insight. 2024;9(21). doi:10.1172/jci.insight.180081
55. Yin L, Bao F, Wu J, Li K. NLRP3 inflammasome-dependent pyroptosis is proposed to be involved in the mechanism of age-dependent isoflurane-induced cognitive impairment. J Neuroinflammation. 2018;15(1):266. doi:10.1186/s12974-018-1299-x
56. Wang Y, Lv MN, Zhao WJ. Research on ferroptosis as a therapeutic target for the treatment of neurodegenerative diseases. Ageing Res Rev. 2023;91:102035. doi:10.1016/j.arr.2023.102035
57. von Krusenstiern AN, Robson RN, Qian N, et al. Identification of essential sites of lipid peroxidation in ferroptosis. Nat Chem Biol. 2023;19(6):719–730. doi:10.1038/s41589-022-01249-3
58. Yan B, Ai Y, Sun Q, et al. Membrane damage during ferroptosis is caused by oxidation of phospholipids catalyzed by the oxidoreductases POR and CYB5R1. Mol Cell. 2021;81(2):355–369.e10. doi:10.1016/j.molcel.2020.11.024
59. Ajoolabady A, Aslkhodapasandhokmabad H, Libby P, et al. Ferritinophagy and ferroptosis in the management of metabolic diseases. Trends Endocrinol Metab. 2021;32(7):444–462. doi:10.1016/j.tem.2021.04.010
60. HKC C, Wu CC, Lee YC, Chen SH. Emergence of large-scale cell death through ferroptotic trigger waves. Nature. 2024;631(8021):654–662. doi:10.1038/s41586-024-07623-6
61. Shui S, Zhao Z, Wang H, Conrad M, Liu G. Non-enzymatic lipid peroxidation initiated by photodynamic therapy drives a distinct ferroptosis-like cell death pathway. Redox Biol. 2021;45:102056. doi:10.1016/j.redox.2021.102056
62. Tomitsuka Y, Imaeda H, Ito H, et al. Gene deletion of long-chain acyl-CoA synthetase 4 attenuates xenobiotic chemical-induced lung injury via the suppression of lipid peroxidation. Redox Biol. 2023;66:102850. doi:10.1016/j.redox.2023.102850
63. Jalil A, Pilot T, Bourgeois T, et al. Plasmalogen remodeling modulates macrophage response to cytotoxic oxysterols and atherosclerotic plaque vulnerability. Cell Rep Med. 2025;6(5):102131. doi:10.1016/j.xcrm.2025.102131
64. Gan B. ACSL4, PUFA, and ferroptosis: new arsenal in anti-tumor immunity. Signal Transduct Target Ther. 2022;7(1):128. doi:10.1038/s41392-022-01004-z
65. Chen J, Shi Z, Chen Y, Xiong K, Wang Y, Zhang H. A CoQ10 analog ameliorates cognitive impairment and early brain injury after subarachnoid hemorrhage by regulating ferroptosis and neuroinflammation. Redox Biol. 2025;84:103684. doi:10.1016/j.redox.2025.103684
66. Li W, Liang L, Liu S, Yi H, Zhou Y. FSP1: a key regulator of ferroptosis. Trends Mol Med. 2023;29(9):753–764. doi:10.1016/j.molmed.2023.05.013
67. Mao C, Liu X, Zhang Y, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593(7860):586–590. doi:10.1038/s41586-021-03539-7
68. Kraft VAN, Bezjian CT, Pfeiffer S, et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci. 2020;6(1):41–53. doi:10.1021/acscentsci.9b01063
69. Wang B, Jin Y, Liu J, et al. EP1 activation inhibits doxorubicin-cardiomyocyte ferroptosis via Nrf2. Redox Biol. 2023;65:102825. doi:10.1016/j.redox.2023.102825
70. Xi C, Pang J, Xue W, et al. Transsulfuration pathway activation attenuates oxidative stress and ferroptosis in sickle primary erythroblasts and transgenic mice. Commun Biol. 2025;8(1):15. doi:10.1038/s42003-024-07424-7
71. Da Costa Caiado MJ, Dolga AM, den Dunnen WFA. Iron(ing) out parkinsonisms: the interplay of proteinopathy and ferroptosis in Parkinson’s disease and tau-related parkinsonisms. Redox Biol. 2025;79:103478. doi:10.1016/j.redox.2024.103478
72. Lopes KP, Snijders GJL, Humphrey J, et al. Genetic analysis of the human microglial transcriptome across brain regions, aging and disease pathologies. Nat Genet. 2022;54(1):4–17. doi:10.1038/s41588-021-00976-y
73. Song W, Kothari V, Velly AM, et al. Evaluation of salivary heme oxygenase-1 as a potential biomarker of early Parkinson’s disease. Mov Disord. 2018;33(4):583–591. doi:10.1002/mds.27328
74. Liu S, Yao S, Yang H, Liu S, Wang Y. Autophagy: regulator of cell death. Cell Death Dis. 2023;14(10):648. doi:10.1038/s41419-023-06154-8
75. King KE, Losier TT, Russell RC. Regulation of autophagy enzymes by nutrient signaling. Trends Biochem Sci. 2021;46(8):687–700. doi:10.1016/j.tibs.2021.01.006
76. Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18(4):571–580. doi:10.1038/cdd.2010.191
77. Fu J, Zhao L, Pang Y, et al. Apicoplast biogenesis mediated by ATG8 requires the ATG12-ATG5-ATG16L and SNAP29 complexes in Toxoplasma gondii. Autophagy. 2023;19(4):1258–1276. doi:10.1080/15548627.2022.2123639
78. Tian X, Teng J, Chen J. New insights regarding SNARE proteins in autophagosome-lysosome fusion. Autophagy. 2021;17(10):2680–2688. doi:10.1080/15548627.2020.1823124
79. Tu HY, Yuan BS, Hou XO, et al. α-synuclein suppresses microglial autophagy and promotes neurodegeneration in a mouse model of Parkinson’s disease. Aging Cell. 2021;20(12):e13522. doi:10.1111/acel.13522
80. Gao Y, Wang C, Jiang D, et al. New insights into the interplay between autophagy and oxidative and endoplasmic reticulum stress in neuronal cell death and survival. Front Cell Dev Biol. 2022;10:994037. doi:10.3389/fcell.2022.994037
81. Kuo SH, Tasset I, Cuervo AM, Sulzer D. Misfolded GBA/β-glucocerebrosidase impairs ER-quality control by chaperone-mediated autophagy in Parkinson disease. Autophagy. 2022;18(12):3050–3052. doi:10.1080/15548627.2022.2071383
82. Ballesteros-álvarez J, Andersen JK. mTORC2: the other mTOR in autophagy regulation. Aging Cell. 2021;20(8):e13431. doi:10.1111/acel.13431
83. Bonello F, Hassoun SM, Mouton-Liger F, et al. LRRK2 impairs PINK1/Parkin-dependent mitophagy via its kinase activity: pathologic insights into Parkinson’s disease. Hum Mol Genet. 2019;28(10):1645–1660. doi:10.1093/hmg/ddz004
84. Bellomo G, Paciotti S, Gatticchi L, Parnetti L. The vicious cycle between α-synuclein aggregation and autophagic-lysosomal dysfunction. Mov Disord. 2020;35(1):34–44. doi:10.1002/mds.27895
85. Moors TE, Hoozemans JJ, Ingrassia A, et al. Therapeutic potential of autophagy-enhancing agents in Parkinson’s disease. Mol Neurodegener. 2017;12(1):11. doi:10.1186/s13024-017-0154-3
86. Gao C, Jiang J, Tan Y, Chen S. Microglia in neurodegenerative diseases: mechanism and potential therapeutic targets. Signal Transduct Target Ther. 2023;8(1):359. doi:10.1038/s41392-023-01588-0
87. Candelario-Jalil E, Dijkhuizen RM, Neuroinflammation MT. Stroke, Blood-Brain Barrier Dysfunction, and Imaging Modalities. Stroke. 2022;53(5):1473–1486. doi:10.1161/strokeaha.122.036946
88. Wu A, Zhang J. Neuroinflammation, memory, and depression: new approaches to hippocampal neurogenesis. J Neuroinflammation. 2023;20(1):283. doi:10.1186/s12974-023-02964-x
89. Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol. 2020;20(2):95–112. doi:10.1038/s41577-019-0215-7
90. Zhang W, Xiao D, Mao Q, Xia H. Role of neuroinflammation in neurodegeneration development. Signal Transduct Target Ther. 2023;8(1):267. doi:10.1038/s41392-023-01486-5
91. Cheng J, Zhang R, Xu Z, et al. Early glycolytic reprogramming controls microglial inflammatory activation. J Neuroinflammation. 2021;18(1):129. doi:10.1186/s12974-021-02187-y
92. Rostami J, Mothes T, Kolahdouzan M, et al. Crosstalk between astrocytes and microglia results in increased degradation of α-synuclein and amyloid-β aggregates. J Neuroinflammation. 2021;18(1):124. doi:10.1186/s12974-021-02158-3
93. Backman EA, Gardberg M, Luntamo L, et al. Nigral neuroinflammation and dopaminergic neurons in Parkinson’s disease and atypical Parkinsonisms. Ann Neurol. 2025;97(6):1096–1109. doi:10.1002/ana.27202
94. Miyauchi E, Shimokawa C, Steimle A, Desai MS, Ohno H. The impact of the gut microbiome on extra-intestinal autoimmune diseases. Nat Rev Immunol. 2023;23(1):9–23. doi:10.1038/s41577-022-00727-y
95. Tan AH, Chong CW, Lim SY, et al. Gut microbial ecosystem in parkinson disease: new clinicobiological insights from multi-omics. Ann Neurol. 2021;89(3):546–559. doi:10.1002/ana.25982
96. Douglass AM, Resch JM, Madara JC, et al. Neural basis for fasting activation of the hypothalamic-pituitary-adrenal axis. Nature. 2023;620(7972):154–162. doi:10.1038/s41586-023-06358-0
97. Ye L, Chen H, Wang J, et al. Aflatoxin B(1)-induced liver pyroptosis is mediated by disturbing the gut microbial metabolites: the roles of pipecolic acid and norepinephrine. J Hazard Mater. 2024;474:134822. doi:10.1016/j.jhazmat.2024.134822
98. Kim CS. Roles of diet-associated gut microbial metabolites on brain health: cell-to-cell interactions between gut bacteria and the central nervous system. Adv Nutr. 2024;15(1):100136. doi:10.1016/j.advnut.2023.10.008
99. Chatterjee P, Dassama LMK. Unveiling of a messenger: gut microbes make a neuroactive signal. Cell. 2024;187(12):2903–2904. doi:10.1016/j.cell.2024.05.014
100. Cignarella F, Cantoni C, Ghezzi L, et al. Intermittent fasting confers protection in CNS autoimmunity by altering the gut microbiota. Cell Metab. 2018;27(6):1222–1235.e6. doi:10.1016/j.cmet.2018.05.006
101. Sharon G, Sampson TR, Geschwind DH, Mazmanian SK. The central nervous system and the gut microbiome. Cell. 2016;167(4):915–932. doi:10.1016/j.cell.2016.10.027
102. Chandra R, Sokratian A, Chavez KR, et al. Gut mucosal cells transfer α-synuclein to the vagus nerve. JCI Insight. 2023;8(23). doi:10.1172/jci.insight.172192
103. O’Riordan KJ, Moloney GM, Keane L, Clarke G, Cryan JF. The gut microbiota-immune-brain axis: therapeutic implications. Cell Rep Med. 2025;6(3):101982. doi:10.1016/j.xcrm.2025.101982
104. Osadchiy V, Martin CR, Mayer EA. The gut-brain axis and the microbiome: mechanisms and clinical implications. Clin Gastroenterol Hepatol. 2019;17(2):322–332. doi:10.1016/j.cgh.2018.10.002
105. Sun MF, Shen YQ. Dysbiosis of gut microbiota and microbial metabolites in Parkinson’s disease. Ageing Res Rev. 2018;45:53–61. doi:10.1016/j.arr.2018.04.004
106. Zhang X, Tang B, Guo J. Parkinson’s disease and gut microbiota: from clinical to mechanistic and therapeutic studies. Transl Neurodegener. 2023;12(1):59. doi:10.1186/s40035-023-00392-8
107. Pan W, Zhao J, Wu J, et al. Dimethyl itaconate ameliorates cognitive impairment induced by a high-fat diet via the gut-brain axis in mice. Microbiome. 2023;11(1):30. doi:10.1186/s40168-023-01471-8
108. Wu J, Li CS, Huang WY, et al. Gut microbiota promote the propagation of pathologic α-syn from gut to brain in a gut-originated mouse model of Parkinson’s disease. Brain Behav Immun. 2025;128:152–169. doi:10.1016/j.bbi.2025.04.001
109. Ohno K, Hirayama M. [Ascendance of abnormal α-synuclein fibrils through the vagal nerve in Parkinson’s disease]. Brain Nerve. 2022;74(8):979–984. doi:10.11477/mf.1416202164
110. Chen J, Xiang P, Duro-Castano A, et al. Rapid amyloid-β clearance and cognitive recovery through multivalent modulation of blood–brain barrier transport. Signal Transduct Target Ther. 2025;10(1):331. doi:10.1038/s41392-025-02426-1
111. Han EL, Tang S, Kim D, et al. Peptide-functionalized lipid nanoparticles for targeted systemic mRNA delivery to the brain. Nano Lett. 2025;25(2):800–810. doi:10.1021/acs.nanolett.4c05186
112. Khoury N, Pizzo ME, Discenza CB, et al. Fc-engineered large molecules targeting blood-brain barrier transferrin receptor and CD98hc have distinct central nervous system and peripheral biodistribution. Nat Commun. 2025;16(1):1822. doi:10.1038/s41467-025-57108-x
113. Meng Q, Zhang X, Chen Y, et al. Nanoparticles modified with glucose analogs to enhance the permeability of the blood-brain barrier and their accumulation in the epileptic brain. J Mater Chem B. 2025;13(8):2810–2819. doi:10.1039/d4tb02476a
114. Pan -T-T, Sun -Y-Y, Shi Y-F, et al. Endothelial delivery of simvastatin by LRP1-targeted nanoparticles ameliorates pathogenesis of Alzheimer’s disease in a mouse model. Alzheimer’s Res Ther. 2025;17(1):193. doi:10.1186/s13195-025-01840-5
115. Zhang C, Zhang X, Zhao G. Ferritin nanocage: a versatile nanocarrier utilized in the field of food, nutrition, and medicine. Nanomaterials. 2020;10(9). doi:10.3390/nano10091894
116. Flinterman M, Farzaneh F, Habib N, Malik F, Gäken J, Tavassoli M. Delivery of therapeutic proteins as secretable TAT fusion products. Mol Ther. 2009;17(2):334–342. doi:doi:10.1038/mt.2008.256
117. Haney MJ, Klyachko NL, Zhao Y, et al. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J Control Release. 2015;207:18–30. doi:10.1016/j.jconrel.2015.03.033
118. Ren J, Jepson CE, Nealy SL, et al. Site-oriented conjugation of poly(2-methacryloyloxyethyl phosphorylcholine) for enhanced brain delivery of antibody. Front Cell Dev Biol. 2023;11:1214118. doi:10.3389/fcell.2023.1214118
119. Perets N, hertz S, London M, Offen D. Intranasal administration of exosomes derived from mesenchymal stem cells ameliorates autistic-like behaviors of BTBR mice. Mol Autism. 2018;9(1):57. doi:10.1186/s13229-018-0240-6
120. Jiang W, Li Q, Zhang R, et al. Chiral metal-organic frameworks incorporating nanozymes as neuroinflammation inhibitors for managing Parkinson’s disease. Nat Commun. 2023;14(1):8137. doi:10.1038/s41467-023-43870-3
121. He M, Zhang X, Ran X, et al. Black phosphorus nanosheets protect neurons by degrading aggregative α-syn and clearing ROS in Parkinson’s disease. Adv Mater. 2024;36(30):e2404576. doi:10.1002/adma.202404576
122. García-Pardo J, Novio F, Nador F, et al. Bioinspired theranostic coordination polymer nanoparticles for intranasal dopamine replacement in Parkinson’s disease. ACS Nano. 2021;15(5):8592–8609. doi:10.1021/acsnano.1c00453
123. Liu Y-Q, Mao Y, Xu E, et al. Nanozyme scavenging ROS for prevention of pathologic α-synuclein transmission in Parkinson’s disease. Nano Today. 2021;36:101027. doi:doi:10.1016/j.nantod.2020.101027
124. Wang Z, Wen H, Zheng C, et al. Synergistic Co–Cu dual-atom nanozyme with promoted catalase-like activity for Parkinson’s disease treatment. ACS Appl Mater Interfaces. 2025;17(1):583–593. doi:10.1021/acsami.4c17416
125. Liu Y, Liu Y, Shi P, et al. Single-atom nanozyme liposome-integrated microneedles for in situ drug delivery and anti-inflammatory therapy in Parkinson’s disease. J Nanobiotechnol. 2024;22(1):643. doi:10.1186/s12951-024-02924-4
126. Ni H, Zhou H, Liang X, et al. Reactive oxygen species-responsive nanoparticle delivery of small interfering ribonucleic acid targeting olfactory receptor 2 for atherosclerosis theranostics. ACS Nano. 2024;18(34):23599–23614. doi:10.1021/acsnano.4c07988
127. Chen H-X, Liang F-C, Gu P, et al. Exosomes derived from mesenchymal stem cells repair a Parkinson’s disease model by inducing autophagy. Cell Death Dis. 2020;11(4):288. doi:10.1038/s41419-020-2473-5
128. Shi X, Yang G, Liu MY, Yuan MT, Wang D, Wang XF. Exosomes derived from human dental pulp stem cells increase flap survival with ischemia-reperfusion injuries. Regener Med. 2023;18(4):313–327. doi:10.2217/rme-2022-0206
129. Menozzi E, Schapira AHV, Borghammer P. The gut-brain axis in Parkinson disease: emerging concepts and therapeutic implications. Mov Disord Clin Pract. 2025;12(7):904–916. doi:10.1002/mdc3.70029
130. Loh JS, Mak WQ, Tan LKS, et al. Microbiota-gut-brain axis and its therapeutic applications in neurodegenerative diseases. Signal Transduct Target Ther. 2024;9(1):37. doi:10.1038/s41392-024-01743-1
131. Forero-Rodríguez LJ, Josephs-Spaulding J, Flor S, Pinzón A, Kaleta C. Parkinson’s disease and the metal-microbiome-gut-brain axis: a systems toxicology approach. Antioxidants. 2021;11(1). doi:10.3390/antiox11010071
132. Kabra A, Puri R, Goyal P, Arora V. Chapter 12 - Novel drug delivery system in the management of Parkinson’s disease. In: Chawla PA, Loebenberg R, Dua K, Parikh V, Chawla V, editors. Novel Drug Delivery Systems in the Management of CNS Disorders. Academic Press; 2025:185–198.
133. Vrijsen S, Houdou M, Cascalho A, Eggermont J, Vangheluwe P. Polyamines in Parkinson’s disease: balancing between neurotoxicity and neuroprotection. Annu Rev Biochem. 2023;92:435–464. doi:10.1146/annurev-biochem-071322-021330
134. Dawson TM, Dawson VL. Neuroprotective and neurorestorative strategies for Parkinson’s disease. Nat Neurosci. 2002;5(Suppl):1058–1061. doi:10.1038/nn941
135. Han L, Jiang C. Evolution of blood-brain barrier in brain diseases and related systemic nanoscale brain-targeting drug delivery strategies. Acta Pharm Sin B. 2021;11(8):2306–2325. doi:10.1016/j.apsb.2020.11.023
136. Nonnekes J, Timmer MH, de Vries NM, Rascol O, Helmich RC, Bloem BR. Unmasking levodopa resistance in Parkinson’s disease. Mov Disord. 2016;31(11):1602–1609. doi:10.1002/mds.26712
137. Mauro-Lizcano M, Esteban-Martínez L, Seco E, et al. New method to assess mitophagy flux by flow cytometry. Autophagy. 2015;11(5):833–843. doi:10.1080/15548627.2015.1034403
138. Singh AP, Biswas A, Shukla A, Maiti P. Targeted therapy in chronic diseases using nanomaterial-based drug delivery vehicles. Signal Transduct Target Ther. 2019;4:33. doi:10.1038/s41392-019-0068-3
139. Ng J, Barral S, Waddington SN, Kurian MA. Gene therapy for dopamine dyshomeostasis: from Parkinson’s to primary neurotransmitter diseases. Mov Disord. 2023;38(6):924–936. doi:10.1002/mds.29416
140. Zheng Q, Gao Y, Han M, et al. Inhibiting immune crosstalk by modulation of the intracellular function and extracellular environment of diseased microglia to boost Parkinson’s disease therapy. ACS Nano. 2025;19(20):19177–19197. doi:10.1021/acsnano.5c01068
141. Henrich MT, Oertel WH, Surmeier DJ, Geibl FF. Mitochondrial dysfunction in Parkinson’s disease - a key disease hallmark with therapeutic potential. Mol Neurodegener. 2023;18(1):83. doi:10.1186/s13024-023-00676-7
142. Wu X, Ma F, Pan BB, et al. Tailoring a nanochaperone to regulate α-synuclein assembly. Angew Chem Int Ed Engl. 2022;61(19):e202200192. doi:10.1002/anie.202200192
143. Cheng G, Liu Z, Yan Z, et al. Minocycline nanoplatform penetrates the BBB and enables the targeted treatment of Parkinson’s disease with cognitive impairment. J Control Release. 2025;377:591–605. doi:10.1016/j.jconrel.2024.11.066
144. Vestuto V, Conte M, Vietri M, et al. Multiomic profiling and neuroprotective bioactivity of salvia hairy root-derived extracellular vesicles in a cellular model of Parkinson’s disease. Int J Nanomed. 2024;19:9373–9393. doi:10.2147/ijn.S479959
145. Yuan J, Xu L, Han Y, et al. Boosting neurite outgrowth and anti-oxidative stress for treatment of Parkinson’s disease by biomimetic ultrasmall nanoparticles. Sustainable Mater Technol. 2024;39:e00807. doi:doi:10.1016/j.susmat.2023.e00807
146. Ma X, Hao J, Wu J, Li Y, Cai X, Zheng Y. Prussian blue nanozyme as a pyroptosis inhibitor alleviates neurodegeneration. Adv Mater. 2022;34(15):e2106723. doi:10.1002/adma.202106723
147. Lei L, Yuan J, Dai Z, et al. Targeting the Labile iron pool with engineered DFO nanosheets to inhibit ferroptosis for Parkinson’s disease therapy. Adv Mater. 2024;36(41):e2409329. doi:10.1002/adma.202409329
148. Lu H, Zhang B, Ge M, et al. Microglial membrane-coated nanoparticles for ACSL4-siRNA delivery in Parkinson’s disease. Chem Eng J. 2025;509:161251. doi:doi:10.1016/j.cej.2025.161251
149. Zhai L, Gao Y, Yang H, et al. A ROS-responsive nanoparticle for nuclear gene delivery and autophagy restoration in Parkinson’s disease therapy. Biomaterials. 2025;321:123345. doi:10.1016/j.biomaterials.2025.123345
150. Vriend J, Reiter RJ. Melatonin and ubiquitin: what’s the connection? Cell Mol Life Sci. 2014;71(18):3409–3418. doi:10.1007/s00018-014-1659-3
151. Li B, Bai Y, Yion C, et al. Single-atom nanocatalytic therapy for suppression of neuroinflammation by inducing autophagy of abnormal mitochondria. ACS Nano. 2023;17(8):7511–7529. doi:10.1021/acsnano.2c12614
152. Liu Z, Xiang S, Chen B, et al. Parkinson disease -targeted nanocapsules for synergistic treatment: combining dopamine replacement and neuroinflammation mitigation. Adv Sci. 2024;11(46):e2404717. doi:10.1002/advs.202404717
153. Li Q, Ding X, Chang Z, et al. Metal-organic framework based nanozyme system for NLRP3 inflammasome-mediated neuroinflammatory regulation in Parkinson’s disease. Adv Healthc Mater. 2024;13(10):e2303454. doi:10.1002/adhm.202303454
154. Zhang C, Shao W, Yuan H, et al. Engineered extracellular vesicle-based nanoformulations that coordinate neuroinflammation and immune homeostasis, enhancing Parkinson’s disease therapy. ACS Nano. 2024;18(34):23014–23031. doi:10.1021/acsnano.4c04674
155. Cui W, Guo Z, Chen X, et al. Targeting modulation of intestinal flora through oral route by an antimicrobial nucleic acid-loaded exosome-like nanovesicles to improve Parkinson’s disease. Sci Bull. 2024;69(24):3925–3935. doi:10.1016/j.scib.2024.10.027
156. Pan H, Sun T, Cui M, et al. Light-sensitive lactococcus lactis for microbe-gut-brain axis regulating via upconversion optogenetic micro-nano system. ACS Nano. 2022;16(4):6049–6063. doi:10.1021/acsnano.1c11536
157. Guo M, Yang C, Li B, et al. Bionic dormant body of timed wake-up for bacteriotherapy in vivo. ACS Nano. 2022;16(1):823–836. doi:10.1021/acsnano.1c08377
158. Lu H, Wei J, Liu K, et al. Radical-scavenging and subchondral bone-regenerating nanomedicine for osteoarthritis treatment. ACS Nano. 2023;17(6):6131–6146. doi:10.1021/acsnano.3c01789
159. Xie A, Cheng G, Wu J, et al. Highly BBB-permeable nanomedicine reverses neuroapoptosis and neuroinflammation to treat Alzheimer’s disease. Biomaterials. 2025;312:122749. doi:10.1016/j.biomaterials.2024.122749
160. Zhang J, Han S, Zhao Z, et al. Ultrasmall black phosphorus quantum dots with robust antioxidative properties for acute kidney and liver injury therapy. Small. 2025;21(1):e2407543. doi:10.1002/smll.202407543
161. Tan S, Li S, Tang C, et al. A regenerable and reducing false-positive fluorescent switch for detection of β-amyloid (1-42) oligomers. Talanta. 2022;246:123461. doi:10.1016/j.talanta.2022.123461
162. Tan Y, Sgobio C, Arzberger T, et al. Loss of fragile X mental retardation protein precedes Lewy pathology in Parkinson’s disease. Acta Neuropathol. 2020;139(2):319–345. doi:10.1007/s00401-019-02099-5
163. Zhang D, Li S, Hou L, et al. Microglial activation contributes to cognitive impairments in rotenone-induced mouse Parkinson’s disease model. J Neuroinflammation. 2021;18(1):4. doi:10.1186/s12974-020-02065-z
164. Oda W, Fujita Y, Baba K, Mochizuki H, Niwa H, Yamashita T. Inhibition of repulsive guidance molecule-a protects dopaminergic neurons in a mouse model of Parkinson’s disease. Cell Death Dis. 2021;12(2):181. doi:10.1038/s41419-021-03469-2
165. van Niel G, G D, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19(4):213–228. doi:10.1038/nrm.2017.125
166. Larrañaga-SanMiguel A, Bengoa-Vergniory N, Flores-Romero H. Crosstalk between mitochondria-ER contact sites and the apoptotic machinery as a novel health meter. Trends Cell Biol. 2025;35(1):33–45. doi:10.1016/j.tcb.2024.08.007
167. García-Revilla J, Ruiz R, Espinosa-Oliva AM, et al. Dopaminergic neurons lacking Caspase-3 avoid apoptosis but undergo necrosis after MPTP treatment inducing a Galectin-3-dependent selective microglial phagocytic response. Cell Death Dis. 2024;15(8):625. doi:10.1038/s41419-024-07014-9
168. Fu J, Wu H. Structural mechanisms of NLRP3 inflammasome assembly and activation. Annu Rev Immunol. 2023;41:301–316. doi:10.1146/annurev-immunol-081022-021207
169. Huang Y, Xu W, Zhou R. NLRP3 inflammasome activation and cell death. Cell Mol Immunol. 2021;18(9):2114–2127. doi:10.1038/s41423-021-00740-6
170. Lin X, Dong Q, Chang Y, Shi P, Zhang S. Transition-metal-based nanozymes for biosensing and catalytic tumor therapy. Anal Bioanal Chem. 2024;416(27):5933–5948. doi:10.1007/s00216-024-05345-2
171. Wu J, Wang X, Wang Q, et al. Nanomaterials with enzyme-like characteristics (nanozymes): next-generation artificial enzymes (II). Chem Soc Rev. 2019;48(4):1004–1076. doi:10.1039/c8cs00457a
172. Huang Y, Ren J, Qu X. Nanozymes: classification, catalytic mechanisms, activity regulation, and applications. Chem Rev. 2019;119(6):4357–4412. doi:10.1021/acs.chemrev.8b00672
173. Doveri L, Dacarro G, Fernandez YAD, et al. Prussian Blue nanoparticles: an FDA-approved substance that may quickly degrade at physiological pH. Colloids Surf B Biointerfaces. 2023;227:113373. doi:10.1016/j.colsurfb.2023.113373
174. Mahoney-Sánchez L, Bouchaoui H, Ayton S, Devos D, Duce JA, Devedjian JC. Ferroptosis and its potential role in the physiopathology of Parkinson’s disease. Prog Neurobiol. 2021;196:101890. doi:10.1016/j.pneurobio.2020.101890
175. Liang X, Liu Y, Wen K, Jiang W, Li Q. Immobilized enzymes in inorganic hybrid nanoflowers for biocatalytic and biosensing applications. J Mater Chem B. 2021;9(37):7597–7607. doi:10.1039/d1tb01476e
176. Brodie MJ. Tiagabine pharmacology in profile. Epilepsia. 1995;36(Suppl 6):S7–s9. doi:10.1111/j.1528-1157.1995.tb06015.x
177. Koochaki Z, Higson SPJ, Mutlu M, Vadgam PM. The diffusion limited oxidase-based glucose enzyme electrode: relation between covering membrane permeability and substrate response. J Membr Sci. 1993;76(2):261–268. doi:doi:10.1016/0376-7388(93)85223-J
178. Liu W, Zhang Y, Wei G, et al. Integrated cascade nanozymes with antisenescence activities for atherosclerosis therapy. Angew Chem Int Ed Engl. 2023;62(33):e202304465. doi:10.1002/anie.202304465
179. Wang X, Wang Z-D, Hu S-Y, Ding X, Hu Y-J. Regulator of oxidative balance: research progress of nanozymes in ROS-related diseases. Mater Today Chem. 2025;44:102540. doi:doi:10.1016/j.mtchem.2025.102540
180. Feng S, Tang D, Wang Y, et al. The mechanism of ferroptosis and its related diseases. Mol Biomed. 2023;4(1):33. doi:10.1186/s43556-023-00142-2
181. Zhou X, Zhao R, Lv M, et al. ACSL4 promotes microglia-mediated neuroinflammation by regulating lipid metabolism and VGLL4 expression. Brain Behav Immun. 2023;109:331–343. doi:10.1016/j.bbi.2023.02.012
182. Zhu L, Yuan Y, Yuan L, et al. Activation of TFEB-mediated autophagy by trehalose attenuates mitochondrial dysfunction in cisplatin-induced acute kidney injury. Theranostics. 2020;10(13):5829–5844. doi:10.7150/thno.44051
183. Kumar A, Tamjar J, Waddell AD, et al. Structure of PINK1 and mechanisms of Parkinson’s disease-associated mutations. Elife. 2017;6. doi:10.7554/eLife.29985
184. Song P, Peng W, Sauve V, et al. Parkinson’s disease-linked parkin mutation disrupts recycling of synaptic vesicles in human dopaminergic neurons. Neuron. 2023;111(23):3775–3788.e7. doi:10.1016/j.neuron.2023.08.018
185. Lee BH, Hille B, Koh DS. Serotonin modulates melatonin synthesis as an autocrine neurotransmitter in the pineal gland. Proc Natl Acad Sci U S A. 2021;118(43). doi:10.1073/pnas.2113852118
186. Yao Y, Liu X, Niu X, Li Y, Han L. Lycopene regulates macrophage immune response through the autophagy pathway mediated by RIPK1. J Agric Food Chem. 2024;72(26):14747–14759. doi:10.1021/acs.jafc.4c02531
187. Xia X, Li H, Xu X, Zhao G, Du M. Facilitating pro-survival mitophagy for alleviating Parkinson’s disease via sequence-targeted lycopene nanodots. ACS Nano. 2023;17(18):17979–17995. doi:10.1021/acsnano.3c04308
188. Shi P, Gustafson JA, MacKay JA. Genetically engineered nanocarriers for drug delivery. Int J Nanomed. 2014;9:1617–1626. doi:10.2147/ijn.S53886
189. Zhang Z, Li J, Wang Y, et al. Angiopep-2 conjugated biomimetic nano-delivery system loaded with resveratrol for the treatment of methamphetamine addiction. Int J Pharm. 2024;663:124552. doi:10.1016/j.ijpharm.2024.124552
190. Jiang L, Zhang X, Wang S, et al. Functional monomers equipped microgel system for managing Parkinson’s disease by intervening chemokine axis-mediated nerve cell communications. Adv Sci. 2025;12(7):e2410070. doi:10.1002/advs.202410070
191. Mridha AR, Wree A, Robertson AAB, et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol. 2017;66(5):1037–1046. doi:10.1016/j.jhep.2017.01.022
192. Masoudifar R, Pouyanfar N, Liu D, et al. Surface engineered metal-organic frameworks as active targeting nanomedicines for mono- and multi-therapy. Appl Mater Today. 2022;29:101646. doi:doi:10.1016/j.apmt.2022.101646
193. Harms AS, Ferreira SA, Romero-Ramos M. Periphery and brain, innate and adaptive immunity in Parkinson’s disease. Acta Neuropathol. 2021;141(4):527–545. doi:10.1007/s00401-021-02268-5
194. Zhang ZX, Zhou YJ, Gu P, et al. Exosomes derived from human umbilical cord mesenchymal stem cells alleviate Parkinson’s disease and neuronal damage through inhibition of microglia. Neural Regen Res. 2023;18(10):2291–2300. doi:10.4103/1673-5374.368300
195. Lu MC, Ji JA, Jiang ZY, You QD. The Keap1-Nrf2-ARE pathway as a potential preventive and therapeutic target: an update. Med Res Rev. 2016;36(5):924–963. doi:10.1002/med.21396
196. Zeng H, Wang L, Zhang J, et al. Activated PKB/GSK-3β synergizes with PKC-δ signaling in attenuating myocardial ischemia/reperfusion injury via potentiation of NRF2 activity: therapeutic efficacy of dihydrotanshinone-I. Acta Pharm Sin B. 2021;11(1):71–88. doi:10.1016/j.apsb.2020.09.006
197. Liang D, Liu H, Jin R, et al. Escherichia coli triggers α-synuclein pathology in the LRRK2 transgenic mouse model of PD. Gut Microbes. 2023;15(2):2276296. doi:10.1080/19490976.2023.2276296
198. Cirstea MS, Sundvick K, Golz E, et al. The gut mycobiome in Parkinson’s disease. J Parkinsons Dis. 2021;11(1):153–158. doi:10.3233/jpd-202237
199. Ma X, Liu Q, Yang G. The multifaceted roles of Akkermansia muciniphila in neurological disorders. Trends Neurosci. 2025;48(6):403–415. doi:10.1016/j.tins.2025.04.004
200. Yang Z, Shi L, Wang Y, Zhou D, Zhang C, Lin Y. Unveiling the potential of tetrahedral DNA frameworks in clinical medicine: mechanisms, advances, and future perspectives. Small. 2025;21(5):e2410162. doi:10.1002/smll.202410162
201. Lin Y, Li Q, Wang L, et al. Advances in regenerative medicine applications of tetrahedral framework nucleic acid-based nanomaterials: an expert consensus recommendation. Int J Oral Sci. 2022;14(1):51. doi:10.1038/s41368-022-00199-9
202. Fan Q, Sun B, Chao J. Advancements in engineering tetrahedral framework nucleic acids for biomedical innovations. Small Methods. 2024;e2401360. doi:10.1002/smtd.202401360
203. Chia N, Lee SY, Tong Y. Optogenetic tools for microbial synthetic biology. Biotechnol Adv. 2022;59:107953. doi:10.1016/j.biotechadv.2022.107953
204. Chen S, Weitemier AZ, Zeng X, et al. Near-infrared deep brain stimulation via upconversion nanoparticle-mediated optogenetics. Science. 2018;359(6376):679–684. doi:10.1126/science.aaq1144
205. Liu X, Chen H, Wang Y, et al. Near-infrared manipulation of multiple neuronal populations via trichromatic upconversion. Nat Commun. 2021;12(1):5662. doi:10.1038/s41467-021-25993-7
206. Watling CZ, Kelly RK, Murphy N, et al. Prospective analysis reveals associations between carbohydrate intakes, genetic predictors of short-chain fatty acid synthesis, and colorectal cancer risk. Cancer Res. 2023;83(12):2066–2076. doi:10.1158/0008-5472.Can-22-3755
207. Kumar A, Toghyani M, Kheravii SK, et al. Potential of blended organic acids to improve performance and health of broilers infected with necrotic enteritis. Anim Nutr. 2021;7(2):440–449. doi:10.1016/j.aninu.2020.11.006
208. Shashni B, Tajika Y, Nagasaki Y. Design of enzyme-responsive short-chain fatty acid-based self-assembling drug for alleviation of type 2 diabetes mellitus. Biomaterials. 2021;275:120877. doi:10.1016/j.biomaterials.2021.120877
209. Kar KPP. Recalibrating the four hour target. BMJ. 2019;367:l5878. doi:10.1136/bmj.l5878
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Exercise-Induced Autophagy Ameliorates Motor Symptoms Progressivity in Parkinson’s Disease Through Alpha-Synuclein Degradation: A Review
Goenawan H, Kiasati S, Sylviana N, Megantara I, Lesmana R
Neuropsychiatric Disease and Treatment 2023, 19:1253-1262
Published Date: 25 May 2023
Programmed Cell Death in Liver Fibrosis
Gao R, Tang H, Mao J
Journal of Inflammation Research 2023, 16:3897-3910
Published Date: 1 September 2023
Quercetin is a Potential Therapy for Rheumatoid Arthritis via Targeting Caspase-8 Through Ferroptosis and Pyroptosis
Zheng Q, Wang D, Lin R, Chen Y, Xu Z, Xu W
Journal of Inflammation Research 2023, 16:5729-5754
Published Date: 1 December 2023
Autophagy, Pyroptosis and Ferroptosis are Rising Stars in the Pathogenesis of Diabetic Nephropathy
Li X, Gao L, Li X, Xia J, Pan Y, Bai C
Diabetes, Metabolic Syndrome and Obesity 2024, 17:1289-1299
Published Date: 13 March 2024
Programmed Cell Death in Rheumatoid Arthritis
Tong L, Qiu J, Xu Y, Lian S, Xu Y, Wu X
Journal of Inflammation Research 2025, 18:2377-2393
Published Date: 18 February 2025