Back to Journals » Neuropsychiatric Disease and Treatment » Volume 19
Exercise-Induced Autophagy Ameliorates Motor Symptoms Progressivity in Parkinson’s Disease Through Alpha-Synuclein Degradation: A Review
Authors Goenawan H ![]() , Kiasati S
, Kiasati S ![]() , Sylviana N
, Sylviana N ![]() , Megantara I
, Megantara I ![]() , Lesmana R
, Lesmana R
Received 15 December 2022
Accepted for publication 20 April 2023
Published 25 May 2023 Volume 2023:19 Pages 1253—1262
DOI https://doi.org/10.2147/NDT.S401416
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Hanna Goenawan,1 Shabrina Kiasati,2 Nova Sylviana,1 Imam Megantara,1 Ronny Lesmana1
1Department of Biomedical Sciences, Faculty of Medicine, Universitas Padjadjaran, Bandung, Indonesia; 2Undergraduate Program, Faculty of Medicine, Universitas Padjadjaran, Bandung, Indonesia
Correspondence: Hanna Goenawan, Department of Biomedical Sciences, Faculty of Medicine, Universitas Padjadjaran, Jalan Raya Bandung-Sumedang KM 21, Bandung, Indonesia, Email [email protected]
Abstract: This study reviews the molecular mechanism of exercise-induced autophagy/mitophagy and its possible mechanism in delaying motor symptoms progressivity in Parkinson’s disease (PD). Relevant articles obtained from PubMed and EBSCOhost were reviewed. After analyzing the articles, it was found that autophagy can be induced by exercise and can possibly be activated through the AMPK-ULK1 pathway. Mitophagy can also be induced by exercise and can possibly be activated through PINK1/Parkin pathway and AMPK-dependent pathway. Moreover, exercise-induced autophagy can decrease the accumulation of toxic α-synuclein aggregates in PD and therefore can delay motor symptoms progressivity.
Keywords: exercise, autophagy, Parkinson’s disease, alpha-synuclein
Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disease after Alzheimer’s disease.1 Over the years, the number of people affected by PD is growing, which is correlated with the increase in life expectancy.2 PD is related to the loss of dopaminergic neurons of the substantia nigra pars compacta (SNc), which causes progressive motor symptoms.3 The cardinal motor manifestations are bradykinesia, rigidity and rest tremor.4 Progressive and worsening mobility impairment in PD has been associated with a decline in the quality of life of a patient.5
The pathological hallmark of PD is the presence of Lewy bodies, which mainly consist of α-synuclein aggregates.6 α-Synuclein aggregates are toxic and could lead to dopaminergic neuron death.6,7 α-Synuclein aggregates may also self-propagate to healthy neurons, resulting in the progression of neuronal loss.6,8 Upregulation of autophagy, a cellular system that transports unneeded components to lysosome for degradation, could be neuroprotective in PD by withholding α-synuclein toxicity.9,10 There are three types of autophagy described in mammals: microautophagy, macroautophagy, and chaperone-mediated autophagy.11 Of the three, macroautophagy (hereafter referred to as autophagy) is best understood.12 Furthermore, there is a selective macroautophagy that has specific cytosolic components for degradation such as mitophagy that targeted mitochondrial removal.11
Presently, levodopa and other pharmacological dopamine substitution therapies are the mainstays of PD symptomatic treatment.3 However, long-term use of it is related to several side effects and wearing-off phenomenon.13,14 Therefore, studies are being done to discover new ways to delay PD progression and improve symptoms using a mechanism different from conventional PD treatment. Among the studied mechanisms are autophagy and mitophagy induction. Autophagy and mitophagy can be induced in several ways; however, this review will only focus on discussing the activation of autophagy and mitophagy via exercise.
Exercise is known to provide a neuroprotective effect in multiple ways such as inducing autophagy, causing neuronal plasticity, employing neurogenesis and decreasing neurodegeneration.15 Studies have shown that exercise improved motor functions in patients with PD compared to sedentary patients with PD and has been proposed to modify the progressive course of PD.13,16 Thus, to understand more about the effect of exercise-induced autophagy in delaying motor symptoms progressivity in PD, a literature review was conducted. This review aims to elucidate the molecular mechanism of exercise-induced autophagy and/or mitophagy and its correlation with PD pathology.
Methods
Works of literature were searched via advanced search in PubMed and EBSCOhost with the combined keywords: “parkinson”, “exercise”, “autophagy”, “mitophagy”, “autophagosome”, “synuclein”, and “synuclein clearance”. The limits apply to the search were original article in vivo studies and those published between 2010 and 2023.
Results and Discussion
PubMed and EBSCOhost were searched for studies published between 2010 and 2023 with the combination of keywords mentioned above. The works of literature were chosen based on the PRISMA 2020 flow diagram as stated in Figure 1. Ten literatures were included.
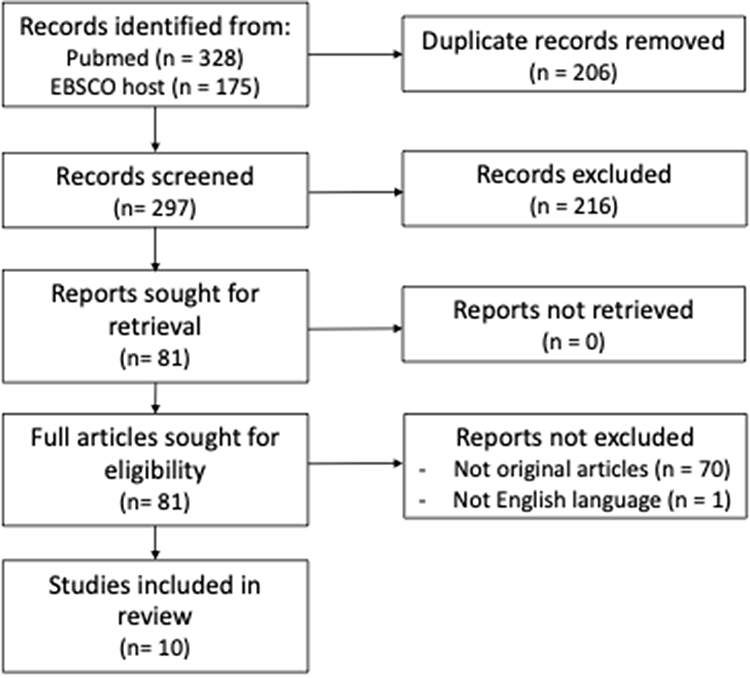 |
Figure 1 PRISMA 2020 flow diagram of the literature search of this review. Notes: PRISMA figure adapted from Haddaway NR, Page MJ, Pritchard CC, McGuinness LA. PRISMA2020: An R package and Shiny app for producing PRISMA 2020-compliant flow diagrams, with interactivity for optimised digital transparency and Open Synthesis. Campbell Systematic Reviews. 2022;18:e1230.17 doi: 10.1002/cl2.1230 |
Molecular Mechanism of Exercise-Induced Autophagy
Exercise resulted in an imbalance of energy demand to energy supply, an increase in the AMP-to-ATP ratio, and an increase in reactive oxygen species (ROS) production.18 During exercise, ATP consumption increases, which causes the accumulation of intracellular AMP; this then leads to an increase in the AMP-to-ATP ratio, which would then activate AMP-activated protein kinase (AMPK).18–20 AMPK regulates various metabolic pathways, one of which is autophagy. Schwalm et al21 found that in human skeletal muscle, exercise-induced autophagy depends on AMPK activation. Similar findings were seen in the studies by Møller et al22 and Fritzen et al,19 which gave insight that AMPK activation in skeletal muscle would then activate Unc-51-like kinase-1 (ULK1) through a site-specific phosphorylation.19,22 ULK1 is a protein kinase that has a significant role in initiating autophagy.22 Another important role of AMPK is its ability to inhibit the mammalian target of the rapamycin complex 1 (mTORC1) pathway through phosphorylation on tuberous sclerosis complex 2 (TSC2) and the mTOR binding partner raptor.23 MTORC1 inhibition is important to initiate the autophagy process.
The next step after the initiation of autophagy is the formation of phagophore, a cytosolic double membrane that will mature into autophagosome. Several in vitro studies showed that upon ULK1 activation, the VPS34 complex is activated. VPS34 complex is a class III phosphoinositide 3-kinase (PI3K) in mammals and can phosphorylate phosphatidylinositol leading to phosphatidylinositol 3-phosphate (PI3P) generation.24–26 PI3P promotes phagophore expansion through WD-repeat domain phosphoinositide-interacting protein 2 (WIPI2).27 The final step of autophagosome maturation is microtubule-associated protein 1A/1b-light chain 3 (LC3) lipidation, which refers to the conjugation of phosphatidylethanolamine (PE) to LC3-I forming LC3-PE/LC3-II.28 Hence, the LC3-II protein is known as a key autophagic marker; it is often measured in studies as indicators of autophagy activity.18,28 Other autophagic markers commonly measured as indicators of autophagy activity are p62, Beclin1, and LAMP2.29,30 Autophagosome will then fuse with lysosome, which results in autophagosome’s cargo degradation by hydrolase enzymes of lysosome.
Molecular Mechanism of Exercise-Induced Mitophagy
As the powerhouse of cell, mitochondria play a crucial role in the metabolism process of cell. Mitochondrial homeostasis and function need to be controlled at all times, therefore there is a pathway called mitochondrial quality control (MQC). One of the critical pathways of MQC is the phosphatase and tensin homolog-induced putative kinase 1 (PINK1) and Parkin pathway.31 In healthy mitochondria, PINK1 is cleared from the outer mitochondrial membrane (OMM) by importing it to the inner mitochondrial membrane before it is cleaved and degraded. In dysfunctional mitochondria, PINK1 accumulates in OMM due to its inability to translocate to the inner membrane.32 Accumulation of PINK1 will then recruit Parkin and initiate the cascade of mitophagy.31 Exercise could induce mitophagy with the activation of PINK1/Parkin pathway stated above or independently from PINK1/Parkin pathway.
Exercise induces the secretion of irisin, a peptide that is cleaved from fibronectin type III domain-containing protein 5 (FNDC-5). Recently, it has been reported that there is an increase of irisin in the brain tissue of a mice after it was given exercise.33 Irisin and PINK1/Parkin pathway correlate positively as shown by the study of He et al34 and Li et al.35 Therefore, mitophagy is known to be inducible by exercise through the FNDC5/irisin-PINK1/Parkin pathway. However, other study has shown that mitophagy can be induced by exercise independently from the PINK1/Parkin pathway. Seabright et al36 and Drake et al37 reported mitophagy regulation via the AMPK-dependent pathway. This pathway is similar to the activation of autophagy mentioned above. The rise of AMP-to-ATP ratio inside the mitochondria will cause mitochondrial energetic stress which then activates mitoAMPK and phosphorylates ULK1 causing the activation of mitophagy cascade.37
The Relationship Between Autophagy/Mitophagy and PD Pathology
The progression of PD symptoms is suspected due to the progression of α-synuclein pathology in SNc. α-Synuclein is a small, 140-amino acid presynaptic protein. Physiologically, α-synuclein has a structure of an unfolded monomer or a helix form, with the function of regulating neurotransmitter release in the synaptic terminals.38 Due to disease-associated mechanisms, both isoforms can misfold and aggregate forming a higher-molecular-weight protofibrils, which can polymerize forming an amyloid fibril found in Lewy bodies of patients with PD.6 α-Synuclein aggregates are toxic and can lead to dopaminergic neuron death.6,7
α-Synuclein aggregates neurotoxicity in PD occurs intracellularly and extracellularly. Intracellularly, α-synuclein aggregates disrupt membrane integrity by creating a pore in the lipid bilayer of the cell membrane which causes in an influx of ions from extracellular space that could lead to cell death.7 Extracellularly, α-synuclein aggregates are known to activate microglial cells that could then result in an abundant ROS production causing oxidative stress and eventually neuronal loss. Another thing α-synuclein is believed to be able to do extracellularly is propagate between cells. This implies that the neurotoxicity brought on by α-synuclein aggregate can propagate from one neuron to another, explaining the progression of PD.7 Furthermore, α-synuclein aggregates can cause disturbance in synaptic transport, causing further dopamine homeostasis disturbance.39
Another main cause of neuronal death in PD is mitochondrial dysfunction, which occurs both in sporadic PD and familial PD. In sporadic PD, mitochondrial dysfunction occurs due to the overproduction of ROS as well as the disruptions in the mitochondrial complex activity.7 In familial PD, mitochondrial dysfunction happens early in the onset of the disease since most of the proteins encoded by PD-related genes are correlated with mitochondrial function.40
Therefore, autophagy as a system involved in dysfunctional protein degradation can be beneficial in PD by degrading α-synuclein aggregates. With fewer α-synuclein aggregates, there might be a potential PD motor symptoms’ progression delay. In vivo and in vitro studies have shown that autophagy activation contributed to the decrease in α-synuclein aggregates accumulation.8,41,42 Gao et al8 demonstrated the improvements of α-synuclein aggregate accumulation in human neural cells treated with α-synuclein fibrils after the intervention. Cells were given small-molecule AMPK activators, and afterward, there were significantly reduced α-synuclein inclusions levels, associated with the changes of key autophagic markers indicating autophagy involvement in the clearance process induced via AMPK activation.8 By contrast, inhibition of autophagy halted α-synuclein aggregate clearance. This was shown from the studies of Gao et al8 and Masaracchia et al,42 which reported that α-synuclein-treated neural cells that were given autophagy inhibitor substances, such as bafilomycin or chloroquine, resulted in the inhibition of α-synuclein aggregate degradation resulting in its accumulation intracellularly.
Besides autophagy, the activation of mitophagy may also be beneficial in delaying PD progression considering that the degradation of dysfunctional mitochondria will reduce the burden of mitochondrial stress inside SNc cells, thus decreasing the accumulation of ROS and increasing the turnover of mitochondrial cells to support mitochondrial homeostasis. Ivankovic et al43 reported that after the induction of PINK1/Parkin mediated mitophagy in human neuroblastoma SH-SY5Y, transcription factors Nrf2 and TFEB were activated. Nrf2 and TFEB are known to be involved in mitochondrial biogenesis, lysosomal biogenesis, and exert antioxidant effects. Therefore, the induction of mitophagy in PD could possibly protect mitochondria from further oxidative stress damage through its antioxidant effects, and replace dysfunctional mitochondria with newer functional mitochondria through its mitochondrial biogenesis ability.43
Because of its effects, autophagy and mitophagy activation becomes a promising therapeutic potential in PD treatment by decreasing α-synuclein aggregates and improving mitochondrial homeostasis. Studies have shown that exercise can induce autophagy/mitophagy in the brain, which leads to neuroprotective effects.44–47 This review focused on exercise-induced autophagy and mitophagy as a nonpharmacological approach of PD treatment. Figures 2 and 3 conclude a possible mechanism of how exercise-induced autophagy and mitophagy led to motor improvement in PD.
 |
Figure 2 Possible mechanism of exercise-induced autophagy through the AMPK-ULK1 pathway to slow PD progression by decreasing synuclein accumulation. |
 |
Figure 3 Possible mechanism of exercise-induced mitophagy through the PINK1/Parkin pathway and AMPK-dependent pathway. |
Current Perspective of Exercise-Induced Autophagy and Mitophagy from Experimental Studies
It was evident from animal studies that exercise can lead to a decrease of α-synuclein accumulation, and it was associated with motor function improvements. Although still limited, in recent years, studies are emerging to support the notion that autophagy or mitophagy activation is involved in exerting this beneficial effect. Table 1 summarizes the study findings from five articles.
 |
Table 1 Summary of Studies about Exercise Modulate Autophagy and Mitophagy in PD model |
A study by Koo and Cho in 2017 found that treadmill exercise alleviates dopaminergic neuron loss in animals with PD and it was correlated to autophagy activation. In the study, mice were injected with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and probenecid to induce PD; afterward, the mice were given mild-to-moderate-intensity treadmill exercise for 8 weeks (5 days/week).48 The results were a significant reduction in α-synuclein levels and dopaminergic neuron loss, along with motor deficits improvements in the PD model mouse compared to the sedentary control. It was also noted that the reduction of α-synuclein is likely due to the increase of autophagic flux, as this was evidenced by the increase of autophagic marker, LC3-II. Afterward, studies by Jang et al,49 Liu et al,50 and Jang et al51 also come up with similar results, in which aerobic exercise increased motoric function, as well as decreased α-synuclein and increased autophagic marker. Although several important autophagy-inducing proteins (eg, AMPK, ULK1) are not measured, these studies are still in line with our propose mechanism of exercise-induced autophagy (Figure 2) because the end-result of autophagy are measured by quantifying several autophagic markers such as LC3-II, Beclin and p62. We also found four other studies that reported the beneficial effect of exercise in PD animal model, ie, alleviating motor deficits and decreasing α-synuclein accumulation; however, the role of autophagy in causing this effect cannot be proven yet from these studies because the autophagic marker was not assessed. The findings of the studies are summarized in Table 2.
 |
Table 2 Summary of Exercise Effects on Motor Function in PD Model |
As for exercise-induced mitophagy involvement in PD, we found only two studies that discuss this matter. In 2017, a study by Almeida et al52 found that moderate exercise decreased the accumulation of α-synuclein in rotenone-injected rats and it was associated with the improvement of mitophagy activity. In 2018, Hwang et al41 also found similar findings, which showed significant improvement of motor behaviour and decreased of α-synuclein accumulation in rotenone-injected rats after exercise, associated with the improvement of mitophagy flux.
Interestingly, there are previous studies that fail to observe an increase in tyrosine hydroxylase (TH) expression in the PD animal model after given exercise, although there is a significant improvement in motoric function.53,54 These result differences are maybe due to the variation in exercise duration and intensity. It is suggested that exercise with mild intensity and shorter duration might not be sufficient to induce neuroprotective effects. This result emphasizes how crucial it is to select the most suitable exercise program in order to realize its benefits. The analysis of animal studies listed in Table 1 shows that the possible exercise regimen to exert neuroprotective effects through exercise-induced autophagy is a mild-to-moderate- or moderate-intensity exercise, 5 days/week, 40 min for each session, and with a minimum of 6-week duration.51 Nevertheless, to find the best regimen, further studies are still necessary considering that our recommendation is only based on animal studies and very few articles.
To our knowledge, there has not been any human study regarding exercise-induced autophagy/mitophagy in PD. However, there have been several human studies that analyzed the effect of exercise on PD motor symptoms. A Cochrane systematic review by Mehrholz et al55 stated that treadmill exercise in patients with PD may improve gait speed. A recent meta-analysis and systematic review by Choi et al13 also showed that aerobic exercise showed a significant effect on motor symptoms and balance in patients with PD in comparison with patients with no exercise regimen. A review by Kim et al56 identified the recommended exercise regimen for adults with PD from existing literature and guidelines. The exercise regimen recommendation is moderate-intensity exercise, 3–5 days/week, with a duration of 20 min for each session that should gradually progress to 60 min over time. The type of activity should be tailored to the patient’s condition and preference; several activity options include walking (overground or treadmill), ergometry, and aquatics (swimming or other aquatic exercises). Overall, this exercise recommendation is similar to the ones we proposed above.
Intriguingly, exercise training given to patients prior to PD onset also resulted in delayed motor symptoms when compared to sedentary control. In a study by Liu et al,50 rats were given 8 weeks of moderate-intensity treadmill exercise and afterward were administered with 6-OHDA. In the analysis, it was noted that the exercise group in comparison with the sedentary group has an improvement in motor function. Although still inconsistent, several studies involving humans also support the notion that exercise not only improves the symptoms of PD but may also reduce the risk of PD.57
Conclusions
Our review shows that autophagy can be induced by exercise and can possibly be activated through the AMPK-ULK1 pathway; moreover, there has been evidence in vivo and in vitro that exercise-induced autophagy could decrease the accumulation of toxic α-synuclein aggregates in PD. Mitophagy can also be induced by exercise via PINK1/Parkin pathway and AMPK-dependent pathway, and can improve mitochondrial homeostasis in PD. There has been evidence of beneficial exercise effects on PD in humans. However, there is currently no human study to back up our proposed molecular mechanism of exercise-induced autophagy/mitophagy, which ameliorates PD motor symptoms. Thus, future research is needed to assess the changes of autophagy and mitophagy process in brain during exercise.
Acknowledgment
This work was supported by Internal Research Grant UNPAD no 1549/UN6.3.1/PT.00/2023 to HG.
Disclosure
No author has any financial interest or received any financial benefit from this research. The authors state no conflict of interest in this work.
References
1. Skogar O, Lokk J. Pain management in patients with Parkinson’s disease: challenges and solutions. J Multidiscip Healthc. 2016;9:469–479. doi:10.2147/JMDH.S105857
2. Ray Dorsey E, Elbaz A, Nichols E, et al. Global, regional, and national burden of Parkinson’s disease, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018;17:939–953. doi:10.1016/S1474-4422(18)30295-3
3. Sarkar S, Raymick J, Imam S. Neuroprotective and therapeutic strategies against Parkinson’s disease: recent perspectives. Int J Mol Sci. 2016;17:904. doi:10.3390/ijms17060904
4. Postuma RB, Berg D, Stern M, et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord. 2015;30:1591–1601. doi:10.1002/mds.26424
5. Moreira RC, Zonta MB, Araújo APS, Israel VL, Teive HAG. Quality of life in Parkinson’s disease patients: progression markers of mild to moderate stages. Arq Neuropsiquiatr. 2017;75:497–502. doi:10.1590/0004-282x20170091
6. Dehay B, Bourdenx M, Gorry P, et al. Targeting α-synuclein for treatment of Parkinson’s disease: mechanistic and therapeutic considerations. Lancet Neurol. 2015;14:855–866. doi:10.1016/S1474-4422(15)00006-X
7. Ingelsson M. Alpha-synuclein oligomers-neurotoxic molecules in Parkinson’s disease and other Lewy body disorders. Front Neurosci. 2016;10:1–10. doi:10.3389/fnins.2016.00408
8. Gao J, Perera G, Bhadbhade M, Halliday GM, Dzamko N. Autophagy activation promotes clearance of α-synuclein inclusions in fibril-seeded human neural cells. J Biol Chem. 2019;294:14241–14256. doi:10.1074/jbc.RA119.008733
9. Karabiyik C, Lee MJ, Rubinsztein DC. Autophagy impairment in Parkinson’s disease. Essays Biochem. 2017;61:711–720. doi:10.1042/EBC20170023
10. Rivero-Ríos P, Madero-Pérez J, Fernández B, et al. Targeting the autophagy/lysosomal degradation pathway in Parkinsons disease. Curr Neuropharmacol. 2016;14:238–249. doi:10.2174/1570159X13666151030103027
11. Scrivo A, Bourdenx M, Pampliega O, Cuervo AM. Selective autophagy as a potential therapeutic target for neurodegenerative disorders. Lancet Neurol. 2018;17:802–815. doi:10.1016/S1474-4422(18)30238-2
12. Vainshtein A, Hood DA. The regulation of autophagy during exercise in skeletal muscle. J Appl Physiol. 2016;120:664–673. doi:10.1152/japplphysiol.00550.2015
13. Choi HY, Cho KH, Jin C, et al. Exercise therapies for Parkinson’s disease: a systematic review and meta-analysis. Parkinsons Dis. 2020;2020. doi:10.1155/2020/2565320
14. Marinus J, Zhu K, Marras C, Aarsland D, van Hilten JJ. Risk factors for non-motor symptoms in Parkinson’s disease. Lancet Neurol. 2018;17:559–568. doi:10.1016/S1474-4422(18)30127-3
15. Mahalakshmi B, Maurya N, Lee S-D, Bharath Kumar V, Kumar VB. Possible neuroprotective mechanisms of physical exercise in neurodegeneration. Int J Mol Sci. 2020;21:1–17. doi:10.3390/ijms21165895
16. Paillard T, Rolland Y, de Barreto PS. Protective effects of physical exercise in Alzheimer’s disease and Parkinson’s disease: a narrative review. J Clin Neurol. 2015;11:212–219. doi:10.3988/jcn.2015.11.3.212
17. Haddaway NR, Page MJ, Pritchard CC, McGuinness LA. PRISMA2020: An R package and Shiny app for producing PRISMA 2020-compliant flow diagrams, with interactivity for optimised digital transparency and Open Synthesis. Campbell Systematic Reviews. 2022;18(2):e1230. doi:10.1002/cl2.1230
18. Tarawan VM, Gunadi JW, Setiawan LR, et al. Alteration of autophagy gene expression by different intensity of exercise in gastrocnemius and soleus muscles of Wistar rats. J Sport Sci Med. 2019;18:146–154.
19. Fritzen AM, Madsen AB, Kleinert M, et al. Regulation of autophagy in human skeletal muscle: effects of exercise, exercise training and insulin stimulation. J Physiol. 2016;594:745–761. doi:10.1113/JP271405
20. Kjøbsted R, Hingst JR, Fentz J, et al. AMPK in skeletal muscle function and metabolism. FASEB J. 2018;32:1741–1777. doi:10.1096/fj.201700442R
21. Schwalm C, Jamart C, Benoit N, et al. Activation of autophagy in human skeletal muscle is dependent on exercise intensity and AMPK activation. FASEB J. 2015;29:3515–3526. doi:10.1096/fj.14-267187
22. Møller AB, Vendelbo MH, Christensen B, et al. Physical exercise increases autophagic signaling through ULK1 in human skeletal muscle. J Appl Physiol. 2015;118:971–979. doi:10.1152/japplphysiol.01116.2014
23. Kim J, Kundu M, Viollet B, Guan K-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi:10.1038/ncb2152
24. Karanasios E, Stapleton E, Manifava M, et al. Dynamic association of the ULK1 complex with omegasomes during autophagy induction. J Cell Sci. 2013;126:5224–5238. doi:10.1242/jcs.132415
25. Russell RC, Tian Y, Yuan H, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013;15:741–750. doi:10.1038/ncb2757
26. Fracchiolla D, Chang C, Hurley JH, Martens S. A PI3K-WIPI2 positive feedback loop allosterically activates LC3 lipidation in autophagy. J Cell Biol. 2020;219(9);e201912098. doi:10.1083/JCB.201912098
27. Wan W, You Z, Zhou L, et al. mTORC1-regulated and HUWE1-mediated WIPI2 degradation controls autophagy flux. Mol Cell. 2018;72:303–315.e6. doi:10.1016/j.molcel.2018.09.017
28. Dooley HC, Razi M, Polson HEJ, Girardin SE, Wilson MI, Tooze SA. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12–5-16L1. Mol Cell. 2014;55:238–252. doi:10.1016/j.molcel.2014.05.021
29. Yoshii SR, Mizushima N. Monitoring and measuring autophagy. Int J Mol Sci. 2017;18:1–13. doi:10.3390/ijms18091865
30. Jung G, Roh J, Lee H, et al. Autophagic markers BECLIN 1 and LC3 are associated with prognosis of multiple myeloma. Acta Haematol. 2015;134:17–24. doi:10.1159/000368848
31. Matsuda N, Sato S, Shiba K, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189:211–221. doi:10.1083/jcb.200910140
32. Wu W, Xu H, Wang Z, et al. PINK1-parkin-mediated mitophagy protects mitochondrial integrity and prevents metabolic stress-induced endothelial injury. PLoS One. 2015;10:e0132499. doi:10.1371/journal.pone.0132499
33. Guo P, Jin Z, Wang J, Sang A, Wu H. Irisin rescues blood-brain barrier permeability following traumatic brain injury and contributes to the neuroprotection of exercise in traumatic brain injury. Oxidative Med Cell Long. 2021;2021:154 doi:10.1155/2021/1118981
34. He W, Wang P, Chen Q, Li C. Exercise enhances mitochondrial fission and mitophagy to improve myopathy following critical limb ischemia in elderly mice via the PGC1a/FNDC5/irisin pathway. Skelet Muscle. 2020;10:25. doi:10.1186/s13395-020-00245-2
35. Li H, Qin S, Liang Q. Exercise training enhances myocardial mitophagy and improves cardiac function via Irisin / FNDC5-PINK1 / Parkin. Biomedicines. 2021;9:701. doi:10.3390/biomedicines9060701
36. Seabright AP, Fine NHF, Barlow JP, et al. AMPK activation induces mitophagy and promotes mitochondrial fission while activating TBK1 in a PINK1-Parkin independent manner. FASEB J. 2020;34:6284–6301. doi:10.1096/fj.201903051R
37. Drake JC, Wilson RJ, Laker RC, Guan Y, Spaulding HR. Mitochondria-localized AMPK responds to local energetics and contributes to exercise and energetic stress-induced mitophagy. Proc Natl Acad Sci. 2021;118:1–10 doi:10.1073/pnas.2025932118
38. Bellucci A, Mercuri NB, Venneri A, et al. Parkinson’s disease: from synaptic loss to connectome dysfunction. Neuropathol Appl Neurobiol. 2016;42:77–94. doi:10.1111/nan.12297
39. Hu Q, Wang G. Mitochondrial dysfunction in Parkinson’s disease. Transl Neurodegener. 2016;5:1–8. doi:10.1186/s40035-016-0060-6
40. Komilova NR, Angelova PR, Berezhnov AV, et al. Metabolically induced intracellular pH changes activate mitophagy, autophagy, and cell protection in familial forms of Parkinson’s disease. FEBS J. 2022;289:699–711. doi:10.1111/febs.16198
41. Hwang D-J-J, Koo J-H-H, Kwon K-C-C, et al. Neuroprotective effect of treadmill exercise possibly via regulation of lysosomal degradation molecules in mice with pharmacologically induced Parkinson’s disease. J Physiol Sci. 2018;68:707–716. doi:10.1007/s12576-017-0586-0
42. Masaracchia C, Hnida M, Gerhardt E, et al. Membrane binding, internalization, and sorting of alpha-synuclein in the cell. Acta Neuropathol Commun. 2018;6:79. doi:10.1186/s40478-018-0578-1
43. Ivankovic D, Chau K-Y, Schapira AHV, Gegg ME. Mitochondrial and lysosomal biogenesis are activated following PINK1/parkin‐mediated mitophagy. J Neurochem. 2016;136:388–402. doi:10.1111/jnc.13412
44. Bayod S, Del Valle J, Pelegri C, et al. Macroautophagic process was differentially modulated by long-term moderate exercise in rat brain and peripheral tissues. J Physiol Pharmacol. 2014;65:229–239.
45. Huang J, Wang X, Zhu Y, et al. Exercise activates lysosomal function in the brain through AMPK‐SIRT1‐TFEB pathway. CNS Neurosci Ther. 2019;25:796–807. doi:10.1111/cns.13114
46. Luo L, Dai J-R, Guo -S-S, et al. Lysosomal proteolysis is associated with exercise-induced improvement of mitochondrial quality control in aged hippocampus. J Gerontol Ser A. 2017;72:1342–1351. doi:10.1093/gerona/glw242
47. Kwon I, Jang Y, Lee Y. Endurance exercise-induced autophagy/mitophagy coincides with a reinforced anabolic state and increased mitochondrial turnover in the cortex of young male mouse brain. J Mol Neurosci. 2020;71:42–54. doi:10.1007/s12031-020-01624-6
48. Koo J-H, Cho J-Y. Treadmill exercise attenuates α-synuclein levels by promoting mitochondrial function and autophagy possibly via SIRT1 in the chronic MPTP/P-induced mouse model of Parkinson’s disease. Neurotox Res. 2017;32:473–486. doi:10.1007/s12640-017-9770-5
49. Jang Y, Hwang DJ, Koo JH, et al. Association of exercise-induced autophagy upregulation and apoptosis suppression with neuroprotection against pharmacologically induced Parkinson’s disease. J Exerc Nutr Biochem. 2018;22:1–8. doi:10.20463/jenb.2018.0001
50. Liu W, Fu R, Wang Z, et al. Regular aerobic exercise-alleviated dysregulation of CAMKIIα carbonylation to mitigate parkinsonism via homeostasis of apoptosis with autophagy. J Neuropathol Exp Neurol. 2020;79:46–61. doi:10.1093/jnen/nlz106
51. Jang Y, Kwon I, Song W, Cosio-Lima LM, Lee Y. Endurance exercise mediates neuroprotection against MPTP-mediated Parkinson’s disease via enhanced neurogenesis, antioxidant capacity, and autophagy. Neuroscience. 2018;379:292–301. doi:10.1016/j.neuroscience.2018.03.015
52. Almeida MF, Silva CM, Chaves RS, et al. Effects of mild running on substantia nigra during early neurodegeneration. J Sports Sci. 2017;36:1363–1370. doi:10.1080/02640414.2017.1378494
53. Hood RL, Liguore WA, Moore C, Pflibsen L, Meshul CK. Exercise intervention increases spontaneous locomotion but fails to attenuate dopaminergic system loss in a progressive MPTP model in aged mice. Brain Res. 2016;1646:535–542. doi:10.1016/j.brainres.2016.06.032
54. Sconce MDD, Churchill MJJ, Greene REE, Meshul CKK. Intervention with exercise restores motor deficits but not nigrostriatal loss in a progressive MPTP mouse model of Parkinson’s disease. Neuroscience. 2015;299:156–174. doi:10.1016/j.neuroscience.2015.04.069
55. Mehrholz J, Kugler J, Storch A, Pohl M, Elsner, B, Hirsch, K. Treadmill training for patients with Parkinson’s disease. Cochrane Database Syst Rev. 2015;2015(8):1465–1858. doi:10.1002/14651858.CD007830.pub3
56. Kim Y, Lai B, Mehta T, et al. Exercise training guidelines for multiple sclerosis, stroke, and Parkinson disease. Am J Phys Med Rehabil. 2019;98:613–621. doi:10.1097/PHM.0000000000001174
57. Fan B, Jabeen R, Bo B, et al. What and how can physical activity prevention function on Parkinson’s disease? Oxid Med Cell Longev. 2020;2020:4293071. doi:10.1155/2020/4293071
58. Koo J-H, Jang Y-C, Hwang D-J, et al. Treadmill exercise produces neuroprotective effects in a murine model of Parkinson’s disease by regulating the TLR2/MyD88/NF-κB signaling pathway. Neuroscience. 2017;356:102–113. doi:10.1016/j.neuroscience.2017.05.016
59. Shin M-S, Kim T-W, Lee J-M, Ji E-S, Lim B-V. Treadmill exercise alleviates nigrostriatal dopaminergic loss of neurons and fibers in rotenone-induced Parkinson rats. J Exerc Rehabil. 2017;13:30–35. doi:10.12965/jer.1734906.453
60. Jang Y, Koo J-H-H, Kwon I, et al. Neuroprotective effects of endurance exercise against neuroinflammation in MPTP-induced Parkinson’s disease mice. Brain Res. 2017;2016:186–193. doi:10.1016/j.brainres.2016.10.029
61. Tuon T, Valvassori SSS, Lopes-Borges J, et al. Physical training exerts neuroprotective effects in the regulation of neurochemical factors in an animal model of Parkinson’s disease. Neuroscience. 2012;227:305–312. doi:10.1016/j.neuroscience.2012.09.063
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Shisandra Decoction Alleviates Parkinson’s Disease Symptoms in a Mouse Model Through PI3K/AKT/mTOR Signalling Pathway
Pan Y, Chen M, Pan L, Tong Q, Cheng Z, Lin S, Pan R, Chen M, Zhi Y
Neuropsychiatric Disease and Treatment 2024, 20:2011-2027
Published Date: 23 October 2024
Differential Effects of Cycling Exercise on Pain Types in Patients with Early Parkinson’s Disease: A Subgroup Analysis of a Randomized Pilot Trial
Jun JS, Choi S, Kang N, Park K, Jeon B, Byun K, Kim R
Journal of Pain Research 2025, 18:7225-7232
Published Date: 28 December 2025
Nanotherapeutic Interventions for Parkinson’s Disease: Modulating Pathogenic Mechanisms and Overcoming Therapeutic Obstacles
Yin X, Wang C, Lin W, Huang S, Chen T
International Journal of Nanomedicine 2026, 21:570452
Published Date: 7 February 2026