Back to Journals » Journal of Inflammation Research » Volume 16
Programmed Cell Death in Liver Fibrosis
Received 27 June 2023
Accepted for publication 23 August 2023
Published 1 September 2023 Volume 2023:16 Pages 3897—3910
DOI https://doi.org/10.2147/JIR.S427868
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Adam Bachstetter
Ruoyu Gao,1 Haiying Tang,2 Jingwei Mao1
1Department of Gastroenterology, First Affiliated Hospital of Dalian Medical University, Dalian, 116011, People’s Republic of China; 2Department of Respiratory and Critical Care Medicine, First Affiliated Hospital of Dalian Medical University, Dalian, 116011, People’s Republic of China
Correspondence: Jingwei Mao, Department of Gastroenterology, First Affiliated Hospital of Dalian Medical University, 222 Zhongshan Road, Dalian, 116011, Liaoning Province, People’s Republic of China, Email [email protected]
Abstract: Programmed cell death (PCD) is a comprehensive term that encompasses various forms of cell death, such as apoptosis, necroptosis, pyroptosis, ferroptosis, and autophagy, which play a crucial role in the pathogenesis of liver fibrosis. PCD facilitates the elimination of aberrant cells, particularly activated hepatic stellate cells (HSCs), which are the primary producers of extracellular matrix (ECM). The removal of HSCs may impede ECM synthesis, thereby mitigating liver fibrosis. As such, PCD has emerged as a promising therapeutic target for the development of novel drugs to treat liver fibrosis. Numerous studies have been conducted to investigate the underlying mechanisms of PCD in the elimination of activated HSCs and other aberrant liver cells in fibrotic liver tissue, including hepatocytes, hepatic sinusoid endothelial cells (LSECs), and Kupffer cells (KCs). The induction of PCD, the interplay between different forms of PCD, and the potential harm or benefit of PCD in liver fibrosis are topics of ongoing research. Evidences suggest that PCD is a complex process with dual effects on liver fibrosis. The purpose of this review is to summarize the most recent advances in PCD and liver fibrosis research.
Keywords: liver fibrosis, necroptosis, pyroptosis, ferroptosis, autophagy, apoptosis
Introduction
Despite extensive research into the mechanisms of liver fibrosis and the identification of several signal pathways involving numerous proteins, non-coding RNAs, lipids metabolism, and cells in the liver, including hepatic stellate cells (HSCs), hepatocytes, liver sinusoidal endothelial cells (LSECs), and Kupffer cells (KCs), effective treatments for patients with liver fibrosis remain elusive. Non-alcoholic fatty liver disease (NAFLD) is the predominant etiological factor contributing to liver fibrosis, owing to its global prevalence and the substantial mortality associated with this condition. Furthermore, NAFLD is also associated with an elevated risk of extrahepatic malignancies, including bladder cancer.1 Although liver fibrosis is a reversible process when the underlying etiologic agents are removed, failure to intervene early in the process may result in irreversible damage. To date, there are no treatments that have proven to be sufficiently effective for patients with diverse forms of liver injury and varying degrees of liver fibrosis. In the realm of therapeutic interventions for NAFLD, the available options are limited, primarily consisting of dietary recommendations and physical activity. However, there exists a notable dearth in effective treatments, prompting numerous physicians to explore the potential of natural products as a means to ameliorate or even cure NAFLD.2 Among them, berberine is commonly acknowledged as a highly promising therapeutic agent for the treatment of cancer including hepatocellular carcinoma (HCC).3 Failure to intervene in the process of liver fibrosis in its early stages may result in its gradual progression to irreversible cirrhosis, cirrhosis decompensation, and HCC.4,5
Liver fibrosis is a reparative process that involves the mediation of HSCs through the production ECM proteins to replace damaged tissue. While an appropriate repair response is advantageous, the continuous production of collagen fibers due to chronic injury and inflammation can lead to the destruction of normal liver structure.6 Programmed cell death (PCD) appears to play a pivotal role in both the initiation and regression of liver fibrosis. PCD has the ability to induce the death of abnormal cells in the liver, particularly HSCs, which are crucial cells in liver fibrosis. This results in the inhibition of danger signals that are transmitted from injured or dying hepatocytes to HSCs, and the suppression of adjacent hepatocytes undergoing epithelial-mesenchymal transition (EMT) in fibrotic liver tissue. Furthermore, aside from its significant involvement in liver fibrosis, PCD also assumes a crucial function in the immune tolerance of various liver diseases or conditions, including liver allograft transplantation. As highlighted in a review conducted by Manuel Muro et al,7 the interaction between naive CD8+ T cells and LSECs leads to the partial activation of T cells, subsequently resulting in passive cell death within the unique state of tolerance towards liver graft transplantation. Consequently, it is plausible to suggest that PCD may be implicated in the mechanism of immune regulation.
Hence, inducing the demise of these aberrant cells may potentially serve as a favorable approach for ameliorating liver fibrosis. It is noteworthy that mere cell death is insufficient, as the subsequent clearance of these deceased cells, which are attributed to various forms of PCD, by macrophages in a timely manner through efferocytosis can effectively suppress tissue inflammation. Failure to do so may result in the release of various danger signals by the dying cells, leading to more severe responses that can further exacerbate liver injury and fibrosis8–11 Therefore, the identification of PCD in intrahepatic cells holds significant importance in the treatment of liver fibrosis.
PCD Participates in the Pathogenesis of Liver Fibrosis
Necroptosis and Liver Fibrosis
Necroptosis is a form of programmed lytic cell death that shares similarities with necrosis, both of which exhibit analogous features such as increased cell volume, organelle swelling, and loss of membrane integrity, ultimately resulting in the release of Damage-associated molecular patterns (DAMPs) that trigger inflammation and secondary injury.12–14 Figure 1 provides a detailed description of the process of necroptosis. Several studies have demonstrated a correlation between necroptosis and the progression of liver injury induced by various conditions, including NAFLD, NASH, ALD, HBV related liver diseases, primary biliary cholangitis (PBC), and liver cancer.15–20 Thus, necroptosis appears to be a stimulatory factor for fibrosis, as has been demonstrated in experiments.21
 |
Figure 1 The process of necroptosis. The interaction between TNF and TNFR stimulates the assembly of the TNFR complex, which consists of TRADD, TRAF2, RIPK1, CIAP1/2, and LUBAC. This complex has the ability to facilitate the phosphorylation of RIPK1, which subsequently results in the phosphorylation of RIPK3. Ultimately, the activation of RIPK3 can also occur via the LPS-TLR signaling pathway in the presence of TRIF. The phosphorylation of RIPK3 is known to facilitate the phosphorylation of MLKL, which in turn can lead to its oligomerization and translocation to the cellular membrane. This event is responsible for the disruption of membrane integrity and cytosolic osmolarity, ultimately resulting in membrane permeabilization. Inhibition of NF-κB activity driven by complex I can result in the induction of apoptosis through the involvement of complex II, comprising TRADD, TRAF2, RIPK1, FADD, casepase8, and cFLIP, which cleaves RIPK1 to prevent necroptosis. Additionally, the activation of MLKL can stimulate the NLRP3-caspase1 inflammasome, leading to the release of IL-1β and IL-18. |
The process of necroptosis requires the involvement of receptor-interacting serine-threonine kinase 1, 3 (RIPK1, RIPK3), and mixed lineage kinase (MLKL), which is facilitated through the phosphorylation of these proteins. Studies have shown that the knockdown of MLKL in hepatocytes can significantly reduce liver injury inflammation and the activation of HSCs. Similarly, the downregulation of RIPK1 and RIPK3 has been identified as an effective therapeutic strategy, as it can alleviate liver fibrosis by inhibiting hepatocyte necroptosis and reducing inflammation.9,22,23 In the context of hepatocytes, necroptosis serves to exacerbate inflammation.24 The release of DAMPs from necroptotic hepatocytes plays a crucial role in stimulating inflammation and activating HSCs. SA1009, a DAMP associated with necroptosis.12,25 Exhibits a reduction in liver tissue levels that corresponds to a decrease in the extent of liver fibrosis.26 However, the function of necroptosis in HSCs appears to be distinct from its impact on hepatocytes. A previous investigation has demonstrated that necroptosis induced in HSCs can significantly mitigate liver fibrosis by promoting HSC death and inhibiting their proliferation.27
Macrophages play a crucial role in mitigating liver fibrosis by phagocytosing damaged and deceased hepatocytes, thereby inhibiting the release of DAMPs, other harmful substances, and exosomes containing fibrotic non-coding RNAs; and the latter can exacerbate liver injury and activate HSCs.16,28 Necroptosis is a vital process for macrophages, as evidenced by the induction of necroptosis in liver-resident macrophages (KCs) during Listeria monocytogenes (LM) infection, and the death of KCs can attract monocyte-derived macrophages to the liver to eliminate LM.29 There is limited evidence to demonstrate the occurrence of necroptosis in KCs during liver fibrosis. M2 macrophages play a crucial anti-inflammatory role in acute-on-chronic liver failure by suppressing necroptosis in hepatocytes. It is imperative to promptly clear dying cells through macrophages to prevent liver fibrosis following hepatocyte necroptosis. However, macrophages are unable to eliminate necroptotic hepatocytes, and research has indicated that phagocytosis is impaired in NASH.16 The upregulation of the ligand CD47 on necrotic hepatocytes and its corresponding receptor signal regulatory protein ɑ (SIRPɑ) on liver macrophages serves as a “Don’t Eat Me” signal for the macrophages; repression of either CD47 or SIRPɑ can restore the phagocytic activity of macrophages, leading to the elimination of necrotic hepatocytes and the mitigation of liver fibrosis.16 MLKL plays a significant role in the phagocytosis of KCs, as demonstrated by the reduced uptake of bio-particles in KCs deficient in MLKL compared to those with normal MLKL levels in liver tissue injured by alcohol consumption.30 The loss of RIP3 in macrophages also inhibits the release of pro-inflammatory cytokines to alleviate liver fibrosis by suppressing the NF-κB pathway.31
In brief, necroptosis plays a significant role in liver fibrosis, with varying effects depending on the type of cell undergoing necroptosis. Specifically, necroptosis exacerbates inflammation and worsens fibrosis in hepatocytes, while in HSCs, it appears to alleviate liver fibrosis. Although research on the occurrence of necroptosis in KCs during liver fibrosis is limited, it is evident that their ability to clear necroptotic hepatocytes is diminished.
Pyroptosis and Liver Fibrosis
Pyroptosis, a form of lytic cell death, is characterized by the release of intracellular molecules that serve as a critical cue for neighboring cells to respond to infection and combat pathogens. Figure 2 illustrates the process and mechanism of pyroptosis. Pyroptosis has been identified as a potential anti-cancer therapy by promoting tumor cell death.32,33 However, excessive pyroptosis can lead to severe inflammation and contribute to the development of liver fibrosis.34 The inflammasome is characterized by a disc-shaped molecular platform that functions in proptosis.35 It serves as an initiator for activating pyroptosis by maturing caspase-1, a key effector of pyroptosis, and as a pro-inflammatory molecule released from the cell after pyroptosis to cause an inflammatory response.36,37 The inflammasome is a multi-protein complex comprised of three constituents, including leucine-repeat-containing protein (NLRP), AIM2-like receptor protein (ALRP), and apoptosis-associated speck-like protein containing a CARD (ASC).38 NLRP particularly NLRP3, inflammasome drives liver injury and fibrosis. Inhibition of its activity can significantly decrease liver fibrosis induced by NAFLD.39,40
 |
Figure 2 The process of pyroptosis. DAMPs or PAMPs generated by diverse pathogens have the potential to stimulate TLR4, which subsequently initiates the activation of the NLRP3 inflammasome. This disc-shaped structure facilitates the conversion of inactivated pro-caspase1 to activated caspase, necessitating the involvement of ASC. Caspase1 performs two primary roles. Firstly, it cleaves GSDMD into an N-terminal and a C-terminal fragment. The N-terminal fragment is then recruited to the cell membrane, where it forms transmembrane pores that result in potassium efflux and water influx, ultimately leading to cell lysis. Secondly, caspase1 promotes the conversion of pro-IL-1β and pro-IL-18 into the bioactive inflammatory IL-1β and IL-18. Furthermore, caspase1 signaling induces proptosis, while intracellular LPS directly cleaves pro-caspase4/5 into activated caspase4/5, which in turn cleaves GSDMD to promote membrane pores. In comparison to caspase 1, caspase 4/5 lacks the ability to cleave pro-IL-1β and pro-IL-18, and its expression is dependent on NF-κB. Upon activation by DAMPs and PAMPs, TLR4 initiates a cascade of events involving the adaptor protein MYD88, which subsequently triggers the activation of IRAK1 and TAK1. This process ultimately leads to the activation of IKK, which phosphorylates IκB, the inhibitor of NF-κB. The phosphorylation of IκB promotes its ubiquitination and subsequent degradation by proteasome, thereby facilitating the activation of NF-κB. |
The activation of HSCs by inflammasomes released from hepatocytes undergoing pyroptosis is found to be a key mechanism contributing to liver fibrosis.8 The release of DAMPs from dying cells via pyroptosis is shown to play a significant role in liver fibrosis. Specifically, High mobility group-1 (HMGB1), a DAMP, is positively correlated with pyroptosis and is released from macrophages and dying cells, thereby exacerbating liver injury.41,42 Furthermore, dying hepatocytes induced by pyroptosis can release HMGB1, which promotes HSC activation.43 Suppression of hepatocyte pyroptosis is found to reduce liver fibrosis.44,45
Pyroptosis appears to occur with greater frequency in macrophages during liver injury induced by various etiologies, and is involved in liver fibrosis.46 SA1008, a DAMP that is elevated in fibrotic liver tissue, can induce pyroptosis in KCs. Evidence showed that SA1008 was released from dead macrophages to activate HSCs and promote liver fibrosis.47 Suppression of pyroptosis in KCs can effectively ameliorate liver fibrosis by repressing the nicotinamide adenine dinucleotide phosphate oxidase (NOX2)/NLRP3 pathway, which drives NLRP3 inflammasome activation.48 Pyroptosis in HSCs is mediated by ROS augmentation, which increases caspase1 and leads to HSC activation and aggravation of fibrosis.49
The present study has demonstrated a significant increase in Angiotensin II levels in patients with NAFLD, which acts as a stimulator for HSCs by inducing proptosis.50 NLRP6, a member of the NLR family, has been reported to promote inflammasome assembly, leading to the cleavage of pro-caspase-1 and activation of caspase1, ultimately resulting in pyroptosis.51 Interestingly, Enforced expression of NLRP6 can effectively suppress the expression of various fibrotic markers (eg, ɑ-SMA, MMP2, MMP9) and proliferation of HSCs.52 Unfortunately, the present study failed to investigate the correlation between NLRP6 and pyroptosis in HSCs, as well as the potential anti-fibrotic effects of pyroptotic HSCs through the promotion of their death. It is possible to hypothesize that timely clearance of pyroptotic hepatocytes by macrophages could prevent liver fibrosis by preventing the release of fibrotic factors from damaged hepatocytes. However, impaired efferocytosis of macrophages, whether induced by pyroptosis or recruited to fibrotic sites, may contribute to inflammation. Therefore, the use of pyroptosis inhibitors may increase macrophage efferocytosis while reducing inflammation.53 Additionally, the demise of invariant natural killer T (NKT) cells stimulated by pyroptosis can also contribute to the liver injury via release potent pro-inflammatory cytokines.54
In aggregate, the occurrence of excessive pyroptosis has the potential to elicit significant inflammation and facilitate the development of liver fibrosis. Specifically, pyroptosis of hepatocytes can result in the release of inflammasomes, which in turn triggers the activation of HSCs, ultimately contributing to the progression of liver fibrosis. Mitigation of hepatocyte pyroptosis has the potential to ameliorate liver fibrosis. In the case of KCs, timely elimination of pyroptotic hepatocytes may forestall liver fibrosis by preventing the release of fibrotic factors from dying hepatocytes. However, the process of liver fibrosis impairs macrophage efferocytosis, and inhibition of pyroptosis in KCs may serve to prevent the development of liver fibrosis.
Autophagy and Liver Fibrosis
Autophagic cell death, also known as autophagy-dependent cell death, is a form of cell death that requires the process of autophagy.55 Autophagy is a crucial mechanism for maintaining cellular homeostasis by eliminating harmful cargos, including damaged organelles and protein aggregates, and by inhibiting inflammasome activation through the formation of double-membrane structures known as autophagosomes. These structures are responsible for encapsulating toxic cargos and transporting them to lysosomes for degradation, a process that involves several autophagy-related proteins (ATGs).56,57 Autophagy serves as a beneficial mechanism for cell survival by safeguarding against stress and injury-induced cell death. Nevertheless, excessive autophagy triggered by various stressors can result in cellular demise, rendering autophagy ineffective in maintaining basic survival.
While the association between autophagy and fibrosis exists, the correlation between autophagy and hepatic fibrosis remains inconclusive and may vary depending on the cell type. The preceding investigation demonstrated that the promotion of autophagy in HSCs can alleviate liver fibrosis by decreasing the release of extracellular vesicles that contain fibrotic substances derived from activated HSCs. In addition to autophagy, mitophagy also plays a role in liver fibrosis. It has been reported that inhibiting the mitophagy of HSCs can significantly activate HSCs, exacerbating hepatic fibrosis.58 Conversely, another study demonstrated that enhancing the autophagy of HSCs can expedite HSCs activation and liver fibrosis.59
The induction of mitophagy in KCs is a prerequisite for the production of TGF-β1 and subsequent activation of HSCs leading to liver fibrosis.60 Proper autophagy is crucial in inhibiting the inflammatory effects of macrophages.61 A recent study has suggested that macrophages deficient in autophagy release higher levels of IL-1α and IL-1β, resulting in increased apoptosis and necrosis of hepatocytes, liver inflammation, and fibrotic gene expression when exposed to carbon tetrachloride (CCL4).62 The impairment of autophagy in LSECs may potentially worsen sinusoidal fibrosis, as evidenced by previous studies.63
As we know, proper autophagy serves as a protective mechanism in various liver diseases, ranging from NAFLD and NASH to fibrosis, particularly in hepatocytes, HSCs, KCs and LSECs. The impact of autophagy on HSCs activation remains a topic of debate in the scientific community. To further elucidate this relationship, researchers must consider the various types of autophagy, including mitophagy, lipophagy, and Endoplasmic reticulum (ER)-phagy. It is imperative to determine which specific types of autophagy exert beneficial or detrimental effects on liver fibrosis in order to identify novel targets for reversing this condition.
Ferroptosis and Liver Fibrosis
Ferroptosis is a form of cell death that is reliant on autophagy.64 It is widely accepted that autophagy and ferroptosis have a symbiotic relationship, with autophagy inducing ferroptosis and ferroptosis, in turn, enhancing autophagy.65 As a distinct type of PCD, ferroptosis differs from apoptosis, necroptosis, and pyroptosis, and holds significant implications for anti-cancer therapies.66 Lipid peroxidation at polyunsaturated fatty acyl moieties and the generation of reactive oxygen species (ROS) are closely associated with ferroptosis. However, the potent antioxidant glutathione (GSH) effectively suppresses excessive oxidative responses, thereby inhibiting ferroptosis. The synthesis of GSH is of paramount importance, as it is involved in the cysteine, cystine-glutamate antiporter system Xc- and glutathione peroxidases (GPXs), particularly GPX4, which act as scavengers of ROS. This process is critical in increasing GSH production by enhancing the activity of system Xc- and GPXs, thereby protecting cells from ferroptosis-induced death.67–69 Iron plays a crucial role in the process of ferroptosis by upregulating the levels of iron through the mediation of ferritin, an iron-storage protein, and transferrin, which can promote ferroptosis by facilitating ferritinophagy and transporting iron into the cell, respectively.70,71 Figure 3 illustrates the process of ferroptosis.
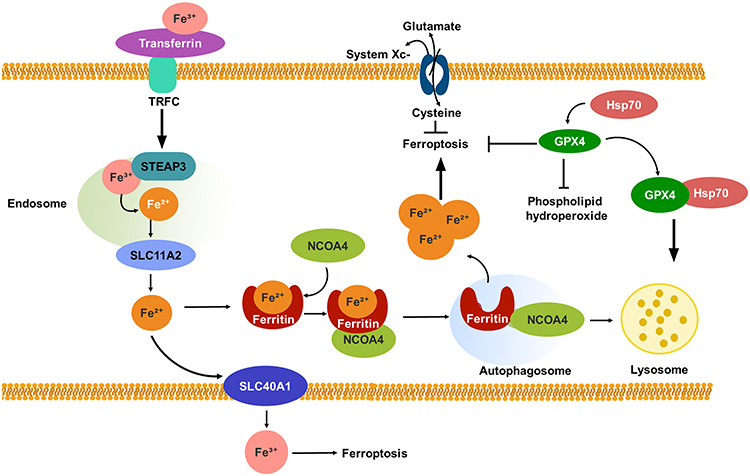 |
Figure 3 The process of ferroptosis. The ferric form of iron present in serum is transported by transferrin and recognized by TFRC. Upon binding to TFRC, STEAP3 metalloreductase mediates the reduction of ferric iron to ferrous iron within the endosome. Subsequently, SLC11A2 facilitates the release of ferrous iron into the cytoplasm, where it can bind to ferritin. Ferritin serves as the primary means of storing iron within cells. The release of ferric iron can be achieved through the degradation of ferritin by ferritinophagy, a form of selective autophagy that is regulated by NCOA4. Intracellular ferrous iron can be exported to the extracellular environment by SLC40A1, where it is converted to ferric iron. The accumulation of excess ferrous iron can trigger ferroptosis. In addition to autophagy-dependent ferroptosis, two antioxidant systems are associated with this process. One system, known as system Xc-, functions as an antiporter that facilitates the importation of cysteine into cells while simultaneously countertransporting glutamate. This process promotes the production of glutathione. Another system involves GPX4, which contains selenium and is primarily responsible for catalyzing the reduction of phospholipid hydroperoxides to decrease their production. Both of these systems are capable of inhibiting ferroptosis. GPX4 can be degraded through chaperone-mediated autophagy, with Hsp70 acting as a molecular chaperone to promote GPX4 degradation in lysosomes. |
The impact of ferroptosis on liver fibrosis, like other forms of PCD, remains a topic of debate. Previous research has demonstrated that excessive iron-induced ferroptosis is heightened in both cirrhotic patients and fibrotic animal models, suggesting a potential association between ferroptosis and cirrhosis.10,72 The majority of research on the anti-fibrotic role of ferroptosis has been conducted in HSCs. The findings from these studies suggest that inducing ferroptosis to eliminate activated HSCs can effectively alleviate liver fibrosis.73,74 Additionally, ferroptosis has been linked to inflammation, as dead hepatocytes resulting from ferroptosis can release DMAPs that promote macrophage activation. HGMB1, a positive regulator of ferroptosis, has been shown to contribute to immune cell infiltration and the production of inflammatory cytokines such as IL-1β and IL-6 by macrophages.75,76 The degradation of GPX4 can occur subsequent to its import into mitochondria via mitophagy, which is facilitated by the mitophagy receptor FUNDC1. This process results in the reduction of GPX4 and an increase in ROS, both of which can instigate ferroptosis in hepatocytes, leading to liver injury and fibrosis.77 Additionally, macrophages are susceptible to ferroptosis induction, which can prompt their polarization into the M1 subtype, also known as pro-inflammatory macrophages.78,79 Although this conclusion has not been directly confirmed in the context of liver fibrosis, it is sufficient to shed light on the significance of ferroptosis in macrophages by augmenting pro-inflammatory activity, which exacerbates liver injury and fibrosis.
General speaking, the unique characteristics of ferroptosis in liver fibrosis may be contingent upon the specific cell types undergoing ferroptosis. Induction of ferroptosis in hepatocytes is likely to exacerbate liver fibrosis, whereas ferroptosis of HSCs appears to inhibit hepatic fibrosis by eliminating the primary source of collagens. The occurrence of ferroptosis as a form of cell death is dependent on autophagy, specifically through ferritinophagy. This may explain why some researchers have concluded that increased autophagy can suppress HSCs activation and exert an anti-fibrotic effect.
Apoptosis and Liver Fibrosis
Apoptosis is a form of non-lytic cell death whereby the remnants of expired cells are enclosed within membrane structures known as apoptosis bodies. These structures are subsequently engulfed by phagocytes, and the contents of the apoptosis bodies remain contained. The process of apoptosis is regulated by cascades of caspases and the tumor necrosis factor (TNF) receptor family, which includes caspase-9, caspase-3, caspases-7, caspase-8, FAS, and TNFR1. These factors can initiate apoptosis through two distinct pathways: the intrinsic pathway (also referred to as the mitochondrial pathway) and the extrinsic pathway (known as the death receptor pathway).80–82 Figure 4 provides a succinct overview of the process of apoptosis.
 |
Figure 4 The process of apoptosis. There exist two distinct pathways that can result in apoptosis, namely the intrinsic pathway (also known as the mitochondrial pathway) and the extrinsic pathway (also known as the death receptor pathway). Despite their differences, these pathways can converge and trigger apoptosis. The intrinsic pathway is activated by various cytotoxic agents, growth factors, or nutrient deficiencies, leading to an increase in the expression of BH3-only proteins that exhibit a strong affinity for BCL2, a pro-survival protein that plays a role in anti-apoptosis. The interaction between NH3-only proteins and BCL-2 results in the release of BAX and BAK, which subsequently oligomerize and assemble into structures that breach the outer membrane of mitochondria, leading to MOMP and the release of cytochrome C into the cytoplasm. The cytoplasmic cytochrome C then binds to APF1, promoting apoptosome formation and inducing apoptosis. This process recruits the precursor of apoptosis initiator pro-caspase9 to the apoptosome, where it is cleaved and becomes bioactive caspase 9. Apoptosis may be induced by TNF/FASL through binding to TNFR/FAS, which leads to the activation of pro-caspase8 to caspase8 via FADD or TRADD. The resulting bioactive caspase8 can directly proteolyze effector caspase3/7. In addition to its direct role in apoptosis, caspase8 can also induce apoptosis indirectly. Specifically, caspase8 activates Bid, which is then transformed into tBID and, in conjunction with the intrinsic pathway, initiates apoptosis. |
Apoptosis exhibits a dual impact on liver fibrosis, whereby it serves as a beneficial mechanism for mitigating liver fibrosis by facilitating the death of HSCs and interrupting collagen production. The induction of HSCs apoptosis is attributed to CD8+ tissue-resident memory T cells (CD8+ Trm), which can effectively reduce the extent of liver fibrosis and promote its resolution.83 The transcription factor STAT1 is associated with the induction of apoptosis and its activity is observed to increase in activated HSCs, thereby significantly attenuating liver fibrosis through the enhancement of HSCs apoptosis.84
The differential effects of apoptosis in HSCs and hepatocytes are noteworthy. Specifically, hepatocyte apoptosis is associated with liver injury, inflammation, and fibrosis.85,86 Additionally, macrophages may undergo apoptosis as a protective mechanism, potentially preventing severe hepatic injury. In fact, apoptotic cell death of macrophages may promote the apoptosis of M1 KCs by M2 KCs, thereby alleviating liver injury.87 Moreover, the induction of activated macrophage apoptosis can significantly reduce inflammation and effectively mitigate steatohepatitis88 In addition to the occurrence of apoptosis within macrophages themselves, an important function of macrophages is their efferocytosis. Apoptotic vesicles, including apoptotic bodies, apoptotic macrovesicles, and apoptotic exosomes, which are generated from apoptotic cells, are vital molecules that can be phagocytized by macrophages. The absorption of these vesicles by macrophages can regulate their transformation into an anti-inflammatory type.89 M2 macrophages have been observed to phagocytose apoptotic bone marrow derived mesenchymal stem cells (BM-MSC), thereby inducing the expression of anti-fibrotic genes and facilitating the resolution of hepatic fibrosis.90 Conversely, it has been reported that the ingestion of apoptotic bodies derived from dead hepatocytes by macrophages can promote their transdifferentiation into pro-inflammatory macrophages, thereby exacerbating liver inflammation and fibrosis.91 M1 macrophages have been observed to exert an anti-fibrotic effect by means of recruiting natural killer (NK) cells, which in turn contribute to the enhancement of apoptosis in HSCs.92 The promotion of HSCs apoptosis by NK cells, through the TNF ligand induced apoptosis pathway, has been shown to alleviate hepatic fibrosis.93 Similar to NK cells, NKT cells have antifibrotic effects by directly promoting activated HSCs apoptosis and by producing IFN-γ.94
In summary, the induction of apoptosis in HSCs and macrophages presents a potential mechanism for mitigating liver fibrosis by promoting the death of these activated cells, thereby reducing collagen production and inflammation, respectively. The appropriate induction of apoptosis in hepatocytes is beneficial in alleviating liver injury. However, in cases where more severe damage, such as steatosis induced by NASH, occurs, necroptosis may be a more likely outcome. The induction of necroptosis in hepatocytes can lead to subsequent inflammation, exacerbating hepatic damage and fibrosis. Therefore, it may be prudent to increase the induction of apoptosis in the early stages of liver injury to prevent the occurrence of more severe and uncontrollable injury.
The Crosstalk Between Different Types of PCD
Necroptosis and Apoptosis
There exists a correlation between necroptosis and apoptosis in the context of liver injury. Specifically, in NASH, the upregulation of activating transcription factor 3 (ATF3) can result in an increase in RIPK3 expression. This, in turn, leads to a shift in the mode of cell death of hepatocytes from apoptosis to necroptosis, thereby exacerbating hepatic steatosis.95 A study has shown that in CCL4-induced liver injury, both necroptosis and apoptosis occur simultaneously, and inducing one can suppress the other. Additionally, the study found that promoting apoptosis in hepatocytes contributed to inhibiting necroptosis, effectively alleviating liver injury, including fibrogenesis.96 There appears to be a correlation between the fundamental constituents of apoptosis and the controllers of necroptosis, with caspase-8 being the preeminent factor. RIPK1 has the ability to impede caspase-8, thereby hindering apoptosis.97 Additionally, caspase-8 has a unique role in cleaving RIPK1, which effectively restricts necroptosis.98
Necroptosis and Pyroptosis
Necroptosis and pyroptosis are two distinct forms of PCD characterized by unique initiators and morphological features.99 Despite their differences, both forms share common features such as cell membrane rupture and induction of inflammation. Additionally, both forms are regulated by caspase-10, which cleaves RIPK1 and inhibits NLRP3 inflammasome assembly in macrophages. This regulation significantly reduces the occurrence of necroptosis and pyroptosis in macrophages, leading to a reduction in the release of inflammatory cytokines and ultimately alleviating cirrhosis and suppressing HSCs activation.100 Moreover, necroptosis has the ability to trigger inflammasome activation, leading to pyroptosis and subsequent propagation of inflammation through the release of cytokines. The phosphorylation of MLKL, facilitated by RIPK3, or the upregulation of MLKL can also contribute to the activation of NLRP3 inflammasome, thereby initiating pyroptosis and ultimately resulting in the release of IL-1β.101,102
Apoptosis, Necroptosis and Pyroptosis
PANoptosis is a pro-inflammatory form of PCD that encompasses three distinct modes of cellular demise, namely apoptosis, necroptosis, and pyroptosis. This type of cell death is subject to regulation by a diverse array of molecules, with caspase-6 and caspase-8 representing the most significant substances involved in PANoptosis. Caspase-6 is classified as an apoptotic caspase and serves as a significant effector caspase in PANoptosis through a ZBP1-dependent mechanism. In contrast, caspase-8 is implicated not only in apoptosis but also in necroptosis and pyroptosis, whereby it can facilitate apoptosis and suppress necroptosis; notably, when both necroptosis and apoptosis are impeded, pyroptosis is induced.103–105 The phenomenon of PANoptosis has been linked to liver fibrosis, whereby RIPK1 is responsible for driving PANoptosis in KCs, thereby exacerbating the NASH phenotype, including steatosis, inflammation, and fibrosis.105 It has been demonstrated that inhibiting PANoptosis in hepatocytes can effectively alleviate the liver injury and fibrosis that are induced by fatty liver disease associated with metabolic dysfunctions.106
Ferroptosis with Other Forms of PCD
One characteristic of ferroptosis is the amplification of intracellular ROS. Additionally, the elevation of ROS levels is a shared feature among various forms of PCD, such as apoptosis, necroptosis, and pyroptosis, suggesting the existence of interactive regulatory relationships between ferroptosis and other PCD pathways.107–109 A prior investigation demonstrated that the utilization of GPX4 inhibitor can elicit ferroptosis and concomitantly activate apoptosis by elevating the early growth response-1 (EGR1) level.110 Ferroptosis instigates necroptosis by generating ROS.
It has been reported that the inhibition of ferroptosis can exert a therapeutic effect in NAFLD by ameliorating apoptosis, necroptosis, and proptosis.106 The co-induction of liver fibrosis, ferroptosis, and necroptosis through Yes-associated protein (YAP) overexpression, a transcription factor in the Hippo pathway associated with liver fibrosis, can be alleviated by exosomes derived from human umbilical cord mesenchymal stem cells (huMSC-ex).111 These exosomes have the ability to secrete BECN1, an autophagy regulator, through mediating ferritinophagy, which reduces system Xc-/GPX4. The decrease in system Xc-/GPX4 can significantly increase ferroptosis. Additionally, MSC-ex can induce necroptosis in HSCs, leading to their inactivation.112 Previous research has suggested that ferroptosis can induce pyroptosis, and that MLKL3 serves as both an executor of necroptosis and an initiator of ferroptosis and pyroptosis in myocardial fibrosis induced by pressure overload.112 However, it remains unclear whether these findings can be extrapolated to liver fibrosis.
Autophagy and Other Forms of PCDs
The process of autophagy in hepatocytes induces apoptosis, which in turn stimulates transdifferentiation of HSCs into myofibroblasts by promoting Fas/FasL and elevating the level of P53-upregulated modulator of apoptosis.113 Conversely, reducing expression of NF-κBp65 can induce apoptosis in hepatocytes. Promoting mitophagy can increase apoptosis in HSCs, thereby alleviating liver fibrosis by constraining expression of the anti-apoptotic protein BCL-B, a member of the BCL-2 family.114 Carvedilol significantly reduces fibrotic markers by inhibiting autophagy and promoting apoptosis in activated HSCs.115 The induction of necroptosis, which is dependent on the RIP1/RIP3 pathway, also requires autophagy. In the presence of Atg5, necroptosis can eliminate activated HSCs and improve liver fibrosis.116 Although the relationship between autophagy and pyroptosis in liver fibrosis remains insufficiently understood, insights into their interaction can be gleaned from other liver diseases. For example, in arsenic-induced NAFLD, enhancing autophagy promotes the pyroptosis of hepatocytes.117 However, in free fatty acid induced NASH, suppressing mitophagy can attenuate pyroptosis in hepatocytes.118
The interplay between various forms of PCD remains unclear, with existing data indicating that multiple forms of PCD are concurrently induced during liver fibrosis. Therefore, it is imperative for researchers to investigate their interactions. Of particular note, medications targeting apoptosis and ferroptosis have received more attention than those targeting other forms of PCD, highlighting the need for further in-depth exploration of necroptosis and pyroptosis. However, current research is insufficient to demonstrate the interplay between different forms of PCD in liver fibrosis, necessitating increased investment in this area in the future. Figure 5 illustrates the correlation between PCD and liver fibrosis.
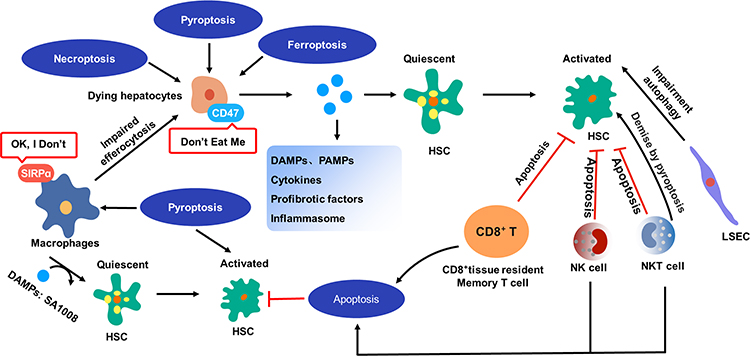 |
Figure 5 The relation between PCD and liver fibrosis. Lytic cell death of hepatocytes results in the release of various substances, including DAMPs, PAMPs, cytokines, profibrotic factors, and inflammasome. These substances have the potential to induce transdifferentiation of quiescent HSCs into activated myofibroblast-like cells, which produce collagens (collagen I and collagen III). Pyroptotic hepatocytes exhibit an increased expression of CD47 on their cytomembrane, while the receptor of CD47 on macrophages, SIRPα, is also upregulated. This upregulation of CD47 and SIRPα facilitates the evasion of macrophage pursuit and clearance. Pyroptotic macrophages possess the capacity to release DAMPs, including SA1008, which can activate HSCs and promote fibrosis. Additionally, proptosis has been found to play a significant role in activating HSCs. The attenuation of liver fibrosis has been observed through the initiation of HSC apoptosis by CD8⁺ tissue resident memory T cells and NK cells. Furthermore, NK cells have been found to constrain liver fibrosis by promoting the senescence of HSCs. Similar to NK cells, NKT cells have antifibrotic effects by directly promoting activated HSCs apoptosis. It has also been observed that impaired autophagy in LSECs can activate HSCs transdifferentiation. |
Conclusions and Perspectives
Liver fibrosis is a complex process that involves various forms of PCD. Currently, there is a dearth of sufficient research to elucidate the interplay between these forms of PCD in liver fibrosis, necessitating further investigation in the future. Whatever, as a multi-cell death pathway, PANoptosis plays a significant role in the development of liver fibrosis, and inhibiting this pathway in different specific cell types can potentially alleviate liver fibrosis. There is little doubt that various forms of cell death in hepatocytes contribute to the progression of liver damage and fibrosis. Consequently, the inhibition of PANoptosis represents a promising therapeutic target for hepatocytes. By limiting hepatocyte death and reducing the release of DAMPs and inflammation, this approach may prove effective in treating liver fibrosis. Additionally, inducing PANoptosis in HSCs may offer a means of reversing fibrosis by eliminating activated HSCs and reducing ECM accumulation.
Further investigation is necessary to determine the predominant form of cell death during different stages of liver injury or fibrosis. This is because the suppression of hepatocellular death may compromise the therapeutic efficacy of cell death. Therefore, more precise treatment options are required for patients with liver injury or fibrosis. Such a therapeutic strategy may yield better treatment outcomes than the use of a simple inhibitor of hepatocyte death alone. In addition, MLKL3, a regulator of necroptosis, has been observed to stimulate pyroptosis and ferroptosis, despite not being a direct molecule in either process. This highlights the need for further investigation into the interplay between ferroptosis and other forms of PCD beyond their direct targets. Such investigation may reveal additional PCD pathways, similar to the discovery of necroptosis as a target in the study of ferroptosis and pyroptosis.
Abbreviations
PCD, programmed of cell death; HSCs, hepatic stellate cells; ECM, extracellular matrix; LSECs, hepatic sinusoid endothelial cells; KCs, kupper cells; HCC, hepatocellular carcinoma; EMT, epithelial-mesenchymal transition; DAMPs, damaged-associated molecular patterns; PBC, primary biliary cholangitis; HBV, hepatitis B virus; TNF, tumor necrosis factor; TNFR, tumor necrosis factor receptor; TRADD, TNF-associated death domain; TRAF2, TNFR-associated factor 2; RIPK, receptor-interacting serine-threonine kinase; CIAP1/2, cellular inhibitor of apoptosis 1/2; LUBAC, linear ubiquitin chain assembly complex; TLR, toll-like receptor; TRIF, domain-containing adapter-inducing interferon-β; MLKL, mixed lineage; NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis; ALD, alcoholic fatty liver disease; HMGB1, high mobility group 1; PAMPs, pathogen-associated molecular patterns; TLR4, toll-like receptor 4; NLRP3, nod-, lrp-, and pyrin domain-comtaning 3; ɑ-SMA, alpha smooth muscle actin; MMP2, matrix metalloproteinase 2; MMP9, matrix metalloproteinase 9; NF-κB, nuclear factor kappa-beta; ASC, apoptosis-associated speck-like protein containing a CARD; GSDMD, gasdermin-D; IRAK1, IL-1 receptor associated kinase 1; TAK1, TGF-β-activated kinase 1; IκB, inhibitor of NF-κB; TFRC, transferrin receptor; STEAP3, six-transmembrane epithelial antigen of prostate 3; ALRP, AIM2-like receptor protein; NOX2, nicotinamide adenine dinucleotide phosphate oxidase; ATGs, autophagy-related proteins; GPXs, cystine-glutamate antiporter system Xc- and glutathione peroxidases; SLC11A2, solute carrier family 11 member 2; NCOA4, cargo receptor nuclear receptor coactivator 4; SLC40A1, solute carrier family 40 member 1; GPX4, glutathione peroxidase 4; Hsp70, heat shock protein 70; BH3-only proteins, BCL-2 homology domain 3 only proteins; MOMP, mitochondrial outer membrane permeabilization; APF-1, apoptotic peptidase activating factor 1; FASL, FAS ligand; FADD, FAS associated via death domain; SIRPα, signal regulatory protein ɑ; NK cells, natural killer cells; NKT cells, natural killer T cells; ROS, reactive oxygen species; GSH, glutathione; BM-MSCs, bone marrow derived mesenchymal stem cells; ATF3, activating transcription factor 3; hucMSC-ex, exosomes derived from human umbilical cord mesenchymal stem cells; CD8+ Trm, CD8+ tissue-resident memory T cells; EGR1, early growth response 1; YAP, yes-associated protein.
Additional Information
Correspondence and requests for materials should be addressed to the corresponding author.
Data Sharing Statement
All data generated or analyzed during this study are included in this published article.
Disclosure
The authors declare that they have no conflicts of interest.
References
1. Tarantino G, Crocetto F, Di Vito C, et al. Association of NAFLD and insulin resistance with non metastatic bladder cancer patients: a cross-sectional retrospective study. J Clin Med. 2021;10(2):346. doi:10.3390/jcm10020346
2. Tarantino G, Balsano C, Santini SJ, et al. It is high time physicians thought of natural products for alleviating NAFLD. Is there sufficient evidence to use them? Int J Mol Sci. 2021;22(24):13424. doi:10.3390/ijms222413424
3. Rauf A, Abu-Izneid T, Khalil AA, et al. Berberine as a potential anticancer agent: a comprehensive review. Molecules. 2021;26(23):7368. doi:10.3390/molecules26237368
4. Pol S. Similar 5-year HCC occurrence in Tenofovir- and Entecavir-treated HBV chronic infection in the French AFEF/ANRS CO22 Hepather cohort. Aliment Pharmacol Ther. 2021;53(5):616–629. doi:10.1111/apt.16197
5. Sanyal AJ, Anstee QM, Trauner M, et al. Cirrhosis regression is associated with improved clinical outcomes in patients with nonalcoholic steatohepatitis. Hepatology. 2022;75(5):1235–1246. doi:10.1002/hep.32204
6. Kisseleva T, Brenner D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastroenterol Hepatol. 2021;18(3):151–166. doi:10.1038/s41575-020-00372-7
7. Muro M, Moya-Quiles MR, Mrowiec A. Humoral response in liver allograft transplantation: a review of the role of anti-Human Leukocyte Antigen (HLA) antibodies. Curr Protein Pept Sci. 2016;17(8):776–784. doi:10.2174/1389203717666160226145101
8. Gaul S, Leszczynska A, Alegre F, et al. Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. J Hepatol. 2021;74(1):156–167. doi:10.1016/j.jhep.2020.07.041
9. Guo R, Jia X, Ding Z, et al. Loss of MLKL ameliorates liver fibrosis by inhibiting hepatocyte necroptosis and hepatic stellate cell activation. Theranostics. 2022;12(11):5220–5236. doi:10.7150/thno.71400
10. Wu A, Feng B, Yu J, et al. Fibroblast growth factor 21 attenuates iron overload-induced liver injury and fibrosis by inhibiting ferroptosis. Redox Biol. 2021;46:102131. doi:10.1016/j.redox.2021.102131
11. An P, Wei -L-L, Zhao S, et al. Hepatocyte mitochondria-derived danger signals directly activate hepatic stellate cells and drive progression of liver fibrosis. Nat Commun. 2020;11(1):2362. doi:10.1038/s41467-020-16092-0
12. Bai L, Kong M, Duan Z, Liu S, Zheng S, Chen Y. M2-like macrophages exert hepatoprotection in acute-on-chronic liver failure through inhibiting necroptosis-S100A9-necroinflammation axis. Cell Death Dis. 2021;12(1):93. doi:10.1038/s41419-020-03378-w
13. Zille M, Karuppagounder SS, Chen Y, et al. Neuronal death after hemorrhagic stroke in vitro and in vivo shares features of ferroptosis and necroptosis. Stroke. 2017;48(4):1033–1043. doi:10.1161/STROKEAHA.116.015609
14. Lin S-Y, Hsieh S-Y, Fan Y-T, et al. Necroptosis promotes autophagy-dependent upregulation of DAMP and results in immunosurveillance. Autophagy. 2018;14(5):778–795. doi:10.1080/15548627.2017.1386359
15. Qian LL, Ji JJ, Jiang Y, et al. Serpina3c deficiency induced necroptosis promotes non-alcoholic fatty liver disease through β-catenin/Foxo1/TLR4 signaling. FASEB J. 2022;36(5):e22316. doi:10.1096/fj.202101345RRR
16. Shi H, Wang X, Li F, et al. CD47-SIRPα axis blockade in NASH promotes necroptotic hepatocyte clearance by liver macrophages and decreases hepatic fibrosis. Sci Transl Med. 2022;14(672):eabp8309. doi:10.1126/scitranslmed.abp8309
17. Chen H, McKeen T, Chao X, et al. The role of MLKL in hepatic ischemia-reperfusion injury of alcoholic steatotic livers. Int J Biol Sci. 2022;18(3):1096–1106. doi:10.7150/ijbs.67533
18. Chen L, Cao Z, Yan L, et al. Circulating receptor-interacting protein kinase 3 are increased in HBV patients with acute-on-chronic liver failure and are associated with clinical outcome. Front Physiol. 2020;11:526. doi:10.3389/fphys.2020.00526
19. Afonso MB, Rodrigues PM, Simão AL, et al. miRNA-21 ablation protects against liver injury and necroptosis in cholestasis. Cell Death Differ. 2018;25(5):857–872. doi:10.1038/s41418-017-0019-x
20. Seehawer M, Heinzmann F, D’Artista L, et al. Necroptosis microenvironment directs lineage commitment in liver cancer. Nature. 2018;562(7725):69–75. doi:10.1038/s41586-018-0519-y
21. Mohammed S, Thadathil N, Selvarani R, et al. Necroptosis contributes to chronic inflammation and fibrosis in aging liver. Aging Cell. 2021;20(12):e13512. doi:10.1111/acel.13512
22. Yan L, Zhang T, Wang K, et al. SENP1 prevents steatohepatitis by suppressing RIPK1-driven apoptosis and inflammation. Nat Commun. 2022;13(1):7153. doi:10.1038/s41467-022-34993-0
23. Afonso MB, Rodrigues PM, Mateus-Pinheiro M, et al. RIPK3 acts as a lipid metabolism regulator contributing to inflammation and carcinogenesis in non-alcoholic fatty liver disease. Gut. 2021;70(12):2359–2372. doi:10.1136/gutjnl-2020-321767
24. Koschel J, Nishanth G, Just S, et al. OTUB1 prevents lethal hepatocyte necroptosis through stabilization of c-IAP1 during murine liver inflammation. Cell Death Differ. 2021;28(7):2257–2275. doi:10.1038/s41418-021-00752-9
25. Wu R, Zhang Y, Xiang Y, et al. Association between serum S100A9 levels and liver necroinflammation in chronic hepatitis B. J Transl Med. 2018;16(1):83. doi:10.1186/s12967-018-1462-2
26. Liu Q, Lei X, Cao Z, et al. TRPM8 deficiency attenuates liver fibrosis through S100A9-HNF4α signaling. Cell Biosci. 2022;12(1):58. doi:10.1186/s13578-022-00789-4
27. Jia Y, Wang F, Guo Q, et al. Curcumol induces RIPK1/RIPK3 complex-dependent necroptosis via JNK1/2-ROS signaling in hepatic stellate cells. Redox Biol. 2018;19:375–387. doi:10.1016/j.redox.2018.09.007
28. Hu M, Wang Y, Liu Z, et al. Hepatic macrophages act as a central hub for relaxin-mediated alleviation of liver fibrosis. Nat Nanotechnol. 2021;16(4):466–477. doi:10.1038/s41565-020-00836-6
29. Blériot C, Dupuis T, Jouvion G, Eberl G, Disson O, Lecuit M. Liver-resident macrophage necroptosis orchestrates type 1 microbicidal inflammation and type-2-mediated tissue repair during bacterial infection. Immunity. 2015;42(1):145–158. doi:10.1016/j.immuni.2014.12.020
30. Wu X, Fan X, McMullen MR, et al. Macrophage-derived MLKL in alcohol-associated liver disease: regulation of phagocytosis. Hepatology. 2023;77(3):902–919. doi:10.1002/hep.32612
31. Wei S, Zhou H, Wang Q, et al. RIP3 deficiency alleviates liver fibrosis by inhibiting ROCK1-TLR4-NF-κB pathway in macrophages. FASEB J. 2019;33(10):11180–11193. doi:10.1096/fj.201900752R
32. Yuan R, Zhao W, Wang -Q-Q, et al. Cucurbitacin B inhibits non-small cell lung cancer in vivo and in vitro by triggering TLR4/NLRP3/GSDMD-dependent pyroptosis. Pharmacol Res. 2021;170:105748. doi:10.1016/j.phrs.2021.105748
33. Kayagaki N, Kornfeld OS, Lee BL, et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature. 2021;591(7848):131–136. doi:10.1038/s41586-021-03218-7
34. Wree A, Eguchi A, McGeough MD, et al. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology. 2014;59(3):898–910. doi:10.1002/hep.26592
35. Xiao L, Magupalli VG, Wu H. Cryo-EM structures of the active NLRP3 inflammasome disc. Nature. 2023;613(7944):595–600. doi:10.1038/s41586-022-05570-8
36. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10(2):417–426. doi:10.1016/S1097-2765(02)00599-3
37. Shi J, Zhao Y, Wang K, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526(7575):660–665. doi:10.1038/nature15514
38. von Moltke J, Trinidad NJ, Moayeri M, et al. Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature. 2012;490(7418):107–111. doi:10.1038/nature11351
39. Mridha AR, Wree A, Robertson AAB, et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol. 2017;66(5):1037–1046. doi:10.1016/j.jhep.2017.01.022
40. Wree A, McGeough MD, Inzaugarat ME, et al. NLRP3 inflammasome driven liver injury and fibrosis: roles of IL-17 and TNF in mice. Hepatology. 2018;67(2):736–749. doi:10.1002/hep.29523
41. Wang Y, Zhang H, Chen Q, et al. TNF-α/HMGB1 inflammation signalling pathway regulates pyroptosis during liver failure and acute kidney injury. Cell Prolif. 2020;53(6):e12829. doi:10.1111/cpr.12829
42. Hernandez C, Huebener P, Pradere J-P, Antoine DJ, Friedman RA, Schwabe RF. HMGB1 links chronic liver injury to progenitor responses and hepatocarcinogenesis. J Clin Invest. 2018;128(6):2436–2451. doi:10.1172/JCI91786
43. Wu J, Zhang M, Xia S, et al. Hepatic HRC induces hepatocyte pyroptosis and HSCs activation via NLRP3/caspase-1 pathway. J Mol Med. 2022;100(12):1787–1799. doi:10.1007/s00109-022-02270-8
44. Zhang Y, Zhangdi H, Nie X, et al. Exosomes derived from BMMSCs mitigate the hepatic fibrosis via anti-pyroptosis pathway in a cirrhosis model. Cells. 2022;11(24):4004. doi:10.3390/cells11244004
45. Chen P, Zhou Y-K, Han C-S, et al. Stem cells from human exfoliated deciduous teeth alleviate liver cirrhosis via inhibition of gasdermin D-executed hepatocyte pyroptosis. Front Immunol. 2022;13:860225. doi:10.3389/fimmu.2022.860225
46. Shu B, Zhou Y-X, Li H, Zhang R-Z, He C, Yang X. The METTL3/MALAT1/PTBP1/USP8/TAK1 axis promotes pyroptosis and M1 polarization of macrophages and contributes to liver fibrosis. Cell Death Discov. 2021;7(1):368. doi:10.1038/s41420-021-00756-x
47. Liu Y, Kong X, You Y, et al. S100A8-mediated NLRP3 inflammasome-dependent pyroptosis in macrophages facilitates liver fibrosis progression. Cells. 2022;11(22):3579. doi:10.3390/cells11223579
48. Wan Y, Zhang W, Huang C, et al. Ursolic acid alleviates Kupffer cells pyroptosis in liver fibrosis by the NOX2/NLRP3 inflammasome signaling pathway. Int Immunopharmacol. 2022;113(Pt A):109321. doi:10.1016/j.intimp.2022.109321
49. Kong D-L, Kong F-Y, Liu X-Y, et al. Soluble egg antigen of Schistosoma japonicum induces pyroptosis in hepatic stellate cells by modulating ROS production. Parasit Vectors. 2019;12(1):475. doi:10.1186/s13071-019-3729-8
50. Xie Z-Y, Xu Y-X, Yao L. Angiotensin II can trigger HSC-LX2 pyroptosis through both classical and non-classical pathways. Life Sci. 2022;307:120878. doi:10.1016/j.lfs.2022.120878
51. Liu W, Liu J, Wang W, Wang Y, Ouyang X. NLRP6 induces pyroptosis by activation of caspase-1 in gingival fibroblasts. J Dent Res. 2018;97(12):1391–1398. doi:10.1177/0022034518775036
52. Zhu Y, Ni T, Deng W, et al. Effects of NLRP6 on the proliferation and activation of human hepatic stellate cells. Exp Cell Res. 2018;370(2):383–388. doi:10.1016/j.yexcr.2018.06.040
53. Jin Y, Liu Y, Xu L, et al. Novel role for caspase 1 inhibitor VX765 in suppressing NLRP3 inflammasome assembly and atherosclerosis via promoting mitophagy and efferocytosis. Cell Death Dis. 2022;13(5):512. doi:10.1038/s41419-022-04966-8
54. Lan P, Fan Y, Zhao Y, et al. TNF superfamily receptor OX40 triggers invariant NKT cell pyroptosis and liver injury. J Clin Invest. 2017;127(6):2222–2234. doi:10.1172/JCI91075
55. Galluzzi L, Vitale I, Aaronson SA, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018;25(3):486–541. doi:10.1038/s41418-017-0012-4
56. Ma X, Lu C, Chen Y, et al. CCT2 is an aggrephagy receptor for clearance of solid protein aggregates. Cell. 2022;185(8):1325–1345.e22. doi:10.1016/j.cell.2022.03.005
57. Liu T, Wang L, Liang P, et al. USP19 suppresses inflammation and promotes M2-like macrophage polarization by manipulating NLRP3 function via autophagy. Cell Mol Immunol. 2021;18(10):2431–2442. doi:10.1038/s41423-020-00567-7
58. Gao J, Wei B, De Assuncao TM, et al. Hepatic stellate cell autophagy inhibits extracellular vesicle release to attenuate liver fibrosis. J Hepatol. 2020;73(5):1144–1154. doi:10.1016/j.jhep.2020.04.044
59. Cao Y, Mai W, Li R, et al. Macrophages evoke autophagy of hepatic stellate cells to promote liver fibrosis in NAFLD mice via the PGE2/EP4 pathway. Cell Mol Life Sci. 2022;79(6):303. doi:10.1007/s00018-022-04319-w
60. Wu H, Chen G, Wang J, Deng M, Yuan F, Gong J. TIM-4 interference in Kupffer cells against CCL4-induced liver fibrosis by mediating Akt1/Mitophagy signalling pathway. Cell Prolif. 2020;53(1):e12731. doi:10.1111/cpr.12731
61. Liu K, Zhao E, Ilyas G, et al. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy. 2015;11(2):271–284. doi:10.1080/15548627.2015.1009787
62. Lodder J, Denaës T, Chobert M-N, et al. Macrophage autophagy protects against liver fibrosis in mice. Autophagy. 2015;11(8):1280–1292. doi:10.1080/15548627.2015.1058473
63. Hammoutene A, Biquard L, Lasselin J, et al. A defect in endothelial autophagy occurs in patients with non-alcoholic steatohepatitis and promotes inflammation and fibrosis. J Hepatol. 2020;72(3):528–538. doi:10.1016/j.jhep.2019.10.028
64. Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26(9):1021–1032. doi:10.1038/cr.2016.95
65. Park E, Chung SW. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. 2019;10(11):822. doi:10.1038/s41419-019-2064-5
66. Chen P, Wu Q, Feng J, et al. Erianin, a novel dibenzyl compound in Dendrobium extract, inhibits lung cancer cell growth and migration via calcium/calmodulin-dependent ferroptosis. Signal Transduct Target Ther. 2020;5(1):51. doi:10.1038/s41392-020-0149-3
67. Xu Y, Li Y, Li J, Chen W. Ethyl carbamate triggers ferroptosis in liver through inhibiting GSH synthesis and suppressing Nrf2 activation. Redox Biol. 2022;53:102349. doi:10.1016/j.redox.2022.102349
68. Wang L, Liu Y, Du T, et al. ATF3 promotes erastin-induced ferroptosis by suppressing system Xc. Cell Death Differ. 2020;27(2):662–675. doi:10.1038/s41418-019-0380-z
69. Yang WS, SriRamaratnam R, Welsch ME, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1–2):317–331. doi:10.1016/j.cell.2013.12.010
70. Qin X, Zhang J, Wang B, et al. Ferritinophagy is involved in the zinc oxide nanoparticles-induced ferroptosis of vascular endothelial cells. Autophagy. 2021;17(12):4266–4285. doi:10.1080/15548627.2021.1911016
71. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59(2):298–308. doi:10.1016/j.molcel.2015.06.011
72. Yu Y, Jiang L, Wang H, et al. Hepatic transferrin plays a role in systemic iron homeostasis and liver ferroptosis. Blood. 2020;136(6):726–739. doi:10.1182/blood.2019002907
73. Luo P, Liu D, Zhang Q, et al. Celastrol induces ferroptosis in activated HSCs to ameliorate hepatic fibrosis via targeting peroxiredoxins and HO-1. Acta Pharm Sin B. 2022;12(5):2300–2314. doi:10.1016/j.apsb.2021.12.007
74. Li L, Wang K, Jia R, et al. Ferroportin-dependent ferroptosis induced by ellagic acid retards liver fibrosis by impairing the SNARE complexes formation. Redox Biol. 2022;56:102435. doi:10.1016/j.redox.2022.102435
75. Tsurusaki S, Tsuchiya Y, Koumura T, et al. Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis. 2019;10(6):449. doi:10.1038/s41419-019-1678-y
76. Ye F, Chai W, Xie M, et al. HMGB1 regulates erastin-induced ferroptosis via RAS-JNK/p38 signaling in HL-60/NRASQ61L cells. Am J Cancer Res. 2019;9(4):730–739.
77. Bi Y, Liu S, Qin X, et al. FUNDC1 interacts with GPx4 to govern hepatic ferroptosis and fibrotic injury through a mitophagy-dependent manner. J Adv Res. 2023. doi:10.1016/j.jare.2023.02.012
78. Wu C, Shen Z, Lu Y, Sun F, Shi H. p53 promotes ferroptosis in macrophages treated with Fe3O4 nanoparticles. ACS Appl Mater Interfaces. 2022;14(38):42791–42803. doi:10.1021/acsami.2c00707
79. Gu Z, Liu T, Liu C, et al. Ferroptosis-strengthened metabolic and inflammatory regulation of tumor-associated macrophages provokes potent tumoricidal activities. Nano Lett. 2021;21(15):6471–6479. doi:10.1021/acs.nanolett.1c01401
80. Batoon L, Koh AJ, Kannan R, McCauley LK, Roca H. Caspase-9 driven murine model of selective cell apoptosis and efferocytosis. Cell Death Dis. 2023;14(1):58. doi:10.1038/s41419-023-05594-6
81. Jiang S, Song MJ, Shin EC, Lee MO, Kim SJ, Park JH. Apoptosis in human hepatoma cell lines by chemotherapeutic drugs via Fas-dependent and Fas-independent pathways. Hepatology. 1999;29(1):101–110. doi:10.1002/hep.510290102
82. Inoue S, Browne G, Melino G, Cohen GM. Ordering of caspases in cells undergoing apoptosis by the intrinsic pathway. Cell Death Differ. 2009;16(7):1053–1061. doi:10.1038/cdd.2009.29
83. Koda Y, Teratani T, Chu P-S, et al. CD8+ tissue-resident memory T cells promote liver fibrosis resolution by inducing apoptosis of hepatic stellate cells. Nat Commun. 2021;12(1):4474. doi:10.1038/s41467-021-24734-0
84. Martí-Rodrigo A, Alegre F, Moragrega ÁB, et al. Rilpivirine attenuates liver fibrosis through selective STAT1-mediated apoptosis in hepatic stellate cells. Gut. 2020;69(5):920–932. doi:10.1136/gutjnl-2019-318372
85. Ding K, Li X, Ren X, et al. GBP5 promotes liver injury and inflammation by inducing hepatocyte apoptosis. FASEB J. 2022;36(1):e22119. doi:10.1096/fj.202101448R
86. Mohs A, Otto T, Schneider KM, et al. Hepatocyte-specific NRF2 activation controls fibrogenesis and carcinogenesis in steatohepatitis. J Hepatol. 2021;74(3):638–648. doi:10.1016/j.jhep.2020.09.037
87. Wan J, Benkdane M, Teixeira-Clerc F, et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: a protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology. 2014;59(1):130–142. doi:10.1002/hep.26607
88. Malhi H, Kropp EM, Clavo VF, et al. C/EBP homologous protein-induced macrophage apoptosis protects mice from steatohepatitis. J Biol Chem. 2013;288(26):18624–18642. doi:10.1074/jbc.M112.442954
89. Zheng C, Sui B, Zhang X, et al. Apoptotic vesicles restore liver macrophage homeostasis to counteract type 2 diabetes. J Extracell Vesicles. 2021;10(7):e12109. doi:10.1002/jev2.12109
90. Li L, Shen S, Shao T, et al. Mesenchymal stem cells attenuate liver fibrosis by targeting Ly6Chi/lo macrophages through activating the cytokine-paracrine and apoptotic pathways. Cell Death Discov. 2021;7(1):239. doi:10.1038/s41420-021-00584-z
91. Ganesan M, Poluektova LY, Enweluzo C, Kharbanda KK, Osna NA. Hepatitis C virus-infected apoptotic hepatocytes program macrophages and hepatic stellate cells for liver inflammation and fibrosis development: role of ethanol as a second hit. Biomolecules. 2018;8(4):113. doi:10.3390/biom8040113
92. Ma P-F, Gao -C-C, Yi J, et al. Cytotherapy with M1-polarized macrophages ameliorates liver fibrosis by modulating immune microenvironment in mice. J Hepatol. 2017;67(4):770–779. doi:10.1016/j.jhep.2017.05.022
93. Radaeva S, Sun R, Jaruga B, Nguyen VT, Tian Z, Gao B. Natural killer cells ameliorate liver fibrosis by killing activated stellate cells in NKG2D-dependent and tumor necrosis factor-related apoptosis-inducing ligand-dependent manners. Gastroenterology. 2006;130(2):435–452. doi:10.1053/j.gastro.2005.10.055
94. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14(7):397–411. doi:10.1038/nrgastro.2017.38
95. Inaba Y, Hashiuchi E, Watanabe H, et al. The transcription factor ATF3 switches cell death from apoptosis to necroptosis in hepatic steatosis in male mice. Nat Commun. 2023;14(1):167. doi:10.1038/s41467-023-35804-w
96. Lu K, Shen S-Y, Luo O-Y, et al. Manipulating PP2Acα-ASK-JNK signaling to favor apoptotic over necroptotic hepatocyte fate reduces the extent of necrosis and fibrosis upon acute liver injury. Cell Death Dis. 2022;13(11):985. doi:10.1038/s41419-022-05353-z
97. Dillon CP, Weinlich R, Rodriguez DA, et al. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell. 2014;157(5):1189–1202. doi:10.1016/j.cell.2014.04.018
98. Newton K, Wickliffe KE, Dugger DL, et al. Cleavage of RIPK1 by caspase-8 is crucial for limiting apoptosis and necroptosis. Nature. 2019;574(7778):428–431. doi:10.1038/s41586-019-1548-x
99. Chen X, He W-T, Hu L, et al. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res. 2016;26(9):1007–1020. doi:10.1038/cr.2016.100
100. Cho M, Dho SH, Shin S, et al. Caspase-10 affects the pathogenesis of primary biliary cholangitis by regulating inflammatory cell death. J Autoimmun. 2022;133:102940. doi:10.1016/j.jaut.2022.102940
101. Conos SA, Chen KW, De Nardo D, et al. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. Proc Natl Acad Sci USA. 2017;114(6):E961–E969. doi:10.1073/pnas.1613305114
102. Wu Q, He X, L-M W, et al. MLKL aggravates Ox-LDL-induced cell pyroptosis via activation of NLRP3 inflammasome in human umbilical vein endothelial cells. Inflammation. 2020;43(6):2222–2231. doi:10.1007/s10753-020-01289-8
103. Zheng M, Karki R, Vogel P, Kanneganti T-D. Caspase-6 is a key regulator of innate immunity, inflammasome activation, and host defense. Cell. 2020;181(3):674–687.e13. doi:10.1016/j.cell.2020.03.040
104. Zheng M, Williams EP, Malireddi RKS, et al. Impaired NLRP3 inflammasome activation/pyroptosis leads to robust inflammatory cell death via caspase-8/RIPK3 during coronavirus infection. J Biol Chem. 2020;295(41):14040–14052. doi:10.1074/jbc.RA120.015036
105. Tao L, Yi Y, Chen Y, et al. RIP1 kinase activity promotes steatohepatitis through mediating cell death and inflammation in macrophages. Cell Death Differ. 2021;28(4):1418–1433. doi:10.1038/s41418-020-00668-w
106. Tong J, Lan X-T, Zhang Z, et al. Ferroptosis inhibitor liproxstatin-1 alleviates metabolic dysfunction-associated fatty liver disease in mice: potential involvement of PANoptosis. Acta Pharmacol Sin. 2023;44(5):1014–1028. doi:10.1038/s41401-022-01010-5
107. Liu H, Lai W, Liu X, et al. Exposure to copper oxide nanoparticles triggers oxidative stress and endoplasmic reticulum (ER)-stress induced toxicology and apoptosis in male rat liver and BRL-3A cell. J Hazard Mater. 2021;401:123349. doi:10.1016/j.jhazmat.2020.123349
108. Weindel CG, Martinez EL, Zhao X, et al. Mitochondrial ROS promotes susceptibility to infection via gasdermin D-mediated necroptosis. Cell. 2022;185(17):3214–3231.e23. doi:10.1016/j.cell.2022.06.038
109. Evavold CL, Hafner-Bratkovič I, Devant P, et al. Control of gasdermin D oligomerization and pyroptosis by the Ragulator-Rag-mTORC1 pathway. Cell. 2021;184(17):4495–4511.e19. doi:10.1016/j.cell.2021.06.028
110. Ding Y, Chen X, Liu C, et al. Identification of a small molecule as inducer of ferroptosis and apoptosis through ubiquitination of GPX4 in triple negative breast cancer cells. J Hematol Oncol. 2021;14(1):19. doi:10.1186/s13045-020-01016-8
111. Zhao W, Lei M, Li J, et al. Yes-associated protein inhibition ameliorates liver fibrosis and acute and chronic liver failure by decreasing ferroptosis and necroptosis. Heliyon. 2023;9(4):e15075. doi:10.1016/j.heliyon.2023.e15075
112. Tan Y, Huang Y, Mei R, et al. HucMSC-derived exosomes delivered BECN1 induces ferroptosis of hepatic stellate cells via regulating the xCT/GPX4 axis. Cell Death Dis. 2022;13(4):319. doi:10.1038/s41419-022-04764-2
113. Tan S, Liu X, Chen L, et al. Fas/FasL mediates NF-κBp65/PUMA-modulated hepatocytes apoptosis via autophagy to drive liver fibrosis. Cell Death Dis. 2021;12(5):474. doi:10.1038/s41419-021-03749-x
114. Ding Q, Xie X-L, Wang -M-M, et al. The role of the apoptosis-related protein BCL-B in the regulation of mitophagy in hepatic stellate cells during the regression of liver fibrosis. Exp Mol Med. 2019;51(1):1–13. doi:10.1038/s12276-018-0199-6
115. Meng D, Li Z, Wang G, Ling L, Wu Y, Zhang C. Carvedilol attenuates liver fibrosis by suppressing autophagy and promoting apoptosis in hepatic stellate cells. Biomed Pharmacother. 2018;108:1617–1627. doi:10.1016/j.biopha.2018.10.005
116. Sun S, Li Z, Huan S, et al. Modification of lysine deacetylation regulates curcumol-induced necroptosis through autophagy in hepatic stellate cells. Phytother Res. 2022;36(6):2660–2676. doi:10.1002/ptr.7483
117. Qiu T, Pei P, Yao X, et al. Taurine attenuates arsenic-induced pyroptosis and nonalcoholic steatohepatitis by inhibiting the autophagic-inflammasomal pathway. Cell Death Dis. 2018;9(10):946. doi:10.1038/s41419-018-1004-0
118. Gao X, Ruan Y, Zhu X, et al. Deoxycholic acid promotes pyroptosis in free fatty acid-induced steatotic hepatocytes by inhibiting PINK1-mediated mitophagy. Inflammation. 2022;45(2):639–650. doi:10.1007/s10753-021-01573-1
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
PANoptosis: A Cell Death Characterized by Pyroptosis, Apoptosis, and Necroptosis
Shi C, Cao P, Wang Y, Zhang Q, Zhang D, Wang Y, Wang L, Gong Z
Journal of Inflammation Research 2023, 16:1523-1532
Published Date: 12 April 2023
The Emerging Roles of Pyroptosis, Necroptosis, and Ferroptosis in Non-Malignant Dermatoses: A Review
Teng Y, Xu D, Yang X, Tang H, Tao X, Fan Y, Ding Y
Journal of Inflammation Research 2023, 16:1967-1977
Published Date: 6 May 2023
Autophagy, Pyroptosis and Ferroptosis are Rising Stars in the Pathogenesis of Diabetic Nephropathy
Li X, Gao L, Li X, Xia J, Pan Y, Bai C
Diabetes, Metabolic Syndrome and Obesity 2024, 17:1289-1299
Published Date: 13 March 2024
Regulated Cell Death of Alveolar Macrophages in Acute Lung Inflammation: Current Knowledge and Perspectives
Xia S, Gu X, Wang G, Zhong Y, Ma F, Liu Q, Xie J
Journal of Inflammation Research 2024, 17:11419-11436
Published Date: 21 December 2024
Programmed Cell Death in Rheumatoid Arthritis
Tong L, Qiu J, Xu Y, Lian S, Xu Y, Wu X
Journal of Inflammation Research 2025, 18:2377-2393
Published Date: 18 February 2025