Back to Journals » ImmunoTargets and Therapy » Volume 14
Myeloid Cells in the Immunosuppressive Microenvironment as Immunotargets in Osteosarcoma
Authors Sholevar CJ ![]() , Liu NM, Mukarrama T, Kim J
, Liu NM, Mukarrama T, Kim J ![]() , Lawrence J, Canter RJ
, Lawrence J, Canter RJ
Received 31 December 2024
Accepted for publication 11 March 2025
Published 19 March 2025 Volume 2025:14 Pages 247—258
DOI https://doi.org/10.2147/ITT.S485672
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Michael Shurin
Cyrus J Sholevar,1 Natalie M Liu,1 Tasneem Mukarrama,2 Jinhwan Kim,2 Jessica Lawrence,3 Robert J Canter1
1Department of Surgery, Division of Surgical Oncology, University of California Davis, Sacramento, CA, USA; 2Biomedical Engineering, University of California Davis, Sacramento, CA, USA; 3Department of Surgical & Radiological Sciences, School of Veterinary Medicine, University of California Davis, Davis, CA, USA
Correspondence: Robert J Canter, Research Building III, University of California Davis, 4645 Second Ave Room 2200D, Sacramento, CA, 95817, USA, Email [email protected]
Abstract: Osteosarcoma is an aggressive primary malignant bone tumor associated with high rates of metastasis and poor 5-year survival rates with limited improvements in approximately 40 years. Standard multimodality treatment includes chemotherapy and surgery, and survival rates have remained stagnant. Overall, response rates to immunotherapy like immune checkpoint inhibitors have been disappointing in osteosarcoma despite exciting results in other epithelial tumor types. The poor response of osteosarcoma to current immunotherapies is multifactorial, but a key observation is that the tumor microenvironment in osteosarcoma is profoundly immunosuppressive, and increasing evidence suggests a significant role of suppressive myeloid cells in tumor progression and immune evasion, particularly by myeloid-derived suppressor cells. Targeting suppressive myeloid cells via novel agents are attractive strategies to develop novel immunotherapies for osteosarcoma, and combination strategies will likely be important for durable responses. In this review, we will examine mechanisms of the immunosuppressive microenvironment, highlight pre-clinical and clinical data of combination strategies including colony-stimulating factor 1 (CSF-1) receptor, phosphoinositide 3-kinase (PI3K), CXCR4, and checkpoint inhibition, as well as the role of canine models in elucidating myeloid cells as targets in osteosarcoma immunotherapy.
Keywords: MDSC, macrophage, immunotherapy, sarcoma, tumor microenvironment
Biological Characteristics and Treatment Resistance of Osteosarcoma
Osteosarcoma (OSA) is the most common primary malignant bone tumor in children and adolescent patients. It is often characterized by a frequently aggressive growth pattern and propensity for metastasis, leading to significant morbidity and mortality. Approximately 20% of OSA patients are found to have metastatic spread at the time of diagnosis.1–3 Furthermore, the presence of metastases is the most significant predictor of survival, and metastatic OSA has a significantly lower 5-year survival rate of 24% when compared to 76% for localized disease.4
Primary OSA typically occurs in the metaphysis of long bones. It has a clear predilection for metastatic dissemination to the lungs.5 The second most common site of metastasis is other bones (20%).5 Other less common sites include brain, lymph nodes, liver, peritoneum, and adrenal glands.5–11 Additionally, OSA often displays synchronous metastases, in which metastases develop concurrently with the primary tumor.12 In contrast, metachronous metastases may occur after initial treatment, and the clinical setting of relapse/recurrence after prior combined modality treatment typically portends aggressive tumor biology and a poor prognosis.12 Over the past 40 years, while there has been increased research in novel therapeutic targets including insulin-like growth factor (IGF) and vascular endothelial growth factor (VEGF), survival rates for OSA have remained stagnant, and despite the clear success of traditional multiagent chemotherapy, which includes high-dose methotrexate, doxorubicin, and cisplatin/platinum commonly referred to as MAP regimen, patients develop resistance leading to limited treatment options.13–15 A greater understanding of OSA biology and its immunosuppressive impact is therefore required to understand the immunobiology of these tumors and foster the development of novel therapies to improve long-term patient outcomes.
Myeloid Cells and the Immunosuppressive Tumor Microenvironment in Osteosarcoma
Mounting evidence indicates a significant role of myeloid cells in disease progression and metastasis that may be targeted for therapeutic benefit. The tumor microenvironment (TME) of OSA consists of a complex network of osteoblasts, osteocytes, stromal cells, vascular cells, immune cells, and an extracellular matrix.16 Myeloid cells are the predominant cell type with sequencing analysis showing they comprise approximately 44–53% of all cells and the majority of immune cells in the TME, with moderate variability between patients and across studies.17–20 Myeloid cells (as described below) are a significant component of both adaptive and innate immune responses through a diversity of functions including phagocytosis, activation of bactericidal mechanisms, killing of antibody-coated cells or parasites, promotion of allergic responses, antigen presentation, and many more.21,22 Myeloid cells including monocytes, dendritic cells (DCs), macrophages, neutrophils, and others are involved in a host of activities related to tumor surveillance including phagocytosis, antigen presentation, and regulation of tumor inflammation.19,23,24 There is significant heterogeneity across the many different functional states of myeloid cells, particularly macrophages and neutrophils, and specifically in the tumor microenvironment, where these cells can promote tumor growth, cancer cell maintenance, and impair immunosurveillance.25,26 An overview of the anti-tumor and pro-tumorigenic dichotomous effects of various myeloid cells is provided in Table 1.
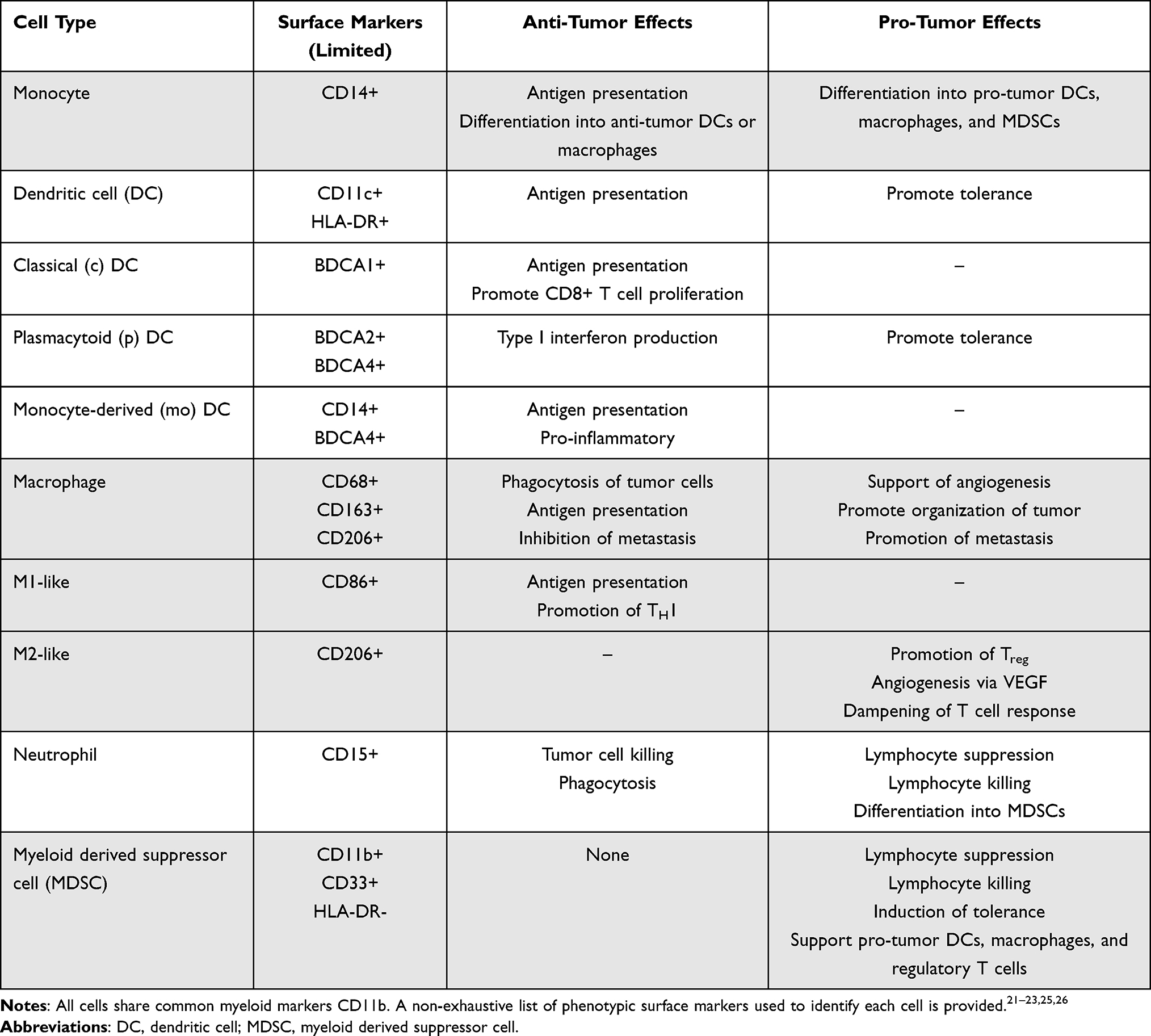 |
Table 1 Overview of Myeloid Cell Function in Cancer |
Myeloid derived suppressor cells (MDSC) are a diverse group of pathologically activated myeloid cells with potent immunosuppressive function, identified in a variety of inflammatory conditions including trauma, sepsis, auto-immunity, surgery, obesity, and malignancy.27–29 MDSCs can be broadly grouped as granulocytic (G-)/polymorphonuclear (PMN-) or monocytic (M-) based on their origin of either granulocytic or monocytic myeloid precursor cells; a third group of myeloid progenitor cells with neither lineage has also been described as “early” eMDSCs.28 MDSCs suppress natural killer (NK) and T cell proliferation and function both directly and indirectly through a multitude of distinct mechanisms including generation of reactive oxygen species (ROS) and nitric oxide (NO), depletion of L-arginine by arginase-1, secretion of TGFβ, and induction of T regulatory cells by indoleamine 2,3-dioxygenase (IDO) and IL-10. MDSCs also promote exhaustion and growth arrest of effector NK and T cells via binding of PD-1-PD-L1 and TIGIT-CD155 receptor ligand interactions, among other negative regulatory pathways.28–31 Considering the heterogeneity of myeloid cells and their diverse functions, significant effort has been made to better characterize the immune TME.
One study utilizing single-cell RNA sequencing (scRNAseq) analysis sought to create an archive of myeloid cells in OSA and further elucidate their potential roles.19 By characterizing OSA cells into high and low copy number variation (CNV) cells, investigators demonstrated that OSA cells utilize unique methods of immune evasion to promote immunosuppression, including repression of MHC-I on tumor cells as a method to evade macrophages and thus killing by cytotoxic T cells.19 Additional RNAseq data have supported the complexity of OSA cell types, pointing at evidence for potential modifiers of tumor associated M1 and M2 macrophages.20 Macrophages show significant diversity in their functions and have been described by multiple phenotypes including M1, classically described as anti-tumor, and M2, classically described as pro-tumor.32,33 M1 and M2 phenotypes were initially described as dichotomous states of macrophages activated in vitro after exposure to microbial components, resulting in either enhanced microbial elimination driven by M1 activation or tolerance driven by M2 activation.34,35 While key transcription factor families have been associated with both phenotypes, tolerance does not suppress all macrophage responses, and transcriptional studies of macrophages across tissues and pathologic states have shown a spectrum of activation states with M1 and M2 polarization serving as extremes.23,26 Furthermore, epigenetic regulation and microRNAs have been identified as mechanisms that modulate macrophage activation states in various conditions.36–38
Previous attempts to target myeloid cells in OSA included the use of Liposomal Muramyl tri-peptide phosphatidylethanolamine (L-MTP-PE) as an immune modulator. L-MTP-PE can activate monocytes and macrophages to target tumor cells.39 In high-grade OSA, the incorporation of L-MTP-PE into the therapeutic regimen has demonstrated favorable outcomes and thereby received regulatory approval in the European Union, but not in the US.40 Despite increased macrophage activity, overcoming macrophage evasion by OSA will likely also require additional targeting. For example, messenger RNA and proteomic profiling have suggested that CD24 expression on OSA cells repress phagocytosis and macrophage activity, potentially contributing to aggressive biologic behavior.41 Future strategies to address both macrophage phagocytic activity and avoidance of myeloid suppressive tactics may be promising. Efforts to identify specific new targets directly contributing to myeloid-orchestrated immune evasion methods will therefore shape novel drug development.
Standard treatment for high-grade resectable OSA for the last 40 years includes neoadjuvant chemotherapy, resection, and adjuvant chemotherapy. While systemic chemotherapy has traditionally been viewed as immunosuppressive, several agents utilized against OSA have been shown to alter the tumor microenvironment in ways that may support anti-tumor immune responses. Cisplatin upregulates MHC class I expression on tumor cells as well as increases the frequency of tumor infiltrating lymphocytes (TILs).42 Doxorubicin can decrease the frequencies of circulating and intratumoral MDSCs in mouse models of breast cancer, concurrently increasing frequencies of effector lymphocytes including CD8+ T cells and NK cells, and induced apoptosis of circulating human MDSCs from patients with breast cancer.43 One trial studying induction of doxorubicin followed by anti-PD-1 checkpoint therapy (nivolumab) versus doxorubicin alone showed increases in intratumoral T cell clonality with doxorubicin followed by nivolumab compared to nivolumab alone.44 Exploiting potential immunomodulatory effects of chemotherapy with specific immunotherapy agents may be a thoughtful approach to incorporating novel strategies, as immunotherapy alone may not provide sustained responses.45
Another recent study analyzed RNA sequencing data from the Therapeutically Applicable Research to Generate Effective Treatments (TARGET) database to assess infiltrating immune cell populations in OSA. Consistent with the known immunosuppressive TME, M2 and M0 macrophages were the most abundant infiltrating immune cell in naïve OSA.46 M0 macrophages were inversely correlated with cytotoxic CD8+ T cells, and higher frequencies of regulatory T cells were associated with higher likelihood of necrosis following neoadjuvant therapy. Following neoadjuvant chemotherapy (methotrexate, doxorubicin, cisplatin, and ifosfamide) and tumor resection, investigators compared immune populations in surgical sections compared to patient-matched biopsies utilizing immunohistochemistry and immunofluorescence microscopy. Macrophages were roughly 8 times more prevalent than the next most common cell type, T cells, prior to treatment; macrophage subsets were unable to be identified in the tissue microarray. There was also a significant increase in CD8+ T cells and decrease in MDSCs after neoadjuvant chemotherapy. Of note, low MDSC infiltration in biopsy sections was associated with strong pathological response to neoadjuvant therapy. Interestingly, PD-L1 expression on immune cells significantly increased after neoadjuvant chemotherapy, suggesting possible synergy when these therapies are combined, with relevant attention to how these therapies are sequenced. Importantly, increased density of MDSCs both at diagnosis and after neoadjuvant chemotherapy was associated with an increased likelihood of progressive disease compared to stable disease or partial response by RECIST 1.1 criteria; survival analysis was not performed in this study.46 Collectively, these results suggest that chemotherapy may be, at least transiently, capable of modulating the TME to be more immunogenic, and the addition of immune engagers at specific time-points might sustain this response.
Myeloid Cells in Metastasis
The metastatic capability of OSA is multifactorial, and undoubtedly influenced by various genetic mutations. Studies have associated metastatic OSA with germline mutations in TP53 (Li-Fraumeni Syndrome) and protein tyrosine phosphatase receptor type Q (PTPRQ).47 Additional somatic mutations in signaling pathways such as PI3K-AKT and MAPK have also been implicated in OSA metastasis.48 Likely due in part to the genetic heterogeneity of metastatic OSA, this disease is known to be highly resistant to standard chemotherapy regimens, especially in the setting of relapse/recurrence after standard MAP chemotherapy regimen.49,50 While clearly important to mechanisms of metastasis and immune evasion, genetic mutations in the tumor do not seem to be directly related to the phenotype, function, or frequency of myeloid cells in the TME, although rigorous data exploring these aspects of OSA biology are not available at this time.
Targeting Suppressive Myeloid Cells
Tumor-associated macrophages (TAMs) are a heterogeneous group of macrophages that play significant roles in tumor growth and progression through actions in the TME. TAMs may arise from circulating bone marrow-derived monocytes, M-MDSCs, or local yolk-sac progenitors.21,51,52 Considering their heterogeneity and plasticity, as well as the context-dependent gene expression and function of macrophages and specifically TAMs, nomenclature surrounding subsets of these cells continues to evolve.26,53 Thus, although TAMs can clearly display opposing pro-tumorigenic and anti-tumorigenic roles, specifically assigning one or the other of these states in specific pre-clinical or clinical studies may be imprecise given macrophage heterogeneity and plasticity both biologically and how these categories are assessed experimentally. These nuances may impact study results across studies and experimental readouts. Nevertheless, TAMs directly support tumor growth by promoting angiogenesis through VEGF signaling, improving tumor cell interaction with the extracellular matrix via osteonectin secretion, and promoting epithelial–mesenchymal transition.26,52 TAMs also indirectly promote tumor growth by suppressing effector cells including T and NK cells.47,51 TAMs express the ligands for inhibitory receptors PD-1 and CTLA-4, secrete inhibitory cytokines IL-10 and TGFβ, and suppress T cell activity by depleting L-arginine, similar to the M-MDSCs they can arise from (discussed below).52
One preclinical evaluation of myeloid cells in a mouse model of OSA showed that differentiation of TAMs is mediated by colony-stimulating factor 1 (CSF-1) receptor signaling.54 CSF-1 recruits monocytes from the periphery to the TME, drives differentiation into macrophages, and further induces polarization of macrophages into the M2-like phenotype as opposed to the M1-like phenotype induced by CSF-2.52,55 Bone-marrow derived macrophages showed high expression of CD206, associated with the M2-like phenotype, when exposed to CSF-1 or tumor-conditioned media generated from the OSA cell line LM8 compared to control media. CSF-1 also induced chemotaxis of macrophages in transwell experiments, demonstrating its role in recruitment of macrophages to the OSA TME. These in vitro experiments suggest that interactions between tumor cells and the immune system can promote a feed forward loop ultimately favoring tumor progression and immune evasion. The investigators further showed that inhibition of CSF-1R with pexidartinib reduced both this M2-like polarization and chemotaxis. In another pre-clinical study, pexidartinib showed decreased tumor growth and prolonged time to spontaneous lung metastases in a mouse model of OSA utilizing LM8 tumors with a dose-dependent response.54
Clinical trials have shown overall weak evidence supporting the use of pexidartinib in malignancy despite these encouraging pre-clinical results; we have not identified any completed clinical trials to date investigating CSF-1/CSF-1R inhibition in OSA specifically, although some Phase I and II trials included small numbers of OSA as advanced solid tumors or advanced sarcomas (NCT01004861, NCT05093322, NCT02584647), and many Phase II trials evaluating pexidartinib did not show significant efficacy for a variety of solid tumors.56,57 Conversely, the ENLIVEN trial, a completed Phase III trial including patients with tenosynovial giant cell tumor (TGCT), showed that pexidartinib led to significantly higher overall response than placebo, leading to the first FDA-approved systemic therapy for this rare mesenchymal tumor.58 TGCT cells overexpress CSF-1, recruiting the monocytes and macrophages that physically compose most of the tumor and promoting polarization to pro-tumor M2-like phenotypes. As such, inhibition of the CSF-1/CSF-1R axis is likely to have unique benefit in this tumor type compared to other solid tumors, evidenced by these mixed clinical results. Combination therapies that target both TAMs and their interactions with other immune cells in the TME, such as T cells and MDSCs, may have synergistic anti-tumor effects. One such strategy involves CSF-1/CSF-1R inhibition combined with checkpoint inhibition, and the results of exciting ongoing clinical trials in sarcoma (NCT04242238) as well as other solid tumors (NCT05438420) are eagerly anticipated.
Myeloid Derived Suppressor Cells
The numerous functions of MDSCs are mediated by their effects on many cellular processes. T cell activation is inhibited by the depletion of usable L-arginine via hydrolysis by arginase-1 produced by MDSCs and via the glycation of L-arginine by methylglyoxal accumulated by MDSCs and directly transferred to T cells.29 Chemotaxis and recognition of antigens are both suppressed by the nitration of the T cell receptor, chemokines, and MHC molecules by ROS and NOS2. T cell exhaustion is induced by PD-L1 expression on MDSCs.59 The induction of Tregs is mediated by IDO produced by MDSCs, and myeloid differentiation is further skewed towards MDSCs and away from dendritic cells with anti-tumor effect by synthesis of S100A9, resulting in indirect suppression of T cell activation.60 NK cell function is suppressed by the inhibition of IFNγ and TNFα production as a result of TIGIT binding and ROS production by MDSCs.61,62 MDSCs additionally drive macrophage polarization towards pro-tumor TAMs via IL-10 production, reinforcing an immunosuppressive milieu.62 Strategies to target MDSCs include interrupting the above mechanisms as well as preventing their differentiation and recruitment into the TME (see Figure 1). Combination therapies are likely necessary for significant clinical benefit considering the impact of MDSCs on so many diverse cellular processes.
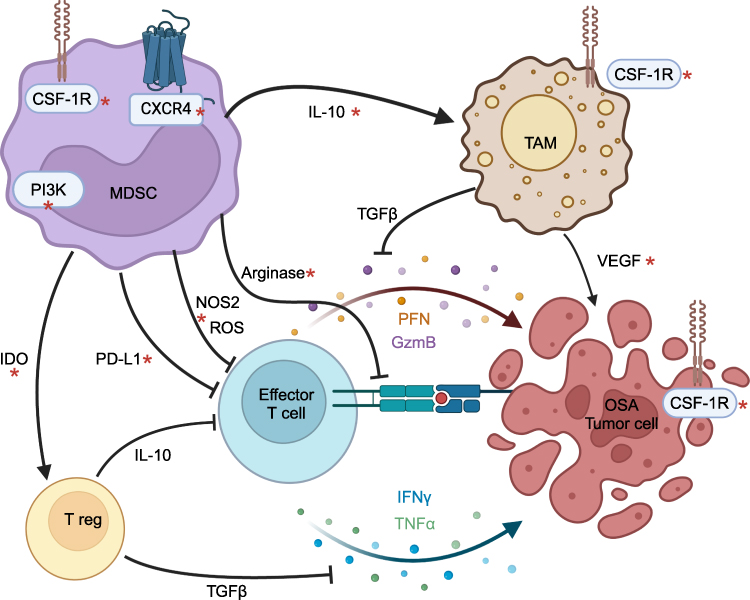 |
Figure 1 Mechanisms of myeloid-derived suppressor cell function as therapeutic targets in osteosarcoma. Notes: Schematic showing mechanisms of myeloid-derived suppressor cell (MDSC) suppression through direct effects on effector lymphocytes and indirectly through regulatory T cells and tumor-associated macrophages (TAMs). Effector T cells are activated by binding of the TCR to MHC class I on tumor cells and secrete perforin (PFN), granzyme B (GzmB), interferon gamma (IFNγ), and tumor necrosis factor alpha (TNFα) to induce apoptosis of osteosarcoma (OSA) tumor cells. MDSCs inhibit T cell activation by depleting L-arginine and prevent the activation, proliferation, and secretion of these T cell soluble factors by production of reactive oxygen species (ROS) and nitric oxide. Natural killer cell function is suppressed through similar mechanisms (not shown). MDSCs induce T cell exhaustion via PD-L1 expression. MDSCs produce IL-10 and indoleamine 2.3-dioxygenase (IDO), promoting differentiation of tumor-associated macrophages (TAMs) and regulatory T cells (T regs), respectively, that both subsequently suppress T cell function via TGFβ and other mechanisms not shown. TAMs also exhibit direct tumorigenic effects including supporting angiogenesis via VEGF. MDSC survival and function are driven by the G protein coupled receptor CXCR4, colony-stimulating factor 1 receptor (CSF-1R), and phosphatidylinositide 3-kinases (PI3K). TAM differentiation and OSA growth are also driven by CSF-1R signaling. Mechanisms with potential for therapeutic targeting, particularly in combination for synergistic effect, are marked by a red asterisk*. Created in BioRender. Sholevar, C. (2025) https://BioRender.com/n14l999. |
Phosphatidylinositide 3-kinases (PI3Ks) have been shown to activate MDSCs in mice.63 A mouse model utilizing the K7M2 OSA line showed increased frequencies of G-MDSCs in tumor-bearing mice compared to control, and MSDC function was verified by demonstrating decreased T cell proliferation, interferon (IFN) γ production, and cytotoxicity in killing assays. G-MDSCs showed increased expression of the PI3K δ/γ isoforms compared to tumor-infiltrating lymphocytes (TILs), and inhibition of the PI3K δ/γ isoforms with (S)-(-)-N-[2-(3-Hydroxy-1H-indol-3-yl)-methyl]-acetamide (SNA) reversed MDSC suppression of T cell proliferation as well as IFNγ production. While SNA alone did not affect tumor growth nor MDSC infiltration, it increased PD-L1 expression on infiltrating MDSCs and CD8+ T cells, promoting immune suppression. Subsequent co-administration of anti-PD1 with SNA showed significant increases in survival, decreased tumor growth, increased CD8+ T cell infiltration, and increased IFNγ production on CD8+ T cells. Depletion of CD8+ T cells reversed these effects, suggesting the pro-tumor effects of MDSCs in this model were T cell dependent.63
Overall, immune checkpoint inhibition has not shown significant clinical benefit for patients with OSA in clinical trials.64 One murine model of OSA utilizing the K7M2 cell line in immune competent BALB/C mice found significant PD-L1 expression on stromal cells in the majority of osteosarcoma tissues.65 Expression of the chemokine receptor CXCR4 in tumor tissues was associated with decreased CD8+ T cell infiltration as well as significantly shorter overall survival. CXCR4 was found to be expressed on most tumor infiltrating MDSCs, at higher frequencies than splenic MDSCs. Combination blockade with anti-PD-1 and the CXCR4 antagonist AMD3100 significantly decreased tumor growth and prolonged survival compared to either monotherapy. CXCR4 antagonism increased CD8+ T cell infiltration in the tumor microenvironment, and combination blockade significantly increased activation of effector T cells as measured by Ki67, granzyme B, IFNγ, and TNFα expression. Checkpoint inhibition did not show an effect on MDSC infiltration, while AMD3100 and combination blockade both resulted in lower frequencies of tumor infiltrating MDSCs. MDSC migration was found to be dependent on CXCL12/stromal cell-derived factor-1, the ligand for CXCR4, and was inhibited by AMD3100. MDSCs exposed to AMD3100 showed increased apoptosis and decreased levels of phosphorylated Akt.65 This murine model overall demonstrated that AMD3100 inhibits MDSC migration mediated by the CXCL12/CXCR4 axis, decreased MDSC survival through CXCR4 downstream signaling via the Akt pathway, and ultimately increased effector T cell infiltration in the tumor microenvironment.
Barriers to Targeting Immunosuppressive Myeloid Cells
Many of the in vitro and in vivo pre-clinical studies discussed above, including checkpoint inhibition, CSF-1/CSF-1R blockade, PI3K inhibition, and CXCR4 targeting, showed profound anti-tumor effects while subsequent clinical trials did not demonstrate meaningful clinical benefit. As previously discussed, immune populations in the OSA TME show significant heterogeneity between patients, across timepoints, and in response to chemotherapy, and macrophage polarization as well as MDSC suppressive capability can be influenced by tumor cells.19,24,26 The dynamic nature of myeloid cells suggests an inherent resistance to single-agent therapies targeting an individual functional mechanism or pathway, and changes in myeloid cells over time may contribute to the negative clinical trials completed thus far targeting individual mechanisms of suppression by myeloid cells. An additional challenge in effectively targeting myeloid cells lies in the multitude of mechanisms that pro-tumor macrophages and MDSCs use to dampen immune responses and promote tumor survival. Many of these mechanisms are either directly or indirectly connected, as shown in Figure 1, and inhibiting a single pathway is unlikely to produce durable effects in patients. These difficulties are compounded by the fundamental limitations of using transgenic mouse models to study complex immune interactions.
Leveraging Canine Models to Study Immunosuppressive Myeloid Infiltration
When evaluating the role of immunosuppressive MDSC and M2 macrophages within the OSA TME with the intent to identify novel drug targets, it is essential to have a spontaneous mammalian tumor model in which tumor immunodynamics are analogous. Pre-clinical murine models have traditionally been used to evaluate the complex biological pathways that contribute to local tumor progression and metastasis.66 Mouse models also provide an opportunity to examine therapeutic intervention in vivo; however, the natural development of OSA in mice is rare, thus most models involve xenograft or transgenic mouse models that lack an intact immune system and are contained within controlled environmental conditions.67–70 Collectively, these factors likely contribute to the lack of success when translating novel immunotherapy agents to human clinical trials.68–70 The incorporation of pet dogs with naturally-occurring OSA provides an opportunity to evaluate the immune system in an outbred, patient-based population that is complementary to humans with OSA. Importantly, dogs are a heterogeneous group of large animals with an intact immune system that share the same complex environment as humans. Canine OSA further shows significant homology to human OSA, and occurs in relatively high frequency compared to humans, contributing tremendous value in the development of novel therapeutic approaches.71–74 Indeed, preclinical success of the incorporation of L-MTP with surgery and adjuvant doxorubicin-cyclophosphamide in canine osteosarcoma aided in its translation to the clinic.75,76
Similar to human OSA, macrophages and MDSCs have gained significant traction as potential immunotherapy targets in pet dogs with OSA, as their recruitment and activation may contribute to progression and therapeutic resistance.77,78 Increased pre-treatment peripheral monocyte concentrations have been correlated with shorter disease-free intervals in dogs with OSA compared to dogs with low peripheral monocyte concentrations, which has been demonstrated in pediatric OSA as well.79,80 One primary limitation in evaluating peripheral monocyte counts is that subtypes are not evaluated, which could alter the potential OSA TME. For example, in mouse models of OSA and naturally-occurring pet dogs with OSA, animals with infections (eg, postoperative infection) have delayed time to metastasis compared to animals without infections.81,82 Importantly, depletion of macrophages/monocytes or natural killer cells reversed the positive effects of concurrent infection and OSA, supporting that macrophages are not purely immunosuppressive and can contribute to upregulated anti-tumor immunity.81 Another canine study utilized flow cytometry and quantitative RT-PCR to evaluate monocyte phenotype while also evaluating migration ability.77 Investigators found that, when compared to healthy dogs, OSA-bearing dogs had depressed chemokine receptors and reduced chemotactic function.
Finally, in a recent study that evaluated lung tissue from dogs with treatment-naïve OSA or dogs without cancer, authors highlighted altered monocyte/macrophage dynamics that may shed light on the metastatic cascade.83 Investigators compared multiplex immunofluorescence detection of M2 macrophages (CD204+ and CD206+ macrophages) and CD11d+ bone marrow-derived cells (presumptive bone marrow-derived monocyte origin) in lung tissue of OSA-bearing dogs and non-cancer bearing dogs. In OSA-bearing dogs without pulmonary metastasis, a significantly higher population of M2 macrophages and CD11d+ bone marrow-derived monocytes was measured compared to control dogs. However, in OSA-bearing dogs with established pulmonary metastasis, the proportion of CD11d+ bone marrow-derived monocytes was significantly lower compared to dogs without metastasis, leading the authors to postulate that these bone-marrow derived cells aid in the establishment of the pre-metastatic niche. Therapies that limit infiltration of M2 macrophages and bone marrow-derived cells may hinder distant metastasis. Collectively, these data serve to highlight the significance of simultaneously evaluating the peripheral monocyte compartment in the context of the OSA TME to better understand monocyte/macrophage dynamics. As novel therapeutic approaches are tested, study design could incorporate similar techniques to evaluate M2 macrophage and MDSC populations within lung tissue.
MDSCs are increased in dogs with OSA and show similar suppression of T cell proliferation as human MDSCs.84 Additionally, M-MDSC, PMN-MDSC, and eMDSC populations that are similar to human MDSC subtypes have been identified in several canine tumor types.84,85 One canine clinical trial in metastatic OSA showed significant clinical benefit in blocking monocyte recruitment and MDSC migration with the combination of losartan, an angiotensin II receptor blocker (ARB) widely used as an antihypertensive agent in human patients, and toceranib, a tyrosine kinase inhibitor.86 Losartan, in addition to its ARB activity, has also been observed by Regan et al to significantly inhibit signaling of the chemokine CCL2 through its receptor CCR2, an axis implicated in OSA tumor progression.87 Toceranib is a canine tyrosine kinase inhibitor that targets KIT, VEGFR-2, PDGFR and Flt-3 to modulate the TME, similar to human sunitinib. Interestingly, sunitinib has been shown to decrease MDSC accumulation in some solid tumors, and may synergize well with traditional immunotherapy approaches, thus supporting the rationale to combine myeloid targeting agents.86,88,89
Leveraging this powerful pet dog model is an exciting opportunity to address several knowledge gaps regarding MDSCs and immunosuppressive M2 macrophages. Because pet dogs are large, repeated peripheral blood and tumor sampling is feasible, and may shed light on the immunodynamics of myeloid populations migrating from blood to tumor. These immune responses can be directly tied to objective tumor responses through standard advanced imaging approaches used in humans, as novel combination immunotherapy approaches continue development.
Summary and Conclusions
Osteosarcoma is an aggressive malignancy that has not responded to current T cell-based immunotherapies. While OSA shows relatively poor lymphocytic infiltration, the immune TME is populated by suppressive myeloid cells, particularly pro-tumor TAMs and MDSCs. Challenges in targeting these cells for therapeutic benefit include their phenotypic and functional plasticity as well as the variety of connected but distinct suppressive mechanisms. Strategies targeting individual suppressive pathways based on murine models have not yet demonstrated clinical benefit in osteosarcoma as they have in other tumors. Combination therapies targeting multiple suppressive pathways are likely to have translational relevance, and canine models for cancer immunotherapy have high potential to benefit both dog and human patients with this devastating disease.
Acknowledgments
This work was supported by the V Foundation and the following National Institutes of Health/National Cancer Institute Grants: R03CA270854, R03CA252793, and NIH Training Grant 1T32CA251007.
Disclosure
Robert Canter reports advisory board fees from Replimune, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Huang JF, Shen J, Li X, et al. Incidence of patients with bone metastases at diagnosis of solid tumors in adults: a large population-based study. Ann Transl Med. 2020;8(7):482. PMID: 32395526; PMCID: PMC7210217. doi:10.21037/atm.2020.03.55
2. Harris MA, Hawkins CJ. Recent and ongoing research into metastatic osteosarcoma treatments. Int J Mol Sci. 2022;23(7):3817. PMID: 35409176; PMCID: PMC8998815. doi:10.3390/ijms23073817
3. Silva JAM, Marchiori E, Macedo FC, Silva PRGD, Amorim VB. Pulmonary metastasis of osteosarcoma: multiple presentations in a single patient. J Bras Pneumol. 2022;48(2):e20210478. PMID: 35475867; PMCID: PMC9064642. doi:10.36416/1806-3756/e20210478
4. SEER*Explorer: an interactive website for SEER cancer statistics [Internet]. Surveillance Research Program, National Cancer Institute; 2024 [updated November 5, 2024; cited December 26, 2024]. Data source(s): SEER Incidence Data, November 2023 Submission (1975–2021), SEER 22 registries. Available from: https://seer.cancer.gov/statistics-network/explorer/.
5. Silva JAM, Marchiori E, Amorim VB, Barreto MM. CT features of osteosarcoma lung metastasis: a retrospective study of 127 patients. J Bras Pneumol. 2023;49(2):e20220433. PMID: 37132704; PMCID: PMC10171270. doi:10.36416/1806-3756/e20220433
6. Sheng G, Gao Y, Yang Y, Wu H. Osteosarcoma and metastasis. Front Oncol. 2021;11:780264. PMID: 34956899; PMCID: PMC8702962. doi:10.3389/fonc.2021.780264
7. Kokkali S, Andriotis E, Katsarou E, et al. Cerebral metastasis from osteosarcoma: “Bone” in the brain. Radiol Case Rep. 2020;15(6):780–783. PMID: 32322331; PMCID: PMC7171257. doi:10.1016/j.radcr.2020.03.020
8. Wang T, Liao S, Ding X, et al. Intraperitoneal extraosseous osteosarcoma: a case report and literatures review. BMC Musculoskelet Disord. 2020;21(1):452. PMID: 32653041; PMCID: PMC7353749. doi:10.1186/s12891-020-03429-5
9. Di QY, Long XD, Ning J, Chen ZH, Mao ZQ. Relapsed primary extraskeletal osteosarcoma of liver: a case report and review of literature. World J Clin Cases. 2023;11(3):662–668. PMID: 36793644; PMCID: PMC9923861. doi:10.12998/wjcc.v11.i3.662
10. Hattori H, Yamamoto K. Lymph node metastasis of osteosarcoma. J Clin Oncol. 2012;30(33):e345–9. PMID: 23032623. doi:10.1200/JCO.2012.42.3384
11. Siddiqui NH, Jani J. Osteosarcoma metastatic to adrenal gland diagnosed by fine-needle aspiration. Diagn Cytopathol. 2005;33(3):201–204. PMID: 16078243. doi:10.1002/dc.20335
12. Odri GA, Tchicaya-Bouanga J, Yoon DJY, Modrowski D. Metastatic progression of osteosarcomas: a review of current knowledge of environmental versus oncogenic drivers. Cancers. 2022;14(2):360. doi:10.3390/cancers14020360
13. Anderson PM, Bielack SS, Gorlick RG, et al. A phase II study of clinical activity of SCH 717454 (robatumumab) in patients with relapsed osteosarcoma and Ewing sarcoma. Pediatric Blood Cancer. 2016;63:1761–1770. doi:10.1002/pbc.26087
14. Fernandes I, Melo-Alvim C, Lopes-Brás R, Esperança-Martins M, Costa L. Osteosarcoma pathogenesis leads the way to new target treatments. Int J Mol Sci. 2021;22(2):813. PMID: 33467481; PMCID: PMC7831017. doi:10.3390/ijms22020813
15. Tippett VL, Tattersall L, Ab Latif NB, et al. The strategy and clinical relevance of in vitro models of MAP resistance in osteosarcoma: a systematic review. Oncogene. 2023;42:259–277. doi:10.1038/s41388-022-02529-x
16. Corre I, Verrecchia F, Crenn V, Redini F, Trichet V. The osteosarcoma microenvironment: a complex but targetable ecosystem. Cells. 2020;9(4):976. PMID: 32326444; PMCID: PMC7226971. doi:10.3390/cells9040976
17. Chen F, Liu J, Yang T, et al. Analysis of intercellular communication in the osteosarcoma microenvironment based on single cell sequencing data. J Bone Oncol. 2023;41:100493. PMID: 37501717; PMCID: PMC10368934. doi:10.1016/j.jbo.2023.100493
18. Cascini C, Chiodoni C. The immune landscape of osteosarcoma: implications for prognosis and treatment response. Cells. 2021;10(7):1668. PMID: 34359840; PMCID: PMC8304628. doi:10.3390/cells10071668
19. Liu W, Hu H, Shao Z, et al. Characterizing the tumor microenvironment at the single-cell level reveals a novel immune evasion mechanism in osteosarcoma. Bone Res. 2023;11(1):4. PMID: 36596773; PMCID: PMC9810605. doi:10.1038/s41413-022-00237-6
20. Liu Y, Feng W, Dai Y, et al. Single-cell transcriptomics reveals the complexity of the tumor microenvironment of treatment-naive osteosarcoma. Front Oncol. 2021;11:709210. Erratum in: Front Oncol. 2022 Nov 15;12:1077067. doi: 10.3389/fonc.2022.1077067. PMID: 34367994; PMCID: PMC8335545. doi:10.3389/fonc.2021.709210
21. van Vlerken-Ysla L, Tyurina YY, Kagan VE, Gabrilovich DI. Functional states of myeloid cells in cancer. Cancer Cell. 2023;41(3):490–504. PMID: 36868224; PMCID: PMC10023509. doi:10.1016/j.ccell.2023.02.009
22. Murphy K, Weaver C. Janeway’s Immunobiology. New York, NY: Garland Science/Taylor & Francis Group, LLC; 2017.
23. Jahchan NS, Mujal AM, Pollack JL, et al. Tuning the tumor myeloid microenvironment to fight cancer. Front Immunol. 2019;10:1611. PMID: 31402908; PMCID: PMC6673698. doi:10.3389/fimmu.2019.01611
24. Binnewies M, Roberts EW, Kersten K, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24(5):541–550. PMID: 29686425; PMCID: PMC5998822. doi:10.1038/s41591-018-0014-x
25. Quail DF, Amulic B, Aziz M, et al. Neutrophil phenotypes and functions in cancer: a consensus statement. J Exp Med. 2022;219(6):e20220011. doi:10.1084/jem.20220011
26. Locati M, Curtale G, Mantovani A. Diversity, mechanisms, and significance of macrophage plasticity. Annu Rev Pathol. 2020;15:123–147. PMID: 31530089; PMCID: PMC7176483. doi:10.1146/annurev-pathmechdis-012418-012718
27. Gabrilovich DI, Nagaraj S. Myeloid-derived-suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–174. doi:10.1038/nri2506
28. Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol. 2021;21(8):485–498. doi:10.1038/s41577-020-00490-y
29. Ostrand-Rosenberg S, Lamb TJ, Pawelec G. Here, there, and everywhere: myeloid-derived suppressor cells in immunology. J Immunol. 2023;210(9):1183–1197. PMID: 37068300; PMCID: PMC10111205. doi:10.4049/jimmunol.2200914
30. Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol. 2018;19(2):108–119. PMID: 29348500; PMCID: PMC5854158. doi:10.1038/s41590-017-0022-x
31. Sarhan D, Cichocki F, Zhang B, et al. Adaptive NK cells with low TIGIT expression are inherently resistant to myeloid-derived suppressor cells. Cancer Res. 2016;76(19):5696–5706. PMID: 27503932; PMCID: PMC5050142. doi:10.1158/0008-5472.CAN-16-0839
32. Gao J, Liang Y, Wang L. Shaping polarization of tumor-associated macrophages in cancer immunotherapy. Front Immunol. 2022;13:888713. PMID: 35844605; PMCID: PMC9280632. doi:10.3389/fimmu.2022.888713
33. Zhang H, Liu L, Liu J, et al. Roles of tumor-associated macrophages in anti-PD-1/PD-L1 immunotherapy for solid cancers. Mol Cancer. 2023;22(1):58. PMID: 36941614; PMCID: PMC10029244. doi:10.1186/s12943-023-01725-x
34. Evans R, Alexander P. Cooperation of immune lymphoid cells with macrophages in tumour immunity. Nature. 1970;228:620–622. doi:10.1038/228620a0
35. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889–896. doi:10.1038/ni.1937
36. Ghisletti S, Barozzi I, Mietton F, et al. Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity. 2010;32:317–328. doi:10.1016/j.immuni.2010.02.008
37. Zhong Y, Yi C. MicroRNA-720 suppresses M2 macrophage polarization by targeting GATA3. Biosci Rep. 2016;36. doi:10.1042/BSR20160105
38. Curtale G, Mirolo M, Renzi TA, Rossato M, Bazzoni F, Locati M. Negative regulation of Toll-like receptor 4 signaling by IL-10-dependent microRNA-146b. Proc Natl Acad Sci USA. 2013;110:11499–11504. doi:10.1073/pnas.1219852110
39. Chou AJ, Kleinerman ES, Krailo MD, et al. Addition of muramyl tripeptide to chemotherapy for patients with newly diagnosed metastatic osteosarcoma: a report from the Children’s Oncology Group. Cancer. 2009;115(22):5339–5348. PMID: 19637348; PMCID: PMC2783515. doi:10.1002/cncr.24566
40. Barnes DJ, Dutton P, Bruland Ø, et al. Outcomes from a mechanistic biomarker multi-arm and randomised study of liposomal MTP-PE (Mifamurtide) in metastatic and/or recurrent osteosarcoma (EuroSarc-Memos trial). BMC Cancer. 2022;22:629. doi:10.1186/s12885-022-09697-9
41. Tang J, Cai H, Lin L, et al. Increased expression of CD24 is associated with tumor progression and prognosis in patients suffering osteosarcoma. Clin Transl Oncol. 2013;15:541–547. doi:10.1007/s12094-012-0961-5
42. de Biasi AR, Villena-Vargas J, Adusumilli PS. Cisplatin-induced antitumor immunomodulation: a review of preclinical and clinical evidence. Clin Cancer Res. 2014;20(21):5384–5391. PMID: 25204552; PMCID: PMC4216745. doi:10.1158/1078-0432.CCR-14-1298
43. Alizadeh D, Trad M, Hanke NT, et al. Doxorubicin eliminates myeloid-derived suppressor cells and enhances the efficacy of adoptive T-cell transfer in breast cancer. Cancer Res. 2014;74(1):104–118. PMID: 24197130; PMCID: PMC3896092. doi:10.1158/0008-5472.CAN-13-1545
44. Voorwerk L, Slagter M, Horlings HM, et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: the TONIC trial. Nat Med. 2019;25(6):920–928. Erratum in: Nat Med. 2019 Jul;25(7):1175. doi: 10.1038/s41591-019-0520-5. PMID: 31086347. doi:10.1038/s41591-019-0432-4
45. Wood GE, Meyer C, Petitprez F, D’Angelo SP. Immunotherapy in Sarcoma: current data and promising strategies. Am Soc Clin Oncol Educ Book. 2024;44(3):e432234. PMID: 38781557. doi:10.1200/EDBK_432234
46. Deng C, Xu Y, Fu J, et al. Reprograming the tumor immunologic microenvironment using neoadjuvant chemotherapy in osteosarcoma. Cancer Sci. 2020;111(6):1899–1909. Erratum in: Cancer Sci. 2020 Jul;111(7):2657. doi: 10.1111/cas.14543. PMID: 32232912; PMCID: PMC7293104. doi:10.1111/cas.14398
47. Pires SF, Barros JS, Costa SSD, et al. Analysis of the mutational landscape of osteosarcomas identifies genes related to metastasis and prognosis and disrupted biological pathways of immune response and bone development. Int J Mol Sci. 2023;24(13):10463. PMID: 37445641; PMCID: PMC10342084. doi:10.3390/ijms241310463
48. Wang D, Niu X, Wang Z, et al. Multiregion sequencing reveals the genetic heterogeneity and evolutionary history of osteosarcoma and matched pulmonary metastases. Cancer Res. 2019;79(1):7–20. doi:10.1158/0008-5472.CAN-18-1086
49. Du X, Wei H, Zhang B, et al. Molecular mechanisms of osteosarcoma metastasis and possible treatment opportunities. Front Oncol. 2023;13:1117867. doi:10.3389/fonc.2023.1117867
50. Hattinger CM, Pasello M, Ferrari S, Picci P, Serra M. Emerging drugs for high-grade osteosarcoma. Expert Opin Emerg Drugs. 2010;15:615–634. doi:10.1517/14728214.2010.505603
51. Dong J, Chai X, Xue Y, et al. ZIF-8-encapsulated pexidartinib delivery via targeted peptide-modified M1 macrophages attenuates MDSC-mediated immunosuppression in osteosarcoma. Small. 2024;20(29):e2309038. PMID: 38456768. doi:10.1002/smll.202309038
52. Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41(1):49–61. Erratum in: Immunity. 2014 Nov 20;41(5):866. PMID: 25035953; PMCID: PMC4137410. doi:10.1016/j.immuni.2014.06.010
53. Mantovani A. Reflections on immunological nomenclature: in praise of imperfection. Nat Immunol. 2016;17:215–216. doi:10.1038/ni.3354
54. Fujiwara T, Yakoub MA, Chandler A, et al. CSF1/CSF1R signaling inhibitor pexidartinib (PLX3397) reprograms tumor-associated macrophages and stimulates T-cell infiltration in the sarcoma microenvironment. Mol Cancer Ther. 2021;20(8):1388–1399. PMID: 34088832; PMCID: PMC9336538. doi:10.1158/1535-7163.MCT-20-0591
55. Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol. 2008;8(7):533–544. PMID: 18551128. doi:10.1038/nri2356
56. Benner B, Good L, Quiroga D, et al. Pexidartinib, a novel small molecule CSF-1R inhibitor in use for tenosynovial giant cell tumor: a systematic review of pre-clinical and clinical development. Drug Des Devel Ther. 2020;14:1693–1704. PMID: 32440095; PMCID: PMC7210448. doi:10.2147/DDDT.S253232
57. Monestime S, Lazaridis D. Pexidartinib (TURALIO™): the first FDA-indicated systemic treatment for tenosynovial giant cell tumor. Drugs R D. 2020;20(3):189–195. PMID: 32617868; PMCID: PMC7419392. doi:10.1007/s40268-020-00314-3
58. Tap WD, Gelderblom H, Palmerini E, et al. ENLIVEN investigators. Pexidartinib versus placebo for advanced tenosynovial giant cell tumour (ENLIVEN): a randomised Phase 3 trial. Lancet. 2019;394(10197):478–487. PMID: 31229240; PMCID: PMC6860022. doi:10.1016/S0140-6736(19)30764-0
59. Wang JC, Sun L. PD-1/PD-L1, MDSC pathways, and checkpoint inhibitor therapy in Ph(-) myeloproliferative neoplasm: a review. Int J Mol Sci. 2022;23(10):5837. PMID: 35628647; PMCID: PMC9143160. doi:10.3390/ijms23105837
60. Cheng P, Corzo CA, Luetteke N, et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med. 2008;205(10):2235–2249. PMID: 18809714; PMCID: PMC2556797. doi:10.1084/jem.20080132
61. Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J Immunol. 2009;182(1):240–249. PMID: 19109155. doi:10.4049/jimmunol.182.1.240
62. Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol. 2007;179(2):977–983. PMID: 17617589. doi:10.4049/jimmunol.179.2.977
63. Shi X, Li X, Wang H, Yu Z, Zhu Y, Gao Y. Specific inhibition of PI3Kδ/γ enhances the efficacy of anti-PD1 against osteosarcoma cancer. J Bone Oncol. 2018;16:100206. PMID: 31334002; PMCID: PMC6617297. doi:10.1016/j.jbo.2018.11.001
64. Yu S, Yao X. Advances on immunotherapy for osteosarcoma. Mol Cancer. 2024;23(1):192. PMID: 39245737; PMCID: PMC11382402. doi:10.1186/s12943-024-02105-9
65. Jiang K, Li J, Zhang J, et al. SDF-1/CXCR4 axis facilitates myeloid-derived suppressor cells accumulation in osteosarcoma microenvironment and blunts the response to anti-PD-1 therapy. Int Immunopharmacol. 2019;75:105818. PMID: 31437795. doi:10.1016/j.intimp.2019.105818
66. Beck J, Ren L, Huang S, et al. Canine and murine models of osteosarcoma. Vet Pathol. 2022;59(3):399–414. PMID: 35341404; PMCID: PMC9290378. doi:10.1177/03009858221083038
67. Kavirayani AM, Sundberg JP, Foreman O. Primary neoplasms of bones in mice: retrospective study and review of literature. Vet Pathol. 2012;49(1):182–205. PMID: 21343597; PMCID: PMC3151475. doi:10.1177/0300985811398252
68. Chuprin J, Buettner H, Seedhom MO, et al. Humanized mouse models for immuno-oncology research. Nat Rev Clin Oncol. 2023;20(3):192–206. doi:10.1038/s41571-022-00721-2
69. Olson B, Li Y, Lin Y, et al. Mouse models for cancer immunotherapy research. Cancer Discov. 2018;8(11):1358–1365. doi:10.1158/2159-8290.CD-18-0044
70. Park JS, Withers SS, Modiano JF, et al. Canine cancer immunotherapy studies: linking mouse and human. J Immunother Cancer. 2016;4:97. doi:10.1186/s40425-016-0200-7
71. Canter RJ, Grossenbacher SK, Foltz JA, et al. Radiotherapy enhances natural killer cell cytotoxicity and localization in pre-clinical canine sarcomas and first-in-dog clinical trial. J Immunother Cancer. 2017;5(1):98. doi:10.1186/s40425-017-0305-7
72. Rebhun RB, York D, Cruz SM, et al. Inhaled recombinant human IL-15 in dogs with naturally occurring pulmonary metastases from osteosarcoma or melanoma: a Phase 1 study of clinical activity and correlates of response. J Immunother Cancer. 2022;10(6):e004493. doi:10.1136/jitc-2022-004493
73. LeBlanc AK, Mazcko CN. Improving human cancer therapy through the evaluation of pet dogs. Nat Rev Cancer. 2020;20(12):727–742. doi:10.1038/s41568-020-0297-3
74. Mason NJ. Comparative Immunology and Immunotherapy of Canine Osteosarcoma. In: Kleinerman ES, Gorlick R, editors. Current Advances in the Science of Osteosarcoma: Research Perspectives: Tumor Biology, Organ Microenvironment, Potential New Therapeutic Targets, and Canine Models. Advances in Experimental Medicine and Biology. Springer International Publishing; 2020:199–221. doi:10.1007/978-3-030-43085-6_14
75. Kurzman ID, MacEwen EG, Rosenthal RC, et al. Adjuvant therapy for osteosarcoma in dogs: results of randomized clinical trials using combined liposome-encapsulated muramyl tripeptide and cisplatin. Clin Cancer Res. 1995;1(12):1595–1601. PMID: 9815961.
76. Gardner HL, Fenger JM, London CA. Dogs as a model for cancer. Annu Rev Anim Biosci. 2016;4:199–222. PMID: 26566160; PMCID: PMC6314649. doi:10.1146/annurev-animal-022114-110911
77. Tuohy JL, Lascelles BD, Griffith EH, Fogle JE. Association of canine osteosarcoma and monocyte phenotype and chemotactic function. J Vet Intern Med. 2016;30(4):1167–1178. PMID: 27338235; PMCID: PMC5094498. doi:10.1111/jvim.13983
78. Wycislo KL, Fan TM. The immunotherapy of canine osteosarcoma: a historical and systematic review. J Vet Intern Med. 2015;29(3):759–769. PMID: 25929293; PMCID: PMC4895426. doi:10.1111/jvim.12603
79. Sottnik JL, Rao S, Lafferty MH, et al. Association of blood monocyte and lymphocyte count and disease-free interval in dogs with osteosarcoma. J Vet Intern Med. 2010;24(6):1439–1444. PMID: 20840314. doi:10.1111/j.1939-1676.2010.0591.x
80. Hingorani P, Maas ML, Gustafson MP, et al. Increased CTLA-4(+) T cells and an increased ratio of monocytes with loss of class II (CD14(+) hLA-DR(lo/neg)) found in aggressive pediatric sarcoma patients. J Immunother Cancer. 2015;3:35. PMID: 26286851; PMCID: PMC4539889. doi:10.1186/s40425-015-0082-0
81. Sottnik JL, U’Ren LW, Thamm DH, Withrow SJ, Dow SW. Chronic bacterial osteomyelitis suppression of tumor growth requires innate immune responses. Cancer Immunol Immunother. 2010;59(3):367–378. PMID: 19701748; PMCID: PMC11030164. doi:10.1007/s00262-009-0755-y
82. Lascelles BD, Dernell WS, Correa MT, et al. Improved survival associated with postoperative wound infection in dogs treated with limb-salvage surgery for osteosarcoma. Ann Surg Oncol. 2005;12(12):1073–1083. PMID: 16252138. doi:10.1245/ASO.2005.01.011
83. Kerboeuf M, Anfinsen KP, Koppang EO, et al. Immunological pre-metastatic niche in dogs with naturally occurring osteosarcoma. Vet Comp Oncol. 2024;23:62–72. PMID: 39526499. doi:10.1111/vco.13026
84. Goulart MR, Hlavaty SI, Chang YM, et al. Phenotypic and transcriptomic characterization of canine myeloid-derived suppressor cells. Sci Rep. 2019;9(1):3574. doi:10.1038/s41598-019-40285-3
85. Sherger M, Kisseberth W, London C, Olivo-Marston S, Papenfuss TL. Identification of myeloid derived suppressor cells in the peripheral blood of tumor bearing dogs. BMC Vet Res. 2012;8:209. PMID: 23110794; PMCID: PMC3557223. doi:10.1186/1746-6148-8-209
86. Regan DP, Chow L, Das S, et al. Losartan blocks osteosarcoma-elicited monocyte recruitment, and combined with the kinase inhibitor toceranib, exerts significant clinical benefit in canine metastatic osteosarcoma. Clin Cancer Res. 2022;28(4):662–676. PMID: 34580111; PMCID: PMC8866227. doi:10.1158/1078-0432.CCR-21-2105
87. Qian BZ, Li J, Zhang H, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475(7355):222–225. PMID: 21654748; PMCID: PMC3208506. doi:10.1038/nature10138
88. Ko JS, Zea AH, Rini BI, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res. 2009;15(6):2148–2157. PMID: 19276286. doi:10.1158/1078-0432.CCR-08-1332
89. Draghiciu O, Nijman HW, Hoogeboom BN, Meijerhof T, Daemen T. Sunitinib depletes myeloid-derived suppressor cells and synergizes with a cancer vaccine to enhance antigen-specific immune responses and tumor eradication. Oncoimmunology. 2015;4(3):e989764. PMID: 25949902; PMCID: PMC4404834. doi:10.4161/2162402X.2014.989764
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Overexpression of TREM1 is Associated with the Immune-Suppressive Microenvironment and Unfavorable Prognosis in Pan-Cancer
Zhou X, Lin K, Fu L, Liu F, Lin H, Chen Y, Zhuang B, Liang H, Deng Q, Wang Z, Chen W, Luo J, Cao J, Li P
Journal of Inflammation Research 2023, 16:1375-1391
Published Date: 27 March 2023
Exploring the Correlation Between GPR176, a Potential Target Gene of Gastric Cancer, and Immune Cell Infiltration
Gu X, Shen H, Xiang Z, Li X, Zhang Y, Zhang R, Su F, Wang Z
Pharmacogenomics and Personalized Medicine 2023, 16:519-535
Published Date: 1 June 2023
Integrating Bulk and Single-Cell RNA Sequencing Reveals Heterogeneity, Tumor Microenvironment, and Immunotherapeutic Efficacy Based on Sialylation-Related Genes in Bladder Cancer
Tan Z, Chen X, Zuo J, Fu S, Wang J, Wang H
Journal of Inflammation Research 2023, 16:3399-3417
Published Date: 14 August 2023
NOTCH1 Mutations Predict Superior Outcomes of Immune Checkpoint Blockade in Non-Small Cell Lung Cancer
Huang Q, Cao H, Yao Q, Zhou X, Li H, Bai Q, Hu H
ImmunoTargets and Therapy 2023, 12:165-173
Published Date: 5 December 2023
Gut Microbiota–Tumor Microenvironment Interactions: Mechanisms and Clinical Implications for Immune Checkpoint Inhibitor Efficacy in Cancer
Said SS, Ibrahim WN
Cancer Management and Research 2025, 17:171-192
Published Date: 25 January 2025