Back to Journals » Journal of Hepatocellular Carcinoma » Volume 13
Machine Learning-Based Sialylation-Associated Gene Signature Predicts Prognosis and Immune Landscape in Hepatocellular Carcinoma: Validation via Multi-Omics Analysis and in vitro Assays
Authors Zheng Z, Wang Y, Zhang W ![]() , Du G, Luo B
, Du G, Luo B
Received 22 October 2025
Accepted for publication 31 January 2026
Published 18 February 2026 Volume 2026:13 575986
DOI https://doi.org/10.2147/JHC.S575986
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr David Gerber
Zijie Zheng,* Yuening Wang,* Wenyue Zhang,* Guoqing Du, Baoming Luo
Department of Ultrasound, Sun Yat-Sen Memorial Hospital of Sun Yat-Sen University, Guangzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Guoqing Du, Department of Ultrasound, Sun Yat-Sen Memorial Hospital of Sun Yat-Sen University, 107 Yanjiangxi Road, Guangzhou, 510120, People’s Republic of China, Email [email protected] Baoming Luo, Department of Ultrasound, Sun Yat-Sen Memorial Hospital of Sun Yat-Sen University, 107 Yanjiangxi Road, Guangzhou, 510120, People’s Republic of China, Email [email protected]
Background: Sialylation, an important post-translational modification crucial for protein activity, affects tumor development and spread by altering immune response. Nevertheless, the roles they play in the microenvironment of Hepatocellular Carcinoma (HCC) and their clinical implications remain unclear. The purpose of this research was to investigate the function of genes involved in sialylation concerning tumor immunity and their clinical implications in HCC.
Methods: We intended to build a prognostic prediction model called the sialylation score through the application of sialylation-associated genes and a machine learning integrative approach.
Results: Our findings show that the sialylation score independently affects overall survival of HCC patients, with trustworthy and stable outcomes. Sialylation scores are notably more accurate than conventional clinical variables and previously published signatures. Moreover, patients with low sialylation scores had significant immune infiltration. Further analysis of single-cell cohorts indicates that patients with high sialylation scores have an immunosuppressive microenvironment, where T and NK cells improve their interactions with myeloid cells via signaling pathways like MHC-II, CLEC and COLLAGEN pathways. To validate the biological significance of this signature, we targeted the key gene ST3GAL4 in vitro, revealing that its knockdown significantly reduces the proliferation, invasion, and migration capabilities of HCC cells.
Conclusion: The sialylation score could serve as a dependable method for anticipating immune response, thereby improving clinical results in patients with HCC.
Keywords: sialylation, hepatocellular carcinoma, machine learning, tumor immune microenvironment, prognosis
Introduction
Primary liver cancer is the sixth most prevalent cancer worldwide and the third leading cause of cancer-associated deaths, posing a significant threat to human health and life.1 With 75–90% of cases, Hepatocellular Carcinoma (HCC) is the most common type of liver cancer.2 Surgical removal is a highly effective treatment option. However, as many as 70% of patients might face a recurrence within five years after the procedure.3 Moreover, patients with advanced HCC have few treatment choices and usually survive only six months.4 Improving clinical outcomes for HCC patients is hindered by the high rate of recurrence and decreased survival rates.5 Although immunotherapy is showing promising clinical benefits for patients with advanced HCC, some Phase II/III trials have shown disappointing results, posing ongoing challenges.6,7
Sialylation is essential for cancer cells to proliferation and spread.8 In various forms of cancer, such as breast, lung and ovarian, heightened sialyltransferase activity results in excessive sialylation, affecting over 60% of the surfaces of tumor cells.9–12 This mechanism boosts immune evasion, promotes tumor movement and invasion, and interferes with interactions between cells.13 Importantly, hypersialylation helps cancer cells evade apoptosis by obstructing essential receptors like Fas and TNF, thus hindering cell death signaling.14 Moreover, sialylation changes how cells adhere to the extracellular matrix, promoting cancer progression and metastasis.15 Our results emphasize the importance of sialylation in cancer development and immune system evasion, pointing to its potential as a target for therapy. Considering the important role of sialylation in antitumor immunity, we propose that the expression patterns of sialylation genes might affect tumor immune infiltration and clinical results, leading to the development of a new predictive model for HCC patients.
Although models like the Barcelona Clinic Liver Cancer (BCLC) system are well-established, recent systematic reviews reveal that they are inadequate when subjected to external validation.16–18 While five new models appear promising for predicting overall survival (OS), they do not fully consider immune characteristics and etiological differences.17–21 The research performed an in-depth examination of bulk transcriptome data and single-cell sequencing to pinpoint genes related to sialylation in HCC. Through an integrated machine learning framework, a signature related to sialylation was created. Additional examination of multi-omics data showed promising connections between this signature and the prognosis of HCC, along with the status of the immune microenvironment. These results may act as a basis for examining the significance of sialylation in HCC and for developing clinical applications that involve sialylation. The workflow is depicted in Figure 1.
 |
Figure 1 Flowchart of this study. *p < 0.05, **p < 0.01. |
Methods
Data Acquisition and Analysis
Two different cohorts provided clinical data and RNA sequencing for HCC patients: The Cancer Genome Atlas (TCGA-LIHC, https://portal.gdc.cancer.gov) and the International Cancer Genome Consortium (ICGC-LIRI-JP, https://dcc.icgc.org).The GEO database (GSE151530, https://doi.org/10.1101/2020.08.18.254748) was used to obtain single-cell level data of HCC samples. Afterward, 351 genes associated with sialylation were retrieved from Genecard database (Relevance score ≥1) (Table S1).
Consistency Cluster Analysis
To explore molecular cancer subtypes through sialylation gene expression, HCC samples were randomly selected for repeated analysis. Consistency analysis was carried out with the Consensus Cluster Plus package of R, with the maximum number of clusters capped at 6 to select the best cluster. The pheatmap package in R was subsequently utilized to generate cluster heatmaps, focusing on genes whose variance were exceeding 0.1. In gene sets containing over 1000 targets, the top 25% of genes ranked by variance were shown. To evaluate survival variations among different subgroups, a Kaplan-Meier survival analysis was carried out.
Sialylation Prognostic Score System
To achieve a stable and accurate sialylation score, 117 combinations of 10 machine-learning algorithms were utilized (https://github.com/l-magnificence/Mime). The extensive algorithm combinations included CoxBoost, Enet, GBM, Lasso, plsRcox, Ridge, RSF, stepwise Cox, SuperPC, and survival-SVM. The process of generating the signature involved. (a) Identifying sialylation genes related to prognosis through univariate Cox regression in the TCGA-LIHC cohort. Genes exhibiting a p-value less than 0.05 were recognized as having prognostic significance. (b) Using 117 different algorithm combinations to fit prediction models for these genes in the TCGA-LIHC. All subsequent processes, including feature selection, hyperparameter tuning, and model fitting, were conducted exclusively within the TCGA-LIHC cohort and each process were performed independently within each cross-validation loop. K-fold cross-validation was employe to evaluate the performance (C-index) of each algorithm combination, guiding hyperparameter optimization. (c) Using the ICGC-LIRI-JP datasets to validate all models. During the whole process of developing and selecting the model, the ICGC-LIRI-JP datasets were kept constant and served only for an objective evaluation of the final optimal model. (d) Choosing the model with the highest average C-index. The patients were split into groups with high or low scores according to the median sialylation score. The prognostic scoring system’s predictive accuracy was evaluated using a ROC (Receiver Operating Characteristic) curve analysis with the time ROC package in R.
Clinical Significance of Sialylation Score
Clinical features like gender, age, T, M, and N stages, stage and grade, along with sialylation scores, were examined using multivariate and univariate regression to determine prognostic indicators. The association between sialylation scores and clinical characteristics was further examined using T-tests. To contrast different published prediction models, Decision curve analysis (DCA) was employed, using “ALL” and “None” as benchmarks.
Immune Cell Infiltration Analysis
Various computational tools like xCell, CIBERSORT, EPIC, MCPcounter, and Quantiseq were employed to examine the distribution and composition of immune cells populations in tumor samples from different sialylation groups.22–25 ESTIMATE score, tumor purity, Immune score, and stromal score were also explored. The relationship between immune activity and the sialylation score was examined through ssGSEA.
Analysis of RNA Sequencing Data at the Single-Cell Level
Single-cell quality control and cell clustering analysis were conducted using the Seurat package in R, while the calculation of the sialylation score utilized the “AllModuleScore” function. To determine diverse ligand-receptor pairs and cell-cell communication networks, CellChat package was utilized in R.
Drug Sensitivity Analysis
The R package pRRophetic and the Genomics of Drug Sensitivity in Cancer database were applied to forecast the chemotherapy sensitivity of two groups based on sialylation scores.
Cell Culture
Cell lines SK-HEP-1 and SNU449, obtained from the Cell Bank of the Chinese Academy of Sciences in Shanghai, China, were grown in DMEM medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin at 37°C and 5% CO2.
Quantitative Real-Time PCR (qRT‒PCR)
Total RNA was extracted from cells using the RNA pure Tissue & Cell Kit (ESscience) and then served as a template for reverse transcription reactions with the Color Reverse Transcription Kit (EZBioscience). Using the LightCycler® 96 System, the quantitative amplification employed 2×SYBR Green qPCR Master mix (EZBioscience) and primers from IGE Biotechnology Co., Ltd (Guangzhou, China). The ST3GAL sequences for the primers were as follows: Forward: CTTCCTGCGGCTTGAGGATTA; Reverse: CTCACTCCCCTTGGTCCCATA. The β-actin sequences for the primers were as follows: Forward: CATGTACGTTGCTATCCAGGC; Reverse: CTCCTTAATGTCACGCACGAT.
Western Blot
RIPA cell lysis buffer, supplemented with phosphatase and proteinase inhibitors, was used to lyse protein from each cell line, and the BCA method was employed to determine the total protein concentration. A 10% SDS-PAGE gel was used to run the samples, which were then transferred to a PVDF membrane and blocked with 5% skim milk at 4 °C overnight. After washing, the membrane was treated with primary antibodies and then with secondary antibodies. An enhanced chemiluminescence substrate was used to identify bands. The primary antibodies used in this study are listed in Table S3.
siRNA Transfection
IGE Biotechnology Co., Ltd. (Guangzhou, China) designed and synthesized ST3GAL4 siRNA and sequences are listed in Table S2. According to Thermo Fisher Scientific’s guidelines (Shanghai, China), the RNAiMAX and siRNA reagent were mixed. After 24 hours, 2×105 cells were placed into a 6-well plate and transfected at a confluence level of 60–70%.
Colony Formation Assay
A density of 800 cells per well was used to seed into six-well plates. The medium was carefully removed after 10 days of cultivation, and the cells were fixed for 20 minutes at room temperature with 4% paraformaldehyde (PFA). Subsequently, the cells were stained at room temperature for 20 minutes using a 0.1% crystal violet solution. The surplus stain was carefully rinsed off with running water and the plates were left dry naturally. The colonies were subsequently counted and analyzed.
The Migration and Invasion of Cells
Experiments on cell invasion were performed using a Transwell chamber and a 24-well plate. In the lower chamber had a medium containing 20% fetal bovine serum, while the upper chamber, transfected cells were evenly spread without serum. Cells were counted using crystal violet staining. For experiments on cell migration, cells that received siRNA treatment were placed in six wells without serum. A wound was created using a 100 μL plastic pipette tip. Then migration distance was measured after 24 hours to evaluate migration capability.
5-Ethynyl-2’-Deoxyuridine (EdU) Assay
The proliferative capacity of HCC cells was evaluated by the EdU Imaging Kit (APExBio, USA), as per the manufacturer’s instructions. A fluorescence microscope (Olympus IX83, Japan) was employed to randomly capture fields in order to determine the fraction of EdU-positive (green) to Hoechst-positive (blue) cells.
Statistical Analysis
All statistical analyses were conducted using R software (version 4.4.2) and GraphPad Prism 8.0. The Student’s t-test, Mann–Whitney U-test, or Log rank test were employed for comparisons between two groups, whereas one-way ANOVA was applied to analyze data from experiments with three or more groups. Kaplan-Meier survival curves were compared using the Log rank test, and a P-value below 0.05 was considered statistically significant. Statistical significance levels were set at *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Results
Construction of Sialylation-Associated Gene Consensus Clusters in HCC Patients
A consensus clustering analysis was carried out using 351 genes linked to sialylation. Initially, k clusters were formed from all HCC samples, with k values ranging between 2 and 9. The ideal cluster number was determined to be k = 2, as shown by the heatmap from Consensus Cluster Plus results and the cumulative distribution function(CDF) curve (Figure 2A and B). There was no significant difference in the expression levels of genes associated with sialylation between Clusters1 (C1) and Clusters2 (C2) (Figure 2C), with the majority of showing high expression in C1 and low expression in C2. Using Principal Component Analysis (PCA), patients were divided into two distinct clusters (Figure 2D). Moreover, a substantial difference in survival outcomes was observed between C1 and C2, with C2 showing a significantly higher OS rate compared to C1 (Figure 2E). As a result, we labeled C1 as “sialylation activated” and C2 as “sialylation-suppressed” group, respectively.
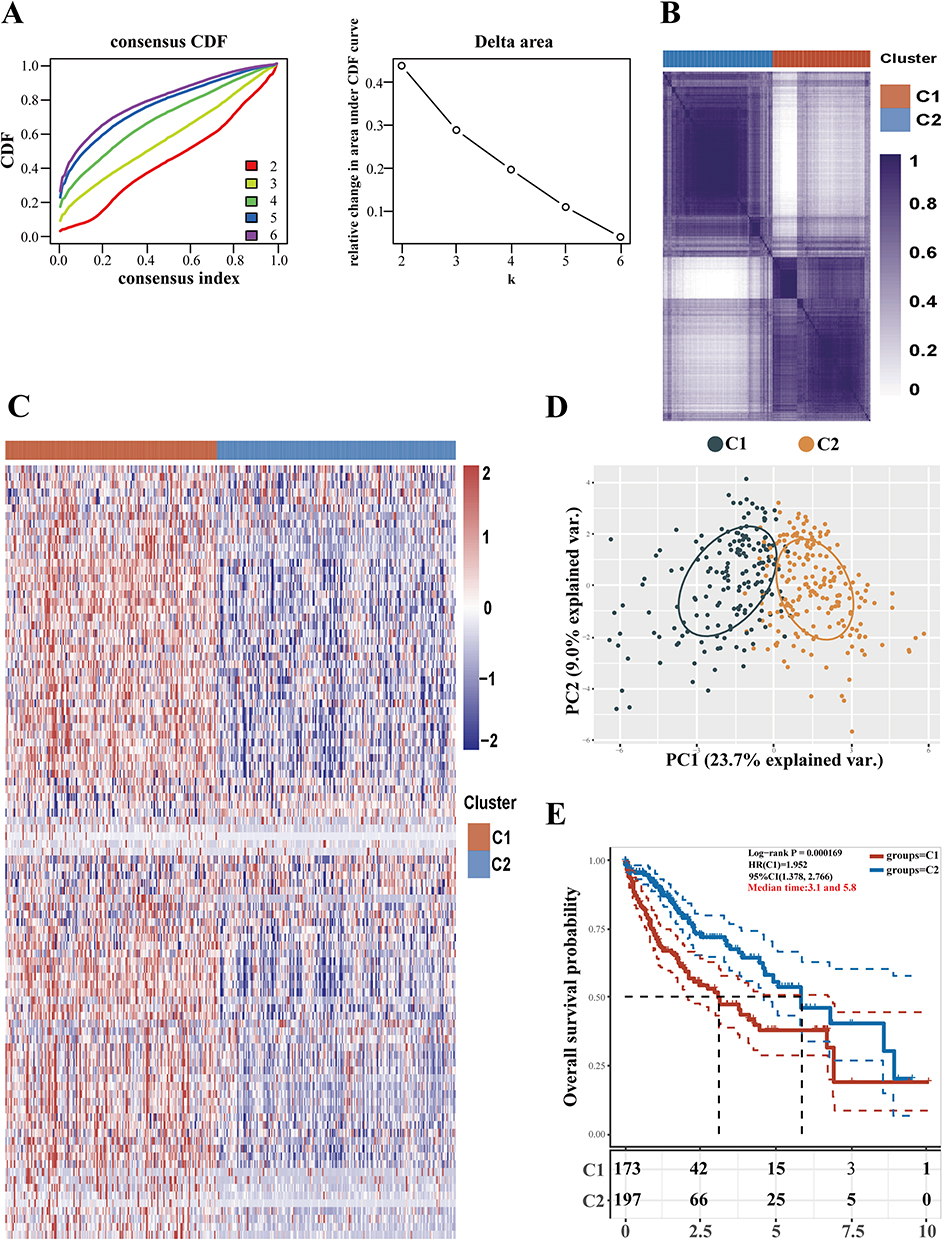 |
Figure 2 Classification of HCC patients according to genes linked to sialylation. (A) HCC samples from TCGA were analyzed using consistency clustering with 351 sialylation-associated genes. The CDF curve achieves its maximum area under the curve when K is set to 2. (B) The results from Consensus Cluster Plus include a heatmap for consistency clustering with K = 2, where samples are represented by rows and columns, and unique cluster categories are shown in different colors. (C) The expression of sialylation-associated genes across different subgroups is depicted in a heatmap, with high expression marked in red and low expression marked in blue. (D) PCA subgroup analysis. (E) Various subgroups were analyzed using Kaplan-Meier survival curves, and Log rank tests were conducted to compare these groups. |
HCC Patients with Activated Sialylation Associated with Increased Immune Evasion and Poorer Response to Immune Therapy
Eight algorithms were applied to determine the differences of immune cells between HCC subgroups. The analysis with the X cell showed that C1 subgroup had a notable increase in immune-suppressive cells, like CD4 + Th2 T cells, in comparison to C2 (Figure 3A). In the C1 subgroup, there was a notable decrease in antitumor immune cells like T cell CD8+ naive and T cell CD4+ central memory cells, indicating a tumor immune suppressive microenvironment. The EPIC, TIMER, CIBERSORT, QUANTISEQ, and MCPCOUNTER algorithms also validated this phenomenon (Figure 3B–F). Additionally, The C1 subgroup showed much higher expression levels of genes related to immune checkpoints than the C2, pointing to a marked immunosuppressive state in the C1 (Figure 3G). In addition, the TIDE algorithm was employed to characterize the immune landscape and generate hypotheses regarding potential immunotherapy response in HCC patients. The C2 subgroup had a notably lower TIDE score than the C1, which suggesting a reduced response to immunotherapy and an increased immune evasion (Figure 3H). It implies that tumors with sialylation activation may be more increased immune evasion.
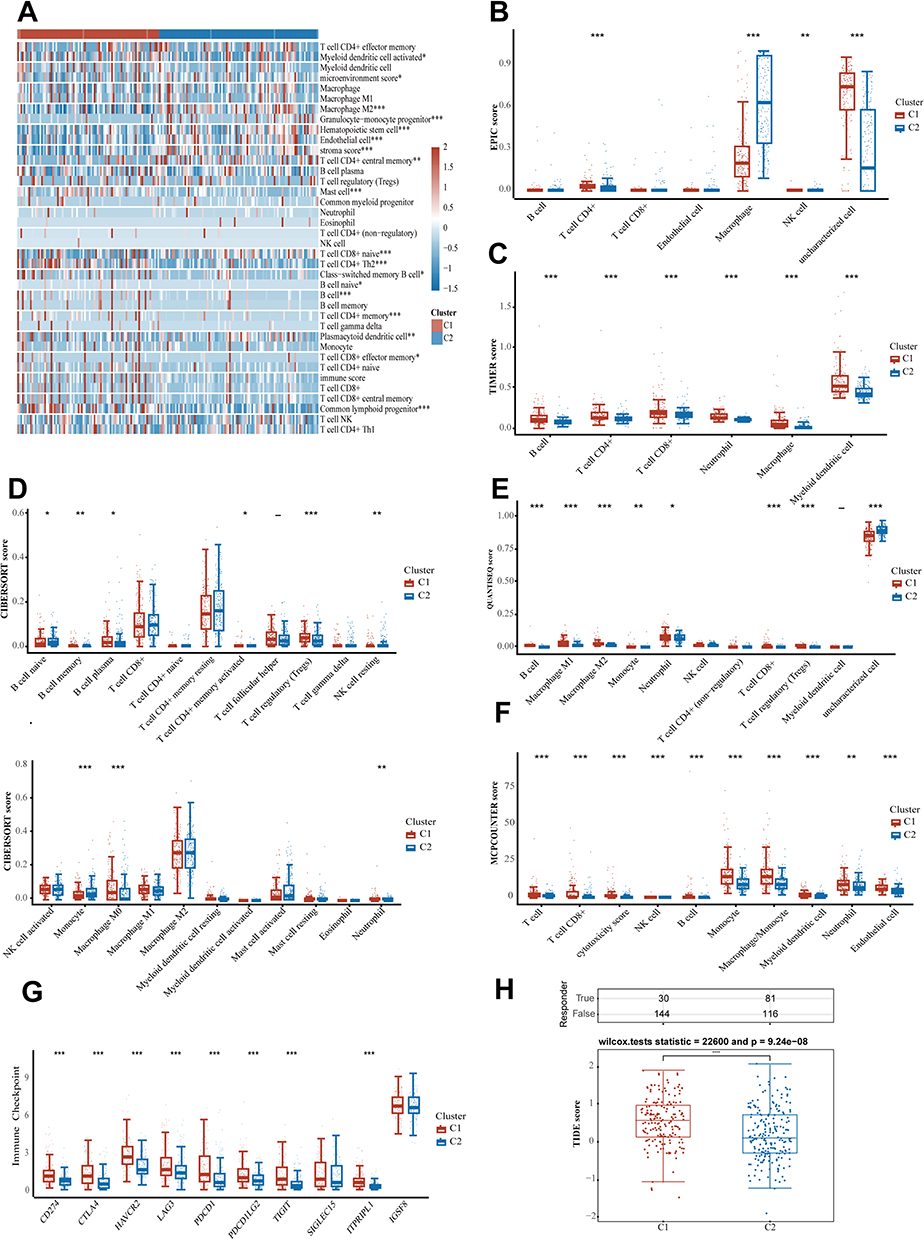 |
Figure 3 Association between sialylation associated gene subtypes and immune profiles. Immune cell comparison across various groups using algorithms like X cell, EPIC, TIMER, CIBERSORT, QUANTISEQ, and MCPCOUNTER (A–F). Investigation into the expression of genes linked to immune checkpoints within various subgroups of samples (G). Prediction of the response to immune checkpoint inhibitors with TIDE (H). *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. |
Constructing a Prognostic Model Through the Application of a Machine Learning Ensemble Method Focused on Sialylation for HCC Patients
From 351 genes associated with sialylation, 22 prognostic genes were identified through univariate regression analysis. In order to develop an ideal sialylation associated prognostic model for patients with HCC, we applied 117 predictive model frameworks using K-fold cross-validation and computed the C-index of each. The best model of the 8 sialylation genes combined StepCox[forward] and Ridge (Figure 4A), reaching the top average C-index of 0.645 and consistently showing better values of C-index across two datasets. Then we determined the sialylation score for each patient by applying the regression coefficients from the StepCox[forward] and Ridge model. Based on the median sialylation score, we categorized all patients into high or low sialylation groups. Both the TCGA-LIHC training and ICGC-LIRI-JP validation datasets revealed that the high sialylation group had notably poorer overall survival compared to the low sialylation group (Figure 4B–E). The ROC analysis was used to estimate the sialylation score discrimination, resulting in AUCs of 0.69, 0.62, and 0.62 for ICGC-LIRI-JP and 0.71, 0.72, and 0.71 for TCGA-LIHC over one, two, and three years, respectively. Our results showed that the prognostic model for HCC, which is based on the 8 sialylation genes, exhibits strong and reliable capabilities for prognostic prediction (Figure 4F). Comparisons of clinical data revealed significant differences in OS, Tumor stage and grade, T-staging, and M-staging between high sialylation and low sialylation groups. We examined the regulatory impact of sialylation in HCC by identifying the differentially expressed genes between groups with high and low sialylation levels in the TCGA-LIHC dataset. In total, 163 DEGs were found, with 77 genes being upregulated and 86 downregulated (Figure 4G). Further enrichment analyses revealed notable enrichment in pathways involved organic acid biosynthetic process regulation, carboxylic acid biosynthetic process, organic anion transmembrane transporter activity, fatty acid binding, collagen-containing extracellular matrix, Biosynthesis of amino acids, Phenylalanine metabolism (Figure 4H).
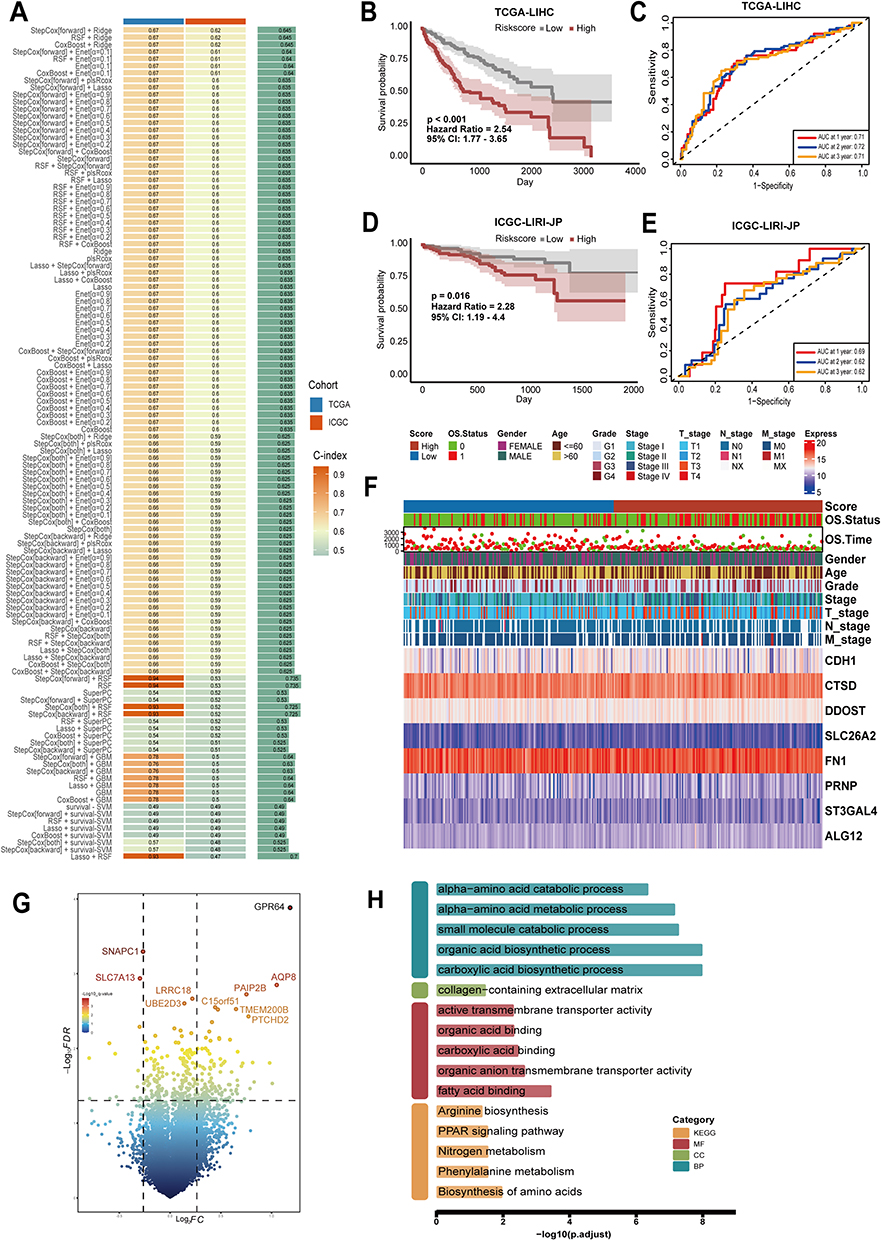 |
Figure 4 Design and confirmation of a sialylation associated prognostic model using an ensemble strategy in machine learning. (A) Generally, 117 predictive models were built and C index was then calculated. (B–E) The sialylation score was utilized in Kaplan–Meier curves and time-related ROC analysis to predict 1, 2, and 3-year overall survival in the TCGA-LIHC (n = 365) and ICGC-LIRI-JP (n = 224) datasets. (F) Heat map depicting the expression profiles of the 8 crucial prognosis genes alongside a comparison of clinical features of high-sialylation versus low- sialylation groups in the TCGA. (G) Different expressed genes (DEGs, fold change > 1.2, p < 0.05) between the high- and low- sialylation groups in the TCGA-OS dataset. (H) Enrichment analysis outcomes of DEGs across different risk groups in GO and KEGG pathways. |
Assessment of the Sialylation Score Model
Independent prognostic factors were determined through univariate and multivariate Cox regression analyses. Our findings indicated that the sialylation score, T-staging, M-staging, and tumor stage were important indicators of overall survival (Figure 5A). Next, we investigated the relationship between other clinical characteristics and the sialylation score. Furthermore, we observed notable differences between various grades (such as Grade 1 compared to Grade 3) and stages (like Stage I versus Stage III), indicating a possible link between the progression of HCC and sialylation score (Figure 5B). The AUC of the sialylation score outperformed other clinical factors in predictive performance (1,3,5-year survival AUC were 0.700, 0.690, 0.695, respectively, Figure 5C). Furthermore, the DCA results showed that risk Model 1, which focuses on sialylation signaling-associated gene features, exhibited higher 3- and 5-year values of OS across a broader range of thresholds (Figure 5D) in comparison to other existing models,26–28 suggesting its improved clinical relevance.29
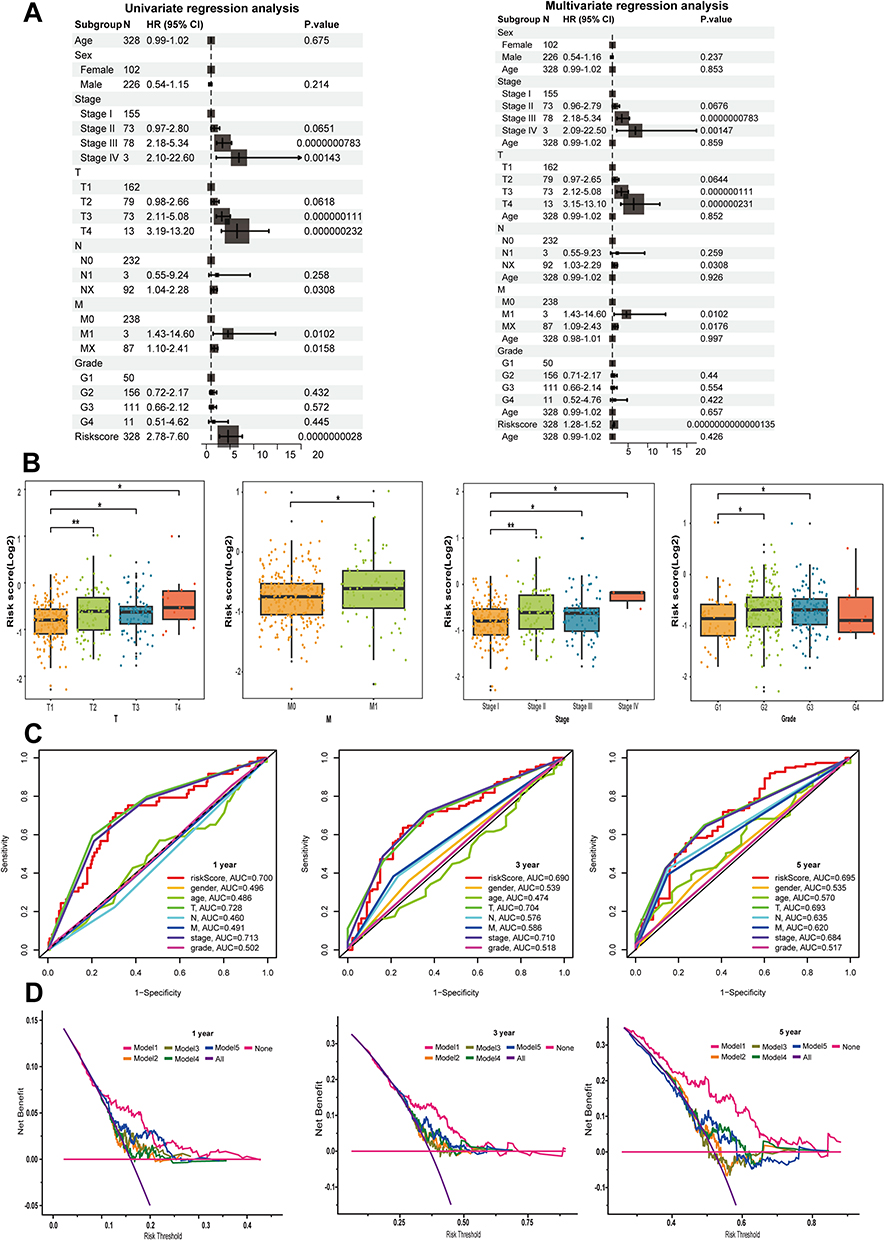 |
Figure 5 Clinical significance evaluation of the sialylation score. (A) The correlation between the sialylation score and clinical prognostic factors was analyzed through univariate and multivariate regression methods. (B) Assessment of the association between the sialylation score and different clinical parameters. (C) Time-base ROC curves for 1, 3, and 5-year OS. (D) DCA is used to compare the effectiveness of the sialylation score with other clinical prediction models. The models are as follows: 1 is sialylation, 2 is pyroptosis, 3 is ferroptosis, 4 is cuproptosis, and 5 is immune. *p < 0.05, **p < 0.01. |
A High Sialylation Score in Patients is Associated with a More Immunosuppressive Tumor Microenvironment
To explore the immune infiltration profiles of the two groups with different sialylation scores in more detail, the levels of immune and non-immune cells in tissues were assessed using gene expression data from TCGA. Six algorithms were employed to analyze the types and distribution of immune cells in tumor samples from groups with differing scores. Figure 6A shows that CD8+ T-cells displayed a decreasing trend in the group with high expression of sialylation genes, as revealed by several algorithms. Moreover, MCPCOUNTER analysis results indicated a similar trend for natural killer cells (NK) and cytotoxic lymphocytes (CTLs). Patients with higher sialylation scores have notably lower immune, stromal, and ESTIMATE scores, suggesting reduced immune and fibroblast infiltration compared to those with lower scores (Figure 6B). Furthermore, we conducted an analysis of pathway correlations and immune cell infiltration levels using the sialylation scores (Figure 6C). The sialylation score showed a positive correlation with Macrophage, Th1, and Th2 cells. Moreover, there was a notable correlation between the recruitment of dendritic cells, NK cells, Th1 cells and the sialylation score. Additionally, we applied the ssGSEA algorithm to gauge the 15 pathways linked to immunity. Patients with decreased sialylation scores demonstrated significantly heightened activity in CD8+ T cells, cytolytic activity, and Mast cells (Figure 6D).
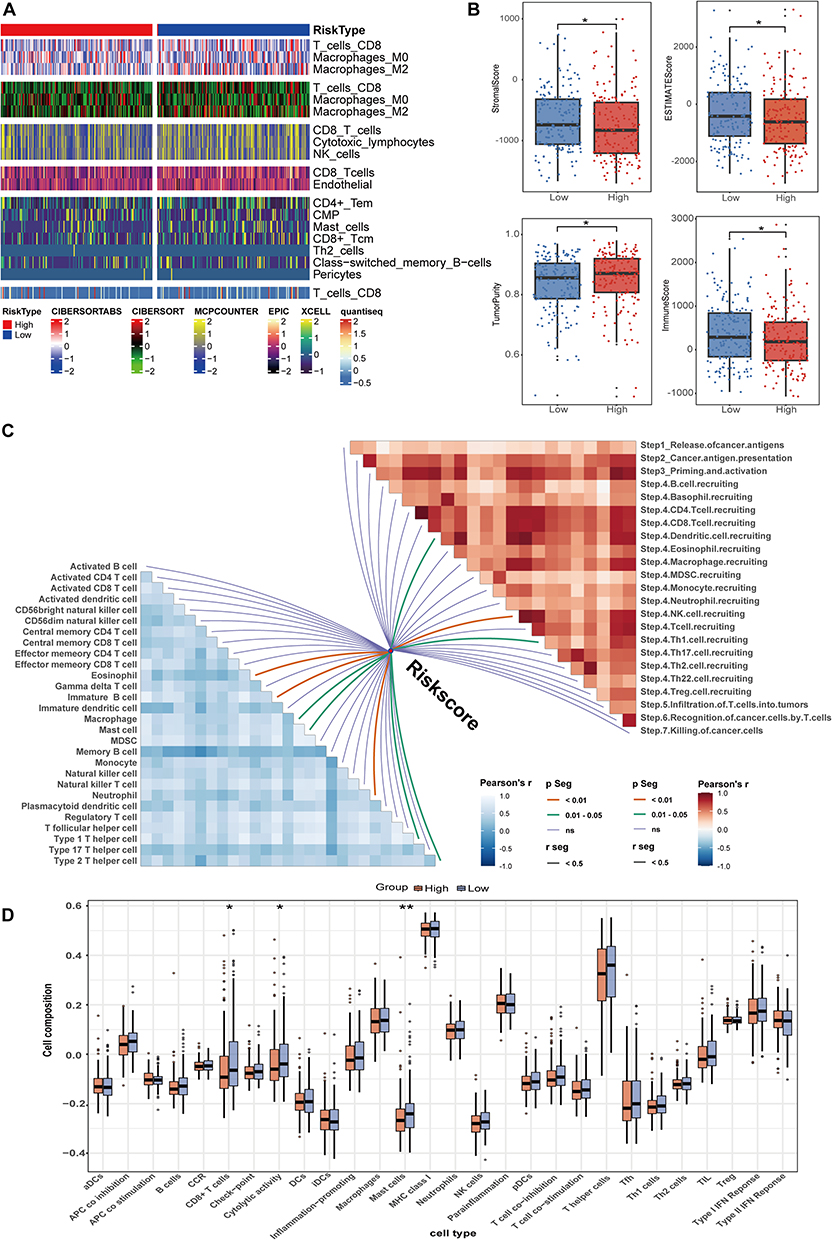 |
Figure 6 Immune landscape of tumors based on sialylation scores in the TCGA database. (A) Infiltration of different immune cells were examined using CIBERSORTABS, CIBERSORT, MCPcounter, EPIC, xCell, and Quantiseq algorithms. (B) Immunity, stromal status, and tumor purity were calculated. (C) Correlation between sialylation score and immunotherapy pathways (right) and immune cells (left). (D) Using the ssGSEA algorithm, scoring of immune-related pathways and immunophenotyping were conducted for the two groups. *p < 0.05, **p < 0.01. |
A Link Between Sialylation Phenotypes and the Heterogeneous Nature of the Tumor Immune Microenvironment is Revealed Through Single-Cell Analysis
Our next step was to study how alterations in the composition of cells affect sialylation traits at level of single cell. From the GSE151530 dataset, we collected 46 HCC samples, performed quality control, and normalized them. By employing standard marker genes, we sorted the filtered cells into 6 primary clusters: hepatocytes, NK cells, T cells, B cells, macrophages, and fibroblasts (Figure 7A). Cells were assessed for their sialylation activity based on sialylation score model related gene. Higher scores in Macrophage cells, NK cells, and T cells suggest that genes related to sialylation are actively expressed in these cells (Figures 7B). Figure 7C illustrates the expression of 8 sialylation-associated genes across various cell types. Based on the sialylation score distribution, we categorized the 46 HCC patients into groups with high and low sialylation scores. A comparative study of risk groups revealed unique cellular compositions, with the high sialylation group showing an abundance of macrophage cells and the low sialylation group having a predominance of immune cells (Figure 7D). Moreover, we scrutinized the intercellular interactions and intensity to further determine the primary agents involved in sialylation subtypes within the immune tumor microenvironment (Figure 7E). Our findings suggest that patients with elevated sialylation scores showed stronger and more intricate cellular interactions than those with lower scores (Figure 7H and I). Next, we compared the communication intensities strength and number between high and low sialylation subtypes. There was a marked increase in the connections between macrophage cells in the High subtype (Figure 7F).
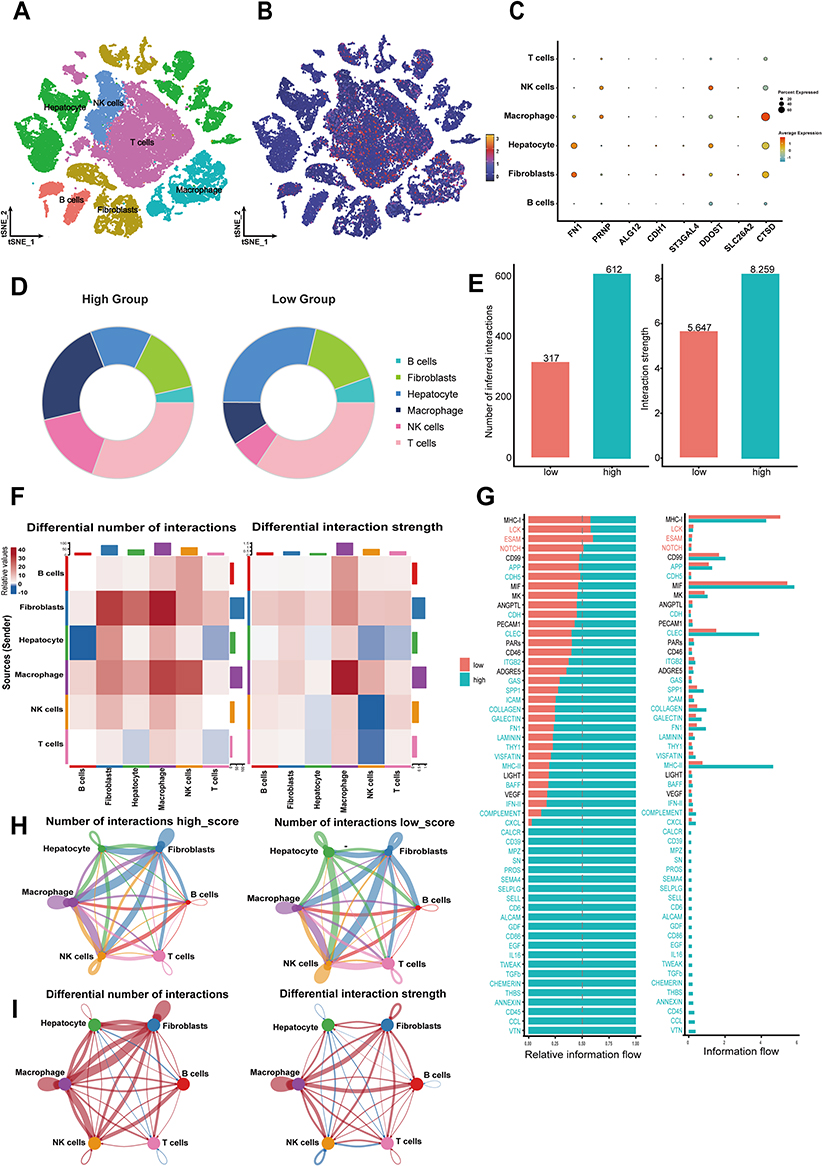 |
Figure 7 A high sialylation score suggests anti-immune behavior in the tumor microenvironment at the single-cell level (A) The t-SNE plot shows the composition of six major subtypes in primary HCC samples. (B) The distribution of high- and low- sialylation score cells. (C) Expression levels of the eight genes in the sialylation prognostic model across primary cell types. (D) Different proportions of cell types in different sialylation score groups. (E) Variations in both the number and the strength of interactions between the two groups with differing sialylation scores. (F) Variations in Intercellular Communication. (G) Histogram showing the absolute information flow in the signal path between the two groups. Histogram depicting the relative information flow of particular signaling pathways between the two groups. Paths with greater information flow in the high-risk score group were highlighted in blue, while those in the low-risk score group were highlighted in red. (H) The Circos plot shows the interactions between cells across two groups. (I) The Circos plot illustrates changes in interaction strength among various sialylation score categories, where blue signifies weaker interactions relative to the low sialylation score group, and red denotes stronger interactions. |
At the level of global signaling pathways, the high sialylation subtype mainly showed increased signaling in the MHC-II, CLEC, and COLLAGEN pathways (Figure 7G). In the high sialylation subgroup, macrophage cells primarily send enhanced signals to B cells via the MHC-II pathway, whereas signals from macrophage and fibroblast cells are mainly directed to NK and T cells through the CLEC pathway. Furthermore, in the CLLAGEN signaling pathway, the signals sent by fibroblast cells to other cells are generally stronger in the high sialylation subgroup (Figure 8A–C). The complexity of the network was also visible in the communication patterns, whether incoming or outgoing, across different cell types (Figure 8D and E). Significantly, NK and T cells had robust incoming interaction patterns, whereas fibroblast cells had marked outgoing interaction signatures. These findings revealed that patients with elevated sialylation scores had notably greater molecular complexity, stronger signaling networks, and better cellular interactions, which may facilitate immune escape. Moreover, the group with a high sialylation score had a notably higher count of ligand-receptor pairs than the group with a low sialylation score. Analysis of ligand-receptor pairs showed that in patients with high sialylation scores, myeloid cells mainly strengthened their interaction with NK and T cells via the CLEC2B-KLRB1, COL1A1-CD44 and HLA-DRA-CD4 pathways, demonstrating a stronger communication effect on high sialylation score group compared to low (Figure 8F and G). Overall, these findings reinforce the diversity of the immune tumor microenvironment among different sialylation score groups, indicating that the tumor microenvironment is immunosuppressive in the High sialylation score group.
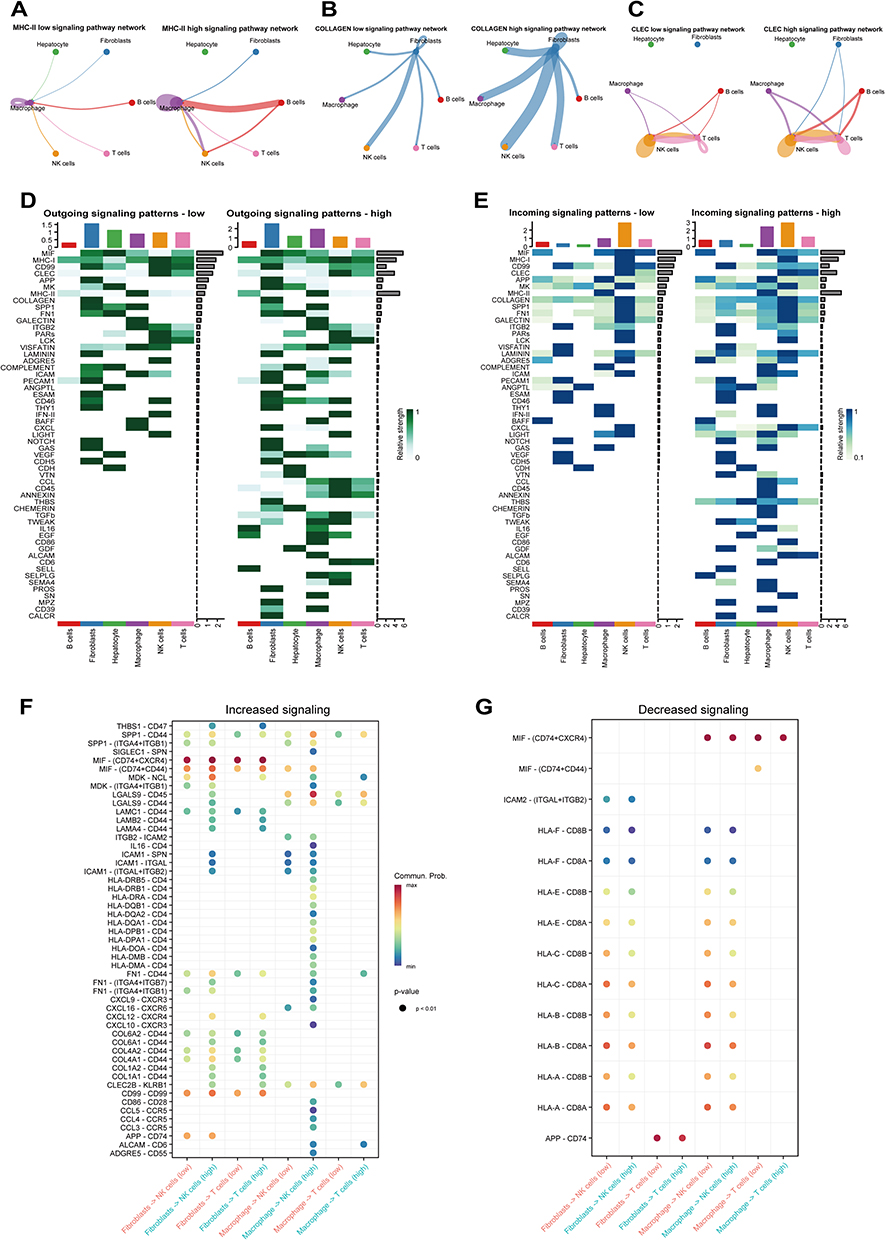 |
Figure 8 Variations in Ligand-Receptor Pairs among samples with high versus low sialylation scores. (A–C) Network of cell-cell interactions involving MHC-II, COLLAGEN, and CLEC in groups with high and low risk scores. (D and E) Heat maps showing the pattern and strength of outgoing and incoming signals for each cell type in the high-risk score group (D) and the low-risk score group (E). (F and G) Comparison of ligand-receptor pairs between groups with high and low risk scores. |
The Sialylation Scoring Model Forecasts Potential Targeted Treatments for Patients with HCC
We utilized the GDSC database to analyze how different sialylation subtypes respond to various drugs, aiming to comprehend the link between sialylation phenotypes and drug resistance. Differences in IC50 values for 15 potential drugs were observed on different sialylation scores group, implying a potential correlation between drug sensitivity and sialylation scores. Patients who had high sialylation scores demonstrated increased responsiveness to medications such as Elephantin_1835, AZ6102_2109, Nutlin_3a 1047, Dactinomycin_1911, Epirubicin 1511, AZD5153 1706, Dactolisib 1057, GNE-317_1926, Ipatasertib_1924, PRIMA-1MET_1131, Oxaliplatin 1089, Oxaliplatin 1806, Acetalax 1804, Sabutoclax_1849, WZ4003 1614 (Figure 9A and B). These results demonstrate a significant link between sialylation phenotypes and drug resistance, offering important insights for choosing targeted cancer therapies. The variation in drug responses between groups with high and low sialylation scores highlights the importance of considering sialylation phenotypes in personalized treatment plans for HCC patients.
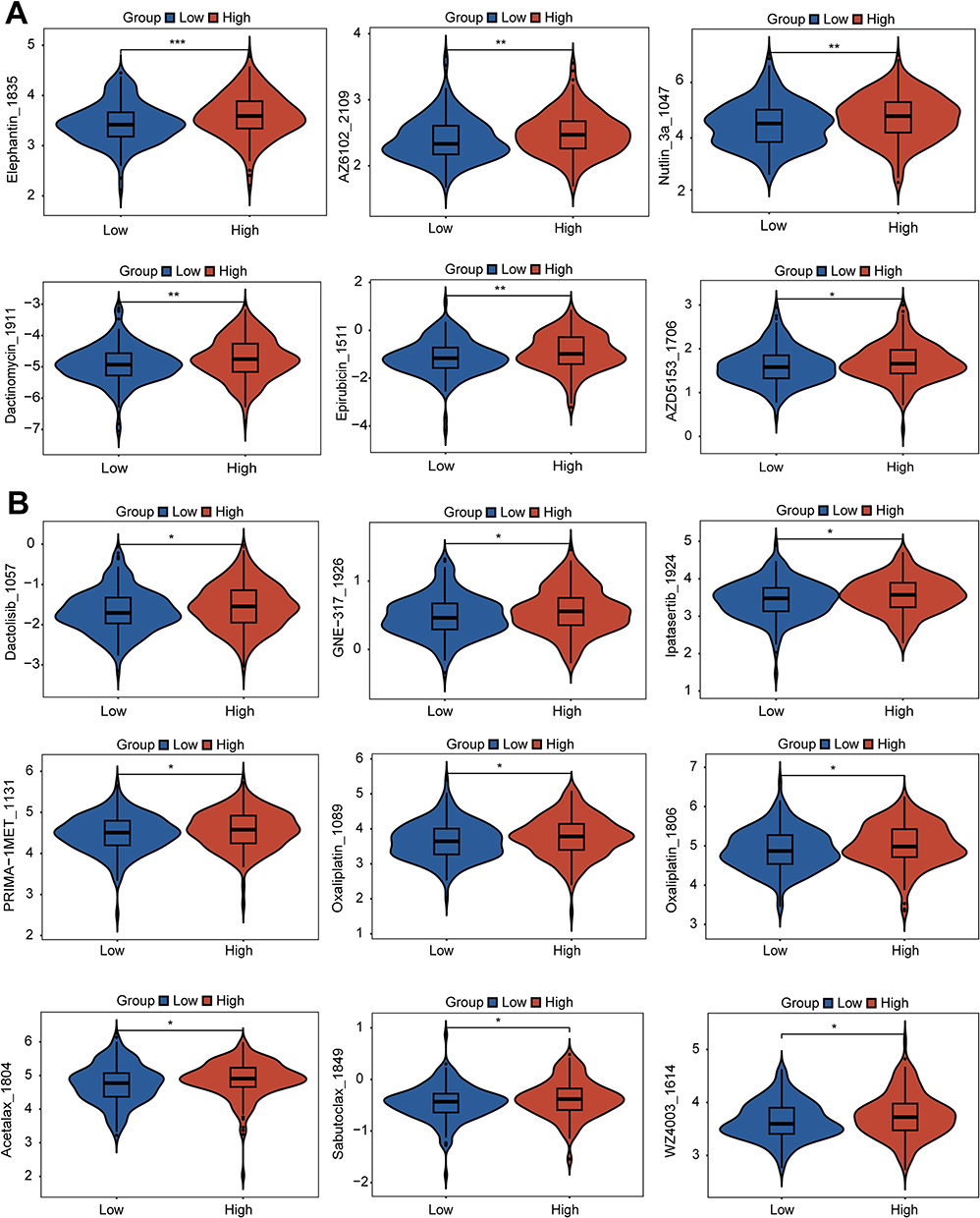 |
Figure 9 Potential targeted drugs’ IC50 values across two clusters based on sialylation scores (A and B) The top 15 drugs exhibit increased sensitivity in patients with high sialylation scores. *p < 0.05, **p < 0.01, ***p < 0.001. |
Targeting the Sialylation Signaling Gene ST3GAL4 May Offer an Effective Approach for HCC Therapy
The expression of eight sialylation genes was investigated in HCC patients compared to normal tissues to identify those with biological significance and potential roles. First, eight genes showed varying expression levels in tissues from HCC patients compared to normal controls. CTSD was downregulated in HCC, whereas FN1, PRNP, ALG12, CDH1, ST3GAL4, DDOST, and SLC26A2 were significantly upregulated (Figure S1A). Second, there was a marked increase in the expression of DDOST, PRNP, and ST3GAL4 with advancing grades (Figure 10A). Apart from ST3GAL4, DDOST, PRNP have been extensively studied in HCC.30,31 Third, ST3GAL4 has the highest relevance score, among these 8 genes in Genecards database (Table S1). As a result, studying the function of ST3GAL4 may offer new perspectives on HCC biology and therapeutic approaches. Previous studies have shown that ST3GAL4 exhibits a unique expression pattern in sialylation32 and influences sialylation in fibroblasts, promoting immune suppression through the modulation of myeloid cells.33 Moreover, higher levels of ST3GAL4 expression were closely linked to poor outcomes in HCC patients in various datasets (Figure S1B-C). To assess the functional relevance of ST3GAL4, siRNA plasmids were designed to knock down its expression in SK-Hep-1 and SNU-449 cells. As shown in Figure 10B, the expression levels of ST3GAL4 were effectively decreased by both siRNAs. The results of the Western blot also indicated a significant downregulation of ST3GAL4 expression in the two distinct Si-ST3GAL4 groups (Figure 10C). Next, a detailed array of functional analyses was performed on the cell lines to understand the biological outcomes of ST3GAL4 reduction. Compared to the control group, The ST3GAL4-knockdown group exhibited a notable reduction in the number of cell colonies (Figure 10D). Furthermore, the Transwell and wound healing assays demonstrated that silencing ST3GAL4 notably diminished the invasive and migratory potential of HCC cells (Figure 10E and F). Furthermore, A notable decrease in HCC cell proliferation was observed in the EdU assay following the knockdown of ST3GAL4. (Figure 10G). The results indicate that ST3GAL4 is crucial in the regulation of tumorigenesis, progression, and migration in HCC.
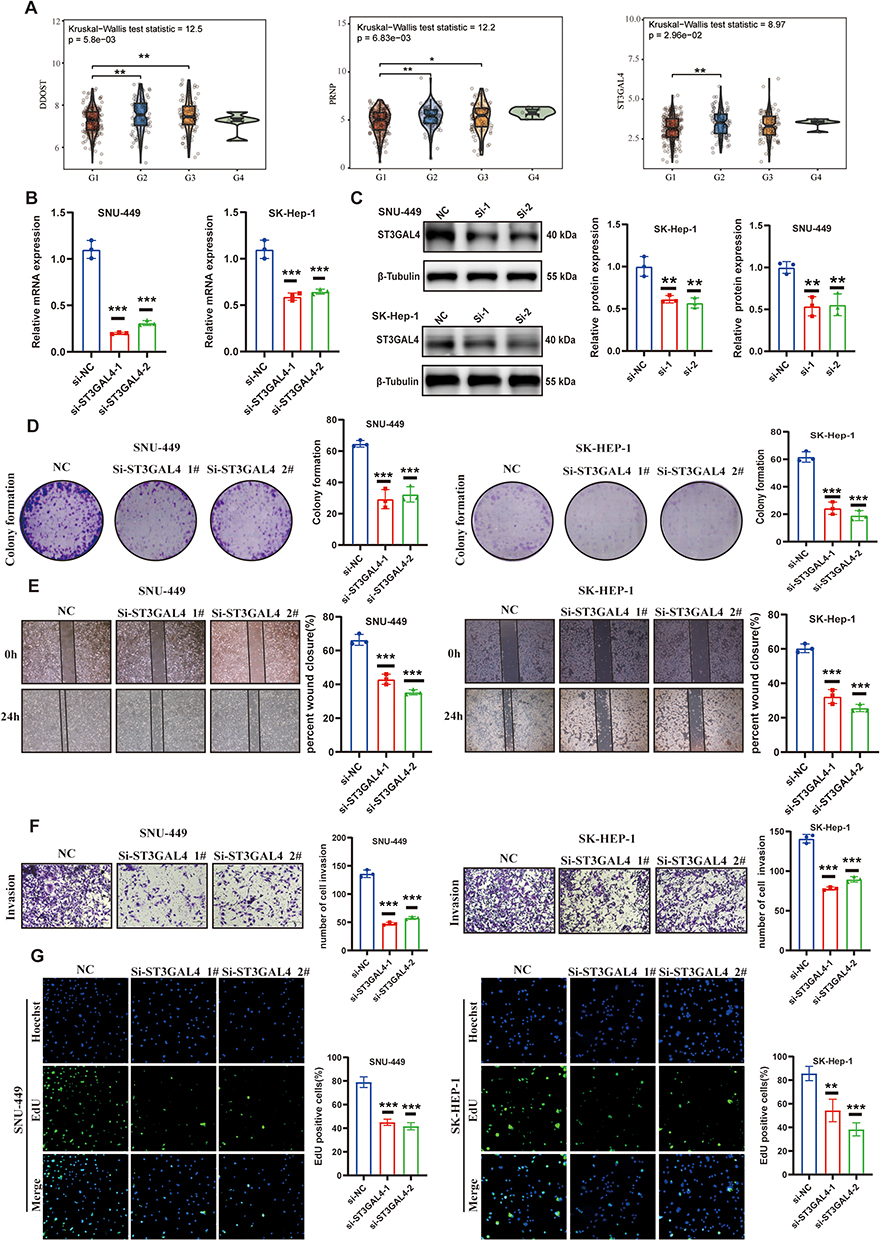 |
Figure 10 The role of the sialylation gene ST3GAL4 in the biology of HCC. (A) Three genes involved in sialylation showed a significant correlation with tumor grade. (B) The expression of ST3GAL4 mRNA was significantly decreased in SK-HEP-1 and SNU-449 cells transfected with si-ST3GAL4-1 and si-ST3GAL4-2 compared to those transfected with the negative control (si-NC). (C) WB analysis to assess ST3GAL4 protein levels in SK-HEP-1 and SNU-449 cells transfected with si-NC, si-ST3GAL4-1, and si-ST3GAL4-2. (D) Assay for colony formation to evaluate the clonogenic potential of SK-HEP-1 and SNU-449 cells. (E) Wound healing assay to evaluate the effect of si-ST3GAL4 transfection on the migratory ability of SK-HEP-1 and SNU-449 cells. (F) Transwell assay to assess the invasive abilities of SK-HEP-1 and SNU-449 cells after transfection. (G) Ethynyl deoxyuridine (EdU) incorporation assay to analysis the proliferation of SK-HEP-1 and SNU-449 cells after transfection. *p < 0.05, **p < 0.01, ***p < 0.001. |
Discussion
The BCLC, TNM, and Child-Pugh systems are key prognostic tools for HCC, offering vital direction for the clinical management of patients with this condition.34,35 The heterogeneity of HCC patients presents a challenge for any one predictive model or staging system to accurately predict prognosis, possibly causing undertreatment or over-treatment.5 Progress in molecular biology and immunology is expanding treatment choices for HCC.36–41 Hypersialylation can facilitate tumor metastasis through various mechanisms, such as boosting immune evasion, increasing tumor cell survival, and encouraging tumor invasion and migration.42 However, the lack of dependable prognostic biomarkers complicates personalized evaluation. Moreover, we investigated the relationship between gene expression profiles related to sialylation, prognosis, response to immunotherapy, and drug efficacy to provide a theoretical basis for creating new therapeutic approaches.
In this study, we uncovered several significant findings. First, we performed the initial transcriptome analysis of genes associated with sialylation and identified novel molecular subtypes in HCC. Furthermore, two groups of HCC patients were formed according to 351 sialylation genes. Cluster 2, characterized by inhibited sialylation, showed reduced expression of sialylation genes and higher overall survival rates, while Cluster 1, with activated sialylation, exhibited lower overall survival rates. A higher overall survival rate in the group with low sialylation could be linked to their increased susceptibility to T cell-mediated tumor cell killing due to sialylation suppression.43 Additional examination revealed that HCC patients with activated sialylation are linked to a poorer response to immunotherapy and increased immune evasion, indicating a significant connection between sialylation subtypes and unfavorable outcomes. Consequently, different sialylation subtypes might be utilized as a novel prognostic indicator for evaluating HCC progression. Significantly, past previous research has discovered analogous subgroups through Post-translational Modification (PTM),44,45 lending support to the credibility of our molecular subtypes.
Additional validation on two different datasets using 117 machine-learning algorithms affirmed that StepCox[forward] combined with Ridge was the most effective model. Our sialylation prognostic model offered more dependable prediction accuracy compared to TNM staging and other prediction models,26–29 with most AUC values exceeding 0.690. The potential for clinical application of the sialylation score is significant, as both multivariate and univariate regression analyses demonstrated its independent association with HCC prognosis. Additionally, the DCA method was utilized to aid doctors in choosing the most suitable clinical prediction model among several options.26–29 The 8-gene prognostic model we developed outperformed previously published models in clinical application. Assessing the sialylation score in patients with HCC provides researchers and clinicians with important information about the chances of advancement. This prognostic model could aid in making therapeutic decisions, assessing prognosis, and developing targeted treatments.
Third, using various immune scoring methods, our research discovered that patients with high sialylation scores have a significantly immunosuppressive microenvironment at single-cell level, delivering novel clinical understandings of the immune features related to sialylation in HCC. Patients with high sialylation scores exhibited a notable decrease in immune cell infiltration levels across the eight immune algorithms. The reason is that tumors in patients with elevated sialylation scores possess enhanced immune evasion tactics, which help them avoid being detected and attacked by the immune system.46 By emitting immunosuppressive cytokines or recruiting immunosuppressive cells like Tregs, these tumors may significantly improve immune evasion and promote tumor progression.47 Patients with low sialylation scores, in contrast, show significantly elevated levels of CD4+ central memory T and CD8+ T cells, compared to patients with high sialylation scores. The variations in survival rates and immunotherapy success rates among different sialylation phenotypes can be attributed to this. Higher levels of immune cell infiltration are frequently linked to better outcomes.48 This is consistent with our findings, which further imply that patients with decreased sialylation scores possess more effective antitumor actions and enhanced immune responses. Furthermore, we investigated the underlying reasons for this immune heterogeneity at the level of individual cells. The findings showed that patients with elevated sialylation scores have notably more cellular interactions, stronger signaling networks, and greater molecular complexity than those with lower scores. This complex network of intercellular communication indicates a more elaborate regulatory system in tumor microenvironment. We proposed that these complex signaling networks mainly convey signals that suppress the immune system and regulate its functions, possibly aiding in immune evasion, increasing resistance to treatments, and boosting tumor aggressiveness.49 For patients with low sialylation scores, analysis of ligand-receptor pairs showed that myeloid cells increase interaction with NK and T cells via the CLEC2B-KLRB1, COL1A1-CD44, and HLA-DRA-CD4 pathways. This complex network of intercellular communication indicates a more elaborate regulatory system within the tumor microenvironment. Hence, modulating sialylation expression and immune cell interactions could help develop novel treatment strategies to improve the prognosis for those with HCC.
Finally, we examined 8 sialylation signature genes and their impact on the progression of HCC.
From these, ST3GAL4 was chosen for subsequent in vitro experiments. ST3GAL4, an α2,3-sialyltransferase involved in sialylation, specifically alters the integrin β1 subunit,50 which is linked to cancer metastasis and the tumor immune microenvironment (TIME).51 ST3GAL4 dysregulation can promote the differentiation of monocytes into immunosuppressive tumor-associated macrophages,33 induce eosinophil recruitment,52 and accelerate cancer progression.53 Our experiments demonstrated that reducing ST3GAL4 levels impaired the invasion and migration capabilities of HCC cells, pointing to ST3GAL4 as a possible therapeutic target.
Although the findings are positive, there are several limitations in our study that require attention. To ensure its applicability to various patient groups, the sialylation score model must be validated in large-scale, multi-center clinical cohorts. The strength of our results may be compromised by biases in the datasets, like demographic imbalances or sampling bias. Moreover, the precise immune modulation mechanism and biological processes related to sialylation gene patterns in HCC remain to be experimentally verified. In addition, Although the role of ST3GAL4 in cell lines of HCC has been confirmed, additional in vivo studies are needed to fully understand its mechanisms in regulating ST3GAL4 and the progression of HCC. Overcoming these limitations is essential for enhancing the clinical usefulness of our model and comprehending its significance for various patient groups.
Conclusion
Our research emphasizes the importance of gene expression patterns associated with sialylation as important prognostic indicators for HCC. Developing a prognostic model based on sialylation and identifying crucial genes like ST3GAL4 may offer new ideas for targeted therapy. Further studies could investigate their clinical application value and precise mechanisms to offer more detailed precision medicine solutions for HCC.
Abbreviations
HCC, Hepatocellular Carcinoma; OS, Overall survival; ROC, Receiver Operating Characteristic; DCA, Decision curve analysis; PFA, Paraformaldehyde; EdU, 5-Ethynyl-2’- deoxyuridine; CDF, Cumulative distribution function; PCA, Principal Component Analysis; NK, Natural killer cells; CTLs, Cytotoxic lymphocytes; PTM, Post-translational Modification; TIME, Tumor immune microenvironment.
Data Sharing Statement
This study analyzed publicly available datasets. The data were derived from TCGA database, ICGC-LIRI-JP database and the GSE151530 in the GEO database, respectively. All data generated or analyzed during this study are available from the corresponding author upon reasonable request.
Ethics Approval and Consent to Participate
This study is exempt from ethical review in accordance with item 1 of Article 32 of the Measures for Ethical Review of Life Science and Medical Research Involving Human Subjects dated February 18, 2023, China. The specific provision is as follows: Article 32: Ethical review may be exempted for human life science and medical research involving the use of personal information data or biological samples under the following circumstances, provided that the research causes no harm to human subjects and involves no sensitive personal information or commercial interests, so as to reduce unnecessary burdens on researchers and facilitate such studies: Research utilizing legally obtained publicly available data or data generated through observation of public behavior without interference.
Acknowledgments
Thanks to Figdraw (https://www.figdraw.com/) for providing a platform for us to create some flowchart materials.
Funding
This work was supported by the grants from the National Natural Science Foundation of China (No. 82171944, 81873899 and 82472003), the Natural Science Foundation of Guangdong Province (No 2021A1515012611 and 2023A1515010579), and the Scientific Research Fund for Hundred Talents Program Talent Introduction of Sun Yat-sen University (1320323001).
Disclosure
The authors declare no competing interests.
References
1. Rumgay H, Arnold M, Ferlay J, et al. Global burden of primary liver cancer in 2020 and predictions to 2040. J Hepatol. 2022;77:1598–21. doi:10.1016/j.jhep.2022.08.021
2. Gordan JD, Kennedy EB, Abou-Alfa GK, et al. Systemic therapy for advanced hepatocellular carcinoma: ASCO guideline update. J Clin Oncol. 2024;42:1830–1850. doi:10.1200/JCO.23.02745
3. Gao Y-X, Ning -Q-Q, Yang P-X, et al. Recent advances in recurrent hepatocellular carcinoma therapy. World J Hepatol. 2023;15:460. doi:10.4254/wjh.v15.i4.460
4. Singal AG, Sanduzzi-Zamparelli M, Nahon P, et al. International Liver Cancer Association (ILCA) white paper on hepatocellular carcinoma risk stratification and surveillance. J Hepatol. 2023;79:226–239. doi:10.1016/j.jhep.2023.02.022
5. Vogel A, Meyer T, Sapisochin G, Salem R, Saborowski A. Hepatocellular carcinoma. Lancet. 2022;400:1345–1362. doi:10.1016/S0140-6736(22)01200-4
6. Llovet JM, Castet F, Heikenwalder M, et al. Immunotherapies for hepatocellular carcinoma. Nat Rev Clin. 2022;19:151–172. doi:10.1038/s41571-021-00573-2
7. Greten TF, Sangro B. Targets for immunotherapy of liver cancer. J Hepatol. 2018;68:157–166. doi:10.1016/j.jhep.2017.09.007
8. Boelaars K, Goossens-Kruijssen L, Wang D, et al. Unraveling the impact of sialic acids on the immune landscape and immunotherapy efficacy in pancreatic cancer. J ImmunoTher Cancer. 2023;11:e007805. doi:10.1136/jitc-2023-007805
9. Cao K, Zhang G, Yang M, et al. Attenuation of sialylation augments antitumor immunity and improves response to immunotherapy in ovarian cancer. Cancer Res. 2023;83:2171–2186. doi:10.1158/0008-5472.CAN-22-3260
10. Dashzeveg NK, Jia Y, Zhang Y, et al. Dynamic glycoprotein hyposialylation promotes chemotherapy evasion and metastatic seeding of quiescent circulating tumor cell clusters in breast cancer. Cancer Discovery. 2023;13:2050–2071. doi:10.1158/2159-8290.CD-22-0644
11. Wang Z, Wu Z, Wu H, et al. The pan-cancer multi-omics landscape of key genes of sialylation combined with RNA-sequencing validation. Comput Biol. 2023;166:107556. doi:10.1016/j.compbiomed.2023.107556
12. Huang X, Zhang S, Tang J, et al. A self-propagating c-met–SOX2 axis drives cancer-derived IgG signaling that promotes lung cancer cell stemness. Cancer Res. 2023;83:1866–1882. doi:10.1158/0008-5472.CAN-22-2733
13. Scott E, Archer Goode E, Garnham R, et al. ST6GAL1‐mediated aberrant sialylation promotes prostate cancer progression. J Pathol. 2023;261:71–84. doi:10.1002/path.6152
14. Wang Y, Khan A, Antonopoulos A, et al. Loss of α2-6 sialylation promotes the transformation of synovial fibroblasts into a pro-inflammatory phenotype in arthritis. Nat Commun. 2021;12:2343. doi:10.1038/s41467-021-22365-z
15. Natoni A, Cerreto M, De Propris MS, et al. Sialylation regulates migration in chronic lymphocytic leukemia. Haematologica. 2023;108:1851. doi:10.3324/haematol.2022.281999
16. Beumer BR, Buettner S, Galjart B, et al. Systematic review and meta-analysis of validated prognostic models for resected hepatocellular carcinoma patients. Eur J Surg Oncol. 2022;48:492–499. doi:10.1016/j.ejso.2021.09.012
17. Li J, Zhou J, Yang PH, et al. Nomograms for survival prediction in patients undergoing liver resection for hepatitis B virus related early stage hepatocellular carcinoma. Eur J Cancer. 2016;62:86–95. doi:10.1016/j.ejca.2016.04.011
18. Wang Y, Sun K, Shen J, et al. Novel prognostic nomograms based on inflammation-related markers for patients with hepatocellular carcinoma underwent hepatectomy. Cancer Res. 2019;51:1464–1478.
19. Xia Y, Li Y, Li J, et al. Prognostic nomograms for pre-and postoperative predictions of long-term survival for patients who underwent liver resection for huge hepatocellular carcinoma. J Am College Surg. 2015;221(962–974):e964.
20. Yang P, Qiu J, Li J, et al. Nomograms for pre-and postoperative prediction of long-term survival for patients who underwent hepatectomy for multiple hepatocellular carcinomas. Ann Surg. 2016;263:778–786. doi:10.1097/SLA.0000000000001339
21. Sun HC, Xie L, Yang XR, et al. Shanghai Score: a Prognostic and Adjuvant Treatment-evaluating System Constructed for Chinese Patients with Hepatocellular Carcinoma after Curative Resection. Chin Med J. 2017;130:2650–2660. doi:10.4103/0366-6999.218019
22. Newman AM, Liu CL, Green MR, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Meth. 2015;12:453–457. doi:10.1038/nmeth.3337
23. Racle J, De Jonge K, Baumgaertner P, Speiser DE, Gfeller D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. elife. 2017;6:e26476. doi:10.7554/eLife.26476
24. Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18:220. doi:10.1186/s13059-017-1349-1
25. Becht E, Giraldo NA, Lacroix L, et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016;17:218. doi:10.1186/s13059-016-1070-5
26. Zhang Z, Zeng X, Wu Y, Liu Y, Zhang X, Song Z. Cuproptosis-related risk score predicts prognosis and characterizes the tumor microenvironment in hepatocellular carcinoma. Front Immunol. 2022;13:925618. doi:10.3389/fimmu.2022.925618
27. Wang H, Yang C, Jiang Y, Hu H, Fang J, Yang F. A novel ferroptosis-related gene signature for clinically predicting recurrence after hepatectomy of hepatocellular carcinoma patients. Am J Cancer Res. 2022;12:1995.
28. Gao X, Wang WX, Zhang XL. A novel pyroptosis risk model composed of NLRP6 effectively predicts the prognosis of hepatocellular carcinoma patients. Cancer Med. 2023;12:808–823. doi:10.1002/cam4.4898
29. Wang Z, Zhu J, Liu Y, et al. Development and validation of a novel immune-related prognostic model in hepatocellular carcinoma. J Transl Med. 2020;18:67. doi:10.1186/s12967-020-02255-6
30. Su D, Zhang Z, Xia F, et al. ICD-related risk model predicts the prognosis and immunotherapy response of patients with liver cancer. Front Pharmacol. 2023;14:1202823. doi:10.3389/fphar.2023.1202823
31. Zhu C, Xiao H, Jiang X, Tong R, Guan J. Prognostic Biomarker DDOST and Its Correlation With Immune Infiltrates in Hepatocellular Carcinoma. Front Genet. 2021;12:819520. doi:10.3389/fgene.2021.819520
32. Rodriguez E, Boelaars K, Brown K, et al. Sialic acids in pancreatic cancer cells drive tumour-associated macrophage differentiation via the Siglec receptors Siglec-7 and Siglec-9. Nat Commun. 2021;12:1270. doi:10.1038/s41467-021-21550-4
33. Boelaars K, Rodriguez E, Huinen ZR, et al. Pancreatic cancer-associated fibroblasts modulate macrophage differentiation via sialic acid-Siglec interactions. Commun Biol. 2024;7:430. doi:10.1038/s42003-024-06087-8
34. Reig M, Forner A, Rimola J, et al. BCLC strategy for prognosis prediction and treatment recommendation: the 2022 update. J Hepatol. 2022;76:681–693. doi:10.1016/j.jhep.2021.11.018
35. Fan Z, Zhou P, Jin B, et al. Recent therapeutics in hepatocellular carcinoma. Am J Cancer Res. 2023;13:261–275.
36. Luo X-Y, Wu K-M, He -X-X. Advances in drug development for hepatocellular carcinoma: clinical trials and potential therapeutic targets. J Exp Clin Cancer Res. 2021;40:172. doi:10.1186/s13046-021-01968-w
37. Jiang Y, Su K, Li H, et al. Efficacy and safety of the combination of envafolimab and lenvatinib in unresectable hepatocellular carcinoma: a single-arm, multicentre, exploratory Phase II clinical study. Invest New Drugs. 2025;43:18–29. doi:10.1007/s10637-024-01468-6
38. Li H, Zhou C, Wang C, et al. Lasso-Cox interpretable model of AFP-negative hepatocellular carcinoma. Clin Transl Oncol. 2025;27:309–318. doi:10.1007/s12094-024-03588-0
39. Su K, Liu X, Zeng YC, et al. Machine Learning Radiomics for Predicting Response to MR-Guided Radiotherapy in Unresectable Hepatocellular Carcinoma: a Multicenter Cohort Study. J Hepatocell Carcinoma. 2025;12:933–947. doi:10.2147/JHC.S521378
40. Xu K, Gu T, Su K, et al. Stereotactic body radiation therapy (SBRT) increases anti-PD-1 antitumor activity by enhancing the tumor immune microenvironment in mice with metastatic hepatocellular carcinoma. Discov Oncol. 2025;16:1081. doi:10.1007/s12672-025-02914-4
41. Jiang Y, Guo L, Han L, et al. Thymidine kinase 1 appears to be a marker for the prognosis of hepatocellular carcinoma based on a large-scale, multicenter study. J Cancer Res Clin Oncol. 2023;149:14271–14282. doi:10.1007/s00432-023-05089-z
42. Dobie C, Skropeta D. Insights into the role of sialylation in cancer progression and metastasis. Br J Cancer. 2021;124:76–90. doi:10.1038/s41416-020-01126-7
43. Appadurai MI, Chaudhary S, Shah A, et al. ST6GalNAc-I regulates tumor cell sialylation via NECTIN2/MUC5AC-mediated immunosuppression and angiogenesis in non–small cell lung cancer. J Clin Invest. 2025;135. doi:10.1172/JCI186863
44. Zhu Z, Feng S, Zeng A, Song L. Advances in Palmitoylation: a key Regulator of liver cancer development and therapeutic targets. Biochem Pharmacol. 2025;116810. doi:10.1016/j.bcp.2025.116810
45. Li M, Li H, Liu L, et al. Integrated multi-omics analysis and machine learning refine molecular subtypes and prognosis in hepatocellular carcinoma through O-linked glycosylation genes. Funct Integr Genomics. 2025;25:162. doi:10.1007/s10142-025-01669-z
46. Jhunjhunwala S, Hammer C, Delamarre L. Antigen presentation in cancer: insights into tumour immunogenicity and immune evasion. Nat Rev Cancer. 2021;21:298–312. doi:10.1038/s41568-021-00339-z
47. Shen KY, Zhu Y, Xie SZ, Qin LX. Immunosuppressive tumor microenvironment and immunotherapy of hepatocellular carcinoma: current status and prospectives. J Hematol Oncol. 2024;17:25. doi:10.1186/s13045-024-01549-2
48. Melssen MM, Sheybani ND, Leick KM, Slingluff CL Jr. Barriers to immune cell infiltration in tumors. J Immunother Cancer. 2023;11. doi:10.1136/jitc-2022-006401
49. Zhang M, Liu C, Tu J, et al. Advances in cancer immunotherapy: historical perspectives, current developments, and future directions. Mol Cancer. 2025;24:136. doi:10.1186/s12943-025-02305-x
50. Qi F, Isaji T, Duan C, et al. ST3GAL3, ST3GAL4, and ST3GAL6 differ in their regulation of biological functions via the specificities for the α2,3-sialylation of target proteins. FASEB j. 2020;34:881–897. doi:10.1096/fj.201901793R
51. Wu C, Tan J, Shen H, et al. Exploring the relationship between metabolism and immune microenvironment in osteosarcoma based on metabolic pathways. J Biomed Sci. 2024;31:4. doi:10.1186/s12929-024-00999-7
52. Immler R, Nussbaumer K, Doerner A, et al. CCR3-dependent eosinophil recruitment is regulated by sialyltransferase ST3Gal-IV. Proc Natl Acad Sci U S A. 2024;121:e2319057121. doi:10.1073/pnas.2319057121
53. Han R, Lin C, Lu C, et al. Sialyltransferase ST3GAL4 confers osimertinib resistance and offers strategies to overcome resistance in non-small cell lung cancer. Cancer Lett. 2024;588:216762. doi:10.1016/j.canlet.2024.216762
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Decaprenyl Diphosphate Synthase Subunit 1 (PDSS1): A Potential Prognostic Biomarker and Immunotherapy-Target for Hepatocellular Carcinoma
Yang Y, Li J, Tang M, Nie B, Huang W
Cancer Management and Research 2022, 14:1627-1639
Published Date: 3 May 2022
Identification of KRBA1 as a Potential Prognostic Biomarker Associated with Immune Infiltration and m6A Modification in Hepatocellular Carcinoma
Liu Y, Fu B, Yu Z, Song G, Zeng H, Gong Y, Ding Y, Huang D
Journal of Hepatocellular Carcinoma 2022, 9:497-516
Published Date: 31 May 2022
A Machine Learning Model Based on Health Records for Predicting Recurrence After Microwave Ablation of Hepatocellular Carcinoma
An C, Yang H, Yu X, Han Z, Cheng Z, Liu F, Dou J, Li B, Li Y, Li Y, Yu J, Liang P
Journal of Hepatocellular Carcinoma 2022, 9:671-684
Published Date: 28 July 2022
Prognosis and Personalized Treatment Prediction in Different Mutation-Signature Hepatocellular Carcinoma
Zhang Y, Liu Z, Li J, Li X, Duo M, Weng S, Lv P, Jiang G, Wang C, Li Y, Liu S, Li Z
Journal of Hepatocellular Carcinoma 2023, 10:241-255
Published Date: 15 February 2023
Machine Learning-Based Radiopatho-Clinical Model Integrating Ultrasound Radiomics and Kleiner Score for Prognosis Prediction in NAFLD-Related Hepatocellular Carcinoma
Li CL, Jia Z, Yao ZY, Cui XT, Sun A, Hao AY, Chen ZY, Chen F, Cao JY, Wang ZS
Journal of Hepatocellular Carcinoma 2026, 13:611041
Published Date: 19 June 2026