Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 21
Identification of ERN1 as a Potential Context-Dependent Biomarker in Chronic Obstructive Pulmonary Disease Based on Bioinformatics Analysis of GSE57148 Dataset
Authors Peng Q, Yang M, Fan D, Zhou P
Received 20 March 2026
Accepted for publication 15 June 2026
Published 17 June 2026 Volume 2026:21 610706
DOI https://doi.org/10.2147/COPD.S610706
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Zijing Zhou
Qing Peng, Mi Yang, Du Fan, Pei Zhou
Department of Respiratory Medicine, The Third Hospital of Changsha (The Affiliated Changsha Hospital of Hunan University), Changsha, Hunan, 410015, People’s Republic of China
Correspondence: Du Fan, Email [email protected] Pei Zhou, Email [email protected]
Purpose: To identify an endoplasmic-reticulum-stress-related candidate gene in chronic obstructive pulmonary disease (COPD) lung tissue and assess its internal discriminative performance and cross-cohort reproducibility.
Patients and Methods: This bioinformatics study used the GSE57148 lung tissue dataset (98 COPD and 91 subjects with normal-spirometry; all male smokers undergoing lung resection). Differential expression was analyzed using limma on log2 (fragments per kilobase of transcript per million mapped reads [FPKM] + 1), followed by enrichment and protein-protein interaction analyses. Endoplasmic reticulum to nucleus signaling 1 (ERN1) was prioritized using a literature-informed post hoc multi-criteria framework. Internal discrimination was evaluated by receiver operating characteristic (ROC) analysis with repeated stratified 10-fold cross-validation and bootstrap optimism correction. External sensitivity analyses were performed in independent cohorts.
Results: A total of 308 differentially expressed genes were identified. ERN1 was significantly upregulated in COPD (log2FC = 0.75, adjusted P = 1.98 x 10^-15). In the discovery cohort, ERN1 showed internal discrimination (area under the ROC curve [AUC] = 0.853; cross-validated AUC = 0.848). However, external replication was heterogeneous; in the largest mixed-sex cohort (GSE47460), discrimination was limited (AUC = 0.477), and adjusted external models remained non-significant.
Conclusion: ERN1 is upregulated in COPD lung tissue in GSE57148 and represents an endoplasmic-reticulum-stress-related, context-dependent candidate signal. Current evidence is preliminary and requires prospective validation in independent, sex-balanced cohorts and clinically accessible biospecimens.
Keywords: chronic obstructive pulmonary disease, COPD, ERN1, GSE57148, biomarker, bioinformatics analysis, endoplasmic reticulum stress, lung tissue
Introduction
Chronic Obstructive Pulmonary Disease (COPD) is a common, preventable, and treatable chronic respiratory disease characterized by persistent airflow limitation and chronic airway inflammation.1 It is a global public health problem with high morbidity and mortality—according to the Global Burden of Disease Study, COPD affects approximately 384 million people worldwide, accounting for 6.4 million deaths annually, bringing a heavy burden to families and society. The pathogenesis of COPD is complex, involving multiple factors such as genetic susceptibility, environmental exposure (smoking, air pollution), inflammatory response, oxidative stress, and endoplasmic reticulum stress.2 Currently, the diagnosis of COPD mainly relies on pulmonary function tests, but there are limitations in early diagnosis and severity assessment—pulmonary function changes are often not obvious in the early stage of COPD, leading to missed diagnosis and delayed treatment. Therefore, screening for reliable molecular biomarkers is crucial for improving the early diagnosis, treatment effect, and prognosis of COPD.
With the development of high-throughput sequencing technology and bioinformatics analysis methods, gene expression profile datasets have become an important tool for screening disease-related biomarkers and exploring disease pathogenesis.3 The Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) is a public database that stores a large number of gene expression profile data, providing convenient conditions for bioinformatics research on various diseases. The GSE57148 dataset was submitted by Kim et al in 2015,4 containing RNA-seq gene expression data from lung tissues of 98 COPD subjects and 91 subjects with normal-spirometry, sequenced on the GPL11154 platform (Illumina HiSeq 2000) All subjects were male smokers who underwent lung surgical resection. The detailed clinical characteristics of the subjects are shown in Table 1. The large sample size (n = 189) of this dataset provides robust statistical power for screening COPD-related differentially expressed genes and candidate biomarkers.
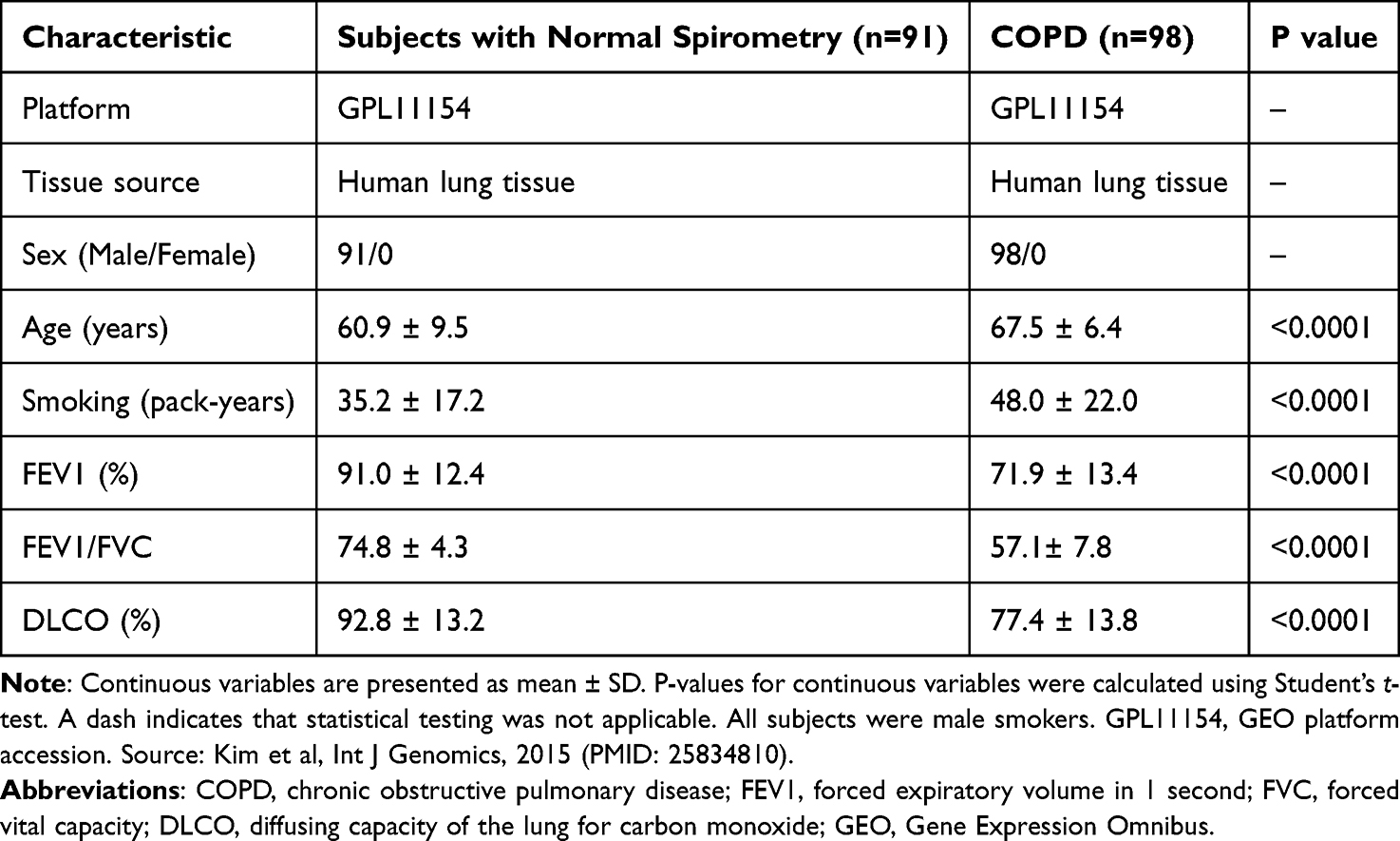 |
Table 1 Clinical Characteristics of Study Subjects |
ERN1 (Endoplasmic Reticulum To Nucleus Signaling 1), also known as IRE1α, is a key gene involved in the endoplasmic reticulum stress response.5 It plays an important role in regulating cell survival, apoptosis, and inflammatory response by activating downstream signaling pathways such as the unfolded protein response (UPR) and the endoplasmic reticulum stress sensor IRE1α/X-box binding protein 1 (XBP1) axis. Studies have shown that ERN1 is closely related to the occurrence and development of various diseases such as cancer, diabetes, and neurodegenerative diseases,6–8 and accumulating evidence suggests that endoplasmic reticulum stress and UPR activation play critical roles in COPD pathogenesis, including cigarette smoke-induced epithelial cell apoptosis, inflammatory responses, and mucus hypersecretion. However, the role of ERN1 as a biomarker in COPD has not been fully clarified. In this study, we used bioinformatics methods to analyze the GSE57148 dataset, screen differentially expressed genes in COPD lung tissues, identify hub genes through protein-protein interaction (PPI) network analysis, and focus on evaluating the expression level and within-cohort discriminative signal of ERN1, aiming to propose a mechanistically plausible candidate for subsequent validation rather than to establish a clinically validated diagnostic biomarker.
Notably, hub genes with the highest topological centrality in inflammatory networks (for example, IL6, STAT3, and CXCL8) are biologically important but are also broadly activated across many inflammatory conditions, which may limit pathway specificity for COPD-focused biomarker prioritization. In contrast, ERN1 was one of the most statistically upregulated genes in the discovery dataset and directly maps to the endoplasmic reticulum stress/UPR axis, an emerging mechanism in COPD pathobiology. Therefore, this study reports topological hubs and mechanistic candidates separately and prioritizes ERN1 as a mechanism-focused candidate for downstream evaluation.
Because the discovery samples were surgically resected lung tissue, candidate signals should be interpreted as tissue-level mechanistic evidence rather than immediately deployable early-diagnosis biomarkers in routine clinical screening. Translation to early diagnosis will require prospective validation in more accessible biospecimens (for example, blood, sputum, or exhaled airway samples).
Ethics Statement
This study was a secondary analysis of publicly available, de-identified transcriptomic data from GEO and did not involve direct participant contact or new human sample collection. According to Article 32 (Items 1 and 2) of the Measures for Ethical Review of Life Science and Medical Research Involving Human Subjects (China, issued February 18, 2023), use of de-identified public data may be exempt from additional ethical review when data use remains within original consent scope. Therefore, no additional institutional ethics approval was required for this secondary analysis.
Materials and Methods
Data Source
Sex composition was not predefined by our study but inherited from the source dataset (GSE57148), which enrolled male smokers undergoing lung resection. This relatively homogeneous design may reduce heterogeneity and confounding from sex-related biological variation, but it limits population representativeness.
The discovery cohort was retrieved from GEO (GSE57148, GPL11154). The exact discovery files used in this study were: GSE57148_COPD_FPKM_Normalized.txt.gz (processed gene-level FPKM matrix), GSE57148_series_matrix.txt.gz (sample-level metadata), and GSE57148_family.soft.gz (metadata cross-check). The processed expression matrix contained 16739 genes and 189 samples (98 COPD and 91 subjects with normal-spirometry). All subjects were male smokers who underwent lung surgical resection. Clinical characteristics were referenced from the original publication4 and are summarized in Table 1. The COPD group (n = 98) had mean age 67.5±6.4 years, smoking history 48.0 ±22.0 pack-years, forced expiratory volume in one second (FEV1)% 71.9±13.4, and FEV1/forced vital capacity (FVC) ratio 57.1±7.8. The normal-spirometry group (n = 91) had mean age 60.9±9.5 years, smoking history 35.2 ±17.2 pack-years, FEV1% 91.0±12.4, and FEV1/FVC 74.8±4.3.
Data Preprocessing and Differential Expression Analysis
Data preprocessing followed a predefined reproducible pipeline. First, the processed matrix (GSE57148_COPD_FPKM_Normalized.txt.gz) and sample metadata (GSE57148_series_matrix.txt.gz) were imported and sample IDs were matched to ensure one-to-one alignment between metadata and expression columns (189/189 matched). Group labels were assigned as COPD vs normal-spirometry according to GEO metadata and cross-checked against sample naming suffixes. Second, gene annotation for discovery data used the provided GeneName field (gene symbol level); no additional Ensembl-to-symbol remapping was required. Third, filtering and quality-control steps were applied in fixed order: (i) remove rows with empty gene symbols (removed n = 0), (ii) collapse duplicated gene symbols by retaining the row with the largest mean expression across samples (duplicated symbols detected n = 0), and (iii) scan for missing expression values across the full matrix (missing values n = 0), so no imputation was performed. Fourth, expression values were transformed as log2 (FPKM + 1). According to GEO submitter processing notes, genes with all-zero FPKM across samples had already been excluded and upper-quantile normalization had been applied before public release. Because the publicly released discovery files did not provide a directly matched raw-count matrix for all analyzed samples in this workflow, count-based pipelines (DESeq2/edgeR) or limma-voom could not be implemented as the primary analysis. Therefore, limma with empirical Bayes moderation was applied to log2-transformed processed expression as an exploratory analysis, and all diagnostic claims were interpreted conservatively. Methodological benchmark studies indicate that empirical-Bayes linear modeling on log-scale expression can provide stable and reproducible differential-expression ranking in RNA-seq-related analyses, and the limma-trend/voom framework is widely adopted for this purpose when suitable inputs are available. Accordingly, when raw counts are not retrievable from public releases, analysis of log2-transformed, normalized expression matrices with moderated t-statistics is an acceptable and conservative exploratory strategy, while count-based workflows remain the preferred first-line option when raw counts are available.9–11 Genes with |log2FC| > 0.5 and Benjamini-Hochberg adjusted P < 0.05 were defined as differentially expressed genes (DEGs).
Functional Enrichment Analysis of DEGs
To explore the biological functions of upregulated DEGs, functional enrichment analysis—including Gene Ontology (GO) biological process (BP), cellular component (CC), and molecular function (MF) enrichment analyzes, as well as Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis—was performed using the Enrichr tool. Adjusted P-value < 0.05 was considered statistically significant. Dot plots and bar plots were drawn to visualize the enrichment results, with the top 10 enriched terms selected for each category.
Construction of PPI Network and Identification of Key Genes
Hub-gene ranking by network topology (degree centrality) was pre-specified for unbiased screening, whereas ERN1-focused candidate prioritization was not pre-specified before analysis. After global differentially expressed gene (DEG) and PPI analyses were completed, ERN1 was selected using a literature-informed post hoc multi-criteria strategy integrating differential-expression significance, mechanistic relevance, and translational interpretability. The upregulated DEGs were uploaded to the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database (https://string-db.org/) to construct a PPI network, with the minimum interaction score set to 0.4 (medium confidence). The PPI network was visualized using the NetworkX package in Python, where node size was proportional to degree centrality. The degree centrality of each node was calculated, and the top 10 hub genes (highest degree centrality) were identified.
Verification of ERN1 Expression Level
The expression level of ERN1 in COPD lung tissues and tissues from subjects with normal-spirometry was extracted from the preprocessed GSE57148 expression matrix. A boxplot with overlaid strip plot was drawn to compare the expression difference of ERN1 between the two groups, and a Welch t-test was performed for statistical analysis. P < 0.05 was considered statistically significant. The expression level of ERN1 was presented as mean ± standard deviation (SD).
Analysis of Internal Discriminative Performance of ERN1
Internal discriminative performance of ERN1 in the discovery cohort was evaluated by drawing a receiver operating characteristic (ROC) curve. The area under the curve (AUC) and the 95% confidence interval (95% CI) were calculated. Bootstrap resampling (n = 2000) was used to estimate the 95% CI. The optimal cutoff value was determined using Youden’s J statistic (maximum of sensitivity + specificity - 1). The ROC result was interpreted as within-cohort discrimination because discovery and ROC assessment used the same dataset.
Statistical Analysis
Measurement data were expressed as mean ± standard deviation (SD). A Welch t-test was used for two-group comparisons. P < 0.05 was considered statistically significant. All statistical analyses were performed using R and Python.
External Validation and Evidence Synthesis Strategy
To improve interpretability beyond a single discovery cohort, we predefined external validation criteria before analysis: (1) respiratory tissue relevance (lung tissue preferred), (2) explicit COPD/control grouping for case-control validation, (3) sample size and metadata completeness, and (4) measurable ERN1 probe availability in released matrices. External files downloaded and analyzed included GSE47460-GPL14550_series_matrix.txt.gz and GSE38974-GPL4133_series_matrix.txt.gz for main-text validation; GSE37768_series_matrix.txt.gz, GSE76925_non-normalized.txt.gz, and GSE69818_series_matrix.txt.gz were used as supplementary cohorts. Platform annotation files were used for probe-to-gene mapping where needed (for example, GPL4133.annot.gz and GPL570.annot.gz). Probe-handling rules were predefined: if multiple ERN1 probes existed, the primary probe was selected by canonical transcript annotation when available (GSE38974 primary probe 27584, NM_001433), and a probe-averaged sensitivity analysis was additionally reported; for platforms with multiple ERN1 matches but without canonical-transcript priority, the probe with the highest mean expression was used in primary analysis. For covariate-adjusted logistic models, complete-case analysis was used (samples with missing age/sex were excluded from adjusted models). In parallel, we performed structured literature triangulation of ERN1/ER-stress evidence in COPD and summarized this in Supplementary Table S2 (Supplementary File 1). Individual-level covariates for age and smoking exposure (for example, pack-years) were not uniformly available in the public GEO metadata for GSE57148 (series matrix/pData), so joint covariate adjustment for these factors could not be performed in the discovery cohort. Therefore, age- and sex-adjusted models were applied as sensitivity analyses only in external cohorts with complete covariates (GSE47460 and GSE76925 non-normalized). For the discovery cohort, age and smoking information was mainly available as group-summary statistics in the original publication.
Reproducibility and Software Environment
Discovery and validation workflows were implemented in R 4.5.3 and Python 3.14.3. Key R packages included limma 3.66.0 (differential expression), GEOquery 2.78.0 (GEO parsing), and base stats functions for Welch t-test and logistic regression. Python scripts were used for matrix parsing, PPI topological statistics, and report generation. All major validation scripts used a fixed random seed (20260508) for bootstrap reproducibility. Detailed software and package versions are provided in Supplementary Table S3 (Supplementary File 1). Reproducibility script inventory and executable code appendix are provided in Supplementary Table S5 and the Supplementary Code Appendix (Supplementary File 1). All supplementary information cited in the manuscript is consolidated in Supplementary File 1 (Supplementary Tables S1–S5).
Sensitivity Analysis for Expression-Modeling Choice
To evaluate whether key findings depended on analytical choices and to quantify potential optimism from same-cohort ROC evaluation, we performed two internal validation procedures. First, model-choice sensitivity analysis used limma-trend (eBayes with trend = TRUE) on the same log2 (FPKM + 1) matrix, and ERN1 group difference was additionally tested by non-parametric Wilcoxon rank-sum test. Second, discrimination robustness was evaluated by repeated stratified 10-fold cross-validation and bootstrap optimism correction (B = 2000) for the ERN1-based logistic model in the discovery cohort.
Results
Identification of DEGs in COPD Lung Tissues
After data preprocessing and differential expression analysis of the GSE57148 dataset (98 COPD subjects and 91 subjects with normal-spirometry), a total of 308 DEGs were identified between COPD lung tissues and tissues from subjects with normal-spirometry, including 183 upregulated DEGs and 125 downregulated DEGs (|log2FC| > 0.5, padj < 0.05). The complete DEG list is provided in Supplementary Table S4 (Supplementary File 1). The distribution of all DEGs was visualized by a volcano plot (Figure 1A), where red dots represented upregulated DEGs, blue dots represented downregulated DEGs, and gray dots represented non-differentially expressed genes. A heatmap was drawn to show the expression pattern of the top 100 DEGs (50 upregulated and 50 downregulated), which showed only a partial group-separation trend between COPD samples and subjects with normal-spirometry, with substantial overlap (Figure 1B). Among the upregulated DEGs, the most statistically significant genes included RAD54L2 (log2FC = 0.60, padj = 2.80 × 10−18), STAT3 (log2FC = 0.53, padj = 3.88 × 10−18), and ERN1 (log2FC = 0.75, padj = 1.98 × 10−15). Among the downregulated DEGs, the most significant genes included COX6A1 (log2FC = −0.52, padj = 1.94 × 10−21), LSM7 (log2FC = −0.72, padj = 9.94 × 10−20), and STRA13 (log2FC = −0.72, padj = 1.79 × 10−19).
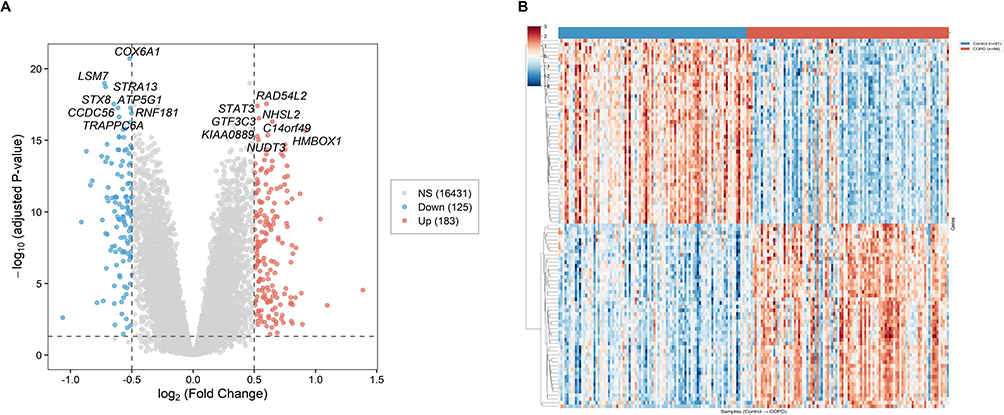 |
Figure 1 Differential expression landscape in COPD lung tissue. (A) Volcano plot of differentially expressed genes (DEGs) between COPD and normal-spirometry groups. Red dots represent upregulated genes (log2FC > 0.5, padj < 0.05), blue dots represent downregulated genes (log2FC < −0.5, padj < 0.05), and gray dots represent non-significant genes. (B) Heatmap of the top 100 DEGs between COPD and normal-spirometry groups. Rows represent genes and columns represent samples. |
Functional Enrichment Analysis of Upregulated DEGs
GO enrichment analysis revealed that the 183 upregulated DEGs were significantly enriched in multiple biological process (BP), cellular component (CC), and molecular function (MF) terms. Representative BP terms included positive regulation of transcription by RNA polymerase II (GO:0045944, 30 genes, p-adjust = 1.76 × 10−6), positive regulation of DNA-templated transcription (GO:0045893, 35 genes, p-adjust = 1.76 × 10−6), regulation of extrinsic apoptotic signaling pathway via death domain receptors (GO:1902041, 7 genes, p-adjust = 5.59 × 10−6), response to lipopolysaccharide (GO:0032496, 10 genes, p-adjust = 4.86 × 10−4), and regulation of cell population proliferation (GO:0042127, 22 genes, p-adjust = 4.86 × 10−4) (Figure 2A). Representative CC terms included collagen-containing extracellular matrix (GO:0062023, 16 genes, p-adjust = 4.61 × 10−5), platelet alpha granule lumen (GO:0031093, 7 genes, p-adjust = 1.54 × 10−4), and secretory granule lumen (GO:0034774, 11 genes, p-adjust = 5.35 × 10−3) (Figure 2B). Representative MF terms included DNA-binding transcription activator activity, RNA polymerase II-specific (GO:0001228, 12 genes, p-adjust = 1.54 × 10−2), protease binding (GO:0002020, 7 genes, p-adjust = 1.54 × 10−2), cytokine activity (GO:0005125, 7 genes, p-adjust = 3.81 × 10−2), and growth factor activity (GO:0008083, 5 genes, p-adjust = 3.75 × 10−2) (Figure 2C).
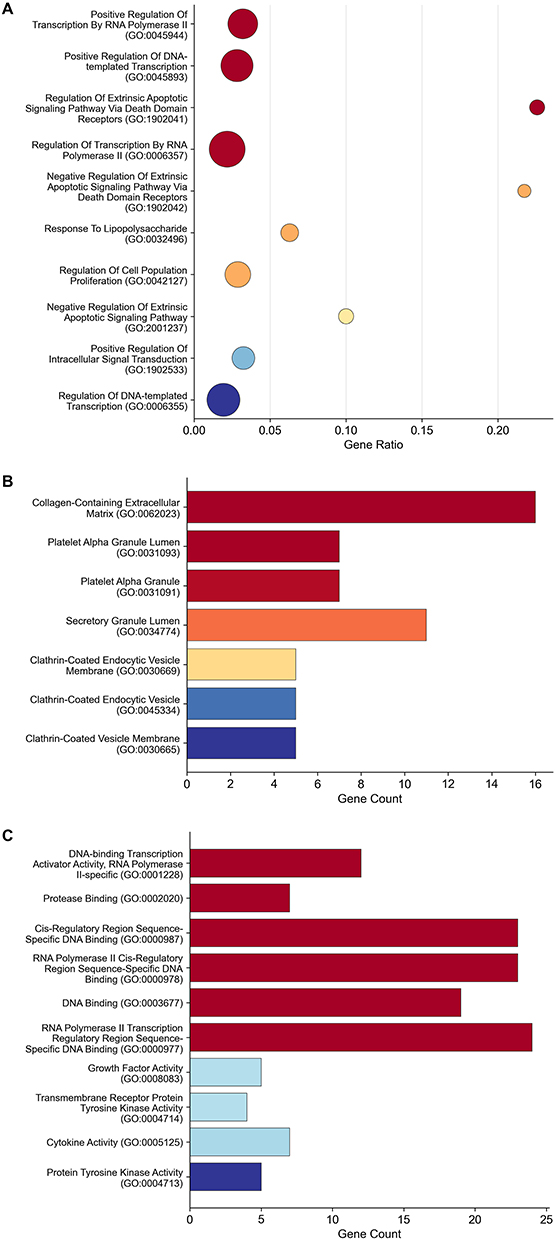 |
Figure 2 Gene Ontology enrichment profile of upregulated DEGs. (A) GO biological process (GO-BP) enrichment analysis of upregulated DEGs. (B) GO cellular component (GO-CC) enrichment analysis of upregulated DEGs. (C) GO molecular function (GO-MF) enrichment analysis of upregulated DEGs. |
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis showed that the upregulated DEGs were mainly involved in signaling pathways closely associated with COPD, including the interleukin-17 (IL-17) signaling pathway (10 genes, p-adjust = 2.95 × 10−6), tumor necrosis factor (TNF) signaling pathway (10 genes, p-adjust = 8.00 × 10−6), complement and coagulation cascades (7 genes, p-adjust = 5.10 × 10−4), Janus kinase-signal transducer and activator of transcription (JAK-STAT) signaling pathway (9 genes, p-adjust = 6.16 × 10−4), and phosphoinositide 3-kinase (PI3K)-Akt signaling pathway (12 genes, p-adjust = 2.63 × 10−3) (Figure 3). Collectively, these results indicate that the upregulated DEGs are closely related to the core pathological processes of COPD, such as inflammation, transcriptional regulation, and extracellular matrix remodeling.
 |
Figure 3 KEGG pathway enrichment of upregulated DEGs. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of upregulated DEGs. |
Construction of PPI Network and Identification of Key Genes
To further explore the functional relationships among the upregulated DEGs, the 183 upregulated DEGs were uploaded to the STRING database to construct a PPI network. After filtering with a minimum interaction score of 0.4 (medium confidence), the PPI network contained 134 nodes and 557 edges. The largest connected component included 124 nodes and 552 edges, which was visualized in Figure 4A, where node size was proportional to degree centrality. The degree centrality of each node was calculated, and the top 10 hub genes were identified, including IL6 (degree = 50), STAT3 (degree = 38), CXCL8 (degree = 38), PTGS2 (degree = 36), EGFR (degree = 35), EGR1 (degree = 30), SOCS3 (degree = 29), THBS1 (degree = 28), ATF3 (degree = 28), and CXCL1 (degree = 24) (Figure 4B, Table 2). These hub genes are primarily involved in inflammatory signaling and transcriptional regulation, reflecting the core pathological mechanisms of COPD.
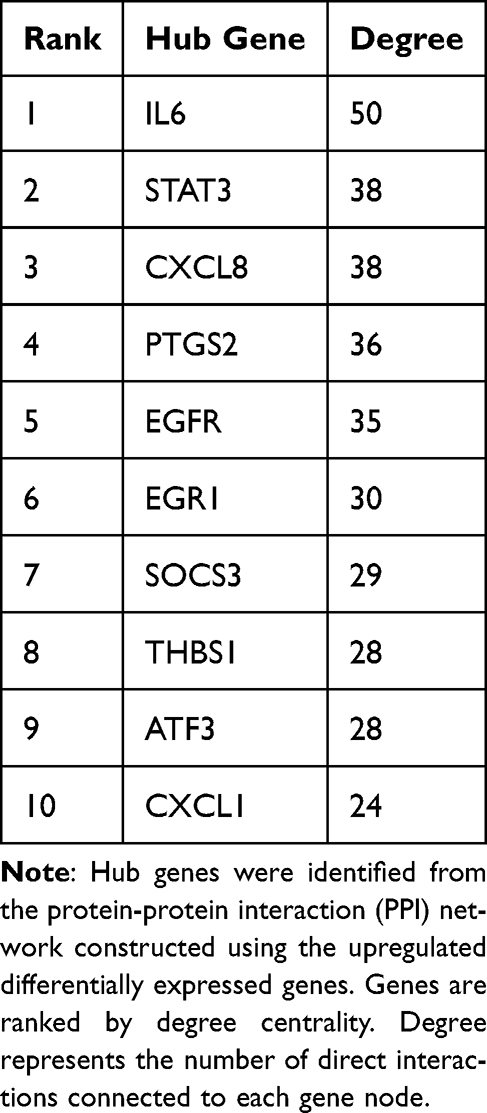 |
Table 2 Top 10 Hub Genes Ranked by Degree Centrality |
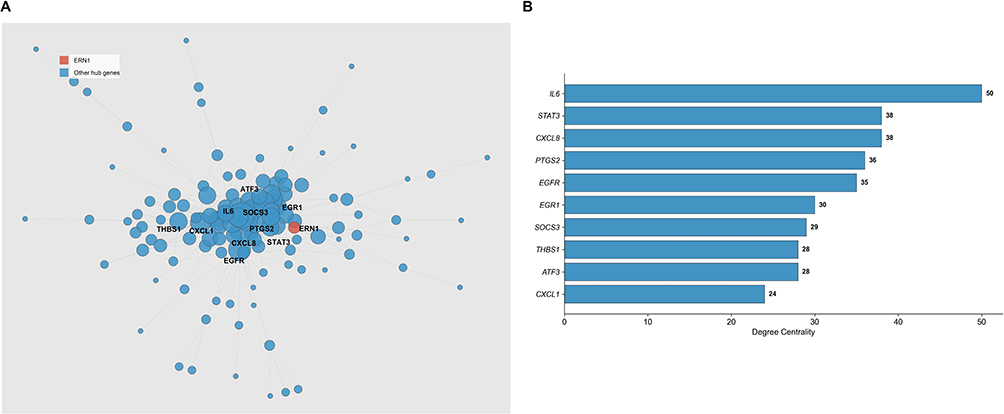 |
Figure 4 Protein-protein interaction network and hub-gene topology of upregulated DEGs. (A) PPI network of upregulated DEGs constructed using STRING database. Node size represents degree centrality. (B) Top 10 hub genes ranked by degree centrality in the PPI network. ERN1 is not included in this topology-based hub list because it was evaluated separately as a literature-informed post hoc mechanistic candidate. |
In addition to hub-gene screening based on network topology, ERN1 was examined as a literature-informed post hoc candidate rather than a pre-defined primary target. ERN1 (also known as IRE1alpha), ranked 57th in degree centrality (degree = 6) in the PPI network, was one of the most significantly upregulated DEGs (log2FC = 0.75, padj = 1.98 x 10^-15, ranking 9th among all upregulated DEGs by statistical significance). ERN1 encodes inositol-requiring enzyme 1alpha, the most conserved sensor of the UPR and a master regulator of endoplasmic reticulum stress signaling. Accumulating evidence has shown that endoplasmic reticulum stress and UPR activation are involved in COPD pathogenesis, including cigarette-smoke-induced epithelial cell apoptosis, inflammatory responses, and mucus hypersecretion.
Although ERN1 was not among the top nodes by degree centrality in the PPI network (ranked 57th), candidate prioritization in this study was not based on topology alone. We used a multi-criteria strategy integrating: (1) statistical robustness in differential expression, (2) mechanistic relevance to COPD pathobiology, and (3) translational interpretability as a biologically interpretable signaling node. Compared with several high-degree inflammatory hub genes (for example, IL6, CXCL8, and STAT3), which are often upregulated across diverse inflammatory and neoplastic contexts (including asthma, pneumonia, and tumors),12–14 ERN1 may offer greater pathway specificity for COPD-oriented mechanistic prioritization by representing the endoplasmic reticulum stress/UPR axis. As the most conserved UPR sensor, ERN1 upregulation may better reflect a defined organelle-stress state and therefore provides a concrete mechanism-focused candidate for future ER-stress-targeted pharmacologic exploration and precision mechanistic studies. Notably, ATF3 (degree = 28; included in the topology-defined top-10 hub set) is a well-recognized UPR-responsive stress transcription factor downstream of endoplasmic reticulum-stress signaling. Therefore, the coexistence of a topological hub signal (ATF3) and a mechanism-focused sensor candidate (ERN1) provides convergent support for the involvement of ER-stress biology in COPD lung-tissue remodeling.15
To avoid confusion, topological hub genes (ranked by degree centrality) are reported separately from ERN1, which was selected as a literature-prioritized mechanistic candidate under a literature-informed post hoc multi-criteria strategy.
Verification of ERN1 Expression Level in COPD Lung Tissues
To verify the differential expression of ERN1 in COPD, its expression levels in COPD lung tissues and tissues from subjects with normal-spirometry were extracted from the preprocessed GSE57148 expression matrix. The normalized log2 expression level of ERN1 in the COPD group was 2.89 ± 0.55, which was significantly higher than that in the normal-spirometry group (2.12 ± 0.52). A Welch t-test showed that the expression level of ERN1 in COPD lung tissues was significantly higher than that in tissues from subjects with normal-spirometry (P = 8.9 x 10^-18) (Figure 5). This result was consistent with the differential expression analysis, confirming that ERN1 is significantly upregulated in COPD lung tissues.
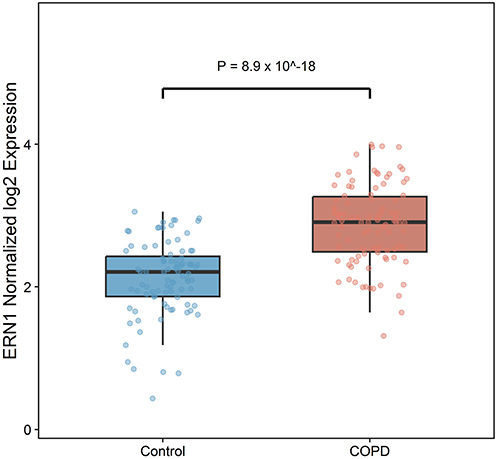 |
Figure 5 Differential expression of ERN1 in discovery cohort lung tissue. Expression levels of ERN1 (literature-informed post hoc candidate) in COPD lung tissues and tissues from subjects with normal spirometry. Statistical comparison was performed using Welch t-test. |
Internal Discriminative Performance of ERN1 in Discovery Cohort
Internal discriminative performance of ERN1 in the discovery cohort was evaluated by ROC analysis. The area under the curve (AUC) in GSE57148 was 0.853 (95% CI: 0.796–0.903, p < 0.001) (Figure 6). At the Youden-optimized cutoff (2.45), sensitivity was 81.6% and specificity was 76.9%. Because gene discovery, candidate prioritization, and ROC evaluation were conducted in the same cohort, this AUC should be interpreted as within-cohort discrimination rather than independent diagnostic validation.
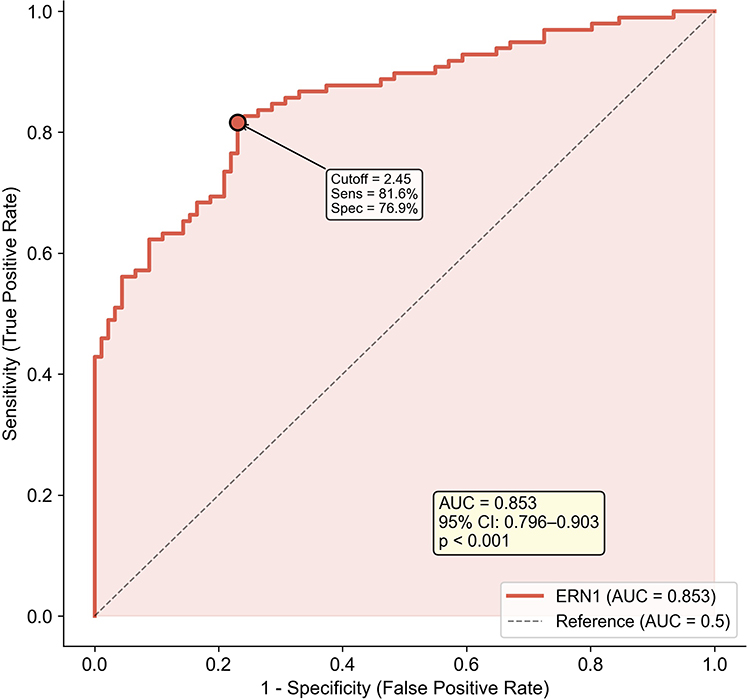 |
Figure 6 Internal discrimination performance of ERN1 in the discovery cohort. Receiver operating characteristic (ROC) curve for ERN1 in the discovery cohort. AUC = 0.853 (95% CI: 0.796–0.903). This ROC reflects internal dataset discrimination only and does not constitute external diagnostic validation. |
External Cohort Validation in GSE47460
Primary external validation was performed in GSE47460 (GPL14550), using the COPD-vs-Control subset (N = 236; COPD n = 145, control n = 91; male n = 119, female n = 117). ERN1 probe A_23_P164042 showed no significant case-control difference (COPD: 5.131 ± 0.569 vs control: 5.173 ± 0.556; Welch t = −0.568, p = 0.571). Diagnostic discrimination was limited (AUC = 0.477, 95% CI: 0.400–0.552; sensitivity = 40.7%, specificity = 65.9% at cutoff 5.268). In sex-adjusted logistic regression (COPD ~ ERN1 + Sex), ERN1 remained non-significant (OR = 0.897, 95% CI: 0.562–1.431, p = 0.647).
Supplementary External Case-Control Validation in GSE38974
In the supplementary cohort GSE38974 (GPL4133; N = 32; COPD n = 23, control n = 9), four ERN1 probes were available (3173, 27,584, 32,118, 43,270). Single-probe ERN1 (27584, NM_001433) showed no significant difference and poor discrimination (p = 0.785; AUC = 0.459, 95% CI: 0.266–0.662). Probe-averaged ERN1 showed directionally positive but non-significant separation (p = 0.132), with moderate discrimination (AUC = 0.681, 95% CI: 0.478–0.870). These findings suggest probe-level sensitivity and limited reproducibility across cohorts.
Age-confounding sensitivity analysis was performed in cohorts with complete covariates. In GSE47460, the age- and sex-adjusted model remained non-significant for ERN1 (OR = 0.896, 95% CI: 0.561–1.432, p = 0.646; n = 236). In GSE76925 non-normalized data, the age- and sex-adjusted ERN1 association was also non-significant (OR = 0.343, 95% CI: 0.088–1.335, p = 0.123; n = 151). These external adjusted analyses suggest that age imbalance alone is unlikely to explain cross-cohort inconsistency; however, they do not replace unavailable age adjustment within the discovery cohort itself.
Additional External Cohorts in Supplementary Materials
Additional cohorts (GSE37768, GSE76925 non-normalized, and COPD-only GSE69818) are reported in Supplementary File 1 (Supplementary Table S1 and Supplementary Results) and are not detailed in the main text.
Cross-Study Evidence Context for Generalizability
Across the two main-text external cohorts, ERN1 replication remained inconsistent: the largest mixed-sex cohort (GSE47460) was negative, while GSE38974 showed model-dependent partial support only after probe averaging; supplementary cohorts did not provide stronger support (Supplementary Table S1).
Literature-Based Mechanistic Triangulation
Because external diagnostic replication was heterogeneous, we further evaluated cross-study biological coherence at the pathway level. Prior studies consistently implicate endoplasmic reticulum stress and unfolded protein response signaling in COPD-related epithelial injury, inflammatory amplification, and airway remodeling. These convergent findings support mechanistic plausibility of the ERN1/IRE1 axis even when standalone diagnostic performance is unstable across cohorts. A structured evidence comparison is provided in Supplementary Table S2 (Supplementary File 1).
Sensitivity Analysis of Modeling Strategy
In sensitivity analysis using limma-trend on the same processed discovery matrix, ERN1 remained significantly upregulated in COPD (log2FC = 0.775, raw P = 5.10 x 10^-19, BH-adjusted P = 3.16 x 10^-16). A non-parametric Wilcoxon test also supported the ERN1 group difference (P = 4.53 x 10^-16). For internal discrimination robustness, repeated stratified 10-fold cross-validation yielded AUC = 0.848 (95% empirical interval 0.844–0.852), compared with apparent AUC = 0.853 in the same cohort. Bootstrap optimism-corrected AUC was 0.853 (B = 2000; mean optimism approximately 0.000). These analyses suggest limited measurable optimism in internal discrimination estimates; however, internal validation cannot replace truly independent external validation. This consistency supports the robustness of the signal under an alternative limma-based variance-trend specification and is directionally concordant with established limma/voom methodological evidence.9,10
Discussion
Consistent with established COPD inflammatory and stress-related biology, our findings reproduced an inflammatory transcriptomic background and identified an endoplasmic reticulum-stress-associated signal centered on ERN1. The innovation of this study is therefore incremental and hypothesis-generating: we define ERN1 as a testable mechanistic candidate and delineate its reproducibility boundaries, rather than claiming a confirmed universal diagnostic marker.
From a translational perspective, the value of this study is not only in identifying a discovery-cohort signal, but also in delineating boundary conditions for ERN1 reproducibility through predefined external validation plus literature triangulation. This combined framework narrows overgeneralization risk and reframes ERN1 as a context-dependent mechanistic candidate linked to endoplasmic reticulum stress biology, which is testable in future harmonized cohorts and experimental models.
A total of 308 DEGs were identified in COPD lung tissues compared with subjects with normal-spirometry, including 183 upregulated and 125 downregulated DEGs (|log2FC| > 0.5, padj < 0.05). Functional enrichment analysis showed that the upregulated DEGs were mainly enriched in biological processes such as transcriptional regulation, apoptotic signaling, and response to lipopolysaccharide, and were involved in signaling pathways including the IL-17, TNF, JAK-STAT, and PI3K-Akt pathways. These results are consistent with the known pathogenesis of COPD: inflammatory response is the core pathological feature of COPD, and the IL-17 and TNF signaling pathways are key pathways regulating inflammatory response and cell survival, whose abnormal activation is closely related to COPD development.16–20 The enrichment of extracellular matrix-related terms in the CC category is also consistent with the airway remodeling observed in COPD.21
PPI network analysis identified 10 hub genes with the highest degree centrality, including IL6 (degree = 50), STAT3 (degree = 38), CXCL8 (degree = 38), PTGS2 (degree = 36), and EGFR (degree = 35). IL6 and CXCL8 are well-known inflammatory cytokines, and their elevated expression in COPD has been confirmed by numerous studies.13,14 STAT3, a key transcription factor mediating inflammatory signaling, and PTGS2 (COX-2), a rate-limiting enzyme in prostaglandin synthesis, are also closely associated with COPD pathogenesis. The identification of these hub genes further validates the reliability of our analysis. Of note, ATF3 (degree = 28; ninth in our listed hub set) is biologically linked to downstream UPR stress-response transcriptional programs, which complements the ERN1-centered signal and supports a convergent endoplasmic reticulum -stress-related interpretation from both topology and mechanism.15
In addition to hub gene screening based on network topology, we adopted a combined strategy integrating differential-expression significance, biological function, and literature evidence to identify mechanistic candidates. ERN1 (IRE1alpha), while ranked 57th in degree centrality in the PPI network (degree = 6), was one of the most significantly upregulated DEGs (log2FC = 0.75, padj = 1.98 x 10^-15, ranking 9th by statistical significance among all upregulated DEGs). ERN1 encodes a dual-function transmembrane kinase/endoribonuclease that serves as the most conserved sensor of the UPR. Upon endoplasmic reticulum stress, ERN1 activates downstream signaling through the IRE1alpha/XBP1 axis, regulating cell survival, apoptosis, and inflammatory responses.5,22 Accordingly, ERN1 was prioritized as a mechanism-focused post hoc candidate (not as a topological hub), while IL6/STAT3/CXCL8 were reported as core inflammatory hubs rather than the primary candidate in this study. From a translational-selection perspective, high-degree inflammatory hubs are biologically important but relatively broad and can be elevated across multiple inflammatory disease settings (for example, asthma and pneumonia) and neoplastic contexts,12–14 whereas ERN1 more directly indexes an organelle-stress process (endoplasmic reticulum-stress/UPR) that is mechanistically coherent with COPD injury biology.23 Therefore, ERN1 was treated as a specific pathway-oriented candidate for downstream validation rather than as proof of a confirmed therapeutic target. Importantly, the current study demonstrates expression-level association rather than causal mechanism, and does not confirm ERN1 as a therapeutic target.
ROC analysis in the discovery cohort showed internal discrimination (AUC = 0.853, 95% CI: 0.796–0.903; sensitivity = 81.6%; specificity = 76.9%). However, this should be interpreted as dataset-internal case-control discrimination only, because discovery and ROC evaluation were performed in the same dataset, and therefore it does not constitute independent diagnostic validation.24 Under current spirometry-centered guideline frameworks, biomarker-oriented claims should be interpreted cautiously until externally validated.25
This study has several key limitations. First, the discovery cohort (GSE57148) was a single-center cohort composed of male smokers undergoing lung resection, which limits generalizability to female patients and broader COPD populations. Second, individual-level covariates for age and smoking exposure were not uniformly available in public metadata for GSE57148, so direct covariate-adjusted modeling in the discovery cohort could not be performed. Third, candidate selection and ROC evaluation were conducted in the same cohort; therefore, the reported AUC reflects internal discrimination rather than independent diagnostic validation. Fourth, external replication was heterogeneous across datasets, and platform/probe differences may contribute to cross-cohort inconsistency. Fifth, this is a bioinformatics association study without in vitro/in vivo functional experiments, so causal inference and therapeutic-target claims cannot be made.
Independent validation in one primary cohort (GSE47460) and one supplementary cohort (GSE38974) refined interpretation of ERN1 and reduced single-dataset bias. The primary cohort did not replicate diagnostic performance, and the supplementary cohort showed probe-dependent partial support only after averaging. Additional age- and sex-adjusted analyses in covariate-complete cohorts (GSE47460 and GSE76925 non-normalized) remained non-significant, suggesting that age imbalance is unlikely to be the sole explanation for cross-cohort inconsistency. Furthermore, variation in baseline smoking status across control groups represents another critical dimension of cross-cohort heterogeneity. In the discovery cohort (GSE57148), the normal-spirometry control group was composed of heavy smokers with substantial tobacco exposure (35.2 ± 17.2 pack-years).4 Because cigarette smoke is a well-established chronic inducer of organelle stress,18,23 the significant ERN1 upregulation observed in discovery cases is more plausibly interpreted as an incremental, progression-related acceleration of endoplasmic-reticulum stress occurring on top of a high smoke-induced baseline, rather than a simple healthy-versus-disease contrast. Conversely, if external control groups include never-smokers or long-term former smokers with lower cumulative exposure, baseline endoplasmic reticulum stress activity may be lower, potentially attenuating apparent case-control contrasts and contributing to reduced external reproducibility.
Beyond being a limitation, the contrast between a male-only discovery cohort (GSE57148) showing a positive within-cohort ERN1 signal and a non-significant mixed-sex external cohort (GSE47460) may suggest a potential sex- or context-dependent background for ERN1-related endoplasmic reticulum-stress biology in COPD, rather than a uniform cross-population effect. Prior transcriptomic studies have reported sex-biased smoking response programs and sexually dimorphic molecular targeting in airway/COPD datasets, which supports sex-stratified validation of the ERN1/UPR axis in future cohorts.26,27
Although the analytical workflow was technically robust, biomarker generalizability remains constrained by sex restriction in the discovery cohort and inconsistent replication across external datasets. Therefore, ERN1 should currently be interpreted as a context-dependent candidate in male smoking-associated COPD, pending prospective multi-center validation with harmonized assay platforms.
Conclusion
In conclusion, ERN1 is highly expressed in COPD lung tissue in the GSE57148 dataset and can be reasonably interpreted as an endoplasmic-reticulum-stress-related candidate gene. Given the single-cohort discovery design and inconsistent external replication, the current evidence is preliminary and requires further validation before confirmatory interpretation.
Disclosure
The author report no conflicts of interest in this work.
References
1. Global Initiative for Chronic Obstructive Lung Disease. Global strategy for prevention, diagnosis and management of COPD: 2026 report. Global Initiative for Chronic Obstructive Lung Disease, 2026.
2. Barnes PJ. Pathophysiology of chronic obstructive pulmonary disease. Eur Respir J. 2016;48(3):831–14.
3. Wang Y, Li J, Zhang H. Bioinformatics analysis of hub genes and signaling pathways in COPD based on GEO datasets. Comput Biol Chem. 2022;100:107568.
4. Kim WJ, Lim JH, Lee JS, Lee SD, Kim JH, Oh YM. Comprehensive analysis of transcriptome sequencing data in the lung tissues of COPD subjects. Int J Genomics. 2015;2015:206937. doi:10.1155/2015/206937
5. Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334(6059):1081–1086. doi:10.1126/science.1209038
6. Li X, Zhang Y, Wang L. ERN1 promotes proliferation and invasion of lung cancer cells by activating the PI3K/Akt signaling pathway. Oncol Rep. 2020;44(3):1027–1038.
7. Zhang H, Li J, Chen Y. ERN1 regulates endoplasmic reticulum stress and apoptosis in diabetic nephropathy. J Cell Mol Med. 2019;23(11):7345–7356.
8. Liu Y, Wang H, Zhang J. ERN1-mediated endoplasmic reticulum stress contributes to neurodegeneration in Alzheimer’s disease. Neurobiol Aging. 2021;102:187–198.
9. Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi:10.1093/nar/gkv007
10. Law CW, Chen Y, Shi W, Smyth GK. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15(2):R29. doi:10.1186/gb-2014-15-2-r29
11. Conesa A, Madrigal P, Tarazona S, et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016;17:13. doi:10.1186/s13059-016-0881-8
12. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi:10.1016/j.cell.2011.02.013
13. Brusselle GG, Joos GF, Bracke KR. Cytokines in chronic obstructive pulmonary disease. Eur Respir J. 2013;41(3):679–694.
14. Barnes PJ, Paquet A, Sanfiorenzo C. Interleukin-6 in chronic obstructive pulmonary disease. Eur Respir J. 2018;52(3):1800437. doi:10.1183/13993003.00437-2018
15. Jiang HY, Wek SA, McGrath BC, et al. Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol Cell Biol. 2004;24(3):1365–1377. doi:10.1128/MCB.24.3.1365-1377.2004
16. Barnes PJ, Parikh SM. Inflammation in chronic obstructive pulmonary disease. J Allergy Clin Immunol. 2017;140(3):663–671. doi:10.1016/j.jaci.2016.10.042
17. Rahman I, Vizio B, Mascia C. Oxidative stress in chronic obstructive pulmonary disease. Free Radic Biol Med. 2006;41(4):443–450. doi:10.1016/j.freeradbiomed.2006.04.005
18. Lee JS, Park JH, Kim YH. Endoplasmic reticulum stress in chronic obstructive pulmonary disease: a new therapeutic target. Int J Chron Obstruct Pulmon Dis. 2020;15:2877–2888.
19. Takahashi K, Nakamura Y, Tanaka T, Giersig M, Rybka JD. TNF-α signaling pathway in chronic obstructive pulmonary disease. J Clin Med. 2019;8(11):1865. doi:10.3390/jcm8111865
20. Wang C, Li Y, Zhang L. PI3K-Akt signaling pathway in chronic obstructive pulmonary disease: a review. Cell Physiol Biochem. 2018;49(3):927–938.
21. Lee JS, Kim YH, Julich-Gruner KK, Behl M, Lendlein A. Role of endoplasmic reticulum stress in airway remodeling in chronic obstructive pulmonary disease. Int J Mol Sci. 2021;22(11):5892. doi:10.3390/ijms22115892
22. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8(7):519–529. doi:10.1038/nrm2199
23. Chen Y, Wang H, Li J. Endoplasmic reticulum stress in airway epithelial cells contributes to airway inflammation in COPD. Am J Physiol Lung Cell Mol Physiol. 2018;315(3):L433–L443.
24. Sin DD, Man SF. Biomarkers in chronic obstructive pulmonary disease. Lancet. 2006;368(9533):716–728. doi:10.1016/S0140-6736(06)69266-0
25. Global Initiative for Chronic Obstructive Lung Disease. Pocket guide to COPD diagnosis, management and prevention: 2026 report. Global Initiative for Chronic Obstructive Lung Disease, 2026.
26. Yang CX, Shi H, Ding I, et al. Widespread sexual dimorphism in the transcriptome of human airway epithelium in response to smoking. Sci Rep. 2019;9:17600. doi:10.1038/s41598-019-54051-y
27. Glass K, Quackenbush J, Silverman EK, et al. Sexually-dimorphic targeting of functionally-related genes in COPD. BMC Syst Biol. 2014;8:118. doi:10.1186/s12918-014-0118-y
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Plasma miR-150-5p as a Biomarker for Chronic Obstructive Pulmonary Disease
Ding Y, Tang S, Zhou Z, Wei H, Yang W
International Journal of Chronic Obstructive Pulmonary Disease 2023, 18:399-406
Published Date: 23 March 2023
LARS1 is a Prognostic Biomarker and Exhibits a Correlation with Immune Infiltrates in Hepatocellular Carcinoma
Fan L, Qin Z, Wu D, Yang Y, Zhang Y, Xie B, Qian J, Wei J, Wang Z, Yang P, Qian Z, Yuan M, Zhu Z, Tan Y, Tan Y
International Journal of General Medicine 2024, 17:2203-2221
Published Date: 17 May 2024
PPIH Expression Correlates with Tumor Aggressiveness and Immune Dysregulation in Hepatocellular Carcinoma
Bei J, Sun Z, Fu R, Huang X, Huang J, Luo Y, Li Y, Chen Y, Wei Z
Journal of Hepatocellular Carcinoma 2024, 11:2453-2470
Published Date: 11 December 2024
TRPC6 is a Biomarker for Prognosis and Immunotherapy of Stomach Adenocarcinoma Based on Bioinformatic Analysis and Experimental Validation
Hu X, Wang H, Sun H, Zhang J, Ye Z, Huang Z
ImmunoTargets and Therapy 2024, 13:735-748
Published Date: 12 December 2024
The Association Between the CALLY Index and All-Cause Mortality in Patients with COPD: Results from the Cohort Study of NHANES 2007–2010
Ding Y, Liu Y, Yu J, Cai C, Fu L, Zhu J, Yang S, Jiang Y, Wang J
International Journal of Chronic Obstructive Pulmonary Disease 2025, 20:159-169
Published Date: 22 January 2025