Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 21
Identification and Functional Characterization of COPD Molecular Subtypes Based on Oxeiptosis-Related Genes via WGCNA and Machine Learning
Authors Chen W, Zhao J, Sun Z, Zhou X, Zhang X, Yan Z
Received 16 January 2026
Accepted for publication 22 May 2026
Published 5 June 2026 Volume 2026:21 596477
DOI https://doi.org/10.2147/COPD.S596477
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Zijing Zhou
Wenlong Chen,1 Jie Zhao,1,2 Zhigang Sun,1 Xingye Zhou,1,3 Xingzi Zhang,1 Zhen Yan1
1Key Laboratory of Tropical Translational Medicine of Ministry of Education, School of Public Health, Hainan Academy of Medical Sciences, Hainan Medical University, Haikou, 571199, People’s Republic of China; 2Department of Pulmonary and Critical Care Medicine, Hainan General Hospital, Hainan Affiliated Hospital of Hainan Medical University, Haikou, 571199, People’s Republic of China; 3Hospital Infection Control Department, The Second Affiliated Hospital of Hainan Medical University, Haikou, 571199, People’s Republic of China
Correspondence: Zhen Yan, Key Laboratory of Tropical Translational Medicine of Ministry of Education, School of Public Health, Hainan Academy of Medical Sciences, Hainan Medical University, Haikou, 571199, People’s Republic of China, Email [email protected]
Background: This study establishes the first molecular stratification framework for chronic obstructive pulmonary disease (COPD) based on oxeiptosis biology, exploring how this reactive oxygen species (ROS)-induced cell death pathway shapes disease heterogeneity. The precise role of oxeiptosis in COPD pathogenesis remains poorly understood.
Methods: Transcriptomic profiles from two independent cohorts, GSE47460 (220 COPD cases and 108 controls) and GSE76925 (111 COPD cases and 40 controls), were systematically evaluated using a fully in silico computational pipeline. Oxeiptosis-related differentially expressed genes (ORDEGs) were identified through correlation and differential expression analyses, prioritized using machine learning, and then applied for unsupervised clustering. Subtypes were externally validated and functionally characterized through pathway enrichment and network analysis.
Results: The support vector machine (SVM) model prioritized four of the seven ORDEGs for further analysis. Two reproducible subtypes were identified: C1, characterized by diminished ORDEGs activity and significantly worse pulmonary function (FEV1% predicted: C1 vs. C2, 44% vs 59%, P < 0.05), and C2, marked by heightened activity and less severe dysfunction. Weighted gene coexpression network analysis (WGCNA) revealed a black module associated with subtype classification, enriched in cytokine signaling and extracellular matrix remodeling pathways. A predictive model was constructed to investigate clinical applicability.
Conclusion: This computational discovery framework introduces an oxeiptosis-specific molecular taxonomy of COPD. The results underscore oxidative stress and the interaction between immune regulation and matrix remodeling as pivotal elements linked to disease heterogeneity, presenting potential pathways for precise diagnosis and treatment.
Keywords: COPD, oxeiptosis, oxidative stress, molecular subtypes, machine learning
Introduction
Chronic obstructive pulmonary disease (COPD) is the fourth leading cause of mortality worldwide, accounting for approximately 3.5 million deaths, nearly 5% of all global deaths in 2021.1 The disease is characterized by progressive and irreversible airflow limitation, mainly due to chronic inflammation and structural changes in the airways. COPD is closely associated with prolonged exposure to deleterious particulates and gases, especially tobacco smoke, which expedites the deterioration of pulmonary function.2,3 Despite advances in therapeutic strategies, current interventions remain insufficient to halt or reverse disease progression. The molecular basis for COPD heterogeneity is still not well understood.
The pathogenesis of COPD is characterized by a complex interaction among chronic inflammation, oxidative stress dysregulation, and the protease-antiprotease system.4,5 Reactive oxygen species (ROS), originating from exogenous sources such as cigarette smoke and endogenous sources including activated inflammatory cells, surpass antioxidant defense mechanism in the lung.6,7 This oxidative imbalance damages cellular macromolecules, activates pro-inflammatory signaling pathways, and breaks down extracellular matrix, which perpetuates the cycle of tissue injury and repair going.8 While conventional frameworks frequently conceptualize radical-induced parenchymal damage as a passive, unprogrammed necrotic event, accumulating evidence indicates that prolonged oxidative distress actively engages highly regulated, genetically encoded cell death programs that dictate tissue loss.9,10
Oxeiptosis is a reactive oxygen species (ROS)-induced cellular death pathway resulting in a caspase-independent, non-inflammatory cell demise.11 Distinct from lytic death modalities like pyroptosis (driven by gasdermins) or necroptosis (driven by MLKL phosphorylation), oxeiptosis preserves cell membrane integrity, thereby preventing the catastrophic leakage of pro-inflammatory damage-associated molecular patterns (DAMPs).11,12 Furthermore, while ferroptosis is fundamentally driven by iron overload and lipid peroxidation, oxeiptosis is governed by an intracellular radical-sensing relay mediated through the KEAP1-PGAM5-AIFM1 signaling axis.13,14 This network operates as a sophisticated, threshold-dependent molecular rheostat. Under basal conditions, KEAP1 targets the master antioxidant transcription factor NRF2 for proteasomal degradation, whereas intermediate ROS triggers NRF2 nuclear translocation to activate cytoprotective responses.15 However, upon breaching a toxic threshold of severe ROS, this protective feedback loop is dismantled; excessive radicals selectively suppress the NRF2 survival program, driving KEAP1 to instead bind and activate the mitochondrial phosphatase PGAM5. Once activated, PGAM5 dephosphorylates the mitochondrial flavoprotein AIFM1 at its highly conserved Serine 116 residue, irreversibly committing the cell to non-inflammatory death.11 Given that the COPD lung microenvironment experiences sustained, oxidative distress that routinely breaches upstream antioxidant thresholds, epithelial barrier fate is intimately tied to this linear execution cascade. Consequently, decoding individual variations within this specific death pathway provides a critical, pathologically relevant lens to understand parenchymal destruction. To date, the specific pathophysiological involvement of oxeiptosis within the context of COPD remains virtually unexplored, with definitive mechanistic studies remaining exceptionally scarce. Nevertheless, a rigorous rationale for this exploration can be established by drawing a direct pathogenic and toxicological analogy to the benchmark in vivo lung model utilized in the seminal study that first defined the oxeiptosis pathway.11 In pulmonary pathophysiology, acute ozone (O3) inhalation and chronic cigarette smoke (CS) exposure are highly established functional analogues; both etiological insults act through an identical primary mechanism by flooding the respiratory tract with massive amounts of exogenous oxidants, depleting local antioxidant defenses, generating severe mitochondrial ROS overload, and subsequently provoking robust secondary airway inflammation.16,17 Functional validation in animal models has demonstrated that wild-type mice tolerate this oxidative-inflammatory insult via normal oxeiptosis activation to silently clear damaged epithelial cells, whereas genetic ablation of the core machinery (Pgam5−/−) results in a catastrophic, overshooting cascade of lung inflammation, massive neutrophil infiltration, and profound airway tissue damage.11 Thus, oxeiptosis functions as an indispensable, genetically programmed “immunological firewall” designed specifically to prevent severe ROS-induced airway inflammation from escalating into uncontrolled tissue necrosis, and its individual integrity or exhaustion may directly dictate human COPD heterogeneity.
In recent years, transcriptomic subtyping based on alternate programmed cell death pathways (such as ferroptosis and pyroptosis) has mapped clinical heterogeneity in respiratory diseases.18,19 However, while these existing classifications effectively capture iron-dependent or inflammasome-mediated processes, they frequently overlook the immediate, threshold-dependent death signature induced by the primary trigger of COPD: extreme oxygen radical stress. Establishing a molecular taxonomy based on oxeiptosis directly addresses this critical gap. This study sought to investigate the molecular heterogeneity of COPD within the context of oxeiptosis. We aimed to delineate molecular subtypes of COPD by identifying oxeiptosis-related differentially expressed genes (ORDEGs) and employing unsupervised clustering. Furthermore, WGCNA and single-sample gene set enrichment analysis (ssGSEA) were utilized to characterize the functional relevance of these genes and to measure pathway activity at the individual sample level, providing mechanistic insights into the possible role of oxeiptosis in COPD pathogenesis. Based on these considerations, we explicitly hypothesized that the expression profiles of downstream oxeiptosis-execution machinery exhibit significant molecular heterogeneity among COPD patients, and that these distinct patterns can define unique molecular endotypes characterized by divergent clinical severities and immune microenvironments.
Materials and Methods
Overview of the Study Population
The overall workflow of this study is illustrated in Figure 1. Two publicly available gene expression datasets (GSE47460 and GSE76925) were retrieved from the Gene Expression Omnibus (GEO) database.20,21 To ensure biological homogeneity, both datasets utilized whole lung tissue as the transcriptomic source. The GSE47460 cohort, consisting of 220 COPD patients and 108 controls, was employed as the discovery dataset, whereas 111 COPD patients from the GSE76925 cohort were utilized as the external validation dataset, baseline clinical and demographic characteristics are summarized in Supplementary Table S1. To ensure dataset integrity prior to downstream analyses, sample hierarchical clustering was performed on the discovery cohort to verify data homogeneity and exclude potential outliers (Supplementary Figure S1).
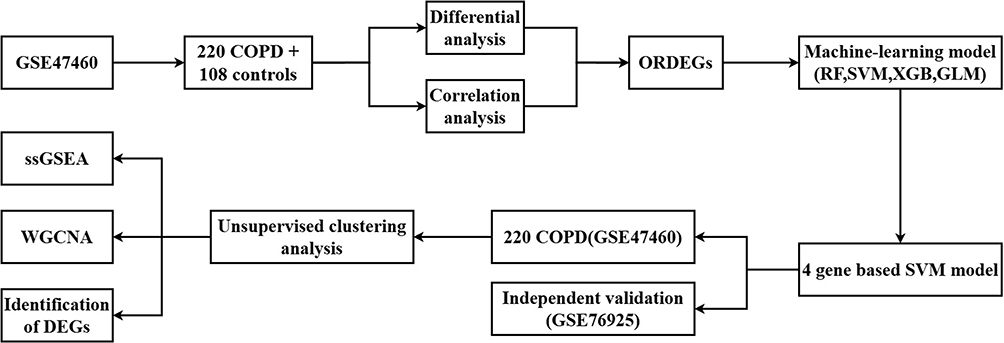 |
Figure 1 Schema of the study. |
Identification of ORDEGs (Oxeiptosis-Related Differentially Expressed Genes)
Four canonical regulators of the oxeiptosis pathway (KEAP1, PGAM5, AIFM1, and CUL3) were designated as core seed markers based on established literature.11,15 Starting from a global expression matrix of 15,180 annotated genes in the GSE47460 cohort, pairwise Pearson correlation analysis was performed between all global transcripts and each of the four seed markers. Transcripts displaying a strong correlation with any of these seed markers, defined by a Pearson correlation coefficient |r| > 0.5 and a Benjamini-Hochberg adjusted False Discovery Rate (FDR < 0.05), were considered oxeiptosis-related genes (ORGs).22 Concurrently, differentially expressed genes (DEGs) between COPD patients and healthy controls were identified using the “limma” R package with thresholds of |log2FC| > 0.5 and P < 0.05. The intersection of the union of ORGs and these DEGs was subsequently defined as ORDEGs, which were utilized as the final input features for downstream machine learning model construction.
Machine Learning for Key ORDEGs Selection
Multiple machine learning algorithms, including Random Forest (RF), Generalized Linear Model (GLM), Support Vector Machine (SVM), and XGBoost (XGB), were applied to identify robust predicted markers for COPD. The dataset was randomly split into training (70%) and validation (30%) cohorts in a stratified manner. Model construction and cross-validation were conducted using the “caret” R package (version 7.0–1), with up-sampling employed to address class imbalance. To ensure objective and systematic optimization, hyperparameters for each algorithm were tuned via a grid search strategy within a K-fold cross-validation framework. Specifically, a 5-fold cross-validation was utilized for the SVM model to balance computational efficiency with parameter stability, while a repeated 10-fold cross-validation was applied to the other algorithms. The grid search evaluated key tuning parameters, including the cost parameter (C) for SVM, the number of randomly selected predictors (mtry) for RF, and the α and λ parameters for the L1-regularized GLM. Model performance was primarily assessed by the area under the ROC curve (AUC), which was calculated using the “pROC” R package (version 1.18.5), while residual distribution analysis was employed to evaluate calibration. To further enhance interpretability, feature importance was estimated through the “DALEX” R package (version 2.4.3). Based on comparative performance, the top four genes derived from the best-performing algorithm were selected as candidate predicted markers for downstream analyses. SVM was selected as the final model due to its recognized strength in high-dimensional classification tasks.
Construction and Validation of Predictive Model Based on ORDEGs
Key ORDEGs identified through machine learning were incorporated into a clinical nomogram for COPD risk prediction. Using the “rms” R package (version 8.0–0), a logistic regression model was constructed based on the expression levels of key ORDEGs. The nomogram assigned a score to each predictor, and a total score was calculated to represent the overall probability of COPD. Model calibration was assessed using calibration curves with 1000 bootstrap resamples, ensuring consistency between predicted and observed outcomes. Discriminative performance was further evaluated by ROC curve analysis with the “pROC” R package (version 1.18.5), yielding the area under the curve (AUC) and its 95% confidence interval. Together, these evaluations demonstrated the predictive accuracy and clinical utility of the constructed nomogram.
Subtype Identification and Functional Scoring
Molecular subtypes of COPD were identified in the GSE47460 dataset using k-means clustering based on the expression of the key ORDEGs. The stability of clustering was assessed by silhouette analysis, and cluster-specific gene expression patterns were illustrated with heatmaps. To further confirm the reliability of subtype assignments, the Nearest Template Prediction (NTP) algorithm was applied, and consistency between k-means and NTP classifications was evaluated using a confusion matrix and Spearman correlation analysis. In addition, the oxeiptosis-related pathway activity of each sample was quantified via ssGSEA implemented in the “GSVA” R package (version 2.2.0), and enrichment scores were compared across the identified clusters.
Network and Functional Analyses
Subtype-associated DEGs were subjected to Weighted Gene Co-expression Network Analysis (WGCNA) to identify key co-expression modules. Functional annotation was conducted via Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) using the DAVID database.23
Statistical Analysis
Statistical analyses were performed using R software (version 4.5.0). Continuous variables were presented as mean ± standard deviation (SD) or median [interquartile range (IQR)]. The Shapiro–Wilk test was performed to assess the normality of all continuous clinical variables prior to analysis. For comparisons between the identified subtypes, the Student’s t-test or Wilcoxon rank-sum test was employed for continuous variables, while the Fisher’s exact test was used for categorical variables. To control for Type I error inflation in multiple comparisons, all P-values for clinical parameters were adjusted using the Benjamini-Hochberg (BH) method. P < 0.05 (or Padj < 0.05) was considered statistically significant.
Results
Identification of ORDEGs and DEGs in GSE47460
A total of 291 DEGs were identified in GSE47460 between COPD samples and controls (|log2FoldChange| > 0.5, P < 0.05), and 2357 ORGs were obtained via correlation analysis. Notably, none of the four canonical upstream seed genes (KEAP1, PGAM5, AIFM1, and CUL3) were contained within the 291 DEG list. Seven overlapping genes were identified between DEGs and ORGs (Figure 2A and B).
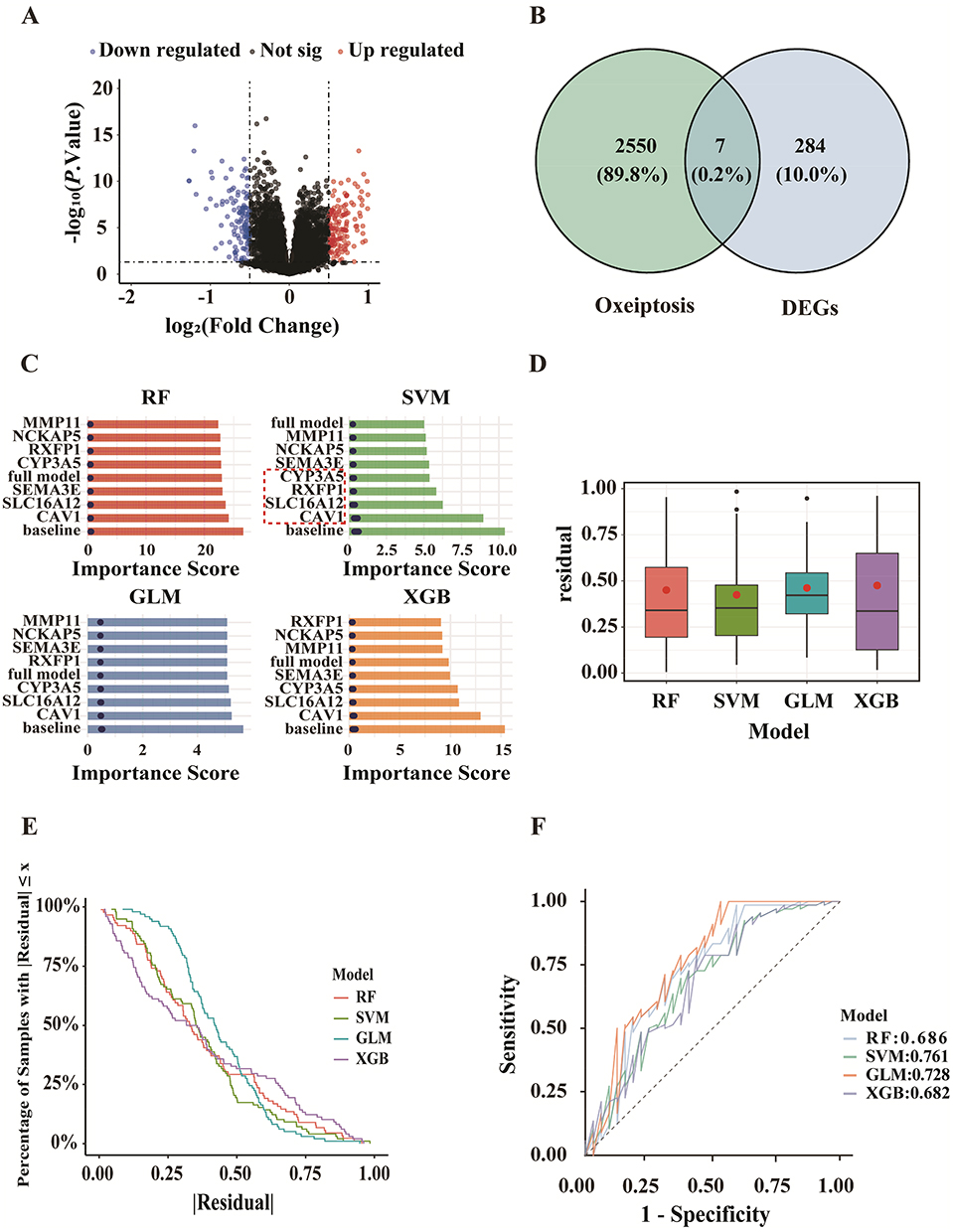 |
Figure 2 Identification and selection of oxeiptosis-related DEGs (ORDEGs) for COPD classification. (A) 291 DEGs. (B) 7 overlapping genes between DEGs and Oxeiptosis-related genes (ORGs). (C) Variable importance of 7 ORDEGs as ranked by machine learning algorithms. The red dotted box highlights the top four most important genes prioritized by the Support Vector Machine (SVM) model for downstream analysis. (D–E) Residual distributions of four machine learning models. (F) ROC curves of four machine learning models. |
The Identification of Four Key ORDEGs by the Machine Learning Method
The seven ORDEGs were ranked by RF, GLM, SVM, and XGB models according to their discriminative capacity between COPD patients and controls (Figure 2C). SVM yielded the best performance with the lowest residuals and highest AUC (0.761), as confirmed by residual distribution and ROC analysis (Figure 2D–F). The top four ORDEGs (CAV1, SLC16A12, RXFP1, and CYP3A5) were selected for further analysis.
Figure 3A presented the ROC curves for the selected ORDEGs, each evaluated for its ability to discriminate between COPD patients and controls. Among the selected ORDEGs, CAV1 demonstrated the highest predicted performance (AUC = 0.755), followed by SLC16A12 (AUC = 0.706), CYP3A5 (AUC = 0.675), and RXFP1 (AUC = 0.638). To provide a composite risk score for COPD prediction, a clinical nomogram was constructed integrating key ORDEGs (Figure 3C). The predictive accuracy was assessed via ROC analysis, which yielded an AUC of 0.777 (95% CI: 0.720–0.834) and indicated favorable predictive performance (Figure 3B). Furthermore, the calibration curve of the diagnostic nomogram was evaluated via bootstrap resampling with 1000 repetitions (Figure 3D).
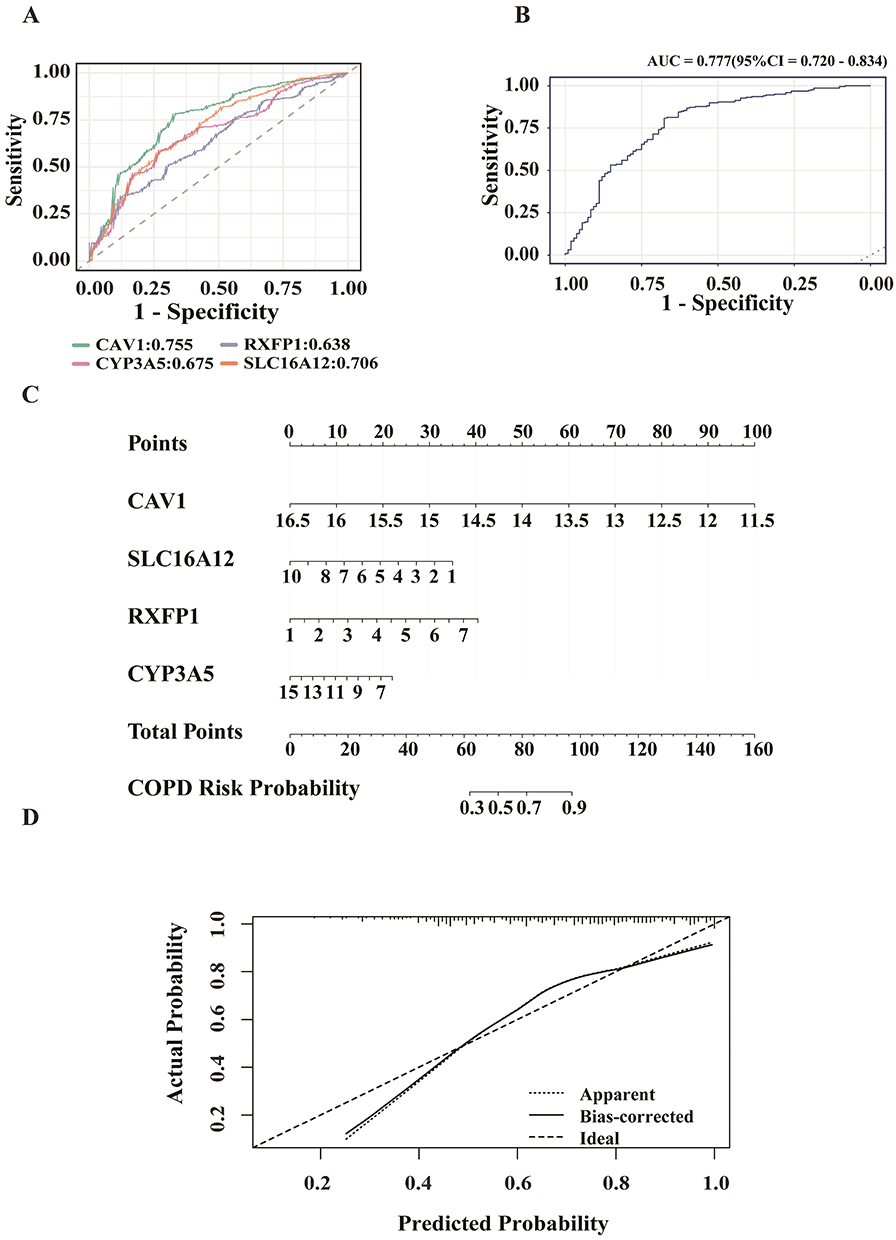 |
Figure 3 Predicted efficacy of the four-gene signature in COPD prediction. (A) ROC curves of ORDEGs in distinguishing COPD patients from controls. (B) ROC curve for the integrated ORDEGs model. (C) Nomogram combining the expression levels of ORDEGs. (D) Calibration curve of nomogram. |
To further validate the structural adaptation of the 7 candidate features within the canonical oxeiptosis pathway, a pairwise Pearson correlation analysis was executed between these candidates and the 4 upstream seed markers (Supplementary Figure S2). The correlation matrix revealed that all 7 identified ORDEGs are robustly and tightly anchored to the scaffold seed marker CUL3, with absolute correlation coefficients consistently exceeding the pre-specified threshold of 0.5 (CAV1: r = 0.59; NCKAP5: r = 0.57; SEMA3E: r = 0.58; MMP11: r = −0.53; all adjusted FDR < 0.05).
Consensus Clustering of COPD Cases in GSE47460
To investigate intra-disease heterogeneity, we carried out molecular subtyping on the GSE47460 cohort. Based on consensus clustering of the key ORDEGs, two stable clusters were identified as the optimal solution (Figure 4A). The selection of k=2 was quantitatively justified by the consensus cumulative distribution function (CDF) curves and cluster-consensus scores (Supplementary Figure S3). COPD samples were subsequently assigned to subtypes C1 and C2 via K-means clustering. Figure 4B showed strong concordance between k-means and NTP (P = 0.90).24 ROC curve analysis demonstrated that RXFP1 had the highest discriminative power between the two subtypes, while SLC16A12 showed the lowest (Figure 4D). Figure 4C indicated that key ORDEGs were significantly upregulated in subtype C2. To further explore functional differences, oxeiptosis enrichment scores were calculated using ssGSEA, based on expression of key ORDEGs. Subtype C2 exhibited significantly higher scores, indicating enhanced activation of oxeiptosis-related processes (Figure 4E).
 |
Figure 4 Subtype identification and molecular characterization in GSE47460. (A) K-means clustering heatmap based on key ORDEGs. (B) Concordance heatmap between K-means clustering and nearest template prediction (NTP). (C) Expression levels of key ORDEGs across subtypes. (D) ROC curves of key ORDEGs distinguishing C1 vs C2. (E) Oxeiptosis enrichment scores calculated via ssGSEA (****P < 0.0001, ***P < 0.001). |
Clinical characteristics of the two subtypes were summarized in Table 1. Despite comparable age, sex distribution, and smoking status (all P > 0.05), C1 was associated with lower pulmonary function indices. Specifically, patients in C1 displayed significantly reduced predicted Diffusing Capacity for Carbon Monoxide (DLCO), forced expiratory volume in 1 s (FEV1), and forced vital capacity (FVC) in both pre- and post-bronchodilator conditions (all P < 0.01), indicative of more severe functional impairment. The post-bronchodilator FEV1/FVC ratio displayed a trend toward reduction in the subtype C1, although this difference did not achieve statistical significance (P = 0.18).
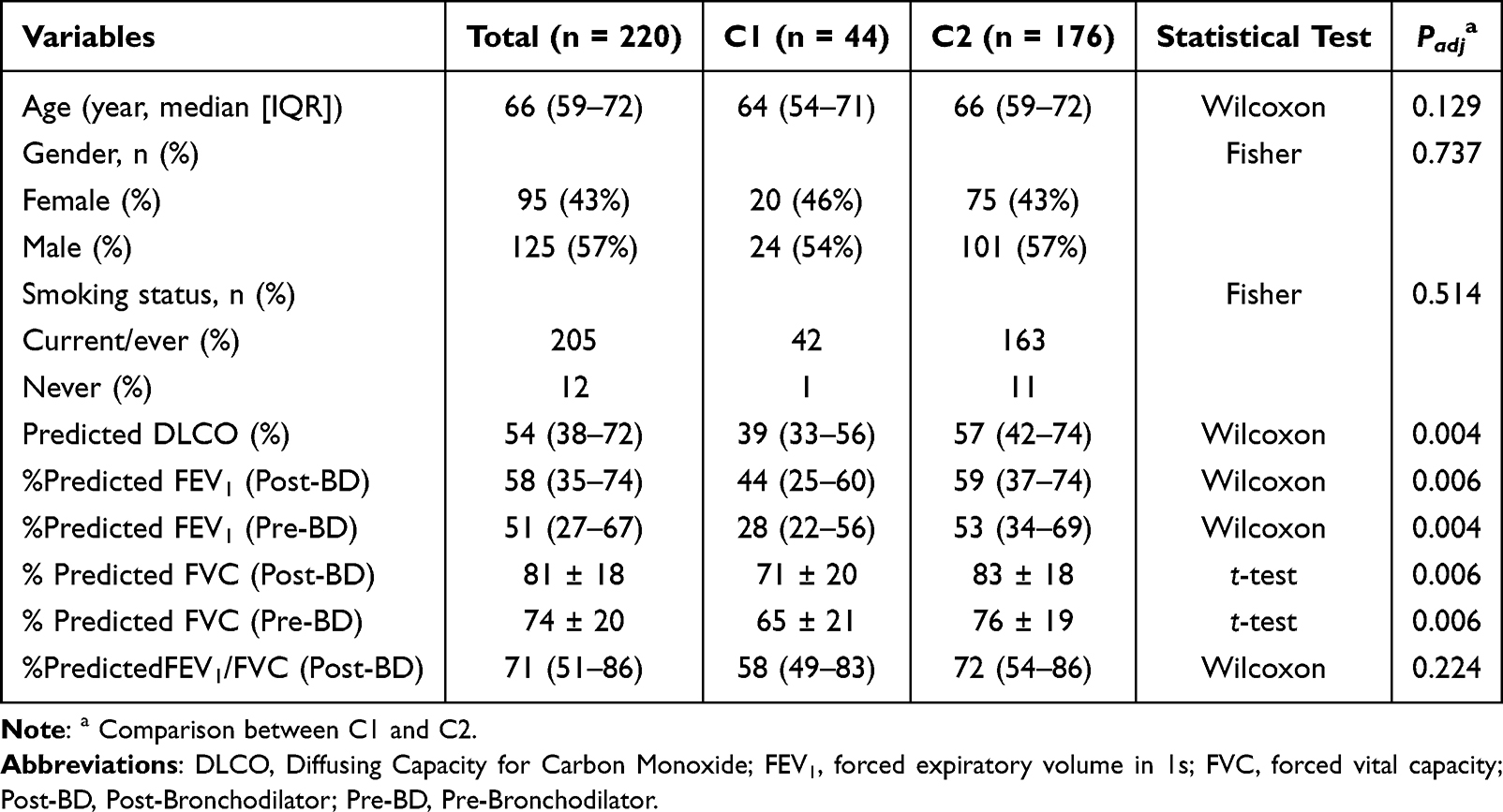 |
Table 1 Baseline Characteristics of COPD Patients by Clusters |
Validation of COPD Subtypes in GSE76925 Based on Four ORDEGs
Validation of the molecular subtypes identified in the GSE47460 cohort was achieved by applying key ORDEGs to classify COPD patients in the independent GSE76925 dataset. K-means clustering, based on the expression profiles of these genes, yielded two subtypes (C1 and C2), which were consistent with those identified in the discovery dataset (Figure 5A).
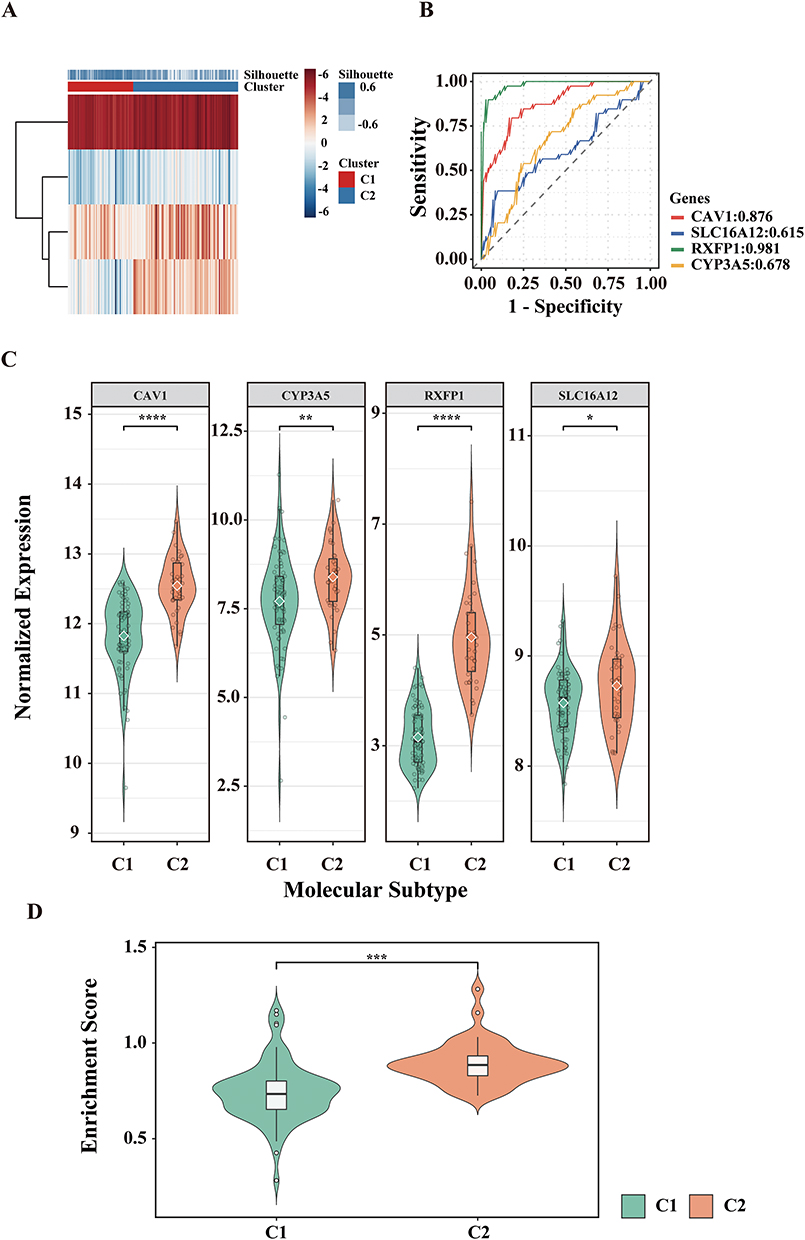 |
Figure 5 Validation of COPD subtypes in the GSE76925 cohort. (A) K-means clustering heatmap. (B) ROC curves of key ORDEGs for subtype classification. (C) Expression levels of key ORDEGS across subtypes. (D) Oxeiptosis ssGSEA scores between C1 and C2 (****P<0.0001, ***P < 0.001; **P < 0.01; *P < 0.05). |
The expression profiles of the key ORDEGs in GSE76925 recapitulated the molecular patterns observed in the discovery cohort (GSE47460), with all key ORDEGs consistently upregulated in subtype C2. Among the key ORDEGs, RXFP1 exhibited the highest discriminative performance as determined by ROC analysis, whereas SLC16A12 demonstrated comparatively lower classification efficacy (Figure 5B). In addition, ssGSEA-derived oxeiptosis scores, computed based on the key ORDEGs, were significantly elevated in subtype C2, recapitulating the enrichment trends observed in the training cohort and further supporting the robustness of functional stratification (Figure 5C and D).
These results reinforce the biological relevance and reproducibility of the identified COPD molecular subtypes across independent population. The subtypes identified in GSE76925 through key ORDEGs and the clustering strategy exhibited consistent gene expression and enrichment trends, thereby reinforcing the robustness and reproducibility of the stratification scheme.
WGCNA: Identification of Key Modules
As illustrated in Figure 6A, differential expression analysis between the two molecular subtypes yielded a total of 387 significant DEGs, comprising 187 upregulated and 200 downregulated genes, based on the criteria of |log2FoldChange| > 1 and P value < 0.05.
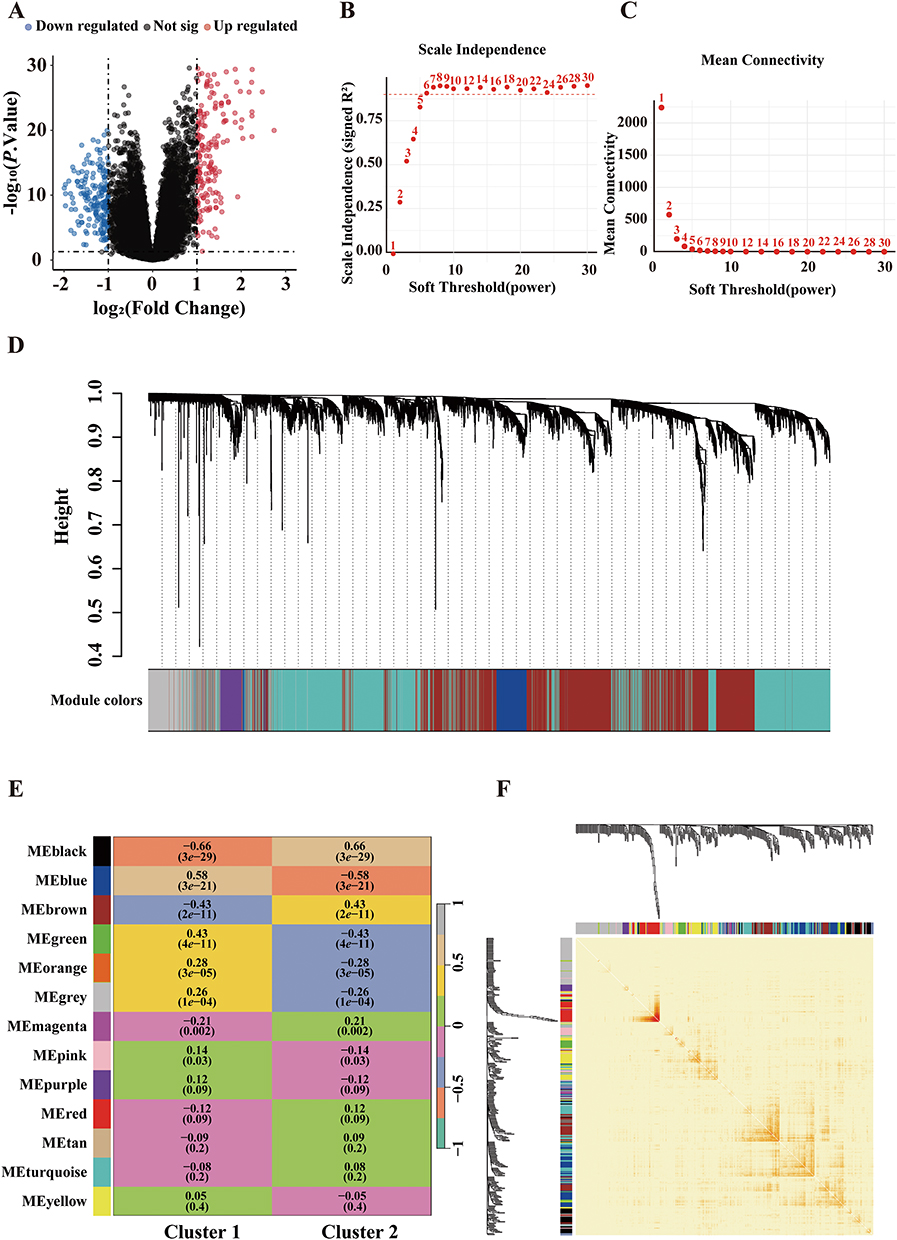 |
Figure 6 Weighted gene co-expression network analysis (WGCNA) of COPD molecular subtypes. (A) 387 DEGs betweenC1 and C2. (B and C) Selection of soft-thresholding power based on scale-free topology and mean connectivity. (D) Gene dendrogram and module identification. (E) Heatmap of module-trait correlations. (F) Topological overlap matrix heatmap. |
Transcriptomic variation between the two COPD subtypes was further explored using WGCNA based on the expression profiles of DEGs. To ensure construction of a scale-free network, we evaluated the scale-free topology fit index (R2) and mean connectivity across soft-thresholding powers ranging from 1 to 30. A power of 6 was selected as the optimal threshold, at which the network reached an approximate scale-free topology (R2 = 0.9) while maintaining acceptable mean connectivity, and this resulted in the identification of 13 distinct gene co-expression modules (Figure 6B–D). Each module was assigned a unique color label (black, blue, brown, green, orange, grey, magenta, pink, purple, red, tan, turquoise, and yellow). A module-trait correlation heatmap was constructed to assess the relationship between these modules and the two molecular subtypes. As shown in Figure 6E, eight modules (black, blue, brown, green, orange, grey, magenta, and pink) were significantly associated with subtype classification (P < 0.05). Among them, the black module exhibited the highest correlation coefficient (|r| = 0.66) and was selected for further analysis. Additionally, a topological overlap matrix (TOM) heatmap further visualized gene-gene interconnectedness within the network, with darker red indicating stronger co-expression (Figure 6F).
Functional Enrichment Analysis of the Black Module
The functional landscape of the black module was explored through GO and KEGG pathway analysis, as illustrated in Figure 7. GO molecular function enrichment analysis revealed that genes in this module were predominantly associated with activities such as heparin binding, transmembrane signaling receptor activity, and extracellular matrix (ECM) structural constituents (Figure 7A). KEGG pathway analysis indicated significant enrichment in cytokine-cytokine receptor interaction, neuroactive ligand-receptor interaction, and ECM-receptor interaction pathways (Figure 7B). Notably, many of these pathways were related to immune modulation and cell adhesion, highlighting the potential role of the black module in inflammatory signaling and tissue remodeling processes implicated in COPD pathogenesis. Taken together, these results highlighted the black module as the most relevant co-expression cluster associated with COPD subtype stratification.
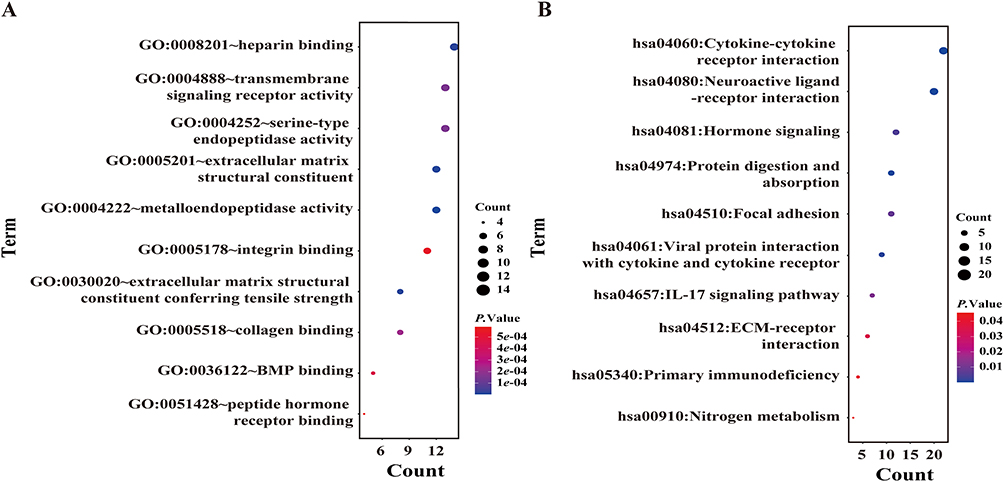 |
Figure 7 Functional Enrichment Analysis of the Black Module Genes. (A) Gene Ontology (GO) enrichment analysis of black module genes. (B) KEGG pathway enrichment of black module genes. |
Discussion
Our study identifies two distinct molecular subtypes (C1 and C2) of COPD, driven by the differential expression of four key oxeiptosis-related genes (ORDEGs: CAV1, SLC16A12, RXFP1, and CYP3A5). This molecular stratification, validated across two independent cohorts, represents the most significant finding of this work, offering a novel redox-based framework for understanding the profound clinical heterogeneity in COPD patients. This stratification’s robustness is supported by the application of machine learning techniques to identify oxeiptosis-related biomarkers associated with the oxidative stress pathway, which is central to COPD pathogenesis. This methodology enabled the identification of distinct molecular signatures associated with disease severity, thereby providing a novel perspective on the contribution of oxeiptosis to COPD progression.
Our results revealed that ORDEGs, including CAV1, SLC16A12, RXFP1, and CYP3A5, are significantly associated with the disease phenotype. While these genes have previously been implicated in inflammation, oxidative regulation, and metabolic adaptation, their identification within an oxeiptosis-related framework points to a potential role of this ROS-triggered cell death pathway in shaping the molecular heterogeneity of COPD.25–29
The relationship between key ORDEG expression and pulmonary function provided critical mechanistic insight. Compared with controls, COPD patients consistently presented with reduced expression of CAV1, SLC16A12, RXFP1, and CYP3A5, corresponding with impaired lung function. This association remained pronounced between subgroups; patients classified as subtype C1 exhibited lower ORDEG activity and more severe functional decline than those in subtype C2. Based on foundational biology, this paradox likely reflects a state of pathological pathway exhaustion rather than a lack of involvement. While physiological oxeiptosis operates as an immunologically silent buffer to clear damaged cells, genetic ablation of its core machinery paradoxically unleashes severe necro-inflammation and airway damage.11 Consequently, the preserved ORDEG expression in Cluster 2 may represent a compensatory defense, whereas the advanced disease stage in Cluster 1 is marked by the functional collapse or exhaustion of this protective clearing mechanism, forcing injured cells to default to destructive, lytic death modalities.
Network-level analyses further reinforced these findings. WGCNA identified a black module moderately associated with subtype classification. Functional enrichment revealed significant involvement in cytokine-cytokine receptor interaction as well as extracellular matrix (ECM) remodeling. This finding is particularly critical as it unifies the oxeiptosis-related findings with established mechanisms of COPD pathogenesis. While oxeiptosis itself is described as a non-inflammatory cell death pathway, the strong correlation between the ORDEG-defined subtypes and a gene network enriched for inflammatory signaling provides a crucial link. It suggests that the differential expression of the ORDEGs acts as a molecular indicator of the cellular environment, which in turn influences the broader inflammatory cascade. For example, CAV1, one of the key ORDEGs, is a known negative regulator of inflammation, and its reduced expression in the more severe C1 subtype may contribute to unchecked inflammatory signaling.26,30 Similarly, the enrichment of ECM-receptor interaction pathways provides a compelling biological explanation for the differences in pulmonary function. Aberrant ECM remodeling is a principal factor of irreversible airflow limitation in COPD, and the identified correlation suggests that that the ORDEG-defined subtypes represent unique profiles of tissue repair and destruction.31,32
Collectively, these findings substantiate a multi-faceted pathogenic model for COPD. In this model, an initial oxidative stress insult initiates a differential cellular response, regulated by the expression of the four key ORDEGs. This varied response then spreads through the gene network, leading to distinct downstream patterns of immune regulation and tissue remodeling. Ultimately, this multi-layered pathogenic state shows up as the clinical heterogeneity we see, with the C1 subtype being a more severe, decompensated state marked by chronic inflammation and extensive ECM degradation, and the C2 subtype exhibiting a more preserved or compensatory state. This systems-level perspective offers a more comprehensive and biologically coherent understanding of how genetic predisposition and environmental factors converge to shape the diverse clinical presentations of COPD.
While recent molecular taxonomies successfully stratify COPD patients based on downstream transcriptomic signatures of ferroptosis or pyroptosis,18,19,33 our framework distinctively builds upon these models by offering unparalleled proximity to the primary insult. Oxeiptosis serves as a dedicated, genetically encoded execution cascade responsive specifically to initial hyper-oxidative thresholds via the KEAP1-PGAM5-AIFM1 axis. While alternate pathways predominantly capture secondary metabolic adaptations or macroscale tissue inflammation, our oxeiptosis-specific taxonomy addresses a critical gap in existing literature. It provides a disease-specific lens that directly aligns with the upstream oxidative stress profiles of these patients, delivering a layer of pathophysiological precision unachievable through generalized cell-death markers.
Notably, the machine learning model demonstrated a high capacity for predicted discrimination. The SVM algorithm outperformed the other models in our analysis, showing relatively higher accuracy and better residual performance, consistent with its recognized strength in high-dimensional classification tasks.34 The construction of a nomogram based on the most informative ORDEGs further demonstrates the translational potential of this approach.
The present study, while providing a compelling framework, is subject to several limitations that necessitate further investigation. Foremost, the reliance on publicly available, retrospective gene expression data establishes a strong associational link but cannot, by its nature, establish direct causality. Additionally, because our analysis was conducted on bulk lung tissue transcriptomic matrices, the observed profiles reflect an average signal across multiple cell types; thus, cell-type-specific oxeiptosis signatures within the respiratory epithelium might be partially masked or diluted by infiltrating inflammatory cells. The findings generate a robust hypothesis that the differential expression of the identified ORDEGs modulates the cellular response to oxidative stress; however, this requires functional validation. The study itself acknowledges that direct mechanistic evidence linking these genes to oxeiptosis is yet to be established. Consequently, a primary area for future research must be the direct experimental validation of this proposed mechanism.
Future work should prioritize targeted in vitro and in vivo studies to fully elucidate the functional roles of CAV1, SLC16A12, RXFP1, and CYP3A5 within the context of the KEAP1-PGAM5-AIFM1 signaling axis. Techniques such as gene knockdown or knockout in pulmonary cell lines or animal models of COPD could directly test whether modulating the expression of these genes influences cell viability, inflammatory cytokine production, and extracellular matrix remodeling. Furthermore, while the validation in an independent cohort is a strength, both datasets originate from similar research settings, and the generalizability of these findings to broader, more genetically and environmentally diverse global patient populations remains to be confirmed. Future studies in larger, multi-ethnic cohorts are essential to ensure the clinical utility of this stratification scheme. Finally, while WGCNA provided valuable network-level insights, a more comprehensive understanding of COPD pathogenesis will require the integration of additional data modalities, such as proteomics, metabolomics, and single-cell sequencing, to build a higher-resolution map of the dynamic processes that distinguish these two molecular subtypes.
Conclusion
This study identified two molecular subtypes (C1 and C2) of COPD, characterized by distinct oxidative and functional profiles, which are driven by the differential expression of four key oxeiptosis-related genes (ORDEGs: CAV1, SLC16A12, RXFP1, and CYP3A5). Although direct functional evidence is pending, the established roles of these ORDEGs in ROS regulation and immune-metabolic modulation strongly support their biological plausibility as potential regulators of this pathway. These findings highlight the significant molecular heterogeneity of COPD and propose a new framework for redox-based subtype stratification. The identified gene signature and classification scheme hold significant translational potential, providing a foundation for developing a patient-centric precision medicine approach that could lead to improved diagnosis, more accurate prognosis, and the eventual development of subtype-specific therapeutic interventions for this complex and devastating disease. Targeted functional studies are warranted to experimentally validate these proposed mechanisms and translate this redox-based framework into clinical practice.
Abbreviations
COPD, chronic obstructive pulmonary disease; ORDEGs, Oxeiptosis-Related Differentially Expressed Genes; SVM, Support Vector Machine; WGCNA, Weighted Gene Coexpression Network Analysis; ROS, Reactive Oxygen Species; ssGSEA, single-sample Gene Set Enrichment Analysis; GEO, Gene Expression Omnibus; FDR, False Discovery Rate; ORGs, Oxeiptosis-Related Genes; DEGs, Differentially Expressed Genes; RF, Random Forest; GLM, Generalized Linear Model; XGB, Extreme Gradient Boosting; ROC, Receiver Operating Characteristic; AUC, Area Under the Curve; NTP, Nearest Template Prediction; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; DLCO, Diffusing Capacity for Carbon Monoxide; FEV1, Forced Expiratory Volume in 1s; FVC, Forced Vital Capacity; R2, scale-free topology fit index; ECM, Extracellular Matrix.
Data Sharing Statement
The primary gene expression data analyzed in this study are fully available in the NCBI Gene Expression Omnibus (GEO) repository under accession numbers GSE47460 and GSE76925. The code used for WGCNA analysis, machine learning model implementation, and molecular subtyping in this manuscript has been deposited in the Zenodo repository and is publicly accessible via the Digital Object Identifier (DOI):10.5281/zenodo.17284955.
Ethics Approval and Informed Consent
The transcriptomic data analyzed in this study were retrieved from the publicly available Gene Expression Omnibus (GEO) database (GSE47460 and GSE76925). In accordance with Items 1 and 2 of Article 32 of the “Measures for Ethical Review of Life Science and Medical Research Involving Human Subjects”, this study is officially exempt from local Institutional Review Board (IRB) review. Specifically, Item 1 exempts research utilizing information already obtained through public channels, and Item 2 exempts research using fully anonymized data that cannot identify specific natural persons. Since this study strictly involves the secondary bioinformatic analysis of public, de-identified datasets, no direct human intervention or patient recruitment was conducted, thereby fully satisfying the national criteria for ethical exemption.
Acknowledgments
We gratefully acknowledge the invaluable support and assistance received from all individuals and institutions throughout the course of this research.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This project was supported by the grants from National Natural Science Foundation of China (NO. 82160628, 82360636) and Hainan Provincial Natural Science of Foundation of China (NO. 825RC768).
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Chronic obstructive pulmonary disease (COPD). Available from: https://www.who.int/news-room/fact-sheets/detail/chronic-obstructive-pulmonary-disease-copd.
2. Wang M, Aaron CP, Madrigano J, et al. Association between long-term exposure to ambient air pollution and change in quantitatively assessed emphysema and lung function. JAMA. 2019;322(6):546. doi:10.1001/jama.2019.10255
3. Soriano JB, Kendrick PJ, Paulson KR, et al. Prevalence and attributable health burden of chronic respiratory diseases, 1990–2017: a systematic analysis for the global burden of disease study 2017. Lancet Respir Med. 2020;8(6):585–16. doi:10.1016/S2213-2600(20)30105-3
4. Barnes PJ. Oxidative stress-based therapeutics in COPD. Redox Biol. 2020;33:101544. doi:10.1016/j.redox.2020.101544
5. Kwak N, Lee KH, Woo J, Kim J, Lee CH, Yoo CG. Synergistic cycles of protease activity and inflammation via PPARγ degradation in chronic obstructive pulmonary disease. Exp Mol Med. 2021;53(5):947–955. doi:10.1038/s12276-021-00626-7
6. Hou Y, Wang H, Wu J, Guo H, Chen X. Dissecting the pleiotropic roles of reactive oxygen species (ROS) in lung cancer: from carcinogenesis toward therapy. Med Res Rev. 2024;44(4):1566–1595. doi:10.1002/med.22018
7. Albano GD, Gagliardo RP, Montalbano AM, Profita M. Overview of the mechanisms of oxidative stress: impact in inflammation of the airway diseases. Antioxidants. 2022;11(11):2237. doi:10.3390/antiox11112237
8. Wang C, Zhou J, Wang J, et al. Progress in the mechanism and targeted drug therapy for COPD. Signal Transduct Target Ther. 2020;5(1):248. doi:10.1038/s41392-020-00345-x
9. Xue J, Wang C, Fan H. The role of programmed cell death in chronic obstructive pulmonary disease: from pathogenesis to treatment. Front Immunol. 2026;17:1715665. doi:10.3389/fimmu.2026.1715665
10. Ni FX, Wang HX, Hu J, et al. Pyroptosis-driven immune dysregulation in COPD: molecular mechanisms and therapeutic implications. Front Immunol. 2025;16:1686175. doi:10.3389/fimmu.2025.1686175
11. Holze C, Michaudel C, Mackowiak C, et al. Oxeiptosis, a ROS-induced caspase-independent apoptosis-like cell-death pathway. Nat Immunol. 2018;19(2):130–140. doi:10.1038/s41590-017-0013-y
12. Sendtner N, Seitz R, Brandl N, Müller M, Gülow K. Reactive oxygen species across death pathways: gatekeepers of apoptosis, ferroptosis, pyroptosis, paraptosis, and beyond. Int J Mol Sci. 2025;26(20):10240. doi:10.3390/ijms262010240
13. Park SY, Gurung R, Hwang JH, et al. Development of KEAP1-targeting PROTAC and its antioxidant properties: in vitro and in vivo. Redox Biol. 2023;64:102783. doi:10.1016/j.redox.2023.102783
14. Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 2016;26(3):165–176. doi:10.1016/j.tcb.2015.10.014
15. Kobayashi A, Kang MI, Okawa H, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-basedE3ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24(16):7130–7139. doi:10.1128/mcb.24.16.7130-7139.2004
16. Russo RC, Togbe D, Couillin I, et al. Ozone-induced lung injury and inflammation: pathways and therapeutic targets for pulmonary diseases caused by air pollutants. Environ Int. 2025;198:109391. doi:10.1016/j.envint.2025.109391
17. Sunil VR, Vayas KN, Radbel J, et al. Lung injury, oxidative stress, and impaired functioning in a model of prolonged ozone exposure in female mice are associated with macrophage proinflammatory and profibrotic activation and altered bioenergetics. Toxicol Sci. 2026;209(2):kfaf173. doi:10.1093/toxsci/kfaf173
18. Luan X, Xie J, Zhang L, Ma X, Jiao L, Zhu J. Identification of ferroptosis-related genes involved in chronic obstructive pulmonary disease based on bioinformatics analysis. Anim Models Exp Med. 2025;8(12):2232–2252. doi:10.1002/ame2.70040
19. Shu HM, Lin CQ, He B, et al. Pyroptosis-related genes as diagnostic markers in chronic obstructive pulmonary disease and its correlation with immune infiltration. Int J Chron Obstruct Pulmon Dis. 2024;19:1491–1513. doi:10.2147/COPD.S438686
20. Tan J, Tedrow JR, Dutta JA, et al. Expression of RXFP1 is decreased in idiopathic pulmonary fibrosis. Implications for relaxin-based therapies. Am J Respir Crit Care Med. 2016;194(11):1392–1402. doi:10.1164/rccm.201509-1865oc
21. Morrow JD, Zhou X, Lao T, et al. Functional interactors of three genome-wide association study genes are differentially expressed in severe chronic obstructive pulmonary disease lung tissue. Sci Rep. 2017;7(1):44232. doi:10.1038/srep44232
22. Huang J, Zhang J, Zhang F, et al. Identification of a disulfidptosis-related genes signature for prognostic implication in lung adenocarcinoma. Comput Biol Med. 2023;165:107402. doi:10.1016/j.compbiomed.2023.107402
23. Sherman BT, Panzade G, Imamichi T, Chang W. DAVID ortholog: an integrative tool to enhance functional analysis through orthologs. Bioinformatics. 2024;40(10). doi:10.1093/bioinformatics/btae615
24. Zhou X, Ba Y, Xu N, et al. Pharmacogenomics-based subtype decoded implications for risk stratification and immunotherapy in pancreatic adenocarcinoma. Mol Med. 2025;31(1):62. doi:10.1186/s10020-024-01049-6
25. Tan J, Li X, Dou N. Insulin resistance triggers atherosclerosis: caveolin 1 cooperates with PKCzeta to block insulin signaling in vascular endothelial cells. Cardiovasc Drugs Ther. 2024;38(5):885–893. doi:10.1007/s10557-023-07477-6
26. Shivshankar P, Halade GV, Calhoun C, et al. Caveolin-1 deletion exacerbates cardiac interstitial fibrosis by promoting M2 macrophage activation in mice after myocardial infarction. J Mol Cell Cardiol. 2014;76:84–93. doi:10.1016/j.yjmcc.2014.07.020
27. Hirai K, Kimura T, Suzuki Y, Shimoshikiryo T, Shirai T, Itoh K. Gene polymorphisms of NLRP3 associated with plasma levels of 4β-hydroxycholesterol, an endogenous marker of <span>CYP3A</span> activity, in patients with asthma. Clin Pharmacol Ther. 2024;116(1):147–154. doi:10.1002/cpt.3254
28. Bouchez C, Devin A. Mitochondrial biogenesis and mitochondrial reactive oxygen species (ROS): a complex relationship regulated by the cAMP/PKA signaling pathway. Cells. 2019;8(4):287. doi:10.3390/cells8040287
29. Felmlee MA, Jones RS, Rodriguez-Cruz V, Follman KE, Morris ME. Monocarboxylate transporters (SLC16): function, regulation, and role in health and disease. Pharmacol Rev. 2020;72(2):466–485. doi:10.1124/pr.119.018762
30. Fan J, Zheng S, Wang M, Yuan X. The critical roles of caveolin-1 in lung diseases. Front Pharmacol. 2024;15. doi:10.3389/fphar.2024.1417834
31. Karakioulaki M, Papakonstantinou E, Stolz D. Extracellular matrix remodelling in COPD. Eur Respir Rev. 2020;29(158):190124. doi:10.1183/16000617.0124-2019
32. Herro R, Miki H, Sethi GS, et al. TL1A promotes lung tissue fibrosis and airway remodeling. J Immunol. 2020;205(9):2414–2422. doi:10.4049/jimmunol.2000665
33. Qi X, Cao S, Chen J, Yin X. Integrated analysis of ferroptosis- and cellular senescence-related biomarkers in atherosclerosis based on machine learning and single-cell sequencing data. In Review. 2024. doi:10.21203/rs.3.rs-5239772/v1
34. El-Mageed AAA, Elkhouli AE, Abohany AA, Gafar M. Gene selection via improved nuclear reaction optimization algorithm for cancer classification in high-dimensional data. J Big Data. 2024;11(1):46. doi:10.1186/s40537-024-00902-z
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Using Machine Learning to Predict Likelihood and Cause of Readmission After Hospitalization for Chronic Obstructive Pulmonary Disease Exacerbation
Bonomo M, Hermsen MG, Kaskovich S, Hemmrich MJ, Rojas JC, Carey KA, Venable LR, Churpek MM, Press VG
International Journal of Chronic Obstructive Pulmonary Disease 2022, 17:2701-2709
Published Date: 20 October 2022
Can Blood Biomarkers Be Used to Assess Oxidative Stress in COPD Patients After Pulmonary Rehabilitation
Beykumül A, Ersoy Y, Gülbaş G, Neselioglu S
International Journal of Chronic Obstructive Pulmonary Disease 2023, 18:2179-2186
Published Date: 5 October 2023
A Macrophage-Related Gene Signature for Identifying COPD Based on Bioinformatics and ex vivo Experiments
Zhang Z, Yu H, Wang Q, Ding Y, Wang Z, Zhao S, Bian T
Journal of Inflammation Research 2023, 16:5647-5665
Published Date: 29 November 2023
Identification of Biomarkers Associated with Oxidative Stress in Aortic Dissection Based on Bulk Transcriptome Analyses
Li Z, Li C, Shao Y, Ran H, Shi H, Zhou R, Liu X, Wu Q, Zhang C
International Journal of General Medicine 2024, 17:5633-5650
Published Date: 28 November 2024
Identification and Validation of Feature Genes Related to Mitochondrial Dysfunction and Oxidative Stress in Ulcerative Colitis
Wu Y, Tan Z, Lan Q, Liu Y, Liu C, He H, Zhang J, Dong W
Journal of Inflammation Research 2025, 18:5835-5850
Published Date: 30 April 2025