Back to Journals » Journal of Inflammation Research » Volume 18
Ferroptosis in Hypertriglyceridemic Acute Pancreatitis: Mechanisms and Therapeutic Implications
Authors Zou K, Liu W, Xia W, Zhao Y ![]()
Received 30 September 2025
Accepted for publication 4 December 2025
Published 24 December 2025 Volume 2025:18 Pages 18115—18135
DOI https://doi.org/10.2147/JIR.S567900
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Nadia Andrea Andreani
Ke Zou,1,2 Wenzhen Liu,3 Wenwen Xia,2 Yan Zhao1,2
1Department of Gastroenterology, Shanghai Tenth People’s Hospital Chongming Branch, Shanghai, 202150, People’s Republic of China; 2Department of Gastroenterology, Shanghai Tenth People’s Hospital, School of Medicine, Tongji University, Shanghai, 200072, People’s Republic of China; 3Guangzhou University of Chinese Medicine, Guangzhou, 510006, People’s Republic of China
Correspondence: Yan Zhao, Department of Gastroenterology, Shanghai Tenth People’s Hospital of Tongii University, 30L Middle Yanchang Road, Jing’an, Shanghai, 200072, People’s Republic of China, Email [email protected]
Abstract: Ferroptosis, a iron-dependent programmed cell death characterized by iron-dependent accumulation of lipid peroxidation to lethal levels, is closely related to the pathogenesis of hypertriglyceridemic acute pancreatitis, a condition marked by lipid metabolism disorders. This paper summarizes the latest research progress in understanding the mechanistic contributions on the mechanisms of ferroptosis in HTG-AP, with a particular focus on the roles of lipid peroxidation and iron-catalyzed reactive oxygen species generation in the pathogenesis and progression of HTG-AP. It further elaborates on critical molecules-including the GPX4, ACSL4, SLC7A11 and FSP1-CoQ10-NAD(P)H and key cellular signaling pathways-including the HIF pathways, JAK-STAT pathways, PI3K/ Akt pathways closely linked to ferroptosis in HTG-AP. Understanding the pathophysiological role of hypertriglyceridemia in pancreatic injury is essential for unraveling the complex interplay between lipid and iron metabolic homeostasis. Additionally, by integrating evidence from preclinical models and human studies, this review emphasizes the importance of ferroptosis mechanisms in the treatment of HTG-AP, with the goal of identifying potential therapeutic targets and proposing innovative intervention strategies aimed at mitigating ferroptosis, potentially improving outcomes in HTG-AP.
Keywords: ferroptosis, hypertriglyceridemic acute pancreatitis, lipid peroxidation, therapeutic targets
Introduction
Hypertriglyceridemic acute pancreatitis (HTG-AP) is a distinct subtype of acute pancreatitis (AP), characterized by markedly elevated serum triglyceride (TG) levels, typically exceeding 11.3 mmol/L (1000 mg/dL), in the absence of other common etiologies such as gallstones or alcohol abuse.1,2 In recent years, with the global epidemic of obesity, diabetes, and metabolic syndrome, the incidence of HTG-AP has risen substantially. It has now become the third leading cause of AP, following gallstones and alcohol. Epidemiological studies indicate that HTG-AP accounts for approximately 8–20% of all AP cases, with a significantly higher prevalence in Eastern populations, where the proportion can reach nearly 30% in some Asian cohorts.3–5 Recent researches have reported that the incidence of HTG-AP is substantially higher in Eastern countries than in Western populations, and that its mortality rate is approximately three times higher than that of pancreatitis caused by other etiologies.4 Moreover, HTG-AP tends to affect younger individuals. The mean age of onset is approximately 10 years earlier than that of biliary pancreatitis, and men are disproportionately affected. Patients frequently have metabolic risk factors such as obesity, diabetes mellitus, excessive alcohol consumption, or familial hyperlipidemia.3,6 Clinically, the presentation of HTG-AP resembles that of other forms of AP, with persistent epigastric pain, nausea, vomiting, and elevated pancreatic enzymes. However, HTG-AP is often more severe in nature, characterized by rapid onset, extensive pancreatic necrosis, and a heightened systemic inflammatory response that predisposes to multi-organ dysfunction. Respiratory failure, acute kidney injury, and circulatory collapse occur more frequently in this subtype.1,7 Several studies have reported that the proportion of severe cases can reach 30–40%, with an overall mortality rate ranging from 5% to 15%, significantly higher than that of biliary or alcoholic pancreatitis.1,8 Furthermore, serum TG concentration at admission correlates closely with the extent of pancreatic necrosis, systemic inflammation, and clinical outcome. When TG levels exceed 22.6 mmol/L (2000 mg/dL), the risk of developing severe pancreatitis and organ failure increases markedly.9,10 Consequently, HTG-AP represents a major challenge in the management of severe pancreatitis.11 In terms of prognosis, patients with HTG-AP often experience impaired metabolic recovery and incomplete restoration of pancreatic function. Follow-up studies have shown that approximately 20–30% of patients develop pancreatic exocrine insufficiency within six months after the initial episode, manifesting as steatorrhea, weight loss, and malnutrition.12,13 Some patients also develop impaired glucose tolerance, secondary diabetes mellitus and pancreatic cancer14,15 with a markedly increased risk of recurrent pancreatitis.16 In addition, 15–25% of patients progress to pancreatogenic diabetes, primarily due to sustained β-cell dysfunction and persistent insulin resistance.17,18 The incidence of chronic pancreatitis is also elevated; structural changes and pancreatic fibrosis can occur within one to three years after the initial episode in a subset of patients.16,19
Recent advancements have highlighted the role of ferroptosis, a novel type of controlled cell death distinguished by iron-reliant lipid peroxidation, as a key element in the pathogenesis of HTG-AP.11 This review explores the underlying mechanisms of ferroptosis in HTG-AP, with a focus on lipid peroxidation, iron catalysis, and the synergistic effects of these processes in pancreatic injury related to hypertriglyceridemia. By doing so, it sheds light on the emerging links between metabolic disorders and cell death pathways.
In terms of treatment, this review discusses current therapeutic approaches and their limitations, while emphasizing the potential of ferroptosis inhibitors as promising therapeutic agents that could fundamentally alter the treatment landscape. However, challenges such as treatment specificity and adverse effects remain significant barriers.20
In conclusion, this paper aims to synthesize existing research findings and propose future directions for HTG-AP research, recognizing the potential of ferroptosis as a therapeutic target. By doing so, it seeks to enhance our understanding of clinical treatment strategies for HTG-AP.
Pathophysiology and Etiology of HTG-AP
HTG-AP is an acute form of pancreatitis caused by elevated TG levels in the blood. The diagnostic threshold for HTG-AP is usually a serum TG concentration above 1000 mg/dL, which significantly increases the risk of pancreatitis developing. This type of pancreatitis is etiologically different from other types of pancreatitis due to the disorder of lipid metabolism. Up to now, the definite pathophysiological mechanisms by which HTG-AP remain uncertain and are still a subject of controversy. Several studies using animal models have put forward hypothesis that are generally agreed. According to the theory originally proposed by Havel, when TG are injected into the isolated pancreas of dogs, pancreatic lipase within the pancreas hydrolyzed excess TG,21,22 leading to the production of dense accumulations of free fatty acids (FFAs). When the binding capacity of albumin is surpassed, these FFAs naturally assemble micelle-like structures, demonstrating detergent-like properties. The micelles aggressively target platelets, vascular endothelial cells, and acinar cells, leading to the destruction of acinar cells and pancreatic capillaries. This subsequently induces pancreatic ischemia and creates an acidic milieu within the pancreas.23 Acidosis can activate trypsinogen, which further exacerbates the toxicity of FFAs, leading to pancreatic congestion, edema, and the onset of HTG-AP.24,25 Additionally, acidosis may promote the activation of cathepsin B109, which further activates trypsinogen, triggering or exacerbating inflammation.23 The inflammatory cascade can culminate in cellular death, fluid accumulation, and pancreatitis, potentially evoking a widespread inflammatory reaction.26 Furthermore, heightened TG-rich lipoprotein levels increased blood viscosity, and the accumulated concentration of chylomicrons, especially large-diameter ones, can occlude capillaries and compromise pancreatic microcirculation, which diminishes pancreatic blood flow, resulting in additional ischemic damage.25
Severe HTG-AP often lies a complex interplay between inherent genetic susceptibility, derangements in metabolic homeostasis, and, in some cases, the compounding factor of excessive alcohol intake. The genetic backgrounds of HTG-AP are intricate. Mild to moderate hypertriglyceridemia-induced pancreatitis is often polygenic in origin, while severe cases, especially in younger patients, are more likely due to a combination of monogenic factors and secondary triggers that, when uncontrolled, significantly elevate TG levels.27 Therefore, HTG-AP may be influenced by other metabolic factors associated with AP, including poorly controlled diabetes, obesity, and excessive alcohol consumption.
In recent years, ferroptosis has been identified as a form of programmed cell death closely associated with lipid peroxidation, disrupted iron homeostasis, and oxidative stress. Lipid peroxidation and iron dysregulation can form a feedback loop that exacerbates cellular damage and inflammation. These characteristics show significant overlap with the pathological context of HTG-AP, making this area highly worthy of in-depth investigation.
Cellular Iron Dynamics and Lipid Peroxidation: Unveiling the Pathway of Ferroptosis
As a distinctive iron-dependent programmed cell death pathway, ferroptosis is marked by the progressive collection of peroxidized phospholipids (PLs). Unlike apoptosis or necrosis, ferroptosis is intrinsically linked to the metabolic processes of iron and lipids, making it a distinct pathway of cell death that is associated with a variety of diseases, including neurodegenerative disorders, cancer, and pancreatitis.11,28 At the heart of this process lies the role of iron as a catalyst, facilitating the generation of reactive oxygen species (ROS). If the ROS generated are of a specific type—such as phospholipid hydroperoxides (PLOOH)—and cannot be effectively neutralized, they accumulate and compromise the integrity of the plasma membrane, leading to ferroptosis.
In cells, the substrates for phospholipid peroxidation are PLs that contain polyunsaturated fatty acids (PUFAs) chains at the sn-2 position. In the presence of bioactive iron, these PUFA-PLs can undergo both enzymatic and non-enzymatic lipid peroxidation, resulting in the formation of PLOOH. Since all mammalian cells contain baseline levels of PUFA-PLs and bioactive iron, specific monitoring or protective mechanisms exist under physiological conditions to prevent unnecessary ferroptosis. One of the primary surveillance mechanisms is mediated by glutathione peroxidase 4(GPX4),29 which is a singular enzyme in the mammalian, catalyzes the reduction of PLOOH to alcohols, thus detoxifying lipid peroxides. Inhibition or downregulation of GPX4 renders cells more susceptible to ferroptosis.28,30 The Xc- cystine/glutamate antiporter system also plays a critical role in this process, as it facilitates the import of cystine, which is essential for the synthesis of glutathione (GSH). Disruption of this pathway may trigger iron-catalyzed lipid peroxidation, which is a defining characteristic of ferroptosis. Alongside GPX4-reliant mechanisms, several GPX4-independent pathways also mitigate ferroptosis. For instance, proteins such as ferroptosis suppressor protein 1(FSP1), GCH1, and DHODH generate metabolites with radical-trapping activity, which can terminate the Fenton radical chain reaction and prevent the propagation of lipid peroxidation.29,31–34
The Link Between Hypertriglyceridemia and Ferroptosis: A Critical Role in HTG-AP
Elevated levels of TG in the blood disrupt the delicate balance of lipid metabolism, rendering the pancreas vulnerable to oxidative stress and cellular dysfunction.21,27 Compared with non-lipotoxic forms of pancreatitis, HTG-AP generates a distinct biochemical milieu characterized by synergistic amplification of toxic lipid metabolites, dysregulation of iron homeostasis, and a pro-inflammatory microenvironment. These factors collectively initiate or exacerbate ferroptosis, thereby triggering a destructive cycle of oxidative damage and inflammation.
Role of Lipid Peroxidation in HTG-AP
HTG-AP is a complex, multifactorial condition defined by increased TG levels, which accelerate lipid peroxidation. Research indicates that enhanced lipid peroxidation is closely associated with increased acinar cell death.25 Gaining insight into the factors driving lipid peroxidation in HTG-AP is essential for devising targeted therapeutic interventions. Lipid peroxidation begins with the oxidative degradation of lipids, which occurs when reactive ROS target PUFAs in cellular membranes. This interaction results in the generation of lipid radicals that initiate a chain reaction, ultimately producing lipid hydroperoxides. Elevated lipid hydroperoxide levels not only serve as indicators of oxidative stress but also directly contribute to tissue damage and inflammation.
From the perspective of substrate availability, HTG-AP provides an exceptionally rich pool of oxidizable lipids. Hypertriglyceridemia markedly expands lipid stores in the circulation and peripancreatic tissues. Upon pancreatic injury—such as premature leakage of digestive enzymes—lipases (eg, pancreatic lipase) hydrolyze these TG and release large amounts of FFAs.35 These FFAs are heterogeneous but are typically enriched in PUFAs. Due to their multiple double bonds, PUFAs are highly prone to spontaneous auto-oxidation or iron-catalyzed peroxidation, effectively serving as the direct “fuel” for ferroptosis. Ferroptosis is driven by iron-dependent peroxidation of PUFA-containing PLs; thus, when antioxidant defenses such as the GPX4–GSH axis are compromised, membrane lipid damage is easily triggered.36 The abundant PUFA substrates provided by HTG-AP therefore significantly enhance ferroptotic potential, in contrast to etiologies lacking extensive TG hydrolysis, such as purely biliary pancreatitis.
Beyond substrate overload, the enzymology of lipid metabolism and membrane phospholipid remodeling also shapes ferroptotic susceptibility. FFAs do not dictate cell fate solely in their free form; instead, they undergo enzymatic activation and incorporation into membrane PLs, thereby determining membrane composition and oxidation vulnerability. A key enzyme in this process is acyl-CoA synthetase long-chain family member 4 (ACSL4), which displays substrate specificity for long-chain PUFAs. In HTG-AP, abundant PUFAs such as arachidonic acid (AA) and eicosapentaenoic acid serve as preferential substrates for ACSL4, promoting its activation and expression.37 Following conversion to PUFA-CoA, these lipids are incorporated into phosphatidylcholine or phosphatidylethanolamine (PE) by enzymes such as lysophosphatidylcholine acyltransferase 3 (LPCAT3), markedly increasing susceptibility to iron-catalyzed lipid peroxidation38,39—a hallmark of ferroptosis. Compared with non-HTG forms of AP, the ACSL4/LPCAT3 pathway is induced or upregulated in HTG-AP, leading to rapid esterification of abundant PUFAs into membranes and increasing the density of peroxidation-sensitive PLs.40 At the molecular level, HTG-AP therefore strengthens the “loading” phase that primes cells for ferroptosis.
Moreover, dysregulated lipid metabolism in HTG-AP indirectly enhances ferroptotic sensitivity by altering cellular energy metabolism and mitochondrial function. The mitochondrial membrane exhibits a high concentration of PUFAs, making it particularly vulnerable to oxidative damage. The presence of oxidative stress results in diminished mitochondrial membrane potential, consequently compromising ATP synthesis capacity.41,42 Excessive FFAs entering mitochondria lead to intensified β-oxidation, mitochondrial membrane depolarization, and dysfunction of the electron transport chain.43 These changes increase ROS production while depleting NADPH and GSH, weakening antioxidant capacity. The impairment of mitochondrial function further triggers the release of apoptotic factors, particularly cytochrome c, enlightening a cascade of apoptotic signaling.44 Mitochondrial ROS synergize with membrane lipid peroxidation, lowering the threshold for ferroptosis and promoting iron-dependent oxidation of phospholipid acyl chains into toxic aldehydes such as malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE),45 ultimately compromising membrane integrity and leading to cell death. Navina et al similarly reported that unsaturated fatty acids can inhibit mitochondrial respiratory complexes and trigger Ca2⁺ overload and necrotic injury in models of obesity-related pancreatic lipotoxicity.36 Studies utilizing mitochondria-targeted antioxidants, such as mitoquinone(MitoQ), have demonstrated the potential to mitigate oxidative damage by restoring respiratory chain function and energy production. These findings highlight the necessity of safeguarding mitochondria from ROS to maintain cellular energy homeostasis and promote cell survival.46
In addition, inflammation and immunometabolic reprogramming in HTG-AP further amplify ferroptosis. The lipid-rich microenvironment alters the metabolic phenotype of infiltrating macrophages and neutrophils, enhancing the release of pro-inflammatory mediators and perturbing iron homeostasis. Upregulation of ACSL4 in immune cells also increases their intrinsic susceptibility to ferroptosis.47 Once immune cells undergo ferroptosis, they release damage-associated molecular patterns, which intensify inflammation and propagate ferroptotic death to neighboring acinar and endothelial cells.48,49 This self-reinforcing loop among immunity, lipid metabolism, and ferroptosis is markedly more pronounced in HTG-AP than in non-lipotoxic pancreatitis.48
Moreover, oxidative events also affect the endoplasmic reticulum and plasma membrane, leading to disruptions in calcium homeostasis, which exacerbates cellular injury. Experimental evidence suggests that increased lipid peroxidation is associated with elevated intracellular calcium levels, thereby promoting cell death pathways.50,51 Calcium dysregulation is also recognized as a fundamental mechanism in the development of pancreatitis.
Role of Iron Catalysis in Hypertriglyceridemic AP
Iron plays a paradoxical role in biological systems, functioning as both an essential mediator and a potential toxicant in various physiological processes. While it is an essential trace mineral for various life functions, excessive iron can catalyze oxidative stress in pancreatic cells, leading to ferroptosis and tissue damage.52 Therefore, it is crucial to investigate the role of iron in HTG-AP and identify potential therapeutic targets. A growing body of research has explored the cellular and molecular mechanisms by which iron metabolism promotes the development of HTG-AP. Clinical data indicate that elevated iron levels are commonly observed in patients with pancreatitis and are associated with markers of oxidative damage, such as MDA and HNE.53 This suggests a close relationship between iron metabolism dysregulation and oxidative damage in pancreatic tissue. In animal models of pancreatitis with excessive iron intake, increased production of pro-inflammatory cytokines and pancreatic necrosis have been observed, further supporting the role of iron in oxidative stress-driven pancreatic injury.54,55 Iron’s pro-oxidant nature enhance the accumulation of ROS, which are central to oxidative stress, a well-known contributor to pancreatic tissue damage.52
The fundamental elements of ferroptosis can be distilled into two essential components: (i) an expansion of the labile iron pool, which provides catalytic iron for Fenton chemistry, and (ii) the oxidation of PUFAs within membrane PLs, leading to loss of membrane integrity. Consequently, any pathological condition that simultaneously increases bioavailable iron and enhances membrane susceptibility to oxidation—or weakens antioxidant defenses—significantly lowers the threshold for ferroptotic cell death.48 HTG-AP uniquely promotes both processes: massive lipolysis supplies abundant PUFAs substrates, while inflammation, hemorrhage, and autophagy-driven ferritinophagy disrupt iron homeostasis, rapidly increasing local iron availability and thus predisposing the pancreas to ferroptosis more strongly than non-HTG etiologies.
In HTG-AP, the presence of abundant peripancreatic adipose tissue results in extensive lipolysis when pancreatic lipase leaks into surrounding areas. The hydrolysis of TG induces marked fat necrosis and structural tissue injury, which often accompanies microvascular leakage, focal hemorrhage, or erythrocyte lysis—events that release heme and free iron into the interstitial space.56 Moreover, phagocytosis and clearance of necrotic tissue by macrophages can further liberate iron or alter its recycling dynamics, causing a transient surge in the local labile iron pool within the pancreatic microenvironment.35,57 By contrast, typical non-HTG etiologies—such as simple biliary obstruction or mild alcohol-induced pancreatitis—usually lack extensive fat necrosis or hemorrhage, resulting in less iron release and a slower accumulation of iron-driven lipid peroxidation. This “iron‐source differential” is a major reason why HTG-AP is more prone to ferroptosis.
Inflammatory stimuli such as IL-6 upregulate hepatic hepcidin, which promotes degradation of the iron exporter ferroportin and thereby reshapes systemic iron distribution.58 In HTG-AP, lipotoxicity is frequently accompanied by more intense systemic and local inflammation, leading to dynamic changes in hepcidin and redistribution of iron between organs. This results in a paradoxical pattern—transient hepatic iron sequestration alongside intracellular iron retention within pancreatic tissues owing to phagocytosis, cellular injury, and activated autophagy. Animal studies demonstrate that hepcidin deficiency or impaired hepcidin signaling causes intrapancreatic iron accumulation and chronic pancreatic injury,59 underscoring the essential role of the liver–pancreas iron regulatory axis in maintaining pancreatic tolerance to iron load.59 Disruption of this axis is generally more severe in HTG-AP than in non-HTG diseases, predisposing the pancreas to a ferroptosis-permissive iron microenvironment.36 Ferritin serves as a major iron-storage complex that limits the labile iron pool; however, when autophagic activity increases, selective ferritinophagy—mediated by NCOA4—degrades ferritin and rapidly releases stored iron.60 Under HTG conditions, pancreatic acinar cells experience FFAs overload, mitochondrial and ER stress, and typically display enhanced autophagic flux.61 Recent pancreatitis models show that autophagy-dependent ferritin degradation is tightly coupled to ferroptosis.57 In HTG-AP, this pathway is more robustly activated, leading to faster and greater liberation of intracellular iron than in non-HTG etiologies, thereby accelerating the lipid peroxidation cascade.
Iron and PUFAs are critical determinants of ferroptosis, but the integrity of antioxidant systems—such as GPX4/GSH and the FSP1–CoQ10 axis—ultimately dictates whether cells cross the ferroptotic threshold.31,62 In HTG-AP, NEFA-induced mitochondrial ROS, NADPH depletion, and ER stress collectively weaken these defensive mechanisms.57 Simultaneously, increased local iron availability amplifies peroxidation reactions, exhausting antioxidant reserves at an accelerated rate. In other words, the convergence of “more iron and more oxidizable substrates and weaker antioxidant defenses” makes HTG-AP far more prone to rapidly triggering ferroptosis, whereas non-HTG causes typically lack at least one of these components (most commonly PUFAs overload or iron release), rendering ferroptosis less likely.
Overall, from the perspective of iron homeostasis, the heightened susceptibility to ferroptosis in HTG-AP compared with non-HTG pancreatitis reflects not a single mechanism but rather the combined effects of increased iron sources, disruption of the liver–pancreas iron regulatory axis, enhanced autophagy-dependent ferritinophagy, and coordinated failure of antioxidant systems. These insights highlight the need to incorporate “iron homeostasis” into both research and therapeutic strategies—on par with lipid metabolism—to develop mechanism-driven, precision interventions specifically tailored to HTG-AP.
Key Cellular Signaling and Molecule Between HTG-AP and Ferroptosis
GPX4
GPX4 is a pivotal intracellular antioxidant enzyme, playing an indispensable role in defending against lipid peroxidation and regulating ferroptosis. Unlike other GPX isoforms that broadly reduce hydrogen peroxides, GPX4 contains a unique selenocysteine (Sec) residue that confers exceptionally high catalytic activity and substrate specificity. A distinguishing feature of GPX4 is its ability to directly associate with cellular membranes and reduce PLOOH embedded within the lipid bilayer to their corresponding phospholipid alcohols, thereby interrupting the lipid peroxidation chain reaction and preserving membrane integrity.63–65 Mechanistically, GPX4 utilizes reduced GSH as an electron donor to catalyze the reduction of PLOOH. The Sec moiety first attacks PLOOH to form a selenenic acid intermediate, which is subsequently reduced by two molecules of GSH to regenerate the catalytically active selenol, yielding oxidized GSH in the process.62 This two-electron reduction mechanism enables GPX4 to terminate peroxidation reactions in a non-radical manner, preventing further propagation of damaging reactive species.66 GSH reductase then regenerates GSH using NADPH, thereby sustaining this essential antioxidant cycle.67
GPX4 exhibits remarkable selectivity toward both its substrates and the surrounding membrane environment.68 Evidence indicates that hydrophobic residues near its active site interact with the membrane interface, allowing the enzyme to access esterified PUFAs within PLs. Structural and computational studies support a model in which GPX4 is strategically positioned on the membrane surface, enabling efficient recognition and reduction of membrane-embedded PLOOH, thus mitigating membrane damage.69 GPX4 is particularly crucial for detoxifying oxidized PE, especially those enriched in AA or adrenic acid (AdA). When AA- or AdA-containing PE-OOH are not efficiently removed, they readily accumulate and trigger cell death. Indeed, studies demonstrate that inhibition or genetic ablation of GPX4 leads to a rapid buildup of these oxidized PEs, which constitute the key lethal lipid species driving ferroptosis.
The functional status of GPX4 critically shapes cellular fate under both physiological and pathological conditions. In settings of heightened metabolic stress or increased iron burden—such as pancreatic tissue exposed to HTG—oxidative pressure intensifies, membrane lipid composition is remodeled (eg, elevated PUFA content), and iron-catalyzed oxidation reactions such as the Fenton reaction become more prevalent. Under these circumstances, any compromise in GPX4 activity—whether due to GSH depletion, inadequate selenium supply, or direct enzyme inhibition—impairs timely reduction of lipid hydroperoxides, leading to progressive membrane disruption and ultimately ferroptotic cell death.70,71 This mechanism has been validated in multiple tissue injury models and underscores the crucial importance of preserving GPX4 function in HTG-AP.
Experimental evidence further supports the central role of the GPX4–GSH axis as a frontline defense against ferroptosis. Direct inhibition of GPX4 using small-molecule inhibitors such as RSL3, or depletion of GSH through blockade of the system Xc− cystine/glutamate antiporter (eg, by erastin), reliably induces ferroptosis.72 Moreover, in vivo GPX4 knockout models—including those targeting embryonic tissues, liver, heart, and retina—exhibit profound tissue damage due to uncontrolled lipid peroxidation. Supplementation with lipid-soluble antioxidants such as vitamin E partially ameliorates these phenotypes,62 further underscoring the irreplaceable role of GPX4 in maintaining membrane stability.
In summary, GPX4 plays a decisive role in preventing the propagation of lipid peroxidation and maintaining membrane integrity through its highly specific two-electron reduction mechanism, membrane-targeting capacity, and stringent substrate selectivity. In pathological states characterized by dysregulated lipid and iron metabolism—such as HTG-AP—disruption of GPX4 activity directly precipitates fatal membrane damage and ferroptosis. Therefore, a comprehensive understanding of GPX4 structure–function relationships, regulatory mechanisms (including selenium availability and GSH metabolism), and the ways in which these processes are perturbed in pancreatic pathology is essential for advancing both mechanistic insight and therapeutic development.
Within the pathological milieu of HTG-AP, GPX4 function faces dual challenges (Table 1). Firstly, the high FFA environment significantly increases the PUFA content while decreasing MUFA levels in membrane PLs. PUFAs, due to their bis-allylic structures, are highly susceptible to peroxidation. This substantially elevates the substrate load for GPX4-mediated detoxification.73 Furthermore, under sustained high-lipid stress, lipid droplets—the primary PUFA storage depots—become saturated. This saturation triggers massive PUFA release and their subsequent incorporation into membrane PLs, becoming major substrates for peroxidation.63 Additionally, ACSL4 catalyzes the activation of long-chain PUFAs to PUFA-CoAs, promoting their integration into membrane PLs and further amplifying lipid peroxidation.74 Secondly, exacerbated oxidative stress leads to a burst of ROS, which further consumes GPX4 activity.63,73 GPX4 activity is critically dependent on a continuous GSH supply. In HTG-AP, glucose metabolism dysregulation induced by the hyperlipidemic microenvironment restricts the pentose phosphate pathway. This limitation reduces NADPH generation, consequently impairing GSH regeneration capacity and indirectly suppressing GPX4 function.63 During the initial phase of HTG-AP, pancreatic acinar cells initiate an adaptive antioxidant response to high-lipid stress. Animal model studies indicate that GPX4 expression undergoes transient upregulation in the early stages of sodium taurocholate-induced AP. This represents a compensatory response mediated by nuclear factor erythroid 2-related factor 2(Nrf2) pathway activation.75 At this stage, despite elevated intracellular ROS levels, enhanced GPX4 activity effectively maintains the clearance of lipid peroxidation products, delaying the progression of cell death.63 Notably, specific knockout of the pancreatic GPX4 gene in mice resulted in significantly higher serum amylase and lipase levels, along with increased tissue necrosis area, compared to wild-type mice in cerulein-induced pancreatitis models.65 As high-lipid stress persists, the GPX4 defense system gradually collapses, entering an irreversible phase of functional exhaustion. In canine HTG-AP models, GPX4 protein expression was reduced compared to healthy controls. Transmission electron microscopy revealed characteristic ferroptotic ultrastructural alterations, including mitochondrial cristae breakdown and loss of membrane integrity.67 Concomitantly, the decline in GPX4 enzymatic activity was closely associated with GSH depletion and insufficient NADPH supply.63,76,77 In summary, GPX4, as a core regulator of ferroptosis, acts as a critical “molecular gatekeeper” in the pathological evolution of HTG-AP. Its dynamic trajectory—from early compensatory adaptation to late-stage functional collapse—profoundly influences the fate determination of pancreatic acinar cells.
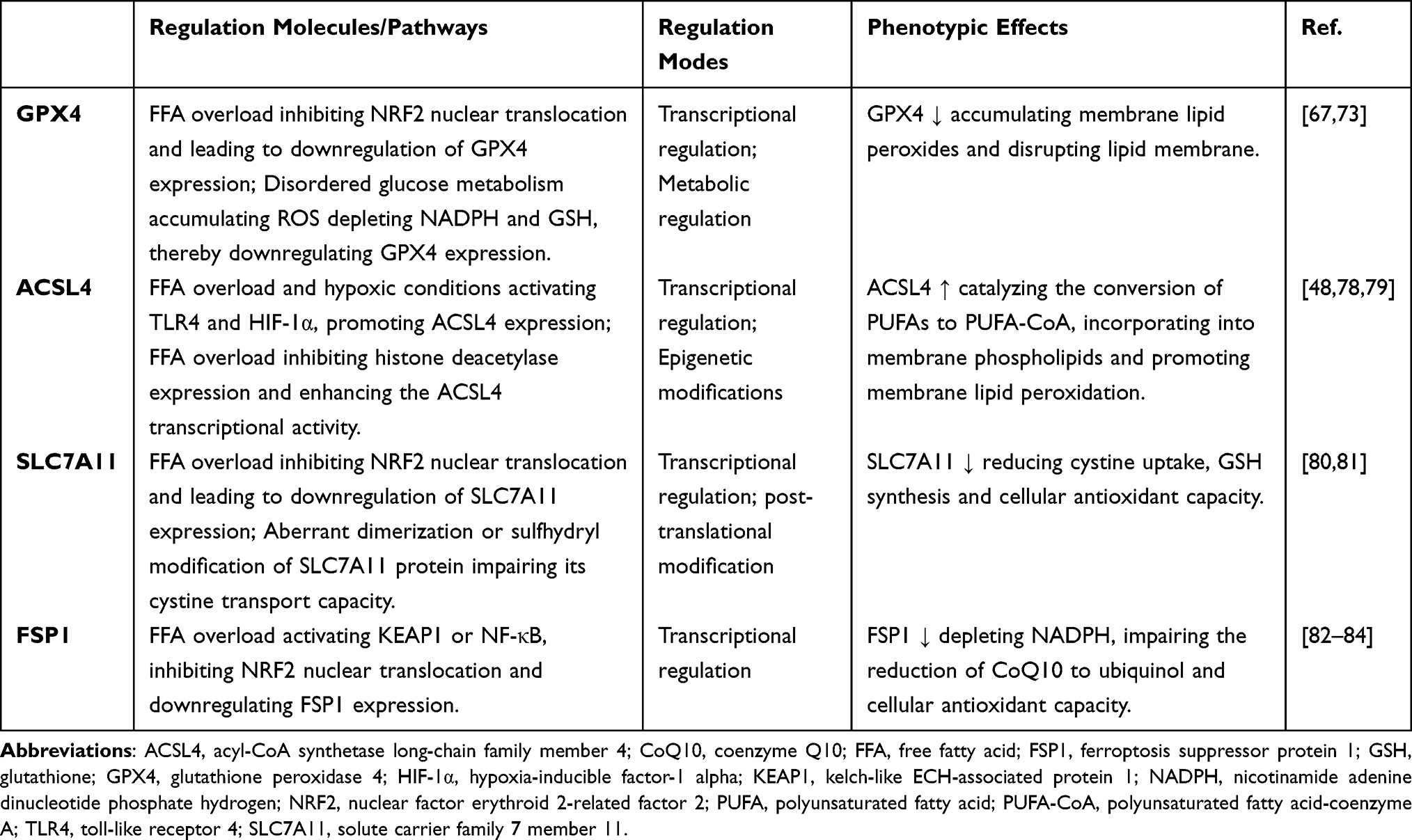 |
Table 1 Key Regulation Molecule Between HTG-AP and Ferroptosis |
Acsl4
ACSL4, a key enzyme in lipid metabolic reprogramming, plays a central role in ferroptosis during HTG-AP by driving lipid peroxidation through its promotion of PUFA incorporation into membrane PLs (Table 1). ACSL4 catalyzes the activation of long-chain PUFAs, particularly ω-6 fatty acids (eg, AA, AdA), to acyl-coa (eg, AA-coa, ADA-coa). These activated PUFAs are subsequently incorporated into membrane PLs—particularly PEs—via LPCAT3. Oxidation by lipoxygenases (LOXs) or the Fenton reaction then generates lipid peroxides, forming the lipid substrates essential for ferroptosis.48,85,86 Mechanistically, ACSL4 initiates the pathway by catalyzing an ATP-dependent ligation of PUFAs with coenzyme A to generate PUFA-CoAs. These activated PUFA-CoAs are subsequently incorporated by LPCAT3 into the sn-2 position of PEs, producing species such as AA-PE and AdA-PE.40 PUFA-enriched PEs serve as preferential substrates for subsequent oxidation by LOXs or iron-dependent radical reactions. Once oxidized to PE-OOH,87 these PLOOH readily initiate membrane damage and trigger ferroptosis. In this context, ACSL4 functions as a “substrate hub”, orchestrating the membrane PUFA-PE composition and thereby dictating cellular susceptibility to ferroptotic death.
The strong association between ACSL4 expression/activity and ferroptosis sensitivity has been validated across multiple studies. CRISPR-based loss-of-function screens and pharmacological inhibition experiments demonstrate that deletion or suppression of ACSL4 markedly diminishes ferroptotic cell death.40 Additionally, high ACSL4 expression predicts heightened sensitivity to GPX4 inhibitors such as RSL3 in several cell types. Beyond its role in phospholipid remodeling, ACSL4 is also linked to mitochondrial dysfunction and ROS production. Specifically, overexpression of ACSL4 and LPCAT2 renders cells more vulnerable to ferroptosis inducers like RSL3, accompanied by reduced mitochondrial membrane potential, impaired respiratory capacity, and elevated mitochondrial ROS. Notably, selective scavenging of mitochondrial ROS using MitoQ partially rescues this cell death phenotype,88 indicating that mitochondrial ROS act as a key mediator in ACSL4-driven ferroptosis.
Under pathological conditions such as inflammation, lipid overload, or pancreatic injury, ACSL4 likely acts as a central determinant of ferroptosis sensitivity by reshaping membrane phospholipid composition and supplying oxidizable substrates.89 In HTG-AP, elevated FFAs, inflammatory signaling, and oxidative stress synergistically enhance ACSL4-mediated synthesis of PUFA-PEs, thereby increasing the availability of substrates for LOX-dependent or iron-catalyzed oxidation.87 When GPX4 is inhibited or functionally compromised, these newly formed PE-OOH species accumulate rapidly, precipitating membrane destruction and subsequent ferroptotic death.
In HTG-AP, elevated levels of saturated fatty acids (eg, palmitic acid) upregulate ACSL4 transcription through the TLR4/NF-κB pathway. Consistent with this, ACSL4 expression is elevated in pancreatic acinar cells within HTG-AP mouse models.78,90 Furthermore, FFAs inhibit histone deacetylases, increasing H3K27 acetylation at the ACSL4 promoter region. This epigenetic modification enhances ACSL4 transcriptional activity,79,91 further amplifying ACSL4 function. Hypoxia induced by hyperlipidemia activates HIF-1α, which directly binds to the ACSL4 promoter and upregulates its expression.92 Supporting this regulatory link, clinical samples reveal a significant positive correlation between HIF-1α and ACSL4 expression in the pancreas of HTG-AP patients.93 ACSL4 also potentiates the inflammatory microenvironment characteristic of HTG-AP. Lipid peroxidation products generated via ACSL4 activity (eg, 4-HNE) activate the NLRP3 inflammasome, promoting IL-1β release and recruiting macrophage infiltration.94 Accordingly, ACSL4 inhibitors reduce pancreatic IL-1β levels in HTG-AP models.48 These lipid peroxides further activate macrophages via TLR2/TLR4 signaling, promoting M1 polarization95 and establishing a detrimental “ferroptosis-inflammation” vicious cycle. Currently, pharmacological inhibitors targeting ACSL4 are being explored as potential therapeutic strategies to mitigate cell death in ferroptosis-associated pathologies, including liver disease and cancer.96,97 Notably, suppressing the ACSL4/p38 MAPK/GPX4 axis reverses silica nanoparticle-induced hepatic ferroptosis.78 This mechanism may be extrapolated to HTG-AP-associated liver injury. Therefore, modulating ACSL4 activity represents a promising therapeutic target for managing hypertriglyceridemia-associated pancreatitis.98
SLC7A11
SLC7A11 (also known as xCT) serves as the key functional subunit of the system xc− transporter located on the plasma membrane. It forms a heterodimeric complex with SLC3A2, responsible for the 1:1 exchange transport of extracellular cystine into the cell and intracellular glutamate out of the cell. This process constitutes the core metabolic hub of the cellular antioxidant system: imported cystine is rapidly reduced to cysteine, the rate-limiting precursor for GSH synthesis80 (Table 1). GSH, one of the most crucial intracellular antioxidant molecules, acts as an essential substrate for GPX4, protecting cells against lipid peroxidation and ferroptosis.99 GPX4 utilizes GSH to reduce toxic PLOOH into harmless, thereby halting the lipid peroxidation chain reaction and maintaining plasma membrane integrity.100 Under physiological conditions, SLC7A11 expression is finely regulated by multiple transcription factors, with NRF2 being the most critical activator. During oxidative stress, NRF2 translocates into the nucleus and binds to the antioxidant response element (ARE) within the SLC7A11 promoter, significantly upregulating its transcription and enhancing cellular antioxidant defenses.81,100 RNA sequencing analysis of human pancreatic tissue and murine AP models revealed significant enrichment of cell survival-related pathways during the early disease stage, including transient upregulation of SLC7A11 expression.80 SLC7A11 specifically protects acinar cells from ferroptosis by promoting the GSH pool and maintaining ROS balance. Conversely, acinar cells with impaired or reduced SLC7A11 function exhibit GSH depletion, increased ferroptosis associated with lipid peroxidation, and significantly exacerbated inflammation and pancreatic tissue necrosis. Pharmacological activation of the NRF2-SLC7A11 axis significantly alleviates pancreatic injury.80,99 Within the pathological context of HTG-AP, this intricate regulatory network is particularly susceptible to disruption, leading to SLC7A11 dysfunction. The abundant FFAs generated in HTG-AP markedly induce the expression of nicotinamide N-methyltransferase (NNMT). NNMT depletes S-adenosylmethionine to generate methylated products, resulting in a global reduction in cellular methylation levels and consequently impacting the expression regulation of both GPX4 and SLC7A11.101 Experimental evidence demonstrates that NNMT inhibitor treatment significantly elevates GPX4 and SLC7A11 expression, reduces lipid peroxidation levels and Fe2⁺ content, and mitigates ferroptosis.101 Furthermore, abundant lipid metabolites activate the aryl hydrocarbon receptor (AhR) signaling pathway. AhR, a ligand-activated transcription factor, transcriptionally represses SLC7A11 expression, thereby enhancing cellular susceptibility to ferroptosis. This regulatory role of AhR is likely amplified within the pancreatic inflammatory milieu.81
FSP1-CoQ10-NAD(P)H Antioxidant Axis
FSP1 is a core ferroptosis inhibitor independent of the GPX4 pathway. FSP1 is a flavoprotein that utilizes NAD(P)H as an electron donor to catalyze the reduction of CoQ to its antioxidant form, ubiquinol (CoQH2).102 CoQH2 acts as a potent lipophilic radical-trapping antioxidant that terminates the propagation of lipid peroxyl radicals within membranes, thereby preventing the lethal spread of lipid peroxides.31,82,83 N-terminal myristoylation facilitates the membrane association of FSP1, enabling the enzyme to execute its reductive function in close proximity to the phospholipid bilayer.32 Expression-based screening and CRISPR-mediated synthetic lethal analyses have identified FSP1 as a powerful ferroptosis-resistance factor capable of compensating for the loss or inhibition of GPX4 and suppressing otherwise fatal lipid peroxidation.31
Beyond its canonical role in CoQ reduction, FSP1 is involved in additional regulatory mechanisms with antioxidant and cytoprotective functions. Emerging evidence suggests that FSP1 may also participate in the vitamin K reduction cycle to confer antioxidant protection,103 and can promote membrane repair through ESCRT-III–mediated mechanisms, thereby providing resistance to ferroptotic injury.104 Both in vitro and in vivo studies demonstrate that inhibition of FSP1 markedly sensitizes cells to ferroptosis induced by GPX4 inhibitors, whereas elevated FSP1 expression correlates positively with ferroptosis resistance across multiple cancer cell lines.31 When the GPX4–GSH axis is suppressed, the FSP1–CoQ pathway appears to become rapidly activated to maintain localized membrane redox homeostasis.105
In summary, FSP1 constitutes a GPX4-independent and GSH-independent defensive pathway that suppresses the propagation of lipid peroxidation through CoQ reduction and strategic membrane localization. Under conditions of heightened oxidative stress and metabolic dysregulation, FSP1 may serve as a crucial compensatory mechanism to maintain membrane integrity. Elucidating its molecular functions and developing small-molecule modulators targeting FSP1 will advance our understanding of ferroptosis regulation, particularly within the pathological landscape of HTG-AP.
FSP1 exerts a central protective role against ferroptosis in HTG-AP through two interconnected mechanisms: the CoQ10-NAD(P)H antioxidant axis and PPARα/CPT1A-mediated lipid metabolism regulation (Table 1). Under these conditions, a burst of reactive ROS activates NRF2, leading to a transient upregulation of FSP1 expression. This increase facilitates enhanced ubiquinol generation, buffering lipid peroxidation damage106 and delaying acinar cell death. Animal studies demonstrate that FSP1 expression initially rises compensatorily in mice fed a high-fat diet. However, with sustained oxidative stress, FSP1 protein degradation accelerates, ultimately leading to functional exhaustion.107 Notably, FSP1 overexpression improves survival rates in cerulein-induced pancreatitis models.108 FSP1 function is dynamically regulated by the hyperlipidemic microenvironment. Under persistent high-lipid stress, FFAs activate KEAP1, inhibiting NRF2 nuclear translocation. This cascade results in the downregulation of FSP1 expression, NADPH depletion, and diminished FSP1 reductase activity, impairing its capacity to effectively regenerate ubiquinol.84 Concurrently, FSP1 may undergo nuclear translocation, where its C-terminal fragment can induce caspase-independent apoptosis, exacerbating tissue damage.109 Furthermore, continuous high-lipid stimulation can activate NF-κB via the TLR4/MyD88 pathway, suppressing NRF2 transcriptional activity and consequently reducing FSP1 expression.84 Clinical investigations have revealed that serum FSP1 levels in severe HTG-AP patients positively correlate with the extent of pancreatic necrosis and serve as an independent risk factor for predicting multiple organ failure.110 Current research efforts are exploring FSP1 as a therapeutic target for HTG-AP, particularly in combination with lipid metabolism modulation. For instance, the natural compound carvacrol activates the NRF2/FSP1 axis, reducing ferroptosis in acinar cells.84 However, significant challenges remain, including the lack of molecular probes for real-time detection of pancreatic FSP1 activity and the difficulty in precisely determining the optimal therapeutic window. Therefore, future studies should integrate single-cell spatial transcriptomics to decipher the FSP1 regulatory networks within distinct pancreatic cell subpopulations across HTG-AP stages, thereby addressing the critical hurdles of tissue-specific drug delivery and dynamic monitoring.
HIF Pathway
Hypoxia-inducible factors (HIFs) are transcription factors composed of an oxygen-sensitive α subunit (HIF-1α or HIF-2α) and a constitutively expressed β subunit. Under normoxic conditions, HIF-α undergoes prolyl hydroxylation by prolyl hydroxylase domain (PHD) enzymes, leading to its ubiquitination and degradation. Conversely, under hypoxia or metabolic stress, HIF-α stabilizes, translocates into the nucleus, and regulates downstream target genes (eg, LDHA, VEGF, GLUT1), thereby promoting glycolysis, angiogenesis, and iron metabolism adaptation.111 The HIF signaling pathway is intricately linked to the ferroptosis process. HIF-1α and HIF-2α have been identified as promoters of ferroptosis, partly through downregulation of genes involved in lipid metabolism (Table 2).112 HTG-AP is characterized by severe dyslipidemia, increased lipid droplet synthesis, and excessive FFA accumulation. This milieu promotes iron accumulation (via upregulation of the transferrin receptor TFRC), exacerbates lipid peroxidation via the Fenton reaction, and stabilizes HIF-1α.48 HIF-2α contributes by directly upregulating the expression of the lipid droplet-associated protein HILPDA, enhancing the incorporation of PUFAs into triacylglycerols and PLs. HILPDA binds to and inhibits adipose triglyceride lipase, potentially stimulating the specific conversion of PUFAs into PLs.113 Furthermore, HIF-1α upregulates hepcidin and the TFRC, promoting intracellular iron accumulation and establishing a deleterious “iron overload-hypoxia” positive feedback loop.48 Notably, chelating intracellular free iron using the ferroptosis inhibitor deferoxamine (DFO) suppresses PHD activity and consequently inhibits HIF-1α activation.114 Under hypoxic conditions, HIF signaling alters the expression of genes governing iron and lipid metabolism, profoundly impacting lipid peroxidation and ferroptosis susceptibility.112 Specifically, HIF-1α suppresses the expression of ferroptosis-related genes GLUT1, SCD1, and FADS2, promoting the desaturation and conversion of LA to AA115 – a lipid molecule highly susceptible to peroxidation. Concurrently, HIF-1α upregulates lactate dehydrogenase A (LDHA), facilitating the conversion of pyruvate to lactate while inhibiting pyruvate entry into mitochondrial oxidative phosphorylation. This metabolic shift reduces ATP production and depletes NADPH, thereby weakening GPX4-mediated antioxidant defense.116 HIF-1α also induces the expression of fatty acid synthase and ACSL4, promoting the incorporation of PUFAs into membrane PLs and enhancing cellular sensitivity to ferroptosis.48 Moreover, HIF-1α directly binds to the NLRP3 promoter, promoting inflammasome assembly, caspase-1 activation, and subsequent IL-1β release, driving pyroptosis. This process synergizes with ferroptosis, as HIF-1α concurrently suppresses GPX4 expression.116 Targeting this axis, xanthine oxidase inhibitors like febuxostat indirectly modulate the HIF-1α/LDHA/NLRP3 pathway and effectively reduce the incidence of SAP.116 In summary, the HIF pathway, particularly HIF-1α, acts as a central integrator of hypoxic stress, iron dysmetabolism, and lipid peroxidation, positioning it as a critical hub for ferroptosis in HTG-AP.
 |
Table 2 Key Regulation Pathways Between HTG-AP and Ferroptosis |
PI3K/ Akt Pathway
The phosphoinositide 3-kinase (PI3K) family comprises lipid and protein kinases that play pivotal roles in intracellular signal transduction, critically regulating cell survival, growth, metabolism, and inflammation. Following its phosphorylation, Akt activates Nrf2, facilitating its nuclear translocation. This initiates the transcription of a suite of antioxidant genes, including GPX4, SLC7A11, and FTH1, thereby conferring cellular resistance to ferroptosis.122,123 Sustained activation of the PI3K/Akt/mTORC1 pathway upregulates stearoyl-CoA desaturase-1 (SCD1) expression via sterol regulatory element-binding protein 1 (SREBP1), a master transcriptional regulator of lipid metabolism (Table 2). SCD1 catalyzes the generation of MUFAs, altering membrane lipid composition by reducing the PUFA/MUFA ratio. This shift may potentially diminish cellular susceptibility to ferroptosis.117 Conversely, inhibition of the PI3K/Akt pathway can enhance cellular sensitivity to ferroptosis by augmenting iron-dependent lipid peroxidation. For instance, the unsaturated fatty acid ω-3 PUFA activates NF-κB by suppressing PI3K/Akt signaling, promoting the release of pro-inflammatory cytokines such as IL-1β and TNF-α.124,125 This effect is particularly pronounced in cancer cells characterized by hyperactive PI3K/Akt signaling.126 In contrast, normal cells, owing to their distinct basal metabolic state, are less prone to ferroptosis induction upon PI3K/Akt inhibition. Silencing Akt using small interfering RNA increases ROS production by disrupting metabolic flux and diminishing intracellular antioxidant levels,39,126 thereby heightening susceptibility to ferroptosis. This underscores a direct mechanistic link between PI3K/Akt signaling and ferroptotic regulation. Furthermore, comprehensive proteomic and functional analyses have revealed that inhibiting specific targets within the PI3K/Akt pathway, such as mTORC1, can promote GPX4 protein synthesis. This paradoxically increases tumor cell sensitivity to ferroptosis, and co-treatment with ferroptosis inducers synergistically enhances cell death in refractory cancers.127 This discovery lays the groundwork for novel therapeutic strategies targeting the PI3K/Akt axis to modulate ferroptosis in oncology. The PI3K/Akt pathway also exerts significant influence on lipid metabolism. Its activation mitigates oxidative stress and promotes fatty acid β-oxidation.128,129 In the context of AP, enhanced Akt signaling facilitates pancreatic tissue regeneration and attenuates inflammation,130 thereby potentially reducing the incidence of HTGP.
JAK-STAT Pathway
The JAK-STAT pathway is indispensable for cytokine signal transduction. Its dysregulation in HTG-AP can lead to excessive cytokine production, thereby triggering an inflammatory cascade.131 Persistent activation of JAK2/STAT3 inhibits the nuclear translocation of Nrf2, consequently downregulating the expression of ferroptosis suppressor proteins such as GPX4 and SLC7A11 (Table 2).131 FFAs promote JAK2 autophosphorylation, activating STAT3 for nuclear translocation, where it regulates the transcription of pro-inflammatory genes, culminating in a “cytokine storm”131 Saturated fatty acids (eg, palmitic acid) and ω-6 PUFAs (eg, linoleic acid) also induce JAK2/STAT3 phosphorylation by stimulating receptors on pancreatic acinar cell membranes. Studies demonstrate that treating pancreatic exocrine cells with linoleic acid significantly increases p-JAK2 and p-STAT3 expression, concomitant with elevated release of inflammatory cytokines IL-6 and TNF-α.132,133 STAT3 competitively binds DNA with Nrf2, leading to reduced GPX4 transcription134 and impairing the cell’s ability to clear lipid peroxides. In severe acute pancreatitis (SAP) rat models, JAK2/STAT3 activation positively correlates with the degree of lipid peroxidation in pancreatic tissue.133,135 Furthermore, the JAK2/STAT3 pathway directly regulates hepcidin expression: activated STAT3 binds the hepcidin promoter, enhancing its transcription. This results in decreased serum iron and increased intracellular iron accumulation, providing a source of free iron for ferroptosis.119–121 Inhibition of this pathway (eg, via AG490 treatment) reduces p-STAT3 levels and diminishes characteristic ferroptotic ultrastructural changes in acinar cells, such as plasma membrane rupture and mitochondrial cristae breakdown.136 This hyperactive inflammatory response damages not only pancreatic tissue but also leads to systemic complications. Activated STAT3 promotes TNF-α release from alveolar macrophages, increasing pulmonary vascular permeability137 and facilitating the deposition of lipid peroxidation end-products (eg, 4-HNE) in lung tissue.138 Excessive STAT3 activation also contributes to SAP-induced hepatocyte injury.139,140 PPAR-γ agonists attenuate pancreatitis-associated renal injury involving ferroptosis (eg, elevated levels of the lipid peroxidation marker MDA) by inhibiting HMGB1 and downregulating JAK2/STAT3 phosphorylation.141,142 Similarly, Picroside II and the herbal formulation Da-Huang-Fu-Zi-Tang ameliorate SAP-induced liver injury and exhibit anti-inflammatory and antioxidant properties by modulating JAK2/STAT3 phosphorylation signaling.140,143 Therefore, modulating JAK-STAT signaling through targeted inhibitors represents a potential therapeutic strategy to control the inflammatory response and improve disease outcomes.
Therapeutic Implications and Challenges for HTG-AP
HTG-AP represents a multifaceted pathological condition wherein elevated circulating TG levels trigger inflammatory cascades culminating in pancreatic injury. Current treatment strategies primarily focus on reducing TG levels and controlling inflammation to prevent further pancreatic damage and systemic complications. Initial therapeutic approaches typically include fasting to diminish pancreatic enzyme secretion and intravenous fluid administration to maintain hydration and electrolyte balance. These treatments are often combined with analgesics to manage pain; in severe cases, insulin therapy may be employed due to its lipid-lowering effects, which can enhance the breakdown of TG.144 Additionally, plasma exchange, which removes TG and inflammatory mediators from the bloodstream, is utilized for rapid TG level reduction. Further interventions include the use of lipid-lowering agents, such as fibrates or omega-3 fatty acids, which help reduce TG synthesis and promote fatty acid oxidation. These treatments are beneficial in maintaining lower TG levels over the long term and preventing the recurrence of pancreatitis.145
While these therapies have proven beneficial, they do not address the underlying mechanisms associated with ferroptosis—a regulated form of cell death involved in HTG-AP. Recent insights into the critical role of ferroptosis in pancreatitis provide a novel therapeutic avenue. For instance, lipid-lowering therapies may attenuate lipid peroxidative stress, potentially decreasing iron reactivity and subsequent cellular injury.146 In various organ injury models, ferroptosis inhibitors such as ferrostatin-1 and liproxstatin-1 have shown promising preclinical results in reducing lipid peroxidation and cell death.147 Administering ferroptosis inhibitors in mouse models alleviates pancreatic necrosis and markedly decreases systemic inflammatory markers.11,148 These promising preclinical data have stimulated growing interest in the clinical translation of ferroptosis-targeted therapies.
ACSL4 and FSP1 occupy opposite regulatory axes of ferroptosis, functioning as pro-ferroptotic and anti-ferroptotic hubs, respectively. They are not only key molecular determinants for understanding the pathological mechanisms of diseases, including HTG-AP, but also represent promising therapeutic targets. ACSL4 is a pivotal enzyme that activates PUFAs and incorporates them into membrane PLs.39 Upregulation of ACSL4 increases the abundance of peroxidation-prone PUFA substrates within membranes. Since ACSL4 activity dictates the cellular “loading capacity” for lipid peroxidation, its inhibition can fundamentally reduce the number of oxidizable phospholipid sites and thereby lower the likelihood of ferroptosis. In HTG-AP, elevated ACSL4/LPCAT3 expression has been reported,40 and treatment with the ferroptosis inhibitor Liproxstatin-1 attenuates pancreatic injury by suppressing ACSL4 expression and lipid peroxidation.90 In contrast, FSP1 exerts a ferroptosis-protective function through the FSP1–CoQ10–NAD(P)H axis, regenerating reduced coenzyme Q10 (CoQ10H2) at the plasma membrane and thereby scavenging radical intermediates to prevent lipid peroxidation.149 Acting independently of GPX4, FSP1 confers an additional layer of cellular resistance to ferroptosis. Together, these pathways represent complementary strategies—one reducing oxidizable substrates, the other enhancing membrane-associated antioxidant defense—and may serve as independent or synergistic intervention points for blocking ferroptosis.
From a therapeutic development perspective, several candidate ACSL4 inhibitors—including small-molecule and covalent inhibitors—have emerged, demonstrating the ability to reduce lipid peroxidation and ferroptotic cell death in vitro and in animal models. Recent studies (2024–2025) have identified multiple specific ACSL4 inhibitors, such as AS-series molecules and LIBX-A401, which effectively suppress lipid peroxidation and inhibit ferroptosis in cultured cells and mouse models.150,151 These compounds have exhibited tissue-protective effects in models of kidney injury, liver damage, fibrosis, and enhanced chemosensitivity in cancer models. In HTG-AP specifically, Liproxstatin-1–mediated ferroptosis blockade markedly improves histopathological damage and inflammatory responses.40 Conversely, enhancing FSP1 activity—or supplementing CoQ10 as an upstream metabolic cofactor—represents an additional promising strategy for mitigating ferroptosis.31,108 Notably, studies in AP have shown that activation of the IDH2–NADPH metabolic pathway can elevate CoQ10 levels, attenuate ferroptosis, and protect pancreatic acinar cells.108
However, to date, no ferroptosis-specific inhibitor has entered registered clinical trials for HTG-AP or other forms of AP. The safety and efficacy of these agents in humans remain untested, and additional preclinical validation and early-phase clinical development are therefore warranted.152,153 Before these therapies can be widely applied in clinical practice, several challenges must be addressed. Systemic administration may lead to unintended off-target effects: long-term inhibition of ACSL4 could disrupt lipid metabolic processes in other tissues—such as those involved in tumor suppression, neuronal function, or cardiomyocyte homeostasis154—while enhancing FSP1 activity may interfere with plasma membrane repair or other CoQ10-dependent cellular functions.31 Thus, one critical challenge is achieving selective targeting of pathological ferroptosis without interfering with the physiological processes in which ferroptosis plays a protective role, such as tissue modifications and neurogenesis-associated processes.147,155 Another challenge in advancing ferroptosis-targeted therapeutic strategies lies in the identification and validation of predictive biomarkers, such as MDA, 4-HNE, GPX4, CoQ10 and free iron,156 that could stratify patients according to their potential therapeutic responsiveness.
Therefore, the clinical translation of these strategies will likely require a gradual and structured approach—beginning with drug optimization and pancreas-targeted delivery, followed by rigorous preclinical evaluation of toxicity and metabolic/pharmacokinetic profiles in animal models, progressing to small-scale first-in-human studies focused on safety and pharmacokinetics, and ultimately advancing to efficacy trials with predefined clinical endpoints.
Based on emerging mechanistic insights, ferroptosis-targeted interventions may be incorporated into clinical management as an adjunct to standard therapy, with treatment pathways adapted according to patient risk stratification and biomarker monitoring. Specifically, ferroptosis-related biomarkers may be assessed within the first 24–48 hours after hospital admission in patients with HTG-AP156 to evaluate the degree of ferroptotic activation. For high-risk individuals—such as those with markedly elevated TG levels, early severe inflammation, or evolving organ dysfunction—additional therapy with ferroptosis inhibitors or coenzyme CoQ10 supplementation may be considered alongside standard lipid-lowering measures.157,158 Evidence from animal models and early clinical observations suggests that such combination approaches can simultaneously reduce membrane lipid peroxidation, limit necrotic progression, and attenuate inflammatory amplification.90 During the intermediate phase (48 hours to 7 days), ferroptosis-targeted therapy may be continued, particularly in patients with persistent inflammation, necrosis, or organ impairment. Adjunctive iron chelation (eg, DFO) may also be employed to decrease iron-dependent Fenton-driven ROS production and lipid peroxidation.159 However, iron chelators require careful monitoring due to potential adverse effects, including hypotension and neurotoxicity.160 In the later phase (≥7 days), especially when infection or complications arise, clinicians must balance the benefits of ferroptosis inhibition with its potential impact on host immunity and tissue repair.
Furthermore, an “HTG-AP–ferroptosis rapid-access pathway” should be established. A multidisciplinary team—including specialists in pancreatology, metabolism, hematology, and critical care—should evaluate candidates for ferroptosis-targeted therapy at the time of diagnosis36 and incorporate these considerations into clinical trial design. Priority should be given to small-scale Phase I/II randomized controlled trials using an add-on design to evaluate safety, biomarker modulation, and short-term clinical outcomes such as necrotic burden, inflammatory mediators, and organ function.152 In the long term, multicenter studies will be needed to determine the effects of ferroptosis-targeted interventions on recurrence, progression to severe disease, and mortality. Ultimately, ferroptosis monitoring may be incorporated into supplementary recommendations for HTG-AP clinical guidelines such as suggesting evaluation of ferroptosis-targeted therapy in high risk patients, to lay the foundation for future precision medicine strategies.
In summary, while the exploration of ferroptosis inhibitors expands the therapeutic landscape for HTG-AP, ongoing scientific investigations and rigorous clinical trials hold promise for establishing tailored therapeutic approaches that may revolutionize both the clinical treatment and prognostic outcomes of hypertriglyceridemia-induced AP.161 Furthermore, the application of ferroptosis-targeted therapies may potentially extend to other inflammation-related diseases.
Conclusion and Future Directions
Research on ferroptosis in HTG-AP has revealed the underlying mechanisms of this novel cell death pathway. Emerging scientific data increasingly demonstrates that ferroptotic cell death represents a fundamental mechanism in the progression of hypertriglyceridemia-induced pancreatic injury. Central to this process is the accumulation of lipid peroxides, caused by impaired antioxidant defenses and iron-catalyzed oxidative stress. These findings suggest a complex interplay between abnormal lipid metabolism and iron homeostasis, which promotes pancreatic cell death.162 Based on the body of research accumulated so far, targeting ferroptosis appears to be a viable therapeutic strategy for mitigating the inflammation and necrotic damage associated with HTG-AP. This review highlights the diverse roles of lipid peroxidation and its mediators in pancreatic injury, offering insights into cellular vulnerabilities during hyperlipidemic events.163
Subsequent investigations should prioritize elucidating the complex interplay between ferroptotic mechanisms and other controlling cascades that collectively modulate pancreatic pathophysiology. Developing multi-target therapies aimed at both hypertriglyceridemia and ferroptosis holds promise, especially with the use of cutting-edge molecular techniques such as CRISPR genetic modification to validate the roles of various genes involved in ferroptosis. Furthermore, the integration of advanced proteomic and metabolomic approaches holds promise for elucidating the complex metabolic alterations associated with ferroptotic cellular demise.
Data Sharing Statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
Author Contributions
KZ – Conceptualization, Writing–original draft, Visualization, Writing–review and editing; WZL – Conceptualization, Writing–original draft; WWX – Conceptualization, Writing–original draft; YZ - Conceptualization, Supervision, Writing-review and editing.
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
No funding was received.
Disclosure
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. De Pretis N, Amodio A, Frulloni L. Hypertriglyceridemic pancreatitis: epidemiology, pathophysiology and clinical management. United Eur Gastroenterol J. 2018;6(5):649–655. doi:10.1177/2050640618755002
2. Tsuang W, Navaneethan U, Ruiz L, et al. Hypertriglyceridemic pancreatitis: presentation and management. Am J Gastroenterol. 2009;104(4):984–991. doi:10.1038/ajg.2009.27
3. Kiss L, Fűr G, Pisipati S, et al. Mechanisms linking hypertriglyceridemia to acute pancreatitis. Acta Physiol. 2023;237(3):e13916. doi:10.1111/apha.13916
4. Fan Z, Zhang Y, Li J, et al. Global burden and characterization of hypertriglyceridemia-induced acute pancreatitis: results from a systematic review and a multi-center cohort study. Sci China Life Sci. 2025;68(10):3010–3020. doi:10.1007/s11427-024-2900-6
5. Lu J, Wang Z, W MEI, et al. A systematic review of the epidemiology and risk factors for severity and recurrence of hypertriglyceridemia-induced acute pancreatitis. BMC Gastroenterol. 2025;25(1):374. doi:10.1186/s12876-025-03954-4
6. Sacks FM, Stanesa M, Hegele RA. Severe hypertriglyceridemia with pancreatitis: thirteen years’ treatment with lomitapide. JAMA Intern Med. 2014;174(3):443–447. doi:10.1001/jamainternmed.2013.13309
7. Wu C, Ke L, Tong Z, et al. Hypertriglyceridemia is a risk factor for acute kidney injury in the early phase of acute pancreatitis. Pancreas. 2014;43(8):1312–1316. doi:10.1097/MPA.0000000000000180
8. Thong VD, Mong Trinh NT, Phat HT. Factors associated with the severity of hypertriglyceridemia induced acute pancreatitis. Medicine. 2021;100(21):e25983. doi:10.1097/MD.0000000000025983
9. Scherer J, Singh VP, Pitchumoni CS, et al. Issues in hypertriglyceridemic pancreatitis: an update. J Clin Gastroenterol. 2014;48(3):195–203. doi:10.1097/01.mcg.0000436438.60145.5a
10. Chaudhary A, Iqbal U, Anwar H, et al. Acute pancreatitis secondary to severe hypertriglyceridemia: management of severe hypertriglyceridemia in emergency setting. Gastroenterology Res. 2017;10(3):190–192. doi:10.14740/gr762e
11. Meng YT, Zhou Y, Han PY, et al. Ferroptosis inhibition attenuates inflammatory response in mice with acute hypertriglyceridemic pancreatitis. World J Gastroenterol. 2023;29(15):2294–2309. doi:10.3748/wjg.v29.i15.2294
12. Huang W, De La Iglesia-García D, Baston-Rey I, et al. Exocrine pancreatic insufficiency following acute pancreatitis: systematic review and meta-analysis. Dig Dis Sci. 2019;64(7):1985–2005. doi:10.1007/s10620-019-05568-9
13. Hollemans RA, Hallensleben NDL, Mager DJ, et al. Pancreatic exocrine insufficiency following acute pancreatitis: systematic review and study level meta-analysis. Pancreatology. 2018;18(3):253–262. doi:10.1016/j.pan.2018.02.009
14. Gu A, Li J, Qiu S, et al. Pancreatic cancer environment: from patient-derived models to single-cell omics. Mol Omics. 2024;20(4):220–233. doi:10.1039/D3MO00250K
15. Gurav S, Singh GP, Ostwal V, et al. Metastatic pancreatic cancer mimicking medication related osteonecrosis of the jaw—A rare clinical presentation. Medicine Advances. 2023;1(4):408–412. doi:10.1002/med4.42
16. Hegyi PJ, Soós A, Tóth E, et al. Evidence for diagnosis of early chronic pancreatitis after three episodes of acute pancreatitis: a cross-sectional multicentre international study with experimental animal model. Sci Rep. 2021;11(1):1367. doi:10.1038/s41598-020-80532-6
17. Zhi M, Zhu X, Lugea A, et al. Incidence of new onset diabetes mellitus secondary to acute pancreatitis: a systematic review and meta-analysis. Front Physiol. 2019;10(637). doi:10.3389/fphys.2019.00637
18. Hart PA, Bradley D, Conwell DL, et al. Diabetes following acute pancreatitis. Lancet Gastroenterol Hepatol. 2021;6(8):668–675. doi:10.1016/S2468-1253(21)00019-4
19. Gagyi EB, Teutsch B, Veres DS, et al. Incidence of recurrent and chronic pancreatitis after acute pancreatitis: a systematic review and meta-analysis. Therap Adv Gastroenterol. 2024;17(17562848241255303). doi:10.1177/17562848241255303
20. Trikudanathan G, Yazici C, Evans Phillips A, et al. Diagnosis and management of acute pancreatitis. Gastroenterology. 2024;167(4):673–688. doi:10.1053/j.gastro.2024.02.052
21. Saharia P, Margolis S, Zuidema GD, et al. Acute pancreatitis with hyperlipemia: studies with an isolated perfused canine pancreas. Surgery. 1977;82(1):60–67.
22. Carr RA, Rejowski BJ, Cote GA, et al. Systematic review of hypertriglyceridemia-induced acute pancreatitis: a more virulent etiology? Pancreatology. 2016;16(4):469–476. doi:10.1016/j.pan.2016.02.011
23. Sakorafas GH, Tsiotos GG, Sarr MG. Ischemia/reperfusion-induced pancreatitis. Dig Surg. 2000;17(1):3–14. doi:10.1159/000018793
24. Valdivielso P, Ramírez-Bueno A, Ewald N. Current knowledge of hypertriglyceridemic pancreatitis. Eur J Intern Med. 2014;25(8):689–694. doi:10.1016/j.ejim.2014.08.008
25. Yang AL, Mcnabb-Baltar J. Hypertriglyceridemia and acute pancreatitis. Pancreatology. 2020;20(5):795–800. doi:10.1016/j.pan.2020.06.005
26. Bosques-Padilla FJ, Vázquez-Elizondo G, González-Santiago O, et al. Hypertriglyceridemia-induced pancreatitis and risk of persistent systemic inflammatory response syndrome. Am J Med Sci. 2015;349(3):206–211. doi:10.1097/MAJ.0000000000000392
27. Hegele RA, Ginsberg HN, Chapman MJ, et al. The polygenic nature of hypertriglyceridaemia: implications for definition, diagnosis, and management. Lancet Diabetes Endocrinol. 2014;2(8):655–666. doi:10.1016/S2213-8587(13)70191-8
28. Stoyanovsky DA, Tyurina YY, Shrivastava I, et al. Iron catalysis of lipid peroxidation in ferroptosis: regulated enzymatic or random free radical reaction? Free Radic Biol Med. 2019;133:153–161. doi:10.1016/j.freeradbiomed.2018.09.008
29. Soula M, Weber RA, Zilka O, et al. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat Chem Biol. 2020;16(12):1351–1360. doi:10.1038/s41589-020-0613-y
30. Lei G, Mao C, Horbath AD, et al. BRCA1-mediated dual regulation of ferroptosis exposes a vulnerability to GPX4 and PARP co-inhibition in BRCA1-deficient cancers. Cancer Discov. 2024;14(8):1476–1495. doi:10.1158/2159-8290.CD-23-1220
31. Bersuker K, Hendricks JM, Li Z, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575(7784):688–692. doi:10.1038/s41586-019-1705-2
32. Doll S, Freitas FP, Shah R, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575(7784):693–698. doi:10.1038/s41586-019-1707-0
33. Kraft VAN, Bezjian CT, Pfeiffer S, et al. GTP cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci. 2020;6(1):41–53. doi:10.1021/acscentsci.9b01063
34. Mao C, Liu X, Zhang Y, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593(7860):586–590. doi:10.1038/s41586-021-03539-7
35. De Oliveira C, Khatua B, Noel P, et al. Pancreatic triglyceride lipase mediates lipotoxic systemic inflammation. J Clin Invest. 2020;130(4):1931–1947. doi:10.1172/JCI132767
36. Navina S, Acharya C, Delany JP, et al. Lipotoxicity causes multisystem organ failure and exacerbates acute pancreatitis in obesity. Sci Transl Med. 2011;3(107):107ra10. doi:10.1126/scitranslmed.3002573
37. Kepp O, Kroemer G. Pro-ferroptotic fatty acid metabolism renders cancer cells immunogenic. Trends Cancer. 2022;8(10):785–787. doi:10.1016/j.trecan.2022.04.002
38. Reed A, Ichu TA, Milosevich N, et al. LPCAT3 inhibitors remodel the polyunsaturated phospholipid content of human cells and protect from ferroptosis. ACS Chem Biol. 2022;17(6):1607–1618. doi:10.1021/acschembio.2c00317
39. Liang D, Minikes AM, Jiang X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol Cell. 2022;82(12):2215–2227. doi:10.1016/j.molcel.2022.03.022
40. Doll S, Proneth B, Tyurina YY, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91–98. doi:10.1038/nchembio.2239
41. Miranda ER, Shahtout JL, Funai K. Chicken or egg? Mitochondrial phospholipids and oxidative stress in disuse-induced skeletal muscle atrophy. Antioxid Redox Signal. 2023;38(4–6):338–351. doi:10.1089/ars.2022.0151
42. Nanadikar MS, Vergel Leon AM, Guo J, et al. IDH3γ functions as a redox switch regulating mitochondrial energy metabolism and contractility in the heart. Nat Commun. 2023;14(1):2123. doi:10.1038/s41467-023-37744-x
43. Egnatchik RA, Leamy AK, Noguchi Y, et al. Palmitate-induced activation of mitochondrial metabolism promotes oxidative stress and apoptosis in H4IIEC3 rat hepatocytes. Metabolism. 2014;63(2):283–295. doi:10.1016/j.metabol.2013.10.009
44. Bock FJ, Tait SWG. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol. 2020;21(2):85–100. doi:10.1038/s41580-019-0173-8
45. Ly LD, Xu S, Choi SK, et al. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp Mol Med. 2017;49(2):e291. doi:10.1038/emm.2016.157
46. Tusa NV, Abuelo A, Levy NA, et al. Peripheral biomarkers of oxidative stress in dogs with acute pancreatitis. J Vet Intern Med. 2022;36(6):1958–1965. doi:10.1111/jvim.16535
47. Cao D, Zheng J, Li Z, et al. ACSL4 inhibition prevents macrophage ferroptosis and alleviates fibrosis in bleomycin-induced systemic sclerosis model. Arthritis Res Ther. 2023;25(1):212. doi:10.1186/s13075-023-03190-9
48. Gu X, Huang Z, Ying X, et al. Ferroptosis exacerbates hyperlipidemic acute pancreatitis by enhancing lipid peroxidation and modulating the immune microenvironment. Cell Death Discov. 2024;10(1):242. doi:10.1038/s41420-024-02007-1
49. Wen Q, Liu J, Kang R, et al. The release and activity of HMGB1 in ferroptosis. Biochem Biophys Res Commun. 2019;510(2):278–283. doi:10.1016/j.bbrc.2019.01.090
50. Angelova PR, Choi ML, Berezhnov AV, et al. Alpha synuclein aggregation drives ferroptosis: an interplay of iron, calcium and lipid peroxidation. Cell Death Differ. 2020;27(10):2781–2796. doi:10.1038/s41418-020-0542-z
51. Wang X, Chen X, Cao L, et al. Mechanism of deoxynivalenol-induced neurotoxicity in weaned piglets is linked to lipid peroxidation, dampened neurotransmitter levels, and interference with calcium signaling. Ecotoxicol Environ Saf. 2020;194(110382):110382. doi:10.1016/j.ecoenv.2020.110382
52. Arumugam G, Padmanaban M, Krishnan D, et al. Influence of copper, iron, zinc and fe (3) (+) haemoglobin levels on the etiopathogenesis of chronic calcific pancreatitis--a study in patients with pancreatitis. Biol Trace Elem Res. 2011;142(3):424–434. doi:10.1007/s12011-010-8822-y
53. Silva-Vaz P, Rato L, Abrantes AMC, et al. Characterization of oxidative stress in acute pancreatitis. Pancreatology. 2019;19:S150. doi:10.1016/j.pan.2019.05.403
54. Esrefoglu M. Experimental and clinical evidence of antioxidant therapy in acute pancreatitis. World J Gastroenterol. 2012;18(39):5533–5541. doi:10.3748/wjg.v18.i39.5533
55. Tandon RK, Garg PK. Oxidative stress in chronic pancreatitis: pathophysiological relevance and management. Antioxid Redox Signal. 2011;15(10):2757–2766. doi:10.1089/ars.2011.4115
56. Patel K, Trivedi RN, Durgampudi C, et al. Lipolysis of visceral adipocyte triglyceride by pancreatic lipases converts mild acute pancreatitis to severe pancreatitis independent of necrosis and inflammation. Am J Pathol. 2015;185(3):808–819. doi:10.1016/j.ajpath.2014.11.019
57. Li H, Wu D, Zhang H, et al. Autophagy-mediated ferroptosis is involved in development of severe acute pancreatitis. BMC Gastroenterol. 2024;24(1):245. doi:10.1186/s12876-024-03345-1
58. Wrighting DM, Andrews NC. Interleukin-6 induces hepcidin expression through STAT3. Blood. 2006;108(9):3204–3209. doi:10.1182/blood-2006-06-027631
59. Lunova M, Schwarz P, Nuraldeen R, et al. Hepcidin knockout mice spontaneously develop chronic pancreatitis owing to cytoplasmic iron overload in acinar cells. J Pathol. 2017;241(1):104–114. doi:10.1002/path.4822
60. Hou W, Xie Y, Song X, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12(8):1425–1428. doi:10.1080/15548627.2016.1187366
61. Lee S, Hwang N, Seok BG, et al. Autophagy mediates an amplification loop during ferroptosis. Cell Death Dis. 2023;14(7):464. doi:10.1038/s41419-023-05978-8
62. Friedmann Angeli JP, Schneider M, Proneth B, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16(12):1180–1191. doi:10.1038/ncb3064
63. Ursini F, Maiorino M. Lipid peroxidation and ferroptosis: the role of GSH and GPx4. Free Radic Biol Med. 2020;152:175–185. doi:10.1016/j.freeradbiomed.2020.02.027
64. Huang JQ, Jiang YY, Ren FZ, et al. Novel role and mechanism of glutathione peroxidase-4 in nutritional pancreatic atrophy of chicks induced by dietary selenium deficiency. Redox Biol. 2022;57(102482). doi:10.1016/j.redox.2022.102482
65. Dai E, Han L, Liu J, et al. Ferroptotic damage promotes pancreatic tumorigenesis through a TMEM173/STING-dependent DNA sensor pathway. Nat Commun. 2020;11(1):6339. doi:10.1038/s41467-020-20154-8
66. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi:10.1016/j.cell.2012.03.042
67. Ge Y, Chen M, Li M, et al. Adipose-derived stem cells alleviate acute pancreatitis by inhibiting ferroptosis and oxidative damage in canines. Stem Cell Res Ther. 2025;16(1):355. doi:10.1186/s13287-025-04466-4
68. Yang WS, Stockwell BR. Ferroptosis: death by Lipid Peroxidation. Trends Cell Biol. 2016;26(3):165–176. doi:10.1016/j.tcb.2015.10.014
69. Conrad M, Friedmann Angeli JP. Glutathione peroxidase 4 (Gpx4) and ferroptosis: what’s so special about it? Mol Cell Oncol. 2015;2(3):e995047. doi:10.4161/23723556.2014.995047
70. Stockwell BR, Friedmann Angeli JP, Bayir H, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273–285. doi:10.1016/j.cell.2017.09.021
71. Bayir H, Anthonymuthu TS, Tyurina YY, et al. Achieving life through death: redox biology of lipid peroxidation in ferroptosis. Cell Chem Biol. 2020;27(4):387–408. doi:10.1016/j.chembiol.2020.03.014
72. Yang WS, Sriramaratnam R, Welsch ME, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1–2):317–331. doi:10.1016/j.cell.2013.12.010
73. Liu Z, Cheng S, Zheng X, et al. Paclitaxel attenuates atherosclerosis by suppressing macrophage ferroptosis and improving lipid metabolism via the Sirt1/Nrf2/GPX4 pathway. FASEB j. 2025;39(15):e70917. doi:10.1096/fj.202501047RR
74. Gan B. ACSL4, PUFA, and ferroptosis: new arsenal in anti-tumor immunity. Signal Transduct Target Ther. 2022;7(1):128. doi:10.1038/s41392-022-01004-z
75. Wen RJ, Dong X, Zhuang HW, et al. Baicalin induces ferroptosis in osteosarcomas through a novel Nrf2/xCT/GPX4 regulatory axis. Phytomedicine. 2023;116(154881):154881. doi:10.1016/j.phymed.2023.154881
76. Ye L, Wen X, Qin J, et al. Metabolism-regulated ferroptosis in cancer progression and therapy. Cell Death Dis. 2024;15(3):196. doi:10.1038/s41419-024-06584-y
77. Ściskalska M, Milnerowicz H. Association of genetic variants in the GPX1 and GPX4 genes with the activities of glutathione-dependent enzymes, their interaction with smoking and the risk of acute pancreatitis. Biomed Pharmacother. 2022;146(112591):112591. doi:10.1016/j.biopha.2021.112591
78. Sun M, Sun Q, Li T, et al. Silica nanoparticles induce liver lipid metabolism disorder via ACSL4-mediated ferroptosis. Environ Pollut. 2024;359(124590):124590. doi:10.1016/j.envpol.2024.124590
79. Yan YX, Zhou HM, Shang XG, et al. Dynamic regulators of ferroptosis: post-translational modifications in ferroptotic cell death. Pharmacol Res. 2025;217(107815):107815. doi:10.1016/j.phrs.2025.107815
80. Pan Z, Van Den Bossche JL, Rodriguez-Aznar E, et al. Pancreatic acinar cell fate relies on system x(C)(-) to prevent ferroptosis during stress. Cell Death Dis. 2023;14(8):536. doi:10.1038/s41419-023-06063-w
81. Kou Z, Tran F, Colon T, et al. AhR signaling modulates ferroptosis by regulating SLC7A11 expression. Toxicol Appl Pharmacol. 2024;486(116936):116936. doi:10.1016/j.taap.2024.116936
82. Koppula P, Lei G, Zhang Y, et al. A targetable CoQ-FSP1 axis drives ferroptosis- and radiation-resistance in KEAP1 inactive lung cancers. Nat Commun. 2022;13(1):2206. doi:10.1038/s41467-022-29905-1
83. Li W, Liang L, Liu S, et al. FSP1: a key regulator of ferroptosis. Trends Mol Med. 2023;29(9):753–764. doi:10.1016/j.molmed.2023.05.013
84. Hu Y, Zou W, Zhang L, et al. TRPV3 facilitates lipolysis and attenuates diet-induced obesity via activation of the NRF2/FSP1 signaling axis. Free Radic Biol Med. 2024;221:155–168. doi:10.1016/j.freeradbiomed.2024.05.035
85. Zhang HL, Hu X, Li ZL, et al. PKCβII phosphorylates ACSL4 to amplify lipid peroxidation to induce ferroptosis. Nat Cell Biol. 2022;24(1):88–98. doi:10.1038/s41556-021-00818-3
86. Stockwell BR. Ferroptosis turns 10: emerging mechanisms, physiological functions, and therapeutic applications. Cell. 2022;185(14):2401–2421. doi:10.1016/j.cell.2022.06.003
87. Li D, Li Y. The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct Target Ther. 2020;5(1):108. doi:10.1038/s41392-020-00216-5
88. Merkel M, Goebel B, BOLL M, et al. Mitochondrial reactive oxygen species formation determines ACSL4/LPCAT2-mediated ferroptosis. Antioxidants. 2023;12(8):1590. doi:10.3390/antiox12081590
89. Ding K, Liu C, Li L, et al. Acyl-CoA synthase ACSL4: an essential target in ferroptosis and fatty acid metabolism. Chin Med J. 2023;136(21):2521–2537. doi:10.1097/CM9.0000000000002533
90. Xiang X, Xu M, Liu L, et al. Liproxstatin-1 attenuates acute hypertriglyceridemic pancreatitis through inhibiting ferroptosis in rats. Sci Rep. 2024;14(1):9548. doi:10.1038/s41598-024-60159-7
91. Fan Y, Ma L, Fang X, et al. Role of hypoxia-inducible-factor-1α (HIF-1α) in ferroptosis of adipose tissue during ketosis. J Dairy Sci. 2024;107(12):10611–10627. doi:10.3168/jds.2024-24822
92. Gao D, Wu Y, Zhan Y, et al. Chronic hypoxia drives the occurrence of ferroptosis in liver of fat greening (Hexagrammos otakii) by activating HIF-1α and promoting iron production. Ecotoxicol Environ Saf. 2024;285(117135):117135. doi:10.1016/j.ecoenv.2024.117135
93. Zhang T, Huang X, Feng S, et al. Lactate-dependent HIF1A transcriptional activation exacerbates severe acute pancreatitis through the ACSL4/LPCAT3/ALOX15 pathway induced ferroptosis. J Cell Biochem. 2025;126(1):e30687. doi:10.1002/jcb.30687
94. LI Z, Yao YX, Lu X, et al. Short-term respiratory cadmium exposure partially activates pulmonary NLRP3 inflammasome by inducing ferroptosis in mice. Ecotoxicol Environ Saf. 2024;285(117106):117106. doi:10.1016/j.ecoenv.2024.117106
95. Wang J, Li R, Peng Z, et al. HMGB1 participates in LPS‑induced acute lung injury by activating the AIM2 inflammasome in macrophages and inducing polarization of M1 macrophages via TLR2, TLR4, and RAGE/NF‑κB signaling pathways. Int J Mol Med. 2020;45(1):61–80. doi:10.3892/ijmm.2019.4402
96. Lei G, Horbath A, Li Z, et al. PKCβII-ACSL4 pathway mediating ferroptosis execution and anti-tumor immunity. Cancer Commun. 2022;42(7):583–586. doi:10.1002/cac2.12319
97. Mortensen MS, Ruiz J, Watts JL. Polyunsaturated fatty acids drive lipid peroxidation during ferroptosis. Cells. 2023;12(5):804. doi:10.3390/cells12050804
98. Yang Y, Zhu T, Wang X, et al. ACSL3 and ACSL4, distinct roles in ferroptosis and cancers. Cancers. 2022;14(23):5896. doi:10.3390/cancers14235896
99. Dixon SJ, Olzmann JA. The cell biology of ferroptosis. Nat Rev Mol Cell Biol. 2024;25(6):424–442. doi:10.1038/s41580-024-00703-5
100. Zheng J, Conrad M. The Metabolic Underpinnings of Ferroptosis. Cell Metab. 2020;32(6):920–937. doi:10.1016/j.cmet.2020.10.011
101. Huang W, Song C, Hua Y, et al. NNMT is involved in deoxynivalenol-induced hepatocyte toxicity via promoting ferroptosis. Toxicology. 2025;513(154084):154084. doi:10.1016/j.tox.2025.154084
102. Anderson T. Communicating science-based messages on vaccines. Bull World Health Organ. 2017;95(10):670–671. doi:10.2471/BLT.17.021017
103. Mishima E, Ito J, Wu Z, et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature. 2022;608(7924):778–783. doi:10.1038/s41586-022-05022-3
104. Dai E, Meng L, Kang R, et al. ESCRT-III-dependent membrane repair blocks ferroptosis. Biochem Biophys Res Commun. 2020;522(2):415–421. doi:10.1016/j.bbrc.2019.11.110
105. Lv Y, Liang C, Sun Q, et al. Structural insights into FSP1 catalysis and ferroptosis inhibition. Nat Commun. 2023;14(1):5933. doi:10.1038/s41467-023-41626-7
106. Müller F, Lim JKM, Bebber CM, et al. Elevated FSP1 protects KRAS-mutated cells from ferroptosis during tumor initiation. Cell Death Differ. 2023;30(2):442–456. doi:10.1038/s41418-022-01096-8
107. Lin G, Liu Q, Xie C, et al. Upregulated FSP1 by GPD1/1L mediated lipid droplet accumulation enhances ferroptosis resistance and peritoneal metastasis in gastric cancer. Cell Commun Signal. 2025;23(1):132. doi:10.1186/s12964-025-02126-x
108. Peng Q, Li B, Song P, et al. IDH2-NADPH pathway protects against acute pancreatitis via suppressing acinar cell ferroptosis. Br J Pharmacol. 2024;181(20):4067–4084. doi:10.1111/bph.16469
109. Zhang S, Gou S, Zhang Q, et al. FSP1 oxidizes NADPH to suppress ferroptosis. Cell Res. 2023;33(12):967–970. doi:10.1038/s41422-023-00879-z
110. Venkatesh D, O’brien NA, Zandkarimi F, et al. MDM2 and MDMX promote ferroptosis by PPARα-mediated lipid remodeling. Genes Dev. 2020;34(7–8):526–543. doi:10.1101/gad.334219.119
111. Liu Y, Cui H, Mei C, et al. Sirtuin4 alleviates severe acute pancreatitis by regulating HIF-1α/HO-1 mediated ferroptosis. Cell Death Dis. 2023;14(10):694. doi:10.1038/s41419-023-06216-x
112. Miess H, Dankworth B, Gouw AM, et al. The glutathione redox system is essential to prevent ferroptosis caused by impaired lipid metabolism in clear cell renal cell carcinoma. Oncogene. 2018;37(40):5435–5450. doi:10.1038/s41388-018-0315-z
113. Zou Y, Palte MJ, Deik AA, et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun. 2019;10(1):1617. doi:10.1038/s41467-019-09277-9
114. Wu Y, Zhang S, Gong X, et al. The epigenetic regulators and metabolic changes in ferroptosis-associated cancer progression. Mol Cancer. 2020;19(1):39. doi:10.1186/s12943-020-01157-x
115. Jiang Y, Mao C, Yang R, et al. EGLN1/c-Myc induced lymphoid-specific helicase inhibits ferroptosis through lipid metabolic gene expression changes. Theranostics. 2017;7(13):3293–3305. doi:10.7150/thno.19988
116. Rong J, Han C, Huang Y, et al. Inhibition of xanthine oxidase alleviated pancreatic necrosis via HIF-1α-regulated LDHA and NLRP3 signaling pathway in acute pancreatitis. Acta Pharm Sin B. 2024;14(8):3591–3604. doi:10.1016/j.apsb.2024.04.019
117. Yi J, Zhu J, Wu J, et al. Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proc Natl Acad Sci U S A. 2020;117(49):31189–31197. doi:10.1073/pnas.2017152117
118. Yu Y, Xie X, Zhang Y, et al. Salvigenin mitigates neuronal ferroptosis by binding to PI3K and enhancing the interaction between VCP and PI3K in the repair of spinal cord injury. Phytomedicine. 2025;147(157181):157181. doi:10.1016/j.phymed.2025.157181
119. Yang L, Wang H, Yang X, et al. Auranofin mitigates systemic iron overload and induces ferroptosis via distinct mechanisms. Signal Transduct Target Ther. 2020;5(1):138. doi:10.1038/s41392-020-00253-0
120. Zhang F, Zhao P, Qian Z, et al. Central nervous system inflammation induced by lipopolysaccharide up-regulates hepatic hepcidin expression by activating the IL-6/JAK2/STAT3 pathway in mice. Front Nutr. 2021;8:649640.
121. Zheng J, Wang JJ, Ma HM, et al. Norcantharidin down-regulates iron contents in the liver and spleen of lipopolysaccharide-treated mice. Redox Rep. 2022;27(1):119–127. doi:10.1080/13510002.2022.2088011
122. Sarker RS, Steiger K. A critical role for Akt1 signaling in acute pancreatitis progression(†). J Pathol. 2020;251(1):1–3. doi:10.1002/path.5391
123. Deng X, Yu T, Gao M, et al. Sodium selenite (Na(2)SeO(3)) attenuates T-2 toxin-induced iron death in LMH cells through the ROS/PI3K/AKT/Nrf2 pathway. Food Chem Toxicol. 2023;182(114185):114185. doi:10.1016/j.fct.2023.114185
124. Liu B, Zhang Y, Yang Z, et al. ω-3 DPA protected neurons from neuroinflammation by balancing microglia M1/M2 polarizations through inhibiting NF-κB/MAPK p38 signaling and activating neuron-BDNF-PI3K/AKT pathways. Mar Drugs. 2021;19(11):587. doi:10.3390/md19110587
125. Lupia E, Pigozzi L, Goffi A, et al. Role of phosphoinositide 3-kinase in the pathogenesis of acute pancreatitis. World J Gastroenterol. 2014;20(41):15190–15199. doi:10.3748/wjg.v20.i41.15190
126. Yang L, Chen L, Chen T, et al. The crosstalk between classic cell signaling pathways, non-coding RNAs and ferroptosis in drug resistance of tumors. Cell Signal. 2023;102(110538):110538. doi:10.1016/j.cellsig.2022.110538
127. Zhang Y, Swanda RV, Nie L, et al. mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat Commun. 2021;12(1):1589. doi:10.1038/s41467-021-21841-w
128. Fan H, Ma X, Lin P, et al. Scutellarin prevents Nonalcoholic Fatty Liver Disease (NAFLD) and hyperlipidemia via PI3K/AKT-dependent activation of nuclear Factor (Erythroid-Derived 2)-like 2 (Nrf2) in rats. Med Sci Monit. 2017;23:5599–5612. doi:10.12659/MSM.907530
129. Zhou YJ, Xu N, Zhang XC, et al. Chrysin improves glucose and lipid metabolism disorders by regulating the AMPK/PI3K/AKT signaling pathway in insulin-resistant HepG2 Cells and HFD/STZ-induced C57BL/6J mice. J Agric Food Chem. 2021;69(20):5618–5627. doi:10.1021/acs.jafc.1c01109
130. Wu J, Zhang L, Shi J, et al. Macrophage phenotypic switch orchestrates the inflammation and repair/regeneration following acute pancreatitis injury. EBioMedicine. 2020;58(102920):102920. doi:10.1016/j.ebiom.2020.102920
131. Lytrivi M, Castell AL, Poitout V, et al. Recent insights into mechanisms of β-cell lipo- and glucolipotoxicity in type 2 diabetes. J Mol Biol. 2020;432(5):1514–1534. doi:10.1016/j.jmb.2019.09.016
132. Yang J, Han F, Wu G, et al. Dysregulated B7H4/JAK2/STAT3 pathway involves in hypertriglyceridemia acute pancreatitis and is attenuated by baicalin. Dig Dis Sci. 2023;68(2):478–486. doi:10.1007/s10620-022-07606-5
133. Yang X, Geng H, You L, et al. Rhein protects against severe acute pancreatitis in vitro and in vivo by regulating the JAK2/STAT3 pathway. Front Pharmacol. 2022;13:778221.
134. Xu Z, Li J, Zhou K, et al. Exocarpium Citri Grandis ameliorates LPS-induced acute lung injury by suppressing inflammation, NLRP3 inflammasome, and ferroptosis. J Ethnopharmacol. 2024;329(118162):118162. doi:10.1016/j.jep.2024.118162
135. Xia T, Gu Y, Shen J, et al. Limonin ameliorates acute pancreatitis by suppressing JAK2 / STAT3 signaling pathway. Environ Toxicol: Int J. 2021;36(12):2392–2403. doi:10.1002/tox.23352
136. Wang G, Zhang J, Dui D, et al. High mobility group box 1 induces the activation of the Janus kinase 2 and signal transducer and activator of transcription 3 (JAK2/STAT3) signaling pathway in pancreatic acinar cells in rats, while AG490 and rapamycin inhibit their activation. Bosn J Basic Med Sci. 2016;16(4):307–312. doi:10.17305/bjbms.2016.1442
137. Yang S, Song Y, Wang Q, et al. Daphnetin ameliorates acute lung injury in mice with severe acute pancreatitis by inhibiting the JAK2-STAT3 pathway. Sci Rep. 2021;11(1):11491. doi:10.1038/s41598-021-91008-6
138. Wang L, Yuan N, Li Y, et al. Stellate ganglion block relieves acute lung injury induced by severe acute pancreatitis via the miR-155-5p/SOCS5/JAK2/STAT3 axis. Eur J Med Res. 2022;27(1):231. doi:10.1186/s40001-022-00860-3
139. Feng Y, Jin X, Xu H, et al. Pancreatic elastase affects liver injury by activating pro-inflammatory cytokines in kupffer cells via the JAK2/STAT3 signaling pathway. Curr Mol Med. 2025;25. doi: 10.2174/0115665240363959250624052201
140. Piao X, Sui X, Liu B, et al. Picroside II improves severe acute pancreatitis-induced hepatocellular injury in rats by affecting JAK2/STAT3 phosphorylation signaling. Biomed Res Int. 2021;2021(9945149). doi:10.1155/2021/9945149
141. Yu JH, Kim KH, Kim H. SOCS 3 and PPAR-gamma ligands inhibit the expression of IL-6 and TGF-beta1 by regulating JAK2/STAT3 signaling in pancreas. Int J Biochem Cell Biol. 2008;40(4):677–688. doi:10.1016/j.biocel.2007.10.007
142. Huang F, Wang Y, Lv X, et al. WTAP-mediated N6-methyladenosine modification promotes the inflammation, mitochondrial damage and ferroptosis of kidney tubular epithelial cells in acute kidney injury by regulating LMNB1 expression and activating NF-κB and JAK2/STAT3 pathways. J Bioenerg Biomembr. 2024;56(3):285–296. doi:10.1007/s10863-024-10015-0
143. Wu L, Li H, Zheng SZ, et al. Da-Huang-Fu-Zi-Tang attenuates liver injury in rats with severe acute pancreatitis. J Ethnopharmacol. 2013;150(3):960–966. doi:10.1016/j.jep.2013.09.051
144. Inayat F, Zafar F, Riaz I, et al. Hypertriglyceridemic pancreatitis: is insulin monotherapy a feasible therapeutic option? Cureus. 2018;10(10):e3461. doi:10.7759/cureus.3461
145. García Díaz JD. Novel therapeutics in hypertriglyceridaemia and chylomicronaemia. Med Clin. 2020;154(8):308–314. doi:10.1016/j.medcli.2019.11.003
146. Zhirong Z, Li H, Yi L, et al. Ferroptosis in pancreatic diseases: potential opportunities and challenges that require attention. Hum Cell. 2023;36(4):1233–1243. doi:10.1007/s13577-023-00894-7
147. Zou Y, Schreiber SL. Progress in understanding ferroptosis and challenges in its targeting for therapeutic benefit. Cell Chem Biol. 2020;27(4):463–471. doi:10.1016/j.chembiol.2020.03.015
148. Luo M, Yan J, Hu X, et al. Targeting lipid metabolism for ferroptotic cancer therapy. Apoptosis. 2023;28(1–2):81–107. doi:10.1007/s10495-022-01795-0
149. Dai E, Zhang W, Cong D, et al. AIFM2 blocks ferroptosis independent of ubiquinol metabolism. Biochem Biophys Res Commun. 2020;523(4):966–971. doi:10.1016/j.bbrc.2020.01.066
150. Huang Q, Ru Y, Luo Y, et al. Identification of a targeted ACSL4 inhibitor to treat ferroptosis-related diseases. Sci Adv. 2024;10(13):eadk1200. doi:10.1126/sciadv.adk1200
151. Mazhari Dorooee D, Ravez S, Vertommen D, et al. LIBX-A401: a novel selective inhibitor of Acyl-CoA Synthetase long chain family member 4 (ACSL4) and its binding mode. Angew Chem Int Ed Engl. 2025;64(19):e202500518. doi:10.1002/anie.202500518
152. Maremonti F, Tonnus W, Gavali S, et al. Ferroptosis-based advanced therapies as treatment approaches for metabolic and cardiovascular diseases. Cell Death Differ. 2024;31(9):1104–1112. doi:10.1038/s41418-024-01350-1
153. Scarpellini C, Klejborowska G, Lanthier C, et al. Beyond ferrostatin-1: a comprehensive review of ferroptosis inhibitors. Trends Pharmacol Sci. 2023;44(12):902–916. doi:10.1016/j.tips.2023.08.012
154. Wu M, Zhao Y, Li D, et al. GSTP1-mediated inhibition of ACSL4-dependent ferroptosis via JNK pathway in DOX-induced cardiomyopathy. Chin Med J. 2025;138(19):2498–2510. doi:10.1097/CM9.0000000000003758
155. Chen X, Kang R, Kroemer G, et al. Targeting ferroptosis in pancreatic cancer: a double-edged sword. Trends Cancer. 2021;7(10):891–901. doi:10.1016/j.trecan.2021.04.005
156. Chen X, Comish PB, Tang D, et al. Characteristics and biomarkers of ferroptosis. Front Cell Dev Biol. 2021;9:637162.
157. Shin JY, Choi JW, Kim DG, et al. Protective effects of Coenzyme Q10 against acute pancreatitis. Int Immunopharmacol. 2020;88(106900):106900. doi:10.1016/j.intimp.2020.106900
158. Hao X-T, Peng R, Guan M, et al. The food and medicinal homological resources benefiting patients with hyperlipidemia: categories, functional components, and mechanisms. Food Med Homol. 2024;1(2):9420003. doi:10.26599/FMH.2024.9420003
159. Liang Y, Wang Y, Sun C, et al. Deferoxamine reduces endothelial ferroptosis and protects cerebrovascular function after experimental traumatic brain injury. Brain Res Bull. 2024;207(110878):110878. doi:10.1016/j.brainresbull.2024.110878
160. Farr AC, Xiong MP. Challenges and opportunities of deferoxamine delivery for treatment of alzheimer’s disease, parkinson’s disease, and intracerebral hemorrhage. Mol Pharm. 2021;18(2):593–609. doi:10.1021/acs.molpharmaceut.0c00474
161. Yang Y, Zhang ZJ, Wen Y, et al. Novel perspective in pancreatic cancer therapy: targeting ferroptosis pathway. World J Gastrointest Oncol. 2021;13(11):1668–1679. doi:10.4251/wjgo.v13.i11.1668
162. Hung W, Dolmans D, van Merriënboer JJG. A review to identify key perspectives in PBL meta-analyses and reviews: trends, gaps and future research directions. Adv Health Sci Educ Theory Pract. 2019;24(5):943–957. doi:10.1007/s10459-019-09945-x
163. Kimberley TJ, Novak I, Boyd L, et al. Stepping up to rethink the future of rehabilitation: IV STEP considerations and inspirations. J Neurol Phys Ther. 2017;41(Suppl 3):S63–s72. doi:10.1097/NPT.0000000000000182
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Shikonin Could Be Used to Treat Tubal Pregnancy via Enhancing Ferroptosis Sensitivity
Lai Y, Zeng F, Chen Z, Feng M, Huang Y, Qiu P, Zeng L, Ke Y, Deng G, Gao J
Drug Design, Development and Therapy 2022, 16:2083-2099
Published Date: 1 July 2022
The Emerging Roles of Ferroptosis in Neonatal Diseases
Chen W, Zheng D, Yang C
Journal of Inflammation Research 2023, 16:2661-2674
Published Date: 26 June 2023
The Emerging Role of Ferroptosis in Sepsis, Opportunity or Challenge?
Huang Q, Ding Y, Fang C, Wang H, Kong L
Infection and Drug Resistance 2023, 16:5551-5562
Published Date: 23 August 2023
The Chains of Ferroptosis Interact in the Whole Progression of Atherosclerosis
Wan X, Zhang H, Tian J, Hao P, Liu L, Zhou Y, Zhang J, Song X, Ge C
Journal of Inflammation Research 2023, 16:4575-4592
Published Date: 16 October 2023
Targeting Ferroptosis in Bone-Related Diseases: Facts and Perspectives
Chen H, Han Z, Wang Y, Su J, Lin Y, Cheng X, Liu W, He J, Fan Y, Chen L, Zuo H
Journal of Inflammation Research 2023, 16:4661-4677
Published Date: 18 October 2023