Back to Journals » Journal of Inflammation Research » Volume 19
Circulating and Respiratory Biomarkers in Sepsis-Induced ARDS: Diagnostic and Prognostic Insights – A Narrative Review
Authors Muhoza BG, Liu S, Niyonkuru E ![]() , Sun T
, Sun T
Received 24 October 2025
Accepted for publication 5 February 2026
Published 25 March 2026 Volume 2026:19 571504
DOI https://doi.org/10.2147/JIR.S571504
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Xin Du
Bertrand-Geoffrey Muhoza,1,2 Shaohua Liu,1,2 Emery Niyonkuru,3 Tongwen Sun1,2
1General Intensive Care Unit, Department of Emergency Medicine, The First Affiliated Hospital of Zhengzhou University, Henan Engineering Research Center for Critical Care Medicine, Zhengzhou, 450052, People’s Republic of China; 2Henan Key Laboratory of Critical Care Medicine, Henan Key Laboratory of Sepsis in Health Commission, Zhengzhou Key Laboratory of Sepsis, Henan Sepsis Diagnosis and Treatment Center, Zhengzhou, 450052, People’s Republic of China; 3Department of Anesthesiology, Pain and Perioperative Medicine, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, 450052, People’s Republic of China
Correspondence: Tongwen Sun, Email [email protected]
Abstract: Sepsis affects around 50 million people annually and is a significant contributor to acute respiratory distress syndrome (ARDS), leading to high morbidity and mortality. Sepsis-induced ARDS is the leading cause of ARDS globally and has worse outcomes compared to other causes, with mortality rates up to 40% in severe instances. Geographic variability in ARDS prevalence is notable, with rates of 27% among septic patients in China, 6– 7% in Western countries, and up to 31% in sub-Saharan African Intensive Care Units, highlighting significant disparities in disease burden and outcomes. Management of sepsis-induced ARDS generally necessitates mechanical ventilation, yet extended ventilatory support can lead to negative outcomes. Weaning from ventilation is complicated by the inflammatory response and multiorgan dysfunction seen in sepsis. Traditional weaning predictors, like the rapid shallow breathing index, show inadequate sensitivity and specificity, indicating a requirement for more effective predictive tools. Recent studies have identified several biomarkers, including Pancreatic Stone Protein, soluble receptor for advanced glycation end-products, and soluble urokinase plasminogen activator receptor, as promising tools for enhancing the prediction of mechanical ventilation outcomes in sepsis-induced ARDS. Despite the identification of multiple biomarkers, a major clinical gap remains: there is currently no consensus on their routine use to guide weaning, largely due to inconsistent findings, heterogeneity in study designs, and limited large-scale validation. This review explores the role of circulating and respiratory biomarkers in improving outcomes for patients with sepsis-induced ARDS, particularly in predicting successful mechanical ventilation weaning. It appraises the evidence surrounding these biomarkers against traditional weaning indices and identifies gaps in existing research. The review emphasizes the strengths and limitations of current studies, suggesting that validated biomarker-guided strategies could significantly enhance clinical management by reducing ventilation duration, preventing extubation failures, and improving survival rates in this vulnerable patient group.
Keywords: sepsis, ARDS, biomarker, mechanical ventilation, weaning
A Letter to the Editor has been published for this article.
Introduction
Sepsis is a serious illness that affects approximately 50 million people worldwide. It is defined by sudden organ failure caused by an inappropriate immune response to infection,1,2 with fatality rates ranging from 12.5% to 31.8%.3,4 The majority of sepsis cases are identified in hospitalized patients upon admission, particularly in emergency departments.5 This condition can lead to significant organ impairment, especially acute renal (kidney) and pulmonary (lung) damage, frequently exacerbating Acute Respiratory Distress Syndrome (ARDS). Sepsis is the leading cause of ARDS, accounting for about 32% of cases, and it is associated with more severe outcomes compared to ARDS caused by other factors.6,7
According to a study conducted in China from January 2017 to May 2019, ARDS developed in 27% of 150 sepsis patients.8 In contrast, in Western nations, the prevalence of ARDS among adult sepsis patients is approximately 6% to 7%, with a specific rate of 6.8% reported in Korea. Patients with sepsis often experience a rapid progression to ARDS, significantly increasing their risk of in-hospital mortality.9 A study investigating national incidence rates and cause-specific variables for ARDS in the United States from 2006 to 2014 analyzed nearly 69 million hospital discharges, identifying 1,151,969 ARDS cases, of which 969,567 had an associated risk factor.10
The most common associations with ARDS were sepsis (46.7%), pneumonia (44.9%), and shock (44.4%), along with less common factors such as pancreatitis (3.4%), pulmonary contusion (1.4%), and drowning (0.2%). ARDS linked to shock and sepsis exhibited higher fatality rates compared to other etiologies. The death rate for patients with severe ARDS, defined as a ratio of arterial oxygen partial pressure to fractional inspired oxygen (PaO2/FIO2) of less than 100, was 40%, with a frequency of 10% in critical care units, affecting approximately 1 in 4 patients on mechanical ventilation (MV). Recent global research indicates that sepsis accounts for over 75% of ARDS cases, with 59% attributed to pneumonia and 16% to extrapulmonary sepsis. Each year in the United States, sepsis-induced ARDS results in around 210,000 cases, which have a higher fatality rate than other forms of ARDS.11
A comprehensive evaluation of 14 observational studies conducted from 2009 to 2022 in eight sub-Saharan African nations examined the prevalence and mortality rates of sepsis. In nine comprehensive hospital investigations, the aggregated prevalence was 17%, accompanied by a fatality rate of 15%.12 The aggregated frequency in five intensive care unit (ICU) based investigations was 31%, with a notably elevated death rate of 46%. Subgroup studies revealed a hospital-wide frequency of 18% in East Africa and 20% in Southern Africa, while the prevalence in ICUs was lower, at 14% for East Africa and 13% for Southern Africa.12 These data underscore the significant burden of sepsis and the associated elevated death rates across sub-Saharan Africa.
ARDS is a complex condition that is not primarily caused by heart failure or fluid overload. It is characterized by rapid hypoxemic respiratory failure with bilateral infiltrates and often necessitates mechanical ventilation assistance.13–20 Although prolonged mechanical ventilation can be life-saving, it is associated with considerable complications, including ventilator-associated pneumonia (VAP), barotrauma, and diaphragm dysfunction, leading to extended ICU stays.21,22 In patients with sepsis-induced ARDS, the timing and efficacy of discontinuing mechanical ventilation are particularly challenging due to the multifaceted pathophysiology, which encompasses systemic inflammation, multiorgan failure, and diverse patterns of pulmonary recovery.23
Traditional clinical weaning predictions, such as the Rapid Shallow Breathing Index (RSBI), oxygenation metrics, and spontaneous breathing trials (SBT), often exhibit limited sensitivity and specificity, particularly among the diverse cohort of septic patients.24 Recent research has indicated that while the RSBI is a valuable tool for informing weaning choices, exclusive reliance on it may mislead clinicians.25 The RSBI demonstrates modest sensitivity and inadequate specificity as an independent predictor of successful extubation.24 Recently, several circulating and respiratory biomarkers have been investigated in relation to mechanical ventilation outcomes in sepsis-induced ARDS.26–29 Biomarkers, defined as quantifiable biological substances indicative of physiological or pathological processes, have emerged as valuable tools to enhance clinical decision-making.30–32 These biomarkers may provide real-time insights into the severity of lung damage, infection management, systemic inflammation, and organ failure,33,34 including inflammatory cytokines35,36 and indicators of epithelial and endothelial damage.13,37–43
Despite the identification of multiple biomarkers, a significant clinical gap remains: there is currently no consensus on their routine use to guide weaning, largely due to inconsistent findings, heterogeneity in study designs, and limited large-scale validation. This review explores how circulating and respiratory biomarkers may improve outcomes in patients with sepsis-induced ARDS, particularly in predicting successful liberation from mechanical ventilation. Importantly, it critically appraises the existing evidence to evaluate the value of these biomarkers in comparison to traditional weaning indices and to identify key gaps in current research. By highlighting the strengths and limitations of current studies, this review underscores the potential impact of validated biomarker-guided strategies, which could transform clinical management by reducing ventilation days, preventing extubation failures, and improving survival in this high-risk population.
Core Concepts of Pathophysiology
Sepsis is the primary factor that causes ARDS and can initiate its onset within a week, leading to a dire prognosis for affected patients.44 ARDS is a severe condition with a fatality rate of approximately 40%, and septic ARDS demonstrates a poorer prognosis and higher than average death rates.44 The mechanisms underlying sepsis-induced ARDS are complex and multifaceted, involving various injurious elements such as bacterial and viral infections, trauma, and peritonitis. These factors stimulate immune cells and generate inflammatory mediators, which damage the lung endothelium and exacerbate inflammation, ultimately leading to lung injury.6 Septic ARDS is characterized by epithelial and endothelial damage, cellular necrosis, oxidative stress, microcirculatory dysfunction, and pulmonary edema. Sepsis itself is a complex condition marked by systemic inflammation, immune dysfunction, and organ failure, with causes that include imbalances in inflammatory responses, mitochondrial damage, coagulopathy, neuroendocrine anomalies, endoplasmic reticulum stress, and autophagy, all of which contribute to organ dysfunction.45
Inflammation and Compromised Barrier Integrity
ARDS is a severe inflammatory disease characterized by rapid vascular damage and increased permeability of lung cells, leading to multiple organ failure.46–48 Invasive infections stimulate immune cell activation, resulting in the release of inflammatory mediators that aggravate vascular endothelial dysfunction, thereby establishing a pro-inflammatory cycle. This process causes hyperpermeability of the pulmonary microvasculature, leading to alveolar edema and the formation of hyaline membranes.46,49 The host’s initial response involves macrophages phagocytosing pathogens and releasing pro-inflammatory cytokines. This stimulation of the innate immune system relies on pattern recognition receptors (PRRs), which are essential for recognizing damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs).50,51 PRRs detect pathogens and damaged cells,52,53 playing a crucial role in the immunological reaction to sepsis. Ultimately, this response can lead to mortality by triggering the release of additional inflammatory cytokines.54,55
Oxidative Stress
Oxidative stress occurs when there is an imbalance between antioxidants and reactive oxygen species (ROS), resulting in cellular damage.56–59 Significant contributors to oxidative stress include air pollution and inflammation, with ROS compromising immunological function during sepsis.60–63 Sepsis affects the innate immune system, including neutrophils and macrophages, which produce free radicals that impact cellular health.64 Understanding free radicals as both damaging agents and signaling molecules is essential.65
In sepsis-induced acute lung injury (ALI), oxidants released from activated pulmonary macrophages and enzymes exacerbate inflammation and lung damage.66 Increased peroxynitrite concentrations and oxidative stress impair immunological responses, as high mobility group box 1 (HMGB1) recruits neutrophils and macrophages, further exacerbating inflammation.67 Alterations in T-cell metabolism also lead to increased ROS and cytokine production.68 Interventions aimed at mitigating oxidative stress, such as overexpression of MUC1, can facilitate recovery from acute lung injury.69 Additionally, the actions of catalase and superoxide dismutase can reduce the effects of harmful enzymes. Mice with acute lung damage caused by sepsis can recover more easily due to the Gly-Pro-Ala protein’s ability to lower ROS levels and enhance superoxide dismutase activity.70
Vascular Endothelium
Individuals with sepsis exhibit immunological dysfunction that triggers the release of inflammatory agents, compromising capillary endothelial integrity and leading to increased pulmonary and systemic inflammation.71 This mechanism initiates the activation and apoptosis of endothelial cells, generating pro-inflammatory signaling molecules and facilitating leukocyte accumulation, which attracts neutrophils, macrophages, and T cells.72 The accumulation of leukocytes, particularly neutrophil-platelet aggregates, demonstrates complex thrombo-inflammatory characteristics.73 Alveolar macrophages play a vital role in lung inflammation; their loss exacerbates ARDS by promoting neutrophil migration and increasing cytokine levels.74–78 Vascular endothelial (VE)-cadherin and endothelial receptor kinase (TIE2), regulated by vascular endothelial protein tyrosine phosphatase (VE-PTP), are crucial for maintaining the integrity of vascular endothelial cells.79,80 Cytoskeletal connections, guanosine triphosphate (GTP)ases, and phosphorylation influence their functionality.81 Inflammatory responses can undermine VE-cadherin, but actin filaments and stress fibers help preserve cell junction integrity.82–84
Injury to Alveolar Epithelium
ARDS is primarily triggered by damage to the alveolar epithelium, with its severity influenced by the exudative phase.13 This damage results in immune cell-mediated pulmonary injury,72 generating pro-inflammatory mediators and necessitating lung-protective breathing.11,85 The alveolar epithelium, composed of type I and type II cells, is essential for fluid clearance and pulmonary gas exchange.86 In ARDS, alveolar injury is predominantly caused by the death of these cells. Type I cells, which are crucial for gas exchange, establish tight junctions,87 while type II cells promote repair and regeneration of lung tissue.88,89 Strategies aimed at attenuating apoptosis and employing mesenchymal stem cells may enhance therapeutic outcomes for ARDS.90–92
Cellular Mortality
Defective neutrophil-macrophage efferocytosis in ARDS increases secondary necrosis and the generation of inflammatory mediators,93 while oxidative stress and mitochondrial dysfunction exacerbate lung disorders by inducing apoptosis and necroptosis.94–96 Excessive apoptosis pathways may contribute to the disease’s pathogenesis.97 Cell death, which can be categorized into apoptosis and necrosis, is essential for maintaining the equilibrium of multicellular organisms and their defense against pathogens or nutrient deficiency.98 Apoptosis is characterized by cytoplasmic vacuolation, nuclear condensation, and plasma membrane blebbing.99
Programmed Cell Death
Apoptosis occurs through intrinsic and extrinsic mechanisms, leading to the activation of caspases.100 Intrinsic apoptosis in the lung may be initiated by the withdrawal of growth factors, oxidative stress, endoplasmic reticulum stress, and DNA damage, thereby enhancing pro-survival signaling pathways, including those mediated by protein kinase B.101 Anoikis is caused by the loss of integrin-mediated adhesion,102 whereas DNA damage, oxidative stress, and death receptor signaling lead to intrinsic and extrinsic apoptosis, respectively.103,104 Necroptosis is a controlled necrotic process triggered by tumor necrosis factor receptor 1 (TNFR1)-mediated receptor-interacting protein kinase 1 (RIPK1)-RIPK3 activation and necrosome formation.105,106
Pyroptosis
Programmed cell death mediated by inflammation has been well investigated for its role in the progression from ARDS to pulmonary fibrosis.107 Pyroptosis is a type of programmed necrotic cell death driven by gasdermins, whose proteolytic cleavage facilitates the formation of pores.108 Gasdermin D is activated by inflammasomes, although other family members may be activated independently of this process. While pyroptosis aids in pathogen defense, its excessive activation can lead to cytokine storms, inflammation, and tissue damage, resulting in conditions such as sepsis, ARDS, atherosclerosis, and neurodegeneration.109 The NLRP3 inflammasome-mediated pyroptosis in ARDS creates membrane pores and releases pro-inflammatory cytokines after infections or trauma activate alveolar macrophages.110–116
Ferroptosis
Sepsis impairs iron metabolism and lipid peroxidation, potentially leading to metabolic disorders, including ferroptosis, a cell death pathway dependent on iron associated with ARDS.117,118 In cases of acute pancreatitis, sepsis can induce ferroptosis in intestinal epithelial cells, weakening the barrier and allowing the movement of bacteria and toxins.119 This process is exacerbated by respiratory infections caused by Pseudomonas aeruginosa.120 Early immunological responses lead to iron and lipid peroxidation during macrophage activation, initiating ferroptosis.121 Additionally, infection with Mycobacterium tuberculosis causes glutathione depletion, iron overload, and downregulation of glutathione peroxidase 4 (GPX4),122 all of which affect the survival and functionality of CD8 T cells.123
Oxidative stress is associated with the pathophysiology of ARDS, as infections may stimulate ROS generation, altering the balance between oxidative and antioxidant capabilities. Redox imbalance can induce ferroptosis,124 leading to immune cell activation and the generation of pro-inflammatory cytokines, which aggravate lung injury and render ferroptosis an immunogenic form of cell death.125 Lipopolysaccharide (LPS) treatment may inhibit the expression of GPX4 and SLC7A11 in sepsis-induced ARDS while elevating levels of malondialdehyde (MDA), 4-hydroxynonenal (4-HNE), and total iron, thus reversing these alterations.126
Immunologic Mechanism
Sepsis-induced ARDS results from a complex interplay of systemic infection, immunological dysregulation, and pulmonary damage. The immunological pathways are characterized by an initial hyperinflammatory phase induced by innate immune activation, followed by an immunological response that is either suppressed or dysregulated, leading to ongoing tissue damage and organ failure.
Initiation by Pathogen Recognition
The innate immune system’s identification of pathogens is the first step in the development of sepsis-induced ARDS.127 Macrophages phagocytose pathogens, release pro-inflammatory cytokines, and trigger a cytokine storm, thereby initiating the innate immune response.50,128 Immune cell stimulation by pattern recognition receptors damages the alveolar space, resulting in the accumulation of edema fluid.51,86 Negative feedback systems and immunological fatigue can lead to immune paralysis in later phases, impacting the host’s hyperimmune state.129
Cytokine Storm and Inflammatory Cascade
Severe sepsis results from a cytokine storm.130 Most rat models primarily focus on general sepsis and fail to accurately represent severe sepsis, which is characterized by increased early mortality within the initial 72 hours.131 Tumor necrosis factor-alpha (TNF-α) and interleukin-6 (IL-6) are important indicators of the degree of inflammation.132 Immunological complexes, bacterial infections, and endotoxins can cause alveolar epithelial cells to undergo apoptosis133 and cease proliferating,134,135 accelerating the progression of sepsis-associated ALI.136 Pharmacological suppression of alveolar epithelial cell apoptosis has shown effectiveness in reducing acute lung injury in animal studies, thereby enhancing survival outcomes.137 The distinction between normal and dysregulated inflammation in severe infections can be ambiguous, as many cytokines exert both protective and detrimental effects.138,139 When cytokine levels exceed a certain threshold, reversing the cytokine storm becomes challenging, and the intricate interactions of mediators further complicate this limit.139 A severe inflammatory cascade could be triggered by the dysregulated cytokine storm, leading to either irreversible or reversible end-organ damage and possibly death.140 Chronic illnesses may precipitate sepsis via inflammatory cascade responses and cytokine storms. IL-6, a pivotal factor in the cytokine storm, and its associated signaling pathways have been recognized as essential factors in sepsis.141 The inflammatory cascade progressively escalates, resulting in sepsis driven by a cytokine storm. Activation of the IL-6-associated signaling system renders some chronic illnesses susceptible to sepsis due to persistent low-grade inflammation. Conversely, sepsis may exacerbate these chronic conditions, resulting in multiple organ failure.141
Physiological Role and Pathophysiological Significance of Emerging Biomarkers
Interleukin-6 (IL-6)
IL-6 regulates numerous physiological processes, including cell proliferation, differentiation, survival, and apoptosis. It is essential for the functioning of the immune and hematopoietic systems, influencing various other systems such as neurological, endocrine, and bone metabolism.142 Synthesized primarily by myeloid cells, including macrophages and dendritic cells, IL-6 is produced in response to pathogen recognition via toll-like receptors(TLRs).143 It is vital for the proliferation of myeloma cells and the survival of plasma blasts during the adaptive immune response, promoting plasma cell differentiation and enhancing antibody synthesis.143
Originally identified as B-cell stimulatory factor 2 (BSF-2), IL-6 also stimulates immunoglobulin production in activated B cells.144,145 Rapidly synthesized in reaction to infections or tissue damage, IL-6 facilitates host defense by regulating acute-phase responses and hematopoiesis.146–149 Furthermore, the IL-6 family of cytokines helps maintain the balance between pro-inflammatory and anti-inflammatory responses, augments thermogenesis, inhibits diet-induced obesity, and ameliorates insulin resistance through interactions with adipocytes and activation of the p38 mitogen-activated protein kinase (MAPK) and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) signaling pathway.150
Despite its essential physiological functions, excessive production of IL-6 can lead to significant pathophysiological consequences.151 Overproduction of IL-6 is associated with systemic inflammatory response syndrome (SIRS) and cytokine-release syndrome (CRS), conditions characterized by hyperinflammation.148,152 In sepsis, IL-6 contributes to a cytokine storm, which entails dysregulated immune responses marked by the simultaneous activation of numerous cytokine networks.153 This hyperactive state can exacerbate feedback loops, resulting in tissue damage, multi-organ failure, and increased mortality.151 Additionally, IL-6 plays a critical role in endothelial dysfunction during sepsis-induced ARDS by activating endothelial cells and enhancing vascular permeability. This leads to the effusion of fluid and proteins into the alveolar space, contributing to respiratory failure.154,155 Ultimately, while IL-6 is vital for normal immune function and metabolic regulation, its dysregulation can significantly impact health, underscoring its dual role in both protective and pathological processes.153
Angiopoietin-2 (Ang-2)
Ang-2 is a growth factor that is part of the angiopoietin/Tie signaling pathway, which is crucial for angiogenesis. It was discovered after the identification of angiopoietin-1 (Ang1) through complementary DNA (cDNA) library screening.156 Structurally, Ang-2 is a 496-amino-acid protein that shares approximately 60% similarity with Ang1, though it lacks one of the nine cysteine residues present in Ang1. Ang2 features a secretion signal peptide, an NH2-terminal coiled-coil domain, and a COOH-terminal fibrinogen-like domain.157 Under physiological conditions, Ang2 functions in an autocrine manner with tightly regulated expression. It binds to the Tie2 receptor with an affinity similar to that of Ang1 but typically acts as an antagonist, inhibiting Ang1-mediated phosphorylation of Tie2 in blood endothelial cells (BECs).157 Interestingly, Ang2 can also weakly phosphorylate Tie2, enhancing endothelial functions.158,159 The Tie1 receptor modulates Ang2’s activity, converting it from an antagonist to an agonist during inflammation following Tie1 cleavage.160 While Ang2 is not essential for embryonic blood vessel development, it plays a significant role in modulating angiogenic sprouting and vascular regression, particularly in neonates.161
In pathological contexts, the dysregulation of Ang2 has significant implications. Its expression is induced by inflammatory mediators, such as thrombin, as well as by conditions like hypoxia and malignancy.162–166 Ang2 compromises endothelial integrity, although this effect can be countered by Ang1 or vascular endothelial growth factor (VEGF). In pathological settings, Ang2 facilitates angiogenesis independently of Tie2 or Ang1, promoting endothelial cell migration and tube formation.167,168 This is particularly evident in conditions associated with increased vascular permeability and angiogenesis, where overexpression of Ang2 can lead to larger tumors and increased metastasis, while inhibition of Ang2 has the opposite effect.169–171
Ang2 is also implicated in various inflammatory disorders, including autoimmune diseases and acute lung injury.172,173 It induces vascular permeability through interactions with cell junction proteins such as integrins and vascular endothelial cadherin (VE-cadherin). Ang2 is essential for initiating the inflammatory response, as shown by the inability of Ang2-deficient mice to mount such responses in infection models. Elevated levels of Ang2 in the blood correlate with inflammatory markers, suggesting its potential as an inflammatory biomarker.174 For example, patients with pneumonia exhibit increased Ang2 levels, particularly among those who do not survive 28 days post-diagnosis.175 Furthermore, Ang2 plays a critical role in endothelial function and cardiovascular remodeling. Increased levels of Ang2 are associated with conditions such as coronary heart disease and congestive heart failure.176–179 It serves as a biomarker and influences post-stroke recovery by initially increasing detrimental vascular permeability, which later correlates with microvessel maturation after one week.180 Ang2 also facilitates the differentiation and migration of brain progenitor cells independently of Tie2.181 Additionally, a reduction in Ang2 alongside an increase in Ang1 promotes vascular remodeling, indicating potential therapeutic targets for post-stroke interventions and improved cardiac allograft outcomes by mitigating endothelial cell activation and leukocyte infiltration.182
Pancreatic Stone Protein (PSP)
PSP, also known as lithostathine or Reg protein, is a glycoprotein composed of 144 residues, primarily synthesized by the exocrine pancreas.183,184 It is also detected in the endocrine component of the regenerating pancreas and in certain tumors, including pancreatic ductal cell carcinoma and colorectal tumors.185–187Although the precise physiological role of PSP remains somewhat ambiguous, it has been shown to promote islet regeneration in partially depancreatized rats,188 suggesting its potential function as a β-cell growth factor that helps preserve β-cell mass and functionality.189,190 Additionally, PSP regulates pancreatic enzyme secretion, thereby assisting in digestion and nutrient absorption, which contributes to overall gastrointestinal health.183 In pancreatic juice, the presence of lithostathine indicates a potential role in inhibiting stone formation by modulating the nucleation, development, and aggregation of calcite crystals. Specifically, lithostathine impedes crystal nucleation and restricts the development of pre-existing calcium carbonate (CaCO3) from supersaturated solutions, while also functioning as a modulator of calcite crystal habit, leading to the formation of smaller crystals.191,192
In pathological contexts, dysregulation of PSP is observed, particularly in conditions such as sepsis. The inflammatory response associated with sepsis may compromise PSP function, as elevated pro-inflammatory cytokines can lead to pancreatic dysfunction and negatively impact pulmonary health.193 Sepsis often precipitates acute pancreatitis, which can further exacerbate lung damage. In this setting, dysregulation of PSP may contribute to the formation of pancreatic stones, resulting in duct obstruction and an increased risk of inflammation affecting other organs, including the lungs.194 Similarly, decreased levels of PSP in chronic pancreatitis may promote pancreatic stone development, duct obstruction, and heightened inflammation and discomfort.194
Pancreatic duct obstruction leads to elevated duct pressure and permeability, facilitating the passage of macromolecules, such as pancreatic enzymes, through the typically impermeable duct. This increased permeability, along with morphological alterations, is likely linked to elevated secretory pressure during blockage, fostering inflammation and injury.195 Consequently, PSP may modulate the inflammatory response associated with pancreatic disorders, where its dysregulation could exacerbate chronic inflammation in diseases such as pancreatitis and pancreatic cancer.196
Syndecan-1 (Sdc-1)
Sdc-1 is a heparan sulfate proteoglycan that plays a crucial role in various physiological processes, including host defense, angiogenesis, leukocyte recruitment, microbial interactions, and matrix remodeling.197 It is particularly significant in regulating protein leakage in the intestinal epithelium, smooth muscle cell proliferation during vascular injury, keratinocyte proliferation in dermal injury, and lipoprotein metabolism.198–201 Sdc-1 is generally absent from most leukocytes, except for active macrophages, but serves as a hallmark for plasma cells and B cell malignancies.202,203
During inflammation, Sdc-1 negatively regulates leukocyte adhesion and migration by inhibiting integrin interactions with endothelial intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1). This function is vital for controlling the inflammatory response and ensuring appropriate immune cell trafficking. Additionally, research has shown that syndecan-1 shedding can facilitate the resolution of neutrophil inflammation by aiding in the removal of sequestered CXC chemokines in a heparan sulfate-dependent manner.204 The pathophysiological significance of Sdc-1 is highlighted by the severe symptoms observed in syndecan-1 null animals during various illness challenges.197 The absence of Sdc-1 can lead to detrimental outcomes in multiple contexts. For example, in cardiac tissue remodeling following myocardial infarction, Sdc-1 deficiency results in poor collagen cross-linking, impaired cardiac function, disorganized collagen matrices, increased inflammatory cell infiltration, and elevated matrix metalloproteinases (MMP-2 and MMP-9) activity.205,206 These changes contribute to adverse cardiac remodeling and poorer healing outcomes. Conversely, the overexpression of syndecan-1 has been shown to be critical for effective cardiac tissue remodeling by reducing inflammation and protecting against heart degeneration. This illustrates the dual role of Sdc-1, where both its presence and absence can significantly impact disease outcomes.206
Presepsin (P-SEP)
P-SEP is a soluble form of CD14, a co-receptor that plays a critical role in the immune response to bacterial infections. It is released into the bloodstream when bacteria and antigens interact, remaining stable in circulation. P-SEP serves as a readily measurable biomarker for systemic infections, particularly in their early stages.207 The immune system relies on its ability to recognize and respond to infections through both innate and adaptive immunity. While adaptive immunity takes time to develop, innate immunity provides immediate defense through mechanisms such as phagocytes and antimicrobial peptides. Innate immunity utilizes PRRs,208 including TLRs, to identify conserved PAMPs and trigger an immediate immune response.209 CD14, as a co-receptor, binds specifically to ligands like bacterial lipopolysaccharides (LPS).210,211 For effective recognition of LPS, CD14 requires interaction with lipoprotein binding protein (LBP), which facilitates its presentation. This interaction activates TLRs, leading to the generation of cytokines and the initiation of phagocytosis by monocyte-macrophages. Although CD14 itself lacks a transmembrane domain and cannot independently transmit signals, it is essential for forming complexes with LPS and TLRs, enhancing intracellular signaling and host defenses.209,212
The pathophysiological significance of P-SEP is underscored by its role as a biomarker for sepsis and other systemic infections. Following the proteolytic cleavage of CD14, soluble CD14 fragments, particularly sCD14-ST, enter the bloodstream and serve as indicators of an immunological response.213 Elevated levels of circulating soluble CD14 (sCD14) correlate with the severity of sepsis in both adults and neonates, making it a valuable tool for detecting systemic illnesses associated with Enterobacteriaceae.214,215 P-SEP was first identified as a diagnostic biomarker for septic patients in 2005 due to its significant increases in blood plasma during the early stages of sepsis.216 Its potential as a sepsis biomarker has been further validated by the development of a one-step test for routine evaluation of presepsin levels. The measurement of presepsin in clinical settings can aid in early diagnosis and management of sepsis, ultimately improving patient outcomes.213,217
Endocan
Endocan is a recently identified dermatan sulfate proteoglycan primarily found in serum and expressed in activated endothelial cells.218 Its expression is influenced by various growth factors, including vascular endothelial growth factor (VEGF-A and VEGF-C), hepatocyte growth factor (HGF/MET), fibroblast growth factor (FGF-2), and epidermal growth factor receptor (EGFR). These factors act through several signaling pathways, such as protein kinase C (PKC)/nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), phosphoinositide 3-kinase (PI3K)/Akt, and Janus kinase/signal transducer and activator of transcription 3 (JAK/STAT3), to elevate endocan expression.219,220 In healthy physiological conditions, endocan plays a role in promoting angiogenesis and vascular integrity. It facilitates leukocyte recruitment and adhesion, which are essential for angiogenesis and inflammatory responses.221 Endocan increases the release of adhesion molecules such as VCAM-1, ICAM-1, and E-selectin from inflammatory endothelial cells, contributing to the regulation of immune responses and vascular homeostasis. Additionally, endocan may serve as a biomarker for endothelial activation.222–225
The pathophysiological significance of endocan is highlighted by its correlation with various conditions, including vascular disorders, systemic inflammation, and several types of cancer. Elevated levels of endocan are associated with atherogenesis, as it promotes the synthesis of nitric oxide, reactive oxygen species, and inflammatory mediators like inducible nitric oxide synthase (iNOS) and C-reactive protein (CRP).226 Proinflammatory cytokines, such as TNF-α and IL-1β, enhance endocan expression by activating inflammatory pathways like NF-κB.227,228 This upregulation contributes to vascular inflammation and the progression of various diseases.221 Endocan also plays a role in modulating leukocyte behavior; it can inhibit leukocyte migration by binding to integrin CD11a/CD18 (LFA-1), potentially mitigating acute lung inflammation.229 The P14 fragment of endocan, generated by cathepsin G, facilitates leukocyte extravasation by competing for LFA-1 binding, with elevated levels observed in sepsis patients, indicating its potential as a marker of neutrophil activation.230,231 Research has shown that endocan reduces inflammation markers such as TNF-α and IL-6 and decreases LPS-induced apoptosis in pulmonary epithelial cells. Lower levels of endocan in individuals with ARDS and acute lung injury suggest its significant role in respiratory inflammation and its potential as a therapeutic target.232
Soluble Receptor for Advanced Glycation Endproducts (sRAGE)
sRAGE is a member of the immunoglobulin superfamily located in the Class III region of the major histocompatibility complex.233,234 Initially recognized for its affinity for advanced glycation endproducts (AGEs), RAGE has since been reclassified as a PRR due to the discovery of several additional ligands, including DAMPs.235,236 Under normal physiological conditions, RAGE is typically expressed at low levels in differentiated adult cells. However, its expression is significantly elevated in mature lung type I pneumocytes, where it may play a role in maintaining lung function and responding to injury.237
RAGE is associated with a variety of inflammation-related disorders, including vascular disease, cancer, neurodegeneration, and diabetes.238,239 Its involvement in these conditions is primarily due to its role in mediating inflammatory responses. When activated by its ligands, RAGE can trigger pro-inflammatory signaling pathways, leading to increased inflammation and tissue damage. Interestingly, RAGE expression is significantly reduced in lung tumors and idiopathic pulmonary fibrosis compared to levels found in healthy tissue, suggesting a complex role in these diseases. The downregulation of RAGE in such contexts may indicate a loss of its protective functions or a response to chronic inflammatory states.
Soluble Urokinase Plasminogen Activator Receptor (suPAR)
suPAR is a soluble variant of the membrane-bound receptor uPAR, which is primarily found on the surfaces of immune cells, including monocytes, macrophages, activated T-lymphocytes, endothelial cells, and certain cancer cells.240 The uPAR plays a crucial role in plasminogen activation and fibrinolysis, influencing key cellular processes such as proliferation, migration, adhesion, angiogenesis, and the inflammatory response.241 The expression of the PLAUR gene, which encodes uPAR, is typically low but increases in response to immunological activation, inflammation, and tumor growth. This upregulation is driven by the activation of innate immune receptors that induce transcription factors like nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and activator protein 1 (AP-1), promoting PLAUR gene expression.240 suPAR is released into the bloodstream through the proteolytic cleavage of uPAR and can be detected in various bodily fluids, including blood plasma, serum, cerebrospinal fluid, saliva, and urine.242–244 Its levels in the bloodstream correlate positively with total white blood cell counts and are associated with both innate and adaptive immune cells,245,246 particularly neutrophils, monocytes, and eosinophils.245,247–249
In pathological contexts, elevated levels of suPAR are associated with various inflammatory conditions and diseases. The expression of uPAR and the subsequent release of suPAR can be stimulated by inflammatory mediators such as LPS, IL-8, TNF-α, granulocyte colony-stimulating factor (G-CSF), and formyl-methionyl-leucyl-phenylalanine (fMLP).250,251 For instance, LPS injection in healthy individuals leads to increased uPAR expression on monocytes and elevated suPAR levels in the bloodstream.252,253 Additionally, interleukin-1 beta (IL-1β) promotes suPAR release from endothelial and vascular smooth muscle cells, although IL-6 can inhibit LPS-induced suPAR release.254–257
The binding of uPAR to its ligand, urokinase plasminogen activator (uPA), on immune cells is critical for cell migration during inflammation and immune responses. Active uPA catalyzes the conversion of plasminogen to plasmin, initiating a cascade that activates matrix metalloproteinases, which degrade extracellular matrix components.241,258 This process accelerates extracellular matrix disintegration, activates growth factors, enhances cell migration, facilitates fibrinolysis, causes vasodilation, increases vascular permeability, and aids in the opsonization and phagocytosis of pathogens.241,259,260
Elevated suPAR levels have been linked to various inflammatory diseases, including infections, autoimmune disorders, and cancer. In particular, suPAR is predominantly expressed in pro-inflammatory monocyte subsets during acute liver failure and is released from monocytes following LPS stimulation, highlighting its role in the immune response.261 The correlation between suPAR levels and innate immune cells underscores its potential as a biomarker for inflammation and disease progression.
Clinical Challenges in Weaning from Mechanical Ventilation
Mechanical ventilation (MV) is an essential life-saving intervention for severely ill patients. However, 20% to 40% of these individuals encounter problems during the weaning process, experiencing varying levels of strain (Table 1).262 Although MV is associated with potential complications, prompt weaning from ventilation is crucial once the underlying disease has fully recovered and the patient demonstrates the ability for spontaneous, unassisted respiration.263 Despite its advantages, this treatment approach may lead to several types of harm, including ventilator-induced lung Injury(VILI),VAP, tracheal injuries, and neurodevelopmental abnormalities (Table 1).264,265
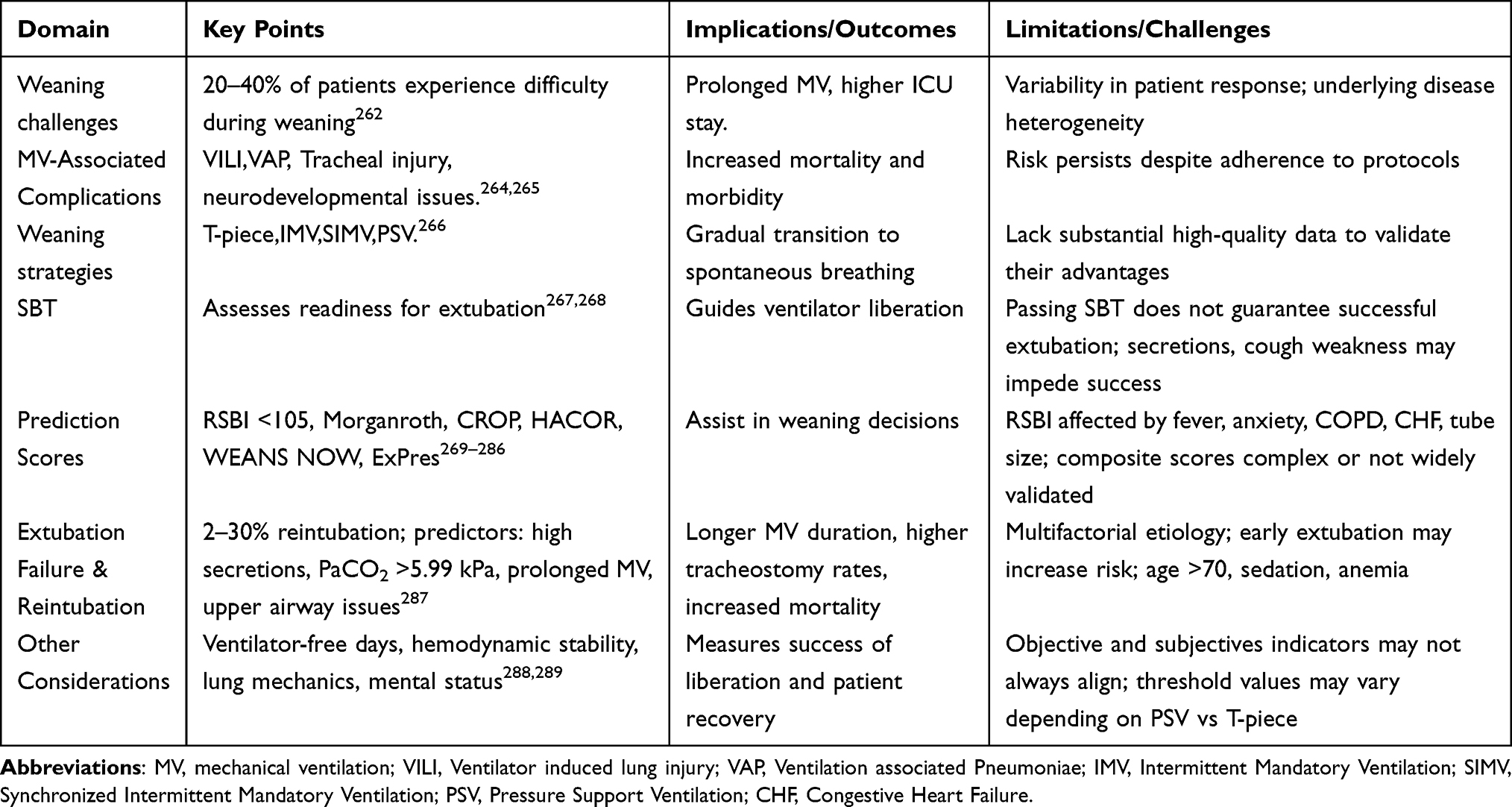 |
Table 1 Clinical Challenges in Weaning from Mechanical Ventilation |
Weaning from mechanical ventilation involves the systematic reduction of ventilatory assistance, while emancipation refers to the complete cessation of support for patients deemed ready. The weaning method began in the 1960s, initially employing a T-piece connected to the endotracheal tube for oxygen administration to progressively increase the duration of spontaneous breathing.266 Subsequently,Intermittent Mandatory Ventilation (IMV) and Synchronized Intermittent Mandatory Ventilation (SIMV) were introduced. These methods have become fundamental to prevalent weaning procedures, although they lack substantial high-quality data to validate their effectiveness. SIMV allows for a gradual decrease in the number of mandatory breaths, enabling patients to engage in more spontaneous breathing, thereby facilitating a smoother transition. The introduction of Pressure Support Ventilation (PSV) provided enhanced assistance for spontaneous breaths during SIMV; alternatively, PSV may serve as the sole mode of support, with weaning achieved by gradually reducing the pressure support level.266
The optimal outcome of weaning is successful extubation; however, some patients may require a tracheostomy before being liberated from ventilator support. It is important to distinguish between ventilator release (the discontinuation of all invasive support) and extubation (the removal of the endotracheal tube), as these are separate procedures. The count of ventilator-free days, typically assessed over a 28-day period, reflects the duration during which patients are not reliant on artificial ventilation.290
A SBT evaluates a patient’s ability to breathe with minimal or no ventilatory assistance. It is recommended that SBTs commence whenever the primary cause of respiratory failure appears to be addressed; however, there is no consensus on the criteria for assessing the reversal of the underlying condition.267 Clinicians often utilize a combination of subjective evaluations and objective indicators, including improved gas exchange, cognitive state, neuromuscular function, and imaging results. It is acknowledged that some patients may be effectively weaned even if they do not meet all criteria completely.267 Successfully passing an SBT does not guarantee effective extubation or complete ventilator release, as factors such as excessive secretions and reduced cough reflexes may compromise the results. Research indicates that only 55% of patients who successfully completed an SBT were extubated without requiring another trial.268 Re-intubation is necessary in approximately 2% to 30% of patients following a scheduled extubation, which correlates with prolonged ventilation, an increased likelihood of tracheostomy, a higher incidence of VAP, and elevated mortality rates.291 The standard protocol for SBT failure includes assessing reversible factors, optimizing the patient’s physiology, and conducting another SBT the following day.288 Failure often arises from cardiovascular issues or problems with the respiratory pump.292,293 Extubation failure, commonly due to an inability to manage respiratory demands, is associated with higher mortality rates from complications such as aspiration and pneumonia.294,295 Although mortality is minimal (11%) in cases of upper airway obstruction, it can increase significantly (36%) in other circumstances.296
Key predictors of extubation failure include high secretions, arterial carbon dioxide tension exceeding 5.99 kPa, prolonged mechanical ventilation lasting more than 72 hours, upper airway complications, and previous unsuccessful attempts at weaning from mechanical ventilation.287 The failure rates following a single SBT range from 26% to 42%.288 Objective measures of weaning readiness include hemodynamic stability, adequate gas exchange, and favorable lung mechanics, such as a RSBI of less than 105. Subjective measures encompass improvements in the underlying disease, airway patency, cough strength, and mental state.288,289 The timing and criteria for initiating weaning are critical, as early extubation may increase the risk of re-intubation, which is linked to higher mortality rates. Re-intubation within 48 hours after extubation is associated with negative outcomes. A study involving 336 critically ill patients, all of whom were extubated using a consistent technique, provides insight into this risk. The cohort included 121 medical patients (36%), 85 surgical patients (25.2%), and 53 trauma patients (15.7%). Extubation was unsuccessful in 52 patients (15.4%), all of whom required re-intubation within 48 hours, with most occurring between 12 and 24 hours. Factors contributing to re-intubation include ineffective cough, laryngeal stridor, cardiogenic pulmonary edema, gasping, bronchospasm, muscular weakness, sensory alterations, bronchoaspiration, cardiorespiratory arrest, shock, and other complications.291 Risk factors for extubation failure include patients with medical and interdisciplinary conditions, individuals over 70 years of age, prolonged mechanical ventilation, continuous intravenous sedation, and anemia. The causes of failure may vary, potentially including weaning failure, upper airway obstruction, insufficient cough, excessive respiratory secretions, encephalopathy, or cardiac dysfunction. Extubation failure leads to extended mechanical ventilation, prolonged ICU and hospital admissions, increased tracheostomy rates, and higher mortality rates within the hospital setting.294
Prediction scores, such as the Morganroth scale (Table 1) and RSBI, provide systematic guidance for ventilator weaning, although their accuracy varies among different patient demographics.269–271 RSBI values below 105 indicate a heightened sensitivity for successful extubation, while higher values suggest a greater likelihood of weaning failure.270 A meta-analysis has shown reasonable overall accuracy, with a sensitivity of 0.60 and a specificity of 0.68.272
Despite its simplicity and widespread use, the RSBI can be influenced by several factors, including fever, anxiety, discomfort, illness, or anemia, which may elevate respiratory rates and result in inaccurately high readings, thus hindering ventilator liberation. Technical and patient-related variables, such as reduced endotracheal tube diameter, female gender, and suctioning, may also affect measurements.273 Conversely, conditions like Chronic Obstructive Pulmonary Disease (COPD) can yield inaccurately low RSBI values, especially when patients fail to trigger the ventilator during COPD exacerbations or benefit from positive pressure ventilation, as seen in Congestive Heart Failure (CHF) patients.274,275 Neurological disorders that cause bradypnea may also lead to inaccurately low RSBI measurements.276 The ideal threshold value may differ based on whether PSV or T-piece trials are employed, with prevailing standards recommending the use of PSV during weaning.277,278 Research indicates that RSBI remains largely unchanged during SBTs.279 Moreover, successive RSBI measurements taken over a 120-minute interval have not consistently identified liberation failure.280 Fluctuations in RSBI during SBT may more accurately predict weaning success, showing high sensitivity and specificity.281 Composite indices, such as CROP, Gluck-Corgian, and HACOR scores, combine various respiratory indicators to enhance predictive accuracy.270,282,283 RSBI and maximal inspiratory pressure are critical indicators for successful extubation and weaning failure. The HACOR score evaluates cardiac and diaphragmatic function; however, calculating it at different times during SBT poses practical challenges, necessitating additional validation through extensive research due to existing data limitations. The WEANS NOW score, developed in 2020, comprises eight elements related to the risk of extubation failure. Despite its thoroughness, concerns have been raised regarding its complexity and usability in bedside settings.284 In 2021, Baptistella et al established the Extubation Predicting Score (ExPreS), which integrates both respiratory and non-respiratory factors. A score of 59 or higher indicates a substantial likelihood of successful extubation. While promising, the ExPreS requires further validation through extensive studies to confirm its efficacy as a comprehensive clinical tool (Table 1).285
RSBI alone has demonstrated limited diagnostic efficacy in predicting effective extubation under current weaning protocols. These findings highlight the need to revise RSBI threshold values and suggest that integrating RSBI with other predictors may enhance its utility in clinical decision-making.286
Weaning Prediction of Biomakers
The complex and heterogeneous nature of ARDS, which arises from both pulmonary and non-pulmonary etiologies, presents significant challenges in improving patient outcomes through randomized controlled trials. This inherent variability is frequently cited as a major factor contributing to the failure of such trials.297 Consequently, biomarkers offer a promising strategy for addressing this heterogeneity and enhancing therapeutic success. By aiding in the understanding of ARDS variability and enriching trial populations, biomarkers facilitate the identification of appropriate patient subgroups for targeted therapies.86
For instance, a study investigating the sRAGE in 164 COVID-19 patients, along with convalescent individuals, non-COVID pneumonia cases, and healthy controls, demonstrated its high prognostic value. Serum sRAGE levels correlated positively with critical indicators of disease severity, including the need for oxygen therapy, the use of high-flow nasal oxygen (HFNO), MV, ARDS severity, requirements for dialysis and catecholamines, 30-day mortality, and elevated Sequential Organ Failure Assessment (SOFA) and quick SOFA (qSOFA) scores.298 Notably, sRAGE exhibited robust predictive capability for the requirement of MV Area Under the Curve (AUC 0.871) and mortality (AUC 0.903). Even after adjusting for confounding variables such as age, sex, comorbidities, and a SOFA score of 3 or higher, elevated sRAGE remained an independent predictor for the need for HFNO or MV.161,298 Furthermore, analyses utilizing sRAGE and other biomarkers helped differentiate ARDS based on its origin. Patients whose ARDS resulted from direct lung injury exhibited higher concentrations of lung epithelial injury markers (plasma sRAGE and surfactant protein D (SP-D) and, simultaneously, lower levels of lung endothelial injury markers (Ang-2) and inflammatory markers (Von Willebrand factor (VWF), IL-6, and IL-8), compared to patients whose ARDS stemmed from indirect causes.299
PCT serves as a biomarker for the early detection of bacterial infections and sepsis across various infection sites, including pneumonia, soft tissue infections, intra-abdominal infections, and urinary tract infections, particularly within emergency and ICU settings.300 However, its kinetic profile and overall utility require further investigation across diverse patient populations and clinical contexts.300 In the context of ARDS specifically caused by Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2), research on circulating biomarkers of endothelial dysfunction, particularly in patients receiving anti-inflammatory treatments, revealed that conventional markers of inflammation, coagulation, and epithelial injury did not show a significant association with clinical outcomes.301 In contrast, endothelial biomarkers, particularly plasma Neural Precursor Cell Expressed Developmentally Downregulated 9(NEDD9), were significantly linked to 60-day mortality, suggesting they act as stable indicators of immune activation.301
Recent studies, including those involving large COVID-19 cohorts, indicate that early levels of suPAR can predict the progression to respiratory failure and the subsequent necessity for mechanical ventilation. Elevated suPAR levels upon admission are correlated with the development of severe or critical COVID-19 and extended hospital stays. Furthermore, suPAR has been established as an independent predictor of disease severity in COVID-19.302 While meta-analyses affirm suPAR’s efficacy in prognosticating sepsis outcomes, its specific capacity to predict weaning success or duration in classical sepsis-induced ARDS necessitates dedicated future studies.
Endothelial markers such as Ang-2 and VWF are valuable predictors of mortality in patients with ARDS and those at risk. Specifically, elevated Ang-2 levels are associated with the development of ARDS in trauma-related cases, and Ang-2 levels correlate with VWF levels in predicting mortality.27 Inflammatory biomarkers, including IL-6, CRP, and PCT, serve as sensitive indicators of systemic inflammation. These markers can predict disease severity and the need for mechanical ventilation, especially in COVID-19 patient cohorts. However, because these inflammatory markers lack lung specificity, their utility in predicting weaning outcomes is limited when used alone.303 Early measurement of biomarkers such as sRAGE and suPAR within the first 24 hours of ICU admission can identify patients at higher risk of progressing to invasive and prolonged mechanical ventilation. Elevated levels of sRAGE are associated with increased ARDS severity and higher mortality. Meanwhile, suPAR levels correlate with disease severity and the need for organ support, including VV-ventilation.298 Serial monitoring of these biomarkers provides dynamic insights into a patient’s evolving condition, allowing clinicians to track injury progression or recovery in real-time, thereby enhancing prognostic accuracy and supporting timely treatment modifications.304
The integration of biomarker assessment early in the clinical course, coupled with serial measurements, holds significant promise for individualized patient management, optimization of ventilatory support, and improved outcomes in ARDS, particularly in the context of sepsis and LI.304 While SBT are fundamental for assessing extubation readiness, elevated levels of specific biomarkers can offer insights into persistent respiratory impairment that might not be evident through traditional physiological parameters. For example, the combined elevation of SP-D and sRAGE has shown a strong association with pulmonary edema, suggesting that epithelial injury may be a common pathway underlying alveolar-capillary barrier dysfunction in pulmonary edema. In this context, Ang-2 did not improve the predictive model, indicating that biomarkers of epithelial injury (SP-D, sRAGE) and endothelial dysfunction remain important as they are also linked to pulmonary edema in invasively ventilated patients.305
Elevated biomarkers indicative of ongoing lung injury, specifically those reflecting endothelial or epithelial damage, suggest residual pulmonary compromise that may not be immediately detectable through SBT. These biomarkers can assist clinicians in making more informed decisions regarding whether to delay extubation, thereby promoting individualized patient care.306 Furthermore, biomarkers are crucial in identifying distinct ARDS subphenotypes, which allows for personalized treatment approaches.306 The use of machine learning and advanced computational techniques enhances the understanding of these biomarkers, leading to more accurate models for predicting ARDS risk and patient outcomes.307 For instance, a biomarker panel consisting of lung epithelium-derived proteins SP-D, sRAGE, and Clara cell secretory protein (CC-16) has shown excellent diagnostic discrimination for ARDS in patients with severe sepsis.308
Altered plasma levels of these biomarkers can serve as biological confirmation of an ARDS diagnosis in septic patients and may also aid in selecting appropriate candidates for clinical trials focused on reducing lung epithelial injury.308 These multi-marker panels provide a more nuanced understanding of the underlying pathophysiological processes, enabling clinicians to tailor interventions more precisely. Integrating such biomarker panels into clinical practice holds the potential for identifying ARDS subphenotypes and facilitating personalized treatment strategies that target specific mechanisms of lung injury.6
Clinical Relevance of Biomakers
Interleukin-6 (IL-6)
IL-6 is a crucial pro-inflammatory cytokine that significantly contributes to the pathophysiology of sepsis and its sequelae, including ALI, ARDS, and multiple organ dysfunction syndrome (MODS) (Table 2).309,310 In numerous studies, increased plasma IL-6 levels have been demonstrated to distinguish sepsis from non-infectious SIRS and to forecast the advancement to ARDS, indicating its potential as a novel biomarker for prompt diagnosis and management, which may decrease patient morbidity and mortality.311 The predictive value of IL-6 as an independent marker has been questioned due to overlapping levels between survivors and non-survivors, indicating poor specificity. Additionally, inconsistencies in reported IL-6 concentrations across different studies have created uncertainty regarding appropriate therapeutic thresholds.312 This ambiguity is heightened by evidence showing that cytokine levels, such as IL-6, are not consistently elevated in severe COVID-19 cases compared to moderate cases or critically ill ARDS/sepsis patients. Therefore, the routine use of broad immunosuppressive treatments, which have largely been ineffective in unselected ARDS patients in Phase 3 trials, remains controversial.312
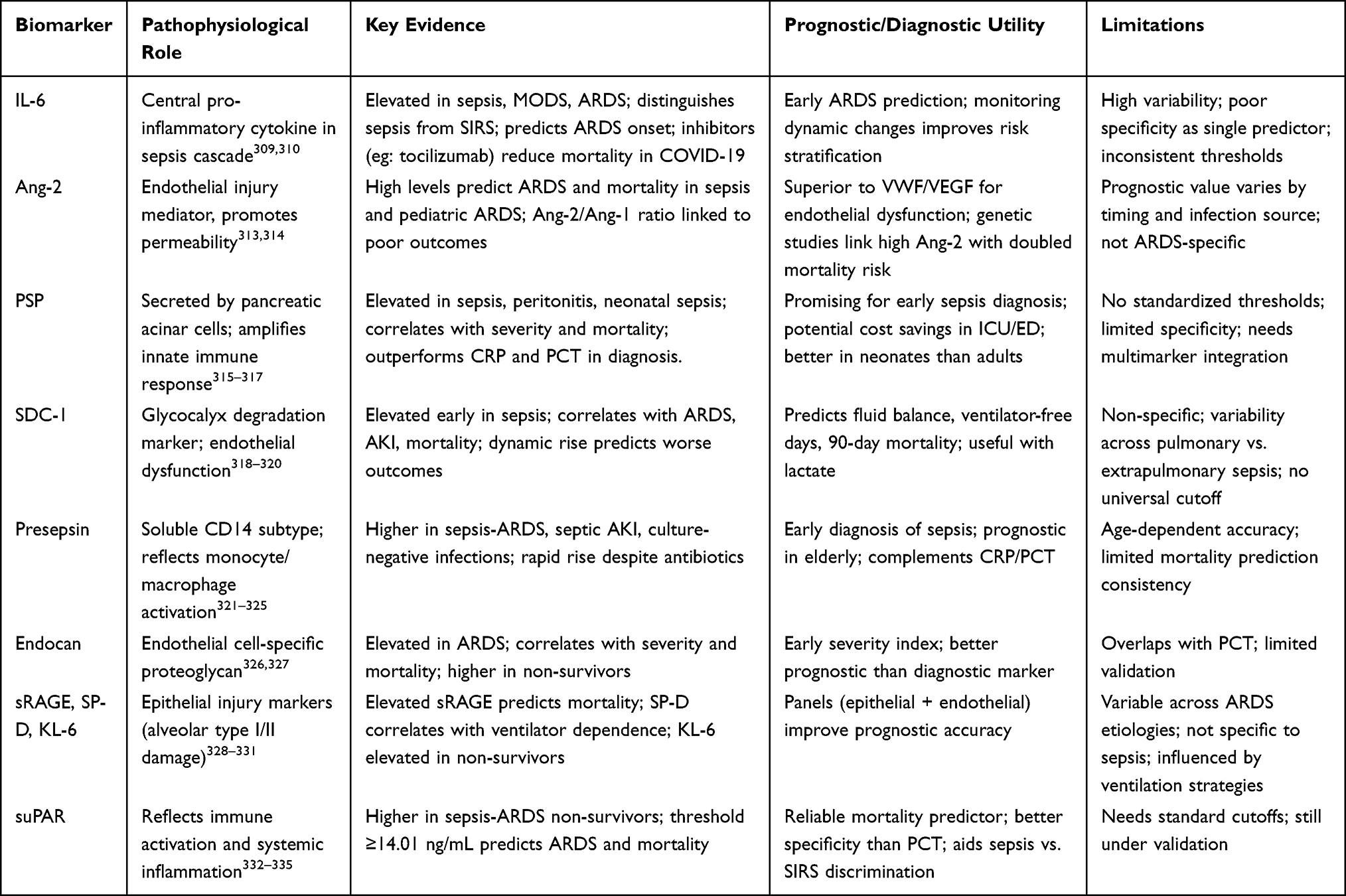 |
Table 2 Clinical Relevance of Emerging Biomarkers |
Numerous studies emphasize the importance of serial monitoring of IL-6 levels, particularly in COVID-19 patients, where high IL-6 concentrations are identified as independent risk factors for greater disease severity and in-hospital mortality, thus indicating the cytokine’s role in predicting aggravated lung injury.336 Research on ARDS patients indicated that serum levels of IL-6 and IL-8 were significantly elevated at presentation compared to healthy controls. Survivors showed a decrease toward baseline by Day 7, while non-survivors retained high levels.337 Moreover, heightened plasma IL-6 levels in critically ill septic patients have been associated with an augmented risk of developing ALI and MODS, with concentrations correlating with elevated Acute Physiology and Chronic Health Evaluation (APACHE) II and SOFA scores, serving as a biomarker for the severity and onset of lung injury.309 Baseline IL-6 levels are weak predictors of long-term outcomes when adjusted for confounding factors, but the temporal variation in IL-6 remains a significant predictor of death, supporting the need for serial monitoring in high-risk patients.338
Due to the constraints of utilizing IL-6 alone, researchers advocate for its integration with additional biomarkers, including PCT. The integrated dynamic evaluation of these markers is seen as more efficacious for the early identification of ARDS in individuals with MODS and for forecasting overall outcomes.339 Moreover, clinical efficacy findings endorse targeting IL-6; a meta-analysis of individual patient data from randomized trials verified that IL-6 inhibitors markedly decreased death and enhanced intubation rates and patient discharge relative to standard care in COVID-19 patients.340
Angiopoietin-2
Prior research indicates that elevated early plasma Ang-2 levels serve as a predictor for ARDS and mortality in sepsis patients. Higher Ang-2 levels are linked to increased illness severity, a greater likelihood of ARDS development, and higher mortality rates, especially in patients with both ARDS and sepsis compared to those with sepsis alone.313 Plasma Ang-2 levels have been found to be superior to other markers of endothelial damage, such as VWF and VEGF, in predicting mortality in pediatric ARDS. Elevated plasma Ang-2 is independently linked to a higher mortality risk, with a significant trend noted from days 1 to 3, particularly among pediatric hematopoietic stem cell transplant patients, highlighting ongoing endothelial injury (Table 2).314
Fluctuations in Ang-2 levels over time show varying relationships depending on assessment dates, with serial evaluations failing to effectively distinguish ARDS from non-ARDS conditions, thus limiting the reliability of a single measurement for diagnostics. In critically ill septic patients, levels of sRAGE, Ang-2, and SP-D were similar between ARDS and non-ARDS groups but were higher in non-survivors than in survivors.28 Early plasma Ang-2 levels are linked to overall disease severity, the onset of ARDS, and mortality risk, indicating that increased Ang-2 reflects general disease severity rather than ARDS-specific issues.313 This perplexing consequence complicates the interpretation of Ang-2 elevations as exclusively symptomatic of ARDS pathobiology. Conversely, new genetic research on individuals of European descent has revealed that the primary genetic factor influencing Ang-2 levels correlates with an increased risk of ARDS, indicating that plasma Ang-2 may have a causative involvement in the onset of ARDS.341 In cases of infection-related acute lung injury, initial Ang-2 levels were similar in survivors and non-survivors. However, a rise in plasma Ang-2 levels from day 0 to day 3 increased the likelihood of mortality by over twofold compared to declining levels, emphasizing the importance of tracking dynamic changes.342 The prognostic significance of plasma Ang-2, in contrast to VWF, is contingent upon the presence or absence of infection as a precipitating factor for acute lung damage.342
Moreover, numerous studies indicate that elevated baseline Ang-2 levels correlate with a heightened risk of mortality in ARDS patients. Increased levels of circulating Ang-2 may act as an independent predictor of mortality; nevertheless, additional extensive prospective and interventional investigations are required to elucidate its diagnostic significance and causative influence on ARDS outcomes.343 Although the research about Ang-2 is intriguing, the intricate and diverse characteristics of sepsis diminish the probability of appropriately categorizing sepsis severity using a singular biomarker.344 Canonical Correlation Analysis, alongside Forward Selection and Random Forest techniques, identified Ang-1, Ang-2, and bicarbonate (HCO3−) as significant biomarkers for sepsis severity. Their diagnostic reliability was confirmed through a linear Support Vector Machine (SVM) classifier. Furthermore, these biomarkers can predict the progression of sepsis severity in pediatric patients.344 In their study, Calfee et al measured Ang-2 levels in preserved samples from 931 participants in the ARDS Network Fluid and Catheter Treatment Trial (FACTT). They found that higher Ang-2 concentrations over time correlated significantly with worse clinical outcomes in ALI. Additionally, subgroup analyses revealed that individuals with infection-related ALI, such as pneumonia and sepsis, had elevated baseline Ang-2 levels compared to those with non-infectious ALI.342 The research by Agrawal et al indicated that elevated plasma Ang-2 levels measured upon admission to the emergency room or ICU can effectively predict the onset of ARDS an average of 22 hours before clinical symptoms appear in critically ill patients, primarily those with infections like pneumonia or sepsis.345
The role of Ang-2 as a diagnostic marker for ARDS progression is not fully elucidated, with some researchers urging caution in its use due to possible confounding variables.346 Ong et al demonstrated that the Ang-2 to Ang-1 ratio, in conjunction with established clinical and physiological variables, markedly enhanced risk prediction in patients with ALI.347 Ongoing research is essential to elucidate the role of Ang-2 and its potential applications in clinical practice, especially considering the conflicting findings related to its diagnostic and prognostic capabilities.346
Pancreatic Stone Protein (PSP)
PSP levels are highly effective in diagnosing the severity of peritonitis and predicting mortality in ICU patients with sepsis-related complications, highlighting their potential importance for clinical decision-making in critical care.315 Recent studies indicate that PSP may be effective in diagnosing sepsis, but further research is needed to determine its role in guiding early antibiotic treatment, particularly for newborns.316 Diagnosing newborn sepsis is problematic due to the vague clinical manifestations associated with the illness (Table 2).317 Schlapbach et al conducted an early evaluation of PSP in neonatal sepsis, revealing that infected infants exhibited significantly elevated PSP levels compared to their uninfected counterparts, with an area under the receiver operating characteristic curve (ROC) of 0.69.348 In a study of 156 emergency department patients, PSP levels were notably elevated in those with sepsis. Multivariate regression showed PSP as an independent sepsis predictor with an AUC of 0.69. While PSP outperformed traditional markers like white blood cell (WBC) counts and CRP, its diagnostic accuracy was insufficient for it to serve as a standalone biomarker, indicating a limitation in its clinical application.349 A systematic study and individual-patient-level meta-analysis evaluating the accuracy of PSP for diagnosing infections in hospitalized individuals determined that PSP is a potential biomarker. With a cutoff value of 44.18 ng/mL, PSP demonstrated superior performance compared to CRP and PCT in the analyzed studies.350
PSP levels are elevated in various inflammatory disorders and correlate with overall illness severity and organ dysfunction, complicating the differentiation between lung-specific ARDS and systemic sepsis, which reduces the specificity of PSP as a diagnostic tool in certain clinical contexts.351 In comparison to other biomarkers like PCT and CRP, PSP demonstrates superior performance and reliability in the diagnosis of sepsis in adults, providing potential advantages for patient management, doctors, and healthcare organizations. However, additional studies are required to define its ideal therapeutic applications and elucidate its function in conjunction with current biomarkers.352 The potential of PSP to provide additional prognostic or predictive value beyond established severity measures like SOFA and APACHE for ARDS outcomes remains uncertain.
Meta-analysis subgroup studies show that while PSP has adequate pooled sensitivity for newborn sepsis, it has limited specificity. This suggests that PSP should be used in combination with other biomarkers, emphasizing the need for rigorous prospective studies to support this approach.316 The expedited PSP test has shown promise in decreasing healthcare expenditures, with projected savings of $1688 per patient in the emergency room and $3315 per patient in the ICU relative to conventional care. The savings are mainly due to PSP’s specificity in the emergency department and its sensitivity in the ICU.353 Pooled sensitivity in adults is lower than in neonates, while specificity is higher in adults. This difference may be due to lower peak PSP levels in neonates, affecting diagnostic efficacy. Further research is needed to explore this disparity.316
Syndecan-1 (Sdc-1)
Sdc-1 serves as a biomarker for endothelial glycocalyx breakdown and is linked to poor outcomes in sepsis and septic shock. In patients experiencing septic shock, elevated plasma Sdc-1 levels during the first week correlated with the development of ARDS, particularly within the first 72 hours. Increased Sdc-1 was associated with fewer ventilator-free days, lower arterial oxygen partial pressure to fractional inspired oxygen ratio (PaO2/FiO2), and a greater positive fluid balance.318 Research on sepsis patients in the emergency department indicated that baseline and 6-hour Sdc-1 levels correlate with higher SOFA scores, increased fluid administration over 24–72 hours, septic shock diagnosis, the need for vasopressors or renal replacement therapy, and greater 90-day mortality rates (Table 2).319 The specificity of Sdc-1 as a biomarker for ARDS varies with the source of infection. In a study of 262 septic patients, elevated Sdc-1 levels were linked to ARDS mainly in cases of non-pulmonary sepsis, but not in pulmonary sepsis, showing that its prognostic relevance depends on the infection’s origin.320 The situation is exacerbated by variable cutoff thresholds, as there is no established standard for predicting ARDS or ventilator outcomes, and significant overlap in Sdc-1 levels is observed between patients who develop ARDS and those who do not.354
Comprehensive data substantiates the predictive value of Sdc-1 in sepsis. A meta-analysis involving roughly 2318 patients indicated that elevated Sdc-1 was substantially correlated with negative composite outcomes, including septic shock, acute renal injury, and heightened mortality.355 In pneumonia-induced sepsis, Sdc-1 showed strong correlations with neutrophil activation, respiratory failure, the necessity for MV, and 30-day mortality, in contrast to endocan.356 Repeated assessments of Sdc-1 on days 1, 2, and 7 in the ALBIOS substudy demonstrated independent correlations with the requirement for new renal replacement therapy, coagulation failure, and 90-day mortality.357 This indicates that the early presence of endothelial degradation products like hyaluronic acid (HA), Sdc-1, and heparan sulfate (HS) correlates positively with later organ dysfunction, allowing clinicians to use these changes for timely prediction and prevention.358
Sdc-1 demonstrates strong predictive capability for survival outcomes in sepsis patients. A study found that baseline Sdc-1 levels of ≥121 ng/mL were linked to reduced survival (p=0.001), and an increase of over 8 ng/mL within two days indicated a higher mortality risk (p=0.0075). The predictive performance of Sdc-1 (AUC = 0.706) improved when combined with blood lactate levels (AUC = 0.773), confirming that elevated or rising plasma Sdc-1 levels correlate with worse prognosis.321
Presepsin (P-SEP)
P-SEP is becoming a significant biomarker for distinguishing and forecasting outcomes in sepsis-induced ARDS. Evidence indicates that P-SEP levels are markedly higher in individuals with sepsis-related ARDS than in those with non-septic ARDS, aiding in the early differentiation between these causes.321,322 Moreover, in patients with sepsis-related ARDS, P-SEP levels were significantly elevated in non-survivors compared to survivors (P<0.001), establishing P-SEP as an independent predictor of in-hospital death and highlighting its potential as a prognostic instrument.322 The prognostic value of P-SEP is inconsistent. In a trial with 30 patients, 35% died before leaving the ICU, with no significant differences in levels of P-SEP, CRP, PCT, or IL-6 between survivors and non-survivors (Table 2).323 The ROC curve analysis did not identify reliable mortality predictions.
However, P-SEP, alongside PCT, CRP, and IL-6, was significantly higher in patients with positive microbiological cultures, suggesting its potential as a biomarker for detecting microbiological positivity in suspected sepsis, despite poor reliability in predicting in-hospital mortality (Table 2).323 The investigation into P-SEP’s effectiveness in sepsis patients indicates that it may be a better predictor of septic acute kidney injury and ARDS in those aged 75 and older, highlighting age-related differences in its applicability.324 P-SEP’s ability to quickly rise in the bloodstream while remaining accurate even after antibiotic treatment is crucial for early sepsis identification, particularly in culture-negative cases. This characteristic underscores its importance for antimicrobial stewardship. Future studies are expected to include profiles of polymicrobial and multidrug-resistant bloodstream infections to improve the therapeutic relevance of this biomarker.325
Endocan
Increased endocan levels correlate with adverse outcomes in patients suffering from sepsis-induced ARDS, indicating its potential as an early marker of severity. In research, endocan concentrations assessed the day after ARDS diagnosis were markedly elevated in patients with adverse clinical trajectories compared to those with more favorable outcomes (Table 2).326 Furthermore, at the time of ARDS admission, non-survivors demonstrated markedly elevated endocan levels compared to survivors. The diagnostic accuracy of endocan for identifying or assessing the severity of sepsis was equivalent to that of PCT; however, its predictive value for ARDS outcomes was notably superior.359
The investigation into the role of endocan in assessing ARDS severity suggests inconsistent results across three studies. Only one study found elevated endocan levels in patients experiencing deteriorating respiratory failure by Day 15, while the other two did not show significant correlations with ARDS severity. However, endocan levels were notably higher in all ARDS patients compared to non-ARDS individuals, with non-survivors consistently exhibiting greater levels than survivors. These findings imply that endocan may be relevant for evaluating both the severity and prognosis of ARDS, warranting further research into its therapeutic potential (Table 2).327
Soluble RAGE (sRAGE) and Epithelial Injury Markers (SP-D, KL-6)
A comprehensive meta-analysis found that high baseline levels of sRAGE are independently linked to increased 90-day mortality in ARDS patients, supporting the idea that alveolar epithelial injury is a key prognostic factor (Table 2).328 The role of epithelial and endothelial damage indicators in predicting ARDS is unclear. In a study of septic patients, levels of sRAGE, SP-D, and Ang-2 showed no significant differences between those who developed ARDS and those who did not, suggesting these biomarkers may indicate mortality risk rather than predict ARDS onset in sepsis.28
In critically ill septic patients, sRAGE, Ang-2, and SP-D levels did not significantly differ between ARDS and non-ARDS groups but were higher in non-survivors. ARDS patients showed a trend of elevated sRAGE and SP-D levels, indicating epithelial damage in sepsis-induced ARDS. However, changes in these biomarker levels over time did not effectively distinguish between the outcomes of the groups.28 Increased levels of the epithelial injury biomarker SP-D were observed in direct ARDS cases caused by viral and atypical pathogens, unlike ARDS from conventional bacterial pneumonia or non-pulmonary sepsis. sRAGE levels showed no significant changes based on the pathogen type. Patients with atypical pathogen-related ARDS had lower Ang-2 levels compared to those with viral or indirect ARDS. Additionally, serum IL-6 concentrations were significantly lower in patients with viral and atypical pneumonia-related ARDS than in those with bacterial pneumonia, with atypical cases reporting the lowest levels. These findings suggest that ARDS biomarker profiles vary by underlying illness and pathogen etiology.329
In mechanically ventilated patients with ALI or ARDS, SP-D levels had a tendency to rise with time, especially when potentially harmful ventilation methods were employed. Baseline SP-D levels correlated with extended ventilator reliance and increased mortality rates(Table).330 In the intrapulmonary ARDS subgroup, patients who succumbed exhibited markedly elevated serum KL-6 levels in the seven days post-diagnosis compared to survivors. In the extrapulmonary ARDS cohort, the disparity in KL-6 levels between survivors and non-survivors was significant solely on the first day following diagnosis. Patients with intra- and extrapulmonary ARDS in the mortality group demonstrated significantly elevated peak blood KL-6 levels relative to survivors.331 Baseline plasma concentrations of CC16 and KL-6 were significantly higher in ALI/ARDS patients than in those without lung injury, while SP-D and sRAGE levels showed no significant differences. Over time, SP-D and KL-6 levels increased in ALI/ARDS patients, with this increase being mitigated by lung-protective mechanical ventilation using lower tidal volumes.330
Soluble Urokinase Plasminogen Activator Receptor (suPAR)
In patients with sepsis-induced ARDS, serum suPAR levels correlate significantly with illness severity and prognosis, showing positive relations with clinical severity indices like APACHE II and SOFA scores, as well as inflammatory biomarkers such as CRP, TNF-α, IL-1β, and IL-8.332 Additionally, serum suPAR levels predict a higher risk of ARDS onset and were found to be lower in survivors compared to non-survivors.332 In a study of 104 patients with sepsis-induced ARDS, elevated plasma suPAR concentrations in the non-survivor group demonstrated substantial prognostic value for mortality, with an AUC of 0.81 (95% confidence interval: 0.73–0.89) in ROC curve analysis.333 A separate investigation identified suPAR as a reliable predictor of death, demonstrating significant discriminative ability through its AUC. Beyond prognostic use, suPAR has diagnostic potential; ROC analysis indicates it can differentiate between patients with sepsis plus ARDS and those with sepsis alone, achieving a sensitivity of 63.2% and specificity of 63.8% at a cut-off of 14.01 ng/mL.332 Additionally, suPAR helps in distinguishing sepsis from SIRS (Table 2).334 A study indicated that plasma suPAR levels remained stable during the first week of illness in both survivors and non-survivors.335 Regression analysis showed that an APACHE II score of ≥15 and suPAR levels of ≥10.82 ng/mL were independently associated with adverse outcomes, aiding in patient classification by severity with different fatality rates.335 Compared to PCT, suPAR not only offers similar clinical guidance but also exhibits superior specificity.334 Therefore, combining the APACHE II score and suPAR levels may provide a more effective predictive approach for assessing mortality risk in septic patients.335
Guideline-Baseline Methodologies
These treatments are underpinned by rigorous clinical studies and established protocols, providing a core framework for incorporating new medications. MV employs low tidal volume techniques, typically around 6 mL/kg of anticipated body weight, with a plateau pressure limit of ≤30 cm H2O. It also advocates for elevated positive end-expiratory pressure (PEEP) in mild to severe cases to prevent volutrauma and barotrauma.360 In moderate to severe ARDS, the prompt implementation of prone positioning for at least 12 to 16 hours daily in ventilated patients has demonstrated a considerable reduction in mortality.361
Neuromuscular blockade (NMB) is recommended in the initial stages of severe ARDS to enhance patient-ventilator synchronization and mitigate ventilator-induced lung injury (VILI). Data support the preference for occasional boluses over continuous infusion, accompanied by meticulous monitoring.362 In moderate to severe ARDS, recruitment maneuvers and PEEP titration procedures tailored to individual lung mechanics are recommended to enhance alveolar recruitment. In milder cases, excessive PEEP should be avoided (Table 3).363
 |
Table 3 Guideline-Based Core Strategies Management |
Fluid management is essential; a conservative fluid strategy following initial resuscitation may reduce pulmonary edema and improve outcomes. Dynamic parameters are favored over static measurements to guide treatment.360 Prompt treatment with corticosteroids, such as dexamethasone, in certain settings of ARDS, particularly moderate-to-severe or COVID-19-related ARDS, has shown a reduction in mechanical ventilation duration and may lower mortality rates, although the precise types, dosages, and timing of administration can vary.362
Ultimately, for patients with severe ARDS who do not respond to conventional ventilation, venovenous extracorporeal membrane oxygenation (ECMO) may be considered in facilities with sufficient expertise. These evidence-based techniques collectively endorse a holistic approach to ARDS management, combining contemporary best practices with ongoing innovations (Table 3).362
Novel Therapeutic and Experimental Targets
In sepsis and ARDS, damage to the pulmonary microvascular endothelium leads to capillary leakage, alveolar flooding, and impaired gas exchange. Therapeutic approaches designed to stabilize the endothelial barrier have shown potential in preclinical research. One strategy involves modulating the Ang-1/Ang-2–Tie2 axis, which has demonstrated efficacy in reducing vascular permeability and inflammation in experimental models (Table 4).364 Sphingosine-1-phosphate (S-1P), through its G-protein-coupled receptor S1P1, is essential for maintaining lung endothelial integrity. It triggers signaling pathways that engage Rho and Rac1, reorganizing the endothelial cytoskeleton to create cortical actin rings. This process strengthens focal adhesions and intercellular junctions, including cadherins and catenins. Consequently, this coordinated mechanism reduces vascular permeability and preserves the integrity of the alveolar–capillary barrier, thereby preventing alveolar flooding in ARDS models (Table 4).365,366 Moreover, statins have shown the ability to protect the lungs by maintaining endothelial integrity, reducing vascular permeability, and providing anti-inflammatory and antithrombotic effects.367 In endotoxin-induced lung damage models, simvastatin decreased protein permeability and pulmonary edema, restricted neutrophil migration, and reduced cytokine production from endothelial cells, including IL-6, IL-8, monocyte chemoattractant protein-1 (MCP-1), and regulated upon activation, normal T cell expressed and secreted (RANTES)(Table 4).368 These data suggest the efficacy of endothelial-targeted treatments in alleviating lung damage associated with sepsis-induced ARDS.
 |
Table 4 Novel Therapeutic and Experimental Targets |
The modulation of neutrophil migration and activation is a crucial therapeutic target in the treatment of ARDS. In patients with ARDS, a notable concentration of neutrophils is observed in bronchoalveolar lavage (BAL) fluid, and the sustained elevation of these neutrophil levels is closely correlated with impaired gas exchange and worse clinical outcomes.369 A primary mechanism governing neutrophil survival in hypoxic environments is the hypoxia-inducible factor (HIF) oxygen-sensing pathway. HIF has shown the potential to prolong neutrophil viability under hypoxic conditions, which are prevalent in inflamed pulmonary tissue. The function and expression of HIF are meticulously controlled by oxygen-sensitive prolyl hydroxylases (PHD1-3), which regulate HIF stability and transcriptional activity in response to oxygen levels.370 Understanding these processes underscores the importance of focusing on neutrophil activation and survival pathways to potentially mitigate inflammation and tissue injury in ARDS.
Immunomodulation and anti-cytokine medications have become significant in the treatment of sepsis and ARDS, particularly during the hyperinflammatory phase characterized by elevated concentrations of cytokines such as IL-6, IL-1, and TNF-α. The overproduction of cytokines, along with immunological dysregulation, substantially contributes to lung damage. Targeted inhibition of these inflammatory mediators presents a potential approach to reduce tissue damage. In the context of COVID-19-related ARDS, immunomodulatory treatments have become essential adjuncts, exhibiting the ability to mitigate the cytokine storm and reduce lethal lung injury. Early administration of tocilizumab, an IL-6 receptor antagonist, has been shown to dramatically decrease mortality (Table 1)371 however, the effectiveness of baricitinib remains less certain.372 Moreover, additional cytokine inhibitors, including sarilumab, canakinumab, adalimumab, and itolizumab, as well as agents such as dexamethasone and colchicine, have demonstrated potential advantages in decreasing mortality or altering disease progression; however, further validation through comprehensive studies is required (Table 4).373,374 Furthermore, combination therapies targeting both viral replication and immunological dysregulation, such as remdesivir paired with baricitinib or repurposed medication combinations like sirolimus and dactinomycin, are currently under investigation to enhance clinical outcomes.383
Mesenchymal stem cell (MSC) treatment has garnered attention for its immunomodulatory capabilities, capacity to facilitate epithelial and endothelial repair, regulation of inflammation, and potential to improve alveolar fluid clearance. Initial clinical studies have primarily focused on evaluating the safety and efficacy of MSCs in patients with ARDS. Zheng et al first conducted a trial using allogeneic adipose-derived MSCs in 12 patients, revealing no therapeutic effect while confirming safety.375 Subsequently, Wilson et al conducted a Phase 1 study using bone marrow-derived MSCs in nine patients with moderate-to-severe ARDS, reaffirming the intervention’s safety but acknowledging specific limitations. Recently, Matthay et al published findings from a multicenter, double-blind, Phase 2a study comprising 60 ventilated patients with ARDS. This research indicated that a single intravenous infusion of MSCs was safe and exhibited initial indications of clinical benefit, including improvements in APACHE II scores and ventilatory parameters, suggesting prospective therapeutic efficacy (Table 4).375
ARDS often manifests with elements of coagulopathy, microthrombosis, and fibrin accumulation in the alveoli. Medications that regulate coagulation and fibrinolysis may enhance alveolar gas exchange, whereas nebulized medications provide localized distribution to the lungs. Tissue factor (TF), a glycoprotein that increases during pulmonary inflammation, significantly contributes to fibrin deposition and exacerbates inflammation in ARDS. Increased blood levels of TF have been associated with higher mortality rates.376 In a Phase 1 placebo-controlled study, the anti-TF antibody ALT-836 demonstrated safety in patients with ARDS.377 A phase 2 study involving 150 septic ARDS patients was completed in 2013, although effectiveness data remain unreported. Moreover, heparin and antithrombin have demonstrated the ability to mitigate inflammation and ALI in preclinical animal studies without negatively impacting systemic coagulation.378 A study of 50 critically ill patients demonstrated that nebulized heparin reduced the duration of mechanical ventilation and improved alveolar perfusion and carbon dioxide removal in patients undergoing heart surgery.379,380 A phase 3 study indicated that nebulized streptokinase enhanced oxygenation and lung compliance in 60 patients with late-phase severe ARDS, suggesting its potential as a rescue treatment (Table 4).381 Adjunct therapy, encompassing drugs for diverse conditions, may affect the development of ARDS. Studies indicate that alpha-1 adrenergic receptor (α1-AR) antagonists can mitigate hyperinflammation and decrease mortality in murine models.382 Patients using α1-AR antagonists exhibited a 34% decrease in the likelihood of requiring mechanical ventilation and mortality. Additional trials are required to determine whether this medication effectively mitigates hyperinflammation and mortality in lower respiratory tract infections (Table 4).382
Futures Guidance And Individualized Medication
The future treatment of sepsis-induced ARDS is progressively emphasizing biomarker-guided therapy and precision medicine, moving away from the conventional “one-size-fits-all” approach. Circulating and respiratory biomarkers, including Ang-2, sRAGE, suPAR, and PSP, show promise in risk stratification, the identification of subphenotypes (hyperinflammatory vs. hypoinflammatory), and the prediction of important events, such as ventilator weaning success or mortality rates. The amalgamation of these biomarkers with clinical assessments and sophisticated computer models may enable tailored therapeutic approaches, optimizing the timing and selection of therapies such as corticosteroids, statins, MSCs, and immune modulators.
Notwithstanding these promising developments, many research deficiencies remain. Numerous biomarker investigations are limited by inadequate sample sizes, varied demographics, and insufficient validation across diverse clinical settings. The integration of potential biomarkers into standard clinical practice is impeded by inconsistencies in testing techniques, ambiguous cutoff values, and questionable cost-effectiveness. Future investigations should prioritize extensive, multicenter trials to validate biomarker panels, develop point-of-care tests, and explore the integration of genomics, proteomics, and machine learning into bedside decision-making. Such efforts might facilitate the development of truly personalized medicine in the treatment of sepsis-induced ARDS, ultimately enhancing outcomes in this potentially fatal condition.
Conclusion
Sepsis poses a significant global health challenge, impacting millions and serving as a major contributor to the development of ARDS. The intricate relationship between sepsis and ARDS results in severe outcomes, including elevated mortality rates and extended mechanical ventilation, highlighting the urgent need for effective management strategies. While advancements have been made in understanding the underlying mechanisms, notable gaps persist in the clinical application of biomarkers for predicting weaning success in this vulnerable population. The use of circulating and respiratory biomarkers presents a promising opportunity to enhance clinical decision-making and improve patient outcomes. By incorporating these indicators into standard practice, healthcare providers may more effectively navigate the complexities of weaning protocols, ultimately decreasing the duration of mechanical ventilation and lowering the risk of extubation failures. Ultimately, the implementation of validated biomarker-guided strategies could transform clinical management, leading to reduced ventilation days, fewer extubation failures, and improved survival rates in this high-risk population.
Abbreviations
BSF-2, B-cell stimulatory factor 2 malondialdehyde; LPS, Lipopolysaccharide; VE-PTP, Vascular Endothelial Protein Tyrosine Phosphatase; VE, Vascular Endothelial; ROS, reactive oxygen species; PRR, Pattern Recognition Receptors; DAMP, Damage-Associated Molecular Patterns; PAMPs, Pathogen-Associated Molecular Patterns; AUC, Area Under the Curve; ALI, Acute Lung Injury; Ang-1, Angiopoietin-1; Ang-2, Angiopoietin-2; APACHE, Acute Physiology and Chronic Health Evaluation; AR, Acute Respiratory; ARDS, Acute Respiratory Distress Syndrome; BAL, Bronchoalveolar Lavage; CHF, Congestive Heart Failure; COPD, Chronic Obstructive Pulmonary Disease; COVID-19, Coronavirus Disease 2019; CRP, C-Reactive Protein; DAMPs, Damage-Associated Molecular Patterns; ECMO, Extracorporeal Membrane Oxygenation; ExPreS, Extubation Predicting Score; G-CSF, Granulocyte Colony-Stimulating Factor; GTP, Guanosine Triphosphate; GPX4, Glutathione Peroxidase 4; HIF, Hypoxia-Inducible Factor; HMGB1, High Mobility Group Box 1; IL-1, Interleukin-1; IL-1β, Interleukin-1 Beta; IL-6, Interleukin-6; IMV, Intermittent Mandatory Ventilation; MCP-1, Monocyte Chemoattractant Protein-1; MSC, Mesenchymal Stem Cell; NMB, Neuromuscular Blockade; NEDD9, Neural Precursor Cell Expressed Developmentally Downregulated 9; PAMPs, Pathogen-Associated Molecular Patterns; PaO2/FiO2, Arterial Oxygen Partial Pressure to Fractional Inspired Oxygen Ratio; PEEP, Positive End-Expiratory Pressure; PCT, Procalcitonin; PSP, Pancreatic Stone Protein; PSV, Pressure Support Ventilation; RANTES, Regulated upon Activation, Normal T Cell Expressed and Secreted; RSBI, Rapid Shallow Breathing Index; sCD14, Soluble CD14; sRAGE, Soluble Receptor for Advanced Glycation End-products; S-1P, Sphingosine-1-Phosphate; SBT, Spontaneous Breathing Trial; SIRS, Systemic Inflammatory Response Syndrome; SOFA, Sequential Organ Failure Assessment; SP-D, Surfactant Protein D; suPAR, Soluble Urokinase Plasminogen Activator Receptor; TF, Tissue Factor; TNF-α, Tumor Necrosis Factor Alpha; TLR, Toll-like Receptor; VAP, Ventilator-Associated Pneumonia; VEGF, Vascular Endothelial Growth Factor; VILI, Ventilator-Induced Lung Injury; COPD, Chronic Obstructive Pulmonary Disease; SIRS, Systemic Inflammatory Response Syndrome; MODS, Multiple Organ Dysfunction Syndrome; VWF, von Willebrand factor; VEGF, Vascular Endothelial Growth Factor; sRAGE, soluble receptor for advanced glycation end-products; PSP, Pancreatic stone protein; ED, Emergency Department; SOFA, Sequential Organ Failure Assessment; APACHE, Acute Physiology and Chronic Health Evaluation; SDC-1, Syndecan-1; SP-D, surfactant protein D; SuPAR, Soluble urokinase plasminogen activator receptor; IMV, Intermittent Mandatory Ventilation; SIMV, Synchronized Intermittent Mandatory Ventilation; SBT, Spontaneous Breathing Trial; CC16, Clara Cell protein16; KL-6, Krebs von den Lungen-6; PSV, Pressure Support Ventilation; ExPreS, Extubation Predicting Score; HFNO, High-Flow Nasal Oxygen; PEEP, Positive End-Expiratory Pressure; NMB, Neuromuscular blockade; VILI, Ventilator-Induced Lung Injury; MSC, Mesenchymal stem cell.
Data Sharing Statement
As this review relies solely on previously published data, data availability does not apply.
Ethical Approval
This study did not include original research involving humans or animals; hence, ethical approval was not required.
Author Contributions
Professor Sun and Professor Liu contributed to the conceptualization and supervision of the study. Dr. Emery contributed to the study design and execution, while Dr. Geoffrey contributed to the study design and the writing of the original draft. All authors participated in writing, review, and editing of the manuscript. Each author has given final approval for the version to be published, agreed on the journal to which the article has been submitted, and accepts accountability for all aspects of the work.
Funding
This study was supported by The Noncommunicable Chronic Diseases-National Science and Technology Major Project (Grant No. 2023ZD0506504), National Natural Science Foundation of China (Grant No. 8217212), Scientific Research and Innovation Excellence Team of First Affiliated Hospital of Zhengzhou University (Grant No. ZYCXTD2023006).
Disclosure
The authors declare that there are no conflicts of interest.
References
1. Srzić I, Nesek Adam V, Tunjić Pejak D. Sepsis definition: what’s new in the treatment guidelines. Acta Clin Croat. 2022;61(Suppl 1):67–35. doi:10.20471/acc.2022.61.s1.11
2. Gauer R, Forbes D, Boyer N. Sepsis: diagnosis and management. Am Family Phys. 2020;101(7):409–418.
3. Sun Q, Jian J, Zhou X, et al. Population pharmacokinetics of teicoplanin and dosage optimization in sepsis patients based on continuous renal replacement therapy. Front Pharmacol. 2025;16:1621959. doi:10.3389/fphar.2025.1621959
4. Bauer M, Gerlach H, Vogelmann T, Preissing F, Stiefel J, Adam D. Mortality in sepsis and septic shock in Europe, North America and Australia between 2009 and 2019—results from a systematic review and meta-analysis. Critical Care. 2020;24(1):239. doi:10.1186/s13054-020-02950-2
5. Bolanaki M, Kurland L, Brabrand M, et al. Current sepsis management practices in European emergency departments: the ISG-emergency department European Survey. Eur J Emerg Med. 2025;32(5):368–376. doi:10.1097/mej.0000000000001255
6. Xu H, Sheng S, Luo W, Xu X, Zhang Z. Acute respiratory distress syndrome heterogeneity and the septic ARDS subgroup. Front Immunol. 2023;14:1277161. doi:10.3389/fimmu.2023.1277161
7. Hendrickson KW, Peltan ID, Brown SM. The epidemiology of acute respiratory distress syndrome before and after coronavirus disease 2019. Crit Care Clin. 2021;37(4):703–716. doi:10.1016/j.ccc.2021.05.001
8. Li S, Zhao D, Cui J, Wang L, Ma X, Li Y. Prevalence, potential risk factors and mortality rates of acute respiratory distress syndrome in Chinese patients with sepsis. J Int Med Res. 2020;48(2):300060519895659. doi:10.1177/0300060519895659
9. Kim WY, Hong SB. Sepsis and acute respiratory distress syndrome: recent update. Tuberc Respir Dis. 2016;79(2):53–57. doi:10.4046/trd.2016.79.2.53
10. Eworuke E, Major JM, Gilbert McClain LI. National incidence rates for Acute Respiratory Distress Syndrome (ARDS) and ARDS cause-specific factors in the United States (2006-2014). J Crit Care. 2018;47:192–197. doi:10.1016/j.jcrc.2018.07.002
11. Englert JA, Bobba C, Baron RM. Integrating molecular pathogenesis and clinical translation in sepsis-induced acute respiratory distress syndrome. JCI Insight. 2019;4(2):e124061. doi:10.1172/jci.insight.124061
12. Kiya GT, Mekonnen Z, Melaku T, et al. Prevalence and mortality rate of sepsis among adults admitted to hospitals in sub-Saharan Africa: a systematic review and meta-analysis. J Hosp Infect. 2024;144:1–13. doi:10.1016/j.jhin.2023.11.012
13. Gong H, Chen Y, Chen M, et al. Advanced development and mechanism of sepsis-related acute respiratory distress syndrome. Front Med. 2022;9:1043859. doi:10.3389/fmed.2022.1043859
14. Vassiliou AG, Kotanidou A, Dimopoulou I, Orfanos SE. Endothelial damage in acute respiratory distress syndrome. Int J Mol Sci. 2020;21(22):8793. doi:10.3390/ijms21228793
15. Gorman EA, O’Kane CM, McAuley DF. Acute respiratory distress syndrome in adults: diagnosis, outcomes, long-term sequelae, and management. Lancet. 2022;400(10358):1157–1170. doi:10.1016/S0140-6736(22)01439-8
16. Huppert LA, Matthay MA, Ware LB. Pathogenesis of acute respiratory distress syndrome. Semin Respir Crit Care Med. 2019;40(1):31–39. doi:10.1055/s-0039-1683996
17. Matthay MA, Arabi Y, Arroliga AC, et al. A new global definition of acute respiratory distress syndrome. Am J Respir Crit Care Med. 2024;209(1):37–47. doi:10.1164/rccm.202303-0558WS
18. Matthay MA, Aldrich JM, Gotts JE. Treatment for severe acute respiratory distress syndrome from COVID-19. Lancet Respir Med. 2020;8(5):433–434. doi:10.1016/S2213-2600(20)30127-2
19. Grotberg JC, Reynolds D, Kraft BD. Management of severe acute respiratory distress syndrome: a primer. Critical Care. 2023;27(1):289. doi:10.1186/s13054-023-04572-w
20. Fujishima S. Guideline-based management of acute respiratory failure and acute respiratory distress syndrome. J Intens Care. 2023;11(1):10. doi:10.1186/s40560-023-00658-3
21. Ha TS, Oh DK, Lee HJ, et al. Liberation from mechanical ventilation in critically ill patients: Korean Society of critical care medicine clinical practice guidelines. Tuberc Respir Dis. 2024;87(4):415–439. doi:10.4046/trd.2024.0039
22. Mandyam S, Qureshi M, Katamreddy Y, et al. Neurally Adjusted ventilatory assist versus pressure support ventilation: a comprehensive review. J Intens Care Med. 2024;39(12):1194–1203. doi:10.1177/08850666231212807
23. Swenson KE, Swenson ER. Pathophysiology of acute respiratory distress syndrome and COVID-19 lung injury. Crit Care Clin. 2021;37(4):749–776. doi:10.1016/j.ccc.2021.05.003
24. Trivedi V, Chaudhuri D, Jinah R, et al. The Utility of the rapid shallow breathing index in predicting successful extubation: a systematic review and meta-analysis. Chest. 2021;161(1):97–111. doi:10.1016/j.chest.2021.06.030
25. Fadaii A, Amini SS, Bagheri B, Taherkhanchi B. Assessment of rapid shallow breathing index as a predictor for weaning in respiratory care unit. Tanaffos. 2012;11(3):28–31.
26. García-Laorden MI, Lorente JA, Flores C, Slutsky AS, Villar J. Biomarkers for the acute respiratory distress syndrome: how to make the diagnosis more precise. Ann Translat Med. 2017;5(14):283. doi:10.21037/atm.2017.06.49
27. Spadaro S, Park M, Turrini C, et al. Biomarkers for Acute Respiratory Distress syndrome and prospects for personalised medicine. J Inflamm. 2019;16(1):1. doi:10.1186/s12950-018-0202-y
28. Yang P, Iffrig E, Harris F, Holder AL, Martin GS, Esper AM. Serial measurements of protein biomarkers in sepsis-induced acute respiratory distress syndrome. Crit Care Explor. 2022;4(10):e0780. doi:10.1097/cce.0000000000000780
29. Do Nascimento MF, Ferreira LRP, Vieira Junior JM, et al. Circulating extracellular vesicles as potential biomarkers and mediators of acute respiratory distress syndrome in sepsis. Sci Rep. 2025;15(1):5512. doi:10.1038/s41598-025-89783-7
30. Park JE, Kim TY, Jung YJ, et al. Biosignal-Based digital biomarkers for prediction of ventilator weaning success. Int J Environ Res Public Health. 2021;18(17). doi:10.3390/ijerph18179229
31. Stivi T, Padawer D, Dirini N, Nachshon A, Batzofin BM, Ledot S. Using artificial intelligence to predict mechanical ventilation weaning success in patients with respiratory failure, including those with acute respiratory distress syndrome. J Clin Med. 2024;13(5):1505. doi:10.3390/jcm13051505
32. Raventós AA, Serpa Neto A. Biomarkers to guide ventilation management and readiness for extubation. Am J Respir Crit Care Med. 2021;203(10):1211–1212. doi:10.1164/rccm.202101-0093ED
33. Bodaghi A, Fattahi N, Ramazani A. Biomarkers: promising and valuable tools towards diagnosis, prognosis and treatment of Covid-19 and other diseases. Heliyon. 2023;9(2):e13323. doi:10.1016/j.heliyon.2023.e13323
34. Ahmad A, Imran M, Ahsan H. Biomarkers as biomedical bioindicators: approaches and techniques for the detection, analysis, and validation of novel biomarkers of diseases. Pharmaceutics. 2023;15(6):1630. doi:10.3390/pharmaceutics15061630
35. Patel P, Walborn A, Rondina M, Fareed J, Hoppensteadt D. Markers of inflammation and infection in sepsis and disseminated intravascular coagulation. Clin Appl Thromb Hemost. 2019;25:1076029619843338. doi:10.1177/1076029619843338
36. McKenzie C, Mumin M, Page V, McAuley DF. 42nd international symposium on intensive care & emergency medicine. Critical Care. 2023;27(1):119. doi:10.1186/s13054-023-04377-x
37. Bajwa EK, Boyce PD, Januzzi JL, Gong MN, Thompson BT, Christiani DC. Biomarker evidence of myocardial cell injury is associated with mortality in acute respiratory distress syndrome. Crit Care Med. 2007;35(11):2484–2490. doi:10.1097/01.CCM.0000281852.36573.22
38. Alladina JW, Levy SD, Hibbert KA, et al. Plasma concentrations of soluble suppression of tumorigenicity-2 and interleukin-6 are predictive of successful liberation from mechanical ventilation in patients with the acute respiratory distress syndrome*. Crit Care Med. 2016;44(9):1735–1743. doi:10.1097/ccm.0000000000001814
39. Alladina J, Levy SD, Cho JL, et al. Plasma soluble suppression of tumorigenicity-2 associates with ventilator liberation in acute hypoxemic respiratory failure. Am J Respir Crit Care Med. 2021;203(10):1257–1265. doi:10.1164/rccm.202005-1951OC
40. Raventós AA, Serpa Neto A. Biomarkers to guide ventilation management and readiness for extubation. Am Thorac Soc. 2021;2021:1211–1212.
41. Delaunay E, Poussard N, Gourjon G, et al. Role of InterLeukin-6 monitoring during weaning from volume-controlled ventilation in patients with COVID-19 acute respiratory distress syndrome. Sci Prog. 2025;108(2):00368504251335850. doi:10.1177/00368504251335850
42. Liu J, Wang C-J, Ran J-H, et al. The predictive value of brain natriuretic peptide or N-terminal pro-brain natriuretic peptide for weaning outcome in mechanical ventilation patients: evidence from SROC. J Renin Angiotensin Aldosterone Syst. 2021;22(1):1470320321999497. doi:10.1177/1470320321999497
43. Son E, Cho WH, Jang JH, et al. Neutrophil gelatinase-associated lipocalin as a prognostic biomarker of severe acute respiratory distress syndrome. Sci Rep. 2022;12(1):7909. doi:10.1038/s41598-022-12117-4
44. Bellani G, Laffey JG, Pham T, et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA. 2016;315(8):788–800. doi:10.1001/jama.2016.0291
45. Huang M, Cai S, Su J. The pathogenesis of sepsis and potential therapeutic targets. Int J Mol Sci. 2019;20(21):5376. doi:10.3390/ijms20215376
46. Hu Q, Hao C, Tang S. From sepsis to acute respiratory distress syndrome (ARDS): emerging preventive strategies based on molecular and genetic researches. Biosci Rep. 2020;40(5). doi:10.1042/bsr20200830
47. Reiss LK, Schuppert A, Uhlig S. Inflammatory processes during acute respiratory distress syndrome: a complex system. Curr Opinion Crit Care. 2018;24(1):1–9. doi:10.1097/MCC.0000000000000472
48. Sharp C, Millar AB, Medford AR. Advances in understanding of the pathogenesis of acute respiratory distress syndrome. Respiration. 2015;89(5):420–434. doi:10.1159/000381102
49. Chavez A, Smith M, Mehta D. New insights into the regulation of vascular permeability. Int Rev Cell Mol Biol. 2011;290:205–248.
50. Zhou A, Chen K, Gao Y, et al. Bioengineered neutrophil extinguisher targets cascade immune pathways of macrophages for alleviating cytokine storm in pneumonia. ACS Nano. 2023;17(17):16461–16477. doi:10.1021/acsnano.3c00227
51. Raymond SL, Holden DC, Mira JC, et al. Microbial recognition and danger signals in sepsis and trauma. BBA. 2017;1863(10):2564–2573. doi:10.1016/j.bbadis.2017.01.013
52. Rathinam VA, Fitzgerald KA. Inflammasome complexes: emerging mechanisms and effector functions. Cell. 2016;165(4):792–800. doi:10.1016/j.cell.2016.03.046
53. Wang Y, Zhang S, Li H, et al. Small-molecule modulators of toll-like receptors. Acc Chem Res. 2020;53(5):1046–1055. doi:10.1021/acs.accounts.9b00631
54. Deng M, Tang Y, Li W, et al. The endotoxin delivery protein HMGB1 mediates caspase-11-dependent lethality in sepsis. Immunity. 2018;49(4):740–753.e7. doi:10.1016/j.immuni.2018.08.016
55. Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science. 2013;341(6151):1250–1253. doi:10.1126/science.1240988
56. Goode HF, Cowley HC, Walker BE, Howdle PD, Webster NR. Decreased antioxidant status and increased lipid peroxidation in patients with septic shock and secondary organ dysfunction. Crit Care Med. 1995;23(4):646–651. doi:10.1097/00003246-199504000-00011
57. Galley HF, Davies MJ, Webster NR. Xanthine oxidase activity and free radical generation in patients with sepsis syndrome. Crit Care Med. 1996;24(10):1649–1653. doi:10.1097/00003246-199610000-00008
58. Cowley HC, Bacon PJ, Goode HF, Webster NR, Jones JG, Menon DK. Plasma antioxidant potential in severe sepsis: a comparison of survivors and nonsurvivors. Crit Care Med. 1996;24(7):1179–1183. doi:10.1097/00003246-199607000-00019
59. Sies H. Oxidative stress: a concept in redox biology and medicine. Redox Biol. 2015;4:180–183. doi:10.1016/j.redox.2015.01.002
60. Joffre J, Hellman J. Oxidative stress and endothelial dysfunction in sepsis and acute inflammation. Antioxid Redox Signal. 2021;35(15):1291–1307. doi:10.1089/ars.2021.0027
61. Chow C-W, Herrera Abreu MT, Suzuki T, Downey GP. Oxidative stress and acute lung injury. Am J Respir Cell Mol Biol. 2003;29(4):427–431. doi:10.1165/rcmb.F278
62. Qin H, Fan J, Mao S. Exploring the mechanism of the Fe (iii)-activated Fenton-like reaction based on a quantitative study. New J Chem. 2020;44(21):8952–8959. doi:10.1039/C9NJ06104E
63. Bargagli E, Olivieri C, Bennett D, Prasse A, Muller-Quernheim J, Rottoli P. Oxidative stress in the pathogenesis of diffuse lung diseases: a review. Respir Med. 2009;103(9):1245–1256. doi:10.1016/j.rmed.2009.04.014
64. Bertozzi G, Ferrara M, Di Fazio A, et al. Oxidative stress in sepsis: a focus on cardiac pathology. Int J Mol Sci. 2024;25(5):2912. doi:10.3390/ijms25052912
65. Kumar S, Saxena J, Srivastava VK, et al. The interplay of oxidative stress and ROS scavenging: antioxidants as a therapeutic potential in sepsis. Vaccines. 2022;10(10):1575. doi:10.3390/vaccines10101575
66. Fu X, Dong J, Yang J, et al. Identification and validation of oxidative stress-related genes for the diagnosis of sepsis-induced acute lung injury. PLoS One. 2025;20(7):e0327945. doi:10.1371/journal.pone.0327945
67. Li R, Shang Y, Yu Y, Zhou T, Xiong W, Zou X. High-mobility group box 1 protein participates in acute lung injury by activating protein kinase R and inducing M1 polarization. Life Sci. 2020;246:117415. doi:10.1016/j.lfs.2020.117415
68. Speer CP, Pabst MJ, Hedegaard HB, Rest RF, Johnston RB. Enhanced release of oxygen metabolites by monocyte-derived macrophages exposed to proteolytic enzymes: activity of neutrophil elastase and cathepsin G. J Immunol. 1984;133(4):2151–2156. doi:10.4049/jimmunol.133.4.2151
69. Ye C, Xu B, Yang J, et al. Mucin1 relieves acute lung injury by inhibiting inflammation and oxidative stress. Eur J Histochem. 2021;65(4). doi:10.4081/ejh.2021.3331
70. Liu Y, Zhang Y, Feng Q, et al. GPA peptide attenuates sepsis-induced acute lung injury in mice via inhibiting oxidative stress and pyroptosis of alveolar macrophage. Oxid Med Cell Longev. 2021;2021(1):5589472. doi:10.1155/2021/5589472
71. Mehta Y, Dixit SB, Zirpe K, et al. Therapeutic approaches in modulating the inflammatory and immunological response in patients with sepsis, acute respiratory distress syndrome, and pancreatitis: an expert opinion review. Cureus. 2021;13(9):e18393. doi:10.7759/cureus.18393
72. Thompson BT, Chambers RC, Liu KD, Drazen JM. Acute respiratory distress syndrome. N Engl J Med. 2017;377(6):562–572. doi:10.1056/NEJMra1608077
73. Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest. 2012;122(8):2731–2740. doi:10.1172/jci60331
74. Fan EKY, Fan J. Regulation of alveolar macrophage death in acute lung inflammation. Respir Res. 2018;19(1):50. doi:10.1186/s12931-018-0756-5
75. Yang J, Zhao Y, Zhang P, et al. Hemorrhagic shock primes for lung vascular endothelial cell pyroptosis: role in pulmonary inflammation following LPS. Cell Death Dis. 2016;7(9):e2363. doi:10.1038/cddis.2016.274
76. Li Z, Scott MJ, Fan EK, et al. Tissue damage negatively regulates LPS-induced macrophage necroptosis. Cell Death Differ. 2016;23(9):1428–1447. doi:10.1038/cdd.2016.21
77. Franklin BS, Bossaller L, De Nardo D, et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat Immunol. 2014;15(8):727–737. doi:10.1038/ni.2913
78. He X, Qian Y, Li Z, et al. TLR4-Upregulated IL-1β and IL-1RI promote alveolar macrophage pyroptosis and lung inflammation through an autocrine mechanism. Sci Rep. 2016;6(1):31663. doi:10.1038/srep31663
79. Juettner VV, Kruse K, Dan A, et al. VE-PTP stabilizes VE-cadherin junctions and the endothelial barrier via a phosphatase-independent mechanism. J Cell Biol. 2019;218(5):1725–1742. doi:10.1083/jcb.201807210
80. Frye M, Dierkes M, Küppers V, et al. Interfering with VE-PTP stabilizes endothelial junctions in vivo via Tie-2 in the absence of VE-cadherin. J Exp Med. 2015;212(13):2267–2287. doi:10.1084/jem.20150718
81. Giannotta M, Trani M, Dejana E. VE-cadherin and endothelial adherens junctions: active guardians of vascular integrity. Dev Cell. 2013;26(5):441–454. doi:10.1016/j.devcel.2013.08.020
82. Jones JH, Minshall RD. Endothelial Transcytosis in acute lung injury: emerging mechanisms and therapeutic approaches. Front Physiol. 2022;13:828093. doi:10.3389/fphys.2022.828093
83. Schnoor M, García Ponce A, Vadillo E, Pelayo R, Rossaint J, Zarbock A. Actin dynamics in the regulation of endothelial barrier functions and neutrophil recruitment during endotoxemia and sepsis. Cell Mol Life Sci. 2017;74(11):1985–1997. doi:10.1007/s00018-016-2449-x
84. Broermann A, Winderlich M, Block H, et al. Dissociation of VE-PTP from VE-cadherin is required for leukocyte extravasation and for VEGF-induced vascular permeability in vivo. J Exp Med. 2011;208(12):2393–2401. doi:10.1084/jem.20110525
85. Curley GF, Laffey JG, Zhang H, Slutsky AS. Biotrauma and Ventilator-induced lung injury: clinical implications. Chest. 2016;150(5):1109–1117. doi:10.1016/j.chest.2016.07.019
86. Matthay MA, Zemans RL, Zimmerman GA, et al. Acute respiratory distress syndrome. Nature Reviews Disease Primers. 2019;5(1):18. doi:10.1038/s41572-019-0069-0
87. Weibel ER. On the tricks alveolar epithelial cells play to make a good lung. Am J Respir Crit Care Med. 2015;191(5):504–513. doi:10.1164/rccm.201409-1663OE
88. John AE, Joseph C, Jenkins G, Tatler AL. COVID-19 and pulmonary fibrosis: a potential role for lung epithelial cells and fibroblasts. Immunol Rev. 2021;302(1):228–240. doi:10.1111/imr.12977
89. Liberti DC, Kremp MM, Liberti WA, et al. Alveolar epithelial cell fate is maintained in a spatially restricted manner to promote lung regeneration after acute injury. Cell Rep. 2021;35(6):109092. doi:10.1016/j.celrep.2021.109092
90. Tojo K, Yamamoto N, Tamada N, et al. Early alveolar epithelial cell necrosis is a potential driver of COVID-19-induced acute respiratory distress syndrome. iScience. 2023;26(1):105748. doi:10.1016/j.isci.2022.105748
91. Qin H, Zhao A. Mesenchymal stem cell therapy for acute respiratory distress syndrome: from basic to clinics. Protein Cell. 2020;11(10):707–722. doi:10.1007/s13238-020-00738-2
92. Cui Y, Wang X, Lin F, et al. MiR-29a-3p improves acute lung injury by reducing alveolar epithelial cell PANoptosis. Aging Dis. 2022;13(3):899–909. doi:10.14336/ad.2021.1023
93. Hussain N, Wu F, Zhu L, Thrall RS, Kresch MJ. Neutrophil apoptosis during the development and resolution of oleic acid-induced acute lung injury in the rat. Am J Respir Cell Mol Biol. 1998;19(6):867–874. doi:10.1165/ajrcmb.19.6.3118
94. Pokharel MD, Garcia-Flores A, Marciano D, et al. Mitochondrial network dynamics in pulmonary disease: bridging the gap between inflammation, oxidative stress, and bioenergetics. Redox Biol. 2024;70:103049. doi:10.1016/j.redox.2024.103049
95. Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9(3):231–241. doi:10.1038/nrm2312
96. Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res. 2019;29(5):347–364. doi:10.1038/s41422-019-0164-5
97. newton K, Strasser A, Kayagaki N, Dixit VM. Cell death. Cell. 2024;187(2):235–256. doi:10.1016/j.cell.2023.11.044
98. Johnson AG, Wein T, Mayer ML, et al. Bacterial gasdermins reveal an ancient mechanism of cell death. Science. 2022;375(6577):221–225. doi:10.1126/science.abj8432
99. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26(4):239–257. doi:10.1038/bjc.1972.33
100. Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281(5381):1312–1316. doi:10.1126/science.281.5381.1312
101. Portt L, Norman G, Clapp C, Greenwood M, Greenwood MT. Anti-apoptosis and cell survival: a review. BBA. 2011;1813(1):238–259. doi:10.1016/j.bbamcr.2010.10.010
102. Horowitz JC, Rogers DS, Sharma V, et al. Combinatorial activation of FAK and AKT by transforming growth factor-β1 confers an anoikis-resistant phenotype to myofibroblasts. Cell Signalling. 2007;19(4):761–771. doi:10.1016/j.cellsig.2006.10.001
103. Ryter SW, Kim HP, Hoetzel A, et al. Mechanisms of cell death in oxidative stress. Antioxid Redox Signaling. 2007;9(1):49–89. doi:10.1089/ars.2007.9.49
104. Galani V, Tatsaki E, Bai M, et al. The role of apoptosis in the pathophysiology of Acute Respiratory Distress Syndrome (ARDS): an up-to-date cell-specific review. Pathol Res Pract. 2010;206(3):145–150. doi:10.1016/j.prp.2009.12.002
105. Weinlich R, Oberst A, Beere HM, Green DR. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol. 2017;18(2):127–136. doi:10.1038/nrm.2016.149
106. Li D, Xu T, Cao Y, et al. A cytosolic heat shock protein 90 and cochaperone CDC37 complex is required for RIP3 activation during necroptosis. Proc Natl Acad Sci. 2015;112(16):5017–5022. doi:10.1073/pnas.1505244112
107. Liao J, Liang Y, Liu Z, et al. Pyroptosis in acute respiratory distress syndrome and pulmonary fibrosis. Biomed Pharmacother. 2025;189:118286. doi:10.1016/j.biopha.2025.118286
108. Mitra S, Exline M, Habyarimana F, et al. Microparticulate caspase 1 regulates gasdermin D and pulmonary vascular endothelial cell injury. Am J Respir Cell Mol Biol. 2018;59(1):56–64. doi:10.1165/rcmb.2017-0393OC
109. Vasudevan SO, Behl B, Rathinam VA. Pyroptosis-induced inflammation and tissue damage. Semin Immunol. 2023;69:101781. doi:10.1016/j.smim.2023.101781
110. Jones HD, Crother TR, Gonzalez-Villalobos RA, et al. The NLRP3 inflammasome is required for the development of hypoxemia in LPS/mechanical ventilation acute lung injury. Am J Respir Cell Mol Biol. 2014;50(2):270–280. doi:10.1165/rcmb.2013-0087OC
111. Song C, Li H, Li Y, et al. NETs promote ALI/ARDS inflammation by regulating alveolar macrophage polarization. Exp Cell Res. 2019;382(2):111486. doi:10.1016/j.yexcr.2019.06.031
112. Fujimura K, Karasawa T, Komada T, et al. NLRP3 inflammasome-driven IL-1β and IL-18 contribute to lipopolysaccharide-induced septic cardiomyopathy. J Mol Cell Cardiol. 2023;180:58–68. doi:10.1016/j.yjmcc.2023.05.003
113. Peukert K, Sauer A, Seeliger B, et al. Increased alveolar epithelial damage markers and inflammasome-regulated cytokines are associated with pulmonary superinfection in ARDS. J Clin Med. 2023;12(11):3649. doi:10.3390/jcm12113649
114. Huang Q, Le Y, Li S, Bian Y. Signaling pathways and potential therapeutic targets in acute respiratory distress syndrome (ARDS). Respir Res. 2024;25(1):30. doi:10.1186/s12931-024-02678-5
115. Neu C, Thiele Y, Horr F, et al. DAMPs released from proinflammatory macrophages induce inflammation in cardiomyocytes via activation of TLR4 and TNFR. Int J Mol Sci. 2022;23(24):15522. doi:10.3390/ijms232415522
116. Cicco S, Cicco G, Racanelli V, Vacca A. Neutrophil extracellular traps (NETs) and damage‐associated molecular patterns (DAMPs): two potential targets for COVID‐19 treatment. Mediators Inflammation. 2020;2020(1):7527953. doi:10.1155/2020/7527953
117. Xl L, Gy Z, G R, C N. Ferroptosis in sepsis: the mechanism, the role and the therapeutic potential. Front Immunol. 2022;13:956361. doi:10.3389/fimmu.2022.956361
118. Zheng Y, Huang Y, Xu Y, Sang L, Liu X, Li Y. Ferroptosis, pyroptosis and necroptosis in acute respiratory distress syndrome. Cell Death Discovery. 2023;9(1):91. doi:10.1038/s41420-023-01369-2
119. Ma D, Jiang P, Jiang Y, Li H, Zhang D, Cipak Gasparovic A. Effects of lipid peroxidation‐mediated ferroptosis on severe acute pancreatitis‐induced intestinal barrier injury and bacterial translocation. Oxid Med Cell Longev. 2021;2021(1):6644576. doi:10.1155/2021/6644576
120. Dar HH, Tyurina YY, Mikulska-Ruminska K, et al. Pseudomonas aeruginosa utilizes host polyunsaturated phosphatidylethanolamines to trigger theft-ferroptosis in bronchial epithelium. J Clin Invest. 2019;128(10):4639–4653. doi:10.1172/JCI99490
121. Ma R, Fang L, Chen L, Wang X, Jiang J, Gao L. Ferroptotic stress promotes macrophages against intracellular bacteria. Theranostics. 2022;12(5):2266. doi:10.7150/thno.66663
122. Amaral EP, Costa DL, Namasivayam S, et al. A major role for ferroptosis in Mycobacterium tuberculosis–induced cell death and tissue necrosis. J Exp Med. 2019;216(3):556–570. doi:10.1084/jem.20181776
123. Matsushita M, Freigang S, Schneider C, Conrad M, Bornkamm GW, Kopf M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med. 2015;212(4):555–568. doi:10.1084/jem.20140857
124. Liu Q, Gao Y, Ci X. Role of Nrf2 and its activators in respiratory diseases. Oxid Med Cell Longev. 2019;2019(1):7090534. doi:10.1155/2019/7090534
125. Sun Y, Chen P, Zhai B, et al. The emerging role of ferroptosis in inflammation. Biomed Pharmacother. 2020;127:110108. doi:10.1016/j.biopha.2020.110108
126. Liu P, Feng Y, Li H, et al. Ferrostatin-1 alleviates lipopolysaccharide-induced acute lung injury via inhibiting ferroptosis. Cell Mol Bio Lett. 2020;25(1):10. doi:10.1186/s11658-020-00205-0
127. Kumar V, Sharma A. Innate immunity in sepsis pathogenesis and its modulation: new immunomodulatory targets revealed. J Chemother. 2008;20(6):672–683. doi:10.1179/joc.2008.20.6.672
128. Zhang J, Yan W, Dong Y, et al. Early identification and diagnosis, pathophysiology, and treatment of sepsis-related acute lung injury: a narrative review. J Thoracic Dis. 2024;16(8):5457–5476. doi:10.21037/jtd-24-1191
129. Yao RQ, Ren C, Zheng LY, Xia ZF, Yao YM. Advances in Immune Monitoring Approaches for Sepsis-Induced Immunosuppression. Front Immunol. 2022;13:891024. doi:10.3389/fimmu.2022.891024
130. Chousterman BG, Swirski FK, Weber GF. Cytokine storm and sepsis disease pathogenesis. Semin Immunopathol. 2017;39(5):517–528. doi:10.1007/s00281-017-0639-8
131. Zingarelli B, Coopersmith CM, Drechsler S, et al. Part I: Minimum Quality Threshold in Preclinical Sepsis Studies (MQTiPSS) for study design and humane modeling endpoints. Review. Shock. 2019;51(1):10–22. doi:10.1097/SHK.0000000000001243
132. Gong M, Qi S, Wu Z, et al. A novel therapeutic approach to modulate the inflammatory cascade: a timely exogenous local inflammatory response attenuates the sepsis-induced cytokine storm. Cytokine. 2024;176:156533. doi:10.1016/j.cyto.2024.156533
133. Gong Y, Lan H, Yu Z, et al. Blockage of glycolysis by targeting PFKFB3 alleviates sepsis-related acute lung injury via suppressing inflammation and apoptosis of alveolar epithelial cells. Biochem Biophys Res Commun. 2017;491(2):522–529. doi:10.1016/j.bbrc.2017.05.173
134. Yang Y, Peng J, She Q, et al. miR-153 aggravates lung injury induced by lipopolysaccharide via inhibiting activated protein C (APC) in rats with sepsis and its mechanism. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 2022;38(10):911–917.
135. Pei Q, Ni W, Yuan Y, Yuan J, Zhang X, Yao M. HSP70 Ameliorates Septic Lung Injury via Inhibition of Apoptosis by Interacting with KANK2. Biomolecules. 2022;12(3):410. doi:10.3390/biom12030410
136. Shen Y, Zhao S, Hua M. Long non-coding RNA LINC01194 promotes the inflammatory response and apoptosis of lipopolysaccharide-treated MLE-12 cells through the miR-203a-3p/MIP-2 axis. Can J Physiol Pharmacol. 2022;100(5):402–411. doi:10.1139/cjpp-2021-0255
137. Chen X, Dumbuya JS, Du J, Xue L, Zeng Q. Bovine pulmonary surfactant alleviates inflammation and epithelial cell apoptosis in the early phase of lipopolysaccharide-induced acute lung injury in rats. Biotechnol Genet Eng Rev. 2024;40(4):4361–4379. doi:10.1080/02648725.2023.2210452
138. Lee DW, Santomasso BD, Locke FL, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant. 2019;25(4):625–638. doi:10.1016/j.bbmt.2018.12.758
139. de Jong HK, van der Poll T, Wiersinga WJ. The systemic pro-inflammatory response in sepsis. J Innate Immun. 2010;2(5):422–430. doi:10.1159/000316286
140. Baddam S, Burns B. Systemic inflammatory response syndrome. In: StatPearls. StatPearls Publishing LLC; 2025.
141. Yang J, Yang L, Wang Y, et al. Interleukin-6 related signaling pathways as the intersection between chronic diseases and sepsis. Mol Med. 2025;31(1):34. doi:10.1186/s10020-025-01089-6
142. Kamimura D, Ishihara K, Hirano T. IL-6 signal transduction and its physiological roles: the signal orchestration model. Rev Physiol Biochem Pharmacol. 2003;149:1–38. doi:10.1007/s10254-003-0012-2
143. Kang S, Tanaka T, Narazaki M, Kishimoto T. Targeting Interleukin-6 Signaling in Clinic. Immunity. 2019;50(4):1007–1023. doi:10.1016/j.immuni.2019.03.026
144. Kishimoto T. The Biology of Interleukin-6. Blood. 1989;74(1):1–10. doi:10.1182/blood.V74.1.1.1
145. Akira S, Taga T, Kishimoto T. Interleukin-6 in biology and medicine. In: Dixon FJ, editor. Advances in Immunology. Academic Press; 1993:1–78.
146. Poplutz MK, Wessels I, Rink L, Uciechowski P. Regulation of the Interleukin-6 gene expression during monocytic differentiation of HL-60 cells by chromatin remodeling and methylation. Immunobiology. 2014;219(8):619–626. doi:10.1016/j.imbio.2014.03.016
147. Garbers C, Hermanns HM, Schaper F, et al. Plasticity and cross-talk of Interleukin 6-type cytokines. Cytokine Growth Factor Rev. 2012;23(3):85–97. doi:10.1016/j.cytogfr.2012.04.001
148. Mauer J, Denson JL, Brüning JC. Versatile functions for IL-6 in metabolism and cancer. Trends Immunol. 2015;36(2):92–101. doi:10.1016/j.it.2014.12.008
149. Osborne J, Moore PS, Chang Y. KSHV-encoded viral IL-6 activates multiple human IL-6 signaling pathways. Hum Immunol. 1999;60(10):921–927. doi:10.1016/S0198-8859(99)00083-X
150. Wang Q, Li D, Cao G, et al. IL-27 signalling promotes adipocyte thermogenesis and energy expenditure. Nature. 2021;600(7888):314–318. doi:10.1038/s41586-021-04127-5
151. Wiersinga WJ, Leopold SJ, Cranendonk DR, van der Poll T. Host innate immune responses to sepsis. Virulence. 2014;5(1):36–44. doi:10.4161/viru.25436
152. Kishimoto T, Taga T, Akira S. Cytokine signal transduction. Cell. 1994;76(2):253–262. doi:10.1016/0092-8674(94)90333-6
153. Pignataro G, Gemma S, Petrucci M, et al. Unraveling NETs in sepsis: from cellular mechanisms to clinical relevance. Int J Mol Sci. 2025;26(15):7464. doi:10.3390/ijms26157464
154. Desai T, Leeper N, Hynes K, Gewertz B. Interleukin-6 causes endothelial barrier dysfunction via the protein kinase C pathway. J Surg Res. 2002;104(2):118–123. doi:10.1006/jsre.2002.6415
155. Klein D. The vascular endothelium as decision maker in lung injury. Front Cell Dev Biol. 2025;13:1564627. doi:10.3389/fcell.2025.1564627
156. Davis S, Aldrich TH, Jones PF, et al. Isolation of angiopoietin-1, a ligand for the TIE2 receptor, by secretion-trap expression cloning. Cell. 1996;87(7):1161–1169. doi:10.1016/s0092-8674(00)81812-7
157. Maisonpierre PC, Suri C, Jones PF, et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. 1997;277(5322):55–60. doi:10.1126/science.277.5322.55
158. Yuan HT, Khankin EV, Karumanchi SA, Parikh SM. Angiopoietin 2 is a partial agonist/antagonist of Tie2 signaling in the endothelium. Mol Cell Biol. 2009;29(8):2011–2022. doi:10.1128/mcb.01472-08
159. Teichert-Kuliszewska K, Maisonpierre PC, Jones N, et al. Biological action of angiopoietin-2 in a fibrin matrix model of angiogenesis is associated with activation of Tie2. Cardiovasc Res. 2001;49(3):659–670. doi:10.1016/s0008-6363(00)00231-5
160. Kim M, Allen B, Korhonen EA, et al. Opposing actions of angiopoietin-2 on Tie2 signaling and FOXO1 activation. J Clin Invest. 2016;126(9):3511–3525. doi:10.1172/jci84871
161. Gale NW, Thurston G, Hackett SF, et al. Angiopoietin-2 is required for postnatal angiogenesis and lymphatic patterning, and only the latter role is rescued by Angiopoietin-1. Dev Cell. 2002;3(3):411–423. doi:10.1016/s1534-5807(02)00217-4
162. Huang YQ, Li JJ, Hu L, Lee M, Karpatkin S. Thrombin induces increased expression and secretion of angiopoietin-2 from human umbilical vein endothelial cells. Blood. 2002;99(5):1646–1650. doi:10.1182/blood.v99.5.1646
163. Kelly BD, Hackett SF, Hirota K, et al. Cell type-specific regulation of angiogenic growth factor gene expression and induction of angiogenesis in nonischemic tissue by a constitutively active form of hypoxia-inducible factor 1. Circ Res. 2003;93(11):1074–1081. doi:10.1161/01.Res.0000102937.50486.1b
164. Wang Q, Lash GE. Angiopoietin 2 in placentation and tumor biology: the yin and yang of vascular biology. Placenta. 2017;56:73–78. doi:10.1016/j.placenta.2017.03.021
165. Sfiligoi C, de Luca A, Cascone I, et al. Angiopoietin-2 expression in breast cancer correlates with lymph node invasion and short survival. Int, J, Cancer. 2003;103(4):466–474. doi:10.1002/ijc.10851
166. Hu B, Cheng SY. Angiopoietin-2: development of inhibitors for cancer therapy. Curr Oncol Rep. 2009;11(2):111–116. doi:10.1007/s11912-009-0017-3
167. Kim I, Kim JH, Moon SO, Kwak HJ, Kim NG, Koh GY. Angiopoietin-2 at high concentration can enhance endothelial cell survival through the phosphatidylinositol 3’-kinase/Akt signal transduction pathway. Oncogene. 2000;19(39):4549–4552. doi:10.1038/sj.onc.1203800
168. Oliner J, Min H, Leal J, et al. Suppression of angiogenesis and tumor growth by selective inhibition of angiopoietin-2. Cancer Cell. 2004;6(5):507–516. doi:10.1016/j.ccr.2004.09.030
169. Mazzieri R, Pucci F, Moi D, et al. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell. 2011;19(4):512–526. doi:10.1016/j.ccr.2011.02.005
170. Lewis CE, Ferrara N. Multiple effects of angiopoietin-2 blockade on tumors. Cancer Cell. 2011;19(4):431–433. doi:10.1016/j.ccr.2011.03.016
171. Holopainen T, Saharinen P, D’Amico G, et al. Effects of angiopoietin-2-blocking antibody on endothelial cell-cell junctions and lung metastasis. J Natl Cancer Inst. 2012;104(6):461–475. doi:10.1093/jnci/djs009
172. Scholz A, Plate KH, Reiss Y. Angiopoietin-2: a multifaceted cytokine that functions in both angiogenesis and inflammation. Ann N Y Acad Sci. 2015;1347(1):45–51. doi:10.1111/nyas.12726
173. Sajib S, Zahra FT, Lionakis MS, German NA, Mikelis CM. Mechanisms of angiogenesis in microbe-regulated inflammatory and neoplastic conditions. Angiogenesis. 2018;21(1):1–14. doi:10.1007/s10456-017-9583-4
174. Schuldt EA, Lieb W, Dörr M, et al. Circulating angiopoietin-2 and its soluble receptor Tie-2 concentrations are related to inflammatory markers in the general population. Cytokine. 2018;105:1–7. doi:10.1016/j.cyto.2018.02.003
175. Gutbier B, Neuhauß AK, Reppe K, et al. Prognostic and Pathogenic Role of Angiopoietin-1 and −2 in Pneumonia. Am J Respir Crit Care Med. 2018;198(2):220–231. doi:10.1164/rccm.201708-1733OC
176. Lee KW, Lip GY, Blann AD. Plasma angiopoietin-1, angiopoietin-2, angiopoietin receptor tie-2, and vascular endothelial growth factor levels in acute coronary syndromes. Circulation. 2004;110(16):2355–2360. doi:10.1161/01.Cir.0000138112.90641.7f
177. David S, Kümpers P, Hellpap J, et al. Angiopoietin 2 and cardiovascular disease in dialysis and kidney transplantation. Am J Kidney Dis. 2009;53(5):770–778. doi:10.1053/j.ajkd.2008.11.030
178. Patel JV, Lim HS, Varughese GI, Hughes EA, Lip GY. Angiopoietin-2 levels as a biomarker of cardiovascular risk in patients with hypertension. Ann Med. 2008;40(3):215–222. doi:10.1080/07853890701779586
179. Chong AY, Caine GJ, Freestone B, Blann AD, Lip GY. Plasma angiopoietin-1, angiopoietin-2, and angiopoietin receptor tie-2 levels in congestive heart failure. J Am Coll Cardiol. 2004;43(3):423–428. doi:10.1016/j.jacc.2003.08.042
180. Moisan A, Favre IM, Rome C, et al. Microvascular plasticity after experimental stroke: a molecular and MRI study. Cerebrovasc Dis. 2014;38(5):344–353. doi:10.1159/000368597
181. Liu XS, Chopp M, Zhang RL, et al. Angiopoietin 2 mediates the differentiation and migration of neural progenitor cells in the subventricular zone after stroke. J Biol Chem. 2009;284(34):22680–22689. doi:10.1074/jbc.M109.006551
182. Syrjälä SO, Tuuminen R, Nykänen AI, et al. Angiopoietin-2 inhibition prevents transplant ischemia-reperfusion injury and chronic rejection in rat cardiac allografts. Am J Transplant. 2014;14(5):1096–1108. doi:10.1111/ajt.12672
183. De Caro AM, Bonicel JJ, Rouimi P, De Caro JD, Sarles H, Rovery M. Complete amino acid sequence of an immunoreactive form of human pancreatic stone protein isolated from pancreatic juice. Eur J Biochem. 1987;168(1):201–207. doi:10.1111/j.1432-1033.1987.tb13405.x
184. De Reggi M, Capon C, Gharib B, Wieruszeski JM, Michel R, Fournet B. The glycan moiety of human pancreatic lithostathine. Structure characterization and possible pathophysiological implications. Eur J Biochem. 1995;230(2):503–510. doi:10.1111/j.1432-1033.1995.tb20589.x
185. Terazono K, Yamamoto H, Takasawa S, et al. A novel gene activated in regenerating islets. J Biol Chem. 1988;263(5):2111–2114. doi:10.1016/S0021-9258(18)69176-8
186. Watanabe T, Yonekura H, Terazono K, Yamamoto H, Okamoto H. Complete nucleotide sequence of human reg gene and its expression in normal and tumoral tissues. The reg protein, pancreatic stone protein, and pancreatic thread protein are one and the same product of the gene. J Biol Chem. 1990;265(13):7432–7439. doi:10.1016/S0021-9258(19)39132-X
187. Kimura N, Yonekura H, Okamoto H, Nagura H. Expression of human regenerating gene mRNA and its product in normal and neoplastic human pancreas. Cancer. 1992;70(7):1857–1863. doi:10.1002/1097-0142(19921001)70:7<1857::aid-cncr2820700708>3.0.co;2-8
188. Watanabe T, Yonemura Y, Yonekura H, et al. Pancreatic beta-cell replication and amelioration of surgical diabetes by Reg protein. Proc Natl Acad Sci U S A. 1994;91(9):3589–3592. doi:10.1073/pnas.91.9.3589
189. Perfetti R, Egan JM, Zenilman ME, Shuldiner AR. Expression of the regenerating gene in the pancreas of aging mice. Transplant Proc. 1994;26(2):733.
190. Moriscot C, Renaud W, Bouvier R, Figarella-Branger D, Figarella C, Guy-Crotte O. Absence of correlation between reg and insulin gene expression in pancreas during fetal development. Pediatr Res. 1996;39(2):349–353. doi:10.1203/00006450-199602000-00026
191. Bernard JP, Adrich Z, Montalto G, et al. Inhibition of nucleation and crystal growth of calcium carbonate by human lithostathine. Gastroenterology. 1992;103(4):1277–1284. doi:10.1016/0016-5085(92)91516-7
192. Geider S, Baronnet A, Cerini C, et al. Pancreatic lithostathine as a calcite habit modifier. J Biol Chem. 1996;271(42):26302–26306. doi:10.1074/jbc.271.42.26302
193. Hu P, Lu YH, Deng W, et al. The critical role of pancreatic stone protein/regenerating protein in sepsis-related multiorgan failure. Front Med. 2023;10:1172529. doi:10.3389/fmed.2023.1172529
194. Wu S, Zheng Y, Mou JW, Zhang YJ, Zhu KX. Mechanism and treatment of pancreatic duct stones. World J Gastrointest Surg. 2025;17(8):108312. doi:10.4240/wjgs.v17.i8.108312
195. Austin JL, Peick AL, Wipfler J, Sieckman G. Effect of chronic partial pancreatic duct obstruction on pancreatic duct secretory pressure and permeability in cats. J Surg Res. 1988;44(6):772–780. doi:10.1016/0022-4804(88)90113-8
196. Hassan HM, Liu R, Lv Y, et al. Plasma pancreatic stone protein is a promising biomarker for prognosis and severity outcome in sepsis patients with COVID-19. Sci Rep. 2025;15(1):34859. doi:10.1038/s41598-025-09750-0
197. Teng YH-F, Aquino RS, Park PW. Molecular functions of syndecan-1 in disease. Matrix Biol. 2012;31(1):3–16. doi:10.1016/j.matbio.2011.10.001
198. Bode L, Salvestrini C, Park PW, et al. Heparan sulfate and syndecan-1 are essential in maintaining murine and human intestinal epithelial barrier function. J Clin Invest. 2008;118(1):229–238. doi:10.1172/jci32335
199. Fukai N, Kenagy RD, Chen L, Gao L, Daum G, Clowes AW. Syndecan-1: an inhibitor of arterial smooth muscle cell growth and intimal hyperplasia. Arterioscler Thromb Vasc Biol. 2009;29(9):1356–1362. doi:10.1161/atvbaha.109.190132
200. Stanford KI, Bishop JR, Foley EM, et al. Syndecan-1 is the primary heparan sulfate proteoglycan mediating hepatic clearance of triglyceride-rich lipoproteins in mice. J Clin Invest. 2009;119(11):3236–3245. doi:10.1172/jci38251
201. Stepp MA, Gibson HE, Gala PH, et al. Defects in keratinocyte activation during wound healing in the syndecan-1-deficient mouse. J Cell Sci. 2002;115(Pt 23):4517–4531. doi:10.1242/jcs.00128
202. Sanderson RD, Børset M. Syndecan-1 in B lymphoid malignancies. Ann Hematol. 2002;81(3):125–135. doi:10.1007/s00277-002-0437-8
203. Yeaman C, Rapraeger AC. Post-transcriptional regulation of syndecan-1 expression by cAMP in peritoneal macrophages. J Cell Biol. 1993;122(4):941–950. doi:10.1083/jcb.122.4.941
204. Hayashida K, Parks WC, Park PW. Syndecan-1 shedding facilitates the resolution of neutrophilic inflammation by removing sequestered CXC chemokines. Blood. 2009;114(14):3033–3043. doi:10.1182/blood-2009-02-204966
205. Lorand L, Graham RM. Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat Rev Mol Cell Biol. 2003;4(2):140–156. doi:10.1038/nrm1014
206. Vanhoutte D, Schellings MW, Götte M, et al. Increased expression of syndecan-1 protects against cardiac dilatation and dysfunction after myocardial infarction. Circulation. 2007;115(4):475–482. doi:10.1161/circulationaha.106.644609
207. Okamura Y, Yokoi H. Development of a point-of-care assay system for measurement of presepsin (sCD14-ST). Clin Chim Acta. 2011;412(23–24):2157–2161. doi:10.1016/j.cca.2011.07.024
208. Chenevier-Gobeaux C, Borderie D, Weiss N, Mallet-Coste T, Claessens Y-E. Presepsin (sCD14-ST), an innate immune response marker in sepsis. Clin Chim Acta. 2015;450:97–103. doi:10.1016/j.cca.2015.06.026
209. Hancock RE. Cationic peptides: effectors in innate immunity and novel antimicrobials. Lancet Infect Dis. 2001;1(3):156–164. doi:10.1016/s1473-3099(01)00092-5
210. Wright SD, Ramos RA, Tobias PS, Ulevitch RJ, Mathison JC. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science. 1990;249(4975):1431–1433. doi:10.1126/science.1698311
211. Jack RS, Fan X, Bernheiden M, et al. Lipopolysaccharide-binding protein is required to combat a murine gram-negative bacterial infection. Nature. 1997;389(6652):742–745. doi:10.1038/39622
212. Modlin RL, Brightbill HD, Godowski PJ. The toll of innate immunity on microbial pathogens. N Engl J Med. 1999;340(23):1834–1835. doi:10.1056/nejm199906103402312
213. Mussap M, Noto A, Fravega M, Fanos V. Soluble CD14 subtype presepsin (sCD14-ST) and lipopolysaccharide binding protein (LBP) in neonatal sepsis: new clinical and analytical perspectives for two old biomarkers. J Matern Fetal Neonatal Med. 2011;24 Suppl 2(sup2):12–14. doi:10.3109/14767058.2011.601923
214. Landmann R, Zimmerli W, Sansano S, et al. Increased circulating soluble CD14 is associated with high mortality in gram-negative septic shock. J Infect Dis. 1995;171(3):639–644. doi:10.1093/infdis/171.3.639
215. Pavcnik-Arnol M, Hojker S, Derganc M. Lipopolysaccharide-binding protein, lipopolysaccharide, and soluble CD14 in sepsis of critically ill neonates and children. Intensive Care Med. 2007;33(6):1025–1032. doi:10.1007/s00134-007-0626-y
216. Yaegashi Y, Shirakawa K, Sato N, et al. Evaluation of a newly identified soluble CD14 subtype as a marker for sepsis. J Infect Chemother. 2005;11(5):234–238. doi:10.1007/s10156-005-0400-4
217. Shirakawa K, Naitou K, Hirose J, Takahashi T, Furusako S. Presepsin (sCD14-ST): development and evaluation of one-step ELISA with a new standard that is similar to the form of presepsin in septic patients. Clin Chem Lab Med. 2011;49(5):937–939. doi:10.1515/cclm.2011.145
218. Bechard D, Meignin V, Scherpereel A, et al. Characterization of the secreted form of endothelial-cell-specific molecule 1 by specific monoclonal antibodies. J Vasc Res. 2000;37(5):417–425. doi:10.1159/000025758
219. Abid MR, Yi X, Yano K, Shih SC, Aird WC. Vascular endocan is preferentially expressed in tumor endothelium. Microvasc Res. 2006;72(3):136–145. doi:10.1016/j.mvr.2006.05.010
220. Yang YC, Pan KF, Lee WJ, et al. Circulating Proteoglycan Endocan Mediates EGFR-Driven Progression of Non-Small Cell Lung Cancer. Cancer Res. 2020;80(16):3292–3304. doi:10.1158/0008-5472.Can-20-0005
221. Chen J, Jiang L, Yu XH, et al. Endocan: a Key Player of Cardiovascular Disease. Front Cardiovasc Med. 2021;8:798699. doi:10.3389/fcvm.2021.798699
222. Tayman MA, Önder C, Kurgan Ş, Serdar MA, Günhan M. Endocan (ESM-1) levels in gingival crevicular fluid correlate with ICAM-1 and LFA-1 in periodontitis. Braz Oral Res. 2020;35:e005. doi:10.1590/1807-3107bor-2021.vol35.0005
223. Pawlak K, Mysliwiec M, Pawlak D. Endocan--the new endothelial activation marker independently associated with soluble endothelial adhesion molecules in uraemic patients with cardiovascular disease. Clin Biochem. 2015;48(6):425–430. doi:10.1016/j.clinbiochem.2015.01.006
224. Lee W, Ku SK, Kim SW, Bae JS. Endocan elicits severe vascular inflammatory responses in vitro and in vivo. J Cell Physiol. 2014;229(5):620–630. doi:10.1002/jcp.24485
225. Sun H, Zhang H, Li K, et al. ESM-1 promotes adhesion between monocytes and endothelial cells under intermittent hypoxia. J Cell Physiol. 2019;234(2):1512–1521. doi:10.1002/jcp.27016
226. Kartik kumar S, Muthusamy S, Kadhirvel S, Mani KP. In-silico and in-vitro analysis of endocan interaction with statins. Int J Biol Macromol. 2020;146:1087–1099. doi:10.1016/j.ijbiomac.2019.09.235
227. Scuruchi M, Aliquò F, Avenoso A, et al. Endocan knockdown down-regulates the expression of angiogenesis-associated genes in Il-1ß activated chondrocytes. Biomolecules. 2023;13(5):851. doi:10.3390/biom13050851
228. Scuruchi M, D’Ascola A, Avenoso A, Mandraffino G, Campo S, Campo GM. Endocan, a novel inflammatory marker, is upregulated in human chondrocytes stimulated with IL-1 beta. Mol Cell Biochem. 2021;476(3):1589–1597. doi:10.1007/s11010-020-04001-4
229. Gaudet A, Portier L, Prin M, et al. Endocan regulates acute lung inflammation through control of leukocyte diapedesis. J Appl Physiol. 2019;127(3):668–678. doi:10.1152/japplphysiol.00337.2019
230. Kali A, Shetty KS. Endocan: a novel circulating proteoglycan. Indian J Pharmacol. 2014;46(6):579–583. doi:10.4103/0253-7613.144891
231. Zhang X, Zhuang R, Wu H, et al. A novel role of endocan in alleviating LPS-induced acute lung injury. Life Sci. 2018;202:89–97. doi:10.1016/j.lfs.2018.04.005
232. Kechagia M, Papassotiriou I, Gourgoulianis KI. Endocan and the respiratory system: a review. Int J Chron Obstruct Pulmon Dis. 2016;11:3179–3187. doi:10.2147/copd.S118692
233. Yan SF, Ramasamy R, Schmidt AM. Mechanisms of disease: advanced glycation end-products and their receptor in inflammation and diabetes complications. Nat Clin Pract Endocrinol Metab. 2008;4(5):285–293. doi:10.1038/ncpendmet0786
234. Bierhaus A, Humpert PM, Morcos M, et al. Understanding RAGE, the receptor for advanced glycation end products. J Mol Med. 2005;83(11):876–886. doi:10.1007/s00109-005-0688-7
235. Schmidt AM, Yan SD, Yan SF, Stern DM. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest. 2001;108(7):949–955. doi:10.1172/jci14002
236. Xie J, Reverdatto S, Frolov A, Hoffmann R, Burz DS, Shekhtman A. Structural basis for pattern recognition by the receptor for advanced glycation end products (RAGE). J Biol Chem. 2008;283(40):27255–27269. doi:10.1074/jbc.M801622200
237. Demling N, Ehrhardt C, Kasper M, Laue M, Knels L, Rieber EP. Promotion of cell adherence and spreading: a novel function of RAGE, the highly selective differentiation marker of human alveolar epithelial type I cells. Cell Tissue Res. 2006;323(3):475–488. doi:10.1007/s00441-005-0069-0
238. Hudson BI, Carter AM, Harja E, et al. Identification, classification, and expression of RAGE gene splice variants. FASEB j. 2008;22(5):1572–1580. doi:10.1096/fj.07-9909com
239. Yan SF, Ramasamy R, Schmidt AM. Receptor for AGE (RAGE) and its ligands-cast into leading roles in diabetes and the inflammatory response. J Mol Med. 2009;87(3):235–247. doi:10.1007/s00109-009-0439-2
240. Thunø M, Macho B, Eugen-Olsen J. suPAR: the molecular crystal ball. Dis Markers. 2009;27(3):157–172. doi:10.3233/dma-2009-0657
241. Smith HW, Marshall CJ. Regulation of cell signalling by uPAR. Nat Rev Mol Cell Biol. 2010;11(1):23–36. doi:10.1038/nrm2821
242. Mustjoki S, Sidenius N, Sier CF, et al. Soluble urokinase receptor levels correlate with number of circulating tumor cells in acute myeloid leukemia and decrease rapidly during chemotherapy. Cancer Res. 2000;60(24):7126–7132.
243. Kjellman A, Akre O, Gustafsson O, et al. Soluble urokinase plasminogen activator receptor as a prognostic marker in men participating in prostate cancer screening. J Intern Med. 2011;269(3):299–305. doi:10.1111/j.1365-2796.2010.02284.x
244. Gustafsson A, Ajeti V, Ljunggren L. Detection of suPAR in the saliva of healthy young adults: comparison with plasma levels. Biomark Insights. 2011;6:119–125. doi:10.4137/bmi.S8326
245. Eugen-Olsen J, Ladelund S, Sørensen LT. Plasma suPAR is lowered by smoking cessation: a randomized controlled study. Eur J Clin Invest. 2016;46(4):305–311. doi:10.1111/eci.12593
246. Haupt TH, Petersen J, Ellekilde G, et al. Plasma suPAR levels are associated with mortality, admission time, and Charlson Comorbidity Index in the acutely admitted medical patient: a prospective observational study. Crit Care. 2012;16(4):R130. doi:10.1186/cc11434
247. Zimmermann HW, Koch A, Seidler S, Trautwein C, Tacke F. Circulating soluble urokinase plasminogen activator is elevated in patients with chronic liver disease, discriminates stage and aetiology of cirrhosis and predicts prognosis. Liver Int. 2012;32(3):500–509. doi:10.1111/j.1478-3231.2011.02665.x
248. Ostrowski SR, Ullum H, Goka BQ, et al. Plasma concentrations of soluble urokinase-type plasminogen activator receptor are increased in patients with malaria and are associated with a poor clinical or a fatal outcome. J Infect Dis. 2005;191(8):1331–1341. doi:10.1086/428854
249. Wrotek A, Jackowska T, Pawlik K. Soluble urokinase plasminogen activator receptor: an indicator of pneumonia severity in children. Adv Exp Med Biol. 2015;835:1–7. doi:10.1007/5584_2014_40
250. Pliyev BK. Activated human neutrophils rapidly release the chemotactically active D2D3 form of the urokinase-type plasminogen activator receptor (uPAR/CD87). Mol Cell Biochem. 2009;321(1–2):111–122. doi:10.1007/s11010-008-9925-z
251. Selleri C, Montuori N, Ricci P, et al. Involvement of the urokinase-type plasminogen activator receptor in hematopoietic stem cell mobilization. Blood. 2005;105(5):2198–2205. doi:10.1182/blood-2004-06-2424
252. Dekkers PE, ten Hove T, te Velde AA, van Deventer SJ, van Der Poll T, Moore RN. Upregulation of monocyte urokinase plasminogen activator receptor during human endotoxemia. Infect Immun. 2000;68(4):2156–2160. doi:10.1128/iai.68.4.2156-2160.2000
253. Ostrowski SR, Plomgaard P, Fischer CP, et al. Interleukin-6 infusion during human endotoxaemia inhibits in vitro release of the urokinase receptor from peripheral blood mononuclear cells. Scand J Immunol. 2005;61(2):197–206. doi:10.1111/j.0300-9475.2005.01547.x
254. Chavakis T, Willuweit AK, Lupu F, Preissner KT, Kanse SM. Release of soluble urokinase receptor from vascular cells. Thromb Haemost. 2001;86(2):686–693. doi:10.1055/s-0037-1616105
255. Zimmermann HW, Reuken PA, Koch A, et al. Soluble urokinase plasminogen activator receptor is compartmentally regulated in decompensated cirrhosis and indicates immune activation and short-term mortality. J Intern Med. 2013;274(1):86–100. doi:10.1111/joim.12054
256. Bhandary YP, Velusamy T, Shetty P, et al. Post-transcriptional regulation of urokinase-type plasminogen activator receptor expression in lipopolysaccharide-induced acute lung injury. Am J Respir Crit Care Med. 2009;179(4):288–298. doi:10.1164/rccm.200712-1787OC
257. Matsumoto H, Ueshima S, Fukao H, Mitsui Y, Matsuo O. Effects of lipopolysaccharide on the expression of fibrinolytic factors in an established cell line from human endothelial cells. Life Sci. 1996;59(2):85–96. doi:10.1016/0024-3205(96)00265-2
258. Carmeliet P, Moons L, Lijnen R, et al. Urokinase-generated plasmin activates matrix metalloproteinases during aneurysm formation. Nat Genet. 1997;17(4):439–444. doi:10.1038/ng1297-439
259. Houck KA, Leung DW, Rowland AM, Winer J, Ferrara N. Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J Biol Chem. 1992;267(36):26031–26037. doi:10.1016/S0021-9258(18)35712-0
260. Lyons RM, Gentry LE, Purchio AF, Moses HL. Mechanism of activation of latent recombinant transforming growth factor beta 1 by plasmin. J Cell Biol. 1990;110(4):1361–1367. doi:10.1083/jcb.110.4.1361
261. Koch A, Zimmermann HW, Gassler N, et al. Clinical relevance and cellular source of elevated soluble urokinase plasminogen activator receptor (suPAR) in acute liver failure. Liver Int. 2014;34(9):1330–1339. doi:10.1111/liv.12512
262. Song F, Liu H, Ma H, et al. AI model based on diaphragm ultrasound to improve the predictive performance of invasive mechanical ventilation weaning: prospective cohort study. JMIR Form Res. 2025;9:e72482. doi:10.2196/72482
263. Schmidt GA, Girard TD, Kress JP, et al. Liberation From mechanical ventilation in critically ill adults: executive summary of an Official American College of Chest Physicians/American Thoracic Society Clinical Practice Guideline. Chest. 2017;151(1):160–165. doi:10.1016/j.chest.2016.10.037
264. Miller JD, Carlo WA. Pulmonary complications of mechanical ventilation in neonates. Clin Perinatol. 2008;35(1):273–81,x–xi. doi:10.1016/j.clp.2007.11.004
265. Vliegenthart RJ, Van Kaam AH, Aarnoudse-Moens CS, Van Wassenaer AG, Onland W. Duration of mechanical ventilation and neurodevelopment in preterm infants. Arch Dis Childhood-Fetal Neonatal Ed. 2019;104(6):F631–F635. doi:10.1136/archdischild-2018-315993
266. Roberts KJ, Goodfellow LT, Battey-Muse CM, et al. AARC clinical practice guideline: spontaneous breathing trials for liberation from adult mechanical ventilation. Respir Care. 2024;69(7):891–901. doi:10.4187/respcare.11735
267. Zein H, Baratloo A, Negida A, Safari S. Ventilator weaning and spontaneous breathing trials; an educational review. Emerg. 2016;4(2):65–71.
268. Robertson TE, Mann HJ, Hyzy R, et al. Multicenter implementation of a consensus-developed, evidence-based, spontaneous breathing trial protocol. Crit Care Med. 2008;36(10):2753–2762. doi:10.1097/ccm.0b013e3181872833
269. Morganroth ML, Morganroth JL, Nett LM, Petty TL. Criteria for weaning from prolonged mechanical ventilation. Arch Intern Med. 1984;144(5):1012–1016. doi:10.1001/archinte.1984.00350170172028
270. Yang KL, Tobin MJ. A prospective study of indexes predicting the outcome of trials of weaning from mechanical ventilation. N Engl J Med. 1991;324(21):1445–1450. doi:10.1056/nejm199105233242101
271. Gaddam M, Gullapalli D, Adrish ZA, Reddy AY, Adrish M. Predicting weaning failure from invasive mechanical ventilation: the promise and pitfalls of clinical prediction scores. World J Crit Care Med. 2025;14(3):108272. doi:10.5492/wjccm.v14.i3.108272
272. Jia D, Wang H, Wang Q, et al. Rapid shallow breathing index predicting extubation outcomes: a systematic review and meta-analysis. Intens Crit Care Nurs. 2024;80:103551. doi:10.1016/j.iccn.2023.103551
273. Epstein SK, Ciubotaru RL. Influence of gender and endotracheal tube size on preextubation breathing pattern. Am J Respir Crit Care Med. 1996;154(6 Pt 1):1647–1652. doi:10.1164/ajrccm.154.6.8970349
274. Purro A, Appendini L, De Gaetano A, Gudjonsdottir M, Donner CF, Rossi A. Physiologic determinants of ventilator dependence in long-term mechanically ventilated patients. Am J Respir Crit Care Med. 2000;161(4 Pt 1):1115–1123. doi:10.1164/ajrccm.161.4.9812160
275. Siegel MD. Technique and the rapid shallow breathing index. Respir Care. 2009;54(11):1449–1450.
276. Vidotto MC, Sogame LC, Calciolari CC, Nascimento OA, Jardim JR. The prediction of extubation success of postoperative neurosurgical patients using frequency-tidal volume ratios. Neurocrit Care. 2008;9(1):83–89. doi:10.1007/s12028-008-9059-x
277. Zhang B, Qin YZ. Comparison of pressure support ventilation and T-piece in determining rapid shallow breathing index in spontaneous breathing trials. Am J Med Sci. 2014;348(4):300–305. doi:10.1097/maj.0000000000000286
278. Schmidt GA, Girard TD, Kress JP, et al. Official executive summary of an American Thoracic Society/American College of Chest Physicians Clinical Practice Guideline: liberation from mechanical ventilation in critically ill adults. Am J Respir Crit Care Med. 2017;195(1):115–119. doi:10.1164/rccm.201610-2076ST
279. Shah NG, Lee B, Colice G. Analysis of rapid shallow breathing index as a predictor for successful extubation from mechanical ventilation. Chest. 2004;126(4):756S. doi:10.1378/chest.126.4_MeetingAbstracts.756S-b
280. Teixeira C, Zimermann teixeira PJ, Hohër JA, de Leon PP, Brodt SF, da Siva Moreira J. Serial measurements of f/VT can predict extubation failure in patients with f/VT < or = 105? J Crit Care. 2008;23(4):572–576. doi:10.1016/j.jcrc.2007.12.011
281. Segal LN, Oei E, Oppenheimer BW, et al. Evolution of pattern of breathing during a spontaneous breathing trial predicts successful extubation. Intensive Care Med. 2010;36(3):487–495. doi:10.1007/s00134-009-1735-6
282. Gluck EH, Corgian L. Predicting eventual success or failure to wean in patients receiving long-term mechanical ventilation. Chest. 1996;110(4):1018–1024. doi:10.1378/chest.110.4.1018
283. Duan J, Han X, Bai L, Zhou L, Huang S. Assessment of heart rate, acidosis, consciousness, oxygenation, and respiratory rate to predict noninvasive ventilation failure in hypoxemic patients. Intensive Care Med. 2017;43(2):192–199. doi:10.1007/s00134-016-4601-3
284. Lin FC, Kuo YW, Jerng JS, Wu HD. Association of weaning preparedness with extubation outcome of mechanically ventilated patients in medical intensive care units: a retrospective analysis. PeerJ. 2020;8:e8973. doi:10.7717/peerj.8973
285. Baptistella AR, Mantelli LM, Matte L, et al. Prediction of extubation outcome in mechanically ventilated patients: development and validation of the Extubation Predictive Score (ExPreS). PLoS One. 2021;16(3):e0248868. doi:10.1371/journal.pone.0248868
286. Albashir SM, Robert RC, Jairath NN, Raub CB, Alzumai OA, Salem SS. The rapid shallow breathing index (RSBI) as a predictor for extubation success in medical and surgical ICU patients: a retrospective cohort study. Heart Lung. 2025;70:321–328. doi:10.1016/j.hrtlng.2025.01.007
287. Carlucci A, Richard JC, Wysocki M, Lepage E, Brochard L. Noninvasive versus conventional mechanical ventilation. An epidemiologic survey. Am J Respir Crit Care Med. 2001;163(4):874–880. doi:10.1164/ajrccm.163.4.2006027
288. Boles JM, Bion J, Connors A, et al. Weaning from mechanical ventilation. Eur Respir J. 2007;29(5):1033–1056. doi:10.1183/09031936.00010206
289. Peñuelas Ó, Thille AW, Esteban A. Discontinuation of ventilatory support: new solutions to old dilemmas. Curr Opin Crit Care. 2015;21(1):74–81. doi:10.1097/mcc.0000000000000169
290. Girard TD, Kress JP, Fuchs BD, et al. Efficacy and safety of a paired sedation and ventilator weaning protocol for mechanically ventilated patients in intensive care (Awakening and Breathing Controlled trial): a randomised controlled trial. Lancet. 2008;371(9607):126–134. doi:10.1016/s0140-6736(08)60105-1
291. Dadam MM, Pereira AB, Cardoso MR, Carnin TC, Westphal GA. Effect of reintubation within 48 hours on mortality in critically ill patients after planned extubation. Respir Care. 2024;69(7):829–838. doi:10.4187/respcare.11077
292. Jubran A, Tobin MJ. Pathophysiologic basis of acute respiratory distress in patients who fail a trial of weaning from mechanical ventilation. Am J Respir Crit Care Med. 1997;155(3):906–915. doi:10.1164/ajrccm.155.3.9117025
293. Capdevila X, Perrigault PF, Ramonatxo M, et al. Changes in breathing pattern and respiratory muscle performance parameters during difficult weaning. Crit Care Med. 1998;26(1):79–87. doi:10.1097/00003246-199801000-00020
294. Epstein SK. Decision to extubate. Intensive Care Med. 2002;28(5):535–546. doi:10.1007/s00134-002-1268-8
295. Torres A, Gatell JM, Aznar E, et al. Re-intubation increases the risk of nosocomial pneumonia in patients needing mechanical ventilation. Am J Respir Crit Care Med. 1995;152(1):137–141. doi:10.1164/ajrccm.152.1.7599812
296. Esteban A, Alía I, Tobin MJ, et al. Effect of spontaneous breathing trial duration on outcome of attempts to discontinue mechanical ventilation. Spanish Lung Failure Collaborative Group. Am J Respir Crit Care Med. 1999;159(2):512–518. doi:10.1164/ajrccm.159.2.9803106
297. Beitler JR, Goligher EC, Schmidt M, et al. Personalized medicine for ARDS: the 2035 research agenda. Intensive Care Med. 2016;42(5):756–767. doi:10.1007/s00134-016-4331-6
298. Lim A, Radujkovic A, Weigand MA, Merle U. Soluble receptor for advanced glycation end products (sRAGE) as a biomarker of COVID-19 disease severity and indicator of the need for mechanical ventilation, ARDS and mortality. Ann Intensive Care. 2021;11(1):50. doi:10.1186/s13613-021-00836-2
299. Jabaudon M, Blondonnet R, Ware LB. Biomarkers in acute respiratory distress syndrome. Curr Opin Crit Care. 2021;27(1):46–54. doi:10.1097/mcc.0000000000000786
300. Michailides C, Paraskevas T, Demiri S, et al. Diagnostic and prognostic ability of pancreatic stone protein: a scoping review. Int J Mol Sci. 2024;25(11):6046. doi:10.3390/ijms25116046
301. Alladina JW, Giacona FL, Haring AM, et al. Circulating biomarkers of endothelial dysfunction associated with ventilatory ratio and mortality in ARDS resulting from SARS-CoV-2 infection treated with antiinflammatory therapies. Chest Crit Care. 2024;2(2):100054. doi:10.1016/j.chstcc.2024.100054
302. Enocsson H, Idoff C, Gustafsson A, et al. Soluble Urokinase Plasminogen Activator Receptor (suPAR) independently predicts severity and length of hospitalisation in patients with COVID-19. Front Med. 2021;8:791716. doi:10.3389/fmed.2021.791716
303. Herold T, Jurinovic V, Arnreich C, et al. Elevated levels of IL-6 and CRP predict the need for mechanical ventilation in COVID-19. J Allergy Clin Immunol. 2020;146(1):128–136.e4. doi:10.1016/j.jaci.2020.05.008
304. Andrijevic A, Batranovic U, Nedeljkov D, et al. sRAGE as a prognostic biomarker in ARDS: insights from a clinical cohort study. Medicina. 2025;61(2):229. doi:10.3390/medicina61020229
305. Atmowihardjo LN, Heijnen NFL, Smit MR, et al. Biomarkers of alveolar epithelial injury and endothelial dysfunction are associated with scores of pulmonary edema in invasively ventilated patients. Am J Physiol Lung Cell Mol Physiol. 2023;324(1):L38–l47. doi:10.1152/ajplung.00185.2022
306. Wilson JG, Calfee CS. ARDS subphenotypes: understanding a heterogeneous syndrome. Crit Care. 2020;24(1):102. doi:10.1186/s13054-020-2778-x
307. Matthay MA, Arabi YM, Siegel ER, et al. Phenotypes and personalized medicine in the acute respiratory distress syndrome. Intensive Care Med. 2020;46(12):2136–2152. doi:10.1007/s00134-020-06296-9
308. Ware LB, Koyama T, Zhao Z, et al. Biomarkers of lung epithelial injury and inflammation distinguish severe sepsis patients with acute respiratory distress syndrome. Crit Care. 2013;17(5):R253. doi:10.1186/cc13080
309. Liu Y, Chen L. Impact of interleukin 6 levels on acute lung injury risk and disease severity in critically ill sepsis patients. World J Clin Cases. 2024;12(23):5374–5381. doi:10.12998/wjcc.v12.i23.5374
310. Meduri GU, Headley S, Kohler G, et al. Persistent elevation of inflammatory cytokines predicts a poor outcome in ARDS. Plasma IL-1 beta and IL-6 levels are consistent and efficient predictors of outcome over time. Chest. 1995;107(4):1062–1073. doi:10.1378/chest.107.4.1062
311. Molano Franco D, Arevalo-Rodriguez I, Roqué IFM, Montero Oleas NG, Nuvials X, Zamora J. Plasma interleukin-6 concentration for the diagnosis of sepsis in critically ill adults. Cochrane Database Syst Rev. 2019;4(4):Cd011811. doi:10.1002/14651858.CD011811.pub2
312. Wilson JG, Simpson LJ, Ferreira AM, et al. Cytokine profile in plasma of severe COVID-19 does not differ from ARDS and sepsis. JCI Insight. 2020;5(17). doi:10.1172/jci.insight.140289
313. Rosenberger CM, Wick KD, Zhuo H, et al. Early plasma angiopoietin-2 is prognostic for ARDS and mortality among critically ill patients with sepsis. Critical Care. 2023;27(1):234. doi:10.1186/s13054-023-04525-3
314. Zinter MS, Spicer A, Orwoll BO, et al. Plasma angiopoietin-2 outperforms other markers of endothelial injury in prognosticating pediatric ARDS mortality. Am J Physiol Lung Cell Mol Physiol. 2016;310(3):L224–31. doi:10.1152/ajplung.00336.2015
315. Gukasjan R, Raptis DA, Schulz HU, Halangk W, Graf R. Pancreatic stone protein predicts outcome in patients with peritonitis in the ICU. Crit Care Med. 2013;41(4):1027–1036. doi:10.1097/CCM.0b013e3182771193
316. Mai B, Zhou L, Wang Q, et al. Diagnostic accuracy of pancreatic stone protein in patients with sepsis: a systematic review and meta-analysis. BMC Infect Dis. 2024;24(1):472. doi:10.1186/s12879-024-09347-4
317. Cantey JB, Lee JH. biomarkers for the diagnosis of neonatal sepsis. Clin Perinatol. 2021;48(2):215–227. doi:10.1016/j.clp.2021.03.012
318. Kajita Y, Terashima T, Mori H, et al. A longitudinal change of syndecan-1 predicts risk of acute respiratory distress syndrome and cumulative fluid balance in patients with septic shock: a preliminary study. J Intensive Care. 2021;9(1):27. doi:10.1186/s40560-021-00543-x
319. Saoraya J, Wongsamita L, Srisawat N, Musikatavorn K. Plasma syndecan-1 is associated with fluid requirements and clinical outcomes in emergency department patients with sepsis. Am J Emerg Med. 2021;42:83–89. doi:10.1016/j.ajem.2021.01.019
320. Murphy LS, Wickersham N, McNeil JB, et al. Endothelial glycocalyx degradation is more severe in patients with non-pulmonary sepsis compared to pulmonary sepsis and associates with risk of ARDS and other organ dysfunction. Ann Intensive Care. 2017;7(1):102. doi:10.1186/s13613-017-0325-y
321. Qian X, Lui KY, Hu X, et al. Dynamic changes and prognosis value of plasma syndecan-1 and different microcirculatory parameters in sepsis: a prospective observational study. World J Surg. 2025;49(2):353–363. doi:10.1002/wjs.12452
322. Zhao J, Tan Y, Wang L, Shi Y. Discriminatory ability and prognostic evaluation of presepsin for sepsis-related acute respiratory distress syndrome. Sci Rep. 2020;10(1):9114. doi:10.1038/s41598-020-66121-7
323. Pluta MP, Czempik PF, Kwiatkowska M, et al. Presepsin does not predict risk of death in sepsis patients admitted to the intensive care unit: a prospective single-center study. Biomedicines. 2024;12(10):2313. doi:10.3390/biomedicines12102313
324. Shimoyama Y, Kadono N, Umegaki O. Presepsin is a more useful predictor of septic AKI and ARDS for very-old sepsis patients than for young sepsis patients in ICUs: a pilot study. BMC Res Notes. 2024;17(1):53. doi:10.1186/s13104-024-06719-6
325. Park S, Kym D, Yoon J, Cho YS, Hur J. Diagnostic accuracy of presepsin and its impact on early antibiotic de-escalation in burn-related sepsis. Antibiotics. 2025;14(8):822. doi:10.3390/antibiotics14080822
326. Orbegozo D, Rahmania L, Irazabal M, et al. Endocan as an early biomarker of severity in patients with acute respiratory distress syndrome. Ann Intensive Care. 2017;7(1):93. doi:10.1186/s13613-017-0311-4
327. Behnoush AH, Khalaji A, Ghasemi H, et al. Endocan as a biomarker for acute respiratory distress syndrome: a systematic review and meta-analysis. Health Sci Rep. 2024;7(9):e70044. doi:10.1002/hsr2.70044
328. Jabaudon M, Blondonnet R, Pereira B, et al. Plasma sRAGE is independently associated with increased mortality in ARDS: a meta-analysis of individual patient data. Intensive Care Med. 2018;44(9):1388–1399. doi:10.1007/s00134-018-5327-1
329. Peukert K, Seeliger B, Fox M, et al. SP-D serum levels reveal distinct epithelial damage in direct human ARDS. J Clin Med. 2021;10(4):737. doi:10.3390/jcm10040737
330. Determann RM, Royakkers AA, Haitsma JJ, et al. Plasma levels of surfactant protein D and KL-6 for evaluation of lung injury in critically ill mechanically ventilated patients. BMC Pulm Med. 2010;10(1):6. doi:10.1186/1471-2466-10-6
331. Han L, Wang S, Ma J, Zhao Z. Expression and significance of serum KL-6 in patients with acute respiratory distress syndrome. J Thorac Dis. 2023;15(12):6988–6995. doi:10.21037/jtd-23-1787
332. Chen D, Wu X, Yang J, Yu L. Serum plasminogen activator urokinase receptor predicts elevated risk of acute respiratory distress syndrome in patients with sepsis and is positively associated with disease severity, inflammation and mortality. Exp Ther Med. 2019;18(4):2984–2992. doi:10.3892/etm.2019.7931
333. Wu X, Hu K, Yu L, Wang H, Long D. Correlation of plasma suPAR expression with disease risk and severity as well as prognosis of sepsis-induced acute respiratory distress syndrome. Int J Clin Exp Pathol. 2017;10(12):11378–11383.
334. Huang Q, Xiong H, Yan P, et al. The diagnostic and prognostic value of suPAR in patients with sepsis: a systematic review and meta-analysis. Shock. 2020;53(4):416–425. doi:10.1097/shk.0000000000001434
335. Liu X, Shen Y, Li Z, et al. Prognostic significance of APACHE II score and plasma suPAR in Chinese patients with sepsis: a prospective observational study. BMC Anesthesiol. 2016;16(1):46. doi:10.1186/s12871-016-0212-3
336. Liu Z, Li J, Chen D, et al. Dynamic interleukin-6 level changes as a prognostic indicator in patients with COVID-19. Front Pharmacol. 2020;11:1093. doi:10.3389/fphar.2020.01093
337. Swaroopa D, Bhaskar K, Mahathi T, et al. Association of serum interleukin-6, interleukin-8, and Acute Physiology and Chronic Health Evaluation II score with clinical outcome in patients with acute respiratory distress syndrome. Indian J Crit Care Med. 2016;20(9):518–525. doi:10.4103/0972-5229.190369
338. Nguyen CV, Luong CQ, Dao CX, et al. Predictive validity of interleukin 6 (IL-6) for the mortality in critically ill COVID-19 patients with the B.1.617.2 (Delta) variant in Vietnam: a single-centre, cross-sectional study. BMJ Open. 2024;14(12):e085971. doi:10.1136/bmjopen-2024-085971
339. Hui L, Zhang X, An X, et al. Higher serum procalcitonin and IL-6 levels predict worse diagnosis for acute respiratory distress syndrome patients with multiple organ dysfunction. Int J Clin Exp Pathol. 2017;10(7):7401–7407.
340. Tasoudis PT, Arvaniti CK, Adamou AT, et al. Interleukin-6 inhibitors reduce mortality in coronavirus disease-2019: an individual patient data meta-analysis from randomized controlled trials. Eur J Intern Med. 2022;101:41–48. doi:10.1016/j.ejim.2022.04.004
341. Reilly JP, Wang F, Jones TK, et al. Plasma angiopoietin-2 as a potential causal marker in sepsis-associated ARDS development: evidence from Mendelian randomization and mediation analysis. Intensive Care Med. 2018;44(11):1849–1858. doi:10.1007/s00134-018-5328-0
342. Calfee CS, Gallagher D, Abbott J, Thompson BT, Matthay MA. Plasma angiopoietin-2 in clinical acute lung injury: prognostic and pathogenetic significance. Crit Care Med. 2012;40(6):1731–1737. doi:10.1097/CCM.0b013e3182451c87
343. Li F, Yin R, Guo Q. Circulating angiopoietin-2 and the risk of mortality in patients with acute respiratory distress syndrome: a systematic review and meta-analysis of 10 prospective cohort studies. Ther Adv Respir Dis. 2020;14:1753466620905274. doi:10.1177/1753466620905274
344. Wang K, Bhandari V, Giuliano JS, CS OH, Shattuck MD, Kirby M. Angiopoietin-1, angiopoietin-2 and bicarbonate as diagnostic biomarkers in children with severe sepsis. PLoS One. 2014;9(9):e108461. doi:10.1371/journal.pone.0108461
345. Agrawal A, Matthay MA, Kangelaris KN, et al. Plasma angiopoietin-2 predicts the onset of acute lung injury in critically ill patients. Am J Respir Crit Care Med. 2013;187(7):736–742. doi:10.1164/rccm.201208-1460OC
346. Kümpers P, Lukasz A. The curse of angiopoietin-2 in ARDS: on stranger TI(E)des. Crit Care. 2018;22(1):44. doi:10.1186/s13054-018-1978-0
347. Ong T, McClintock DE, Kallet RH, Ware LB, Matthay MA, Liu KD. Ratio of angiopoietin-2 to angiopoietin-1 as a predictor of mortality in acute lung injury patients. Crit Care Med. 2010;38(9):1845–1851. doi:10.1097/CCM.0b013e3181eaa5bf
348. Schlapbach LJ, Graf R, Woerner A, et al. Pancreatic stone protein as a novel marker for neonatal sepsis. Intensive Care Med. 2013;39(4):754–763. doi:10.1007/s00134-012-2798-3
349. de Hond TAP, Oosterheert JJ, van Hemert-Glaubitz SJM, Musson REA, Kaasjager KAH. Pancreatic stone protein as a biomarker for sepsis at the emergency department of a large tertiary hospital. Pathogens. 2022;11(5):559. doi:10.3390/pathogens11050559
350. Prazak J, Irincheeva I, Llewelyn MJ, et al. Accuracy of pancreatic stone protein for the diagnosis of infection in hospitalized adults: a systematic review and individual patient level meta-analysis. Crit Care. 2021;25(1):182. doi:10.1186/s13054-021-03609-2
351. Klein HJ, Niggemann P, Buehler PK, et al. Pancreatic stone protein predicts sepsis in severely burned patients irrespective of trauma severity: a monocentric observational study. Ann Surg. 2021;274(6):e1179–e1186. doi:10.1097/sla.0000000000003784
352. Permana SA, Hartono EJ, Hartono EJ. Measurement of pancreatic stone protein compared with c-reactive protein and procalcitonin in the diagnosis of sepsis in an intensive care unit: a systematic review. Malays J Med Sci. 2024;31(5):32–40. doi:10.21315/mjms2024.31.5.3
353. Schneider JE, Dick K, Cooper JT, Chami N. Pancreatic stone protein point-of-care testing can reduce healthcare expenditure in sepsis. Health Econ Rev. 2022;12(1):39. doi:10.1186/s13561-022-00381-z
354. Puskarich MA, Cornelius DC, Tharp J, Nandi U, Jones AE. Plasma syndecan-1 levels identify a cohort of patients with severe sepsis at high risk for intubation after large-volume intravenous fluid resuscitation. J Crit Care. 2016;36:125–129. doi:10.1016/j.jcrc.2016.06.027
355. Sun T, Wang Y, Wu X, Cai Y, Zhai T, Zhan Q. Prognostic value of syndecan-1 in the prediction of sepsis-related complications and mortality: a meta-analysis. Front Public Health. 2022;10:870065. doi:10.3389/fpubh.2022.870065
356. Smart L, Bosio E, Macdonald SPJ, et al. Glycocalyx biomarker syndecan-1 is a stronger predictor of respiratory failure in patients with sepsis due to pneumonia, compared to endocan. J Crit Care. 2018;47:93–98. doi:10.1016/j.jcrc.2018.06.015
357. Piotti A, Novelli D, Meessen J, et al. Endothelial damage in septic shock patients as evidenced by circulating syndecan-1, sphingosine-1-phosphate and soluble VE-cadherin: a substudy of ALBIOS. Crit Care. 2021;25(1):113. doi:10.1186/s13054-021-03545-1
358. Li Z, Jiang X, Li J, Wang Y. Correlation of Hyaluronic Acid (HA), Syndecan-1 (SDC-1), Heparan Sulfate (HS) with early stage end organ dysfunction in sepsis patients. J Cardiovasc Pharmacol. 2025;85(2):129–136. doi:10.1097/fjc.0000000000001654
359. De Freitas Caires N, Gaudet A, Portier L, Tsicopoulos A, Mathieu D, Lassalle P. Endocan, sepsis, pneumonia, and acute respiratory distress syndrome. Crit Care. 2018;22(1):280. doi:10.1186/s13054-018-2222-7
360. Evans L, Rhodes A, Alhazzani W, et al. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock 2021. Crit Care Med. 2021;49(11):e1063–e1143. doi:10.1097/ccm.0000000000005337
361. Cabrera M, Bharil S, Chin M, Yohannes S, Clark P. Impact of proning with and without inhaled pulmonary vasodilators and neuromuscular blocking agents in COVID acute respiratory distress syndrome. World J Crit Care Med. 2025;14(3):101327. doi:10.5492/wjccm.v14.i3.101327
362. Ma W, Tang S, Yao P, et al. Advances in acute respiratory distress syndrome: focusing on heterogeneity, pathophysiology, and therapeutic strategies. Signal Transduct Target Ther. 2025;10(1):75. doi:10.1038/s41392-025-02127-9
363. Fan E, Del Sorbo L, Goligher EC, et al. An Official American Thoracic Society/European Society of intensive care medicine/society of critical care medicine clinical practice guideline: mechanical ventilation in adult patients with acute respiratory distress syndrome. Am J Respir Crit Care Med. 2017;195(9):1253–1263. doi:10.1164/rccm.201703-0548ST
364. Jacobson JR. Pharmacologic therapies on the horizon for acute lung injury/acute respiratory distress syndrome. J Investig Med. 2009;57(8):870–873. doi:10.2310/JIM.0b013e3181c04681
365. Abbasi T, Garcia JG. Sphingolipids in lung endothelial biology and regulation of vascular integrity. Handb Exp Pharmacol. 2013;216:201–226. doi:10.1007/978-3-7091-1511-4_10
366. Uhlig S, Gulbins E. Sphingolipids in the lungs. Am J Respir Crit Care Med. 2008;178(11):1100–1114. doi:10.1164/rccm.200804-595SO
367. Chalmers JD, Short PM, Mandal P, Akram AR, Hill AT. Statins in community acquired pneumonia: evidence from experimental and clinical studies. Respir Med. 2010;104(8):1081–1091. doi:10.1016/j.rmed.2010.04.005
368. Chen W, Sammani S, Mitra S, Ma SF, Garcia JG, Jacobson JR. Critical role for integrin-β4 in the attenuation of murine acute lung injury by simvastatin. Am J Physiol Lung Cell Mol Physiol. 2012;303(4):L279–85. doi:10.1152/ajplung.00361.2011
369. Steinberg KP, Milberg JA, Martin TR, Maunder RJ, Cockrill BA, Hudson LD. Evolution of bronchoalveolar cell populations in the adult respiratory distress syndrome. Am J Respir Crit Care Med. 1994;150(1):113–122. doi:10.1164/ajrccm.150.1.8025736
370. Walmsley SR, Chilvers ER, Thompson AA, et al. Prolyl hydroxylase 3 (PHD3) is essential for hypoxic regulation of neutrophilic inflammation in humans and mice. J Clin Invest. 2011;121(3):1053–1063. doi:10.1172/jci43273
371. Vu CA, DeRonde KJ, Vega AD, et al. Effects of Tocilizumab in COVID-19 patients: a cohort study. BMC Infect Dis. 2020;20(1):964. doi:10.1186/s12879-020-05701-4
372. Trøseid M, Arribas JR, Assoumou L, et al. Efficacy and safety of baricitinib in hospitalized adults with severe or critical COVID-19 (Bari-SolidAct): a randomised, double-blind, placebo-controlled phase 3 trial. Crit Care. 2023;27(1):9. doi:10.1186/s13054-022-04205-8
373. Ng WH, Tang PCH, Mahalingam S, Liu X. Repurposing of drugs targeting the cytokine storm induced by SARS-CoV-2. Br J Pharmacol. 2023;180(2):133–143. doi:10.1111/bph.15987
374. Furci F, Murdaca G, Allegra A, Gammeri L, Senna G, Gangemi S. IL-33 and the cytokine storm in COVID-19: from a potential immunological relationship towards precision medicine. Int J Mol Sci. 2022;23(23):14532. doi:10.3390/ijms232314532
375. Zhang LS, Yu Y, Yu H, Han ZC. Therapeutic prospects of mesenchymal stem/stromal cells in COVID-19 associated pulmonary diseases: from bench to bedside. World J Stem Cells. 2021;13(8):1058–1071. doi:10.4252/wjsc.v13.i8.1058
376. Bastarache JA, Fremont RD, Kropski JA, Bossert FR, Ware LB. Procoagulant alveolar microparticles in the lungs of patients with acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol. 2009;297(6):L1035–41. doi:10.1152/ajplung.00214.2009
377. Morris PE, Steingrub JS, Huang BY, et al. A Phase I study evaluating the pharmacokinetics, safety and tolerability of an antibody-based tissue factor antagonist in subjects with acute lung injury or acute respiratory distress syndrome. BMC Pulm Med. 2012;12(1):5. doi:10.1186/1471-2466-12-5
378. Camprubí-Rimblas M, Tantinyà N, Guillamat-Prats R, et al. Effects of nebulized antithrombin and heparin on inflammatory and coagulation alterations in an acute lung injury model in rats. J Thromb Haemost. 2020;18(3):571–583. doi:10.1111/jth.14685
379. Dixon B, Schultz MJ, Smith R, Fink JB, Santamaria JD, Campbell DJ. Nebulized heparin is associated with fewer days of mechanical ventilation in critically ill patients: a randomized controlled trial. Crit Care. 2010;14(5):R180. doi:10.1186/cc9286
380. Dixon B, Smith R, Santamaria JD, et al. A trial of nebulised heparin to limit lung injury following cardiac surgery. Anaesth Intensive Care. 2016;44(1):28–33. doi:10.1177/0310057x1604400106
381. Abdelaal Ahmed Mahmoud A, Mahmoud HE, Mahran MA, Khaled M. Streptokinase versus unfractionated heparin nebulization in patients with severe Acute Respiratory Distress Syndrome (ARDS): a randomized controlled trial with observational controls. J Cardiothorac Vasc Anesth. 2020;34(2):436–443. doi:10.1053/j.jvca.2019.05.035
382. Koenecke A, Powell M, Xiong R, et al. Alpha-1 adrenergic receptor antagonists to prevent hyperinflammation and death from lower respiratory tract infection. Elife. 2021;10. doi:10.7554/eLife.61700
383. Zhou Y, Wang F, Tang J, Nussinov R, Cheng F. Artificial intelligence in COVID-19 drug repurposing. Lancet Digit Health. 2020;2(12):e667–e676. doi:10.1016/s2589-7500(20)30192-8
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Identifying Potential Effective Diagnostic and Prognostic Biomarkers in Sepsis by Bioinformatics Analysis and Validation
Huang X, Tan J, Chen X, Zhao L
International Journal of General Medicine 2022, 15:6055-6071
Published Date: 6 July 2022
Impaired Circulating Antibody-Secreting Cells Generation Predicts the Dismal Outcome in the Elderly Septic Shock Patients
Xu H, Li T, Zhang X, Li H, Lv D, Wang Y, Huo F, Bai J, Wang C
Journal of Inflammation Research 2022, 15:5293-5308
Published Date: 13 September 2022
SPINK1 is a Potential Diagnostic and Prognostic Biomarker for Sepsis
Chen D, Shi Z, Gao X, Yang Y, Lei X, Hu Y
Infection and Drug Resistance 2024, 17:875-884
Published Date: 8 March 2024
Integrated Analysis Identified TGFBI as a Biomarker of Disease Severity and Prognosis Correlated with Immune Infiltrates in Patients with Sepsis
Shi M, Wei Y, Guo R, Luo F
Journal of Inflammation Research 2024, 17:2285-2298
Published Date: 15 April 2024
Impact of High Troponin Level on the Outcome in COVID-19 Positive Patients
Abohamr SI, Kattea MO, Abazid RM, Aldossari MA, Al Asiri N, Alhussini AU, Al Hussaini KI, Alasiri GA, Ali A, Elsheikh E
Journal of Multidisciplinary Healthcare 2024, 17:4989-5000
Published Date: 1 November 2024