Back to Journals » International Journal of Nanomedicine » Volume 20
Cholesterol-Rich Antibiotic-Loaded Liposomes as Efficient Antimicrobial Therapeutics
Authors Natsaridis E ![]() , Gkartziou F, Mouzoura P, Mourtas S
, Gkartziou F, Mouzoura P, Mourtas S ![]() , Papadia K, Kolonitsiou F, Klepetsanis P, Dermon CR, Spiliopoulou I, Antimisiaris SG
, Papadia K, Kolonitsiou F, Klepetsanis P, Dermon CR, Spiliopoulou I, Antimisiaris SG ![]()
Received 14 January 2025
Accepted for publication 21 March 2025
Published 17 April 2025 Volume 2025:20 Pages 4943—4965
DOI https://doi.org/10.2147/IJN.S513553
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Kamakhya Prakash Misra
Evangelos Natsaridis,1,2,* Foteini Gkartziou,2,* Panagiota Mouzoura,1 Spyridon Mourtas,3 Konstantina Papadia,1 Fevronia Kolonitsiou,4 Pavlos Klepetsanis1,2 ,† Catherine R Dermon,5 Iris Spiliopoulou,4,6 Sophia G Antimisiaris1,2
1Laboratory of Pharmaceutical Technology, Department of Pharmacy, School of Health Sciences, University of Patras, Rion, Patras, 26504, Greece; 2Institute of Chemical Engineering Sciences, FORTH/ICE-HT, Platani, Patras, 26504, Greece; 3Department of Chemistry, University of Patras, Rion, Patras, 26510, Greece; 4Department of Microbiology, School of Medicine, University of Patras, Rion, Patras, 26504, Greece; 5Laboratory of Human and Animal Physiology, Department of Biology, University of Patras, Rion, Patras, 26504, Greece; 6National Reference Centre for Staphylococci, School of Medicine, University of Patras, Rion, Patras, 26504, Greece
†Pavlos Klepetsanis passed away on October 23, 2024
*These authors contributed equally to this work
Correspondence: Sophia G Antimisiaris, Laboratory Pharm Technology, University of Patras, Pharmacy Building, Rio Campus, Rio Patras, 26504, Greece, Tel +30-2610-962332, Email [email protected]
Introduction: Liposomal antibiotics have demonstrated higher bacteriostatic and bactericidal activities than free drugs. In this study, we investigated the effects of cholesterol (Chol) content of liposomes, liposome concentration, and surface coating with polyethylene glycol (PEG) on the antimicrobial activity of moxifloxacin (MOX) liposomes against Staphylococcus epidermidis (ATCC 35984) (S.e).
Methods: MOX-liposome compositions with increasing Chol content were evaluated for their susceptibility to planktonic S.e (growth inhibition, killing, and live-dead staining), as well as against pre-formed biofilms (crystal violet, MTT assay, and confocal microscopy). The MOX-liposomes prepared by active loading were characterized in terms of loading, size distribution, and zeta potential.
Results-Discussion: All liposomes had nano-dimensions ranging in diameter from 92nm to 114nm, with zeta-potential values from − 2.30mV to − 4.50mV. Planktonic bacteria and established biofilms are significantly more susceptible to MOX-liposomes with higher Chol-content than other liposome-types, and the same MOX dose encapsulated in 10 times higher lipids demonstrated higher antimicrobial activity. Coating the MOX liposomes with PEG did not affect their activity. Flow cytometry showed higher binding of Chol-rich liposomes to bacteria, explaining the higher antimicrobial activity. Interestingly, the integrity of calcein-loaded Chol-rich liposomes was much lower than that of liposomes with low or no Chol during incubation with various strains of S. epidermidis. In vivo results in a zebrafish infection model (bacteremia) confirmed the superior activity of Chol-rich MOX-liposomes compared to the free drug.
Conclusion: The current in vitro and in vivo findings demonstrated the potential of PEGylated and Chol-rich liposomal antibiotics as highly efficient therapeutics for the treatment of S. epidermidis infections.
Keywords: liposomes, cholesterol, antimicrobial, in vivo, antibiotic, moxifloxacin, Staphylococcus epidermidis
Graphical Abstract:

Introduction
Liposomes are well known for their advantages as drug carriers, because liposomal drug products have been used as therapeutics for the treatment of life-threatening diseases for more than 30 years.1,2 Concerning the applications of liposomal-drugs in the treatment of bacterial infections, several studies have been conducted up-to-date, with the aim to examine the potential advantages of liposomal antibiotics for the treatment of multi-resistant bacterial infections that are currently considered as high priority unmet medical needs.3–6
The superior antimicrobial and/or antibiofilm activity of liposomal drugs has been proven in several in vitro and in vivo investigations,7 and the reduced penetration of antibiotics through biofilm and low accumulation at infected sites can be overcome using nanotechnological approaches.7 The surface charge and liposome surface coating with polyethylene glycol (PEG) have been reported to affect the antimicrobial efficacy of liposomes. For example, neutral gentamicin liposomes reduced the minimum inhibitory concentration (MIC) and bactericidal concentration (MBC) against planktonic Pseudomonas aeruginosa and Klebsiella oxytoca bacteria significantly more than negatively charged liposomes.8 The antibacterial efficacy of curcumin was augmented by encapsulation in liposomes with positive charge,9 and negatively charged tobramycin liposomes demonstrated an increased antibiofilm effect against Burkholderia cepacia complex (Bcc) compared to free drug and neutral liposomes.10 Neutral daptomycin liposomes exhibited dramatically enhanced inhibition of planktonic bacterial growth (compared to the free drug) in MRSE and MRSA bacterial strains, whereas negatively charged liposomes were completely inactive. Neutral liposomes are also more effective for the prevention of biofilms, whereas negatively charged liposomes are used to treat established biofilms.11 Positively charged (cationic) liposomes demonstrated a high reduction in P. aeruginosa biofilm, while anionic liposomes had high biofilm permeability; PEG coating increased the antibiofilm effect but reduced the retention of the liposomes on the biofilms.12 However, the cytotoxicity of cationic liposomes is an aspect that could not be overlooked. In one case, although cationic rifampicin-loaded liposomes had the highest interaction with S. aureus biofilms, non-cytotoxic negatively charged liposomes were selected as a rifampicin formulation against S. aureus infections because of the toxicity of the cationic liposomes.13
In addition to the surface charge of liposomes, other physicochemical characteristics such as drug loading and retention have been reported to affect the activity of liposomal drugs towards bacteria, such as the drug loading and retention in liposomes.5,7,14 Recently, the manufacturing method applied to load moxifloxacin (MOX) into liposomes was found to affect their antimicrobial activity.14 Indeed, MOX-liposomes (MLIP) prepared by active drug loading demonstrated substantially higher antimicrobial activity against planktonic and biofilm Staphylococcus epidermidis bacteria, compared to liposomes (with same MOX content) prepared by a passive drug loading method (where the drug is loaded into liposomes during their preparation). The high antimicrobial activity of the active-loaded liposomes was attributed to the prolonged retention of MOX in the vesicles, suggesting that liposomal drugs that retain higher amounts of their drug content during interaction with bacteria demonstrate increased activity.
In recent years, bacterial toxins have been reported to trigger the rapid release of liposomal contents from liposomes with high cholesterol (Chol) levels,15 and the Chol content of liposomal drug formulations has been proposed as an important factor that determines their antimicrobial activity. In addition, coating of liposome surface with polyethylene glycol, up to a certain amount, increased the sensitivity of Chol-rich liposomes towards bacterial toxins, while at higher amounts, liposome–toxin interactions were substantially reduced.15
Based on previous findings about preferred interaction between bacterial toxins and Chol-rich membranes and liposomes,15 Henry et al reported in 2015,16 that empty “artificial liposomes” bind bacterial toxins decoying the lysis of mammalian cells (in vitro) and rescuing mice from septicemia (by S. aureus or S. pneumonia) infections (in vivo). Empty sphingomyelin (SM)/Chol liposomes protected cells from Chol-dependent cytolysins such as phospholipase C and α-hemolysin, whereas Chol contents above 50 mol% were required for SM/Chol liposomes to fully protect the cells from bacterial toxins. The need for high Chol content was attributed to the fact that liposomes with Chol microdomains fuse better with bacterial membranes; such lipid rafts demonstrate a stronger affinity for bacterial virulence factors. In another report, Clostridium perfringens delta-toxin (a β-pore-forming toxin) caused the release of carboxyfluorescein (CF) from SM/Chol liposomes at a Chol-content-dependent rate (release increased with Chol content, in liposomes with 30–50 mol% Chol).17 More recently, the Chol-content of berberine-loaded liposomes was found to be directly related to their ability to reduce S. aureus biofilms,18 proving that the above strategy (related to the affinity of Chol for cholesterol-dependent cytolysins, secreted from bacteria) is also effective for biofilm eradication and may be applied for bacteria (component)-triggered release of antibiotics from Chol-rich liposomes.
Based on the above-mentioned findings, we evaluated herein the effect of lipid composition (Chol and PEG content) and lipid concentration of active-loaded MOX-liposomes (MLIP) on their antimicrobial properties towards S.e planktonic bacteria and biofilms. S.e bacteria have not been previously investigated for the effect of such liposome preparative parameters (and especially liposomal Chol content) on their susceptibility to liposomal antibiotics. In addition to in vitro studies, we performed in vivo experiments to verify the enhanced antimicrobial potential of Chol-rich MLIP formulations in comparison to the free drug in a zebrafish model of bacteremia.
Materials and Methods
Egg phosphatidylcholine (PC), 1.2-Distearoyl-sn-glycerol−3-phosphatidyl-ethanolamine-N- [methoxy (polyethylene glycol) −2000] (PEG) were purchased from Lipoid (Germany). Lissamine-rhodamine B phosphatidylethanolamine (RHO) was purchased from Avanti Polar Lipids (Alabaster, AL). Cholesterol (99%) (Chol), Triton X-100, calcein, and Sephadex G-50 were obtained from–Aldrich (Darmstadt, Germany). Moxifloxacin was supplied by COOPER (Athens, Greece). Agar was obtained from Sigma-Aldrich (Darmstadt, Germany). All solvents used were of analytical (or HPLC) grade and purchased from Merck (Germany). All other materials, such as salts used for buffer preparation and reagents for lipid concentration determination, were of analytical grade and purchased from Sigma–Aldrich (Darmstadt, Germany).
Preparation of MOX-Loaded Liposomes (MLIP)
A recently reported active loading protocol was used to prepare MLIPs.14 Knowing the range of drug loadings achieved for liposomes with different lipid compositions, from our previous study,14 we adjusted the drug loading or final Drug/Lipid (D/L) (mol:mol) ratio using a specific initial D/L ratio in the drug loading step. In brief, empty small unilamellar vesicle (SUV) liposomes were prepared as described previously in ammonium sulfate (120mM) at pH 5.60, and eluted through a Sephadex G−50 column (1 × 30 cm) with Phosphate Buffered Saline (PBS) pH 7.40 buffer, to replace the external ammonium sulfate solution with PBS buffer and attain a pH gradient between the interior and exterior of vesicles. MOX was then remotely loaded into the liposomes by incubating the empty SUV liposomes (LIPs) with different concentrations of MOX (dissolved in PBS) in a total volume of 1mL, at 40°C for 1h. Specifically, in order to prepare liposomes with a given MOX concentration (0.15µM) and different lipid concentrations, the Drug/Lipid (D/L) ratio of the MLIPs was adjusted during MOX loading in order to attain two types of MLIPs, having either “High” lipid concentration (25.1µM) (or low drug/lipid (D/L) ratio; D/L = 0.005, mol/mol), where 10 mg of empty liposomes were incubated with 0.05 mg of MOX during MOX loading, or “Low” lipid concentration (2.51µM) (high D/L = 0.05) where 10 mg of empty liposomes were incubated with 0.5 mg of MOX.
For the study of the effect of MLIP Chol-content on their antimicrobial activity, the lipid compositions prepared were as follows: PC [referred to as MLIP-0], PC/Chol 4:1 (mole/mole) [referred to as MLIP-20%] and PC/Chol 1:1 (mole/mole) [referred to as MLIP-50%]; for these liposomes the initial D/L ratio was adjusted to attain MOX concentration of 0.15µM in 9.5µM liposomal lipid. In all cases, empty liposomes (LIP) [LIP-0, LIP-20 and LIP-50%] were additionally prepared as control liposomes. Additionally, PEGylated MLIP-50% with compositions PC/Chol/PEG (0.50:0.50:0.04, mole/mole) [MLIP-50% +4% PEG] and PC/Chol/PEG (0.50:0.50:0.08, mole/mole) [MLIP-50% +8% PEG] were prepared.
In all cases, MLIPs were purified from the non-encapsulated drug by size-exclusion chromatography on a Sephadex G−50 column (1×30cm), eluted with PBS (pH 7.40), and characterized as described below.
Physicochemical Characterization of MLIPs
MOX Loading in Liposomes
As previously reported, the MOX Loading Efficiency (D/L%) was calculated from purified (from non-encapsulated MOX) liposomes by quantifying the liposome lipid concentration and MOX concentration. Phospholipid content was measured by the Stewart assay,19 which is a colorimetric method used for phospholipid quantification. The MOX concentration in the liposomes was quantified by isocratic high-performance liquid chromatography (HPLC) using a Shimadzu 20A5 Gradient HPLC system coupled to aSPD−20A Prominence UV/VIS detector operating at 292 nm. A Luna® 5 µm C18 (2) 100Å, LC Column (250×4.6mm) was used, the mobile phase was a mixture of acidified water (0.1% trifluoroacetic acid) and acetonitrile at 77:23 v/v. The column was eluted at a flow rate of 1 mL/min at 25°C, and MOX was eluted at 12.3 min. The sample injection volume was 50μL. Liposomes were analyzed after disruption in methanol. One volume of liposomes was mixed with nine volumes of methanol, and the mixture was agitated by vortex until it was clear. A calibration curve in the range of 1–80μg/mL was constructed by preparing standard solutions of MOX in media with a composition similar to that of the samples.14
Vesicle Physicochemical Properties
The particle size distribution (mean hydrodynamic diameter and polydispersity index) of liposomes dispersed at 0.4 mg/mL lipid, in phosphate-buffered saline (10mM) with pH 7.40, was measured by dynamic light scattering (DLS) (Malvern Nano-Zs, Malvern Instruments, Malvern, UK) at 25°C and a 173° angle.20 Each sample was measured 11 times using three independent measurements. The polydispersity index (PDI) was used to measure liposomal dispersion homogeneity. Dispersions with a PDI less than 0.200 or 0.250 are generally considered to have a narrow size distribution. Zeta potential was measured in the same dispersions at 25°C using the Doppler electrophoresis technique, as previously reported.20
We did not characterize the MLIPs in this study for morphology and MOX release kinetics, since such studies were previously carried out for active-loaded MLIPs with varying lipid compositions, and results have been reported.14
Antimicrobial Activity or MLIPs (In Vitro)
Bacterial Strains and Growth Conditions
All strains of S.e were grown aerobically in tryptic soy broth (TSB, Oxoid CM0129, Oxoid Ltd., Wade Road, Basingstoke, Hants, RG24 8PW, UK) and on tryptic soy agar (TSA, Oxoid) plates at 37°C overnight.
Most experiments were conducted with S.e reference strain ATCC 35984, which is a slime-positive/ica-positive (slime+) biofilm-forming strain. In some studies, the S.e reference strain ATCC 12228 (slime negative/ica-negative (slime-) and non-biofilm-forming strain) was used. In the calcein-liposome bacteria interaction study, two clinical S.e strains (previously characterized for slime production and ica operon carriage) were also used, the GRE4388 (slime-positive/ica positive) (CLIN+) and GRE2264 (slime-negative/ica-positive) (CLIN-), in order to investigate potential differences between reference and clinical strains.21 Furthermore, in liposome bacteria interaction studies, in addition to S.e ATCC 35984 strain, two nosocomial strains, S.e 762 (quinolone-susceptible) and S.e 11465 (quinolone-resistant), were used.22
Bacterial Growth Inhibition
Bacterial growth rates were monitored by spectrophotometry22,23 in the presence and absence of different liposomal or drug solutions (MLIPs, empty LIP, mixtures of free MOX and LIPs and free MOX) at the MIC50 of free MOX (0.15μM).14 Briefly, bacterial cells grown overnight on TSA agar plates were grown in fresh TSB broth (without glucose) until the early exponential phase. The broth containing bacteria was inoculated into 96-well flat-bottomed polystyrene plates with an initial absorbance at λmax of 570–0.01. The change in the absorbance of each well was monitored every hour for 24h using a Fluostar microplate reader (BMG LABTECH).
For time–kill studies, a fresh culture of S.e in a final inoculum of ~4 × 107cfu/mL (in TSB broth) was incubated at 37°C for 2, 4, 6, and 24 h with 0.15μM (MIC 50 of free MOX) of free MOX or MLIP. A culture of the same strain without antibiotics was used as the positive control. At each time interval, bacteria were harvested in serial dilutions, and CFU/mL was determined by counting the single colonies that emerged in the appropriate dilution on TSA plates. The experiments were performed in triplicates.
Biofilm Susceptibility Assays
Antibiofilm activity was evaluated at concentrations corresponding to the MIC of MOX ie, 0.3μM, for all formulations, according to previously reported methods.22–25 Crystal violet (CV) staining and the validated MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] cell viability assay were used to assess the biofilm susceptibility to each free or MLIP formulation. Briefly, a single bacterial colony isolated from fresh agar plates was inoculated into a tube filled with 5mL sterile TSB and incubated at 37°C for 24h. Fresh bacterial suspensions were prepared in TSB with 1% glucose from overnight cultures and adjusted to 0.5 MacFarland turbidity standard, followed by 1:10 dilution in fresh media. Then, 200μL of the suspension was added to 96-well sterile polystyrene plates and incubated at 37°C for 24h. Following overnight incubation, the plates were gently washed with 1x phosphate-buffered saline (PBS pH 7.4) to remove planktonic cells, and well-formed biofilms were incubated with free MOX or MLIP at 37°C for 24h. The next day, the bacterial suspension from each well was gently spent, washed three times with phosphate buffer saline (pH 7.2) and stained with 195μL of 0.1% Crystal Violet (Sigma-Aldrich, St. Louis, MO) for 15min at room temperature. Excess crystal violet was removed by washing with tap water, and the biofilm was quantified by measuring the corresponding OD−570 nm of the supernatant after dissolving CV in 95% ethanol. For each tested sample (free or liposomal), biofilm assays were performed in triplicate, and the mean biofilm absorbance value was determined.
For the MTT assay, the biofilms were incubated with MTT (0.5 mg/mL) at 37°C for 1h. After washing, the purple formazan crystals formed inside the bacterial cells were dissolved in acidified isopropanol and measured using a microplate reader by setting the detection and reference wavelengths at 570 nm and 630 nm, respectively.
Flow Cytometry Studies
Flow cytometry studies were performed to verify the bactericidal activity of the various liposomal formulations and to quantify liposome-bacterial interactions, as described in detail below.
Bacteria Viability
To evaluate bacterial viability either in planktonic bacteria or in biofilms, the LIVE/DEAD® BacLight™ Bacterial Viability Kit was used, and analysis was carried out by flow cytometry (for planktonic bacteria) or confocal microscopy for the biofilms (see details in Antibiofilm Activity by Confocal Microscopy). This assay employs two nucleic acid stains: green-fluorescent SYTO®9 stain, which labels both live and dead bacteria, and red-fluorescent propidium iodide stain, which penetrates only bacteria with damaged membranes, reducing SYTO®9 fluorescence when both dyes are present. In brief, a fresh culture of S.e in a final inoculum of 4×107cfu/mL (in TSB broth) was incubated at 37°C for 24h with 0.15µM of free MOX or MLIP (MIC50 of free MOX). A culture of the same strain without antibiotics was used as the positive control. After 24h of incubation, the bacteria were washed with 1mL of PBS by centrifugation at 4000–6000 rpm for 5 min. The bacterial pellet was re-suspended in 1 mL of PBS and stained with the LIVE/DEAD® BacLight™ Bacterial Viability Kit (L7007, Invitrogen) according to the manufacturer’s instructions and analyzed by flow cytometry25,26 using a BD Accuri C6 Plus Flow Cytometer (BD Biosciences). Data were analyzed using Accuri C6 Plus Flow Cytometer Software ν1.0.34.1. build 2018111.34.1. The experiments were performed in triplicates.
Liposomes–Bacteria Interaction (Binding)
To evaluate liposome interaction/binding to bacterial cells, liposomes were labelled with 0.1 mol% RHO, and the analysis was carried out by flow cytometry. Three bacterial strains were used in the experiment: ATCC 35984 and two nosocomial strains, Se 762 (quinolone-susceptible) and Se 11465 (quinolone-resistant). RHO-Liposomes at a final lipid concentration of 0.25 mg/mL in PBS were mixed with the bacterial suspension (4.5×107cfu/mL) at a 1:1 ratio (v/v) and incubated at 37°C under agitation. Free RHO micelles were used as controls. After 24h of incubation, the bacteria were washed three times with 1mL PBS to remove free liposomes by centrifugation at 4000–6000 rpm for 5 min. The bacterial pellet was finally suspended in 1 mL of PBS for flow cytometry analysis.25,26
Antibiofilm Activity by Confocal Microscopy
Antibiofilm activity was evaluated at concentrations corresponding to the MIC of free MOX, ie 0.3μM, for all the formulations. The biofilm was visualized using confocal fluorescence microscopy after live/dead staining. For confocal imaging, ~1.5x108cfu/mL bacteria were seeded on glass coverslips and incubated at 37°C for 24h. Following overnight incubation, the cells were gently washed with 1x phosphate buffered saline (PBS pH 7.4) to remove planktonic cells, and the well-formed biofilms were incubated with free MOX or MLIP at 37°C for 24h. The biofilm was stained post-treatment, using a LIVE/DEADTM BacLight Bacterial Viability Kit. Intact cells were stained green, whereas dead or dying cells were stained red. After staining, the biofilms were fixed with 4% formaldehyde diluted in saline for 10 min and mounted on microscope slides using mounting oil (supplied with the kit). The slides were observed using fluorescence microscopy with an SP5 confocal microscope (Leica, Heidelberg, Germany) at 40x magnification. To estimate the percentages of dead bacteria, the program Image J 1.47 was used to count the propidium-stained cells (given as cell area). Biofilms were analyzed using a series of images along the z-axis, followed by three-dimensional reconstruction images.24–28
Liposome Integrity in the Presence of Bacteria
The integrity of the calcein-loaded liposomes during incubation in the presence of bacteria was used to evaluate the effect of potential interactions between different liposome types and bacteria, as previously reported.29 For this PC (LIP-0), PC/Chol 2:1 (LIP-33%), and PC/Chol 1:1 (LIP-50%) calcein-encapsulating liposomes were prepared using the thin-lipid film hydration method. Lipid film hydration was performed using a 100mM isotonic calcein solution at pH 7.40, at which calcein fluorescence is quenched. The integrity of calcein-LIPs was evaluated in the presence of four S.e strains, two clinical strains [CLIN+ (GRE4388, slime+) and CLIN- (GRE2264, slime-)] and two reference strains [ATCC+ (ATCC 35984, slime+) and ATCC- (ATCC 12228, slime-)], under conditions (lipid and bacterial concentrations) that were found to confer substantial decreases in liposome integrity in a previous study from our laboratory.29 In more detail liposome calcein latency (%) was measured at various time-points during their incubation at 37°C, in either PBS 7.4 (control) or in presence of bacteria (final concentration of 4×108 cfu/mL), with a final lipid concentration of 0.25 mg/mL; samples were incubated in pre-sterilized closed test tubes in a thermostated orbital incubator (SI500, Stuart, UK).
For calcein latency (%) measurement, samples (20µL) were retrieved from the incubation tubes, diluted with 4mL buffer pH 7.40, and measured for fluorescence intensity (FI) at EM 470 nm, EX 520 nm, and slits of 5nm before and after addition of Triton X-100 at a final concentration of 2% v/v (which ensures total liposome disruption and release of all encapsulated dye). Calcein Latency (%latency)30 was calculated using Eq. (1):

Where FBT and FAT are the calcein fluorescence intensities before and after addition of Triton X-100, respectively (for FAT, dilution due to Triton addition was considered). Liposome Integrity (%) for each time point was calculated using Eq. (2):

In Vivo Study in Zebrafish
Adult male zebrafish (three-month-old) (Cyprinidae, Teleostei, Danio rerio) were purchased from a local supplier. Male animals of similar age and body size were selected to avoid individual variations and to test the antibacterial activity of MLIP after staphylococcal infection. All experimental procedures were performed in accordance with the European Community’s Council Directive (86/609/EEC) for the care and use of laboratory animals, and efforts were made to minimize the number of animals used and their suffering. The protocol was approved by the Western Greece Regional General Administration of Agricultural Economy & Veterinary Medicine (approval number 375893/1582, 22-12-2021, to be carried out in approved installation EL13 BIOexp-06).
Bacteremia Model
Following a period of 10 days in a 30 L tank according to standard procedures for acclimatization and maintenance (12hr light/12hr dark, 26 ± 2),31 the zebrafish were anesthetized with 0.02% ethyl 3-aminobenzoate methanesulfonate (Tricaine MS-222)32,33 and subsequently 10μL of the reference strain S.e (ATCC 35984) with a final density of 108cfu/mL was administered by intraperitoneal (i.p.) injection for establishment of infection.34,35 For the injection, the anesthetized fish were placed supine (ie, abdomen facing upward) and supported by a moistened foam bed to ensure that they remained in an upright position. The injection was performed carefully in the intraperitoneal cavity and at a specific angle to avoid injury of internal organs.36 The first study was carried out to evaluate the bacterial growth profile in zebrafish blood post-infection over a period of 24h, in order to determine the optimal time-point for administration of antimicrobial treatments. At specific time-points (2, 4, 6 and 24h post-infection), blood samples were collected from the zebrafish, and the bacterial load was quantified. For blood sample collection, zebrafish were anesthetized and 2μL blood sample was collected from the posterior cardinal vein of each fish. Blood samples were transferred to Eppendorf tubes containing 45μL of 0.2Μ KCl as an anti-coagulant. Then, three ten-fold serial dilutions of each recovered blood sample were prepared, plated onto TSA (tryptone soy agar) plates, and incubated overnight (at 37°C/5% CO2) to enumerate the bacterial colonies produced (cfu).37 At the end of the study period, the zebrafish were euthanized by applying 300 mg/L MS-222.
Antibacterial Efficacy Study (In Vivo)
In order to investigate the antibacterial effect of Chol-rich MLIPs, three experimental groups were studied: (i) Control group (PBS solution); (ii) Free-MOX group (MOX solution), and (iii) Pegylated MLIP-50% (PC/Chol/PEG (5:5:0.8 mol/mol/mol; D/L = 0.5 mol/mol)).
Zebrafish were infected with S.e as described above, and 4h post-infection, 5μL of each MOX formulation, was administered to the zebrafish i.p. (as described above). PBS was administered to the fish in the control group. The dose of free MOX or MLIP selected was 5 mg/kg, as this dose of MOX was previously demonstrated to reduce bacteremia,38 and the corresponding liposomal lipid dose (in MLIP) was ~19 mg/kg. Four hours after administration of each treatment (according to the corresponding group), blood samples were drawn, and bacteremia was quantified as described above.
Statistical Analysis
The IBM SPSS statistics package was used for statistical analysis of the results. All experiments were performed in triplicate. All data are presented as the mean ± standard deviation of independent experiments. Statistical significance was evaluated by one-way analysis of variance (ANOVA) or two-way ANOVA and least significant difference (LSD) post-hoc tests with a significance level of p < 0.05.
Results
MLIP Properties
The physicochemical properties of the MLIPs prepared for the current study are reported in Table 1 (liposomes prepared for evaluation of the effect of Chol-content on MLIP antimicrobial activity) and Table 2 (liposomes prepared for evaluation of the effect of lipid concentration (or D/L ratio), or PEGylation (of liposomes), on MLIP antimicrobial activity). In all cases, the MLIPs had dimensions within the nanoscale, with diameters between 92 and 114 nm, and were monodisperse with PDIs ranging between 0.082 and 0.250, while their zeta potential values were neutral to slightly negative (ranging from −2.39mV to −4.50mV), as anticipated because of their lipid composition (consisting of non-charged zwitterionic phospholipids). The current MLIP properties are consistent with those reported before for the same type of MOX-loaded liposomes prepared using the same active-loading method.14
 |
Table 1 Physicochemical Properties of the MLIPs That Were Prepared for Evaluation of Liposome Chol Content on Antimicrobial Activity |
 |
Table 2 Physicochemical Properties of the MLIPs That Were Prepared for Evaluation of Liposomal Lipid Content (Concentration) (Low or High) and PEG Coating (of Liposomes) on Antimicrobial Activity |
At this point, it should be mentioned that MLIPs were also characterized for their morphology and for the release of MOX.14 As reported, the vesicles are unilamellar with round shape, while cryo-EM micrographs revealed that MOX is probably precipitated in the aqueous core of the (active-loaded liposomes, MLIPs) as also indicated by XRD study results. Concerning MOX release, MLIP-50% (non-pegylated and pegylated) vesicles release MOX significantly slower compared to MLIP-20%.14
Effect of Chol-Content of MLIP on Their Antimicrobial Efficacy
The effect of the Chol-content of MLIPs on their antimicrobial activity was evaluated in planktonic bacteria and biofilms of S.e reference strain ATCC 35984. For this purpose, the liposomes listed in Table 1 were used.
Inhibition of Planktonic Bacterial Growth, Time–Kill Curves, Flow Cytometry
Planktonic ATCC 35984 S.e bacteria growth is presented in Figure 1A, and the percent of bacteria growth inhibition after 24h incubation with MLIPs or free-MOX is seen in Figure 1B. Although free MOX (at the MIC of 0.15µM) inhibited bacterial growth by 30% after 24h, MLIPs demonstrated substantially higher bacterial growth inhibition, especially in the case of MLIP-50% that exhibited 78.8% inhibition (at 24h) (Figure 1B). The later inhibition value is lower than the inhibition reported previously from our laboratory for the same MLIP type, which was 93%,14 however although the previous studies were carried out using MLIPs with the same MOX concentration (0.15µM) as here, an approximately double lipid concentration was used before (21.5µM lipid vs 9.5µM in the current study), suggesting a potential effect of the liposomal lipid concentration. Furthermore, as shown (Figure 1B), as the Chol-content of the liposomes decreased, so did the inhibitory activity of MLIPs towards Se bacteria. Indeed, MLIP-20% exhibited 46% inhibition and MLIP-0 exhibited 36.5% bacterial growth inhibition (both after 24h); in fact, bacterial growth inhibition was demonstrated to be linearly related to the Chol-content of MLIPs (R2 = 0.9265) (see Supplementary Data file, Figure S1a).
 |
Figure 1 (A) Growth curves of ATCC 35984 S.e in absence and presence of 0.15µM MOX as free or liposomal drug (MLIP) with varying Chol-contents; empty liposomes (LIP) and mixtures (mix) of empty liposomes (9.5µM lipid) with free MOX were used as control groups. (B) Bacteria growth inhibition (%) by free MOX or MLIPs after 24h incubation. Individual differences are marked by ** for p≤ 0.01, *** for p≤ 0.001, and **** for p ≤ 0.0001. |
In agreement with previous results,14 empty liposomes (LIP-0, LIP-20%, and LIP-50%) (with the same lipid composition and at the same lipid concentration (9.5µM) as those of MLIPs) did not have any significant effect on bacterial growth, whereas the mixtures (mix) of free drug (0.15µM) and LIPs (9.5µM) demonstrated a slightly lower effect on bacterial growth compared to that of the free drug, in the case of the two Chol-containing MLIP types (LIP-20%_mix and LIP-50%_mix) (Figure 1A). The latter result was previously attributed to the potential use of LIPs as nutrients for bacterial growth; however, the current results show that Chol is required for this effect (since the same effect was not observed in the case of the mixture consisting of LIPs without Chol (LIP-0_mix)).
The bacterial growth and growth inhibition results shown in Figure 1 were further verified by bacterial killing experiments performed under identical conditions. As seen in the corresponding time–kill curves (Figure 2A), all MLIPs demonstrated significantly higher bacterial killing activity than the free drug, while the highest activity of MLIP-50% compared to the other liposome types was already evident after 4h. In fact, the higher killing activity of MLIP-50% is seen to persist (more or less) for (at least) 24h. In contrast, the MLIP-20% had bactericidal activity similar to that of MLIP-0, up to the 6h time-point. However, at the 24h time-point, the activity of MLIP-0 was dramatically decreased but MLIP-20% retained their bacteria killing activity. In summary, MLIP-50% demonstrated the highest bactericidal activity, resulting in a bacterial Log Reduction of 4.17 (compared to control) after 24h, compared to MLIP-20% and MLIP-0 with corresponding log reductions of 3.36 and 1.54 (after 24h), respectively (Figure 2B). The correlation between Bacteria Log Reduction and MLIP Chol-content is not linear, however it is clear that Chol-content has a substantial effect on Bacterial reduction (see Supplementary Data file, Figure S1b). In fact, according to the log reduction values, viable bacteria are highly reduced after treatment with MLIP-20% (<0.1%) and MLIP-50% (<0.01%) revealing that the corresponding bacterial growth inhibition demonstrated by OD value reduction (Figure 1A) is highly due to bacteria killing. Representative images of single plate-serial dilutions are seen in Figure 2C.
 |
Figure 2 (A) Time–kill curves of ATCC 35984 S.e, in absence and presence of MOX (0.15µM) as free or liposomal drug (MLIP), with varying Chol-contents. The corresponding total lipid concentration (in each well) is 9.5µM. (B) Effect of MLIP Chol-content on bacteria log Reduction after 24h incubation. (C) Representative images of single plate-serial dilution spotting with S.e (ATCC 35984) corresponding to 101–106 dilutions after 24h incubation with free-MOX or MLIP from the time-killing studies for each case. Individual differences are marked by ** for p≤ 0.01, and **** for p ≤ 0.0001. |
The effect of the MLIP Chol-content on their ability to kill planktonic ATCC 35984 bacteria was further verified by flow cytometry using the LIVE/DEAD BacLight staining kit (Figure 3A). After 24h of incubation with bacteria, all MLIPs resulted in a statistically significant decrease of live bacterial, compared to the corresponding values acquired by treatment with free MOX (Figure 3B). The ability of MOX-liposomes to reduce live bacterial cells increased with MLIP Chol-content. As expected, the opposite result was demonstrated for dead bacterial (the number of dead cells increased with MLIP Chol-content) (Figure 3C).
 |
Figure 3 (A) Flow cytometry images obtained after 24h incubation of ATCC 35984 S.e planktonic bacteria with MOX (0.15µM) as free-MOX or MLIPs (with varying Chol-content); stained live cells (green) and dead cells (red). Total lipid concentration (in each well) is 9.5µM. (B) Live bacterial cells (%) and (C) Dead bacterial cells (%). Asterisks on top of columns denote significant differences from control (plain bacteria) and individual differences are also marked as asterisks. In both cases * is for p≤ 0.05, ** for p≤ 0.01, *** for p≤ 0.001, and **** for p ≤ 0.0001. |
Effect of Liposome Chol-Content of MLIPs on Antibiofilm Activity
The effect of MLIP Chol-content on their antibiofilm activity was also studied by performing bacterial biofilm susceptibility assays with the same bacterial strain and MLIPs, using a MOX concentration of 0.3µM (equal to MIC), as used in a previous study.14 Herein, a lipid concentration of 19µM, was used to keep the D/L ratio constant with that used in the studies on planktonic bacteria, and also to obtain some information about the potential effect of liposomal lipid concentration on the antibiofilm activity of MLIPs (by comparing current results with previous ones,14 where MLIP-50% with approx. double lipid concentration (43µM) was studied). As shown in Figure 4, in established biofilms, the antibiofilm effect of free MOX (at MIC) was minimal, in accordance with previous reports;14,39,40 however, all MLIPs conferred significantly higher reductions in biomass (Figure 4A). Furthermore, MLIP-50% conferred higher antibiofilm activity (biomass/viability reduction) compared to MLIP-20% and MLIP-0 (Figure 4A and B). When comparing the current results for MLIP-50% with previous results where the same MOX concentration but almost double lipid amount was used,14 the effect of liposomal lipid concentration on biofilm formation is evident (as also observed for planktonic bacteria). Indeed, when the lipid concentration is increased (from 19µM to 43µM) the biofilm mass reduction is significantly increased from 61.8±1.4% to 75.3±7.1%, while the viability reduction was not affected (71.4% and 70.3%, respectively).
 |
Figure 4 Antibiofilm activity on pre-establish biofilms: (A) Biofilm mass and (B) biofilm cell viability of ATCC 35984 S.e bacteria in absence and presence of 0.30µM MOX as free-MOX or MLIPs (liposomes with varying Chol-contents are tested); Empty liposomes (LIP) and Mixtures of empty liposomes (19µM lipid) with free MOX (Mix) are reported as control groups. Individual differences are marked by * for p≤ 0.05), *** for p≤ 0.001, and **** for p ≤ 0.0001. |
The results of the biofilm susceptibility assays were additionally confirmed by confocal microscopy of pre-established biofilms (after 24h incubation with free-MOX or MLIPs under identical experimental conditions) (Figure 5A and B). Untreated biofilms and biofilms treated with free-MOX were almost completely green, indicating practically no antibiofilm activity of the free drug; conversely, the appearance of red areas (indicative of dead bacterial cell presence) was clear in biofilms treated with MLIPs (Figure 5A). Furthermore, the percentage of red (dead) cells in the biofilm planes increased as the Chol-content of MLIPs increased, as verified by the quantification of red areas in the micrographs (Figure 5B).
 |
Figure 5 Antibiofilm activity towards pre-establish biofilms: (A) Confocal microscope images of ATCC 35984 S.e biofilms after 24h incubation with MOX (0.30µM) as free-MOX or MLIPs (with varying Chol-content); stained live cells (green) and dead cells (red). Total lipid concentration (in each well) is 19µM. (B) Dead bacterial cells (%) in total area. Individual differences are marked by ** for p≤ 0.01, *** for p≤ 0.001, and **** for p ≤ 0.0001. |
Effect of Liposomal Lipid Content and PEGylation of MLIPs on Antimicrobial Efficacy
As mentioned above, the effects of liposomal lipid amount (or lipid concentration) of MLIPs on their antimicrobial efficacy were observed by comparing the current results for MLIP-50% with the results of a previous study,14 where an approx. double amount of lipid was used. In order to verify this observation, we carried out a series of experiments (bacterial growth inhibition, time killing assay, and live-dead assay), using MLIP-50%, with either high lipid concentration (High – 21.5µM) or a 10 times lower lipid concentration (Low – 2.15µM); a constant MOX concentration of 0.15µM applied in both cases. The properties of the liposomes used in these studies are reported in Table 2 and the results are presented in Figures 6 and 7 (values for no-PEG samples).
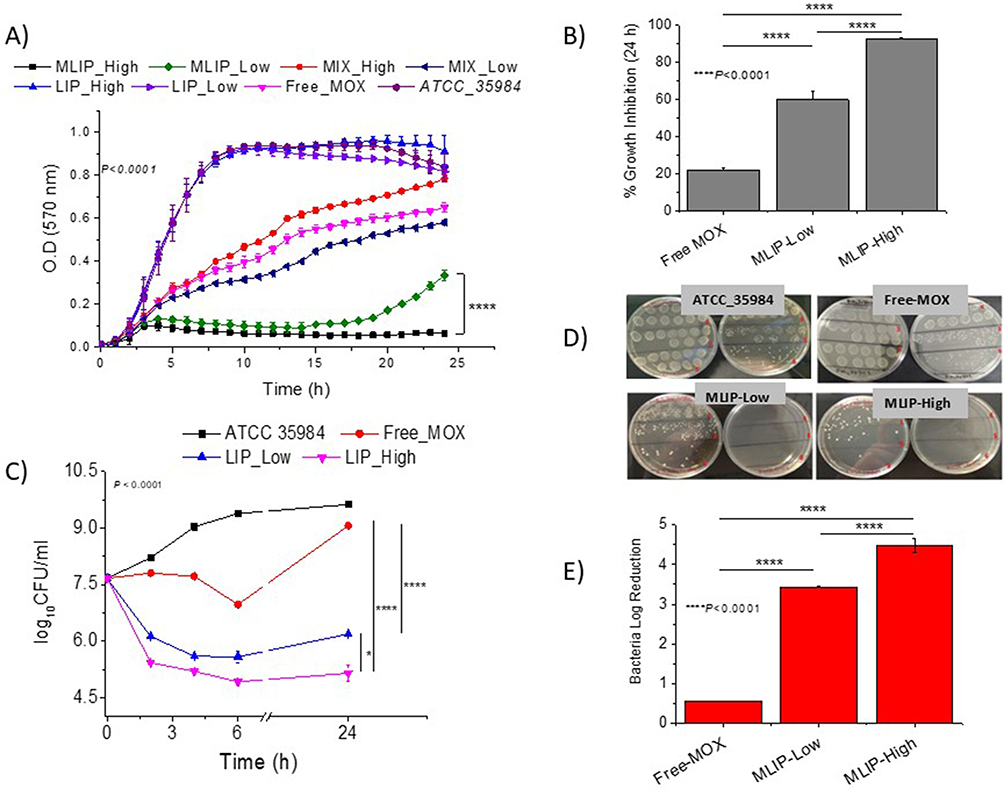 |
Figure 6 (A) Growth curves, (B) Bacteria growth inhibition (%) after 24h incubation, (C) Time–kill curves, (D) Representative images of plates (of time-kill study), and (E) Bacteria log Reduction values after 24h incubation, of ATCC 35984 S.e in absence and presence of 0.15µM MOX as free-MOX or MLIP-50% with varying lipid concentrations (Low 2.15µM and High 21.5µM); empty liposomes (LIP-50%) and mixtures (MIX) of LIP-50% with free MOX were also used as controls (in A); Individual differences are marked by * symbol when p≤ 0.05, and **** when p ≤ 0.0001. |
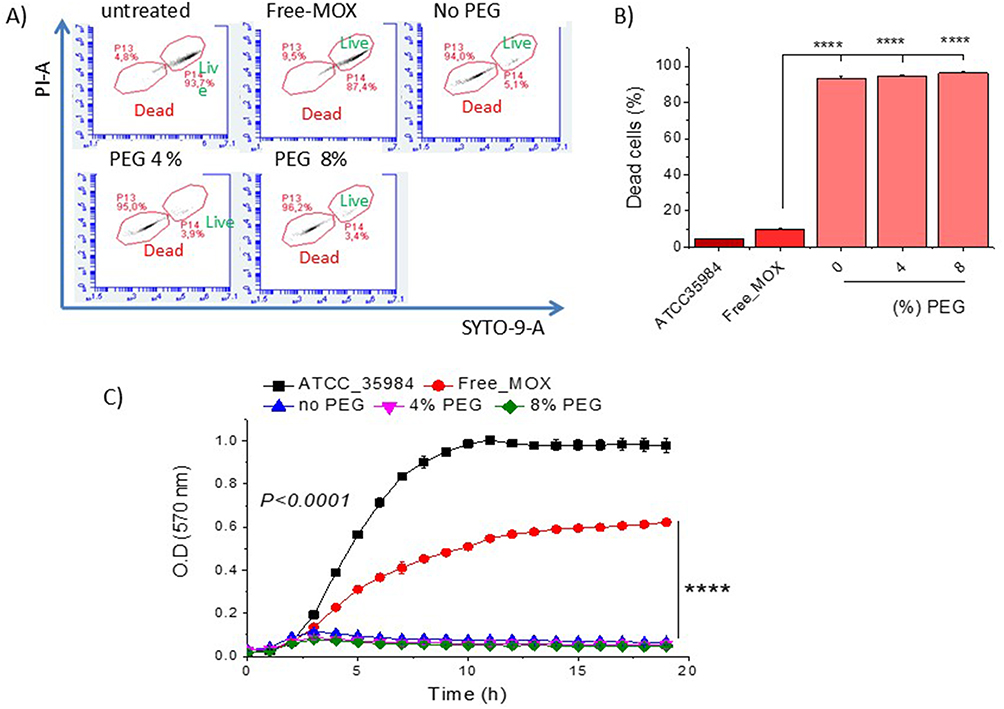 |
Figure 7 (A) Flow cytometric dot plots of ATCC 35984 S.e planktonic bacteria after 24h incubation with MOX (0.15µM) as free-MOX or MLIP-50% (with 21.5 µM lipid [High]) with varying PEG contents; Stained live cells (green) and dead cells (red). (B) Dead bacterial cells (%). (C) Growth curves for bacteria in presence and absence of free-MOX or MLIP-50% (High) with varying PEG contents; Symbols **** denote differences where p ≤ 0.0001. |
The bacterial growth in the presence of MLIPs and controls is seen in Figure 6A. Regarding inhibition of planktonic bacterial growth, it is seen in Figure 6B that MLIP-High (21.5µM lipid) demonstrated almost 100% (92.1 ± 4.3) bacterial growth inhibitory activity at 24h, in good agreement with previous results (similar experiment) (93%),14 while the MLIP-Low (lipid content 2.15µM) exhibited a significantly lower inhibition of bacterial growth equal to 59.6±2.5% (at the same MOX concentration). As presented above, for lipid concentration of 9.5µM (intermediate to high and low concentrations) and the same MOX concentration (0.15µM), the bacterial growth inhibition (at 24h) is 78.8±3.0% (Figure 1B – MLIP-50%). Similar conclusions regarding the effect of the liposomal lipid content of MLIPs on their activity are drawn from the results of the time-killing study (Figure 6C–6E). In more detail, the Bacterial Log Reduction values (after 24h) where 4.48 for MLIP-50%-High and 3.34 for MLIP-20%-Low (Figure 6E); while at intermediate lipid content (of 9.5µM) MLIP-50% conferred a log reduction equal to 4.17 (Figure 2B). Good linear correlation between MLIP-50% lipid concentration and growth inhibition (%) is observed, while the linearity of the correlation of liposomal lipid and bacterial log reduction is lower (see supplementary Data, Figure S1c and d, respectively) as also observed for liposome Chol-content (Figure S1b).
The very high bacterial killing activity of MLIP-50%-High (0.15µM MOX and 21.5µM lipid concentration) was also verified by flow cytometry measurements of live (and dead) bacterial cells, following 24h incubation of planktonic bacteria with the specific liposome formulation, as seen in Figure 7A and 7B (no PEG formulation).
In the same set of experiments, we evaluated the effect of PEGylation of the MLIP-50% formulation (with addition of 4 mol% or 8 mol% PEG in liposomes) on bacterial killing (Figure 7A and 7B) and bacterial growth inhibition (Figure 7C). As observed, PEGylation (at both concentrations) did not modulate the antimicrobial activity of MLIP-50%.
Despite these encouraging results, it needs to be pointed out that it will be extremely difficult from a practical point of view, to use liposomal drugs at such “High” lipid concentrations that correspond to very low D/L ratios, since very high liposomal formulation volumes will need to be administered, making systemic administration by parenteral routes very difficult (since a high volume of liposome dispersion would have to be injected). Furthermore, it is extremely difficult, if not impossible, to perform in vivo studies in small animals, where injection volumes could not exceed 100µL (in the best case), with such formulations. Therefore, we decided to focus on the Chol-content of liposomal drugs in subsequent experiments.
Interactions of Bacterial Cells with Liposomes
To understand whether the effect of Chol-content of MLIPs in increasing their antimicrobial activity is linked to increased interaction/binding between Chol-rich liposome-types and bacteria, two sets of experiments were performed.
In the first set, the binding of liposomes with varying Chol-contents to different types of bacterial cells was measured by flow cytometry. For these experiments, the liposomes were labelled with RHO (a lipid conjugate of Rhodamine) which is known for its high retention in liposomes and for the fact that it does not migrate to other lipid membranes.41,42 Three bacterial strains were used to determine whether any effect was bacterial strain-specific. As seen in Figure 8A and 8B, the binding of LIP-50% was significantly higher compared to that of LIP-0 and LIP-20% for all S.e strains evaluated. Nevertheless, the binding of Chol-containing liposomes was substantially higher to the reference S.e strain compared to the two nosocomial strains, indicating that the phenomenon may potentially be bacterial strain-dependent. The highest binding percent (16.92±0.45) was observed between LIP-50% and ATCC_35984 bacteria (which were used herein for antimicrobial efficacy studies). For this bacterial strain, LIP-20% was also efficient in conferring a significant binding (9.52±0.14) to bacteria (more than 10 times higher than of LIP-0). Conversely, the LIP-20% did not bind to the two nosocomial S.e strains (11465 and 762) significantly more than LIP-0, indicating that the Chol-content required to induce a significant interaction with bacteria is probably bacterial strain-dependent. However, interestingly, the LIP-50% binding to the quinolone-resistant S.e 11465 bacteria was significantly higher (3.50±0.30) compared to their binding to the quinolone-sensitive S.e 762 bacteria (2.75±0.33), a finding that may be worth for future exploitation of MLIP-50% efficacy towards quinolone resistant nosocomial/clinical S.e strains.
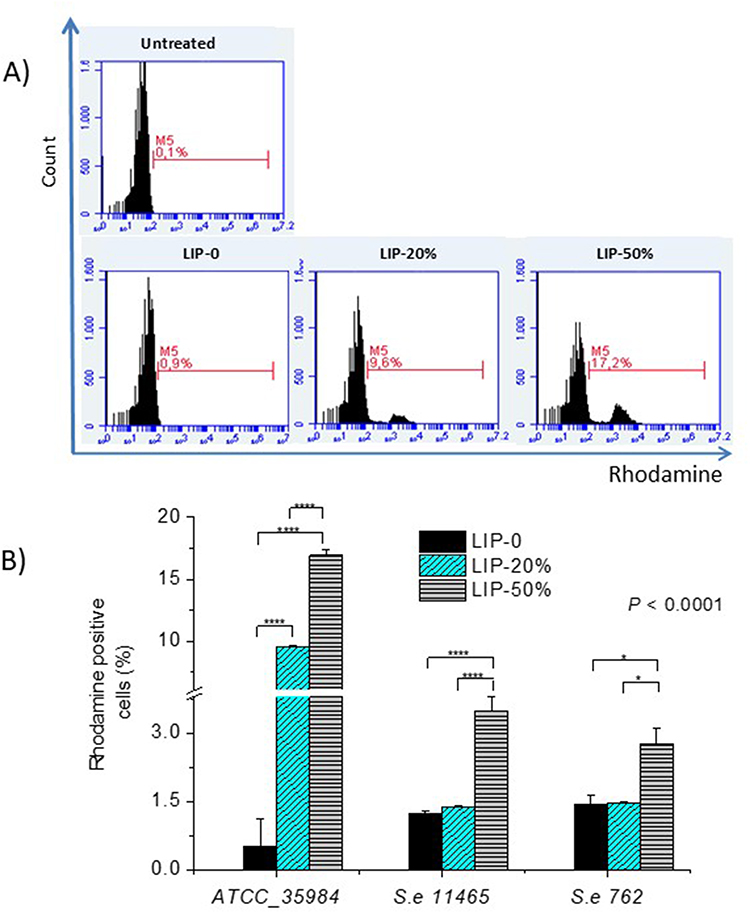 |
Figure 8 Liposome-Bacteria binding (interaction) (A) FACS histograms representing Rhodamine-positive ATCC 35984 bacterial cells following 24h incubation with LIPs (Rhodamine-lipid incorporating LIPs with varying Chol-contents) at 37°C. Bacterial strains: Se ATCC 35984, Se 11465 (quinolones resistant), Se 762 (quinolones sensitive). Lipid concentration is 0.25mg/mL; bacterial suspension is 4.5x107cfu/mL. (B) Quantitative results. Individual differences are marked by * for p≤ 0.05; and **** for p ≤ 0.0001. |
In the second set of experiments, LIP-0, LIP-33% and LIP-50% were loaded with 100mM calcein in order to evaluate the integrity of the liposomes in the presence of bacteria as a measure of their interaction with bacteria (or bacterial components). For this, the liposomes were incubated with four different S.e strains (two reference and two clinical strains) and tested for their integrity (release of calcein) during incubation in the presence of bacteria or in plain PBS (control study). As seen in Figure 9, the LIP-50% (Figure 9C) were found to have very low integrity during incubation in the presence of bacteria (all bacterial strains tested) compared to LIP-0 and LIP-33% (Figure 9A and B). This result is particularly surprising since it is well known that cholesterol increases the rigidity of liposomal membranes and that Chol-rich liposomes demonstrate high integrity in plasma proteins and the blood.2,30,43 Therefore, it is suggested that Chol-rich liposomes somehow “attract” bacteria or interact with bacterial components, resulting in higher leakage of calcein from the particular liposomes, compared to liposomes with no or low cholesterol levels in their membranes (despite the reduced rigidity of the latter liposomes, compared to Chol-rich liposomes). This suggestion is supported by the highest binding of LIP-50% to S.e (Figure 8B).
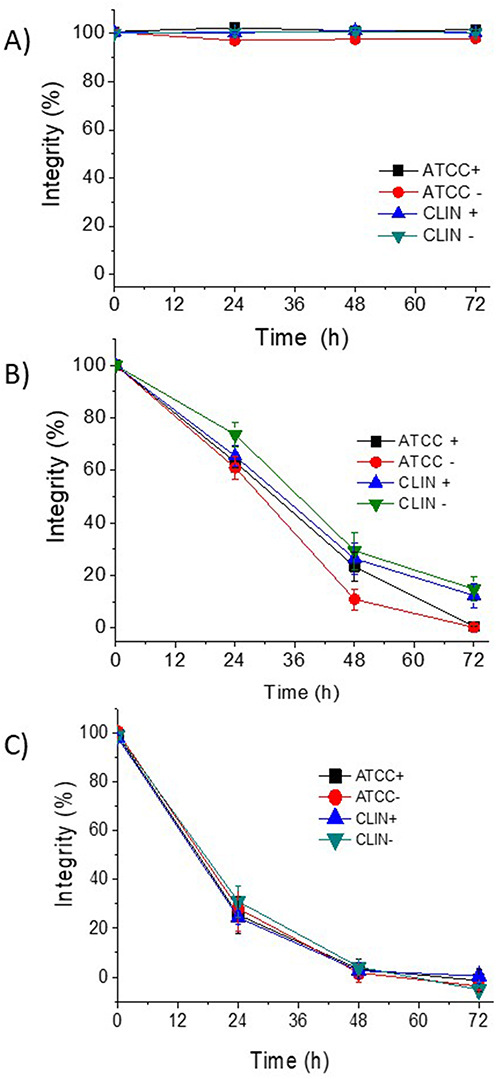 |
Figure 9 Integrity (%) of Calcein-loaded liposomes during incubation with S.e strains CLIN+ (GRE4388, slime+), CLIN- (GRE2264, slime-), ATCC+ (ATCC 35984, slime+) and ATCC- (ATCC 12228, slime-) at 37°C for a period of 72h. (A) LIP-0; (B) LIP-33%; (C) LIP-50%. |
In Vivo Antibacterial Efficacy of MLIP in Zebrafish Model
For in vivo evaluation of the antimicrobial efficacy of Chol-rich liposomes, Danio Rerio (zebrafish) was infected by intraperitoneal injection of S.e (ATCC 35984) and treated with PEGylated MLIP-50% or free MOX (Figure 10A). As shown by bacterial growth in zebrafish blood (Figure 10B), the 4h post-infection time-point was considered optimal for administration of antibiotic formulations because by that time the bacterium adapted to the interior of the organism, passed the lag phase, and entered the logarithmic growth phase. Finally, 4h post-treatment (and 8h post-staphylococcal infection), the bacterial cfu/mL in the untreated group (control/PBS) was high (similar to the value measured in the infection model development study (Figure 10B)), but both free MOX and MLIP-50% were highly effective and significantly reduced bacteremia (Figure 10C). However, MLIP-50% exhibited significantly higher antimicrobial activity than free MOX, providing in vivo evidence of the high antibacterial efficacy of MLIP-50%. In more detail, 4h post-treatment with free MOX, the bacteremia in zebrafish was reduced by 29.5±4.9 times (compared to the no-treatment group), while the same dose in MLIP-50% resulted in 78±4.9 times reduction in bacteria, at the same time-point. In fact, this is the first in vivo proof for the enhanced antibacterial efficacy of Chol-rich PEGylated liposomes in blood (compared to free drug) following systemic administration of liposomes, since in vivo studies (to verify the activity of Chol-rich liposomes) were conducted only in two previous reports, and the experimental conditions differed. In the first case, empty non-PEGylated liposomes or empty liposome/antibiotic mixtures (but not liposomal antibiotic formulations) were evaluated in vivo (also after systemic administration),16 In the second case, PEGylated berberine-loaded liposomes were studied, but the liposomal antimicrobial was administered topically by inhalation of an aerosolized colloidal dispersion.18
 |
Figure 10 In vivo study in zebrafish. (A) Protocol of studies; (B) Kinetics of infection following zebrafish i.p. injection with Se (reference strain); (C) Bacteremia (cfu/mL) 4h post-treatment with free-MOX and MLIPs. Individual differences are marked by ** for p≤ 0.01, and *** for p ≤ 0.001. |
Discussion
Herein, the effect of Chol-content, liposomal lipid concentration, and PEGylation of MLIPs on their antimicrobial activity against S.e (planktonic and biofilm) was proven by in vitro and in vivo studies. MLIP-50% demonstrated highest antimicrobial efficacy against both planktonic bacteria (Figures 1–3) and biofilms (Figures 4 and 5), compared to MLIP-20% or MLIP-0, as well as free MOX. Interestingly, MLIP-50% were the only liposome-type that significantly reduced biofilm viability compared to the free drug (Figure 4B), whereas in all other experiments all MLIPs (MLIP-0, MLIP-20%, and MLIP-50%) performed better than the free drug (Figures 1–3, Figures 4A and 5).
MLIP-50% activity was further enhanced when their lipid concentration was increased to 21.5µM, keeping the MOX concentration constant (Figure 6). This suggests a direct interaction between liposomes and bacteria, since when more liposome particles (being the case when lipid concentration is increased and liposome size is unchanged) are incubated with a given bacteria population, the interaction is increased. Nevertheless, although interesting as a finding, the application of liposomal drugs with “high” lipid concentrations (or low D/L ratios) is practically impossible, as mentioned above, a fact that shifted our focus to the effect of liposome Chol-content on their antimicrobial efficacy.
Potential effects of coating MLIP-50% with PEG (at 4 or 8 mol% of total lipid) on their activity were investigated, since it is well known that PEGylated liposomal drugs demonstrate higher integrity in blood and avoid uptake by the macrophages of the reticuloendothelial system (RES) following liposome absorption into the bloodstream (compared to non-PEGylated),1–3 being thus optimal liposome types for systemic pharmacological action. PEGylation did not modulate the bacteriostatic nor the bactericidal activity of MLIP-50% (Figure 7); therefore, Pegylated-MLIP-50% were tested for treatment of S.e -infected zebrafish (in vivo). The in vivo study results confirmed the superiority of PEGylated MLIP-50% to substantially reduce bacteremia 4h post-infection, to a much higher degree than the same dose of free MOX.
Concerning the potential mechanism(s) involved for enhanced antimicrobial activity of Chol-rich liposomes, some possible explanations can be drawn from the bacteria/liposome interaction (binding) investigation (Figure 8) as well as the experiments in which the integrity of liposomes with different Chol contents was monitored during incubation with bacteria (Figure 9).
Indeed, LIP-50% was found to have very low integrity during incubation with bacteria, indicating that perhaps some substances released from the bacteria enhance the leakage of liposome-entrapped calcein (Figure 9). In the latter study, two reference and two clinical strains of S.e were used, two being slime – and two slime +; however, under the specific conditions applied, no differences between the different strains were observed with respect to the integrity of the various liposome types, indicating that perhaps the Chol-dependent bacterial substance(s) responsible for the decreased integrity of LIP-50% (and to a lower degree of LIP-33%) were present in all the bacterial cultures tested at sufficient amounts. The affinity (binding) of liposomes to S.e bacterial cells was also found to be (liposome) Chol-content dependent (Figure 8), correlating well with the very low integrity of LIP-50% during incubation with bacteria, since increased liposome/bacteria affinity is expected to lead to disruption or perhaps fusion of liposomes, and consequently to the release of encapsulated substances.
Our liposome integrity study results (Figure 9) agree with other studies where liposome sensitivity towards specific toxins, such as α-toxin,15 delta-toxin,17 leukotoxin (LtxA),44 and pneumolysin,45 or towards homogenates of S. aureus biofilm,18 was found to be (liposome) Chol-content dependent. Furthermore, liposome Chol-content-dependent affinity towards bacteria cells or specific bacteria produced toxins was reported before towards S. aureus bacteria46 or alpha-toxin.47 Table 3 summarizes similarities of previous results with our findings. As seen, in most of the previous studies, the bacteria secreted and Chol-dependent component that is responsible for increased antimicrobial activities of Chol-rich liposomes (compared to other liposomes) has been identified, and the specific toxins were used in the corresponding studies. Furthermore, in all cases, the Chol-content of liposomes determines the degree of liposome interactions (with) and/or liposome integrity in the presence of different bacteria or bacterial secreted substances.
 |
Table 3 Summarized Similarities of Previous Reports with Current Findings for Chol-Content Dependent Liposome Interaction with Bacteria |
With the existing knowledge about S.e bacteria we cannot identify potential Chol-dependent bacterial component(s) implicated with the current findings, since research concerning biofilm and/or other virulence determinants in S.e has been slow (compared to other bacteria), highly contributing to the establishment of S.e-related chronic infections with high morbidity or mortality.48 Today, it is known that S.e strains produce several types of toxins,48 and that clinical peri-prosthetic joint infection-associated biofilm forming strains are less likely to produce such toxins,49 providing a potential explanation for Chol-rich liposome lower binding to the clinical S.e strains (compared to the reference strain) (Figure 8). However, no differences between clinical and reference S.e strains were observed in the bacteria-liposome interaction studies (Figure 9). Unfortunately, the S.e strains used in the two later sets of experiments were not identical (for practical reasons), hindering extraction of conclusions about S.e strain effects. Nevertheless, the initial bacterial cell loads used in liposome integrity studies (4x108cfu/mL) were 1 order of magnitude higher than those used in the binding studies (4.5×107cfu/mL [diluted to half]). Thereby, we may postulate that perhaps any causative bacterial substances for Chol-dependent effects were present at the required amounts in all bacterial strain cultures during the integrity studies, but not in (or during) the binding studies, explaining the different results.
The effect of Chol content of liposomal antibiotics (or other antimicrobials) on S.e has not been evaluated before, in vitro or in vivo, with the exception of one study where S.e (Fussel, 14990™) was used as a control bacteria during evaluation of nisin-liposome Chol-content effect on their antimicrobial activity against S. pneumoniae. Chol-rich nisin-liposomes did not demonstrate Chol-content dependent antimicrobial activity towards S. e, as observed for S. pneumoniae.45 However, in that study, a much lower initial bacterial load (8.5x105cfu/mL) was used compared to the initial load used in the current study (4x108 cfu/mL), indicating a potential role of the bacterial load, in agreement with the point raised above. In other words, perhaps S.e cultures require higher bacterial cell numbers to reach the required concentrations of particular causative substances for realization of Chol-responsive bacteria/liposome interactions (compared to other bacteria).
Conclusions
The current finding that Chol-rich liposomal antibiotics demonstrate enhanced antimicrobial activity towards S.e bacteria (planktonic and biofilm), compared to liposomes with lower or no Chol, is extremely interesting since it provides novel insights towards the development of improved liposomal therapeutic solutions for multidrug-resistant S.e strains. Such S.e strains are known as leading causative agents of bloodstream infections49–52 (representing 31% of reported cases50) and are extremely difficult to treat with available therapeutics, indicating the need to identify novel solutions with enhanced and more specific activity. A proposal for future exploitation of such Chol-rich liposomal antibiotics for the treatment of S.e infections is to further increase their specificity for S.e by attaching targeting ligands on their surface, such as aptamers46 or other ligand types.
Data Sharing Statement
Data are available upon request, from S.G.A.
Acknowledgments
All authors highly recognize Professor Klepetsanis’ valuable contributions to the study, especially in the initial setup of the methodologies and protocols applied, and in the supervision of students.
The help provided in the Confocal Microscopy studies by Dr Konstantina Kaplani and Prof. S. Taraviras, Laboratory of Physiology, Dept. of Medicine, School of Health Sciences, University of Patras, is highly acknowledged.
Funding
This research was funded by the following grants: (1). Operational Program “Human Resources Development, Education and Lifelong Learning 2014–2020“, co-financed by Greece and the European Union (European Social Fund-ESF) through in the context of the subproject “Innovative lipid nanoparticles for treatment of staphylococcal biomembranes” (MIS 5049223), grant number 39540000 awarded to S.G.A and I.S, and (2). Operational Program “Competitiveness, Entrepreneurship and Innovation” (NSRF 2014–2020), co-financed by Greece and the European Union (European Regional Development Fund) subproject “Preclinical development of INNOvative FORmulations of antibiotics for intraocular administration for the treatment/prevention of postoperative endophthalmIitis (ΙΝΝO-FOR-I) (MIS 5031792)”, awarded to S.G.A.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Antimisiaris SG, Marazioti A, Kannavou M. et al. Overcoming barriers by local drug delivery with liposomes. Adv. Drug Delivery Rev. 2021;174:53–86. doi:10.1016/j.addr.2021.01.019
2. Antimisiaris SG. Introduction to liposome assisted drug delivery. In: Antimisiaris Sophia G., editor. Liposomes in Drug Delivery: What, Where, How and When to Deliver. Academic Press; 2024:1–17.
3. Gkartziou F, Antimisiaris SG. Liposomes for infectious diseases. In: Antimisiaris Sophia G., editor. Liposomes in Drug Delivery: What, Where, How and When to Deliver (Academic Press). 2024:363–404.
4. Shao H, Zhou J, Lin X, et al. Bio-inspired peptide-conjugated liposomes for enhanced planktonic bacteria killing and biofilm eradication. Biomaterials. 2023;300:122183. doi:10.1016/j.biomaterials.2023.122183
5. Panthi VK, Fairfull-Smith KE, Islam N. Liposomal drug delivery strategies to eradicate bacterial biofilms: challenges, recent advances, and future perspectives. Int J Pharm. 2024;655:124046. doi:10.1016/j.ijpharm.2024.124046
6. Gkartziou F, Giormezis N, Spiliopoulou I, Antimisiaris SG. Nanobiosystems for antimicrobial drug-resistant infections. Nanomaterials. 2021;11(5):1075. doi:10.3390/nano11051075
7. Makhlouf Z, Ali AA, Al-Sayah MH. Liposomes-based drug delivery systems of antibiofilm agents to combat bacterial biofilm formation. Antibiotics. 2023;12(5):875. doi:10.3390/antibiotics12050875
8. Alhariri M, Majrashi MA, Bahkali AH, et al. Efficacy of neutral and negatively charged liposome-loaded gentamicin on planktonic bacteria and biofilm communities. Int J Nanomed. 2017;12:6949–6961. PMID: 29075113; PMCID: PMC5609801.doi:10.2147/IJN.S141709
9. Battista S, Maggi MA, Bellio P, et al. Curcuminoids-loaded liposomes: influence of lipid composition on their physicochemical properties and efficacy as delivery systems. Colloids Surf a Physicochem Eng Asp. 2020;597:124759. doi:10.1016/j.colsurfa.2020.124759
10. Messiaen AS, Forier K, Nelis H, Braeckmans K, Coenye T. Transport of nanoparticles and tobramycin-loaded liposomes in Burkholderia cepacia complex biofilms. PLoS One. 2013;8(11):e79220. doi:10.1371/journal.pone.0079220
11. Gkartziou F, Plota M, Kypraiou C, et al. Daptomycin liposomes exhibit enhanced activity against staphylococci biofilms compared to free drug. Pharmaceutics. 2024;16(4):459.PMID: 38675120; PMCID: PMC11054717. doi:10.3390/pharmaceutics16040459
12. Ibaraki H, Kanazawa T, Chien W-Y, et al. The effects of surface properties of liposomes on their activity against Pseudomonas aeruginosa PAO−1 biofilm. J Drug Deliv Sci Technol. 2020;57:101754. doi:10.1016/j.jddst.2020.101754
13. Ferreira M, Pinto SN, Aires-da-Silva F, Bettencourt A, Aguiar SI, Gaspar MM. Liposomes as a nanoplatform to improve the delivery of antibiotics into staphylococcus aureus biofilms. Pharmaceutics. 2021;13(3):321. doi:10.3390/pharmaceutics13030321
14. Natsaridis E, Gkartziou F, Mourtas S, et al. Moxifloxacin liposomes: effect of liposome preparation method on physicochemical properties and antimicrobial activity against staphylococcus epidermidis. Pharmaceutics. 2022;14(2):370.PMID: 35214102; PMCID: PMC8875207. doi:10.3390/pharmaceutics14020370
15. Pornpattananangkul D, Zhang L, Olson S, et al. Bacterial toxin-triggered drug release from gold nanoparticle-stabilized liposomes for the treatment of bacterial infection. J Am Chem Soc. 2011;133(11):4132–4139. doi:10.1021/ja111110e
16. Henry BD, Neill DR, Becker KA, et al. Engineered liposomes sequester bacterial exotoxins and protect from severe invasive infections in mice. Nat Biotechnol. 2015;33(1):81–88. doi:10.1038/nbt.3037
17. Seike S, Miyamoto K, Kobayashi K, Takehara M, Nagahama M. Clostridium perfringens delta-toxin induces rapid cell necrosis. PLoS One. 2016;11(1):e0147957.PMID: 26807591; PMCID: PMC4726729. doi:10.1371/journal.pone.0147957
18. Xie J, Meng Z, Han X, et al. Cholesterol microdomain enhances the biofilm eradication of antibiotic liposomes. Adv Healthc Mater. 2022;11(8):e2101745. doi:10.1002/adhm.202101745
19. Stewart JC. Colorimetric determination of phospholipids with ammonium ferrothiocyanate. Analy Biochem. 1980;104(1):10–14. doi:10.1016/0003-2697(80)90269-9
20. Pefani-Antimisiari K, Athanasopoulos DK, Marazioti A, et al. Synergistic effect of cold atmospheric pressure plasma and free or liposomal doxorubicin on melanoma cells. Sci Rep. 2021;11(1):14788. doi:10.1038/s41598-021-94130-7
21. Foka A, Katsikogianni MG, Anastassiou ED, Spiliopoulou I, Missirlis YF. The combined effect of surface chemistry and flow conditions on Staphylococcus epidermidis adhesion and ica operon expression. Eur Cell Mater. 2012;24:386–402. doi:10.22203/ecm.v024a28
22. Plota M, Sazakli E, Giormezis N, et al. In Vitro Antibiofilm Activity of Bacteriophage K (ATCC 19685-B1) and Daptomycin against Staphylococci. Microorganisms. 2021;9(9):1853.PMID: 34576751; PMCID: PMC8468654. doi:10.3390/microorganisms9091853
23. Ghafelehbashi R, Akbarzadeh I, Tavakkoli Yaraki M, Lajevardi A, Fatemizadeh M, Heidarpoor Saremi L. Preparation, physicochemical properties, in vitro evaluation and release behavior of cephalexin-loaded niosomes. Int J Pharm. 2019;569:118580. doi:10.1016/j.ijpharm.2019.118580
24. Bhatia E, Banerjee R. Hybrid silver-gold nanoparticles suppress drug resistant polymicrobial biofilm formation and intracellular infection. J Mater Chem B. 2020;8(22):4890–4898. doi:10.1039/D0TB00158A
25. Bhatia E, Sharma S, Jadhav K, Banerjee R. Combinatorial liposomes of berberine and curcumin inhibit biofilm formation and intracellular methicillin resistant Staphylococcus aureus infections and associated inflammation. J Mater Chem B. 2021;9(3):864–875. doi:10.1039/d0tb02036b
26. Halwani M, Mugabe C, Azghani AO, Lafrenie RM, Kumar A, Omri A. Bactericidal efficacy of liposomal aminoglycosides against Burkholderia cenocepacia. J Antimicrob Chemother. 2007;60(4):760–769.PMID: 17673475. doi:10.1093/jac/dkm289
27. Zhang X, Zhang G, Chai M, Yao X, Chen W, Chu PK. Synergistic antibacterial activity of physical-chemical multi-mechanism by TiO2 nanorod arrays for safe biofilm eradication on implant. Bioact Mater. 2020;6(1):12–25.PMID: 32817910; PMCID: PMC7417618. doi:10.1016/j.bioactmat.2020.07.017
28. Wu X, Santos RR, Fink-Gremmels J. Cadmium modulates biofilm formation by Staphylococcus epidermidis. Int J Environ Res Public Health. 2015;12(3):2878–2894.PMID: 25749322; PMCID: PMC4377938. doi:10.3390/ijerph120302878
29. Mourtas S, Diamanti G, Foka A, et al. Inhibition of bacterial attachment on surfaces by immobilization of tobramycin-loaded liposomes. J Biomed Nanotechnol. 2015;11(12):2186–2196. doi:10.1166/jbn.2015.2160
30. Kokkona M, Kallinteri P, Fatouros D, Antimisiaris SG. Stability of SUV liposomes in the presence of cholate salts and pancreatic lipases: effect of lipid composition. Eur J Pharm Sci. 2000;9(3):245–252. doi:10.1016/s0928-0987(99)00064-0
31. Westerfield M. The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish (Danio Rerio). University of Oregon Press; 2000.
32. Lidster K, Readman GD, Prescott MJ, Owen SF. International survey on the use and welfare of zebrafish Danio rerio in research. J Fish Biol. 2017;90(5):1891–1905. doi:10.1111/jfb.13278
33. Jorge S, Ferreira JM, Olsson IAS, Valentim AM. Adult zebrafish anesthesia: a study of efficacy and behavioral recovery of different anesthetics. Zebrafish. 2021;18(5):330–337. doi:10.1089/zeb.2021.0023
34. Neely MN, Pfeifer JD, Caparon M. Streptococcus-zebrafish model of bacterial pathogenesis. Infect Immun. 2002;70(7):3904–3914.PMID: 12065534; PMCID: PMC128100. doi:10.1128/IAI.70.7.3904-3914.2002
35. Chen L, Yan J, Sun W, et al. A zebrafish intelectin ortholog agglutinates both Gram-negative and Gram-positive bacteria with binding capacity to bacterial polysaccharide. Fish Shellfish Immunol. 2016;55:729–736. PMID: 27329687.doi:10.1016/j.fsi.2016.06.023
36. Chaudhari GH, Chennubhotla KS, Chatti K, Kulkarni P. Optimization of the adult zebrafish ECG method for assessment of drug-induced QTc prolongation. J Pharmacol Toxicological Methods. 2013;67(2):115–120. doi:10.1016/j.vascn.2013.01.007
37. Patterson H, Saralahti A, Parikka M, et al. Adult zebrafish model of bacterial meningitis in Streptococcus agalactiae infection. Dev Comp Immunol. 2012;38(3):447–455.PMID: 22867759. doi:10.1016/j.dci.2012.07.007
38. Sridevi JP, Anantaraju HS, Kulkarni P, Yogeeswari P, Sriram D. Optimization and validation of Mycobacterium marinum-induced adult zebrafish model for evaluation of oral anti-tuberculosis drugs. Int J Mycobacteriology. 2014;3(4):259–267. doi:10.1016/j.ijmyco.2014.10.001
39. Soriano F, Huelves L, Naves P, et al. In vitro activity of ciprofloxacin, moxifloxacin, vancomycin and erythromycin against planktonic and biofilm forms of Corynebacterium urealyticum. J Antimicrob Chemother. 2009;63(2):353–356. doi:10.1093/jac/dkn491
40. Nguyen TK, Argudín MA, Deplano A, et al. Antibiotic resistance, biofilm formation, and intracellular survival as possible determinants of persistent or recurrent infections by staphylococcus aureus in a Vietnamese tertiary hospital: focus on bacterial response to moxifloxacin. Microb Drug Resist. 2020;26(6):537–544. doi:10.1089/mdr.2019.0282
41. Chazotte B. Labeling membranes with fluorescent phosphatidylethanolamine. Cold Spring Harb Protoc. 2011;2011(5). doi:10.1101/pdb.prot5621
42. Markoutsa E, Papadia K, Giannou AD, et al. Mono and dually decorated nanoliposomes for brain targeting, in vitro and in vivo studies. Pharm Res. 2014;31(5):1275–1289. doi:10.1007/s11095-013-1249-3
43. Mourtas S, Duraj S, Fotopoulou S, Antimisiaris SG. Integrity of liposomes in presence of various formulation excipients, when dispersed in aqueous media and in hydrogels. Colloids Surf B Biointerfaces. 2008;61(2):270–276. doi:10.1016/j.colsurfb.2007.09.003
44. Li Z, Baidoun R, Brown AC. Toxin-triggered liposomes for the controlled release of antibiotics to treat infections associated with the gram-negative bacterium, Aggregatibacter actinomycetemcomitans. Colloids Surf B Biointerfaces. 2024;238:113870. doi:10.1016/j.colsurfb.2024.113870
45. watt E, Andriescu I, Ho EA. Pneumolysin-responsive liposomal platform for selective treatment of Streptococcus pneumoniae. Drug Deliv Transl Res. 2024;15(5):1739–1754. doi:10.1007/s13346-024-01708-5
46. Benariba MA, Hannachi K, Rhouati A, Al-Ansi W, Cai R, Zhou N. Enhanced sensitivity in Staphylococcus aureus detection: unveiling the impact of lipid composition on the performance of carboxyfluorescein (CF)-Loaded liposome-based assay. Talanta. 2024;270:125577. doi:10.1016/j.talanta.2023.125577
47. Nagahama M, Michiue K, Sakurai J. Membrane-damaging action of Clostridium perfringens alpha-toxin on phospholipid liposomes. Biochim Biophys Acta. 1996;1280(1):120–126. doi:10.1016/0005-2736(95)00288-x
48. Burke Ó, Zeden MS, O’Gara JP. The pathogenicity and virulence of the opportunistic pathogen Staphylococcus epidermidis. Virulence. 2024;15(1):2359483. doi:10.1080/21505594.2024.2359483
49. Vuong C, Durr M, Carmody AB, Peschel A, Klebanoff SJ, Otto M. Regulated expression of pathogen-associated molecular pattern molecules in staphylococcus epidermidis: quorum-sensing determines pro-inflammatory capacity and production of phenol-soluble modulins. Cell Microbiol. 2004;6(8):753–759. doi:10.1111/j.1462-5822.2004.00401.x
50. Rosenthal VD, Chaparro GJ, Servolo-Medeiros EA. An eight-year multicenter study on short-term peripheral intravenous catheter–related bloodstream infection rates in 100 intensive care units of 9 countries in Latin America: Argentina, Brazil, Colombia, Costa Rica, Dominican Republic, Ecuador, Mexico, Panama and Venezuela. Findings of the International Nosocomial Infection Control Consortium (INICC). Infect Control Hosp Epidemiol. 2021;42(9):1098–1104. doi:10.1017/ice.2020.1373
51. Simon D, Fischer S, Grossman A. Left ventricular assist device–related infection: treatment and outcome. Clin Infect Dis. 2005;40(8):1108–1115. doi:10.1086/428728
52. Post V, Harris LG, Morgenstern M. Comparative genomics study of staphylococcus epidermidis isolates from orthopedic-device-related infections correlated with patient outcome. J Clin Microbiol. 2017;55(10):3089–3103. doi:10.1128/JCM.00881-17
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Non-Traditional Blood Lipid Indices for Metabolism Dysfunction-Associated Fatty Liver Disease Prediction in Non-Obese Type 2 Diabetes Mellitus
Gao Q, Feng L, Zhou W, Li X, Yin L, Wang Y
Diabetes, Metabolic Syndrome and Obesity 2023, 16:2345-2354
Published Date: 7 August 2023
Antibiotic Use, Awareness of Antimicrobial Resistance and Residue in Veterinary Professionals and Farmers in Selected Districts of Kellem Wollega Zone, Ethiopia
Ragassa S, Berhanu G
Veterinary Medicine: Research and Reports 2023, 14:159-175
Published Date: 26 September 2023
Detection of Klebsiella Pneumoniae and Antibiotic Resistance in Burn Wards in China From 2019 to 2023
Sun X, Li R, Xiang J
Infection and Drug Resistance 2025, 18:1595-1604
Published Date: 24 March 2025