Back to Journals » Journal of Pain Research » Volume 19
Central Sensitization and Nociplastic Pain: Shared Mechanisms in Fibromyalgia, Osteoarthritis, and Inflammatory Arthritis
Authors Hladkykh FV ![]() , Liadova TI, Matvieienko MS
, Liadova TI, Matvieienko MS ![]() , Komorovsky R
, Komorovsky R ![]() , Smiyan S, Student V
, Smiyan S, Student V ![]()
Received 3 October 2025
Accepted for publication 14 March 2026
Published 26 March 2026 Volume 2026:19 571311
DOI https://doi.org/10.2147/JPR.S571311
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor King Hei Stanley Lam
Fedir Volodymyrovych Hladkykh,1,2 Tetiana Ivanivna Liadova,3 Mariia Serhiivna Matvieienko,1 Roman Komorovsky,4 Svitlana Smiyan,4 Volodymyr Student5,6
1Department of General Surgery, Anesthesiology and Palliative Care, School of Medicine, V. N. Karazin Kharkiv National University, Kharkiv, Ukraine; 2Department of Radiation Pathology and Palliative Medicine, Grigoriev Institute for Medical Radiology and Oncology, Kharkiv, Ukraine; 3Department of Infectious Diseases and Clinical Immunology, School of Medicine, V. N. Karazin Kharkiv National University, Kharkiv, Ukraine; 4Department of Internal Medicine II, Ivan Horbachevsky Ternopil National Medical University, Ternopil, Ukraine; 5Department of Specialist Training and Postgraduate Education, Lviv Medical Professional College of Postgraduate Education, Lviv, Ukraine; 6Medical 3D Diagnostics Centre, Lviv, Ukraine
Correspondence: Roman Komorovsky, Department of Internal Medicine II, Ivan Horbachevsky Ternopil National Medical University, Maidan Voli, 1, Ternopil, 46001, Ukraine, Email [email protected]
Introduction: Central sensitization explains the mismatch between structural damage or inflammation and pain intensity in chronic musculoskeletal diseases. It defines the phenotype of fibromyalgia and contributes to persistent pain in osteoarthritis, rheumatoid arthritis, and psoriatic arthritis.
Objective: To characterize the role of central sensitization in nociplastic pain in fibromyalgia, osteoarthritis, rheumatoid arthritis, and psoriatic arthritis.
Materials and Methods: A structured search of PubMed, Scopus, Web of Science, and Google Scholar (1990– 2025) identified open-access, evidence-based publications addressing pain pathophysiology, diagnosis, and treatment in these conditions.
Results: Central sensitization manifests as hyperalgesia, allodynia, expanded receptive fields, and impaired endogenous pain inhibition. It predominates in fibromyalgia and contributes to persistent pain in osteoarthritis and rheumatoid arthritis that may not correlate with inflammation or structural damage. Screening tools such as the Widespread Pain Index and Symptom Severity Scale, together with quantitative sensory testing and algometry, help identify nociplastic pain features. Neuroimmune mechanisms, including microglial activation and imbalance between excitatory and inhibitory neurotransmission, may contribute to the mismatch between pain intensity and clinical findings.
Conclusion: Chronic pain reflects inflammatory, mechanical, and nociplastic mechanisms in rheumatoid arthritis, osteoarthritis, and fibromyalgia, respectively. Recognition of central sensitization improves assessment and supports mechanism-based management.
Keywords: central sensitization, nociplastic pain, fibromyalgia, osteoarthritis, rheumatoid arthritis, psoriatic arthritis, quantitative sensory testing, algometry, allodynia, hyperalgesia
Introduction
Central sensitization (CS) is the predominant mechanism underlying nociplastic pain, manifested by hyperalgesia and allodynia. This phenomenon arises from persistent alterations within the central nervous system (CNS). According to the definition proposed by the International Association for the Study of Pain (IASP), CS is “an increased responsiveness of nociceptive neurons in the central nervous system to their normal or subthreshold afferent input”.1 This means that neurons involved in the transmission of nociceptive stimuli become hyperreactive even when the intensity of peripheral stimulation is insufficient to evoke a pain sensation in healthy individuals. The CS phenomenon has major pathophysiological significance, explaining the transition from acute to chronic pain. Clinically, CS often accounts for pain that persists after tissue healing or becomes markedly amplified in pre-existing chronic conditions.1 Prolonged activation of central nociceptive pathways is accompanied by remodeling of neuronal networks, lowered activation thresholds, and exaggerated responses to minimal stimuli. Consequently, patients with CS often exhibit a marked dissociation between the absence of a clear source of peripheral nociceptive input and high pain intensity, which clinically manifests as increased disability and greater disease severity.
The World Health Organization (WHO) officially recognizes pain as one of the leading causes of disability worldwide, making CS an exceptionally important target of current research.2,3 Understanding its mechanisms opens avenues for innovative diagnostic and therapeutic strategies aimed not only at symptom relief but also at correcting central pathophysiological processes.
Features of CS have been documented in a wide range of clinical conditions, including fibromyalgia (FM), osteoarthritis (OA), rheumatoid arthritis (RA) and psoriatic arthritis (PsA), upper-limb tendinopathies, various forms of headache, and chronic spinal pain.4–6 These disorders share a common feature – the lack of proportionality between the extent of structural damage or degree of inflammation and the intensity of pain. This phenomenon explains why patients with similar objective tissue changes can report markedly different levels of subjective pain.7 Accordingly, CS emerges as a key factor shaping not only the clinical presentation but also the prognosis of musculoskeletal diseases.
The introduction of the IASP nociplastic pain concept in 2017 marked a major advance.8,9 This category emphasizes pain arising from altered pain modulation within the CNS and is defined as “Pain that arises from altered nociception despite no clear evidence of actual or threatened tissue damage causing the activation of peripheral nociceptors or evidence for disease or lesion of the somatosensory system causing the pain”. The term is relevant because not all chronic pain states can be explained solely by structural or tissue damage. Recognizing nociplastic pain enables clinicians and researchers to broaden the mechanistic understanding of chronic pain, refine diagnosis, and optimize therapeutic approaches.10
Nociplastic conditions remain challenging to diagnose and treat because they lack clear objective biomarkers and are often driven by multifactorial mechanisms. The role of CS is most prominent in FM, OA, RA, and PsA, underscoring the need for an in-depth analysis of its diagnostic and therapeutic dimensions.
Despite substantial clinical advances in studying FM, OA, RA, and PsA, fundamental knowledge of the pathophysiological mechanisms of pain in these disorders remains fragmentary and insufficiently systematized. Comparative analysis of the nociplastic component caused by CS, hyperalgesia, and allodynia requires special attention. The absence of an integrated understanding of shared and distinct pathogenetic patterns across these conditions limits the integration of novel diagnostic criteria and the development of effective therapeutic strategies. A foundational appraisal of the common mechanisms of CS and nociplastic pain is therefore essential to deepen contemporary concepts and to shape promising directions for interdisciplinary research.
Scope of the Review
This review synthesizes current evidence on central sensitization (CS) as a driver of nociplastic pain across four high-prevalence conditions – fibromyalgia (FM), osteoarthritis (OA), rheumatoid arthritis (RA), and psoriatic arthritis (PsA), with a focus on how CS explains the dissociation between tissue pathology/inflammation and pain intensity. We compare inter-disease similarities and differences in mechanisms (descending inhibition failure, facilitated temporal summation, maladaptive cortical/limbic plasticity), clinical phenotypes (hyperalgesia, allodynia, pain catastrophizing, sleep/mood comorbidity), and measurement paradigms including quantitative sensory testing (QST), temporal summation (TS), conditioned pain modulation (CPM), the Central Sensitization Inventory (CSI), and neurophysiologic and neuroimaging indicators.
To assess the current state of knowledge, we retrieved publications (1990–2025) from the bibliographic/scientometric databases PubMed (n=732), Scopus (n=513), Web of Science (n=312), and Google Scholar (n=300), covering nociplastic pain, central sensitization, and diagnostic and therapeutic approaches to chronic musculoskeletal pain.
In stage one, we conducted a search (Figure 1) using the keywords: “nociplastic pain”, “central sensitization”, “musculoskeletal pain”, “fibromyalgia”, “osteoarthritis”, “rheumatoid arthritis”, “psoriatic arthritis”, “diagnosis”, and “treatment.”
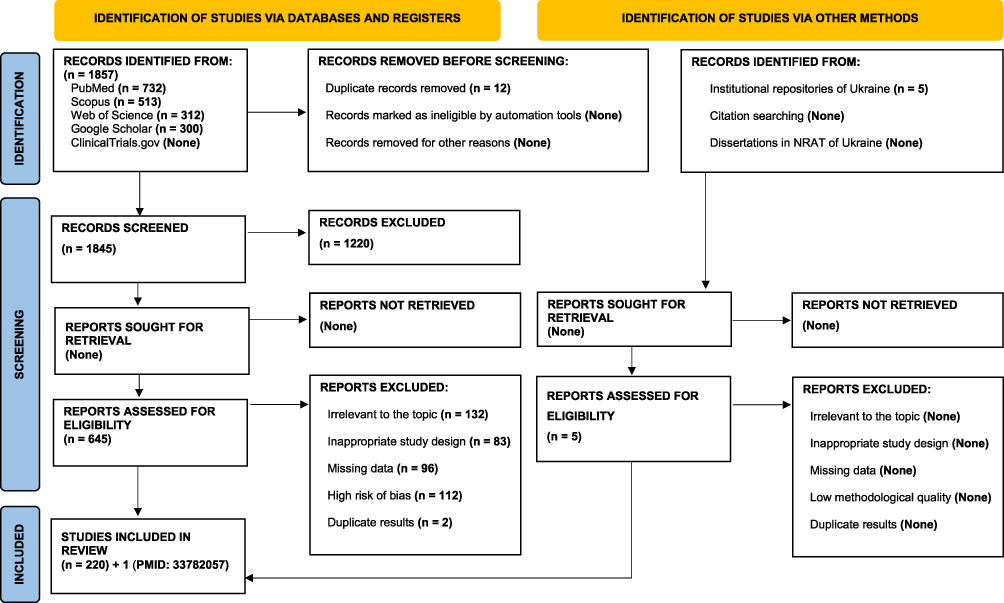 |
Figure 1 PRISMA 2020 flow diagram for the selection of publications in systematic reviews and meta-analyses. Abbreviation: NRAT, national repository of academic texts. |
In stage two, article abstracts were screened and records not meeting the study criteria were excluded.
In stage three, full texts of the selected articles were assessed for eligibility and relevance to the review aims.
Inclusion criteria for content analysis were: (1) reporting contemporary data on the pathophysiology of central sensitization, its role in nociplastic pain, diagnostic methods, and treatment; (2) alignment with core principles of evidence-based medicine; and (3) open-access availability of the full text.
The review was prepared in accordance with key elements of the PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) guideline.11
Neurobiological Mechanisms of Pain Chronification
Pain is a complex clinical phenomenon that can be classified by pathogenesis, course, etiology, and anatomical location. By mechanism, pain is divided into nociceptive, neuropathic, and nociplastic; by course, into acute and chronic; by etiology, into malignant and non-malignant; and by topography, into region- or organ-specific pain.12
Chronic pain syndromes frequently lack a clear nociceptive source or demonstrate a mismatch between structural changes and pain intensity, disability, and associated symptoms such as fatigue, sleep disturbance, and cognitive impairment. In these cases, central sensitization (CS) represents a key mechanism of chronic pain and is defined by altered nociceptive processing in the absence of sufficient tissue damage or somatosensory system pathology.10 Accordingly, pain is classified into nociceptive, neuropathic, and nociplastic types, reflecting distinct but overlapping pathogenetic mechanisms relevant for diagnosis and treatment.
Nociceptive pain is the most prevalent type and results from activation of nociceptors by mechanical injury, inflammation, or ischemia. Nociceptors are located on free nerve endings of primary afferent fibers and transduce peripheral stimuli into neural impulses perceived by the central nervous system (CNS) as pain.13,14 Primary afferents include Aβ fibers mediating touch and vibration, Aδ fibers transmitting fast, sharp pain, and unmyelinated C fibers conveying slow, diffuse, burning pain.15 Typical clinical examples include acute trauma, peptic ulcer disease, and arthritis.13,14 Pain generation involves transduction, transmission, perception, and modulation. Tissue injury induces release of substance P (SP), prostaglandins (PGs), bradykinin, and histamine, while modulation is mediated by endogenous endorphins, norepinephrine, and serotonin.16
Neuropathic pain arises from damage to or disease of the somatosensory nervous system and is defined by the International Association for the Study of Pain (IASP) as pain caused by a lesion or disease of this system. It is usually chronic, treatment-resistant, and presents as burning, shooting, or electric shock-like pain. Etiologies include post-stroke pain, spinal cord injury, postherpetic neuralgia, and carpal tunnel syndrome.17 Diabetic polyneuropathy is a common example, resulting from hyperglycemia-induced peripheral nerve damage with distal sensory deficits and chronic pain.18
Nociplastic pain arises from altered nociceptive processing within the CNS and is mediated by CS.19 In 2017, the IASP defined nociplastic pain as pain arising from altered nociception without evidence of tissue damage or somatosensory system lesions.9,13 Common examples include fibromyalgia (FM), irritable bowel syndrome, chronic low back pain, migraine, chronic fatigue syndrome, and non-traumatic neck pain.20 FM represents the paradigmatic nociplastic disorder, characterized by widespread pain, fatigue, sleep disturbance, and depressive symptoms.13
The pathophysiology of nociplastic pain reflects an imbalance between excitatory and inhibitory neurotransmission within the CNS. Elevated SP, excessive glutamate release, and hyperactivation of N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors induce sustained neuronal depolarization and CS.21,22 Excessive activation of astrocytes and microglia promotes chronic neuroinflammation and amplifies spinal nociceptive transmission, while dysfunction of descending serotonergic and noradrenergic antinociceptive pathways further enhances pain signaling. Reduced γ-aminobutyric acid (GABA) weakens inhibitory control and sustains neuronal hyperexcitability.21,22 Elevated glutamate correlates with FM severity, whereas reduced GABA correlates with pain intensity.23 Increased cerebrospinal fluid SP levels further confirm the role of neuropeptides in maintaining CS and pain chronification.13,14 A comparative overview of pain mechanisms is presented in Table 1.
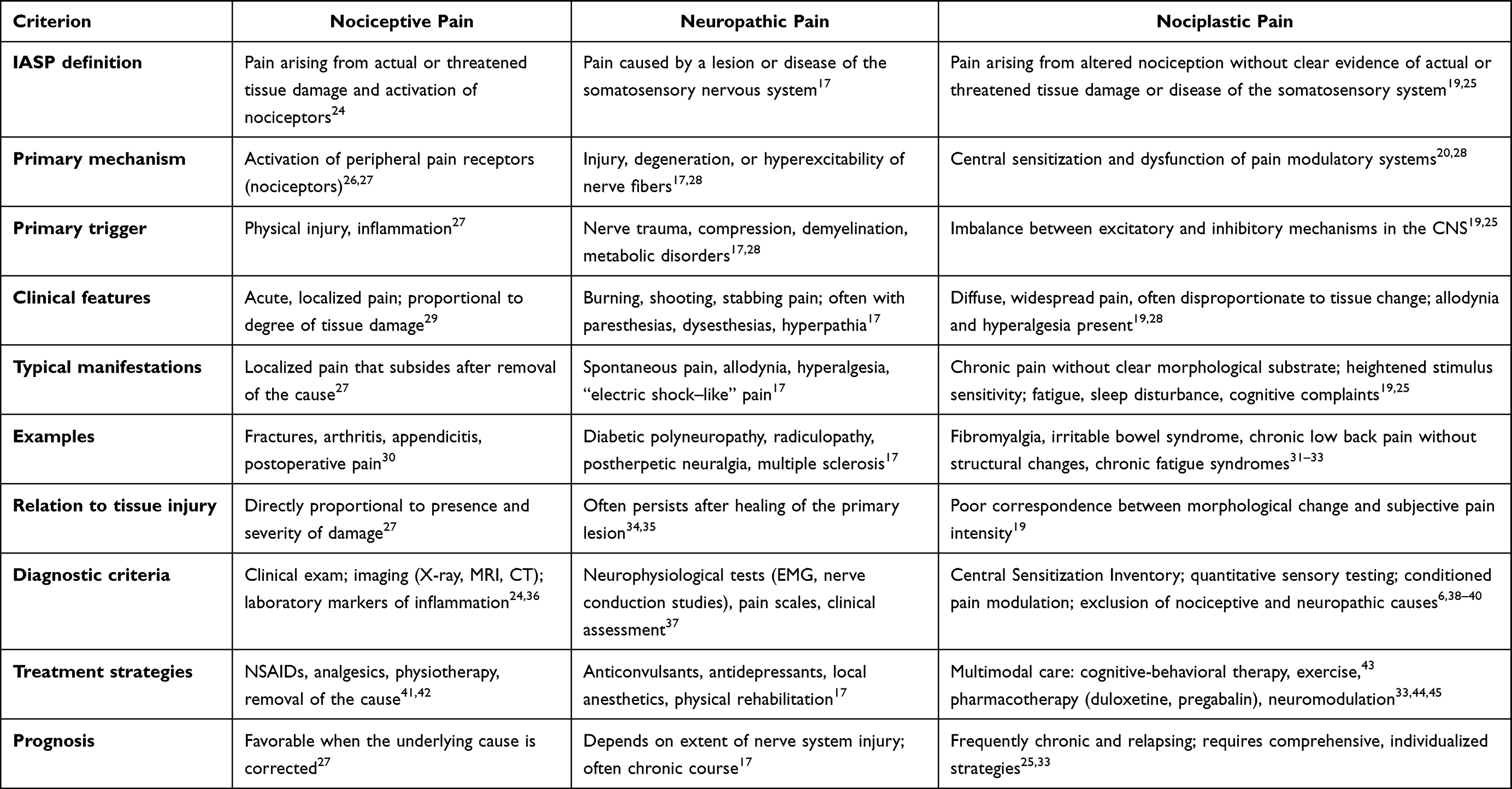 |
Table 1 Comparative Characteristics of the Main Types of Pain |
The principal clinical manifestations of CS are hyperalgesia and allodynia. Hyperalgesia is defined as increased pain intensity or reduced thresholds to noxious stimuli and initially develops as primary hyperalgesia at the injury site, serving a protective function.14,46 PGs enhance nociceptor sensitivity and promote secondary hyperalgesia via central amplification. CS consolidates pain through increased excitability of dorsal horn and supraspinal neurons, expansion of receptive fields, and failure of inhibitory mechanisms.
Allodynia refers to pain elicited by normally non-painful stimuli and reflects pathological sensory reorganization. Following nerve injury, tactile Aβ afferents form aberrant synapses in lamina II of the dorsal horn. Prostaglandin E2 (PGE2), generated via cyclooxygenase activity, stabilizes neuronal hyperexcitability and maintains CS.14,46,47
Glial activation is central to chronic pain maintenance. Microglia and astrocytes are activated by adenosine triphosphate (ATP), chemokines, SP, and calcitonin gene–related peptide (CGRP), releasing mediators that amplify neuronal excitability.48 After nerve injury, ATP-sensitive receptor upregulation promotes microglial hyperactivity and mechanical allodynia. The fractalkine pathway is critical: cathepsin S-mediated cleavage of CX3CL1 (fractalkine) activates microglia via CX3CR1, which is overexpressed after nerve trauma. Mechanical allodynia is attenuated by CX3CR1 blockade and abolished in Cx3cr1 knockout models.49,50 Similar microglial activation occurs after fractures and orthopedic surgery.51–53 Astrocytes further contribute through impaired glutamate clearance, maladaptive synaptogenesis, and sustained release of TNF, interleukins, and neurotrophins.
Peripheral sensitization develops at sites of tissue injury and inflammation due to mediators including ATP, bradykinin, serotonin, norepinephrine, PGE2, nerve growth factor, SP, and local acidosis. Key ion channels include Transient Receptor Potential Vanilloid 1 (TRPV1), voltage-gated sodium channels Nav1.7–Nav1.9, and mechanosensitive Piezo channels.14,54–58 Neurogenic inflammation mediated by SP, CGRP, and neurokinins induces vasodilation, vascular permeability, and immune-cell infiltration, contributing to migraine and complex regional pain syndrome.59–61
Bidirectional nociceptor–immune interactions further amplify sensitization; for example, neuronal CCl2 regulates macrophage activation in dorsal root ganglia after chemotherapy, promoting neuropathic pain.62,63 Sustained nociceptor input induces CS, characterized by glutamate and SP release, NMDA and AMPA receptor activation, and reduced inhibitory control mediated by GABA, glycine, adenosine, endogenous opioids, and cannabinoids.65 Proinflammatory cytokines such as tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) activate intracellular cascades including protein kinase A (PKA), protein kinase C (PKC), and mitogen-activated protein kinases (MAPKs). Activation of p38 MAPK enhances TRPV1 activity and stabilizes chronic hyperexcitability.54
Inflammation, neurogenic inflammation, and neuroinflammation differ in localization but synergistically drive chronic pain, as summarized in Table 2.
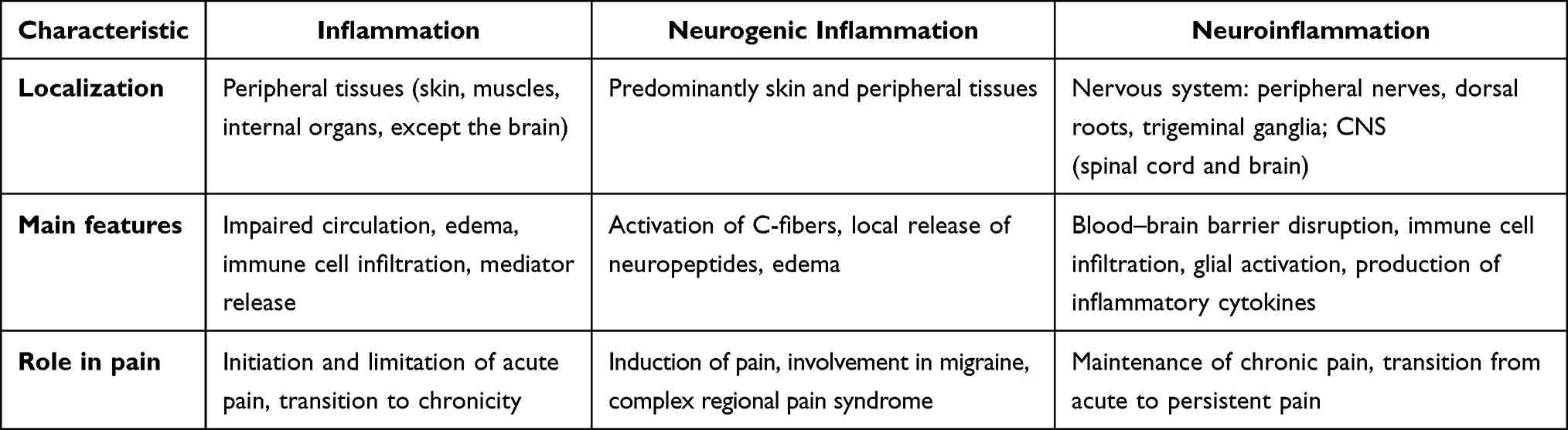 |
Table 2 Comparison of Inflammation, Neurogenic Inflammation, and Neuroinflammation (Adapted from Ji et al48) |
Fibromyalgia (FM) as a Model of Nociplastic Pain in the Context of Comorbid Syndromes
Early clinical descriptions resembling FM date back to the 19th century, when physicians began noting chronic musculoskeletal pain without overt morphological changes. A pivotal step was the introduction in 1904 of the term “fibrositis” by the British neurologist William Richard Gowers (1845–1915), who posited local inflammatory processes in fibrous tissue.64,65 This term was widely used in the medical literature for several decades, until the 1970s–1980s, when accumulating clinical observations called its pathogenetic accuracy into question. At that time, the hypothesis emerged that the central nervous system plays a leading role in the genesis of chronic pain syndromes.65
Further conceptual development occurred in the mid-20th century. In 1950, the British rheumatologist Graham Harvey (1904–1977) described fibrositis as a “pain syndrome” existing in the absence of specific organic changes.66 This stance effectively stripped the “fibrositis” concept of a localist, inflammatory basis and emphasized the systemic and functional nature of the disorder. That perspective laid the groundwork for subsequently viewing FM as chronic pain with pronounced neuropathic and neuroendocrine components.
A landmark advance came in the mid-1970s, when Canadian researchers Hugh Smythe (1915–1989) and Harvey Moldofsky (b. 1937; https://orcid.org/0000-0002-5234-8864) introduced the term “fibromyalgia” and first described “tender points” (areas of heightened pain sensitivity that became important diagnostic criteria).67 Their work was pivotal, enabling abandonment of the term “fibrositis” and shifting the focus from hypothetical inflammation to the clinical pain phenotype that integrates both somatic and neuropsychological mechanisms.
The culmination of the modern diagnostic paradigm was the adoption in 1990 by the American College of Rheumatology (ACR) of standardized diagnostic criteria for FM.68 These were the first official recommendations that unified diagnostic approaches and provided a robust foundation for subsequent epidemiological and clinical research. The criteria remained influential for decades and were recently updated in line with contemporary concepts of FM pathogenesis.64,68 The same group defined FM pain as widespread if present in the left and right sides of the body, above and below the waist, with pain in the axial skeleton (cervical spine, anterior chest wall, thoracic and lumbar spine) for a continuous duration of at least 3 months. Clinical confirmation requires pain in 11 of 18 specifically defined tender points, as shown in Figure 2A.69
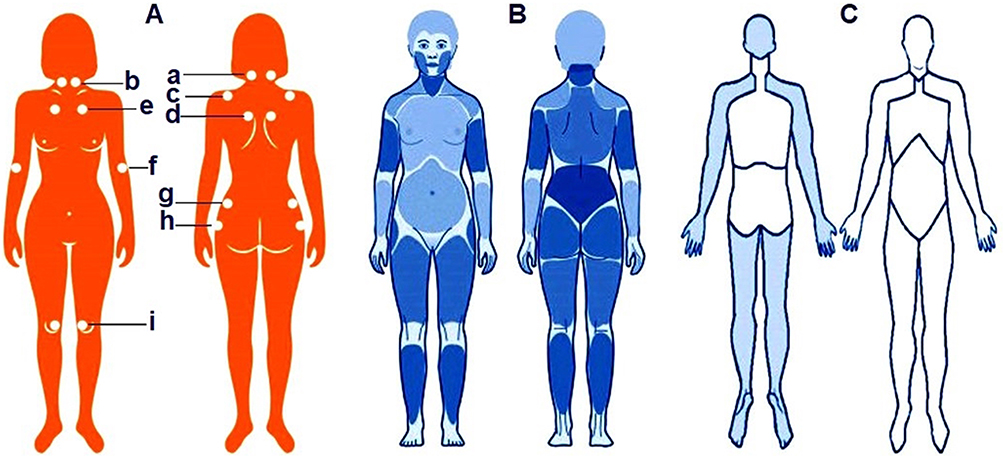 |
Figure 2 Evolution of diagnostic criteria for fibromyalgiaLettered panels: (A) – ACR (American College of Rheumatology) 1990 criteria: required pain at 11 of 18 specified tender points, numbered on the schematic. (B) – ACR 2010, 2011, 2016 and FAS 2019 criteria: the emphasis progressively shifted from tender points to pain widespreadness (Widespread Pain Index, WPI) and symptom severity (Symptom Severity Scale, SSS). ACR 2010: first replaced “tender points” with a count of painful sites (WPI; 19 areas) plus the SSS. ACR 2011: refined questionnaire wording and introduced a self-report questionnaire to avoid reliance on clinician palpation. ACR 2016: fully standardized WPI (19 regions) + SSS, and added the requirement of pain in ≥4 of 5 anatomical regions (left, right, upper, lower, axial). FAS 2019 (Fibromyalgia Assessment Status): the body is divided into 19 regions – jaw, shoulder, arm, thigh, lower leg, Hip/gluteal area, and chest (all left and right), plus neck, thoracic spine, lumbar spine, abdomen, and breasts/sternum. (C) – AAPT (ACTTION – American Pain Taxonomy) criteria: for FM, apply Multisite Pain (MSP) across 9 body regions; FM is considered probable if pain lasts ≥3 months and involves ≥4 of the 9 regions. Numerical labels for Panel 2A (tender points): (a) Occiput: bilaterally at the insertions of the suboccipital muscles (mm. suboccipitales). (b) Cervical region: bilaterally at the anterior intertransverse spaces at C5–C7 (vertebrae cervicales V–VII). (c) Trapezius muscle: bilaterally at the midpoint of the upper border (m. trapezius). (d) Supraspinatus muscle: bilaterally above the scapula near the midline (m. supraspinatus). (e) Second rib: bilaterally at the second costochondral junction (articulatio costochondralis II). (f) Lateral epicondyle of the humerus: bilaterally 2 cm distal to the epicondyle (epicondylus lateralis humeri). (g) Gluteal region (gluteus maximus): bilaterally in the upper outer quadrant (m. gluteus maximus). (h) Greater trochanter: bilaterally posterior to the greater trochanter (trochanter major). (i) Knee joint: bilaterally on the medial aspect, proximal to the joint line (articulatio genus). |
Further advances in understanding the pathophysiological mechanisms of FM have come from research in the neurobiology of pain. The hypothesis that CS predominates in patients with FM rests primarily on findings from functional neuroimaging and biochemical analyses of cerebrospinal fluid, which demonstrate disturbances in pain neurotransmission systems.70
Accumulating evidence from functional magnetic resonance imaging (fMRI) provides compelling neurobiological confirmation of CS as a core mechanism underlying chronic musculoskeletal pain syndromes, particularly FM, and as an important contributor to persistent pain in osteoarthritis OA and inflammatory arthritis. Task-based, resting-state, and spinal cord fMRI studies converge in demonstrating abnormal amplification and dysregulation of nociceptive processing at multiple levels of the central nervous system.
In fibromyalgia, task-based fMRI studies using pressure or thermal pain paradigms consistently reveal exaggerated blood oxygen level–dependent (BOLD) responses in classical pain-processing regions, including the insular cortex, anterior cingulate cortex (ACC), primary and secondary somatosensory cortices (S1/S2), and the thalamus, even when stimuli are of low intensity or normally non-noxious. Importantly, Hubbard et al demonstrated abnormal salience attribution to pain onset and offset, with disproportionate cortical activation relative to stimulus intensity, supporting the concept of lowered central pain thresholds and amplified nociceptive gain in FM71 These findings directly substantiate CS as a mechanism exceeding normal physiological pain responses and explain the pronounced hyperalgesia and allodynia observed clinically.
Resting-state fMRI further refines this model by showing altered intrinsic brain network organization in FM. Specifically, increased functional connectivity between the default mode network (DMN) and the insular cortex has been reported, with the magnitude of these changes closely dependent on current clinical pain intensity.72 Such aberrant coupling suggests that pain-related processing becomes embedded into baseline brain activity, generating a persistent “pain background” even in the absence of external stimuli. This persistent representation of pain is accompanied by impaired descending inhibitory control, reinforcing the central sensitization phenotype.
Beyond cortical and subcortical structures, spinal cord fMRI provides evidence that CS in FM involves early amplification of nociceptive signaling at the spinal level. During paradigms of temporal summation, patients with FM exhibit enhanced neural activity in the dorsal horn and ascending spinal pathways compared with healthy controls.73 These findings indicate that abnormal nociceptive facilitation is not confined to supraspinal networks but begins at the earliest stages of central pain transmission, contributing to sustained hyperexcitability throughout the neuraxis.
In osteoarthritis, fMRI studies demonstrate a different but related pattern. Both task-based and resting-state paradigms reveal altered activation of the insula, ACC, and sensorimotor cortex, reflecting central pain amplification that correlates only weakly with the extent of structural joint damage.74,75 These observations support the concept of secondary central sensitization, whereby prolonged peripheral nociceptive input from degenerative joint pathology induces maladaptive plasticity within central pain networks. Clinically, this explains why pain severity in OA often persists or escalates despite relatively modest radiographic changes and why pain may remain refractory even after successful surgical intervention.
Evidence in RA, although more limited, points in the same direction. Task-based and resting-state fMRI studies reveal altered pain processing in insular, limbic, and prefrontal regions, particularly in patients who continue to experience pain despite low inflammatory activity or clinical remission.76,77 These neurofunctional alterations provide objective support for the dissociation between pain intensity and inflammatory markers observed in a substantial subset of RA patients and indicate that CS contributes meaningfully to residual, non-inflammatory pain.
 |
Table 3 Functional Magnetic Resonance Imaging (fMRI) Evidence of Central Sensitization Across Chronic Musculoskeletal Pain Conditions |
Taken together, the fMRI data summarized in Table 3 demonstrate that CS is not a phenomenon restricted to fibromyalgia but represents a shared neurobiological mechanism across chronic musculoskeletal pain conditions. In FM, CS is primary and dominant, shaping the entire clinical phenotype. In OA and RA, CS develops secondarily, driven by prolonged peripheral nociceptive input and sustained immune-mediated inflammation, yet it becomes a critical determinant of pain persistence and treatment resistance.
Finally, emerging neuroimaging data in psoriatic arthritis indicate involvement of the insular cortex and affective pain networks, with neurofunctional alterations associated with a persistent pain burden. Although still limited, these findings suggest a potential overlap between inflammation-driven nociceptive mechanisms and centrally mediated nociplastic processes, positioning central sensitization as a contributory factor in pain chronification in psoriatic arthritis.78 These convergent neuroimaging findings provide a robust mechanistic framework for the concept of nociplastic pain and underscore the necessity of phenotyping pain mechanisms to avoid misinterpretation of disease activity and unjustified escalation of anti-inflammatory therapy.
Synthesizing these findings, the convergent neuroimaging and neurochemical evidence substantiates the reality of pain in patients with FM, countering any presumption of a psychogenic or “imagined” origin. At the same time, current data do not definitively identify the primary source of pathological nociception, since similar patterns of heightened neuronal activity in brain regions responsible for pain processing are also observed in patients with neuropathic pain, both in humans and in animal models.78,79 This underscores shared pathophysiological mechanisms across chronic pain conditions of diverse etiology while leaving open the question of changes specific to FM.
In recent years, increasing attention has focused on the role of autoantibodies in FM pathogenesis. This line of inquiry gained momentum following an experimental study convincingly demonstrating, in an animal model, that passive transfer of immunoglobulin G (IgG) from the serum of patients with FM, but not from controls, can induce pain hypersensitivity. Administration of this IgG to healthy mice produced sensory hypersensitivity via sensitization of nociceptive afferent neurons in the dorsal root ganglia.80,81 These results support a direct effect of antibodies on peripheral nervous system structures, marking a fundamentally new step in our understanding of FM pathogenesis.
Thus, the evolution of concepts regarding FM reflects a shift from a localized inflammation model to recognition of the leading role of central sensitization mechanisms. This change has not only altered terminology but also substantially influenced contemporary approaches to the pathogenesis of chronic pain, paving the way for novel therapeutic strategies and multidisciplinary management. At present, the prevailing view considers FM a central sensitization syndrome.20,82
The clinical presentation of FM is heterogeneous, with chronic widespread musculoskeletal pain as its defining feature, commonly accompanied by severe fatigue, sleep disturbances, cognitive dysfunction, and emotional lability. Epidemiological data indicate that FM has substantial medico-social significance, given its pronounced negative impact on productivity and quality of life.83,84
Prevalence estimates, however, vary considerably across countries and study settings. Such variability is expected for a nociplastic condition in which case identification relies heavily on self-reported multisymptom burden closely linked to CS. In a cross-culturally adapted German validation of the Central Sensitization Inventory (CSI-GE), mean CSI scores were markedly higher in individuals with fibromyalgia (54.9 ± 11.7) compared with healthy controls (18.4 ± 9.3), demonstrating a quantifiable population-level “СІ symptom load” that may differ across cultural contexts.85
Consistently, in a large clinical cohort of patients with fibromyalgia (n = 562), 35.4% exhibited CSI scores ≥40, and CSI showed moderate-to-strong correlations with established fibromyalgia severity indices (FIQR ρ = 0.542; modFAS ρ = 0.580; PDS ρ = 0.518), indicating that epidemiological estimates are sensitive to how different populations perceive, express, and report CS-related symptoms.38
This phenomenon may partly explain extremely low prevalence estimates reported in certain regions, such as Mainland China (0.03–0.12%), where clinical impact profiles appear milder and therefore less likely to reach diagnostic thresholds. In a multicenter Chinese study, patients with fibromyalgia showed comparatively lower functional impairment and symptom burden, which the authors explicitly discuss as a potential contributor to under-recognition at the population level.84
Additional support for the epidemiological relevance of CS comes from population-based studies demonstrating that CS-related symptoms extend beyond formally diagnosed fibromyalgia. In the original CSI validation, clinically relevant CSI scores (≥40) were observed not only in fibromyalgia but also in 10–20% of individuals with chronic musculoskeletal pain without an FM diagnosis, indicating that epidemiological prevalence estimates capture only the upper tail of a broader CS spectrum.86
Cross-national comparisons further suggest that differences in diagnostic uptake may reflect culturally shaped thresholds for reporting fatigue, sleep disturbance, and cognitive complaints rather than a true absence of CS. For example, comparative studies of Portuguese and Brazilian fibromyalgia cohorts revealed similar pain intensity but significantly different levels of fatigue, emotional distress, and cognitive symptoms – core features of CS – highlighting the modulatory role of sociocultural context on the clinical phenotype detected in epidemiological surveys.87
Finally, longitudinal population-based evidence supports a temporal relationship between CS and the development of chronic pain. In a prospective cohort study, higher baseline CS scores were shown to precede and predict the onset of chronic musculoskeletal pain, providing mechanistic support for a causal framework in which social stressors and contextual vulnerability promote CS, which subsequently shapes future prevalence patterns of fibromyalgia and related nociplastic pain conditions.88
As noted by Sluka K.A. et al,70 the diagnosis assigned to a patient with FM often depends on the specialist first consulted; the same clinical profile may receive different nosological labels, leading to fragmented care pathways and multiple diagnoses Gastroenterologists typically invoke categories of functional gastrointestinal disorders, chiefly irritable bowel syndrome, non-ulcer dyspepsia, and esophageal motility disorders to explain abdominal pain, bloating, postprandial discomfort, and transit disturbances Urologists commonly manage patients with predominant pelvic pain and dysuric symptoms under the umbrellas of interstitial cystitis/bladder pain syndrome, chronic prostatitis/chronic pelvic pain syndrome, as well as vulvodynia or vestibulitis of the vulva. Dentists, in turn, most often encounter temporomandibular disorders characterized by orofacial pain, myofascial dysfunction, and palpation-induced hyperalgesia. Despite differences in nosological “labels”, these clinical clusters substantially overlap, forming a continuum of chronic comorbid pain syndromes and explaining frequent patient movement across specialties.
The concept of these comorbid conditions rests on a shared neurobiological substrate: enhanced nociceptive processing together with insufficiency of descending inhibitory pathways in the CNS. In practice, this manifests as lowered pain thresholds, allodynia, widespread pain, and a stable cluster of accompanying symptoms: fatigue, fragmented sleep, cognitive dysfunction, and mood lability. This symptom organization supports viewing FM as a “model” of nociplastic pain capable of integrating gastroenterological, urological, and dental phenotypes within a single CNS-mediated pathophysiological framework.89
Epidemiological studies indicate that FM prevalence varies substantially by geographic region. In Europe the average prevalence is approximately 2.64%, in North America 2.41%, and in some parts of Asia it does not exceed 1.62%.90 Notably, wide discrepancies reflect methodological differences in case definition, heterogeneity of age groups included, and sociocultural factors that influence diagnosis and case ascertainment. For example, a large population-based study in Spain reported a prevalence of about 2.4%, whereas in the United States it was somewhat lower (around 2.0%).91 Thus, comparative analyses suggest that the true scope of the problem likely exceeds official statistics. Across studies, women account for 80% to 96% of FM cases.91 However, recent systematic reviews and meta-analyses refine this paradigm: pooled international data estimate global FM prevalence at roughly 3.98% in women versus 2.40% in men, an apparently modest but statistically significant difference.92 Sex differences in FM prevalence and diagnosis are likely influenced by social stigma surrounding a predominantly “female” condition and by sociocultural features of Western countries, where men less often seek care for chronic pain symptoms, limiting accurate diagnosis.83
A distinct clinical subset comprises individuals with FM in whom the disorder coexists with conditions characterized by a persistent peripheral nociceptive drive for example, autoimmune inflammatory diseases, sickle cell anemia, or advanced degenerative joint disease. Such cases have traditionally been described as “secondary” FM.70,84
Accordingly, in patients with suspected FM it is essential, first, to purposefully identify co-occurring phenotypes within the cluster of chronic comorbid pain syndromes: functional gastrointestinal disorders (irritable bowel syndrome, non-ulcer dyspepsia, esophageal motility disorders), interstitial cystitis/bladder pain syndrome, chronic prostatitis/chronic pelvic pain syndrome, vulvodynia or vulvar vestibulitis, and temporomandibular disorders. The presence of these phenotypes supports a nociplastic pain mechanism and justifies a multimodal treatment strategy. Second, in the subgroup with a prominent peripheral nociceptive component, priority should be given to reducing or eliminating the peripheral driver, which not only diminishes afferent input but may also “off-load” the CNS, attenuating CS manifestations and improving rehabilitative potential.70 Third, regardless of the clinical scenario, management must move beyond organ-centric thinking. FM rarely occurs in isolation, so the therapeutic program should integrate control of peripheral triggers with interventions targeting central mechanisms: normalizing sleep, addressing affective-cognitive factors, prescribing graded physical activity, and employing rational pharmacotherapy with explicit consideration of the nociplastic component.70
Thus, within the spectrum of chronic comorbid pain conditions, FM emerges less as a discrete nosology and more as an integrative syndrome dominated by central pain mechanisms. Recognition of comorbid phenotypes, systematic identification of peripheral nociceptive sources, and targeted modulation of central processes provide the foundation for personalized management, mitigation of symptom burden, and prevention of the chronification of disabling pain.84
Evolution and Refinement of the Diagnostic Criteria for Fibromyalgia
After prolonged debate and successive revisions of diagnostic approaches to FM, the most widely accepted criteria today are those proposed by the ACR in 2016 (Table 1).93,94 They represent the culmination of years of refinement of earlier recommendations (1990, 2010, 2011), each of which revealed strengths as well as practical limitations.
As noted above, the clinical diagnosis of FM rests on clearly defined criteria and, in particular, requires detection of increased tenderness on palpation at ≥11 of 18 standardized tender points.68 It is crucial to emphasize that these are tender points, not the so-called trigger points, which are often treated as analogues in clinical practice but are not diagnostically identical. This distinction is fundamental, as it influences both the diagnostic process and the selection of therapeutic strategies.
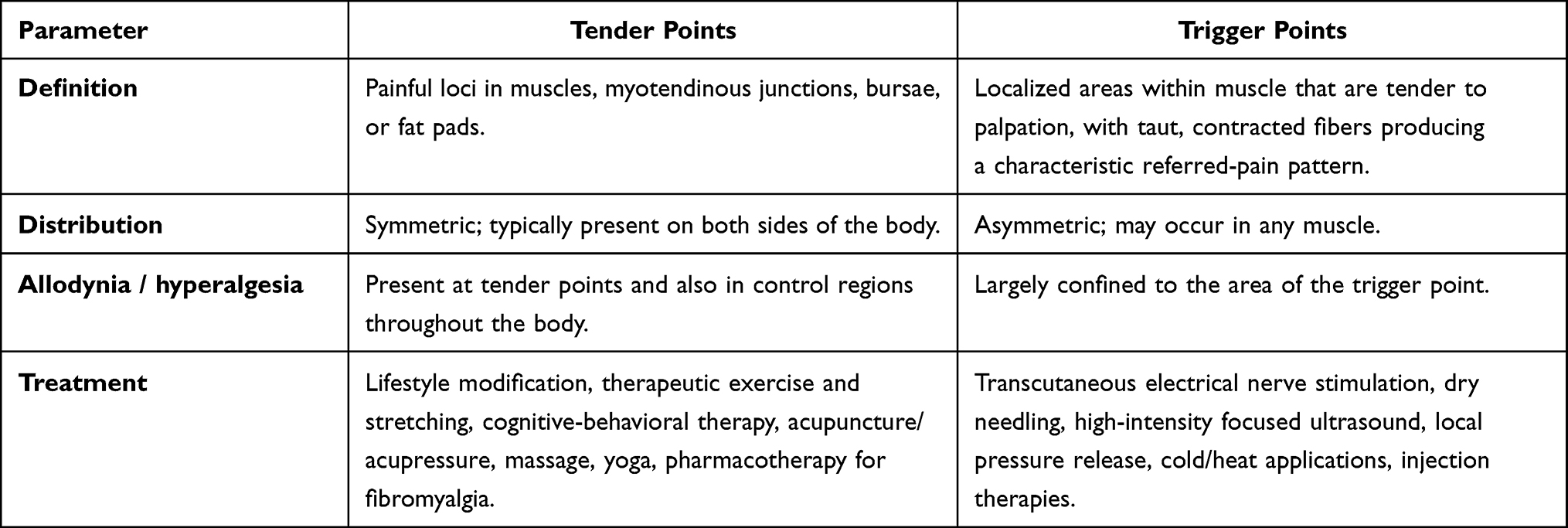 |
Table 4 Similarities and Differences Between Tender Points and Trigger points69,95,96 |
Differences between tender and trigger points are summarized in Table 4. Tender points are located predominantly at myotendinous junctions, bursae, or fat pads, whereas trigger points arise within muscle tissue and are characterized by palpable, taut muscle bands. Clinically, tender points show a symmetric distribution, while trigger points may be asymmetric and occur in any muscle. An important diagnostic consideration is that in FM, allodynia and hyperalgesia extend beyond the tender points to control regions of the body, whereas with trigger points these phenomena are confined to the local site.69
It is noteworthy that the presence of another somatic or neurological disorder does not preclude a diagnosis of FM, underscoring the comorbid nature of this syndrome. The 1990 ACR criteria were repeatedly criticized and subsequently revised because of the subjective components involved in clinical examination and the high risk of assessment variability by physician and patient.
The updated criteria issued in 2010 and 2011 substantially reduced the emphasis on palpation of tender points and proposed a more comprehensive approach based on assessing pain widespreadness and the severity of accompanying symptoms. This transformation aimed to create a universal, clinically practical diagnostic framework that accounts not only for localized tenderness but also for the full spectrum of clinical manifestations shaping the overall disease picture.93,94,97
According to the 2016 ACR criteria (Figure 2B),98 a diagnosis of FM is established when the following three requirements are met:
- Generalized chronic pain persisting for at least 3 months;
- Generalized pain is defined as pain in at least 4 of 5 body regions (left upper, right upper, left lower, right lower, axial);99
- To objectify clinical manifestations, two standardized assessment tools are used – the Widespread Pain Index (WPI) and the Symptom Severity Scale (SSS). The diagnosis is considered substantiated if either of the following is present:
a. WPI ≥ 7 in combination with SSS ≥ 5, reflecting a high number of painful body sites together with substantial symptom intensity;
b. WPI 4–6 in combination with SSS ≥ 9, indicating less extensive pain but a markedly higher burden of systemic symptoms such as fatigue, sleep disturbance, or cognitive dysfunction.99
In 2019, the Fibromyalgia Assessment Status (FAS) – a modified set of criteria, was developed to simplify diagnosis and enhance its usability in clinical practice. In this system, the body is partitioned into 19 regions (Figure 2B): jaw, shoulder, arm, thigh, lower leg, hip/gluteal area, and chest (each left and right), plus the neck, thoracic and lumbar spine, abdomen, and breasts/sternum. This framework permits quantitative assessment of pain distribution and its integration with key symptom severity, making it suitable for both clinical research and routine care. In parallel, alternative diagnostic criteria were proposed under AAPT (ACTTION–American Pain Taxonomy; Figure 2C), grounded in the concept of multisite pain (MSP). These criteria grade pain by the number of affected regions (0–9) and consider FM probable when pain persists for ≥3 months and involves four or more of the nine regions. AAPT also incorporates concurrent features – particularly sleep disturbance and marked fatigue – highlighting the disorder’s multidimensional nature.93,99–101
Over the past three decades, diagnostic approaches to FM have evolved from a localized assessment of tender points to multicomponent systems that account for pain distribution, symptom intensity, and their impact on patients’ quality of life. Each iteration of the criteria has had strengths and limitations, prompting further modifications and the search for the most practical clinical tool. The consolidated differences among the ACR-1990, ACR-2010/2011, ACR-2016, FAS-2019, and AAPT criteria are summarized in Table 5.
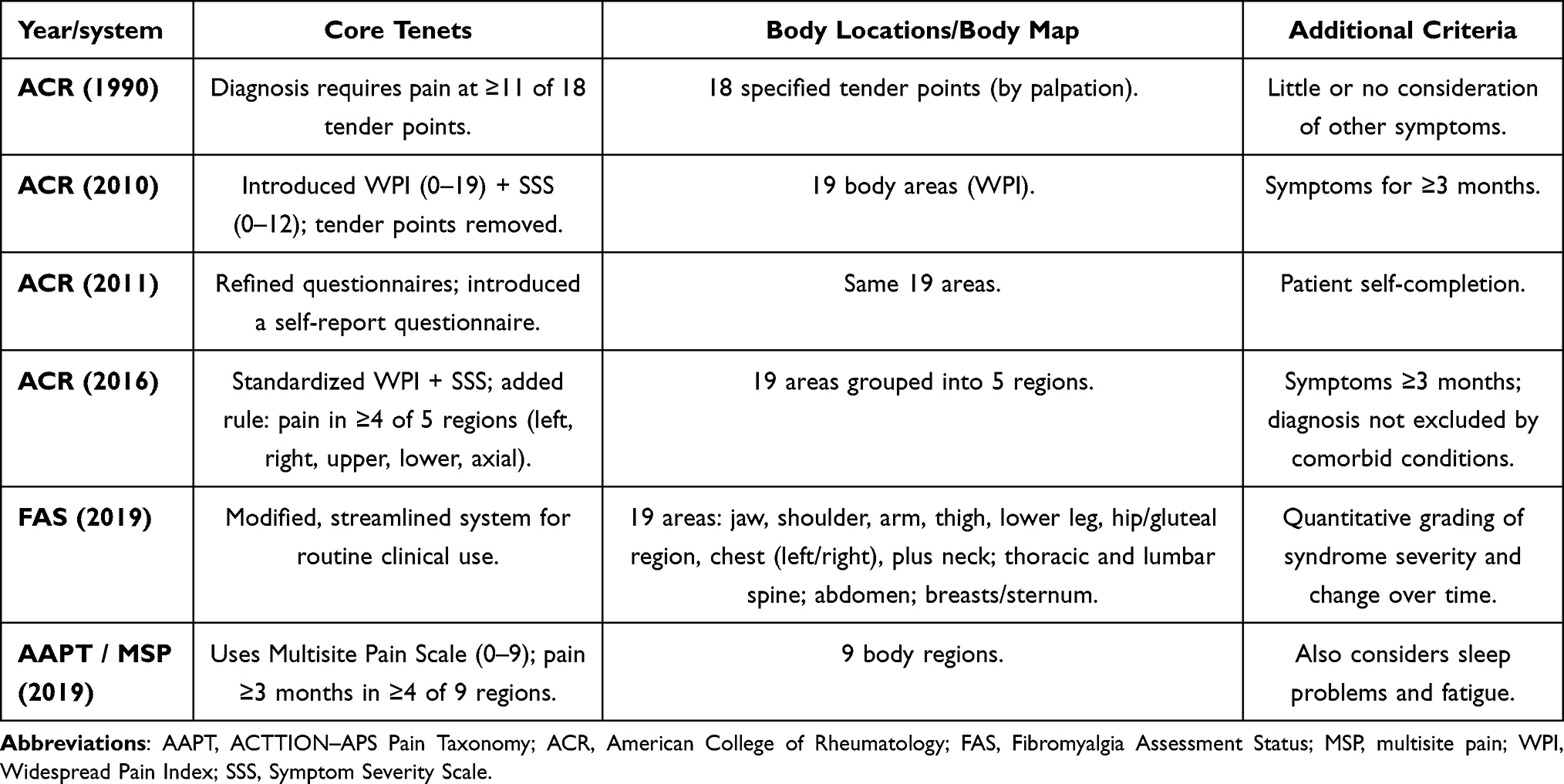 |
Table 5 Comparison of Major Diagnostic Criteria for Fibromyalgia |
In 2023, the Neuropathic Pain Study Group (NPSG) of the Italian Society of Neurology102 proposed a set of practical recommendations reflecting an innovative, contemporary approach to FM diagnosis. This approach integrates classical clinical examination with state-of-the-art tools for assessing sensory system function, thereby broadening opportunities both to refine diagnostic workflows and to deepen understanding of the disorder’s pathophysiology.103 Particular emphasis is placed on the view that FM is increasingly considered not only a central sensitization syndrome but also a condition that may exhibit features of peripheral involvement, notably affecting small nerve fibers.104
In parallel, questionnaire-based approaches are widely used to screen for a central sensitization symptom load, particularly when comprehensive QST batteries are impractical. The most widely used instrument is the Central Sensitization Inventory (CSI; 25 items), which was developed to quantify symptoms associated with central sensitivity syndromes (including fibromyalgia). A cutoff value of approximately ≥40 is commonly applied to indicate a clinically relevant CS-related symptom burden.86,105 In musculoskeletal disorders, questionnaires assessing neuropathic-like pain symptoms are also used as indirect indicators of centrally amplified pain. A prominent example is the (modified) painDETECT questionnaire in knee osteoarthritis, where higher scores correlate with QST findings consistent with central sensitization.106
Across the four diseases (FM, OA, RA, PsA), questionnaire-based assessments of central sensitization reveal a broadly consistent pattern. Fibromyalgia typically shows the highest CSI scores, reflecting CS as a primary and defining pain mechanism. In contrast, OA, RA, and PsA exhibit elevated CSI and painDETECT-type scores mainly in specific subgroups, particularly in patients with pain disproportionate to structural damage or inflammatory activity, greater disability, and poorer patient-reported outcomes.78,106–108 In rheumatoid arthritis, CSI-defined central sensitivity syndrome is associated with distinct clinical characteristics and increased reporting of neuropathic-like symptoms, supporting the concept that persistent pain may be partially uncoupled from inflammatory activity in a subset of patients.107
In psoriatic arthritis, CSI-defined central sensitization has been linked to greater disease impact, reduced physical function, and impaired quality of life. It may also complicate the interpretation of composite disease activity indices that rely heavily on patient-reported components.78,108 Thus, although these questionnaires do not provide a mechanistic diagnosis of central sensitization, they consistently identify a cross-disease pattern: a gradient ranging from primary nociplastic pain dominance in fibromyalgia to secondary or overlay central sensitization phenotypes in OA, RA, and PsA, which track with pain persistence, disability, and a potential overestimation of inflammatory disease activity when symptom-driven indices are used.78,107,108
Algometry for Assessing Central Sensitization to Pain in Patients with Fibromyalgia
Mechanical hyperalgesia, defined as a reduction in pressure pain thresholds, is commonly associated with pain.109 Algometry has proven highly effective as a standardized tool for quantifying both central allodynia and various forms of hyperalgesia in patients with FM, as well as across a broad range of other chronic pain syndromes. The method detects even minimal changes in pain threshold, conferring a significant advantage over cruder clinical tests. In the literature, algometry is often imprecisely referred to as dolorimetry or equated with quantitative sensory testing (QST). However, such terminology is partly inaccurate. While QST shares methodological foundations, it encompasses a broader evaluation of sensory function, whereas algometry focuses primarily on the standardized measurement of pressure pain threshold under mechanical loading.110
Algometry enables assessment of individual sensitivity to nociceptive stimuli across multiple body regions, substantially improving the precision of detecting abnormalities in pain processing. Various stimulus modalities can be employed including pressure, heat, cold, and electrical stimuli, allowing a more comprehensive characterization of nociceptive mechanisms. This multiparametric evaluation provides an in-depth, exhaustive analysis of pathological sensory responses, which is particularly pertinent in FM, where central sensitization plays a leading role.110
Clinical studies have also confirmed that algometry-captured pain responses correlate closely with the intensity of clinical pain experienced by patients with FM.110
Consensus statements from leading experts emphasize the utility of a standardized QST protocol, for which normative reference data are already available.111,112 This is particularly important to ensure between-center reproducibility and enable data comparison in large multicenter studies. Among the various QST parameters, two nociceptive stimuli are considered most clinically informative and can be incorporated into an abbreviated protocol (the pressure pain threshold (PPT) and the cold pain threshold (CPT)).113,114 Using these tests markedly reduces participant burden in large-scale investigations without sacrificing informativeness.
According to the QST consensus statement, at least two parameters should be employed to adequately assess the functional state of the nociceptive system. Specifically, CPT reflects the functional activity of thin afferent fibers, whereas PPT primarily characterizes the function of larger sensory fibers.112 This approach provides a more comprehensive interrogation of the pain system and allows for more precise identification of the mechanisms underlying sensory dysfunction.
Particular attention should be paid to data derived from large population-based cohorts. Such studies minimize selection bias and encompass a broader spectrum of clinical and demographic characteristics, making them valuable for examining individual variability in pain sensitivity.115 Access to reliable reference values for pain sensitivity stratified by sex and age enables more precise identification of hypersensitivity or lowered pain thresholds, facilitates deeper phenotyping of pain mechanisms and prognosis,113,116 and supports optimization of therapeutic strategies.117,118 Accordingly, QST is regarded not only as a research tool but also as an important component of personalized medicine, allowing targeted interventions based on the identified sensory phenotype.
Pressure pain sensitivity is most commonly assessed using an electronic algometer. The participant is carefully instructed to press a stop button as soon as the sensation changes from pressure to pain, defined as the pressure pain threshold (PPT) or when the pressure becomes intolerable (pressure/pain tolerance). Typically, the mean of three trials is calculated for each test site, with 30–60 seconds of rest between trials to avoid temporal summation.105 Recent studies, however, have shown high measurement reliability (intraclass correlation coefficient ICC 0.86–0.99) can also be achieved using two series of three repetitions or a single block of four repetitions.119,120
Recording PPT values enables mapping of individual sites as well as entire muscles or anatomical regions. This laid the groundwork for a new approach to visualizing pain sensitivity the construction of topographical pressure pain sensitivity maps.121 This method vividly depicts spatial variability in pain thresholds across regions, allowing a more precise and comprehensive assessment of the spatial characteristics of the nociceptive response.
The use of topographical pressure-sensitivity maps has substantially influenced current understanding of pain perception and processing. These maps make it possible to identify areas of increased or decreased sensitivity, trace the spread of hyperalgesia, and differentiate local from generalized sensory dysfunction. This approach has significantly expanded both clinical diagnostic tools and experimental assessment of pain states, opening new avenues for investigating pathophysiological mechanisms and developing more personalized treatment strategies.122
Particular attention is warranted for a study that set a high methodological benchmark for spatial analysis of mechanical hyperalgesia, the 2018 review by researchers from Spain, Denmark, and Brazil, “Spotlight on topographical pressure pain sensitivity maps: a review”.105 This work systematically describes the construction and clinical application of PPT maps, demonstrating how multipoint measurements on predefined grids capture the spatial heterogeneity of pressure sensitivity within muscles and anatomical areas. The review is especially valuable for summarizing grid-design methodology and interpolation algorithms (Shepard and Franke–Nielsen) used to generate intuitive sensitivity maps. It therefore supports the use of exemplar images from this work as models of successful mapping of mechanical hyperalgesia – including in central sensitization and, specifically, in FM.122–125
Among the most compelling demonstrations are trapezius muscle maps (Figure 3) constructed with grids ranging from 11 to 48 points. These maps reproducibly reveal higher sensitivity in the muscle belly compared with myotendinous zones, as well as a sensitivity gradient in the upper portion of the muscle. Using cluster analysis, investigators partitioned the trapezius into three functionally distinct spatial subregions with characteristic PPT profiles, findings that are important for phenotyping pain mechanisms in the cervico-shoulder region.122,123
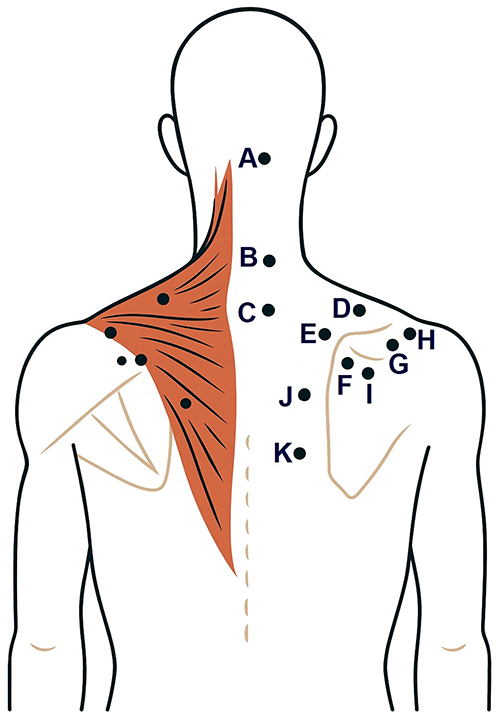 |
Figure 3 Sampling points for topographical pressure pain sensitivity maps in the trapezius muscle. The brown shaded area schematically represents trapezius muscle. Black dots on the left represent mirrored counterparts of the landmark points labeled on the right (A–K). Notes: For the m. trapezius, 11 landmark points with precise anatomical referencing were used: (А) Suboccipital region (regio suboccipitalis); (B) Transverse process of C5 (processus transversus C5); (C) Transverse process of C7 (processus transversus C7); (D) Midpoint between the C7 spinous process (processus spinosus C7) and the acromion (acromion); (E) 2 cm cranial to the superior angle of the scapula (angulus superior scapulae); (F) Superior angle of the scapula (angulus superior scapulae); (G) 1 cm medial to the acromioclavicular joint (articulatio acromioclavicularis); (H) 3 cm cranial to the midpoint of the scapular spine (spina scapulae); (I) 2 cm caudal to the midpoint of the scapular spine (spina scapulae); (J) Midpoint between the T4 spinous process (processus spinosus Th4) and the medial border of the scapula (margo medialis scapulae) in the plane of the scapular spine; (K) Midpoint between the T6 spinous process (processus spinosus Th6) and the medial border of the scapula (margo medialis scapulae) in the plane of the scapular spine. |
An important issue considered in previous work105 is whether the approach can be scaled from a single muscle to an entire anatomical region. Maps have been constructed for the lumbar region using the spinous processes L1–L5 as landmarks, for the scalp employing the international 10–20/10–10 system,126,127 and for the shoulder, elbow, hand, and lower limb (Figure 4) with precise reference points and point matrices. Collectively, these examples clearly show that the spatial distribution of mechanical sensitivity is inherently non-uniform.
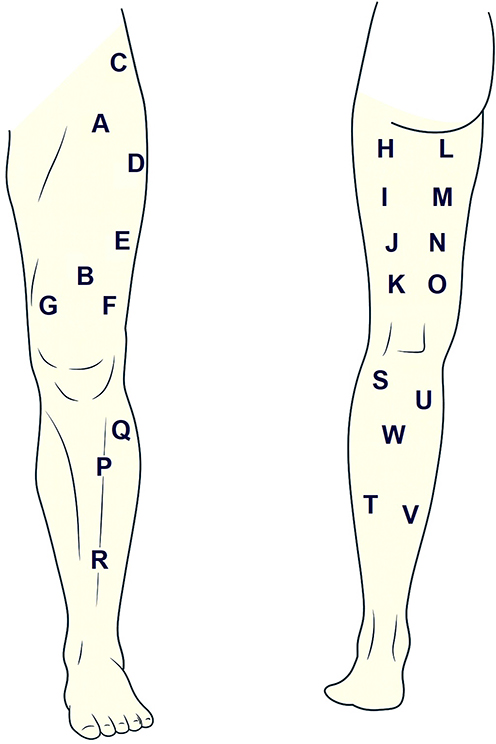 |
Figure 4 Measurement sites for topographic maps of pressure pain sensitivity of the lower extremity. (A and B–M) rectus femoris. (C–M) tensor fasciae latae. (D, E and F–M) vastus lateralis. (G–M) vastus medialis. (H, I, J and K–M) biceps femoris. (L, M, N and O–Mm) semimembranosus and semitendinosus. (P–M) tibialis anterior. (Q and R) – Peroneal muscles. (S and T) – Lateral head of the gastrocnemius muscle. (U and V) – Medial head of the gastrocnemius muscle. (W–M) soleus. Notes: M = singular muscle; Mm. = plural muscles. |
For the entire lower limb, topographical maps were generated from 23 points distributed as follows: two for the rectus femoris muscle; one for the tensor fasciae latae; three for the vastus lateralis; one for the vastus medialis; four for the biceps femoris muscle; four in total for the semimembranosus and semitendinosus muscles; one for the tibialis anterior; two for the fibular (peroneal) muscles; four for the gastrocnemius; and one for the soleus.105,128
Topographical pressure-sensitivity maps of the lumbar region (Figure 5) were constructed using the spinous processes of L1–L5 (processus spinosi vertebrarum lumbalium L1–L5) as reliable anatomical landmarks.122 The total distance between the L1 and L5 spinous processes served as the baseline metric for calculating intervals between adjacent points. The grid comprised 14 test positions arranged as follows: on each side of the midline (linea mediana), a vertical column of five points was placed at the levels L1, L2, L3, L4, and L5; each column was shifted laterally by ¼ of the L1–L5 distance from the imaginary line connecting the spinous processes of these vertebrae. Additionally, on each side a second column with two points was marked at the L2 and L3 levels, shifted laterally by ½ of the L1–L5 distance. This grid geometry provides adequate spatial resolution along the craniocaudal axis and symmetric coverage of the region of interest, enabling accurate depiction of mechanical sensitivity gradients within the lumbar spine (pars lumbalis columnae vertebralis).
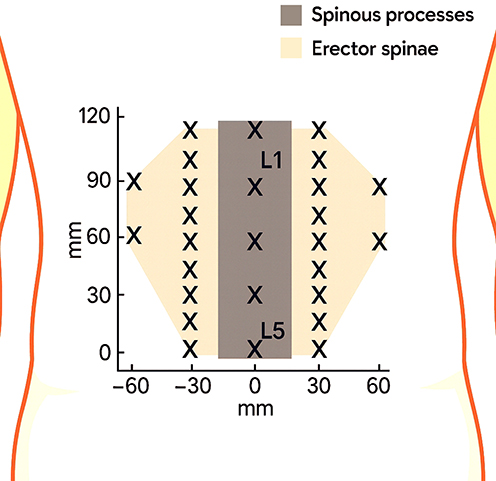 |
Figure 5 Measurement sites for topographic mapping of pressure pain sensitivity in the lumbar region. “X” indicates pressure measurement sites. L1 and L5 denote the first and fifth lumbar vertebrae, respectively. |
The Role of Synovitis, Cytokines, and Sensitization in the Development of Osteoarthritic Pain
OA is the most prevalent degenerative joint disease, integrating mechanical, metabolic, and immune factors. At its pathogenetic core lies synovitis. A low-grade inflammation present in most patients that determines both the rate of structural progression and the severity of clinical symptoms.129–131
The inflammatory process is sustained by a broad array of mediators, among which cytokines, lipid derivatives, and reactive oxygen species (ROS) play key roles. These mediators are produced by chondrocytes, synoviocytes, and osteoblasts, establishing a self-perpetuating cascade of pathological signaling. Proinflammatory cytokines disrupt anabolic processes in cartilage and stimulate the release of proteolytic enzymes, leading to extracellular matrix degradation and cartilage loss.132–135 Consequently, the balance shifts toward catabolism, with proinflammatory factors predominating over anti-inflammatory mechanisms.129,133
Chronic pain in OA is among the most pressing challenges in contemporary medicine, as it persists in most patients even in the absence of active inflammation or overt structural joint damage. This dissociation between structural changes and clinical manifestations indicates a complex pathogenesis in which nociceptive mechanisms coexist with nociplastic phenomena.120 Nociplastic pain is now regarded as a key driver of persistent pain in OA, as it accounts for the hyperalgesia and allodynia frequently observed in practice.136,137
Nociplastic pain arises from plastic remodeling within the CNS that alters excitability thresholds and the quality of pain perception. CS develops against a background of prolonged afferent input from the affected joint and manifests as hyperreactivity of neurons in the spinal cord and higher CNS centers. In the dorsal horn, there is excessive release of glutamate, ATP, and substance P, leading to activation of NMDA receptors and sustained enhancement of postsynaptic potentials.2 At the same time, descending inhibitory mechanismsб notably those mediated by γ-aminobutyric acid (GABA) are weakened, further shifting the balance toward hyperexcitability.138 This drives an increased flow of nociceptive signals to the thalamus, hypothalamus, amygdala, and prefrontal cortex, where both the sensory and affective components of pain are formed.139
Peripheral sensitization also plays an important role. Under normal conditions, articular nociceptors have high activation thresholds and remain quiescent at rest. In OA, they become overly sensitive to mechanical and chemical stimuli. Cartilage destruction, synovitis, and degradation products of the extracellular matrix create an environment rich in proinflammatory mediators. Prostaglandins, bradykinin, tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), interleukin-6 (IL-6), and damage-associated molecular patterns (DAMPs) interact with nociceptive endings via transient receptor potential (TRP) channels and voltage-gated sodium channels, lowering their activation thresholds.140 This generates a pathological afferent drive that initiates mechanisms of central sensitization.
Experimental models corroborate these findings. In rats with monosodium iodoacetate–induced OA, increased firing frequency of afferent fibers was documented and correlated directly with the severity of tissue damage. Similar results were observed in animals with spontaneous age-related OA, where higher action-potential firing rates coincided with lower mechanical activation thresholds.141 This helps explain the clinical phenomenon of more intense, persistent pain in older patients. It is now established that bidirectional crosstalk between the immune and nervous systems is a decisive pathogenetic factor in the development of chronic pain in OA.142,143 In the synovium, nociceptors are in close apposition to macrophages, forming microfoci of persistent inflammation. In the dorsal root ganglia, macrophage infiltration alters the electrophysiological properties of sensory neurons. Within the dorsal horn, microglia play a key role: driven by nociceptive input, they modify synaptic contacts between primary afferents and second-order neurons, thereby amplifying CS.144
Clinical studies corroborate these mechanisms. Patients with OA exhibit heightened sensitivity to experimental pain stimuli in areas remote from the affected joints, indicating central sensitization beyond the local pathological focus.145 Notably, this phenomenon does not correlate with radiographic findings, underscoring the leading role of neuroplastic changes in pain pathogenesis.146 Consequently, severe pain may persist despite stable joint structure, justifying a comprehensive approach to assessment.
Particular attention has been drawn to the role of mast cells. They contribute to the neuropathic component of pain, promote microglial activation, and sustain CS.143 Injury to nerve fibers induces mast-cell degranulation via neuropeptides. The release of histamine and nerve growth factor (NGF) facilitates remodeling of sensory endings and lowers their activation thresholds.147 These findings affirm the active role of immune cells in modulating neural excitability.
Persistent peripheral sensitization constitutes the initial stage in the development of central hyperexcitability. Chronic peripheral afferent drive leads to excessive neurotransmitter release at dorsal horn synapses, producing hyperactivity of second-order neurons and secondary activation of microglia.129,143 This establishes a self-perpetuating pathological loop: peripheral inflammation initiates sensitization that is transmitted to central structures, while CS amplifies responses to peripheral inputs.
Accordingly, pain in OA results from an integrated interplay between peripheral and central sensitization. Persistent inflammatory changes generate a stream of nociceptive signals that maintain neuroplastic processes within the CNS, whereas CS heightens sensitivity to these inputs. This comprehensive view of pathogenesis opens therapeutic avenues aimed not only at reducing inflammation but also at modulating neuronal and glial mechanisms. Contemporary strategies include NGF blockade, regulation of mast-cell and microglial function, and restoration of the balance between excitatory and inhibitory processes in the CNS.142
Immunoinflammatory and Neuroimmune Pathways of Peripheral and Central Sensitization in the Pathogenesis of Chronic Pain in Rheumatoid Arthritis
In the early stages of RA, morphological changes develop rapidly, affecting the vast majority of patients within the first year of disease: bone erosions, joint-space narrowing, and periarticular osteoporosis. Persistent immunoinflammatory activity of the synovial membrane against bone tissue drives irreversible structural damage that sustains chronic pain and promotes a shift toward a nociplastic phenotype. Notably, destruction of cartilage and the subchondral plate not only generates peripheral nociceptive inputs but also creates conditions for long-lasting neuroplastic changes within somatosensory pathways, which partly explains the dissociation between arthritis activity and pain intensity observed in some patients.148,149
There is no doubt that inflammation plays a key role in the generation of pain in RA.150 However, contemporary clinical observations and multicenter studies reveal a substantial discordance between physician-assessed inflammation measured by standardized indices and patients’ self-reported pain intensity. Specifically, in 64% of patients, pain flares did not coincide temporally with increases in the Disease Activity Score based on 28 joints (DAS28), whereas in 60% the converse occurred increases in DAS28 were not accompanied by marked intensification of pain.151 This underscores the complexity of the pathophysiological mechanisms underpinning pain in RA and highlights the need to move beyond the traditional “inflammation equals pain” paradigm.
During acute synovitis, the relationship between pain intensity and the severity of inflammation in affected joints is more evident and statistically robust. Outside periods of active inflammation, particularly preceding clinical flares and after successful symptom control with anti-inflammatory therapy, this association substantially weakens or disappears altogether.152–156 A key corroborating observation is the prevalence of clinically significant pain among patients who meet DAS28 remission criteria: nearly 11.9% continue to experience persistent pain despite the absence of objective signs of active inflammation. Thus, even under DAS28-defined remission, immunoinflammatory activity shows no statistically significant association with pain severity.157 This indicates the presence of additional, non-inflammatory nociceptive and nociplastic mechanisms that sustain pain in RA.
Contemporary rheumatology increasingly recognizes that non-inflammatory mechanisms including CS, dysfunction of nociceptive pathways, and psychological/affective factors may be predominant drivers of chronic pain in a subset of patients with RA. In this group, escalating disease-modifying antirheumatic drug (DMARD) therapy solely to suppress inflammation may be not only ineffective but potentially harmful. Patients are exposed to unwarranted risks of adverse effects, treatment-related complications, and diminished quality of life without meaningful analgesic benefit.150,158,159 Such scenarios lead to unjustified changes in therapeutic strategy and foster a perception of treatment failure, which in turn undermines adherence.
In RA, peripheral sensitization is initiated by close interactions between immune cells and primary afferent Aδ and C fibers under the influence of inflammatory mediators. Key factors include TNF-α, IL-1β, IL-6, IL-17, various chemokines, NGF and PGs.14 Some mediators are produced by resident and infiltrating immune cells within the synovium; others arrive via the systemic circulation (notably bradykinin). And additional signals arise secondarily from cellular injury and tissue acidosis (H⁺ ions).160 Binding of these mediators to nociceptor receptors activates intracellular phosphorylation cascades, modulates the expression and function of ion channels, and, at the membrane level, produces a sustained lowering of excitation thresholds.161
The changes are especially noticeable in the Nav group of sodium channels and TRP channels. Under inflammatory signaling, sodium channels become hyperexcitable, facilitating action potential generation, while TRP receptors, especially TRPV1 and TRPA1 (transient receptor potential ankyrin 1), develop hypersensitivity to thermal, chemical, and mechanical stimuli.162 An important modulatory element is the mast cell, which degranulates under synovial inflammation with the release of histamine and serotonin.163 Engagement of H1 and H2 histamine receptors on nociceptors upregulates Nav1.8 expression and deepens sensory-fiber hyperexcitability, lowering thresholds to noxious inputs. The combined effect of these mediators, together with a local pH shift toward acidosis, acts synergistically: primary afferent activity rises and firing thresholds fall, which clinically corresponds to peripheral hyperalgesia.148
Subsequently, the repetitiveness and intensity of peripheral impulses induce neuroplastic shifts at the level of the dorsal horns of the spinal cord, thalamocortical, and associative networks, establishing CS. As detailed in integrative reviews, sustained nociceptive afferentation is accompanied by increased release of glutamate, SP and CGRP, together with diminished efficacy of inhibitory mechanisms culminating in persistent hyperexcitability of second-order neurons.164,165 Clinically, this manifests as spatial and temporal summation of pain, allodynia, altered pain-thresholds, and a reduced response to anti-inflammatory therapy alone.
A distinct layer of pain mechanisms involves neuroinflammation - activation of glial cells with subsequent release of proinflammatory mediators within the CNS.166 Transmission of the peripheral inflammatory signal into the CNS occurs both via mediator permeation across the blood–brain barrier and via immune signaling that activates resident myeloid cells, chiefly microglia and perivascular macrophages.167 Under sustained nociceptive drive, spinal microglia and astrocytes enter a reactive state and produce IL-6 and IL-1β, which directly sensitize nociceptive pathways and maintain chronic pain.168,169 At the brain level, glial reactivity in the prefrontal cortex, primary somatosensory cortex, and anterior cingulate cortex is accompanied by dysregulation of excitatory glutamatergic and inhibitory GABA-ergic influences, creating a persistent imbalance of network activity that amplifies pain perception.148,168,170
In clinical practice, increasing attention is paid not only to nociceptive and nociplastic mechanisms but also to the possible involvement of a neuropathic pain component in RA. This is especially relevant in chronic courses marked by refractoriness to DMARD therapy or inadequate inflammatory control.171,172 Such patients develop a difficult-to-treat phenotype characterized by persistent pain that does not always correlate with inflammatory activity and requires a comprehensive analysis of the mechanisms sustaining it.
A review of several studies173–176 indicates that the proportion of RA patients with neuropathic pain features is substantial ranging from 17% to 36%, depending on the assessment instruments. Although methodological differences account for variability, the trend is consistent: neuropathic pain can emerge even when inflammation is controlled. This underscores the need for a comprehensive approach to pain assessment in RA, since conventional anti-inflammatory therapy is often insufficient under these circumstances.148
The cornerstone of RA pain management is the treat-to-target (T2T) strategy, aimed at achieving clinical remission and preventing structural damage and disability. Clinical examination remains paramount, although overlapping joint pain symptoms, stiffness, and fluctuating manifestations can complicate diagnosis.177
Pain as a Multifactorial Symptom in Psoriatic Arthritis
PsA is a heterogeneous, chronic, immune-mediated disease that together with RA belongs to the group of inflammatory arthritides and is characterized by painful articular and periarticular inflammation of the musculoskeletal system.178 Its manifestations include peripheral arthritis involving both small and large joints; dactylitis with typical spindle-shaped swelling of the digits; enthesitis reflecting inflammation at tendon and ligament insertions; and axial disease with spinal pain and morning stiffness.179,180 The clinical picture ranges from predominantly asymmetric oligoarthritis to erosive polyarticular forms with progressive structural damage, resulting in reduced functional status and quality of life. In addition to musculoskeletal symptoms, a subset of patients exhibit significant cutaneous and nail psoriasis; in some series, up to 30% present with concomitant skin or nail involvement.181 Nail psoriasis frequently coexists with distal interphalangeal joint disease and enthesitis, which has additional diagnostic and prognostic value. Taken together, these features underscore the multisystem nature of PsA and justify early phenotypic stratification to guide optimal diagnostic and therapeutic strategies.
Recognition of PsA as a distinct nosological entity was a pivotal milestone in rheumatology. Historically, this occurred in 1964, when the American Rheumatism Association (now the American College of Rheumatology (ACR)) formally distinguished PsA from other chronic arthritides, placing it within the spectrum of spondyloarthropathies.180 The first detailed scientific definitions came from John M. Moll and Verna Wright of the University of Leeds (UK), who in 1973 proposed that the condition be regarded as “an inflammatory arthritis in patients with psoriasis, usually associated with absence of rheumatoid factor”.182 At the time, this definition was crucial for differentiating PsA from RA, with which it was often confused due to overlapping clinical features. The absence of rheumatoid factor was considered a decisive diagnostic criterion, facilitating more accurate clinical identification of affected patients.
However, the accumulation of clinical and laboratory data and the advent of new immunologic and imaging methods revealed the limitations of the original diagnostic approaches. This necessitated a revision of the criteria and the development of more reliable, universally applicable classification schemes capable of capturing the clinical heterogeneity of PsA, its variable course, and pathogenetic complexity. In 2006, the CASPAR (ClASsification criteria for Psoriatic ARthritis) criteria were introduced. However, they prioritize specificity over sensitivity and are less suited to frontline diagnosis. Patients frequently experience a prolonged diagnostic journey, with delays in disease recognition and timely referral to secondary care.179,183 In addition to EULAR and ACR treatment recommendations for PsA, the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA) published treatment guidelines in 2015, updated in 2021 following the emergence of new evidence and therapeutics.180
Despite advances in PsA therapy, many patients continue to suffer from pain. Inflammation and joint damage caused by PsA can activate pain pathways within the nervous system, leading to heightened pain sensitivity.184 Patients with PsA report pain as their top priority and major health concern, whereas clinicians may underestimate this symptom.185–188 Pain represents the principal burden in this population, adversely affecting quality of life and everyday functioning.189
Pain is the dominant and persistent symptom in PsA and correlates heterogeneously with standard measures of inflammatory activity. Investigating the underlying mechanisms responsible for this phenomenon will help clinicians understand the causes of persistent pain, interpret composite disease activity indices, optimize patient counseling, and select the most appropriate treatment strategies.185 Pain in inflammatory arthritis depends on joint inflammation, but it is also closely linked to a wide spectrum of psychological factors among which pain catastrophizing appears to play a decisive role.190 The concept of catastrophizing was first introduced by the American psychologist Albert Ellis (1913–2007)191 and later adapted by the “father” of cognitive therapy and cognitive-behavioral therapy, Aaron T. Beck (1921–2021),192 to describe a maladaptive cognitive style in patients with anxiety and depressive disorders. Catastrophizing amplifies negative cognitive and emotional reactions during real or anticipated painful situations.193–195
Although active PsA is predominantly characterized by inflammation-driven nociceptive pain, clinical observations suggest the coexistence of nociplastic mechanisms. However, to date, objective neurobiological evidence confirming this observation in PsA is lacking.178
In most cases, the pain syndrome in PsA has a nociceptive nature, driven by the activation of afferent nerve fibers in the inflamed synovial tissue. The inflammatory process accompanied by the release of cytokines, inflammatory mediators, and activation of the cellular infiltrate initiates the transmission of pain signals from the periphery to the central nervous system. However, clinical observations indicate that even after regression of inflammation, a significant proportion of patients continue to experience intense and persistent pain, which cannot be explained solely by synovitis activity. In such cases, pain acquires the characteristics of complex neuropathic pain, involving distinct pathophysiological mechanisms.196,197
Researchers pay particular attention to the phenomenon of CS. This process develops as a result of repeated stimulation of peripheral nociceptors, leading to increased excitability of neuronal membranes and dysfunction of central pain control mechanisms.198,199 Consequently, in patients with PsA, pain may persist even in the absence of active inflammation, complicating the clinical picture and necessitating a multifactorial approach to diagnosis and therapy.
The clinical manifestations of CS in PsA, as in RA, include hyperalgesia and allodynia. These phenomena may mimic the clinical picture of FM, which is frequently observed as a comorbid condition in PsA patients. Despite the similarity in symptoms, it is important to emphasize that pain in FM is classified as nociplastic, whereas in PsA it most often involves a neuropathic component.200 This distinction is crucial for the appropriate selection of diagnostic and therapeutic strategies.
Evidence from other rheumatic diseases, particularly RA, shows that the phenomenon of CS is not limited to patients with comorbid FM. Several studies confirm that even in the absence of FM, RA patients may exhibit signs of pathological central pain processing.98 This provides grounds to assume that similar mechanisms may also occur in PsA, regardless of the presence or absence of comorbid FM.
Despite this, the current evidence base on the mechanisms of pain generation in PsA remains limited. Only a small number of studies have specifically investigated the mechanisms underlying pain in this condition and their impact on disease activity assessment. Most of these works emphasize that comorbid FM is the principal driver of central sensitization in PsA.201–203 By contrast, evidence supporting other mechanisms that could directly account for neuropathic pain in patients with a chronic inflammatory process is currently insufficient.
At the same time, in a broader clinical context, numerous interrelationships are observed between neuropathic pain and other pathological conditions that are not confined to rheumatic diseases. In patients with chronic pain, associations have been identified between neuropathic pain and affective disorders, including depression and anxiety, as well as between neuropathic pain and sleep disturbances.204 Moreover, several studies have described a link between neuropathic pain and obesity-related metabolic processes, underscoring the systemic nature of alterations in pain-sensitivity regulation.205 These findings indicate that neuropathic pain in PsA should be considered within a multidimensional biopsychosocial framework that accounts for inflammatory, neuropsychological, and metabolic factors.196
Thus, pain in PsA has a multifactorial origin: it is predominantly nociceptive due to synovial inflammation, yet it frequently persists after inflammatory regression, acquiring neuropathic or nociplastic features. CS with manifestations of hyperalgesia and allodynia, comorbid FM, psych emotional disorders, and metabolic factors amplify its intensity and promote chronification. Accordingly, pain in PsA extends beyond a simple inflammatory response and requires a comprehensive biopsychosocial approach to diagnosis and treatment.
Fibromyalgia in Combination with Rheumatoid Arthritis, Psoriatic Arthritis, and Osteoarthritis: Pathogenetic Interrelationships and Therapeutic Strategies
FM rarely occurs in isolation, as its clinical manifestations frequently co-exist with a broad spectrum of other medical and psychiatric disorders.206 This multifactorial interconnectedness is explained by the syndrome’s complex pathophysiology, which integrates immune, neurobiological, and psychosocial factors.207 In a substantial proportion of patients, FM develops against the background of rheumatic diseases accompanied by systemic inflammation, for example, RA or systemic lupus erythematosus. Comorbidity with psychiatric disorders is also typical (depression, anxiety disorders, and post-traumatic stress disorder (PTSD)) which substantially enhance pain perception and exacerbate cognitive and affective symptoms. Chronic fatigue syndrome occupies an important place within the spectrum of co-existing conditions, presenting with reduced endurance, cognitive impairment, and prolonged exhaustion even after minimal exertion. Additionally, affected patients often have pronounced sleep disturbances that not only diminish quality of life but also potentiate the development of CS, regarded as one of the key mechanisms sustaining chronic pain in FM.208 Consequently, clinical management should focus not only on reducing pain intensity but also on correcting comorbid conditions, since only a comprehensive approach can improve overall well-being and increase functional capacity.
Contemporary guidelines emphasize the importance of a multidisciplinary approach that combines pharmacologic therapies, cognitive-behavioral therapy, physical rehabilitation, and patient education programs. This integrated strategy can provide long-term stabilization and improve social adaptation in patients with FM. Particular attention is drawn to the close relationship between FM and obesity, which carries both clinical and pathogenetic significance. Excess body weight has been shown to correlate with an increased risk of FM and to aggravate its course, leading to more severe pain, worsening fatigue, and higher rates of affective disorders.206,209
The comorbidity of RA with FM remains a pressing concern for both clinicians and researchers, as it substantially influences disease assessment and therapeutic decision-making. Patients with RA exhibit a markedly higher prevalence of multimorbidity, which, according to epidemiologic studies, ranges from 31% to 86%, whereas among individuals without RA this figure is 18–71%.210 Across epidemiologic observations, the prevalence of FM in patients with RA varies between 4.9% and 52.4%, which markedly exceeds the prevalence in the general population, reported at only 1–5%.148,211,212
Clinicians should maintain a high index of suspicion for FM, particularly when patients report severe pain, fatigue, and other symptoms indicative of CNS dysfunction that are discordant with physical examination findings and laboratory parameters.177 Timely identification of patients with comorbid FM and implementation of management in line with contemporary European Alliance of Associations for Rheumatology (EULAR) recommendations could substantially reduce the risk of unwarranted intensification of antirheumatic therapy and mitigate the problem of apparent refractoriness.213
Of particular concern is that patients with RA and FM rarely achieve remission or even low disease activity according to composite indices.214,215 This is explained by the fact that, in such patients, a substantial component of pain is of non-inflammatory (non-immune-mediated) origin; however, the disease activity indices widely used in clinical practice do not always adequately distinguish inflammation-driven pain from pain related to CS. Consequently, patients with comorbid FM more often exhibit a lack of treatment response, leading to repeated changes of therapeutic regimens and escalation of treatment intensity, including the use of biologic DMARDs, which is not always justified.216 Thus, the clinician’s task is not only to control inflammation in RA but also to account for non-inflammatory pain mechanisms, which substantially complicates the attainment of therapeutic targets. In such cases, it is crucial to incorporate routine screening for FM into the comprehensive assessment of the patient’s condition, especially when remission cannot be achieved despite adequate anti-inflammatory therapy.
In clinical practice, FM has been shown to occur markedly more often among patients with PsA than in the general population. Across studies, the prevalence of FM in PsA spans a wide range – from 9.3% to 38.3%,92,178,217–221 whereas among individuals without inflammatory rheumatic diseases it is only about 1.78%.173 This indicates the presence of specific pathogenetic and clinical overlaps between these two conditions, which considerably complicates diagnosis and influences treatment strategies.
One of the key challenges in managing such patients is the difficulty of clearly distinguishing between pain driven by active inflammation and nociplastic pain, which is characteristic of FM. PsA is often accompanied by widespread, poorly localized pain that is closely linked to enthesitis. This pain phenotype tends to mimic the clinical picture of FM, as patients report diffuse pain and generalized tenderness similar to the symptoms of classic FM.178,222
Taken together, these observations indicate that FM should not be viewed solely as an isolated comorbidity but rather as a nociplastic pain phenotype capable of modifying symptom expression, disease activity assessment, and therapeutic decision-making across inflammatory and degenerative joint diseases. While the coexistence of FM with RA has been most extensively studied, accumulating evidence demonstrates similar clinical and pathogenetic overlaps in PsA and OA. In these conditions, widespread pain, fatigue, and hypersensitivity may persist despite adequate control of inflammation or limited structural damage, reflecting central sensitization–driven nociplastic mechanisms rather than ongoing peripheral pathology. Failure to recognize this overlap may lead to misinterpretation of disease activity, apparent treatment refractoriness, and unjustified escalation of anti-inflammatory therapy.
Accordingly, a cross-disease perspective is essential for distinguishing inflammation-driven pain from nociplastic pain and for guiding mechanism-based management strategies. To clarify the clinical relevance of FM comorbidity across major rheumatic conditions, Table 6 summarizes its impact on disease assessment, typical sources of diagnostic distortion, and practical considerations for clinical practice and research reporting.212,223–225
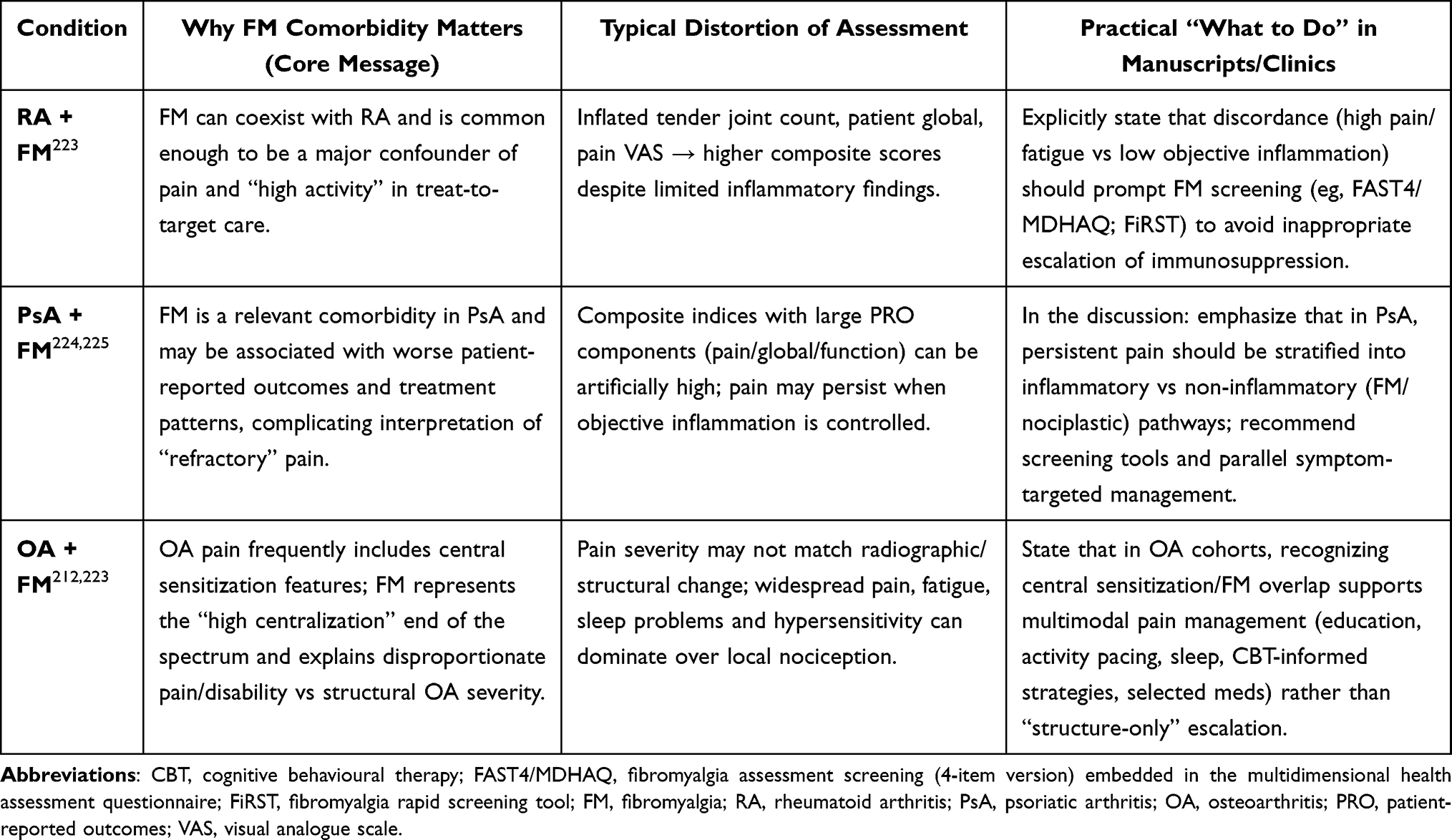 |
Table 6 Fibromyalgia Comorbidity Across Four Conditions (RA, PsA, OA, and FM) |
An additional complication is that pain, as one of the key components of subjective scales and summary indicators of disease activity (eg, DAPSA, PASDAS), can significantly increase their values in patients with PsA and concomitant FM. In such cases, higher disease activity indices do not necessarily reflect the true intensity of the inflammatory process and are often the result of a predominance of nociplastic pain mechanisms. This, in turn, creates a risk of misinterpreting the clinical situation and unjustifiably escalating anti-inflammatory therapy, which fails to deliver the expected benefit.178,222
It is also important to emphasize that diagnostic difficulties may arise in clinical practice due to potential misinterpretation of cases in which patients with primary FM also have symptoms of cutaneous psoriasis. In such individuals, a false impression of PsA may be created because the two conditions share substantial similarity in clinical features.202,226 This is particularly relevant in the context of pain, since patients with a neuropathic pain component and patients with FM often describe sensory experiences similar in quality, including burning and tingling, as well as the development of allodynia, where even minimal touch or pressure elicits pronounced pain.227
Another challenge is the lack of clear stratification of such patients in most studies: if patients with PsA and concomitant FM are not analyzed as a separate subgroup or excluded from analyses, it becomes impossible to adequately assess the contribution of FM to the development of neuropathic pain. The question remains unresolved as to whether the combination of these two conditions in a single patient may have an additive effect, leading to intensification of neuropathic pain and refractoriness to standard anti-inflammatory interventions. This scenario is of particular clinical importance, as it may affect both therapeutic outcomes and the interpretation of disease activity indices. Accordingly, accurate identification of patients with concomitant PsA and FM should become a priority in research and clinical practice, as it can elucidate the true nature of pain and optimize the selection of therapeutic strategies.202
Thus, the coexistence of PsA and FM creates a complex clinical phenotype in which accurate identification of the pain source becomes critical for selecting an appropriate therapeutic strategy. This underscores the need for an integrated approach that incorporates both precise tools for assessing inflammation and consideration of the neuropsychological dimensions of pain.
An equally relevant clinical problem is the combination of RA with OA, which also exerts a substantial impact on disease course and the patient’s pain perception. Comorbidity of inflammatory and degenerative joint diseases is relatively common, particularly in older patients with RA.228 In such cases, pain in weight-bearing joints (knees, hips, ankles) is more often related to mechanical factors rather than active immune-mediated inflammation. This constitutes a distinct pathogenetic mechanism of chronic pain that should be evaluated independently of synovitis. Accordingly, management of comorbid OA should be considered a separate avenue, encompassing both non-pharmacological interventions and the use of specific symptom-modifying agents.229
Clinical studies confirm that in patients with concomitant RA and OA, the proportion of chronic refractory pain is substantially higher than in those with RA alone (59% vs. 29%), even in the absence of objective signs of inflammation.230 This indicates that mechanical factors and degenerative changes play a role in shaping the pain phenotype no less important than the activity of the immune-inflammatory process. Studies have also shown that late-onset RA is associated with an increased risk of joint damage driven by pre-existing degenerative changes characteristic of OA. Therefore, differential diagnosis of the mechanical versus inflammatory components of pain is crucial for determining the optimal therapeutic strategy.
Particular interest lies in the findings of systematic reviews, including the work by Beswick A.D. et al,231 which showed that even after large-joint arthroplasty a substantial proportion of patients report unsatisfactory long-term pain outcomes: 9% after total hip arthroplasty and 20% after total knee arthroplasty This indicates that pain in OA cannot be explained solely by structural joint damage. An accumulating body of evidence supports CS as the predominant pathogenetic mechanism in a considerable subset of patients, shaping a distinct chronic pain phenotype.232 This mechanism accounts for rheumatoid-like pain in patients without clear signs of progressive joint damage. Recognizing the role of CS opens avenues for multimodal therapeutic strategies that include not only anti-inflammatory measures but also neuromodulatory approaches aimed at reducing pathological excitability within central pain pathways.
Given the above data, it becomes evident that FM constitutes a complex clinical phenotype in which inflammatory, mechanical, and centrally mediated pain mechanisms converge. Of particular interest is its interaction with RA and OA, as these combinations most clearly manifest the phenomenon of chronic refractory pain that does not respond to conventional anti-inflammatory therapy. CS, which underlies the nociplastic component, not only complicates differentiation between inflammatory and mechanical pain but also substantially shifts therapeutic priorities. Accordingly, optimal management of patients with FM requires a reorientation away from standard anti-inflammatory strategies toward methods capable of modulating CNS function. In this context, the contemporary approach to FM treatment is shaped by the use of agents that act on neurotransmitter systems, alongside the integration of pharmacological and non-pharmacological therapies.
Treatment of FM has traditionally focused primarily on the CNS, given that the key mechanisms driving symptomatology are linked to disrupted central nociceptive modulation.233 The most studied and widely used agents are antidepressants and anticonvulsants which, by modulating neurotransmitter systems, can partially reduce pain intensity and improve overall patient status (Table 7). However, accumulated clinical experience and multicenter study results indicate that even these medications do not invariably provide sufficient clinical efficacy, and their effects may vary substantially according to individual patient characteristics.
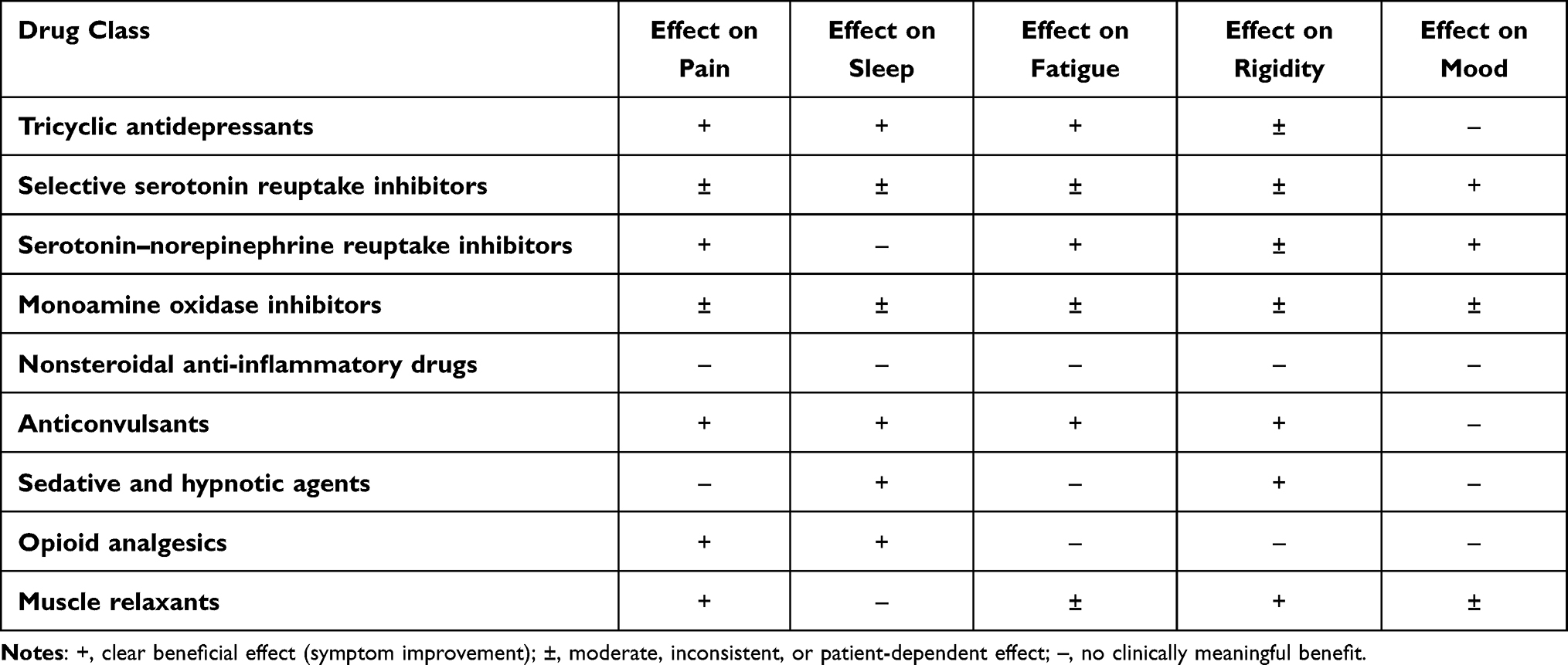 |
Table 7 Pharmacological Agents Used in the Treatment of Various Symptoms of fibromyalgia233 |
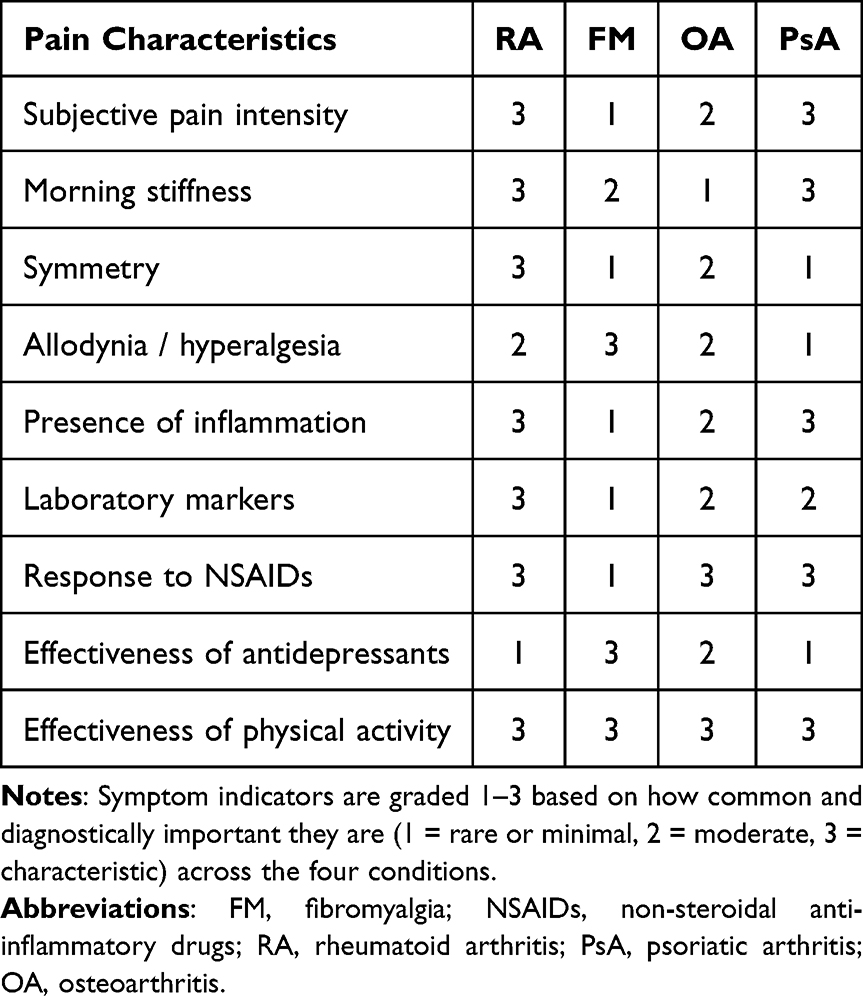 |
Table 8 Comparative Matrix of Pain Characteristics in Rheumatoid Arthritis, Psoriatic Arthritis, Fibromyalgia, and Osteoarthritis |
The use of analgesics and nonsteroidal anti-inflammatory drugs (NSAIDs) in FM has proven to be of limited effectiveness, supporting the absence of a primary inflammatory component in the syndrome’s pathogenesis. Tricyclic antidepressants, despite some ability to reduce pain and improve sleep, provide only limited therapeutic benefit, particularly with long-term use. Greater attention has shifted to more contemporary agents, notably the serotonin–norepinephrine reuptake inhibitors duloxetine and milnacipran, as well as the anticonvulsant pregabalin, which targets neuronal voltage-gated calcium channels. These agents have shown promising results in reducing pain, alleviating fatigue, and improving quality of life.234
Despite some progress, it should be noted that the overall effectiveness of pharmacologic approaches in the treatment of FM remains relatively modest. Recent data indicate the absence of a radical breakthrough in this field: newly marketed drugs largely do not possess mechanisms of action fundamentally different from existing agents and therefore do not provide a substantial improvement in treatment efficacy. At the same time, interest is growing in combined therapeutic strategies that integrate pharmacologic methods with cognitive-behavioral therapy, physical exercise, and other non-pharmacological interventions aimed at modifying central mechanisms of pain sensitization. This underscores the need for a multidisciplinary approach to FM management that extends beyond pharmacotherapy alone.233
A comparative analysis of therapeutic outcomes across FM, OA, RA, and PsA demonstrates substantial divergence in treatment targets, expected results, and determinants of refractoriness. In RA and PsA, early initiation of DMARDs, including biologic and targeted synthetic agents, enables achievement of remission or low disease activity in approximately 40–60% of patients under treat-to-target strategies, with significant inhibition of radiographic progression and improvement in functional indices.235,236
Structural control of inflammation is therefore an attainable goal in a substantial proportion of cases. However, persistent pain despite adequate inflammatory control occurs in approximately 20–30% of patients and is increasingly attributed to CS, which does not respond sufficiently to further immunosuppression.237
In contrast, OA therapy remains predominantly symptomatic. NSAIDs and intra-articular interventions improve pain and function but do not modify structural progression. Even after total joint arthroplasty, persistent postoperative pain is reported in 9–20% of patients, indicating that structural correction alone does not eliminate centrally mediated pain mechanisms. Subgroups exhibiting pressure hyperalgesia or widespread pain demonstrate poorer analgesic outcomes, supporting the contribution of nociplastic mechanisms.231,238
FM differs fundamentally in its therapeutic approach. Because inflammation is not the primary driver, anti-inflammatory agents demonstrate minimal efficacy. Centrally acting agents such as serotonin–norepinephrine reuptake inhibitors and pregabalin provide clinically meaningful pain reduction only in a subset of patients, typically with modest effect sizes. Exercise therapy and cognitive-behavioral interventions yield comparable or greater improvements in function and quality of life than pharmacotherapy alone, underscoring the central role of neuromodulation rather than immunomodulation.213
Thus, across all four conditions, the presence of CS predicts poorer response to peripheral anti-inflammatory or structural interventions and necessitates a multimodal strategy integrating neuromodulatory and behavioral approaches. Recognizing these mechanistic differences helps avoid ineffective escalation in predominantly nociplastic pain.
Summary of the Differential Characteristics of Pain in Rheumatoid Arthritis, Osteoarthritis, Psoriatic Arthritis, and Fibromyalgia
In the preceding sections, we systematized the key mechanisms underlying chronic pain (nociceptive, nociplastic, and neuropathic) and demonstrated their roles across different clinical entities. The overview matrix (Table 8) compares the most relevant features of pain in RA, PsA, OA, and FM. They were selected for their diagnostic relevance, frequency in guidelines and reviews, and ability to reflect differences between inflammatory, degenerative, and CS–related pain. The matrix and its scoring are based on analysis of over 250 sources included in this review, providing a clear overview of key clinical and pathophysiological distinctions across the four conditions. This approach not only characterizes pain intensity but also elucidates its pathogenetic origins, which is of crucial importance for designing personalized treatment strategies.239
The subjective intensity of pain is high across all four groups; however, the qualitative characteristics differ substantially. In RA, the leading driver remains inflammatory nociceptive pain, which produces prolonged morning stiffness, diurnal variability of symptoms, and marked improvement with mobilization.240 OA is characterized by a mechanical profile: pain intensifies toward the end of the day or after physical exertion, whereas morning stiffness is brief. FM exhibits a fundamentally different pattern diffuse, generalized pain without clear temporal dynamics closely linked to CS.241 In PsA, pain reaches high levels and is accompanied by morning stiffness lasting more than 30 minutes, frequently in combination with enthesitis, dactylitis, and axial manifestations.242 Thus, inflammation is key in RA and PsA, mechanical mechanisms dominate in OA, and a nociplastic process with prominent CS underlies FM.243
Spatial characteristics also differ markedly. RA typically presents with symmetric involvement of the small joints of the hands and feet, which is a classic diagnostic feature.244 OA is characterized by an asymmetric, regional distribution of pathology, predominantly affecting large joints. FM lacks precise localization – pain is widespread across at least four anatomical regions, consistent with ACR diagnostic criteria. PsA commonly presents as asymmetric oligoarthritis with a tendency toward polyarthritis in some patients, a combination of peripheral and axial disease, as well as enthesitis and characteristic cutaneous manifestations. This spatial pattern clearly distinguishes PsA from RA and aligns it more closely with systemic inflammatory arthropathies.
Sensory phenomena vary in importance. In FM, allodynia and hyperalgesia are key diagnostic features, reflecting the role of CS in symptom generation.245 In RA, similar manifestations are seen in some patients with residual pain persisting after suppression of inflammation. In OA, they are usually localized to the affected joint and do not become generalized.246 In PsA, sensory phenomena are not predominant; however, with long-standing disease, CS may develop, amplifying pain intensity and complicating therapeutic control.
The magnitude of objective inflammatory manifestations and laboratory markers also clearly differentiates these conditions. RA is characterized by maximal indices: synovitis, swelling, hyperemia, and elevations in ESR and CRP. In patients with OA, such changes are minimal and associated with secondary synovitis. In FM, laboratory and clinical markers of inflammation are absent, confirming the functional nature of the disturbances. PsA is marked by a high level of objective inflammation, including enthesitis and dactylitis; however, in a subset of patients, laboratory indices remain within normal limits, reflecting the heterogeneity of the disease course.
The response to pharmacologic interventions corroborates the pathogenetic mechanisms. RA is typified by high sensitivity to NSAIDs and glucocorticoids, as well as the effectiveness of DMARDs.247 OA responds well to NSAIDs but does not demonstrate a response to DMARDs or biologic therapy. FM shows little to no response to NSAIDs or glucocorticoids; instead, antidepressants, agents acting on NMDA and AMPA receptors, and cognitive-behavioral therapy are effective. In PsA, the effectiveness of NSAIDs is high, but DMARDs and biologic agents – targeting TNF-α, IL-17, and IL-23 – play the central role. The use of antidepressants in PsA has no meaningful impact on the disease course.
Non-pharmacological approaches, particularly physical activity, are an essential component of management across all four nosological entities. In RA, exercise reduces stiffness and improves functional status. In OA, it helps decrease mechanical loading, supports weight control, and enhances cartilage metabolism. In FM, physical activity exerts a pronounced modulatory effect on CS, improves neuroendocrine regulation, and lowers levels of SP, PGs, and CGRP. In PsA, exercise helps maintain mobility, attenuate inflammatory manifestations, and prevent disability, especially in axial disease.248
Thus, the comparative analysis indicates that despite similarly high subjective pain intensity, the pathogenetic mechanisms and clinical characteristics differ substantially. In RA and PsA, the dominant component is inflammatory; in OA, it is mechanical; whereas in FM, it is nociplastic with predominant CS. These differences determine the therapeutic approach: inflammatory arthropathies require DMARDs and biologic agents, OA calls for optimization of mechanical load and pain control, and FM necessitates multimodal therapy with an emphasis on CS modulation.249–253
Clinical Implications
Routine screening for central sensitization and comorbid fibromyalgia should be incorporated into the assessment of patients who report disproportionate or refractory pain. Pain phenotyping according to predominant mechanisms – inflammatory, mechanical, or nociplastic – should guide therapeutic decisions. Escalation of anti-inflammatory or immunosuppressive therapy should be avoided when nociplastic mechanisms are likely to predominate. Quantitative sensory testing and algometry may support a more objective evaluation of altered pain processing in clinical practice.
Future Research Directions
The development and validation of objective biomarkers for nociplastic pain represent a critical unmet need. Future studies should focus on integrating clinical indices with neuroimaging, neurophysiological, and molecular markers of central sensitization. Longitudinal research is required to clarify the transition from nociceptive to nociplastic pain and to identify predictors of treatment response.
Conclusions
Chronic pain in rheumatic diseases arises from different mechanisms. In RA, inflammatory nociceptive pain predominates and correlates with synovitis and laboratory markers. In OA, pain is mainly mechanical due to cartilage degeneration, while discordance between structural damage and symptoms is often explained by CS. PsA combines inflammatory and mechanical features (enthesitis, axial involvement). FM is characterized by dominant CS with diffuse pain, allodynia, hyperalgesia, and systemic symptoms; treatment should target the leading pain mechanism.
Allodynia and hyperalgesia are key markers of CS. In RA and OA they signal transition from peripheral nociception to persistent pain; in PsA they appear with chronicity; in FM they are diagnostically central. CS involves expanded receptive fields, reduced pain thresholds, and poor response to anti-inflammatory therapy, requiring pain phenotyping and targeting of central mechanisms.
FM comorbidity in RA, OA, and PsA complicates assessment by creating discordance between inflammation and pain severity, producing refractory phenotypes and potential overtreatment. Screening for FM is warranted when pain does not match laboratory or imaging findings.
Modern ACR-2016, FAS-2019, and AAPT criteria, together with QST, algometry, and pressure pain mapping, allow objective CS assessment. WPI and SSS improve phenotyping and help distinguish inflammatory from nociplastic pain components.
Management should be multimodal and personalized: FM responds best to duloxetine, milnacipran, pregabalin, CBT, and graded exercise; RA and PsA require anti-inflammatory therapy plus neuromodulatory strategies; OA management should emphasize load control and physical activity. Phenotype-based treatment improves outcomes and helps avoid unnecessary escalation of immunosuppression.
Date Sharing Statement
This is a narrative review without statistical analysis of the raw medical record data. If necessary, more data can be provided by the corresponding author upon reasonable request.
Ethics Statement
Ethical review and approval were not required for this submission.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was not supported by any external funds.
Disclosure
All the authors declare that they have no conflicts of interest in this work.
References
1. Tomašević-Todorović S, Spasojević T. Central Sensitization In Patients With Chronic Musculoskeletal Pain. Acta Clin Croat. 2023;62(Suppl 4):102–36. doi:10.20471/acc.2023.62.s4.15
2. Cayrol T, van den Broeke EN. Central sensitisation: causes, therapies, and terminology. Lancet Rheumatol. 2021;3(8):e548. doi:10.1016/S2665-9913(21)00176-4
3. Williams A, Kamper SJ, Wiggers JH, et al. Musculoskeletal conditions may increase the risk of chronic disease: a systematic review and meta-analysis of cohort studies. BMC Med. 2018;16(1):167. doi:10.1186/s12916-018-1151-2
4. Zhuang J, Mei H, Fang F, Ma X. What is new in classification, diagnosis and management of chronic musculoskeletal pain: a narrative review. Front Pain Res. 2022;3:937004. doi:10.3389/fpain.2022.937004
5. Garcia-Hernandez A, de la Coba P, Reyes DEl Paso GA. Central sensitisation pain and autonomic deficiencies in fibromyalgia. Clin Exp Rheumatol. 2022;40(6):1202–1209. doi:10.55563/clinexprheumatol/n280oi
6. Nijs J, George SZ, Clauw DJ, et al. Central sensitisation in chronic pain conditions: latest discoveries and their potential for precision medicine. Lancet Rheumatol. 2021;3(5):e383–e392. doi:10.1016/S2665-9913(21)00032-1
7. Bjurström MF, Blennow K, Zetterberg H, et al. Central nervous system monoaminergic activity in Hip osteoarthritis patients with disabling pain: associations with pain severity and central sensitization. Pain Rep. 2022;7(1):e988. doi:10.1097/PR9.0000000000000988
8. Ablin JN. Nociplastic pain: a critical paradigm for multidisciplinary recognition and management. J Clin Med. 2024;13(19):5741. doi:10.3390/jcm13195741
9. Kosek E, Clauw D, Nijs J, et al. Chronic nociplastic pain affecting the musculoskeletal system: clinical criteria and grading system. Pain. 2021;162(11):2629–2634. doi:10.1097/j.pain.0000000000002324
10. Nijs J, Malfliet A, Nishigami T. Nociplastic pain and central sensitization in patients with chronic pain conditions: a terminology update for clinicians. Braz J Phys Ther. 2023;27(3):100518. doi:10.1016/j.bjpt.2023.100518
11. Page MJ, McKenzie JE, Bossuyt PM, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. Br Med J. 2021;372:n71. doi:10.1136/bmj.n71
12. Abd-Elsayed A, Deer TR. Different types of pain. In: Pain. Cham: Springer;2019:15–16. doi:10.1007/978-3-319-99124-5_3
13. Atta AA, Ibrahim WW, Mohamed AF, Abdelkader NF. Microglia polarization in nociplastic pain: mechanisms and perspectives. Inflammopharmacology. 2023;31(3):1053–1067. doi:10.1007/s10787-023-01216-x
14. Yam MF, Loh YC, Tan CS, Khadijah Adam S, Abdul Manan N, Basir R. General pathways of pain sensation and the major neurotransmitters involved in pain regulation. Int J Mol Sci. 2018;19(8):2164. doi:10.3390/ijms19082164
15. Doody O, Bailey ME. Understanding pain physiology and its application to person with intellectual disability. J Intellect Disabil. 2019;23(1):5–18. doi:10.1177/1744629517708680
16. Ossipov MH. The perception and endogenous modulation of pain. Scientifica. 2012;2012:561761. doi:10.6064/2012/561761
17. Finnerup NB, Kuner R, Jensen TS. Neuropathic pain: from mechanisms to treatment. Physiol Rev. 2021;101(1):259–301. doi:10.1152/physrev.00045.2019
18. Abdelkader NF, Elbaset MA, Moustafa PE, Ibrahim SM. Empagliflozin mitigates type 2 diabetes-associated peripheral neuropathy: a glucose-independent effect through AMPK signaling. Arch Pharm Res. 2022;45(7):475–493. doi:10.1007/s12272-022-01391-5
19. Fitzcharles MA, Cohen SP, Clauw DJ, Littlejohn G, Usui C, Häuser W. Nociplastic pain: towards an understanding of prevalent pain conditions. Lancet. 2021;397(10289):2098–2110. doi:10.1016/S0140-6736(21)00392-5
20. Nijs J, Lahousse A, Kapreli E, et al. Nociplastic pain criteria or recognition of central sensitization? Pain phenotyping in the past, present and future. J Clin Med. 2021;10(15):3203. doi:10.3390/jcm10153203
21. Williams DA. Phenotypic features of central sensitization. J Appl Biobehav Res. 2018;23(2):e12135. doi:10.1111/jabr.12135
22. Rekatsina M, Paladini A, Piroli A, Zis P, Pergolizzi JV, Varrassi G. Pathophysiologic approach to pain therapy for complex pain entities: a narrative review. Pain Ther. 2020;9(1):7–21. doi:10.1007/s40122-019-00147-2. Erratum in: Pain Ther. 2020;9(1):23. doi: 10.1007/s40122-020-00152-w.
23. Cruz-Almeida Y, Forbes M, Cohen RC, et al. Brain gamma-aminobutyric acid, but not glutamine and glutamate levels are lower in older adults with chronic musculoskeletal pain: considerations by sex and brain location. Pain Rep. 2021;6(3):e952. doi:10.1097/PR9.0000000000000952
24. Bonezzi C, Fornasari D, Cricelli C, Magni A, Ventriglia G. Not all pain is created equal: basic definitions and diagnostic work-up. Pain Ther. 2020;9(Suppl 1):1–15. doi:10.1007/s40122-020-00217-w
25. Clauw DJ. From fibrositis to fibromyalgia to nociplastic pain: how rheumatology helped get us here and where do we go from here? Ann Rheum Dis. 2024;83(11):1421–1427. doi:10.1136/ard-2023-225327
26. Middleton SJ, Barry AM, Comini M, et al. Studying human nociceptors: from fundamentals to clinic. Brain. 2021;144(5):1312–1335. doi:10.1093/brain/awab048
27. Karcz M, Abd-Elsayed A, Chakravarthy K, et al. Pathophysiology of pain and mechanisms of neuromodulation: a narrative review (a Neuron Project). J Pain Res. 2024;17:3757–3790. doi:10.2147/JPR.S475351
28. Leone CM, Truini A. Understanding neuropathic pain: the role of neurophysiological tests in unveiling underlying mechanisms. J Anesth Analg Crit Care. 2024;4(1):77. doi:10.1186/s44158-024-00212-z
29. Tedeschi R, Giorgi F, Platano D, Berti L. Classifying low back pain through pain mechanisms: a scoping review for physiotherapy practice. J Clin Med. 2025;14(2):412. doi:10.3390/jcm14020412
30. Mitchell SAT, Majuta LA, Mantyh PW. New insights in understanding and treating bone fracture pain. Curr Osteoporos Rep. 2018;16(4):325–332. doi:10.1007/s11914-018-0446-8
31. Lim EJ, Son CG. Review of case definitions for myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). J Transl Med. 2020;18(1):289. doi:10.1186/s12967-020-02455-0
32. Mayer EA, Ryu HJ, Bhatt RR. The neurobiology of irritable bowel syndrome. Mol Psychiatry. 2023;28(4):1451–1465. doi:10.1038/s41380-023-01972-w
33. Jones EA, Asaad F, Patel N, Jain E, Abd-Elsayed A. Management of fibromyalgia: an update. Biomedicines. 2024;12(6):1266. doi:10.3390/biomedicines12061266
34. Kim J, Kim MK, Choi GJ, Shin HY, Kim BG, Kang H. Pharmacological and non-pharmacological strategies for preventing postherpetic neuralgia: a systematic review and network meta-analysis. Korean J Pain. 2021;34(4):509–533. doi:10.3344/kjp.2021.34.4.509
35. Tsubaki T, Kodaka E, Kitano Y, et al. Effective pain management of postherpetic neuralgia using a combination of analgesics and conservative measures. Cureus. 2024;16(11):e73132. doi:10.7759/cureus.73132
36. Perrot S, Cohen M, Barke A, Korwisi B, Rief W, Treede RD. IASP Taskforce for the Classification of Chronic Pain. The IASP classification of chronic pain for ICD-11: chronic secondary musculoskeletal pain. Pain. 2019;160(1):77–82. doi:10.1097/j.pain.0000000000001389
37. Jensen TS, Karlsson P, Gylfadottir SS, et al. Painful and non-painful diabetic neuropathy, diagnostic challenges and implications for future management. Brain. 2021;144(6):1632–1645. doi:10.1093/brain/awab079
38. Salaffi F, Farah S, Mariani C, Sarzi-Puttini P, Di Carlo M. Validity of the central sensitization inventory compared with traditional measures of disease severity in fibromyalgia. Pain Pract. 2022;22(8):702–710. doi:10.1111/papr.13162
39. Orr NL, Huang AJ, Liu YD, et al. Association of central sensitization inventory scores with pain outcomes after endometriosis surgery. JAMA Network Open. 2023;6(2):e230780. doi:10.1001/jamanetworkopen.2023.0780
40. Weaver KR, Griffioen MA, Klinedinst NJ, et al. Quantitative sensory testing across chronic pain conditions and use in special populations. Front Pain Res. 2022;2:779068. doi:10.3389/fpain.2021.779068
41. Ma CH, Tworek KB, Kung JY, et al. Systemic nonsteroidal anti-inflammatories for analgesia in postoperative critical care patients: a systematic review and meta-analysis of randomized control trials. Crit Care Explor. 2023;5(7):e0938. doi:10.1097/CCE.0000000000000938
42. Paladini A, Rawal N, Coca Martinez M, et al. Advances in the management of acute postsurgical pain: a review. Cureus. 2023;15(8):e42974. doi:10.7759/cureus.42974
43. Kuzu S, Canli M, Valamur I, Ozudogru A, Alkan H, Hartavi A. Effects of aerobic exercise in addition to core stabilisation exercises on functional capacity, physical performance and fall risk in geriatric individuals with chronic non-specific low back pain. BMC Sports Sci Med Rehab. 2025;17(1):218. doi:10.1186/s13102-025-01271-7
44. Migliorini F, Maffulli N, Eschweiler J, Baroncini A, Bell A, Colarossi G. Duloxetine for fibromyalgia syndrome: a systematic review and meta-analysis. J Orthop Surg Res. 2023;18(1):504. doi:10.1186/s13018-023-03995-z
45. Moore A, Bidonde J, Fisher E, et al. Effectiveness of pharmacological therapies for fibromyalgia syndrome in adults: an overview of Cochrane Reviews. Rheumatology. 2025;64(5):2385–2394. doi:10.1093/rheumatology/keae707
46. Lu R, Bausch AE, Kallenborn-Gerhardt W, et al. Slack channels expressed in sensory neurons control neuropathic pain in mice. J Neurosci. 2015;35(3):1125–1135. doi:10.1523/JNEUROSCI.2423-14.2015
47. Boyce-Rustay JM, Honore P, Jarvis MF. Animal models of acute and chronic inflammatory and nociceptive pain. Methods Mol Biol. 2010;617:41–55. doi:10.1007/978-1-60327-323-7_4
48. Ji RR, Nackley A, Huh Y, Terrando N, Maixner W. Neuroinflammation and central sensitization in chronic and widespread pain. Anesthesiology. 2018;129(2):343–366. doi:10.1097/ALN.0000000000002130
49. Clark AK, Yip PK, Grist J, et al. Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. Proc Natl Acad Sci USA. 2007;104(25):10655–10660. doi:10.1073/pnas.0610811104
50. Zhuang ZY, Kawasaki Y, Tan PH, Wen YR, Huang J, Ji RR. Role of the CX3CR1/p38 MAPK pathway in spinal microglia for the development of neuropathic pain following nerve injury-induced cleavage of fractalkine. Brain Behav Immun. 2007;21(5):642–651. doi:10.1016/j.bbi.2006.11.003
51. Zhang MD, Barde S, Yang T, et al. Orthopedic surgery modulates neuropeptides and BDNF expression at the spinal and hippocampal levels. Proc Natl Acad Sci USA. 2016;113(43):E6686–E6695. doi:10.1073/pnas.1614017113
52. Li WW, Guo TZ, Shi X, et al. Substance P spinal signaling induces glial activation and nociceptive sensitization after fracture. Neuroscience. 2015;310:73–90. doi:10.1016/j.neuroscience.2015.09.036
53. Yang Y, Li H, Li TT, et al. Delayed activation of spinal microglia contributes to the maintenance of bone cancer pain in female Wistar rats via P2X7 receptor and IL-18. J Neurosci. 2015;35(20):7950–7963. doi:10.1523/JNEUROSCI.5250-14.2015
54. Ji RR, Berta T, Nedergaard M. Glia and pain: is chronic pain a gliopathy? Pain. 2013;154(Suppl 1):S10–S28. doi:10.1016/j.pain.2013.06.022
55. Gold MS, Gebhart GF. Nociceptor sensitization in pain pathogenesis. Nat Med. 2010;16(11):1248–1257. doi:10.1038/nm.2235
56. Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139(2):267–284. doi:10.1016/j.cell.2009.09.028
57. Hucho T, Levine JD. Signaling pathways in sensitization: toward a nociceptor cell biology. Neuron. 2007;55(3):365–376. doi:10.1016/j.neuron.2007.07.008
58. Pan HL, Liu BL, Lin W, Zhang YQ. Modulation of Nav1.8 by lysophosphatidic acid in the induction of bone cancer pain. Neurosci Bull. 2016;32(5):445–454. doi:10.1007/s12264-016-0060-7
59. Hladkykh FV. Modern pharmacotherapeutic strategies in the treatment of migraine: from calcitonin gene-related peptide (CGRP) signaling pathways to novel therapeutic agents. Psychiatry Neurol Med Psychol. 2024;11(4):447–475. doi:10.26565/2312-5675-2024-26-10
60. Chiu IM, von Hehn CA, Woolf CJ. Neurogenic inflammation and the peripheral nervous system in host defense and immunopathology. Nat Neurosci. 2012;15(8):1063–1067. doi:10.1038/nn.3144
61. Wei T, Li WW, Guo TZ, et al. Post-junctional facilitation of Substance P signaling in a tibia fracture rat model of complex regional pain syndrome type I. Pain. 2009;144(3):278–286. doi:10.1016/j.pain.2009.04.020
62. Liu XJ, Liu T, Chen G, et al. TLR signaling adaptor protein MyD88 in primary sensory neurons contributes to persistent inflammatory and neuropathic pain and neuroinflammation. Sci Rep. 2016;6:28188. doi:10.1038/srep28188
63. Zhang H, Boyette-Davis JA, Kosturakis AK, et al. Induction of monocyte chemoattractant protein-1 (MCP-1) and its receptor CCR2 in primary sensory neurons contributes to paclitaxel-induced peripheral neuropathy. J Pain. 2013;14(10):1031–1044. doi:10.1016/j.jpain.2013.03.012
64. Gyorfi M, Rupp A, Abd-Elsayed A. Fibromyalgia pathophysiology. Biomedicines. 2022;10(12):3070. doi:10.3390/biomedicines10123070
65. Gowers WR. A Lecture on lumbago: its lessons and analogues: delivered at the national hospital for the paralysed and epileptic. Br Med J. 1904;1(2246):117–121. doi:10.1136/bmj.1.2246.117
66. Graham W. The fibrosits syndrome. Bull Rheum Dis. 1953;3(8):33–34.
67. Smythe HA, Moldofsky H. Two contributions to understanding of the “fibrositis” syndrome. Bull Rheum Dis. 1977–1978;28(1):928–931.
68. Wolfe F, Smythe HA, Yunus MB, et al. The American College of Rheumatology 1990 Criteria for the classification of fibromyalgia. Report of the Multicenter Criteria Committee. Arthritis Rheum. 1990;33(2):160–172. doi:10.1002/art.1780330203
69. Filipovic T, Filipović A, Nikolic D, et al. Fibromyalgia: understanding, diagnosis and modern approaches to treatment. J Clin Med. 2025;14(3):955. doi:10.3390/jcm14030955
70. Sluka KA, Clauw DJ. Neurobiology of fibromyalgia and chronic widespread pain. Neuroscience. 2016;338:114–129. doi:10.1016/j.neuroscience.2016.06.006
71. Hubbard CS, Lazaridou A, Cahalan CM, et al. Aberrant Salience? Brain Hyperactivation in Response to Pain Onset and Offset in Fibromyalgia. Arthritis Rheumatol. 2020;72(7):1203–1213. doi:10.1002/art.41220
72. Сeko M, Frangos E, Gracely J, et al. Default mode network changes in fibromyalgia patients are largely dependent on current clinical pain. Neuroimage. 2020;216:116877. doi:10.1016/j.neuroimage.2020.116877
73. Staud R, Boissoneault J, Lai S, et al. Spinal cord neural activity of patients with fibromyalgia and healthy controls during temporal summation of pain: an fMRI study. J Neurophysiol. 2021;126(3):946–956. doi:10.1152/jn.00276.2021
74. Fauchon C, Binvignat M, Berenbaum F, Conaghan PG, Peyron R, Sellam J. Going Inside Osteoarthritis-Related Pain Phenotyping (GO-PAIN) network members. Brain functional imaging contributions in osteoarthritis-related pain: a viewpoint. Osteoarthr Cartil Open. 2024;7(1):100554. doi:10.1016/j.ocarto.2024.100554
75. Soni A, Wanigasekera V, Mezue M, et al. Central Sensitization in Knee Osteoarthritis: relating Presurgical Brainstem Neuroimaging and PainDETECT-Based Patient Stratification to Arthroplasty Outcome. Arthritis Rheumatol. 2019;71(4):550–560. doi:10.1002/art.40749
76. Flodin P, Martinsen S, Altawil R, et al. Intrinsic Brain Connectivity in Chronic Pain: a Resting-State fMRI Study in Patients with Rheumatoid Arthritis. Front Hum Neurosci. 2016;10:107. doi:10.3389/fnhum.2016.00107
77. Sandström A, Ellerbrock I, Jensen KB, et al. Altered cerebral pain processing of noxious stimuli from inflamed joints in rheumatoid arthritis: an event-related fMRI study. Brain Behav Immun. 2019;81:272–279. doi:10.1016/j.bbi.2019.06.024
78. Salaffi F, Farah S, Bianchi B, Di Carlo M. Central Sensitization in Psoriatic Arthritis: relationship With Composite Measures of Disease Activity, Functional Disability, and Health-Related Quality of Life. J Rheumatol. 2024;51(2):144–149. doi:10.3899/jrheum.2023-0177
79. Hsieh PC, Tseng MT, Chao CC, et al. Imaging signatures of altered brain responses in small-fiber neuropathy: reduced functional connectivity of the limbic system after peripheral nerve degeneration. Pain. 2015;156(5):904–916. doi:10.1097/j.pain.0000000000000128
80. Inami C, Tanihira H, Kikuta S, et al. Visualization of brain activity in a neuropathic pain model using quantitative activity-dependent manganese magnetic resonance imaging. Front Neural Circuits. 2019;13:74. doi:10.3389/fncir.2019.00074
81. Goebel A, Krock E, Gentry C, et al. Passive transfer of fibromyalgia symptoms from patients to mice. J Clin Invest. 2021;131(13):e144201. doi:10.1172/JCI144201
82. Martínez-Lavín M. Centralized nociplastic pain causing fibromyalgia: an emperor with no cloths? Clin Rheumatol. 2022;41(12):3915–3917. doi:10.1007/s10067-022-06407-5
83. Ruschak I, Montesó-Curto P, Rosselló L, Aguilar Martín C, Sánchez-Montesó L, Toussaint L. Fibromyalgia syndrome pain in men and women: a scoping review. Healthcare. 2023;11(2):223. doi:10.3390/healthcare11020223
84. Jiao J, Cheng Z, Wang W, Zhao Y, Jiang Q. Demographic characteristics and clinical features of fibromyalgia in China: a cross-sectional study. Rheumatol Ther. 2021;8(2):817–831. doi:10.1007/s40744-021-00303-1. Erratum in: Rheumatol Ther. 2021;8(2):833. doi: 10.1007/s40744-021-00320-0.
85. Klute M, Laekeman M, Kuss K, et al. Cross-cultural adaptation and validation of the German Central Sensitization Inventory (CSI-GE). BMC Musculoskelet Disord. 2021;22(1):708. doi:10.1186/s12891-021-04481-5
86. Neblett R, Cohen H, Choi Y, et al. The Central Sensitization Inventory (CSI): establishing clinically significant values for identifying central sensitivity syndromes in an outpatient chronic pain sample. J Pain. 2013;14(5):438–445. doi:10.1016/j.jpain.2012.11.012
87. Alvarez MC, Albuquerque MLL, Neiva HP, et al. Differences between Portuguese and Brazilian Patients with Fibromyalgia Syndrome: exploring the Associations across Age, Time of Diagnosis, and Fatigue-Related Symptoms. Medicina. 2021;57(4):322. doi:10.3390/medicina57040322
88. Kelleher E, Kaplan CM, Kheirabadi D, et al. The number of central nervous system-driven symptoms predicts subsequent chronic primary pain: evidence from UK Biobank. Br J Anaesth. 2025;134(3):772–782. doi:10.1016/j.bja.2024.12.009
89. Clauw DJ. Fibromyalgia: a clinical review. JAMA. 2014;311(15):1547–1555. doi:10.1001/jama.2014.3266
90. Working Group Proyecto EPISER2016, Font Gayà T, Bordoy Ferrer C, Juan Mas A, et al. Prevalence of fibromyalgia and associated factors in Spain. Clin Exp Rheumatol. 2020;38(1):47–52.
91. Cabo-Meseguer A, Cerdá-Olmedo G, Trillo-Mata JL. Fibromyalgia: prevalence, epidemiologic profiles and economic costs. Med Clin. 2017;149(10):441–448. doi:10.1016/j.medcli.2017.06.008
92. Heidari F, Afshari M, Moosazadeh M. Prevalence of fibromyalgia in general population and patients, a systematic review and meta-analysis. Rheumatol Int. 2017;37(9):1527–1539. doi:10.1007/s00296-017-3725-2
93. Cuyul-Vásquez I, Araya-Quintanilla F, Gutiérrez-Espinoza H. Comment on Siracusa et al. Fibromyalgia: pathogenesis, Mechanisms, Diagnosis and Treatment Options Update. Int. J. Mol. Sci. 2021, 22, 3891. Int J Mol Sci. 2021;22(16):9075. doi:10.3390/ijms22169075
94. Salaffi F, Di Carlo M, Farah S, et al. Diagnosis of fibromyalgia: comparison of the 2011/2016 ACR and AAPT criteria and validation of the modified Fibromyalgia Assessment Status. Rheumatology. 2020;59(10):3042–3049. doi:10.1093/rheumatology/keaa061
95. Motaqi M, Ghanjal A. Trigger and tender points (definitions, similarities, differences, treatments). International Journal of Musculoskeletal Pain Prevention. 2020;5(4):393–395. doi:10.52547/ijmpp.5.4.393
96. Borg-Stein J, Stein J. Trigger points and tender points: one and the same? Does injection treatment help? Rheum Dis Clin North Am. 1996;22(2):305–322. doi:10.1016/s0889-857x(05)70274-x
97. Berwick R, Barker C, Goebel A. guideline development group. The diagnosis of fibromyalgia syndrome. Clin Med Lond. 2022;22(6):570–574. doi:10.7861/clinmed.2022-0402
98. Wolfe F, Michaud K, Busch RE, et al. Polysymptomatic distress in patients with rheumatoid arthritis: understanding disproportionate response and its spectrum. Arthritis Care Res. 2014;66(10):1465–1471. doi:10.1002/acr.22300
99. Salaffi F, Di Carlo M, Di Franco M, et al. Società Italiana di Reumatologia (SIR). Determining the PASS cut-off points for the FIQR, FASmod and PSD in patients with fibromyalgia: a registry-based study. Clin Exp Rheumatol. 2023;41(6):1275–1282. doi:10.55563/clinexprheumatol/on8j9a
100. Jurado-Priego LN, Cueto-Ureña C, Ramírez-Expósito MJ, Martínez-Martos JM. Fibromyalgia: a review of the pathophysiological mechanisms and multidisciplinary treatment strategies. Biomedicines. 2024;12(7):1543. doi:10.3390/biomedicines12071543
101. Salaffi F, Di Carlo M, Farah S, et al. Società Italiana di Reumatologia (SIR). The measurement of fibromyalgia severity: converting scores between the FIQR, the PSD and the FASmod. Clin Exp Rheumatol. 2023;41(6):1225–1229. doi:10.55563/clinexprheumatol/31gsnd
102. Devigili G, Di Stefano G, Donadio V, et al. Clinical criteria and diagnostic assessment of fibromyalgia: position statement of the Italian Society of Neurology-Neuropathic Pain Study Group. Neurol Sci. 2023;44(7):2561–2574. doi:10.1007/s10072-023-06836-3
103. Gemignani F. The diagnostic assessment of fibromyalgia and the role of small fiber pathology. Neurol Sci. 2024;45(4):1789–1790. doi:10.1007/s10072-023-07207-8
104. Marshall A, Elshafei M, Preston FG, et al. Small fibre pathology in fibromyalgia: a review. Pain Ther. 2025;14(2):461–478. doi:10.1007/s40122-024-00696-1
105. Mayer TG, Neblett R, Cohen H, et al. The development and psychometric validation of the central sensitization inventory. Pain Pract. 2012;12(4):276–285. doi:10.1111/j.1533-2500.2011.00493.x
106. Hochman JR, Davis AM, Elkayam J, Gagliese L, Hawker GA. Neuropathic pain symptoms on the modified painDETECT correlate with signs of central sensitization in knee osteoarthritis. Osteoarthritis Cartilage. 2013;21(9):1236–1242. doi:10.1016/j.joca.2013.06.023
107. Saitou M, Noda K, Matsushita T, Ukichi T, Kurosaka D. Central sensitisation features are associated with neuropathic pain-like symptoms in patients with longstanding rheumatoid arthritis: a cross-sectional study using the central sensitisation inventory. Clin Exp Rheumatol. 2022;40(5):980–987. doi:10.55563/clinexprheumatol/msy022
108. Adami G, Gerratana E, Atzeni F, et al. Is central sensitization an important determinant of functional disability in patients with chronic inflammatory arthritides? Ther Adv Musculoskelet Dis. 2021;13:1759720X21993252. doi:10.1177/1759720X21993252
109. Alburquerque-Sendín F, Madeleine P, Fernández-de-Las-Peñas C, Camargo PR, Salvini TF. Spotlight on topographical pressure pain sensitivity maps: a review. J Pain Res. 2018;11:215–225. doi:10.2147/JPR.S135769
110. de la Coba P, Montoro CI, Reyes DEl Paso GA, Galvez-Sánchez CM. Algometry for the assessment of central sensitisation to pain in fibromyalgia patients: a systematic review. Ann Med. 2022;54(1):1403–1422. doi:10.1080/07853890.2022.2075560
111. Waller R, Brown E, Lim J, et al. Pressure and cold pain threshold reference values in a pain-free older adult population. Br J Pain. 2024:20494637241276104. doi:10.1177/20494637241276104.
112. Gierthmühlen J, Enax-Krumova EK, Attal N, et al. Who is healthy? Aspects to consider when including healthy volunteers in QST--based studies-a consensus statement by the EUROPAIN and NEUROPAIN consortia. Pain. 2015;156(11):2203–2211. doi:10.1097/j.pain.0000000000000227
113. Waller R, Smith AJ, O’Sullivan PB, et al. Pressure and cold pain threshold reference values in a large, young adult, pain-free population. Scand J Pain. 2016;13:114–122. doi:10.1016/j.sjpain.2016.08.003
114. Waller R, Smith AJ, O’Sullivan PB, Slater H, Sterling M, Straker LM. Associations between musculoskeletal pain experience and pressure and cold pain sensitivity: a community-based cross-sectional study of young adults in the raine study. Clin J Pain. 2019;35(1):56–64. doi:10.1097/AJP.0000000000000650
115. Hafner J, Lee G, Joester J, et al. Thermal quantitative sensory testing: a study of 101 control subjects. J Clin Neurosci. 2015;22(3):588–591. doi:10.1016/j.jocn.2014.09.017
116. Arendt-Nielsen L, Morlion B, Perrot S, et al. Assessment and manifestation of central sensitisation across different chronic pain conditions. Eur J Pain. 2018;22(2):216–241. doi:10.1002/ejp.1140
117. Briggs AM, Page CJ, Shaw BR, et al. A model of care for osteoarthritis of the Hip and knee: development of a system-wide plan for the health sector in Victoria, Australia. Healthc Policy. 2018;14(2):47–58. doi:10.12927/hcpol.2018.25686
118. Mallick-Searle T, Sharma K, Toal P, Gutman A. Pain and function in chronic musculoskeletal pain-treating the whole person. J Multidiscip Healthc. 2021;14:335–347. doi:10.2147/JMDH.S288401
119. Balaguier R, Madeleine P, Vuillerme N. Is one trial sufficient to obtain excellent pressure pain threshold reliability in the low back of asymptomatic individuals? A test-retest study. PLoS One. 2016;11(8):e0160866. doi:10.1371/journal.pone.0160866
120. Balaguier R, Madeleine P, Vuillerme N. Intra-session absolute and relative reliability of pressure pain thresholds in the low back region of vine-workers: effect of the number of trials. BMC Musculoskelet Disord. 2016;17(1):350. doi:10.1186/s12891-016-1212-7
121. Binderup A, Arendt-Nielsen L, Madeleine P. Pressure pain threshold mapping: a new imaging modality of muscle sensitivity to pain. IEEExplore. 2008;2008:1–4. doi:10.1109/AISPC.2008.4460549
122. Binderup AT, Arendt-Nielsen L, Madeleine P. Cluster analysis of pressure pain threshold maps from the trapezius muscle. Comput Methods Biomech Biomed Engin. 2010;13(6):677–683. doi:10.1080/10255840903446979
123. Nie H, Kawczynski A, Madeleine P, Arendt-Nielsen L. Delayed onset muscle soreness in neck/shoulder muscles. Eur J Pain. 2005;9(6):653–660. doi:10.1016/j.ejpain.2004.12.009
124. Fernández-de-las-Peñas C, Madeleine P, Cuadrado ML, Ge HY, Arendt-Nielsen L, Pareja JA. Pressure pain sensitivity mapping of the temporalis muscle revealed bilateral pressure hyperalgesia in patients with strictly unilateral migraine. Cephalalgia. 2009;29(6):670–676. doi:10.1111/j.1468-2982.2008.01831.x
125. Tornero-Caballero MC, Salom-Moreno J, Cigarán-Méndez M, Morales-Cabezas M, Madeleine P, Fernández-de-Las-Peñas C. Muscle trigger points and pressure pain sensitivity maps of the feet in women with fibromyalgia syndrome. Pain Med. 2016;17(10):1923–1932. doi:10.1093/pm/pnw090
126. Cuadrado ML, Valle B, Fernández-de-las-Peñas C, et al. Pressure pain sensitivity of the scalp in patients with nummular headache: a cartographic study. Cephalalgia. 2010;30(2):200–206. doi:10.1111/j.1468-2982.2009.01895.x
127. Barón J, Ruiz M, Palacios-Ceña M, et al. Differences in topographical pressure pain sensitivity maps of the scalp between patients with migraine and healthy controls. Headache. 2017;57(2):226–235. doi:10.1111/head.12984
128. Domínguez-Martin MA, López-Ruiz F, Valenza G, Fernández-de-las-Peñas C, Madeleine P. Differences in pressure pain sensitivity of elite male soccer players on artificial turf and natural grass. Sports Tech. 2013;6(1):22–28.
129. Coaccioli S, Sarzi-Puttini P, Zis P, Rinonapoli G, Varrassi G. Osteoarthritis: new insight on its pathophysiology. J Clin Med. 2022;11(20):6013. doi:10.3390/jcm11206013
130. van den Bosch MHJ. Osteoarthritis year in review 2020: biology. Osteoarthritis Cartilage. 2021;29(2):143–150. doi:10.1016/j.joca.2020.10.006
131. Barreto G, Manninen M, Eklund K. Osteoarthritis and toll-like receptors: when innate immunity meets chondrocyte apoptosis. Biology. 2020;9(4):65. doi:10.3390/biology9040065
132. Zahan OM, Serban O, Gherman C, Fodor D. The evaluation of oxidative stress in osteoarthritis. Med Pharm Rep. 2020;93(1):12–22. doi:10.15386/mpr-1422
133. Jang S, Lee K, Ju JH. Recent updates of diagnosis, pathophysiology, and treatment on osteoarthritis of the knee. Int J Mol Sci. 2021;22(5):2619. doi:10.3390/ijms22052619
134. Kulkarni P, Martson A, Vidya R, Chitnavis S, Harsulkar A. Pathophysiological landscape of osteoarthritis. Adv Clin Chem. 2021;100:37–90. doi:10.1016/bs.acc.2020.04.002
135. Hladkykh FV, Liadova TI. Analgesic potential of cryoextracts of biological tissues and conditioned media of mesenchymal stem cells in the treatment of experimental autoimmune arthritis. Odesa Medl J. 2024;186(1):35–41. doi:10.32782/2226-2008-2024-1-6
136. Lluch E, Torres R, Nijs J, Van Oosterwijck J. Evidence for central sensitization in patients with osteoarthritis pain: a systematic literature review. Eur J Pain. 2014;18(10):1367–1375. doi:10.1002/j.1532-2149.2014.499.x
137. Arendt-Nielsen L, Nie H, Laursen MB, et al. Sensitization in patients with painful knee osteoarthritis. Pain. 2010;149(3):573–581. doi:10.1016/j.pain.2010.04.003
138. Mease PJ, Hanna S, Frakes EP, Altman RD. Pain mechanisms in osteoarthritis: understanding the role of central pain and current approaches to its treatment. J Rheumatol. 2011;38(8):1546–1551. doi:10.3899/jrheum.100759
139. Fu K, Robbins SR, McDougall JJ. Osteoarthritis: the genesis of pain. Rheumatology. 2018;57(suppl_4):iv43–iv50. doi:10.1093/rheumatology/kex419
140. Liu-Bryan R, Terkeltaub R. Emerging regulators of the inflammatory process in osteoarthritis. Nat Rev Rheumatol. 2015;11(1):35–44. doi:10.1038/nrrheum.2014.162
141. Schuelert N, McDougall JJ. Grading of monosodium iodoacetate-induced osteoarthritis reveals a concentration-dependent sensitization of nociceptors in the knee joint of the rat. Neurosci Lett. 2009;465(2):184–188. doi:10.1016/j.neulet.2009.08.063
142. Garland EL. Pain processing in the human nervous system: a selective review of nociceptive and biobehavioral pathways. Prim Care. 2012;39(3):561–571. doi:10.1016/j.pop.2012.06.013
143. Fusco M, Skaper SD, Coaccioli S, et al. Degenerative joint diseases and neuroinflammation. Pain Pract. 2017;17(4):522–532. doi:10.1111/papr.12551
144. Geraghty T, Winter DR, Miller RJ, Miller RE, Malfait AM. Neuroimmune interactions and osteoarthritis pain: focus on macrophages. Pain Rep. 2021;6(1):e892. doi:10.1097/PR9.0000000000000892
145. Arendt-Nielsen L. Pain sensitisation in osteoarthritis. Clin Exp Rheumatol. 2017;35(5):68–74.
146. Imamura M, Imamura ST, Kaziyama HH, et al. Impact of nervous system hyperalgesia on pain, disability, and quality of life in patients with knee osteoarthritis: a controlled analysis. Arthritis Rheum. 2008;59(10):1424–1431. doi:10.1002/art.24120
147. Nieto FR, Clark AK, Grist J, Chapman V, Malcangio M. Calcitonin gene-related peptide-expressing sensory neurons and spinal microglial reactivity contribute to pain states in collagen-induced arthritis. Arthritis Rheumatol. 2015;67(6):1668–1677. doi:10.1002/art.39082
148. Motyl G, Krupka WM, Maślińska M. The problem of residual pain in the assessment of rheumatoid arthritis activity. Reumatologia. 2024;62(3):176–186. doi:10.5114/reum/189779
149. Hladkykh FV. Current understanding of the immunological basis of rheumatoid arthritis: from post-translational modification of proteins to the use of disease-modifying antirheumatic drugs. East Ukrainian Med J. 2023;11(4):326–336. doi:10.21272/eumj.2023
150. Cao Y, Fan D, Yin Y. Pain mechanism in rheumatoid arthritis: from cytokines to central sensitization. Mediators Inflamm. 2020;2020:2076328. doi:10.1155/2020/2076328
151. McWilliams DF, Rahman S, James RJE, et al. Disease activity flares and pain flares in an early rheumatoid arthritis inception cohort; characteristics, antecedents and sequelae. BMC Rheumatol. 2019;3:49. doi:10.1186/s41927-019-0100-9
152. Ebbinghaus M, Müller S, Segond von Banchet G, et al. Contribution of inflammation and bone destruction to pain in arthritis: a study in murine glucose-6-phosphate isomerase-induced arthritis. Arthritis Rheumatol. 2019;71(12):2016–2026. doi:10.1002/art.41051
153. McWilliams DF, Walsh DA. Pain mechanisms in rheumatoid arthritis. Clin Exp Rheumatol. 2017;35(5):94–101.
154. Malcangio M. Translational value of preclinical models for rheumatoid arthritis pain. Pain. 2020;161(7):1399–1400. doi:10.1097/j.pain.0000000000001851
155. Karlsson ML, Elkan AC, Hafström I. Widespread pain and pain intensity in patients with early rheumatoid arthritis. A cross-sectional comparison between smokers and non-smokers. Nurs Open. 2019;6(3):942–947. doi:10.1002/nop2.284
156. Hladkykh FV. Non-Steroidal Anti-Inflammatory Drugs: Therapeutic and Adverse Effects, Ways of Their Optimization. Vinnytsia: Tvory; 2022. 216. doi:10.46879/2022.1
157. Lee YC, Cui J, Lu B, et al. Pain persists in DAS28 rheumatoid arthritis remission but not in ACR/EULAR remission: a longitudinal observational study. Arthritis Res Ther. 2011;13(3):R83. doi:10.1186/ar3353
158. Boyden SD, Hossain IN, Wohlfahrt A, Lee YC. Non-inflammatory causes of pain in patients with rheumatoid arthritis. Curr Rheumatol Rep. 2016;18(6):30. doi:10.1007/s11926-016-0581-0
159. Chakr RM, Brenol C, Ranzolin A, et al. Rheumatoid arthritis seems to have DMARD treatment decision influenced by fibromyalgia. Rev Bras Reumatol. 2016;57:403–411. doi:10.1016/j.rbr.2016.11.004
160. Zhang A, Lee YC. Mechanisms for joint pain in rheumatoid arthritis: from cytokines to central sensitization. Curr Osteoporos Rep. 2018;16(5):603–610. doi:10.1007/s11914-018-0473-5
161. Kathryn B, Nidhi S. Understanding the mechanisms of pain in rheumatoid arthritis. In: Reem Hamdy AM, editor. Rheumatoid Arthritis. IntechOpen; 2020.
162. Vergne-Salle P, Bertin P. Chronic pain and neuroinflammation. Joint Bone Spine. 2021;88(6):105222. doi:10.1016/j.jbspin.2021.105222
163. Yue JX, Wang RR, Yu J, et al. Histamine upregulates Nav1.8 expression in primary afferent neurons via H2 receptors: involvement in neuropathic pain. CNS Neurosci Ther. 2014;20(10):883–892. doi:10.1111/cns.12305
164. Biddle K, Sofat N. Understanding the mechanisms of pain in rheumatoid arthritis. Rheumatoid arthritis – other perspectives towards a better practice. IntechOpen. 2020. doi:10.5772/intechopen.93829
165. Buch MH, Eyre S, McGonagle D. Persistent inflammatory and non-inflammatory mechanisms in refractory rheumatoid arthritis. Nat Rev Rheumatol. 2021;17(1):17–33. doi:10.1038/s41584-020-00541-7
166. DiSabato DJ, Quan N, Godbout JP. Neuroinflammation: the devil is in the details. J Neurochem. 2016;139(Suppl 2):136–153. doi:10.1111/jnc.13607
167. Süß P, Rothe T, Hoffmann A, Schlachetzki JCM, Winkler J. The joint-brain axis: insights from rheumatoid arthritis on the crosstalk between chronic peripheral inflammation and the brain. Front Immunol. 2020;11:612104. doi:10.3389/fimmu.2020.612104
168. Malange KF, Navia-Pelaez JM, Dias EV, et al. Macrophages and glial cells: innate immune drivers of inflammatory arthritic pain perception from peripheral joints to the central nervous system. Front Pain Res. 2022;3:1018800. doi:10.3389/fpain.2022.1018800
169. Sebba A. Pain: a review of interleukin-6 and its roles in the pain of rheumatoid arthritis. Open Access Rheumatol. 2021;13:31–43. doi:10.2147/OARRR.S291388
170. Mahmoud S, Gharagozloo M, Simard C, Gris D. Astrocytes maintain glutamate homeostasis in the cns by controlling the balance between glutamate uptake and release. Cells. 2019;8(2):184. doi:10.3390/cells8020184
171. Li T, Chen X, Zhang C, Zhang Y, Yao W. An update on reactive astrocytes in chronic pain. J Neuroinflammation. 2019;16(1):140. doi:10.1186/s12974-019-1524-2
172. Hladkykh FV. The role of disease-modifying antirheumatic drugs in the implementation of the «treat-to-target» strategy for rheumatoid arthritis. Karazin J Immunol. 2025;8(15):136–159. doi:10.26565/3083-5615-2025-15-10
173. Koop SM, ten Klooster PM, Vonkeman HE, Steunebrink LM, van de Laar MA. Neuropathic-like pain features and cross-sectional associations in rheumatoid arthritis. Arthritis Res Ther. 2015;17(1):237. doi:10.1186/s13075-015-0761-8
174. Ahmed S, Magan T, Vargas M, Harrison A, Sofat N. Use of the painDETECT tool in rheumatoid arthritis suggests neuropathic and sensitization components in pain reporting. J Pain Res. 2014;7:579–588. doi:10.2147/JPR.S69011
175. Perrot S, Dieudé P, Pérocheau D, Allanore Y. Comparison of pain, pain burden, coping strategies, and attitudes between patients with systemic sclerosis and patients with rheumatoid arthritis: a cross-sectional study. Pain Med. 2013;14(11):1776–1785. doi:10.1111/pme.12213
176. Hallström H, Norrbrink C. Screening tools for neuropathic pain: can they be of use in individuals with spinal cord injury? Pain. 2011;152(4):772–779. doi:10.1016/j.pain.2010.11.019
177. Minhas D, Murphy A, Clauw DJ. Fibromyalgia and centralized pain in the rheumatoid arthritis patient. Curr Opin Rheumatol. 2023;35(3):170–174. doi:10.1097/BOR.0000000000000929
178. Sunzini F, Stefanov K, Al-Wasity S, et al. The insula represents a key neurobiological pain hub in psoriatic arthritis. Arthritis Res Ther. 2025;27(1):70. doi:10.1186/s13075-025-03526-7
179. Hackett S, Ogdie A, Coates LC. Psoriatic arthritis: prospects for the future. Ther Adv Musculoskelet Dis. 2022;14:1759720X221086710. doi:10.1177/1759720X221086710
180. Coates LC, Helliwell PS. Psoriatic arthritis: state of the art review. Clin Med Lond. 2017;17(1):65–70. doi:10.7861/clinmedicine.17-1-65
181. Fitzgerald O, Ogdie A, Chandran V, et al. Psoriatic arthritis. Nat Rev Dis Primers. 2021;7(1):59. doi:10.1038/s41572-021-00293-y
182. Moll JM, Wright V. Psoriatic arthritis. Semin Arthritis Rheum. 1973;3(1):55–78. doi:10.1016/0049-0172(73)90035-8
183. Taylor W, Gladman D, Helliwell P, Marchesoni A, Mease P, Mielants H, CASPAR Study Group. Classification criteria for psoriatic arthritis: development of new criteria from a large international study. Arthritis Rheum. 2006;54(8):2665–2673. doi:10.1002/art.21972
184. Alp G, Kurut Aysin İ, Cinakli H, Solmaz D, Akar S. Prevalence of central sensitization and neuropathic pain in patients with psoriatic arthritis: a cross-sectional study. Arch Rheumatol. 2025;40(1):87–97. doi:10.46497/ArchRheumatol.2025.11011
185. Højgaard P, Christensen R, Dreyer L, et al. Pain mechanisms and ultrasonic inflammatory activity as prognostic factors in patients with psoriatic arthritis: protocol for a prospective, exploratory cohort study. BMJ Open. 2016;6(4):e010650. doi:10.1136/bmjopen-2015-010650
186. Bagnato G, De Andres I, Sorbara S, et al. Pain threshold and intensity in rheumatic patients: correlations with the Hamilton Depression Rating scale. Clin Rheumatol. 2015;34(3):555–561. doi:10.1007/s10067-013-2477-y
187. Lee YC, Nassikas NJ, Clauw DJ. The role of the central nervous system in the generation and maintenance of chronic pain in rheumatoid arthritis, osteoarthritis and fibromyalgia. Arthritis Res Ther. 2011;13(2):211. doi:10.1186/ar3306
188. Gossec L, de Wit M, Kiltz U, et al. EULAR PsAID Taskforce. A patient-derived and patient-reported outcome measure for assessing psoriatic arthritis: elaboration and preliminary validation of the Psoriatic Arthritis Impact of Disease (PsAID) questionnaire, a 13-country EULAR initiative. Ann Rheum Dis. 2014;73(6):1012–1019. doi:10.1136/annrheumdis-2014-205207
189. Sumpton D, Kelly A, Tunnicliffe DJ, et al. Patients’ perspectives and experience of psoriasis and psoriatic arthritis: a systematic review and thematic synthesis of qualitative studies. Arthritis Care Res. 2020;72(5):711–722. doi:10.1002/acr.23896
190. Currado D, Berardicurti O, Saracino F, et al. The relationship between metabolic syndrome and pain catastrophizing in psoriatic arthritis. Rheumatol Ther. 2025;12(3):581–592. doi:10.1007/s40744-025-00758-6
191. Ellis A. Reason and Emotion in Psychotherapy. NY, USA: Lyle Stuart; 1962.
192. Beck AT, Rush AJ, Shaw BF, Emery G. Cognitive Therapy of Depression. NY, USA: Guilford Press; 1979.
193. Wilk M, Łosińska K, Pripp AH, Korkosz M, Haugeberg G. Pain catastrophizing in rheumatoid arthritis, psoriatic arthritis and axial spondyloarthritis: biopsychosocial perspective and impact on health-related quality of life. Rheumatol Int. 2022;42(4):669–682. doi:10.1007/s00296-021-05070-4
194. Frede N, Hiestand S, Schauer F, et al. Psoriasis and psoriatic arthritis have a major impact on quality of life and depressive symptoms: a cross-sectional study of 300 patients. Rheumatol Ther. 2023;10(6):1655–1668. doi:10.1007/s40744-023-00602-9
195. Frede N, Rieger E, Lorenzett R, et al. Sleep behaviour differs in women and men with psoriatic arthritis and axial spondyloarthritis with impact on quality of life and depressive symptoms. RMD Open. 2023;9(2):e002912. doi:10.1136/rmdopen-2022-002912
196. Toledano E, Queiro R, Gómez-Lechón L, et al. Influence of comorbidities not associated with fibromyalgia on neuropathic pain in patients with psoriatic arthritis: relationship with clinical parameters. Front Med Lausanne. 2024;11:1331761. doi:10.3389/fmed.2024.1331761
197. Schaible H-G, von Banchet GS, Boettger MK, et al. The role of proinflammatory cytokines in the generation and maintenance of joint pain. Ann N Y Acad Sci. 2010;1193(1):60–69. doi:10.1111/j.1749-6632.2009.05301.x
198. Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain. 2009;10(9):895–926. doi:10.1016/j.jpain.2009.06.012
199. Woolf CJ. Central sensitization: implications for the diagnosis and treatment of pain. Pain. 2011;152(3):S2–S15. doi:10.1016/j.pain.2010.09.030
200. Sarzi-Puttini P, Giorgi V, Marotto D, Atzeni F. Fibromyalgia: an update on clinical characteristics, aetiopathogenesis and treatment. Nat Rev Rheumatol. 2020;16(11):645–660. doi:10.1038/s41584-020-00506-w
201. Rifbjerg-Madsen S, Christensen AW, Christensen R, et al. Pain and pain mechanisms in patients with inflammatory arthritis: a Danish nationwide cross-sectional DANBIO registry survey. PLoS One. 2017;12(7):e0180014. doi:10.1371/journal.pone.0180014
202. Ramjeeawon A, Choy E. Neuropathic-like pain in psoriatic arthritis: evidence of abnormal pain processing. Clin Rheumatol. 2019;38(11):3153–3159. doi:10.1007/s10067-019-04656-5
203. Di Carlo M, Muto P, Benfaremo D, Luchetti MM, Atzeni F, Salaffi F. The neuropathic pain features in psoriatic arthritis: a cross-sectional evaluation of prevalence and associated factors. J Rheumatol. 2020;47(8):1198–1203. doi:10.3899/jrheum.190906
204. Finan PH, Goodin BR, Smith MT. The association of sleep and pain: an update and a path forward. J Pain. 2013;14(12):1539–1552. doi:10.1016/j.jpain.2013.08.007
205. Chin S-H, Huang W-L, Akter S, Binks M. Obesity and pain: a systematic review. Int J Obes. 2020;44(5):969–979. doi:10.1038/s41366-019-0505-y
206. Al Sharie S, Varga SJ, Al-Husinat L, et al. Unraveling the complex web of fibromyalgia: a narrative review. Medicina. 2024;60(2):272. doi:10.3390/medicina60020272
207. Galvez-Sánchez CM, Duschek S, Reyes DEl Paso GA. Psychological impact of fibromyalgia: current perspectives. Psychol Res Behav Manag. 2019;12:117–127. doi:10.2147/PRBM.S178240
208. Hudson JI, Goldenberg DL, Pope HG, Keck PE, Schlesinger L. Comorbidity of fibromyalgia with medical and psychiatric disorders. Am J Med. 1992;92(4):363–367. doi:10.1016/0002-9343(92)90265-d
209. Meresh E, Khieu K, Krupa J, et al. Correlation of psychological factors, obesity, serum cortisol, and c-reactive protein in patients with fibromyalgia diagnosed with obstructive sleep apnea and other comorbidities. Biomedicines. 2024;12(6):1265. doi:10.3390/biomedicines12061265
210. Katz J, Bartels CM. Multimorbidity in rheumatoid arthritis: literature review and future directions. Curr Rheumatol Rep. 2024;26(1):24–35. doi:10.1007/s11926-023-01121-w
211. Jones GT, Atzeni F, Beasley M, Flüß E, Sarzi-Puttini P, Macfarlane GJ. The prevalence of fibromyalgia in the general population: a comparison of the American College of Rheumatology 1990, 2010, and modified 2010 classification criteria. Arthritis Rheumatol. 2015;67(2):568–575. doi:10.1002/art.38905
212. Duffield SJ, Miller N, Zhao S, Goodson NJ. Concomitant fibromyalgia complicating chronic inflammatory arthritis: a systematic review and meta-analysis. Rheumatology. 2018;57(8):1453–1460. doi:10.1093/rheumatology/key112
213. Macfarlane GJ, Kronisch C, Dean LE, et al. EULAR revised recommendations for the management of fibromyalgia. Ann Rheum Dis. 2017;76(2):318–328. doi:10.1136/annrheumdis-2016-209724
214. Durán J, Combe B, Niu J, Rincheval N, Gaujoux-Viala C, Felson DT. The effect on treatment response of fibromyalgic symptoms in early rheumatoid arthritis patients: results from the ESPOIR cohort. Rheumatology. 2015;54(12):2166–2170. doi:10.1093/rheumatology/kev254. Erratum in: Rheumatology (Oxford). 2017;56(7):1244. doi: 10.1093/rheumatology/kew472.
215. Salaffi F, Gerardi MC, Atzeni F, et al. The influence of fibromyalgia on achieving remission in patients with long-standing rheumatoid arthritis. Rheumatol Int. 2017;37(12):2035–2042. doi:10.1007/s00296-017-3792-4
216. Durmaz Y, Ilhanli I. Rheumatoid arthritis activity scores in patients with and without fibromyalgia syndrome. Ann Saudi Med. 2021;41(4):246–252. doi:10.5144/0256-4947.2021.246
217. Kancharla H, Jain S, Mishra S, et al. Fibromyalgia influences health-related quality of life and disease activity in psoriatic arthritis. Rheumatol Int. 2022;42(3):511–517. doi:10.1007/s00296-021-04925-0
218. Brikman S, Furer V, Wollman J, et al. The effect of the presence of fibromyalgia on common clinical disease activity indices in patients with psoriatic arthritis: a cross-sectional study. J Rheumatol. 2016;43(9):1749–1754. doi:10.3899/jrheum.151491. Erratum in: J Rheumatol. 2019;46(1):119. doi: 10.3899/jrheum.151491.
219. Iannone F, Nivuori M, Fornaro M, Venerito V, Cacciapaglia F, Lopalco G. Comorbid fibromyalgia impairs the effectiveness of biologic drugs in patients with psoriatic arthritis. Rheumatology. 2020;59(7):1599–1606. doi:10.1093/rheumatology/kez505
220. Elsawy NA, Helal AH, Abd ElHamid HA, Abdel-Fattah YH. Fibromyalgia in patients with psoriatic arthritis: impact on disease activity indices, fatigue and health-related quality of life. Int J Rheum Dis. 2021;24(2):189–196. doi:10.1111/1756-185X.13987
221. Falasinnu T, Nguyen T, Jiang TE, et al. The problem of pain in rheumatology: clinical profiles associated with concomitant diagnoses with chronic overlapping pain conditions. ACR Open Rheumatol. 2022;4(10):890–896. doi:10.1002/acr2.11488
222. Lubrano E, Scriffignano S, Morelli R, Perrotta FM. Assessment of widespread and extraarticular pain in psoriatic arthritis: a case-control study. J Rheumatol. 2021;48(9):1405–1409. doi:10.3899/jrheum.201163
223. El-Kasmi H, Amine B, Kabbaj A, et al. The Prevalence of Fibromyalgia in Rheumatoid Arthritis Patients Using the Fibromyalgia Assessment Screening Tool (FAST 4) Based on the Multidimensional Health Assessment Questionnaire (MDHAQ). Cureus. 2024;16(7):e64011. doi:10.7759/cureus.64011. PMID: 39109111; PMCID: PMC11302173.
224. Elizur Y, Amital M, Ben-Shabat N, et al. The Association Between Psoriasis, Psoriatic Arthritis, and Fibromyalgia Syndrome: effects on Treatment-A Population-Based Study. Medicina. 2025;61(10):1809. doi:10.3390/medicina61101809. PMID: 41155795; PMCID: PMC12566094.
225. Paroli M, Gioia C, Accapezzato D, Caccavale R. Demographic Characteristics and Inflammatory Biomarker Profile in Psoriatic Arthritis Patients with Comorbid Fibromyalgia: a Cross-Sectional Study. Medicina. 2025;61(6):1050. doi:10.3390/medicina61061050. PMID: 40572738; PMCID: PMC12194970.
226. Marchesoni A, Atzeni F, Spadaro A, et al. Identification of the clinical features distinguishing psoriatic arthritis and fibromyalgia. J Rheumatol. 2012;39(4):849–855. doi:10.3899/jrheum.110893
227. Koroschetz J, Rehm SE, Gockel U, et al. Fibromyalgia and neuropathic pain--differences and similarities. A comparison of 3057 patients with diabetic painful neuropathy and fibromyalgia. BMC Neurol. 2011;11:55. doi:10.1186/1471-2377-11-55
228. Mohammed A, Alshamarri T, Adeyeye T, Lazariu V, McNutt LA, Carpenter DO. A comparison of risk factors for osteo- and rheumatoid arthritis using NHANES data. Prev Med Rep. 2020;20:101242. doi:10.1016/j.pmedr.2020.101242
229. Mathias K, Amarnani A, Pal N, et al. Chronic pain in patients with rheumatoid arthritis. Curr Pain Headache Rep. 2021;25(9):59. doi:10.1007/s11916-021-00973-0
230. Ruiz-Medrano E, Espinosa-Ortega HF, Arce-Salinas CA. The effect of concomitant hand osteoarthritis on pain and disease activity in patients with rheumatoid arthritis. Clin Rheumatol. 2019;38(10):2709–2716. doi:10.1007/s10067-019-04574-6
231. Beswick AD, Wylde V, Gooberman-Hill R, Blom A, Dieppe P. What proportion of patients report long-term pain after total Hip or knee replacement for osteoarthritis? A systematic review of prospective studies in unselected patients. BMJ Open. 2012;2(1):e000435. doi:10.1136/bmjopen-2011-000435
232. Wylde V, Sayers A, Odutola A, Gooberman-Hill R, Dieppe P, Blom AW. Central sensitization as a determinant of patients’ benefit from total Hip and knee replacement. Eur J Pain. 2017;21(2):357–365. doi:10.1002/ejp.929
233. Giorgi V, Sarzi-Puttini P, Pellegrino G, et al. Pharmacological treatment of fibromyalgia syndrome: a practice-based review. Curr Pain Headache Rep. 2024;28(12):1349–1363. doi:10.1007/s11916-024-01277-9
234. Häuser W, Walitt B, Fitzcharles MA, Sommer C. Review of pharmacological therapies in fibromyalgia syndrome. Arthritis Res Ther. 2014;16(1):201. doi:10.1186/ar4441
235. Smolen JS, Landewé RBM, Bergstra SA, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2022 update. Ann Rheum Dis. 2023;82(1):3–18. doi:10.1136/ard-2022-223356
236. Gossec L, Kerschbaumer A, Ferreira RJO, et al. EULAR recommendations for the management of psoriatic arthritis with pharmacological therapies: 2023 update. Ann Rheum Dis. 2024;83(6):706–719. doi:10.1136/ard-2024-225531
237. Pollard LC, Ibrahim F, Choy EH, Scott DL. Pain thresholds in rheumatoid arthritis: the effect of tender point counts and disease duration. J Rheumatol. 2012;39(1):28–31. doi:10.3899/jrheum.110668
238. Arendt-Nielsen L, Skou ST, Nielsen TA, Petersen KK. Altered central sensitization and pain modulation in the CNS in chronic joint pain. Curr Osteoporos Rep. 2015;13(4):225–234. doi:10.1007/s11914-015-0276-x
239. Sofat N, Lambarth A. Can we achieve pain stratification in musculoskeletal conditions? Implications for clinical practice. Front Pain Res. 2024;5:1362757. doi:10.3389/fpain.2024.1362757
240. D’Orazio A, Cirillo AL, Greco G, et al. Pathogenesis of rheumatoid arthritis: one year in review 2024. Clin Exp Rheumatol. 2024;42(9):1707–1713. doi:10.55563/clinexprheumatol/0307ed
241. Fuggle NR, Beaudart C, Bruyère O, et al. Evidence-Based Guideline for the management of osteoporosis in men. Nat Rev Rheumatol. 2024;20(4):241–251. doi:10.1038/s41584-024-01094-9
242. Kharouf F, Gladman DD. Advances in the management of psoriatic arthritis in adults. BMJ. 2024;387:e081860. doi:10.1136/bmj-2024-081860
243. Kosek E. The concept of nociplastic pain-where to from here? Pain. 2024;165(11S):S50–S57. doi:10.1097/j.pain.0000000000003305
244. DI Matteo A, Emery P. Rheumatoid arthritis: a review of the key clinical features and ongoing challenges of the disease. Panminerva Med. 2024;66(4):427–442. doi:10.23736/S0031-0808.24.05272-8
245. Dydyk AM, Chiebuka E, Stretanski MF, Givler A. Central Pain Syndrome. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2025. PMID: 31971703.
246. Batchelor V, Perry TA, Cader MZ, Vincent TL. Peripheral neuronal sensitization and neurovascular remodelling in osteoarthritis pain. Nat Rev Rheumatol. 2025;21(9):526–545. doi:10.1038/s41584-025-01280-3
247. Brown P, Pratt AG, Hyrich KL. Therapeutic advances in rheumatoid arthritis. BMJ. 2024;384:e070856. doi:10.1136/bmj-2022-070856
248. Teuwen MMH, van Weely SFE, Vliet Vlieland TPM, et al. Effectiveness of longstanding exercise therapy compared with usual care for people with rheumatoid arthritis and severe functional limitations: a randomised controlled trial. Ann Rheum Dis. 2024;83(4):437–445. doi:10.1136/ard-2023-224912
249. Kaplan CM, Kelleher E, Irani A, Schrepf A, Clauw DJ, Harte SE. Deciphering nociplastic pain: clinical features, risk factors and potential mechanisms. Nat Rev Neurol. 2024;20(6):347–363. doi:10.1038/s41582-024-00966-8
250. Coates LC, Soriano E, Corp N, et al. OP0229 THE GROUP FOR RESEARCH AND ASSESSMENT OF PSORIASIS AND PSORIATIC ARTHRITIS (GRAPPA) TREATMENT RECOMMENDATIONS 2021. Ann Rheum Dis. 2021;80:139–140. doi:10.1136/annrheumdis-2021-eular.4091
251. Wolfe F, Clauw DJ, Fitzcharles M-A, et al. 2016 Revisions to the 2010/2011 fibromyalgia diagnostic criteria. Semin Arthritis Rheum. 2016;46(3):319–329. doi:10.1016/j.semarthrit.2016.08.012
252. Binderup AT, Arendt-Nielsen L, Madeleine P. Pressure pain sensitivity maps of the neck-shoulder and the low back regions in men and women. BMC Musculoskelet Disord. 2010;11:234. doi:10.1186/1471-2474-11-234
253. Ohashi Y, Uchida K, Fukushima K, Inoue G, Takaso M. Mechanisms of Peripheral and Central Sensitization in Osteoarthritis Pain. Cureus. 2023;15(2):e35331. doi:10.7759/cureus.35331
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
The Psychosocial Characteristics and Somatosensory Function of Children and Adolescents Who Meet the Criteria for Chronic Nociplastic Pain
Ocay DD, Ross BD, Moscaritolo L, Ahmed N, Ouellet JA, Ferland CE, Ingelmo PM
Journal of Pain Research 2023, 16:487-500
Published Date: 15 February 2023
The m6A/m1A/m5C-Related Methylation Modification Patterns and Immune Landscapes in Rheumatoid Arthritis and Osteoarthritis Revealed by Microarray and Single-Cell Transcriptome
Zheng H, Aihaiti Y, Cai Y, Yuan Q, Yang M, Li Z, Xu K, Xu P
Journal of Inflammation Research 2023, 16:5001-5025
Published Date: 1 November 2023
The Hippo-YAP Signaling Pathway in Osteoarthritis and Rheumatoid Arthritis
Li M, Zhang FJ, Bai RJ
Journal of Inflammation Research 2024, 17:1105-1120
Published Date: 19 February 2024
Health Disparities in Rheumatology in the United States
Wright GC, Zueger PM, Copley-Merriman C, Khan S, Costello J, Krumbach A, Reddy P, Tanjinatus O, Wells AF
Open Access Rheumatology: Research and Reviews 2025, 17:1-12
Published Date: 9 January 2025
The Emotional Allodynia Questionnaire: Preliminary Validation and Clinical Phenotyping in Fibromyalgia
Corriero A, Giglio M, Pilolla A, Galdini F, Mucci O, Vurro M, Fornarelli F, Di Venosa C, Trerotoli P, Puntillo F
Journal of Pain Research 2026, 19:607309
Published Date: 3 June 2026