Back to Journals » Journal of Inflammation Research » Volume 19
A Cross-Layer Inflammatory Signature Links Residual Inflammatory Risk to Heart Failure Risk in Metabolic Syndrome: Network Mapping and Brown Adipose Transcriptomic Support
Authors Chen C ![]() , Mao X, Wang Y, Wang Y, Li Y, Shi S, Song Q
, Mao X, Wang Y, Wang Y, Li Y, Shi S, Song Q
Received 25 January 2026
Accepted for publication 4 May 2026
Published 10 July 2026 Volume 2026:19 595888
DOI https://doi.org/10.2147/JIR.S595888
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Chengming Fan
Chunmei Chen,1,2 Xinxin Mao,1,2 Yuxin Wang,1,2 Yajiao Wang,1 Yumeng Li,1 Shuqing Shi,1 Qingqiao Song1
1Department of General Internal Medicine, Guang’anmen Hospital, China Academy of Chinese Medical Sciences, Beijing, People’s Republic of China; 2Graduate School of China Academy of Chinese Medical Sciences, Beijing, People’s Republic of China
Correspondence: Qingqiao Song, Department of General Internal Medicine, Guang’anmen Hospital, China Academy of Chinese Medical Sciences, Beijing, People’s Republic of China, Email [email protected]
Background: Residual inflammatory risk (RIR), commonly indexed by high-sensitivity C-reactive protein (hs-CRP), reflects a persistent inflammatory burden that may not be captured by conventional metabolic indices. Whether RIR is associated with a higher heart failure (HF)-risk phenotype in metabolic syndrome (MetS), and whether it aligns with a coherent cross-level inflammatory signature, remains unclear.
Objective: To determine the association between RIR and a higher HF-risk phenotype in MetS, and to characterize related inflammatory signatures across clinical, bioinformatic, and experimental datasets.
Methods: This study integrated three complementary components. First, a propensity score-matched cohort of hospitalized patients with MetS was used to evaluate the association between RIR and a higher HF-risk phenotype. Second, curated disease-gene resources and GEO transcriptomic datasets were integrated for enrichment and network analyses to identify shared inflammatory features between MetS and HF. Third, an HFpEF mouse model induced by high-fat diet (HFD) plus transverse aortic constriction (TAC), together with brown adipose tissue (BAT) bulk RNA sequencing, was used to assess tissue-level transcriptomic context.
Results: In the propensity score-matched MetS cohort, high RIR (hs-CRP ≥ 2 mg/L) was associated with higher odds of HF-risk phenotype (NT-proBNP > 125 pg/mL) within matched pairs (conditional OR 1.70, 95% CI 1.15– 2.51; P = 0.0078). The association remained after additional adjustment (OR 1.59, 95% CI 1.06– 2.37; P = 0.02) and was also observed using an alternative NT-proBNP threshold (> 300 pg/mL; adjusted OR 1.55, 95% CI 1.14– 2.10; P = 0.005). Cross-disease analyses converged on inflammatory programs involving cytokine and chemokine activity, IL-17 and TNF signaling, and extracellular matrix (ECM) remodeling, with six shared genes (CCL2, FOSL1, THBS1, ATP2A2, FASN, and CFH) identified at the MetS-HF interface. In vivo, HFD plus TAC produced an HFpEF phenotype with preserved ejection fraction, diastolic dysfunction, and myocardial fibrosis. BAT transcriptomics showed enrichment of immune and inflammatory pathways, including chemotaxis pathways, which overlapped with the discovery-level inflammatory signature.
Conclusion: In MetS, RIR was associated with higher odds of a HF-risk phenotype and aligned with a coherent inflammatory network characterized by cytokine-chemokine signaling and remodeling-related pathways. BAT transcriptomic changes provide supportive tissue-level context for this inflammatory signature. These findings are associative and hypothesis-generating, and further mechanistic studies are needed to define causal tissue-to-heart pathways.
Keywords: metabolic syndrome, residual inflammatory risk, heart failure risk phenotype, brown adipose tissue, transcriptomics
Introduction
Heart failure with preserved ejection fraction (HFpEF) has become an increasingly prevalent HF phenotype, particularly among older individuals with multiple cardiometabolic comorbidities. Obesity, hypertension, type 2 diabetes, and dyslipidemia are highly overrepresented in HFpEF, and the syndrome is characterized by concentric left ventricular remodeling, diastolic dysfunction, and chronic low-grade systemic inflammation.1 In this context, MetS is increasingly recognized as an important upstream substrate for HFpEF. However, the multilevel pathways through which MetS-related metabolic disturbance and inflammatory burden align with a higher HF-risk phenotype remain incompletely understood.2,3
A growing body of evidence supports inflammation as a key non-metabolic dimension of cardiometabolic progression. Even when conventional metabolic indices are broadly similar, a substantial proportion of patients exhibit persistently elevated inflammatory burden, a phenomenon conceptualized as RIR.4–6 In clinical practice, RIR is commonly indexed by hs-CRP, which reflects a pragmatic but incomplete measure of inflammatory burden and has been associated with adverse cardiovascular outcomes.7 It remains unclear whether RIR is associated with a higher HF-risk phenotype in MetS, and which tissue-level processes may contribute to this residual inflammatory state.
Adipose tissue dysfunction represents one plausible upstream contributor to RIR in MetS. Beyond quantitative expansion, adipose tissue undergoes immunometabolic remodeling, including impaired lipid storage, increased lipolysis, ectopic fat deposition, and dysregulated secretion of adipokines and cytokines. BAT, traditionally viewed as a thermogenic organ, is increasingly recognized as a metabolically active tissue with endocrine and immune features relevant to systemic energy homeostasis and cardiometabolic regulation.8,9
Recent advances in transcriptomics and network biology have enabled identification of shared molecular signatures across cardiometabolic conditions.10 Several bioinformatic studies suggest that MetS and HF may converge on pathways related to lipid metabolism, oxidative stress, extracellular matrix remodeling, and immune activation.11 However, such findings have seldom been integrated with clinically phenotyped MetS populations and organ-specific remodeling. To address these gaps, we designed an integrated translational study spanning four complementary levels of evidence: a propensity score-matched MetS cohort to assess the association between RIR and a higher HF-risk phenotype; discovery-level bioinformatic analyses to identify shared genes and regulatory networks between MetS and HF; an HFpEF mouse model to evaluate cardiac structure and function in vivo; and BAT transcriptomic profiling to assess its concordance with the discovery-level molecular signatures. Collectively, this study aimed to evaluate whether hs-CRP-defined RIR, cross-disease inflammatory network features, and BAT transcriptomic remodeling converge in the setting of MetS with higher HF risk.
Methods
Human Cohort and Definition of Residual Inflammatory Risk
This retrospective observational study was conducted at Guang’anmen Hospital, China Academy of Chinese Medical Sciences (Beijing, China). Consecutive hospitalized patients with MetS were identified from the department of general internal medicine between September 2018 and September 2021. MetS was defined according to the 2009 harmonized criteria (≥3 of 5 components; detailed diagnostic criteria are provided in Supplementary Table S1). Eligible participants had complete clinical and laboratory data required for HF-risk phenotype. Patients with conditions likely to substantially confound cardiometabolic status or biomarker interpretation, including severe systemic disease and missing key exposure, outcome, or covariate data, were excluded (Figure S1). RIR was defined as hs-CRP ≥2 mg/L. In sensitivity analysis, an alternative NT-proBNP threshold of >300 pg/mL was additionally evaluated.
Data Collection, Missing Data, and Propensity Score Matching
Demographic characteristics, lifestyle factors, comorbidities, medication history, and laboratory measurements were extracted from electronic medical records at the index visit. Missingness was quantified for all variables. Variables with >30% missing data were excluded, and remaining missing values were handled using multiple imputation by chained equations, with pooled estimates using Rubin’s rules. To reduce baseline imbalance, propensity score matching (PSM) was performed using 1:1 nearest-neighbor matching without replacement with a caliper of 0.2 standard deviations of the logit of the propensity score. The propensity score model included age, sex, smoking, alcohol use, chronic obstructive pulmonary disease, pancreatitis, chronic kidney disease, liver cirrhosis, renal insufficiency, ulcerative colitis, Alzheimer’s disease, and other prespecified major clinical confounders available in the electronic medical record. Covariate balance was assessed using standardized mean differences, with SMD <0.1 considered adequate balance.
Systems Bioinformatics and Network Analyses
Genes associated with MetS and HF were collected from the Comparative Toxicogenomics Database (CTD), GeneCards, and DisGeNET and were complemented with GEO transcriptomic datasets for MetS (GSE98895) and HF (GSE21610 and GSE84796). To standardize input size across disease-gene resources, if the number of retrieved genes from an individual database exceeded 500, the top 500 ranked genes according to the native database ranking system were retained; otherwise, all retrieved genes were included. Detailed selection rules and access dates for each resource are provided in the Supplementary Methods. Differential expression analysis of GEO datasets was performed using the limma package, and genes with adjusted P <0.05 and absolute fold change >1.5-fold were defined as differentially expressed genes. Filtered gene lists were then deduplicated and merged within each disease, and overlapping genes between the MetS and HF gene sets were carried forward for enrichment and network analyses. Functional enrichment was performed using Enrichr, including Gene Ontology and KEGG pathway analyses, with FDR <0.05 considered statistically significant. Protein-protein interaction networks were constructed using STRING with a minimum interaction score of 0.4 and a maximum of 50 first-shell interactors, and disconnected nodes were excluded. Networks were visualized in Cytoscape (v3.9.1), and hub genes were identified using cytoHubba based on degree centrality. TF-gene and TF-miRNA co-regulatory networks were generated using NetworkAnalyst 3.0 with prespecified filtering parameters as described in the Supplementary Methods S5–S8.
HFpEF Mouse Model and Phenotyping
Male SPF C57BL/6J mice (8 weeks old, 22–25 g) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China) and housed under specific pathogen-free conditions with free access to food and water. A total of 20 mice were used. After 1 week of acclimatization, mice were fed a high-fat diet (60% kcal from fat; Research Diets, D12492) for 4 weeks and were then randomized to sham or HFpEF groups using a computer-generated random sequence by an investigator not involved in outcome assessment. Sham surgery consisted of thoracotomy and aortic dissection without constriction, whereas the HFpEF model was induced by transverse aortic constriction (TAC) using a 26-gauge needle. After surgery, high-fat feeding was continued for an additional 4 weeks before endpoint phenotyping and tissue collection.
For TAC surgery, mice were anesthetized with 0.5% sodium pentobarbital (10 mL/kg, intraperitoneally), and adequate anesthesia depth was confirmed by loss of tail-pinch reflex. Postoperative analgesia was provided with meloxicam (2 mg/kg, subcutaneously) once every 24 h for 72 h. Transthoracic echocardiography was performed using a VINNO 6 LAB system equipped with a high-frequency linear probe. M-mode and pulsed Doppler images were acquired to measure left ventricular ejection fraction (EF), early diastolic mitral inflow velocity (E), early diastolic mitral annular velocity (E′), and the E/E′ ratio. Blood pressure and heart rate were measured in conscious mice using a noninvasive tail-cuff system after at least 3 days of acclimatization, and at least five consecutive measurements with stable waveforms were averaged. Exercise capacity was evaluated using an incremental treadmill protocol to exhaustion. At sacrifice, heart weight-to-tibia length ratio, lung wet-to-dry ratio, and histological changes were assessed. Investigators performing echocardiography and histological quantification were blinded to group allocation, and measurements were obtained in randomized order across animals.
Inclusion criteria for animal analyses were successful completion of the assigned procedures and availability of analyzable data. Exclusion criteria were defined a priori and included perioperative death, unsuccessful TAC, and inadequate RNA sample quality for sequencing. At study endpoint, mice were deeply anesthetized with sodium pentobarbital as described above, followed by terminal blood collection and tissue harvest after confirmation of death by cessation of respiration and absence of reflexes. Additional ARRIVE-related details are provided in the Supplementary Methods S1–S4. No mice met a reduced ejection fraction threshold (<50%) at endpoint assessment.
BAT Collection, RNA Sequencing, and Differential Expression
Interscapular brown adipose tissue (BAT) was collected from individual mice immediately after euthanasia, snap-frozen in liquid nitrogen, and stored at −80°C until RNA extraction. For RNA sequencing, five biological replicates per group were analyzed (n=5 per group). Total RNA was extracted using TRIzol reagent, and RNA purity and concentration were assessed with a NanoDrop 2000 spectrophotometer, while RNA integrity was evaluated using an Agilent 2100 Bioanalyzer. RNA-seq libraries were prepared using the VAHTS Universal V10 RNA-seq Library Prep Kit and sequenced on an Illumina NovaSeq X Plus platform to generate 150-bp paired-end reads. Raw FASTQ reads were processed with fastp to remove adapters and low-quality bases. Clean reads were aligned to the mouse reference genome using HISAT2, and read counts were obtained using HTSeq-count. Principal component analysis and hierarchical clustering were performed in R to assess sample similarity. Differential expression analysis between sham and HFpEF groups was performed using DESeq2, and genes with adjusted Q <0.05 and absolute fold change >2 were defined as differentially expressed genes. GO and KEGG enrichment analyses were then performed for differentially expressed genes, with additional pathway analyses performed when appropriate as described in the Supplementary Methods S9–S12.
Statistical Analysis
Clinical analyses were performed in R (4.3.0). Continuous variables are presented as mean±SD or median (IQR), and categorical variables as n (%). Normality was assessed using the Shapiro–Wilk test before selecting parametric or nonparametric methods. Comparisons between two clinical groups were performed using the independent-samples t test or Mann–Whitney U-test, as appropriate, and categorical variables were compared using the chi-square test or Fisher’s exact test. All statistical tests were two-sided, and P <0.05 was considered statistically significant.
Ethics
The clinical study was approved by the Ethics Committee of Guang’anmen Hospital, China Academy of Chinese Medical Sciences (2023–123-KY), which also granted the waiver of informed consent because of the retrospective design and the use of de-identified data. The clinical study was conducted in accordance with the Declaration of Helsinki and followed the RECORD reporting guideline for studies using routinely collected health data. All animal procedures were approved by Institutional Animal Care and Use Committee (IACUC) of Guang’anmen Hospital, China Academy of Chinese Medical Sciences (IACUC No. IACUC-GAMH-2023-029), and were conducted in accordance with the Regulations for the Administration of Affairs Concerning Experimental Animals, the Beijing Municipality Regulations on the Administration of Experimental Animals, and the Guiding Opinions on the Kind Treatment of Laboratory Animals. The study is reported in accordance with ARRIVE 2.0.
Results
Residual Inflammatory Risk Is Associated with a Higher HF-Risk Phenotype in MetS
Demographic and Clinical Characteristics of the Study Population
A total of 4481 hospitalized individuals were screened, of whom 1,991 met diagnostic criteria for MetS. A higher HF-risk phenotype was operationally defined as NT-proBNP >125 pg/mL. After propensity score matching, 938 patients remained for analysis (469 per group). Baseline characteristics are summarized in Table 1.
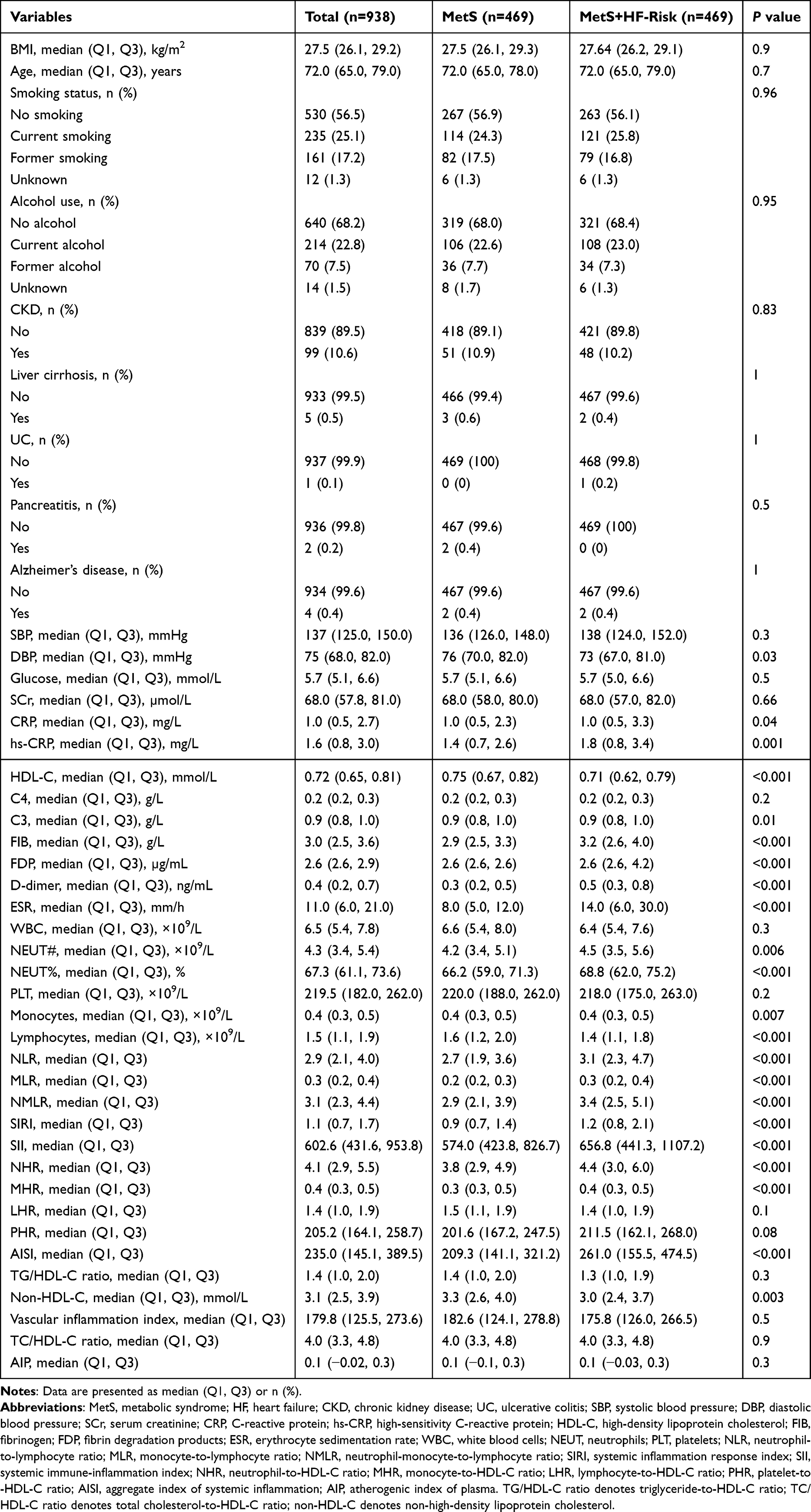 |
Table 1 Baseline Characteristics of Metabolic Syndrome Patients with and Without Heart Failure Risk After Propensity Score Matching |
Conventional metabolic indices, including BMI, SBP, and fasting glucose, were broadly comparable between groups (P >0.05). In conditional logistic regression accounting for matched pairs, hs-CRP ≥2 mg/L was associated with higher odds of higher HF-risk phenotype compared with hs-CRP <2 mg/L (OR 1.70, 95% CI 1.15–2.51; P = 0.0078). This association remained after further adjustment for age, sex, and conventional metabolic indices (OR 1.59, 95% CI 1.06–2.37; P = 0.02, Supplementary Table S2). In addition, representative leukocyte-derived indices and thromboinflammatory markers were higher in the higher HF-risk phenotype group. Using an alternative HF-risk definition (NT-proBNP >300 pg/mL), the association persisted (adjusted OR 1.55, 95% CI 1.14–2.10; P = 0.005; Supplementary Table S3).
Cross-Disease Bioinformatic Analyses Reveal a Limited but Convergent Inflammatory Signature Between MetS and HF
Collection of MetS and HF Gene Sets
We first compiled candidate gene sets for MetS and HF. After merging and removing duplicates, the knowledge-based gene sets contained 1,227 genes related to MetS and 1,141 genes associated with HF. In parallel, GEO transcriptomic datasets provided an additional 4,666 and 423 protein-coding genes, respectively. Cross-disease comparison identified six overlapping genes (CCL2, FOSL1, THBS1, ATP2A2, FASN, and CFH) (Figure 1a). Given the relatively small overlap, these genes were interpreted as a limited but potentially informative point of convergence between MetS and HF and were carried forward for downstream enrichment, interaction, and regulatory analyses.
 |
Figure 1 Cross-disease bioinformatic analyses of MetS and HF. (a) Venn diagram showing overlap between MetS and HF genes. (b) GO biological process enrichment of the six shared genes. (c) Protein-protein interaction network with first-shell expansion. (d) Pathway enrichment analysis of the shared-gene signature. (e) Hub-node identification based on degree centrality in the expanded interaction network. (f) TF-gene regulatory network of the shared-gene signature. Abbreviations: MetS, metabolic syndrome; HF, heart failure; GO, Gene Ontology; TF, transcription factor. |
Functional Enrichment Analyses
To explore the biological context of the six shared genes, we performed GO and pathway enrichment analyses. The top enriched biological process terms were mainly related to regulation of endothelial and epithelial apoptosis, endothelial cell proliferation, inflammatory response, and cellular response to endoplasmic reticulum stress (Figure 1b). Cellular component terms highlighted the sarcoplasmic reticulum, extracellular matrix, and secretory granule compartments (Figure S2), whereas molecular function terms were enriched for chemokine and cytokine activity and growth factor binding (Figure S3). Pathway enrichment further implicated IL-17 signaling, complement and coagulation cascades, fatty acid metabolism, and cellular stress-response programs involving p53 signaling and transforming growth factor beta signaling (Figure 1d).
Protein-Protein Interaction Network and Hub Identification
To place the six shared genes within a broader interaction context, we constructed a protein-protein interaction network with first-shell expansion. The resulting network contained 52 nodes and 352 edges (Figure 1c). Topological ranking identified THBS1, PLG, CXCL8, and SERPINE1 as the highest-degree hub nodes (Figure 1e). As the expanded network included first-shell interactors in addition to the six shared genes, these hub nodes were derived from the expanded interaction network rather than from the direct MetS-HF overlap set.
Upstream Regulatory Networks
The TF-gene regulatory network comprised 165 nodes and 242 edges, indicating that the shared-gene signature may be subject to extensive transcriptional regulation (Figure 1f). Among the six shared genes, FOSL1 and FASN showed relatively high connectivity, being regulated by 114 and 72 transcription factors, respectively. The TF-miRNA coregulatory network contained 182 nodes and 226 edges, integrating 61 TFs and 116 miRNAs across five of the six shared genes (Figure S4). ATP2A2 displayed relatively dense connectivity in this co-regulatory network, suggesting that multilayer regulation may contribute to the observed cross-disease convergence.
Animal Validation: The HFpEF Model Shows Preserved Ejection Fraction, Diastolic Dysfunction, and Cardiometabolic Remodeling
As illustrated in Figure 2a, echocardiography showed preserved ejection fraction (Figure 2e), together with reduced E′ velocities (Figure 2c and d), elevated E/e′ ratios (Figure 2f), and increased LV mass and wall thickness (Figure S5a and b), consistent with concentric hypertrophy and impaired relaxation. Representative gross cardiac morphology is shown in Figure 2b. Consistently, tail-cuff measurements showed higher systolic and diastolic blood pressure as well as an increased heart rate in HFpEF mice compared with sham group (Figure S5d–f). The HFpEF group also developed a typical metabolic-syndrome profile, with elevated fasting glucose, triglycerides, and LDL-C, together with reduced HDL-C (Figure S5c, g, h and i). Morphometric indices showed increased heart weight-to-tibia length ratio and a higher lung wet-to-dry weight ratio, accompanied by reduced exercise capacity, indicating cardiac hypertrophy, pulmonary congestion, and exercise intolerance (Figure S6a–c). Hematoxylin-eosin staining showed enlarged and disorganized cardiomyocytes (Figure S7), and Masson trichrome staining demonstrated increased interstitial and perivascular fibrosis in the HFpEF group compared with the sham group (Figure S8). Together, these findings indicate that the HFD plus TAC model recapitulated key features of an HFpEF phenotype, including preserved ejection fraction, diastolic dysfunction, structural remodeling, and cardiometabolic disturbance.
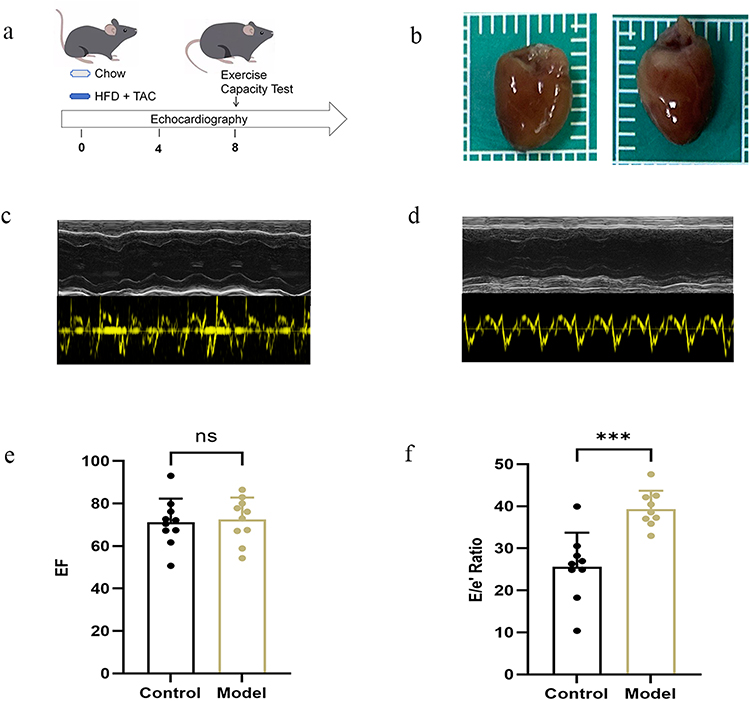 |
Figure 2 HFpEF phenotype induced by HFD plus TAC. (a) Experimental timeline. (b) Representative gross heart morphology from sham and model mice. (c and d) Representative echocardiographic images. (e) Left ventricular ejection fraction (EF). (f) E/E′ ratio. Data are presented as mean±SD, with each dot representing one mouse (n = 10 per group). Statistical comparisons were performed using Mann–Whitney U-test, as appropriate. ***P < 0.001 vs sham. Abbreviation: ns, not significant. |
BAT Transcriptomic Profiling in the HFpEF Model
BAT transcriptomic profiling revealed marked remodeling in HFpEF mice compared with sham mice. Differential expression analysis showed substantial transcriptional changes in BAT from HFpEF mice (Figure 3a and b). Functional enrichment analyses showed overlap with the inflammatory and stress-response programs identified in the cross-disease discovery analyses. At the biological process level, top GO terms were enriched for immune and inflammatory processes, including inflammatory response, immune response, chemotaxis, chemokine-mediated signaling, cell migration, cellular response to interleukin-1, positive regulation of tumor necrosis factor production, and positive regulation of the MAPK cascade (Figure 3c). In parallel, KEGG analysis prioritized cytokine-cytokine receptor interaction, IL-17 signaling pathway, TNF signaling pathway, antigen processing and presentation, MAPK signaling pathway, and cell adhesion molecules (Figure 3d). At the single-gene level, three shared genes prioritized by co-expression analysis were downregulated in BAT of HFpEF mice relative to sham mice (Figure 3e). Grouping differentially expressed genes into immune, mito-energy, inflammatory, and ECM-stress themes revealed distinct category-specific expression patterns across the two groups (Figure 3f).
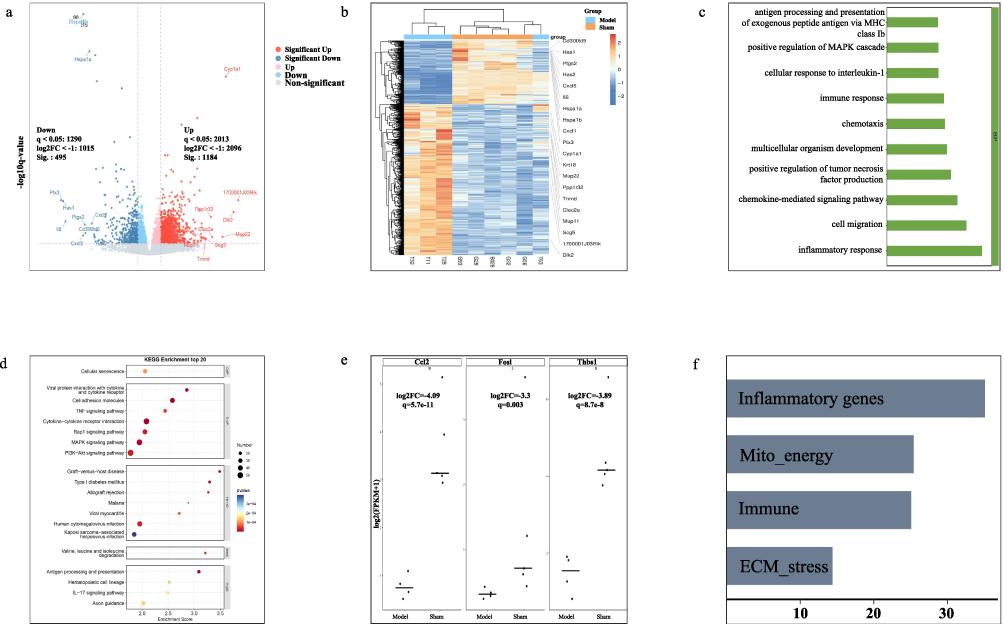 |
Figure 3 BAT transcriptomic remodeling in the HFpEF model. (a) Volcano plot of differentially expressed genes in BAT. (b) Heatmap of representative differentially expressed genes. (c) GO biological process enrichment analysis. (d) KEGG pathway enrichment analysis. (e) Expression of three shared genes prioritized by co-expression analysis in BAT. (f) Category-specific summary of differentially expressed genes grouped into immune, mito-energy, inflammatory, and ECM-stress themes. RNA sequencing was performed using individual BAT samples (n = 5 per group). |
Discussion
In this study, we used RIR as a clinically observable inflammatory phenotype and found that it was associated with a higher HF-risk state within MetS despite broadly comparable conventional metabolic indices. Cross-level analyses further showed that this phenotype aligned with a coherent inflammatory framework characterized by cytokine and chemokine signaling, enrichment of IL-17 and TNF programs, immune cell migration, and signatures linked to tissue remodeling. At the tissue level, BAT transcriptomic profiling revealed marked inflammatory and stress-response remodeling in the HFpEF model. Taken together, these findings support the interpretation that RIR reflects coordinated inflammatory signaling coupled with remodeling-related pathways, and suggest that this inflammatory dimension may be relevant to cardiometabolic HF risk and HFpEF remodeling.
Prior clinical evidence supports RIR as an important dimension of residual cardiovascular risk. RIR remains prognostically relevant among patients undergoing percutaneous coronary intervention, including those achieving low on-treatment LDL-C, and it also predicts unfavorable outcomes in cerebrovascular atherosclerotic disease settings.12 Clinical evidence further suggests that residual inflammatory risk may contribute to adverse cardiovascular outcomes beyond LDL-C-related risk.13 Beyond atherothrombosis, inflammatory biomarkers including hs-CRP and interleukin-6 have been associated with incident HF, worse outcomes among those with established HF, and adverse cardiac remodeling, supporting the concept that a residual inflammatory milieu is relevant to HF phenotypes, including HFpEF, in cardiometabolic populations.14 In this context, our findings extend the RIR framework to MetS by showing that an inflammatory phenotype is associated with higher odds of a HF-risk state. Because echocardiographic data were unavailable in the clinical cohort, this phenotype should be interpreted as a higher HF-risk state rather than confirmed HF or HFpEF.
A central implication of our multi-level results is that RIR aligns with an inflammatory network characterized by cytokine and chemokine signaling, IL-17 and TNF programs. In the discovery-level analysis, the shared MetS-HF signature was enriched for inflammatory response and cytokine or chemokine activity and implicated IL-17 signaling among the top pathways. In BAT transcriptomics, the dominant enrichment pattern likewise concentrated on inflammatory response, chemokine-mediated signaling, chemotaxis and cell migration, and cytokine and chemokine activity. KEGG enrichment further prioritized cytokine-cytokine receptor interaction, IL-17 signaling pathway, TNF signaling, together with MAPK signaling and cell adhesion molecules. These convergent findings support the interpretation that RIR corresponds to an integrated cytokine and chemokine network with IL-17 and TNF inflammatory amplification, rather than being fully explained by conventional metabolic indices alone.15 This interpretation is consistent with current mechanistic understanding of HFpEF as a syndrome with prominent systemic and myocardial inflammatory activation, in which immune cell recruitment, cytokine signaling, and downstream stress pathways contribute to remodeling and functional impairment.15
Within the shared MetS-HF signature, several genes provide mechanistic plausibility for an RIR interpretation. CCL2 is a prototypical chemokine that promotes monocyte recruitment through CCR2 pathways, thereby sustaining inflammatory cell infiltration and remodeling. Monocyte and macrophage recruitment is a recognized driver of cardiac inflammation and fibrosis across injury contexts, supporting CCL2 as a biologically plausible upstream node linking residual inflammation to downstream cardiac risk.16 THBS1 is a matricellular protein at the interface of inflammation, thrombosis signaling, and tissue remodeling. One important mechanism is its capacity to activate latent transforming growth factor beta, providing a plausible route by which chronic inflammatory activation can couple to fibrotic remodeling; in addition, THBS1 has been increasingly linked to obesity, diabetes, and cardiometabolic disease contexts.17 CFH is a key regulator of the alternative complement pathway. Complement dysregulation is increasingly recognized as interwoven with lipid-inflammatory states and chronic inflammatory diseases, supporting a potential role for complement regulation as part of a residual inflammatory milieu in MetS-related HF risk.18 FOSL1 is a stress-responsive AP-1 family transcription factor with documented roles in inflammatory gene regulation and cardiovascular injury responses. AP-1 factors are important regulators of injury-induced gene programs in cardiovascular tissues, providing a plausible transcriptional regulatory layer that can integrate inflammatory stimuli with broader remodeling responses.19 Together, these nodes support a parsimonious RIR-centered interpretation, in which hs-CRP captures a residual inflammatory state corresponding to coordinated cytokine and chemokine signaling, immune activation, and stress-responsive transcriptional regulation.
The HFpEF model provided physiological context for interpreting this inflammatory framework. In vivo, HFD plus TAC reproduced preserved ejection fraction, diastolic dysfunction, concentric remodeling, fibrosis, pulmonary congestion, exercise intolerance, and cardiometabolic disturbance, thereby offering an experimental setting in which adipose and cardiac remodeling coexist. Within this context, BAT transcriptomic profiling showed marked remodeling, with enrichment of immune activation, cytokine and chemokine signaling, and inflammatory amplification pathways. These findings suggest that BAT may represent one tissue-level window into adipose inflammatory remodeling relevant to cardiometabolic HF risk, rather than direct proof that BAT alone drives myocardial remodeling.
This interpretation is biologically plausible in light of the emerging concept of adipose-heart crosstalk. Beyond thermogenesis, BAT is increasingly recognized as a metabolically active endocrine and secretory tissue capable of influencing peripheral organs, including the heart, through batokines, lipid mediators, and extracellular vesicle signals.20–22 Previous studies suggest that BAT factors such as FGF21 and 12,13-diHOME may attenuate pathological cardiac remodeling or improve cardiac function, and BAT extracellular vesicles have also been implicated in cardiomyocyte protection and interorgan communication.23–25 In addition, the heart and BAT may interact bidirectionally, in that cardiac stress signals such as sympathetic activation and natriuretic peptide pathways can modulate BAT activity, whereas BAT dysfunction in HF models has been linked to disturbed metabolic homeostasis and to accumulation of cardiotoxic intermediates that may further aggravate myocardial stress.25–28 Therefore, incorporating BAT into the present study is biologically reasonable, not because BAT has already been established as the dominant driver of HFpEF, but because BAT remodeling may reflect one component of a broader adipose-cardiac inflammatory-metabolic axis.
This framework is also relevant to the apparent directionality of our BAT results. In the present study, several shared genes were downregulated in BAT despite strong enrichment of inflammatory and chemokine-related programs at the pathway level. This pattern does not necessarily contradict an adipose-cardiac inflammatory framework. Instead, it may indicate that BAT remodeling in the HFpEF state includes loss of normal metabolic or secretory homeostasis alongside immune activation. Previous studies suggest that BAT status in HF may be dynamic rather than uniform, with possible early compensatory activation followed by thermogenic dysfunction, metabolic exhaustion, or altered secretory behavior during disease progression.29–32 In HFpEF models in particular, BAT mass and BAT-specific functional markers may change in different directions, suggesting that BAT remodeling may reflect a transition from adaptive activation to functional impairment rather than a single stable phenotype.33 Accordingly, the BAT transcriptomic changes observed here are better interpreted as evidence of tissue remodeling within an inflammatory-metabolic network than as proof of a unidirectional BAT-to-heart proinflammatory pathway.
Conclusion
By integrating discovery-level bioinformatics, a propensity score–matched MetS cohort, and experimental tissue-level analyses, this study identifies RIR as a clinically relevant inflammatory dimension associated with a HF-risk phenotype in metabolic syndrome. Elevated hs-CRP was independently associated with higher odds of HF-risk state despite broadly comparable conventional metabolic indices, supporting the view that cardiometabolic inflammatory burden is not fully captured by standard metabolic measures alone. Cross-level analyses further converged on cytokine and chemokine inflammatory programs, including IL-17 and TNF signaling. In an HFpEF mouse model, BAT transcriptomic profiling showed inflammatory and stress-response remodeling that overlapped with these discovery-level signatures. Overall, these findings support the interpretation that residual inflammation is linked to a broader inflammatory network relevant to MetS-related HF risk and provide a rationale for further mechanistic studies of adipose-cardiac inflammatory communication in HFpEF.
Clinical and Translational Implications
Clinically, these findings suggest that RIR may help contextualize a higher HF-risk phenotype despite similar conventional metabolic indices. Translationally, the observed convergence on cytokine and chemokine programs, particularly IL-17 signaling, highlights candidate inflammatory processes for further investigation rather than definitive therapeutic targets. These results support future studies incorporating longitudinal phenotyping, broader inflammatory profiling, and targeted mechanistic interventions to determine whether modulation of residual inflammation or adipose-associated inflammatory remodeling can improve risk stratification or inform preventive strategies for HF and HFpEF.
Limitations and Future Directions
This study has several limitations. First, the clinical analysis was retrospective and association-based, and therefore does not permit causal inference. Second, the clinical endpoint represented a biomarker-defined higher HF-risk phenotype based on NT-proBNP rather than confirmed HF or HFpEF, because echocardiographic data were unavailable in the human cohort. In addition, NT-proBNP levels may be influenced by renal function and obesity. Although major renal comorbidities were considered in the matching and adjustment strategy, residual confounding related to biomarker interpretation cannot be fully excluded. Third, RIR was defined using hs-CRP alone; additional inflammatory mediators such as IL-6 and TNF were not available in the clinical dataset, and thus the inflammatory phenotype cannot be considered comprehensive. Fourth, the cross-disease molecular overlap between MetS and HF was modest, with only six shared genes identified. Although the shared genes yielded biologically coherent enrichment patterns, the limited overlap indicates that the observed convergence should be interpreted cautiously and may be sensitive to the disease-gene selection strategy. Fifth, the multi-level integration in this study was primarily concordance-based. BAT was selected as a tissue-level window for adipose remodeling, but adult human HFpEF is also strongly influenced by other adipose depots, particularly visceral adipose tissue and possibly epicardial fat, and the shared-gene signature was not directly assessed in myocardial tissue. Finally, the animal study used only male mice, which may limit translational relevance given the recognized sex heterogeneity of HFpEF.
Future studies should combine longitudinal clinical phenotyping with broader inflammatory profiling to refine the temporal relationship between residual inflammation and HF risk in MetS. In parallel, multi-tissue validation, including visceral adipose tissue, epicardial fat, and myocardium, will be important to better define tissue specificity across the adipose-cardiac inflammatory-metabolic axis. Most importantly, targeted genetic or pharmacological perturbation experiments will be required to determine the causal directionality between BAT inflammatory remodeling and cardiac phenotypes and to clarify whether adipose inflammatory remodeling is a driver, amplifier, or bystander in MetS-related HF and HFpEF. Such studies may help define the causal architecture of this axis and evaluate its potential as a therapeutic target.
Abbreviations
MetS, metabolic syndrome; HF, heart failure; DEGs, differentially expressed genes; GEO, gene expression omnibus; GO, gene ontology; KEGG, kyoto encyclopedia of genes and genomes; PPI, protein-protein interaction; TF, transcription factor; miRNAs, microRNAs; CKD, chronic kidney disease; UC, ulcerative colitis; SD, standard deviation; SBP, systolic blood pressure; DBP, diastolic blood pressure; NLR, neutrophil-to-lymphocyte ratio; SIRI, systemic inflammation response index; SII, systemic immune-inflammation index; NHR, neutrophil-to-HDL-C ratio; MHR, monocyte-to-HDL-C ratio; LHR, lymphocyte-to-HDL-C ratio; PHR, platelet-to-HDL-C ratio; AISI, aggregate index of systemic inflammation; non-HDL-C, non-high-density lipoprotein cholesterol; NMLR, neutrophil-monocyte-to-lymphocyte ratio; AIP, atherogenic index of plasma; ER, endoplasmic reticulum; HDL-C, high-density lipoprotein cholesterol.
Data Sharing Statement
Available upon reasonable request to the authors.
Ethics Approval and Consent to Participate
The clinical study was approved by the Ethics Committee of Guang’anmen Hospital, China Academy of Chinese Medical Sciences (2023-123-KY), which also granted the waiver of informed consent because of the retrospective design and the use of de-identified data. All animal procedures were approved by the Animal Care and Use Committee of Guang’anmen Hospital (IACUC-GAMH-2023-029).
Acknowledgment
The research design flowchart was created using Figdraw software. The authors acknowledg the Beijing Traditional Chinese Medicine Science and Technology Development Fund Project for supporting the article processing charge for this publication.
Author Contributions
Chunmei Chen: Conceptualization, Methodology, Formal analysis, Data curation, Writing-original draft. Xinxin Mao: Formal analysis, Validation, Writing-review & editing. Yuxin Wang: Investigation, Data curation, Writing-review & editing. Yajiao Wang: Investigation, Visualization, Writing-review & editing. Yumeng Li: Resources, Validation, Writing-review & editing. Shuqing Shi: Methodology, Supervision, Writing-review & editing. Qingqiao Song: Conceptualization, Supervision, Funding acquisition, Project administration, Writing-review & editing. All authors gave final approval of the version to be published; agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Beijing Traditional Chinese Medicine Science and Technology Development Fund Project (BJZYZD-2025-16) and the Project of Improving the Ability of Clinical Research and Achievement Transformation in Central High-level Chinese Medicine Hospital (HLCMHPP20230).
Disclosure
The authors declare that they have no conflict of interest.
References
1. Tilg H, Zmora N, Adolph TE, Elinav E. The intestinal microbiota fuelling metabolic inflammation. Nat Rev Immunol. 2020;20(1):40–14. doi:10.1038/s41577-019-0198-4
2. Stern MP, Williams K, González-Villalpando C, Hunt KJ, Haffner SM. Does the metabolic syndrome improve identification of individuals at risk of type 2 diabetes and/or cardiovascular disease? Diabetes Care. 2004;27(11):2676–2681. doi:10.2337/diacare.27.11.2676
3. Mottillo S, Filion KB, Genest J, et al. The metabolic syndrome and cardiovascular risk a systematic review and meta-analysis. J Am Coll Cardiol. 2010;56(14):1113–1132. doi:10.1016/j.jacc.2010.05.034
4. Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011;11(2):98–107. doi:10.1038/nri2925
5. Tilg H, Moschen AR. Insulin resistance, inflammation, and non-alcoholic fatty liver disease. Trends Endocrinol Metab. 2008;19(10):371–379. doi:10.1016/j.tem.2008.08.005
6. Bay B, Tanner R, Gao M, et al. Residual cholesterol and inflammatory risk in statin-treated patients undergoing percutaneous coronary intervention†. Eur Heart J. 2025;46(32):3167–3177. doi:10.1093/eurheartj/ehaf196
7. Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. New Engl J Med. 2008;359(21):2195–2207. doi:10.1056/NEJMoa0807646
8. Martins FF, Souza-Mello V, Aguila MB, Mandarim-de-Lacerda CA. Brown adipose tissue as an endocrine organ: updates on the emerging role of batokines. Hormone Mol Biol Clin Invest. 2023;44(2):219–227. doi:10.1515/hmbci-2022-0044
9. Wang GX, Zhao XY, Lin JD. The brown fat secretome: metabolic functions beyond thermogenesis. Trends Endocrinol Metab. 2015;26(5):231–237. doi:10.1016/j.tem.2015.03.002
10. Norzagaray-Valenzuela CD, Valdez-Flores MA, Camberos-Barraza J, et al. Shared transcriptional regulators and network rewiring identify therapeutic targets linking type 2 diabetes mellitus and hypertension. Front Mol Biosci. 2025;12:1621413. doi:10.3389/fmolb.2025.1621413
11. Gorica E, Geiger MA, Di Venanzio L, et al. Cardiometabolic heart failure with preserved ejection fraction: from molecular signatures to personalized treatment. Cardiovasc Diabetol. 2025;24(1):265. doi:10.1186/s12933-025-02774-w
12. Li J, Pan Y, Xu J, et al. Residual inflammatory risk predicts poor prognosis in acute ischemic stroke or transient ischemic attack patients. Stroke. 2021;52(9):2827–2836. doi:10.1161/STROKEAHA.120.033152
13. Yang L, Yue Q, Fang F, et al. Effect of dual residual risk of cholesterol and inflammation on all-cause mortality in patients with cardiovascular disease. Cardiovasc Diabetol. 2023;22(1):96. doi:10.1186/s12933-023-01826-3
14. Ridker PM, Rane M. Interleukin-6 signaling and anti-interleukin-6 therapeutics in cardiovascular disease. Circ res. 2021;128(11):1728–1746. doi:10.1161/CIRCRESAHA.121.319077
15. Daou D, Gillette TG, Hill JA. Inflammatory mechanisms in heart failure with preserved ejection fraction. Physiology. 2023;38(5):217–230. doi:10.1152/physiol.00004.2023
16. Sager HB, Hulsmans M, Lavine KJ, et al. Proliferation and Recruitment Contribute to Myocardial Macrophage Expansion in Chronic Heart Failure. Circ Res. 2016;119(7):853–864. doi:10.1161/CIRCRESAHA.116.309001
17. Schultz-Cherry S, Lawler J, Murphy-Ullrich JE. The type 1 repeats of thrombospondin 1 activate latent transforming growth factor-beta. J Biol Chem. 1994;269(43):26783–26788. doi:10.1016/S0021-9258(18)47087-1
18. Meri S, Haapasalo K. Function and dysfunction of complement factor H during formation of lipid-rich deposits. Front Immunol. 2020;11:611830. doi:10.3389/fimmu.2020.611830
19. Beisaw A, Kuenne C, Guenther S, et al. AP-1 contributes to chromatin accessibility to promote sarcomere disassembly and cardiomyocyte protrusion during zebrafish heart regeneration. Circ res. 2020;126(12):1760–1778. doi:10.1161/CIRCRESAHA.119.316167
20. Becher T, Palanisamy S, Kramer DJ, et al. Brown adipose tissue is associated with cardiometabolic health. Nature Med. 2021;27(1):58–65. doi:10.1038/s41591-020-1126-7
21. Orava J, Nuutila P, Lidell ME, et al. Different metabolic responses of human brown adipose tissue to activation by cold and insulin. Cell Metab. 2011;14(2):272–279. doi:10.1016/j.cmet.2011.06.012
22. Picca A, Guerra F, Calvani R, et al. Mitochondrial-derived vesicles: the good, the bad, and the ugly. Int J Mol Sci. 2023;24(18):13835. doi:10.3390/ijms241813835
23. Martí-Pàmies Í, Thoonen R, Morley M, et al. Brown adipose tissue and BMP3b decrease injury in cardiac ischemia-reperfusion. Circ res. 2023;133(4):353–365. doi:10.1161/CIRCRESAHA.122.322337
24. Ruan CC, Kong LR, Chen XH, et al. A(2A) receptor activation attenuates hypertensive cardiac remodeling via promoting brown adipose tissue-derived FGF21. Cell Metab. 2018;28(3):476–489.e475. doi:10.1016/j.cmet.2018.06.013
25. Cereijo R, Gavaldà-Navarro A, Cairó M, et al. CXCL14, a brown adipokine that mediates brown-fat-to-macrophage communication in thermogenic adaptation. Cell Metab. 2018;28(5):750–763.e756. doi:10.1016/j.cmet.2018.07.015
26. Bartelt A, Bruns OT, Reimer R, et al. Brown adipose tissue activity controls triglyceride clearance. Nature Med. 2011;17(2):200–205. doi:10.1038/nm.2297
27. Wang GX, Zhao XY, Meng ZX, et al. The brown fat-enriched secreted factor Nrg4 preserves metabolic homeostasis through attenuation of hepatic lipogenesis. Nature Med. 2014;20(12):1436–1443. doi:10.1038/nm.3713
28. Kong X, Yao T, Zhou P, et al. Brown adipose tissue controls skeletal muscle function via the secretion of myostatin. Cell Metab. 2018;28(4):631–643.e633. doi:10.1016/j.cmet.2018.07.004
29. Ahmed A, Aboshady I, Munir SM, et al. Decreasing body temperature predicts early rehospitalization in congestive heart failure. J Card Fail. 2008;14(6):489–496. doi:10.1016/j.cardfail.2008.02.008
30. Valero-Muñoz M, Li S, Wilson RM, et al. Heart failure with preserved ejection fraction induces beiging in adipose tissue. Circ Heart Fail. 2016;9(1):e002724. doi:10.1161/CIRCHEARTFAILURE.115.002724
31. Collins HL, Drazul-Schrader D, Sulpizio AC, et al. L-Carnitine intake and high trimethylamine N-oxide plasma levels correlate with low aortic lesions in ApoE(-/-) transgenic mice expressing CETP. Atherosclerosis. 2016;244:29–37. doi:10.1016/j.atherosclerosis.2015.10.108
32. Tahara A, Tahara N, Maeda-Ogata S, et al. Brown adipose tissue activation in severe heart failure. Eur Heart J. 2020;41(25):2415. doi:10.1093/eurheartj/ehaa241
33. Yoshida Y, Shimizu I, Shimada A, et al. Brown adipose tissue dysfunction promotes heart failure via a trimethylamine N-oxide-dependent mechanism. Sci Rep. 2022;12(1):14883. doi:10.1038/s41598-022-19245-x
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Clinical Value of Inflammatory Cytokines in Patients with Aneurysmal Subarachnoid Hemorrhage
Luo C, Yao J, Bi H, Li Z, Li J, Xue G, Li K, Zhang S, Zan K, Meng W, Zhang Z, Chen H
Clinical Interventions in Aging 2022, 17:615-626
Published Date: 26 April 2022
Diagnostic Value of a Novel Eosinophil Cationic Protein-Myeloperoxidase Test Paper Before and After Treatment for Allergic Rhinitis
Xi Y, Deng YQ, Li HD, Jiao WE, Chen J, Chen JJ, Tao ZZ
Journal of Asthma and Allergy 2022, 15:1005-1019
Published Date: 4 August 2022
Literature Overview of the Relation Between Psoriasis and Alzheimer
Yang Q, Wang J, Mi N, Zou Y
Neuropsychiatric Disease and Treatment 2023, 19:461-468
Published Date: 28 February 2023
Anti-Psoriatic Efficacies of Psorogrit and Divya-Taila, in Murine Models of Imiquimod and TPA-Induced Psoriasis-Like Inflammation are Driven by Modulation in IL-17RA/IL-23 and IL-8/TNF-α Signaling Axes

Balkrishna A, Sharma S, Dey T, Maity M, Shukla S, Kumari A, Tomer M, Dev R, Sinha S, Varshney A
Journal of Inflammation Research 2025, 18:5235-5259
Published Date: 18 April 2025
JAK-STAT Signaling in Autoimmunity and Cancer
Parveen S, Fatma M, Mir SS, Dermime S, Uddin S
ImmunoTargets and Therapy 2025, 14:523-554
Published Date: 11 May 2025