Back to Journals » ImmunoTargets and Therapy » Volume 13
Unveiling Dynamic Changes and Regulatory Mechanisms of T Cell Subsets in Sepsis Pathogenesis
Authors Jiang C, Chen J, Sun T ![]() , Xu J, Zhu H, Chen J
, Xu J, Zhu H, Chen J
Received 7 November 2023
Accepted for publication 17 January 2024
Published 1 February 2024 Volume 2024:13 Pages 29—44
DOI https://doi.org/10.2147/ITT.S448691
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Michael Shurin
Chunhui Jiang,1– 3 Jiani Chen,1– 3 Tong Sun,2,3 Jiaqin Xu,2,3 Hongguo Zhu,2,3 Jiaxi Chen1– 3
1School of Basic Medical Sciences & Forensic Medicine, Hangzhou Medical College, Hangzhou, People’s Republic of China; 2Clinical Laboratory, Taizhou Hospital of Zhejiang Province Affiliated to Wenzhou Medical University, Linhai, People’s Republic of China; 3Department of Laboratory Medicine, Enze Hospital, Taizhou Enze Medical Center (Group), Taizhou, People’s Republic of China
Correspondence: Jiaxi Chen, Clinical Laboratory, Taizhou Hospital of Zhejiang Province Affiliated to Wenzhou Medical University, 150 Ximen Street, Linhai, Zhejiang Province, 317000, People’s Republic of China, Tel +86-13606652901, Email [email protected]
Purpose: The pathogenesis of T cell subsets in sepsis during the body’s resistance to infection is currently unknown. We aimed to investigate the dynamics and molecular mechanisms of T cells during the development of sepsis.
Patients and Methods: Perform single-cell data analysis on peripheral blood mononuclear cells (PBMCs) specimen samples from seven healthy controls, five early-stage sepsis patients, and four late sepsis patients, and the atlas were mapped and analyzed using reference mapping to identify the T cell subpopulations specific to early sepsis. Expression network upstream to investigate the changes of regulatory transcription factors and pathways by pySCENIC.
Results: Twenty-two CD4+ T-cell subpopulations and 10 CD8+ T-cell subpopulations were identified by mapping analysis. At the early stage of sepsis, we observed altered ratios of multiple immune cells in PBMCs. Three cell types CD4 Tn cells, CD8 (GZMK+ early Tem), and CD8 (ZNF683+CXCR6− Tm) showed an upward trend (p < 0.05) in the early stages of sepsis compared to normal and returned to normal levels after two weeks. In addition, we found the presence of four sepsis-specific transcription factors (MXI1, CHD1, ARID5A, KLF9) in these three types of cells, which were validated in two external datasets. The differentially expressed genes in CD4 Tn cells, CD8 (GZMK+ early Tem), and CD8 (ZNF683+CXCR6− Tm) cells between the healthy group and the early-stage sepsis group are commonly enriched in the allograft rejection pathway. In addition, it was found that CD8 cells exhibit a trend towards differentiation into CD8 Temra cells in sepsis.
Conclusion: We successfully depicted the dynamic changes of T cell subsets during sepsis onset and progression, which provides important clues for an in-depth understanding of T cells’ function and regulatory mechanisms during sepsis pathogenesis.
Keywords: scRNA-seq, pySCENIC, GRN, reference mapping
Introduction
Sepsis, defined as life-threatening organ dysfunction caused by a dysregulated host response to infection, has become a major global health crisis.1 Globally, sepsis affects nearly 19 million people each year and may cause up to 5 million deaths.2 In 2017, the World Health Assembly and the World Health Organization listed sepsis as a global health priority.3 By delving into the complex immunological pathomechanisms of sepsis, we hope to provide more precise early diagnosis and treatment strategies to minimize the threat of sepsis to patients’ health.
T-cell exhaustion is not only involved in chronic viral infections and cancer,4 there have also been numerous articles demonstrating the impairment of T lymphocyte response in sepsis, accompanied by a rapid onset of T-cell exhaustion.5,6 However, the mechanisms underlying T-cell alterations in sepsis are not fully understood. Some viewpoints suggest that sepsis is characterized by lymphocyte reduction and immune dysfunction,5 with T-cell subsets playing a central role in the body’s immune response against infections. Therefore, T cells may be an essential aspect of the complex pathological mechanisms of sepsis, and understanding the mechanisms leading to T-cell dysfunction is crucial for preventing septic infections.
Recently, Zhang’s team constructed a pan-cancer single-cell landscape of tumor-infiltrating T cells,7 which divides T cells into subpopulations and searches for commonalities and differences among these T cells, revealing in more detail the broad-spectrum characteristics of T cells in terms of state, dynamics, and regulation. Based on this finding, we mapped T cells from sepsis single-cell data to the CD4 and CD8 T cell atlas created by Zhang’s team, yielding 22 CD4 T cell subpopulations and 10 CD8 T cell subpopulations. Previous studies have not categorized sepsis T cells into such detailed subpopulations. Our study deepens the understanding of T cell dynamics in sepsis and provides a new perspective on sepsis. In addition, our study also broadens the understanding of the dynamic changes of T cell subsets in the context of sepsis, revealing the molecular mechanisms and pathway changes of sepsis-associated T cell subsets. In conclusion, our work builds on existing studies to provide a more nuanced and comprehensive understanding of T cell changes in sepsis, thereby providing valuable insights for future therapeutic strategies and drug development.
Materials and Methods
Datasets
Single-cell RNA sequencing (scRNA-seq) data were downloaded from the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/.) database and subsequently analyzed using the Seruat8 package in R (Version 4.3.0.1). Our aim is to integrate two sets of data to examine the dynamic changes in various stages of sepsis. The first set of data is derived from sepsis patients diagnosed within 0 and 6 hours from GSE1673639. The second set of data originates from late-stage sepsis patients diagnosed 14–21 days post-sepsis from GSE17545310. Furthermore, we have utilized control groups of healthy individuals from these two datasets. To validate the expression differences of the selected genes in sepsis and healthy individuals, we have chosen GSE134347 and GSE65682 for validation, both of which are also derived from whole blood samples. All data, publicly disclosed in the GEO database, are summarized in Table 1, including details of all datasets utilized in this study. The study received approval from the Enze Hospital Ethics Committee (ethical number: K20230315), and informed consent was obtained from all participating individuals. The overall study design is illustrated in Figure 1.
 |
Table 1 The Details Pertaining to the Datasets Utilized in This Study |
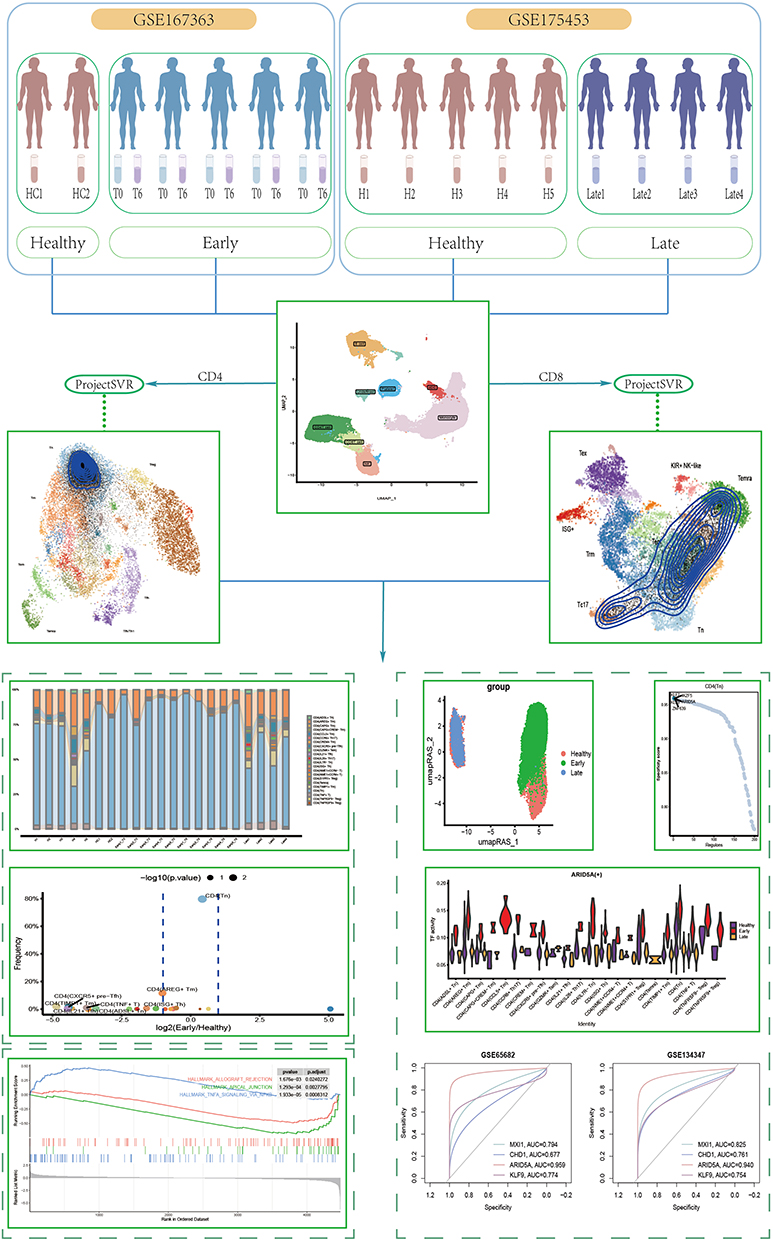 |
Figure 1 Scheme of the overall study design. |
Quality Control
Four quality control (QC) measures were used to screen cells before further analysis. Cell exclusion criteria were (1) several expressed genes less than 500 or more than 4000, (2) more than 15% UMIs of mitochondrial genes, (3) more than 3% UMIs of ribosomal genes, and (4) more than 25% hemoglobin percentage. (Supplementary Figure 1A and B) Through these screening steps, 91,616 cells that met the quality requirements were finally obtained and included in the subsequent analysis.
Subsequently, these cells were imported into the Seurat, and the expression matrix was normalized and scaled by applying the “NormalizeData”, “FindVariableFeatures”, and “ScaleData” functions sequentially. Principal Component Analysis (PCA) was performed with dim=29 set to explain 90% of the variance, with the remaining 10% of the variance attributed to noise. Harmony adjustment was used to remove the batch effect in the two datasets, and uniform manifold approximation and projection (UMAP) were calculated from the resulting Harmony matrix. Next, the graph-based clustering method constructed a shared nearest-neighbor graph using the “FindNeighbors” and identified clusters in the datasets using the “FindClusters” with the resolution of the clusters set to 0.26 (Supplementary Figure 1C).
Cell Annotation
In cell type annotation, different cell types are identified by selected markers. The following cell types are annotated and their corresponding markers: T cell (CD3D, CD3E, IL7R, CD8A, CD8B); B cell (CD79A, MS4A1); Macrophage (CD68, CD163, MS4A7, CD14); Monocyte (S100A9, S100A8, LYZ); cDC (CD1C, FCER1A); NK (GNLY, NKG7); Platelet (PPBP). Note that we manually removed cell clusters expressing two or more major lineage markers on the UMAP when performing cell clustering. This was done to avoid including mixed cells not recognized by Seurat in clusters of specific cell types.
Cell Fraction Calculation
Cell fractions for each significant cell lineage were calculated and compared across groups and individuals. Subsequently, subpopulation the relative abundance of each cell type in each sample was calculated based on statistical information about the sample or group and the percentage of CD4/CD8 T cell subpopulation types. We calculated normalized count matrices for T-cell subpopulations in the healthy and early sepsis groups. Data containing missing values were excluded during data processing. Statistical significance was determined using one-sided Wilcoxon rank-sum tests, and values with p-values < 0.05 and fold change (FC) values > 1 were screened. Finally, significance markers were added to the VolcanoPlot plots based on the statistical analysis results using the ggsignif13 package.
CD4 and CD8 T Cell Mapping
After cell annotation was completed, CD4 T cells and CD8 T cells were extracted individually, and genes overlapping with the reference gene set were selected from our data, followed by gene set score matrices based on own gene counts and the information of the reference gene set, which were calculated using the “ScoreSignatures_UCell” function in the UCell14 package. Finally, the resulting feature matrix was mapped to the reference model of Zhang’s team7 using the “ProjectNewdata” in the ProjectSVR15 package and normalized by applying the L2 paradigm.
Phenotypic Characterization of Exhausted Cells
To characterize the phenotypic profile of the cells, a set of markers was selected as reference including Exhaustion makers (PDCD1, TOX, CXCL13, TIGIT, CTLA4, TNFRSF9, HAVCR, LAG3), Cytokines and effector molecules (CST7, GZMK, GZMA, NKG7, IFNG, PRF1, GZMB, GNLY), Higher in GZMK+ Tex (CD28, EOMES, CCR5, CD200R1, TMEM155, CD27), Higher in terminal Tex (TOX2, TSHZ2, BATF, GEM Higher in terminal Tex (TOX2, TSHZ2, BATF, GEM, CD200, SNX9, ENTPD1, LAYN, ETV1, PRDM1, CSF1, MYO7A, MYO1E, GNG4), Higher in TCF7+ Tex (BTLA, FOXP3, TNFRSF4, TCF7, EEF1A1, SELL, CCR7, IL6R, IGFBP4, IGFL2). IGFL2).
Gene Regulatory Network Inference
Downscaling clustering was performed using the Seurat object, and the resolution of the clusters was specified as 50; the gene expression matrices of all meta cells were merged into a single matrix, and meta cells containing at least 10 cells were retained to construct the metacell. The metacells were filtered to exclude genes that were lowly expressed and not in the cisTargetDB. Reference data were downloaded from the cisTarget database (https://resources.aertslab.org/cistarget/), including (hg38_500bp_up_100bp_down_full_tx_v10_clust.genes_vs_motifs. rankings, hg38_10kbp_up_10kbp_down_full_tx_v10_clust.genes_vs_motifs.rankings, motifs-v10nr_clust-nr.hgnc-m0.001-o0.0.tbl). pySCENIC16 was used to evaluate the gene regulatory network (GRN). Finally, the activity of each regulon was scored using AUCell17 package. The Early group of cells was selected, and the results of pySCENIC were used to calculate the cell type indicated matrix, followed by the philentropy18 package to calculate the Jensen-Shannon divergence to identify cell-specific regulons.
KEGG and Differential Expression Enrichment Analysis (GSEA)
The “FindAllMarkers” function was used to find differences between the three groups of cells and identify CD4/CD8 subpopulation marker genes that were differentially expressed in the healthy and early sepsis groups. Subsequently, an analysis of KEGG pathways was performed using “clusterProfiler”19 Bioconductor.
In addition, differential gene expression analysis between the early sepsis and healthy groups was performed to identify marker genes and examine quantitative changes in expression levels between samples. CD4 Tn, CD8 (GZMK+ early Tem), and CD8 (ZNF683+CXCR6− Tm) differentially expressed genes (DEGs) were calculated using the “FindMarkers” function (Wilcoxon rank sum test). The clusterProfiler package assigned pathway activity estimates to individual cells. A gene set file (h.all.v7.2.symbols.gmt) containing 50 marker pathways was downloaded from the GSEA website (https://www.gsea-msigdb.org/gsea/index.jsp). By filtering the Benjamini-Hochberg corrected p-values, we identified pathways with significant differences.
Statistical Analysis
To assess the predictive sensitivity and specificity of the selected genes for sepsis progression, we used subject work characteristics (ROC) curves generated by the R. All statistical analyses were performed using R studio (version 4.2.3). The test methods used are described in the figure legends and the method, with P values provided in the figures, where P < 0.05 indicates statistical significance.
Results
Characterization of Immune Cell Response in Sepsis
In analyzing the changes in PBMCs during the development of sepsis, two sets of data were integrated and processed for quality control. Based on the expression of the corresponding typical marker genes (Figure 2A), the cells were classified into eight main categories: CD4 T cell, CD8 T cell, NK cell, B cell, platelet, monocyte, cDC, and an uncharacterized cell (Figure 2B). The expression of these cell categories was further elucidated by showing the expression of some characterized marker genes in the cells (Figure 2C). Each group has different cell clustering frequencies (Supplementary Figure 1D).
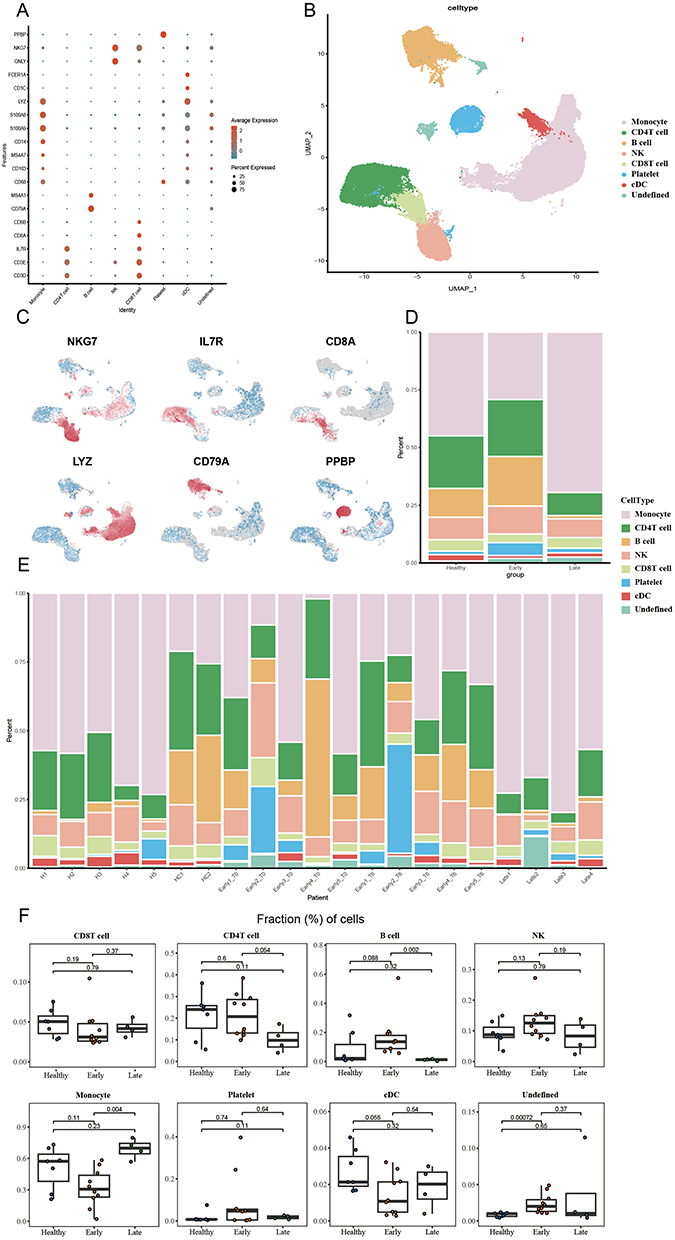 |
Figure 2 ScRNA-seq analysis. (A) Characteristic markers for all cells. (B) UMAP plots for all cells. (C) Projection of significant markers on UMAP plots. (D) Bar graph showing the proportion of major cell lineages in each group. (E) bar graph showing the proportion of significant cell lineages in each patient. (F) Proportion of cells between groups. A one-sided unpaired Wilcoxon test was used to calculate p-values. The center line indicates the median value, and the upper and lower hinge lines indicate the 75th and 25th percentiles, respectively. |
Calculations were performed for each patient and the proportions between the different cell types in the early and late sepsis and healthy group to further understand this difference. The results showed that CD4 T cells continued to decline in the early sepsis, whereas Monocyte, CD8 T cells, and Platelet tended to rebound after an early decline. On the other hand, Platelet, B cell, and NK cells rose in the early sepsis but showed a decline in the late stage. (Figure 2D-F).
Characterization of T Cell Subsets in Response to Sepsis
T cells play a central immune role in the body’s fight against infection, so a mapping study of CD4 T cell populations was carried out (Figure 3A), mapping 22 cell populations. By comparing the abundance of cells represented by each subpopulation (Figure 3B and C), eight cells of significance were screened out in the healthy and early sepsis groups (Figure 3D, Supplementary Figure 2): CD4 (ADSL+ Tn), CD4 (AREG+ Tm), CD4 (CXCR5+pre− Tfh), CD4 (IL21+ Tfh), CD4 (ISG+ Th), CD4 (TIMP1+ Tm), CD4 (Tn), CD4 (TNF+ T). Subsequently, these eight cells were compared between the three groups (Figure 3E). Surprisingly, in the healthy, the percentage of CD4 Tn cells was 76.90%. However, in patients with early sepsis, we observed an abnormally elevated CD4 Tn cell percentage of 91% (n=8559). As the disease progressed, the number of CD4 Tn cells gradually decreased. After two weeks of disease onset, the percentage of CD4 Tn cells decreased to 59% (n=693). In contrast, the number of the other seven cell types (CD4 (ADSL+ Tn), CD4 (ISG+ Th), CD4 (IL21+ Tfh), CD4 (AREG+ Tm), CD4 (TIMP1+ Tm), CD4 (CXCR5+Pre− Tfh), CD4 (TNF+ T)) decreased significantly in early sepsis. In late sepsis, the levels of cell count of all species gradually returned to a state similar to that of the healthy group.
 |
Figure 3 CD4 T cell subpopulation analysis. (A) Reference mapping of CD4 T cells. (B) Bar graph showing the proportion of CD4 cell subpopulations in each group. (C) Bar graph showing the proportion of CD4 T cell subpopulations in each patient. (D) Volcanic were used to compare the differences in CD4 T cell subsets between the early sepsis group and the healthy group with unilateral unpaired Wilcoxon test, and P-values were calculated. Points with p < 0.05 and FC value > 1 were marked on the atlas. (E) Proportions of CD4 cell subsets significantly differed between the Healthy and Early groups, and p-values were calculated using a one-sided unpaired Wilcoxon test. The center line indicates the median value, and the upper and lower hinge lines indicate the 75th and 25th percentiles, respectively. |
For CD8 T cells, a similar approach to that used for CD4 T cells was adopted, using the CD8 T cell atlas for mapping (Figure 4A), which successfully mapped 10 subpopulations of CD8 T cells and compared the abundance of each subpopulation relative to the total number of cells in each patient or between groups (Figure 4B and C). In early sepsis, we observed significant alterations in the abundance of different CD8 cell types relative to healthy individuals (Supplementary Figure 3). Specifically, CD8 (GZMK+ early Tem), CD8 (ZNF683+CXCR6− Tm), and CD8 (uncharacterized) cells were significantly up-regulated (p < 0.05), while CD8 (IL7R+ Tm), CD8 (MAIT/Tc17), CD8 (Tn), and CD8 (KIR+EOMES+ NK-like) cells exhibited significant down-regulation (p< 0.05) (Figure 4D). Comparison of the six cell types in the healthy group with those in the early and late stages of sepsis (Figure 4E) revealed that the median CD8 (GZMK+ Early Tem) and CD8 (ZNF683+CXCR6− Tm) cells in the early stage of sepsis were higher than those in the healthy group (CD8 (GZMK+ Early Tem): 42.82% vs 35.10%, p < 0.05; CD8 (ZNF683+CXCR6− Tm): 24.56% vs 12.91%, p < 0.05).Moreover, as the sepsis progressed to the late stage, cell proportions were gradually restored. Given that CD8 (uncharacterized) has no obvious characteristic markers, the focus will next be on CD8 (GZMK+ early Tem) and CD8 (ZNF683+CXCR6− Tm).
 |
Figure 4 CD8 T cell subpopulation analysis. (A) Reference mapping of CD8 T cells. (B) Bar graph showing the proportion of CD8 cell subpopulations in each group. (C) Bar graph showing the proportion of CD8 T cell subpopulations in each patient. (D) Volcanic maps were used to compare the differences in CD8 T cell subsets between the early sepsis group and the healthy group with unilateral unpaired Wilcoxon test, and P-values were calculated. Points with p < 0.05 and FC value > 1 were marked on the map. € Proportions of CD8 cell subsets that were significantly different between the Healthy and Early groups, and p-values were calculated using a one-sided unpaired Wilcoxon test. The center line indicates the median value, and the upper and lower hinge lines indicate the 75th and 25th percentiles, respectively. (F) Expression of Exhaustion markers in CD4 Tn, CD8 (GZMK+ early Tem), and CD8 (ZNF683+CXCR6− Tm) cells. |
Exhaustion Markers Reveal the Functional Potential of Three T Cell Subsets
Given the specificity of CD4 Tn, CD8 (GZMK+ early Tem), and CD8 (ZNF683+CXCR6− Tm) cells, we speculated whether these cells would be associated with sepsis T cell exhaustion. Therefore, the expression of exhaustion genes in these cells was observed by screening several exhausted cell markers. The expression of many markers of cytokines and effector molecules was significantly enriched in CD8 (GZMK+ early Tem) and CD8 (ZNF683+CXCR6− Tm) cells, as well as partially high expression of Higher in GZMK+ Tex markers such as CCL4, CCL5. In all three cells, we observed high expressions of both TCF7 and EEF1A1, while we did not see high expressions of terminal exhausted markers (PDCD1, TIGIT, and CTLA4 etc.). However, these results demonstrate that CD4 Tn cells may function similarly to TCF7+ Tex cells, play an essential role in sepsis, and regulate immune responses. (Figure 4F)
Transcription Factors and Pathway Regulation of T Cells in Sepsis
Calculation of regulon activity scores on pySCENIC results revealed that the CD4 T cell subset showed significant intergroup differences between healthy and early sepsis at the level of transcription factor activity, with more minor differences between late sepsis and healthy groups (Figure 5A). Five transcription factors, KLF9, IKZF5, ARID5A, ZNF639, and KLF7, were identified as key transcription factors in CD4 Tn cells. (Figure 5B). Violin plots revealed the expression of these five transcription factors, where KLF9, ARID5A, ZNF639, and IKZF5 showed high expression in early sepsis relative to healthy and late sepsis, and there was a significant intergroup difference among the three groups (Figure 5C). In contrast, the transcription factor KLF7 differences were insignificant (Supplementary Figure 4A). Further KEGG analysis revealed the pathways involved in the changes occurring in these cells (Supplementary Figure 4B). In particular, in CD4 Tn cells, significant oxidative phosphorylation and chemical carcinogenesis-reactive oxygen species occurred in early sepsis (Figure 5D). Furthermore, GSEA analysis revealed that five pathways, interferon-gamma response, and G2M checkpoint, were significantly activated in CD4 Tn cells. In contrast, apical junction, allograft rejection and PI3K AKT MTOR signaling, were significantly inhibited. (Figure 5E).
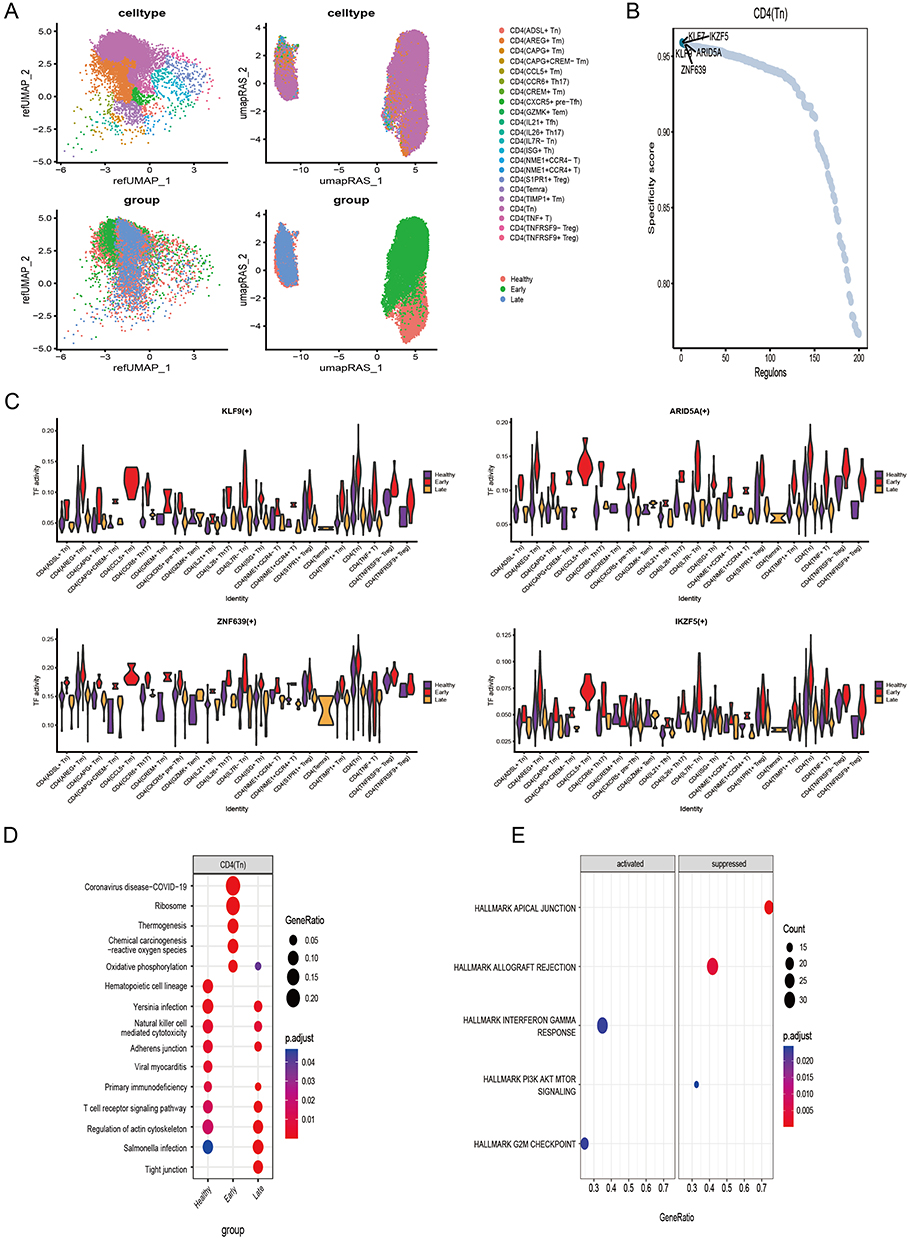 |
Figure 5 Transcription factors and functional analysis of CD4 Tn cells (A) UMAP plot of CD4 T cells with merged TF activity (B) CD4 Tn cell-specific transcription factors, the top five are labeled. (C) Highly expressed CD4 Tn specific transcription factors in the Early group. (D) KEGG analysis of CD4 Tn cells. (E) GSEA analysis of CD4 Tn cells. |
The CD8 T cell subpopulations also showed significant intergroup differences at the level of transcription factor activity. (Figure 6A) MXI1, GABPA, SP3, CHD1, and MAFF were observed to be important transcription factors maintaining CD8 (GZMK+ early Tem) cells; TFEB, TFDP2, SPI1, GFI1, and IKZF2 were observed to be important transcription factors maintaining CD8 (ZNF683+CXCR6− Tm) cells. (Figure 6B) Among these transcription factors, SP3, MXI1, CHD1, MAFF, TFEB, and TFDP2 were significantly different between groups and were highly expressed in the early sepsis (Figure 6C). In contrast, the other transcription factors (GABPA, IKZF2, SPI1, GFI1) did not show significant differences between groups (Supplementary Figure 5A). Some similarity between CD8 (GZMK+ early Tem), CD8 (ZNF683+CXCR6− Tm), and CD4 Tn cells was found by KEGG analysis (Figure 6D, Supplementary Figure 5B). In early sepsis, these cells exhibited significant oxidative phosphorylation and chemical carcinogenesis-reactive oxygen species enrichment. Interestingly, certain common features were found in GSEA analysis (Figure 6E) between CD8 (GZMK+ early Tem) and CD8 (ZNF683+CXCR6− Tm) cells and CD4 Tn cells, which consistently inhibited the allograft rejection pathway and apical junction. Besides, CD8 (GZMK+ early Tem) significantly activated TNFA signaling via the NFKB pathway.
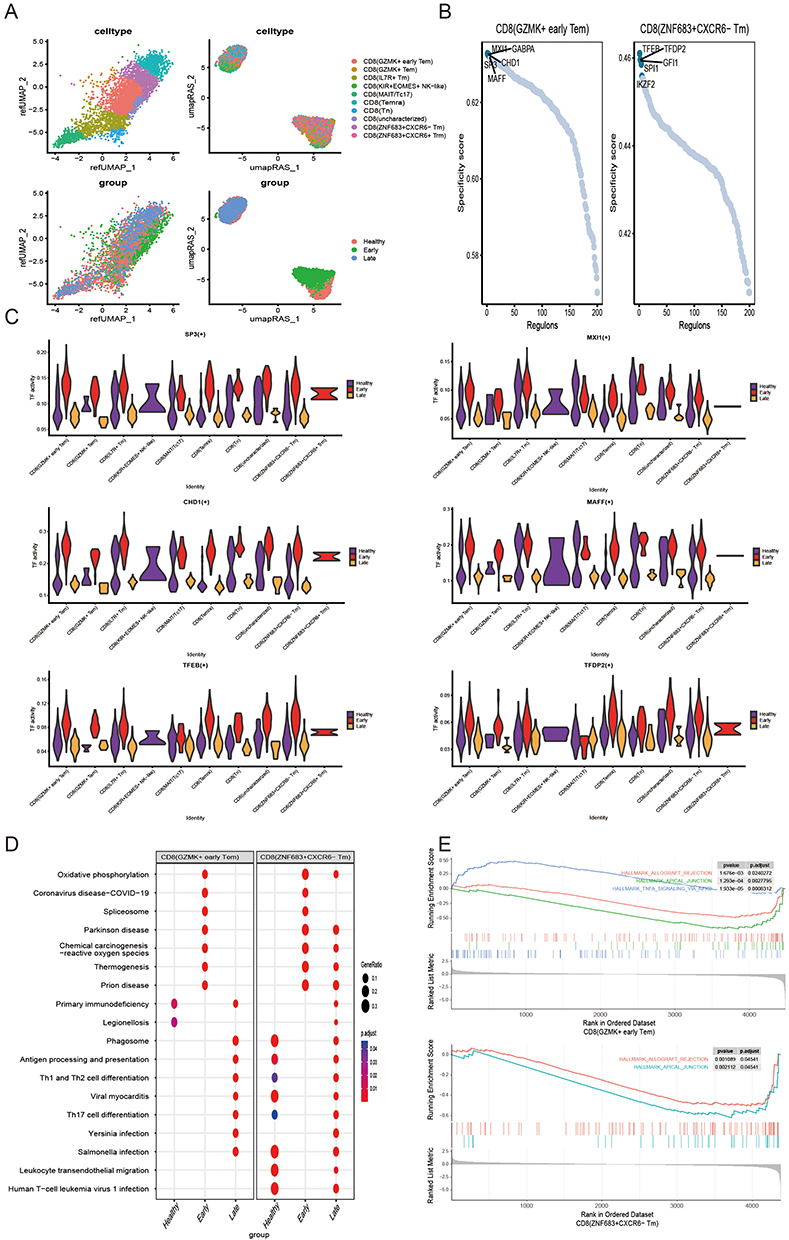 |
Figure 6 Transcription factors and functional analysis of CD8 (GZMK+ early Tem) and CD8 (ZNF683+CXCR6− Tm) cells (A). UMAP plot of CD8 T cells with merged TF activity (B). CD8 (GZMK+ early Tem) and CD8 (ZNF683+CXCR6− Tm) cell-specific transcription factors, the first five are labeled. (C) Early group of highly expressed CD8 (GZMK+ early Tem) and CD8 (ZNF683+CXCR6− Tm) cells specific transcription factors (CD8 (GZMK+ early Tem): SP3, MXI1, CHD1, MAFF; CD8 (ZNF683+CXCR6− Tm): TFEB, and TFDP2). (D) KEGG analysis of CD8 (GZMK+ early Tem) and CD8 (ZNF683+CXCR6− Tm) cells. € GSEA analysis of CD8 (GZMK+ early Tem) and CD8 (ZNF683+CXCR6− Tm) cells by GSEA analysis. |
Validation and ROC Analysis of Gene-Level Expression of Key Transcription Factors in Sepsis
To further investigate the expression patterns of the above ten transcription factors at the gene level, we examined their expression differences in single-cell datasets (Figure 7A, Supplementary Figure 6). We found that at the gene level, MXI1, CHD1, ARID5A, and KLF9 were significantly highly expressed in early sepsis and lowly expressed in the healthy group and late sepsis. These findings were subsequently validated in two other datasets, GSE65682 (Figure 7B) and GSE134347 (Figure 7C). The results showed that all four genes were highly expressed and statistically significant in sepsis. Overall, these findings suggest that these four genes can effectively differentiate sepsis patients among healthy individuals at the level of transcription factor activity and the genetic level.
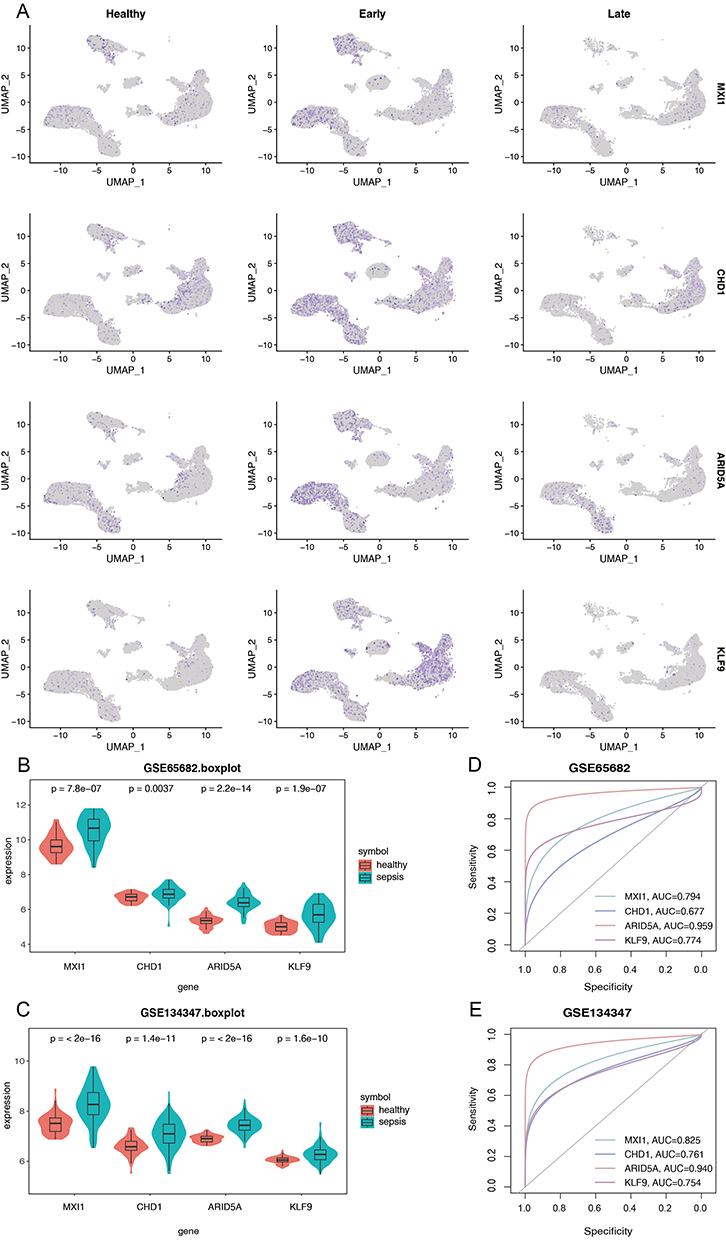 |
Figure 7 Validation of four important transcription factors (MXI1, CHD1, ARID5A, KLF9) at the gene level. (A) UMAP plots of four important transcription factors at the gene level. (B) Gene expression levels of four transcription factors in GSE65685. (C) Gene expression levels of four transcription factors in GSE134347. (D) ROC analysis of gene levels of four transcription factors in GSE65685. (E) ROC analysis of gene levels of four transcription factors in GSE134347. |
Subsequently, we evaluated the diagnostic potential of these four transcription factors as genetic markers for sepsis. (Figure 7D and E) The area under the curve (AUC) for each gene in the GSE65682 dataset was MXI1=0.794, CHD1=0.677, ARID5A=0.959, KLF9=0.774, and in the GSE134347 was MXI1=0.825, CHD1=0.761, ARID5A= 0.940, KLF9=0.754. These results indicate that MXI1, CHD1, ARID5A, and KLF9 have high AUC values differentiating sepsis from healthy, suggesting their potential as biomarkers for sepsis diagnosis.
Discussion
By single-cell sequencing and reference mapping analysis, this study depicted the changes in 22 CD4+ T cell subsets and 10 CD8+ T cell subsets during sepsis development, and CD8+ T cells were found to have the potential for Temra cell differentiation. The GRN analysis unveiled cell-specific transcription factors in CD4 Tn, CD8 (GZMK+ Early Tem), and CD8 (ZNF683+CXCR6−Tm) cells. It further demonstrated that these three cell types exhibit co-enrichment in pathways related to oxidative phosphorylation and the generation of chemically oncogenic reactive oxygen species. These findings shed light on the alterations occurring in T-cell subpopulations, immune function, and molecular mechanisms during the progression of sepsis.
Naïve CD4+ T cells patrolled mainly in lymph nodes, scanning for peptide-MHC-II class II molecular complexes on antigen-presenting cells, and upon activation of antigen-presenting cells, initial CD4+ T cells differentiated into effector T cells, regulatory T cells, or memory T cells.20 We observed abnormally elevated CD4 Tn cells in PBMCs of patients in the early stages of sepsis and a progressive decrease of these cells as the disease progressed, returning to a cellular state similar to that of the healthy group 2 weeks after the onset of the disease. However, there is a discrepancy with the findings of Tomasz Skirecki,21 who characterized T-cell proliferation by injection of BrdU and found a significant increase in the proportion of CD4 T naive cells in the lymph nodes of mice surviving sepsis for 14 days by flow cytometry, of which 72% were found in septic mice compared to 20.3% in normal controls. The reason for the differences may be differences in the timing of CD4 Tn cell proliferation caused by differences in the study population, and further studies are needed to explore these differences.
The present study found a significant reduction in the proportion of CD8 Tn cells in the early stages of sepsis. The authors Kitty P. Cheung found by flow cytometry as early as 2009 that during acute viral infections, when the number of lymphocytes decreases to a certain threshold, the organism undergoes an antigen-independent proliferation of T cells, and that in this case, naïve CD8 T cells progressively acquire the phenotype of memory T cells (CD44, CD122. expression of activation markers such as CD127 and Ly6C) and functional characteristics to provide enhanced protection against infection.22 In 2013, Stephanie A. Condotta found significant reductions in the number of early CD8 T cells in the blood, spleen, and inguinal lymph nodes of mice modeled for sepsis and suggested that sepsis induces changes in the composition of naïve CD8 T cell compartments, with a large proportion of CD8 T cells exhibiting a “memory-like” phenotype, thus contributing to the “memory” phenotype of CD8 T cells, and they suggested that sepsis induces changes in the composition of naïve CD8 T cell compartments, with many CD8 T cells displaying a “memory-like” phenotype, contributing to a reduction in the CD8 T cell response to new infection.23 In addition, we found that the proportion of CD8 (GZMK+ early Tem) and CD8 (ZNF683+CXCR6− Tm) cells tended to increase in the early stages of sepsis and returned to normal levels after two weeks. According to the findings of cell trajectory analysis in the literature, CD8 (GZMK+ early Tem) is an intermediate state cell of CD8 T cell with the potential to differentiate into both CD8 terminally depleted T cells and memory cells; in contrast to CD8 (GZMK+ early Tem), CD8 (ZNF683+CXCR6− Tm) is more inclined to differentiate into terminal memory cells.7 Thus, we found that CD8 T cells differentiated toward Temra cells in patients with early sepsis. According to other reports in the literature, T cell exhaustion is present in sepsis.24 However, the CD4 Tn, CD8 (GZMK+ early Tem), and CD8 (ZNF683+CXCR6− Tm) cells we focused on do not express markers associated with exhaustion. Furthermore, we identified upregulation of two highly expressed genes (TCF7, EEF1A1) in these three cell types, which are markers of TCF7+ Tex cells. Therefore, these three T-cell subgroups may have similar functionality to TCF7+ Tex cells, but they are not the primary drivers of T cell exhaustion in sepsis. The driving factors for T cell exhaustion in sepsis likely originate from other cell types.
Four important transcription factors were identified by analyzing the gene regulatory network of CD4 T and CD8 T cells: MXI1, CHD1, ARID5A, and KLF9. These four transcription factors differed significantly at the level of transcription factor activity and gene level in the early sepsis and healthy. Also, they are key transcription factors in maintaining the function of CD4 Tn, CD8 (GZMK+ early Tem), and CD8 (ZNF683+CXCR6− Tm) cells. Zaman suggested by mouse experiments that ARID5A regulates the enhancement of IL-6 and IFN-γ in response to endotoxin, which may act synergistically to amplify various other cytokines, ultimately leading to septic shock in mice, and that its antagonists targeting ARID5A expression may have therapeutic potential for preventing/controlling some of the pathologies of sepsis.25 KLF9 belongs to the evolutionarily conserved family of KLF transcriptional regulators of transcription factors associated with different types of cancer. It acts by altering participation in ROS metabolism. In advanced stages of melanoma, KLF9-dependent ROS signaling functions as a tumor suppressor, but it has rarely been reported in sepsis.26 MXI1 and CHD1 are tumor suppressors,27,28 but sepsis’s specific functions and regulatory mechanisms must be explored in depth.
KEGG analysis of CD4 Tn cells and CD8 (GZMK+ early Tem) and CD8 (ZNF683+CXCR6− Tm) revealed a statistically significant difference in the co-enrichment of these cells into oxidative phosphorylation and chemical carcinogenesis-reactive oxygen species generating pathways during the early stages of sepsis. Clinical and experimental evidence has amply demonstrated that sepsis patients exhibit overwhelming oxidative stress due to uncontrolled reactive oxygen species (ROS) and reactive nitrogen species (RNS).29 In addition, antioxidants have been reported to be beneficial in sepsis. In mice, injection of vitamin C prevented endotoxin-induced edema and hypotension.30 In a randomized controlled trial of 226 critically ill patients, patients receiving a combination of vitamins C and E had increased 28-day survival.31 Therefore, it is possible that CD4 Tn cells and CD8 (GZMK+ early Tem) and CD8 (ZNF683+CXCR6− Tm) trigger changes in oxidative phosphorylation and chemical carcinogenesis-reactive oxygen species pathways in sepsis through the mechanisms described above, and that antioxidants are effective in mitigating the development of sepsis. In addition, the results of GSEA showed that CD4 Tn cells and CD8 (GZMK+ early Tem) and CD8 (ZNF683+CXCR6− Tm) cells inhibited allograft rejection pathway activation. Consistent with our conclusions, it has been noted that both allograft rejection and autoimmune diseases are characterized by pathogenic responses triggered by naïve and memory T cells.32 Thus, the possibility exists that these three cells trigger suppression of the allograft rejection pathway in the immune response to sepsis. However, further experimental studies on these cells’ specific mechanism of action in sepsis are still needed.
In addition, there are some limitations to this study. First, the early sepsis samples were derived from patients who had just been diagnosed with sepsis and those who had been diagnosed with sepsis for 6 hours. Cell differentiation has not yet entirely occurred at this point; therefore, we did not observe the presence of terminally exhausted T cells. On the other hand, due to the limitation of current experimental technology, we need help to accurately distinguish different cell subpopulations by flow cytometry, which also limits the completion of subsequent experimental validation.
In summary, by analyzing the single-cell transcriptome, we found that CD4 Tn, CD8 (GZMK+ early Tem), and CD8 (ZNF683+CXCR6−Tm) cell subsets were abnormally increased in the early stage of sepsis and produced a series of pathological changes, which successfully depicted the dynamic changes of the T cell subsets in the process of sepsis onset and progression, and contributed to the in-depth understanding of T cell function and regulation in the pathogenesis of sepsis.
Conclusion
In this study, we aimed to investigate the molecular mechanisms of T-cell subsets in the development of sepsis and the body’s resistance to infection. By analyzing single-cell data, we determined that CD4 Tn cells, CD8 (GZMK+ early Tem) cells, and CD8 (ZNF683+ CXCR6- Tm) cells tended to be elevated from normal levels in the early stages of sepsis and returned to normal after two weeks. Furthermore, we identified sepsis-specific transcription factors (MXI1, CHD1, ARID5A, KLF9) in these T-cell subsets. In conclusion, our study successfully characterized the dynamic changes of T cell subsets during sepsis onset and progression and revealed the function and regulatory mechanisms of T cells in sepsis pathogenesis.
Acknowledgments
The authors gratefully acknowledge all participants and staff for their contribution to the study.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Singer M, Deutschman CS, Seymour CW., et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315(8):801–810. doi:10.1001/jama.2016.0287
2. Fleischmann C, Scherag A, Adhikari NK, et al. Assessment of Global Incidence and Mortality of Hospital-treated Sepsis. Current Estimates and Limitations. Am J Respir Crit Care Med. 2016;193(3):259–272. doi:10.1164/rccm.201504-0781OC
3. Reinhart K, Daniels R, Kissoon N, Machado FR, Schachter RD, Finfer S. Recognizing Sepsis as a Global Health Priority - A WHO Resolution. New Engl J Med. 2017;377(5):414–417. doi:10.1056/NEJMp1707170
4. Yao C, Lou G, Sun HW, et al. BACH2 enforces the transcriptional and epigenetic programs of stem-like CD8(+) T cells. Nature Immunology. 2021;22(3):370–380. doi:10.1038/s41590-021-00868-7
5. Boomer JS, To K, Chang KC, et al. Immunosuppression in patients who die of sepsis and multiple organ failure. JAMA. 2011;306(23):2594–2605. doi:10.1001/jama.2011.1829
6. Chang K, Svabek C, Vazquez-Guillamet C, et al. Targeting the programmed cell death 1: programmed cell death ligand 1 pathway reverses T cell exhaustion in patients with sepsis. Critical Care. 2014;18(1):R3. doi:10.1186/cc13176
7. Zheng L, Qin S, Si W, et al. Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science. 2021;374(6574):abe6474. doi:10.1126/science.abe6474
8. Stuart T, Butler A, Hoffman P, et al. Comprehensive Integration of Single-Cell Data. Cell. 2019;177(7):1888–1902.e21. doi:10.1016/j.cell.2019.05.031
9. Qiu X, Li J, Bonenfant J, et al. Dynamic changes in human single-cell transcriptional signatures during fatal sepsis. J Leukocyte Biol. 2021;110(6):1253–1268. doi:10.1002/jlb.5ma0721-825r
10. Darden DB, Dong X, Brusko MA, et al. A Novel Single Cell RNA-seq Analysis of Non-Myeloid Circulating Cells in Late Sepsis. Front Immunol. 2021;12:696536. doi:10.3389/fimmu.2021.696536
11. Scicluna BP, Uhel F, van Vught LA, et al. The leukocyte non-coding RNA landscape in critically ill patients with sepsis. eLife. 2020;(9):57. doi:10.7554/eLife.58597
12. Scicluna BP, Klein Klouwenberg PM, van Vught LA, et al. A molecular biomarker to diagnose community-acquired pneumonia on intensive care unit admission. Am J Respir Crit Care Med. 2015;192(7):826–835. doi:10.1164/rccm.201502-0355OC
13. Patil A-ECaI. ggsignif: r Package for Displaying Significance Brackets for ‘ggplot2’. PsyArxiv. 2021. doi:10.31234/osf.io/7awm6
14. Andreatta M, Carmona SJ. UCell: robust and scalable single-cell gene signature scoring. Comput Struct Biotechnol J. 2021;19:3796–3798. doi:10.1016/j.csbj.2021.06.043
15. Gao J. ProjectSVR: projecting Query scRNA-seq Data Onto A Reference Atlas Via Support Vector Regression. Int J MEd. 2023.
16. Van de Sande B, Flerin C, Davie K, et al. A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nature Protocols. 2020;15(7):2247–2276. doi:10.1038/s41596-020-0336-2
17. Aibar S, González-Blas CB, Moerman T, et al. SCENIC: single-cell regulatory network inference and clustering. Nature Methods. 2017;14(11):1083–1086. doi:10.1038/nmeth.4463
18. Drost H-G. Philentropy: information Theory and Distance Quantification with R. J Open Source Software. 2018;3:765.
19. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. j Integrative Biol. 2012;16(5):284–287. doi:10.1089/omi.2011.0118
20. Dong C. T cells: the usual subsets. Nat Rev Immunol. 2010.
21. Skirecki T, Swacha P, Hoser G, Golab J, Nowis D, Kozłowska E. Bone marrow is the preferred site of memory CD4+ T cell proliferation during recovery from sepsis. JCI Insight. 2020;5(10):447. doi:10.1172/jci.insight.134475
22. Cheung KP, Yang E, Goldrath AW. Memory-like CD8+ T cells generated during homeostatic proliferation defer to antigen-experienced memory cells. J Immunol. 2009;183(5):3364–3372. doi:10.4049/jimmunol.0900641
23. Condotta SA, Rai D, James BR, Griffith TS, Badovinac VP. Sustained and incomplete recovery of naive CD8+ T cell precursors after sepsis contributes to impaired CD8+ T cell responses to infection. J Iimmunol. 2013;190(5):1991–2000. doi:10.4049/jimmunol.1202379
24. Martin-Loeches I, Povoa P, Poulakou G. Focus on infection. Intensive Care Med. 2020;46(4):787–789. doi:10.1007/s00134-020-05995-7
25. Zaman MM, Masuda K, Nyati KK, et al. Arid5a exacerbates IFN-γ-mediated septic shock by stabilizing T-bet mRNA. Proc Natl Acad Sci USA. 2016;113(41):11543–11548. doi:10.1073/pnas.1613307113
26. Arslanbaeva LR, Santoro MM. Adaptive redox homeostasis in cutaneous melanoma. Redox Biol. 2020;37:101753. doi:10.1016/j.redox.2020.101753
27. Huang Y, Hu K, Zhang S, et al. S6K1 phosphorylation-dependent degradation of Mxi1 by β-Trcp ubiquitin ligase promotes Myc activation and radioresistance in lung cancer. Theranostics. 2018;8(5):1286–1300. doi:10.7150/thno.22552
28. Augello MA, Liu D, Deonarine LD, et al. CHD1 Loss Alters AR Binding at Lineage-Specific Enhancers and Modulates Distinct Transcriptional Programs to Drive Prostate Tumorigenesis. Cancer Cell. 2019;35(4):603–617.e8. doi:10.1016/j.ccell.2019.03.001
29. Delano MJ, Ward PA. The immune system’s role in sepsis progression, resolution, and long-term outcome. Immunol Rev. 2016;274(1):330–353. doi:10.1111/imr.12499
30. Shen KP, Lo YC, Yang RC, Liu HW, Chen IJ, Wu BN. Antioxidant eugenosedin-A protects against lipopolysaccharide-induced hypotension, hyperglycaemia and cytokine immunoreactivity in rats and mice. J Pharm Pharmacol. 2005;57(1):117–125. doi:10.1211/0022357055137
31. Crimi E, Liguori A, Condorelli M, et al. The beneficial effects of antioxidant supplementation in enteral feeding in critically ill patients: a prospective, randomized, double-blind, placebo-controlled trial. Anesthesia Analg. 2004;99(3):857–863. doi:10.1213/01.Ane.0000133144.60584.F6
32. Mathews DV, Dong Y, Higginbotham LB, et al. CD122 signaling in CD8+ memory T cells drives costimulation-independent rejection. J Clin Invest. 2018;128(10):4557–4572. doi:10.1172/jci95914
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.