Back to Journals » Open Access Rheumatology: Research and Reviews » Volume 16
Towards Personalized Medicine in Rheumatoid Arthritis
Received 4 January 2024
Accepted for publication 3 May 2024
Published 18 May 2024 Volume 2024:16 Pages 89—114
DOI https://doi.org/10.2147/OARRR.S372610
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Chuan-Ju Liu
Seema D Sharma, James Bluett
Centre for Musculoskeletal Research, Division of Musculoskeletal & Dermatological Sciences, School of Biological Sciences, University of Manchester, Manchester, UK
Correspondence: James Bluett, Centre for Musculoskeletal Research & Dermatological Sciences, Division of Musculoskeletal & Dermatological Sciences, School of Biological Sciences, The University of Manchester, 1.204 AV Hill, Oxford Road, Manchester, M13 9PT, UK, Email [email protected]
Abstract: Rheumatoid arthritis (RA) is a chronic, incurable, multisystem, inflammatory disease characterized by synovitis and extra-articular features. Although several advanced therapies targeting inflammatory mechanisms underlying the disease are available, no advanced therapy is universally effective. Therefore, a ceiling of treatment response is currently accepted where no advanced therapy is superior to another. The current challenge for medical research is the discovery and integration of predictive markers of drug response that can be used to personalize medicine so that the patient is started on “the right drug at the right time”. This review article summarizes our current understanding of predicting response to anti-rheumatic drugs in RA, obstacles impeding the development of personalized medicine approaches and future research priorities to overcome these barriers.
Keywords: genetics, omics, biomarkers, machine learning, precision medicine
Introduction
Rheumatoid arthritis (RA) is a chronic multisystem inflammatory disease affecting up to 1% of the adult population.1 The disease is characterized by synovitis, which leads to joint erosions and disability, and is associated with a reduced life expectancy of 5 years.2 Treatment is based upon a treat-to-target approach, with rapid escalation of therapy when the target is not achieved.3 Therapies that target the underlying pathogenic mechanism of disease, such as the TNF inhibitor (TNFi) adalimumab, have been available since 2002. Since the introduction of the first biologic disease modifying anti-rheumatic drug (bDMARD) targeting TNFα, there has been an exponential growth in the number of targeted RA therapies. Despite the incredible technological progress made in drug development in RA, no disease modifying anti-rheumatic drug (DMARD) is universally effective and TNFi fail to attain remission in up to 70% of patients.4 Current RA management is therefore based on a “trial and error” approach where time on an ineffective medication leads to pain, increased healthcare costs and disease progression.5
RA is a heterogenous disease, but its current management assumes that there are common pathogenic pathways that can be targeted and treated in all patients. There now exists an armamentarium of drugs targeting different aspects of the immune system and, with such a vast choice of treatment, personalized medicine approaches are required to guide therapeutic decisions. Rather than a “one size fits all”, personalized medicine is the targeted treatment of disease based on the individual characteristics of the patient.6 Personalized medicine recognizes that RA is heterogenous and that patients are individuals with a unique biomarker signature that, combined with lifestyle and clinical factors, may be a predictive biomarker for treatment response to enable management of patients according to the “right drug, right patient, right time”.7
RA is a disease that is ideal for personalized medicine discovery research. As a common disease that affects 1% of the population, it is amenable to the recruitment of large cohorts of patients for biomarker discovery studies. RA has a heritability of ~60%,8 with >120 single nucleotide polymorphisms (SNPs)9 associated with disease susceptibility. Progress in understanding the genetics of RA has improved our knowledge of its heterogeneity and the underlying disease mechanisms. For example, HLA is responsible for 18% of the genetic variance of heritability of anti-citrullinated protein antibodies (ACPA)-positive disease but only 2% for ACPA-negative disease.10 This greater understanding of the genetic architecture of RA may assist with pharmacogenomic biomarker discovery.
It is likely that, rather than one RA biomarker that can predict response, and thus aide personalized medicine approaches, a panel of biomarkers exists, with data that is integrated from different sources, eg, genomic, clinical and lifestyle, which can predict drug response. The discovery of predictive biomarkers or a biomarker signature of a drug response would lead to the use of the most effective therapy, a treatment strategy that is more cost-effective compared to time on an ineffective drug. In the future it is hoped that a patient’s unique biomarker signature would be measured and the most clinically effective treatment targeting the underlying pathogenesis would be started early in the course of the disease to prevent joint damage and disability (Figure 1).
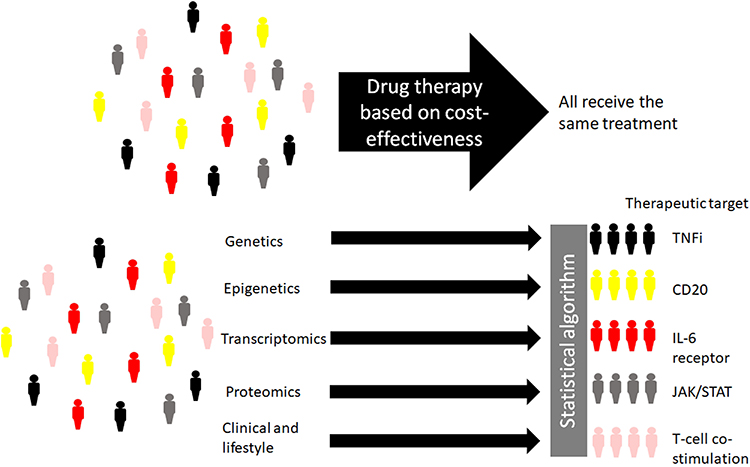 |
Figure 1 Illustration of how personalized medicine approaches using biomarkers and clinical predictors of treatment outcome can be applied to select a therapeutic target with an increased likelihood of response for the individual patient. Abbreviations: TNFi, Tumor Necrosis Factor inhibitor; IL-6, Interleukin-6; JAK/STAT, Janus kinase/signal transducer and activator of transcription proteins. |
The aim of this review article is to summarize the current understanding of predictors of response to anti-rheumatic drugs in RA, obstacles impeding the development of personalized medicine approaches and future research priorities that may overcome these barriers.
Clinical Factors Predicting Drug Response
Age
The incidence of RA increases with age, with a peak incidence of 75–84 years.11,12 Older age has been associated with a reduced response to combination conventional disease-modifying anti-rheumatic drugs (csDMARDs)13 and TNFis14–17 in several studies. One of the largest of the studies was a prospective observational cohort study by Atzeni et al,15 which sought to investigate predictors of response to TNFi therapy in established RA patients (n=1300) with at least moderate disease activity (DAS-28 >3.2). The study analyzed patients starting TNFis available at the time and showed that, whilst younger age was associated with a higher probability of a good EULAR response, its effect was, however, modest (OR=0.98, p=0.002).
Biological Sex
Several observational and clinical trials have consistently demonstrated sex differences that predict drug response, with female sex associated with reduced response to combination csDMARDs13 and TNFi.13,15–21 The prospective observational British Society for Rheumatology Biologics Register analyzed 2879 RA patients commencing etanercept or infliximab and showed that females were 42% less likely to experience EULAR remission by six months compared to males (OR=0.58; 95% CI: 0.39–0.87).
Smoking
Smoking is associated with a two-times increase in the risk of developing RA in people who have a 20-pack per year history compared with non-smokers.22 Several studies have shown that smokers experience worse outcomes compared to non-smokers to methotrexate,23 infliximab19 and combination therapy18 but, interestingly, not rituximab.24 The reason for worse clinical outcomes in smokers is not fully understood but smoking is associated with increased levels of pro-inflammatory cytokines such as TNFα25 that may, in part, explain why TNFi therapy is less effective in smokers compared to non-smokers whilst there appears to be no modifying effect to B-cell depletion therapy.
Disease Activity
High disease activity is associated with poor clinical outcomes, including radiographic progression.26 High baseline disease activity is associated with improved response to bDMARDs, including tocilizumab.14,27,28 It has been hypothesized that the improved response in patients with high disease activity is due to the significant degree of biological inflammation that is amenable to pro-inflammatory cytokine inhibition.
BMI
Obesity has been shown to worsen clinical outcomes in patients commencing DMARD therapy.18,29 The results of the Swedish pharmacotherapy trial (SWEFOT) of early RA patients (n=260) showed that obesity was associated with worse disease activity, functional impairment and pain following DMARD therapy at 24 months compared with non-obese patients. Additionally, obese patients were five times less likely to be in remission at 24 months (ORadj=5.2; 95% CI: 1.8–15.2). There are several potential mechanisms underlying poor clinical outcomes in this patient group. Adipose tissue is biologically active and obesity is associated with an increase in pro-inflammatory cytokines, including TNFα and IL-6,30 as well as an increase in load of weight-bearing joints. Additionally, raised BMI has been associated with reduced bDMARD drug levels through reduced absorption and increased drug clearance.31–33 It may be postulated, therefore, that switching obese patients to weight-based therapies such as intravenous (IV) TOC to personalize therapy may lead to improved disease control. The MUSASHI study by Ogata et al analyzed the efficacy of tocilizumab in patients who switched from weight-adjusted IV tocilizumab to subcutaneous tocilizumab and showed that, whilst serum tocilizumab levels decreased, clinical remission, measured by DAS(ESR)-28, was maintained for the majority of patients; only 9% experienced a recurrence.33 It is not known, however, if this is true for other bDMARDs.
Non-Adherence
Non-adherence is a health behavior that is more common than previously thought, with approximately 1 in 4 patients being non-adherent to methotrexate34 and subcutaneous bDMARDs,35 which is associated with poor treatment response. Clinicians are often unaware of adherence behavior, which can be associated with psychological factors. Directly observed therapy is an intervention whereby a healthcare worker observes medication administration. Whilst typically used for patients with tuberculosis,36 several bDMARDs are available as IV preparations that require healthcare worker administration and can be a treatment strategy that guarantees therapy has been administered.
Summary of Clinical Prediction Guiding Personalized Medicine Approaches
Overall, clinical characteristics remain a modest predictor of response, at best. There are, however, some consistent findings but many are inherent to poor prognostic predictors of RA progression rather than treatment response per se. Additionally, it remains unclear if clinical predictors of a particular therapeutic agent would be similar to an alternative drug with a different mechanism of action that could help to guide personalized therapy. Further real-world studies are required to develop clinical risk prediction tools to individual drugs that can help guide personalized therapy in the future.
Omics
Considerable efforts made to elucidate the molecular pathways that underpin a large spectrum of complex human diseases have led to a rapid explosion of various “omics” technologies. These include genomics, epigenomics, transcriptomics, proteomics, metabolomics and other areas. New methodologies have produced unprecedented resolution and cost-efficient technologies allowing for the generation of large-scale data. We discuss the current landscape in RA for the omics fields and how they have been applied to try and identify indicators of treatment response to pave the way to personalized medicine approaches. The most prominent findings from the literature are summarized in Figure 2.
 |
Figure 2 Summary of potential biomarker candidates for treatment response in RA, as identified in the literature. Rather than an exhaustive review of all previously associated predictors of treatment response, this is an overview. References: Genetics: SLC19A1,37–39 IL10,40,41 PDE3A-SLCO1C1,42,43 PDZD2,44,45 PTPRC,46–49 CD84,50,51 CD69.28,52 References for transcriptomics, proteomics, metabolomics and epigenetics are shown in corresponding tables. |
Pharmacogenomics
Pharmacogenomics is the study of genomic variation and drug response.53 The use of pharmacogenomics to personalize therapeutic drug decisions offers great potential. Our genetics are fixed from birth and can therefore be measured at any time whilst remaining unique, thus offering the potential to truly personalize therapy. Since the human genome was first sequenced in 2003 by the Human Genome Project the cost of sequencing has significantly reduced.
Massey et al explored the heritability of TNFi response from a large (n=1752) UK RA patient cohort enrolled on an observational cohort study.54 The study showed that change in swollen joint count (SJC) and erythrocyte sedimentation rate (ESR) were heritable (restricted maxiumum likelihood [REML] estimates of 0.48 and 0.39, respectively); however, the more subjective measures of tender joint count (TJC) and patient global assessment (visual analog scale [VAS]) that can be inflated by non-inflammatory conditions55 were not (REML estimates of 0.00 and 0.00, respectively). Following the Massey et al study, a two-component DAS-28 was developed which excluded the TJC and VAS and could better predict radiographic progression.56 A study by Gilani et al showed that when utilizing the 2C-DAS28 more single nucleotide polymorphisms (SNPs) were replicated with TNFi response compared to the 4C-DAS-28.57 These observations suggest that development of clinical phenotypes that more closely resemble disease activity may aid discovery of pharmacogenomic biomarkers, leading to stronger associations that could be investigated as part of a stratified clinical trial.
SNP associations with risk of developing severe joint damage or response to B-cell depletion therapy have been discovered in key genes that are targets for therapy such as the IL-6R58 and IL-6 genes,59 demonstrating the power of pharmacogenomics to identify potential treatment targets. To date, genome-wide association studies (GWAS) have identified >30 SNPs associated with TNFi response.40,42,44,46,50,54 One of the strongest effect sizes for being a TNFi non-responder was seen in the rs6028945 SNP of the MAFB gene (p=6x10−3; OR: 11.2),40 a transcription factor regulating macrophage differentiation.60 However, the target gene for many of the variants discovered through pharmacogenomic studies is still uncertain, as DNA folding can lead to associated variants regulating genes far from the SNP identified. Understanding the target gene and underlying mechanism of treatment response could pave the way to more personalized medicine approaches. Genome editing studies may lead to a greater understanding of how these variants affect gene transcription to alter human physiology and drug response.
Whilst various SNP associations with treatment response have been discovered, it is likely that no one SNP will be able to predict in the clinic which drug an individual will respond to. Through the integration of multiple predictive SNPs, a polygenic risk score providing a weighted single score may, in future, guide clinicians to which drug a patient is more likely to respond to.61
Epigenetics
The field of epigenetics explores inheritable changes to the chromatin structure that are not the result of alteration of the nucleotide sequence of DNA. These changes occur through several mechanisms, including DNA methylation, histone modification and RNA-associated silencing. These chemical covalent and noncovalent modifications to DNA molecules and histones are pivotal in influencing gene expression.62
Epigenetic modifications are inheritable, but they are reversible and can be altered by environmental exposure.63 Potentially, epigenetic markers could be monitored over time to assess efficacy of a treatment or to consider individualized dosing adjustment. We summarize studies that investigate epigenetic predictors of response to treatment in Table 1.
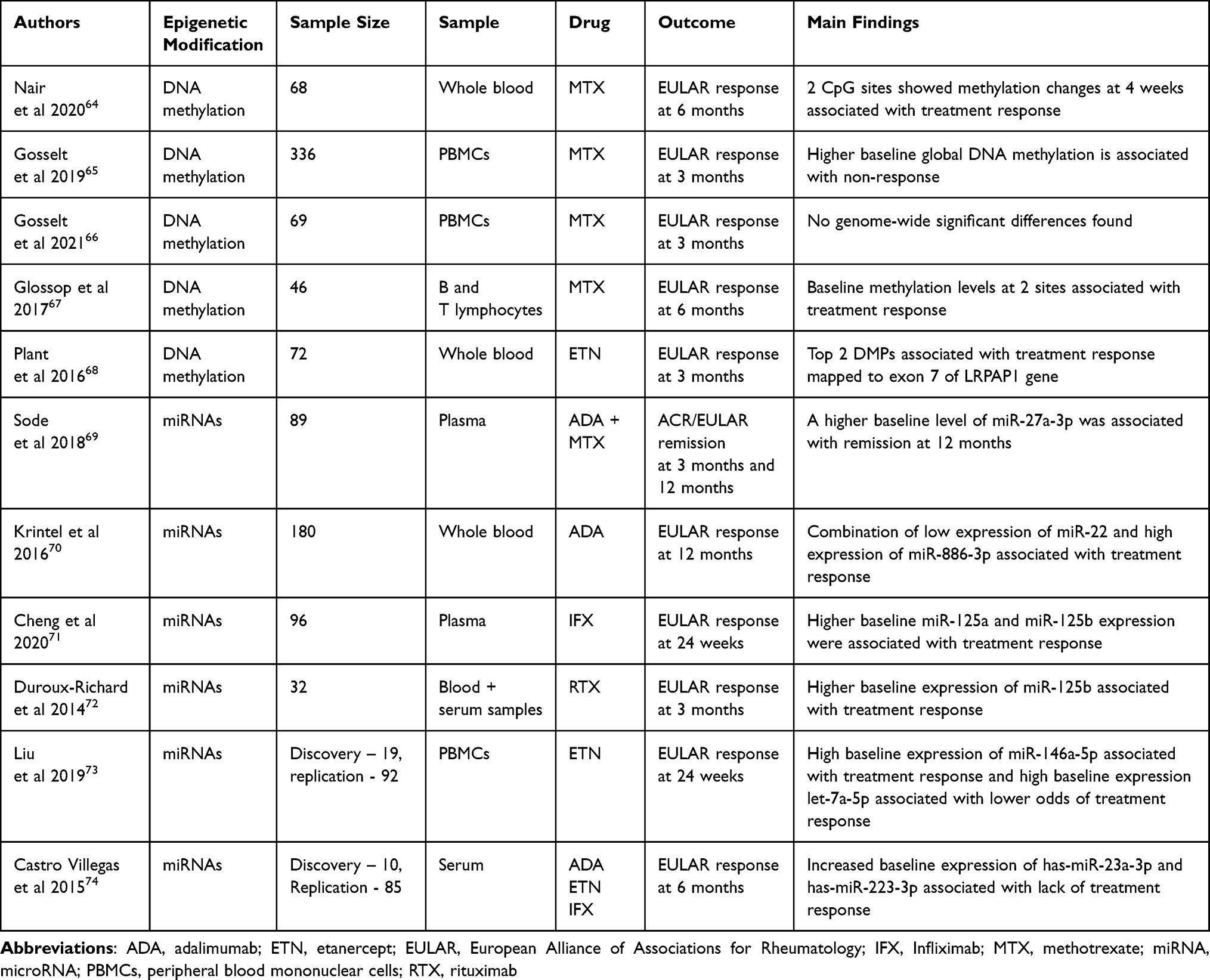 |
Table 1 Summary of Epigenetic Studies Investigating Predictors of Response to Treatment |
Among epigenetic modifications, DNA methylation is the most thoroughly investigated. This process is the addition of a methyl group to cytosine–uanine dinucleotides, referred to as CpGs. Approximately 70–80% of CpGs in the human genome are methylated. There are areas in the genome where CpGs tend to occur together in clusters, known as CpG islands. Methylation is thought to impact on the interaction between transcription factors and DNA. Therefore, in regions where there are active genes, CpGs tend to be hypomethylated. However, in those regions where CpGs are hypermethylated, there tends to be a decrease in gene expression.75 DNA methylation has been proposed in several studies to be a predictor of treatment response in RA, both prior to treatment and within 1–3 months of treatment, the purpose of which would be to allow earlier therapeutic changes.
From research arising from the MAximising Therapeutic Utility in Rheumatoid Arthritis (MATURA) consortium, Nair et al 2020 hypothesized that differential methylation patterns could help identify patients recently started on methotrexate therapy who would be unlikely to respond to therapy. Two CpG sites (cg21040096 [RPH2AL] and cg09894276 [WDR27]) showed methylation changes at 4 weeks and were associated with EULAR response at 6 months (p=2.79×10−7 and 3.62×10−7, respectively.64 However, there were no significant associations at baseline. In contrast, Glossop et al found a correlation between baseline methylation patterns at cg03018489 and cg14345882 in T-lymphocytes and EULAR response at 6 months. These sites were located near the ADAMTSL2 and BTN3A2 genes. Most patients (93.5%) were commencing methotrexate, however 65.2% were commencing at least one other DMARD in addition (sulfasalazine/hydroxychloroquine).67 The associations failed to replicate, however, in another study of patients commencing DMARD therapy.66
In a study of 72 patients, Plant et al identified two differentially methylated regions (DMPs) at baseline associated with response to etanercept. These mapped to the LRPAP1 gene, and methylation levels correlated with the rs3468 genotype. This SNP was also associated with EULAR response in 1204 patients treated with TNFi.68
Another category of epigenetic changes are histone modifications. Histone proteins bind to DNA influencing the accessibility of gene promotors for transcription factor binding. Modifications include acetylation, methylation, phosphorylation and citrullination. These processes intricately regulate gene expression and some are associated with chromatin structure.76 Acetylation and methylation are associated with an aggressive phenotype of synovial fibroblasts in RA, through increased expression of matrix metalloproteinases (MMP-1, MMP-3, MMP9, MMP-13), which cause degradation of proteoglycans and damage to collagen.77–79 However, to date no studies have investigated whether histone modifications may predict future treatment response. Indeed, they are not ideal biomarkers in view of current technical challenges, thus translational potential is likely limited.77
MicroRNAs (miRNAs) are small non-coding RNA molecules that bind to messenger RNAs (mRNAs) and therefore can suppress gene expression at the post-transcriptional stage. These epigenetic modifications are thought to be integral players in the regulation of diverse cellular responses. Immune dysregulation due to expression of specific miRNAs in RA has been observed, and some studies have explored whether changes in miRNA profiles can be used to predict treatment response.69–74
Most studies evaluating the predictive utility of miRNAs have focused on TNFi. Associations found include miR-27a-3p,69 miR-22,70 miR-886-3p,70 miR-125a,71 miR-125b,71 miR-146a-5p,73 let-7a-5p,73 miR-23a-3p,74 and miR-223-3p.74 Although further validation is required, there does appear to be some agreement across certain studies, which is promising. For example, high levels of miR-146a-5p were associated with EULAR response at 24 weeks to etanercept using PBMCs from 92 patients at baseline (OR: 1.508 [1.127, 2.018]; p=0.006).73 Higher levels of miR-146a-5p were also found in the serum of patients after 3 months of TNFi.74,80 Interestingly, miR-125b has also been associated with response to rituximab.72 Both miRNA-146-5p and miR-125b are thought to be involved in the NK-κB signaling pathway, which, through downstream effects, induces cytokines and chemokines such as TNF-α and IL-6 that are key drivers of RA inflammation.81,82
In summary, the potential of epigenetic markers as biomarkers of future treatment response may be limited due to their relative instability in the face of environmental factors in comparison to other omics fields. However, studies have highlighted important potential mechanisms of pathogenesis.
Transcriptomics
Transcriptomics refers to the examination of global gene expression profiles to uncover associations with a diverse array of traits. Gene expression is highly dynamic and can provide valuable insights into disease pathways as well as response to drug treatments. The total sum of all RNA molecules expressed at any one time in a biological specimen is termed the transcriptome. Various techniques are used to investigate differential gene expression and include RNA sequencing (RNA-Seq), reverse transcription polymerase chain reaction (RT-PCR) and microarray analysis.
Studies exploring whether transcriptomic biomarkers may predict response to treatment, either at baseline or very early in treatment, are outlined in Table 2.
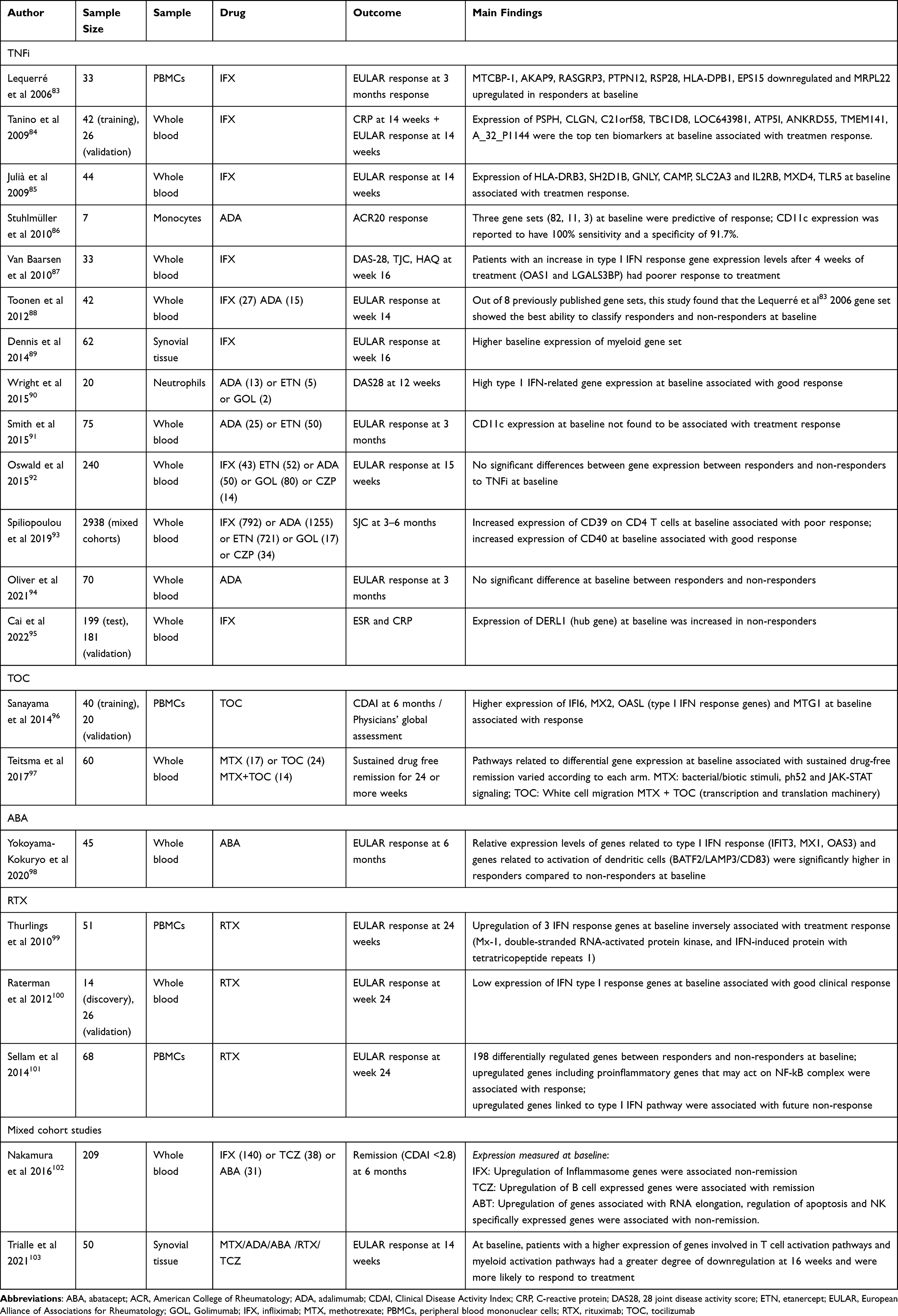 |
Table 2 Summary of Transcriptomic Studies Investigating Predictors of Response to Treatment |
Much of the literature has so far focused on TNFi therapy.83–93
Oswald et al did not find any significant differences in gene expression between responders and non-responders to TNFi-treatment at baseline in a cohort of 240 patients.92 However, after 14 weeks of therapy, responders to treatment did show changes in multiple gene expression modules whereas, despite exposure to TNFi treatments, the non-responder group showed minimal changes across modules. Gene expression modules were mainly related to immunological components, for example B cells, T cells, MHC and inflammation, as well as platelets and downregulation of myeloid lineage. These findings were consistent across three independent RA cohorts.
Oliver et al used whole blood to assess differential gene expression in good responders (n=50) versus non-responders (n=20) to adalimumab therapy.94 They did not identify any significant differential expression between these groups at either timepoint. They did however find that, within the good responder group, there were 813 differentially expressed transcripts pre-treatment compared to at 3 months; 17 transcripts were validated in a cohort of good responders (n=11). The authors suggest that it is possible that transcriptomic signatures may change earlier than at 3 months, which potentially could help identify non-responders earlier than in current clinical practice. One of these 17 transcripts was CD39 (ENTPD1) expression. The authors observed a higher expression of CD39 pre-treatment compared to post-treatment in good responders.94 However, a large study of 2936 patients conducted by Spiliopoulou et al 2019 found that a higher genetic score for expression of CD39 on CD4+ T cells was associated with poor response to TNFi, using SJC as the outcome measure. This study also found that increased CD40 transcription was associated with good response.93
Baseline expression of CD11c was associated with response to adalimumab in a small study using monocytes.86 The CD11c protein, also known as integrin, is an established marker for dendritic cells but is also expressed in monocytes and macrophages. It is thought to have a role in cell adhesion.86 This association was however not replicated in a subsequent study of patients commencing adalimumab (n=25) or etanercept (n=50).91 It is possible that this is due to different tissue types or use of different outcome measures.
DERL1 is a gene involved in regulation of autophagy. Higher expression of this gene at baseline was associated with non-response to infliximab in RA.95 Wright et al suggest that high expression of type I interferon (IFN) response genes at baseline is associated with subsequent response to TNF blockade.87,90 Van Baarsen et al evaluated whole blood samples for differential gene expression after 4 weeks of exposure to infliximab (IFX) and found that patients with an increase in type I IFN response gene expression levels after 4 weeks’ treatment (OAS1 and LGALS3BP) had poorer response to treatment.87 These studies suggest that a high baseline expression of IFN I response genes may indicate future response to therapy, and that failure to suppress type I IFN response at 4 weeks could provide some support for a change in therapy.87,90
Type I IFN response gene expression may also have a role in prediction of response to tocilizumab. Sanayama et al found higher expression of IFI6, MX2 and OASL (type I IFN response genes) at baseline was associated with 6-month CDAI improvement.96 There may also be a role of these genes in predicting abatacept response. In a study of 45 bDMARD-naïve patients, the type I IFN score was significantly higher in patients at baseline who went on to achieve a response compared to those who went on to fail to respond. Furthermore, IFN scores decreased by 15% (p <0.0005) in responders after exposure to abatacept at 6 months, but not in those who did not respond.98 Interestingly, in this study, expression levels of genes related to activation of dendritic cells (BATF2/LAMP3/CD83) showed significant differences between responders and non-responders at baseline. Conversely, to TNFi, tocilizumab and abatacept, upregulation of 3 IFN response genes was associated with poor treatment response to rituximab across multiple studies.99–101
Teitsma et al analyzed data from 60 patients treated with tocilizumab from the U-Act-early study to identify clusters of differentially expressed genes associated with sustained drug-free remission. Pathways correlating with achieving sustained drug-free remission varied according to arm: in the tocilizumab arm, important pathways were related to white cell migration; in the combined methotrexate/tocilizumab arm, modules identified were related to transcription and translation machinery.97
Triaille et al sought to compare transcriptomic effects of abatacept to other commonly used disease modifying agents, namely, tocilizumab, rituximab, methotrexate and adalimumab. Using synovial tissue of 50 RA patients taken at baseline, they identified that patients who had a higher expression of genes involved in T cell and myeloid activation pathways were more likely to respond to treatment. This suggests that common downstream pathways are affected by drugs despite different modes of action.103 Another study investigated the transcriptomic profile of patients who had failed to respond to methotrexate therapy and were started on tocilizumab, abatacept or infliximab instead. This study did show that different signatures across different treatments were associated with remission. Using gene set enrichment analysis, they found upregulation of genes involved in inflammasome in poor responders to infliximab. Conversely, B cell expressed genes were associated with response to tocilizumab. Finally, NK cell expressed genes were associated with non-response to abatacept.102
In the largest biopsy-driven trials to date, the R4RA and STRAP trials sought to combine information related to gene expression and histomorphology using synovial biopsies of participants with RA.104,105 The R4RA trial, in a cohort of inadequate responders to TNFi, demonstrated the superiority of tocilizumab compared to rituximab in participants classified as having low or absent B cell lineage expression signatures.105 The objective of the recent STRAP (Stratification of Biological Therapies by Pathobiology in Biologic-Naive Patients With Rheumatoid Arthritis) trial was to determine whether etanercept or tocilizumab (grouped together) were superior to rituximab in biologic-naïve patients with a B cell-poor synovial pathotype at 16 weeks. However, this trial failed to meet its primary endpoint, suggesting that a dichotomic classification of patients into B cell poor or B cell rich may be insufficient for stratification in biologic-naïve patients. However, the trial did demonstrate lower response rates to rituximab in those with a pauci-immune phenotype and increased radiographic progression in participants who were classified as B cell rich.104
In summary, many transcriptomic biomarkers of treatment response have been suggested in RA. The IFN pathway in particular shows promise in differentiating patients who may respond better to TNFi/CTLA-4/IL-6 inhibition compared to B cell inhibition; however, it remains to be seen whether this will add predictive value in practice. Evaluating patterns of gene expression in synovial tissue also shows promise, as suggested by clinical trials. The advent of spatial transcriptomics to gain a further in-depth understanding of gene expression at the level of single cells may increase the predictive value of these approaches in future.
Proteomics
The proteome is the tapestry of proteins in a cell or tissue for which the human genome provides the blueprint. The proteome has inherent added layers of complexity due to variable abundance, post-translational modifications and interactions between individual proteins and molecules. Proteins may be dysregulated in diseases such as RA and often the efficacy of a drug is related to binding with these proteins. As proteins are not predictable based on genetic sequences alone, the advent of proteomic technology provides an exciting opportunity for personalized medicine. This may be through discovery of dynamic biomarkers of disease activity and of treatment response, both general and drug specific. Proteomic methodology can be broadly placed in one of two methodologies: mass spectrometry or immunoassays. Immunoassays involve the use of antibodies to bind specific target proteins. Conversely, mass spectrometry, conversely through electron ionization, exploits the difference in mass/charge of different proteins to separate and analyze samples to identify proteins.106
We summarize studies investigating whether measurement of proteins at baseline can predict response to treatment in Tables 3 and 4. In discussions of proteomic biomarkers to personalize treatment of RA, one must discuss the role of autoantibodies. Rheumatoid factor (RF) and cyclic citrullinated peptide antibodies (anti-CCP) are used by clinicians in routine practice to aid diagnosis.107 Several studies have found an association between anti-CCP/RF and treatment response, including TNFi, abatacept, IL-6 inhibitors and JAK inhibitors.108–111 The largest study was a registry study of 2583 patients in 2021, which used drug retention as a surrogate measure of treatment response. This found that seropositivity was positively associated with treatment response to rituximab and abatacept, but no association was seen with TNFi.109 This was in contrast to Julia et al, who found that the presence of both RF and CCP was associated with treatment response to TNFi at three months.108 Treatment response to tofacitinib and sarilumab has also been associated with seropositivity.110,111 Overall, it does appear that seropositive patients have improved EULAR responses after rituximab therapy compared to seronegative patients. This factor may be taken into consideration when making treatment decisions.112
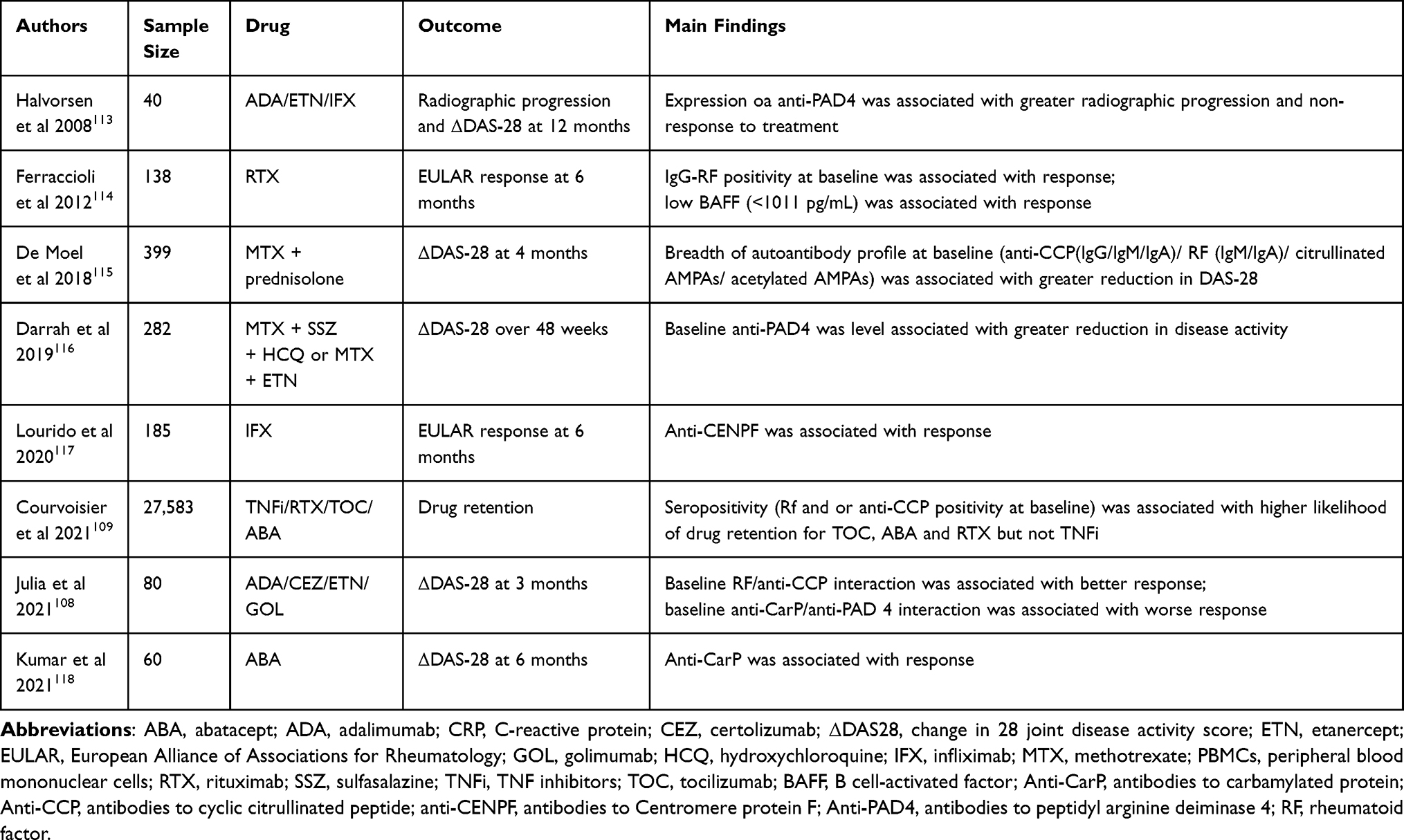 |
Table 3 Summary of Proteomic (Autoantibody) Studies Investigating Predictors of Response to Treatment |
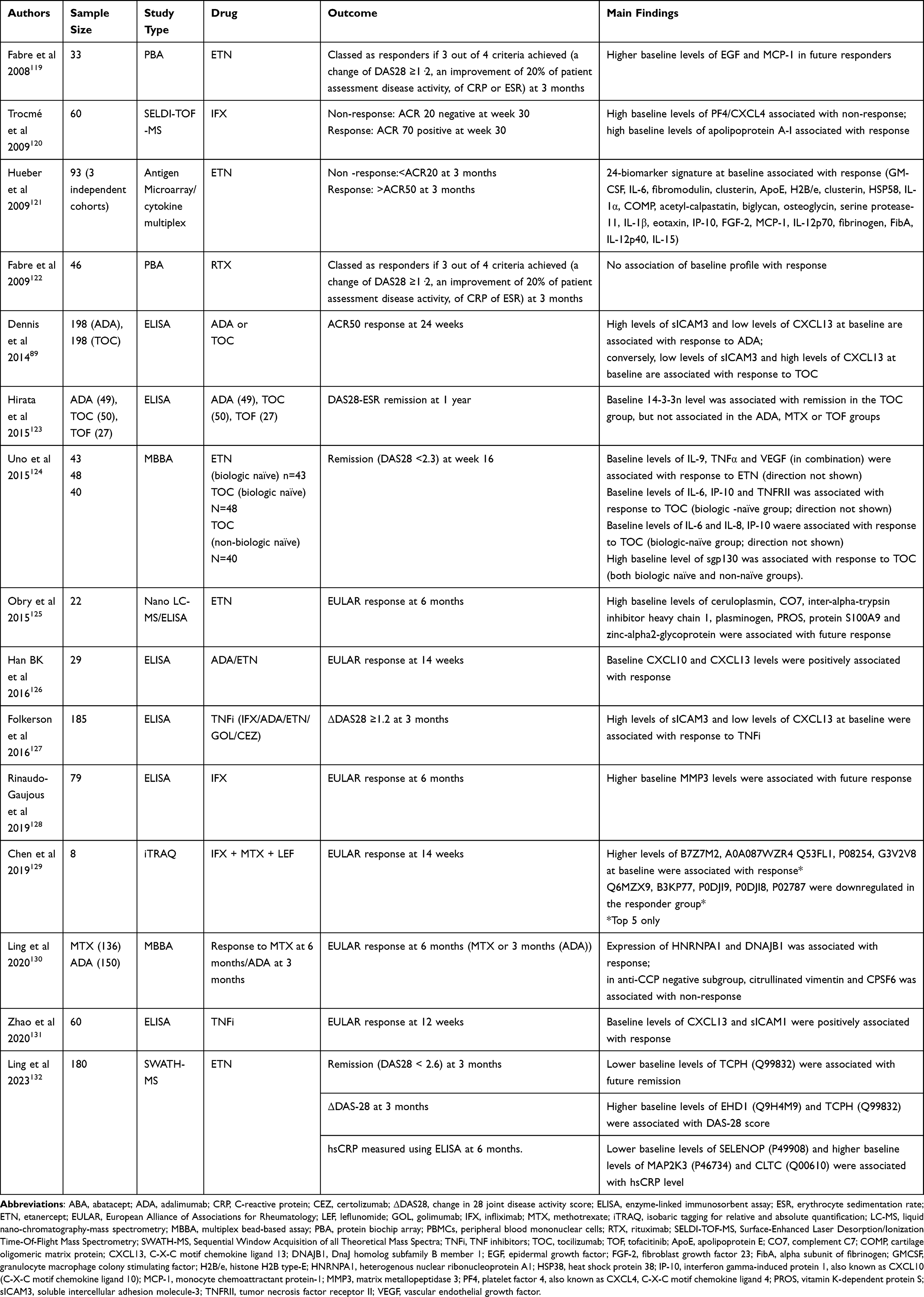 |
Table 4 Summary of Proteomic (Non-Autoantibody) Studies Investigating Predictors of Response to Treatment |
Using a multiplex bead-based assay approach, Ling et al found other citrullinated autoantibodies to be associated with treatment response to methotrexate/adalimumab in ACPA-negative patients.130 In this study, antibodies to citrullinated vimentin and CPSF6 were both found to be negatively associated with response to treatment, whilst antibodies to HNRNPA1 and DNAJB1 were positively associated. However, this added nothing over and beyond commercial CCP2 testing.130
Dennis et al found differentiating proteomic markers of treatment response to tocilizumab and adalimumab. High levels of sICAM3 and low levels of CXCL13 at baseline were associated with response to ADA. Conversely, low levels of sICAM3 and high levels of CXCL13 at baseline were associated with response to TOC.89 Folkerson et al replicated findings for adalimumab in a cohort of 185 patients on various TNFis.127
Obry et al found an association of 7 proteins with treatment response to etanercept/methotrexate therapy, whereby two of these proteins, PROS (vitamin K–dependent protein S) and CHIP (E3 ubiquitin-protein ligase carboxyl terminus of heat shock cognate 70-interacting protein), in combination demonstrated high sensitivity (88.9%) and high specificity (100%).125 A more recent, much larger study used sequential window acquisition of all theoretical fragment ion spectra mass spectrometry (SWATH-MS) in a study of 180 patients on etanercept therapy and identified proteins associated with treatment response.132 Lower baseline levels of TCPH (Q99832) were associated with future remission and higher baseline levels of EHD1 (Q9H4M9) and TCPH (Q99832) were associated with DAS-28 score. In addition, lower baseline levels of SELENOP (P49908) and higher baseline levels of MAP2K3 (P46734) and CLTC (Q00610) were associated with hsCRP level at 6 months. Notably, however, this study failed to replicate proteomic biomarkers found to be associated with response to etanercept in other cohorts.
Although proteomic studies have clearly found a wealth of potential biomarkers that may help to tailor treatment in RA, lack of validation in large cohorts is a frequent occurrence.
Metabolomics
Metabolomics refers to the quantitative measurement of all small molecular weight metabolites present in a biological specimen. Metabolites can be thought of as the end products formed as a consequence of the interplay between DNA, transcription factors and proteins.133 The two most common methods used to conduct metabolomic research are mass spectrometry (MS) and nuclear magnetic resonance (NMR).133 A variety of different biological specimens can be used, such as blood products, urine and synovial fluid.133 Numerous studies have investigated the metabolic profiles of RA patients to identify metabolites associated with pathogenesis, disease severity, efficacy and a small number have looked specifically at extra-articular manifestations. We focus here on attempts to use metabolomic profiles to predict response to treatment, as summarized in Table 5.
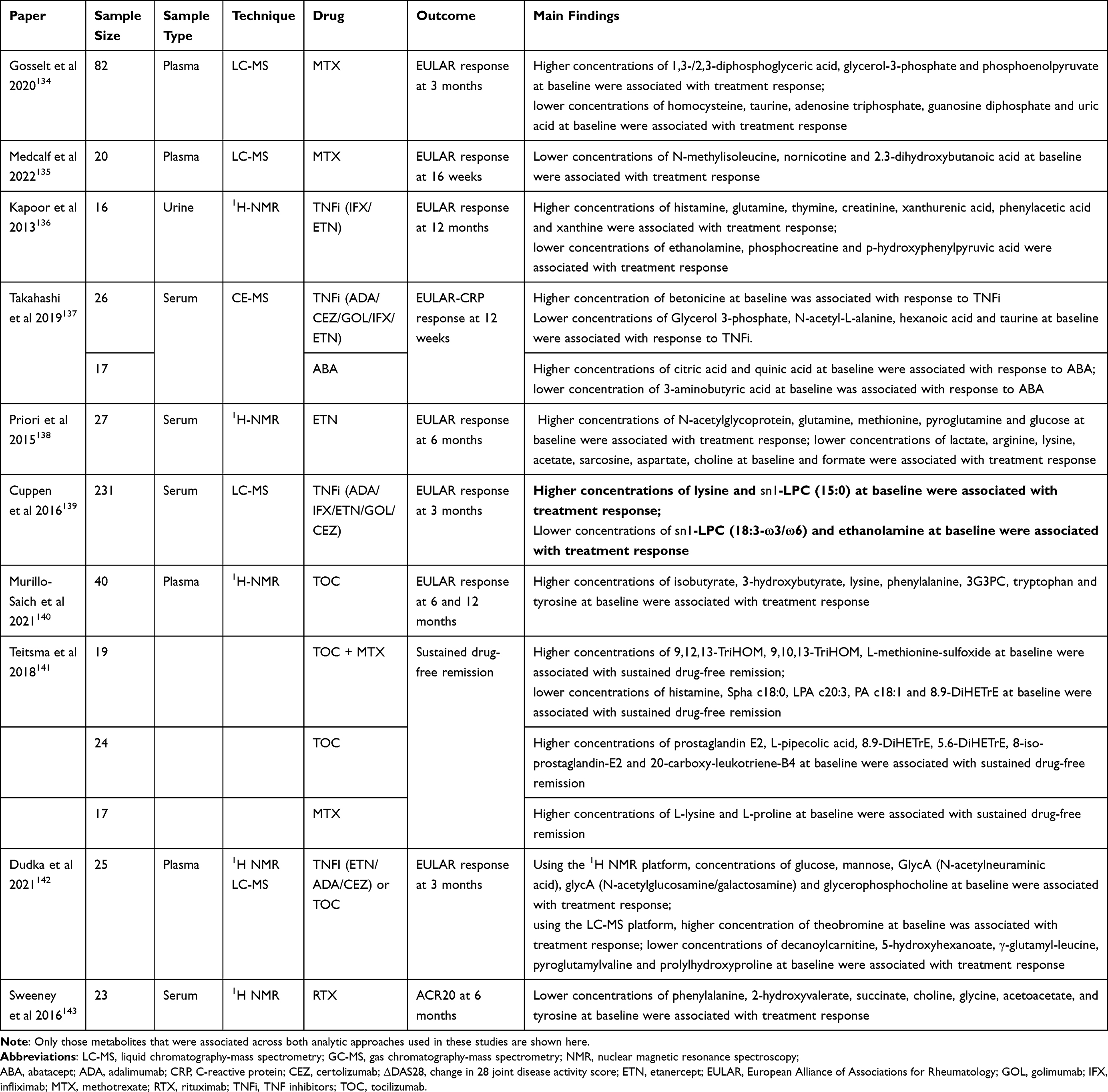 |
Table 5 Summary of Metabolomic Studies Investigating Predictors of Response to Treatment |
The majority of these studies focus on TNFi.136–139 The largest of these studies, by Cuppen et al, enrolled 231 patients with RA commencing a TNFi and used LC-MS to evaluate metabolomic profiles of serum samples. Higher baseline levels of lysine and sn1-LPC (15:0), and lower levels of sn1-LPC (18:3-ω3/ω6) and ethanolamine, contributed to a model to predict treatment response with a high degree of accuracy (AUC-ROC=0.841) and outperformed a model using clinical predictors alone (p=0.01). The model showed particular utility in identification of patients unlikely to respond (NRI=0.23).139 Kapoor et al had also previously found low levels of ethanolamine to be associated with response to TNFi (infliximab/etanercept)136 but Priori et al found the opposite result for lysine.138 In addition to these amino acids, Kapoor et al and Priori et al both found that higher baseline levels of glutamine are associated with response to TNFi.136,138 Low levels of taurine have been associated with treatment response to methotrexate134 and TNFi.137
Some studies have explored prediction of response to non-TNFi therapies.134,135,137,140,141,143 Sweeney et al found that metabolite profiles related to pathways involving glycerophospholipid, amino acid and energy metabolism can distinguish responders and non-responders to rituximab.143 In this study, low levels of tyrosine and phenylalanine were associated with treatment response whereas high levels of these amino acids have been associated with response to tocilizumab.140 Low levels of choline were also associated with response to rituximab and TNFi.138,143
Some studies have suggested differences in baseline predictors of treatment response between different modes of action. Takashi et al found that three amino acid metabolites (citric acid, quinic acid and 3-aminobutyric acid) were associated with response to abatacept. These were different metabolites compared to the TNFi arm of the study.137 Findings by Teitsma et al found that metabolic profiles of early RA patients were associated with sustained drug-free remission. Profiles predictive of response were different in the tocilizumab group, the methotrexate group and the tocilizumab and methotrexate combination group.141
Distinct metabolite patterns that may be predictive of treatment response are starting to emerge. Amino acids (eg, lysine, glutamine, tyrosine, phenylalanine) and lipid-related compounds (eg, sn1-LPC, ethanolamine, choline) stand out as potential predictors; however, further validation is required.
Therapeutic Drug Monitoring
A wide variability in serum concentrations of biopharmaceutical drugs exists. Therapeutic drug monitoring is the measurement of drug concentration with the aim of personalizing dosing and therefore optimizing drug efficacy and/or to reducing the risk of adverse effects by maintaining drug concentrations within the therapeutic window.144 Several studies suggest that the higher drug levels in advanced therapies are associated with clinical efficacy in the case of adalimumab,31,145 infliximab,146–148 etanercept149 and tocilizumab.150,151
Anti-drug antibodies (ADAs) have garnered much attention in recent years. Patients sometimes develop these ADAs in response to biologic medications which may contribute to diminished drug concentration through drug neutralization or enhanced drug elimination.152 It is noted that ADAs are largely not detected for etanercept.153 The prevalence estimates have been suggested to be up to 67% for infliximab and adalimumab.154 The association between ADAs and low drug levels for particular drugs is well established for TNFi: adalimumab153,155 and infliximab.153,154,156,157 It has also been found for IL-6 receptor inhibitors: tocilizumab and sarilumab158,159 and, most recently, rituximab.160
The presence of ADAs has been associated with reduced clinical efficacy for adalimumab31,145,154,155,157,161–163 and infliximab.147,154,157,163 However, this association has appeared to be less clear for tocilizumab,164,165 sarilumab,158,164,166 abatacept154 and rituximab.154,167 The prospective ABIRISK study aimed to determine whether ADAs were associated with therapeutic response in RA for TNFi, tocilizumab and rituximab. It confirmed a high prevalence of ADAs for those on TNFi therapy (26/68, 38.2%), tocilizumab (10/50, 20%) and rituximab (15/30, 50%). The study found a significant association between presence of ADAs and clinical response for all drugs combined, but it lacked power to detect a difference for each drug class. The biggest difference in response rates was noted in the TNFi group, but results for all drug classes do appear to show the same pattern of results.168
Another potential use of measurement of ADAs stems from the hypothesis that, if non-response is caused by immunogenicity, this will inform whether a second TNFi agent will be effective. This theory was tested in a cohort of 292 patients treated with etanercept, 70% of whom had been previously TNFi naïve and the remainder were TNFi inadequate responders (adalimumab or infliximab). Patients who switched from previous TNFi therapy with ADAs were significantly less likely to respond compared to those without ADAs and those who were naïve to TNFi.169
A limited number of prospective studies have assessed the use of therapeutic drug monitoring in RA. A study titled INGEBIO recruited a population with different rheumatic diseases, of which 63/169 (37%) had a diagnosis of RA. All patients had been treated with adalimumab and were clinically stable for at least 6 months. ADAs and drug levels were tested in each patient every 2–3 months but only the test results of the intervention group were revealed to clinicians. The risk of flare was reduced in the intervention group compared to the control group (IRR: 0.7252; 95% CI: 0.4997–1.0578), but this did not reach statistical significance.170 A similar, more recent, study, NOR-DRUM, evaluated the use of therapeutic drug monitoring of patients commencing infliximab in a mixed population of 411 patients with different autoimmune conditions in Norway. Of these patients, 84 had a diagnosis of RA. Although the therapeutic drug monitoring group had fewer infusion reactions compared to the control group, there was no significant difference in remission rates between groups.171
Currently, the NICE recommendations (2019) state that there is insufficient evidence to advocate the routine use of therapeutic drug monitoring of TNFi (drug serum levels and ADAs) in RA.172 More recently, EULAR published points-to-consider for therapeutic drug monitoring in inflammatory rheumatic diseases.173 It also did not recommend the routine use of proactive TDM in the management of inflammatory rheumatic diseases and highlighted the lack of an identified optimal range of most drugs. However, it also identified some specific situations in which measurement of drug levels/ADAs may be considered. These are as follows:
- Measurement of drug levels early during treatment with infliximab/adalimumab to guide predictions of future efficacy.
- Measurement of drug levels and ADAs may help to understand the mechanisms underlying non-response and therefore help guide future treatment.
- In cases of non-severe hypersensitivity reaction to infusions, measurement of ADAs may help guide decisions to continue treatment.
- Measurement of drug levels in cases where tapering is being considered.
It appears clear that there does seem to be a role for at least reactive therapeutic drug monitoring. The caveat to all of these points is that further clinical trials and knowledge of the optimum biopharmaceutical therapeutic range and cost-effectiveness are needed to fully appreciate how therapeutic drug monitoring may be used to personalize care in RA in a cost-effective manner.173
Machine Learning
Artificial intelligence (AI) is the capability of a machine to perform functions and tasks one would associate with human cognition.174 In the current technological climate, AI is being increasingly explored and adopted in many fields, including in precision medicine. Machine learning (ML) is thought of as a branch of AI, in which machines can identify patterns and draw inferences of relationships between variables, but without explicit instructions. This process contrasts from traditional hypothesis-driven data analysis approaches.175
The use of various machine-learning approaches, both supervised and unsupervised, has been explored in RA. The exponential rise of data, particularly highly dimensional omics data, has led to the problem of predictors outnumbering observations, a phenomenon referred to as “the curse of dimensionality”.176 How we best integrate all this information to inform prediction of treatment response is a considerable challenge.
A large Swedish study of 5475 patients sought to use a combination of four different machine-learning approaches using clinical data only to create a model predicting persistence of methotrexate therapy at one year. However, despite its large sample size and the wealth of clinical data available, only moderate predictive performance (AUC: 0.67, LASSO model) was reached, with only a marginal gain above traditional hypothesis-based models.177 Miyoshi et al used artificial neural networks to develop a model using clinical covariates alone to predict response to infliximab and reported 92% accuracy. It contained nine variables: ESR/TJC, albumin, monocyte count, red blood cell number, methotrexate dosage, HbA1c, history of bDMARD use and prednisolone dosage.178 Koo et al analyzed clinical data of 1204 patients treated with various biologics (adalimumab, golimumab, infliximab, abatacept and tocilizumab) and used several machine-learning methods to develop models predicting future remission for each drug type. Interestingly, there were differences in relative importance of clinical features for each drug type. Model accuracy ranged from 52.8% to 72.9% and AUROC ranged from 0.512 to 0.694.179 These studies emphasize that clinical data alone is not sufficient to personalize treatment.
There have been some attempts to integrate omics data with clinical data, to try to improve model performance. Gosselt et al, using data from the Dutch REACH cohort, developed models to predict response to methotrexate at 3 months using 3 ML methods (LASSO, random forest and XGboost). The selected features included known clinical predictors of treatment response (RF and ACPA status, baseline DAS-28 components) combined with genetic predictors: SNPs in ATP-binding cassette (ABC) transporter genes and erythrocyte folate. Of the four developed models, LASSO performed best (AUC: 0.76) in predicting lack of response to methotrexate.180 Lim et al aimed to tackle the issue of the high dimensionality of genetics data through identification of likely functional coding haplotypes, to develop a model to predict response to methotrexate. They used supervised machine-learning approaches (neural networks, SVM, logistic regression, elastic nets, random forest and boosted trees). The final model consisted of 100 features, 95 of which were genetic (AUC: 0.828, sensitivity=0.6875, specificity=0.8684). Non-genetic selected features were platelet count, hemoglobin levels, duration of morning stiffness and anti-CCP positivity.181
Plant et al also demonstrated the superiority of a model containing both biological and clinical data over that of a model containing clinical covariates alone. They used whole blood samples of patients initiating methotrexate at baseline and at 4 weeks and found that genes involved in the type I interferon signaling pathway were differentially expressed in patients with insufficient response to treatment at baseline and at 4 weeks post-treatment. The model which included gene expression data achieved an AUC of 0.78±0.11 compared to the model with clinical data alone (AUC: 0.63±0.06).182
There have also been attempts to combine clinical and biological features to predict response to bDMARDs, namely, TNFi. Guan et al used a Gaussian process regression model trained on a large sample size of 1892 patients and demonstrated improved accuracy of prediction of response to TNFi (adalimumab/etanercept/infliximab) when genetic predictors were added to demographic and clinical data. This model could correctly classify 78% of responder patients. The contribution of SNP biomarkers, however, was relatively small in comparison to clinical predictors used, particularly baseline DAS28, which showed the greatest association with future treatment response.183 Similarly, Luque-Tévar et al demonstrated the superiority of prediction to TNFi using both clinical data and molecular data, namely, serum inflammatory profile, oxidative stress markers and NETosis-derived biomolecules. Distinct clinical and molecular profiles were identified through LASSO regression/ridge regression, and integration in a mixed model showed an AUC of 0.91.184
The integration of various levels of omics data poses an even greater challenge due to the complexity of underlying pathways, co-correlation of covariates and highly dimensional data. Youssef et al conducted a study involving 39 female RA patients who had previously had an inadequate response to methotrexate. The authors analyzed transcriptomics, proteomics and cell phenotypes data generated from PBMCs collected before treatment with TNFi and at 3 months. Various supervised machine-learning models were developed to attempt prediction of non-response. Of all models developed, the linear model based on transcriptomic data at baseline displayed the best ability to predict response to treatment, and this was with a higher degree of accuracy compared to models based on clinical data alone.185 Tao et al used a combination of transcriptomic signatures from monocytes, CD4+ T cells and PBMCs and DNA methylation profiling from PBMCs to develop models predictive of treatment response to TNFi (adalimumab and etanercept). Similar to Youssef et al, highest overall accuracy (84.7%) was found for the model based on DEGs of PBMCs.186
Tasaki et al sought to use a multi-omics approach to molecular signatures associated with long-term remission. They incorporated transcriptomics, proteomics and immunophenotype data, as well as clinical information to explore the effect of drug treatments (methotrexate, infliximab and tocilizumab) at the molecular level. Interestingly, all three drugs had a similar effect at the immune cell-type level; however, they found that treatment with biologic therapies significantly normalized the molecular signature at all three levels towards that of healthy controls. However, a residual signature remained.187
Jung et al used a naïve Bayes classifier to investigate predictors of treatment response based on synovial tissue pathotypes. This unsupervised machine-learning approach yielded three RA subtypes based on gene expression data from the synovial tissues of 180 patients. The first two groups (C1 and C2) showed enrichment of fibroblasts and tissue proliferative signaling pathways whereas C3 showed greater activation of immune cells and proinflammatory signaling pathways. These groups showed differences in clinical characteristics and treatment responses to triple DMARDs and infliximab; those in C3, however, were most likely to respond to both treatments.188
Despite much progress in the application of machine-learning approaches to predict RA treatment response, there remain hurdles to overcome before they can be implemented in clinical practice. As has been a theme for many omics fields, there is a lack of external validation. There are differences in cohort study designs, including different platforms for omics data, differences in method and timing of measurement of treatment outcomes and clinical data available. Indeed, one can see that many of the studies report moderate to excellent predictive abilities of models used; however, this may be partly a consequence of “overfitting”. This is a phenomenon observed when machine-learning algorithms are constructed, and statistical models developed fit exactly to training data, but is rendered inaccurate for new data. This is more likely to occur in models of greater complexity.189 Furthermore, consideration of ethical issues that could potentially be associated with the advent of artificial intelligence/machine-learning approaches are important to consider. For example, in some machine-learning algorithms the inner workings of the models may be unclear. This is commonly referred to as the “black box” concept.189 This poses a problem for applications in treatment decision making, as it is paramount that we understand the basis of how such impactful conclusions are reached.
Despite these barriers, with collaboration between large consortia, as well as between data scientists, biologists and rheumatologists, machine-learning methods have shown great promise, and it is hoped further progress will be made in years to come in terms of implementing them in clinical practice.
Future of Personalized Medicine in Rheumatoid Arthritis
Over recent years, advancement of methodologies and substantial investment has led to an explosion of promising potential biomarkers for treatment response in RA. Furthermore, underlying mechanisms of disease and potential disease endotypes are beginning to emerge. However, most potential markers lack validation in independent cohorts and their utility over and beyond clinical predictors and/or seropositivity is unclear. This lack of replication is driven by a combination of factors. A key problem is the preferred outcome measure. Although TJC and VAS capture important information about the impact of the disease on the patient, they likely do not have the same biomarkers as SJC and inflammation. Another confounding factor is non-adherence. Non-adherence correlates with non-response to treatment and therefore non-adherence can result in patient misclassification as non-responders and severely reduce study power.35 Furthermore, use of different points and other unmeasured confounders may contribute to lack of replication. Lastly, few studies directly compare different drugs, which makes it difficult to ascertain whether a biomarker is prognostic or truly theragnostic. Most studies, to date, have focused on TNFi therapy, perhaps due to TNFi being the first advanced therapy available. Therefore, despite considerable progress, although the customization of therapy for individuals with RA is on the horizon it remains an elusive goal. To overcome these obstacles, development of outcome measures that more closely resemble the underlying biological process of synovitis is required. It is likely that no one biomarker will predict response and future studies should aim to integrate the available data and explore if the developed models are more predictive than clinical predictors alone. Machine learning has demonstrated its ability to integrate large amounts of data; however, further development of machine-learning techniques is required to prevent over-fitting.
Conclusion
In conclusion, research to date has revealed several promising predictive biomarkers that may pave the way to personalized medicine approaches in rheumatoid arthritis. Further work is required to validate and integrate these biomarkers to create a predictive biomarker panel. To personalize therapy, future biomarker studies should look to compare responders to different targeted therapies to identify unique treatment-associated biomarkers.
Disclosure
Dr James Bluett reports grants from Pfizer, travel/conference fees from Fresenius Kabi, travel/conference fees from UCB, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Symmons D, Turner G, Webb R, et al. The prevalence of rheumatoid arthritis in the United Kingdom: new estimates for a new century. Rheumatology. 2002;41(7):793–800. doi:10.1093/rheumatology/41.7.793
2. Chiu YM, Lu YP, Lan JL, Chen DY, Wang JD. Lifetime risks, life expectancy, and health care expenditures for rheumatoid arthritis: a nationwide cohort followed up from 2003 to 2016. Arthritis Rheumatol. 2021;73(5):750–758. doi:10.1002/art.41597
3. Smolen JS, Landewé R, Breedveld FC, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs. Ann Rheum Dis. 2010;69(6):964–975. doi:10.1136/ard.2009.126532
4. Nam JL, Takase-Minegishi K, Ramiro S, et al. Efficacy of biological disease-modifying antirheumatic drugs: a systematic literature review informing the 2016 update of the EULAR recommendations for the management of rheumatoid arthritis. Ann Rheum Dis. 2017;76(6):1113–1136. doi:10.1136/annrheumdis-2016-210713
5. guideline NG100 N. Rheumatoid Arthritis in Adults: Diagnosis and Management. London, UK: National Institute for Health and Care Excellence; 2018.
6. England NH. Improving Outcomes through Personalised Medicine; 2016.
7. Abrahams E. Right drug-right patient-right time: personalized medicine coalition. Clin Transl Sci. 2008;1(1):11–12. doi:10.1111/j.1752-8062.2008.00003.x
8. MacGregor AJ, Snieder H, Rigby AS, et al. Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum. 2000;43(1):30–37. doi:10.1002/1529-0131(200001)43:1<30::Aid-anr5>3.0.Co;2-b
9. Yarwood A, Huizinga TW, Worthington J. The genetics of rheumatoid arthritis: risk and protection in different stages of the evolution of RA. Rheumatology. 2016;55(2):199–209. doi:10.1093/rheumatology/keu323
10. van der Woude D, Houwing-Duistermaat JJ, Toes RE, et al. Quantitative heritability of anti-citrullinated protein antibody-positive and anti-citrullinated protein antibody-negative rheumatoid arthritis. Arthritis Rheum. 2009;60(4):916–923. doi:10.1002/art.24385
11. Doran MF, Pond GR, Crowson CS, O’Fallon WM, Gabriel SE. Trends in incidence and mortality in rheumatoid arthritis in Rochester, Minnesota, over a forty-year period. Arthritis Rheum. 2002;46(3):625–631. doi:10.1002/art.509
12. Symmons DP, Barrett EM, Bankhead CR, Scott DG, Silman AJ. The incidence of rheumatoid arthritis in the United Kingdom: results from the Norfolk Arthritis Register. Br J Rheumatol. 1994;33(8):735–739. doi:10.1093/rheumatology/33.8.735
13. Ma MH, Ibrahim F, Walker D, et al. Remission in early rheumatoid arthritis: predicting treatment response. J Rheumatol. 2012;39(3):470–475. doi:10.3899/jrheum.110169
14. Coulthard LR, Taylor JC, Eyre S, et al. Genetic variants within the MAP kinase signalling network and anti-TNF treatment response in rheumatoid arthritis patients. Ann Rheumatic Dis. 2011;70(1):98–103. doi:10.1136/ard.2010.133249
15. Atzeni F, Bongiovanni S, Marchesoni A, et al. Predictors of response to anti-TNF therapy in RA patients with moderate or high DAS28 scores. Joint Bone Spine. 2014;81(1):37–40. doi:10.1016/j.jbspin.2013.04.005
16. Yamanaka H, Harigai M, Ishiguro N, et al. Trend of patient characteristics and its impact on the response to Adalimumab in patients with rheumatoid arthritis: post hoc time-course analysis of an all-case PMS in Japan. Mod Rheumatol. 2015;25(4):495–502. doi:10.3109/14397595.2014.994263
17. Mancarella L, Bobbio-Pallavicini F, Ceccarelli F, et al. Good clinical response, remission, and predictors of remission in rheumatoid arthritis patients treated with tumor necrosis factor-alpha blockers: the GISEA study. J Rheumatol. 2007;34(8):1670–1673.
18. Levitsky A, Brismar K, Hafström I, et al. Obesity is a strong predictor of worse clinical outcomes and treatment responses in early rheumatoid arthritis: results from the SWEFOT trial. RMD Open. 2017;3(2):e000458. doi:10.1136/rmdopen-2017-000458
19. Hyrich KL, Watson KD, Silman AJ, Symmons DP. Predictors of response to anti-TNF-alpha therapy among patients with rheumatoid arthritis: results from the British society for rheumatology biologics register. Rheumatology. 2006;45(12):1558–1565. doi:10.1093/rheumatology/kel149
20. Fernández-Nebro A, Irigoyen MV, Ureña I, et al. Effectiveness, predictive response factors, and safety of anti-tumor necrosis factor (TNF) therapies in anti-TNF-naive rheumatoid arthritis. J Rheumatol. 2007;34(12):2334–2342.
21. Kleinert S, Tony HP, Krause A, et al. Impact of patient and disease characteristics on therapeutic success during Adalimumab treatment of patients with rheumatoid arthritis: data from a German noninterventional observational study. Rheumatol Int. 2012;32(9):2759–2767. doi:10.1007/s00296-011-2033-5
22. Sugiyama D, Nishimura K, Tamaki K, et al. Impact of smoking as a risk factor for developing rheumatoid arthritis: a meta-analysis of observational studies. Ann Rheum Dis. 2010;69(1):70–81. doi:10.1136/ard.2008.096487
23. Saevarsdottir S, Wedrén S, Seddighzadeh M, et al. Patients with early rheumatoid arthritis who smoke are less likely to respond to treatment with methotrexate and tumor necrosis factor inhibitors: observations from the epidemiological investigation of rheumatoid arthritis and the Swedish rheumatology register cohorts. Arthritis Rheum. 2011;63(1):26–36. doi:10.1002/art.27758
24. Chatzidionysiou K, Lukina G, Gabay C, et al. Smoking and response to rituximab in rheumatoid arthritis: results from an international European collaboration. Scand J Rheumatol. 2019;48(1):17–23. doi:10.1080/03009742.2018.1466363
25. Sokolove J, Wagner CA, Lahey LJ, et al. Increased inflammation and disease activity among current cigarette smokers with rheumatoid arthritis: a cross-sectional analysis of US veterans. Rheumatology. 2016;55(11):1969–1977. doi:10.1093/rheumatology/kew285
26. Adami G, Fassio A, Pistillo F, et al. Factors associated with radiographic progression in rheumatoid arthritis starting biological diseases modifying anti-rheumatic drugs (bDMARDs). Ther Adv Musculoskelet Dis. 2023;15:1759720x231174534. doi:10.1177/1759720x231174534
27. Kristensen LE, Bliddal H, Christensen R, et al. Is swollen to tender joint count ratio a new and useful clinical marker for biologic drug response in rheumatoid arthritis? Results from a Swedish cohort. Arthritis Care Res. 2014;66(2):173–179. doi:10.1002/acr.22107
28. Maldonado-Montoro M, Cañadas-Garre M, González-Utrilla A, Plaza-Plaza JC, Calleja-Hernández M. Genetic and clinical biomarkers of tocilizumab response in patients with rheumatoid arthritis. Pharmacol Res. 2016;111:264–271. doi:10.1016/j.phrs.2016.06.016
29. Gremese E, Carletto A, Padovan M, et al. Obesity and reduction of the response rate to anti-tumor necrosis factor α in rheumatoid arthritis: an approach to a personalized medicine. Arthritis Care Res. 2013;65(1):94–100. doi:10.1002/acr.21768
30. de Heredia FP, Gómez-Martínez S, Marcos A. Obesity, inflammation and the immune system. Proc Nutr Soc. 2012;71(2):332–338. doi:10.1017/s0029665112000092
31. Jani M, Chinoy H, Warren RB, et al. Clinical utility of random anti-tumor necrosis factor drug-level testing and measurement of antidrug antibodies on the long-term treatment response in rheumatoid arthritis. Arthritis Rheumatol. 2015;67(8):2011–2019. doi:10.1002/art.39169
32. Mould DR. The Pharmacokinetics of Biologics: a Primer. Dig Dis. 2015;33(Suppl 1):61–69. doi:10.1159/000437077
33. Ogata A, Atsumi T, Fukuda T, et al. Sustainable efficacy of switching from intravenous to subcutaneous tocilizumab monotherapy in patients with rheumatoid arthritis. Arthritis Care Res. 2015;67(10):1354–1362. doi:10.1002/acr.22598
34. Hope HF, Bluett J, Barton A, Hyrich KL, Cordingley L, Verstappen SM. Psychological factors predict adherence to methotrexate in rheumatoid arthritis; findings from a systematic review of rates, predictors and associations with patient-reported and clinical outcomes. RMD Open. 2016;2(1):e000171. doi:10.1136/rmdopen-2015-000171
35. Bluett J, Morgan C, Thurston L, et al. Impact of inadequate adherence on response to subcutaneously administered anti-tumour necrosis factor drugs: results from the Biologics in rheumatoid arthritis genetics and genomics study syndicate cohort. Rheumatology. 2015;54(3):494–499. doi:10.1093/rheumatology/keu358
36. National Institute for Health and Care Excellence. NICE guideline [NG33]: tuberculosis; 2023. Available from: https://www.nice.org.uk/guidance/ng33/chapter/Recommendations.
37. Drozdzik M, Rudas T, Pawlik A, Gornik W, Kurzawski M, Herczynska M. Reduced folate carrier-1 80G> A polymorphism affects methotrexate treatment outcome in rheumatoid arthritis. Pharmacogeno J. 2007;7(6):404–407. doi:10.1038/sj.tpj.6500438
38. Li X, Hu M, Li W, et al. The association between reduced folate carrier-1 gene 80G/A polymorphism and methotrexate efficacy or methotrexate related-toxicity in rheumatoid arthritis: a meta-analysis. Int Immunopharmacol. 2016;38:8–15. doi:10.1016/j.intimp.2016.05.012
39. Kung TN, Dennis J, Ma Y, et al. RFC1 80G> A is a genetic determinant of methotrexate efficacy in rheumatoid arthritis: a human genome epidemiologic review and meta‐analysis of observational studies. Arthritis Rheumatol. 2014;66(5):1111–1120. doi:10.1002/art.38331
40. Liu C, Batliwalla F, Li W, et al. Genome-wide association scan identifies candidate polymorphisms associated with differential response to anti-TNF treatment in rheumatoid arthritis. Mol Med. 2008;14(9–10):575–581. doi:10.2119/2008-00056.Liu
41. Padyukov L, Lampa J, Heimburger M, et al. Genetic markers for the efficacy of tumour necrosis factor blocking therapy in rheumatoid arthritis. Ann Rheumatic Dis. 2003;62(6):526. doi:10.1136/ard.62.6.526
42. Krintel SB, Palermo G, Johansen JS, et al. Investigation of single nucleotide polymorphisms and biological pathways associated with response to TNFα inhibitors in patients with rheumatoid arthritis. Pharmacogenet Genomics. 2012;22(8):577–589. doi:10.1097/FPC.0b013e3283544043
43. Acosta-Colman I, Palau N, Tornero J, et al. GWAS replication study confirms the association of PDE3A–SLCO1C1 with anti-TNF therapy response in rheumatoid arthritis. Pharmacogenomics. 2013;14(7):727–734. doi:10.2217/pgs.13.60
44. Plant D, Bowes J, Potter C, et al. Genome-wide association study of genetic predictors of anti-tumor necrosis factor treatment efficacy in rheumatoid arthritis identifies associations with polymorphisms at seven loci. Arthritis Rheum. 2011;63(3):645–653. doi:10.1002/art.30130
45. Márquez A, Ferreiro-Iglesias A, Dávila-Fajardo CL, et al. Lack of validation of genetic variants associated with anti–tumor necrosis factor therapy response in rheumatoid arthritis: a genome-wide association study replication and meta-analysis. Arthritis Res Therapy. 2014;16(2):1–7. doi:10.1186/ar4504
46. Cui J, Saevarsdottir S, Thomson B, et al. Rheumatoid arthritis risk allele PTPRC is also associated with response to anti–tumor necrosis factor α therapy. Arthritis Rheum. 2010;62(7):1849–1861. doi:10.1002/art.27457
47. Plant D, Prajapati R, Hyrich KL, et al. Replication of association of the PTPRC gene with response to anti–tumor necrosis factor therapy in a large UK cohort. Arthritis Rheum. 2012;64(3):665–670. doi:10.1002/art.33381
48. Ferreiro-Iglesias A, Montes A, Perez-Pampin E, et al. Replication of PTPRC as genetic biomarker of response to TNF inhibitors in patients with rheumatoid arthritis. Pharmacogeno J. 2016;16(2):137–140. doi:10.1038/tpj.2015.29
49. Lee YH, Bae SC. Associations between PTPRC rs10919563 A/G and FCGR2A R131H polymorphisms and responsiveness to TNF blockers in rheumatoid arthritis: a meta-analysis. Rheumatol Int. 2016;36(6):837–844. doi:10.1007/s00296-016-3476-5
50. Cui J, Stahl EA, Saevarsdottir S, et al. Genome-wide association study and gene expression analysis identifies CD84 as a predictor of response to etanercept therapy in rheumatoid arthritis. PLoS Genet. 2013;9(3):e1003394. doi:10.1371/journal.pgen.1003394
51. Ferreiro-Iglesias A, Montes A, Perez-Pampin E, et al. Evaluation of 12 GWAS-drawn SNPs as biomarkers of rheumatoid arthritis response to TNF inhibitors. A potential SNP association with response to etanercept. PLoS One. 2019;14(2):e0213073. doi:10.1371/journal.pone.0213073
52. Wang J, Bansal A, Martin M, et al. Genome-wide association analysis implicates the involvement of eight loci with response to tocilizumab for the treatment of rheumatoid arthritis. Pharmacogeno J. 2013;13(3):235–241. doi:10.1038/tpj.2012.8
53. International Conference on Harmonisation. Guidance on E15 pharmacogenomics definitions and sample coding; availability. Notice. Fed Regist. 2008;73(68):19074–19076.
54. Massey J, Plant D, Hyrich K, et al. Genome-wide association study of response to tumour necrosis factor inhibitor therapy in rheumatoid arthritis. Pharmacogeno J. 2018;18(5):657–664. doi:10.1038/s41397-018-0040-6
55. Cordingley L, Prajapati R, Plant D, et al. Impact of psychological factors on subjective disease activity assessments in patients with severe rheumatoid arthritis. Arthritis Care Res. 2014;66(6):861–868. doi:10.1002/acr.22249
56. Hensor EMA, McKeigue P, Ling SF, et al. Validity of a two-component imaging-derived disease activity score for improved assessment of synovitis in early rheumatoid arthritis. Rheumatology. 2019;58(8):1400–1409. doi:10.1093/rheumatology/kez049
57. Gilani SS, Nair N, Plant D, et al. Pharmacogenetics of TNF inhibitor response in rheumatoid arthritis utilizing the two-component disease activity score. Pharmacogenomics. 2020;21(16):1151–1156. doi:10.2217/pgs-2020-0043
58. Lopez-Lasanta M, Julià A, Maymó J, et al. Variation at interleukin-6 receptor gene is associated to joint damage in rheumatoid arthritis. Arthritis Res Therapy. 2015;17(1):242. doi:10.1186/s13075-015-0737-8
59. Kastbom A, Cöster L, Arlestig L, et al. Influence of FCGR3A genotype on the therapeutic response to rituximab in rheumatoid arthritis: an observational cohort study. BMJ Open. 2012;2(5):e001524. doi:10.1136/bmjopen-2012-001524
60. Hamada M, Tsunakawa Y, Jeon H, Yadav MK, Takahashi S. Role of MafB in macrophages. Exp Anim. 2020;69(1):1–10. doi:10.1538/expanim.19-0076
61. Lewis CM, Vassos E. Polygenic risk scores: from research tools to clinical instruments. Genome Med. 2020;12(1):44. doi:10.1186/s13073-020-00742-5
62. Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell. 2007;128(4):635–638. doi:10.1016/j.cell.2007.02.006
63. Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nature Genet. 2003;33(3):245–254. doi:10.1038/ng1089
64. Nair N, Plant D, Verstappen SM, et al. Differential DNA methylation correlates with response to methotrexate in rheumatoid arthritis. Rheumatology. 2020;59(6):1364–1371. doi:10.1093/rheumatology/kez411
65. Gosselt HR, van Zelst BD, de Rotte MC, Hazes JM, de Jonge R, Heil SG. Higher baseline global leukocyte DNA methylation is associated with MTX non-response in early RA patients. Arthritis Res Ther. 2019;21(1):1–8. doi:10.1186/s13075-019-1936-5
66. Gosselt HR, Vallerga CL, Mandaviya PR, et al. Epigenome wide association study of response to methotrexate in early rheumatoid arthritis patients. PLoS One. 2021;16(3):e0247709. doi:10.1371/journal.pone.0247709
67. Glossop JR, Nixon NB, Emes RD, et al. DNA methylation at diagnosis is associated with response to disease-modifying drugs in early rheumatoid arthritis. Epigenomics. 2017;9(4):419–428. doi:10.2217/epi-2016-0042
68. Plant D, Webster A, Nair N, et al. Differential methylation as a biomarker of response to etanercept in patients with rheumatoid arthritis. Arthritis Rheumatol. 2016;68(6):1353–1360. doi:10.1002/art.39590
69. Sode J, Krintel SB, Carlsen AL, et al. Plasma microRNA profiles in patients with early rheumatoid arthritis responding to Adalimumab plus methotrexate vs methotrexate alone: a placebo-controlled clinical trial. J Rheumatol. 2018;45(1):53–61. doi:10.3899/jrheum.170266
70. Krintel SB, Dehlendorff C, Hetland ML, et al. Prediction of treatment response to Adalimumab: a double-blind placebo-controlled study of circulating microRNA in patients with early rheumatoid arthritis. Pharmacogeno J. 2016;16(2):141–146. doi:10.1038/tpj.2015.30
71. Cheng P, Wang J. The potential of circulating microRNA‐125a and microRNA‐125b as markers for inflammation and clinical response to infliximab in rheumatoid arthritis patients. J Clin Lab Analysis. 2020;34(8):e23329. doi:10.1002/jcla.23329
72. Duroux-Richard I, Pers Y-M, Fabre S, et al. Circulating miRNA-125b is a potential biomarker predicting response to rituximab in rheumatoid arthritis. Med Inflam. 2014;2014:2. doi:10.1155/2014/342524
73. Liu Y, Han Y, Qu H, Fang J, Ye M, Yin W. Correlation of microRNA expression profile with clinical response to tumor necrosis factor inhibitor in treating rheumatoid arthritis patients: a prospective cohort study. J Clin Lab Analysis. 2019;33(7):e22953. doi:10.1002/jcla.22953
74. Castro-Villegas C, Pérez-Sánchez C, Escudero A, et al. Circulating miRNAs as potential biomarkers of therapy effectiveness in rheumatoid arthritis patients treated with anti-TNFα. Arthritis Res Ther. 2015;17(1):1–15. doi:10.1186/s13075-015-0555-z
75. Cavalli G, Heard E. Advances in epigenetics link genetics to the environment and disease. Nature. 2019;571(7766):489–499. doi:10.1038/s41586-019-1411-0
76. Tessarz P, Kouzarides T. Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Biol. 2014;15(11):703–708. doi:10.1038/nrm3890
77. Nair N, Wilson AG. Assessing the potential of epigenetic targets as biomarkers in the diagnosis and treatment of rheumatoid arthritis. Expert Rev Clin Immunol. 2023;19(5):483–488. doi:10.1080/1744666x.2023.2193686
78. Araki Y, Mimura T. The mechanisms underlying chronic inflammation in rheumatoid arthritis from the perspective of the epigenetic landscape. J Immunol Res. 2016;2016:1. doi:10.1155/2016/6290682
79. Wada TT, Araki Y, Sato K, et al. Aberrant histone acetylation contributes to elevated interleukin-6 production in rheumatoid arthritis synovial fibroblasts. Biochem Biophys Res Commun. 2014;444(4):682–686. doi:10.1016/j.bbrc.2014.01.195
80. Bogunia-Kubik K, Wysoczańska B, Piątek D, Iwaszko M, Ciechomska M, Świerkot J. Significance of polymorphism and expression of miR-146a and NFkB1 genetic variants in patients with rheumatoid arthritis. Archivum Immunol Et Ther Experiment. 2016;64(S1):131–136. doi:10.1007/s00005-016-0443-5
81. Evangelatos G, Fragoulis GE, Koulouri V, Lambrou GI. MicroRNAs in rheumatoid arthritis: from pathogenesis to clinical impact. Autoimmunity Rev. 2019;18(11):102391. doi:10.1016/j.autrev.2019.102391
82. Liu T, Zhang L, Joo D, Sun S-C. NF-κB signaling in inflammation. Signal Transd Target Ther. 2017;2(1):1–9. doi:10.1038/sigtrans.2017.23
83. Lequerré T, Gauthier-Jauneau A-C, Bansard C, et al. Gene profiling in white blood cells predicts infliximab responsiveness in rheumatoid arthritis. Arthritis Res Ther. 2006;8(4):1–11. doi:10.1186/ar1990
84. Tanino M, Matoba R, Nakamura S, et al. Prediction of efficacy of anti-TNF biologic agent, infliximab, for rheumatoid arthritis patients using a comprehensive transcriptome analysis of white blood cells. Biochem Biophys Res Commun. 2009;387(2):261–265. doi:10.1016/j.bbrc.2009.06.149
85. Julià A, Erra A, Palacio C, et al. An eight-gene blood expression profile predicts the response to infliximab in rheumatoid arthritis. PLoS One. 2009;4(10):e7556. doi:10.1371/journal.pone.0007556
86. Stuhlmüller B, Häupl T, Hernandez MM, et al. CD11c as a transcriptional biomarker to predict response to anti-TNF monotherapy with Adalimumab in patients with rheumatoid arthritis. Clin Pharmacol Ther. 2010;87(3):311–321. doi:10.1038/clpt.2009.244
87. van Baarsen LG, Wijbrandts CA, Rustenburg F, et al. Regulation of IFN response gene activity during infliximab treatment in rheumatoid arthritis is associated with clinical response to treatment. Arthritis Res Ther. 2010;12(1):1–10. doi:10.1186/ar2912
88. Toonen EJ, Gilissen C, Franke B, et al. Validation study of existing gene expression signatures for anti-TNF treatment in patients with rheumatoid arthritis. PLoS One. 2012;7(3):e33199. doi:10.1371/journal.pone.0033199
89. Dennis G, Holweg CT, Kummerfeld SK, et al. Synovial phenotypes in rheumatoid arthritis correlate with response to biologic therapeutics. Arthritis Res Ther. 2014;16(2):1–18. doi:10.1186/ar4555
90. Wright HL, Thomas HB, Moots RJ, Edwards SW. Interferon gene expression signature in rheumatoid arthritis neutrophils correlates with a good response to TNFi therapy. Rheumatology. 2015;54(1):188–193. doi:10.1093/rheumatology/keu299
91. Smith SL, Eyre S, Yarwood A, et al. Investigating CD11c expression as a potential genomic biomarker of response to TNF inhibitor biologics in whole blood rheumatoid arthritis samples. Arthritis Res Ther. 2015;17(1):1–7. doi:10.1186/s13075-015-0868-y
92. Oswald M, Curran ME, Lamberth SL, et al. Modular analysis of peripheral blood gene expression in rheumatoid arthritis captures reproducible gene expression changes in tumor necrosis factor responders. Arthritis Rheumatol. 2015;67(2):344–351. doi:10.1002/art.38947
93. Spiliopoulou A, Colombo M, Plant D, et al. Association of response to TNF inhibitors in rheumatoid arthritis with quantitative trait loci for CD40 and CD39. Ann Rheumatic Dis. 2019;78(8):1055–1061. doi:10.1136/annrheumdis-2018-214877
94. Oliver J, Nair N, Orozco G, et al. Transcriptome-wide study of TNF-inhibitor therapy in rheumatoid arthritis reveals early signature of successful treatment. Arthritis Res Therapy. 2021;23(1):80. doi:10.1186/s13075-021-02451-9
95. Cai Y, Xu K, Aihaiti Y, et al. Derlin-1, as a potential early predictive biomarker for nonresponse to infliximab treatment in rheumatoid arthritis, is related to autophagy. Front Immunol. 2022;12:795912. doi:10.3389/fimmu.2021.795912
96. Sanayama Y, Ikeda K, Saito Y, et al. Prediction of therapeutic responses to tocilizumab in patients with rheumatoid arthritis: biomarkers identified by analysis of gene expression in peripheral blood mononuclear cells using genome‐wide DNA microarray. Arthritis Rheumatol. 2014;66(6):1421–1431. doi:10.1002/art.38400
97. Teitsma XM, Jacobs JW, Mokry M, et al. Identification of differential co-expressed gene networks in early rheumatoid arthritis achieving sustained drug-free remission after treatment with a tocilizumab-based or methotrexate-based strategy. Arthritis Res Ther. 2017;19(1):1–10. doi:10.1186/s13075-017-1378-x
98. Yokoyama-Kokuryo W, Yamazaki H, Takeuchi T, et al. Identification of molecules associated with response to Abatacept in patients with rheumatoid arthritis. Arthritis Res Ther. 2020;22(1):1–8. doi:10.1186/s13075-020-2137-y
99. Thurlings RM, Boumans M, Tekstra J, et al. Relationship between the type I interferon signature and the response to rituximab in rheumatoid arthritis patients. Arthritis Rheum. 2010;62(12):3607–3614. doi:10.1002/art.27702
100. Raterman HG, Vosslamber S, de Ridder S, et al. The interferon type I signature towards prediction of non-response to rituximab in rheumatoid arthritis patients. Arthritis Res Ther. 2012;14(2):R95. doi:10.1186/ar3819
101. Sellam J, Marion‐Thore S, Dumont F, et al. Use of whole‐blood transcriptomic profiling to highlight several pathophysiologic pathways associated with response to rituximab in patients with rheumatoid arthritis: data from a randomized, controlled, open‐label trial. Arthritis Rheumatol. 2014;66(8):2015–2025. doi:10.1002/art.38671
102. Nakamura S, Suzuki K, Iijima H, et al. Identification of baseline gene expression signatures predicting therapeutic responses to three biologic agents in rheumatoid arthritis: a retrospective observational study. Arthritis Res Ther. 2016;18(1):1–12. doi:10.1186/s13075-016-1052-8
103. Triaille C, Durez P, Sokolova T, et al. Common transcriptomic effects of Abatacept and other DMARDs on rheumatoid arthritis synovial tissue. Front Immunol. 2021;12:724895. doi:10.3389/fimmu.2021.724895
104. Rivellese F, Nerviani A, Giorli G, et al. Stratification of biological therapies by pathobiology in biologic-naive patients with rheumatoid arthritis (STRAP and STRAP-EU): two parallel, open-label, biopsy-driven, randomised trials. Lancet Rheumatol. 2023;5(11):e648–e659. doi:10.1016/S2665-9913(23)00241-2
105. Humby F, Durez P, Buch MH, et al. Rituximab versus tocilizumab in anti-TNF inadequate responder patients with rheumatoid arthritis (R4RA): 16-week outcomes of a stratified, biopsy-driven, multicentre, open-label, Phase 4 randomised controlled trial. Lancet. 2021;397(10271):305–317. doi:10.1016/S0140-6736(20)32341-2
106. Sobsey CA, Ibrahim S, Richard VR, et al. Targeted and untargeted proteomics approaches in biomarker development. Proteomics. 2020;20(9):1900029. doi:10.1002/pmic.201900029
107. Lee AN, Beck CE, Hall M. Rheumatoid factor and anti-CCP autoantibodies in rheumatoid arthritis: a review. Clin Lab Sci. 2008;21(1):15.
108. Julià A, López-Lasanta M, Blanco F, et al. Interactions between rheumatoid arthritis antibodies are associated with the response to anti-tumor necrosis factor therapy. BMC Musculoskel Disorder. 2021;22(1):1–7. doi:10.1186/s12891-021-04248-y
109. Courvoisier DS, Chatzidionysiou K, Mongin D, et al. The impact of seropositivity on the effectiveness of biologic anti-rheumatic agents: results from a collaboration of 16 registries. Rheumatology. 2020;60(2):820–828. doi:10.1093/rheumatology/keaa393
110. Bird P, Hall S, Nash P, et al. Treatment outcomes in patients with seropositive versus seronegative rheumatoid arthritis in Phase III randomised clinical trials of tofacitinib. RMD Open. 2019;5(1):e000742. doi:10.1136/rmdopen-2018-000742
111. Genovese MC, Fleischmann R, Kivitz A, et al. Efficacy and safety of sarilumab in combination with csDMARDs or as monotherapy in subpopulations of patients with moderately to severely active rheumatoid arthritis in three phase III randomized, controlled studies. Arthritis Res Ther. 2020;22(1):139. doi:10.1186/s13075-020-02194-z
112. Isaacs JD, Cohen SB, Emery P, et al. Effect of baseline rheumatoid factor and anticitrullinated peptide antibody serotype on rituximab clinical response: a meta-analysis. Ann Rheum Dis. 2013;72(3):329–336. doi:10.1136/annrheumdis-2011-201117
113. Halvorsen EH, Haavardsholm EA, Pollmann S, et al. Serum IgG antibodies to peptidylarginine deiminase 4 predict radiographic progression in patients with rheumatoid arthritis treated with tumour necrosis factor-alpha blocking agents. Ann Rheum Dis. 2009;68(2):249–252. doi:10.1136/ard.2008.094490
114. Ferraccioli G, Tolusso B, Bobbio-Pallavicini F, et al. Biomarkers of good EULAR response to the B cell depletion therapy in all seropositive rheumatoid arthritis patients: clues for the pathogenesis. PLoS One. 2012;7(7):e40362. doi:10.1371/journal.pone.0040362
115. de Moel EC, Derksen VFAM, Stoeken G, et al. Baseline autoantibody profile in rheumatoid arthritis is associated with early treatment response but not long-term outcomes. Arthritis Res Ther. 2018;20(1):33. doi:10.1186/s13075-018-1520-4
116. Darrah E, Yu F, Cappelli LC, Rosen A, O’Dell JR, Mikuls TR. Association of baseline peptidylarginine deiminase 4 autoantibodies with favorable response to treatment escalation in rheumatoid arthritis. Arthritis Rheumatol. 2019;71(5):696–702. doi:10.1002/art.40791
117. Lourido L, Ruiz-Romero C, Picchi F, et al. Association of Serum Anti-Centromere Protein F Antibodies with Clinical Response to Infliximab in Patients with Rheumatoid Arthritis: A Prospective Study. Elsevier; 2020:1101–1108.
118. Kumar R, Piantoni S, Boldini M, et al. Anti-carbamylated protein antibodies as a clinical response predictor in rheumatoid arthritis patients treated with Abatacept. Clin Exp Rheumatol. 2021;39(1):91–97. doi:10.55563/clinexprheumatol/g8xqxr
119. Fabre S, Dupuy AM, Dossat N, et al. Protein biochip array technology for cytokine profiling predicts etanercept responsiveness in rheumatoid arthritis. Clin Exp Immunol. 2008;153(2):188–195. doi:10.1111/j.1365-2249.2008.03691.x
120. Trocmé C, Marotte H, Baillet A, et al. Apolipoprotein A-I and platelet factor 4 are biomarkers for infliximab response in rheumatoid arthritis. Ann Rheum Dis. 2009;68(8):1328–1333. doi:10.1136/ard.2008.093153
121. Hueber W, Tomooka BH, Batliwalla F, et al. Blood autoantibody and cytokine profiles predict response to anti-tumor necrosis factor therapy in rheumatoid arthritis. Arthritis Res Ther. 2009;11(3):R76. doi:10.1186/ar2706
122. Fabre S, Guisset C, Tatem L, et al. Protein biochip array technology to monitor rituximab in rheumatoid arthritis. Clin Exp Immunol. 2009;155(3):395–402. doi:10.1111/j.1365-2249.2008.03804.x
123. Hirata S, Marotta A, Gui Y, Hanami K, Tanaka Y. Serum 14-3-3η level is associated with severity and clinical outcomes of rheumatoid arthritis, and its pretreatment level is predictive of DAS28 remission with tocilizumab. Arthritis Res Ther. 2015;17(1):280. doi:10.1186/s13075-015-0799-7
124. Uno K, Yoshizaki K, Iwahashi M, et al. Pretreatment Prediction of individual rheumatoid arthritis patients’ response to anti-cytokine therapy using serum cytokine/chemokine/soluble receptor biomarkers. PLoS One. 2015;10(7):e0132055. doi:10.1371/journal.pone.0132055
125. Obry A, Hardouin J, Lequerré T, et al. Identification of 7 Proteins in sera of RA patients with potential to predict ETA/MTX treatment response. Theranostics. 2015;5(11):1214–1224. doi:10.7150/thno.12403
126. Han BK, Kuzin I, Gaughan JP, Olsen NJ, Bottaro A. Baseline CXCL10 and CXCL13 levels are predictive biomarkers for tumor necrosis factor inhibitor therapy in patients with moderate to severe rheumatoid arthritis: a pilot, prospective study. Arthritis Res Ther. 2016;18:93. doi:10.1186/s13075-016-0995-0
127. Folkersen L, Brynedal B, Diaz-Gallo LM, et al. Integration of known DNA, RNA and protein biomarkers provides prediction of anti-TNF response in rheumatoid arthritis: results from the COMBINE study. Mol Med. 2016;22(1):322–328. doi:10.2119/molmed.2016.00078
128. Rinaudo-Gaujous M, Blasco-Baque V, Miossec P, et al. Infliximab induced a dissociated response of severe periodontal biomarkers in rheumatoid arthritis patients. J Clin Med. 2019;8(5):751. doi:10.3390/jcm8050751
129. Chen J, Tang MS, Xu LC, et al. Proteomic analysis of biomarkers predicting the response to triple therapy in patients with rheumatoid arthritis. Biomed Pharmacother. 2019;116:109026. doi:10.1016/j.biopha.2019.109026
130. Ling SF, Nair N, Verstappen SM, et al. Proteomic analysis to define predictors of treatment response to Adalimumab or methotrexate in rheumatoid arthritis patients. Pharmacogeno J. 2020;20(3):516–523. doi:10.1038/s41397-019-0139-4
131. Zhao J, Ye X, Zhang Z. The predictive value of serum soluble ICAM-1 and CXCL13 in the therapeutic response to TNF inhibitor in rheumatoid arthritis patients who are refractory to csDMARDs. Clin Rheumatol. 2020;39(9):2573–2581. doi:10.1007/s10067-020-05043-1
132. Ling SF, Yap CF, Nair N, et al. A proteomics study of rheumatoid arthritis patients on etanercept identifies putative biomarkers associated with clinical outcome measures. Rheumatology. 2023;(Supplement_2). doi:10.1093/rheumatology/kead321
133. Roessner U, Bowne J. What is metabolomics all about? Biotechniques. 2009;46(5):363–365. doi:10.2144/000113133
134. Gosselt HR, Muller IB, Jansen G, et al. Identification of metabolic biomarkers in relation to methotrexate response in early rheumatoid arthritis. J Pers Med. 2020;10(4):271. doi:10.3390/jpm10040271
135. Medcalf MR, Bantis LE, Shi P, et al. Plasma metabolomic profiling as a tool to identify predictive biomarkers of methotrexate efficacy in rheumatoid arthritis. Semin Arthritis Rheumatism. 2022;56:152056. doi:10.1016/j.semarthrit.2022.152056
136. Kapoor SR, Filer A, Fitzpatrick MA, et al. Metabolic profiling predicts response to anti-tumor necrosis factor α therapy in patients with rheumatoid arthritis. Arthritis Rheum. 2013;65(6):1448–1456. doi:10.1002/art.37921
137. Takahashi S, Saegusa J, Onishi A, Morinobu A. Biomarkers identified by serum metabolomic analysis to predict biologic treatment response in rheumatoid arthritis patients. Rheumatology. 2019;58(12):2153–2161. doi:10.1093/rheumatology/kez199
138. Priori R, Casadei L, Valerio M, Scrivo R, Valesini G, Manetti C. ¹H-NMR-Based metabolomic study for identifying serum profiles associated with the response to etanercept in patients with rheumatoid arthritis. PLoS One. 2015;10(11):e0138537. doi:10.1371/journal.pone.0138537
139. Cuppen BV, Fu J, van Wietmarschen HA, et al. Exploring the inflammatory metabolomic profile to predict response to TNF-α inhibitors in rheumatoid arthritis. PLoS One. 2016;11(9):e0163087. doi:10.1371/journal.pone.0163087
140. Murillo-Saich JD, Diaz-Torne C, Ortiz MA, et al. Metabolomics profiling predicts outcome of tocilizumab in rheumatoid arthritis: an exploratory study. Metabolomics. 2021;17(9):74. doi:10.1007/s11306-021-01822-2
141. Teitsma XM, Yang W, Jacobs JW, et al. Baseline metabolic profiles of early rheumatoid arthritis patients achieving sustained drug-free remission after initiating treat-to-target tocilizumab, methotrexate, or the combination: insights from systems biology. Arthritis Res Ther. 2018;20(1):1–11. doi:10.1186/s13075-018-1729-2
142. Dudka I, Chachaj A, Sebastian A, et al. Metabolomic profiling reveals plasma GlycA and GlycB as a potential biomarkers for treatment efficiency in rheumatoid arthritis. J Pharma Biomed Analysis. 2021;197:113971. doi:10.1016/j.jpba.2021.113971
143. Sweeney SR, Kavanaugh A, Lodi A, et al. Metabolomic profiling predicts outcome of rituximab therapy in rheumatoid arthritis. RMD Open. 2016;2(2):e000289. doi:10.1136/rmdopen-2016-000289
144. Kang J-S, Lee M-H. Overview of therapeutic drug monitoring. Korean J Int Med. 2009;24(1):1. doi:10.3904/kjim.2009.24.1.1
145. Rosas J, Llinares-Tello F, de la Torre I, et al. Clinical relevance of monitoring serum levels of Adalimumab in patients with rheumatoid arthritis in daily practice. Clin Exp Rheumatol. 2014;32(6):942–948.
146. van den Bemt BJ, den Broeder AA, Wolbink GJ, et al. The combined use of disease activity and infliximab serum trough concentrations for early prediction of (non-)response to infliximab in rheumatoid arthritis. Br J Clin Pharmacol. 2013;76(6):939–945. doi:10.1111/bcp.12142
147. Jurado T, Plasencia-Rodríguez C, Martínez-Feito A, et al. Predictive value of serum infliximab levels at induction phase in rheumatoid arthritis patients. Open Rheumatol J. 2017;11:75. doi:10.2174/1874312901711010075
148. Takeuchi T, Miyasaka N, Inoue K, Abe T, Koike T. Impact of trough serum level on radiographic and clinical response to infliximab plus methotrexate in patients with rheumatoid arthritis: results from the RISING study. Mod Rheumatol. 2009;19(5):478–487. doi:10.3109/s10165-009-0195-8
149. Jamnitski A, Krieckaert C, Nurmohamed M, et al. Patients non-responding to etanercept obtain lower etanercept concentrations compared with responding patients. Ann Rheumatic Dis. 2012;71(1):88–91. doi:10.1136/annrheumdis-2011-200184
150. Arad U, Elkayam O. Association of serum tocilizumab trough concentrations with clinical disease activity index scores in adult patients with rheumatoid arthritis. J Rheumatol. 2019;46(12):1577–1581. doi:10.3899/jrheum.181431
151. Kneepkens E, van den Oever I, Plasencia C, et al. Serum tocilizumab trough concentration can be used to monitor systemic IL-6 receptor blockade in patients with rheumatoid arthritis: a prospective observational cohort study. Scand J Rheumatol. 2017;46(2):87–94. doi:10.1080/03009742.2016.1183039
152. Ruscitti P, Sinigaglia L, Cazzato M, et al. Dose adjustments and discontinuation in TNF inhibitors treated patients: when and how. A systematic review of literature. Rheumatology. 2018;57(Supplement_7):vii23–vii31. doi:10.1093/rheumatology/key132
153. Balsa A, Sanmarti R, Rosas J, et al. Drug immunogenicity in patients with inflammatory arthritis and secondary failure to tumour necrosis factor inhibitor therapies: the REASON study. Rheumatology. 2018;57(4):688–693. doi:10.1093/rheumatology/kex474
154. Strand V, Goncalves J, Isaacs JD. Immunogenicity of biologic agents in rheumatology. Nat Rev Rheumatol. 2021;17(2):81–97. doi:10.1038/s41584-020-00540-8
155. Bartelds GM, Krieckaert CL, Nurmohamed MT, et al. Development of antidrug antibodies against Adalimumab and association with disease activity and treatment failure during long-term follow-up. JAMA. 2011;305(14):1460–1468. doi:10.1001/jama.2011.406
156. Ducourau E, Mulleman D, Paintaud G, et al. Antibodies toward infliximab are associated with low infliximab concentration at treatment initiation and poor infliximab maintenance in rheumatic diseases. Arthritis Res Ther. 2011;13(3):1–7. doi:10.1186/ar3386
157. Strand V, Balsa A, Al-Saleh J, et al. Immunogenicity of biologics in chronic inflammatory diseases: a systematic review. BioDrugs. 2017;31(4):299–316. doi:10.1007/s40259-017-0231-8
158. Wells AF, Parrino J, Mangan EK, et al. Immunogenicity of sarilumab monotherapy in patients with rheumatoid arthritis who were inadequate responders or intolerant to disease-modifying antirheumatic drugs. Rheumatol Ther. 2019;6(3):339–352. doi:10.1007/s40744-019-0157-3
159. Xu C, Su Y, Paccaly A, Kanamaluru V. Population pharmacokinetics of sarilumab in patients with rheumatoid arthritis. Clin Pharmacokinet. 2019;58(11):1455–1467. doi:10.1007/s40262-019-00765-1
160. Wincup C, Dunn N, Ruetsch-Chelli C, et al. Anti-rituximab antibodies demonstrate neutralizing capacity, associate with lower circulating drug levels and earlier relapse in lupus. Rheumatology. 2022;62(7):2601–2610. doi:10.1093/rheumatology/keac608
161. Moots RJ, Xavier RM, Mok CC, et al. The impact of anti-drug antibodies on drug concentrations and clinical outcomes in rheumatoid arthritis patients treated with Adalimumab, etanercept, or infliximab: results from a multinational, real-world clinical practice, non-interventional study. PLoS One. 2017;12:4.
162. Maid PJ, Xavier R, Real RM, et al. Incidence of antidrug antibodies in rheumatoid arthritis patients from Argentina treated with Adalimumab, etanercept, or infliximab in a real-world setting. JCR. 2018;24(4):177–182.
163. Quistrebert J, Hässler S, Bachelet D, et al. Incidence and risk factors for Adalimumab and infliximab anti-drug antibodies in rheumatoid arthritis: a European retrospective multicohort analysis. Semin Arthritis Rheum. 2019;48(6):967–975. doi:10.1016/j.semarthrit.2018.10.006
164. Burmester GR, Choy E, Kivitz A, et al. Low immunogenicity of tocilizumab in patients with rheumatoid arthritis. Ann Rheum Dis. 2017;76(6):1078–1085. doi:10.1136/annrheumdis-2016-210297
165. Sigaux J, Hamze M, Daien C, et al. Immunogenicity of tocilizumab in patients with rheumatoid arthritis. Joint Bone Spine. 2017;84(1):39–45. doi:10.1016/j.jbspin.2016.04.013
166. Genovese MC, Fleischmann R, Kivitz AJ, et al. Sarilumab plus methotrexate in patients with active rheumatoid arthritis and inadequate response to methotrexate: results of a phase III study. Arthritis Rheumatol. 2015;67(6):1424–1437. doi:10.1002/art.39093
167. Thurlings RM, Teng O, Vos K, et al. Clinical response, pharmacokinetics, development of human anti-chimaeric antibodies, and synovial tissue response to rituximab treatment in patients with rheumatoid arthritis. Ann Rheum Dis. 2010;69(2):409–412. doi:10.1136/ard.2009.109041
168. Bitoun S, Hässler S, Ternant D, et al. Response to biologic drugs in patients with rheumatoid arthritis and antidrug antibodies. JAMA Network Open. 2023;6(7):e2323098–e2323098. doi:10.1001/jamanetworkopen.2023.23098
169. Jamnitski A, Bartelds GM, Nurmohamed MT, et al. The presence or absence of antibodies to infliximab or Adalimumab determines the outcome of switching to etanercept. Ann Rheum Dis. 2011;70(2):284–288. doi:10.1136/ard.2010.135111
170. Ucar E, Gorostiza Í, Gόmez C, et al. SAT0150 Prospective, Intervention, Multicenter Study of Utility of Biologic Drug Monitoring with Respect to the Efficacy and Cost of Adalimumab Tapering in Patients with Rheumatic Diseases: Preliminary Results of INGEBIO Study. BMJ Publishing Group Ltd; 2017.
171. Syversen SW, Goll GL, Jørgensen KK, et al. Therapeutic drug monitoring of infliximab compared to standard clinical treatment with infliximab: study protocol for a randomised, controlled, open, parallel-group, Phase IV study (the NOR-DRUM study). Trials. 2020;21(1):13. doi:10.1186/s13063-019-3734-4
172. (NICE) NIfHaCE. Therapeutic monitoring of TNF-alpha inhibitors in rheumatoid arthritis. Available from: https://www.nice.org.uk/guidance/dg36.
173. Krieckaert CL, van Tubergen A, Gehin JE, et al. EULAR points to consider for therapeutic drug monitoring of biopharmaceuticals in inflammatory rheumatic and musculoskeletal diseases. Ann Rheumatic Dis. 2023;82(1):65–73. doi:10.1136/annrheumdis-2022-222155
174. Johnson KB, Wei WQ, Weeraratne D, et al. Precision medicine, AI, and the future of personalized health care. Clin Transl Sci. 2021;14(1):86–93. doi:10.1111/cts.12884
175. Deo RC. Machine learning in medicine. Circulation. 2015;132(20):1920–1930. doi:10.1161/CIRCULATIONAHA.115.001593
176. Mirza B, Wang W, Wang J, Choi H, Chung NC, Ping P. Machine learning and integrative analysis of biomedical big data. Genes. 2019;10(2):87. doi:10.3390/genes10020087
177. Westerlind H, Maciejewski M, Frisell T, Jelinsky SA, Ziemek D, Askling J. What Is the persistence to methotrexate in rheumatoid arthritis, and does machine learning outperform hypothesis-based approaches to its prediction? ACR Open Rheumatol. 2021;3(7):457–463. doi:10.1002/acr2.11266
178. Miyoshi F, Honne K, Minota S, Okada M, Ogawa N, Mimura T. A novel method predicting clinical response using only background clinical data in RA patients before treatment with infliximab. Mod Rheumatol. 2016;26(6):813–816. doi:10.3109/14397595.2016.1168536
179. Koo BS, Eun S, Shin K, et al. Machine learning model for identifying important clinical features for predicting remission in patients with rheumatoid arthritis treated with biologics. Arthritis Res Ther. 2021;23(1):178. doi:10.1186/s13075-021-02567-y
180. Gosselt HR, Verhoeven MMA, Bulatović-ćalasan M, et al. Complex machine-learning algorithms and multivariable logistic regression on par in the prediction of insufficient clinical response to methotrexate in rheumatoid arthritis. J Pers Med. 2021;11(1):44. doi:10.3390/jpm11010044
181. Lim AJ, Lim LJ, Ooi BN, et al. Functional coding haplotypes and machine-learning feature elimination identifies predictors of methotrexate response in rheumatoid arthritis patients. EBioMedicine. 2022;3:75.
182. Plant D, Maciejewski M, Smith S, et al. Profiling of gene expression biomarkers as a classifier of methotrexate nonresponse in patients with rheumatoid arthritis. Arthritis Rheumatol. 2019;71(5):678–684. doi:10.1002/art.40810
183. Guan Y, Zhang H, Quang D, et al. Machine learning to predict anti-tumor necrosis factor drug responses of rheumatoid arthritis patients by integrating clinical and genetic markers. Arthritis Rheumatol. 2019;71(12):1987–1996. doi:10.1002/art.41056
184. Luque-Tévar M, Perez-Sanchez C, Patiño-Trives AM, et al. Integrative clinical, molecular, and computational analysis identify novel biomarkers and differential profiles of anti-TNF response in rheumatoid arthritis. Front Immunol. 2021;12:631662. doi:10.3389/fimmu.2021.631662
185. Yoosuf N, Maciejewski M, Ziemek D, et al. Early prediction of clinical response to anti-TNF treatment using multi-omics and machine learning in rheumatoid arthritis. Rheumatology. 2022;61(4):1680–1689. doi:10.1093/rheumatology/keab521
186. Tao W, Concepcion AN, Vianen M, et al. Multiomics and machine learning accurately predict clinical response to Adalimumab and etanercept therapy in patients with rheumatoid arthritis. Arthritis Rheumatol. 2021;73(2):212–222. doi:10.1002/art.41516
187. Tasaki S, Suzuki K, Kassai Y, et al. Multi-omics monitoring of drug response in rheumatoid arthritis in pursuit of molecular remission. Nat Commun. 2018;9(1):2755. doi:10.1038/s41467-018-05044-4
188. Jung SM, Park K-S, Kim K-J. Deep phenotyping of synovial molecular signatures by integrative systems analysis in rheumatoid arthritis. Rheumatology. 2021;60(7):3420–3431. doi:10.1093/rheumatology/keaa751
189. MacEachern SJ, Forkert ND. Machine learning for precision medicine. Genome. 2021;64(4):416–425. doi:10.1139/gen-2020-0131
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Towards Personalized Medicine in Psoriasis: Current Progress
Camela E, Potestio L, Ruggiero A, Ocampo-Garza SS, Fabbrocini G, Megna M
Psoriasis: Targets and Therapy 2022, 12:231-250
Published Date: 1 September 2022
Awareness and Predictors of the Use of Bioinformatics in Genome Research in Saudi Arabia
Alomair L, Abolfotouh MA
International Journal of General Medicine 2023, 16:3413-3425
Published Date: 11 August 2023
Updates on Parkinson’s Disease
Bai H, Ma W, Zhu L, Lu Y, Fan J, Chen M, Huang C
Neuropsychiatric Disease and Treatment 2025, 21:1945-1953
Published Date: 4 September 2025
Precision Medicine in Asthma: The Role of Biomarkers
Quek E, Horn N, Siddiqui S
ImmunoTargets and Therapy 2025, 14:1479-1513
Published Date: 28 December 2025