Back to Journals » Journal of Inflammation Research » Volume 19
The Bioinformatics Analysis of Publicly Available Datasets and Validation in a Mouse Model of Myocardial Infarction Through Coronary Artery Ligation
Authors Sun H ![]() , Liu F, Sun M, Wang W, Wang J
, Liu F, Sun M, Wang W, Wang J ![]() , Wang H, Jiang X, Ding X, Wang C
, Wang H, Jiang X, Ding X, Wang C ![]() , Zhong L
, Zhong L
Received 4 August 2025
Accepted for publication 27 December 2025
Published 6 January 2026 Volume 2026:19 558274
DOI https://doi.org/10.2147/JIR.S558274
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Chengming Fan
Han Sun,1,2,* Fujun Liu,3,* Mengchen Sun,1,2,* Wenlong Wang,1,2 Jiahui Wang,3 Hua Wang,2 Xiaoyan Jiang,4 Xiaoning Ding,5 Chunxiao Wang,2 Lin Zhong2
1Qingdao Medical College, Qingdao University, Qingdao, Shandong, 266000, People’s Republic of China; 2Department of Cardiology, Yantai Yuhuangding Hospital, Qingdao University, Yantai, Shandong, 264000, People’s Republic of China; 3Central Laboratory, Yantai Yuhuangding Hospital, Qingdao University, Yantai, Shandong, 264000, People’s Republic of China; 4Department of Cardiology, People’s Hospital of Fushan District, Yantai, Shandong, 264000, People’s Republic of China; 5Shanghai Children’s Medical Center, Shanghai Jiao Tong University School of Medicine, Shanghai, 200000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Chunxiao Wang, Email [email protected] Lin Zhong, Email [email protected]
Background: Macrophages play a crucial role in the inflammatory response and fibrosis after myocardial infarction (MI). CMTM3 exerts important functions in the immune system and cardiovascular system. This study aims to explore the role and mechanism of CMTM3 in regulating macrophage-related inflammation after MI.
Methods: The CMTM3−/− mouse MI model was established. The effects of CMTM3 on MI and macrophage-related inflammation in mice were evaluated by TTC, Masson, echocardiography, flow cytometry and Elisa. In vitro, the effects of CMTM3 on primary macrophages were assessed by flow cytometry, RT-qPCR and Elisa. The mechanism of CMTM3 regulating macrophages was explored by Western blot.
Results: In the mouse MI model, CMTM3 expression was mainly increased in macrophages. CMTM3 deficiency resulted in an enlarged infarct size, increased collagen deposition, and deteriorated cardiac function. Further studies revealed that CMTM3 deficiency promoted macrophage polarization toward M1 types, and increase the production and secretion of inflammatory factors IL-1β, IL-6 and TNF-α. In vitro studies also confirmed CMTM3 deficiency promoted M1 macrophage differentiation and upregulated the expression of inflammatory factors. Mechanistically, CMTM3 can interact with PPARα, CMTM3 deficiency can inhibit PPARα activity, and increase the phosphorylation of NF-κB, thereby promoting macrophage inflammation.
Conclusion: CMTM3 inhibits macrophage-related inflammation after MI by activating PPARα and inhibiting NF-κB phosphorylation. This study highlights the anti-inflammatory effect of CMTM3 in MI, and holds that CMTM3 can serve as a new target for improving cardiac remodeling after MI.
Keywords: CMTM3, myocardial infarction, macrophage, inflammation, NF-κB, PPARα
Introduction
Myocardial infarction (MI) can result in irreversible myocardial cell necrosis and adverse cardiac remodeling, which subsequently progresses to heart failure, malignant arrhythmias, and ultimately death.1 Inflammation represents a key and persistent pathological process influencing MI. Myocardial ischemic injury triggers a robust inflammatory cascade, characterized by extensive infiltration of immune cells into the infarcted myocardium, which participate in the progression of MI.2,3 Among these, mononuclear macrophages, as the primary immune cells, exhibit functional heterogeneity at different stages of MI.4 In the early stage of MI, they are mainly of the pro-inflammatory types (M1 types), and in the late stage, they are mainly of the pro-repair type (M2 types).5 Pro-inflammatory macrophages possess strong phagocytic and pro-inflammatory properties, which can phagocytic necrotic cardiomyocytes and extracellular matrix (ECM) fragments, and to produce inflammatory factors and chemokines, thereby shaping an inflammatory microenvironment.6 Cardiac repair process after MI involves a balance between inflammatory response and tissue repair. Moderate inflammatory response can promote the clearance of necrotic cells and the scar formation, exerting a positive role on tissue repair.7 However, excessive inflammatory response will hinder wound healing, enlarges the infarct size, and leads to deterioration of cardiac function.8 Therefore, clarifying the phenotypic transition of macrophages and the regulatory mechanism underlying inflammatory responses is of great significance for preventing post-MI cardiac rupture and improving cardiac function.
The chemokine-like factor (CKLF)-like MARVEL transmembrane domain-containing (CMTM) family is a gene superfamily associated with human chemotaxis. It comprises 9 members, including CKLF and CMTM1-8.9 CMTMs not only play important roles in the cardiovascular system but also exert critical functions in inflammatory and immune regulation. Accumulating evidence has demonstrated that CMTMs have significant impacts on immune-related pathways and immune cell infiltration in tumors.10 For instance, CMTM4 and CMTM6 can regulate the anti-tumor function of T cells through PD-L1,11 and CMTM4 is associated with the secretion of the inflammatory factor IL-6.12 In a comprehensive analysis of immune infiltration in esophageal cancer, it was found that the expression levels of CMTM1, 3, 5, and 7 are significantly correlated with macrophage expression.13 Moreover, CMTMs are closely linked to various chronic autoimmune inflammatory diseases, such as rheumatoid arthritis.14 Additionally, CMTM3 has been reported to have dual effects on inflammation in different diseases, including pro-inflammatory effects on neutrophil-associated inflammation in sepsis and inhibition of tumor-associated inflammation in the tumor immune microenvironment.15,16 However, despite studies indicating that CMTMs are highly correlated with immune cells expression and play a crucial role in regulating immune responses, their role in macrophage-related inflammation during MI remains unreported.
In this study, CMTM3 was initially selected from the CMTMs via bioinformatics analysis. We conducted studies in the CMTM3−/− mice MI model, detected the expression changes of CMTM3, explored the effects of CMTM3 on MI area, cardiac function and inflammatory microenvironment. Furthermore, the regulation and mechanism of CMTM3 in macrophage differentiation were explored. It provides a novel target for anti-inflammatory therapy following MI.
Materials and Methods
Microarray Data Retrieval
MI data set from a public repository NCBI GEO (http://www.ncbi.nlm.nih.gov/geo). To better analyze the expression changes of CMTMs in MI, we selected the GSE161427 dataset as the analysis set and GSE236374 as the validation set. GSE161427 (Zhang et al, 2020) (Agilent-028005 SurePrint G3 Mouse GE 8x60K Microarray) contains a total of 14 heart samples of mice Sham, MI3d, and MI14d, generated by the GPL10787 platform. To better analyze the differential genes between the MI group and the CON group, we selected five samples from the MI group at 3 days and four samples from the Sham group for analysis. GSE236374 (Yu et al, 2023) (Illumina NovaSeq 6000) contains a total of 9 heart samples of mice Sham, MI7d, and MI28d, generated by the GPL24247 platform.
Validation of Gene Expression Through DEG Analysis
The downloaded gene expression profiles and their matching platform files were analyzed using R (version 4.0) software, and they were converted into gene symbol expression profiles. Differential gene expression (differential expression genes, DEG) was performed with the “LIMMA” package (3.50.0) with |log2 (Fold-change)|≥1, adjust p<0.05.
Next, the correlations between CMTM2a, CMTM3, CMTM7 and IL-1β, IL-6, CD68, NF-κB, PPARα were analyzed using the Pearson algorithm.
Human Samples and Ethical Statement
This study collected blood samples from 20 patients with acute MI within 24 hours of onset at Yantai Yuhuangding Hospital Affiliated to Qingdao University from May 1, 2024 to October 31, 2024. Another 20 blood samples from healthy individuals undergoing physical examinations were taken as the control group. All the subjects gave informed consent. This study was approved by the Ethics Committee of Yantai Yuhuangding Hospital. The studies in this work abide by the Declaration of Helsinki principles.
Animal Care
C57BL/6 mice (Pengyue, Experimental Animal Breeding Co., LTD., Jinan, China) were raised in the SPF-grade experimental animal room, free of specific pathogens, with a constant room temperature (23±1°C), appropriate humidity (45–55%), 12h/12h light and dark circulation, ventilation, and free feeding and drinking, replace the gasket regularly. Each cage is 390 mm×180 mm×180 mm in size, and no more than 6 adult mice in each cage. The animal experiment process was carried out in accordance with the guidelines of the National Institutes of Health of the United States and was approved by the Animal Ethics Committee of Yuhuangding Hospital.
MI Model
MI was induced in C57BL/6 J male mice (6–8 weeks, male) by permanent ligation of the left anterior descending coronary artery (LAD). The specific steps are as follows: Mice were anesthetized by a small animal air anesthesia machine (Shenzhen Ruiwode Lift Technology Co., Ltd., China, R5301E), fixed in the supine position, and the skin, connective tissue, and the third rib space were dissected successively to expose the heart. Ligate the anterior descending branch of the left coronary artery approximately 2mm away from the tip of the left atrial appendage with a 7–0 thread, and then suture it. In the sham operation group, the left anterior descending coronary artery of mice was encircled with needle and thread, but not ligated.
Echocardiography
After the mice were anesthetized with isoflurane, echocardiography was performed using the animal echocardiography system (VINNO 6 LAB, VINNO, China). B-mode ultrasound observe the long axial section beside the sternum in mice, and M-mode ultrasound was used to analyze left ventricular ejection fraction (LVEF), left ventricular shortening rate (LVFS), left ventricular end-systolic diameter (LVIDs), left ventricular end-diastolic diameter (LVIDd).
Bone Marrow‑derived Macrophage (BMDM) Culture
BMDM was derived from C57BL/ 6J mice. After the mice (6–8 weeks old, male) were sacrificed, disinfected, the tibia and femur were isolated, bone marrow was collected, sieving was performed to obtain a single-cell suspension, and the cell precipitate was obtained by centrifugation. The cells were resuspended and spread in DMEM high glucose medium containing 10% bovine serum. Macrophage colony-stimulating factor (M-CSF, 50 ng/mL, peprotech, China) was used for 3–4 days to allow BMDMs maturity. Subsequently, the cells were digested and cultured in six-well plates. LPS+IFN-γ can induce macrophages to differentiate into pro-inflammatory types in vitro. LPS (1ug/mL, servicebio, China) and IFN-γ (10ng/mL, servicebio, China) were added to BMDMs, while the control group cells were added with the same volume of PBS. After stimulation for 24 h, the cells were extracted for the next step of the experiment.
Triphenyl Tetrazolium Chloride (TTC) Staining
Hearts were isolated 7d after MI and stored at − 20 °C for 25 min. Then, hearts were sectioned into 1 mm short axis slices. It was immediately placed in 37°C water bath with TTC solution (Solarbio, China) for 10 min, and then fixed with 4% paraformaldehyde for 2–3 min for photography. Calculate the proportion of the white infarcted area to the entire cardiac area.
Flow Cytometry Analysis
The mouse hearts were cut into 1mm3 small pieces, and type II collagenase was added. They were digested in 37°C water bath for 30 min to obtain a single-cell suspension of the heart. Then, the red blood cell lysate (Elabscience, China) was used for treatment for 2–5 min. Anti-cd16/32 antibody (Elabscience, China) treatment blocks FcγR. The primary antibody was used to avoid light at 4°C for 20–30 min for staining of cell surface markers, including: Anti-F4/80 antibody (PerCP/Cyanine5.5, Elabscience, China), anti-CD11b antibody (Elab Fluor700, Elabscience, China), anti-Ly6C antibody (FITC, Elabscience, China), anti-CD86 antibody (PE, Elabscience, China). All samples were detected by flow cytometry using the MoFlo XDP flow cytometer (Beckman) and analyzed by Flowjo (version 10.0) software. Gating strategy: Cardiac macrophages: CD11b+F4/80+; Cardiac pro-inflammatory macrophages: CD11b+F4/80+ Ly6ChighCD86+.
ELISA
Take serum or cell supernatants and operate to detect the contents of IL-1β (JONLNBIO, Shanghai, China), IL-6 (JONLNBIO, Shanghai, China), TNF-α (JONLNBIO, Shanghai, China), and CMTM3 (Keqiaobio, Shanghai, China) in accordance with the corresponding kit instructions. And the final absorbance value was determined at 450 nm.
Western Blot
The total protein of tissue and cell samples was extracted using RIPA lysis buffer containing protease inhibitors (Sparkjade, China), and the protein concentration was determined using the BCA protein assay kit (Yeasen, China). The protein samples were successively separated by 10–12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to PVDF or NC membranes. They were blocked with 5% skimmed milk powder for 1–2 h. Subsequently, anti-GAPDH antibody (1:8000, Hongheyuan, China) and anti-CMTM3 antibody (1:800, bioss, China), anti-P- NF-κB antibody (1:1000, CST, America), anti-NF-κB antibody (1:1000, CST, America), anti-PPARα antibody (1:1000, proteintech, China). After overnight incubation at 4°C, the membrane was incubated at room temperature in sheep anti-rabbit antibody (1:10000, affinity, China) for 1h. The ECL system was used for development, and the protein expression level was determined using Image J software.
Co-Immunoprecipitation (CO-IP)
BMDMs were cultured, stimulated with LPS+IFN-γ, proteins were extracted using lysate provided by CO-IP kit (absin, abs9649, China), protein concentration was determined by BCA method. CO-IP samples were prepared using the magnetic beads method according to the kit instructions. Specifically, CMTM3 antibody (5μg) and IgG antibody (5μg) were mixed with magnetic beads for 1–2 hours, the magnetic beads were rinsed, the protein samples were mixed with magnetic beads for 1–2 hours, the magnetic beads were rinsed, and the samples were boiled with a loading buffer, and then gel electrophoresis experiments were performed.
Additional Methods
The expanded methods in the Supplementary Material contain Real‑time qPCR (RT–qPCR), immunofluorescence, flow cytometry analysis, immunohistochemistry (IHC) staining, HE, masson, extraction of mononuclear cells from human peripheral blood (PBMCs), the primer sequence (Supplemental Table 1).
Statistical Analysis
The GraphPad Prism 9.5 software was used for analysis, all data were expressed as mean ±standard error (SEM). The differences between the two groups were analyzed using the unpaired Student’s t-test, and the differences between the three or four groups were analyzed using one-way ANOVA and Tukey’s test or two-way ANOVA and Tukey’s test. P value < 0.05 was considered statistically significant.
Results
Bioinformatics Analysis Reveals the Potential Role of CMTM3 in Myocardial Infarction
Analysis of MI samples based on the GSE161427 dataset revealed differential expression of CMTMs through volcano plot results: CMTM2a, CMTM3, and CMTM7 all showed an upregulated trend, while CMTM8 was downregulated. In contrast, CMTM1, CMTM2b, CMTM4, CMTM5, and CMTM6 exhibited no statistically significant differences (Figure 1A). Notably, in the subsequent correlation analysis, CMTM3 displayed the most significant correlation characteristics — it had the highest correlation coefficient with pro-inflammatory factors IL-1β, IL-6, and macrophage marker CD68. Although CMTM2a and CMTM7 were also correlated with IL-1β, IL-6 and CD68, their correlation coefficients were all lower than that of CMTM3 (Figure 1B). Futhermore, the high-throughput sequencing results of the GSE236374 dataset also indicated that CMTM3 was significantly upregulated at 7 days after MI (Figure 1C). In summary, the bioinformatics analysis results clearly indicated that among all upregulated CMTMs, CMTM3 not only showed a significant change in expression in the MI model but also exhibited the optimal performance in terms of correlation with inflammation. Therefore, this study focuses on CMTM3 to explore in depth its role and molecular mechanism in the inflammatory response of MI.
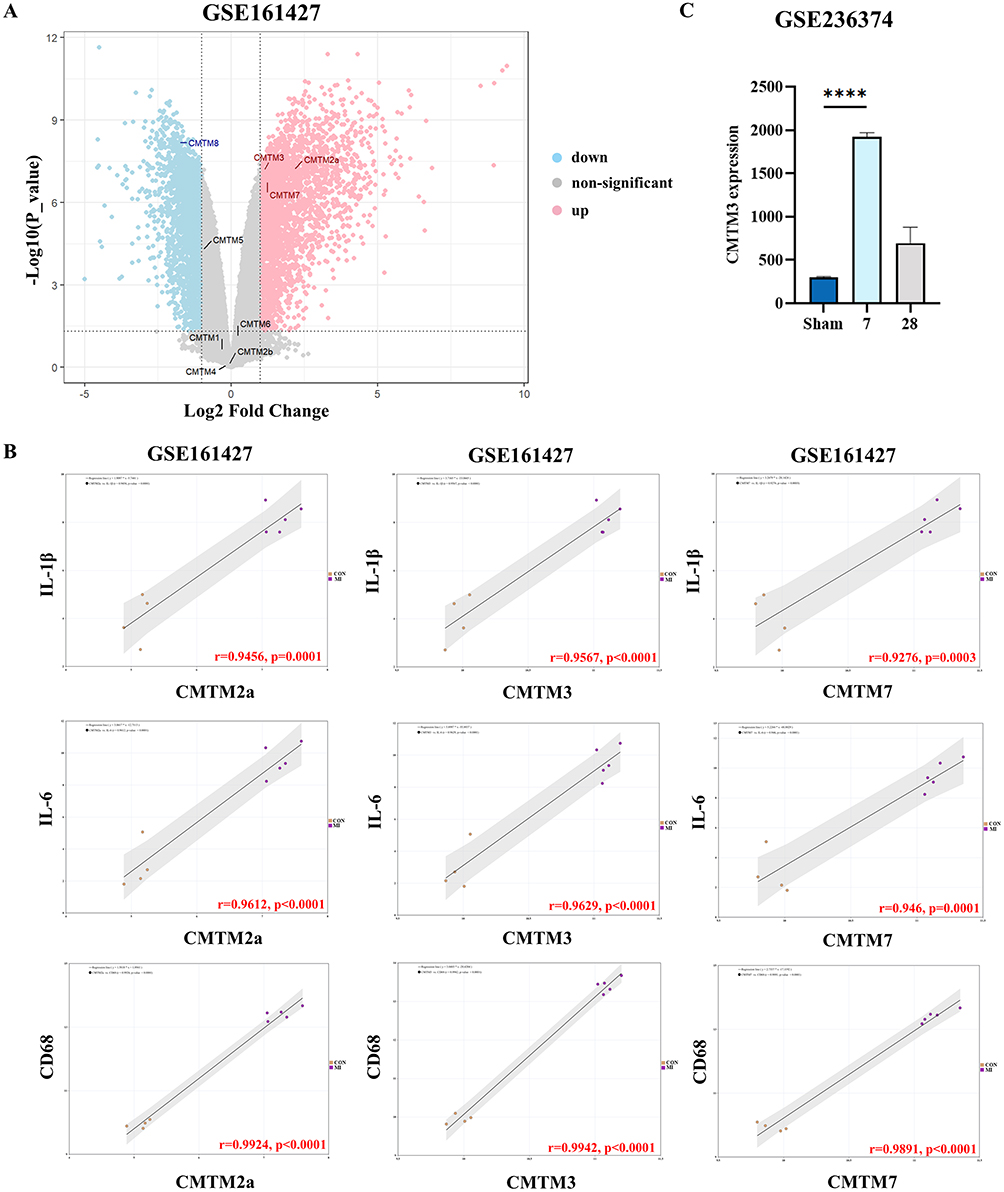 |
Figure 1 In the CMTMs, CMTM3 is upregulated and closely associated with inflammatory factors. (A) Volcano plots of differentially expression genes. Data points in red represent up-regulated, and blue represent down-regulated genes, |log2 (Fold-change)|≥1, adjust p<0.05; (B) Correlation analysis of CMTM2a, CMTM3 and CMTM7 expression with IL-1β, IL-6 and CD68 expression respectively (n=4-5); (C) The expression levels of CMTM3 in the GSE236374 (n=3). ****p<0.0001. |
CMTM3 Is Upregulated After MI
Mice model of MI were established. The expression of CMTM3 in the infarction border zone of mice with MI at 3d, 7d, 14d and 28d was detected by RT-qPCR and Western blot. Results demonstrated that CMTM3 mRNA and protein levels were upregulated as early as 3 days, peaked at 7 days, then decreased slowly, however, expression remained higher than that in the Sham group at 28d (Figure 2A and B). IHC staining further validated the increased expression of CMTM3 in murine hearts following MI (Figure 2C). The secretion of CMTM3 in the serum of patients with MI was detected by Elisa. The results indicated that the secretion of CMTM3 in the serum of patients with MI increased compared with healthy controls (Figure 2D). The above results suggest that CMTM3 is upregulated after MI, and suggest its potential involvement in the pathogenesis and progression of MI.
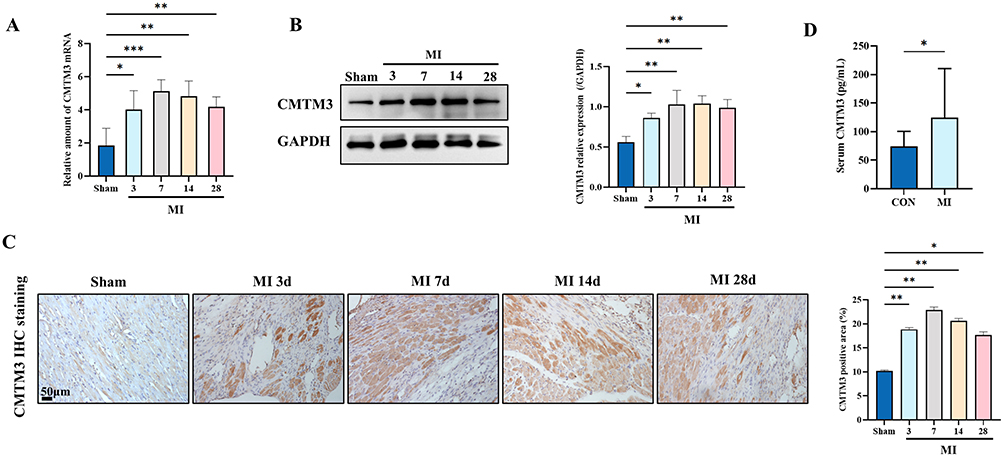 |
Figure 2 CMTM3 is upregulated after MI. (A) RT-qPCR was used to detect the expression of CMTM3 in the infarction border zone on days 0, 3, 7, 14, and 28 of MI (n = 4); (B) Western blot was used to detect the expression of CMTM3 in the infarction border zone on days 0, 3, 7, 14, and 28 of MI (n = 3); (C) Typical IHC staining images and statistics showed the expression of CMTM3 in the hearts of mice on days 0, 3, 7, 14, and 28 of MI (n = 3). Scale bars,50um; (D) Serum CMTM3 levels in patients with MI and the control group (n = 20). *p< 0.05, **p< 0.01, ***p< 0.001. |
CMTM3 Deficiency Exacerbated MI‑induced Cardiac Dysfunction, Increased the Infarction Size and Collagen Deposition
CMTM3 knockout (CMTM3−/−) mice were provided by the Central Laboratory of Yuhuangding Hospital were obtained. Western blot analysis confirmed the absence of CMTM3 expression in the cardiac tissues of CMTM3−/− mice (Supplemental Figure 1A). Baseline assessments, including blood pressure, cardiac weight/tibial length (HW/TL), and TTC staining, revealed no significant differences between wild-type (WT) and CMTM3−/− mice (Supplemental Figure 1B–D), indicating that CMTM3 deficiency does not affect normal cardiac function. To analyse the role of CMTM3 in MI, mice MI model were constructed, TTC staining showed that the infarct size of CMTM3−/− mice was larger than WT mice (Figure 3A). And the HW/TL in CMTM3−/− mice was significantly higher than WT mice (Figure 3B). Cardiac function was evaluated by echocardiography on the 0d, 3d, 7d, and 28d of MI respectively. The results showed, compared with WT mice, LVIDs and LVIDd of CMTM3−/− mice were significantly increased, LVEF and LVFS were decreased (Figure 3C and D). HE staining showed that the myocardium of CMTM3−/− mice was more disordered after MI (Figure 3E). Masson staining indicated an augmented area of blue-stained collagen fibers in infarcted hearts of CMTM3−/− mice (Figure 3F). The mRNA expression of α-SMA, collage I, and collage III in CMTM3−/− MI mice were significantly increased (Figure 3G). In conclusion, CMTM3 deficiency exacerbates post-MI cardiac dysfunction, increases infarct size, and promotes collagen deposition, thereby accelerating adverse cardiac remodeling.
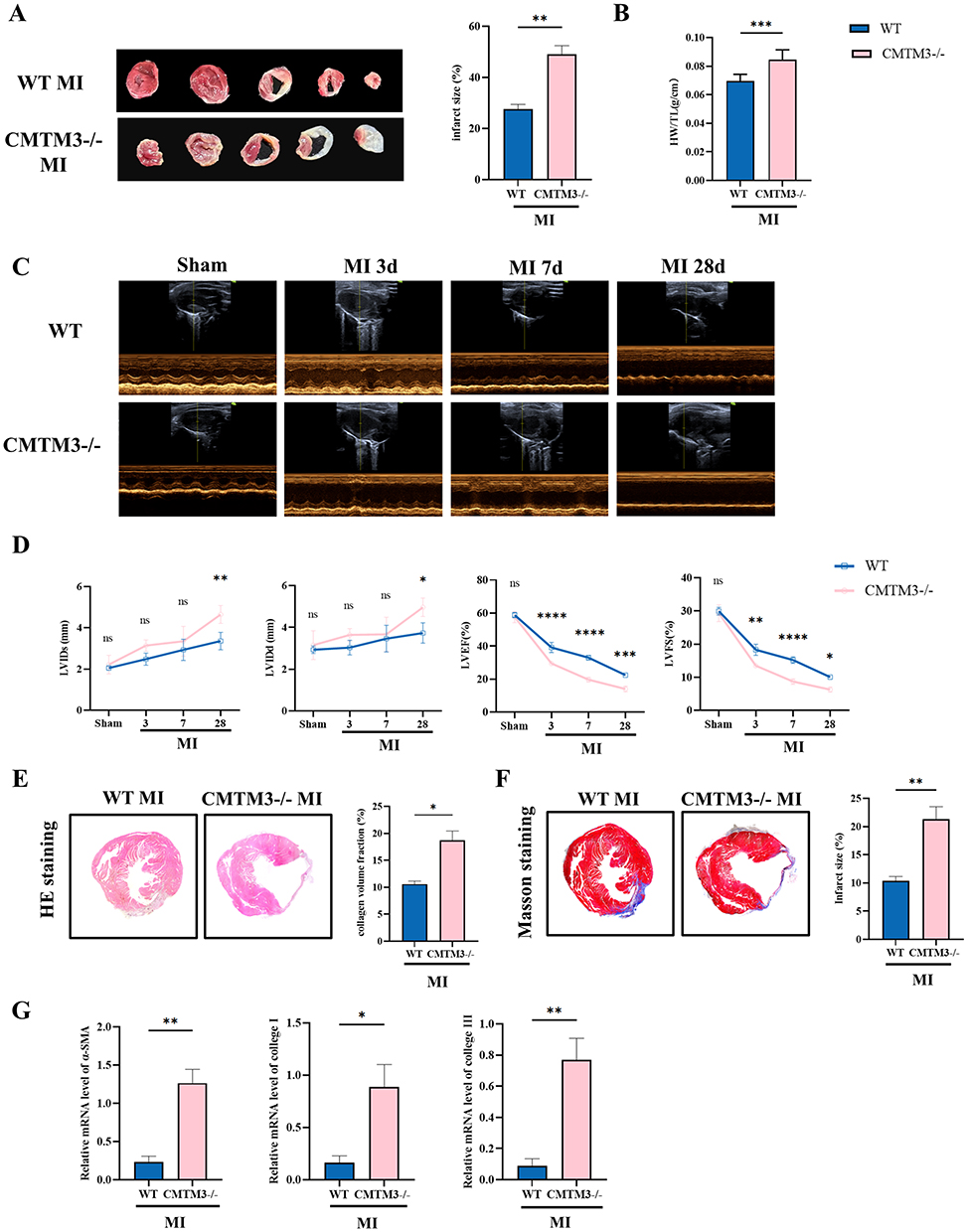 |
Figure 3 CMTM3 absence aggravates cardiac dysfunction in mice with MI. (A) TTC staining evaluate the infarct area in WT and CMTM3−/− mice on the 7d of MI (n = 4); (B) Measure HW/TL on the 28th day after MI (n = 6); (C and D) Evaluation of left ventricular function on days 0, 3, 7, and 28 of MI by echocardiography, including LVIDd, LVIDs, LVEF, LVFS (n = 3); (E) HE staining on the 28th day of MI; (F) Typical plots and statistics of Masson staining on the 28th day of MI (n = 5); (G) RT-qPCR was detected the expressions of α-SMA, collage I and collage III in the cardiac tissues of multiple groups of MI 28d mice (n = 5). *p< 0.05, **p< 0.01, ***p< 0.001, ****p<0.0001. Abbreviation: ns, no significance. |
CMTM3 Is Upregulated in Macrophages After MI
Cardiomyocytes (CM), fibroblasts (CF), endothelial cells (EC) and macrophages (Mø) were isolated from murine heart tissues using density gradient centrifugation and flow cytometry. CMTM3 expression in the above cells was detected, RT-qPCR and the results showed that a marked upregulation of CMTM3 specifically in macrophages following MI, with no significant changes observed in CM, CF and EC (Figure 4A). In pro-inflammatory macrophages induced by LPS+IFN-γ, Western blot analysis demonstrated that CMTM3 protein levels were significantly elevated (Figure 4B). Furthermore, immunofluorescence co-staining of CMTM3 with cell-specific markers—FSP-1 (CF), α-actinin (CM), CD31 (EC) and CD68 (macrophages) was performed respectively—validated that CMTM3 upregulation was restricted to macrophages after MI, with no change in other cell types (Figure 4C and Supplemental Figure 2A). PBMCs were isolated from the blood of patients with MI and the control group. RT-qPCR and results showed increased CMTM3 expression in the MI group (Figure 4D). In addition, since CM are the main cell type of the heart, CMTM3 in H9C2 cells was knocked down in this study to further explore whether CMTM3 affects CM, and the results showed that CMTM3 had no effect on the expression of apoptosis-related protein Bax in H9C2 cells (Supplemental Figure 2B), indicating that CMTM3 had no effect on the apoptosis of hypoxia-treated H9C2 cells, and the possibility of CMTM3 indirectly affecting macrophages by affecting CM was ruled out in this study. The above results support the specific upregulation of CMTM3 in macrophages after MI, and CMTM3 regulates MI by directly affecting macrophage function. These findings indicate that CMTM3 is specifically upregulated in macrophages after MI, suggesting that CMTM3 may contribute to the pathogenesis and progression of MI through its effects on macrophage function.
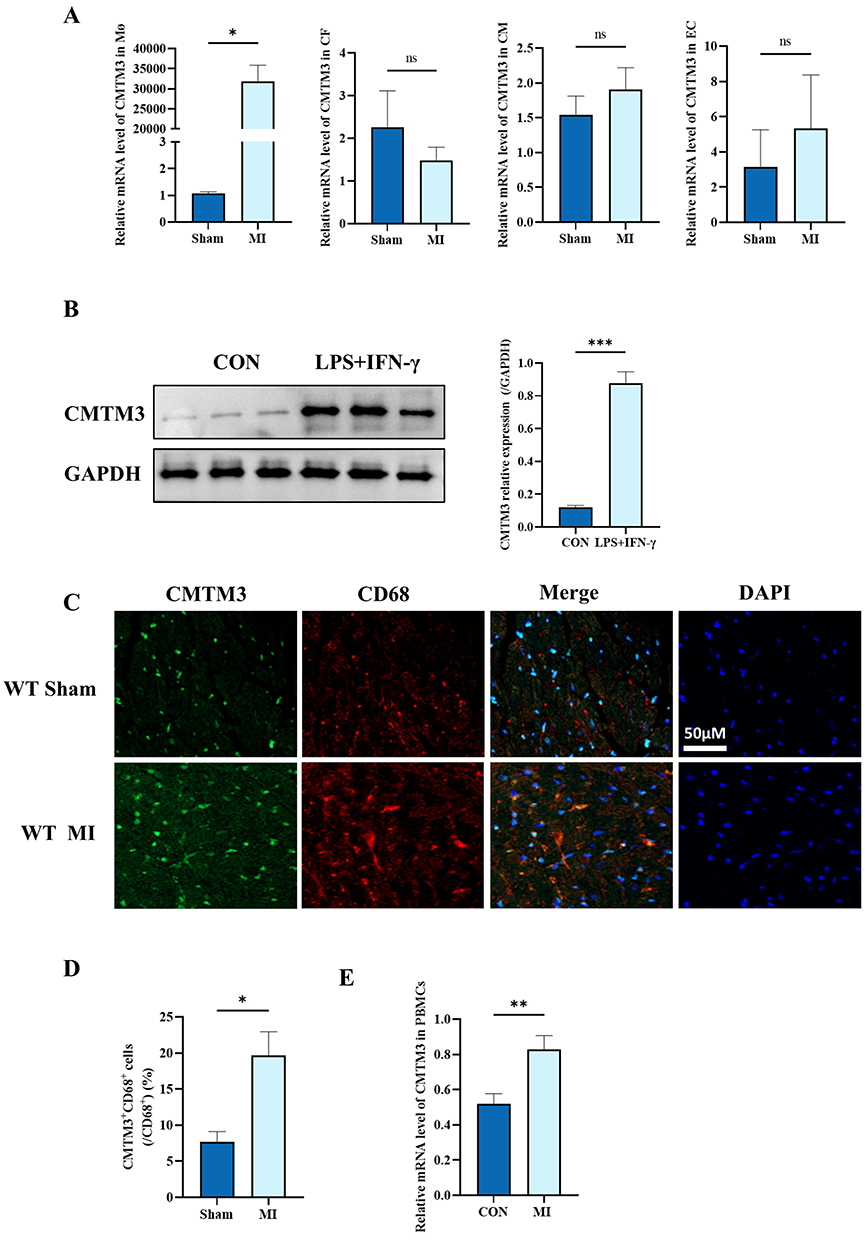 |
Figure 4 CMTM3 is mainly upregulated in macrophages after MI. (A) RT-qPCR was used to detect the expression of CMTM3 in cardiomyocytes (CM), fibroblasts (CF), macrophages (Mφ), and endothelial cells (EC) (n = 3); (B) Western blot analysis of the expression and statistics of CMTM3 in BMDMs (n = 3); (C and D) CMTM3 (green) was co-stained with macrophage markers CD68 (red) and DAPI (blue) for immunofluorescence in cardiac sections of the MI and sham operation groups, and the percentage of CMTM3+CD68+ cells among CD68+ cells was calculated (n = 3). Scale bars, 50μm; (E) RT-qPCR was used to detect the expression of CMTM3 in PBMCs with MI and the control group (n = 10). *p< 0.05, **p< 0.01, ***p< 0.001. Abbreviation: ns, no significance. |
CMTM3 Deficiency Exacerbates the Inflammatory Response After MI
Next, this study focuses on macrophage inflammation. The phenotypes of macrophages in the hearts of MI at 3 days and 7 days were analyzed by flow cytometry, the results showed that compared with the WT group, the proportion of CD11b+F4/80+ macrophages in the hearts of the CMTM3−/− group increased (Figure 5A), and pro-inflammatory macrophages (CD11b+F4/80+Ly6ChighCD86+) in the CMTM3−/− group also increased (Figure 5B). Subsequently, macrophages in the infarcted heart was analyzed by IHC staining, the results showed that CD68+ macrophages in the infarcted hearts of CMTM3−/− mice was higher than WT mice (Figure 5C). The RT-qPCR results showed that at 3 days and 7 days of MI, the pro-inflammatory factors IL-1β, IL-6 and TNF-α of CMTM3−/− mice were more than WT mice (Figure 5D). Elisa results also showed that the secretion of IL-1β, IL-6 and TNF-α in the serum increased after CMTM3 knockout (Figure 5E), indicating that CMTM3 deletion increased the production and secretion of inflammatory factors after MI. The above results indicate that CMTM3 deletion promotes the differentiation of macrophages into pro-inflammatory phenotypes, increases inflammatory factors, aggravates the inflammatory response after MI.
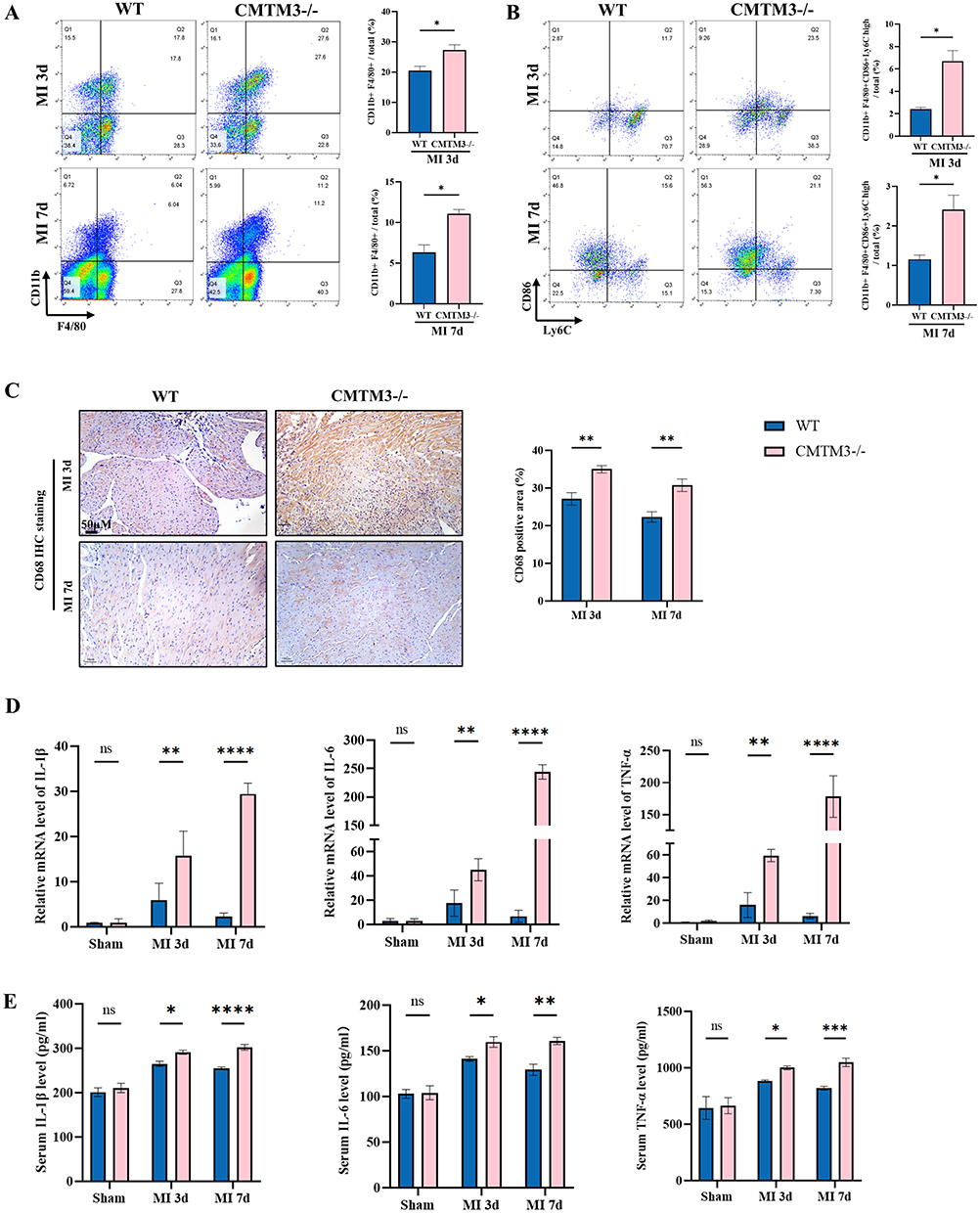 |
Figure 5 CMTM3 deficiency aggravates the macrophage-related inflammation in MI mice. (A) Flow cytometry analyzed the content of CD11b+F4/80+ macrophages in the hearts of mice after MI (n = 3); (B) Taking CD11b+F4/80+ as the cross gating, pro-inflammatory macrophages (CD11b+F4/80+Ly6ChighCD86+) after MI in the two groups were analyzed, and the content of pro-inflammatory macrophages was calculated (n = 3); (C) Typical immunohistochemical staining images and statistics showed that the content of CD68+ macrophages in the hearts of mice on days 3 and 7 of MI. Scale bars,50um (n = 3); (D) RT-qPCR was used to detect the contents of IL-1β, IL-6 and TNF-α in the hearts of mice on days 3 and 7 of MI (n = 3); (E) Elisa kits detected the contents of IL-1β, IL-6 and TNF-α in the serum of mice in the two groups after MI (n = 3). *p< 0.05, **p< 0.01, ***p< 0.001, ****p<0.0001. Abbreviation: ns, no significance. |
CMTM3 Deficiency Exacerbates the Inflammatory Response in BMDMs
Next, the effect of CMTM3 on macrophages was evaluated in BMDMs. Flow cytometry showed that the proportion of CD11b+CD86+ macrophages in the CMTM3−/− group was higher than WT group (Figure 6A). The RT-qPCR results showed that the expressions of IL-1β, IL-6 and TNF-α in BMDMs of the CMTM3−/− group were higher than WT group (Figure 6B). The Elisa results showed that the contents of IL-1β, IL-6 and TNF-α in the supernatant of the CMTM3−/− group were more than WT group (Figure 6C). In conclusion, these results indicate that in vitro, CMTM3 deletion can also exacerbate macrophage-related inflammatory responses.
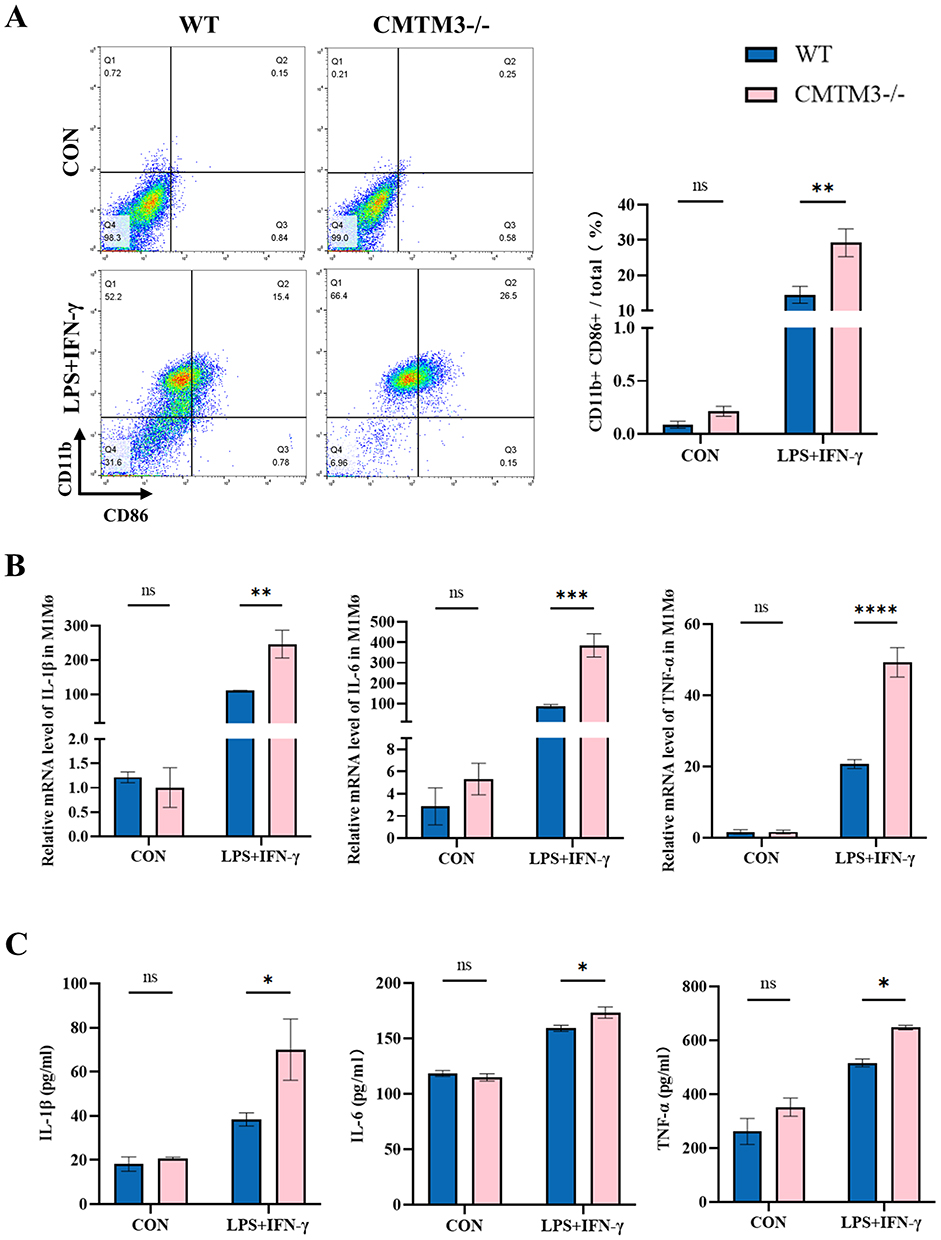 |
Figure 6 CMTM3 deficiency aggravates the macrophage-related inflammation in BMDMs. (A) Flow cytometry analysis of CD11b+CD86+ macrophages in BMDMs (n = 3); (B) The expressions of IL-1β, IL-6 and TNF-α in BMDMs of each group were detected by RT-qPCR (n = 3); (C) The secretion of IL-1β, IL-6 and TNF-α in the supernatants of WT and CMTM3−/− group BMDMs were detected by Elisa kits (n = 3). *p< 0.05, **p< 0.01, ***p< 0.001, ****p<0.0001. Abbreviation: ns, no significance. |
CMTM3 Deficiency Promoted the Inflammatory Response Through NF-κB and PPARα Signaling Pathway
Previous studies have revealed, macrophage-related inflammation is regulated by NF-κB and PPARα.17,18 In addition, the bioinformatics results based on the GSE161427 dataset also indicated that CMTM3 was significantly correlated with NF-κB and PPARα (Supplemental Figure 3A and B). Therefore, in this study, the Western blot method was used to analyze the expressions of NF-κB and PPARα in the hearts and BMDMs of mice in the CMTM3−/− and the WT group. The results showed that in BMDMs and infarcted hearts, after CMTM3 knockout, the phosphorylation level of NF-κB increased, while the non-phosphorylation level showed no difference, the expression of PPARα decreased (Figure 7A–D), suggesting that CMTM3 affect inflammation by regulating the NF-κB and PPARα. Subsequently, the interaction between CMTM3 and NF-κB and PPARα was further explored, and the co-immunoprecipitation results showed that there was a direct interaction between CMTM3 and PPARα, but no interaction with NF-κB (Figure 7E). The results of BMDMs immunofluorescence staining showed that CMTM3 deficiency enhanced the nuclear entry of NF-κB and inhibited the nuclear translocation of PPARα induced by LPS (Figure 8A). Subsequently, the NF-κB inhibitor pyrrolidinedithiocarbamate ammonium (PTDC) (100uM) was added to the CMTM3−/− group BMDMs to observe its effect on inflammation.19 The flow cytometry results showed that, the proportion of CD11b+CD86+ macrophages decreased significantly (Figure 8B). The RT-qPCR results showed that the expressions of IL-1β, IL-6 and TNF-α decreased, and the Elisa results indicated that the release of IL-1β, IL-6 and TNF-α reduced (Figure 8C and D). PPARα is an anti-inflammatory nuclear receptor that inhibits the pro-inflammatory factor NF-κB.20 Combined with the previous literature and the above results, CMTM3 directly interacted with PPARα, and its deletion inhibited the expression of PPARα, increased the activation of NF-κB, the specific mechanism is related to competitive nuclear translocation.
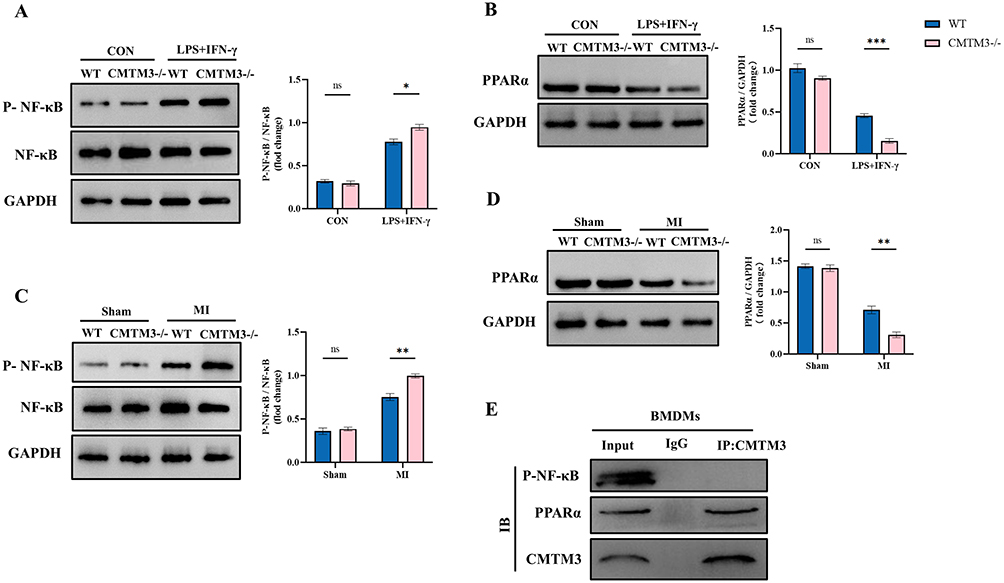 |
Figure 7 CMTM3 deficiency promotes the polarization and inflammation of BMDMs through the NF-κB and PPARα signaling pathway. (A) Western blot typical patterns and statistics showed P-NF-κB and NF-κB levels in BMDMs (n = 3); (B) Western blot analyses of PPARα levels in BMDMs (n = 3); (C) Western blot typical patterns and statistics showed P-NF-κB and NF-κB levels in the hearts of mice in each group (n = 3); (D) Western blot showed PPARα levels in the hearts of mice (n = 3); (E) CO-IP detected the interaction of CMTM3 with PPARα and NF-κB in BMDMs (n = 3). *p< 0.05, **p< 0.01, ***p< 0.001. Abbreviation: ns, no significance. |
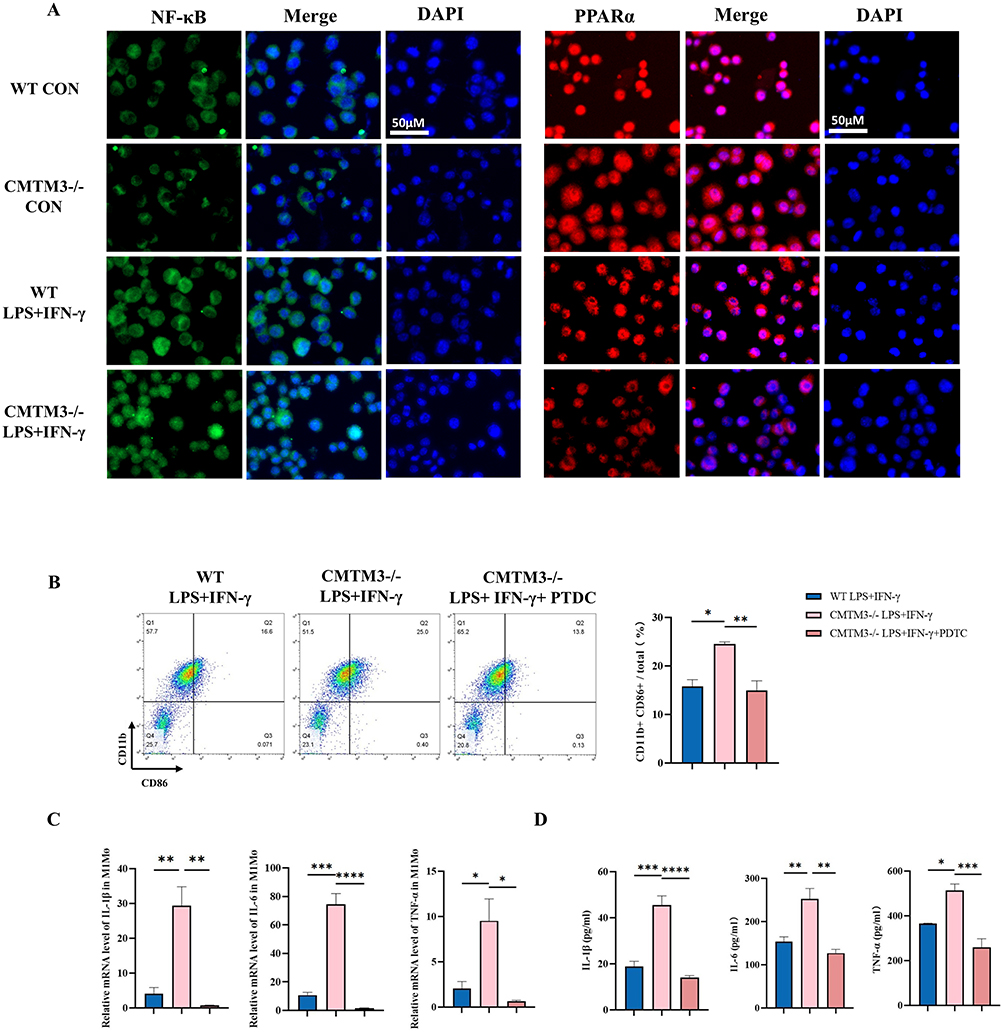 |
Figure 8 CMTM3 deficiency affects BMDMs through the NF-κB and PPARα signaling pathway. (A) Immunofluorescence staining of NF-κB and PPARα in BMDMs of each group (n = 3), Scale bars,50μm; (B) Flow cytometry was used to analyze CD11b+CD86+ macrophages in BMDMs of each group (n = 3); (C) RT-qPCR was used to detect the expressions of IL-1β, IL-6 and TNF-α in BMDMs of each group (n = 3); (D) Elisa kits were used to detect the expressions of IL-1β, IL-6 and TNF-α in the supernatants of BMDMs in each group (n = 3). *p< 0.05, **p< 0.01, ***p< 0.001, ****p<0.0001. |
Discussion
In this study, the expression of CMTM3 was upregulated in the serum of MI patients and the hearts of mice with MI, and the upregulated CMTM3 was mainly expressed in infiltrating macrophages. CMTM3 knockout aggravates the inflammatory response in mice with MI, increases the infarct area, enhances fibrosis and aggravates cardiac dysfunction. In BMDMs, CMTM3 deficiency promotes the differentiation of macrophages into pro-inflammatory phenotypes by activating the NF-κB and inhibiting PPARα, increases the secretion of inflammatory factors, and aggravates the inflammatory response (Figure 9). These results indicate that CMTM3 can reduce the inflammatory response and improve cardiac remodeling in MI, thereby protecting the heart and improving cardiac function.
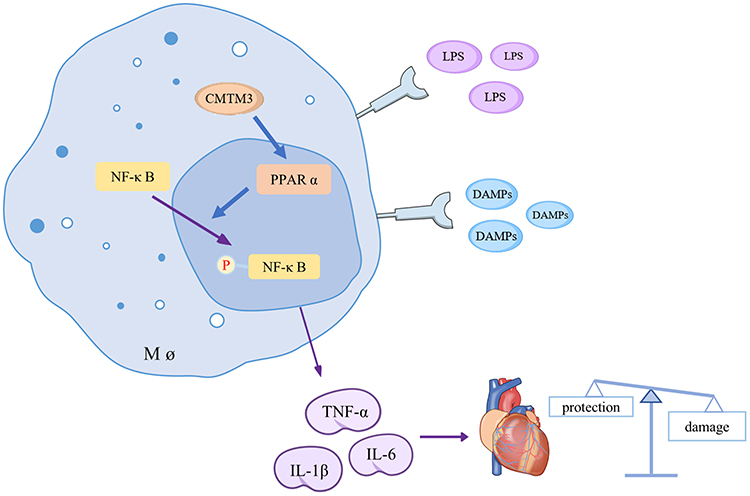 |
Figure 9 Schematic diagram of CMTM3 regulation of macrophages. |
The CMTM family is a class of cytokines with chemotactic effects, consisting of a total of 9 members including CKLF and CMTM1-8, playing an important role in myocardial ischemia. Previous studies have shown that CMTM5 has a certain protective effect in the occurrence and development of coronary heart disease. Overexpression of CMTM5 can inhibit the proliferation and migration of vascular endothelial cells through the PI3K/Akt signaling pathway, thereby exerting an anti-atherosclerotic effect.21–23 CKLF has also been proven to have a protective effect on the hearts of rats with MI, which is related to the fact that CKLF can accelerate the mobilization of CD34+ cells in peripheral blood for migration and differentiation and promote the proliferation of myocardial cells.24 This study found that the absence of CMTM3 would aggravate the cardiac injury in mice with MI, specifically manifested as increased infarction area, aggravated fibrosis, and deteriorated cardiac function, proving the protective effect of CMTM3 on the myocardium of mice with MI. This is consistent with the previously reported protective effects of CKLF and CMTM5 on myocardial ischemia.
CMTM3 is a membrane protein that can participate in immune responses and can be involved in the inflammatory immune responses of various diseases (such as sepsis, gastritis, and various tumor diseases).13,25 CMTM3 has been confirmed to have dual effects of promoting and inhibiting cancer in tumors. Similarly, CMTM3 also has dual regulatory effects on inflammation in different diseases.26 CMTM3 is the chaperone protein of B-cell connexin (BLNK), which can bind to B-cell receptors and activate BLNK-mediated signaling pathways.27 The effect of CMTM3 on neutrophils has also been studied in sepsis models. In sepsis, CMTM3 exerts a pro-inflammatory effect. CMTM3 knockout reduces neutrophil infiltration in the liver, lung and kidney tissues of septic mice. The mechanism is related to the reduction of neutrophil migration from the bone marrow to the bloodstream through the TLR4 pathway by CMTM3.15 Similarly, in the acute respiratory distress syndrome (ARDS) model, CMTM3 can regulate pulmonary vascular endothelial function by promoting pulmonary inflammatory responses.28 However, in various tumor diseases, CMTM3 has been proven to have anti-inflammatory effects, specifically manifested in inhibiting the phosphorylation modification of inflammation-related pathway proteins.16,29,30 In this study, the expression changes of CMTM3 were mainly reflected in the infiltrating mononuclear macrophages of infarcted hearts. The infiltration and phenotypic transformation of macrophages play an important role in MI. CMTM3 deficiency will promote the polarization of infarcted hearts and macrophages treated with LPS towards type M1, aggravating the related inflammatory response. It is suggested that CMTM3 has an inhibitory effect on macrophage-related inflammation in MI.
GO analysis indicated that CMTM3 could be enriched in inflammatory immune-related signaling pathways. Previous studies have also reported that CMTM3 is involved in immune-related pathways, such as MAPK, Erk1/2, JAK2/STAT3, P53, PPARs and NF-κB.16,31–36 Studies have shown that CMTM3 in gastric epithelial cells can regulate the activation of NF-κB by regulating the ubiquitination of the factor NEMO in the NF-κB pathway.26 CMTM3 deletion in hepatocellular carcinoma can inhibit the PPARs pathway.29 NF-κB and PPARα not only participate in the pathological process of MI, but also are important transcription factors regulating LPS-induced inflammatory responses, they are antagonize each other. The activation of NF-κB and the inhibition of PPARα are associated with various chronic inflammatory diseases (such as atherosclerosis).20 The results of the database also indicate that NF-κB and PPARα have a significant correlation with CMTM3. Take together, both in vivo and in vitro experiments of this study demonstrated that CMTM3 deficiency inhibited the expression of PPARα, promoted the phosphorylation modification of NF-κB, enhanced the polarization of macrophages to the pro-inflammatory type, aggravated the inflammatory response.
Myocardial repair and functional reconstruction after MI is an important challenge in the cardiovascular field, and new hydrogel-based myocardial repair technologies have shown great potential in improving myocardial adverse remodeling in recent years. For example, Zhi et al reported that wet-adhesive hydrogels for intramyocardial injections and extracardiac patches are a powerful method for repairing damaged myocardium by inducing polarization and immunomodulation of macrophages.37 Double cross-linked conductive hydrogels effectively prevent infarct scar thinning and expansion by supporting collapsed myocardial structures and conductive connections between normal myocardium and fibrotic islands for electrical signaling.38 This study found that CMTM3 can prevent poor cardiac remodeling after MI and is related to inhibiting the inflammatory cascade, if CMTM3 is loaded in a hydrogel, it is expected to inhibit early inflammatory overactivation while exerting a strong myocardial repair effect, in order to future researchers to combine CMTM3 with cardiac tissue engineering strategies to make more in-depth research to provide new clinical translational benefits.
The limitation of this study is that the use of systemic knockout mice cannot rule out indirect effects caused by CMTM3 deficiency in other tissues and cells. Although NF-kB and PPARα are different signaling pathways, the potential interaction between the two has not been explored in this study, and it is expected that future researchers will explore it further.
Conclusion
This study emphasizes that CMTM3 interacts with PPARα, and its deficiency will inhibit the expression of PPARα, promote the phosphorylation of NF-κB, increase the differentiation of macrophages into the pro-inflammatory phenotype, promote the secretion of inflammatory factors, and aggravate the related inflammatory response, thereby aggravating the damage to infarcted hearts. These findings demonstrate the new functions of CMTM3 in MI and macrophages, which is helpful in providing a new direction for the treatment of MI.
Data Sharing Statement
The data supporting the findings of this study are found in the article and the Supplemental Material. The datasets that support the conclusions of this article are included in the article.
Ethics Approval
All the procedures involving human subjects in this study were carried out in accordance with the approval of the Ethics Committee of Yantai Yuhuangding Hospital. (Approval date: May, 16, 2025, Batch Number: 2025-550). All animal experiments in this study were approved by the Ethics Committee of Yantai Yuhuangding Hospital. (Approval date: June 12, 2025, Batch Number: 2025-579).
Acknowledgments
We sincerely thank Professor Gong Lei and Professor Yang Jun for their valuable opinions on this article.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research was supported by the Natural Science Foundation of Shandong Province (No. ZR2024QH074 and ZR2022QH237) and the National Natural Science Foundation of China (No. 82200503).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Zhang Q, Wang L, Wang S, et al. Signaling pathways and targeted therapy for myocardial infarction. Signal Transduct Target Ther. 2022;7(1):78. doi:10.1038/s41392-022-00925-z
2. Xie W, Gan J, Zhou X, et al. Myocardial infarction accelerates the progression of MASH by triggering immunoinflammatory response and induction of periosti. Cell Metab. 2024;36(6):1269–1286.e1269. doi:10.1016/j.cmet.2024.04.020
3. Kufazvinei TTJ, Chai J, Boden KA, Channon KM, Choudhury RP. Emerging opportunities to target inflammation: myocardial infarction and type 2 diabetes. Cardiovasc Res. 2024;120(11):1241–17. doi:10.1093/cvr/cvae142
4. Wang Y, Zhang Y, Li J, et al. Hypoxia induces M2 macrophages to express VSIG4 and mediate cardiac fibrosis after myocardial infarction. Theranostics. 2023;13(7):2192–2209. doi:10.7150/thno.78736
5. Chen R, Zhang H, Tang B, et al. Macrophages in cardiovascular diseases: molecular mechanisms and therapeutic targets. Signal Transduct Target Ther. 2024;9(1):130. doi:10.1038/s41392-024-01840-1
6. Chang F, Wang C, Zheng P, et al. Malat1 promotes macrophage-associated inflammation by increasing PPAR-γ methylation through binding to EZH2 in acute myocardial infarction. Int Immunopharmacol. 2023;123:110695. doi:10.1016/j.intimp.2023.110695
7. Cai S, Zhao M, Zhou B, et al. Mitochondrial dysfunction in macrophages promotes inflammation and suppresses repair after myocardial infarction. J Clin Invest. 2023;133(4). doi:10.1172/JCI159498
8. Yap J, Irei J, Lozano-Gerona J, Vanapruks S, Bishop T, Boisvert WA. Macrophages in cardiac remodelling after myocardial infarction. Nat Rev Cardiol. 2023;20(6):373–385. doi:10.1038/s41569-022-00823-5
9. Wang Y, Li J, Cui Y, et al. CMTM3, located at the critical tumor suppressor locus 16q22.1, is silenced by CpG methylation in carcinomas and inhibits tumor cell growth through inducing apoptosis. Cancer Res. 2009;69(12):5194–5201. doi:10.1158/0008-5472.CAN-08-3694
10. Wang Z, Zhang J, Zhang H, et al. CMTM family genes affect prognosis and modulate immunocytes infiltration in grade II/III glioma patients by influencing the tumor immune landscape and activating associated immunosuppressing pathways. Front Cell Dev Biol. 2022;10:740822. doi:10.3389/fcell.2022.740822
11. Zhang T, Yu H, Dai X, Zhang X. CMTM6 and CMTM4 as two novel regulators of PD-L1 modulate the tumor microenvironment. Front Immunol. 2022;13:971428. doi:10.3389/fimmu.2022.971428
12. Ding J, Hu H, Zhu Y, et al. Inhibiting CMTM4 reverses the immunosuppressive function of myeloid-derived suppressor cells and augments immunotherapy response in cervical cancer. J Immunother Cancer. 2025;13(6):e011776. doi:10.1136/jitc-2025-011776
13. Xue L, Gou S, Zhang Y, et al. Comprehensive analysis of CMTM family and immune infiltration in esophageal carcinoma. PLoS One. 2025;20(4):e0321037. doi:10.1371/journal.pone.0321037
14. Duan HJ, Li XY, Liu C, Deng XL. Chemokine-like factor-like MARVEL transmembrane domain-containing family in autoimmune diseases. Chin Med J. 2020;133(8):951–958. doi:10.1097/CM9.0000000000000747
15. Xue H, Xiao Z, Zhao X, et al. CMTM3 regulates neutrophil activation and aggravates sepsis through TLR4 signaling. EMBO Rep. 2024;25(12):5456–5477. doi:10.1038/s44319-024-00291-7
16. Yuan W, Wei F, Ouyang H, et al. CMTM3 suppresses chordoma progress through EGFR/STAT3 regulated EMT and TP53 signaling pathway. Cancer Cell Int. 2021;21(1):510. doi:10.1186/s12935-021-02159-5
17. Guo Q, Jin Y, Chen X, et al. NF-κB in biology and targeted therapy: new insights and translational implications. Signal Transduct Target Ther. 2024;9(1):53. doi:10.1038/s41392-024-01757-9
18. Xiong Y, Zhang Z, Liu S, et al. Lupeol alleviates autoimmune myocarditis by suppressing macrophage pyroptosis and polarization via PPARα/LACC1/NF-κB signaling pathway. Phytomedicine. 2024;123:155193. doi:10.1016/j.phymed.2023.155193
19. Németh ZH, Deitch EA, Szabó C, Haskó G. Pyrrolidinedithiocarbamate inhibits NF-kappaB activation and IL-8 production in intestinal epithelial cells. Immunol Lett. 2003;85(1):41–46. doi:10.1016/S0165-2478(02)00208-0
20. Tang Y, Yang LJ, Liu H, et al. Exosomal miR-27b-3p secreted by visceral adipocytes contributes to endothelial inflammation and atherogenesis. Cell Rep. 2023;42(1):111948. doi:10.1016/j.celrep.2022.111948
21. Zhang JW, Liu TF, Chen XH, et al. Validation of aspirin response-related transcripts in patients with coronary artery disease and preliminary investigation on CMTM5 function. Gene. 2017;624:56–65. doi:10.1016/j.gene.2017.04.041
22. Liu TF, Lin T, Ren LH, Li GP, Peng JJ. [Association of CMTM5 gene expression with the risk of in-stent restenosis in patients with coronary artery disease after drug-eluting stent implantation and the effects and mechanisms of CMTM5 on human vascular endothelial cells]. Beijing Da Xue Xue Bao Yi Xue Ban. 2020;52(5):856–862. Dutch. doi:10.19723/j.issn.1671-167X.2020.05.010
23. Liu TF, Lin T, Ren LH, Li GP, Peng JJ. [Association between CMTM5 gene and coronary artery disease and the relative mechanism]. Beijing Da Xue Xue Bao Yi Xue Ban. 2020;52(6):1082–1087. Dutch. doi:10.19723/j.issn.1671-167X.2020.06.015
24. Chen C, Ai Q, Tian H, Wei Y. CKLF1 in cardiovascular and cerebrovascular diseases. Int Immunopharmacol. 2024;139:112718. doi:10.1016/j.intimp.2024.112718
25. Shen Q, Cong Z, Zhou Y, Teng Y, Gao J, Tang W. CMTM3 as a potential new immune checkpoint regulator. J Oncol. 2022;2022:2103515. doi:10.1155/2022/2103515
26. Zhang J, Ning J, Fu W, Shi Y, Zhang J, Ding S. CMTM3 protects the gastric epithelial cells from apoptosis and promotes IL-8 by stabilizing NEMO during Helicobacter pylori infection. Gut Pathog. 2023;15(1):6. doi:10.1186/s13099-023-00533-4
27. Imamura Y, Katahira T, Kitamura D. Identification and characterization of a novel BASH N terminus-associated protein, BNAS2. J Biol Chem. 2004;279(25):26425–26432. doi:10.1074/jbc.M403685200
28. Xiao Z, Zhou G, Xue H, et al. CMTM3 regulates vascular endothelial cell dysfunction by influencing pulmonary vascular endothelial permeability and inflammation in ARDS. Front Immunol. 2025;16:1544610. doi:10.3389/fimmu.2025.1544610
29. Wang J, Chu H, Wang Z, et al. In vivo study revealed pro-tumorigenic effect of CMTM3 in hepatocellular carcinoma involving the regulation of peroxisome proliferator-activated receptor gamma (PPARγ). Cell Oncol. 2022;46(1):49–64. doi:10.1007/s13402-022-00733-1
30. Li S, Gao P, Dai X, Ye L, Wang Z, Cheng H. New prognostic biomarker CMTM3 in low grade glioma and its immune infiltration. Ann Transl Med. 2022;10(4):206. doi:10.21037/atm-22-526
31. Ye J, Yan S, Liu R, et al. CMTM3 deficiency induces cardiac hypertrophy by regulating MAPK/ERK signaling. Biochem Biophys Res Commun. 2023;667:162–169. doi:10.1016/j.bbrc.2023.05.052
32. Fan D, Fan D, Yuan W. CMTM3 suppresses bone formation and osteogenic differentiation of mesenchymal stem cells through inhibiting Erk1/2 and RUNX2 pathways. Genes Dis. 2021;8(6):882–890. doi:10.1016/j.gendis.2020.12.003
33. Shen E, Piao M, Li Y, et al. CMTM3 suppresses proliferation and osteogenic transdifferentiation of C2C12 myoblasts through p53 upregulation. Cells. 2024;13(16):1352. doi:10.3390/cells13161352
34. Li W, Zhang S. CKLF-like MARVEL transmembrane domain-containing member 3 (CMTM3) inhibits the proliferation and tumorigenisis in hepatocellular carcinoma cells. Oncol Res. 2017;25(2):285–293. doi:10.3727/096504016X14732523471442
35. Yuan W, Liu B, Wang X, et al. CMTM3 decreases EGFR expression and EGF-mediated tumorigenicity by promoting Rab5 activity in gastric cancer. Cancer Lett. 2017;386:77–86. doi:10.1016/j.canlet.2016.11.015
36. Su Y, Lin Y, Zhang L, et al. CMTM3 inhibits cell migration and invasion and correlates with favorable prognosis in gastric cancer. Cancer Sci. 2014;105(1):26–34. doi:10.1111/cas.12304
37. Zheng Z, Sun J, Wang J, et al. Enhancing myocardial infarction treatment through bionic hydrogel-mediated spatial combination therapy via mtDNA-STING crosstalk modulation. J Control Release. 2024;371:570–587. doi:10.1016/j.jconrel.2024.06.015
38. Zheng Z, Guo Z, Zhong F, et al. A dual crosslinked hydrogel-mediated integrated peptides and BMSC therapy for myocardial regeneration. J Control Release. 2022;347:127–142. doi:10.1016/j.jconrel.2022.04.010
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Shensong Yangxin Capsule Reduces the Susceptibility of Arrhythmia in db/db Mice via Inhibiting the Inflammatory Response Induced by Endothelium Dysfunction
Zhang J, Li H, Wang D, Gu J, Hou Y, Wu Y
Drug Design, Development and Therapy 2023, 17:313-330
Published Date: 5 February 2023
Associations Between Resolvin D1 and Culprit Plaque Morphologies: An Optical Coherence Tomography Study in Patients with ST-Segment Elevation Myocardial Infarction
Chen R, Li J, Sheng Z, Zhou J, Wang Y, Zhao X, Li N, Liu W, Liu C, Zhou P, Chen Y, Yan S, Song L, Yan H, Zhao H
Journal of Inflammation Research 2023, 16:6457-6467
Published Date: 28 December 2023
Exosomes as Vehicles for Noncoding RNA in Modulating Inflammation: A Promising Regulatory Approach for Ischemic Stroke and Myocardial Infarction
Lai Z, Ye T, Zhang M, Mu Y
Journal of Inflammation Research 2024, 17:7485-7501
Published Date: 21 October 2024
Thymosin β4 Regulates Tissue Inflammatory Response in Mouse Nonalcoholic Fatty Liver Disease by Promoting Macrophage M2-Type Polarization
Zhu Z, Liao Y, Mou Q, Liu H, Shen Y, Zhu L, Cong S
Journal of Inflammation Research 2025, 18:5791-5809
Published Date: 29 April 2025
Immune Mechanisms and Pain in Intervertebral Disc Degeneration
Cheng H, Wang L, Huang C, Yang Z, Wu H, Fang Z
Journal of Inflammation Research 2025, 18:16843-16855
Published Date: 1 December 2025