Back to Journals » Journal of Inflammation Research » Volume 16
Associations Between Resolvin D1 and Culprit Plaque Morphologies: An Optical Coherence Tomography Study in Patients with ST-Segment Elevation Myocardial Infarction
Authors Chen R ![]() , Li J, Sheng Z, Zhou J, Wang Y, Zhao X, Li N, Liu W
, Li J, Sheng Z, Zhou J, Wang Y, Zhao X, Li N, Liu W ![]() , Liu C, Zhou P, Chen Y, Yan S, Song L, Yan H
, Liu C, Zhou P, Chen Y, Yan S, Song L, Yan H ![]() , Zhao H
, Zhao H
Received 1 August 2023
Accepted for publication 21 December 2023
Published 28 December 2023 Volume 2023:16 Pages 6457—6467
DOI https://doi.org/10.2147/JIR.S433404
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tara Strutt
Runzhen Chen,1,2,* Jiannan Li,1,3,* Zhaoxue Sheng,4 Jinying Zhou,1,3 Ying Wang,1 Xiaoxiao Zhao,1,3 Nan Li,1 Weida Liu,5 Chen Liu,1,3 Peng Zhou,1,3 Yi Chen,1,3 Shaodi Yan,2 Li Song,1– 3 Hongbing Yan,2,3 Hanjun Zhao1,3
1Department of Cardiology, Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences, Beijing, People’s Republic of China; 2Fuwai Hospital, Chinese Academy of Medical Sciences, Shenzhen, People’s Republic of China; 3Coronary Heart Disease Center, Fuwai Hospital, Chinese Academy of Medical Sciences, Beijing, People’s Republic of China; 4Department of Cardiology, China-Japan Friendship Hospital, Beijing, People’s Republic of China; 5Medical Research Center, Peking Union Medical College Hospital, Beijing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Hongbing Yan, Fuwai Hospital, Chinese Academy of Medical Sciences, 12 Langshan Road, Nanshan District, Shenzhen, 510000, People’s Republic of China, Email [email protected]; [email protected] Hanjun Zhao, Fuwai Hospital, National Center for Cardiovascular Diseases, Peking Union Medical College and Chinese Academy of Medical Sciences, 167 North Lishi Road, Xicheng District, Beijing, 100037, People’s Republic of China, Email [email protected]
Background: As a specialized pro-resolving lipid mediator, resolvin D1 (RvD1) inhibits atherosclerosis progression in vivo by reducing regional oxidative stress and chronic inflammation. However, it is unclear how RvD1 is involved in human coronary artery disease. This study aims to investigate the association between plasma levels of RvD1 and culprit-plaque characteristics in patients with ST-segment elevation myocardial infarction (STEMI).
Methods: A total of 240 STEMI patients undergoing optical coherence tomography (OCT) examination were analyzed. RvD1 levels were measured in patient plasma samples using an enzyme-linked immunosorbent assay. Logistic regression was performed to assess the association between RvD1 levels and various culprit plaque morphologies, and the receiver operating curve was used to search for an optimal cutoff threshold to predict certain pathological features.
Results: The median RvD1 level was 129.7 (56.6– 297.8) pg/mL. According to multivariable logistic regression, high RvD1 was associated with plaque rupture (≥ 111.5 pg/mL, odds ratio [OR]: 2.09, 95% confidence interval [CI]: 1.20– 3.66, P = 0.010), healed plaques (≥ 246.4 pg/mL, OR: 2.17, 95% CI: 1.11– 4.24, P = 0.023), and calcification (≥ 293.38 pg/mL, OR: 2.10, 95% CI: 1.21– 3.66, P = 0.008) at culprit lesions.
Conclusion: Increased levels of RvD1 were associated with higher instability of coronary atherosclerotic plaques in STEMI patients.
Keywords: resolvin D1, specialized pro-resolving lipid mediator, myocardial infarction, optical coherence tomography, atherosclerosis, inflammation
Introduction
Unresolved chronic inflammation is an important pathogenesis for coronary artery disease.1–3 The continuous uptake of lipoproteins by macrophages in the vessel wall leads to the formation of foam cells, but their apoptosis and defective efferocytosis result in secondary necrosis and chronic regional inflammation, causing increased levels of oxidative stress, persistent release of pro-inflammatory cytokines, infiltration of more macrophages, and progression of atherosclerotic plaques. Specialized pro-resolving lipid mediators (SPMs), a group of intrinsic anti-inflammatory molecules biosynthesized from polyunsaturated fatty acids, have been shown to suppress various types of inflammation.4,5 Basically, SPM can limit the infiltration of leukocytes, enhance efferocytosis of apoptotic cells, and reduce the secretion of various proinflammatory cytokines.4–6 Dysregulation of SPM has been observed in many inflammatory disorders such as atherosclerosis, arthritis, asthma, and gingivitis.7–10
Resolvin is a subgroup of SPM derived from docosahexaenoic acid.4 As a member of the resolvin family, resolvin D1 (RvD1) inhibits the progression of atherosclerosis in ldlr-/- mice by rebalancing pro- and anti-inflammatory lipid mediators, reducing the necrotic core, and stabilizing vulnerable plaques.11 In contrast, the expression of G protein-coupled receptor 32 (GPR32), which is one of the receptors for RvD1, is substantially reduced in human atherosclerotic lesions.12 Although the underlying mechanisms have not been fully determined, successful resolution of vascular inflammation seems to be promoted by RvD1, which rescues macrophages from apoptosis induced by oxidative stress and restores regional efferocytosis of apoptotic cells.13 These findings suggest that RvD1 is involved in the development of atherosclerosis as a pro-resolving antioxidant. However, data is still lacking regarding RvD1 levels in humans, particularly in patients with coronary artery disease or myocardial infarction (MI). In this study, we report for the first time that high levels of RvD1 were associated with unstable coronary plaques in patients with ST-segment elevated MI (STEMI).
Materials and Methods
Study Population
The current research is a sub-study of the ongoing OCTAMI (Optical Coherence Tomography [OCT] Examination in Acute Myocardial Infarction) registry (NCT03593928), which aims to assess culprit lesions using OCT in patients (≥18 years) presented with STEMI. Patients were excluded if they had any one of the following conditions: 1) cardiogenic shock, 2) end-stage renal disease, 3) serious liver dysfunction, 4) allergy to contrast media, or 5) contraindications to aspirin or ticagrelor. STEMI was diagnosed according to key points from up-to-date guidelines, including 1) continuous chest pain lasting >30 min, 2) ST-segment elevation of >0.1 mV in at least two contiguous leads or new left bundle-branch block on the 18-lead electrocardiogram, and 3) elevations of cardiac troponin I (cTnI).14
Coronary Angiography and Percutaneous Coronary Intervention
Coronary angiography was performed via transradial or transfemoral access. All patients received standard care recommended by the relevant guidelines, including initial dual antiplatelet therapy at loading dosages (300 mg aspirin, plus 180 mg ticagrelor or 600 mg clopidogrel) and intravascular infusions of 70–100 IU/kg unfractionated heparin before percutaneous coronary intervention (PCI).
Optical Coherence Tomography Examinations and Image Analysis
OCT examinations were performed after restoration of antegrade blood flow with thrombus aspiration and/or gentle pre-dilatation with balloons. An optical probe of the OCT imaging catheter was placed at 5 mm beyond the distal end of culprit lesion. After confirming the placement of the optical probe, it was automatically pulled back at a rate of 36 mm/s (total length: 75 mm) to acquire continuous cross-sectional images of the culprit plaques, while operators simultaneously flushed the vessel cavity with a continuous injection of contrast media to remove residual blood. In this study, a frequency-domain OCT system (ILUMIEN OPTIS™, St. Jude Medical/Abbott, St. Paul, MN, USA) was used to acquire OCT images using dragonfly catheters (LightLab Imaging, Inc., Westford, MA, USA).
OCT images were stored and analyzed on a St. Jude OCT Offline Review Workstation by investigators who were blinded to the patient information. Each OCT image of the culprit plaque was reviewed by three independent investigators to identify the plaque types and other pathological features. Disagreements were resolved by consensus. A culprit plaque was defined as segments centered on the culprit lesion, extending bilaterally to ≥5 mm of the normal vessel segments.
Identifications of plaque rupture, plaque erosion, and various pathological structures were performed according to established standards (Figure 1).15,16 Plaque rupture was defined as a disrupted fibrous cap with a clear cavity formation. Plaque erosion was identified by the presence of an attached thrombus overlying plaques with an irregular luminal surface, but no evidence of fibrous cap rupture in multiple adjacent frames. Thin-cap fibroatheroma (TCFA) is defined as a lipid-rich plaque (maximum lipid arc > 180°) with the thinnest part of the fibrous cap < 65 μm. Calcifications are defined by well-delineated and low-backscattering heterogeneous regions within the culprit plaques. Macrophage infiltration is identified by signal-rich, distinct, or confluent punctate regions above the intensity of background speckle noise with backward shadowing, usually located at the boundary between the fibrous cap and the inner lipid core. A healed plaque was identified by the presence of at least one layer in the culprit plaque with different optical signal intensities located close to the luminal surface and clearly demarcated from the underlying tissue.
 |
Figure 1 Representative cross-sectional image of various pathological features of culprit plaques acquired through optical coherence tomography. (a) Plaque rupture, identified by disrupted fibrous cap (↑) and underlying cavity (*); (b) Plaque erosion, identified by intact fibrous cap overlayed with thrombus (↑); (c) Thin-cap fibroatheroma, identified by low backscattering area (necrotic core) covered by fibrous cap (↑) with thickness < 65 μm; (d) Calcification, identified by signal-poor or heterogeneous region (*) with a sharply delineated border; (e) Macrophage infiltration, identified by signal-rich, highly backscattering, confluent or punctate focal regions (↑); (f) Healed plaques, identified by one or more heterogeneous signal-rich layer (*) of different optical signal intensities located near by the luminal surface and clearly demarcated from the underlying tissue. |
Blood Samples Collection and Laboratory Tests
Blood samples for RvD1 measurements were collected via radial or femoral access before the initiation of coronary angiography using vacutainer tubes containing EDTA, which were immediately centrifuged at 2000×g for 15 min at room temperature to isolate plasma, and then stored at −80°C until further analysis. The plasma concentration of RvD1 was measured using enzyme-linked immunosorbent assay (ELISA; 500380, Cayman Chemical, Michigan, US) according to the manufacturer’s instructions. Blood samples for routine laboratory tests were collected via the cubital vein at admission (eg, complete blood count, metabolic panel, cTnI, and N-terminal prohormone of brain natriuretic peptide [NT-proBNP]).
Statistical Analysis
All statistical analyses were performed using the Stata 17.0 (StataCorp, College Station, TX, USA). Categorical variables are presented as numbers (%). Continuous variables are presented as mean ± SD if they follow a normal distribution. Otherwise, they are presented as medians with 25th and 75th percentiles. Logistic regression was used to evaluate the associations between RvD1, various clinical risk factors, and plaque morphologies, followed by multiple adjustments for potential confounders. Statistical significance is defined as a two-tailed P-value < 0.05.
Results
Baseline Characteristics and OCT Findings
From March 2017 to March 2019, a total of 426 STEMI patients underwent OCT imaging examinations. Of these, 72 patients were excluded due to unavailable measurement of RvD1, 57 patients were excluded due to poor OCT image quality, and 57 patients were excluded as their culprit plaques were neither plaque rupture nor erosion. A total of 240 patients were included in the final analysis (Figure 2).
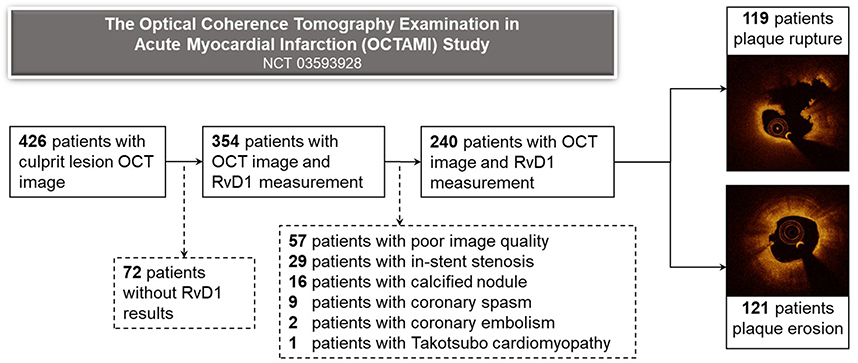 |
Figure 2 Study flow chart. OCT, optical coherence tomography; RvD1, resolvin D1. |
Overall, the median RvD1 level was 129.7 (56.6–297.8) pg/mL. When stratified by the median (Table 1), patients with high levels of RvD1 (≥129.7 pg/mL) acquired lower platelet counts (225.1 ± 59.0 vs 241.2 ± 66.2×109/L, P = 0.049) and cTnI (0.535 [0.125–2.875] vs 1.465 [0.082–7.422] ng/mL, P = 0.043). The demographics, medical history, metabolic profiles, and distribution of diseased vessels were generally similar between the two groups.
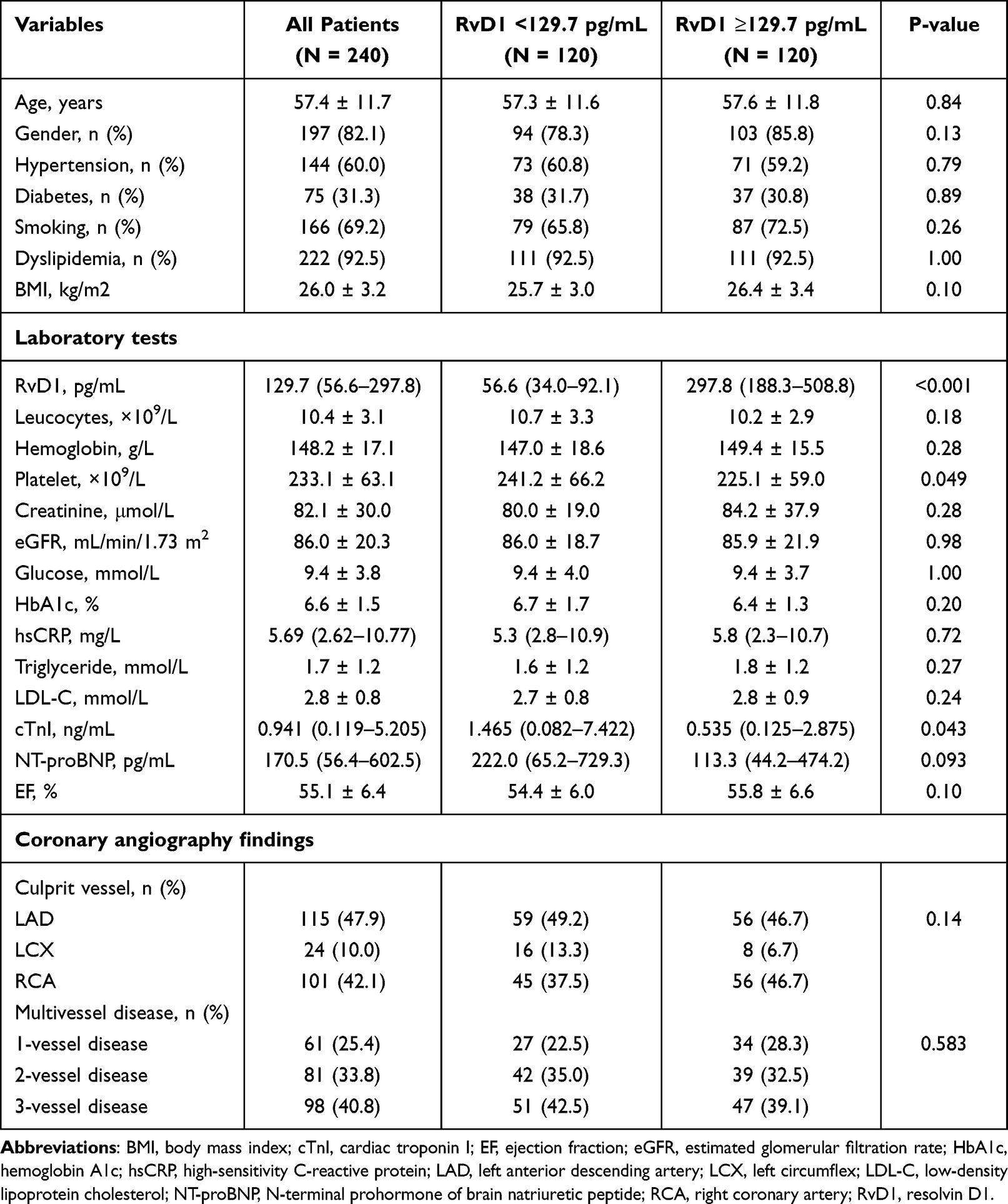 |
Table 1 Baseline Characteristics Stratified by Levels of RvD1 |
Among the 240 patients included, 119 (49.6%) patients presented with plaque rupture at the culprit lesion, and 121 (50.4%) patients presented with plaque erosion. When stratified by pathological findings from the OCT (Figure 3), RvD1 levels were higher for those with plaque rupture (153.7 [71.8–310.6] vs 99.1 [50.5–259.1] pg/mL, P = 0.055), calcification (137.7 [72.9–329.3] vs 116.1 [49.0–218.5] pg/mL, P = 0.045), and healed plaques (203.2 [85.2–433.1] vs 114.1 [53.3–259.1] pg/mL, P = 0.005), while RvD1 levels were similar for those with or without TCFA (159.6 [67.1–312.5] vs 116.1 [53.3–281.6] pg/mL, P = 0.24) or microphage infiltration (136.3 [66.1–304.4] vs 112.2 [49.2–272.0] pg/mL, P = 0.21).
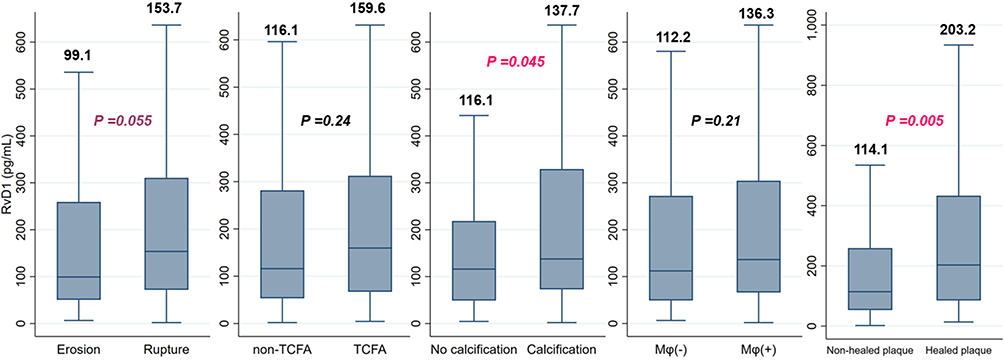 |
Figure 3 Levels of RvD1 according to pathological features identified by OCT. Abbreviations: Mφ, macrophage; OCT, optical coherence tomography; RvD1, resolvin D1; TCFA, thin-cap fibroatheroma. |
When stratified by the median (Table 2), patients with high RvD1 acquired significantly higher prevalence of plaque rupture (57.5% vs 41.7%, P = 0.014) and showed a trend for higher frequency of healed plaques (25.0% vs 15.8%, P = 0.078). However, the prevalence of TCFA (28.3% vs 20.8%, P = 0.18), calcification (55.0% vs 50.8%, P = 0.52), and microphage infiltration (58.3% vs 53.3%, P = 0.44) was similar between the two groups. The fibrous cap thickness was substantially lower in those with high RvD1 (90 [60–120] vs 100 [70–140] μm, P = 0.022), while the maximal lipid arc (360 [253–360] vs 360 [243–360] °, P = 0.58) and minimal lumen area (1.80 [1.45–2.28] vs 1.69 [1.42–2.16] mm2, P = 0.20) were similar between the groups.
 |
Table 2 OCT Findings Stratified by High and Low Levels of RvD1 |
Associations Between RvD1 and Plaque Rupture
Univariable logistic regression analysis showed that RvD1 levels above the median (≥129.7 pg/mL) were associated with plaque rupture (odds ratio [OR]: 1.89, 95% CI: 1.13–3.16, P=0.015) at culprit lesion (Table 3). Other variables associated with plaque rupture included age (OR: 1.03, 95% CI: 1.01–1.05, P=0.012), diabetes (OR: 1.84, 95% CI: 1.06–3.21, P=0.030), platelet (OR: 0.99, 95% CI: 0.99–0.99, P=0.002), estimated glomerular filtration rate (eGFR, OR: 0.99, 95% CI: 0.97–0.99, P=0.030), right coronary artery (RCA) as an infarct-related artery (IRA) (OR: 1.98, 95% CI: 1.15–3.41, P=0.014), and multivessel disease (OR: 2.30, 95% CI: 1.26–4.22, P=0.007). In multivariable analysis, high level of RvD1 was still independently associated with plaque rupture (OR: 1.91, 95% CI: 1.10–3.33, P=0.022). Other independent predictors of plaque rupture were platelets (OR: 0.99, 95% CI: 0.99–0.99, P=0.027) and multivessel disease (OR: 2.21, 95% CI: 1.14–4.27, P=0.019). To determine the best cut-off threshold for identifying plaque rupture, we performed receiver operating curve (ROC) and Youden index analysis. The optimal threshold for RvD1 to predict plaque rupture was 111.5 pg/mL, with an area under the curve (AUC) of 0.59, a sensitivity of 0.62, and a specificity of 0.59. Interestingly, the diagnostic performance of RvD1 was better than that of the conventional inflammatory marker hsCRP (optimal threshold: 11.575 mg/L, sensitivity: 0.20, specificity: 0.85, AUC: 0.53). When stratified by the threshold, high RvD1 (≥111.5 pg/mL) was still an independent predictor of plaque rupture (OR: 2.09, 95% CI: 1.20–3.66, P=0.010).
 |
Table 3 Logistic Regression Analysis of Variables Associated with Plaque Rupture |
Associations Between RvD1 and Healed Plaques
As shown in the baseline characteristics, healed plaques were more frequently observed in patients with high RvD1 (≥129.7 pg/mL). Therefore, we analyzed the association between RvD1 and healed plaques (Table 4). Univariable logistic regression showed that RvD1 above the median level was not a significant predictor for healed plaques (OR: 1.77, 95% CI: 0.93–3.36, P=0.080) at culprit lesions. ROC analysis using Youden index identified 246.4 pg/mL as the optimal threshold for predicting healed plaques with RvD1 (sensitivity: 0.47, specificity: 0.74, AUC: 0.61). When stratified by this cutoff threshold, high RvD1 (≥246.4 pg/mL) was independently associated with healed plaques (OR: 2.17, 95% CI: 1.11–4.24, P=0.023), after adjusting for other variables with P < 0.1 in univariable analysis, including male sex, BMI, eGFR, and LDL-C.
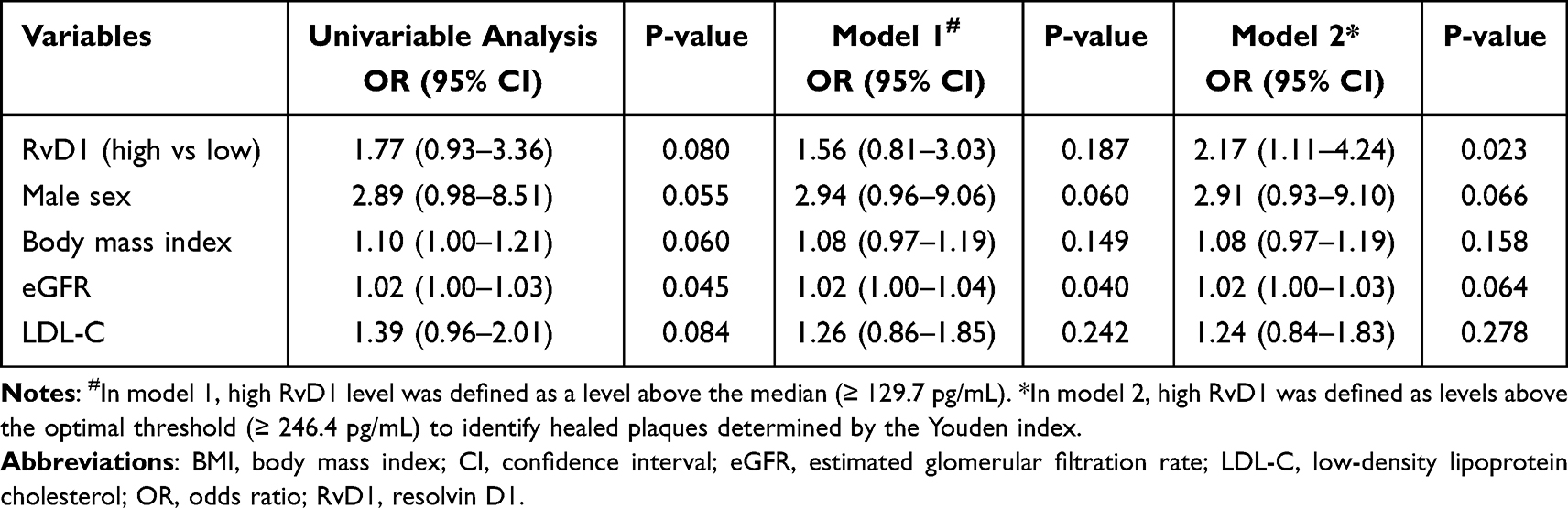 |
Table 4 Logistic Regression Analysis of Variables Associated with Healed Plaques |
Associations Between RvD1 and Calcification
As patients with RvD1 levels above the median (≥129.7 pg/mL) showed a higher prevalence of calcification at culprit plaques, we further analyzed the association between RvD1 and calcification using logistic regression (Table 5). According to univariable analysis, high RvD1 defined by level above the median was not associated with significantly higher odds of calcification at culprit plaques (OR: 1.18, 95% CI: 0.71–1.96, P = 0.518). When stratified by optimal threshold (≥293.4 pg/mL) identified by ROC (sensitivity: 0.32, specificity: 0.81, AUC: 0.57) using Youden index, high RvD1 was an independent predictor of calcification at culprit plaque (OR: 2.10, 95% CI: 1.21–3.66, P = 0.008) after multiple adjustments for age, diabetes, leukocytes, hemoglobin, hemoglobin A1c, and multivessel disease identified by P < 0.1 in univariable analysis.
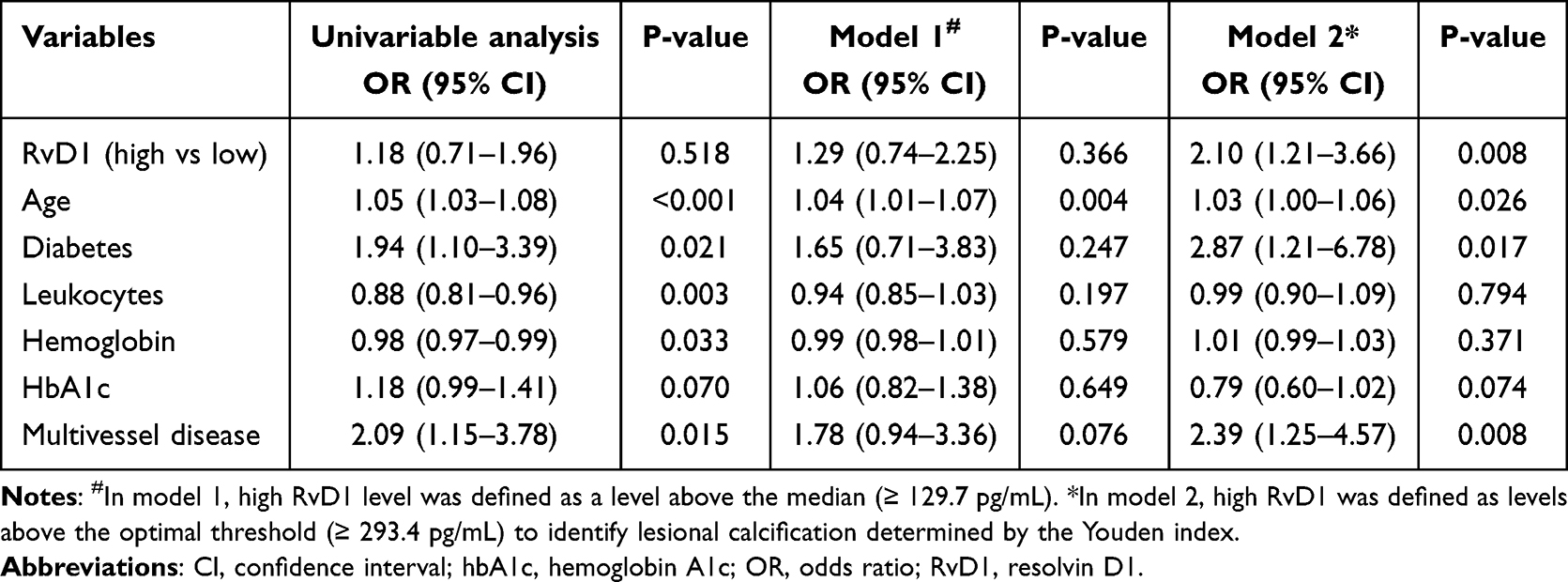 |
Table 5 Logistic Regression Analysis of Variables Associated with Calcification |
Discussion
The major finding of this study was that a high level of RvD1 was associated with plaque rupture, calcification, and healing of culprit lesions in patients with STEMI. To our knowledge, this is the first study to investigate the relationship between RvD1 and culprit plaque morphologies in atherosclerotic patients, which provides essential insights into how pro-resolving lipid mediators are involved in the pathogenesis of atherosclerosis and MI.
RvD1 and High-Risk Ruptured Plaques
According to previous study, nearly 60–70% of STEMI patients present with plaque rupture at culprit lesions, which is generally caused by the chronic predisposition of lipids within the arterial wall, the growing necrotic core as a result of long-lasting regional inflammation, the weakening of fibrous cap due to significant structural instability below, and finally, the rupture of plaques followed by thrombotic occlusion within coronary arteries.1,3,17 Therefore, it is of great clinical interest to search for biomarkers that can predict plaque rupture before coronary events and new therapeutic targets to inhibit the progression of unstable plaques. Previous studies have shown that RvD1 is actively involved in the regulation of atherosclerosis, as RvD1 could effectively reduce the generation of reactive oxygen species (ROS) through suppression of NADPH oxidase, which rescues macrophages from oxidative stress-induced apoptosis, restores regional efferocytosis, and promotes plaque stabilization.11–13 Clinical study also shows that RvD1 levels are substantially lower in STEMI patients compared with healthy controls, while lower levels of RvD1 are correlated with greater thrombus burden within the culprit vessel, poorer cardiac function, more intensive inflammation, and cardiac damage, suggesting a positive role of RvD1 among MI patients.18
However, the current study showed that high RvD1 levels were associated with plaque rupture, which is a pathological feature with the highest risk for coronary lesions. The interpretation of these results could be challenging. Evidence from bench to bedside suggests that the synthesis and release of RvD1 are subject to a positive feedback loop, as the efferocytosis of regionally infiltrated apoptotic polymorphonuclear leukocytes (PMNs) results in increased levels of various SPMs synthesized by macrophages or the microparticles released from the PMN themselves,5,19,20 while the atherogenic oxidized low-density lipoprotein (ox-LDL) within atherosclerotic plaques could also induce the production of RvD1 in endothelial cells.21 Although these inflammatory stimuli could increase the production of SPMs, its circulating levels are far from adequate to suppress the existent inflammation.20,22 Macrophages within or near atherosclerotic plaques continue to become defective in efferocytosis, as the expression of GPR32 receptors for SPM is reduced with constant inflammatory stimuli, further delaying the regional resolution of inflammation.12,23 Taken together, unresolved arterial inflammation continuously recruits inflammatory cells to expanding plaques, which serve as long-lasting stimulators for the synthesis of RvD1, therefore leading to regional and circulatory elevation of RvD1 levels. As a consequence of ongoing inflammation, plaque rupture could therefore be associated with a high circulating level of RvD1 (Figure 4). As is shown in the current study, patients with RvD1 ≥129.7 pg/mL were nearly 90% more likely to have plaque rupture at their culprit lesions. Moreover, RvD1 exhibited a more favorable diagnostic performance for plaque rupture as compared to hsCRP, a classical indicator of systemic inflammatory response.24 In summary, the elevation of RvD1 is an independent predictor of plaque rupture, while future studies are warranted to further determine the biological effect of RvD1 in human atherosclerosis.
 |
Figure 4 Graphical summary of interactions between defective efferocytosis, apoptosis and necrosis of Mφ, plaque damage, and production of RvD1. Phagocytosis of cholesterol particles lead to formation of foam cells in large amount, causing defective efferocytosis of these apoptotic Mφ, which result in the growth of necrotic core, and the later rupture or damage of atherosclerotic plaques. Meanwhile, efferocytes and apoptotic Mφ release RvD1, which is partially released to the circulation and results in an elevated level of circulating RvD1. Abbreviations: Mφ, macrophage; RvD1, resolvin D1. |
RvD1 and Plaque Healing
Characterized by distinct layers of collagen in histopathology, healed plaques reflect the instability of atherosclerotic lesions with episodic rupture or erosion, non-occlusive thrombosis, and subsequent tissue repair.1,25 Therefore, the presence of healed plaques signifies the vulnerability of atherosclerotic lesion, as repeated damage and healing processes could result in the expansion of necrotic core, leading to an increased risk of ischemic events.1,26 In the current study, we reported that patients with elevated RvD1 were more likely to have healed plaques at their culprit lesions. Following discussions regarding RvD1 and high-risk plaque rupture, it is expected that patients with healed plaques would acquire high levels of RvD1 due to an active inflammatory response within the plaques. Previous studies have also shown that RvD1 could suppress platelet function,27 promote clot absorption,28 and attenuate leukocytes infiltration in case of endothelial damage,29 which might have contributed to asymptomatic thrombosis and plaque healing in silent plaque rupture events.
RvD1 and Calcification
In the current study, we found that elevated RvD1 levels were associated with calcification at the culprit lesion of STEMI patients. For a long time, vascular calcification has been considered a result of aging and calcium metabolic disorders due to renal dysfunction.1,25 However, accumulating evidence suggests that hyperlipidemia and inflammation are actively involved in the pathogenesis of coronary calcification.30,31 Previous studies show that ox-LDL and lp(a) could induce osteoblastic differentiation and mineralization of vascular smooth muscle cells by upregulating expressions of BMP-2,32 while proinflammatory cytokines (eg, tumor necrosis factor α [TNF-α], interleukin 6 [IL-6], and IL-1β) actively involved in atherosclerosis can also promote matrix mineralization in extraosseous cells.30 Histological studies show that coronary calcifications usually colocalize with apoptotic macrophages, cholesterol crystals, and large necrotic core within advanced coronary plaques,1 suggesting that calcifications might be an indicator for unresolved inflammation within the coronary plaques, while associations between RvD1 and culprit plaque calcification could be secondary to chronic local arterial inflammation.
Limitations
The major limitations of this study are as follows: First, only circulating levels of RvD1 were measured, and the abundance of RvD1 within the culprit plaque was unknown. Future studies are expected to analyze the correlations between systematic and regional levels of RvD1 with blood samples and atherosclerotic plaques collected during invasive procedures (eg, endarterectomy and CABG). Second, this study acquired a small sample size and was conducted at a single center. Extrapolation of conclusions might be limited owing to variations in ethnicity, region, and clinical practice.
Conclusions
High levels of RvD1 were associated with plaque rupture, calcification, and healed plaques at the culprit lesions in patients with STEMI, indicating chronic inflammation and defective resolution within the coronary atherosclerotic plaques.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committee of Fuwai Hospital, Beijing (protocol code: 2017‐866, date of approval: January 22, 2017).
Informed Consent Statement
Informed consent was obtained from all the subjects involved in the study.
Data Sharing Statement
The data used to support the findings of this study are available from the corresponding authors upon request. The institution (Fuwai Hospital) requires all requests to access any patient data to be applied and processed in a case‐by‐case manner.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval for the version to be published, and agree to be accountable for all aspects of the work.
Funding
This study was supported by the National Natural Science Foundation of China (81970308), the Fund of the “Sanming” Project of Medicine in Shenzhen (SZSM201911017), Fundamental Research Funds for the Central Universities (3332022123), Shenzhen Key Medical Discipline Construction Fund (SZXK001), and the Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (2016‐I2M‐1‐009).
Disclosure
The authors declare no conflict of interest.
References
1. Yahagi K, Kolodgie FD, Otsuka F, et al. Pathophysiology of native coronary, vein graft, and in-stent atherosclerosis. Nat Rev Cardiol. 2015;13(2):79–98. doi:10.1038/nrcardio.2015.164
2. Tabas I. Tabas I: macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol. 2010;10(1):36–46. doi:10.1038/nri2675
3. Crea F, Libby P. Acute coronary syndromes: the way forward from mechanisms to precision treatment. Circulation. 2017;136(12):1155–1166. doi:10.1161/CIRCULATIONAHA.117.029870
4. Serhan CN, Levy BD. Resolvins in inflammation: emergence of the pro-resolving superfamily of mediators. The J Clin Invest. 2018;128(7):2657–2669. doi:10.1172/JCI97943
5. Serhan CN. Pro-resolving lipid mediators are leads for resolution physiology. Nature. 2014;510(7503):92–101. doi:10.1038/nature13479
6. Pirault J, Back M. Lipoxin and resolvin receptors transducing the resolution of inflammation in cardiovascular disease. Front Pharmacol. 2018;9:1273. doi:10.3389/fphar.2018.01273
7. Thul S, Labat C, Temmar M, Benetos A, Back M. Low salivary resolvin D1 to leukotriene B4 ratio predicts carotid intima media thickness: a novel biomarker of non-resolving vascular inflammation. Eu J Prev Cardiol. 2017;24(9):903–906. doi:10.1177/2047487317694464
8. Ozgul Ozdemir RB, Soysal Gunduz O, Ozdemir AT, Akgul O. Low levels of pro-resolving lipid mediators lipoxin-A4, resolvin-D1 and resolvin-E1 in patients with rheumatoid arthritis. Immunol Lett. 2020;227:34–40. doi:10.1016/j.imlet.2020.08.006
9. Lutfioglu M, Aydogdu A, Sakallioglu EE, Alacam H, Pamuk F. Gingival crevicular fluid interleukin-8 and lipoxin A4 levels of smokers and nonsmokers with different periodontal status: a cross-sectional study. J Periodontal Res. 2016;51(4):471–480. doi:10.1111/jre.12324
10. Wu SH, Yin PL, Zhang YM, Tao HX. Reversed changes of lipoxin A4 and leukotrienes in children with asthma in different severity degree. Pediatr Pulmonol. 2010;45(4):333–340. doi:10.1002/ppul.21186
11. Fredman G, Hellmann J, Proto JD, et al. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat Commun. 2016;7:12859. doi:10.1038/ncomms12859
12. Mena HA, Spite M. Spite M: proresolving receptor tames inflammation in atherosclerosis. J Clin Invest. 2021;131(24). doi:10.1172/JCI155240
13. Lee HN, Surh YJ. Resolvin D1-mediated NOX2 inactivation rescues macrophages undertaking efferocytosis from oxidative stress-induced apoptosis. Biochem Pharmacol. 2013;86(6):759–769. doi:10.1016/j.bcp.2013.07.002
14. Ibanez B, James S, Agewall S, et al. 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: the Task Force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society of Cardiology (ESC). Eur Heart J. 2018;39(2):119–177. doi:10.1093/eurheartj/ehx393
15. Tearney GJ, Regar E, Akasaka T, et al. Consensus standards for acquisition, measurement, and reporting of intravascular optical coherence tomography studies: a report from the international working group for intravascular optical coherence tomography standardization and validation. J Am Coll Cardiol. 2012;59(12):1058–1072. doi:10.1016/j.jacc.2011.09.079
16. Fracassi F, Crea F, Sugiyama T, et al. Healed culprit plaques in patients with acute coronary syndromes. J Am Coll Cardiol. 2019;73(18):2253–2263. doi:10.1016/j.jacc.2018.10.093
17. Jia H, Abtahian F, Aguirre AD, et al. In vivo diagnosis of plaque erosion and calcified nodule in patients with acute coronary syndrome by intravascular optical coherence tomography. J Am Coll Cardiol. 2013;62(19):1748–1758. doi:10.1016/j.jacc.2013.05.071
18. Karayiğit O, Nurkoç SG, Başyiğit F, Kızıltunç E. The role of serum resolvin D1 levels in determining the presence and prognosis of ST-segment elevation myocardial infarction. Med Prin Pract. 2022;31(6):548–554. doi:10.1159/000527064
19. Dalli J, Serhan CN. Specific lipid mediator signatures of human phagocytes: microparticles stimulate macrophage efferocytosis and pro-resolving mediators. Blood. 2012;120(15):e60–72. doi:10.1182/blood-2012-04-423525
20. Fredman G. Can inflammation-resolution provide clues to treat patients according to their plaque phenotype? Front Pharmacol. 2019;10:205. doi:10.3389/fphar.2019.00205
21. Tangeten C, Zouaoui Boudjeltia K, Delporte C, Van Antwerpen P, Korpak K. Unexpected Role of MPO-Oxidized LDLs in atherosclerosis: in between inflammation and its resolution. Antioxidants (Basel). 2022;11(5). doi:10.3390/antiox11050874
22. Cai B, Thorp EB, Doran AC, et al. MerTK receptor cleavage promotes plaque necrosis and defective resolution in atherosclerosis. J Clin Invest. 2017;127(2):564–568. doi:10.1172/JCI90520
23. Chiurchiu V, Leuti A, Saracini S, et al. Resolution of inflammation is altered in chronic heart failure and entails a dysfunctional responsiveness of T lymphocytes. FASEB J. 2019;33(1):909–916. doi:10.1096/fj.201801017R
24. Chen R, Liu C, Zhou P, et al. Both Low and High Postprocedural hsCRP associate with increased risk of death in acute coronary syndrome patients treated by percutaneous coronary intervention. Media Inflamm. 2020;2020:9343475. doi:10.1155/2020/9343475
25. Adriaenssens T, Allard-Ratick MP, Thondapu V, et al. Optical coherence tomography of coronary plaque progression and destabilization: JACC Focus Seminar Part 3/3. J Am Coll Cardiol. 2021;78(12):1275–1287. doi:10.1016/j.jacc.2021.07.032
26. Usui E, Mintz GS, Lee T, et al. Prognostic impact of healed coronary plaque in non-culprit lesions assessed by optical coherence tomography. Atherosclerosis. 2020;309:1–7. doi:10.1016/j.atherosclerosis.2020.07.005
27. Eickmeier O, Seki H, Haworth O, et al. Aspirin-triggered resolvin D1 reduces mucosal inflammation and promotes resolution in a murine model of acute lung injury. Mucosal Immunol. 2013;6(2):256–266. doi:10.1038/mi.2012.66
28. Elajami TK, Colas RA, Dalli J, Chiang N, Serhan CN, Welty FK. Specialized proresolving lipid mediators in patients with coronary artery disease and their potential for clot remodeling. FASEB J. 2016;30(8):2792–2801. doi:10.1096/fj.201500155R
29. Makino Y, Miyahara T, Nitta J, et al. proresolving lipid mediators resolvin D1 and Protectin D1 isomer attenuate neointimal hyperplasia in the rat carotid artery balloon injury model. J Surg Res. 2019;233:104–110. doi:10.1016/j.jss.2018.07.049
30. Tintut Y, Honda HM, Demer LL. Biomolecules Orchestrating Cardiovascular Calcification. Biomolecules. 2021;11(10):1482. doi:10.3390/biom11101482
31. Hsu JJ, Tintut Y, Demer LL. Regulation of cardiovascular calcification by lipids and lipoproteins. Curr Opin Lipidol. 2022;33(5):289–294. doi:10.1097/MOL.0000000000000844
32. Su X, Ao L, Shi Y, Johnson TR, Fullerton DA, Meng X. Oxidized low density lipoprotein induces bone morphogenetic protein-2 in coronary artery endothelial cells via Toll-like receptors 2 and 4. J Biol Chem. 2011;286(14):12213–12220. doi:10.1074/jbc.M110.214619
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Study on Relationship Between Carotid Intima-Media Thickness and Inflammatory Factors in Obstructive Sleep Apnea
Ji P, Kou Q, Zhang J
Nature and Science of Sleep 2022, 14:2179-2187
Published Date: 14 December 2022
Identification and Validation of Immune-Related Genes Diagnostic for Progression of Atherosclerosis and Diabetes
Fu Y, Xu L, Zhang H, Ding N, Zhang J, Ma S, Yang A, Hao Y, Gao Y, Jiang Y
Journal of Inflammation Research 2023, 16:505-521
Published Date: 10 February 2023
Insights into the Role of Inflammation in the Management of Atherosclerosis
Rocha VZ, Rached FH, Miname MH
Journal of Inflammation Research 2023, 16:2223-2239
Published Date: 24 May 2023
Resolvin D1 Attenuates Inflammation and Pelvic Pain Associated with EAP by Inhibiting Oxidative Stress and NLRP3 Inflammasome Activation via the Nrf2/HO-1 Pathway
Zhang J, Chen J, Jiang Q, Feng R, Zhao X, Li H, Yang C, Hua X
Journal of Inflammation Research 2023, 16:3365-3379
Published Date: 8 August 2023
Correlations Between Acute Coronary Syndrome and Novel Inflammatory Markers (Systemic Immune-Inflammation Index, Systemic Inflammation Response Index, and Aggregate Index of Systemic Inflammation) in Patients with and without Diabetes or Prediabetes
Tuzimek A, Dziedzic EA, Beck J, Kochman W
Journal of Inflammation Research 2024, 17:2623-2632
Published Date: 29 April 2024