")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 19
Status and Future Directions of Therapeutics and Prognosis of Cardiac Amyloidosis
Authors Zhang W, Ding J, Wang W, Wang D, Pan Y, Xu D
Received 29 March 2023
Accepted for publication 19 June 2023
Published 10 July 2023 Volume 2023:19 Pages 581—597
DOI https://doi.org/10.2147/TCRM.S414821
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Deyun Wang
Wenbing Zhang,1 Jian Ding,2 Wenhai Wang,1 Duo Wang,3 Yinping Pan,4 Dexin Xu5
1Department of Cardiology, Jilin Province FAW General Hospital, Changchun, 130000, People’s Republic of China; 2Department of Electrodiagnosis, Jilin Province FAW General Hospital, Changchun, 130000, People’s Republic of China; 3Department of Geriatrics, Jilin Province FAW General Hospital, Changchun, 130000, People’s Republic of China; 4Department of Pediatrics, Jilin Province FAW General Hospital, Changchun, 130000, People’s Republic of China; 5Department of Orthopedics, Jilin Province FAW General Hospital, Changchun, 130000, People’s Republic of China
Correspondence: Dexin Xu, Department of Orthopedics, Jilin Province FAW General Hospital, Changchun, 130000, People’s Republic of China, Email [email protected]
Abstract: Accumulation of aberrant proteins in the heart causes cardiac amyloidosis, an uncommon and complicated illness. It can be classified into two main types: light chain (AL) and transthyretin (ATTR). The diagnosis of cardiac amyloidosis is challenging due to its non-specific clinical presentation and lack of definitive diagnostic tests. Diagnostic accuracy has increased with the advent of modern imaging methods, including cardiac magnetic resonance imaging (MRI) and positron emission tomography (PET) scans. Depending on the severity of cardiac amyloidosis, a number of treatments may be attempted and specified according to the subtype of amyloidosis and the presence of complications. However, there are still significant challenges in treating this condition due to its complexity and lack of effective treatments. The prognosis for patients with cardiac amyloidosis is poor. Despite recent advances in diagnosis and treatment, there is still a need for more effective treatments to improve outcomes for patients with this condition. Therefore, we aim to review the current and future therapeutics reported in the literature and among ongoing clinical trials recruiting patients with CA.
Keywords: cardiac amyloidosis, ATTR CA, transthyretin amyloidosis, AL CA, light-chain amyloidosis, management, treatment, stem cell transplantation, chemotherapy, immunomodulatory agents
Introduction
Proteins that have not appropriately folded self-aggregate into amyloid fibrils and then deposit in tissues, causing amyloidosis.1,2 As amyloid fibrils accumulate in the myocardial interstitium, a rare but severe condition known as cardiac amyloidosis (CA) develops.1,2 Long diagnostic lags in individuals with CA have been linked to shorter survival after diagnosis, particularly in those with more advanced illnesses.3 Deterioration in quality of life (QoL), more hospitalizations, and mortality are associated with progressive loss of heart structure and efficiency owing to amyloid fibril deposition.1,2,4,5 Heart dysfunction is another direct result of the circulating amyloid precursor in light chain (AL) amyloidosis.6 While the exact causes of CA and heart failure have yet to be determined, it is thought that CA is underreported in the literature.7,8 AL and transthyretin (ATTR) amyloidosis are the commonest types of CA.1–8 AL CA is attributed to clonal B-cell proliferation or plasma cell dyscrasia, both leading to the misfolded immunoglobulin light chains that make up the fibrils.1–8 Misfolded TTR forms fibrils in ATTR amyloidosis, which may be caused by TTR gene mutations (ATTRv) or wild-type protein dissociation (ATTRwt).1–8 The disease is indistinguishable from other cardiomyopathies and characterized by vague symptoms that may make it challenging to identify CA, which can delay the commencement of therapy and worsen the prognosis.9,10
Diagnostic and therapeutic modalities for the different types of CA are continuously updated. In this study, we will shed more light on the diagnosis and identification of CA based on recent updates. We will also review the current and future therapeutics reported in the literature and among ongoing clinical trials recruiting patients with CA.
Methods
The present study is a literature review aiming to discuss the status and future directions of CA therapeutics and prognosis. To obtain relevant original, human, and pre-clinical English articles published until January 2023, we conducted a thorough search strategy with relevant keywords, including “Cardiac amyloidosis”, management, treatment, diagnosis, “red flags”, gastrointestinal, polyneuropathy, cardiac, “hypertrophic cardiomyopathy”, electrocardiograph, ECG, “light-chain amyloidosis”, transthyretin, “AL CA”, “ATTR CA”, ATTRv, “cardiac magnetic resonance imaging”, cMRI, “Tcm99-hydroxymethylene diphosphonate scintigraphy”, “Tcm99-3,3-diphosphono-1,2-propano- dicarboxylic acid”, “Tcm99-pyrophosphate”, “urine or serum protein electrophoresis with immunofixation”, serum-free light chain, FLC, stabilizers, tafamidis, diflunisal, acoramidis, silencers, siRNA, patisiran, Inotersen, vutrisiran, revusiran, “RISPR-Cas9”, “gene editing”, degraders, hyaluronidase, “ursodeoxycholic acid”, doxycycline, GSK2315698, chemotherapy, immunotherapy, daratumumab, “tauroursodeoxycholic acid”, “stem cell”, “Autologous stem cells”, ASCT, prothema, NI006, prognosis, melphalan, “Anti-fibril”, “AT-03”, birtamimab, 6MWT, “CAEL-101”, “Epigallocatechin-3-gallate”, doxycycline, immunomodulatory). Searching different databases, like PubMed, Web of Science, and Embase. Clinicaltrials.gov was also considered to identify published and ongoing relevant trials.
Case Diagnosis and Identification
The first step towards appropriate management of any CA is adequate definition and detection of cases. The diagnosis is initiated by detecting characteristic extracardiac manifestations or red flags inconsistent with cardiac imaging findings. These red flags may include carpal tunnel syndrome, skin bruises, macroglossia, proteinuria, polyneuropathy, dysautonomia, corneal lattice dystrophy, vitreous deposits, lumbar spinal stenosis, ruptured biceps tendon, deafness, cutis laxa, skin discoloration, and gastrointestinal problems. Gastrointestinal problems are frequent extracardiac manifestations among ATTRv, together with unexplained weight loss, and autonomic dysfunction.11 Furthermore, studies have indicated the importance of neurological evaluation for ATTRv patients due to the relatively high prevalence of polyneuropathy among them. For instance, Russo et al demonstrated that 59/113 of their ATTRv population had polyneuropathy and carpal tunnel syndrome was detected in 14 of them.12 Besides, cardiac manifestations might also be found in the affected patients, including heart failure, AV conduction disease and pseudo infarct pattern on ECG, and apical sparing pattern with diminished longitudinal strain, pericardial effusion, increased valve thickness, increased right ventricular wall thickness and myocardial granular sparkling on echocardiography. Moreover, other red flags can be seen in cardiac magnetic resonance imaging, including abnormal gadolinium kinetics, increased extracellular volume, Elevated T1 values, and subendocardial gadolinium enhancement. Cardiac amyloidosis can also be evoked by early conduction system disease, disproportionally low QRS voltage, and persistent troponin elevation.8
Since the diagnosis of CA is mainly made by detecting amyloid fibers within the cardiac tissue, evidence shows the validity of various invasive and non-invasive approaches that can precisely diagnose CA. Using these approaches is based on many factors. For instance, non-invasive ones can diagnose ATTR CA, while invasive ones can be used for all CA subtypes. Invasive approaches are considered the cornerstone in establishing a diagnosis of CA. Reports indicate that regardless of the left ventricular wall thickness, the diagnosis of CA is made by detecting amyloid deposits by endomyocardial biopsy following Congo red staining. Amyloid fibril protein should then be classified by mass spectrometry, which is mainly in this context. However, evidence shows that clinical settings usually use immune-electron microscopy and immunohistochemistry.13 Other diagnostic tools might also include cardiac magnetic resonance imaging (MRI) and electrocardiography in the presence of extracardiac amyloid deposits. Boldrini et al14 suggested that a diagnosis of ATTR or AL CA should be established with extracardiac amyloid deposits, an electrocardiogram score of ≥ 8 points, and a left ventricular wall thickness is ≥ 12 mm. However, such a score is not adequately validated.
Moreover, it has been demonstrated that ATTR can be diagnosed by non-invasive tests for the detection of Grade 2 or 3 myocardial uptake of radiotracer by Tcm99-hydroxymethylene diphosphonate (HMDP) scintigraph, Tcm99-3,3-diphosphono-1,2-propano- dicarboxylic acid (DPD, or Tcm99-pyrophosphate (PYP).15 Moreover, urine and serum protein electrophoresis with immunofixation (UPIE and SPIE) and serum-free light chain (FLC) assay might exclude clonal dyscrasia (with an estimated 99% sensitivity rate for the combination of both modalities for the detection of AL amyloidosis) by detecting monoclonal immunoglobulins (M protein).16 It should be noted that the sensitivity of these modalities might be variable based on the health status of patients. For instance, the sensitivity of FLC might be variable in patients with chronic kidney disease based on the glomerular filtration rate in patients with chronic kidney disease.17 A 100% specificity rate of bone scintigraphy (Grade 2/3) for detecting cardiac ATTR has been estimated when there is abnormal serum FLC ratio with absent monoclonal proteins.15 Myocardium uptake should also be considered when interpreting the positivity of this test.18 It should be considered that cases with ATTRwt CA might have co-existing monoclonal gammopathy of undetermined significance. Therefore, it is essential to demonstrate the specific M protein at the Congo red-positive spot and the kind of immunoglobulin light chain that binds to it. Immunostaining may confirm the presence of ALκ or ALλ (+), ATTR (-), and AA (-) in biopsy tissue samples, or amyloid precursor protein can be detected by liquid chromatography-tandem mass spectrometry (LC-MS/MS) and laser microdissection (LMD). If ATTR is confirmed, it is advisable to conduct genetic counseling, even among elderly patients, to detect ATTR mutations and discriminate ATTRv and ATTRwt.19
Before conducting these tests, CA should be suspected based on the above-mentioned manifestations. Moreover, a diagnosis of the condition should be suspected in systemic conditions, like a chronic systemic inflammatory condition, peripheral neuropathy, nephrotic syndrome, and plasma cell dyscrasia, together with a manifest cardiac disease, especially in imaging-directed cases.8 A significant characteristic of CA is the increased left ventricular wall thickness with no dilatation, particularly in elderly individuals with common cardiac conditions like severe aortic stenosis (especially in individuals having transcatheter aortic valve replacement), heart failure and hypertrophic cardiomyopathy with a non-impacted ejection fraction. In these events (ie, when CA is suspected), attending physicians should seek a definite diagnosis to apply the most appropriate treatment plan.20–22 The diagnosis should be based on a combination of the invasive and non-invasive approaches stated above, as previously described in the literature. Moreover, the diagnostic workup should include a comprehensive approach to detect all the manifestations and associated complications of the affected patients. The diagnostic scheme of CA is summarized in Figure 1A.
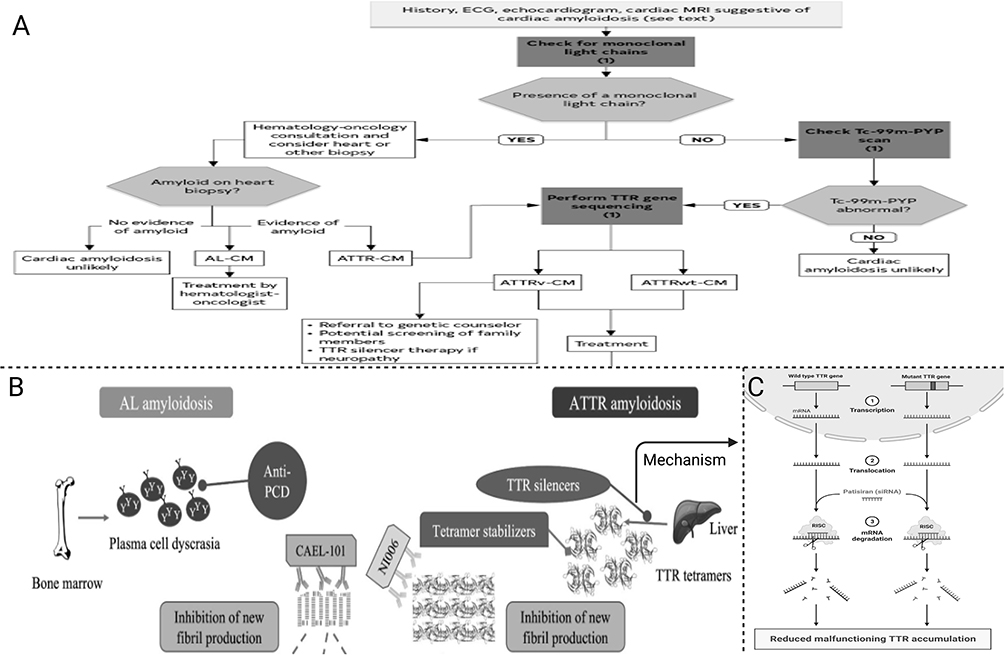 |
Figure 1 A summary of the current management approaches of cardiac amyloidosis. (A) The diagnostic scheme for identifying cardiac amyloidosis and its subtype. (B) Therapeutics for light-chain (Anti-PCD) and transthyretin (tetramers, silencers, stabilizers) amyloidosis. (C) Mechanism of action of TTR silencer patisiran (siRNA). Abbreviations: Anti-PCD, anti-plasma cell dyscrasia; AL, light-chain; ATTR, transthyretin. |
Treatment
The treatment strategy for CA significantly differs based on the type (AL or ATTR) and current disease status (ie, recent or refractory). Historically, CA has been treated by managing the underlying symptoms and related complications. It should be noted that some disease-modifying agents have been reported for their efficacy in managing these symptoms, which should be considered by the healthcare professionals to prescribe the most appropriate treatment modalities rather than managing the symptoms only.12 For instance, the management of neurological complications (polyneuropathy) has been widely investigated by stabilizers for ATTRv CA, indicating the importance of multi-disciplinary care to improve the outcomes of these patients.12,23 Besides, the administration of oral anticoagulants has been advised for managing CA individuals with sinus rhythm to diminish the risk of thromboembolic events.24 Recent disease-modifying agents have been validated as effective therapeutic modalities for the different types of CA (Figures 1B and 2).
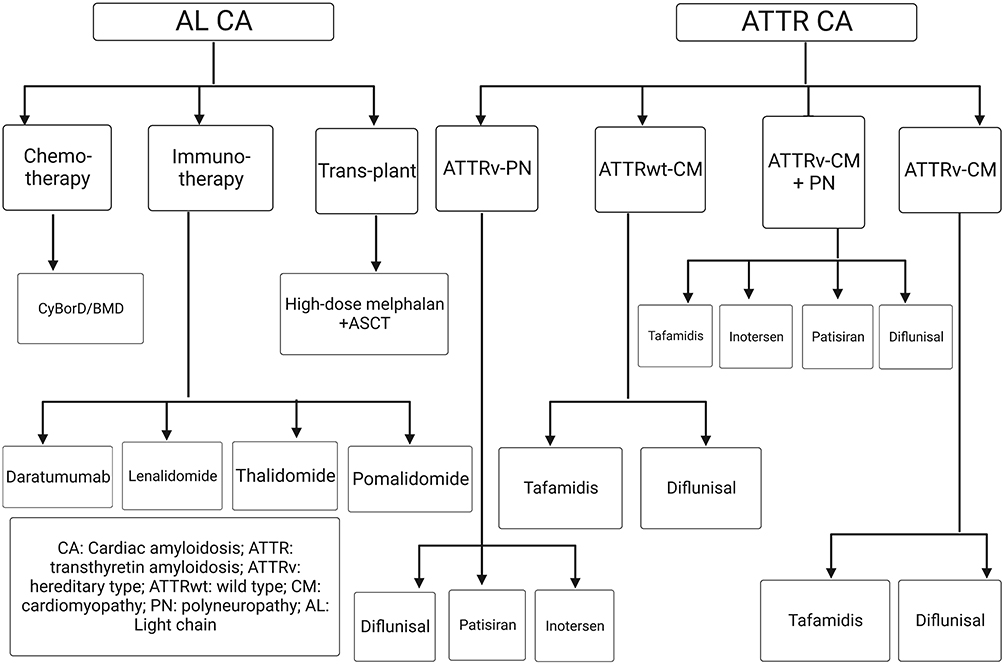 |
Figure 2 A scheme for the current therapeutics for the different subtypes of transthyretin and light-chain cardiac amyloidosis. |
Treatment of ATTR CA
Different mechanisms have been reported for these modalities that target amyloid production in ATTR CA, including degradation of deposited ATTR fibrils aiming at improving morbidity and mortality by restoring cardiac functions and reversing the disease process, inhibiting TTR oligomer aggregation, anti-TTR antibodies, preventing TTR tetramer dissociation by TTR stabilization, and preventing TTR production by hepatocyte that is mediated by gene silencing.25 The current evidence can be obtained from randomized and non-randomized studies. The characteristics of observational studies are presented in Table 1, while randomized trials are discussed in detail in the following text. It should be noted that the treatment of ATTR CA largely depends on the disease phenotype and associated complications (Figure 2). Accordingly, a careful diagnostic work-up is needed to establish the best treatment strategy for the affected patients.
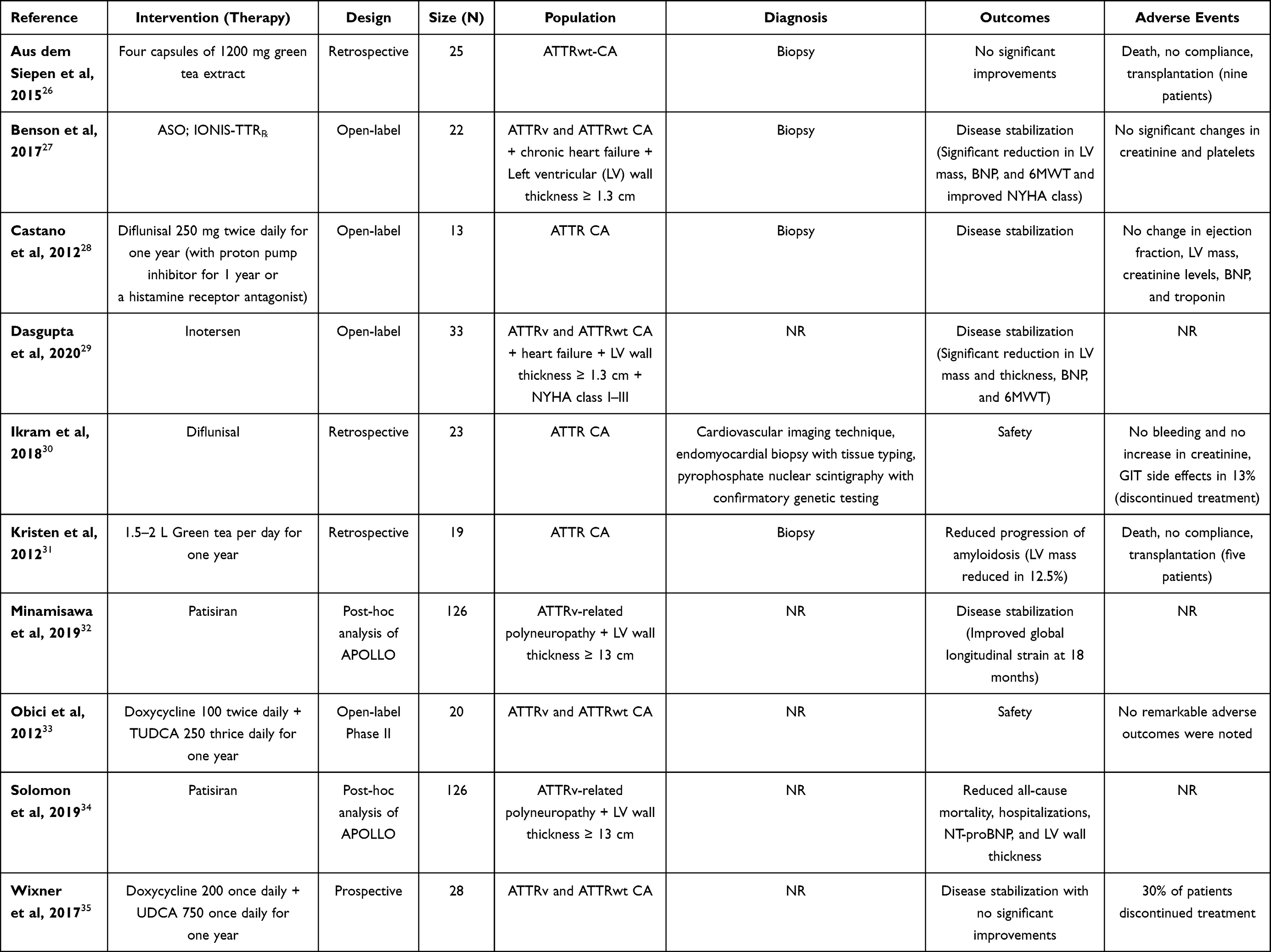 |
Table 1 Observational Studies Reporting Outcomes of Different Interventions for Patients with Non-Complicated ATTR CA |
Stabilizers
Some treatment modalities are currently reported as potentially effective stabilizers of TTR. Some of these are validated, like tafamidis, which was approved in 2019 by the FDA for treating the different types of ATTR CA following the positive outcomes of the ATTR ACT trial, which treated 335 and 106 ATTRwt CA and ATTRv CA patients, respectively, with oral tafamidis in two arms of 20 and 80 mg compared to each other and a placebo group.36 In both treatment arms, the authors showed cardiovascular hospitalizations (HR: 0.68, 95% CI: 0.56–0.81) and all-cause mortality (42.9% and 29.5%; HR: 0.7, 95% CI: 0.51–0.96) were significantly reduced at 30 months. Early administration of tafamidis is recommended for ATTR CA patients because the trial showed that patients with NYHA class III symptoms developed an increased risk of cardiovascular hospitalizations. However, tafamidis is considered a safe and effective agent that should be considered for these patients, mainly due to its proven safety profile.37
On the other hand, other drugs were investigated in vitro,38 with a few human studies reported in the literature, like the nonsteroidal anti-inflammatory drug (NSAID) diflunisal, which has been validated in vitro and single-arm cohort studies of ATTRv CA patients.28,39–41 Accordingly, randomized trials are needed for validation of the potential effectiveness of this modality and its impact on morbidity and mortality of affected patients, although most of the current studies indicate the stabilization of echocardiographic parameters and cardiac biomarkers. Overall, the disease is considered safe. However, patients should be warned about potentially having NSAID-related adverse events, like heart failure exacerbation, bleeding, and gastrointestinal discomfort. Moreover, it is contraindicated in cases of renal dysfunction and significant thrombocytopenia.28 Another drug that is being studied in NYHA I–III ATTR CA cases by the ATTRIBUTE-CM trial (Phase III randomized controlled) NCT03860935) is acoramidis (or AG-10), which has been investigated for its ability to mimic a protective TTR mutation due to its ability to bind to TTR, even more significantly than tafamidis.42
Cost-Effectiveness
Cost-effectiveness is a vital parameter always considered by patients and healthcare authorities and can, therefore, impact compliance to a specific regimen over the other. Not much data can be found in the literature in this regard. In 2020, Kazi et al43 made a cohort Markov modeling study to compare the cost-effectiveness of tafamidis versus no tafamidis, with cost-estimation correlated to all-payer data and effectiveness attributed from the findings of the ATTR-ACT trial. The authors observed that tafamidis medication for ATTR CA cases would further boost quality-adjusted expected lifespan by a mean of 1.29 QALYs (quality-adjusted life year) in a modeling investigation optimized to the findings of the ATTR-ACT trial, but the ICER of $880 000 per QALY gained would be significantly greater than traditional pricing threshold values. To be cost-effective at a commonly accepted standard of $100,000 per gained QALY, the annual cost of tafamidis would need to be reduced by 92.6%, from $225,000 to $16,563.
Silencers
Inotersen and patisiran are two FDA-approved gene silencer therapeutics that can be effectively used for ATTRv CA-associated polyneuropathy based on the findings of different randomized controlled trials. Inotersen is an antisense oligonucleotide administered subcutaneously every week and acts by inhibiting liver-produced transthyretin mRNA. The NEURO-TTR trial investigated the efficacy of Inotersen among 172 patients with TTRv-CA-related polyneuropathy.44 Results from this trial showed that cardiac improvement was observed in 61% of these patients. Furthermore, these patients had significant improvements in the quality of life with a decreased progression in polyneuropathy, but no favorable outcomes were observed in the echocardiographic parameters. However, the study showed that patients treated with Inotersen developed more adverse events, like glomerulonephritis and thrombocytopenia, than patients in the placebo group (32% versus 22%). Another single-center investigation of 33 patients with ATTR-CV without polyneuropathy showed that Inotersen was associated with a significant improvement in exercise tolerance and reduction in left ventricular mass at 2–3 months of follow-up.29 Other studies are also ongoing to investigate the efficacy and safety of this modality among ATTR-CV patients (NCT03702829).
Patisiran, a small interfering RNA (siRNA) that can be employed for TTR mRNA identification and degradation (Figure 1C), is administered every three weeks and works by mediating the degradation of the transthyretin mRNA and binding to an RNA-induced silencing complex to prevent TTR formation. A previous investigation demonstrated the significant favorable impact of patisiran on neuropathy stabilization, leading to remarkable enhancements in body weight and composition, which subsequently reduced the fat and increased the muscle masses, reducing atrophy among ATTRv patients. The authors demonstrated that the favorable impact of patisiran was furtherly attributed to enhancing the gastrointestinal manifestations that are frequently observed with ATTRv.45 Another case series of nine patients with ATTRv CA indicated the remarkable improvement of their cardiovascular and neurological manifestations, with no notable adverse events.23 The efficacy of this drug was investigated among 225 patients with ATTRv-CA-induced polyneuropathy in the APOLLO randomized controlled trial.46 Results from this trial showed that cardiac improvement was observed in 56% of these patients. It should be noted that the safety of patisiran in this trial was comparable to placebo. However, this might be attributed to pre-medicating patisiran patients with H1 and H2 blockers, acetaminophen, and dexamethasone. Moreover, it has been shown that patisiran significantly reduced the progression of polyneuropathy at 18 months from starting the treatment, with a remarkable improvement in NT-proBNP levels, global longitudinal strain, end-diastolic volume, and left ventricular wall thickness.34 Results from a sub-study also showed that the drug significantly reduced mortality and hospitalizations and improved functional capacity compared to the placebo group at 18 months of follow-up.34 The superiority of either patisiran or Inotersen over the other remains controversial. A post hoc analysis of the results of the above-mentioned trials shows that patisiran was significantly associated with a lower risk of adverse outcomes and more favorable symptoms of neuropathy.
Vutrisiran is an RNAi therapy that inhibits the production of both variant and wild-type TTR, hence lowering blood TTR levels.47 By coupling to a triantennary N-acetylgalactosamine (GalNAc) ligand that binds the asialoglycoprotein receptor expressed on the surface of hepatocytes,47–49 the vutrisiran small interfering RNA (siRNA) is targeted to the liver, the principal location of TTR production. With its high metabolic stability and higher efficacy made possible by its enhanced stabilization chemistry design, the vutrisiran siRNA-GalNAc combination may be injected subcutaneously only once every three months. Vutrisiran was well tolerated and produced significant and long-lasting TTR decrease in a Phase 1 research in healthy volunteers.47 Vutrisiran is now a valid option for treating adult patients with ATTRv amyloidosis polyneuropathy. In a Phase 3, open-label, randomized study (HELIOS-A), vutrisiran administration was remarkably correlated with enhanced disability, nutritional status, gait speed, QoL, and neuropathy impairment among ATTRv individuals with polyneuropathy. It has been concluded that the therapeutic is favorable for these patients with a favorable safety profile.50
It is worth mentioning that another silencer therapeutic (revusiran) was investigated and is no longer under consideration because of unfavorable adverse outcomes and mortality.51 Eplontersen is an additional silencer therapeutic currently being investigated for ATTR CA (NCT04153149). Another promising therapeutic is the RISPR-Cas9 system for gene editing, which was validated in some cohort studies and needs further validation by randomized trials.52,53
Degraders
Another category of therapeutic options for ATTR CA is called degraders, which work by degradation of the deposited amyloid fibers and promotion of clearance and breakdown. Evidence from preclinical studies indicates the degradation efficacy of ursodeoxycholic acid (UDCA), doxycycline, and tauroursodeoxycholic acid (TUDCA).54 However, evidence from human studies is not sufficient, and current data can only be obtained from single-arm noncomparative investigations. Moreover, the results of these studies do not support the clinical application of these combinations because of their high attrition and modest efficacy. This has been attributed to doxycycline-related dermatologic and esophageal intolerance, estimated in around 10% of the cases.33,39,55 A clinical trial investigating the efficacy of these combinations is currently active (NCT03481972). Some authors suggested that these degraders can still be considered for clinical use in ATTR CA because of their cost-effectiveness compared to other therapeutics.3,8,56 Such suggestions still need further strengthening by results from randomized controlled trials.
Immunotherapeutics and Others
Immunotherapeutics, like prothema (PRX004), are also currently being investigated as potential therapeutics for ATTR. Evidence from preclinical studies shows that it works by inhibiting the formation of and enhancing the clearance of new and existing amyloid fibrils through immune-mediated mechanisms and phagocytosis. Data from clinical human studies are scarce. Due to COVID-19, a Phase I multicenter randomized trial was terminated. However, the data of seven NYHA I–III ATTRv cases could be analyzed. It has been demonstrated that following 9 months of treatment, a poor enhancement in global longitudinal strain (−1.21%) was noted, with a significant improvement in neuropathic pain and acceptable safety profile (NCT03336580). Preclinical data also show the high affinity of another anti-TTR immunotherapeutic agent (NI006) to ATTRwt and ATTRv in vivo and ex vivo.57 The efficacy and safety of NI006 has been recently reported in a phase I trial which showed that cardiac amyloid uptake was remarkably reduced without developing any serious adverse events.58 Moreover, the drug was associated with a significant reduction in Troponin T and N-terminal pro–B-type natriuretic peptide levels, indicating its efficacy and safety for ATTR CA. As TTR tetramers disintegrate into monomers, a cryptic epitope is revealed where NN6019-0001 binds. Stopping the production of new amyloid fibrils occurs by NN6019- 0001 binding to neutralize the different TTR prefibrillar species.57,59 According to data presented at the XVIII International Conference, NN6019-0001 has been well tolerated across all dosages in phase 1 studies and improved neurologic manifestations and global longitudinal strain. To evaluate the effectiveness and safety of NN6019-0001 at doses of 10 and 60 mg/kg, a Phase 2 trial is presently enrolling cases (NCT05442047).
It is also known that GSK2315698, and serum amyloid P (SAP) component antibody, was also associated with favorable fibril clearance outcomes in a clinical trial.60 However, the clinical application of this modality was not approved due to unfavorable adverse events in a Phase II trial of ATTR CA patients (NCT03044353). Another category was investigated for treating ATTR CA work by inhibiting oligomer disruption and regulation. For instance, data from in vivo and in vitro studies show that TTR amyloid formation can be significantly inhibited by epigallocatechin-3-gallate (EGCG).61,62 However, again, data from clinical comparative studies are scarce, and further studies are needed. Some published studies of patients with ATTRv and ATTRwt CA show that cardiac MRI indicates a slight reduction in the left ventricular mass.26,63 Another study indicated a remarkable decrease in the interventricular septal thickness in 12 out of 14 patients.31 This indicates the urgent need for well-designed studies with larger sample sizes to make a conclusion about the ability to clinically use EGCG for ATTR CA patients, although it might be prescribed because of its favorable safety.
Treatment of AL CA
Treating AL CA is mainly directed at eradicating the underlying plasma cell clone responsible for the excess production of light chains associated with the prognosis and severity of the disease. Accordingly, treatment categories, including autologous stem cell transplantation (ASCT) and chemotherapy, have been investigated for treating AL CA because of their proven capability in treating multiple myeloma, another disease characterized by plasma cell dyscrasia. It should be noted that providing such treatment should be multidisciplinary between cardiologists and hematologist oncologists to intervene against adverse events and enhance clinical efficacy.64,65
Autologous Stem Cell Transplantation (ASCT)
Early treatment is indicated for AL CA patients following adequate risk stratification. Evidence shows that excellent long-term disease remission can be obtained by administering autologous stem cells (ASCs) with high-dose melphalan. However, early administration of ASCs is usually indicated for low-risk patients, including reduced levels of biomarkers below standard thresholds, with no remarkable organ dysfunction (NYHA class < III), good functional status, and patient’s age < 70 years old. Accordingly, only around one-fifth of patients with AL CA receive early ASCT.66,67 Induction therapy might also be needed in cases of expected delays in initiating ASCT or high plasma cell infiltration of bone marrow (> 10%).8,68 Such induction is usually done with bortezomib (most used regimens include bortezomib-melphalan-dexamethasone and cyclophosphamide-bortezomib- dexamethasone (CyBorD)), a proteasome inhibitor, together with an alkylating agent and dexamethasone.66,69 In this context, estimates indicate that such induction can significantly enhance survival, with an 84% overall 5-year survival rate and hematologic response.70,71 Furthermore, using bortezomib can be done following ASCT to boost the response.66
Chemotherapy
Chemotherapy is usually prescribed as the highest dose a patient can safely administer, aiming at increasing survival probability and enhancing cardiac functions. Chemotherapy is usually indicated for patients who cannot tolerate ASCT, particularly those with cardiac stage I–III. A previous study demonstrated that enhanced survival rates were associated with the early administration of chemotherapy at one month in patients with advanced disease stages (Mayo cardiac stage IIIb).72 The efficacy of these regimens is usually determined by the organ-specific (serum-free light chain suppression) and hematologic (improvements in renal and cardiac biomarkers) responses. Evidence also favors assessing such clinical benefits based on combined organ and hematologic responses, which might have a more enhanced prognostic value than isolated either of these parameters.8 It is worth mentioning that, irrespective of noted cardiac parameters, rapid improvement in left ventricular function and thickness is unlikely. Further studies based on measurements of extracellular volume T1 mapping with cardiac MRI and global longitudinal strain give more evidence about the clinical efficacy of these therapeutics.73,74 Historically, high-dose chemotherapy with melphalan was the standard choice for treating intermitted AL CA and is usually administered with dexamethasone whenever ASCT is not tolerated.75 However, the addition of bortezomib was standardized due to its proven efficacy and favorable outcomes in a phase III clinical trial.76 Nevertheless, bortezomib is not used in cases with peripheral neuropathy, pulmonary fibrosis, and in cases of genetic translocation 11; 14 (reported among around 60% of AL patients).77 Accordingly, an alternative medication, venetoclax (an oral selective B-cell lymphoma 2 inhibitor), is currently being investigated in cases with t(11; 14).66,78
Immunotherapy
Daratumumab, an anti-CD38 human IgG-κ monoclonal antibody, was historically applied for treating refractory/relapsed AL CA and can be used as an up-front medication for the disease used as a subcutaneous injection. The FDA approved it with hyaluronidase-fihj (an endoglycosidase) following the results of the phase III ANDROMEDA trial,79 which showed that combined administration of daratumumab with CyBorD for patients with new-onset AL was significantly associated with improved survival rates from hematologic progression or remarkable organ deterioration (HR: 0.58; 95% CI: 0.36–0.93), and more significantly rapid hematologic response than CyBorD alone (53% versus 18%).69 It should be noted that the group treated with daratumumab had a remarkable increase in the rates of severe infections and lymphopenia. However, after adjustment of the analysis and different confounders, the authors demonstrated that the adverse events were not different between the two groups. It was also suggested that daratumumab might be safer and more efficient for cases with higher-grade cardiac involvement. An ongoing phase II trial also investigates the safety and efficacy of daratumumab for AL stage IIIb cases, which do not usually tolerate chemotherapy, requiring an alternative therapeutic regimen (NCT04131309).
New Therapeutics
Anti-fibril agents are currently under investigation for their potential to light chain amyloid fibers deposits from the affected organs to enhance their functions.80–84 Some monoclonal antibodies (mAbs) currently being investigated for their therapeutic potential include AT-03, CAEL-101, and birtamimab. The completely humanized mAb birtamimab was created to detect serum amyloid A protein-related cryptic epitopes. It activates macrophage-mediated fibril breakage and clearance by cross-reacting with amyloid fibrils associated light chains.83 All dosages of birtamimab up to 24 mg/kg were safe and well tolerated in a trial.85 When tested in previously treated AL CA individuals, birtamimab did not increase NT-proBNP levels, heart functions, 6MWT in phase 2b PRONTO study (NCT02632786).86 Additionally, the phase 3 VITAL study of new AL CA cases was discontinued early due to futility analysis results showing that the intervention did not decrease all-cause deaths.87 Nonetheless, encouraging findings were found in a post hoc analysis of stage IV patients.85,87,88 To validate these findings, researchers are enrolling participants in a randomized, placebo-controlled, phase 3 study (AFFIRM-AL; NCT04973137).
Regarding AT-03, which targets serum amyloid P protein through human hybrid mAb since its proven efficacy of binding to all different forms of amyloid proteins,82 is currently under investigation by an open-label, single-arm, biodistribution study for cases with systemic amyloidosis (NCT05201911). The chimeric monoclonal antibody CAEL-101 was designed to detect an immunoglobulin light chains-relevant cryptic epitope and has been shown to bind to amyloid fibrils and free misfolded chains that have been accumulated in end-organs.89–91 Biomarkers of cardiomyopathy and nephropathy were reduced by CAEL-101 in phase 1 studies.92,93 CAEL-101 was well tolerated in an ongoing phase 2 study, whether solely (following anti-PCD treatment) or combined with anti-PCD medication was discontinued. Cases with advanced stages IIIA and IIIB heart disease are now being recruited for two phase III randomized trials (NCT04504825, NCT04512235, and NCT04516236).
Doxycycline
Doxycycline, an antibiotic, has been found to reduce the toxicity of AL amyloid in C elegans and to prevent the development of AL amyloid fibrils in transgenic mice.94,95 Preliminary results from longitudinal studies were encouraging for their potential to enhance hematologic response and overall survival.96,97 While a recent comparative analysis of 140 subjects randomised to CyBorD with doxycycline showed no benefit for hematological or cardiovascular survival progression, the impact of doxycycline is less clear.98 A randomized controlled study examining adding doxycycline to bortezomib-based treatment is underway (NCT03474458), and we look forward to learning its findings.
Epigallocatechin-3-Gallate (EGCG)
Green tea might be a therapeutic option since its most abundant catechin, epigallocatechin-3-gallate (EGCG), is a powerful antioxidant that blocks the development of insoluble amyloid fibrils and stabilizes existing ones, making them less hazardous.99,100 Three phase II studies have looked at using EGCG to treat AL-CA. TAME-AL (NCT02015312) and EpiCardiAL27 (NCT01511263) are two studies that have finished recruiting but have not yet reported their findings. A single-center Japanese trial with 57 patients reported no significant benefits from using EGCG, as well as no noticeable adverse effects.101 It is a common practice to include EGCG in AL-CA treatment plans as an additional therapy due to its low toxicity.
Therapeutics for Relapsing AL CA
Research on the monoclonal antibody NEOD001 in the immunotherapy of refractory AL CA has been conducted. Yet, primary and secondary end goals were not met in a phase III study (the VITAL) for managing AL CA and a phase IIb study (PRONTO) for managing refractory AL.56 One further monoclonal antibody therapeutic, CAEL-101, was associated with favorable safety outcomes in a phase II research and was proven to attack amyloid deposits,102 and is currently tested in other phases III studies in patients with Mayo Stage IIIa24 and IIIb24,25 illness (NCT04504825 and NCT04512235).
Moreover, several encouraging retrospective and phase 2 studies suggest that daratumumab should be used to treat patients who have relapsed or are resistant to initial treatment.103,104 The alternative is immunomodulatory drugs and low-dose steroid treatment (dexamethasone or cyclophosphamide). Although data from retrospective and phase 2 clinical studies suggest that lenalidomide, pomalidomide, and thalidomide are effective, they have not been compared to alternative treatment protocols.105 Treatment with lenalidomide and thalidomide has been linked to thrombotic problems that call for thromboprophylaxis. Heart response evaluation is complicated because immunomodulatory therapeutics have been linked to an asynchronous increase in NT- proBNP values regardless of the enhancements in free light chains.106 While the TOURMALINE-AL1 phase 3 study did not find a remarkable difference in overall hematological response, the proteasome inhibitor ixazomib plus dexamethasone did show a higher complete response rate (26% versus 18%) and longer progression-free survival than physicians’ choice (47% lenalidomide plus dexamethasone).107 In addition to investigating immunotherapeutics-based rescue regimens, a phase II trial looked at an additional therapeutic plan including a combination of dexamethasone and bendamustine (an alkylating therapy) showing a high rate of hematological response (57%).108,109
Prognosis
Overall, studies show that the diagnosis of AL amyloidosis within 6 months of symptoms start was associated with a considerably higher risk of mortality, but survival for cases with a later diagnosis was more enhanced, with >52% living for the whole 5 years. Early detection gives both QoL and functional advantages for CA individuals, but non-diagnostic or delayed diagnosis leads to high morbidity and mortality. ATTR cases have been demonstrated to have a life expectancy of over 5 and 7 years, respectively, when using a disease simulation model to determine the effects of early diagnosis and appropriate treatment.
During the follow-up period, clinicians should always consider the different phenotypes of amyloidosis, considering both extracardiac and cardiac manifestations. These settings should include imaging studies (like cardiac MRI and other evaluative tests), neurophysiological studies (like nerve conduction studies and cardiovascular autonomic testing), and frequent clinical examinations. Various prognostic modalities have been proposed and validated in the literature for CA (Table 2). However, current evidence shows that recent biomarker-based scores are preferred to investigate the prognosis of AL and ATTR CA. Nevertheless, evidence shows that these tools have not been adapted to anticipate the prognostic impact of the different changes during the follow-up period. The exact prognostic rates of CA are not adequately reported in the literature due to the inadequacy of sound longitudinal studies, indicating the need for future investigations, particularly since new updates and management guidelines are continuously added. Adequate follow-up should be offered to these patients. Although there are no current schemes regarding the most optimal follow-up period for these patients, it has been demonstrated that 24-hour Holter ECG, yearly echocardiogram, and evaluation of complete blood tests (troponin and NT-proBNP) and electrocardiogram should be conducted every six months.
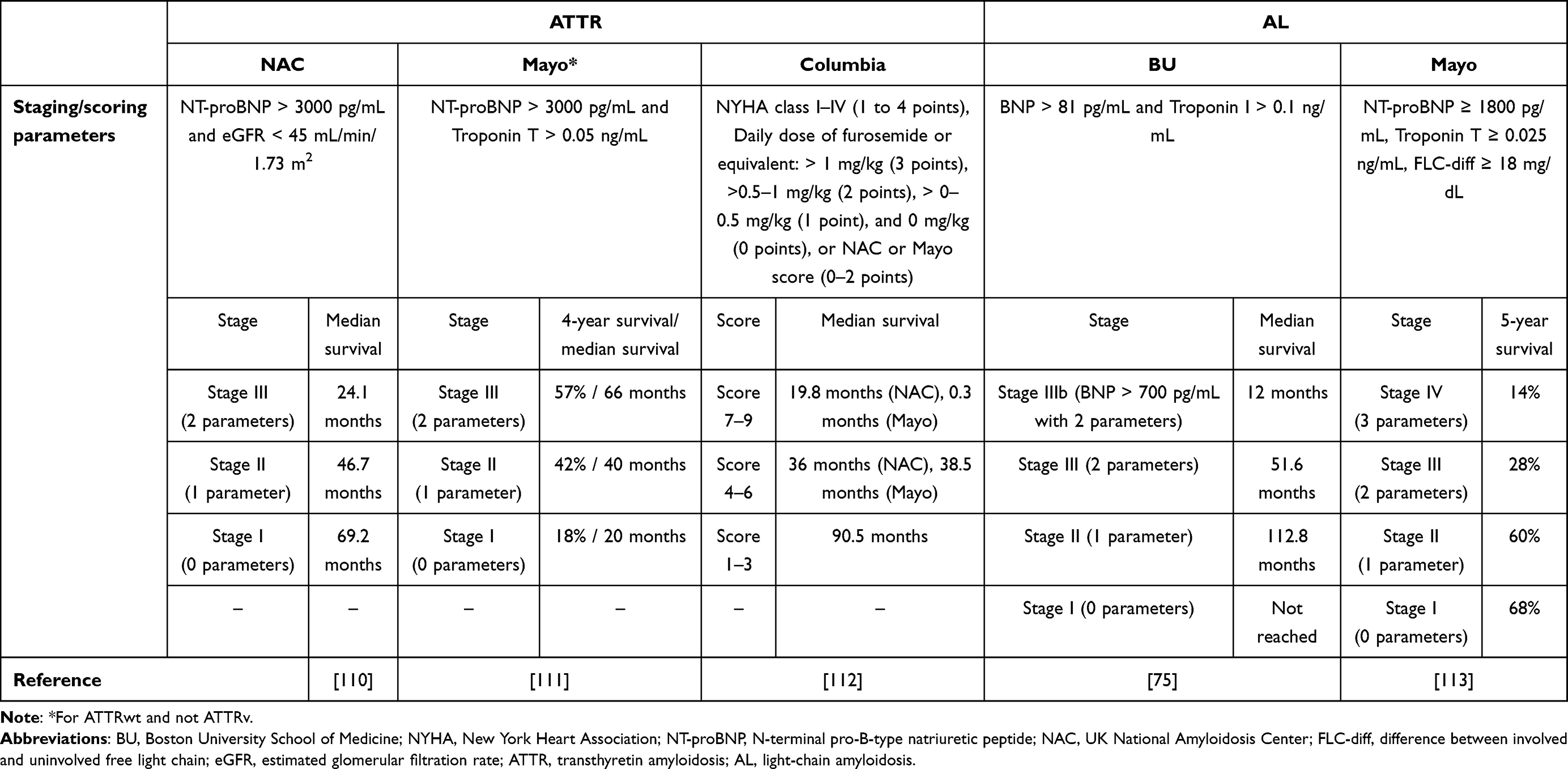 |
Table 2 Current Prognostic Staging Scores for ATTR and AL CA |
It should be noted that the recommended follow-up scheme varies between ATTR CA and AL CA. Clinical and laboratory hematological and cardiological evaluation, serum-free light chain quantification, NT-proBNP, troponin, basic biochemistry, and complete blood count are advisable every month. After completing the initial hematological management, patients are advised to be evaluated by a hematologist together with conducting the above-mentioned tests every 3–4 months. Moreover, echocardiography/CMR should be conducted every 6 months and 24-hour Holter ECG, and a cardiologist should evaluate patients. On the other hand, blood parameters (including troponin and NT-proBNP), ECG, and neurological assessment (in cases with ATTRv) are recommended every 6 months. Moreover, KCCQ and 6MWD are optional during this period. Every year, patients are advised to undergo 24-hour Holter ECG, echocardiography/CMR, and ophthalmological evaluation (in cases with ATTRv). This annual scheme should be done either for diagnosed or asymptomatic genetic carriers. Finally, CMR and scintigraphy should be considered every 3 years in cases of abnormal tests.
It is also important to consider screening for CA in relatives by genetic testing, which should be carried out together with genetic counseling. Current evidence furtherly shows that genetic testing for minors is not recommended since hereditary CA is an adult-onset disease. However, early adulthood genetic counseling might be done in cases of reproductive plans and when counseling can be suggestive of disease risk. The evaluation of penetrance in allele carriers should be initiated as early as CA-suggestive symptoms develop or around ten years before the estimated onset of the condition in family individuals or other corresponding patients and families.
Future Directions
As the field of CA treatment is continuously evolving, and new therapeutics are being investigated, unsolved questions and new challenges arise for future investigations. These challenges can be found in different areas related to CA, including pathophysiology, diagnosis, natural history, disease-modifying drugs, and treatment of complications. Current and future studies are planning to identify the safety and efficacy of novel anti-plasma cell therapeutics and other treatments for AL CA and new gene silencers and stabilizers for ATTR CA. The impact of early treatment initiation on ATTR CA without heart failure and ATTRv mutation carriers without phenotype is yet to be investigated, as well. Other areas might also include identifying the criteria upon which patients can switch from one regimen to another, adequately comparing the efficacy, safety, and cost-effectiveness of different disease-modifying modalities, the impact of combined therapy, genetic editing therapeutics, and the potential applications of antibodies in inducing amyloid deposits removal from tissues.
Conclusion
CA is a very unusual and complicated disease that manifests in the heart and other organs. It develops through an abnormal buildup of proteins in the heart, and it may have devastating effects. Few treatment options exist for CA, which also vary widely depending on the specific form of amyloidosis. Chemotherapy and ASCT are standard treatments for AL amyloidosis, whereas drugs to alleviate symptoms and halt the disease’s development are used in cases of ATTR amyloidosis. The degree of symptoms and the kind of amyloidosis determine the prognosis for CA. The appropriate management of the disease should be done in a multi-disciplinary approach to enhance the prognosis. Clinicians should consider all the extracardiac and cardiac manifestations when evaluating the affected patient to enhance the prognosis. Moreover, a thorough neurological evaluation should be considered during the initial diagnostic setting and the prognostic follow-up procedures. The median survival period for subjects with AL amyloidosis is fewer than two years. Complications from both forms of CA, such as heart failure and arrhythmias, may drastically shorten patients’ expected lifespans.
Several issues remain unresolved in CA care, despite recent breakthroughs in therapy. For instance, CA has no known cure, and therapeutic options are typically inadequate. It is crucial to note that early diagnosis and vigorous treatment are vital in improving outcomes for people with this ailment while research continues into novel therapies and diagnostic methods for this condition. However, the lack of particular diagnostic testing makes diagnosis challenging. There are also numerous unanswered questions concerning the best way to treat this disorder and decelerate its growth. There is optimism that people with cardiac amyloidosis may have better results in the future because of increased access to more effective therapies thanks to ongoing research into potential cures and better diagnostic tools.
Data Sharing Statement
Requested data will be provided on contacting the corresponding author.
Funding
This study was supported by Jilin Provincial Health Commission (No. 2022LC038), for comparative study of ultrasound-guided percutaneous coronary intervention between distal radial artery and radial artery.
Disclosure
The authors declare no conflict of interest.
References
1. Nativi-Nicolau J, Maurer MS. Amyloidosis cardiomyopathy: update in the diagnosis and treatment of the most common types. Curr Opin Cardiol. 2018;33(5):571–579. doi:10.1097/HCO.0000000000000547
2. Griffin JM, Rosenblum H, Maurer MS. Pathophysiology and therapeutic approaches to cardiac amyloidosis. Circ Res. 2021;128(10):1554–1575. doi:10.1161/CIRCRESAHA.121.318187
3. Macedo AVS, Schwartzmann PV, de Gusmão BM, et al. Advances in the treatment of cardiac amyloidosis. Curr Treat Options Oncol. 2020;21(5):36. doi:10.1007/s11864-020-00738-8
4. Lane T, Fontana M, Martinez-Naharro A, et al. Natural history, quality of life, and outcome in cardiac transthyretin amyloidosis. Circulation. 2019;140(1):16–26. doi:10.1161/CIRCULATIONAHA.118.038169
5. Knight DS, Zumbo G, Barcella W, et al. Cardiac structural and functional consequences of amyloid deposition by cardiac magnetic resonance and echocardiography and their prognostic roles. JACC Cardiovasc Imaging. 2019;12(5):823–833. doi:10.1016/j.jcmg.2018.02.016
6. Merlini G, Dispenzieri A, Sanchorawala V, et al. Systemic immunoglobulin light chain amyloidosis. Nat Rev Dis Primers. 2018;4(1):38. doi:10.1038/s41572-018-0034-3
7. Manolis AS, Manolis AA, Manolis TA, et al. Cardiac amyloidosis: an underdiagnosed/underappreciated disease. Eur J Intern Med. 2019;67:1–13. doi:10.1016/j.ejim.2019.07.022
8. Garcia-Pavia P, Rapezzi C, Adler Y, et al. Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J. 2021;42(16):1554–1568. doi:10.1093/eurheartj/ehab072
9. Pour-Ghaz I, Bath A, Kayali S, et al. A review of cardiac amyloidosis: presentation, diagnosis, and treatment. Curr Probl Cardiol. 2022;47(12):101366. doi:10.1016/j.cpcardiol.2022.101366
10. Schulman A, Connors LH, Weinberg J, et al. Patient outcomes in light chain (AL) amyloidosis: the clock is ticking from symptoms to diagnosis. Eur J Haematol. 2020;105(4):495–501. doi:10.1111/ejh.13472
11. Gertz M, Adams D, Ando Y, et al. Avoiding misdiagnosis: expert consensus recommendations for the suspicion and diagnosis of transthyretin amyloidosis for the general practitioner. BMC Fam Pract. 2020;21(1):198. doi:10.1186/s12875-020-01252-4
12. Russo M, Gentile L, Di Stefano V, et al. Use of drugs for ATTRv amyloidosis in the real world: how therapy is changing survival in a non-endemic area. Brain Sci. 2021;11(5):545. doi:10.3390/brainsci11050545
13. Maleszewski JJ. Cardiac amyloidosis: pathology, nomenclature, and typing. Cardiovasc Pathol. 2015;24(6):343–350. doi:10.1016/j.carpath.2015.07.008
14. Boldrini M, Cappelli F, Chacko L, et al. Multiparametric echocardiography scores for the diagnosis of cardiac amyloidosis. JACC Cardiovasc Imaging. 2020;13(4):909–920. doi:10.1016/j.jcmg.2019.10.011
15. Gillmore JD, Maurer MS, Falk RH, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133(24):2404–2412. doi:10.1161/CIRCULATIONAHA.116.021612
16. Palladini G, Russo P, Bosoni T, et al. Identification of amyloidogenic light chains requires the combination of serum-free light chain assay with immunofixation of serum and urine. Clin Chem. 2009;55(3):499–504. doi:10.1373/clinchem.2008.117143
17. Sprangers B, Claes K, Evenepoel P, et al. Comparison of 2 serum-free light-chain assays in CKD patients. Kidney Int Rep. 2020;5(5):627–631. doi:10.1016/j.ekir.2020.01.019
18. Layoun ME, Desmarais J, Heitner SB, et al. Hot hearts on bone scintigraphy are not all amyloidosis: hydroxychloroquine-induced restrictive cardiomyopathy. Eur Heart J. 2020;41(25):2414. doi:10.1093/eurheartj/ehaa091
19. López-Sainz Á, Hernandez-Hernandez A, Gonzalez-Lopez E, et al. Clinical profile and outcome of cardiac amyloidosis in a Spanish referral center. Rev Esp Cardiol. 2021;74(2):149–158. doi:10.1016/j.recesp.2019.12.017
20. Castaño A, Narotsky DL, Hamid N, et al. Unveiling transthyretin cardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoing transcatheter aortic valve replacement. Eur Heart J. 2017;38(38):2879–2887. doi:10.1093/eurheartj/ehx350
21. Damy T, Costes B, Hagège AA, et al. Prevalence and clinical phenotype of hereditary transthyretin amyloid cardiomyopathy in patients with increased left ventricular wall thickness. Eur Heart J. 2016;37(23):1826–1834. doi:10.1093/eurheartj/ehv583
22. González-López E, Gallego-Delgado M, Guzzo-Merello G, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015;36(38):2585–2594. doi:10.1093/eurheartj/ehv338
23. Di Stefano V, Fava A, Gentile L, et al. Italian real-life experience of patients with hereditary transthyretin amyloidosis treated with patisiran. Pharmgenomics Pers Med. 2022;15:499–514. doi:10.2147/PGPM.S359851
24. Di Lisi D, Di Caccamo L, Damerino G, et al. Effectiveness and safety of oral anticoagulants in cardiac amyloidosis: lights and shadows. Curr Probl Cardiol. 2023;48(8):101188. doi:10.1016/j.cpcardiol.2022.101188
25. Di Lisi D, Di Stefano V, Brighina F, et al. Therapy of ATTR cardiac amyloidosis: current indications. Curr Probl Cardiol. 2023;48(2):101487. doi:10.1016/j.cpcardiol.2022.101487
26. Aus Dem Siepen F, Bauer R, Aurich M, et al. Green tea extract as a treatment for patients with wild-type transthyretin amyloidosis: an observational study. Drug Des Devel Ther. 2015;9:6319–6325. doi:10.2147/DDDT.S96893
27. Benson MD, Dasgupta NR, Rissing SM, et al. Safety and efficacy of a TTR specific antisense oligonucleotide in patients with transthyretin amyloid cardiomyopathy. Amyloid. 2017;24(4):219–225. doi:10.1080/13506129.2017.1374946
28. Castaño A, Helmke S, Alvarez J, et al. Diflunisal for ATTR cardiac amyloidosis. Congest Heart Failure. 2012;18(6):315–319. doi:10.1111/j.1751-7133.2012.00303.x
29. Dasgupta NR, Rissing SM, Smith J, et al. Inotersen therapy of transthyretin amyloid cardiomyopathy. Amyloid. 2020;27(1):52–58. doi:10.1080/13506129.2019.1685487
30. Ikram A, Donnelly JP, Sperry BW, et al. Diflunisal tolerability in transthyretin cardiac amyloidosis: a single center’s experience. Amyloid. 2018;25(3):197–202. doi:10.1080/13506129.2018.1519507
31. Kristen AV, Lehrke S, Buss S, et al. Green tea halts progression of cardiac transthyretin amyloidosis: an observational report. Clin Res Cardiol. 2012;101(10):805–813. doi:10.1007/s00392-012-0463-z
32. Minamisawa M, Claggett B, Adams D, et al. Association of patisiran, an RNA interference therapeutic, with regional left ventricular myocardial strain in hereditary transthyretin amyloidosis: the APOLLO Study. JAMA Cardiol. 2019;4(5):466–472. doi:10.1001/jamacardio.2019.0849
33. Obici L, Cortese A, Lozza A, et al. Doxycycline plus tauroursodeoxycholic acid for transthyretin amyloidosis: a phase II study. Amyloid. 2012;19 Suppl 1:34–36.
34. Solomon SD, Adams D, Kristen A, et al. Effects of patisiran, an RNA interference therapeutic, on cardiac parameters in patients with hereditary transthyretin-mediated amyloidosis. Circulation. 2019;139(4):431–443. doi:10.1161/CIRCULATIONAHA.118.035831
35. Wixner J, Pilebro B, Lundgren H-E, et al. Effect of doxycycline and ursodeoxycholic acid on transthyretin amyloidosis. Amyloid. 2017;24(sup1):78–79. doi:10.1080/13506129.2016.1269739
36. Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379(11):1007–1016. doi:10.1056/NEJMoa1805689
37. Elliott P, Drachman BM, Gottlieb SS, et al. Long-term survival with tafamidis in patients with transthyretin amyloid cardiomyopathy. Circ Heart Fail. 2022;15(1):e008193. doi:10.1161/CIRCHEARTFAILURE.120.008193
38. Sekijima Y, Dendle MA, Kelly JW. Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid. 2006;13(4):236–249. doi:10.1080/13506120600960882
39. Wixner J, Westermark P, Ihse E, et al. The Swedish open-label diflunisal trial (DFNS01) on hereditary transthyretin amyloidosis and the impact of amyloid fibril composition. Amyloid. 2019;26(sup1):39–40. doi:10.1080/13506129.2019.1593133
40. Sekijima Y, Tojo K, Morita H, et al. Safety and efficacy of long-term diflunisal administration in hereditary transthyretin (ATTR) amyloidosis. Amyloid. 2015;22(2):79–83. doi:10.3109/13506129.2014.997872
41. Rosenblum H, Castano A, Alvarez J, et al. TTR (Transthyretin) stabilizers are associated with improved survival in patients with TTR cardiac amyloidosis. Circ Heart Fail. 2018;11(4):e004769. doi:10.1161/CIRCHEARTFAILURE.117.004769
42. Ji AX, Wong PW, Betz A, et al. Abstract 13847: differential transthyretin binding, kinetic stability and additive ex vivo stabilization by AG10 compared to tafamidis. Circulation. 2019;140(Suppl_1):A13847.
43. Kazi DS, Bellows BK, Baron SJ, et al. Cost-effectiveness of tafamidis therapy for transthyretin amyloid cardiomyopathy. Circulation. 2020;141(15):1214–1224. doi:10.1161/CIRCULATIONAHA.119.045093
44. Benson MD, Waddington-Cruz M, Berk JL, et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):22–31. doi:10.1056/NEJMoa1716793
45. Di Stefano V, Thomas E, Alonge P, et al. Patisiran enhances muscle mass after nine months of treatment in ATTRv amyloidosis: a study with bioelectrical impedance analysis and handgrip strength. Biomedicines. 2022;11(1):62. doi:10.3390/biomedicines11010062
46. Adams D, Gonzalez-Duarte A, O’Riordan WD, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):11–21. doi:10.1056/NEJMoa1716153
47. Habtemariam BA, Karsten V, Attarwala H, et al. Single-dose pharmacokinetics and pharmacodynamics of transthyretin targeting N-acetylgalactosamine-small interfering ribonucleic acid conjugate, vutrisiran, in healthy subjects. Clin Pharmacol Ther. 2021;109(2):372–382. doi:10.1002/cpt.1974
48. Holmgren G, Steen L, Ekstedt J, et al. Biochemical effect of liver transplantation in two Swedish patients with familial amyloidotic polyneuropathy (FAP-met30). Clin Genet. 1991;40(3):242–246. doi:10.1111/j.1399-0004.1991.tb03085.x
49. Soprano DR, Herbert J, Soprano KJ, et al. Demonstration of transthyretin mRNA in the brain and other extrahepatic tissues in the rat. J Biol Chem. 1985;260(21):11793–11798. doi:10.1016/S0021-9258(17)39100-7
50. Adams D, Tournev IL, Taylor MS, et al. Efficacy and safety of vutrisiran for patients with hereditary transthyretin-mediated amyloidosis with polyneuropathy: a randomized clinical trial. Amyloid. 2023;30(1):18–26. doi:10.1080/13506129.2022.2091985
51. Judge DP, Kristen AV, Grogan M, et al. Phase 3 multicenter study of revusiran in patients with hereditary transthyretin-mediated (hATTR) amyloidosis with cardiomyopathy (ENDEAVOUR). Cardiovasc Drugs Ther. 2020;34(3):357–370. doi:10.1007/s10557-019-06919-4
52. Gillmore JD, Gane E, Taubel J, et al. CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N Engl J Med. 2021;385(6):493–502. doi:10.1056/NEJMoa2107454
53. Lim GB. Gene editing in patients with amyloidosis. Nat Rev Cardiol. 2021;18(9):611.
54. Cardoso I, Martins D, Ribeiro T, et al. Synergy of combined doxycycline/TUDCA treatment in lowering transthyretin deposition and associated biomarkers: studies in FAP mouse models. J Transl Med. 2010;8(1):74. doi:10.1186/1479-5876-8-74
55. Karlstedt E, Jimenez-Zepeda V, Howlett JG, et al. Clinical experience with the use of doxycycline and ursodeoxycholic acid for the treatment of transthyretin cardiac amyloidosis. J Card Fail. 2019;25(3):147–153. doi:10.1016/j.cardfail.2019.01.006
56. Stern LK, Patel J. Cardiac amyloidosis treatment. Methodist Debakey Cardiovasc J. 2022;18(2):59–72. doi:10.14797/mdcvj.1050
57. Michalon A, Hagenbuch A, Huy C, et al. A human antibody selective for transthyretin amyloid removes cardiac amyloid through phagocytic immune cells. Nat Commun. 2021;12(1):3142. doi:10.1038/s41467-021-23274-x
58. Garcia-Pavia P, Aus Dem Siepen F, Donal E, et al. Phase 1 trial of antibody NI006 for depletion of cardiac transthyretin amyloid. N Engl J Med. 2023. doi:10.1056/NEJMoa2303765
59. Galant NJ, Bugyei-Twum A, Rakhit R, et al. Substoichiometric inhibition of transthyretin misfolding by immune-targeting sparsely populated misfolding intermediates: a potential diagnostic and therapeutic for TTR amyloidoses. Sci Rep. 2016;6(1):25080. doi:10.1038/srep25080
60. Richards DB, Cookson LM, Berges AC, et al. Therapeutic clearance of amyloid by antibodies to serum amyloid P component. N Engl J Med. 2015;373(12):1106–1114. doi:10.1056/NEJMoa1504942
61. Ferreira N, Saraiva MJ, Almeida MR, Ferreira ST. Epigallocatechin-3-gallate as a potential therapeutic drug for TTR-related amyloidosis: “in vivo” evidence from FAP mice models. PLoS One. 2012;7(1):e29933. doi:10.1371/journal.pone.0029933
62. Ferreira N, Cardoso I, Domingues MR, et al. Binding of epigallocatechin-3-gallate to transthyretin modulates its amyloidogenicity. FEBS Lett. 2009;583(22):3569–3576. doi:10.1016/j.febslet.2009.10.062
63. Aus Dem Siepen F, Buss SJ, Andre F, et al. Extracellular remodeling in patients with wild-type amyloidosis consuming epigallocatechin-3-gallate: preliminary results of T1 mapping by cardiac magnetic resonance imaging in a small single center study. Clin Res Cardiol. 2015;104(8):640–647. doi:10.1007/s00392-015-0826-3
64. Palladini G, Milani P, Merlini G. Management of AL amyloidosis in 2020. Blood. 2020;136(23):2620–2627. doi:10.1182/blood.2020006913
65. Palladini G, Dispenzieri A, Gertz MA, et al. New criteria for response to treatment in immunoglobulin light chain amyloidosis based on free light chain measurement and cardiac biomarkers: impact on survival outcomes. J Clin Oncol. 2012;30(36):4541–4549. doi:10.1200/JCO.2011.37.7614
66. Wechalekar AD, Fontana M, Quarta CC, et al. AL amyloidosis for cardiologists: awareness, diagnosis, and future prospects: JACC: cardioOncology state-of-the-art review. JACC CardioOncol. 2022;4(4):427–441. doi:10.1016/j.jaccao.2022.08.009
67. Mohty D, Damy T, Cosnay P, et al. Cardiac amyloidosis: updates in diagnosis and management. Arch Cardiovasc Dis. 2013;106(10):528–540. doi:10.1016/j.acvd.2013.06.051
68. Pankuweit S, Dörr R. Diagnosis and treatment of cardiac amyloidosis: a position statement of the ESC working group on myocardial and pericardial diseases 2021. Herz. 2022;47(1):41–47. doi:10.1007/s00059-021-05085-4
69. Kastritis E, Palladini G, Minnema MC, et al. Daratumumab-based treatment for immunoglobulin light-chain amyloidosis. N Engl J Med. 2021;385(1):46–58. doi:10.1056/NEJMoa2028631
70. Sidiqi MH, Aljama MA, Buadi FK, et al. Stem cell transplantation for light chain amyloidosis: decreased early mortality over time. J Clin Oncol. 2018;36(13):1323–1329. doi:10.1200/JCO.2017.76.9554
71. Poczta A, Rogalska A, Marczak A. Treatment of multiple myeloma and the role of melphalan in the era of modern therapies-current research and clinical approaches. J Clin Med. 2021;10(9):1841. doi:10.3390/jcm10091841
72. Manwani R, Foard D, Mahmood S, et al. Rapid hematologic responses improve outcomes in patients with very advanced (stage IIIb) cardiac immunoglobulin light chain amyloidosis. Haematologica. 2018;103(4):e165–e168. doi:10.3324/haematol.2017.178095
73. Martinez-Naharro A, Abdel-Gadir A, Treibel TA, et al. Abstract 14407: regression of cardiac AL amyloid by cardiovascular magnetic resonance. Circulation. 2016;134(suppl_1):A14407.
74. Salinaro F, Meier-Ewert HK, Miller EJ, et al. Longitudinal systolic strain, cardiac function improvement, and survival following treatment of light-chain (AL) cardiac amyloidosis. Eur Heart J Cardiovasc Imaging. 2017;18(9):1057–1064. doi:10.1093/ehjci/jew298
75. Lilleness B, Ruberg FL, Mussinelli R, et al. Development and validation of a survival staging system incorporating BNP in patients with light chain amyloidosis. Blood. 2019;133(3):215–223. doi:10.1182/blood-2018-06-858951
76. Kastritis E, Leleu X, Arnulf B, et al. Bortezomib, melphalan, and dexamethasone for light-chain amyloidosis. J Clin Oncol. 2020;38(28):3252–3260. doi:10.1200/JCO.20.01285
77. Manwani R, Mahmood S, Sachchithanantham S, et al. Carfilzomib is an effective upfront treatment in AL amyloidosis patients with peripheral and autonomic neuropathy. Br J Haematol. 2019;187(5):638–641. doi:10.1111/bjh.16122
78. Premkumar VJ, Lentzsch S, Pan S, et al. Venetoclax induces deep hematologic remissions in t(11;14) relapsed/refractory AL amyloidosis. Blood Cancer J. 2021;11(1):10. doi:10.1038/s41408-020-00397-w
79. Wechalekar AD, Sanchorawala V. Daratumumab in AL amyloidosis. Blood. 2022;140(22):2317–2322. doi:10.1182/blood.2021014613
80. Ruberg FL, Grogan M, Hanna M, et al. Transthyretin amyloid cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol. 2019;73(22):2872–2891. doi:10.1016/j.jacc.2019.04.003
81. Gertz MA. Hereditary ATTR amyloidosis: burden of illness and diagnostic challenges. Am J Manag Care. 2017;23(7 Suppl):S107–S112.
82. Sirac C, Jaccard A, Codo R, et al. Pre-clinical characterization of a novel fusion protein (AT-03), with pan-amyloid binding and removal. Blood. 2021;138(Supplement 1):1207. doi:10.1182/blood-2021-151908
83. Wall JS, Kennel SJ, Williams A, et al. AL amyloid imaging and therapy with a monoclonal antibody to a cryptic epitope on amyloid fibrils. PLoS One. 2012;7(12):e52686. doi:10.1371/journal.pone.0052686
84. O’Nuallain B, Allen A, Kennel SJ, et al. Localization of a conformational epitope common to non-native and fibrillar immunoglobulin light chains. Biochemistry. 2007;46(5):1240–1247. doi:10.1021/bi0616605
85. Gertz MA, Landau H, Comenzo RL, et al. First-in-human Phase I/II study of NEOD001 in patients with light chain amyloidosis and persistent organ dysfunction. J Clin Oncol. 2016;34(10):1097–1103. doi:10.1200/JCO.2015.63.6530
86. Bal S, Landau H. AL amyloidosis: untangling new therapies. Hematology Am Soc Hematol Educ Program. 2021;2021(1):682–688. doi:10.1182/hematology.2021000305
87. Gertz MA, Cohen AD, Comenzo RL, et al. Results of the Phase 3 VITAL study of NEOD001 (Birtamimab) plus standard of care in patients with light chain (AL) amyloidosis suggest survival benefit for mayo stage IV patients. Blood. 2019;134(Supplement_1):3166. doi:10.1182/blood-2019-124482
88. Gertz MA, Landau HJ, Weiss BM. Organ response in patients with AL amyloidosis treated with NEOD001, an amyloid-directed monoclonal antibody. Am J Hematol. 2016;91(12):E506–E508. doi:10.1002/ajh.24563
89. Wall JS, Kennel SJ, Stuckey AC, et al. Radioimmunodetection of amyloid deposits in patients with AL amyloidosis. Blood. 2010;116(13):2241–2244. doi:10.1182/blood-2010-03-273797
90. Solomon A, Weiss DT, Wall JS. Immunotherapy in systemic primary (AL) amyloidosis using amyloid-reactive monoclonal antibodies. Cancer Biother Radiopharm. 2003;18(6):853–860. doi:10.1089/108497803322702824
91. Solomon A, Weiss DT, Wall JS. Therapeutic potential of chimeric amyloid-reactive monoclonal antibody 11-1F4. Clin Cancer Res. 2003;9(10 Pt 2):3831s–8s.
92. Edwards CV, Rao N, Bhutani D, et al. Phase 1a/b study of monoclonal antibody CAEL-101 (11-1F4) in patients with AL amyloidosis. Blood. 2021;138(25):2632–2641. doi:10.1182/blood.2020009039
93. Edwards CV, Gould J, Langer AL, et al. Interim analysis of the phase 1a/b study of chimeric fibril-reactive monoclonal antibody 11-1F4 in patients with AL amyloidosis. Amyloid. 2017;24(sup1):58–59. doi:10.1080/13506129.2017.1292900
94. Diomede L, Rognoni P, Lavatelli F, et al. A caenorhabditis elegans-based assay recognizes immunoglobulin light chains causing heart amyloidosis. Blood. 2014;123(23):3543–3552. doi:10.1182/blood-2013-10-525634
95. Ward JE, Ren R, Toraldo G, et al. Doxycycline reduces fibril formation in a transgenic mouse model of AL amyloidosis. Blood. 2011;118(25):6610–6617. doi:10.1182/blood-2011-04-351643
96. Kumar SK, Dispenzieri A, Lacy MQ, et al. Doxycycline used as post transplant antibacterial prophylaxis improves survival in patients with light chain amyloidosis undergoing autologous stem cell transplantation. Blood. 2012;120(21):3138. doi:10.1182/blood.V120.21.3138.3138
97. Wechalekar AD, Whelan C. Encouraging impact of doxycycline on early mortality in cardiac light chain (AL) amyloidosis. Blood Cancer J. 2017;7(3):e546. doi:10.1038/bcj.2017.26
98. Shen KN, Fu WJ, Wu Y, et al. Doxycycline combined with bortezomib-cyclophosphamide-dexamethasone chemotherapy for newly diagnosed cardiac light-chain amyloidosis: a multicenter randomized controlled trial. Circulation. 2022;145(1):8–17. doi:10.1161/CIRCULATIONAHA.121.055953
99. Ehrnhoefer DE, Bieschke J, Boeddrich A, et al. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat Struct Mol Biol. 2008;15(6):558–566. doi:10.1038/nsmb.1437
100. Zhang N, Yan C, Yin C, et al. Structural remodeling mechanism of the toxic amyloid fibrillary mediated by epigallocatechin-3-gallate. ACS Omega. 2022;7(51):48047–48058. doi:10.1021/acsomega.2c05995
101. Meshitsuka S, Shingaki S, Hotta M, et al. Phase 2 trial of daily, oral epigallocatechin gallate in patients with light-chain amyloidosis. Int J Hematol. 2017;105(3):295–308. doi:10.1007/s12185-016-2112-1
102. Valent J, Silowsky J, Kurman MR, et al. Cael-101 is well-tolerated in AL amyloidosis patients receiving concomitant Cyclophosphamide-Bortezomib-Dexamethasone (CyborD): a phase 2 dose-finding study (NCT04304144). Blood. 2020;136(Supplement 1):26–27. doi:10.1182/blood-2020-139323
103. Sanchorawala V, Sarosiek S, Schulman A, et al. Safety, tolerability, and response rates of daratumumab in relapsed AL amyloidosis: results of a phase 2 study. Blood. 2020;135(18):1541–1547. doi:10.1182/blood.2019004436
104. Roussel M, Merlini G, Chevret S, et al. A prospective phase 2 trial of daratumumab in patients with previously treated systemic light-chain amyloidosis. Blood. 2020;135(18):1531–1540. doi:10.1182/blood.2019004369
105. Warsame R, LaPlant B, Kumar SK, et al. Long-term outcomes of IMiD-based trials in patients with immunoglobulin light-chain amyloidosis: a pooled analysis. Blood Cancer J. 2020;10(1):4. doi:10.1038/s41408-019-0266-9
106. Dispenzieri A, Dingli D, Kumar SK, et al. Discordance between serum cardiac biomarker and immunoglobulin-free light-chain response in patients with immunoglobulin light-chain amyloidosis treated with immune modulatory drugs. Am J Hematol. 2010;85(10):757–759. doi:10.1002/ajh.21822
107. Dispenzieri A, Kastritis E, Wechalekar AD, et al. Primary results from the phase 3 tourmaline-AL1 trial of ixazomib-dexamethasone versus physician’s choice of therapy in patients (Pts) with relapsed/Refractory Primary Systemic AL Amyloidosis (RRAL). Blood. 2019;134:139. doi:10.1182/blood-2019-124409
108. Milani P, Schönland S, Merlini G, et al. Treatment of AL amyloidosis with bendamustine: a study of 122 patients. Blood. 2018;132(18):1988–1991. doi:10.1182/blood-2018-04-845396
109. Lentzsch S, Lagos GG, Comenzo RL, et al. Bendamustine with dexamethasone in relapsed/refractory systemic light-chain amyloidosis: results of a Phase II study. J Clin Oncol. 2020;38(13):1455–1462. doi:10.1200/JCO.19.01721
110. Gillmore JD, Damy T, Fontana M, et al. A new staging system for cardiac transthyretin amyloidosis. Eur Heart J. 2018;39(30):2799–2806. doi:10.1093/eurheartj/ehx589
111. Grogan M, Scott CG, Kyle RA, et al. Natural history of wild-type transthyretin cardiac amyloidosis and risk stratification using a novel staging system. J Am Coll Cardiol. 2016;68(10):1014–1020. doi:10.1016/j.jacc.2016.06.033
112. Cheng RK, Levy WC, Vasbinder A, et al. Diuretic dose and NYHA functional class are independent predictors of mortality in patients with transthyretin cardiac amyloidosis. JACC CardioOncol. 2020;2(3):414–424. doi:10.1016/j.jaccao.2020.06.007
113. Kumar S, Dispenzieri A, Lacy MQ, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol. 2012;30(9):989–995. doi:10.1200/JCO.2011.38.5724
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Updated Perspectives on the Diagnosis and Management of Onychomycosis
Falotico JM, Lipner SR
Clinical, Cosmetic and Investigational Dermatology 2022, 15:1933-1957
Published Date: 15 September 2022
Breast Cancer in Geriatric Patients: Current Landscape and Future Prospects
Abdel-Razeq H, Abu Rous F, Abuhijla F, Abdel-Razeq N, Edaily S
Clinical Interventions in Aging 2022, 17:1445-1460
Published Date: 28 September 2022
Insight into Dysmenorrhea Research from 1992 to 2022: A Bibliometric Analysis
Dong Y, Li MJ, Hong YZ, Li WJ
Journal of Pain Research 2023, 16:3591-3611
Published Date: 27 October 2023