Back to Journals » Journal of Hepatocellular Carcinoma » Volume 13
SPP1+ Macrophages Promote Immunosuppression in Post-TACE Hepatocellular Carcinoma Involving Lactate-Induced Lactylation and Potential SPP1-CD44 Signaling
Authors Wang H ![]() , Liang G
, Liang G ![]() , Liu T, He M, Jiang Y, Li J, Ouyang Y, Hao S, Liu W, Li J
, Liu T, He M, Jiang Y, Li J, Ouyang Y, Hao S, Liu W, Li J
Received 14 February 2026
Accepted for publication 25 June 2026
Published 8 July 2026 Volume 2026:13 603485
DOI https://doi.org/10.2147/JHC.S603485
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr David Gerber
Hongyu Wang,1,2,* Guojun Liang,3,* Ting Liu,3,4 Min He,5 Yang Jiang,2 Jinwei Li,1 Yushu Ouyang,1 Shufang Hao,1 Wendao Liu,1 Jiaping Li2,6
1Department of Interventional Therapy, Second Affiliated Hospital, Guangzhou University of Chinese Medicine, Guangzhou, People’s Republic of China; 2Department of Interventional Oncology, First Affiliated Hospital, Sun Yat-Sen University, Guangzhou, People’s Republic of China; 3Department of Laboratory Medicine, Second Clinical College, Guangzhou University of Chinese Medicine, Guangzhou, People’s Republic of China; 4Clinical Laboratory and State Key Laboratory of Traditional Chinese Medicine Syndrome, Guangdong Provincial Hospital of Chinese Medicine, Guangzhou, People’s Republic of China; 5Department of Laboratory Medicine, Affiliated Foshan Women and Children Hospital, Guangdong Medical University, Foshan, People’s Republic of China; 6Department of Minimally Invasive Interventional Therapy, Guangzhou Institute of Cancer Research, The Affiliated Cancer Hospital, Guangzhou Medical University, Guangzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Wendao Liu; Jiaping Li, Email [email protected]; [email protected]
Background: Transarterial chemoembolization (TACE) treats inoperable liver cancer by inducing tumor ischemia, but subsequent hypoxia can promote an immunosuppressive microenvironment. This study explores how TACE-related hypoxia influences immune cells through lactate metabolism and lactylation.
Methods: Single-cell RNA sequencing of TACE-treated and newly diagnosed liver cancer samples was performed. Scores were assigned to lactate-related gene sets, and differentially expressed genes were identified. Bioinformatics analysis integrated scRNA-seq, TCGA, and GEO data. Machine learning derived a consensus gene signature, and a prognostic nomogram was constructed. The study evaluated immune infiltration and drug sensitivity. Key findings were validated via spatial transcriptomics, in vitro experiments, and immunofluorescence.
Results: We identified seven distinct single-cell types, noting an increase in macrophage infiltration and a decrease in CD8+ T cells within the TACE group. A six-gene consensus signature related to lactate—comprising SPP1, DNASE1L3, HRG, TPX2, LAPTM4B, and G6PD—was identified and found to be significantly associated with poor prognosis. This consensus gene score and T, N staging constructed a prognostic nomogram validated by calibration curves and decision curves. We also found that SPP1+ macrophages were elevated in the TACE treatment group, with increased lactate-related gene set scores, and were associated with hypoxia, glycolysis, and inflammatory pathways. Cell communication analysis suggested a potential regulatory link between SPP1+ macrophages and CD8+ T cells, highlighting the potential involvement of the SPP1-CD44 axis. Cell experiments showed that hypoxic, lactate-rich environments lead to macrophage lactylation, increasing SPP1+ macrophages and decreasing CD8+ IFN-γ+ T cells. Spatial transcriptomics and immunofluorescence staining revealed co-localization of SPP1+ macrophages with CD8+ T cells.
Conclusion: We established a lactate-associated six-gene signature and prognostic nomogram. The immunosuppressive microenvironment following TACE in liver cancer is highly correlated with the activation of lactate-related genes and an increase in SPP1+ macrophages. These macrophages are closely associated with CD8+ T cells dysfunction, with the SPP1-CD44 pathway serving as a candidate narrative for this intercellular crosstalk. These findings suggest that targeting lactate-induced lactylation or the potential SPP1-CD44 interaction may offer promising therapeutic opportunities to counteract immune evasion after TACE therapy.
Keywords: macrophage, lactylation, SPP1, single-cell RNA sequencing, spatial transcriptomics, hepatocellular carcinoma, transarterial chemoembolization
Introduction
Liver cancer is an extremely aggressive tumor with high global incidence and mortality rates, posing a significant threat to patients’ lives and imposing a substantial economic burden on society.1,2 Transarterial chemoembolization (TACE) has become a primary treatment choice for liver cancer that cannot be surgically removed.3 It aims to kill tumor cells by blocking the tumor’s blood supply, leading to ischemia and necrosis. However, the complex blood supply of tumors makes complete embolization difficult. The tumor’s hypoxic microenvironment, worsened by hepatic artery embolization, increases local lactate accumulation, potentially impacting immune cell infiltration and function via metabolic reprogramming or protein post-translational modifications.4,5
Lactate, traditionally viewed as a metabolic byproduct, is now recognized as both an energy source and signaling molecule via the lactate shuttle mechanism.6,7 In 2019, Zhao et al8 first described lactylation—a novel post-translational modification in which L-lactate-derived lactyl-CoA is transferred to histone lysine residues, regulating gene transcription—and demonstrated its role in driving M2 macrophage polarization through a “lactate clock” mechanism. This landmark discovery established lactylation as a key link between metabolism and immune regulation. An extensive study of the lactylome and proteome in a hepatocellular carcinoma cohort identified 9275 lysine lactylation sites, with 9256 on non-histone proteins, indicating that lysine lactylation is a prevalent modification beyond histone proteins.9
Increasing evidence underscores the important role of lactylation, driven by the glycolytic byproduct lactate, in the advancement of liver disease.10 Lactylation, related to hypoxia and lactic acid buildup in the tumor microenvironment, is associated with macrophage polarization and immune suppression.11,12 Our earlier study discovered alterations in immune cell infiltration following TACE, including more macrophage infiltration and less CD8+ T cells infiltration, as well as diminished CD8+ T cells killing and proliferation abilities.13 Nevertheless, there is a significant research gap in understanding how the hypoxic micro-environment caused by TACE treatment affects immune cells from the perspectives of lactic acid metabolism and lactylation. Among tumor-associated macrophages, SPP1+ macrophages have emerged as a functionally distinct subset with potent immunosuppressive properties.14,15 SPP1 (Secreted Phosphoprotein 1, also known as Osteopontin) is a multifunctional matricellular protein implicated in tumor progression, angiogenesis, and immune evasion.16,17 Recent single-cell studies across multiple cancer types have identified SPP1+ macrophages as a conserved pro-tumorigenic population enriched in hypoxic tumor regions,18,19 where they interact with other immune cells — particularly CD8+ T cells — to suppress anti-tumor immunity.20,21 However, whether TACE treatment promotes the expansion or functional reprogramming of SPP1+ macrophages, and whether this process is mediated through lactate-driven lactylation, remains unknown.
We hypothesized that TACE-induced hypoxia and lactate accumulation promote SPP1+ macrophage infiltration and lactylation, which in turn suppress CD8+ T cells function via SPP1-CD44 interactions, contributing to an immunosuppressive microenvironment. To test this hypothesis, this study aims to address this knowledge gap by employing a multi-omics strategy (Figure 1). Elucidating these mechanisms may identify novel therapeutic targets — such as SPP1-CD44 blockade — and provide a rationale for combining TACE with immunotherapy to counteract treatment-induced immunosuppression, ultimately improving outcomes for HCC patients.
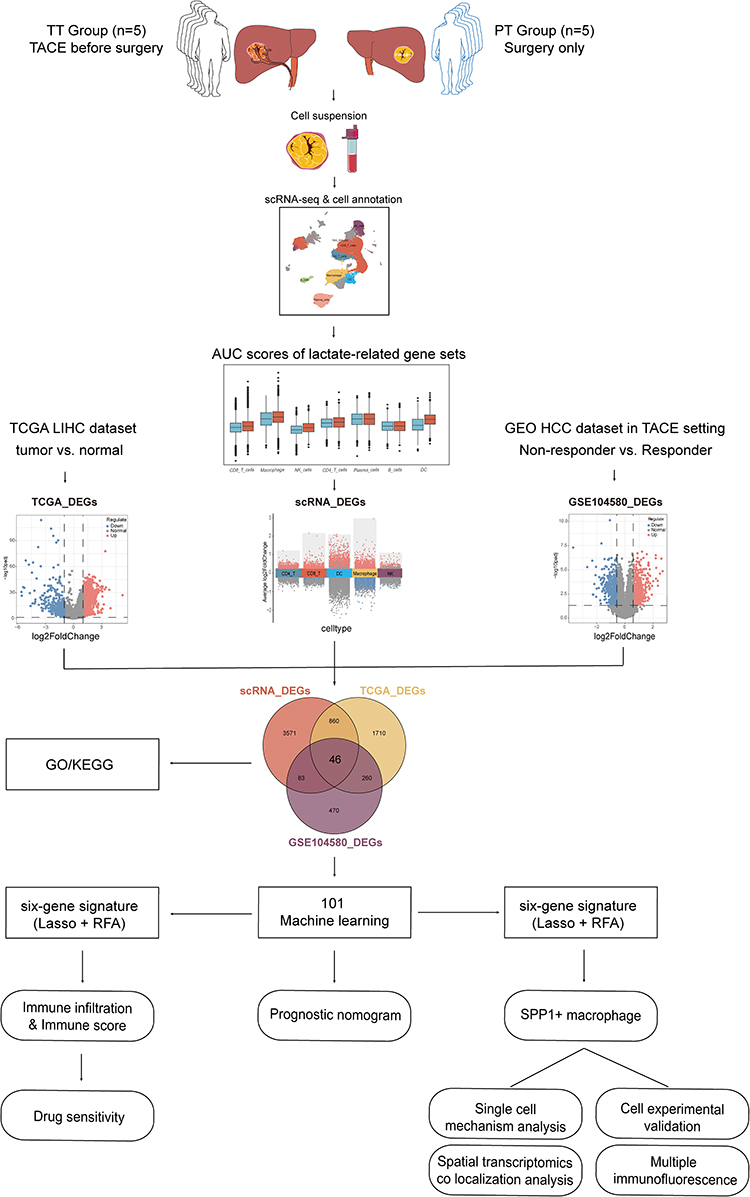 |
Figure 1 Workflow of our study. We annotated immune cells in liver cancer patients undergoing TACE treatment and in primary tumor groups, analyzed lactate-related gene set AUC scores, and identified differentially expressed genes (DEGs) between high and low scoring groups for each cell type. Second, we compared DEGs between the TCGA-LIHC and GSE104580 datasets, preliminarily identifying 46 intersecting genes from these three DEGs. Subsequently, we performed univariate Cox analysis on these 46 genes in the TCGA-LIHC dataset, finding 38 significant prognostic genes. And using 101 machine learning algorithms, we developed and validated a 6-gene signature on the TCGA-LIHC training set and ICGC, GSE14520 validation set. Kaplan–Meier analysis identified the model score as a risk indicator. We developed and validated a prognostic nomogram incorporating T stage, N stage, and risk score using the TCGA-LIHC dataset to demonstrate the clinical prognostic significance of the risk score. Utilizing the TCGA-LIHC dataset, we examined molecular mechanisms and drug screening recommendations for varying risk scores, taking into account immune cell infiltration, immune score, and drug sensitivity. Finally, we investigated molecular mechanisms further, focusing on SPP1+ macrophages in single cells, and conducted preliminary experimental verification. |
Materials and Methods
Sources of Data
Tumor samples from 10 HCC patients were collected for single-cell RNA sequencing (scRNA-seq) at the First Affiliated Hospital of Sun Yat-sen University, comprising five treatment-naive primary (PT) and five post-TACE (TT) patients. PT samples were collected via needle biopsy, and TT samples were acquired through surgical resection. The TT group patients received only DEB-TACE with doxorubicin. Fresh samples were processed into single-cell suspensions, sorted for CD45+ cells, and sequenced using the 10x Chromium Controller. Detailed methods are in our 2023 J Hepatol article.13
Public bulk RNA-seq data were obtained from TCGA, ICGC HCC, and GEO datasets GSE104580, GSE14520, and GSE202069. Spatial transcriptomics and matched scRNA-seq data were obtained from open resource platforms.19 The HCC patients included in this study all received PD-1 treatment and were divided into non-responders and responders based on efficacy. We selected patient #1 (non-responder) and patient #7 (responder) for spatial co-localization analysis. In addition, a lactate-related gene set (LRGS, 484 genes) was compiled by searching the GSEA and Genecards databases with keywords “lactate” and “lactylation” (Supplementary Box 1).
Processing of Single-Cell and Bulk RNA-Seq Data
The scRNA-seq data were preprocessed with the Seurat package (v.5.1.0) in R. Quality control criteria were set as nFeature_RNA between 500 and 6000, and percent.mt below 10%. Each sample matrix was normalized using SCTransform (v2), with mitochondrial percentage regressed out as a confounding variable. For data integration, the top 3000 integration features were selected using SelectIntegrationFeatures, and integration anchors were identified via reciprocal PCA (rPCA) with 30 dimensions using FindIntegrationAnchors. Data integration was performed using IntegrateData to remove batch effects across the 10 individual samples. Integration quality was assessed by visual inspection of UMAP embeddings before and after integration (colored by sample of origin), calculation of the local inverse Simpson’s index (LISI) to quantify sample mixing, and confirmation that known cell-type marker genes were preserved across samples. Post-integration, clustering was driven by cell-type identity rather than sample of origin, confirming successful batch correction. Dimensionality reduction was conducted on the top 2000 variable genes using principal component analysis with 30 components, followed by clustering at a resolution of 0.5. Doublets were removed using DoubletFinder (v2.0.4), and cells expressing multiple marker genes and non-immune cells were removed. Cell clusters were manually annotated based on literature13 and canonical marker genes. Following cell annotation, inter-group differences in cellular composition were evaluated using a generalized linear mixed model (GLMM). Multiple hypothesis testing was corrected using the Benjamini–Hochberg false discovery rate (FDR) procedure. Using the R package TCGAbiolinks (v2.30.4), TPM expression data for TCGA-LIHC was obtained and log2(TPM+1) normalized. Genes with 0 expression in 80% of samples were removed. Normal and tumor samples were identified, and survival and clinical data were matched for tumor samples, excluding those with missing survival information. Liver cancer gene expression data from the ICGC database and GEO database were similarly processed.
AUC Activity Scoring of Feature Gene Sets
The R package AUCell (version 1.26.0) was used to assess feature gene set activity in single-cell or spatial transcriptomics. AUC scores refer to the area under the curve calculated by the AUCell algorithm, which estimates the activity of a gene set in individual cells; higher scores indicate greater gene set activity. The Wilcoxon rank-sum test assesses differences in AUC scores across groups.
Differentially Expressed Genes Analysis and Functional Enrichment
Single-cell types were divided into high and low groups according to the median AUC score of the LRGS. Differentially expressed genes (DEGs) were identified between the groups using the FindMarkers function and the Wilcoxon test. Genes were selected as scRNA_DEGs based on criteria of |avg_log2FC| > 0.25 and p_val_adj < 0.05. In the TCGA-LIHC dataset, differentially expressed genes (DEGs) between tumor and normal groups were identified using the limma package, with criteria of |log2FC| > 1 and adjusted p-value < 0.05, and were labeled as TCGA_DEGs. Similarly, in the GSE104580 dataset, DEGs between treatment response and non-response groups were identified with |log2FC| > 0.585 and adjusted P < 0.05, labeled as GSE104580_DEGs.
Using ggvenn (v0.1.10), we identified overlapping genes among TCGA_DEGs, scRNA_DEGs, and GSE104580_DEGs. Gene Ontology (GO) analysis for Biological Process (BP), Cellular Component (CC), and Molecular Function (MF), along with Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis, was performed using ClusterProfiler (v4.10.1) to explore potential biological pathways. Enrichment results were considered significant if P-values were below 0.05.
Machine Learning & Prognostic Nomogram
In the TCGA-LIHC dataset, intersecting genes were analyzed for prognostic significance using univariate Cox regression from the survival R package (v3.6–4), applying a significance threshold of P<0.05 for overall survival (OS). Prognostic genes were employed to construct prognostic models using a published machine learning integration framework that systematically evaluates 10 algorithms — CoxBoost, elastic net (Enet), survival-SVM, Lasso, plsRcox, Ridge, random survival forest (RSF), stepwise Cox, SuperPC, and gradient boosting machine (GBM) — and their 101 combinations.22 The TCGA-LIHC cohort was used as the training set with 10-fold cross-validation, while the ICGC and GSE14520 cohorts served as fully independent external validation sets. Model performance was evaluated by the time-dependent AUC, and the final model was selected based on the highest average C-index across all three cohorts to ensure generalizability. Wilcoxon rank-sum test evaluated variations in risk scores among clinical features in the TCGA-LIHC training set. The survival R package performed univariate and multivariate Cox regression analyses to assess the prognostic significance of risk scores and clinical characteristics. Factors with a P-value less than 0.05 in the univariate Cox regression were included in the multivariate Cox regression analysis. The rms R package (Version 6.8–0) was utilized to develop a nomogram for predicting liver cancer patient survival rates at 1, 2, and 3 years. The predictive performance of the nomogram was validated using calibration curves and Decision Curve Analysis (DCA).
Immune Cell Infiltration & Immune Score
We analyzed immune cell abundance in TCGA-LIHC samples using CIBERSORT, MCPcounter, and TIMER through the IOBR R package to examine the relationship between risk score and immune infiltration. The tumor immune dysfunction and exclusion (TIDE) algorithm was used to predict responses to immune checkpoint inhibitor (ICI) therapy. Differences in TIDE, immunophenoscore (IPS), interferon-γ (IFN-γ) signature, and microsatellite instability (MSI) scores across risk groups were analyzed using the Wilcoxon test. The prognostic significance of the key genes was confirmed in the GSE202069 immunotherapy cohort using Kaplan–Meier analysis, categorized by risk group.
Drug Sensitivity Analysis
Drug sensitivity was assessed using the Genomics of Drug Sensitivity in Cancer (GDSC) database, with the half-maximal inhibitory concentration (IC50) calculated via the R package pRRophetic (v0.5). The Wilcoxon test was used to assess variations in drug sensitivity between patients in distinct high- and low-risk groups.
Screening Key Single-Cell Type & GSEA Analysis
The AddModuleScore function was used to calculate model gene scores for single-cell types and visualize them with the DotPlot to identify key cell types. Macrophages were extracted from the Seurat object, re-normalized, subjected to dimensionality reduction, and clustered. Subsequently, macrophages were stratified into SPP1+ and SPP1- subsets based on SPP1 expression levels. Differences in the infiltration abundance of SPP1+ macrophages between the TT and PT groups were evaluated using GLMM. Thereafter, the FindMarkers function was used to identify DEGs between the two macrophage subpopulations. Finally, GSEA was performed on these genes using “h.all.v2023.2.Hs.symbols.gmt” as the reference, selecting enriched pathways with p.adjust < 0.05.
Cell Communication Analysis
The R package CellChat (version 1.6.1) was used to analyze ligand-receptor interaction relationships of differentially overexpressed genes in different single-cell subpopulations. Normalized cell gene expression data was input, and intercellular communication probabilities were analyzed by combining gene expression with prior knowledge of interactions. Based on the CellChatDB database, overexpressed ligand-receptor interactions were identified, and cell communication networks were inferred at the ligand-receptor level.
Spatial Transcriptomics Analysis
We redefined eight single-cell types, including “CD8+ T cells” and “SPP1+ macrophage”, using the matched scRNA data (Supplementary Figure 1A and B). We sampled up to 500 cells per subpopulation, keeping genes expressed in at least three cells, and excluded ribosomal and mitochondrial genes to create a reference dataset for spatial cell annotation. We applied SCT normalization, dimensionality reduction clustering, and RCTD deconvolution to determine cell type weights at each spot. Finally, we visualized the spatial distribution and co-localization of “CD8+ T cells” and “SPP1+ macrophage” with SpatialFeaturePlot.
Differential gene sets for “Cytotoxicity CD8+ T cells”, “Exhausted CD8+ T cells”, and “Proliferating CD8+ T cells” were extracted from the cell subpopulation annotation data pertaining to T/NK cells and Myeloid cells through the application of the FindAllMarkers function (Supplementary Figure 1C and D). These differential gene sets, in conjunction with lactate-related, glycolysis, and hypoxia gene sets of interest, underwent ssGSEA enrichment analysis utilizing the GSVA package. The SpatialFeaturePlot function was utilized to depict the spatial distribution and co-localization of the gene sets.
Isolation of Monocytes and T Cells from Peripheral Blood Mononuclear Cells
CD14+ monocytes were isolated from PBMCs using magnetic beads (purity >90%) and differentiated into macrophages for 6 days in medium containing 10% human AB serum. Specifically, the cells were cultured under the following conditions: control medium (10% human AB serum), 30% SK-Hep-1 tumor supernatant (TSN), TSN supplemented with 20 mM sodium L-lactate (Sigma-Aldrich, L7022), TSN with the glycolysis inhibitor 2-deoxyglucose (2-DG), or TSN under 1% hypoxic conditions. T cells were isolated from PBMCs using a Pan T cell isolation kit (Miltenyi Biotec, 130–096-535) for negative selection, achieving a CD3+ enrichment rate exceeding 90% as confirmed by flow cytometry. The isolated T cells were washed and co-cultured with monocytes from each experimental condition at a 1:4 ratio. The co-culture system was enhanced with 2.5 μg/mL of coated anti-CD3 antibody and 1 μg/mL of anti-CD28 antibody, both sourced from Thermo Fisher Scientific. Flow cytometry quantified IFN-γ expression in CD8+ T cells after 3 days.
Flow Cytometry
Monocytes underwent surface staining using anti-CD14 (BioLegend, 301813) and anti-SPP1 antibodies (Santa Cruz Biotechnology, sc-21742), as well as pan-lactylation antibody staining (Jingjie Bioscience Corp, Hangzhou, China). CD8+ T cells from each co-culture were stimulated for 6 hours at 37°C using a leukocyte activation cocktail (BD GolgiPlug™, BD Pharmingen). Subsequently, surface labeling was conducted using anti-CD3 (BioLegend, 300316) and anti-CD8 antibodies (BioLegend, 344722). Cells were fixed and permeabilized with transcription factor buffer (eBioscience, San Diego, CA, USA) and subsequently stained with anti-IFN-γ (BioLegend, 383303).
RNA Isolation and qPCR
Total RNA was extracted using TRIzol reagent (Invitrogen, 15596026) as per the manufacturer’s instructions. RNA was reverse transcribed using the Evo M-MLV reverse transcription premix kit. PCR was conducted with the SYBR Green Pro Taq HS premixed qPCR kit on the Roche LightCycler 480 system. SPP1 Forward: 5′-GGCTAAACCCTGACCCATCT-3′, SPP1 Reverse: 5′-ACTTGGAAGGGTCTGTGGGGG-3′.
Cellular Immunofluorescence
CD14+ cells were fixed with 4% paraformaldehyde at room temperature for 30 minutes, followed by three PBS washes. They were then permeabilized with 0.5% Triton X-100 and blocked with 5% bovine serum albumin, each for 30 minutes. Cells were incubated overnight at 4°C with primary antibodies: anti-CD68 (1:100, Santa Cruz Biotechnology, sc-20060) and anti-Kla (1:200, Jingjie Bioscience Corp, Hangzhou, China). The cells were then incubated with the secondary antibody at room temperature for 1 hour. The cells were rinsed three times with PBS, stained with DAPI (1 μg/mL) in the dark for 5 minutes, and mounted with an anti-fading medium. Images were obtained using a confocal microscope.
Multiple Immunofluorescence Staining
Formalin-fixed tissue sections underwent dewaxing, antigen retrieval via high-temperature and pressure, and a 30-minute immersion in 3% H2O2 to inhibit endogenous peroxidase. Next, blocking was performed with 10% normal serum (half an hour). The TSA method was employed for multiplex immunofluorescence staining. Primary antibodies, including anti-CD68 (abcam ab303565, 1:4000), anti-SPP1 (abcam ab283656, 1:4000), anti-Kla (Jingjie Bioscience PTM-1401RM, 1:400), and anti-CD8 (abcam ab237709, 1:800), were incubated overnight at 4°C. This was followed by a 5-minute incubation with fluorophore-conjugated secondary antibodies (Goat Anti-Rabbit IgG H&L, Abcam ab205718, 1:4000) in the dark. Nuclei were counterstained with DAPI (1 μg/mL) for 5 minutes in the dark, followed by mounting the slides with antifade medium. A confocal microscope was used for imaging.
Results
Significant Activation of Lactate-Related Genes in Macrophages After TACE Treatment
Single-cell data from 10 HCC patients (82,941 high-quality cells) revealed seven immune cell types (Figure 2A and Supplementary Figure 2). Compared to treatment-naïve (PT) samples, post-TACE (TT) samples showed reduced CD8+ T cell and NK cell proportions but increased Macrophage and DC populations (Figure 2B and C, Supplementary Figure 3 and Supplementary Table 1), consistent with prior reports of TACE-induced immunosuppression.13
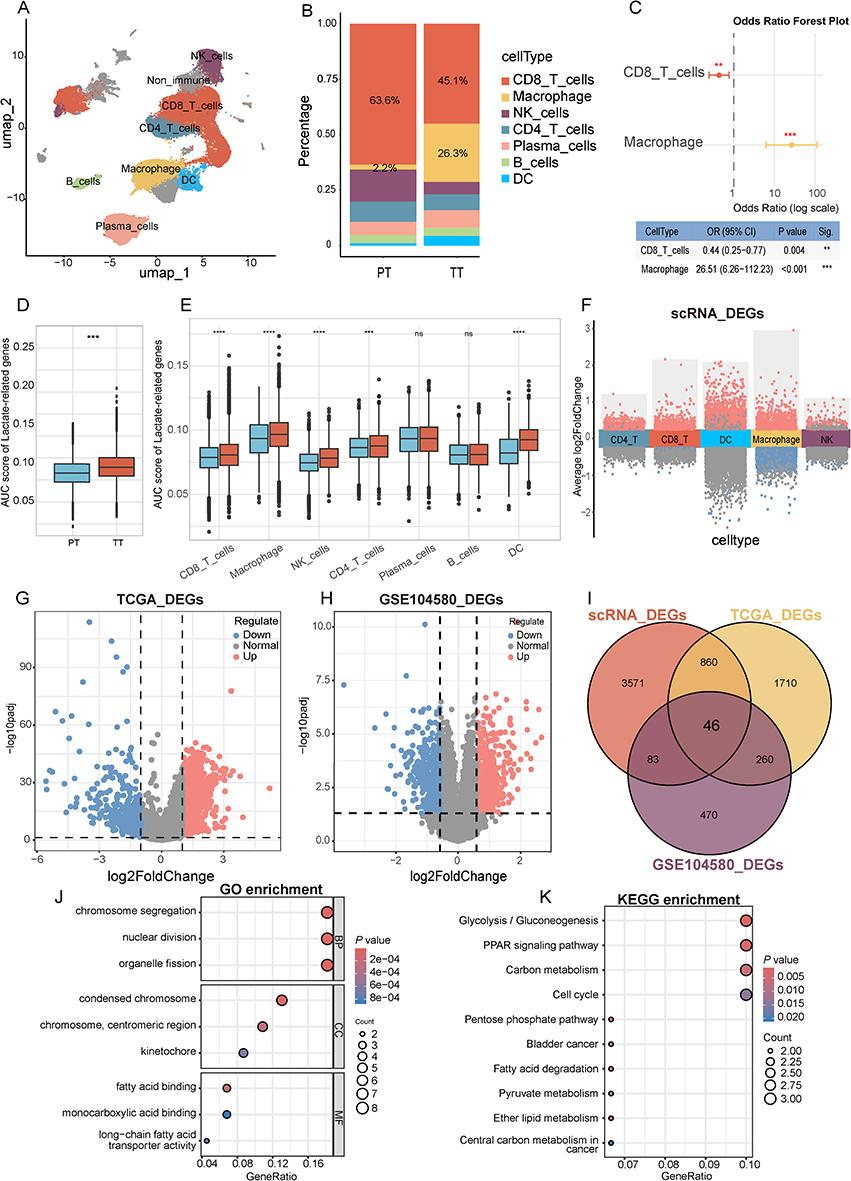 |
Figure 2 Identifying lactate-related genes associated with TACE therapy in hepatocellular carcinoma. (A) UMAP clustering annotated the main immune cells in the TT group (TACE treatment) and PT group (primary tumor). (B) Stacked bar chart of immune cell proportions in TT (TACE treatment) and PT (primary tumor) groups. (C) Forest plot of distribution differences of CD8+ T cells and macrophages between PT group and TT group. P-values were calculated using generalized linear mixed models (GLMM). (D) Box plot of AUCell-based gene set activity scores for the 484 lactate-related gene set in the TT and PT groups. (E) Box plot comparison of AUCell-based gene set activity scores between the TT group (red) and PT group (blue) across seven immune cell types. The Wilcoxon rank-sum test was used for statistical comparisons. (F) Visualization of differentially expressed genes between groups with high and low lactate scores in each cell type. (G) Differential gene volcano map between tumor and normal in TCGA-LIHC dataset. (H) Differential gene volcano map between responder and nonresponder in GSE104580 dataset. (I) Venn diagram identifying lactate-related genes associated with TACE therapy in hepatocellular carcinoma. GO (J) and KEGG (K) enrichment analysis of intersecting genes. **P < 0.01, ***P < 0.001, ****P < 0.0001, ns, not significant (P ≥ 0.05). |
We evaluated the activation of lactate metabolism and lactylation in immune cells post-TACE-induced hypoxia by calculating the AUC scores for 484 characteristic gene sets across different single-cell types. Our findings revealed that the overall AUC score in the TT group was significantly elevated compared to the PT group (Figure 2D). Additionally, the AUC scores for CD8+ T cells, macrophages, natural killer (NK) cells, CD4 T cells, and dendritic cells (DCs) were significantly higher within each single-cell type compared to the PT group (Figure 2E). Notably, the median AUC score for macrophage cells surpassed that of other cell types (Supplementary Figure 4), suggesting that hypoxia post-TACE may exert a more pronounced effect on macrophages.
Identification of DEGs and Enrichment Analysis
To identify key lactate metabolism and lactylation genes, we analyzed DEGs from three datasets. Our single-cell data revealed variations in lactate-related gene expression between PT and TT groups across five immune cell types: CD8+ T cells, macrophages, NK cells, CD4 T cells, and DCs. Cells were categorized into high and low lactate activity groups using the median AUC score of 0.086 as the threshold. Differential expression analysis revealed varying numbers of DEGs across cell types: 1279 (CD8+ T cells), 3100 (macrophages), 283 (NK cells), 536 (CD4+ T cells), and 1591 (DCs). After deduplication, 4560 unique DEGs were retained (Figure 2F). In the TCGA-LIHC dataset, a total of 2876 DEGs were found between Tumor and Normal groups (Figure 2G). In the GSE104580 dataset, 859 DEGs were identified between responder and non-responder groups (Figure 2H).
A total of 46 overlapping genes were identified from the three differential gene sets (Figure 2I). GO and KEGG enrichment analyses of these genes revealed 146 GO_BP, 12 GO_CC, 17 GO_MF pathways (Figure 2J), and 17 KEGG pathways. The KEGG pathways “Glycolysis/Gluconeogenesis,” “Pyruvate metabolism,” “Carbon metabolism,” and “Central carbon metabolism in cancer” are notably linked to lactate metabolism (Figure 2K).
Development and Validation of Prognostic Gene Signatures Using Machine Learning
A univariate Cox regression analysis on 46 intersecting genes from TCGA-LIHC identified 38 prognostic genes with p-values below 0.05. Following this, expression matrices and survival data for 25 common prognostic genes were obtained from the TCGA-LIHC, ICGC, and GSE14520 datasets, and a prognostic model was developed using 101 machine learning algorithms.
The RSF model achieved the highest average C-index of 0.747, but it included 15 model genes. In contrast, the Lasso-RSF combination model, which ranked second, attained an average C-index of 0.737 while incorporating only 6 model genes (SPP1, DNASE1L3, HRG, TPX2, LAPTM4B, G6PD). Consequently, we considered Lasso + RSF to be the optimal prognostic model (Figure 3A). Patients were divided into high-risk and low-risk groups based on the median risk score, and the model’s efficacy was evaluated using Kaplan–Meier and ROC curves. Kaplan–Meier analysis indicated significant prognostic differences between high- and low-risk groups in both training and validation cohorts, with the high-risk group showing poorer outcomes (Figure 3B–F). In the TCGA training set, the AUC values for 1-, 2-, and 3-year survival surpassed 0.95. In contrast, the ICGC and GSE14520 validation sets showed AUC values above 0.7 and 0.6, respectively, indicating the model’s strong predictive performance (Figure 3C–G).
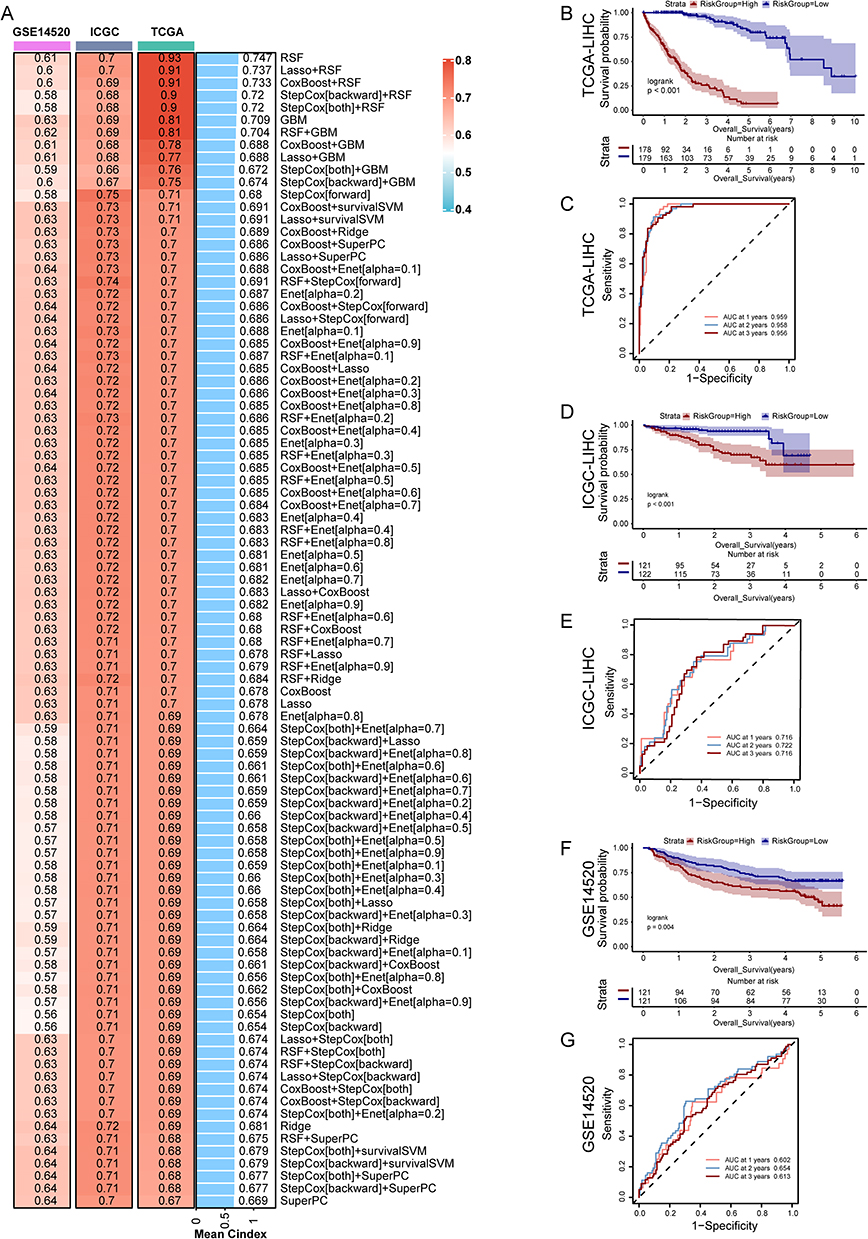 |
Figure 3 Development and validation of prognostic gene signatures using machine learning. (A) The C-index of 101 kinds prognostic models developed by 10 machine learning algorithms in TCGA, ICGC, GSE14520 datasets. (B) Survival curves for high- and low-risk groups in the TCGA cohort. (C) ROC curves (1-, 2-, and 3-year) for the risk model in the TCGA cohort. (D) Survival curves for high- and low-risk groups in the ICGC cohort. (E) ROC curves (1-, 2-, and 3-year) for the risk model in the ICGC cohort. (F) Survival curves for high- and low-risk groups in the GSE14520 cohort. (G) ROC curves (1-, 2-, and 3-year) for the risk model in the GSE14520 cohort. |
Development and Validation of Prognostic Nomogram
The Wilcoxon test was employed to compare risk score differences across clinical subgroups within the TCGA-LIHC cohort. Risk scores significantly differed between the subgroups T1 and T2, T1 and T3, T1 and T4, and T2 and T4 (Figure 4A). Significant differences in risk scores were noted between Stage I and subsequent stages (II, III, and IV) (Figure 4D). No significant differences in risk scores were observed across N-stage, M-stage, age, or gender subgroups (Figure 4B, C and Supplementary Figure 5).
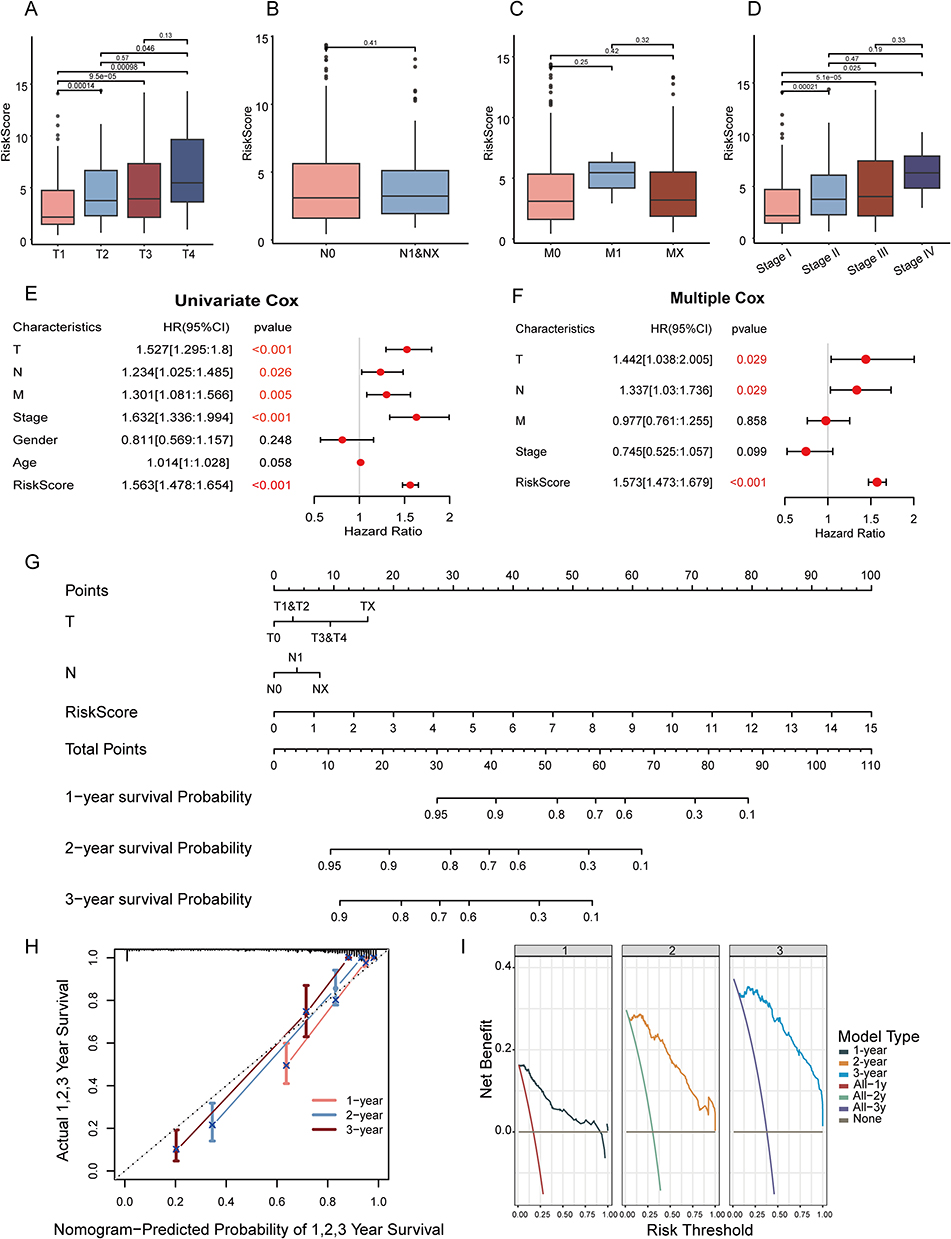 |
Figure 4 Develop and validate of a prognostic nomogram. Comparison of risk scores between T (A), N (B), M (C) and clinical staging subgroups (D). Univariate (E) and multivariate Cox survival analysis (F) and visualization forest map. (G) Nomogram predicting patients’ overall survival at 1-, 2-, and 3- year. (H) Calibration curve of our model in 1-, 2-, and 3- year. (I) Decision curve analysis (DCA) of our model and clinical indexes in 1-, 2-, and 3- year. In panels E and F, the red text corresponding to p-values is less than 0.05, indicating significant statistical differences. |
In the TCGA cohort, univariate Cox analysis indicated that risk scores and clinical features, including T, N, M stages and clinical stage, were significantly linked to prognosis (p < 0.05) (Figure 4E). Multivariate Cox analysis confirmed the statistical significance of T stage, N stage, and risk scores (Figure 4F).
A prognostic nomogram was developed, integrating T stage, N stage, and risk scores (Figure 4G). Calibration and decision curves were plotted to validate the nomogram’s accuracy (Figure 4H and I). The calibration curves indicated that our model closely approximated the ideal scenario at 1-, 2-, and 3-year time points. The decision curves demonstrated that, within the 0–1 threshold range, our model’s curves for 1, 2, and 3 years were consistently above the “All” and “None” lines.
Screening Sensitive Drugs for High-Risk Patients
From the perspective of tumor immunology, the high-risk factors associated with TACE are partially linked to the activation of hypoxia-mediated, lactate-related genes. This raises the question of which pharmacological treatments might be effective for these high-risk populations. Drug sensitivity analysis in the TCGA-LIHC cohort (139 agents) identified 56 compounds with lower IC50 in high-risk patients (Supplementary Table 2). Notably, camptothecin, cisplatin, doxorubicin, and sorafenib are commonly used chemotherapeutic and vascular-targeted agents in clinical practice (Figure 5A–D).
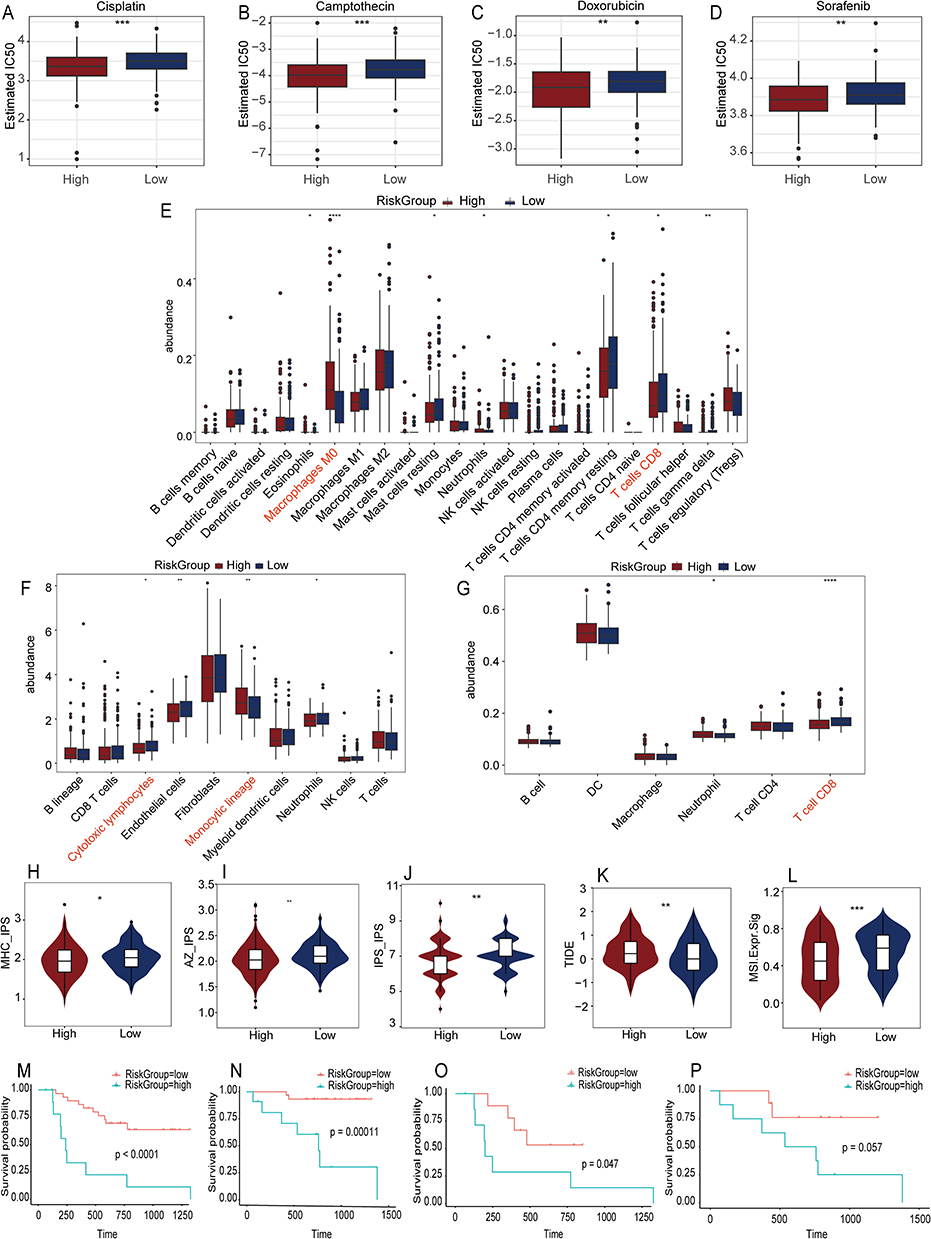 |
Figure 5 Drug sensitivity, Immune cell infiltration, and immune score analysis, and Survival prognosis analysis between high and low risk groups. Drug sensitivity analysis identifies four potentially effective drugs (Cisplatin (A), Camptothecin (B), Doxorubicin (C), Sorafenib (D)) for high-risk groups. Three algorithms – CIBERSORT (E), MCPcounter (F), and TIMER (G) - evaluate immune cell infiltration. Five immune scores (MHC IPS [H], AZ IPS (I), IPI IPS (J), TIDE (K), MSI (L)) showed statistically significant differences between the high and low risk scoring groups. Kaplan-Meier survival curves of OS (M) and PFS (N) for all patients in the external dataset GSE202069, as well as the survival curves of OS (O) and PFS (P) for patients in the PD-1 inhibitor treatment cohort. IPS stands for Immunophenoscore, TIDE refers to Tumor Immune Dysfunction and Exclusion, and MSI denotes microsatellite instability. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. In panels E–G, the red text represents cell subtypes that show significant statistical differences and are of interest. |
Immune Infiltration and Immunotherapy Response Prediction in Risk Subgroups
We employed CIBERSORT, MCPcounter, and TIMER to analyze immune infiltration in the TCGA-LIHC cohort, aiming to identify differences in the immune microenvironment between high- and low-risk patients. CIBERSORT analysis indicated an increased presence of M0 Macrophages and a reduced percentage of CD8+ T cells in the high-risk group (Figure 5E). MCPcounter analysis revealed an increased presence of Monocytic lineage cells and a decreased presence of Cytotoxic lymphocytes in the high-risk group (Figure 5F). According to TIMER, the high-risk group exhibited reduced levels of CD8+ T cells and neutrophils, with no notable difference in macrophage infiltration (Figure 5G). These results align with our single-cell analysis findings.
We analyzed IPS, TIDE, and MSI between high- and low-risk groups to assess the potential immunotherapy benefits for high-risk patients. The high-risk group exhibited notably elevated scores in MHC-IPS, AZ-IPS, IPS-IPS, TIDE, and MSI (Figure 5H–L), suggesting potential advantages from combination immunotherapy. In the GSE202069 immunotherapy dataset, patients were categorized into high-risk and low-risk groups. Survival analysis indicated that low-risk patients experienced notably extended PFS and OS, both in general and among those undergoing anti-PD-1 therapy (Figure 5M–P).
SPP1+ Macrophages: The Cell Type Most Associated with Lactate-Related Gene Activation
We quantified the enrichment scores of six model genes across seven single-cell types, with the results showing the highest scores in Macrophage cells (Figure 6A). Meanwhile, the bubble plot also revealed higher expression of the SPP1 gene in macrophages (Figure 6B), and the AUC score of LRGS initially discovered in macrophages was also higher than that of other cell types (Supplementary Figure 4B). These findings suggest that SPP1+ macrophages may be the cell type most closely associated with lactate-related gene activation in the immune microenvironment following TACE treatment.
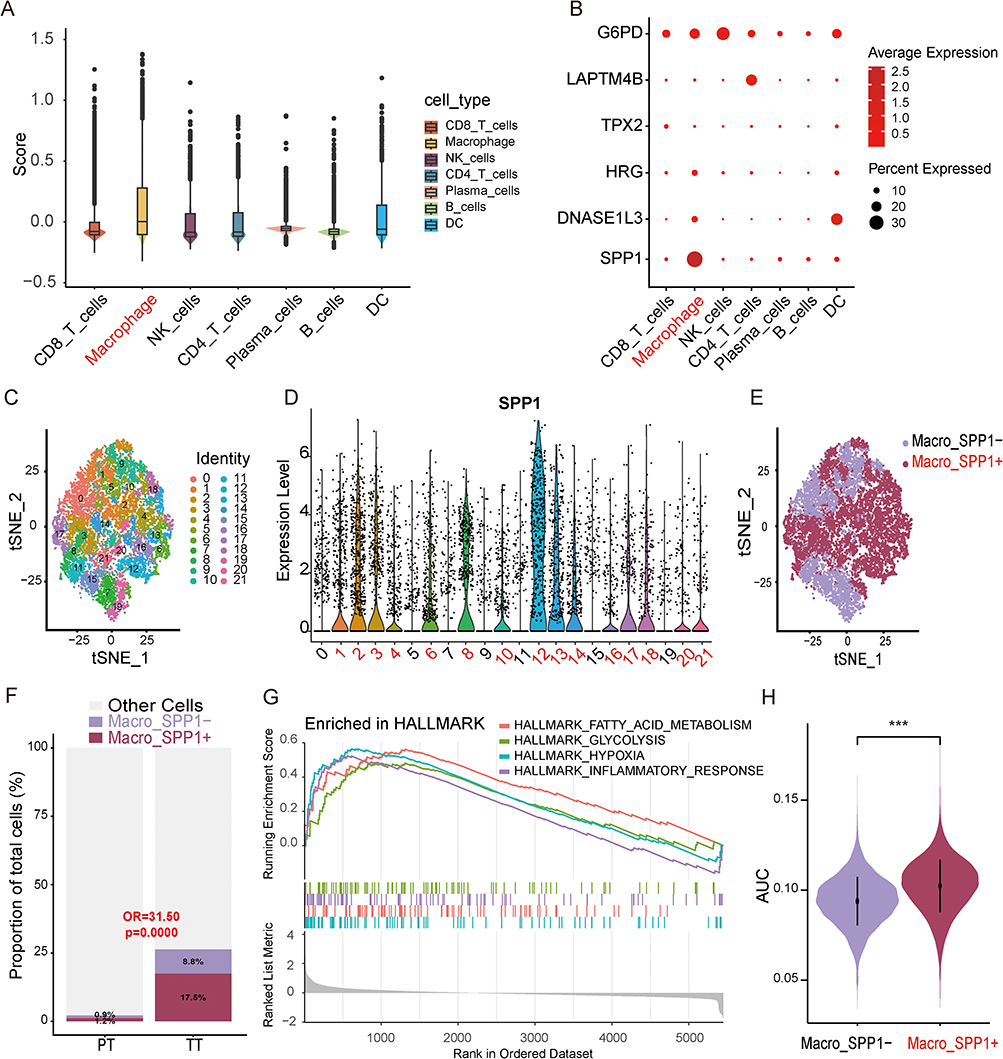 |
Figure 6 Identifying key single-cell subpopulations - SPP1+macrophages. (A) The AddModuleScore analysis of 6 model genes in single-cell types suggests that macrophages have the highest score. (B) The visualization bubble plot of the average gene expression levels of six model genes in different single-cell types suggests that SPP1 is specifically overexpressed in macrophages. (C) t-SNE visualized macrophage dimensionality reduction clustering into 22 clusters. (D) The violin plot visualizes the expression of SPP1 in macrophages, defining clusters 1, 2, 3, 4, 6, 8, 10, 12, 13, 14, 16, 17, 18, 20, and 21 as SPP1+macrophages and the rest as SPP1 macrophages. (E) t-SNE visualization displays SPP1+ and SPP1- macrophages. (F) Comparison of SPP1+ macrophage proportions between the PT and TT groups. A generalized linear mixed model (GLMM) with patient as a random effect was used to account for within-patient cell clustering. (G) GSEA enrichment analysis of HALLMARK pathway in SPP1+ and SPP1- macrophages. (H) Violin plot comparing the AUC scores of the 484 lactate-related gene set between SPP1+ and SPP1− macrophages. The Wilcoxon rank-sum test was used. OR, odds ratio. AUC, area under the curve. ***P < 0.001. In panels A, B, D, E, and H, the highlighted red text indicates macrophage subtypes or SPP1+ macrophage clusters. In panel F, the p-value displayed in red text is less than 0.05, indicating a significant statistical difference. |
Macrophage cells underwent secondary dimensionality reduction and clustering at a resolution of 1.5, yielding 22 clusters. Based on SPP1 expression levels, clusters 1, 2, 3, 4, 6, 8, 10, 12, 13, 14, 16, 17, 18, 20, and 21 were identified as SPP1+ macrophages, while clusters 0, 5, 7, 9, 11, 15, and 19 were SPP1- (Figure 6C–E). Comparing the PT and TT groups by GLMM, the odds of SPP1+ macrophage assignment in the TT group were 31.50 times those in the PT group, indicating that SPP1+ macrophages were enriched in the TT group relative to the PT group (OR = 31.50, 95% CI: 6.41–154.71; P<0.0001, GLMM with patient as random effect) (Figure 6F).
We conducted GSEA analysis on the DEGs between SPP1+ and SPP1- macrophages to explore the potential association between SPP1+ macrophages and immune suppression. Twenty-six significantly enriched pathways were identified, including “HALLMARK HYPOXIA,” “HALLMARK GLYCOLYSIS,” “HALLMARK FATTY ACID METABOLISM,” and “HALLMARK INFLAMMATORY RESPONSE,” all associated with immune suppression (Figure 6G). Additionally, SPP1+ macrophages showed higher AUC scores for 484 LRGS compared to SPP1- macrophages (Figure 6H).
Interaction Between SPP1+ Macrophages and CD8+ T Cells
To further explore the potential molecular mechanisms underlying the association between SPP1+ macrophages and reduced CD8+ T cell proportions, we analyzed the cellular communication between different single-cell types. The results revealed that, compared to SPP1- macrophages, SPP1+ macrophages as signal senders and CD8+ T cells as signal receivers exhibited a closer connection in terms of both the quantity and intensity of communication (Figure 7A–D).
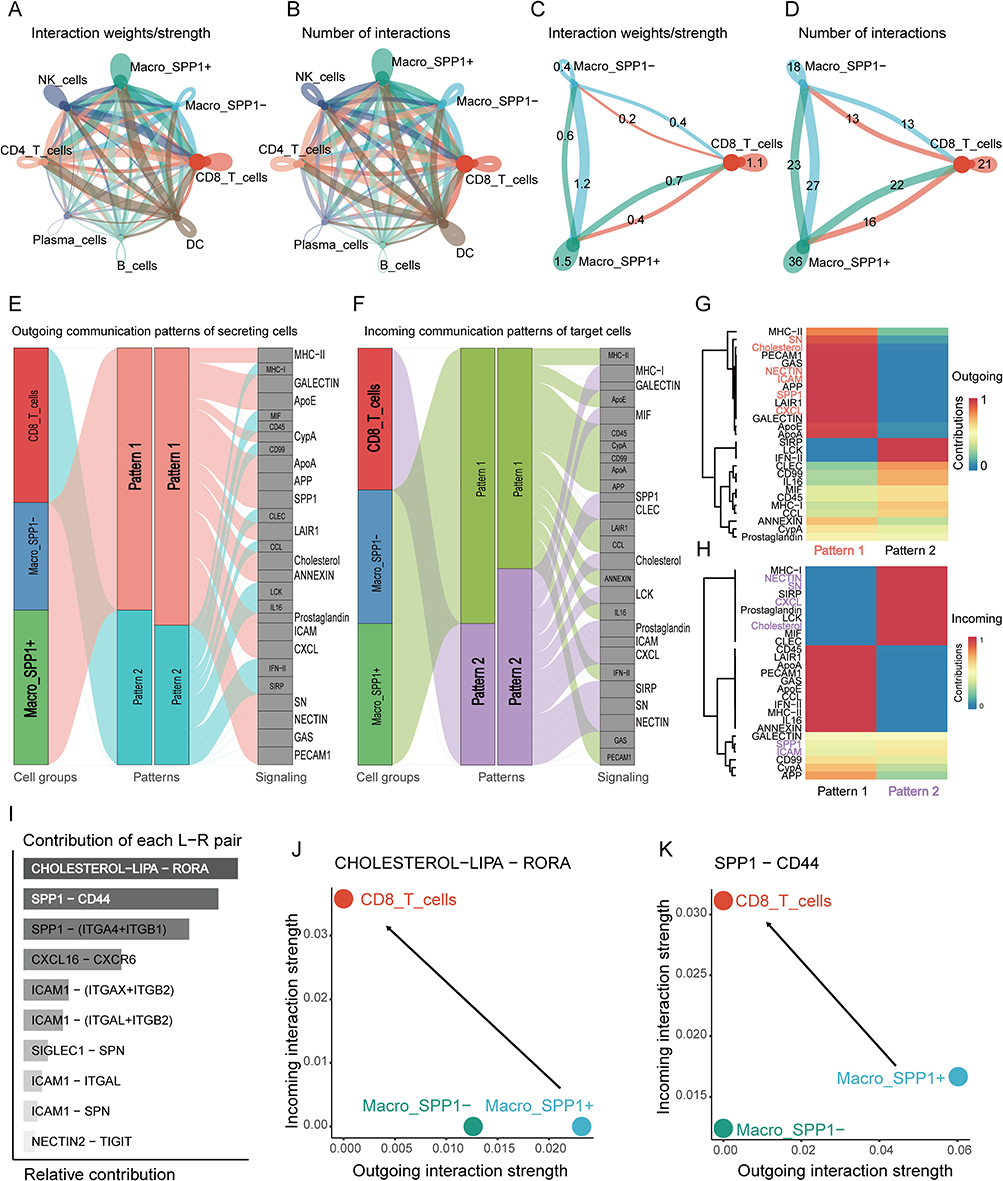 |
Figure 7 Cell communication and ligand pair analysis between single-cell subpopulations. The communication strength (A) and number (B) between all single-cell subtypes, as well as the strength (C) and number (D) of cell communication between SPP1+ macrophages, SPP1− macrophages, and CD8+ T cells. (E) Sankey diagram of pattern 1 signaling pathways originating from macrophages. (F) Sankey diagram of pattern 2 signaling pathways incoming to CD8⁺ T cells. (G) Heatmap displaying the signal intensity of pattern 1 molecular pathways in macrophages. (H) Heatmap displaying the signal intensity of pattern 2 molecular pathways in CD8⁺ T cells. (I) Ranking of contribution of ligand receptor pairs in multiple signaling pathways. The ligand receptor bubble plot of cell communication intensity shows that SPP1+ macrophages are the main signal transmitters and CD8+ T cells are the main signal receivers (J), and the signal intensity emitted by SPP1+macrophages through the SPP1-CD44 signaling pathway is greater than that emitted by the CHOLESTEROL LIPA RORA signaling pathway (K). The red text in panel G and the purple text in panel H highlight the molecular pathways in which the signal sender (pattern 1) and signal receiver (pattern 2) intersect and exhibit strong cellular communication strength. |
We thoroughly analyzed the communication patterns between SPP1+ and SPP1− macrophages and CD8+ T cells. When the communication “pattern” was set to 2, under the outgoing mode, the signaling pathways emitted by SPP1+ macrophages corresponded to pattern 1 (Figure 7E). In the incoming mode, CD8+ T cells received pathways linked to pattern 2 (Figure 7F). Analysis of the signaling pathway heatmaps (Figure 7G and H) revealed six common pathways (SN, Cholesterol, NECTIN, ICAM, SPP1, CXCL) between the main outgoing pathways of SPP1+ macrophages and the primary incoming pathways of CD8+ T cells. These pathways were ranked according to the contribution of various ligand-receptor interactions. The top two contributing pathways were “CHOLESTEROL LIPA RORA” and “SPP1-CD44” (Figure 7I). Furthermore, we quantified the strength of these two signaling pathways using bubble plots. The findings revealed that CD8+ T cells received signals of comparable strength in both pathways, whereas SPP1+ macrophages emitted stronger signals in the “SPP1-CD44” pathway compared to the “CHOLESTEROL LIPA RORA” pathway (Figure 7J–K). This indicates that SPP1-CD44 was identified as the predominant signaling pathway that may mediate the interaction between SPP1+ macrophages and CD8+ T cells.
Lactic Acid and Hypoxia Can Enhance Macrophage Lactylation, SPP1 Expression, and CD8+ T Cells Dysfunction
We established a monocyte-macrophage induction model to study the effects of hypoxic and lactate-enriched microenvironments on macrophage lactylation, SPP1 expression, and CD8+ T cells suppression. Our findings indicate that, in comparison to the medium control group (Med) and the TSN-treated group, both the TSN combined with sodium L-lactate (TSN+L-NA) and the TSN combined with hypoxia groups (TSN+Less O2) exhibited significantly elevated levels of pan-lactylation (Figure 8A and B), an increased proportion of SPP1+ macrophages (Figure 8C and D), and upregulated transcriptional levels of SPP1 (Figure 8E). Co-culturing CD8+ T cells with macrophages for three days significantly decreased the proportion of CD8+IFN-γ+ T cells (Figure 8F and G). Notably, in the TSN combined with 2-DG intervention group, these effects were significantly attenuated. Cellular immunofluorescence imaging more intuitively demonstrated the changes in CD68+ cells and SPP1+ cells across different groups (Figure 8H).
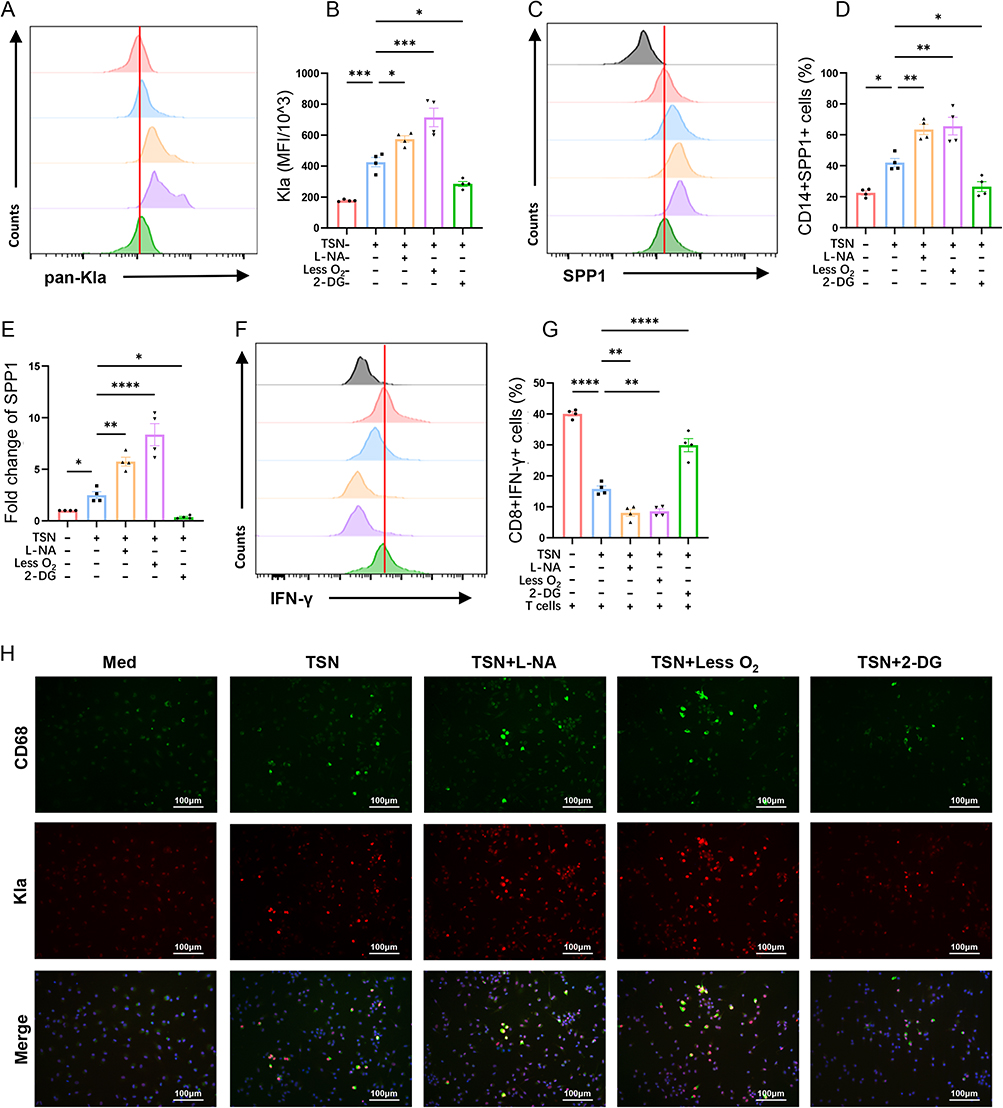 |
Figure 8 The Impact of Lactate and Hypoxic Microenvironments on Macrophages and CD8+ T Cells. Flow cytometry analysis (A) and bar chart (B) of pan-lactylation levels in human mononuclear macrophages under different intervention conditions. Flow cytometry analysis (C) and bar chart (D) of CD14+SPP1+ human monocytes ratio under different intervention conditions. (E) qPCR detection of SPP1 in human mononuclear macrophages under different intervention conditions showed that lactate and hypoxia treatments could increase the expression of SPP1 in macrophages. Flow cytometry analysis (F) and bar graph (G) of IFN-γ+CD8+ T cells co-cultured under various intervention conditions. (H) CD68 expression and pan-lactylation fluorescence staining of human mononuclear macrophages under different intervention conditions. Kla, lysine lactylation; TSN, tumor supernatant; L-NA, sodium L-lactate; 2-DG, 2-deoxyglucose. In panels B, D, E, and G, “+” and “-” respectively indicate the presence or absence of TSN, L-NA, hypoxia, or 2-DG therapy. These results are described using mean ± standard deviation, and inter-group comparisons are analyzed using one-way ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. |
Spatial Localization of SPP1+ Macrophages and CD8+ T Cells
Using single-cell annotation reference datasets and RCTD deconvolution, we detected SPP1+ macrophages and CD8+ T cells in the spatial transcriptome (Figure 9A, B and G, H). In Patient #1 (non-responder), the spatial distribution of different cell subpopulations we annotated, including SPP1+ macrophages, highly recapitulated the results of Liu et al19 indicating that our methods and results for analyzing spatial transcriptome data are reliable (Supplementary Figure 6). Compared with Patient #7, Patient #1 exhibited a smaller area of CD8+ T cells infiltration (Figure 9A and G), and the spatial distribution of SPP1+ macrophages also reproduced the spatial distribution characteristics of the “immune barrier” they proposed (Figure 9B and H).
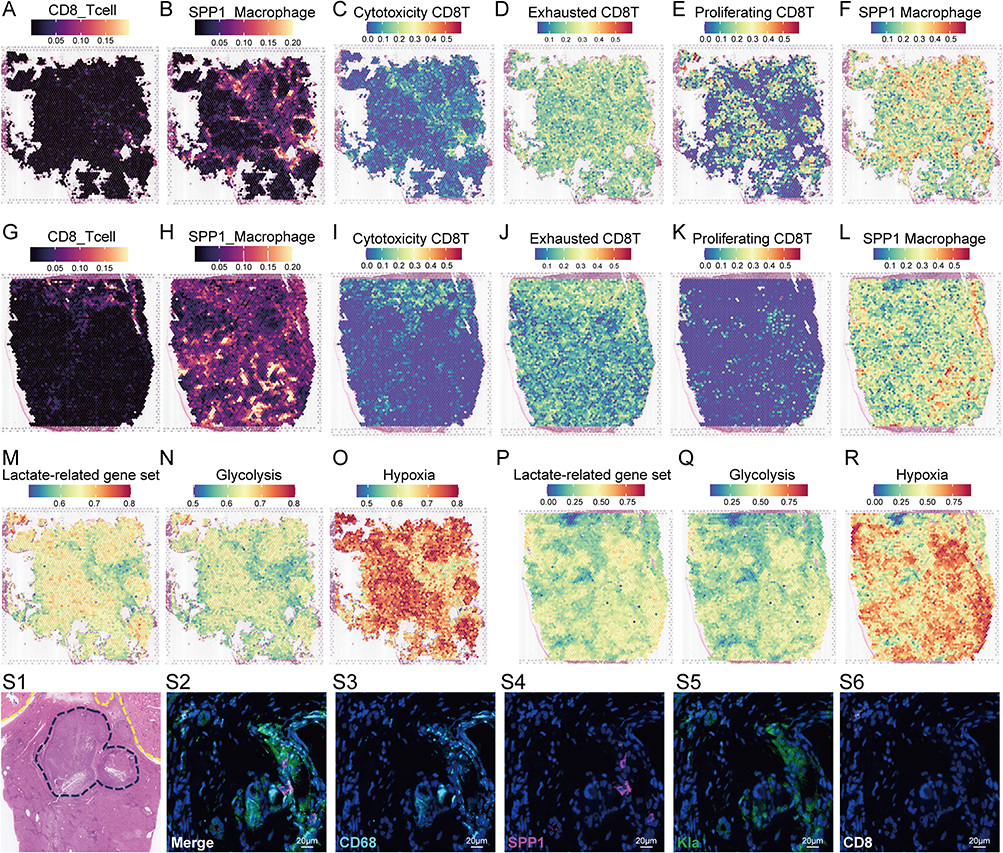 |
Figure 9 Cell annotation, feature gene set enrichment analysis, and multiplex immunofluorescence staining analysis in spatial transcriptome. Spatial distribution of CD8+ T cells (A) and SPP1+ macrophages (B) in patients #1 (non-responder) estimated by RCTD deconvolution. Overlapping regions represent visual co-localization only and were not assessed by quantitative spatial statistics. AUC score based on a single-cell annotated feature gene set, displaying the spatial distribution heatmaps of cytotoxic CD8+T cells (C), exhausted CD8+T cells (D), proliferating CD8+T cells (E), and SPP1+ macrophage (F) subtypes in patient #1. Spatial distribution of CD8+ T cells (G) and SPP1+ macrophages (H) in patients #7 (responder) estimated by RCTD deconvolution. AUC score based on a single-cell annotated feature gene set, displaying the spatial distribution heatmaps of cytotoxic CD8+T cells (I), exhausted CD8+T cells (J), proliferating CD8+T cells (K), and SPP1+ macrophage (L) subtypes in patient #7. AUC score based on a molecular pathway feature gene set, displaying spatial distribution heatmaps of lactate-related gene set (M), glycolysis gene set (N), and hypoxia gene (O) set in patient #1. AUC score based on a molecular pathway feature gene set, displaying spatial distribution heatmaps of lactate-related gene set (P), glycolysis gene set (Q), and hypoxia gene (R) set in patient #7. (S1) HE staining of a liver cancer specimen treated with TACE showed tumor necrosis (outside the yellow dashed line in the upper right corner) and residual active lesions near the liver capsule (inside the blue dashed line). Multiple immunofluorescence staining (S2, merged) demonstrated the spatial co-localization of CD68 (S3), SPP1 (S4), Kla (S5), and CD8 (S6) in liver cancer tissue. |
We conducted AUC scoring and spatial visualization on gene sets of SPP1+ macrophages and three CD8+ T cell types. In patient #1, the activation regions of the cytotoxic and exhausted CD8 T-related gene sets significantly overlap with those of SPP1 macrophage-related genes, while the activation regions of the proliferating CD8 T-related gene set were locked outside the tumor immune barrier (Figure 9C–F). In Patient #7, cytotoxic and exhausted CD8 T-related gene sets showed consistency, but no overlap with SPP1+ macrophages was found (Figure 9I–L). Additionally, both patients exhibited consistent activation regions for lactate, glycolysis, and hypoxia gene sets, with hypoxia showing stronger activation, covering the infiltration area of SPP1+ macrophages (Figure 9M–R).
We examined liver cancer tissue post-TACE resection, finding extensive tumor necrosis and a small active nodule near the liver capsule (Figure 9S1). Using multiplex immunofluorescence, we stained for CD68, Kla, SPP1, and CD8 to visualize macrophages and CD8+ T cells (Figure 9S2–6). Co-localization of CD68+SPP1+ macrophages with Kla was consistent with hypoxia-associated lactylation and high SPP1 expression. Few CD8+ T cells were present, a pattern consistent with the reduced CD8+ T cells infiltration observed in our single-cell analysis of post-TACE samples.
Discussion
Lactylation has become a prominent area of research, especially concerning the tumor immune microenvironment.11,23 It is associated with low oxygen levels, lactate accumulation in the tumor microenvironment, macrophage polarization, and immune suppression.24,25 Our prior research identified alterations in immune cell infiltration following TACE, prompting the current investigation into lactate metabolism and lactylation. To our knowledge, this study is the first to investigate the association between lactylation modification and immune suppression after TACE treatment.
This research used a multi-omics strategy to explore how TACE-induced hypoxia affects immune cells via lactate metabolism and lactylation. Our single-cell data showed that lactate-related genes in immune cells were highly activated post-TACE. We identified 46 intersecting genes and constructed a 6-gene prognostic signature using machine learning. The model score correlated with clinical features and immune infiltration patterns. We further identified SPP1+ macrophages as the key cell type associated with feature genes and explored their interaction with CD8+ T cells through cell communication analysis, in vitro experiments, and spatial transcriptomics. These findings are preliminary and require independent validation.
Our single-cell atlas revealed TACE-induced immune remodeling, characterized by increased macrophages and decreased CD8+ T cells, consistent with an immunosuppressive phenotype.26,27 To evaluate lactylation-related activity, we compiled a 484-gene set from GSEA and Genecards databases. Post-TACE samples showed elevated AUC scores for this gene set across immune cells, with macrophages exhibiting the highest activity. The AUC score quantified the association between cells and lactate-related processes, helping us identify cell types most affected by lactate metabolism and lactylation.
The enrichment analysis results of the 46 initially screened intersecting genes indicate that pathways such as “Glycolysis/Gluconeogenesis,” “Pyruvate metabolism,” and “Carbon metabolism” are closely related to lactate metabolism or lactylation.28,29 Glycolysis, a key metabolic process, converts glucose to pyruvate,30,31 which can turn into lactate anaerobically, enter the TCA cycle aerobically, or be used in gluconeogenesis.32 Tumor cells often exhibit increased glycolysis even with oxygen.33 Post-TACE, the hypoxic environment further enhances glycolysis, boosting lactate production.34,35 In our single-cell data, macrophages exhibit the highest LRGS activity. GSEA analysis revealed that the differential genes between SPP1+ and SPP1- macrophages were enriched in pathways such as hypoxia, glycolysis, fatty acid metabolism, and inflammatory response, which are linked to macrophage-mediated immune suppression.36,37 Additionally, SPP1+ macrophages had a higher AUC score for 484 LRGS, highlighting their strong link to lactate metabolism or lactylation.
In the context of cancer, carbon metabolism is frequently reprogrammed to facilitate rapid cellular proliferation and survival.38,39 Following TACE, the resultant hypoxic and lactate-enriched microenvironment can further modulate carbon metabolism within immune cells.39 The synthesis of fatty acids and other biomolecules in macrophages can be enhanced through carbon metabolism, linked to the activation of the fatty acid metabolism pathway in SPP1+ macrophages.39 This metabolic reprogramming in macrophages may bolster their immunosuppressive role, potentially affecting the CD8+ T cell-mediated anti-tumor immune response.36,38 Collectively, these pathways play crucial roles in immune microenvironment changes after TACE. A thorough grasp of these mechanisms may provide new insights into TACE-induced immunosuppression and identify potential therapeutic targets.
In tumor lactylation research, lactylation occurs in both tumor and immune cells, particularly macrophages.8,10,24 In 2019, Zhao et al8 discovered that macrophages use histone lactylation to promote M2 polarization. Histone lactylation caused by lactic acid is associated with elevated M2 gene expression in alveolar macrophages,40 bone marrow-derived macrophages,41 and human peripheral blood mononuclear cells.42 According to Liu et al, lactate-induced lactylation of pyruvate kinase causes M1 macrophages to transition to anti-inflammatory M2 by suppressing glycolysis.43 Lactate promotes lactylation of the retinoic acid-inducible gene I (RIG-I) in macrophages, disrupting certain signaling pathways and facilitating M2 polarization and liver metastasis in colorectal cancer.44 Hypoxia- and lactate-mediated immunosuppressive macrophages were further validated in our study. Specifically, after TACE treatment, SPP1+ macrophages significantly increased, along with a higher LRGS AUC score. Additional experiments demonstrated that lactate and hypoxia conditions elevated pan-lactylation, SPP1 gene expression, and the proportion of SPP1+ macrophages.
We next sought to clarify the reason for the decreased proportion of CD8+ T cells in the TACE treatment group by investigating the intercellular communication between macrophages and CD8+ T cells. Our research reveals that SPP1+ macrophages strongly interact with CD8+ T cells, primarily through the SPP1-CD44 ligand-receptor signaling pathway. This finding aligns with the work of Shi et al18 who identified cancer stem cells and SPP1+ macrophages in hypoxic regions through single-cell and spatial analysis, highlighting SPP1-CD44 as the primary ligand-receptor pair. Recent studies suggest that elevated SPP1 in macrophages induces T-cell stress, facilitating colon cancer liver metastasis via the SPP1/CD44/PI3K/AKT signaling pathway.45 We examined the interaction between SPP1+ macrophages and CD8+ T cells through various methods, including spatial transcriptomics cell co-localization, spatial distribution analysis of feature gene set AUC scores, cell co-culture experiments, and multiplex immunofluorescence staining. Our results provide preliminary insights into how anoxic or lactic acid microenvironments affect the function and interaction of immune cells in the post-TACE setting, although these observations require confirmation in larger, independent cohorts.
Let our discussion refocus on the 46 intersecting genes. After this step of screening, we constructed a 6-gene signature using machine learning and analyzed the association between model scores and clinical stages. Furthermore, we developed a prognostic nomogram to explore the potential prognostic value of this study. Patients with elevated model scores tend to have a poorer prognosis. The Lactate Metabolism-Related Gene Signature developed by Tu et al46 Patients with elevated scores showed a poor prognosis, along with differences in immune cell infiltration and immune function between high and low score groups. Wang et al47 developed a gene signature associated with hypoxia, glycolysis, and lactate. Patients with higher scores exhibited a poorer prognosis, marked by a higher macrophage proportion, reduced T cell proportion in the high-risk group, and an increased TIDE score. These findings align with our results.
While the 6-gene signature and nomogram demonstrated consistent prognostic stratification across three independent cohorts, their current form should be regarded as a research tool rather than a clinically deployable assay. Several steps are required before clinical translation can be considered: validation in prospective TACE-treated HCC cohorts, head-to-head comparison with established staging systems (eg, BCLC, ALBI grade), and demonstration of incremental predictive value beyond routine clinical variables. If validated, the signature could potentially aid in identifying patients at elevated risk of post-TACE immunosuppression who may benefit from closer surveillance or early combination immunotherapy. The six constituent genes — several of which (eg, SPP1, G6PD) are functionally implicated in glycolysis and lactate metabolism — may also represent biologically relevant candidates for adjuvant targeting.
From the perspective of clinical translational applications, although this study suggests that SPP1+ macrophages are associated with immune suppression related to TACE, treatments targeting the hypoxic microenvironment or SPP1-CD44 axis are still in the experimental stage.5,12,24,45 Based on the risk score stratification of TCGA patients, we conducted sensitivity analysis on multiple drugs and screened several clinically available drugs with potential therapeutic effects on high-risk patients, suggesting promising combination therapies.
Several limitations should be acknowledged. First, the scRNA-seq cohort was modest (n = 10; 5 per group), reflecting the scarcity of fresh post-TACE HCC surgical specimens. Formal a priori power calculation was not performed, and this limits the generalizability of cell-type proportion comparisons. Second, the in vitro experiments used peripheral blood monocyte-derived macrophages stimulated with SK-Hep-1 tumor supernatant, which does not fully recapitulate the complex intrahepatic post-TACE microenvironment; validation in patient-derived organoid co-cultures or in vivo TACE models is warranted. Third, the spatial transcriptomic validation was restricted to two patients with matched scRNA-seq data and should be interpreted as exploratory; future studies employing larger spatial cohorts and quantitative co-localization metrics are needed. Fourth, univariate Cox regression for prognostic gene screening was performed on the full TCGA-LIHC cohort prior to model training, which may introduce optimistic bias; however, consistent external validation across independent ICGC and GSE14520 cohorts partially mitigates this concern. Fifth, while SPP1-CD44 was identified as the predominant ligand–receptor pair, direct functional validation through SPP1-CD44 blockade experiments was not performed; although this pathway has been mechanistically validated by independent groups, future studies employing neutralizing antibodies are warranted to confirm its functional role specifically in the post-TACE context. Sixth, the findings presented herein are correlative and hypothesis-generating in nature; independent prospective validation is required before any clinical translation can be considered.
Conclusion
In summary, this study established a 6-gene signature associated with lactate metabolism and lactylation and constructed a prognostic nomogram. The nomogram in its current form relies on bulk RNA-seq data and should be regarded as a research tool; its translation into a clinically practical assay (eg, immunohistochemistry- or qPCR-based) requires further development and prospective validation. We identified SPP1+ macrophages as the immune cell subpopulation most closely associated with lactate-related gene activation post-TACE, and the SPP1-CD44 pathway was identified as a potential mechanism that may contribute to CD8+ T cells suppression. In vitro experiments demonstrated that hypoxia and lactate are associated with elevated macrophage lactylation and SPP1 expression. These findings collectively generate the hypothesis that TACE-induced hypoxia and lactate accumulation promote SPP1+ macrophage-mediated immunosuppression through lactylation. While this study focused specifically on the post-TACE setting, the hypoxia–lactate–lactylation–SPP1+ macrophage axis may also be relevant to other HCC locoregional treatments that induce tumor ischemia (eg, transarterial embolization or ablation), a possibility that warrants independent investigation. Clinically, if this hypothesis is validated, targeting the SPP1-CD44 axis or lactylation may offer a strategy to counteract TACE-induced immunosuppression and potentially enhance the efficacy of TACE-immunotherapy combinations. Overall, this study provides a foundation for investigating the role of lactylation in post-TACE immune regulation.
Data Sharing Statement
The research datasets can be accessed on the NCBI network under the identifier PRJNA793914 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA793914). The data supporting the study’s findings can be requested from the corresponding author Wendao Liu.
Ethics Approval and Consent to Participate
The single-cell sequencing portion of this study involving HCC patients was approved by the Ethics Committee of the First Affiliated Hospital of Sun Yat-sen University (Approval No. 2021 [204]). The in vitro experiments using human peripheral blood mononuclear cells were approved by the Ethics Committee of Guangdong Provincial Hospital of Chinese Medicine (Approval No. YE2025-147-01). The study was conducted in accordance with the Declaration of Helsinki. All participants were informed of the study’s purpose, assured of confidentiality, and provided written informed consent prior to participation. Participation was voluntary, and respondents could withdraw at any time without penalty.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by Guangdong Basic and Applied Basic Research Foundation (ID: 25201910240001361), the seventh batch of “Guangdong Special Support Plan” Provincial Health Commission (Health Talents) Project (Contract No. 0720240232), the Special Project within Guangdong Provincial Hospital of Chinese Medicine (YN2022MS11), and the Guangzhou University of Traditional Chinese Medicine Postgraduate Innovation Ability Promotion Project (20231110672).
Disclosure
The authors state that the research was carried out without any commercial or financial ties that might present a conflict of interest.
References
1. Liu Z, Jiang Y, Yuan H, et al. The trends in incidence of primary liver cancer caused by specific etiologies: results from the global burden of disease study 2016 and implications for liver cancer prevention. J Hepatol. 2019;70(4):674–22. doi:10.1016/j.jhep.2018.12.001
2. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
3. Lu J, Zhao M, Arai Y, et al. Clinical practice of transarterial chemoembolization for hepatocellular carcinoma: consensus statement from an international expert panel of International Society of Multidisciplinary Interventional Oncology (ISMIO). Hepatobiliary Surg Nutr. 2021;10(5):661–671. doi:10.21037/hbsn-21-260
4. Chen L, Huang M. Oncometabolites in cancer: from cancer cells to the tumor microenvironment. Holist Integ Oncol. 2024;3(1):26. doi:10.1007/s44178-024-00096-7
5. Ni X, Piao LC, Qiang XG, jing MJ. Transcriptional regulation and post-translational modifications in the glycolytic pathway for targeted cancer therapy. Acta Pharmacol Sin. 2024;45(8):1533–1555. doi:10.1038/s41401-024-01264-1
6. Brooks GA. The science and translation of lactate shuttle theory. Cell Metab. 2018;27(4):757–785. doi:10.1016/j.cmet.2018.03.008
7. Zhang W, Wang G, Xu ZG, et al. Lactate is a natural suppressor of RLR Signaling by Targeting MAVS. Cell. 2019;178(1):176–189.e15. doi:10.1016/j.cell.2019.05.003
8. Zhang D, Tang Z, Huang H, et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574:7779):575–580. doi:10.1038/s41586-019-1678-1
9. Yang Z, Yan C, Ma J, et al. Lactylome analysis suggests lactylation-dependent mechanisms of metabolic adaptation in hepatocellular carcinoma. Nat Metab. 2023;5(1):61–79. doi:10.1038/s42255-022-00710-w
10. Xu R, Hao Y, Liu Y, Ji B, Tian W, Zhang W. Functional mechanisms and potential therapeutic strategies for lactylation in liver diseases. Life Sci. 2025;363:123395. doi:10.1016/j.lfs.2025.123395
11. Chen J, Huang Z, Chen Y, et al. Lactate and lactylation in cancer. Signal Transduct Target Ther. 2025;10:38. doi:10.1038/s41392-024-02082-x
12. Jiang M, Wang Y, Zhao X, Yu J. From metabolic byproduct to immune modulator: the role of lactate in tumor immune escape. Front Immunol. 2024;15:1492050. doi:10.3389/fimmu.2024.1492050
13. Tan J, Fan W, Liu T, et al. TREM2+ macrophages suppress CD8+ T-cell infiltration after transarterial chemoembolisation in hepatocellular carcinoma. J Hepatol. 2023;79(1):126–140. doi:10.1016/j.jhep.2023.02.032
14. Reggio A, Fuoco C, Deodati R, Palma A. SPP1 macrophages across diseases: a call for reclassification? FASEB J. 2025;39(5):e70448. doi:10.1096/fj.202403227R
15. Li X, Lian J, Lu H. The role of SPP1+TAMs in cancer: impact on patient prognosis and future therapeutic targets. Int J Cancer. 2025;157(9):1763–1771. doi:10.1002/ijc.70018
16. Matsubara E, Yano H, Pan C, et al. The significance of SPP1 in lung cancers and its impact as a marker for protumor tumor-associated macrophages. Cancers. 2023;15(8):2250. doi:10.3390/cancers15082250
17. Feng Y, Shao X, Xu ZF, et al. SPP1 regulates tumor progression through modulation of signaling pathways and the tumor microenvironment. Discov Oncol. 2025;17(1):115. doi:10.1007/s12672-025-04273-6
18. Fan G, Xie T, Li L, Tang L, Han X, Shi Y. Single-cell and spatial analyses revealed the co-location of cancer stem cells and SPP1+ macrophage in hypoxic region that determines the poor prognosis in hepatocellular carcinoma. NPJ Precis Oncol. 2024;8(1):75. doi:10.1038/s41698-024-00564-3
19. Liu Y, Xun Z, Ma K, et al. Identification of a tumour immune barrier in the HCC microenvironment that determines the efficacy of immunotherapy. J Hepatol. 2023;78(4):770–782. doi:10.1016/j.jhep.2023.01.011
20. Trehan R, Huang P, Zhu XB, et al. SPP1 + macrophages cause exhaustion of tumor-specific T cells in liver metastases. Nat Commun. 2025;16(1):4242. doi:10.1038/s41467-025-59529-0
21. Almgrami RT, Zhang T, Zhao Q, You M, Liu J, Zhang Y. Single-cell transcriptomic analyses provide insights into SPP1+ TAM-mediated immune suppression and CD8+ T cell dysfunction in lung cancer. Cancer Immunol Immunother. 2025;74(10):319. doi:10.1007/s00262-025-04180-3
22. Liu Z, Liu L, Weng S, et al. Machine learning-based integration develops an immune-derived lncRNA signature for improving outcomes in colorectal cancer. Nat Commun. 2022;13(1):816. doi:10.1038/s41467-022-28421-6
23. Yi D, Zhou K, Pan Y, Cai H, Huang P. The lactylation modification of proteins plays a critical role in tumor progression. Front Oncol. 2025;15:1530567. doi:10.3389/fonc.2025.1530567
24. Qu J, Li P, Sun Z. Histone lactylation regulates cancer progression by reshaping the tumor microenvironment. Front Immunol. 2023;14:1284344. doi:10.3389/fimmu.2023.1284344
25. Liu H, Pan M, Liu M, et al. Lactate: a rising star in tumors and inflammation. Front Immunol. 2024;15:1496390. doi:10.3389/fimmu.2024.1496390
26. Stromnes IM, Hulbert A, Pierce RH, Greenberg PD, Hingorani SR. T-cell localization, activation, and clonal expansion in human pancreatic ductal adenocarcinoma. Cancer Immunol Res. 2017;5(11):978–991. doi:10.1158/2326-6066.CIR-16-0322
27. Kikuchi M, Takami H, Kobayashi Y, et al. Dissecting the immunological microenvironment of glioma based on IDH status: implications for immunotherapy. Cells. 2025;14(13):1035. doi:10.3390/cells14131035
28. Sun W, Jia M, Feng Y, Cheng X. Lactate is a bridge linking glycolysis and autophagy through lactylation. Autophagy. 2023;19(12):3240–3241. doi:10.1080/15548627.2023.2246356
29. Zhang Y, Zhai Z, Duan J, et al. Lactate: the mediator of metabolism and immunosuppression. Front Endocrinol. 2022;13:901495. doi:10.3389/fendo.2022.901495
30. Zhang F, Liu H, Duan M, et al. Crosstalk among m6A RNA methylation, hypoxia and metabolic reprogramming in TME: from immunosuppressive microenvironment to clinical application. J Hematol Oncol. 2022;15(1):84. doi:10.1186/s13045-022-01304-5
31. Li C, Liu FY, Shen Y, Tian Y, Han FJ. Research progress on the mechanism of glycolysis in ovarian cancer. Front Immunol. 2023;14:1284853. doi:10.3389/fimmu.2023.1284853
32. Pines D, Ditkovich J, Mukra T, et al. How Acidic Is Carbonic Acid? J Phys Chem B. 2016;120(9):2440–2451. doi:10.1021/acs.jpcb.5b12428
33. Liberti MV, Locasale JW. The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci. 2016;41(3):211–218. doi:10.1016/j.tibs.2015.12.001
34. Amoyav B, Bloom AI, Goldstein Y, et al. Drug-Eluting Porous Embolic microspheres for trans-arterial delivery of dual synergistic anticancer therapy for the treatment of liver cancer. Adv Healthc Mater. 2023;12(30):e2301548. doi:10.1002/adhm.202301548
35. Luo Y, Yang Y, Ye M, Zuo J. Targeting metabolic reprogramming promotes the efficacy of transarterial chemoembolization in the rabbit VX2 liver tumor model. Oncol Lett. 2024;27(3):111. doi:10.3892/ol.2024.14244
36. Liu J, Gao M, Yang Z, et al. Macrophages and metabolic reprograming in the tumor microenvironment. Front Oncol. 2022;12:795159. doi:10.3389/fonc.2022.795159
37. Wang N, Wang B, Maswikiti EP, et al. AMPK–a key factor in crosstalk between tumor cell energy metabolism and immune microenvironment? Cell Death Discov. 2024;10:237. doi:10.1038/s41420-024-02011-5
38. Wang S, Liu G, Li Y, Pan Y. Metabolic reprogramming induces macrophage polarization in the tumor microenvironment. Front Immunol. 2022;13:840029. doi:10.3389/fimmu.2022.840029
39. Goldmann O, Medina E. Metabolic pathways fueling the suppressive activity of myeloid-derived suppressor cells. Front Immunol. 2024;15:1461455. doi:10.3389/fimmu.2024.1461455
40. Cui H, Xie N, Banerjee S, et al. Lung Myofibroblasts promote macrophage profibrotic activity through lactate-induced histone lactylation. Am J Respir Cell Mol Biol. 2021;64(1):115–125. doi:10.1165/rcmb.2020-0360OC
41. Sun S, Xu X, Liang L, et al. Lactic acid-producing probiotic saccharomyces cerevisiae attenuates ulcerative colitis via suppressing macrophage pyroptosis and modulating gut microbiota. Front Immunol. 2021;12:777665. doi:10.3389/fimmu.2021.777665
42. Chu X, Di C, Chang P, et al. Lactylated histone H3K18 as a potential biomarker for the diagnosis and predicting the severity of septic shock. Front Immunol. 2021;12:786666. doi:10.3389/fimmu.2021.786666
43. Wang J, Yang P, Yu T, et al. Lactylation of PKM2 suppresses inflammatory metabolic adaptation in pro-inflammatory macrophages. Int J Biol Sci. 2022;18(16):6210–6225. doi:10.7150/ijbs.75434
44. Gu J, Xu X, Li X, et al. Tumor-resident microbiota contributes to colorectal cancer liver metastasis by lactylation and immune modulation. Oncogene. 2024;43(31):2389–2404. doi:10.1038/s41388-024-03080-7
45. Ding D, Li W, Ren J, et al. SPP1high macrophage-induced T-cell stress promotes colon cancer liver metastasis through SPP1/CD44/PI3K/AKT signaling. J Immunother Cancer. 2025;13(10):e012330. doi:10.1136/jitc-2025-012330
46. Li Y, Mo H, Wu S, Liu X, Tu K. A novel lactate metabolism-related gene signature for predicting clinical outcome and tumor microenvironment in hepatocellular carcinoma. Front Cell Dev Biol. 2021;9:801959. doi:10.3389/fcell.2021.801959
47. Qin X, Sun H, Hu S, Pan Y, Wang S. A hypoxia–glycolysis–lactate-related gene signature for prognosis prediction in hepatocellular carcinoma. BMC Med Genomics. 2024;17:88. doi:10.1186/s12920-024-01867-x
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Effects of Early TACE Refractoriness on Survival in Patients with Hepatocellular Carcinoma: A Real-World Study
Yang C, Luo YG, Yang HC, Yao ZH, Li X
Journal of Hepatocellular Carcinoma 2022, 9:621-631
Published Date: 21 July 2022
Comparison of the Efficacy and Safety of Transarterial Chemoembolization with or without Lenvatinib for Unresectable Hepatocellular Carcinoma: A Retrospective Propensity Score–Matched Analysis
Chen YX, Zhang JX, Zhou CG, Liu J, Liu S, Shi HB, Zu QQ
Journal of Hepatocellular Carcinoma 2022, 9:685-694
Published Date: 1 August 2022
Retrospective Study of the Efficacy and Safety of Chemoembolization with Drug-Eluting Microspheres Combined with Intra-Arterial Infusion of Bevacizumab for Unresectable Hepatocellular Carcinoma
Ueda S, Hori S, Hori A, Makitani K, Wan K, Sonomura T
Journal of Hepatocellular Carcinoma 2022, 9:973-985
Published Date: 12 September 2022
Safety and Effectiveness of Transarterial Chemoembolization in Hepatocellular Carcinoma Patients Aged Greater versus Less Than 80 Years
Zhang L, Hong W, Wang Z, Zheng C, Liang B, Shi H
Clinical Interventions in Aging 2023, 18:1883-1892
Published Date: 14 November 2023
Adjuvant Transarterial Chemoembolization Plus Immunotherapy for Huge Hepatocellular Carcinoma: A Propensity Score Matching Cohort Study
Huang H, Liao W, Zhang K, Wang H, Cheng Q, Mei B
Journal of Hepatocellular Carcinoma 2024, 11:721-735
Published Date: 8 April 2024