Back to Journals » Journal of Inflammation Research » Volume 15
P2X7R-NEK7-NLRP3 Inflammasome Activation: A Novel Therapeutic Pathway of Qishen Granule in the Treatment of Acute Myocardial Ischemia
Authors Li Y ![]() , Sun X
, Sun X ![]() , Liu X
, Liu X ![]() , Li J
, Li J ![]() , Li X
, Li X ![]() , Wang G, Liu Y
, Wang G, Liu Y ![]() , Lu X
, Lu X ![]() , Cui L
, Cui L ![]() , Shao M, Wang Y, Wang W, Li C
, Shao M, Wang Y, Wang W, Li C
Received 9 May 2022
Accepted for publication 26 August 2022
Published 13 September 2022 Volume 2022:15 Pages 5309—5326
DOI https://doi.org/10.2147/JIR.S373962
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Adam Bachstetter
Yanqin Li,1,* Xiaoqian Sun,1,* Xiangning Liu,1,* Junjun Li,2 Xuan Li,1 Gang Wang,1 Yizhou Liu,1 Xiangyu Lu,2 Lingwen Cui,2 Mingyan Shao,3 Yong Wang,1,3,4 Wei Wang,1,4,5 Chun Li2,4
1College of Chinese Medicine, Beijing University of Chinese Medicine, Beijing, 100029, People’s Republic of China; 2Modern Research Center for Traditional Chinese Medicine, School of Chinese Medicine, Beijing University of Chinese Medicine, Beijing, 100029, People’s Republic of China; 3School of Life Sciences, Beijing University of Chinese Medicine, Beijing, 100029, People’s Republic of China; 4Beijing Key Laboratory of TCM Syndrome and Formula, Beijing University of Chinese Medicine, Beijing, 100029, People’s Republic of China; 5Guangzhou University of Chinese Medicine, Guangdong, 510006, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Wei Wang, Guangzhou University of Chinese Medicine, Guangdong, 510006, People’s Republic of China, Tel +86 13910026960, Email [email protected] Chun Li, Modern Research Center for Traditional Chinese Medicine, School of Chinese Medicine, Beijing University of Chinese Medicine, Beijing, 100029, People’s Republic of China, Tel +86 15810068615, Email [email protected]
Background: Acute myocardial ischemia (AMI) is a common heart disease with increasing morbidity and mortality year by year. Persistent and sterile inflammatory infiltration of myocardial tissue is an important factor triggering of acute myocardial ischemia secondary to acute myocardial infarction, and NLRP3 inflammasome activation is an important part of sterile inflammatory response after acute myocardial ischemia. Previous studies have shown that Qishen granule (QSG) can significantly inhibit the inflammatory injury of myocardial tissue caused by ischemia, but its effect and specific mechanism of inhibiting the activation of NLRP3 inflammasome have not been reported. This study was to investigate the specific mechanism of QSG inhibiting inflammation after AMI, and to validate the possible targets.
Methods: The myocardial ischemia model in mice was established by ligation of the left anterior descending coronary artery. Echocardiography was used to evaluate the cardiac function of the mice. Plasma CK-MB and cTnl were detected by ELISA to evaluate the degree of myocardial injury. The extent of myocardial tissue inflammation in mice was assessed by HE staining and immunohistochemistry of IL-18, IL-1β. The expressions of NLRP3, ASC, Caspase-1, and CD86 were detected by immunofluorescence; detection of key pathway proteins P2X7R, NEK7, NLRP3, ASC, Caspase-1, and effector proteins IL-18, IL-1β by Western blot. In vitro experiments, ATP+LPS was used to construct a RAW264.7 macrophage NLRP3 inflammasome activation model. Immunofluorescence and Western blot analysis were performed to detect the expression of NLRP3 pathway activator and effector proteins. Plasmid-transfected P2X7R overexpression and immunoprecipitation assays were used to evaluate the QSG-regulated NLRP3 inflammasome activation pathway.
Results: QSG rescued cardiac function and further reduced inflammatory effects in mice by inhibiting NLRP3 inflammasome activation. In vitro, QSG inhibited LPS combined with ATP-induced NLRP3 inflammasome activation in RAW264.7 macrophages by downregulating the expression of NLRP3 inflammasome key pathway proteins. In addition, inhibition or overexpression of P2X7R in RAW264.7 macrophages and immunoprecipitated protein interactions further confirmed that QSG reduces macrophages inflammasome activation via the P2X7R-NEK7-NLRP3 pathway.
Conclusion: P2X7R-NEK7-NLRP3 inflammasome activation is a novel therapeutic mechanism of QSG in the treatment of acute myocardial ischemia.
Keywords: acute myocardial ischemia, inflammation, macrophages, P2X7R-NEK7, NLRP3 inflammasome, Qishen granule
Introduction
Acute myocardial ischemia (AMI) is a pathological condition that reduces blood perfusion due to the interruption of coronary artery blood circulation, which cannot provide the energy needed for heart work.1–3 AMI-induced myocardial ischemia-hypoxic injury results in the release of various endogenous damage-associated molecular patterns (DAMPs), such as adenosine triphosphate (ATP), mitochondrial proteins and nucleotides, which are recognized by innate pattern recognition receptors (PRRs) expressed by inflammatory cells, leading to increased release of sterile inflammatory responses and pro-inflammatory cytokines.4,5 Studies have shown that when AMI occurs, inflammatory cells are rapidly recruited to the area of myocardial injury, reaching a peak about 3 d after AMI.6,7 Macrophages are the main innate inflammatory cells that respond to DAMPs and promote the activation of multiple inflammatory signaling pathways and lead to the production of pro-inflammatory cytokines such as interleukin-1β (IL-1β) and interleukin-18 (IL-18), which further exacerbating the inflammatory response in myocardial tissue. Research has shown that a moderate inflammatory response is essential for myocardial tissue repair after acute myocardial ischemia, whereas persistent inflammatory responses continue to exacerbate myocardial tissue damage in non-infarcted region and ultimately increases the risk of heart failure.7–9 Thus, regulating inflammation can effectively improve the prognosis of AMI patients.10
Nucleotide-binding oligomerization domain-like receptor protein3 (NLRP3) inflammasome is an intracellular macromolecular complex composed of receptor protein NLRP3, apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) and pro-cysteinyl aspartate specific pro-teinase 1 (pro-Caspase-1), that is activated upon invasion by pathogens or damage to body tissue/cells.11,12 Bacterial and viral products, such as lipopolysaccharide (LPS), act as pathogen associated molecular patterns (PAMPs) of the NLRP3 inflammasome during pathogen invasion; while during tissue/cell injury, intracellularly derived molecules act as DAMPs to activate the NLRP3 inflammasome to induce subsequent inflammatory responses.13 The classical activation of the NLRP3 inflammasome requires two processes: the priming phase and the activation phase. Activation of the TLR4-MyD88-NF-κB signaling pathway during the priming phase resulted in increased transcription of NLRP3, pro-IL −18 and pro-IL-1β genes. The activation stage is based on the priming stage, which promotes the assembly of NLRP3, ASC and pro-Caspar-1 under the continuous stimulation of PAMPs and DAMPs. The purinergic ligand-gated ion channel 7 receptor (P2X7R) channels on the cell membrane open, leading to the outflow of intracellular potassium ions, while NIMA-related kinase7 (NEK7) is sensitive to extracellular potassium concentration and can bind to NLRP3, thereby promoting NLRP3 oligomerization, inducing NLRP3-ASC-pro-Caspase-1 assembly, inflammasome activation, Caspase-1 activation and increased release of pro-inflammatory cytokines IL-18 and IL-1β.14–16 Studies have shown that NLRP3 inflammasome activation in macrophages is associated with initiation of inflammatory responses after AMI17,18.
The Chinese compound QSG is based on the classical formula Zhen Wu Tang and Si Miao Yong An Tang, which consists of six herbs, namely Astragalus camptoceras Bunge [Fabaceae], Salvia miltiorrhiza Bunge [Lamiaceae], Lonicera japonica Thunb. [Caprifoliaceae], Scrophularia ningpoensis Hemsl. [Scrophulariaceae], Aconitum carmichaelii Debeaux [Ranunculaceae] and Glycyrrhiza uralensis Fisch. ex DC. [Fabaceae], in the ratio of 30:15:10:10:10:9:6. It is effective in benefiting Wenyan Yiqi, and Huoxue Jiedu.19,20 The previous experimental studies of the research group confirmed that QSG can improve the cardiac function of mice after acute myocardial infarction, can effectively regulate the release and recruitment of macrophages, reduce the infiltration of macrophages in myocardial tissue in ischemic areas, and reduce inflammation.21 Meanwhile, we found in vivo and in vitro studies that Calycosin, the active ingredient of Astragalus, the main drug in QSG, could reduce inflammation and fibrosis caused by heart failure by inhibiting PI3K and regulating the AKT-IKK/STAT3 axis22.
To further clarify the protective mechanism of QSG against acute myocardial ischemic injury. This experiment focused on the inflammatory response induced by NLRP3 inflammasome activation in macrophages after QSG intervenes in acute myocardial ischemia, combined with NLRP3 inflammasome activation model of RAW264.7 macrophages induced by LPS combined with ATP in vitro, and further used overexpression of P2X7R on RAW264 0.7 macrophages is expected to explore the mechanism by which QSG regulates NLRP3 inflammasome activation, and provide new drug targets and approaches for the treatment of clinical AMI.
Materials and Methods
Drug
QSG consists of six Chinese herbs: Astragalus camptoceras Bunge [Fabaceae], Salvia miltiorrhiza Bunge [Lamiaceae], Lonicera japonica Thunb. [Caprifoliaceae], Scrophularia ningpoensis Hemsl. [Scrophulariaceae], Aconitum carmichaelii Debeaux [Ranunculaceae] and Glycyrrhiza uralensis Fisch. ex DC. [Fabaceae], purchased from Beijing Tong Ren Tang Pharmaceutical Co., Ltd., and processed by the Beijing University of Chinese Medicine. The brief processing is as follows: add the appropriate amount of water and decoct, extract 3 times, combine the extracts and filter, collect the filtrate, concentrate in ethanol, refrigerate and leave for 24 h, filter, collect the filtrate, concentrate in ethanol to a suitable thick paste, dry under reduced pressure, crush and sieve, dry, separate and store. The QSG used in this experiment belonged to the same batch as in the published related study.20
MCC950 (M6164, AbMole, USA), was purchased from Biorain (Beijing) Co., Ltd. It is a specific inhibitor of NLRP3 inflammasome activation and was used as a positive drug in this experiment.
AZ10606120 (AZ) (CAS No. 607378-18-7, Tocris, England), an inhibitor of P2X7R, was used as a positive agent in cellular assays. It was purchased from Biorain (Beijing) Co., Ltd.
Reagents
LPS (l2880, Biodee, China), ATP (10519979001, Roche, Germany), Pentobarbital sodium powder (Tc-p8411, Merck, Germany), 4% tissue cell fixative (paraformaldehyde) (p6148, Biodee, China), Phosphate Buffer Solution (PBS) (yz-bw013004, Solarbio, China), 0.9% normal saline (b020, Jiancheng, Nanjing, China).
Animals and Ethics Statement
All animal experimental protocols in this study were approved by the Animal Ethics Committee of Beijing University of Chinese Medicine (approval number “BUCM-4-2022040605-2006”) and conformed to the “Guidelines for the Care and Use of Laboratory Animals” published by the National Institute of Health (NIH Publication No. Resolution No. 85–23, revised in 1996). Fifty healthy male ICR mice (28 ± 2 g) in Specific Pathogen Free (SPF) grade were purchased from Beijing Spefo Technology Co., Ltd. The animal feeding conditions were 12 h of light/dark cycle, humidity 55 ± 5%, constant temperature 25°C, feeding adaptively for 3 d before the experiment.
Animal Model of Acute Myocardial Ischemia
AMI was induced by ligation of the left anterior descending (LAD) coronary artery. After the mice were anesthetized by intraperitoneal injection of 0.5% sodium pentobarbital (50 mg/kg). Mice were firstly fixed in the supine position on a mouse plate, and then skin preparation, sterilization, tracheal intubation, and connection to a small animal ventilator were performed in the left thoracoabdominal area, followed by open-heart surgery at the third/fourth rib space on the left side. After exposing the heart, the LAD was ligated with a 7–0 sterile suture between the left auricle and the pulmonary artery cone, 1–1.5 mm below the left auricle. Immediately whitening of the heart tissue at the ligation site was observed as a sign of successful surgery. Mice that did not undergo LAD ligation but underwent the same operation were regarded as the sham operation (sham) group. After the mice resumed spontaneous breathing, they were randomly divided into sham group, model group, QSG group, and NLRP3 inflammasome inhibitor (MCC950) group. In vivo, the dose of Qishen granule (QSG) was converted from the clinically administered effective dose, and the ascending dose of lyophilized powder of QSG was 9.33g/kg, with a paste yield of 30.3%. The dose of MCC950 was determined in the animal experiments according to the instructions provided by the manufacturer and the dosing method.
24 h after LAD ligation, QSG freeze-dried powder at a concentration of 566 mg/mL (dose 5.66 g/kg) was given to the QSG group by gavage according to the standard 0.1 mL/10g. MCC950 at a concentration of 1 mg/mL (dose 10 mg/kg) was given to the MCC950 group by intraperitoneal injection according to the same standard. Mice in the sham and model groups were gavaged the same volume of saline was administered for 3 d at a fixed time every day.
Echocardiographic Assessment
As mentioned before,23 after 3 d of the administration, the mice in each group were anesthetized by breathing with isoflurane, and M- and B-mode echocardiograms of the short axis of the heart of each group of mice were obtained at the level of the papillary muscle with the MS-400 probe included in the Vevo 2100 (Vevo TM 2100, Visual Sonics, Canada) next to the left sternum, respectively. During the detection process, the heart rate of mice was ensured to be maintained between 450 and 550. The left ventricular anterior wall; diastole (LVAW; d), left ventricular anterior wall; systole (LVAW; s), left ventricular posterior wall; diastole (LVPW; d), left ventricular posterior wall; systole (LVPW; s) decreased, while left ventricular internal dimension; diastole (LVID; d), and left ventricular internal dimension; systole (LVID; s) values were measured, and the left ventricular ejection fraction (LVEF), left ventricular fractional shortening (LVFS) values were calculated using the ultrasound data analysis software Vevo 2100. The above indicators were used to evaluate the cardiac function of the mice in each group.
Detection of Myocardial Injury Markers
After blood collection from the abdominal aorta of the mice in each group, they were left at room temperature for 2 h, centrifuged at 3000 rpm and 4°C for 10 min using a low-temperature high-speed centrifuge, and the supernatant was taken. Then, according to the instructions of the ELISA kit, the levels of creatine kinase-MB (CK-MB) (SEA479Mu, Cloud-clone, China) and cardiac troponin I (cTnI) (SEA478Mu, Cloud-clone, China) were measured.
Hematoxylin-Eosin Staining and Immunohistochemical Staining
The heart tissues were treated with 4% paraformaldehyde after being removed from mice. Then the myocardial tissue was embedded in paraffin and sectioned. The basic structure of heart tissue was evaluated by Hematoxylin-Eosin (HE) staining to evaluate the degree of myocardial tissue injury.
Heart sections were dewaxed in xylene, rehydrated in an alcohol gradient, and then rinsed with PBS. After blocking with 0.3% hydrogen peroxide for 15 min, the sections were incubated with primary antibodies anti-IL-1β antibody (AF5103, Affinity, USA), anti-IL-18 antibody (10663-1-AP, Proteintech, China) overnight at 4°C. The sections were then incubated with secondary antibodies for 1 h protected from light, stained with hematoxylin for 30s, dehydrated with ethanol and xylene, and fixed with resin gel. Positive areas were clearly stained brownish-yellow.
Immunofluorescence Staining
The antibodies used for immunofluorescence double-staining or triple-staining of mice heart tissue sections in each group are as follows: anti-CD86 antibody (ab119857, Abcam, USA), anti-Caspase-1 antibody (ab1872, Abcam, USA), anti-NLRP3 antibody (ab214185, Abcam, USA), anti-ASC-antibody (sc-514414, Santa Cruz, USA), anti-Caspase-1 antibody (ab1872, Abcam, USA), Alexa Fluor 488 (D001-34, Abcam, USA). DAPI (c1002, Beyotime, China) stained cell nucleus. The brief process is as follows: dewaxing, rehydration, repair, blocking, primary antibody incubation at 4°C overnight, PBS washing, secondary antibody incubation in the dark, DAPI staining for cell nuclei, and mounting.
RAW264.7 macrophages were seeded in confocal dishes, after stimulated administration, fixed with 4% paraformaldehyde for 15 min, and permeabilized with 0.5% TritonX-100 for 20 min. Then, cells were blocked with 1% BSA in the 37°C incubator for 1 h. Incubation with primary antibody at 4°C overnight, followed by incubation with the secondary antibody for 1 h at room temperature away from light. Then, wheat germ agglutinin (WGA) (AC15L012, Life-iLab, China), labeled with AF488, stained cell membrane for 20 min. Finally, nuclei were stained with DAPI for 5 min. The primary antibodies used are as follows: anti-Caspase-1 antibody (ab1872, Abcam, USA), anti-P2X7R antibody (APR-008, Alomone, Israel).
Detection of Inflammatory Cytokines IL-18 and IL-1β by ELISA
The myocardial tissue of each group of mice was lysed, centrifuged at 12,000 rpm, 4°C for 10 min, and the supernatant of myocardial tissue homogenate was extracted. Then the levels of inflammatory factors IL-18 (pi553, Beyotime, China) and IL-1β (ek0394, Boster, China) in myocardial tissue homogenate were measured according to the instructions of EILSA kit.
Cell Culture
RAW264.7 macrophages were obtained from China Infrastructure of Cell Line Resources (Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences). Cells were incubated with complete medium containing 89% high sugar medium DMEM (11995065, Gibco, USA), 10% fetal bovine serum (FBS) (10099141, Gibco, USA) and 1% penicillin/streptomycin (P/S) (15140122, Invitrogen, USA) at 37°C, 5% conditioned cell incubator.
Establishment of NLRP3 Inflammasome Activation Model and Drugs Treatment Options
RAW264.7 macrophages were inoculated at a density of 6×103 in 96-well plates at 100 μL/well. After 24 h of culture, the cell density was observed under a microscope when the cell density reached 70–80%, and subsequent corresponding treatments were carried out. The cells were divided into control group and model group. The control group was not treated, and the model group was given 10 μg/mL of LPS first, before giving 5 mM of ATP stimulation, the CCK-8 (g021-2-1, njjcbio, China) cell viability assay and immunofluorescence were used to verify whether the NLRP3 inflammasome activation model was successfully established.
To screen the non-toxic concentration range of QSG, the concentration gradient of QSG was set to 1–1200 μg/mL based on our previous experience, and then cell viability was determined using the CCK-8 assay kit according to the manufacturer’s instructions. The effective concentration range of QSG in LPS combined with ATP stimulation of RAW264.7 macrophages to induce NLRP3 inflammasome activation was further determined. Similarly, the optimal treatment concentration of the inhibitor MCC950 was screened out by the NO kit assay (s0021s, Beyotime, China), and the optimal treatment concentration of the inhibitor AZ was screened out by the CCK-8 assay. All the above results were read with the enzyme standard for the average optical density value, CCK-8 results were read at 450 nm with the enzyme standard, and NO assay results were read at 540 nm.
Detection of Potassium Ion Concentration in Cell Supernatant
The concentration of free potassium ions in the supernatant of RAW264.7 macrophages was measured using a potassium kit (C001-2-1, njjcbio, China) according to the manufacturer’s protocol. After each group of RAW264.7 cells was treated, 50 μL/well of supernatant was taken for the determination. Control wells, standard wells and sample wells were set up according to the instructions, and deionized water, potassium standard solution, supernatant of the sample to be measured and working solution were added in turn, mixed well, and then placed at room temperature for 5 min, and the absorbance of each well was measured at 450 nm by the enzyme standardizer.
Overexpression of P2X7R Plasmid
Following the manufacturer’s instructions, use mP2X7R pcDNA3.1–3xFlag-T2A-EGFP (211208PC02, Hanbio, China) to overexpress P2X7R. Briefly, RAW264.7 macrophages were plated into 6 mm plates for 24 h, and the medium was replaced with Opti-MEM (31985070, Gibco, USA) 2 h before transfection. Next, cells were transfected with 8 μg plasmid (mP2X7R pcDNA3.1–3xFlag-T2A-EGFP or pcDNA3.1–3xFlag-T2A-EGFP) and 20 μL LipoFiterTM 3.0 (HB-TLRF3-1000, Hanbio, China)/well for 4–6 h. Then, the medium was replaced with DMEM (10% FBS; no P/S). After 48 h, immunofluorescence verification was performed, and proteins were extracted from cells for Western blot analysis.
Western Blot and Immunoprecipitation
Cardiac tissue and cellular proteins were extracted from RIPA buffer (c1053-100, APPLYGEN, China), with a 0.5% protease inhibitor (p1625, APPLYGEN, China) and 1% phosphatase inhibitor (p1260-1, APPLYGEN, China). The protein concentration of each sample was measured using a BCA kit (P1511-1, APPLYGEN, China). Western blot (WB) analysis was conducted using anti-NLRP3 (ab214185, Abcam, USA), anti-ASC (sc-514414, Santa Cruz, USA), anti-Capase-1 (ab1872, Abcam, USA), anti-IL-18 (10663-1-AP, Proteintech, China), anti-IL-1β (AF5103, Affinity, USA), anti-P2X7R (APR-008, Alomone, Israel), anti-NEK7 (ab133514, Abcam, USA) and anti-GAPDH (AB0037, Abways, China) at 4°C overnight. Then the membrane was washed 3 times by TBST solution (1×), finally secondary antibodies Goat Anti-Rabbit IgG (H+L) HRP (AB0101, Abways, China), Goat Anti-Mouse IgG (H+L) HRP (AB0038, Abways, China) were incubated on membrane for 1h, ECL (SQ201, Epizyme, China) was used for detecting the expression. GAPDH was used to normalize the expression content of each protein. Image Lab software was used for band gray analysis.
Immunocomplexes were prepared by incubating RAW264.7 macrophages protein extracts after transfection with P2X7R plasmid with 1–2 μg NEK7 (B-5) antibody (sc-393539, Santa Cruz, USA) overnight at 4°C, the immunocomplexes were then mixed with Protein G Magnetic Beads (70024, CST, USA) and incubated at room temperature for 20 min. After resuspension of the precipitate in 20–40 μL of 3X SDS sample buffer, the samples were denatured in a metal bath at 100°C for 5 min. Finally, the samples were cooled and stored in a −20°C refrigerator for later protein WB experiments. Normal Rabbit IgG (2729, CST, USA) is an isotype control antibody used to detect non-specific binding of target primary antibodies induced by Fc receptor binding or other protein–protein interactions.
Statistical Analysis
All data were analyzed and plotted using GraphPad Prism 7.0 and SPSS software, and the analyzed data were expressed as mean ± standard deviation. If the data conform to a normal distribution and the variance test is homogeneous, the LSD-t-test and one-way analysis of variance were used to compare the differences among multiple groups; if the data are not normally distributed or the variance test is homogeneous among groups, use a nonparametric test. P < 0.05 indicates a statistically significant difference, P < 0.01 and P < 0.001 indicate a statistically significant difference.
Results
Effects of QSG on Cardiac Function in AMI Mice in Each Group
We prepared a model of AMI in mice using LAD ligation after 3 d of adaptive feeding. As shown in (Figure 1A), QSG was administered by oral gavage, and NLRP3 inhibitor (MCC950) by intraperitoneal injection from 4 d - 7 d, subsequent cardiac echocardiography, and acquisition of relevant parameters. After 3 d pharmacological intervention, echocardiography showed that compared with the sham group, the values of LVID; d and LVID; s in the model group increased, and the values of LVEF, LVFS, LVAW; d, LVAW; s, LVPW; d, LVPW; s decreased (P < 0.001), indicating impaired left ventricular systolic and diastolic function in mice after AMI (Figure 1B). After QSG treatment, LVEF and LVFS increased significantly compared with the model group (P<0.001), suggesting that QSG treatment can effectively improve cardiac function in mice with AMI. The positive drug MCC950, an NLRP3 inflammasome inhibitor, was also shown to improve cardiac function in mice with AMI (Figure 1B).
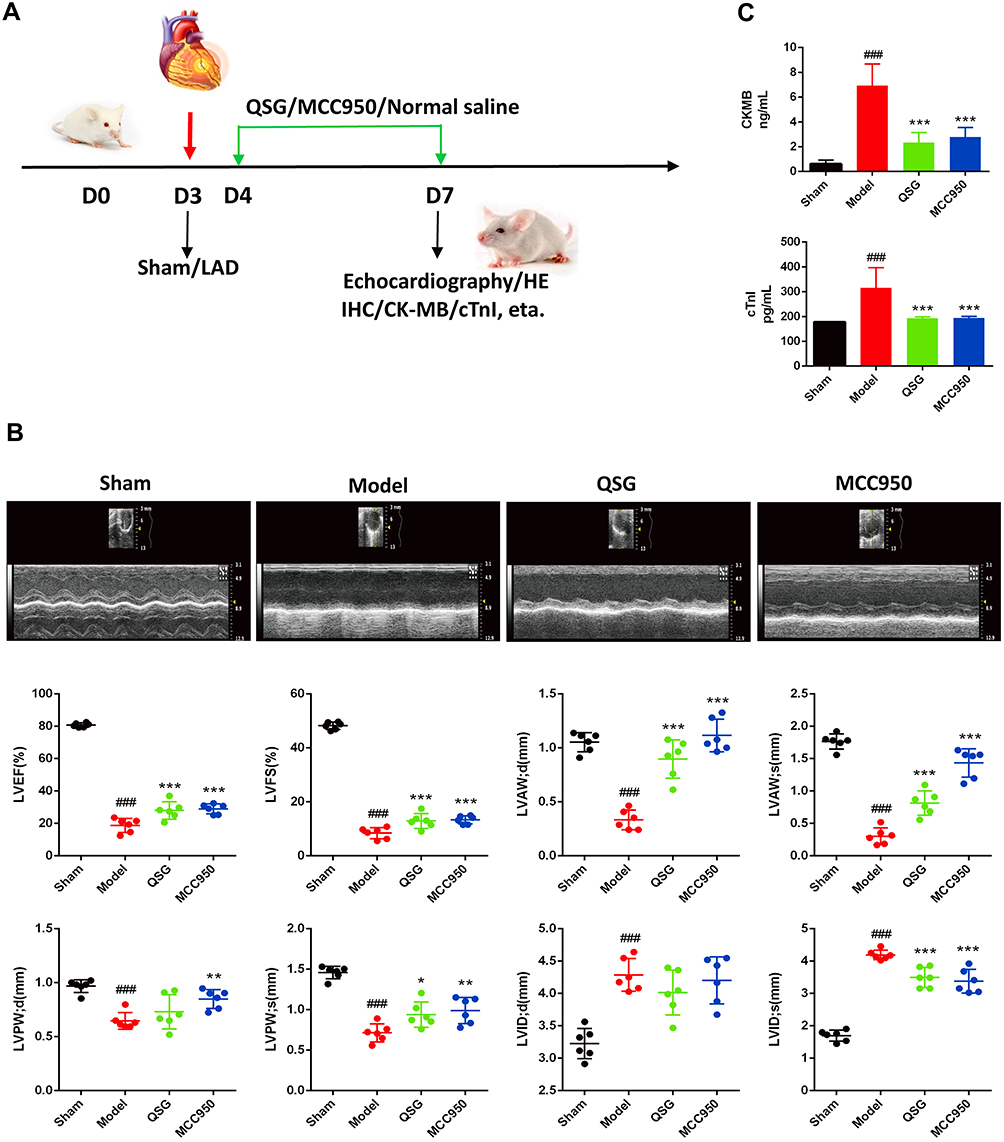 |
Figure 1 QSG improved cardiac function in mice after AMI. (A) Experimental protocol for QSG studies in AMI mice. Experimental protocol for QSG studies in AMI mice, LAD ligation was used to made AMI model in mice after 3 d of adaptive feeding, QSG were administered by oral gavage, and NLRP3 inhibitor (MCC950) by intraperitoneal injection from 4 d - 7 d, subsequent cardiac echocardiography and acquisition of relevant parameters. (B) Representative echocardiographic images of mice in each group and analysis of LVEF, LVFS, LVAW; d, LVAW; s, LVPW; d, LVPW; s, LVID; d, LVID; s levels after QSG treatment. (C) ELISA of plasma CKMB and cTnI levels. N = 6 per group. ###P < 0.001 vs sham group, *P < 0.05, **P < 0.01, ***P < 0.001 vs model group. |
We further examined the effect of QSG on cardiac injury in mice with AMI. The plasma CK-MB and cTnI levels were detected in each group (Figure 1C). Results showed that CK-MB and cTnl levels were increased in the model group compared with the sham group (P < 0.001), suggesting that the myocardial injury in the model group of mice was serious. While compared with the model group, QSG and MCC950 groups reduced the plasma levels of cardiac injury markers CK-MB and cTnI, which indicated that QSG could reduce myocardial injury in mice after AMI.
Effects of QSG on Myocardial Tissue of AMI Mice in Each Group
HE staining was used to evaluate myocardial tissue injury in each group of AMI mice. Results are shown in Figure 2A. In the sham group, myocardial cells were intact, nuclei were normal, and myocardial fibers were neatly arranged, and no inflammatory cell infiltration was seen. Compared with the sham group, the model group myocardial cells were disordered, nuclei were fixed, myocardial fibers were unclear or disappeared, and there was a large amount of inflammatory cell infiltration in the myocardial interstitium. While myocardial tissue of mice in the QSG group showed the integrity of myocardial cells and the neatness of myocardial fiber arrangement, and the degree of inflammatory cell infiltration in the myocardial interstitium was reduced. Meanwhile, MCC950 had the same effect.
 |
Figure 2 QSG attenuated myocardial tissue inflammatory injury in AMI mice. (A) Representative heart images for each group of HE staining, Scale bar=20 µm. N = 3 per group. (B and C) IHC of IL-18 and IL-1β images of AMI mice in each group and analysis, Scale bar=20 µm. N = 3 per group. (D) ELISA results of IL-1β and IL-18 in cardiac tissue homogenates. N = 6 per group. ###P < 0.001 vs sham group, ***P < 0.001 vs model group. |
Furthermore, we detected the expression and distribution of IL-1β and IL-18 in the myocardial tissues of each group by immunohistochemistry (IHC) (Figure 2B and C). Compared with the sham group, the expression of IL-1β and IL-18 in cardiomyocytes was elevated in the model group. It was reduced in the QSG and MCC950 groups compared with the model group. To confirm this phenomenon, the levels of inflammatory factors IL-18 and IL-1β in myocardial tissue homogenates in each group were measured by ELISA (Figure 2D), and the results were similar to those of IHC. Up to this point, we found that QSG protected myocardial tissue from AMI injury with the same pharmacodynamic potency as the NLRP3 inhibitor MCC950, which suggests that QSG protects against inflammatory injury in myocardial tissue may be associated with inhibition of NLRP3 inflammasome.
Effects of QSG on NLRP3 Inflammasome Activation in Myocardial Tissue Macrophages of AMI Mice
To directly clarify the presence of NLRP3 inflammasome activation in myocardial tissue, three antibodies, NLRP3, ASC, and Caspase-1, were used for immunofluorescence triple labeling, which showed red, green, and pink fluorescence, respectively, and DAPI-stained nuclei showed blue fluorescence (Figure 3A). Compared with the sham group, the intensity of red, green and pink fluorescence in myocardial tissues was enhanced in the model group; while compared with the model group, the intensity of red, green and pink fluorescence in myocardial tissues of mice in the QSG and MCC950 groups was decreased. This result showed that NLRP3 inflammasome was activated in myocardial tissues of mice with AMI, and QSG could inhibit NLRP3 inflammasome activation.
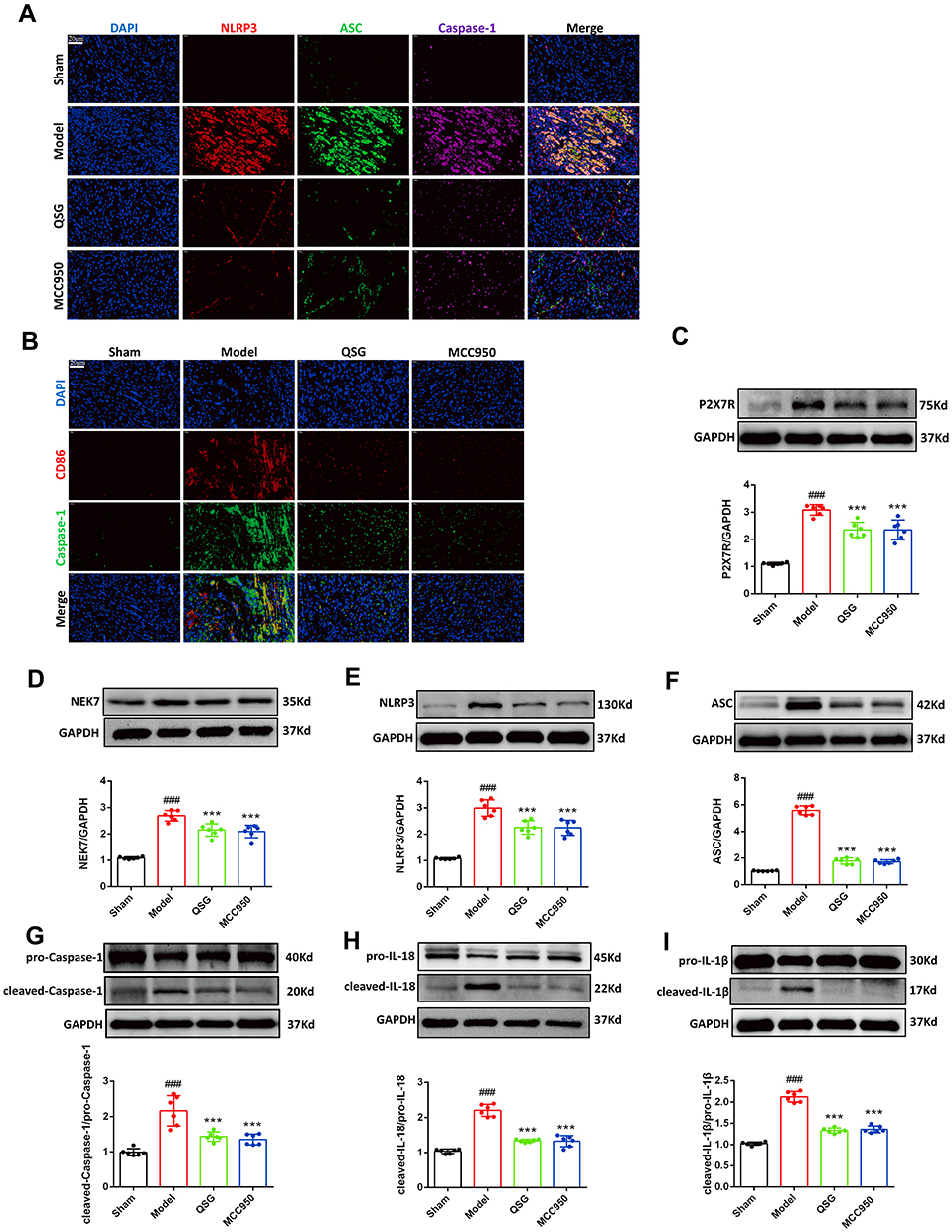 |
Figure 3 QSG inhibited NLRP3 inflamat some activation in cardiac tissue macrophages. (A) Representative NLRP3/ASC/Caspase-1 immunofluorescent staining images of AMI mice in each group. Scale bar=20 µm. (B) Representative CD86/Caspase-1 immunofluorescent staining images of AMI mice in each group. Scale bar=20 µm. (C–I) Western blot analysis showed that QSG treatment reduced the expression of P2X7R, NEK7, NLRP3, ASC, Caspase-1, IL-18, IL-1β in cardiac tissue macrophages. Proteins had been normalized to GAPDH, N = 6 per group. ###P < 0.001 vs sham group, ***P < 0.001 vs model group. |
NLRP3 inflammasome activation induces an inflammatory response after AMI, further aggravating the myocardial injury in the ischemic zone.24,25 Components of NLRP3 inflammasome, such as NLRP3, ASC, and Caspase-1, are mainly expressed in inflammatory cells, especially macrophages26. Therefore, based on the pre-subject basis combined with the literature review, CD86-labeled pro-inflammatory macrophages (red fluorescence) and Caspase-1 (green fluorescence) were used to indirectly represent NLRP3 inflammasome activation. Immunofluorescence double-staining was used to observe the activation of macrophages and NLRP3 inflammasome in myocardial tissue of AMI mice in each group (Figure 3B). Compared with the sham group, the red and green fluorescence intensities in the model group were increased. However, with the administration of QSG, the red and green fluorescence intensities in the myocardial tissue were weakened compared with the model group. MCC950, a specific inhibitor of NLRP3 inflammasome activation, was shown to inhibit NLRP3 inflammasome activation in macrophages.
Next, we detected proteins associated with NLRP3 inflammasome activation in myocardial tissue by WB (Figure 3C–I). P2X7R and NEK7 are key factors in the activation of NLRP3 inflammasome and participate in the activation signal stage of NLRP3 inflammasome activation. The increased protein expression of P2X7R could induce the binding of NEK7 to NLRP3, and promote the assembly of NLRP3, ASC and pro-Caspase-1, resulting in the activation of the NLRP3 inflammasome. The WB results are shown in Figure 3C and D. Compared with the sham group, the expression of P2X7R and NEK7 was increased in the model group (P < 0.001); while the expression of P2X7R and NEK7 was lower in the QSG and MCC950 groups compared with the model group (P < 0.001), indicating that QSG could inhibit the expression of P2X7R and NEK7 protein. The WB results of NLRP3 inflammasome activation proteins NLRP3, ASC, Caspase-1 and effector proteins IL-18 and IL-1β are shown in Figure 3E–I. The levels of NLRP3, ASC, Caspase-1, IL-18 and IL-1β proteins were significantly increased in the model group, while the QSG intervention reduced the expression of the above proteins.
QSG Inhibited NLRP3 Inflammasome Activation in RAW264.7 Cells
Based on the previous experiments combined with literature research, the group selected 10 μg/mL concentration of LPS combined with 5 mM concentration of ATP to stimulate RAW264.7 macrophages to induce NLRP3 inflammasome activation. But since the stimulation time of LPS and ATP was not clear, the intervention time points of 12 h and 24 h for LPS administration and 0.5 h and 1 h for ATP stimulation were set, respectively. The results of CCK8 assay are shown in Figure 4A. Compared with the control group, the model group treated with LPS (10 μg/mL, 24 h) combined with ATP (5 mM, 1 h) significantly reduced cell viability (P < 0.001). Further, immunofluorescence was used to verify the results obtained with CCK8, as shown in Figure 4B. WGA-stained cell membranes showed green fluorescence, Caspase-1 positivity showed red fluorescence, which represents the activation of NLRP3 inflammasome. The results showed that the intensity of Caspase-1 positive red fluorescence was enhanced by LPS (10 μg/mL, 24 h) + ATP (5 mM, 1 h), indicating the activation of a large number of NLRP3 inflammasome. Thus, a model of NLRP3 inflammasome activation in RAW264.7 macrophages induced by LPS combined with ATP was successfully established for the continuation of subsequent experiments.
 |
Figure 4 QSG inhibited LPS combined with ATP-induced NLRP3 inflammasome activation in RAW264.7 macrophages. (A) Establishment of LPS combined with ATP to induce NLRP3 inflammasome activation model. (B) Immunofluorescence determined that LPS in combination with ATP activates NLRP3 inflammasome. Scale bar=75 µm. (C) Safe concentration of QSG in RAW264.7 macrophages. (D) Experimental protocol for QSG in LPS combined with ATP to induce NLRP3 inflammasome activation in RAW264.7 macrophages. (E) Effective concentration of QSG in RAW264.7 macrophages. (F) MCC950 inhibitor concentration range in RAW264.7 macrophages. (G–M) Western blot analysis showed that QSG treatment reduced the expression of P2X7R, NEK7, NLRP3, ASC, Caspase-1, IL-18, IL-1β in RAW264.7 macrophages. N = 3 per group. ##P < 0.01, ###P < 0.001 vs sham group, *P < 0.05, **P < 0.01, ***P < 0.001 vs model group. |
Before evaluating the role of QSG on the regulation of NLPR3 inflammasome activation, CCK8 results showed that QSG was non-toxic when co-treated with RAW264.7 macrophages below 1200 μg/mL (Figure 4C). After successfully establishing a model of NLRP3 inflammasome activation in RAW264.7 macrophages induced by LPS combined with ATP, it is necessary to further determine the effective concentration of QSG under this model condition. The experimental protocol of QSG administration is shown in Figure 4D. The results of CCK8 assay are shown in Figure 4E, after administration of different concentrations of QSG (100, 200, 400, 600, 800, 1000 μg/mL), compared with the model group, the concentration of QSG was effective in the concentration range of 200–1000 μg/mL, and 800 μg/mL was the most effective (P < 0.001). Therefore, the administration concentration of QSG in the later cellular experiments was determined to be 800 μg/mL. At the same time, we determined the optimal concentration of the inhibitor MCC950 on cells at 10 μM (P < 0.001) by detecting NO release from RAW264.7 macrophages (Figure 4F).
Subsequently, under the conditions established above, the ability of QSG to inhibit NLRP3 inflammasome activation in RAW264.7 macrophages has been verified. Consistent with our prediction, WB results showed that the expression levels of NLRP3 inflammasome activation pathway-related proteins P2X7R, NEK7 and inflammasome activation proteins NLRP3, ASC, Caspase-1 and effector proteins IL-18 and IL-1β were significantly increased in the model group compared with the control group, and the expression of the above proteins was downregulated after QSG and MCC950 treatment (Figure 4H–M).
QSG Might Inhibit NLRP3 Inflammasome Activation via the P2X7R-NEK7-NLRP3 Pathway
Since P2X7R is a cell membrane potassium channel, NEK7 is sensitive to changes in intracellular potassium concentration, and thus P2X7R-NEK7-NLRP3 is a key pathway for NLRP3 inflammasome activation when extracellular ATP concentration increases. We measured the potassium ion concentration in RAW264.7 macrophages supernatant, and the results are shown in Figure 5A. Compared with the control group, the potassium ion in the cell supernatant of the model group was higher; after QSG and MCC950 intervention, the potassium ion could be down-regulated compared with the model group, and the effect of QSG combined with MCC950 was better. The above results suggested that QSG might downregulate NLRP3 inflammasome activation by inhibiting P2X7R. Therefore, we choose AZ10606120 (AZ), an inhibitor of P2X7R, as a positive control to further clarify whether QSG could exert an inhibitory effect on inflammasome activation through P2X7R. As shown in Figure 5B and C, the optimal concentration of AZ was first confirmed to be 50 μM by CCK8 and WB methods.
 |
Figure 5 QSG inhibited the P2X7R-NEK7-NLRP3 inflammasome pathway. (A) Determination of potassium ion concentration in RAW264.7 macrophages supernatant. (B) The effective concentration range of CCK8 to detect AZ. (C) Representative graph of optimal concentration of AZ for WB validation. (D) Representative immunofluorescence detection images of RAW264.7 macrophages P2X7R expression. Scale bar=25 µm. (E–H) Western blot analysis showed that both QSG and AZ treatment reduced the expression of P2X7R, Caspase-1, IL-18, IL-1β in RAW264.7 macrophages, N = 3 per group. ###P < 0.001 vs sham group, *P < 0.05, **P < 0.01, ***P < 0.001 vs model group. |
After the concentration of AZ administration was clarified, immunofluorescence and WB were used to detect the effect of QSG on the expression of P2X7R. The results are shown in Figure 5D, compared with the control group, the red fluorescence intensity increased in the model group, indicating that the P2X7R protein expression increased in the model group cells; while compared with the model group, the red fluorescence became weaker after QSG treatment, and similar to the fluorescence intensity of the AZ group. WB results are shown in Figure 5E, compared with the control group, the P2X7R expression increased in the model group (P < 0.001); and after the administration of QSG, the protein content of P2X7R was decreased compared with the model group (P < 0.001). The results showed that QSG could inhibit the expression of P2X7R protein. The activation of NLRP3 inflammasome is accompanied by caspase-1 activation, and the activated caspase-1 cleaved pro-IL-18 and pro-IL-1β, causing the maturation and release of pro-inflammatory cytokines IL-18 and IL-1β. Therefore, detecting the expression of Caspase-1, IL-18, and IL-1β proteins in each group of cells could indirectly indicate the activation level of NLRP3 inflammasome. WB results showed in Figure 5F–H, compared with the control group, Caspase-1, IL-18, and IL-1β protein expression were significantly increased in the model group (P < 0.001), which again demonstrated that LPS combined with ATP stimulation of RAW264.7 macrophages could induce NLRP3 inflammasome activation. The QSG administration group decreased the expression of Caspase-1, IL-18, and IL-1β protein compared with the model group, while the AZ group decreased the expression of Caspase-1, IL-18, and IL-1β protein. The above results suggest that QSG acted like AZ, most likely by inhibiting P2X7R and thus inhibiting LPS combined with ATP-induced NLRP3 inflammasome activation in RAW264.7 macrophages.
QSG Inhibited NLRP3 Inflammasome Activation via the P2X7R-NEK7-NLRP3 Pathway
To further clarify whether QSG could exert a direct down-regulatory effect on NLRP3 inflammasome activation by inhibiting P2X7R, we successfully constructed the P2X7R plasmid. The map of which is shown in Figure 6A, and verified the overexpression effect of P2X7R plasmid at the protein expression level (Figure 6B). The expression of P2X7R protein was higher in the P2X7R pcDNA3.1 group compared with the pcDNA3.1 group (empty plasmid as negative control) (P < 0.001), indicating that the constructed P2X7R plasmid was able to overexpress P2X7R protein on RAW264.7 macrophages. After confirming that P2X7R plasmid could successfully overexpress P2X7R protein, we gave QSG after P2X7R plasmid transfection, and the results showed in Figure 6B. The P2X7R pcDNA3.1+QSG group showed a reduced level of P2X7R protein expression compared with the P2X7R pcDNA3.1 group (P < 0.01). The immunofluorescence results are shown in Figure 6C. The nuclei of DAPI-stained cells showed blue fluorescence; P2X7R pcDNA3.1 with EGFP, when the plasmid was successfully transfected on the cells, the cells could be observed to show green fluorescence under fluorescence microscopy; Caspase-1 positivity showed red fluorescence, indicating increased expression of Caspase-1 in the cells, representing NLRP3 inflammasome activation. At the same time, the expression levels of IL-18 and IL-1β were detected by WB (Figure 6D and E). Compared with the pcNDA3.1 group, the P2X7R pcDNA3.1 group had higher protein expressions of IL-18 and IL-1β (P < 0.001). Compared with the pure P2X7R pcDNA3.1 group, the P2X7R pcDNA3.1+QSG group decreased the protein content of IL-18 and IL-1β (P < 0.001). These results indicated that QSG could inhibit P2X7R expression, thereby downregulating the activation of NLRP3 inflammasome in RAW264.7 macrophages.
 |
Figure 6 QSG regulates the P2X7R-NEK7-NLRP3 pathway. (A) mP2X7R pcDNA3.1–3xFlag-T2A-EGFP map. (B) WB images of images of RAW264.7 macrophages and analysis of P2X7R. (C) Representative immunofluorescence detection images of Caspase-1. Scale bar=75 µm. (D and E) Western blot images of RAW264.7 macrophages and analysis of IL-18 and IL-1β. (F–H) RAW264.7 macrophages were transfected with P2X7R pcDNA3.1 followed by immunoprecipitation and immunoblot using NEK7 antibody to analyze the protein expression levels of NEK7, NLRP3. The Input represents total protein extracts used for immunoprecipitation. GAPDH was used as a loading control. IB, immunoblot. IP, immunoprecipitation. IgG, negative control. ###P < 0.001 vs pcDNA3.1 group, **P < 0.01, ***P < 0.001 vs P2X7R pcDNA3.1 group. |
Furthermore, we clarified whether NLRP3 inflammasome activation in RAW264.7 macrophages transfected with P2X7R pcDNA3.1 was associated with NEK7 by immunoprecipitation and immunoblotting. The results are shown in Figure 6F–H. Transfection of P2X7R pcDNA3.1 caused increased expression of NEK7 and NLRP3 in RAW264.7 macrophages (Figure 6F), and the interaction between NEK7 and NLRP3 was enhanced (Figure 6G). In contrast, QSG decreased NEK7, NLRP3 protein expression levels and attenuated the interaction between NEK7 and NLRP3. Taken together, these data supported that the role of QSG in inhibiting NLRP3 inflammasome activation is related to the P2X7R-NEK7-NLRP3 pathway.
Discussion
Our previous studies elucidated that QSG can modulate the inflammatory phenotype of cardiac macrophages, which in turn affects local inflammatory infiltration of ischemic myocardium. However, the mechanism by which QSG regulates myocardial inflammation mediated by inflammatory activation of cardiac macrophages has not been revealed. The present findings are as follows: 1) QSG ameliorated myocardial injury and inflammatory pathological changes induced by AMI in mice. 2) QSG inhibited the activation of NLPR3 inflammasome in cardiac macrophages of AMI mice, thereby attenuating the inflammatory response in myocardial tissue after AMI. 3) QSG suppressed the activation of NLRP3 inflammasome in RAW264.7 macrophages induced by LPS combined with ATP. 4) QSG inhibited the P2X7R-NEK7-NLRP3 pathway and thereby downregulated NLPR3 inflammasome activation.
The role of NLRP3 inflammasome with clear structure and function in cardiovascular diseases has attracted increasing attention, and it is closely associated with the occurrence and development of various cardiovascular diseases.27 Acute myocardial ischemia-induced myocardial ischemic-hypoxic injury causes the release of various endogenous injury-related molecular patterns such as adenosine triphosphate, mitochondrial proteins, and nucleotides in vivo. In particular, it causes triple assembly of NLRP3/ASC/pro-Caspase-1 leading to NLRP3 inflammasome activation, and self-differentiation of inactive pro-Caspase-1 into shearing-active Caspase-1, which in turn further shears pro-IL-18 and pro-IL-1β, producing the mature inflammatory cytokines IL-18 and IL-1β.4,5,28,29 Activation of inflammasome is accompanied by the activation of caspase-1, which processes and modifies inflammatory cytokines such as pro-IL-1β and pro-IL-18, thereby promoting the maturation and secretion of IL-1β and IL-18, and subsequently causing further inflammatory injury to local tissues.30 P2X7R belongs to the purinergic receptor family, an ATP-dependent gated ion channel that is widely involved in events such as inflammation and cell death.31,32 When tissue injury leads to cell death and releasing of large amounts of ATP, causing an increase in extracellular ATP concentration. P2X7R is sensitive to changes in extracellular ATP concentration as cell membrane receptors. The high concentration of extracellular ATP induces the opening of P2X7R ion channels, causing the transport of potassium, sodium and calcium ions across the membrane inside and outside the cell, resulting in a decrease in intracellular potassium ion concentration. This change in intra- and extracellular ion concentration can cause NEK7 to bind to NLRP3, leading to NLRP3 subunit oligomerization and promoting the recruitment of ASC with pro-Caspase-1, and NLRP3-ASC-pro-Caspase-1 assembly, finally result in NLRP3 inflammasome activation.33–35 Thus, increased expression of P2X7R and NEK7 is closely associated with NLRP3 inflammasome activation.
In this study, starting from the inflammatory burst triggered by AMI, we first identified that AMI in mice induced the activation of NLRP3 inflammasome in cardiac macrophages, accompanied by the activation of downstream IL-1β and IL-18. To elucidate whether QSG regulates NLRP3 through its effect or not, we used MCC950, an inhibitor of NLRP3 inflammasome activation, we confirmed the overall effect of QSG on the activation process. MCC950 is a compound containing a dialkylsulfonylurea structure that inhibits both classical and non-classical activation of NLRP3 inflammasome, regardless of the oligomerization of NLRP3 subunits, which in turn inhibits the recruitment of NLRP3 to ASC and blocks the assembly and activation of NLRP3 inflammasome.36–38 And MCC950 is currently considered to be one of the most effective and selective inhibitors of NLRP3 inflammasome activation, thus it was chosen to be used as a positive agent in the experiments. In vitro, we established a model of LPS combined with ATP-induced NLRP3 inflammasome activation in RAW264.7 macrophages and used the same intervention, ie, administration of the NLRP3 inflammasome activation inhibitor MCC950. The results showed that QSG did inhibit LPS combined with ATP-induced NLRP3 inflammasome activation in RAW264.7 macrophages, and may act through the P2X7R-NEK7-NLRP3 pathway.
After clarifying the inhibitory effect of QSG on NLRP3 inflammasome activation, we focused on its upstream P2X7R-NEK7 receptor signaling axis and found that its expression changes were consistent with the activation trend of inflammasome.
We used the P2X7R inhibitor AZ in combination with QSG and direct QSG administration to intervene in a macrophage P2X7R plasmid overexpression model. As verified by immunoprecipitation and immunoblotting, QSG inhibited the activation of the P2X7R- nek7 -NLRP3 pathway, downregulated the expression of effector proteins Caspase-1, IL-18 and IL-1β, and directly downregulated the activation of NLRP3 inflammasome in RAW264.7 macrophages. Thus, P2X7R is yet to become a new target for myocardial ischemia prevention and treatment (Figure 7).
 |
Figure 7 QSG might protect AMI through the P2X7R-NEK7-NLRP3 pathway. |
Deficiencies and prospects
However, there are still some shortcomings in this study. The traditional Chinese medicine compound QSG is composed of six kinds of traditional Chinese medicines with complex components and diverse functions. It is clear that QSG inhibits NLRP3 inflammasome, the specific active monomer components activated need further experimental exploration. In addition, it is necessary to further explore how the active components of QSG bind to P2X7R to inhibit the activation of NLRP3 inflammasome in macrophages through the interaction between small molecules and proteins, and lay a theory for the secondary optimization and development of the formula.
Conclusions
This study demonstrated that QSG could improve cardiac function by reducing inflammatory response and inhibiting inflammatory injury after AMI. This mechanism may exert anti-inflammatory effects by downregulating NLRP3 inflammasome activation through inhibition of the P2X7R-NEK7-NLRP3 pathway in cardiac macrophages. The current study provides a clue that QSG may be used as an alternative treatment strategy for cardiac impairment in AMI.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (NO. 82174215 and 82174364), Major New Drug Creation of Ministry of Science and Technology (NO. 2019ZX09201004-001-011), the Fundamental Research Funds for the Central Universities (Distinguished project), and Excellent Young Scientist Foundation of BUCM (BUCM-2019-JCRC005).
Disclosure
The authors report no conflicts of interest.
References
1. Bigler MR, Zimmermann P, Papadis A, et al. Accuracy of intracoronary ECG parameters for myocardial ischemia detection. J Electrocardiol. 2021;64:50–57. doi:10.1016/j.jelectrocard.2020.11.018
2. Barstow C. Acute coronary syndrome: presentation and diagnostic evaluation. FP Essent. 2020;490:11–19.
3. Kapur NK, Qiao X, Paruchuri V, et al. Mechanical pre-conditioning with acute circulatory support before reperfusion limits infarct size in acute myocardial infarction. JACC Heart Fail. 2015;3:873–882. doi:10.1016/j.jchf.2015.06.010
4. Gong T, Liu L, Jiang W, et al. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol. 2020;20(2):95–112. doi:10.1038/s41577-019-0215-7
5. Green DR. The coming decade of cell death research: five riddles. Cell. 2019;177(5):1094–1107. doi:10.1016/j.cell.2019.04.024
6. Hua J, Liu Z, Liu Z, et al. Metformin increases cardiac rupture after myocardial infarction via the AMPK-MTOR/PGC-1α signaling pathway in rats with acute myocardial infarction. Med Sci Monit. 2018;24:6989–7000. doi:10.12659/MSM.910930
7. Fei Q, Ma H, Zou J, et al. Metformin protects against ischaemic myocardial injury by alleviating autophagy-ROS-NLRP3-mediated inflammatory response in macrophages. J Mol Cell Cardiol. 2020;145:1–13. doi:10.1016/j.yjmcc.2020.05.016
8. Huang L, Xiang M, Ye P, et al. Beta-catenin promotes macrophage-mediated acute inflammatory response after myocardial infarction. Immunol Cell Biol. 2018;96(1):100–113. doi:10.1111/imcb.1019
9. Fang L, Moore XL, Dart AM, et al. Systemic inflammatory response following acute myocardial infarction. J Geriatr Cardiol. 2015;12(3):305–312. doi:10.11909/j.issn.1671-5411.2015.03.020
10. Zhou R, Yazdi AS, Menu P, et al. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–225. doi:10.1038/nature09663
11. Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi:10.1189/jlb.0306164
12. Toldo S, Abbate A. The NLRP3 inflammasome in acute myocardial infarction. Nat Rev Cardiol. 2018;15:203–214. doi:10.1038/nrcardio.2017.161
13. Toldo S, Mauro AG, Cutter Z, et al. Inflammasome, pyroptosis, and cytokines in myocardial ischemia-reperfusion injury. Am J Physiol. 2018;315:H1553–H1568. doi:10.1152/ajpheart.00158.2018
14. He Y, Zeng MY, Yang D, et al. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature. 2016;530(7590):354–357. doi:10.1038/nature16959
15. Man SM, Kanneganti TD. Regulation of inflammasome activation. Immunol Rev. 2015;265:6–21. doi:10.1111/imr.12296
16. Kelley N, Jeltema D, Duan Y, et al. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. 2019;20(13):3328.
17. Du Y, Gu X, Meng H, et al. Muscone improves cardiac function in mice after myocardial infarction by alleviating cardiac macrophage-mediated chronic inflammation through inhibition of NF-κB and NLRP3 inflammasome. Am J Transl Res. 2018;10:4235–4246.
18. Toldo S, Mezzaroma E, Mauro AG, et al. The inflammasome in myocardial injury and cardiac remodeling. Antioxid Redox Signal. 2015;22(13):1146–1161. doi:10.1089/ars.2014.5989
19. Gao S, Zhang Q, Tian C, et al. The roles of Qishen granules recipes, Qingre Jiedu, Wenyang Yiqi and Huo Xue, in the treatment of heart failure. J Ethnopharmacol. 2020;249:112372. doi:10.1016/j.jep.2019.112372
20. Chen X, Li Y, Li J, et al. Qishen granule (QSG) exerts cardioprotective effects by inhibiting NLRP3 inflammasome and pyroptosis in myocardial infarction rats. J Ethnopharmacol. 2022;285:114841. doi:10.1016/j.jep.2021.114841
21. Lu W, Wang Q, Sun X, et al. Qishen granule improved cardiac remodeling via balancing M1 and M2 macrophages. Front Pharmacol. 2019;10:1399. doi:10.3389/fphar.2019.01399
22. Wang X, Li W, Zhang Y, et al. Calycosin as a novel PI3K activator reduces inflammation and fibrosis in heart failure through AKT-IKK/STAT3 axis. Front Pharmacol. 2022;13:828061. doi:10.3389/fphar.2022.828061
23. Li Y, Li X, Chen X, et al. Qishen granule (QSG) inhibits monocytes released from the spleen and protect myocardial function via the TLR4-MyD88-NF-κB p65 pathway in heart failure mice. Front Pharmacol. 2022;13:850187. doi:10.3389/fphar.2022.850187
24. Rauf A, Shah M, Yellon DM, et al. Role of caspase 1 in ischemia/reperfusion injury of the myocardium. J Cardiovasc Pharmacol. 2019;74(3):194–200. doi:10.1097/FJC.0000000000000694
25. Ren Z, Yang K, Zhao M, et al. Calcium-sensing receptor on neutrophil promotes myocardial apoptosis and fibrosis after acute myocardial infarction via NLRP3 inflammasome activation. Can J Cardiol. 2020;36:893–905. doi:10.1016/j.cjca.2019.09.026
26. Takahashi M. Cell-specific roles of NLRP3 inflammasome in myocardial infarction. J Cardiovasc Pharmacol. 2019;74:188–193. doi:10.1097/FJC.0000000000000709
27. Hwang MW, Matsumori A, Furukawa Y, et al. Neutralization of interleukin-1beta in the acute phase of myocardial infarction promotes the progression of left ventricular remodeling. J Am Coll Cardiol. 2001;38:1546–1553. doi:10.1016/S0735-1097(01)01591-1
28. Takahashi M. Role of NLRP3 inflammasome in cardiac inflammation and remodeling after myocardial infarction. Biol Pharm Bull. 2019;42:518–523. doi:10.1248/bpb.b18-00369
29. Yang Y, Wang H, Kouadir M, et al. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019;10(2):128. doi:10.1038/s41419-019-1413-8
30. Woldbaek PR, Tønnessen T, Henriksen UL, et al. Increased cardiac IL-18 mRNA, pro-IL-18 and plasma IL-18 after myocardial infarction in the mouse; a potential role in cardiac dysfunction. Cardiovasc Res. 2003;59:122–131. doi:10.1016/S0008-6363(03)00339-0
31. Faria RX, Reis RA, Ferreira LG, et al. P2X7R large pore is partially blocked by pore forming proteins antagonists in astrocytes. J Bioenerg Biomembr. 2016;48:309–324. doi:10.1007/s10863-016-9649-9
32. Cao F, Hu LQ, Yao SR, et al. P2X7 receptor: a potential therapeutic target for autoimmune diseases. Autoimmun Rev. 2019;18:767–777. doi:10.1016/j.autrev.2019.06.009
33. Di Virgilio F, Jiang LH, Roger S, et al. Structure, function and techniques of investigation of the P2X7 receptor (P2X7R) in mammalian cells. Methods Enzymol. 2019;629:115–150.
34. Feng L, Chen Y, Ding R, et al. P2X7R blockade prevents NLRP3 inflammasome activation and brain injury in a rat model of intracerebral hemorrhage: involvement of peroxynitrite. J Neuroinflammation. 2015;12:190. doi:10.1186/s12974-015-0409-2
35. Jiang S, Zhang Y, Zheng JH, et al. Potentiation of hepatic stellate cell activation by extracellular ATP is dependent on P2X7R-mediated NLRP3 inflammasome activation. Pharmacol Res. 2017;117:82–93. doi:10.1016/j.phrs.2016.11.040
36. Coll RC, Robertson AA, Chae JJ, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21:248–255. doi:10.1038/nm.3806
37. Coll RC, Hill JR, Day CJ, et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat Chem Biol. 2019;15:556–559. doi:10.1038/s41589-019-0277-7
38. Zahid A, Li B, Kombe AJK, et al. Pharmacological inhibitors of the NLRP3 inflammasome. Front Immunol. 2019;10:2538. doi:10.3389/fimmu.2019.02538
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
The Role of Podoplanin in the Immune System and Inflammation
Zhang Z, Zhang N, Yu J, Xu W, Gao J, Lv X, Wen Z
Journal of Inflammation Research 2022, 15:3561-3572
Published Date: 17 June 2022
CCL20/CCR6 Mediated Macrophage Activation and Polarization Can Promote Adenoid Epithelial Inflammation in Adenoid Hypertrophy
Ye C, Guo X, Wu J, Wang M, Ding H, Ren X
Journal of Inflammation Research 2022, 15:6843-6855
Published Date: 23 December 2022
Resolvin D1 Attenuates Inflammation and Pelvic Pain Associated with EAP by Inhibiting Oxidative Stress and NLRP3 Inflammasome Activation via the Nrf2/HO-1 Pathway
Zhang J, Chen J, Jiang Q, Feng R, Zhao X, Li H, Yang C, Hua X
Journal of Inflammation Research 2023, 16:3365-3379
Published Date: 8 August 2023
HMGB1 Inhibition Alleviates Chronic Nonbacterial Prostatitis by Suppressing M1 Polarization of Macrophages
Zhou J, Ding L, Chen J, Chen C, Jiang P, Mei Z, Jiang Q, Hua X
Journal of Inflammation Research 2025, 18:6735-6748
Published Date: 25 May 2025
The Regulatory Network of Transcription Factors in Macrophage Polarization
Liu J, Wang M, Zhao Y
ImmunoTargets and Therapy 2025, 14:555-575
Published Date: 6 June 2025