Back to Journals » Drug Design, Development and Therapy » Volume 20
Natural Compounds Targeting Coronary Microvascular Dysfunction: Mechanisms and Beyond
Authors Chen M, Xiang X, Yang X ![]() , Fu Y, Wan Z, Yang S, Liu Q, Luo G, Liu M
, Fu Y, Wan Z, Yang S, Liu Q, Luo G, Liu M ![]()
Received 24 December 2025
Accepted for publication 23 March 2026
Published 27 March 2026 Volume 2026:20 591542
DOI https://doi.org/10.2147/DDDT.S591542
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Mingtai Chen,1– 3,* Xinyi Xiang,4,* Xinrui Yang,5,* Youyou Fu,5 Zhenxun Wan,5 Shiyun Yang,5 Qiuyu Liu,6 Gang Luo,5 Mengnan Liu5
1Shenzhen Traditional Chinese Medicine Hospital, Guangzhou University of Chinese Medicine, Shenzhen, 518000, People’s Republic of China; 2Chinese Medicine Guangdong Laboratory (Hengqin Laboratory), Guangdong-Macao In-Depth Cooperation Zone in Hengqin, Zhuhai, 519000, People’s Republic of China; 3Faculty of Chinese Medicine and State Key Laboratory of Quality Research in Chinese Medicine, Macau University of Science and Technology, Macao, 999078, People’s Republic of China; 4The Affiliated Hospital, Southwest Medical University, Luzhou, 646000, People’s Republic of China; 5Affiliated Traditional Chinese Medicine Hospital, Southwest Medical University, Luzhou, 646000, People’s Republic of China; 6School of Pharmacy, Southwest Medical University, Luzhou, 646000, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Gang Luo, Email [email protected] Mengnan Liu, Email [email protected]
Abstract: Coronary microvascular dysfunction (CMD) is increasingly recognized as a major public health burden, independently contributing to myocardial ischemia, adverse cardiovascular events, and elevated healthcare costs. Its pathophysiology is multifactorial, involving endothelial dysfunction, microthrombosis, and metabolic disturbances. Natural compounds, owing to their multi-target effects and favorable safety profiles, have emerged as promising candidates for CMD intervention. This narrative review systematically consolidates current evidence on the key molecular targets and regulatory mechanisms through which natural compounds preserve microvascular homeostasis. It also highlights shared pathological pathways between CMD and other cardiovascular diseases to support cross-disease therapeutic translation. By integrating these findings, this review aims to establish a theoretical foundation for future preclinical and clinical applications of natural compounds in CMD management.
Keywords: coronary microvascular dysfunction, coronary artery disease, natural compounds, inflammation, oxidative stress
Introduction
Coronary microvascular dysfunction (CMD) is a distinct cardiovascular disease subtype characterized by structural and functional abnormalities in coronary microvessels, leading to myocardial ischemia and adverse cardiovascular events. Despite its high prevalence and poor prognosis, the diagnosis and treatment of CMD remain significant clinical challenges. Current therapeutic strategies, largely derived from experience with obstructive coronary artery disease, primarily target macrovascular lesions and fail to address the specific pathophysiological mechanisms underlying microvascular dysfunction. This therapeutic gap represents a critical unmet need in CMD management. Moreover, the complex pathogenesis of CMD—involving intricate interactions among endothelial dysfunction, dysregulated mitophagy, oxidative stress, and chronic inflammation—further complicates the development of effective agents specifically for CMD.1,2 Although insights from other cardiovascular conditions sharing homologous pathogenic pathways have informed CMD research, direct evidence specific to CMD remains scarce. Therefore, identifying therapeutic candidates by focusing on the unique pathological network of CMD itself, rather than relying on extrapolation from macrovascular diseases, has emerged as a more targeted approach.
In recent years, natural compounds have gained attention as potential therapeutics for CMD due to their multi-target effects, low toxicity, and favorable biocompatibility.3,4 While much of the evidence supporting their cardiovascular benefits originates from studies on atherosclerosis or myocardial infarction, emerging research suggests that these compounds may also modulate key CMD-specific pathways, including endothelial homeostasis, mitophagy, oxidative stress, and inflammation.5 However, direct experimental and clinical evidence specifically validating their efficacy in CMD is limited. Preliminary reports indicate that natural compound-derived drugs, when combined with conventional anti-ischemic agents or integrated into secondary prevention regimens, could expand therapeutic options for CMD.6 Nevertheless, robust clinical trials focusing on CMD populations are urgently needed to confirm these potential benefits. With a critical emphasis on distinguishing findings directly obtained from CMD models from those inferred from other cardiovascular diseases, this review systematically evaluates the current evidence on how natural compounds synergistically regulate the CMD pathological network. We aim to provide a theoretical foundation for elucidating their molecular mechanisms in improving endothelial function, mitigating mitophagy dysregulation, remodeling the oxidative-inflammatory microenvironment, and correcting immune-metabolic disturbances, thereby accelerating the translation of natural compounds from bench to bedside for precise CMD intervention. By explicitly linking the therapeutic limitations of current anti-ischemic regimens—largely extrapolated from obstructive coronary artery disease—to the distinct and often overlooked pathophysiological demands of CMD, this review underscores an urgent need for mechanism-driven, CMD-tailored interventions. In particular, we highlight critical evidence gaps specific to CMD, including the paucity of preclinical studies utilizing validated microvascular dysfunction models, the lack of clinical trials with CMD-diagnosed cohorts and endothelial-specific endpoints, and the undercharacterization of natural compounds’ direct effects on coronary microcirculation.
The Pathophysiological Mechanisms of Coronary Microcirculatory Dysfunction
CMD is a pathological state characterized by imbalanced microvascular vasomotor function and inadequate myocardial perfusion. This condition arises from the synergistic interactions among multiple mechanisms, including endothelial dysfunction, microvascular structural remodeling, and dysregulated inflammatory responses. Current research on CMD mechanisms remains extensive but constrained. The interplay among these pathogenic mechanisms are illustrated in Figure 1.
 |
Figure 1 Pathogenesis of coronary microvascular dysfunction. |
CMD Caused by Endothelial Dysfunction
In the coronary microcirculation, endothelial cells (ECs) modulate vascular tone and vasomotor function through the production and release of vasodilatory and vasoconstrictive mediators. Vasodilators include NO, endothelium-derived hyperpolarizing factor (EDHF), and prostaglandins, while the primary vasoconstrictive mediator is endothelin-1 (ET-1).7 As the principal endothelium-dependent vasodilator, NO maintains endothelial integrity through its anti-inflammatory, anti-fibrotic, anti-platelet aggregation, anti-apoptotic, and pro-angiogenic effects.1,8 However, under conditions of oxidative stress, ROS are excessively generated via NADPH oxidase (NOX) and mitochondrial pathways, degrading NO and inhibiting eNOS function. This markedly reduces NO bioavailability and vasodilatory capacity while enhancing the vasoconstrictive action of ET-1, exacerbating microvascular spasm.9 Consequently, disrupted dilation-contraction balance compromises local blood flow regulation and blunts metabolic responsiveness, driving CMD development.
CMD Induced by Structural Abnormalities
Structural remodeling of the coronary microcirculation represents another key mechanism underlying CMD development. These structural abnormalities primarily manifest as hypertrophic remodeling of resistance arteries, luminal narrowing in arterioles and capillaries, perivascular fibrosis, decreased microvascular density, and capillary rarefaction.2 Luminal narrowing and medial wall thickening in arterioles result from smooth muscle cell hypertrophy and collagen deposition, which not only reduces vascular compliance but also directly impairs flow reserve and blood flow distribution.10,11 Decreased capillary density (capillary rarefaction) further restricts blood perfusion. Moreover, these structural alterations frequently interact synergistically with functional abnormalities, collectively compromising myocardial microcirculatory perfusion and contributing to the pathogenesis of CMD.
CMD Induced by Abnormal Inflammatory Factor Levels
Inflammation plays a catalytic and exacerbating role in the pathogenesis of CMD.12 Under pathological conditions, inflammatory mediators such as IL-6, TNF-α, and C-reactive protein (CRP) are released at elevated levels. These factors activate vascular endothelial cells and induce endothelial dysfunction.13 Pro-inflammatory cytokines not only further reduce the production and stability of NO but also directly enhance the sensitivity of vascular smooth muscle cells to vasoconstrictive stimuli, leading to microvascular spasm. Inflammatory responses and oxidative stress mutually reinforce each other: reactive ROS activate the NLRP3 inflammasome, promoting the maturation and release of IL-1β and IL-18.14 Concurrently, inflammatory mediators (eg., TNF-α) increase ROS production by upregulating NOX activity, thereby accelerating endothelial damage and vascular remodeling, ultimately promoting CMD development.15
Sex-Specific CMD with Female Predominance
Females exhibit unique characteristics in the pathogenesis of CMD, primarily associated with estrogen levels and its signaling pathways.16 Current research indicates that estrogen exerts protective effects on the vascular endothelium, promotes NO production, and suppresses inflammation.17 The underlying mechanism involves estrogen upregulating CD39 (ectonucleoside triphosphate diphosphohydrolase) expression, facilitating ATP metabolism into adenosine, thereby enhancing adenosine-mediated vasodilation.18 However, in postmenopausal or estrogen-deficient states, the protective effects of estrogen are significantly diminished. This leads to reduction in NO bioavailability, enhancement of endothelin-1 (ET-1) activity, and decrease in antioxidant capacity, substantially increasing CMD risk. Notably, under other pathological conditions (eg., preeclampsia, collagen vascular diseases, systemic lupus erythematosus), abnormal estrogen receptor signaling may exacerbate endothelial inflammation and microvascular damage, collectively contributing to CMD development.19
Autonomic Nervous Dysregulation and Other Factors
The autonomic nervous system plays a crucial role in maintaining appropriate vasodilation and vasoconstriction in coronary microcirculation, with its balanced state directly influencing vasomotor responses.20 Autonomic imbalance, characterized by increased sympathetic activity and decreased parasympathetic function, leads to sustained vasoconstriction and disrupted blood flow regulation. Additionally, neurotransmitters such as norepinephrine directly induce vasoconstriction when overactive in vascular smooth muscle cells, impairing vasodilation during increased metabolic demands.21 Notably, autonomic dysregulation not only independently contributes to vasomotor imbalance but also interacts synergistically with endothelial dysfunction and inflammatory states, creating a vicious cycle of impaired coronary vascular reactivity.11 Epigenetic mechanisms (eg., DNA methylation and histone modifications) are implicated in CMD pathogenesis by modulating genes involved in oxidative stress, inflammation, and angiogenesis. For instance, the minor G allele at the rs9349379 locus within the ET-1 shared intron region enhances ET-1 expression, promoting microvascular constriction and myocardial ischemia.22,23 CDKN2B-AS1, a long non-coding RNA, may induce endothelial cell apoptosis and senescence through interactions with polycomb repressive complexes, thereby reducing microvascular density.24 VEGF-A, a key regulator in angiogenesis and vascular integrity, may impair endothelial proliferation and microvascular density when functionally deficient, compromising myocardial adaptation to stress.25 Other genes including FOG2, eNOS, and hypoxia-inducible factor 1α (HIF-1α) are also implicated in CMD, modulating angiogenic factor expression, NO synthesis, and vascular development.26–28 Collectively, these epigenetic alterations not only exacerbate endothelial injury and dysfunction but may also contribute to structural abnormalities like microvascular remodeling. They interact with environmental and lifestyle factors, forming a complex pathogenic network.29
Current Therapeutic Agents for CMD
Anti-Atherosclerosis Treatment
The Renin Angiotensin Aldosterone System (RAAS) related pathways play a central role in Ang II-mediated vascular dysfunction. ACE inhibitors (ACEIs) and angiotensin receptor blockers (ARBs) effectively counteract several pathological effects mediated by Ang II through AT1 receptors—including activation of NADPH oxidase, increased O2− production, and elevated oxidative stress levels—by inhibiting ACE to block Ang II generation or by selectively blocking Ang II binding to AT1 receptors. Representative agents include ACEIs such as Captopril, Enalapril, and Ramipril, along with ARBs such as Losartan and Irbesartan. Notably, the classic ACEI Ramipril reduces bradykinin degradation, enhancing its binding to endothelial B2 receptors.30,31 This activates signaling pathways that promote synergistic release of NO and PGI2, directly dilating coronary microvessels and inhibiting platelet aggregation. Collectively, these mechanisms endow ACEIs/ARBs with multifaceted benefits: antihypertensive effects, anti-atherosclerotic properties, anti-vascular remodeling, endothelial protection, and CMD improvement. Clinical trials have shown that ramipril can increase coronary flow reserve by approximately 15–20% in postmenopausal women with CMD.
Calcium channel blockers (CCBs) inhibit voltage-gated calcium channels, leading to reduced influx of extracellular Ca2⁺, decreased intracellular calcium ion concentration, and interference with calmodulin-dependent signaling pathways, ultimately resulting in the relaxation of vascular smooth muscle. CCBs are primarily categorized into non-dihydropyridines (eg., verapamil, diltiazem) and dihydropyridines (eg., nifedipine, amlodipine). Among them, diltiazem exhibits high-affinity binding to L-type calcium channels in coronary microvessels, while amlodipine primarily acts on Cav1.2 channels in peripheral arteries.31 Both agents effectively dilate coronary microvessels and lower blood pressure.
Additionally, diltiazem also inhibits T-type calcium channels in the sinoatrial and atrioventricular nodes, prolonging atrioventricular conduction time and reducing heart rate. In contrast, amlodipine may trigger a baroreceptor reflex-mediated sympathetic activation due to its significant reduction in peripheral vascular resistance, potentially increasing heart rate.30 These effects enable CCBs to exert both antianginal and antihypertensive actions. In patients with coronary slow flow phenomenon, diltiazem can reduce angina attack frequency by 40% to 50%. However, amlodipine often requires co-administration with a β1-blocker to counteract the compensatory tachycardia it may induce.
Statins competitively inhibit 3-Hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase in hepatocytes, thereby reducing circulating cholesterol levels. This class includes agents such as lovastatin, rosuvastatin, and pravastatin. As an example, high-intensity statin rosuvastatin effectively suppresses farnesylation of the RhoA protein, preventing translocation of Rho-associated ROCK to the cell membrane and its degradation of eNOS mRNA. Concurrently, it blocks the phosphorylation of IκB (the inhibitory protein of NF-κB). These mechanisms collectively lead to a significant increase in endothelial nitric oxide synthesis and reduced expression of monocyte chemoattractant protein-1 (MCP-1) and vascular cell adhesion molecule-1 (VCAM-1). Consequently, rosuvastatin exerts pleiotropic effects—improving vascular endothelial function, suppressing inflammatory responses, and stabilizing atherosclerotic plaques.30–32 Clinical data show that rosuvastatin can improve coronary flow reserve by 0.3–0.5 units in patients with diabetes mellitus complicated by CMD.
Thromboxane A2 synthase (TXAS) inhibitors, notably the selective inhibitor ozagrel specifically target TXAS to inhibit thromboxane A2 (TXA2) production. Ozagrel competitively binds to the heme-containing active site of TXAS, preventing the conversion of prostaglandin H2 (PGH2) to TXA2 and resulting in a reduction of TXA2 generation in platelets by over 90%. Additionally, by diminishing thromboxane prostanoid (TP) receptor activation, ozagrel suppresses the phospholipase C beta–diacylglycerol (PLCβ–DAG) signaling cascade. This inhibits protein kinase C (PKC)-mediated assembly of NADPH oxidase, thereby reducing superoxide generation and preserving the junctional integrity of vascular endothelial cadherin. These mechanisms collectively confer potent antiplatelet aggregation and microvascular protective effects.33
The selective estrogen receptor modulator (SERM) raloxifene induces conformational changes in the estrogen receptor alpha (ERα) within vascular endothelial cells, promoting ERα binding to the endothelial nitric oxide synthase (eNOS) promoter and thereby enhancing eNOS transcriptional activity. Concurrently, it also inhibits the ROCK pathway, reducing myosin light chain (MLC) phosphorylation and improving endothelium-dependent vasodilation.30,31,34 A unique advantage of raloxifene is its antagonistic effects on ERα in breast and uterine tissue, resulting in an 80% reduction in endometrial hyperplasia risk. In postmenopausal women with CMD, raloxifene increases CFR by 0.2–0.4 units without elevating cardiovascular event risk.
Anti-Anginal Treatment
β-Blockers exert their mechanism by selective blockade of myocardial β1-adrenergic receptors, thereby inhibiting overactive sympathetic nervous system activity. This action reduces myocardial contractility, slows heart rate, and decreases cardiac oxygen consumption, effectively alleviating myocardial ischemia-induced angina pectoris. Common agents include metoprolol, bisoprolol, and atenolol. Among them, the third-generation highly selective β1-blocker nebivolol exhibits a unique dual mechanism beyond these fundamental cardioprotective effects: it specifically agonizes vascular endothelial β3-receptors, leading to sustained NO release.33,35 This activates the soluble guanylate cyclase–cyclic guanosine monophosphate–protein kinase G (sGC–cGMP–PKG) signaling pathway in vascular smooth muscle, ultimately inducing microvascular dilation. Concurrently, nebivolol upregulates mitochondrial superoxide dismutase-2 (SOD-2) expression via β3-receptor agonism, effectively scavenging excessive ROS and attenuating vascular endothelial oxidative damage. These actions provide comprehensive vascular protection in angina management.
Late sodium current inhibitors exert their core mechanism by selectively inhibiting the late sodium current in cardiomyocytes, reducing abnormal sodium ion influx during the action potential plateau phase. Agents in this category include ranolazine and trimetazidine. Beyond improving myocardial calcium handling and diastolic function, the representative agent ranolazine possesses a unique metabolic regulatory action: it inhibits carnitine palmitoyltransferase-1 (CPT-1), shifting myocardial substrate utilization toward more efficient glucose oxidation.31,36 This metabolic shift is particularly critical under ischemic conditions, where it significantly reduces lactate production and acidosis, optimizes myocardial energy metabolism, and attenuates resulting microvascular endothelial dysfunction. Thus, ranolazine effectively alleviates angina symptoms through this dual mechanism.
Nitrates exert their therapeutic core mechanism by directly or indirectly increasing cyclic guanosine monophosphate (cGMP) levels to induce vasodilation. The classical nitrate nitroglycerin relies on aldehyde dehydrogenase-2 (ALDH-2) for bioactivation to release NO, which subsequently activates soluble guanylate cyclase (sGC) to generate cGMP. It primarily dilates epicardial conduit vessels but has limited effects on microvessels. Moreover, chronic use frequently leads to tolerance due to multiple factors: ALDH-2 activity downregulation, ROS-mediated NO consumption, and increased phosphodiesterase type 5 (PDE5) activity.33 In contrast, the novel sGC stimulator riociguat directly binds to and stabilizes the heme moiety of sGC, substantially enhancing cGMP synthesis. Clinical trials demonstrate that riociguat significantly prolonged exercise tolerance in patients with stable angina. Compared to nitroglycerin, it is associated with a 70% reduction in the risk of tolerance, providing a more effective and better-tolerated therapeutic option, particularly for patients with CMD.
α1-Adrenoceptor blockers exert their therapeutic mechanism by selectively antagonizing α1-adrenoceptors on vascular smooth muscle cells. This blocks the norepinephrine-activated phospholipase C beta–inositol trisphosphate (PLCβ-IP3) signaling pathway, resulting in vascular smooth muscle relaxation, microvascular dilation, and significant reduction in peripheral vascular resistance. Agents include prazosin, terazosin, and doxazosin.33,37 Given its potent microvascular dilatory properties, the representative agent doxazosin is particularly suitable for patients with hypertension and CMD who exhibit sympathetic overactivation. However, this agent may cause orthostatic hypotension (with an incidence of up to 18%) and compensatory tachycardia. Consequently, it is often co-administered with β-blockers (such as metoprolol) in clinical practice to counteract reflex tachycardia, maintain hemodynamic stability, and optimize therapeutic efficacy and safety in hypertensive patients with CMD.
Current therapeutic strategies for CMD predominantly repurpose medications commonly used for pathophysiologically similar conditions such as atherosclerosis and angina pectoris. However, the existing treatment framework still faces challenges, including insufficient target specificity and an incomplete evidence base that requires higher-quality validation. Disease-specific clinical trials are urgently needed to establish definitive therapeutic efficacy. These pharmacological treatment strategies are summarized in Figure 2.
 |
Figure 2 Several common medications used to treat coronary microvascular dysfunction. |
Therapeutic Pathways of Natural Compounds for Coronary Microvascular Dysfunction
CMD is primarily improved through synergistic mechanisms including the suppression of inflammatory factors, reduction of oxidative stress, and endothelial cell protection. Current research indicates that potential therapeutic pathways for CMD are diverse, and these are illustrated in Figure 3 and Table 1.
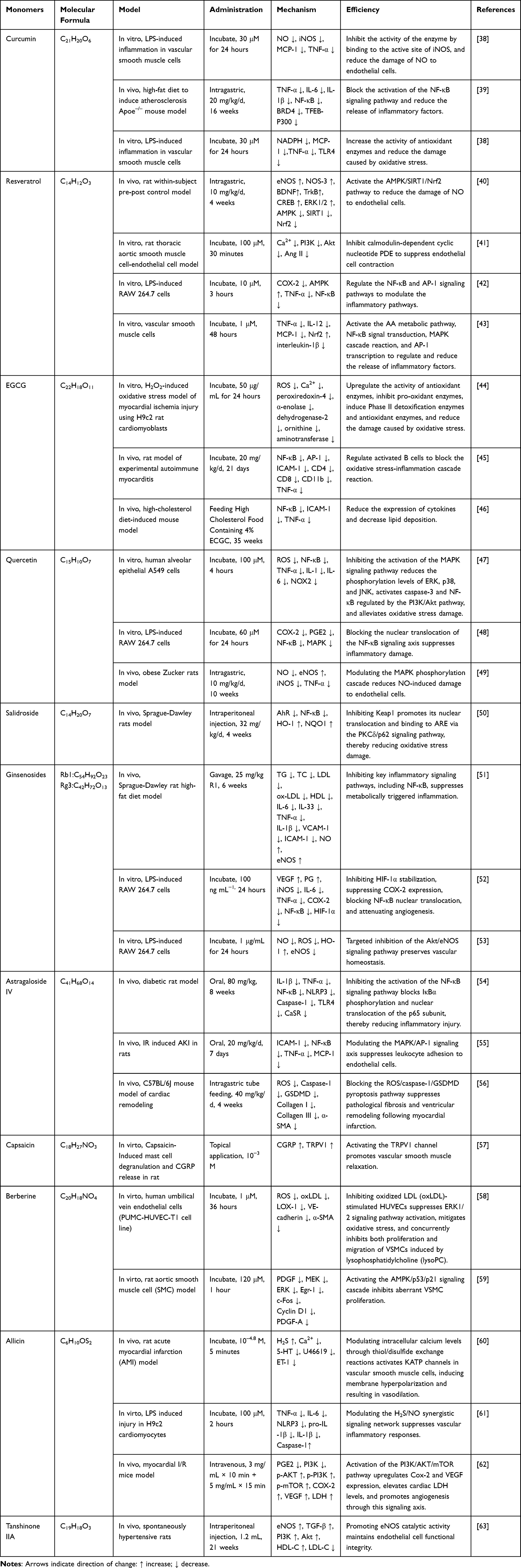 |
Table 1 Pharmacological Profile of Natural Compounds in Treatment of Coronary Microcirculation Disorders |
 |
Figure 3 Schematic diagram of inflammation, vascular smooth muscle, and oxidative stress mechanisms regulated by natural compounds in coronary microcirculation disorders. |
Natural Compounds in Inflammation
Among natural polyphenolic compounds, curcumin, a primary bioactive component of turmeric rhizomes, exhibits potent anti-inflammatory properties. It directly binds to the active site of inducible nitric oxide synthase (iNOS), inhibiting its enzymatic activity and reducing the pathological overproduction of NO under inflammatory conditions. Additionally, it inhibits the activation of the NF-κB signaling pathway, downregulating the release of pro-inflammatory cytokines such as TNF-α, IL-6, and IL-1β.64 This suppresses iNOS expression at both transcriptional and microenvironmental levels, thereby alleviating tissue inflammation. This multi-target anti-inflammatory mechanism has been experimentally validated in an in vitro rat vascular smooth muscle cell inflammation model. Curcumin’s multi-target engagement with both iNOS and NF-κB is conceptually novel, but its clinical translation remains constrained by poor bioavailability and a lack of robust efficacy data in human inflammatory disorders. Resveratrol, a plant-derived metabolite widely present in grapes, red wine, and berries, modulates NO homeostasis in endothelial cells through a multi-target regulatory network, including: (1) Upregulating eNOS expression by activating Estrogen Receptor (ER) and Mitogen-Activated Protein Kinase (MAPK) signaling pathways. (2) Improving endothelial function in ApoE-deficient mice via Sirtuin 1 (SIRT1)-mediated eNOS activation.65 (3) Inducing NOS-3 expression through the AMPK/SIRT1/Nrf2 signaling network.66 (4) Selectively inhibiting COX-1 activity and downregulating COX-2 expression by modulating the NF-κB and AP-1 signaling pathways.67 These mechanisms contribute significantly to the treatment of atherosclerosis. Catechins are a class of biologically important flavonoid polyphenolic compounds widely found in various natural foods and medicinal plants. Tea is the primary source of human catechin intake due to its high content of multiple active catechin derivatives.68 EGCG ((-)-epigallocatechin-3-gallate), the most abundant catechin in green tea, exerts broad health-promoting effects. EGCG attenuates myocardial inflammation by reducing lipid accumulation and decreasing the expression of inflammatory mediators such as NF-κB, ICAM-1, and TNF-α.46,69–71 The combination of its anti-inflammatory mechanism and its ability to enhance endothelium-dependent vasodilation underscores the potential application value of EGCG in the precision treatment of cardiovascular diseases. Quercetin is a typical representative of flavonol polyphenolic compounds, existing widely in free or glycoside-bound forms in apples, berries, onions, tea, and medicinal plants.72 Quercetin ameliorates atherosclerosis by inhibiting the synthesis of inflammatory mediators such as IL-6, IL-1β, MCP-1, and VEGF, regulating macrophage polarization, and suppressing the inflammatory storm via a dual pathway. It blocks the nuclear translocation of the NF-κB signaling axis to inhibit COX-2/PGE2 expression and modulates the MAPK phosphorylation cascade reaction to reduce excessive NO production.48,73,74
Saponin compounds share many similarities with CMD in their therapeutic mechanisms targeting vascular endothelial inflammation. Among saponin compounds, ginsenosides Rb1, Rg1, Rg3, Rh2, and the metabolite Compound K have been confirmed as the key anti-inflammatory components.75 Among them, Rg3 suppresses the inflammatory mediator cascade, encompassing iNOS activity, NO levels, MMP-9, COX-2, and TNF-α/IL-1β/IL-6, through the upregulation of M2 marker genes such as Arg-1.53,76 Another saponin compound—astragaloside IV (AS-IV)—is a main active component of the Chinese medicinal herb Astragalus membranaceus (Huangqi). It is categorized into astragaloside I, II, and IV, with AS-IV being the most biologically active compound.54 The anti-inflammatory properties of AS-IV involve targeted inhibition of the NF-κB signaling pathway, blocking IκBα phosphorylation and the nuclear translocation of the p65 subunit to downregulate pro-inflammatory factor expression. Simultaneously, it modulates the MAPK/AP-1 signaling axis to reduce ICAM-1 synthesis and inhibit leukocyte-endothelial cell adhesion.54,77 Furthermore, it reduces pathological NO concentrations and prevents apoptosis by regulating iNOS activity and intervening in the caspase cascade reaction. In an ISO-induced endothelial dysfunction model, it enhances enzymatic activity by stabilizing the dimeric structure of eNOS, promoting NO bioavailability, while also inhibiting NF-κB p65 nuclear translocation.78 AS-IV can also promote angiogenesis in ischemic tissues by activating the VEGF/FGF signaling pathway. In human umbilical vein endothelial cells (HUVECs) models, it reduces TLR4 membrane expression, inhibits VCAM-1/ICAM-1 generation, and restores eNOS activity, thereby ameliorating hyperglycemia-induced vascular endothelial inflammation-related cardiovascular diseases.79 Their translation is hampered by undefined active metabolites, unclear dose – response relationships, and an over‑reliance on preclinical vascular models.
Natural Compounds in Vascular Smooth Muscle Cells
Regarding the protection of smooth muscle cells, current research indicates that resveratrol may reduce the contractile response of VSMCs by inhibiting Ca2⁺/calmodulin-dependent cyclic nucleotide PDE in a partially PDE1-dependent manner. Additionally, it acts directly on VSMCs through mechanisms including induction of guanylate cyclase, inhibiting protein kinase C (PKC), activating smooth muscle cell K⁺ channels, or modulating Ca2⁺ concentration, thereby promoting pulmonary artery relaxation and thus attenuating atherosclerosis progression in rat models.80,81
In addition to the aforementioned anti-inflammatory activity, ginsenosides also have significant effects in protecting VSMCs. The regulatory role of ginsenoside Rg3 in vascular homeostasis is closely linked to its cross-pathway synergistic mechanism involving the eNOS signaling network. Studies indicate that Rg3 can specifically inhibit VEGF-dependent Akt/eNOS signaling in EPCs, blocking the functional activity of this pro-angiogenic axis, thereby playing a role in the treatment of ischemic diseases.82,83
Berberine (BBR) is an isoquinoline alkaloid extracted from plants such as Coptis chinensis (Huanglian) and Phellodendron amurense (Huangbai).84 In terms of VSMCs protection, BBR effectively inhibits the migration of human aortic VSMCs by downregulating the expression of matrix metalloproteinases MMP-2, MMP-9, and urokinase-type plasminogen activator (u-PA).85 This synergistic action constitutes a key molecular mechanism by which BBR inhibits vascular inflammatory responses, providing an important theoretical basis for treating cardiac fibrosis. Regarding the regulation of proliferation mechanisms, BBR not only antagonizes PDGF-induced abnormal proliferation of VSMCs by activating the AMPK/p53/p21 signaling cascade but also inhibits the dual effects of lysoPC-stimulated VSMC proliferation and migration by reducing ROS levels and modulating the ERK1/2 signaling pathway.86 It thereby treats atherosclerosis by protecting vascular endothelial cells against oxidative stress. Poor oral bioavailability and promiscuous target engagement, however, limit its translatability to cardiovascular fibrosis therapy.
Allicin, an organosulfur compound characteristic of garlic, is a thiosulfinate.60 Allicin regulates blood pressure through multiple metabolic pathways: On one hand, it releases endogenous hydrogen sulfide (H2S) via thiol/disulfide exchange reactions. H2S induces hyperpolarization by activating KATP channels in VSMCs, reducing intracellular calcium ion concentration to trigger vasodilation. On the other hand, it enhances cellular antioxidant capacity by upregulating glutathione (GSH) biosynthesis, thereby exerting protective effects against myocardial infarction in rats.62,87 Notably, allicin also activates the PI3K/AKT/mTOR pathway, increasing levels of Cox-2, VEGF, and myocardial lactate dehydrogenase (LDH). This pathway activation promotes angiogenesis and plays a significant role in treating ischemia-reperfusion injury in mice.88 This multi-level bioactive network supports allicin as a potential natural drug for regulating CMD.
Tanshinone IIA (TsIIA) ameliorates endothelium-dependent vasodilatory dysfunction and thereby treats atherosclerosis by reconstructing the balance of the endothelin system. It achieves this by inhibiting endothelin-1 (ET-1) secretion, upregulating ETB receptor expression, and downregulating ETA receptor expression, forming a protective “ETB/eNOS/NO” axis.89 At the pathological level, this capacity to restore endothelium-dependent vasodilatory dysfunction can effectively alleviate CMD. Future clinical trials are needed to verify its clinical translation potential. Capsaicin is a natural alkaloid and the primary homovanillic acid derivative in Capsicum plants. Its chemical structure features a vanillyl group, an amide group, and a fatty acid chain.90 This unique structure endows it with important biological functions such as inducing tumor cell apoptosis and regulating angiogenesis. In cardiovascular protection, capsaicin primarily exerts its regulatory effects via TRPV1 channels.91 The capsaicin-TRPV1 mechanism exhibits tissue-specific effects: activation of visceral sensory nerve TRPV1 innervating the vascular wall promotes CGRP release, mediating vascular smooth muscle relaxation.92 Conversely, in postganglionic sympathetic fibers, it induces vasoconstriction by promoting NA release.93 This bidirectional regulatory property makes the capsaicin-TRPV1 system an important molecular target for the fine-tuning of vascular tone.
Natural Compounds in Oxidative Stress
Regarding the amelioration of oxidative stress, the polyphenolic compounds previously mentioned, namely curcumin and resveratrol, exert protective effects by enhancing the activity of antioxidant enzymes. Curcumin significantly reduces ROS generation by modulating NADPH oxidase activity and enhancing antioxidant enzyme function, thereby alleviating oxidative stress in RAW264.7 cells and regulating hypertension.94 This represents a potential therapeutic strategy for improving CMD. Resveratrol, on the other hand, elevates the activity of superoxide dismutase (SOD) and glutathione peroxidase (GPx) by activating the Nrf2 transcription factor. Simultaneously, it specifically inhibits the secretion of pro-inflammatory factors such as IL-2 and IFN-γ by lymphocytes and macrophages, reducing oxidative damage in coronary artery endothelial cells.81
In reducing ROS and mitigating oxidative damage, EGCG exerts its antioxidant effects through a dual mechanism. On the one hand, it directly scavenges ROS and chelates metal ions using its phenolic hydroxyl groups. On the other hand, it alleviates elevated blood pressure in stroke-prone spontaneously hypertensive rats by upregulating antioxidant enzyme activity, inhibiting pro-oxidant enzymes, and inducing the phase II detoxifying enzymes. This synergistic effect, confirmed by a 0.2% catechin intervention experiment, significantly enhances the activity of key antioxidant enzymes.95 Additionally, EGCG blocks the oxidative stress-inflammatory cascade reaction by inhibiting NF-κB and AP-1 activity.46,69,71 Quercetin, leveraging its potent antioxidant properties, scavenges ROS in cardiac fibroblasts and inhibits the MAPK pathway (ERK/p38/JNK phosphorylation) to delay myocardial fibrosis.96,97 Simultaneously, it reduces ROS generation by activating the caspase-3 and NF pathways regulated by PI3K/Akt-κB, thereby ameliorating atherosclerosis.48 In a hydrogen peroxide (H2O2)-induced oxidative stress model, quercetin pretreatment significantly lowered ROS levels. It protected the myocardium in rats with acute myocardial infarction by enhancing antioxidant enzyme activity and reducing levels of markers such as MDA and iNOS. Its inhibition of LDL oxidation and downregulation of the ROS/TLR4 signaling pathway can also impede ox-LDL-induced calcification and osteogenic differentiation in vascular smooth muscle cells.50 Their pleiotropy remains un‑druggable without stabilised formulations, and human data on tissue distribution and chronic safety in cardiovascular indications are critically lacking.
Saponin compounds also exhibit potent antioxidant effects. Salidroside (SAL) provides endothelial protection via spatiotemporal precision regulation of the Nrf2/HO-1 signaling pathway. On the one hand, it inhibits Keap1-mediated ubiquitination and degradation of Nrf2 to promote its nuclear translocation. On the other hand, it enhances Nrf2 autophagic activation via the PKCδ/p62 signaling pathway, significantly elevating the expression of phase II detoxifying enzymes such as HO-1 and NQO1 in HUVECs and PAECs. This coordinated action enhances ROS scavenging capacity and reduces MDA levels.98,99 AS-IV inhibits the ROS/caspase-1/GSDMD pyroptosis pathway by directly neutralizing ROS, thereby inhibiting fibrosis and ventricular remodeling post-myocardial infarction.54 Simultaneously, in HUVEC models, it reduces TLR4 expression, inhibits NF-κB p65 nuclear translocation and VCAM-1/ICAM-1 generation, and restores eNOS activity to ameliorate hyperglycemia-related vascular endothelial inflammation.100
In addition to the aforementioned compounds, TsIIA also possesses antioxidant effects. It inhibits the TNF-α signaling network via a triple intervention: blocking IKK complex phosphorylation to inhibit IκB degradation, preventing NF-κB nuclear translocation, and downregulating VCAM-1/ICAM-1 expression. This constitutes a comprehensive regulation of the oxidative stress-inflammatory pathway.38,63
Discussion
This review analyzes the multidimensional pathogenesis of CMD, considering secondary factors such as endothelial dysfunction, microvascular structural abnormalities, inflammatory imbalance, sex-specific regulation, and metabolic disturbances. It proposes an innovative multi-target intervention strategy utilizing natural compounds. Analysis of studies on natural compounds in cardiovascular diseases (such as atherosclerosis and myocardial ischemia-reperfusion injury) that share similar inflammatory pathways and endothelial dysfunction mechanisms with CMD revealed that at the level of endothelial function regulation, natural compounds enhance NO bioavailability through bidirectional regulation of eNOS/iNOS expression while inhibiting the release of platelet-activating factors to maintain endothelial barrier integrity. Regarding oxidative stress injury, they construct a multilayered defense network to clear excess ROS by activating the PI3K/Akt-Nrf2/ARE signaling axis to enhance the activity of endogenous antioxidant enzymes such as SOD and GSH-Px. In terms of inflammation regulation, they inhibit the transcription and expression of pro-inflammatory mediators such as TNF-α and IL-6 by blocking the IKK/IκB/NF-κB pathway and downregulating VCAM-1 level to improve the vascular inflammatory microenvironment. This “mechanistic homology - target sharing” feature across cardiovascular pathologies suggests that the verified effects of natural compounds in multi-target synergistic regulation could potentially be translated into intervention strategies for the specific pathological progression of CMD. This lays a theoretical basis for developing innovative therapeutic drugs targeting microvascular dysfunction.
Current research on CMD, summarized in this review, highlights the following key points: (1) Deepening pharmacological mechanisms research of natural compounds facilitates the elucidation of disease essence. In recent years, driven by the rapid development of cutting-edge technologies, research on the mechanisms of action of natural compounds has entered a stage of multi-level, multi-dimensional systematic exploration. Current studies integrating data science, animal experiments, and multi-omics technologies, are gradually revealing the roles of natural active components in regulating cellular signaling networks, modulating cytokine levels, and maintaining metabolic homeostasis. The research focus has shifted from single-target identification to the analysis of multi-target synergistic regulation. These studies not only deepen the understanding of the molecular essence of disease occurrence and development but also identify various novel biomarkers and therapeutic targets, providing a critical theoretical basis for precision medicine. (2) Broad suppression of inflammatory responses is currently the main research direction for natural compounds in treating CMD. Current research focuses on the comprehensive regulatory capacity of natural components over the entire inflammatory cascade chain, including the source intervention of upstream inflammatory signaling pathways, the dynamic balance regulation of midstream inflammatory mediator release, and the synergistic promotion of downstream tissue repair processes. Through summarization, it has been found that multiple natural components can target key inflammatory pathway nodes such as NF-κB, MAPK, and JAK-STAT, exhibiting unique dose-dependent regulatory characteristics. This multi-target intervention model, compared to traditional anti-inflammatory drugs, can circumvent the limitations of single-target inhibition while achieving the overall restoration of homeostasis in the inflammatory microenvironment. (3) Natural compounds are progressively entering clinical application, gaining recognition by medical institutions and patients as systemic treatments and complementary and alternative therapies. In clinical practice, traditional medication approaches of the past are becoming less prevalent. Natural compounds are gradually being integrated into the application system spanning the entire cycle of disease prevention, treatment, and rehabilitation. Their systematic application is reflected in three dimensions: As independent therapies, they enable holistic regulation of body functions through the synergistic action of multiple components. As adjuvant treatments, they complement conventional drugs, enhancing efficacy while reducing toxicity. As alternative options, they offer options for patients intolerant to standard treatments. Medical institutions can also formulate personalized integrative treatment plans based on patients’ physical conditions and disease characteristics. This promotes the deep integration of traditional medical knowledge with modern medical advances, injecting new momentum into the diversified development of the healthcare system.
Although this review summarizes the pathogenesis of CMD and the therapeutic approaches using related natural compounds, some limitations in current research have been identified: (1) While the pathological mechanisms of CMD are largely understood, it remains extremely difficult to investigate its signs and symptoms through in vivo experiments due to the lack of specific in vivo models. Consequently, evaluating the efficacy of natural compounds in treating CMD is difficult based on disease phenotypes in animals. The highly heterogeneous clinical manifestations of CMD make existing animal models inadequate for accurately simulating pathological features such as vascular endothelial dysfunction and abnormal microcirculatory resistance during human disease progression. This leads to a gap in the efficacy assessment of natural compounds. A unified standard for correlating disease phenotypes in vivo systems with clinical indicators has not yet been established, particularly lacking highly sensitive and specific dynamic monitoring techniques for key evaluation dimensions such as microvascular reactivity testing and quantification of endothelium-dependent vasodilatory function. (2) Comprehensive toxicological studies on natural compounds for CMD treatment have not been widely conducted. Researchers primarily focus on studying effective doses, while long-term toxicology experiments are scarcely reported, hindering further drug translation. The current research paradigm excessively focuses on exploring effective dose ranges, lacking systematic assessment of potential risks such as metabolic toxicity, organ-specific damage, and drug interactions that may arise from long-term medication. This research imbalance results in significant gaps in the toxicological database for natural compounds, failing to meet the regulatory requirements for innovative drug applications and hindering the optimization design of clinical medication regimens. (3) Extensive randomized controlled trials (RCTs) and real-world studies have not been conducted. The efficacy and safety of natural compounds as complementary and alternative medicines, or even conventional therapeutics, have not gained broad acceptance. The clinical evidence generation system for natural compounds requires systematic strengthening. Although therapeutic potential is reported, extant clinical trials generally suffer from small sample sizes, insufficient observation periods, and single endpoint indicators, making it difficult to generate clinically actionable evidence. This evidence gap directly causes medical decision-makers and regulatory agencies to adopt a cautious stance regarding their clinical positioning, constraining their standardized application in modern healthcare systems. Based on the above limitations, future research can explore the following directions: (1) Constructing a multi-dimensional dynamic disease model system. To overcome the limitations of in vivo CMD models, future efforts need to break through traditional animal model construction approaches and establish pathology mechanism-driven precision modeling strategies. Focus on developing specific CMD animal models, coupled with organ-on-a-chip and 3D bioprinting technologies to construct myocardial organoids with functional vascular networks, enabling multi-scale simulation of the CMD pathological microenvironment. Utilize existing technologies to establish dynamic models spanning from molecular mechanisms to organ dysfunction, and develop models capable of real-time monitoring changes in myocardial metabolism, electrophysiology, and contractile function. (2) Establishing a full-cycle integrated toxicological assessment framework. The current fragmented state of toxicology research necessitates a paradigm shift, requiring the construction of a toxicity early-warning system covering the entire drug development chain. Methodologically, high-throughput in vitro toxicity screening platforms for relevant organs should be established, combined with Quantitative Structure-Activity Relationship (QSAR) models to predict potential compound toxicity, and develop chronic toxicity animal models capable of simulating long-term exposure. Technically, integrate metabolomics to track in vivo drug transformation pathways and analyze target organ-specific injury mechanisms, and utilize CRISPR interference (CRISPRi) libraries to screen toxicity-related gene networks. In designing the evaluation system, standardized testing protocols covering genotoxicity, epigenetic effects, and transgenerational influences need to be developed, with a key focus on modeling the correlation between subchronic toxicity endpoints and clinical adverse reactions. (3) Building an evidence-based medicine-driven clinical validation system. To overcome the challenge of low clinical recognition of natural compounds, a hierarchical and progressive evidence-generation system needs to be constructed. Foundationally, prioritize designing international multicenter, adaptive-design RCTs, employing composite endpoints to assess efficacy and embedding pharmacogenomic biomarker modules, while developing blinding implementation and quality control standards tailored to the characteristics of natural compounds. Regarding clinical treatment, integrate electronic medical records, device monitoring, and medical Internet of Things (IoT) data streams to establish a dynamic adverse reaction monitoring network. For evidence integration, create a multi-dimensional evaluation matrix incorporating molecular mechanism data, toxicological information, and clinical evidence, develop a quantitative scoring system to assess their risk-benefit ratio as complementary or alternative therapies, and form an evidence translation pathway that harmonizes traditional medical wisdom with modern scientific standards.
Conclusion
Natural compounds exert multi-target regulatory effects on core pathological pathways shared between CMD and other cardiovascular diseases—exemplified by the NF-κB/NLRP3 inflammasome axis—suggesting a paradigm of “heterogeneous pathologies, homologous mechanisms”. However, direct evidence derived from CMD-specific models remains scarce, with most findings extrapolated from atherosclerosis or ischemia-reperfusion studies. This review systematically consolidates mechanism-based evidence, identifies this critical translational gap, and underscores the urgent need for CMD‑authentic animal models to validate efficacy and enable clinical translation. Bridging this gap will not only advance precision therapies for CMD but also refine cross-disease drug development strategies in cardiovascular medicine.
Abbreviations
CMD, Coronary microvascular dysfunction; MACE, Major adverse cardiovascular events; ECs, Endothelial cells; NO, Nitric oxide; EDHF, Endothelium-derived hyperpolarizing factor; ET-1, Endothelin-1; eNOS, Endothelial nitric oxide synthase; IL-6, Interleukin-6; TNF-α, Tumor necrosis factor-alpha; CRP, C-reactive protein; NLRP3, NACHT, LRR, and PYD domains-containing protein 3; IL-1β, Interleukin-1 beta; IL-18, Interleukin-18; NOX, NADPH oxidase; CD39, Cluster of differentiation 39; ATP, Adenosine triphosphate; CDKN2B-AS1, Cyclin-dependent kinase inhibitor 2B antisense RNA 1; VEGF-A, Vascular endothelial growth factor A; FOG2, Friend of GATA-2; HIF-1α, Hypoxia-inducible factor 1α; ROS, Oxygen species; RAAS, Renin angiotensin aldosterone system; Ang II, Angiotensin II; ACE, Angiotensin-converting enzyme; AT1, Angiotensin II type 1 (receptors); ACEIs, ACE inhibitors; ARBs, Angiotensin receptor blockers; PGI2, Prostacyclin; CCBs, Calcium channel blockers; HMG-CoA, 3-Hydroxy-3-methylglutaryl-coenzyme A; RhoA, Ras homolog gene family, member A; ROCK, Rho-associated kinase; IκB, Inhibitor of kappa B; MCP-1, Monocyte chemoattractant protein-1; VCAM-1, Vascular cell adhesion molecule-1; TXAS, Thromboxane A2 synthase; TXA2, Thromboxane A2; PGH2, Prostaglandin H2; TP, Thromboxane prostanoid; PLCβ–DAG, Phospholipase C beta–diacylglycerol; PKC, Protein kinase C; NADPH, Nicotinamide adenine dinucleotide phosphate; SERM, Selective estrogen receptor modulator; ERα, Estrogen receptor alpha; MLC, Myosin light chain; CFR, Coronary flow reserve; SOD-2, Superoxide dismutase-2; CPT-1, Carnitine palmitoyltransferase-1; cGMP, Cyclic guanosine monophosphate; ALDH-2, Aldehyde dehydrogenase-2; sGc, Soluble guanylate cyclase; PDE5, Phosphodiesterase type 5; PLCβ-IP3, Phospholipase C beta–inositol trisphosphate; iNOS, Inducible nitric oxide synthase; NF-κB, Nuclear factor kappa-B; ER, Estrogen receptor; MAPK, Mitogen-activated protein kinase; ApoE, Apolipoprotein E; SIRT1, Sirtuin 1; NOS-3, Nitric oxide synthase-3; AMPK, AMP-activated protein kinase; Nrf2, Nuclear factor erythroid 2-related factor 2; COX-1, Cyclooxygenase-1; COX-2, Cyclooxygenase-2; AP-1, Activator protein 1; EGCG, Epigallocatechin gallate; ICAM-1, Intracellular adhesion molecule-1; VSMCs, Vascular smooth muscle cells; PDE, Phosphodiesterase type 1; Akt, Ak straintransforming; EPCs, Endothelial progenitor cells; AS-IV, Astragaloside IV; HUVECs, Human umbilical vein endothelial cells; BBR, Berberine; MMP-2, Matrix metalloproteinase-2; MMP-9, Matrix metalloproteinase-9; u-PA, Urokinase-type plasminogen; H2S, Hydrogen sulfide; PDGF, Platelet-derived growth factor; p53, Tumor suppressor protein p53; p21, Cyclin-dependent kinase inhibitor p21; SOD, Superoxide dismutase; GPx, Glutathione peroxidase; IL-2, Interleukin-2; IFN-γ, Interferon-γamma; AP-1, Activator protein 1; ERK, Extracellular signal-regulated kinase; p38, p38 MAP kinase; JNK, Jun n-terminal kinase; caspase-3, Cysteine-aspase 3; PGE-2, Prosta glandin E2; LDH, lactate dehydrogenase; TsIIA, Tanshinone IIA; RCTs, Randomized controlled trials; QSAR, Quantitative structure-activity relationship; CRISPRi, CRISPR interference.
Data Sharing Statement
The references cited in this review were sourced from PubMed. (https://pubmed.ncbi.nlm.nih.gov/).
Acknowledgments
Thanks to Professor Luo Gang and Professor Liu Mengnan for their critical guidance on the design of this review.
Author Contributions
Mingtai Chen, Xinyi Xiang and Xinrui Yang are co-first authors. All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This review was supported by the National Natural Science Foundation of China (No. 82074378), the Project of Science & Technology Department of Sichuan Province (No. 2026NSFSC1823), the Research Fund of Chinese Medicine Guangdong Laboratory (No. HQL2025SU024), Shenzhen High-level Hospital Construction Fund, the Youth Innovation Project of Sichuan Medical Association (No. Q20250014), and the Project of Southwest Medical University (No. 2024ZXYZX30). The funder had no role in the study design, data analysis, or decision to publish.
Disclosure
The authors declare that they have no competing interests.
References
1. Vancheri F, Longo G, Vancheri S, Henein M. Coronary microvascular dysfunction. J Clin Med. 2020;9(9):2880. doi:10.3390/jcm9092880
2. Crea F, Montone RA, Rinaldi R. Pathophysiology of coronary microvascular dysfunction. Circ J. 2022;86(9):1319–19. doi:10.1253/circj.CJ-21-0848
3. Li L, Li L, Chen C, et al. Scutellarin’s cardiovascular endothelium protective mechanism: important role of PKG-Iα. PLoS One. 2015;10(10):e0139570. doi:10.1371/journal.pone.0139570
4. Li W, Sun K, Hu F, et al. Protective effects of natural compounds against oxidative stress in ischemic diseases and cancers via activating the Nrf2 signaling pathway: a mini review. J Biochem Mol Toxicol. 2021;35(3):e22658. doi:10.1002/jbt.22658
5. Del Buono MG, Montone RA, Camilli M, et al. Coronary microvascular dysfunction across the spectrum of cardiovascular diseases: JACC State-of-the-Art review. J Am Coll Cardiol. 2021;78(13):1352–1371. doi:10.1016/j.jacc.2021.07.042
6. Chen J, Wang B, Meng T, et al. Oxidative stress and inflammation in myocardial ischemia-reperfusion injury: protective effects of plant-derived natural active compounds. J Appl Toxicol. 2025;45(7):1103–1123. doi:10.1002/jat.4719
7. Ji B, Patogênese LXB. Avaliação e Tratamento da Disfunção da Microcirculação Coronariana. Arq Bras Cardiol. 2024;121(8):e20230767. doi:10.36660/abc.20230767
8. Shaw J, Anderson T. Coronary endothelial dysfunction in non-obstructive coronary artery disease: risk, pathogenesis, diagnosis and therapy. Vasc Med. 2016;21(2):146–155. doi:10.1177/1358863X15618268
9. Smilowitz NR, Toleva O, Chieffo A, Perera D, Berry C. Coronary microvascular disease in contemporary clinical practice. Circ Cardiovasc Interv. 2023;16(6):e012568. doi:10.1161/CIRCINTERVENTIONS.122.012568
10. Quarta R, Martino G, Romano LR, et al. The role of circulating biomarkers in patients with coronary microvascular disease. Biomolecules. 2025;15(2):177. doi:10.3390/biom15020177
11. Fan Q, Tao R, Zhang H, et al. Dectin-1 contributes to myocardial ischemia/reperfusion injury by regulating macrophage polarization and neutrophil infiltration. Circulation. 2019;139(5):663–678. doi:10.1161/CIRCULATIONAHA.118.036044
12. Kong P, Cui ZY, Huang XF, Zhang DD, Guo RJ, Han M. Inflammation and atherosclerosis: signaling pathways and therapeutic intervention. Signal Transduct Target Ther. 2022;7:131. doi:10.1038/s41392-022-00955-7
13. Guo Z, Yang Z, Song Z, et al. Inflammation and coronary microvascular disease: relationship, mechanism and treatment. Front Cardiovasc Med. 2024;11:1280734. doi:10.3389/fcvm.2024.1280734
14. Masi S, Rizzoni D, Taddei S, et al. Assessment and pathophysiology of microvascular disease: recent progress and clinical implications. Eur Heart J. 2020;42(26):2590–2604. doi:10.1093/eurheartj/ehaa857
15. Cassavaugh J, Longhi MS, Robson SC. Impact of estrogen on purinergic signaling in microvascular disease. Int J Mol Sci. 2025;26(5):2105. doi:10.3390/ijms26052105
16. Tunc E, Eve AA, Madak-Erdogan Z. Coronary microvascular dysfunction and estrogen receptor signaling. Trends Endocrinol Metab. 2020;31(3):228–238. doi:10.1016/j.tem.2019.11.001
17. Mustafa SJ, Morrison RR, Teng B, Pelleg A. Adenosine receptors and the heart: role in regulation of coronary blood flow and cardiac electrophysiology. Handb Exp Pharmacol. 2009;(193):161–188. doi:10.1007/978-3-540-89615-9_6
18. Patel H, Aggarwal NT, Rao A, et al. Microvascular disease and small-vessel disease: the nexus of multiple diseases of women. J Womens Health. 2020;29(6):770–779. doi:10.1089/jwh.2019.7826
19. Duncker DJ, Koller A, Merkus D, Canty JM. Regulation of coronary blood flow in health and ischemic heart disease. Prog Cardiovasc Dis. 2015;57(5):409–422. doi:10.1016/j.pcad.2014.12.002
20. Duncker DJ, Bache RJ, Merkus D. Regulation of coronary resistance vessel tone in response to exercise. J Mol Cell Cardiol. 2012;52(4):802–813. doi:10.1016/j.yjmcc.2011.10.007
21. Ford TJ, Corcoran D, Padmanabhan S, et al. Genetic dysregulation of endothelin-1 is implicated in coronary microvascular dysfunction. Eur Heart J. 2020;41(34):3239. doi:10.1093/eurheartj/ehz915
22. Gupta RM, Hadaya J, Trehan A, et al. A genetic variant associated with five vascular diseases is a distal regulator of endothelin-1 gene expression. Cell. 2017;170(3):522–533.e15. doi:10.1016/j.cell.2017.06.049
23. Li Y, Wang H, Zhang Y. CDKN2B‐AS1 gene rs4977574 A/G polymorphism and coronary heart disease: a meta‐analysis of 40,979 subjects. J Cell Mol Med. 2021;25(18):8877–8889. doi:10.1111/jcmm.16849
24. Leopold JA. Microvascular dysfunction: genetic polymorphisms suggest sex-specific differences in disease phenotype. Coron Artery Dis. 2014;25(4):275–276. doi:10.1097/MCA.0000000000000122
25. Semenza GL. Hypoxia-Inducible factor 1 and cardiovascular disease. Annu Rev Physiol. 2014;76:39–56. doi:10.1146/annurev-physiol-021113-170322
26. Severino P, D’Amato A, Prosperi S, et al. Potential role of eNOS genetic variants in ischemic heart disease susceptibility and clinical presentation. J Cardiovasc Dev Dis. 2021;8(9):116. doi:10.3390/jcdd8090116
27. Zhou B, Ma Q, Kong SW, et al. Fog2 is critical for cardiac function and maintenance of coronary vasculature in the adult mouse heart. J Clin Invest. 2009;119(6):1462–1476. doi:10.1172/JCI38723
28. Wayne N, Singamneni VS, Venkatesh R, Cherlin T, Verma SS, Guerraty MA. Genetic insights into coronary microvascular disease. Microcirculation. 2025;32(1):e12896. doi:10.1111/micc.12896
29. Bairey Merz CN, Pepine CJ, Shimokawa H, Berry C. Treatment of coronary microvascular dysfunction. Cardiovasc Res. 2020;116(4):856–870. doi:10.1093/cvr/cvaa006
30. Ong P, Athanasiadis A, Sechtem U. Pharmacotherapy for coronary microvascular dysfunction. Eur Heart J Cardiovasc Pharmacother. 2025. doi:10.1093/ehjcvp/pvu020
31. Caliskan M, Erdogan D, Gullu H, et al. Effects of atorvastatin on coronary flow reserve in patients with slow coronary flow. Clin Cardiol. 2007;30(9):475–479. doi:10.1002/clc.20140
32. Marinescu MA, Löffler AI, Ouellette M, Smith L, Kramer CM, Bourque J. Coronary microvascular dysfunction and microvascular angina: a systematic review of therapies. JACC Cardiovasc Imaging. 2015;8(2):210–220. doi:10.1016/j.jcmg.2014.12.008
33. Knuuti J, Kalliokoski R, Janatuinen T, et al. Effect of estradiol-drospirenone hormone treatment on myocardial perfusion reserve in postmenopausal women with angina pectoris. Am J Cardiol. 2007;99(12):1648–1652. doi:10.1016/j.amjcard.2007.01.042
34. AlHabeeb W, Mrabeti S, Abdelsalam AAI. Therapeutic properties of highly selective β-blockers with or without additional vasodilator properties: focus on bisoprolol and nebivolol in patients with cardiovascular disease. Cardiovasc Drugs Ther. 2022;36(5):959–971. doi:10.1007/s10557-021-07205-y
35. Mehta PK, Goykhman P, Thomson LEJ, et al. Ranolazine improves angina in women with evidence of myocardial ischemia but no obstructive coronary artery disease. JACC Cardiovasc Imaging. 2011;4(5):514–522. doi:10.1016/j.jcmg.2011.03.007
36. Rosen SD, Lorenzoni R, Kaski JC, Foale RA, Camici PG. Effect of alpha1-adrenoceptor blockade on coronary vasodilator reserve in cardiac syndrome X. J Cardiovasc Pharmacol. 1999;34(4):554–560. doi:10.1097/00005344-199910000-00012
37. Sadeghi M, Dehnavi S, Asadirad A, et al. Curcumin and chemokines: mechanism of action and therapeutic potential in inflammatory diseases. Inflammopharmacology. 2023;31(3):1069–1093. doi:10.1007/s10787-023-01136-w
38. Meng Z, Yan C, Deng Q, feng GD, lin NX. Curcumin inhibits LPS-induced inflammation in rat vascular smooth muscle cells in vitro via ROS-relative TLR4-MAPK/NF-κB pathways. Acta Pharmacol Sin. 2013;34(7):901–911. doi:10.1038/aps.2013.24
39. Li X, Zhu R, Jiang H, et al. Autophagy enhanced by curcumin ameliorates inflammation in atherogenesis via the TFEB–P300–BRD4 axis. Acta Pharm Sin B. 2022;12(5):2280–2299. doi:10.1016/j.apsb.2021.12.014
40. Wiciński M, Malinowski B, Węclewicz MM, Grześk E, Grześk G. Resveratrol increases serum BDNF concentrations and reduces vascular smooth muscle cells contractility via a NOS-3-Independent mechanism. Biomed Res Int. 2017;2017:9202954. doi:10.1155/2017/9202954
41. Wang Y, Lei L, Su Q, et al. Resveratrol inhibits insulin-induced vascular smooth muscle cell proliferation and migration by activating SIRT1. Evid Based Complement Alternat Med. 2022;2022:8537881. doi:10.1155/2022/8537881
42. Yi CO, Jeon BT, Shin HJ, et al. Resveratrol activates AMPK and suppresses LPS-induced NF-κB-dependent COX-2 activation in RAW 264.7 macrophage cells. Anat Cell Biol. 2011;44(3):194–203. doi:10.5115/acb.2011.44.3.194
43. Csiszar A, Sosnowska D, Wang M, Lakatta EG, Sonntag WE, Ungvari Z. Age-Associated proinflammatory secretory phenotype in vascular smooth muscle cells from the non-human primate macaca mulatta: reversal by resveratrol treatment. J Gerontol a Biol Sci Med Sci. 2012;67(8):811–820. doi:10.1093/gerona/glr228
44. Chen WC, Hsieh SR, Chiu CH, Hsu BD, Liou YM. Molecular identification for epigallocatechin-3-gallate-mediated antioxidant intervention on the H2O2-induced oxidative stress in H9c2 rat cardiomyoblasts. J Biomed Sci. 2014;21(1):56. doi:10.1186/1423-0127-21-56
45. ichi SJ, Ogawa M, Futamatsu H, Kosuge H, Sagesaka YM, Isobe M. Tea catechins improve left ventricular dysfunction, suppress myocardial inflammation and fibrosis, and alter cytokine expression in rat autoimmune myocarditis. European Journal of Heart Failure. 2007;9(2):152–159. doi:10.1016/j.ejheart.2006.05.007
46. ichi SJ, Ogawa M, Izawa A, Sagesaka YM, Isobe M. Dietary consumption of green tea catechins attenuate hyperlipidaemia-induced atherosclerosis and systemic organ damage in mice. Acta Cardiol. 2005;60(3):271–276. doi:10.2143/AC.60.3.2005003
47. Sul OJ, Ra SW. Quercetin prevents LPS-Induced oxidative stress and inflammation by modulating NOX2/ROS/NF-kB in lung epithelial cells. Molecules. 2021;26(22):6949. doi:10.3390/molecules26226949
48. Si TL, Liu Q, Ren YF, et al. Enhanced anti-inflammatory effects of DHA and quercetin in lipopolysaccharide-induced RAW264.7 macrophages by inhibiting NF-κB and MAPK activation. Mol Med Rep. 2016;14(1):499–508. doi:10.3892/mmr.2016.5259
49. Rivera L, Morón R, Sánchez M, Zarzuelo A, Galisteo M. Quercetin ameliorates metabolic syndrome and improves the inflammatory status in obese Zucker rats. Obesity. 2008;16(9):2081–2087. doi:10.1038/oby.2008.315
50. Lei W, Hong CM, feng HZ, et al. Salidroside protects pulmonary artery endothelial cells against hypoxia-induced apoptosis via the AhR/NF-κB and Nrf2/HO-1 pathways. Phytomedicine. 2024;128:155376. doi:10.1016/j.phymed.2024.155376
51. Ma L, Gao Y, Yang G, et al. Notoginsenoside R1 ameliorate High-Fat-Diet and vitamin D3-Induced atherosclerosis via alleviating inflammatory response, inhibiting endothelial dysfunction, and regulating gut microbiota. Drug Des Devel Ther. 2024;18:1821–1832. doi:10.2147/DDDT.S451565
52. Yao F, Xue Q, Li K, Cao X, Sun L, Liu Y. Phenolic compounds and ginsenosides in ginseng shoots and their antioxidant and anti-inflammatory capacities in LPS-Induced RAW264.7 mouse macrophages. Int J Mol Sci. 2019;20(12):2951. doi:10.3390/ijms20122951
53. Kim S, Oh MH, Kim BS, et al. Upregulation of heme oxygenase-1 by ginsenoside Ro attenuates lipopolysaccharide-induced inflammation in macrophage cells. J Ginseng Res. 2015;39(4):365. doi:10.1016/j.jgr.2015.03.008
54. Leng B, Zhang Y, Liu X, et al. Astragaloside IV suppresses high glucose-induced NLRP3 inflammasome activation by inhibiting TLR4/NF-κB and CaSR. Mediators Inflammation. 2019;2019:1082497. doi:10.1155/2019/1082497
55. Tan S, Wang G, Guo Y, Gui D, Wang N. Preventive effects of a natural anti-inflammatory agent, astragaloside IV, on ischemic acute kidney injury in rats. Evid Based Complement Alternat Med. 2013;2013:284025. doi:10.1155/2013/284025
56. Zhang X, Qu H, Yang T, Liu Q, Zhou H. Astragaloside IV attenuate MI-induced myocardial fibrosis and cardiac remodeling by inhibiting ROS/caspase-1/GSDMD signaling pathway. Cell Cycle. 2022;21(21):2309–2322. doi:10.1080/15384101.2022.2093598
57. Costa RF, Rosas EP, Paz ST, et al. Topiramate inhibits capsaicin-induced mast cell degranulation and CGRP release in rat dura mater. Brain Sci. 2024;14(11):1070. doi:10.3390/brainsci14111070
58. Zheng Y, Chen B, Zhang M, et al. Autophagic degradation of LOX-1 is involved in the maintenance of vascular integrity injured by oxLDL and protected by Berberine. Int J Biol Sci. 2023;19(6):1813–1830. doi:10.7150/ijbs.80958
59. Liang KW, Ting CT, Yin SC, et al. Berberine suppresses MEK/ERK-dependent Egr-1 signaling pathway and inhibits vascular smooth muscle cell regrowth after in vitro mechanical injury. Biochem Pharmacol. 2006;71(6):806–817. doi:10.1016/j.bcp.2005.12.028
60. Cui T, Liu W, Yu C, et al. Protective effects of allicin on acute myocardial infarction in rats via hydrogen sulfide-mediated regulation of coronary arterial vasomotor function and myocardial calcium transport. Front Pharmacol. 2022;12:752244. doi:10.3389/fphar.2021.752244
61. Sun F, Xu K, Zhou J, Zhang W, Duan G, Lei M. Allicin protects against LPS-induced cardiomyocyte injury by activating Nrf2-HO-1 and inhibiting NLRP3 pathways. BMC Cardiovasc Disord. 2023;23(1):410. doi:10.1186/s12872-023-03442-1
62. Liu M, Yang P, Fu D, et al. Allicin protects against myocardial I/R by accelerating angiogenesis via the miR-19a-3p/PI3K/AKT axis. Aging. 2021;13(19):22843–22855. doi:10.18632/aging.203578
63. Wang J, He X, Chen W, et al. Tanshinone IIA protects mice against atherosclerotic injury by activating the TGF-β/PI3K/Akt/eNOS pathway. Coronary Artery Disease. 2019;31(4):385. doi:10.1097/MCA.0000000000000835
64. Gracia-Sancho J, Villarreal G, Zhang Y, García-Cardeña G. Activation of SIRT1 by resveratrol induces KLF2 expression conferring an endothelial vasoprotective phenotype. Cardiovasc Res. 2010;85(3):514–519. doi:10.1093/cvr/cvp337
65. Avital-Cohen N, Chapnik N, Froy O. Resveratrol induces myotube development by altering circadian metabolism via the SIRT1-AMPK-PP2A axis. Cells. 2024;13(12):1069. doi:10.3390/cells13121069
66. Guo L, Zhang X, Lv N, et al. Therapeutic role and potential mechanism of resveratrol in atherosclerosis: TLR4/NF-κB/HIF-1α. Mediators Inflamm. 2023;2023:1097706. doi:10.1155/2023/1097706
67. Singh BN, Shankar S, Srivastava RK. Green tea catechin, epigallocatechin-3-gallate (EGCG): mechanisms, perspectives and clinical applications. Biochem Pharmacol. 2011;82(12):1807–1821. doi:10.1016/j.bcp.2011.07.093
68. Khan SG, Katiyar SK, Agarwal R, Mukhtar H. Enhancement of antioxidant and phase II enzymes by oral feeding of green tea polyphenols in drinking water to SKH-1 hairless mice: possible role in cancer chemoprevention. Cancer Res. 1992;52(14):4050–4052.
69. Bhardwaj P, Khanna D. Green tea catechins: defensive role in cardiovascular disorders. Chin J Nat Med. 2013;11(4):345–353. doi:10.1016/S1875-5364(13)60051-5
70. Suzuki JI, Ogawa M, Futamatsu H, Kosuge H, Sagesaka YM, Isobe M. Tea catechins improve left ventricular dysfunction, suppress myocardial inflammation and fibrosis, and alter cytokine expression in rat autoimmune myocarditis. Eur J Heart Failure. doi:10.1016/j.ejheart.2006.05.007
71. Li Y, Yao J, Han C, et al. Quercetin, inflammation and immunity. Nutrients. 2016;8(3):167. doi:10.3390/nu8030167
72. Hertog MG, Feskens EJ, Hollman PC, Katan MB, Kromhout D. Dietary flavonoids and cancer risk in the Zutphen Elderly Study. Nutr Cancer. 1994;22(2):175–184. doi:10.1080/01635589409514342
73. Lu XL, Zhao CH, Yao XL, Zhang H. Quercetin attenuates high fructose feeding-induced atherosclerosis by suppressing inflammation and apoptosis via ROS-regulated PI3K/AKT signaling pathway. Biomed Pharmacother. 2017;85:658–671. doi:10.1016/j.biopha.2016.11.077
74. Im DS. Pro-Resolving effect of ginsenosides as an anti-inflammatory mechanism of panax ginseng. Biomolecules. 2020;10(3):444. doi:10.3390/biom10030444
75. Jw L, Yr C, Hj M, et al. Characterization of the changes in eicosanoid profiles of activated macrophages treated with 20(S)-ginsenoside Rg3. J Chromatogr B Anal Technol Biomed Life Sci. 2017:1065–1066. doi:10.1016/j.jchromb.2017.09.002
76. Liang Y, Chen B, Liang D, et al. Pharmacological effects of astragaloside IV: a review. Molecules. 2023;28(16):6118. doi:10.3390/molecules28166118
77. Fu X, Jiao J, Qin T, et al. A new perspective on ameliorating depression-like behaviors: suppressing neuroinflammation by upregulating PGC-1α. Neurotox Res. 2021;39(3):872–885. doi:10.1007/s12640-020-00292-z
78. Du P, Xu L, Wang Y, et al. Astragaloside IV ameliorates pressure overload-induced heart failure by enhancing angiogenesis through HSF1/VEGF pathway. Heliyon. 2024;10(17):e37019. doi:10.1016/j.heliyon.2024.e37019
79. Wiciński M, Malinowski B, Węclewicz MM, Grześk E, Grześk G. Anti-atherogenic properties of resveratrol: 4-week resveratrol administration associated with serum concentrations of SIRT1, adiponectin, S100A8/A9 and VSMCs contractility in a rat model. Exp Ther Med. 2017;13(5):2071–2078. doi:10.3892/etm.2017.4180
80. Zhang Y, Liu H, Tang W, Qiu Q, Peng J. Resveratrol prevents TNF-α-induced VCAM-1 and ICAM-1 upregulation in endothelial progenitor cells via reduction of NF-κB activation. J Int Med Res. 2020;48(9):0300060520945131. doi:10.1177/0300060520945131
81. Jw K, Sy J, Yh K, et al. Ginsenoside Rg3 inhibits endothelial progenitor cell differentiation through attenuation of VEGF-dependent Akt/eNOS signaling. Phytother Res. 2012;26(9). doi:10.1002/ptr.3722
82. Nakhjavani M, Smith E, Townsend AR, Price TJ, Hardingham JE. Anti-Angiogenic properties of ginsenoside Rg3. Molecules. 2020;25(21):4905. doi:10.3390/molecules25214905
83. Feng X, Sureda A, Jafari S, et al. Berberine in cardiovascular and metabolic diseases: from mechanisms to therapeutics. Theranostics. 2019;9(7):1923. doi:10.7150/thno.30787
84. Ai F, Chen M, Yu B, et al. Berberine regulates proliferation, collagen synthesis and cytokine secretion of cardiac fibroblasts via AMPK-mTOR-p70S6K signaling pathway. Int J Clin Exp Pathol. 2015;8(10):12509.
85. Z Q, L J, D H, L R, P W, W C. Activation of Nrf2/HO-1 signaling: an important molecular mechanism of herbal medicine in the treatment of atherosclerosis via the protection of vascular endothelial cells from oxidative stress. J Adv Res. 2021;34. doi:10.1016/j.jare.2021.06.023
86. Borlinghaus J, Albrecht F, Gruhlke MCH, Nwachukwu ID, Slusarenko AJ. Allicin: chemistry and biological properties. Molecules. 2014;19(8):12591. doi:10.3390/molecules190812591
87. Jeong YY, Ryu JH, Shin JH, et al. Comparison of anti-oxidant and anti-inflammatory effects between fresh and aged black garlic extracts. Molecules. 2016;21(4):430. doi:10.3390/molecules21040430
88. Chen Z, Xu H. Anti-Inflammatory and immunomodulatory mechanism of tanshinone IIA for atherosclerosis. Evid Based Complement Alternat Med. 2014;2014:267976. doi:10.1155/2014/267976
89. Chapa-Oliver AM, Mejía-Teniente L. Capsaicin: from plants to a cancer-suppressing agent. Molecules. 2016;21(8):931. doi:10.3390/molecules21080931
90. Caprodossi S, Amantini C, Nabissi M, et al. Capsaicin promotes a more aggressive gene expression phenotype and invasiveness in null-TRPV1 urothelial cancer cells. Carcinogenesis. 2011;32(5):686–694. doi:10.1093/carcin/bgr025
91. Munjuluri S, Wilkerson DA, Sooch G, Chen X, White FA, Obukhov AG. Capsaicin and TRPV1 channels in the cardiovascular system: the role of inflammation. Cells. 2021;11(1):18. doi:10.3390/cells11010018
92. Yang F, Zheng J. Understand spiciness: mechanism of TRPV1 channel activation by capsaicin. Protein and Cell. 2017;8(3):169. doi:10.1007/s13238-016-0353-7
93. Peng Y, Ao M, Dong B, et al. Anti-Inflammatory effects of curcumin in the inflammatory diseases: status, limitations and countermeasures. Drug Des Devel Ther. 2021;15:4503–4525. doi:10.2147/DDDT.S327378
94. Bernatoniene J, Kopustinskiene DM. The role of catechins in cellular responses to oxidative stress. Molecules. 2018;23(4):965. doi:10.3390/molecules23040965
95. Jiang YH, Jiang LY, Wang YC, Ma DF, Li X. Quercetin attenuates atherosclerosis via modulating oxidized LDL-Induced endothelial cellular senescence. Front Pharmacol. 2020;11:512. doi:10.3389/fphar.2020.00512
96. Min Z, Yangchun L, Yuquan W, Changying Z. Quercetin inhibition of myocardial fibrosis through regulating MAPK signaling pathway via ROS. Pak J Pharm Sci. 2019;32(3 Special):1355–1359.
97. Liang Q, Chen Y, Li C, Lu L. Quercetin regulates the ROS/TLR4 signaling pathway and inhibits Ox-LDL-induced vascular smooth muscle cell calcification. Nan Fang Yi Ke Da Xue Xue Bao. 2018;38(8):980–985. doi:10.3969/j.issn.1673-4254.2018.08.13
98. Zhu Y, Zhang YJ, Liu WW, Shi AW, Gu N. Salidroside suppresses HUVECs cell injury induced by oxidative stress through activating the Nrf2 signaling pathway. Molecules. 2016;21(8):1033. doi:10.3390/molecules21081033
99. Zhang WJ, Frei B. Astragaloside IV inhibits NF- κ B activation and inflammatory gene expression in LPS-treated mice. Mediators Inflammation. 2015;2015. doi:10.1155/2015/274314
100. Ren J, Fu L, Nile SH, Zhang J, Kai G. Salvia miltiorrhiza in treating cardiovascular diseases: a review on its pharmacological and clinical applications. Front Pharmacol. 2019;10:753. doi:10.3389/fphar.2019.00753
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Platelet-Activating Factor Promotes the Development of Non-Alcoholic Fatty Liver Disease
Yin H, Shi A, Wu J
Diabetes, Metabolic Syndrome and Obesity 2022, 15:2003-2030
Published Date: 8 July 2022
The Effects and Pathogenesis of PM2.5 and Its Components on Chronic Obstructive Pulmonary Disease

Wang Q, Liu S
International Journal of Chronic Obstructive Pulmonary Disease 2023, 18:493-506
Published Date: 6 April 2023
Quercetin: A Flavonoid with Potential for Treating Acute Lung Injury

Huang M, Liu X, Ren Y, Huang Q, Shi Y, Yuan P, Chen M
Drug Design, Development and Therapy 2024, 18:5709-5728
Published Date: 6 December 2024
Exercise Prescription Training in Chronic Obstructive Pulmonary Disease: Benefits and Mechanisms
Liu S, Yang A, Yu Y, Xu B, Yu G, Wang H
International Journal of Chronic Obstructive Pulmonary Disease 2025, 20:1071-1082
Published Date: 15 April 2025
Current Perspectives on the NF-κB Signaling Axis as a Potential Pharmacological Target in Cardiorenal Syndrome
Liu Q, Wang X, Cheng P, Yang T, Wang C, Zhou H
Drug Design, Development and Therapy 2025, 19:11557-11583
Published Date: 23 December 2025