Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 15
Platelet-Activating Factor Promotes the Development of Non-Alcoholic Fatty Liver Disease
Received 22 March 2022
Accepted for publication 28 June 2022
Published 8 July 2022 Volume 2022:15 Pages 2003—2030
DOI https://doi.org/10.2147/DMSO.S367483
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Antonio Brunetti
Hang Yin, Anhua Shi, Junzi Wu
Key Laboratory of Microcosmic Syndrome Differentiation, Yunnan University of Chinese Medicine, Kunming, People’s Republic of China
Correspondence: Junzi Wu; Anhua Shi, Key Laboratory of Microcosmic Syndrome Differentiation, Yunnan University of Chinese Medicine, Kunming, People’s Republic of China, Tel/Fax +86 187 8855 7524 ; +86 138 8885 0813, Email [email protected]; [email protected]
Abstract: Non-alcoholic fatty liver disease (NAFLD) is a multifaceted clinicopathological syndrome characterised by excessive hepatic lipid accumulation that causes steatosis, excluding alcoholic factors. Platelet-activating factor (PAF), a biologically active lipid transmitter, induces platelet activation upon binding to the PAF receptor. Recent studies have found that PAF is associated with gamma-glutamyl transferase, which is an indicator of liver disease. Moreover, PAF can stimulate hepatic lipid synthesis and cause hypertriglyceridaemia. Furthermore, the knockdown of the PAF receptor gene in the animal models of NAFLD helped reduce the inflammatory response, improve glucose homeostasis and delay the development of NAFLD. These findings suggest that PAF is associated with NAFLD development. According to reports, patients with NAFLD or animal models have marked platelet activation abnormalities, mainly manifested as enhanced platelet adhesion and aggregation and altered blood rheology. Pharmacological interventions were accompanied by remission of abnormal platelet activation and significant improvement in liver function and lipids in the animal model of NAFLD. These confirm that platelet activation may accompany a critical importance in NAFLD development and progression. However, how PAFs are involved in the NAFLD signalling pathway needs further investigation. In this paper, we review the relevant literature in recent years and discuss the role played by PAF in NAFLD development. It is important to elucidate the pathogenesis of NAFLD and to find effective interventions for treatment.
Keywords: non-alcoholic fatty liver disease, platelet-activating factor, oxidative stress, inflammation, insulin resistance
Introduction
Non-alcoholic fatty liver disease (NAFLD) is a clinicopathological syndrome characterised by excessive fat deposition in hepatocytes, leading to steatosis, excluding alcohol and other definite liver injury factors. NAFLD is a continuous disease spectrum from simple hepatic steatosis (hereafter, NAFL) to non-alcoholic steatohepatitis (NASH), liver fibrosis, cirrhosis and even hepatocellular carcinoma (HCC).1 Of these, NAFL has a good prognosis, whereas NASH has a significant fibrotic potential and develops frequently. Approximately 32% of the 53% of patients with NASH are progressing to hepatic fibrosis.2–8 Even 10–25% of patients with NASH may progress to advanced fibrosis or cirrhosis.9–13 In the context of cirrhosis, the incidence of HCC ranges 2–5%.14,15 As an important risk factor for liver cancer (HCC), the incidence of HCC associated with advanced fibrosis is reported to be between 2.4% and 12.8%.16
With the change in people’s lifestyles and eating habits, the prevalence rate of NAFLD is increasing annually. Globally, epidemiological surveys have shown that approximately 25% of the population suffers from NAFLD, of which 10–30% is attributed to NASH.17 The prevalence of NAFLD varies geographically, ie the prevalence is higher in the Middle East and South America, at 32% and 31%, respectively, about 24% in Europe and 14% in Africa. In the United States, the prevalence rate of NAFLD is about 32% and increases with age. In Asia, the overall prevalence rate of NAFLD is about 30%, the onset age is getting younger and the number of patients is increasing annually. Among them, Japan has the lowest, with about 22%. South Korea, Singapore, Malaysia, India and Iran all have prevalence rates of more than 30%, and the prevalence rate in Mainland China is close to 30%18–21 (Figure 1). NAFLD has become the number one chronic liver disease worldwide.22
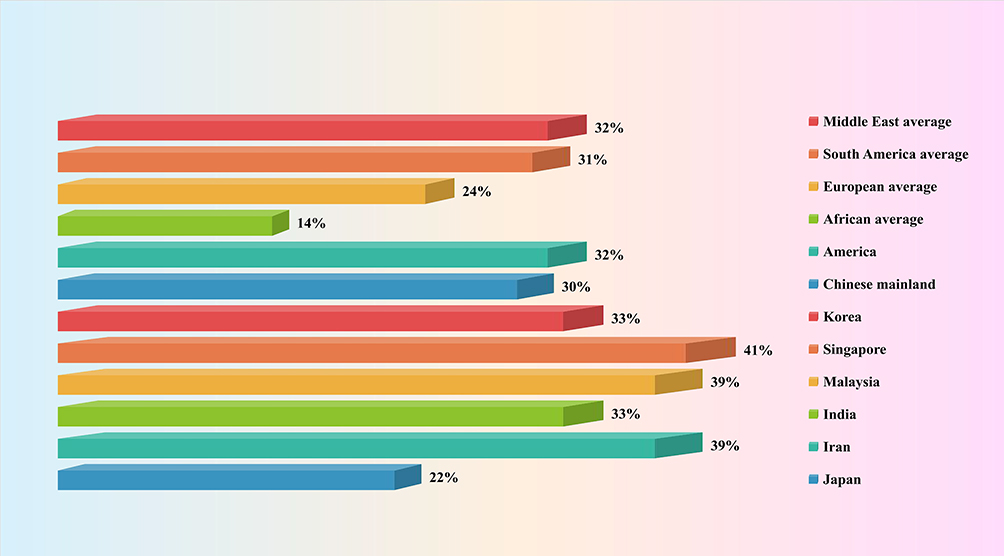 |
Figure 1 Prevalence of non-alcoholic fatty liver disease in different regions. |
The pathogenesis of NAFLD is very complicated. Some risk factors, including lipid metabolism disorder, chronic inflammation and oxidative stress, have been proved to play a key role in the pathogenesis of NAFLD. Currently, the most important pathogenesis of NAFLD is the ‘multiple-hit hypothesis’:23 the “first hit” is mainly insulin resistance (IR), which inhibits lipid β-oxidation, reduces fatty acid consumption and promotes hepatic triglyceride accumulation. Fat deposition enhances glucose-6-phosphatase activity, stimulates gluconeogenesis and insulin secretion and induces IR,24 leading to a vicious circle of IR and fat deposition. In the first attack, lipid peroxidation produces a large number of reactive oxygen species (ROS), induces oxidative stress, causes liver injury, releases inflammatory cytokines [such as tumour necrosis factor-alpha (TNF-α), interleukin-6 (IL-6) and interleukin-1beta (IL-1β)] and causes an inflammatory reaction, resulting in the aggravation of liver injury and even necrosis and fibrosis, forming the “second blow”. Moreover, endoplasmic reticulum stress (ERS), mitochondrial dysfunction, adipose tissue dysfunction and changes in intestinal flora can all exacerbate NAFLD development.
To further improve the pathogenesis of NAFLD and complement the “multiple-hit” theory, we propose the hypothesis that the platelet-activating factor (PAF) contributes to NAFLD development. PAF is a phospholipid that can stimulate platelet activation.25 Recent studies have reported the relationship between platelet activation and NAFLD. For example, Oral et al examined plateletcrit in patients with NAFLD and found that it was significantly elevated and positively correlated with the degree of steatosis.26 Similarly, Ozhan et al compared the mean platelet volume (MPV) between patients with NAFLD and patients with non-fatty liver. Their results showed that MPV in patients with NAFLD was higher and positively correlated with IR, a pathogenic factor of NAFLD, and alanine aminotransferase and aspartate aminotransferase.27 Arslan et al also obtained similar results. Through correlation analysis, they confirmed that the increase in MPV in patients with NAFLD was associated with IR.28 Furthermore, Malehmir et al established platelet glycoprotein receptor I bα (GPIBα) functionally deficient mice fed a choline-deficient high-fat diet (HFD), which resulted in reduced liver damage, such as hepatic steatosis and inflammatory cell infiltration.29 On the contrary, aspirin, a platelet activation antagonist, has become an important drug for delaying NAFLD development. Clinical aspirin can alleviate NAFLD tissue damage and reduce the prevalence of NAFLD and the risk of liver fibrosis.30–33 Consistent with clinical practice, aspirin can also improve NAFLD in animal models. Wang et al used aspirin to treat a mouse model of NAFLD and significantly improved hepatic steatosis, insulin sensitivity and glucose tolerance.34 Similarly, Ford et al used aspirin to improve blood glucose and insulin tolerance in mice with a HFD. Linear regression analysis confirmed that aspirin could down-regulate proinsulin and improve IR in patients with blood glucose disorders.35 Moreover, Fujita et al used aspirin to reduce steatosis and triglyceride accumulation in the liver of choline-deficient rats and reduce liver inflammatory cell infiltration and fibrosis.36 In line with this, Ibrahim et al administered nitroaspirin to NAFLD rats to attenuate oxidative stress, alleviate hepatic steatosis and reduce inflammatory cytokine-induced expressions of nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2).37 This suggests that platelet activation has critical importance in the development and progression of NAFLD. However, how the PAF regulates platelet activation to promote the occurrence and development of NAFLD requires further examination.
To this end, we reviewed a large amount of relevant literature. The results show that the human PAF receptor contains 342 amino acids and belongs to the G protein-coupled receptor family.38,39 PAF receptors are widely distributed in tissues and cells, such as platelets, neutrophils, macrophages, monocytes and endothelial cells, in the brain, lungs, liver and uterus.40 The binding of the PAF to the PAF receptor induces platelet activation that is involved in NAFLD progression through four main pathways.
The first is the PAF–5-hydroxytryptamine (5-HT) signalling pathway. Most 5-HT in the circulation is stored in platelets, and when the PAF binds to the PAF receptor, it stimulates platelet activation and releases 5-HT. Specific binding of 5-HT to the 5-HT receptor on hepatocytes will activate a series of biological effects. The second is the PAF–inositol triphosphate (IP3) signalling pathway. The PAF activates the hydrolysis of phosphatidylinositol 4,5-bisphosphate in hepatocyte membranes to produce IP3 when bound to the PAF receptor. IP3 binds to inositol 1,4,5-trisphosphate receptor (IP3R) in the hepatocytes, regulates Ca2+ release from the endoplasmic reticulum and may trigger ERS. The third is the PAF–arachidonic acid (AA) signalling pathway. Generally, AA is bound to the cell membrane by an ester bond until PLA2 is activated by calcium signalling, at which time it hydrolyses the ester bond and releases AA. Therefore, the PAF, when bound to the PAF receptor, can activate the release of AA from the cell membranes of various cells, including platelets, hepatocytes and inflammatory cells. AA can be further converted into leukotrienes (LTs), prostaglandins (PG) and thromboxane A2 (TXA2). PG mainly includes signalling molecules, such as prostaglandin I2 (PGI2), prostaglandin E2 (PGE2), prostaglandin D2 (PGD2) and prostaglandin F2α (PGF2α). The fourth pathway is the PAF–diacylglycerol (DAG) signalling pathway. The PAF, when combined with the PAF receptor, activates the hydrolysis of phosphatidylinositol 4,5-bisphosphate in the hepatocyte membranes to produce DAG. DAG deposition in hepatocytes may induce the activation of p38-mitogen-activated protein kinases (p38MAPK) and protein kinase C (PKC) (protein kinase Cε and protein kinase Cδ). Protein kinase Cδ can also agonise ERS. These signalling pathways can cause cascading reactions that lead to oxidative stress, inflammatory responses or IR and damage to hepatocytes and even the liver, which may ultimately contribute to NAFLD development and progression (Figure 2).
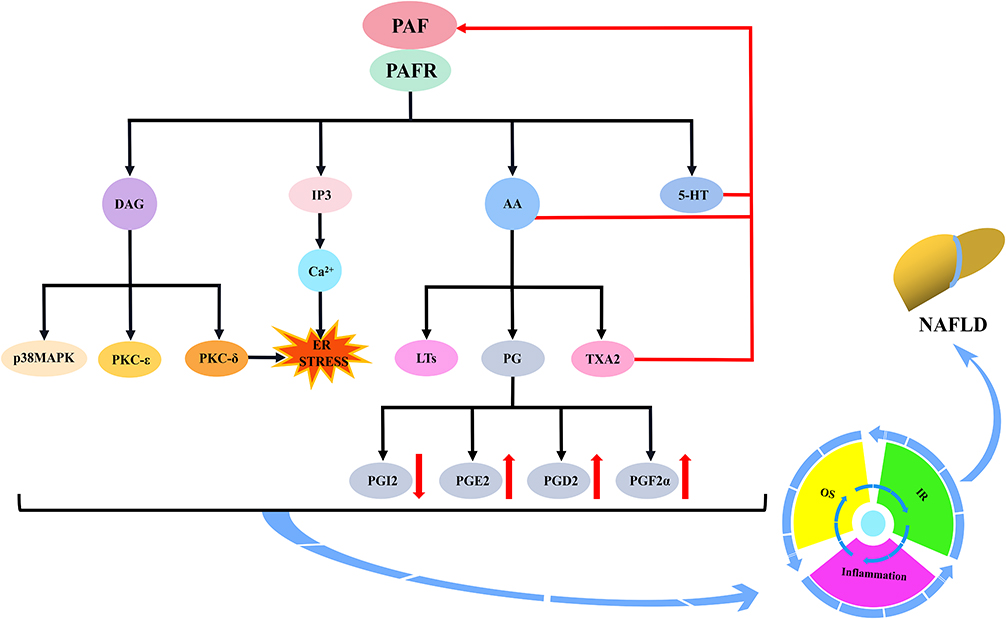 |
Figure 2 Mechanism of action of the PAF via platelet activation to induce NAFLD. The binding of the PAF to the PAFR will induce platelet activation and stimulate the secretion of 5-HT, IP3, AA and DAG by platelets or multiple cells. IP3, by regulating Ca2+ mobilisation, may trigger endoplasmic reticulum stress (ERS). AA can be further converted to LTs, PG and TXA2. PG mainly includes PGI2, PGE2, PGD2 and PGF2α. The activation of DAG induces the activation of p38MAPK and PKC (PKCε and PKCδ), and PKCδ may agonise ERS. Moreover, 5-HT, AA and TXA2 further stimulate the PAF and amplify platelet activation. Subsequently, these substances induce cascade reactions that may be involved in NAFLD development by promoting the development of oxidative stress, inflammatory responses or insulin resistance. Abbreviations: PAF, platelet-activating factor; PAFR, platelet-activating factor receptor; 5-HT, 5-hydroxytryptamine; IP3, inositol triphosphate; AA, arachidonic acid; DAG, diacylglycerol; ER STRESS, endoplasmic reticulum stress; LTs, leukotrienes; PG, prostaglandins; TXA2, thromboxane A2; PGI2, prostaglandin I2; PGE2, prostaglandin E2; PGD2, prostaglandin D2; PGF2α, prostaglandin F2α; p38MAPK, p38-mitogen-activated protein kinases; PKC, protein kinase C; PKCε, protein kinase Cε; PKCδ, protein kinase Cδ; OS, oxidative stress; IR, insulin resistance; NAFLD, non-alcoholic fatty liver disease. |
PAF-Mediated Cytokine Involvement in NAFLD
PAF–5-HT Signalling Pathway and NAFLD
The PAF–5-HT signalling pathway occurs mainly by PAF-induced platelet activation, which stimulates the release of 5-HT from platelet-dense granules. 5-HT is involved in platelet aggregation and other processes by binding to the 5-HT receptor. This phenomenon is supported by clinical data, for example, Greco et al used PAF to treat platelets in patients with type 1 diabetes, resulting in the up-regulation of 5-HT release over time and inducing platelet aggregation.41 Similar results were obtained in the experiments of O’Donnel et al They incubated PAF with human platelets, which significantly increased the release of 5-HT.42 Consistent with clinical data, PAF also has an excitatory effect on 5-HT in animal experiments. This was confirmed in an experiment by Murphy et al, who used PAF to stimulate rabbit platelets and promoted the secretion of 5-HT.43 Similar results were obtained in the experiments by Pédrono et al The incubation of rabbit platelets with increasing PAF concentrations resulted in a concentration-dependent release of 5-HT.44 Moreover, Brooks et al activated horse platelets with PAF to trigger 5-HT release and promote platelet aggregation.45 In line with this, similar results were reported in an in vitro experiment by Bailey et al who used PAF to treat horse platelets, resulting in 5-HT release and platelet activation products, such as thromboxane-B2 (TXB2) and 12-hydroxyeicosatetraenoic acid (12-HETE). Based on this, the PAF–5-HT signalling pathway is formed.46 5-HT is synthesised by tryptophan hydroxylase (TPH) and 5-hydroxytryptophan decarboxylase from tryptophan; it is secreted by intestinal chromaffin cells, widely distributed in the circulation. Most of the 5-HT in the blood circulation will be absorbed and sequestered into platelets. Under PAF stimulation, 5-HT is released from platelets and will enter the liver and other organs for metabolism and participate in the pathological processes of some liver diseases.47–49
5-HT is a well-known modulator of liver functions. During the onset of chronic liver diseases, the elevated plasma levels of 5-HT induce oxidative stress, impair insulin sensitivity and promote the activation of multiple inflammatory signalling molecules in hepatocytes, all of which play a key role in hepatic steatosis, dyslipidaemia and liver injury (Figure 3).50–53
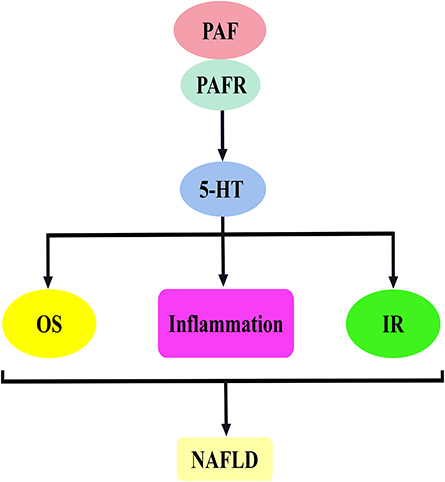 |
Figure 3 PAF activates 5-hydroxytryptamine to promote the mechanism of NAFLD development. The binding of PAF to PAFR stimulates 5-HT release, which is involved in NAFLD development mainly by inducing oxidative stress, inflammatory response and insulin resistance. Abbreviations: PAF, platelet-activating factor; PAFR, platelet-activating factor receptor; 5-HT, 5-hydroxytryptamine; OS, oxidative stress; IR, insulin resistance; NAFLD, non-alcoholic fatty liver disease. |
5-HT needs to bind to its specific receptor for executing biological functions. The presence of 5-HTR2A on the surface of hepatocytes suggested that 5-HT release induced by PAF may be involved in the disease processes in the liver, eg NAFLD.54 In this regard, Osawa et al used 5-HT to treat hepatocytes and, as a result, significantly increased triglyceride levels and lipid droplet accumulation.55 Similarly, Wang et al detected 5-HTR2A expression in liver tissues and hepatocytes (BRL-3A), and they treated hepatocytes with an HTR2A agonist (TCB-2) and an antagonist (ketanserin), respectively. Their results showed that TCB-2 promoted the mRNA expression of peroxisome proliferator-activated receptor γ2 (PPARγ2), sterol regulatory element-binding transcription factor 1c (SREBP-1C) and fatty acid synthase (FAS), genes related to hepatocyte lipid synthesis, and increased hepatocyte neutral lipid and triglyceride levels.54 Consistent with cellular experiments, Wang et al also confirmed in animal models that 5-HT is associated with NAFLD, and they used a high-fat, high-sugar diet to induce a NASH rat model. Their results showed an up-regulation of serum 5-HT levels and a positive correlation with the non-alcoholic fatty liver activity score (NAS).54 Similarly, Osawa et al used L-tryptophan (5-HT raw material) in combination with a high-fat chow to feed mice, which resulted in the steatosis of the liver accompanied by elevated serum 5-HT levels.55 In addition, clinical data confirm the correlation between 5-HT and NALD. For instance, Wang et al compared serum 5-HT levels in patients diagnosed with NAFLD by ultrasonography with those healthy individuals. As shown in the results, the serum 5-HT level was significantly higher in patients with NAFLD than in normal subjects. Moreover, they further analysed and confirmed that serum 5-HT was positively correlated with ultrasound scores. Serum 5-HT may be associated with NAFLD.54 However, the mechanism of 5-HT involvement in NAFLD needs further investigation. In this regard, Nam et al used LP-533401 or p-chlorophenyl alanine (a TPH inhibitor) to treat mice on a HFD. As results showed, hepatic steatosis was improved and the expression of lipid metabolism-related genes, such as Srebp-1c, FAS and apolipoprotein b (Apob) was significantly down-regulated. It was confirmed that 5-HT regulation of lipid metabolism drives NAFLD development.56 Similar results were obtained in a trial by Choi et al, who knocked out the tryptophan hydroxylase 1 (Tph1) or Htr2a genes in mice on a HFD, blocked 5-HT signalling, attenuated hepatic steatosis and reduced the expression of adipogenesis-related genes.57 Notably, 5-HT can induce multiple signals downstream to drive NAFLD development. For example, Wang et al used a free fatty acid (FFA)-induced NASH hepatocyte model and treated hepatocytes with an HTR2A agonist (TCB-2) and an antagonist (ketanserin), respectively. Their results showed that TCB-2 significantly up-regulated the mRNA expressions of Tnf-α, Il-6 and monocyte chemoattractant protein 1 (Mcp-1) compared with ketanserin.54 This indicated that 5-HT-mediated inflammatory responses accelerate NASH progression. Similar results were obtained in a trial by Crane et al, who knocked down the Tph1 gene in mice on a HFD, resulting in a significant reduction in the adipose tissue inflammatory markers cluster of differentiation 68 (Cd68), Tnf-α and Mcp-1.58 In addition, Crane et al further found that Tph1 knockout mice had lower blood glucose and fasting serum insulin. Moreover, glucose tolerance and insulin sensitivity were improved. This indicated that 5-HT is involved in NAFLD via IR.58 Oxidative stress may be another mechanism by which 5-HT is involved in NAFLD. This was verified in a trial by Nocito et al, who used a choline-methionine-deficient diet to induce a mouse model of NASH with significantly elevated liver tissue malondialdehyde (MDA). Interestingly, the knockdown of the Tph1 gene reversed this effect.59
PAF–IP3–Ca2+ Signalling Pathway and NAFLD
The activation of the PAF–IP3–Ca2+ signalling cascade depends on the intracellular signal transduction of PAF. By activating phospholipase C, phosphatidylinositol 4,5-bisphosphate is hydrolysed to induce IP3 production and further mobilisation of intracellular Ca2+ release.60 This effect is present in platelets and cells. For example, Sakon et al used PAF to incubate platelets, which increased the concentration of Ca2+ by stimulating IP3 formation.61 Similarly, Yu et al induced human platelets by using PAF, which significantly promoted the IP3 production and elevated Ca2+ concentration.62 Moreover, the effect of PAF to mobilise Ca2+ is present in monocytes. This was confirmed by Ng et al in their experiments; they used PAF to treat human peripheral blood mononuclear cells by inducing phosphatidylinositol hydrolysis, leading to a dose-dependent release of IP3, which triggered Ca2+ mobilisation.63 However, L-659, L-989 or WEB2086 (PAF receptor antagonists) abolished this effect.63,64 Accordingly, in endothelial cells, Lin et al demonstrated that PAF stimulates phosphatidylinositol metabolism in a dose-dependent manner, generating IP3 and causing Ca2+ elevation, an effect inhibited by WEB2086 (PAF receptor antagonist).65 In neutrophils, Koike et al showed that PAF treatment increased IP3 levels and mobilised the release of intracellular Ca2+ and that SM-12502, WEB-2086 and RP-48740 (PAF receptor antagonists) blocked this effect.66 This effect is also present in hepatocytes. Okayasu et al incubated rat hepatocytes with PAF and detected a down-regulation of phosphatidylinositol-4,5-bis-phosphate, suggesting the synthesis of IP3.67 However, IP3 needs to bind to IP3R to mobilise Ca2+ release. This view is supported by Chu et al; they used PAF pre-treatment of guinea pig-isolated cardiomyocytes, which promoted Ca2+ mobilisation through the activation of the IP3 pathway. Interestingly, this effect was eliminated by the IP3R antagonist 2-aminoethoxydiphenyl borate.68 This suggested that IP3 downstream signalling requires the help of the IP3R. This conclusion was supported by Lautenschläger et al, who used PAF to stimulate Ca2+ release downstream of IP3 and slowed down small intestinal vasoconstriction; in addition, further use of 2-aminoethoxydiphenyl borate to weaken IP3 signalling reversed this effect.69 For this reason, the PAF–IP3–Ca2+ signalling pathway was formed.70 IP3 is a signalling messenger whose main role is to mobilise intracellular Ca2+ release. In response to PAF stimulation, IP3 is produced in large quantities. By binding to the IP3R, IP3 activates the Ca2+ channel and stimulates Ca2+ transport, thereby promoting mitochondrial Ca2+ inward flow;71 which contributes to cellular injury and liver disease development.71
IP3R is mainly located in the endoplasmic reticulum; moreover, binding to IP3 induces receptor conformational changes and channel opening, which mediates Ca2+ release and transfer from the endoplasmic reticulum to mitochondria.72 When Ca2+ release is uncontrolled, it causes mitochondrial calcium overload and functional impairment. In case of serious uncontrolled release, it induces ERS, oxidative stress, inflammatory response and IR as well as damage to hepatocytes, which can lead to various liver diseases, including NAFLD (Figure 4).73,74
 |
Figure 4 PAF activates inositol triphosphate to promote the mechanism of NAFLD development. The binding of PAF to PAFR induces IP3 production, and IP3 binding to the IP3R promotes Ca2+ release, damages mitochondria and induces IR. Moreover, Ca2+ overload may induce ER stress. ER stress is associated with the development of oxidative stress, inflammatory responses and insulin resistance, which contribute to NAFLD development. Abbreviations: PAF, platelet-activating factor; PAFR, platelet-activating factor receptor; IP3, inositol triphosphate; IP3R, inositol 1,4,5-trisphosphate receptors; ER STRESS, endoplasmic reticulum stress; OS, oxidative stress; IR, insulin resistance; NAFLD, non-alcoholic fatty liver disease. |
In hepatocytes, the IP3R is the only intracellular calcium release channel. In this regard, Hirata et al used IP3 to stimulate hepatocytes, and as a result, the expression of the IP3R and Ca2+ waves was detected.75 After induced release by PAF in hepatocytes, IP3 may bind to the IP3R to regulate Ca2+ signalling and be involved in the development of liver disease. Therefore, NAFLD progression is usually accompanied by the dysregulation of IP3R expression.76 This idea is supported by Arruda et al, who examined mitochondrial proteins in obese mouse hepatocytes. As a result, the expression of proteins related to the Ca2+ transport, such as IP3R type 1 (IP3R1), was significantly increased. This causes Ca2+ overload, impairs mitochondrial membrane potential and leads to mitochondrial dysfunction. Subsequently, the phosphorylation of insulin receptors insulin receptor substrate 1 (IRS1) and protein kinase B (AKT) are blocked, resulting in impaired insulin sensitivity and glucose tolerance. This contributes to NAFLD development. However, the knockdown of the IP3R type 1 (Ip3r1) gene significantly improved the Ca2+ flux to avoid these results.77 Accordingly, Feriod et al obtained Ip3r1 gene deletion mice using a hybridisation technique. As a result, hepatic Ip3r1 protein expression was reduced, resulting in impaired Ca2+ signalling; meanwhile, triglycerides were down-regulated, steatosis was improved and expressions of lipogenic genes Srebp and FAS were reduced.78 The suppression of the Ip3r1 gene has good anti-NAFLD activity. The IP3R opens only when it binds to IP3, inducing the Ca2+ release from the endoplasmic reticulum.79 In NAFLD, sustained IP3 stimulation will result in a large loss of Ca2+ from the endoplasmic reticulum, which will activate the calcium channel store-operated calcium channel (SOCC) that mediates the inward flow of extracellular Ca2+80–82 leading to an increase in intracellular Ca2+ concentration and causing calcium imbalance in the endoplasmic reticulum. This was confirmed in the experiments by Zhang et al They established a mouse hepatocyte lipid gradient model using oleic acid, and their results showed a concentration-dependent increase in 1,4,5-trisphosphate receptor and SOCC channel opening and intracellular calcium levels with increasing oleic acid concentration.83 Endoplasmic reticulum calcium imbalance predisposes to ERS.84 Thus, Lai et al used palmitic acid (PA) in L02 cells to induce a hepatic steatosis cell model. Their results showed that reduced sarco-endoplasmic reticulum Ca2+-ATPase (SERCA) activity led to the up-regulation of intracellular calcium ion concentration and increased the expression of ERS-related proteins binding immunoglobulin protein (BIP), C/EBP homologous protein (CHOP), activating transcription factor-6 and inositol-requiring protein-1 (IRE-1). These prompts that ERS is activated.85 Similar results were obtained in the experiments by Zhang et al, who used PA to treat hepatocytes, which impaired SERCA activity and disrupted endoplasmic reticulum Ca2+ homeostasis, leading to ERS.86 Oxidised protein folding releases a single ROS for each disulphide bond formed, whereas the endoplasmic reticulum provides a unique environment for protein folding and disulphide bond generation. Consequently, ERS generates large amounts of ROS, leading to increased oxidative stress, which contributes to NAFLD development.87 Interestingly, ROS is also thought to be one of the main causes of ERS induction, forming a vicious circle with ERS. This was confirmed in an experiment by Zhang et al, who used PA to treat hepatocytes and significantly up-regulated ROS levels, resulting in ERS.86 Further inhibition of ROS using the hepatic stimulating substance was accompanied by the down-regulation of ERS-related marker expression. In addition, by enhancing inflammatory or apoptotic signalling, ERS may be another mechanism to promote NAFLD.88,89 The experiments by Zhang et al confirmed this idea. They administered the ERS-inducing agent clathrin tunicamycin intraperitoneally to mice, which resulted in hepatic steatosis with significant increases in ERS markers, including BIP/glucose-regulated protein 78 (BIP/GRP78), CHOP and IRE1α. Meanwhile, the inflammatory response is enhanced, and IL-1β expression is up-regulated. Further knockdown of the caspase-1 gene significantly improved these results. It is implied that inflammatory vesicle activity is also involved in ERS-induced hepatic steatosis and inflammation.90 Similarly, Xu et al used ginsenoside Rg1 (Rg1) to inhibit the activation of NOD-like receptor family pyrin domain-containing 3 inflammatory vesicles and IL-1β and IL-18 by reducing the expressions of ERS-related proteins GRP78 and CHOP in NAFLD mice.91 Moreover, Ye et al added ERS agonists in a NASH cell model to activate the expressions of nuclear factor kappa B (NF-κB)-related proteins, and the ERS inhibitor taurine deoxycholic acid reverses this phenomenon.92 ERS may also promote NAFLD progression by activating IR. For example, Özcan et al used the ERS inducer clathrin (tunicamycin) to treat hepatocytes. Accordingly, tyrosine phosphorylation of insulin receptor substrate 1 (IRS-1) was decreased, and AKT phosphorylation was inhibited, promoting IR and accelerating NAFLD progression.93 Similarly, Arruda et al utilised a recombinant construct encoding a synthetic linker that increased the endoplasmic reticulum to mitochondrial contact sites and facilitated Ca2+ transport from the endoplasmic reticulum to the mitochondria. In the HFD mice, this linker leads to the impaired phosphorylation of IRS1 and AKT in the liver, activated the expression of c-Jun N-terminal kinase (JNK), activating transcription factor-4 (ATF4) and CHOP proteins as well as activating transcription factor-3 (Atf3) and Atf4 mRNA. ERS may be present during IR development.77
PAF–AA Signalling Pathway and NAFLD
The activation of the PAF–AA signalling pathway is dependent on PAF-driven phospholipase A2 (PLA2) activation, which regulates AA production.94 In platelets and cells, PAF has a strong stimulatory activity against AA. This idea is supported by Catalán et al, as they used PAF to pretreat rabbit platelets. Accordingly, AA sustained a rapid release, and this effect was eliminated by the dihydropyridine derivative PCA-4230, probably because this drug affects a PAF receptor antagonist.95 Analogously, similar results were obtained in the experiments by Oestvang et al; they employed PAF to treat THP-1 monocytes. As a result, PLA2 activation was induced, triggering AA release. However, the PAF antagonist WEB2170 abolished the synthesis of AA.96 Moreover, Hurst et al used PAF to stimulate rabbit corneas, resulting in the up-regulation of AA release with time. However, the addition of BN50727 (PAF receptor antagonist) effectively attenuated PAF signalling, thus reversing this effect.97 The same effect is present in hepatocytes. The incubation of hepatocytes with PAF by Svetlov et al significantly increased the content of free AA.98 From this, the PAF–AA signalling pathway is formed. AA is an essential fatty acid, mainly used for the synthesis of pro-inflammatory mediators. They activate intracellular inflammatory signal transduction after binding to inflammatory cell receptors. This contributes to the occurrence of cardiovascular diseases, liver diseases, etc.99
AA is a polyunsaturated fatty acid, which is closely related to metabolic disorders. The development of NAFLD, as a chronic metabolic liver disease, necessitates the use of AA.100 The increase in AA (an inflammatory regulatory mediator) levels contributes to cellular inflammatory signalling. Interestingly, AA deposition is toxic to cells, which disrupts cellular oxidative stress homeostasis and causes cell damage.101,102 In addition, AA can generate hydroperoxides catalysed by LOX, which induces oxidative stress, inflammatory response and IR, thereby leading to the worsening of NAFLD (Figure 5).103
 |
Figure 5 PAF activates AA to promote the mechanism of NAFLD development. The binding of PAF to PAFR stimulates AA release, which is involved in NAFLD development mainly by inducing oxidative stress, inflammatory response and IR. Abbreviations: PAF, platelet-activating factor; PAFR, platelet-activating factor receptor; AA, arachidonic acid; OS, oxidative stress; IR, insulin resistance; NAFLD, non-alcoholic fatty liver disease. |
PAF is a known phospholipid mediator of inflammation and is a potent chemokine for various inflammatory cells. It activates the inflammatory cascade through G protein-coupled receptors, stimulating inflammatory cell production and releasing inflammatory mediators, such as TNF-α, IL-1 and IL-6, thereby increasing vascular permeability and leading to a vascular inflammatory response.104,105 Normally, AA is bound to the cell membrane as an ester bond until PLA2 is activated through calcium signalling to hydrolyse the ester bond and release AA. AA will synthesise pro-inflammatory mediators, activate intracellular inflammatory signals, promote the synthesis and release of inflammatory factors, such as TNF and ILs, and amplify the inflammatory response.106,107 This suggests that PAF can promote inflammatory responses by activating PLA2, thereby releasing AA. Inflammation is a constant in NAFLD.108 Elevated levels and activation of inflammatory cytokines underlie the pathology of hepatocyte injury.109 PAF–AA signalling may drive NAFLD development by damaging hepatocytes. Thus, NAFLD progression is usually accompanied by the dysregulation of the expression of AA, the inflammatory pathway-related protein PLA2. This view is supported by clinical data that confirm this view. Colak et al compared the levels of PLA2, the key enzyme of AA production, in the serum of patients with NASH and healthy participants. Their results showed that PLA2 levels were elevated in patients with NASH and significantly correlated with steatosis scores.110 Moreover, Zelber et al used liquid chromatography-tandem mass spectrometry to determine AA levels in 105 patients with NAFLD. Their results confirmed that AA levels were elevated in patients with NAFLD and were not associated with obesity.111 Similarly, Tutino et al used aerobic exercise and a hypoglycaemic diet to down-regulate the ratio of pro-inflammatory AA to the anti-inflammatory antioxidant eicosapentaenoic acid (EPA) in patients with NAFLD, resulting in improved steatosis. Furthermore, they developed a linear regression model to confirm that a decrease in the AA/EPA ratio was associated with an improvement in NAFLD.112 Consistent with clinical data, animal experiments have confirmed that with dysregulated PLA2, AA mediates inflammation to promote NAFLD development. Ii et al attenuated HFD induced hepatic steatosis by knocking out the Pla2 gene in HFD mice.113 Furthermore, Sztolsztener et al induced an NAFLD rat model with a HFD. Their results showed that the expressions of inflammation-related proteins NF-κB and IL-6 are elevated with the increase in AA content.114 Similarly, Ma et al used a HFD to establish an NAFLD rat model, and their results showed that PLA2 gene expression, which promoted AA production, led to the expression of pro-inflammatory mediators and exacerbated hepatic steatosis.115 ROS signalling activated by AA may be another mechanism that promotes NAFLD. This was verified in the experiments by Ghazali et al, who treated HepG2 cells with different ratios of AA/docosahexaenoic acid (DHA), and their results showed that a high AA/DHA ratio could inhibit mitochondrial respiration and activity, causing mitochondrial dysfunction, inducing ROS production and thus exacerbating oxidative stress.116 This contributes to NAFLD development. Interestingly, AA can activate cell membrane Ca2+ channels, leading to Ca2+ inward flow,117,118 which activates nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and promotes ROS release.119 Conversely, ROS can trigger AA production signals and enhance AA-related metabolism, creating a cyclic effect.120 The metabolic pathways of AA may also contribute to NAFLD development. For example, 12/15-lipoxygenase (12-LOX) enhances the IR and inflammatory response to accelerate the NAFLD process. This opinion is supported by Nunemaker et al, who knocked out the 12-Lox gene in mice on a HFD by a reverse experiment, resulting in improved IR and macrophage infiltration within adipocytes.121 Similarly, Lazic et al obtained similar results. They knocked out the 12-Lox gene from the whole body of mice on a HFD, resulting in reduced steatosis and macrophage infiltration in the liver, decreased expression of pro-inflammatory cytokine genes (interferon-γ (Ifn-γ), Tnf-α genes and Il-10 mRNA) and decreased immunocyte chemokine microphage inflammatory protein 2 (Cxcl2/3) in the liver.122 Moreover, 5-LOX can accelerate NAFLD progression. This was confirmed in the experiments by Martínez et al 5-LOX is activated in APOE-/- mice, mainly through the NF-κB-induced activation of hepatic pro-inflammatory cytokines (Tnf-α, Mcp-1 and Il-18). This would further stimulate macrophage infiltration and Caspase-3 activation, driving hepatitis development. However, the deletion of the 5-Lox gene mitigates this effect.123 In addition, AA metabolites have been associated with NAFLD development. Puri et al compared the LOX pathway products of patients with NASH with those of healthy individuals. Metabolomics has shown increased LOX pathway products in patients with NASH, including 5-hydroxyeicosatetraenoic acid (5-HETE) and 15-hydroxyeicosatetraenoic acid (15-HETE).124 Similarly, Hall et al used a HFD to establish a NASH mouse model and detected increases in 12-hydroxyeicosatetraenoic acid (12-HETE, 15-HETE and 5-HETE).125 Moreover, Ma et al treated human islets with 12-HETE and showed reduced insulin secretion and β-cell dysfunction, suggesting that 12-HETE is associated with islet stress.126 Similarly, Chakrabarti et al used 12-HETE or 15-HETE to treat 3T3-L1 adipocytes, and their results show that 12-HETE or 15-HETE triggers the secretion of inflammatory adipokines and impairs the action of insulin, suggesting a contribution to NAFLD development.127
PAF–AA–LTs Signalling Pathway and NAFLD
The PAF–AA–LTs signalling cascade is dependent on the activation of the PAF–AA signalling pathway. Then, through the 5-LOX metabolic pathway, it induces the activation of downstream inflammatory factor LTs. This will promote the occurrence of inflammatory reactions.128 Thus, AA is highly agonistic for LTs in platelets and multiple cellular tissues. This idea was verified by the experiments by Lecomte et al who incubated human platelets with AA, resulting in the detection of LTC4-like substances.129 This effect is also present in hepatocytes. Otomo et al used AA to treat rat hepatocytes and showed a concentration-dependent production of LTs.130 AA-induced synthesis of LTs requires the catalysis of 5-LOX. This was confirmed in the experiment by Doskey et al 2,3,7,8-Tetrachlorodibenzo-p-dioxin induced the activation of the 5-LOX pathway via AA, which further stimulated the secretion of leukotriene B4 (LTB4) and leukotriene B3 (LTB3).131 Similarly, Takasugi et al used AA pre-treatment of mast cells, which triggered the release of LTB4. Interestingly, MK-886 (a LOX inhibitor) abolished this process. This indicated that LTB4 is produced via the 5-LOX pathway.132 Moreover, Ito et al used endotoxin lipopolysaccharide (LPS) to induce impaired hepatic microcirculation to explore the role of LTB4 and LTB4 receptor type 1 (BLT1). Consequently, LPS can trigger LTB4 release and cause liver injury by activating the AA-dependent 5-LOX pathway. However, AA-861 (5-LOX synthase inhibitor) or BLT1 knockdown can block AA or LTs signalling and eliminate this effect.133 Accordingly, the PAF–AA–LTs signalling pathway was formed. LTs are a class of highly biologically active inflammatory mediators. They amplify the inflammatory response by acting on the corresponding receptors on inflammatory cells and chemotactic inflammatory cell infiltration and modulating the intensity and duration of inflammation, thus affecting the liver disease process.134
Chronic inflammation is believed to be a key pathophysiological mechanism behind IR. Leukotrienes are potent inflammatory cell chemoattractants that mediate inflammatory responses and promote the development of NAFLD. Moreover, excess leukotrienes induce inflammation-associated IR, reduce insulin sensitivity and exacerbate liver damage (Figure 6).135
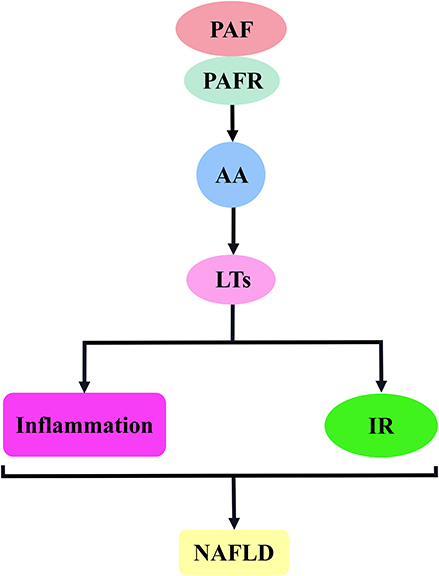 |
Figure 6 PAF activates the AA–LTs signalling pathway to promote the mechanism of NAFLD development. The binding of PAF to PAF receptor stimulates the release of AA, which induces LTs release and is involved in NAFLD development mainly by inducing inflammatory responses and insulin resistance. Abbreviations: PAF, platelet-activating factor; PAFR, platelet-activating factor receptor; AA, arachidonic acid; LTs, leukotrienes; IR, insulin resistance; NAFLD, non-alcoholic fatty liver disease. |
During NAFLD development, AA plays an important role as a regulator of the inflammatory cascade response.34,136 LTs, a downstream product of AA, are usually synthesised under the catalysis of 5-LOX.128 In liver and adipose tissue of experimentally obese mice, the expression and activity of enzymes required for LTs biosynthesis, including 5-LOX and 5-LOX-activated protein, were significantly increased.137–139 This suggests that AA induces the release of large amounts of LTs through the catalysis of 5-LOX; subsequently, the AA–LTs signalling cascade activates an inflammatory response that drives NAFLD.115,140 This view was confirmed by Ma et al They established an NAFLD rat model with a HFD to explore the role of the AA–5-LOX pathway in the pathogenesis of NAFLD. As a result, AA increased significantly as NAFLD progressed, promoting the release of cysteinyl-LTs (CysLTs) through enhanced 5-LOX protein expression. This contributes to NAFLD development. Moreover, they conducted reverse experiments using cysLTs and a 5-LOX pathway inhibitor (zileuton). The inhibition of the AA–5LOX signalling pathway delays NASH progression.115 Indeed, the AA–LTs signalling pathway leads to stable high levels of inflammatory LTB in NAFLD, which contributes to disease progression.136 In this regard, Horrillo et al used reversed-phase high-performance LC to analyse 5-LOX products in obese mice; as a result, the level of the pro-inflammatory 5-LOX product LTB4 was increased.138 In line with this, Chakrabarti et al analysed Zucker rat adipocytes using high-performance LC. Their results showed the up-regulation of LTB4 expression associated with the AA–LTs signalling pathway.141 The presence of inflammation is usually associated with the transcriptional activation of NF-κB. To this end, Horrillo et al examined the effect of LTB4 on NF-κB activity, and their results showed that LTB4 can induce NF-κB activity in adipose tissue in a concentration-dependent manner. Given that NF-κB activation stimulates the production of pro-inflammatory cytokines, further studies found that LTB4 stimulates adipose tissue to produce the pro-inflammatory cytokines MCP-1, IL-6 and TNF-α, which would exacerbate adipose inflammatory damage.138 Furthermore, IR may be another mechanism of LTs’ involvement in NAFLD. This idea is supported by Spite et al who knocked out BLT1 in mice on a HFD, resulting in significant improvements in glucose homeostasis and IR.142 Moreover, Abdallah et al used a cysteinyl-LT1 receptor antagonist (montelukast), which significantly improved fasting blood glucose, fasting insulin level and IR in patients with NASH by blocking the pro-inflammatory effects of LTD4.143
PAF–AA–PG–TXA2 Signalling Pathway and NAFLD
The PAF–AA–PG–TXA2 signalling pathway is dependent on the activation of the PAF–AA signalling pathway; catalysed by COX, AA is converted to the unstable prostaglandin H2 (PGH2). After being induced by various synthases, PG and TXA2 were formed. PG mainly includes PGI2, PGD2, PGE2 and PGF2α.144 This effect is present in platelets and various cells. For example, Son et al induced the production of PGD2 and TXA2 using AA-treated rabbit platelets.145 Similarly, in Srivastava’s experiments, human platelets showed significantly higher levels of PGF2α, PGE2 and TXB2 after AA treatment.146 This effect is also present in hepatocytes. Levine used PAF to treat hepatocytes, which promoted the production of PGI2, PGE2 and PGF2α by inducing AA metabolism.147 Similarly, Levine’s incubation of rat hepatocytes using PAF or AA promoted the production of PGI2.148 The synthesis of PG and TXA2 requires the catalysis of COX enzymes. This was confirmed in the experiments by López-Parra et al Significant expression of COX-1 and COX-2 in rat mesangial cells at rest induced the production of AA metabolites such as PGE2, TXB2 and 8-epi-PGF2α. The use of COX-2 inhibitor celecoxib down-regulated 8-epi-PGF2α and PGE2 levels.149 Similarly, Becker et al used PAF to stimulate neonatal rat cardiomyocytes to see if it could induce AA release. Their results showed a concentration-dependent release of PGI2 and TXA2. Interestingly, this effect was attenuated by the COX inhibitor aspirin (acetylsalicylic acid) and the PAF antagonist Web2086.150 Moreover, Lo et al used AA to induce platelet aggregation to investigate the mechanism of platelet aggregation inhibition by 2-ethoxy-5 -methoxy-2-(5-methylthienyl) chalcone (EMMTC). Their results suggested that AA triggers platelet aggregation by inducing TXB2 activation; however, EMMTC or indomethacin (COX inhibitors) abolished this process.151 Accordingly, PAF activates the AA metabolic pathway to produce various products, such as PG and TXA2, and the PAF–AA–PG/TXA2 signalling pathway is formed. PG and TXA2 are both AA metabolites, and these derivatives have important regulatory roles in inflammatory responses, glucose metabolism and lipid metabolism, which are associated with the development and progression of many diseases, such as diabetes, hypertension and obesity.152–154
Both PG and TXA2 are downstream derivatives of AA, and the catalytic enzymes that induce AA conversion are mainly COX-1 and COX-2.144,155 In the animal model of NAFLD, in addition to increased AA levels, the expressions of COX-1 and COX-2 were significantly up-regulated.34,156 This suggests that PG and TXA2 may have been involved in NAFLD development. Aspirin (acetylsalicylic acid) was originally used as an anti-inflammatory drug. It is also an irreversible inhibitor of the COX enzyme that produces PG and thromboxane precursors.157 Interestingly, in recent years, the clinical use of aspirin has been effective in alleviating NAFLD tissue damage and reducing NAFLD prevalence and the risk of liver fibrosis.30–33 Moreover, in NAFLD animal models, aspirin application significantly improved hepatic steatosis and delayed NAFLD development.34–37 Aspirin is suggested to alleviate NAFLD, probably by inhibiting PG and TXA2 syntheses. In summary, the induction of AA expression by PAF may have promoted the production of PG and TXA2, which are further involved in NAFLD development.
Both PG and TXA2 play an important role in the development of NAFLD. Of these, PG is primarily involved in the progression of NAFLD by regulating oxidative stress, affecting immune cells and interfering with glucose metabolism, whereas TXA2 impairs insulin sensitivity, triggers inflammatory responses and aggravates hepatocyte damage (Figure 7).153,154,158
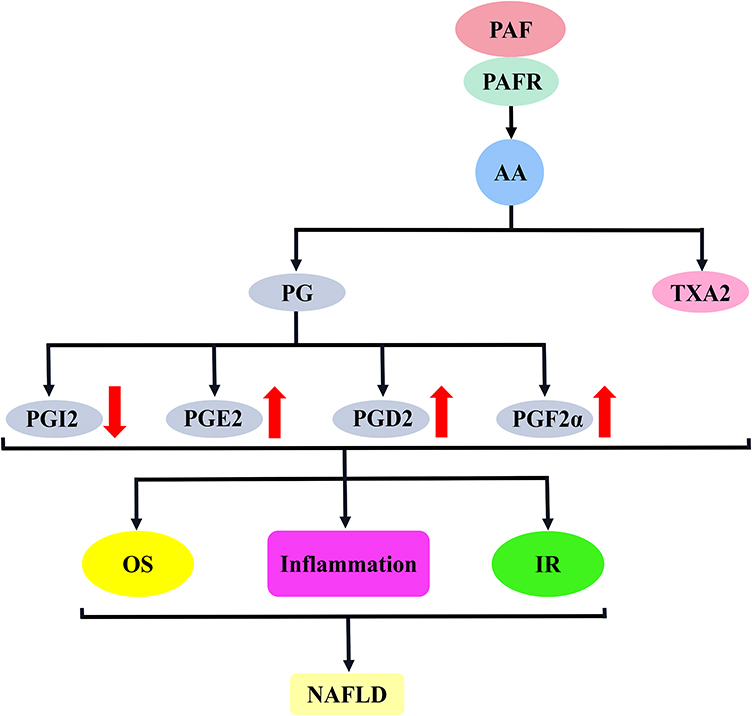 |
Figure 7 PAF activates the AA–PG/TXA2 signalling pathway to promote the mechanism of NAFLD development. The binding of PAF to PAFR stimulates AA release, which induces the synthesis of PG and TXA2; PG mainly included PGI2, PGE2, PGD2 and PGF2α. These substances are involved in NAFLD development mainly through the induction of oxidative stress, inflammatory response and insulin resistance. Abbreviations: PAF, platelet-activating factor; PAFR, platelet-activating factor receptor; PG, prostaglandins; TXA2, thromboxane A2; PGI2, prostaglandin I2; PGE2, prostaglandin E2; PGD2, prostaglandin D2; PGF2α, prostaglandin F2α; OS, oxidative stress; IR, insulin resistance; NAFLD, non-alcoholic fatty liver disease. |
AA, catalysed by COX enzymes, produces large amounts of PG, which drives NAFLD progression by acting on hepatocytes. This view is supported by Pérez et al, who used PGE2, PGD2 and PGF2α to act directly on unstimulated primary rat hepatocytes. As a result, ApoB and lipid secretion are significantly reduced, resulting in decreased triacylglycerol (TAG) and cholesterol transport to the circulation. This may be a cause of steatosis.159 Similarly, Henkel et al used PGE2 to treat obese rat hepatocytes. Their results showed that PGE2 decreased the expression of adipose triglyceride lipase, mitochondrial β-oxidation regulator carnitin–palmitoyltransferase 1 (CPT-1), apolipoprotein B (ApoB) and microsomal transfer protein, via promoted hepatocyte fat accumulation by inhibiting hepatic lipolysis, β-oxidation and very-low-density lipoprotein (VLDL) synthesis.160 Consistent with cellular experiments, Nassir et al confirmed the relevance of PG to NAFLD in animal experiments, and they established a cluster of differentiation 36 (Cd36)-deficient mice to explore the role of the Cd36 gene in hepatic VLDL secretion. Their results showed that Cd36 gene deletion aggravated steatosis by increasing the expressions of PGE2, PGD2 and PGF2α in the liver, impairing the secretions of triglyceride and ApoB in the liver.161 Moreover, clinical data confirm the correlation between PG and NALD. Henkel et al compared the expression of PGE2 synthase (COX-2 and microsomal prostaglandin E synthase 1 [mPGES-1]) in liver samples from patients with NASH and healthy controls. Their results showed that COX-2 and mPGES-1 in the NASH group were much higher than those in the control group; moreover, they were significantly correlated with NASH activity score (NAS).162 Similarly, Loomba et al compared plasma eicosanoids in the NAFL group, NASH group and healthy group quantitatively, and their results showed that PGD2 product levels were up-regulated in the NASH group, significantly correlated with NAFLD development and could even be used as a discriminatory marker between NAFL and NASH.163
PG can be involved in NAFLD development in some ways, such as the inflammatory response. This was demonstrated in animal tests by Kus et al who used a HFD to induce an NAFLD mouse model. To explore the inflammatory response of liver sinusoidal endothelial cells (LSECs) in the early and late stages of NAFLD. Their results suggest that AA metabolism induces the activations of PGD2 and PGI2 via COX-1/2 early in NAFLD, further enhancing the anti-inflammatory response. In the late stage of NAFLD, AA metabolism produces pro-inflammatory factors through COX-2, including PGE2 and PGF2α, thus triggering inflammatory response signals. An imbalance in the expression of anti-inflammatory and pro-inflammatory factors will contribute to NAFLD progression.164 In response, Kumei et al knocked out the mouse Ip gene, which promoted the expression of inflammatory factors (Tnf-α and Mcp-1) by blocking anti-inflammatory PGI2 signalling, leading to hepatocyte injury and driving NASH development. The use of IP-specific agonists (belaprost sodium) enhances PGI2 signalling and has good anti-NAFLD activity.165 Furthermore, Chung et al used a HFD to feed rats, and their results showed that the up-regulation of COX-2 activity promoted inflammatory response-dependent NF-κB and downstream inflammatory factor (MCP-1 and TNF-α) expression through the induction of pro-inflammatory factor PGE2 expression, and these were significantly reversed by green tea extract by decreasing PGE2 expression.166
Oxidative stress may be another mechanism by which PG is involved in NAFLD development. Chung et al used a HFD to feed rats, and their results showed that a HFD increased PGE2 expression through the up-regulation of COX-2 activity and elevated the MDA level, which represents the level of oxidative stress, suggesting that PGE2-mediated oxidative stress is involved in NAFLD development.166 Interestingly, Kumei et al knocked out the Ip gene in mice and promoted the production of lipid peroxidation products (4-hydroxynonenal and thiobarbituric acid reactive substances) by blocking PGI2 signalling and activating oxidative stress, which contributed to NAFLD development. Therefore, the levels of PGI2 and PGE2 expressions may determine the level of oxidative stress in the liver.165
PG may also mediate IR and be involved in NAFLD progression. Clinical data show that PG or PG metabolites are increased in patients with diabetes, including PGE2, PGI2 in islet or blood, 15-keto-dihydro-PGF2α and 8-iso-PGF2α.167–171 IR is closely related to diabetes development. PG may be also involved in IR development. In this regard, Francés et al used a HFD to feed mice, resulting in significant hepatic COX-2 expression, which promoted PGE2 production and led to hepatic IR.172 Furthermore, Hsieh et al significantly down-regulated PGE2 metabolites and improved insulin sensitivity in rats on a HFD using COX-2 inhibitors.173 Similarly, Henkel et al suggested that PGE2, as a COX product, may contribute to IR development. To this end, they incubated rat hepatocytes with PGE2 and insulin to investigate the role of PGE2 in hepatic IR. As a result, glycogen synthesis and AKT phosphorylation were inhibited. To clarify the possible mechanism of this effect, they pre-treated HepG2 cells expressing the EP3 receptor with PGE2. Accordingly, PGE2 induces phosphorylation of the insulin receptor substrate serine, possibly through activation of EP3 receptor-dependent extracellular signal-regulated kinase 1/2. This will lead to the inhibition of AKT phosphorylation and glycogen synthesis, thus promoting IR in the liver.174 After an in-depth study, Henkel et al speculated that PGE2 may affect hepatic IR by regulating the release of other signalling molecules from Kupffer cells. To explore this possibility, they established a NASH rat model and used PGE2 to treat the isolated Kupffer cells. Their results showed that PGE2 stimulated Kupffer cells to produce oncostatin M, which may lead to the phosphorylation of the signal transducer and activator of transcription 3, thereby inducing the activation of the suppressor of cytokine signal transduction 3. Subsequently, this signalling cascade attenuates AKT activation, leading to blocked glucokinase expression, which contributes to IR and NAFLD development.175 Interestingly, circulating PGF2α was also significantly elevated in patients with diabetes. To this end, Wang et al established a PGF2α receptor (FP)-deficient mouse model to investigate whether and how PGF2α regulates hepatic glucose metabolism. Their results indicated that FP receptor deletion in mice inhibits hepatic gluconeogenesis and improves insulin sensitivity and glucose homeostasis. PGF2α is suggested to induce hepatic gluconeogenesis via FP receptors and disrupts glucose homeostasis, which contributes to NAFLD development.176 Moreover, Sato et al significantly improved the serum glucose and insulin levels in obese Zucker rats using the prostacyclin analogue belaprost sodium, suggesting that PGI2 is beneficial in alleviating IR in NAFLD.177
AA generates TXA2 catalysed by COX enzymes, which can drive NAFLD by inducing IR. This hypothesis was confirmed by the animal experiments by Wang et al, who used a HFD to induce NAFLD in a mouse model. As a result, AA-dependent COX-1/2 expression was up-regulated and induced TXA2 release by enhancing thromboxane A2 synthase and thromboxane A2 receptor activities, which affected insulin sensitivity and glucose tolerance. This contributes to NAFLD development. Therefore, the pharmacological inhibition of COX-1/2 and thromboxane A2 receptor expression has a good effect on alleviating NAFLD.34 Moreover, TXA2 can modulate the inflammatory response and drive NAFLD development. To this end, Ryu et al used a HFD-containing persimmon leaf extract fed to rats. As a result, a HFD induced the release of TXA2 or TXB2 and promoted the expression of inflammation-related factors, including TNF-α, C-reactive protein and leptin. This triggers an inflammatory response that drives the onset and progression of NAFLD. Persimmon leaf down-regulation of TXA2 and TXB2 expressions reversed this result.178 However, TXA2 causing steatosis is the most direct evidence of involvement in NAFLD. This idea was supported by Francque et al who used methionine–choline-deficient (MCD) diet-induced steatosis rat model to explore steatosis-associated factors. Their results showed that the expression concentration of hepatic thromboxane synthase was significantly increased, suggesting that the increased production of TXA2 may be an important causative factor for steatosis-related diseases.179 The dynamic balance of PGI2/TXA2 in vivo can maintain the stability of vascular microcirculation. Increased TXA2 or decreased PGI2 induces coagulation and thrombosis through excessive platelet activation. This will further aggravate hepatic microcirculatory disorders, thus promoting hepatocyte degeneration necrosis and fibrosis, which contribute to NAFLD development.180
PAF–DAG Signalling Pathway and NAFLD
The PAF–DAG signalling pathway is activated by PAF by driving phospholipase C activation to produce DAG from phosphatidylinositol catabolism. Therefore, in preclinical studies, PAF has a strong activating activity for DAG. For example, Murphy et al used PAF to activate platelets, which resulted in the rapid increase in DAG levels.181 Furthermore, Catalán et al evaluated the effect of PAF on phosphatidylinositol hydrolysis in rat brain slices, and their results showed that PAF pre-treatment activated phospholipid inositol hydrolysis and induced DAG release in a dose-dependent manner.182 Similarly, Kester et al investigated the effect of PAF treatment on phosphatidylinositol hydrolysis, using glomerular thylakoid cells, and their results showed that PAF stimulated the accumulation of DAG.183 Previously, Uhing et al reported similar results in an assay involving PAF stimulated DAG accumulation in murine peritoneal macrophages. PAF activity was higher and induced PLC activation triggering DAG release.184 In hepatocytes, this effect is also present. This was confirmed in the experiments by Okayasu et al who incubated rat hepatocytes with PAF and showed a down-regulation of phosphatidylinositol content and a progressive increase in DAG content.67 Similarly, Miguel et al showed a rapid and dose- and time-dependent increase in the mass of DAG when PAF was used to treat isolated rat hepatocyte nuclei. However, WEB2086 or PCA-4248 (PAF receptor antagonist) abolished DAG synthesis.185 Accordingly, the PAF–DAG signalling pathway was formed. DAG is an intermediate product of fat metabolism, a second messenger. It can regulate glucose intake and utilisation, as well as the storage and movement of fat and other life activities.186
DAG is a lipid that may be deposited in hepatocytes as a result of PAF-induced abnormal expression, which leads to hepatocyte dysfunction or even injury. When DAG accumulates in large amounts in the liver, it may induce lipotoxicity and affect the glucose metabolism. Moreover, in such cases, the liver, as an insulin-target organ, acts as an IR, which helps to promote NAFLD development.187 In this regard, Preuss et al used liquid chromatography triple quadrupole mass spectrometry to analyse the DAG of the NAFLD mouse model, and their results showed significantly up-regulated type and concentration of DAG.188 Similarly, Gorden et al used mass spectrometry liposomes to analyse DAG changes during NAFLD progression. Significant differences in DAG species were confirmed between the normal and diseased livers of humans and mice.189 In addition, Sanyal et al analysed the changes in liver lipid components, such as DAG and cholesterol, over time in a diet-induced NAFLD model, using a lipidomic approach. Interestingly, the content of monounsaturated fatty acids containing DAG and cholesterol esters was significantly increased along with fatty liver, inflammation and swelling.190 Earlier literature discussed the hypothesis that DAG mediates IR and induces type 2 diabetes (T2D) and NAFLD.191 The reason for this is an excessive accumulation of DAG in hepatocytes, which induces lipotoxicity and impairs hepatic insulin sensitivity.187,192 This was confirmed by Magkos et al who evaluated the effect of hepatic DAG content on insulin sensitivity in patients with obesity. They found that intrahepatic DAG content is positively correlated with steatosis and NAFLD activity scores, suggesting that DAG aggregation causes hepatic lipotoxicity, and that insulin’s inhibitory effect on hepatic gluconeogenesis is negatively correlated with DAG, suggesting that lipotoxicity impairs insulin sensitivity.193 In the liver, insulin plays a role in promoting glycogen synthesis—a process regulated by glycogen synthase—in addition to inhibiting glucose production.194,195 In this regard, Blackmore et al incubated hepatocytes using exogenous phospholipase C, which resulted in a time-dependent accumulation of DAG and inactivation of hepatic glycogen synthase, indicating that DAG may promote IR by inhibiting hepatic glycogen synthesis.196 Under normal conditions, insulin binds to specific receptors on the surface of hepatocytes, and tyrosine kinases in the receptors are activated to induce trans-autophosphorylation of the tyrosine residues. This further confers catalytic activity to the insulin receptor; recruits to catalyse tyrosine phosphorylation of multiple proteins, including insulin receptor substrate (IRS); activates the downstream PI3K/AKT and other pathways; and ultimately regulates the hepatic glucose metabolism.197–199 To elucidate the specific molecular mechanisms of DAG-induced hepatic IR, Lyu et al used antisense oligonucleotides (ASO) to reduce the rat liver DGAT2 protein, which is an enzyme that catalyses the esterification of DAG for producing triglycerides, thereby leading to a significant increase in rat liver DAG content and reducing the phosphorylation level of insulin receptor kinase (IRK)-T1162; this is accompanied by impaired phosphorylation of the downstream insulin-stimulated AKT-S473, glycogen synthase kinase GSK3β-S9 and glycoisomerisation inhibitor enzyme FOXO1-S256.200 In addition, Aroor et al fed a high-fat and high-sugar diet to mice who showed a significant increase in hepatic DAG content, resulting in impaired hepatic insulin sensitivity by reducing AKT phosphorylation, which reduced the effect of insulin inhibition on hepatic glucose output.201 This finding suggests that the down-regulation of hepatic DAG content helps improve insulin sensitivity. This was confirmed by the experiments of Li et al who provided lipocalin treatment to mice on a HFD, which significantly reduced hepatic DAG levels, promoted insulin receptor tyrosine 1162 phosphorylation, increased hepatic IRS-2-associated PI3K kinase activity and AKT-serine phosphorylation levels, improved hepatic insulin sensitivity and increased the inhibitory effect of insulin on hepatic glucose production.202 A hypothesis suggests that a link between hepatic DAG accumulation and IR may be attributed to PKC activation.203 In this regard, Gilijamse et al analysed liver biopsies from patients who were obese, and their results showed increased hepatic cytoplasmic DAG content and PKCε translocation to the plasma membrane in patients with hepatic insulin resistance, suggesting the relevance of hepatic DAG-induced PKCε activation in the pathogenesis of NAFLD-associated hepatic IR.204 Similarly, Kumashiro et al used liver biopsy tissue from patients who were obese and without diabetes and correlated liver and plasma markers with steady-state model assessments of IR indices. Their results showed that the DAG content in hepatocyte plasma lipid droplets was the best predictor of IR; in addition, liver tissue DAG content was positively correlated with hepatocyte PKCε activation, suggesting that DAG-mediated PKCε is involved in hepatic IR.205 In summary, DAG may contribute to NAFLD development.
PAF–DAG–PKC-ε/PKC-δ Signalling Pathway and NAFLD
The PAF–DAG–PKC-ε/PKC-δ signalling pathway is dependent on the activation of the PAF–DAG signalling pathway, and DAG further induces the activation of downstream PKC and its congeners (PKC-ε, PKC-δ).206,207 Therefore, PAF had strong agonistic activity against PKC in preclinical studies. This was confirmed in the experiments by Pelech et al They exposed rabbit platelets to PAF and showed that PAF-induced a significant increase in PKC kinase activity.208 Similarly, Gay et al exposed human neutrophils to PAF and showed a significant increase in PKC activity associated with the granule fraction compared with control cells.209 Furthermore, in the experiments by Hu et al, PAF released from leukocytes enhanced PKC activity, induced platelet activation and promoted platelet P-selectin expression. This effect is time-dependent. Interestingly, SR121566 and C7E3 (GPIIb/IIIα inhibitor) abolished PKC release, suggesting that platelet activation may be associated with glycoprotein IIb/IIIα (GPIIb/IIIα).210 Similarly, Guo et al reported similar results in a study involving PAF-induced inflammatory damage of human bronchial smooth muscle. Elevated PAF activity triggered PKC protein expression and exacerbated bronchial smooth muscle inflammatory injury. However, hydroxysafflor yellow A abolished this effect, probably because of its PAFR antagonist.211 Notably, DAG is a key link in the activation of PKC-ε and PKCδ.212 This hypothesis has been confirmed in several experiments. For example, Sharma et al performed a trial involving the inflammatory response of adipocytes. The study showed that pre-treatment with the DAG analogue 1-Oleoyl-2-acetyl-sn-glycerol would induce PKC-δ activation in adipocytes and further stimulate downstream pro-inflammatory gene expression.213 Moreover, similar results were reported by Balciunaite et al in a trial involving PDGF-activated PKC, and PDGF-pre-treated HepG2 cells with PKC-ε content were up-regulated over time after the addition of DAG.214 Based on this, PAF induces PKC-ε and PKC-δ activation via DAG, and the PAF–DAG–PKC-ε/PKC-δ signalling pathway is formed. PKC is an important intracellular signal transduction molecule that regulates cell motility, adhesion, proliferation, differentiation and apoptosis, and PKC-ε and PKC-δ belong to new isoforms of PKC that regulate activities, such as glucose metabolism, inflammation and apoptosis, and are associated with disease progression.215–218
PKC is a lipid-activated signalling molecule; and its subtypes, PKC-ε and PKCδ, demonstrate a high affinity for DAG.219,220 PKC-ε induces NAFLD by the mechanism of insulin receptor down-regulation and lipid accumulation,221 and PKCδ is closely related to multiple signalling pathways, such as oxidative stress, inflammatory response, ERS and pro-apoptosis, in addition to impaired insulin sensitivity, which plays a key role (Figure 8).222–224
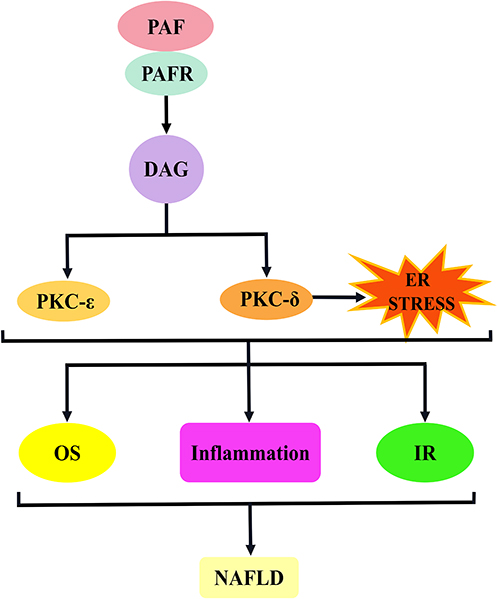 |
Figure 8 PAF activates the DAG–PKCε/PKCδ signalling pathway to promote the mechanism of NAFLD development. The binding of PAF to PAFR induces DAG production. The activation of DAG induces PKC (PKCε and PKCδ) activation, and PKCδ may also agonise endoplasmic reticulum stress. This pathway is involved in NAFLD development mainly through the induction of oxidative stress, inflammatory response and insulin resistance. Abbreviations: PAF, platelet-activating factor; PAFR, platelet-activating factor receptor; DAG, diacylglycerol; PKC, protein kinase C; PKCε, protein kinase Cε; PKCδ, protein kinase Cδ; OS, oxidative stress; IR, insulin resistance; NAFLD, non-alcoholic fatty liver disease. |
In hepatocytes, DAG-induced activation of PKC-ε and PKCδ promoted NAFLD development, possibly through regulation of IR. This idea was confirmed by Kumashiro et al who used liver biopsy tissue from patients who were obese and without diabetes to analyse the association of liver and plasma markers with IR. Their results showed that DAG content in hepatocyte plasma lipid droplets was the best predictor of IR; in addition, liver tissue DAG content was positively correlated with hepatocyte PKCε activation, suggesting that DAG-mediated PKCε is involved in hepatic IR.205 Similarly, to examine the role of PKCδ in cellular IR, Greene et al isolated primary hepatocytes from HFD mice and stimulated them with insulin. Primary hepatocytes from PKCδ-deficient mice improved insulin-stimulated AKT compared with hepatocytes from wild-type mice.225 In line with this, DAG regulation of PKC-ε and PKCδ expression, induction of IR and involvement in NAFLD development were also confirmed in animal experiments. Jornayvaz et al assessed the role of DAG in causing liver IR using diacylglycerol acyltransferase 2 overexpressing mice. Their results showed that a significant increase in hepatic DAG down-regulated IRS-2 tyrosine phosphorylation levels through PKCε activation, leading to IR.226 This is similar to the results of the experiment by Dallak who fed rats with acylated ghrelin, resulting in an up-regulation of hepatic DAG content, leading to PKC-ε and PKC-δ activation, promoting IRS (Ser307) phosphorylation and inducing hepatic IR.227 Conversely, the inhibition of PKCε or PKC-δ signalling improves IR, which helps alleviate NAFLD. For instance, Frangioudakis et al evaluated the effect of Pkc-δ and Pkc-ε gene deletion on abnormal glucose tolerance due to a HFD in a controlled trial in wild-type mice. Their results showed improvements in glucose tolerance, insulin sensitivity and blood glucose levels.228 This result was supported by Zhang et al, who established Pkc-δ-silenced mice to analyse the changes in insulin signalling. By contrast, the phosphorylation levels of AKT and GSK3β in the liver of Pkc-δ-silenced mice were increased, and the liver IR was relieved.229 Analogously, Samuel et al reported similar results in a trial to discern whether PKCε was associated with hepatic IR or not, and they used antisense oligonucleotides targeting PKCε to down-regulate Pkc-ε expression and reverse the defective hepatic insulin signalling.230
In addition to IR, PKC-δ can be involved in NAFLD development in other ways. Greene et al compared the indexes of oxidative stress, inflammation and apoptosis between Pkc-δ-deficient mice and wild-type mice induced by MCD diet to explore the ways of PKC-δ in promoting NASH development. As a result, the inflammation score and NADPH oxidase activity were significantly reduced in Pkc-δ-deficient mice, and the expressions of apoptotic genes caspase-3 and caspase-9 were inhibited, suggesting that the abnormal expression of PKC-δ is involved in the regulation of inflammation, oxidative stress and apoptosis in various aspects, driving NAFLD development.231 Similar to some of the observations, Klymenko et al evaluated the effect of PKC-δ gene silencing using siRNA on endothelial function in diabetic vascular smooth muscle cells using a streptozotocin-induced diabetic rat model. Their results suggest that PKC-δ silencing inhibits oxidative stress-dependent ROS production.232 Interestingly, Pereira et al further investigated that PKC-δ induced IR may be causally related to oxidative stress. They used heparin infusion to up-regulate FFAs and established IR rats, resulting in elevated hepatic PKC-δ levels, along with the detection of a large number of NADPH oxidase and oxidative stress markers and increased c-JNK expression. Interestingly, antioxidants and NADPH oxidase inhibit JNK expression, thereby attenuating IR. PKC-δ is suggested to induce JNK activation through oxidative stress, which contributes to IR development.233 This view is supported by the clinical study of De et al, who determined insulin sensitivity before and after intravenous administration of glutathione with antioxidant effects in 10 patients with non-insulin-dependent diabetes mellitus and 10 healthy individuals. Their results showed a significant improvement in insulin-mediated systemic glucose uptake.234
Evidence shows that ERS plays an important role in the pathological changes of NAFLD.235 For this reason, Lai et al used PA to induce the L02 cell steatosis model to explore whether ERS is another mechanism of PKC-δ promoting NAFLD development. The found that the silent PKC-δ gene can alleviate PA-induced ERS. In addition, siRNA can inhibit the activity of the PKC-δ gene, reduce intracellular calcium overload and restore the function of SERCA, a calcium balance regulator.85 Similarly, Yang et al established a liver ERS model in L02 cells induced by FFA. Similar conclusions were obtained by studying the effect of PKC-δ silencing on ERS: PKC-δ down-regulated SERCA activity, destroyed calcium homeostasis and induced ERS, which was helpful to NAFLD development.236 This conclusion was echoed by Greene et al, who used MCD diet to establish a mouse model of NASH to investigate the causal relationship between PKC-δ and ERS. Their results showed that the PKC-δ protein content was consistent with ERS expression parameters. In vitro induction experiments with the help of MCA cells confirmed that PKC-δ gene silencing down-regulated the expression of ERS markers CHOP and protein kinase-like ER-resident kinase.237
PAF–DAG–p38MAPK Signalling Pathway and NAFLD
The PAF–DAG–p38MAPK signalling pathway depends on the activation of the PAF–DAG signalling pathway, and DAG further stimulates p38MAPK activation. Therefore, PAF demonstrated a strong agonistic activity on p38MAPK in preclinical studies. For example, Wang et al conducted an experiment involving PAF regulating the proliferation and differentiation of colon cancer cells, and their results showed that PAF preconditioning triggered the activation of p38MAPK and induced the growth inhibition and differentiation of colon cancer cells.238 In addition, Yu et al confirmed that this effect also exists in microvessels. They treated mesenteric adipose tissue of mice with PAF and measured microvasculature transport using a computer program. Their results showed that PAF up-regulated microvascular permeability. Interestingly, SB203580 (p38MAPK inhibitor) reversed this effect. This suggests that p38MAPK is a regulatory element in the signalling cascade of PAF-induced microvascular permeability.239 In fact, the mechanism of PAF activating p38MAPK through DAG is unknown. Evidence shows that oxidative stress is an important factor of p38MAPK activation.240 More importantly, DAG-induced PKC expression facilitates ROS production.241,242 Thus, DAG may activate p38MAPK through ROS. Based on the above, the PAF–DAG–p38MAPK signalling pathway is formed.P38MAPK is a member of the mitogen-activated protein kinase (MAPK) family. As a signal transduction molecule, p38MAPK plays a key role in regulating insulin signalling, sugar transport, inflammation and apoptosis.243–245
p38MAPK is an important inflammatory transcription factor that induces the release of inflammatory factors, initiates cellular damage mechanisms and plays a key role in the development of NAFLD. In addition, the activation of p38MAPK can affect insulin signalling and further aggravate NAFLD injury (Figure 9).246,247
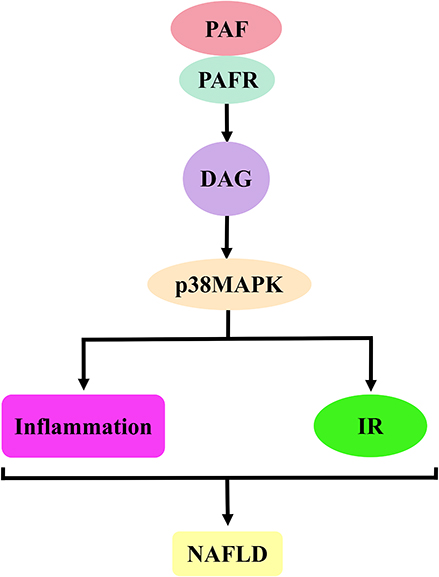 |
Figure 9 PAF activates DAG–p38MAPK signalling pathway to promote the mechanism of NAFLD development. The binding of PAF to PAFR induces DAG production. The activation of DAG induces p38MAPK activation, which is involved in NAFLD development by inducing inflammatory responses and IR. Abbreviations: PAF, platelet-activating factor; PAFR, platelet-activating factor receptor; p38MAPK, p38-mitogen-activated protein kinases; OS, oxidative stress; IR, insulin resistance; NAFLD, non-alcoholic fatty liver disease. |
DAG facilitates ROS generation, and ROS may induce hepatic IR via p38MAPK, which contributes to NAFLD development. This hypothesis was confirmed by Gao et al They used dairy cow hepatocytes and HepG2 cells with non-esterified fatty acids (NEFA) to examine the relationship between mitochondrial dysfunction and IR. Their results showed that NEFA activated p38MAPK by up-regulating ROS content and reduced the phosphorylation levels of IRS-2 and AKT, thus inducing IR. Interestingly, antioxidants lipoic acid and SB203580 (p38MAPK inhibitors) reversed this effect.248 These results were supported by Liu et al They fed rats with a HFD to explore the mechanism of JLD improving insulin sensitivity in IR rats. Studies have shown that a HFD can increase IRS-1 serine phosphorylation, decrease AKT phosphorylation and impair insulin signal transduction by activating p38MAPK. However, JLD reversed IR by down-regulating the expression of ROS, reducing p38MAPK-dependent oxidative stress.249 Zhu et al reported similar results. They adopted HepG2 cells with insulin and established an IR model to study the regulatory effects of pea-derived peptides on IR. Their results showed that pea-derived peptides probably blocked p38MAPK phosphorylation by inhibiting ROS, further reduced IRS-1Ser307 phosphorylation, promoted Ser473 phosphorylation of AKT and improved IR.250
p38MAPK can regulate inflammatory response and promote NAFLD development. Therefore, Gong et al induced the NASH rat model with a HFD to explore the role of inflammation-dependent toll-like receptor 4 (TLR4)–p38MAPK signalling pathway in Kupffer cells and the intervention effect of the Shugan Jianpi recipe. Their results showed that a HFD induced p38MAPK activation through TLR4 and promoted the release of inflammatory cytokines, including TNF-α, IL-1 and IL-6, which could trigger an inflammatory response and promote NAFLD development. The Shugan Jianpi recipe may reverse this effect by inhibiting the TLR4–p38MAPK signalling pathway.251 These results were supported by Zhang et al They used FFA to induce human hepatocyte line L02 to establish a steatosis model and investigate the role of AMP-activated protein kinase α1(AMPKα1) overexpression. Their results have shown that p38MAPK can enhance the inflammatory response by increasing pro-inflammatory cytokines (IL-6, IL-12 and TNF-α) and decreasing anti-inflammatory cytokines (IL-4, IL-10 and IL-13), which is helpful to NAFLD development. However, AMPKα1 overexpression may inactivate p38MAPK and reduce the inflammatory response.252 Notably, p38MAPK expression leads to the activation of many downstream inflammatory regulatory factors, including COX-2253,254 and NF-κB,255,256 which can trigger and maintain the pro-inflammatory response and promote NAFLD development.
Summary and Prospect
NAFLD is a clinicopathological syndrome mediated by multiple factors, which has been attracting increasing attention because of its increasing prevalence and many hazards. The pathogenesis of NAFLD is extremely complex, and the widely recognised “two-hit” theory can only reflect the tip of the iceberg of the disease. Therefore, we clarified the mechanism of the relationship between the signalling pathway of platelet activation induced by PAF and NAFLD (Figure 2) through literature sorting, further enriching the pathogenesis of NAFLD and providing a theoretical basis for the clinical prevention and treatment of NAFLD. Although there is no specific drug for the treatment of NAFLD at present, many studies have shown that drugs that resist platelet activation, such as aspirin, ticlopidine and cilostazol, have a certain effect in delaying NAFLD progression, which also suggests that PAF plays a certain role in NAFLD development. The deficiency of this paper lies in that no literature related to the direct inhibition of PAF by antiplatelet drugs to alleviate NAFLD was found. It only provides a new idea to improve the pathogenesis of NAFLD and find effective intervention measures.
Funding
This investigation was supported by a grant from Yunnan Province Applied Basic Research Project (2019FF002 (−055)).
Disclosure
The authors report no potential conflicts of interest related to this article.
References
1. Friedman SL, Neuschwander-Tetri BA, Rinella M, et al. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018;24(7):908–922. doi:10.1038/s41591-018-0104-9
2. Wong VW, Wong GL, Choi PC, et al. Disease progression of non-alcoholic fatty liver disease: a prospective study with paired liver biopsies at 3 years. Gut. 2010;59(7):969–974. doi:10.1136/gut.2009.205088
3. Adams LA, Sanderson S, Lindor KD, et al. The histological course of nonalcoholic fatty liver disease: a longitudinal study of 103 patients with sequential liver biopsies. J Hepatol. 2005;42(1):132–138. doi:10.1016/j.jhep.2004.09.012
4. Harrison SA, Torgerson S, Hayashi PH. The natural history of nonalcoholic fatty liver disease: a clinical histopathological study. Am J Gastroenterol. 2003;98(9):2042–2047. doi:10.1111/j.1572-0241.2003.07659.x
5. Evans CD, Oien KA, MacSween RN, et al. Non-alcoholic steatohepatitis: a common cause of progressive chronic liver injury?. J Clin Pathol. 2002;55(9):689–692. doi:10.1136/jcp.55.9.689
6. Fassio E, Alvarez E, Domínguez N, et al. Natural history of nonalcoholic steatohepatitis: a longitudinal study of repeat liver biopsies. Hepatology. 2004;40(4):820–826. doi:10.1002/hep.20410
7. Hui AY, Wong VW, Chan HL, et al. Histological progression of non-alcoholic fatty liver disease in Chinese patients. Aliment Pharmacol Ther. 2005;21(4):407–413. doi:10.1111/j.1365-2036.2005.02334.x
8. Argo CK, Northup PG, Al-Osaimi AM, et al. Systematic review of risk factors for fibrosis progression in non-alcoholic steatohepatitis. J Hepatol. 2009;51(2):371–379. doi:10.1016/j.jhep.2009.03.019
9. Matteoni CA, Younossi ZM, Gramlich T, et al. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology. 1999;116(6):1413–1419. doi:10.1016/s0016-5085(99)70506-8
10. Ekstedt M, Franzén LE, Mathiesen UL, et al. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology. 2006;44(4):865–873. doi:10.1002/hep.21327
11. McCullough AJ. The clinical features, diagnosis and natural history of nonalcoholic fatty liver disease. Clin Liver Dis. 2004;8(3):521–533. doi:10.1016/j.cld.2004.04.004
12. Önnerhag K, Nilsson PM, Lindgren S. Increased risk of cirrhosis and hepatocellular cancer during long-term follow-up of patients with biopsy-proven NAFLD. Scand J Gastroenterol. 2014;49(9):1111–1118. doi:10.3109/00365521.2014.934911
13. Cholankeril G, Patel R, Khurana S, et al. Hepatocellular carcinoma in non-alcoholic steatohepatitis: current knowledge and implications for management. World J Hepatol. 2017;9(11):533–543. doi:10.4254/wjh.v9.i11.533
14. Ganne-Carrié N, Chastang C, Chapel F, et al. Predictive score for the development of hepatocellular carcinoma and additional value of liver large cell dysplasia in Western patients with cirrhosis. Hepatology. 1996;23(5):1112–1118. doi:10.1002/hep.510230527
15. Trinchet JC, Bourcier V, Chaffaut C, et al. Complications and competing risks of death in compensated viral cirrhosis (ANRS CO12 CirVir prospective cohort). Hepatology. 2015;62(3):737–750. doi:10.1002/hep.27743
16. White DL, Kanwal F, El-Serag HB. Association between nonalcoholic fatty liver disease and risk for hepatocellular cancer, based on systematic review. Clin Gastroenterol Hepatol. 2012;10(12):1342–1359.e2. doi:10.1016/j.cgh.2012.10.001
17. Mantovani A, Scorletti E, Mosca A, et al. Complications, morbidity and mortality of nonalcoholic fatty liver disease. Metabolism. 2020;111S:154170. doi:10.1016/j.metabol.2020.154170
18. Younossi ZM, Koenig AB, Abdelatif D, et al. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016;64(1):73–84. doi:10.1002/hep.28431
19. Zou B, Yeo YH, Nguyen VH, et al. Prevalence, characteristics and mortality outcomes of obese, nonobese and lean NAFLD in the United States, 1999–2016. J Intern Med. 2020;288(1):139–151. doi:10.1111/joim.13069
20. Le MH, Yeo YH, Cheung R, et al. Ethnic influence on nonalcoholic fatty liver disease prevalence and lack of disease awareness in the United States, 2011–2016. J Intern Med. 2020;287(6):711–722. doi:10.1111/joim.13035
21. Li J, Zou B, Yeo YH, et al. Prevalence, incidence, and outcome of non-alcoholic fatty liver disease in Asia, 1999–2019: a systematic review and meta-analysis. Lancet Gastroenterol Hepatol. 2019;4(5):389–398. doi:10.1016/S2468-1253(19)30039-1
22. Sivell C. nonalcoholic fatty liver disease: a silent epidemic. Gastroenterol Nurs. 2019;42(5):428–434. doi:10.1097/SGA.0000000000000443
23. Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism. 2016;65(8):1038–1048. doi:10.1016/j.metabol.2015.12.012
24. Alkhalidy H, Moore W, Wang A, et al. Kaempferol ameliorates hyperglycemia through suppressing hepatic gluconeogenesis and enhancing hepatic insulin sensitivity in diet-induced obese mice. J Nutr Biochem. 2018;58:90–101. doi:10.1016/j.jnutbio.2018.04.014
25. Lordan R, Tsoupras A, Zabetakis I, et al. Forty years since the structural elucidation of platelet-activating factor (PAF): historical, current, and future research perspectives. Molecules. 2019;24(23):4414. doi:10.3390/molecules24234414
26. Oral A, Sahin T, Turker F, et al. Evaluation of plateletcrit and platelet distribution width in patients with non-alcoholic fatty liver disease: a retrospective chart review study. Med Sci Monit. 2019;25:9882–9886. doi:10.12659/MSM.920172
27. Ozhan H, Aydin M, Yazici M, et al. Mean platelet volume in patients with non-alcoholic fatty liver disease. Platelets. 2010;21(1):29–32. doi:10.3109/09537100903391023
28. Arslan N, Makay B. Mean platelet volume in obese adolescents with nonalcoholic fatty liver disease. J Pediatr Endocrinol Metab. 2010;23(8):807–813. doi:10.1515/jpem.2010.130
29. Malehmir M, Pfister D, Gallage S, et al. Platelet GPIbα is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat Med. 2019;25(4):641–655. doi:10.1038/s41591-019-0379-5
30. Jiang ZG, Feldbrügge L, Tapper EB, et al. Aspirin use is associated with lower indices of liver fibrosis among adults in the United States. Aliment Pharmacol Ther. 2016;43(6):734–743. doi:10.1111/apt.13515
31. Armstrong MJ, Rowe IA. Editorial: would an aspirin a day keep NAFLD and its complications away?. Aliment Pharmacol Ther. 2015;41(1):145. doi:10.1111/apt.12998
32. Shen H, Shahzad G, Jawairia M, et al. Association between aspirin use and the prevalence of nonalcoholic fatty liver disease: a cross-sectional study from the Third National Health and Nutrition Examination Survey. Aliment Pharmacol Ther. 2014;40(9):1066–1073. doi:10.1111/apt.12944
33. Simon TG, Henson J, Osganian S, et al. Daily aspirin use associated with reduced risk for fibrosis progression in patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol. 2019;17(13):2776–2784.e4. doi:10.1016/j.cgh.2019.04.061
34. Wang W, Chen J, Mao J, et al. Genistein Ameliorates Non-alcoholic Fatty Liver Disease by Targeting the Thromboxane A2 Pathway. J Agric Food Chem. 2018;66(23):5853–5859. doi:10.1021/acs.jafc.8b01691
35. Ford RJ, Fullerton MD, Pinkosky SL, et al. Metformin and salicylate synergistically activate liver AMPK, inhibit lipogenesis and improve insulin sensitivity. Biochem J. 2015;468(1):125–132. doi:10.1042/BJ20150125
36. Fujita K, Nozaki Y, Wada K, et al. Effectiveness of antiplatelet drugs against experimental non-alcoholic fatty liver disease. Gut. 2008;57(11):1583–1591. doi:10.1136/gut.2007.144550
37. Ibrahim M, Farghaly E, Gomaa W, et al. Nitro-aspirin is a potential therapy for non alcoholic fatty liver disease. Eur J Pharmacol. 2011;659(2–3):289–295. doi:10.1016/j.ejphar.2011.03.016
38. Kunz D, Gerard NP, Gerard C. The human leukocyte platelet-activating factor receptor. cDNA cloning, cell surface expression, and construction of a novel epitope-bearing analog. J Biol Chem. 1992;267(13):9101–9106.
39. Montrucchio G, Alloatti G, Camussi G. Role of platelet-activating factor in cardiovascular pathophysiology. Physiol Rev. 2000;80(4):1669–1699. doi:10.1152/physrev.2000.80.4.1669
40. Marrache AM, F G, Bernier SG, et al. Proinflammatory gene induction by platelet-activating factor mediated via its cognate nuclear receptor. J Immunol. 2002;169(11):6474–6481. doi:10.4049/jimmunol.169.11.6474
41. Greco NJ, Arnold JH, O’Dorisio TM, et al. Action of platelet-activating factor on type 1 diabetic human platelets. J Lab Clin Med. 1985;105(4):410–416.
42. O’Donnell MC, Henson PM, Fiedel BA. Activation of human platelets by platelet activating factor (PAF) derived from sensitized rabbit basophils. Immunology. 1978;35(6):953–958.
43. Murphy CT, Elmore M, Kellie S, et al. Comparison of the role of protein kinase C in platelet functional responses induced by three different mechanisms, PAF, ionomycin and arachidonic acid. Biochim Biophys Acta. 1991;1133:46–54.
44. Pédrono F, Cheminade C, Legrand AB. Natural 1-O-alkylglycerols reduce platelet-activating factor-induced release of [3H]-serotonin in rabbit platelets. Prostaglandins Leukot Essent Fatty Acids. 2004;71(1):19–23. doi:10.1016/j.plefa.2003.12.003
45. Brooks AC, Menzies-Gow NJ, Wheeler-Jones CP, et al. Regulation of platelet activating factor-induced equine platelet activation by intracellular kinases. J Vet Pharmacol Ther. 2009;32(2):189–196. doi:10.1111/j.1365-2885.2008.01020.x
46. Bailey SR, Andrews MJ, Elliott J, et al. Differential activation of platelets from normal and allergic ponies by PAF and ADP. Inflamm Res. 2000;49(5):224–230. doi:10.1007/s000110050583
47. El-Merahbi R, Löffler M, Mayer A, et al. The roles of peripheral serotonin in metabolic homeostasis. FEBS Lett. 2015;589(15):1728–1734. doi:10.1016/j.febslet.2015.05.054
48. Bajrangee A, Ryan N, Vangjeli C, et al. Impact of genetic variation in the 5-HT transporter and receptor on platelet function in patients with stable CAD taking aspirin. Thromb Res. 2016;146:51–55. doi:10.1016/j.thromres.2016.08.019
49. Qing P, Hao J, Xiquan K, et al.. The role of serotonin in concanavalin A-induced liver injury in mice. Oxid Med Cell Longev. 2020:7504521. doi:10.1155/2020/7504521
50. Lesurtel M, Soll C, Humar B, et al. Serotonin: a double-edged sword for the liver?. Surgeon. 2012;10(2):107–113. doi:10.1016/j.surge.2011.11.002
51. Choi WG, Choi W, Oh TJ, et al. Inhibiting serotonin signaling through HTR2B in visceral adipose tissue improves obesity-related insulin resistance. J Clin Invest. 2021;131(23):e145331. doi:10.1172/JCI145331
52. Fu J, Li C, Zhang G, et al. Crucial roles of 5-HT and 5-HT2 receptor in diabetes-related lipid accumulation and pro-inflammatory cytokine generation in hepatocytes. Cell Physiol Biochem. 2018;48(6):2409–2428. doi:10.1159/000492656
53. Fu J, Ma S, Li X, et al. Long-term Stress with hyperglucocorticoidemia-induced hepatic steatosis with VLDL overproduction is dependent on both 5-HT2 receptor and 5-HT synthesis in liver. Int J Biol Sci. 2016;12(2):219–234. doi:10.7150/ijbs.13062
54. Lulu W, Xiangcheng F, Jichun H, et al. Gut-derived serotonin contributes to the progression of non-alcoholic steatohepatitis the liver HTR2A/PPARγ2 pathway. Front Pharmacol. 2020;11:553. doi:10.3389/fphar.2020.00553
55. Osawa Y, Kanamori H, Seki E, et al. L-tryptophan-mediated enhancement of susceptibility to nonalcoholic fatty liver disease is dependent on the mammalian target of rapamycin. J Biol Chem. 2011;286(40):34800–34808. doi:10.1074/jbc.M111.235473
56. Namkung J, Shong KE, Kim H, et al. Inhibition of serotonin synthesis induces negative hepatic lipid balance. Diabetes Metab J. 2018;42(3):233–243. doi:10.4093/dmj.2017.0084
57. Choi W, Namkung J, Hwang I, et al. Serotonin signals through a gut-liver axis to regulate hepatic steatosis. Nat Commun. 2018;9(1):4824. doi:10.1038/s41467-018-07287-7
58. Crane JD, Palanivel R, Mottillo EP, et al. Inhibiting peripheral serotonin synthesis reduces obesity and metabolic dysfunction by promoting brown adipose tissue thermogenesis. Nat Med. 2015;21(2):166–172. doi:10.1038/nm.3766
59. Nocito A, Dahm F, Jochum W, et al. Serotonin mediates oxidative stress and mitochondrial toxicity in a murine model of nonalcoholic steatohepatitis. Gastroenterology. 2007;133(2):608–618. doi:10.1053/j.gastro.2007.05.019
60. Tintinger GR, Theron AJ, Steel HC, et al. Protein kinase C promotes restoration of calcium homeostasis to platelet activating factor-stimulated human neutrophils by inhibition of phospholipase C. J Inflamm. 2009;6:29. doi:10.1186/1476-9255-6-29
61. Sakon M, Kambayashi J. The regulatory mechanism of free Ca2+ concentration in activated platelets. Nihon Rinsho. 1992;50:249–253.
62. Yu P, Hatakeyama T, Aramoto H, et al. Mitogen-activated protein kinases regulate platelet-activating factor-induced hyperpermeability. Microcirculation. 2005;12(8):637–643. doi:10.1080/10739680500301706
63. Ng DS, Wong K. Platelet-activating factor (PAF) stimulates phosphatidylinositol hydrolysis in human peripheral blood mononuclear leukocytes. Res Commun Chem Pathol Pharmacol. 1989;66(2):219–231.
64. Abebe W, Ali N, Agrawal DK. Platelet-activating factor-induced inositol 1,4,5-trisphosphate generation in undifferentiated and differentiated U937 cells: role of tyrosine kinase. Int J Immunopharmacol. 1996;18(3):173–181. doi:10.1016/0192-0561(96)00004-5
65. Lin AY, Rui YC. Platelet-activating factor induced calcium mobilization and phosphoinositide metabolism in cultured bovine cerebral microvascular endothelial cells. Biochim Biophys Acta. 1994;1224(2):323–328. doi:10.1016/0167-4889(94)90206-2
66. Koike H, Imanishi N, Natsume Y, et al. Effects of platelet activating factor receptor antagonists on intracellular platelet activating factor function in neutrophils. Eur J Pharmacol. 1994;269(3):299–309. doi:10.1016/0922-4106(94)90037-x
67. Okayasu T, Hasegawa K, Ishibashi T. Platelet-activating factor stimulates metabolism of phosphoinositides via phospholipase A2 in primary cultured rat hepatocytes. J Lipid Res. 1987;28(7):760–767.
68. Chu WF, Sun HL, Dong DL, et al. Increasing Intracellular calcium of Guinea pig ventricular myocytes induced by platelet activating factor through IP3 pathway. Basic Clin Pharmacol Toxicol. 2006;98(1):104–109. doi:10.1111/j.1742-7843.2006.pto_313.x
69. Lautenschläger I, Wong YL, Sarau J, et al. Signalling mechanisms in PAF-induced intestinal failure. Sci Rep. 2017;7(1):13382. doi:10.1038/s41598-017-13850-x
70. Pedreño J, Hurt-Camejo E, Wiklund O, et al. Low-density lipoprotein (LDL) binds to a G-protein coupled receptor in human platelets. Evidence that the proaggregatory effect induced by LDL is modulated by down-regulation of binding sites and desensitization of its mediated signaling. Atherosclerosis. 2001;155(1):99–112. doi:10.1016/s0021-9150(00)00545-1
71. Raffaello A, Mammucari C, Gherardi G, et al. Calcium at the center of cell signaling: interplay between endoplasmic reticulum, mitochondria, and lysosomes. Trends Biochem Sci. 2016;41(12):1035–1049. doi:10.1016/j.tibs.2016.09.001
72. Groenendyk J, Agellon LB, Michalak M. Calcium signaling and endoplasmic reticulum stress. Int Rev Cell Mol Biol. 2021;363:1–20. doi:10.1016/bs.ircmb.2021.03.003
73. Szabadkai G, Bianchi K, Várnai P, et al. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol. 2006;175(6):901–911. doi:10.1083/jcb.200608073
74. Marchi S, Bittremieux M, Missiroli S, et al. Endoplasmic reticulum-mitochondria communication through Ca2+ signaling: the importance of mitochondria-associated membranes (MAMs). Adv Exp Med Biol. 2017;997:49–67. doi:10.1007/978-981-10-4567-7_4
75. Hirata K, Pusl T, O’Neill AF, et al. The type II inositol 1,4,5-trisphosphate receptor can trigger Ca2+ waves in rat hepatocytes. Gastroenterology. 2002;122(4):1088–1100. doi:10.1053/gast.2002.32363
76. Khamphaya T, Chukijrungroat N, Saengsirisuwan V, et al. Nonalcoholic fatty liver disease impairs expression of the type II inositol 1,4,5-trisphosphate receptor. Hepatology. 2018;67(2):560–574. doi:10.1002/hep.29588
77. Arruda AP, Pers BM, Parlakgül G, et al. Chronic enrichment of hepatic endoplasmic reticulum-mitochondria contact leads to mitochondrial dysfunction in obesity. Nat Med. 2014;20(12):1427–1435. doi:10.1038/nm.3735
78. Feriod CN, Oliveira AG, Guerra MT, et al. Hepatic Inositol 1,4,5 Trisphosphate Receptor Type 1 Mediates Fatty Liver. Hepatol Commun. 2017;1(1):23–35. doi:10.1002/hep4.1012
79. Alzayady KJ, Wang L, Chandrasekhar R, et al. Defining the stoichiometry of inositol 1,4,5-trisphosphate binding required to initiate Ca2+ release. Sci Signal. 2016;9(422):ra35. doi:10.1126/scisignal.aad6281
80. Barritt GJ, Litjens TL, Castro J, et al. Store-operated Ca2+ channels and microdomains of Ca2+ in liver cells. Clin Exp Pharmacol Physiol. 2009;36(1):77–83. doi:10.1111/j.1440-1681.2008.05095.x
81. Ong HL, Ambudkar IS. STIM-TRP pathways and microdomain organization: contribution of TRPC1 in store-operated Ca2+ entry: impact on Ca2+ signaling and cell function. Adv Exp Med Biol. 2017;993:159–188. doi:10.1007/978-3-319-57732-6_9
82. Thillaiappan NB, Chakraborty P, Hasan G, et al. IP3 receptors and Ca2+ entry. Biochim Biophys Acta Mol Cell Res. 2019;1866(7):1092–1100. doi:10.1016/j.bbamcr.2018.11.007
83. Zhang L, Zhang Y, Jiang Y, et al. Upregulated SOCC and IP3R calcium channels and subsequent elevated cytoplasmic calcium signaling promote nonalcoholic fatty liver disease by inhibiting autophagy. Mol Cell Biochem. 2021;476(8):3163–3175. doi:10.1007/s11010-021-04150-0
84. Hammadi M, Oulidi A, Gackière F, et al. Modulation of ER stress and apoptosis by endoplasmic reticulum calcium leak via translocon during unfolded protein response: involvement of GRP78. FASEB J. 2013;27(4):1600–1609. doi:10.1096/fj.12-218875
85. Lai S, Li Y, Kuang Y, et al. PKCδ silencing alleviates saturated fatty acid induced ER stress by enhancing SERCA activity. Biosci Rep. 2017;37(6):BSR20170869. doi:10.1042/BSR20170869
86. Zhang J, Li Y, Jiang S, et al. Enhanced endoplasmic reticulum SERCA activity by overexpression of hepatic stimulator substance gene prevents hepatic cells from ER stress-induced apoptosis. Am J Physiol Cell Physiol. 2014;306(3):C279–C290. doi:10.1152/ajpcell.00117.2013
87. Zeeshan HM, Lee GH, Kim HR, et al. Endoplasmic reticulum stress and associated ROS. Int J Mol Sci. 2016;17(3):327. doi:10.3390/ijms17030327
88. Lebeaupin C, Vallée D, Hazari Y, et al. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J Hepatol. 2018;69(4):927–947. doi:10.1016/j.jhep.2018.06.008
89. Vallée D, Blanc M, Lebeaupin C, et al. La réponse au stress du réticulum endoplasmique dans la physiopathologie des maladies chroniques du foie [Endoplasmic reticulum stress response and pathogenesis of non-alcoholic steatohepatitis]. Med Sci. 2020;36(2):119–129. doi:10.1051/medsci/2020008
90. Zhang J, Zhang K, Li Z, et al. ER stress-induced inflammasome activation contributes to hepatic inflammation and steatosis. J Clin Cell Immunol. 2016;7(5):457. doi:10.4172/2155-9899.1000457
91. Xu Y, Yang C, Zhang S, et al. Ginsenoside Rg1 protects against non-alcoholic fatty liver disease by ameliorating lipid peroxidation, endoplasmic reticulum stress, and inflammasome activation. Biol Pharm Bull. 2018;41(11):1638–1644. doi:10.1248/bpb.b18-00132
92. Ye L, Zhao D, Xu Y, et al. LncRNA-Gm9795 promotes inflammation in non-alcoholic steatohepatitis via NF-[Formula: see text]B/JNK pathway by endoplasmic reticulum stress. J Transl Med. 2021;19(1):101. doi:10.1186/s12967-021-02769-7
93. Ozcan U, Cao Q, Yilmaz E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306(5695):457–461. doi:10.1126/science.1103160
94. Murakami M, Kudo I. Phospholipase A2. J Biochem. 2002;131(3):285–292. doi:10.1093/oxfordjournals.jbchem.a003101
95. Catalán RE, Martínez AM, Aragonés MD, et al. PAF and thrombin actions in platelets are selectively affected by a new 1,4-dihydropyridine derivative. J Biochem. 1993;113(4):450–455. doi:10.1093/oxfordjournals.jbchem.a124065
96. Oestvang J, Anthonsen MW, Johansen B. LysoPC and PAF trigger arachidonic acid release by divergent signaling mechanisms in monocytes. J Lipids. 2011;2011:532145. doi:10.1155/2011/532145
97. Hurst JS, Bazan HE. Platelet-activating factor preferentially stimulates the phospholipase A2/cyclooxygenase cascade in the rabbit cornea. Curr Eye Res. 1995;14(9):769–775. doi:10.3109/02713689508995798
98. Svetlov SI, Howard KM, Miwa M, et al. Interaction of platelet-activating factor with rat hepatocytes: uptake, translocation, metabolism, and effects on PAF-acetylhydrolase secretion and protein tyrosine phosphorylation. Arch Biochem Biophys. 1996;327(1):113–122. doi:10.1006/abbi.1996.0099
99. Burns JL, Nakamura MT, Ma DWL. Differentiating the biological effects of linoleic acid from arachidonic acid in health and disease. Prostaglandins Leukot Essent Fatty Acids. 2018;135:1–4. doi:10.1016/j.plefa.2018.05.004
100. Sonnweber T, Pizzini A, Nairz M, et al. Arachidonic acid metabolites in cardiovascular and metabolic diseases. Int J Mol Sci. 2018;19(11):3285. doi:10.3390/ijms19113285
101. Innes JK, Calder PC. Omega-6 fatty acids and inflammation. Prostaglandins Leukot Essent Fatty Acids. 2018;132:41–48. doi:10.1016/j.plefa.2018.03.004
102. Di Nunzio M, Valli V, Bordoni A. PUFA and oxidative stress. Differential modulation of the cell response by DHA. Int J Food Sci Nutr. 2016;67(7):834–843. doi:10.1080/09637486.2016.1201790
103. Samala N, Tersey SA, Chalasani N, et al. Molecular mechanisms of nonalcoholic fatty liver disease: potential role for 12-lipoxygenase. J Diabetes Complications. 2017;31(11):1630–1637. doi:10.1016/j.jdiacomp.2017.07.014
104. Garrido D, Chanteloup NK, Trotereau A, et al. Characterization of the phospholipid platelet-activating factor as a mediator of inflammation in chickens. Front Vet Sci. 2017;4:226. doi:10.3389/fvets.2017.00226
105. Dalmaso B, da Silva-Junior IA, Fragel-Madeira L, et al. Platelet activating factor in the eye: physiological roles, diseases and future perspectives. Prostaglandins Other Lipid Mediat. 2021;153:106522. doi:10.1016/j.prostaglandins.2020.106522
106. Evans JH, Spencer DM, Zweifach A, et al. Intracellular calcium signals regulating cytosolic phospholipase A2 translocation to internal membranes. J Biol Chem. 2001;276(32):30150–30160. doi:10.1074/jbc.M100943200
107. Six DA, Dennis EA. Essential Ca(2+)-independent role of the group IVA cytosolic phospholipase A(2) C2 domain for interfacial activity. J Biol Chem. 2003;278(26):23842–23850. doi:10.1074/jbc.M301386200
108. Singh MK, Jayarajan R, Varshney S, et al. Chronic systemic exposure to IL6 leads to deregulation of glycolysis and fat accumulation in the zebrafish liver. Biochim Biophys Acta Mol Cell Biol Lipids. 2021;1866(5):158905. doi:10.1016/j.bbalip.2021.158905
109. Musso G, Gambino R, Cassader M. Non-alcoholic fatty liver disease from pathogenesis to management: an update. Obes Rev. 2010;11(6):430–445. doi:10.1111/j.1467-789X.2009.00657.x
110. Colak Y, Senates E, Ozturk O, et al. Association of serum lipoprotein-associated phospholipase A2 level with nonalcoholic fatty liver disease. Metab Syndr Relat Disord. 2012;10(2):103–109. doi:10.1089/met.2011.0111
111. Zelber-Sagi S, Azar S, Nemirovski A, et al. Serum levels of endocannabinoids are independently associated with nonalcoholic fatty liver disease. Obesity. 2017;25(1):94–101. doi:10.1002/oby.21687
112. Tutino V, De Nunzio V, Caruso MG, et al. Aerobic physical activity and a low glycemic diet reduce the AA/EPA ratio in red blood cell membranes of patients with NAFLD. Nutrients. 2018;10(9):1299. doi:10.3390/nu10091299
113. Ii H, Yokoyama N, Yoshida S, et al. Alleviation of high-fat diet-induced fatty liver damage in group IVA phospholipase A2-knockout mice. PLoS One. 2009;4(12):e8089. doi:10.1371/journal.pone.0008089
114. Sztolsztener K, Chabowski A, Harasim-Symbor E, et al. Arachidonic acid as an early indicator of inflammation during non-alcoholic fatty liver disease development. Biomolecules. 2020;10(8):1133. doi:10.3390/biom10081133
115. Ma K, Chen Y, Liang X, et al. Inhibition of 5-lipoxygenase inhibitor zileuton in high-fat diet-induced nonalcoholic fatty liver disease progression model. Iran J Basic Med Sci. 2017;20(11):1207–1212. doi:10.22038/IJBMS.2017.9482
116. Ghazali R, Mehta KJ, Bligh SA, et al. High omega arachidonic acid/docosahexaenoic acid ratio induces mitochondrial dysfunction and altered lipid metabolism in human hepatoma cells. World J Hepatol. 2020;12(3):84–98. doi:10.4254/wjh.v12.i3.84
117. Fiorio Pla A, Genova T, Pupo E, et al. Multiple roles of protein kinase a in arachidonic acid-mediated Ca2+ entry and tumor-derived human endothelial cell migration. Mol Cancer Res. 2010;8(11):1466–1476. doi:10.1158/1541-7786.MCR-10-0002
118. Thompson J, Mignen O, Shuttleworth TJ. The N-terminal domain of Orai3 determines selectivity for activation of the store-independent ARC channel by arachidonic acid. Channels. 2010;4(5):398–410. doi:10.4161/chan.4.5.13226
119. Tseng CL, Wei JW. Homologous desensitization of histamine-mediated signal transduction system in C6 glioma cells. Chin J Physiol. 2013;56(2):90–100. doi:10.4077/CJP.2013.BAB094
120. Martínez J, Moreno JJ. Role of Ca2+-independent phospholipase A2 on arachidonic acid release induced by reactive oxygen species. Arch Biochem Biophys. 2001;392(2):257–262. doi:10.1006/abbi.2001.2439
121. Nunemaker CS, Chen M, Pei H, et al. 12-Lipoxygenase-knockout mice are resistant to inflammatory effects of obesity induced by Western diet. Am J Physiol Endocrinol Metab. 2008;295(5):E1065–E1075. doi:10.1152/ajpendo.90371.2008
122. Lazic M, Inzaugarat ME, Povero D, et al. Reduced dietary omega-6 to omega-3 fatty acid ratio and 12/15-lipoxygenase deficiency are protective against chronic high fat diet-induced steatohepatitis. PLoS One. 2014;9(9):e107658. doi:10.1371/journal.pone.0107658
123. Martínez-Clemente M, Ferré N, González-Périz A, et al. 5-lipoxygenase deficiency reduces hepatic inflammation and tumor necrosis factor alpha-induced hepatocyte damage in hyperlipidemia-prone ApoE-null mice. Hepatology. 2010;51(3):817–827. doi:10.1002/hep.23463
124. Puri P, Wiest MM, Cheung O, et al. The plasma lipidomic signature of nonalcoholic steatohepatitis. Hepatology. 2009;50(6):1827–1838. doi:10.1002/hep.23229
125. Hall Z, Bond NJ, Ashmore T, et al. Lipid zonation and phospholipid remodeling in nonalcoholic fatty liver disease. Hepatology. 2017;65(4):1165–1180. doi:10.1002/hep.28953
126. Ma K, Nunemaker CS, Wu R, et al. 12-lipoxygenase products reduce insulin secretion and {beta}-cell viability in human islets. J Clin Endocrinol Metab. 2010;95(2):887–893. doi:10.1210/jc.2009-1102
127. Chakrabarti SK, Cole BK, Wen Y, Keller SR, Nadler JL. 12/15-lipoxygenase products induce inflammation and impair insulin signaling in 3T3-L1 adipocytes. Obesity. 2009;17(9):1657–1663. doi:10.1038/oby.2009.192
128. Khan H, Gupta A, Singh TG, et al. Mechanistic insight on the role of leukotriene receptors in ischemic-reperfusion injury. Pharmacol Rep. 2021;73(5):1240–1254. doi:10.1007/s43440-021-00258-8
129. Lecomte M, Lecocq R, Dumont JE, et al. Covalent binding of arachidonic acid metabolites to human platelet proteins. Identification of prostaglandin H synthase as one of the modified substrates. J Biol Chem. 1990;265(9):5178–5187.
130. Otomo Y, Kanda Y, Yoshino Y, Otsuka T. Production of leukotrienes in rat Kupffer cells and hepatocytes by various inducers. Nihon Geka Gakkai Zasshi. 1993;94(3):234–241.
131. Doskey CM, Fader KA, Nault R, et al. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) alters hepatic polyunsaturated fatty acid metabolism and eicosanoid biosynthesis in female Sprague-Dawley rats. Toxicol Appl Pharmacol. 2020;398:115034. doi:10.1016/j.taap.2020.115034
132. Takasugi M, Muta E, Yamada K, et al. A new method to evaluate anti-allergic effect of food component by measuring leukotriene B4 from a mouse mast cell line. Cytotechnology. 2018;70(1):177–184. doi:10.1007/s10616-017-0129-9
133. Ito S, Ito Y, Katagiri H, et al. Leukotriene B4/leukotriene B4 receptor pathway is involved in hepatic microcirculatory dysfunction elicited by endotoxin. Shock. 2008;30(1):87–91. doi:10.1097/shk.0b013e31815d06a1
134. Alvarez ML, Lorenzetti F. Role of eicosanoids in liver repair, regeneration and cancer. Biochem Pharmacol. 2021;192:114732. doi:10.1016/j.bcp.2021.114732
135. Gong M, Duan H, Wu F, et al. Berberine alleviates insulin resistance and inflammation via inhibiting the LTB4-BLT1 axis. Front Pharmacol. 2021;12:722360. doi:10.3389/fphar.2021.722360
136. Gai Z, Visentin M, Gui T, et al. Effects of farnesoid X receptor activation on arachidonic acid metabolism, NF-kB signaling, and hepatic inflammation. Mol Pharmacol. 2018;94(2):802–811. doi:10.1124/mol.117.111047
137. López-Parra M, Titos E, Horrillo R, et al. Regulatory effects of arachidonate 5-lipoxygenase on hepatic microsomal TG transfer protein activity and VLDL-triglyceride and apoB secretion in obese mice. J Lipid Res. 2008;49(12):2513–2523. doi:10.1194/jlr.M800101-JLR200
138. Horrillo R, González-Périz A, Martínez-Clemente M, et al. 5-lipoxygenase activating protein signals adipose tissue inflammation and lipid dysfunction in experimental obesity. J Immunol. 2010;184(7):3978–3987. doi:10.4049/jimmunol.0901355
139. Bäck M, Sultan A, Ovchinnikova O, et al. 5-Lipoxygenase-activating protein: a potential link between innate and adaptive immunity in atherosclerosis and adipose tissue inflammation. Circ Res. 2007;100(7):946–949. doi:10.1161/01.RES.0000264498.60702.0d
140. Gilbert NC, Gerstmeier J, Schexnaydre EE, et al. Structural and mechanistic insights into 5-lipoxygenase inhibition by natural products. Nat Chem Biol. 2020;16(7):783–790. doi:10.1038/s41589-020-0544-7
141. Chakrabarti SK, Wen Y, Dobrian AD, et al. Evidence for activation of inflammatory lipoxygenase pathways in visceral adipose tissue of obese Zucker rats. Am J Physiol Endocrinol Metab. 2011;300(1):E175–E187. doi:10.1152/ajpendo.00203.2010
142. Spite M, Hellmann J, Tang Y, et al. Deficiency of the leukotriene B4 receptor, BLT-1, protects against systemic insulin resistance in diet-induced obesity. J Immunol. 2011;187(4):1942–1949. doi:10.4049/jimmunol.1100196
143. Abdallah MS, Eldeen AH, Tantawy SS, et al. The leukotriene receptor antagonist montelukast in the treatment of non-alcoholic steatohepatitis: a proof-of-concept, randomized, double-blind, placebo-controlled trial. Eur J Pharmacol. 2021;906:174295. doi:10.1016/j.ejphar.2021.174295
144. Beara I, Majkić T, Fioravanti S, et al. The effects of trifluoromethylated derivatives on prostaglandin E2 and thromboxane A2 production in human leukemic U937 macrophages. Med Chem. 2020;16(1):63–68. doi:10.2174/1573406415666190208150253
145. Son DJ, Cho MR, Jin YR, et al. Antiplatelet effect of green tea catechins: a possible mechanism through arachidonic acid pathway. Prostaglandins Leukot Essent Fatty Acids. 2004;71(1):25–31. doi:10.1016/j.plefa.2003.12.004
146. Srivastava KC. Transformations of exogenous arachidonic acid in human platelets in the presence of oleic- and eicosapentaenoic acids. Prostaglandins Leukot Med. 1985;18(1):31–37. doi:10.1016/0262-1746(85)90047-2
147. Levine L. Platelet-activating factor stimulates arachidonic acid metabolism in rat liver cells (C-9 cell line) by a receptor-mediated mechanism. Mol Pharmacol. 1988;34(6):793–799.
148. Levine L. Effects of the protein kinase inhibitors, staurosporine and K-252a, on PGI2 production by rat liver cells (the C-9 cell line). Prostaglandins. 1990;40(3):259–269. doi:10.1016/0090-6980(90)90014-m
149. López-Parra M, Clària J, Titos E, et al. The selective cyclooxygenase-2 inhibitor celecoxib modulates the formation of vasoconstrictor eicosanoids and activates PPARgamma. Influence of Albumin J Hepatol. 2005;42(1):75–81. doi:10.1016/j.jhep.2004.09.011
150. Becker K, Heinroth-Hoffmann I, Giessler C, et al. PAF effects on eicosanoid release in neonatal rat cardiomyocytes. Prostaglandins Leukot Essent Fatty Acids. 1995;53(3):197–200. doi:10.1016/0952-3278(95)90116-7
151. Lo HM, Huang TF, Lin CN, et al. 2’-Ethoxy-5’-methoxy-2-(5-methylthienyl)chalcone inhibits collagen-induced protein tyrosine phosphorylation and thromboxane formation during platelet aggregation and adhesion. Pharmacology. 2009;84(3):145–152. doi:10.1159/000235584
152. Wang W, Zhong X, Guo J. Role of 2‑series prostaglandins in the pathogenesis of type 2 diabetes mellitus and non‑alcoholic fatty liver disease (Review). Int J Mol Med. 2021;47(6):114. doi:10.3892/ijmm.2021.4947
153. Jiang J, Tran L, Vasudevan H, et al. Endothelin-1 blockade prevents COX2 induction and TXA2 production in the fructose hypertensive rat. Can J Physiol Pharmacol. 2007;85(3–4):422–429. doi:10.1139/y06-088
154. Rucker D, Dhamoon AS. Physiology, thromboxane A2. StatPearls; 2021.
155. Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294(5548):1871–1875. doi:10.1126/science.294.5548.1871
156. Tipoe GL, Ho CT, Liong EC, et al. Voluntary oral feeding of rats not requiring a very high fat diet is a clinically relevant animal model of non-alcoholic fatty liver disease (NAFLD). Histol Histopathol. 2009;24(9):1161–1169. doi:10.14670/HH-24.1161
157. Hybiak J, Broniarek I, Kiryczyński G, et al. Aspirin and its pleiotropic application. Eur J Pharmacol. 2020;866:172762. doi:10.1016/j.ejphar.2019.172762
158. Pérez S, Aspichueta P, Ochoa B, et al. The 2-series prostaglandins suppress VLDL secretion in an inflammatory condition-dependent manner in primary rat hepatocytes. Biochim Biophys Acta. 2006;1761(2):160–171. doi:10.1016/j.bbalip.2006.02.003
159. Feingold KR, Doerrler W, Dinarello CA, et al. Stimulation of lipolysis in cultured fat cells by tumor necrosis factor, interleukin-1, and the interferons is blocked by inhibition of prostaglandin synthesis. Endocrinology. 1992;130(1):10–16. doi:10.1210/endo.130.1.1370149
160. Henkel J, Frede K, Schanze N, et al. Stimulation of fat accumulation in hepatocytes by PGE-dependent repression of hepatic lipolysis, β-oxidation and VLDL-synthesis. Lab Invest. 2012;92(11):1597–1606. doi:10.1038/labinvest.2012.128
161. Nassir F, Adewole OL, Brunt EM, et al. CD36 deletion reduces VLDL secretion, modulates liver prostaglandins, and exacerbates hepatic steatosis in ob/ob mice. J Lipid Res. 2013;54(11):2988–2997. doi:10.1194/jlr.M037812
162. Henkel J, Coleman CD, Schraplau A, et al. Augmented liver inflammation in a microsomal prostaglandin E synthase 1 (mPGES-1)-deficient diet-induced mouse NASH model. Sci Rep. 2018;8(1):16127. doi:10.1038/s41598-018-34633-y
163. Loomba R, Quehenberger O, Armando A, et al. Polyunsaturated fatty acid metabolites as novel lipidomic biomarkers for noninvasive diagnosis of nonalcoholic steatohepatitis. J Lipid Res. 2015;56(1):185–192. doi:10.1194/jlr.P055640
164. Kus E, Kaczara P, Czyzynska-Cichon I, et al. LSEC fenestrae are preserved despite pro-inflammatory phenotype of liver sinusoidal endothelial cells in mice on high fat diet. Front Physiol. 2019;10:6. doi:10.3389/fphys.2019.00006
165. Kumei S, Yuhki KI, Kojima F, et al. Prostaglandin I2 suppresses the development of diet-induced nonalcoholic steatohepatitis in mice. FASEB J. 2018;32(5):2354–2365. doi:10.1096/fj.201700590R
166. Chung MY, Mah E, Masterjohn C, et al. Green tea lowers hepatic COX-2 and prostaglandin E2 in rats with dietary fat-induced nonalcoholic steatohepatitis. J Med Food. 2015;18(6):648–655. doi:10.1089/jmf.2014.0048
167. Helmersson J, Vessby B, Larsson A, et al. Association of type 2 diabetes with cyclooxygenase-mediated inflammation and oxidative stress in an elderly population. Circulation. 2004;109(14):1729–1734. doi:10.1161/01.CIR.0000124718.99562.91
168. Kimple ME, Keller MP, Rabaglia MR, et al. Prostaglandin E2 receptor, EP3, is induced in diabetic islets and negatively regulates glucose- and hormone-stimulated insulin secretion. Diabetes. 2013;62(6):1904–1912. doi:10.2337/db12-0769
169. Batchu SN, Majumder S, Bowskill BB, et al. Prostaglandin I2 receptor agonism preserves β-cell function and attenuates albuminuria through nephrin-dependent mechanisms. Diabetes. 2016;65(5):1398–1409. doi:10.2337/db15-0783
170. Arablou T, Aryaeian N, Valizadeh M, et al. The effect of ginger consumption on glycemic status, lipid profile and some inflammatory markers in patients with type 2 diabetes mellitus. Int J Food Sci Nutr. 2014;65(4):515–520. doi:10.3109/09637486.2014.880671
171. Zhu CF, Li GZ, Peng HB, et al. Treatment with marine collagen peptides modulates glucose and lipid metabolism in Chinese patients with type 2 diabetes mellitus. Appl Physiol Nutr Metab. 2010;35(6):797–804. doi:10.1139/H10-075
172. Francés DE, Motiño O, Agrá N, et al. Hepatic cyclooxygenase-2 expression protects against diet-induced steatosis, obesity, and insulin resistance. Diabetes. 2015;64(5):1522–1531. doi:10.2337/db14-0979
173. Hsieh PS, Jin JS, Chiang CF, et al. COX-2-mediated inflammation in fat is crucial for obesity-linked insulin resistance and fatty liver. Obesity. 2009;17(6):1150–1157. doi:10.1038/oby.2008.674
174. Henkel J, Neuschäfer-Rube F, Pathe-neuschäfer-rube A, et al. Aggravation by prostaglandin E2 of interleukin-6-dependent insulin resistance in hepatocytes. Hepatology. 2009;50(3):781–790. doi:10.1002/hep.23064
175. Henkel J, Gärtner D, Dorn C, et al. Oncostatin M produced in Kupffer cells in response to PGE2: possible contributor to hepatic insulin resistance and steatosis. Lab Invest. 2011;91(7):1107–1117. doi:10.1038/labinvest.2011.47
176. Wang Y, Yan S, Xiao B, et al. Prostaglandin F2α facilitates hepatic glucose production through camkiiγ/p38/FOXO1 signaling pathway in fasting and obesity. Diabetes. 2018;67(9):1748–1760. doi:10.2337/db17-1521
177. Sato N, Kaneko M, Tamura M, et al. The prostacyclin analog beraprost sodium ameliorates characteristics of metabolic syndrome in obese Zucker (fatty) rats. Diabetes. 2010;59(4):1092–1100. doi:10.2337/db09-1432
178. Ryu R, Kim HJ, Moon B, et al. Ethanol extract of persimmon tree leaves improves blood circulation and lipid metabolism in rats fed a high-fat diet. J Med Food. 2015;18(7):715–723. doi:10.1089/jmf.2014.3307
179. Francque S, Laleman W, Verbeke L, et al. Increased intrahepatic resistance in severe steatosis: endothelial dysfunction, vasoconstrictor overproduction and altered microvascular architecture. Lab Invest. 2012;92(10):1428–1439. doi:10.1038/labinvest.2012.103
180. Nieuwenhuys CM, Feijge MA, Offermans RF, et al. Modulation of rat platelet activation by vessel wall-derived prostaglandin and platelet-derived thromboxane: effects of dietary fish oil on thromboxane-prostaglandin balance. Atherosclerosis. 2001;154(2):355–366. doi:10.1016/s0021-9150(00)00503-7
181. Murphy CT, Elmore M, Kellie S, et al. The relationship between cytosolic Ca2+, sn-1,2-diacylglycerol and inositol 1,4,5-trisphosphate elevation in platelet-activating-factor-stimulated rabbit platelets. Influence of protein kinase C on production of signal molecules. Biochem J. 1991;278(1):255–261. doi:10.1042/bj2780255
182. Catalán RE, Martínez AM, Aragonés MD, et al. PAF-induced activation of polyphosphoinositide-hydrolyzing phospholipase C in cerebral cortex. Biochem Biophys Res Commun. 1992;183(1):300–305. doi:10.1016/0006-291x(92)91643-5
183. Kester M, Thomas CP, Wang J, et al. Platelet-activating factor stimulates multiple signaling pathways in cultured rat mesangial cells. J Cell Physiol. 1992;153(2):244–255. doi:10.1002/jcp.1041530204
184. Uhing RJ, Prpic V, Hollenbach PW, et al. Involvement of protein kinase C in platelet-activating factor-stimulated diacylglycerol accumulation in murine peritoneal macrophages. J Biol Chem. 1989;264(16):9224–9230.
185. Miguel BG, Calcerrada MC, Martín L, et al. Increase of phosphoinositide hydrolysis and diacylglycerol production by PAF in isolated rat liver nuclei. Prostaglandins Other Lipid Mediat. 2001;65(4):159–166. doi:10.1016/s0090-6980(01)00124-1
186. Kojta I, Zabielski P, Roszczyc-Owsiejczuk K, et al. GPAT gene silencing in muscle reduces diacylglycerols content and improves insulin action in diet-induced insulin resistance. Int J Mol Sci. 2020;21(19):7369. doi:10.3390/ijms21197369
187. Preuss C, Jelenik T, Bódis K, et al. A new targeted lipidomics approach reveals lipid droplets in liver, muscle and heart as a repository for diacylglycerol and ceramide species in non-alcoholic fatty liver. Cells. 2019;8(3):277. doi:10.3390/cells8030277
188. Engin AB. What Is Lipotoxicity?. Adv Exp Med Biol. 2017;960:197–220. doi:10.1007/978-3-319-48382-5_8
189. Gorden DL, Ivanova PT, Myers DS, et al. Increased diacylglycerols characterize hepatic lipid changes in progression of human nonalcoholic fatty liver disease; comparison to a murine model. PLoS One. 2011;6(8):e22775. doi:10.1371/journal.pone.0022775
190. Sanyal AJ, Pacana T. A lipidomic readout of disease progression in A diet-induced mouse model of nonalcoholic fatty liver disease. Trans Am Clin Climatol Assoc. 2015;126:271–288.
191. Erion DM, Shulman GI. Diacylglycerol-mediated insulin resistance. Nat Med. 2010;16(4):400–402. doi:10.1038/nm0410-400
192. Gilijamse PW, Versteeg RI, Ackermans MT, et al. Hepatic diacylglycerol-associated protein kinase Cε translocation links hepatic steatosis to hepatic insulin resistance in humans. Cell Rep. 2017;19(10):1997–2004. doi:10.1016/j.celrep.2017.05.035
193. Yazıcı D, Sezer H. Insulin resistance, obesity and lipotoxicity. Adv Exp Med Biol. 2017;960:277–304. doi:10.1007/978-3-319-48382-5_12
194. Magkos F, Su X, Bradley D, et al. Intrahepatic diacylglycerol content is associated with hepatic insulin resistance in obese subjects. Gastroenterology. 2012;142(7):1444–6.e2. doi:10.1053/j.gastro.2012.03.003
195. Toulis KA, Nirantharakumar K, Pourzitaki C, et al. Glucokinase activators for type 2 diabetes: challenges and future developments. Drugs. 2020;80(5):467–475. doi:10.1007/s40265-020-01278-z
196. Sternisha SM, Miller BG. Molecular and cellular regulation of human glucokinase. Arch Biochem Biophys. 2019;663:199–213. doi:10.1016/j.abb.2019.01.011
197. Blackmore PF, Strickland WG, Bocckino SB, et al. Mechanism of hepatic glycogen synthase inactivation induced by Ca2+-mobilizing hormones. Studies using phospholipase C and phorbol myristate acetate. Biochem J. 1986;237(1):235–242. doi:10.1042/bj2370235
198. Hage hassan R, Bourron O, Hajduch E. Defect of insulin signal in peripheral tissues: important role of ceramide. World J Diabetes. 2014;5(3):244–257. doi:10.4239/wjd.v5.i3.244
199. Titchenell PM, Lazar MA, Birnbaum MJ. Unraveling the regulation of hepatic metabolism by insulin. Trends Endocrinol Metab. 2017;28(7):497–505. doi:10.1016/j.tem.2017.03.003
200. Lewis GF, Carpentier AC, Pereira S, et al. Direct and indirect control of hepatic glucose production by insulin. Cell Metab. 2021;33(4):709–720. doi:10.1016/j.cmet.2021.03.007
201. Lyu K, Zhang Y, Zhang D, et al. A membrane-bound diacylglycerol species induces PKCϵ-mediated hepatic insulin resistance. Cell Metab. 2020;32(4):654–664.e5. doi:10.1016/j.cmet.2020.08.001
202. Aroor AR, Habibi J, Ford DA, et al. Dipeptidyl peptidase-4 inhibition ameliorates Western diet-induced hepatic steatosis and insulin resistance through hepatic lipid remodeling and modulation of hepatic mitochondrial function. Diabetes. 2015;64(6):1988–2001. doi:10.2337/db14-0804
203. Li X, Zhang D, Vatner DF, et al. Mechanisms by which adiponectin reverses high fat diet-induced insulin resistance in mice. Proc Natl Acad Sci U S A. 2020;117(51):32584–32593. doi:10.1073/pnas.1922169117
204. Birkenfeld AL, Shulman GI. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 diabetes. Hepatology. 2014;59(2):713–723. doi:10.1002/hep.26672
205. Kumashiro N, Erion DM, Zhang D, et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc Natl Acad Sci U S A. 2011;108(39):16381–16385. doi:10.1073/pnas.1113359108
206. Zimmerman GA, McIntyre TM, Prescott SM, et al. The platelet-activating factor signaling system and its regulators in syndromes of inflammation and thrombosis. Crit Care Med. 2002;30(5 Suppl):S294–S301. doi:10.1097/00003246-200205001-00020
207. Zaid Y, Senhaji N, Darif Y, et al. Distinctive roles of PKC delta isozyme in platelet function. Curr Res Transl Med. 2016;64(3):135–139. doi:10.1016/j.retram.2016.05.001
208. Pelech SL, Charest DL, Howard SL, et al. Protein kinase C activation by platelet-activating factor is independent of enzyme translocation. Biochim Biophys Acta. 1990;1051(1):100–107. doi:10.1016/0167-4889(90)90179-h
209. Gay JC, Stitt ES. Platelet-activating factor induces protein kinase activity in the particulate fraction of human neutrophils. Blood. 1988;71(1):159–165.
210. Hu H, Zhang W, Li N. Glycoprotein IIb/IIIa inhibition attenuates platelet-activating factor-induced platelet activation by reducing protein kinase C activity. J Thromb Haemost. 2003;1(8):1805–1812. doi:10.1046/j.1538-7836.2003.00324.x
211. Guo X, Zheng M, Pan R, et al. Hydroxysafflor yellow A (HSYA) targets the platelet-activating factor (PAF) receptor and inhibits human bronchial smooth muscle activation induced by PAF. Food Funct. 2019;10(8):4661–4673. doi:10.1039/c9fo00896a
212. Baudel MAS M-A, Shi J, Large WA, et al. Obligatory role for PKCδ in PIP2 -mediated activation of store-operated TRPC1 channels in vascular smooth muscle cells. J Physiol. 2020;598(18):3911–3925. doi:10.1113/JP279947
213. Sharma A, Maurya CK, Arha D, et al. Nod1-mediated lipolysis promotes diacylglycerol accumulation and successive inflammation via PKCδ-IRAK axis in adipocytes. Biochim Biophys Acta Mol Basis Dis. 2019;1865(1):136–146. doi:10.1016/j.bbadis.2018.10.036
214. Balciunaite E, Jones S, Toker A, et al. PDGF initiates two distinct phases of protein kinase C activity that make unequal contributions to the G0 to S transition. Curr Biol. 2000;10(5):261–267. doi:10.1016/s0960-9822(00)00358-4
215. Li M, Vienberg SG, Bezy O, et al. Role of PKCδ in insulin sensitivity and skeletal muscle metabolism. Diabetes. 2015;64(12):4023–4032. doi:10.2337/db14-1891
216. Morgan S, Yamanouchi D, Harberg C, et al. Elevated protein kinase C-δ contributes to aneurysm pathogenesis through stimulation of apoptosis and inflammatory signaling. Arterioscler Thromb Vasc Biol. 2012;32(10):2493–2502. doi:10.1161/ATVBAHA.112.255661
217. Lyu K, Zhang D, Song J, et al. Short-term overnutrition induces white adipose tissue insulin resistance through sn-1,2-diacylglycerol/PKCε/insulin receptor Thr1160 phosphorylation. JCI Insight. 2021;6(4):e139946. doi:10.1172/jci.insight.139946
218. Malavez Y, Voss OH, Gonzalez-Mejia ME, et al. Distinct contribution of protein kinase Cδ and protein kinase Cε in the lifespan and immune response of human blood monocyte subpopulations. Immunology. 2015;144(4):611–620. doi:10.1111/imm.12412
219. Greene MW, Burrington CM, Luo Y, et al. PKCδ is activated in the liver of obese Zucker rats and mediates diet-induced whole body insulin resistance and hepatocyte cellular insulin resistance. J Nutr Biochem. 2014;25(3):281–288. doi:10.1016/j.jnutbio.2013.10.008
220. Newton AC. Protein kinase C: poised to signal. Am J Physiol Endocrinol Metab. 2010;298(3):E395–E402. doi:10.1152/ajpendo.00477.2009
221. Giorgione JR, Lin JH, McCammon JA, et al. Increased membrane affinity of the C1 domain of protein kinase Cdelta compensates for the lack of involvement of its C2 domain in membrane recruitment. J Biol Chem. 2006;281(3):1660–1669. doi:10.1074/jbc.M510251200
222. Samuel VT, Liu ZX, Qu X, et al. Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem. 2004;279(31):32345–32353. doi:10.1074/jbc.M313478200
223. Lee SJ, Kang JH, Choi SY, et al. PKCδ as a regulator for TGFβ1-induced α-SMA production in a murine nonalcoholic steatohepatitis model. PLoS One. 2013;8(2):e55979. doi:10.1371/journal.pone.0055979
224. Badi RM, Mostafa DG, Khaleel EF, et al. Resveratrol protects against hepatic insulin resistance in a rat’s model of non-alcoholic fatty liver disease by down-regulation of GPAT-1 and DGAT2 expression and inhibition of PKC membranous translocation. Clin Exp Pharmacol Physiol. 2019;46(6):545–555. doi:10.1111/1440-1681.13074
225. Samidurai M, Palanisamy BN, Bargues-Carot A, et al. PKC Delta Activation Promotes Endoplasmic Reticulum Stress (ERS) and NLR Family Pyrin Domain-Containing 3 (NLRP3) Inflammasome Activation Subsequent to Asynuclein-Induced Microglial Activation: involvement of Thioredoxin-Interacting Protein (TXNIP)/Thioredoxin (Trx) Redoxisome Pathway. Front Aging Neurosci. 2021;13:661505. doi:10.3389/fnagi.2021.661505
226. Jornayvaz FR, Birkenfeld AL, Jurczak MJ, et al. Hepatic insulin resistance in mice with hepatic overexpression of diacylglycerol acyltransferase 2. Proc Natl Acad Sci U S A. 2011;108(14):5748–5752. doi:10.1073/pnas.1103451108
227. Dallak MA. Acylated ghrelin induces but deacylated ghrelin prevents hepatic steatosis and insulin resistance in lean rats: effects on DAG/ PKC/JNK pathway. Biomed Pharmacother. 2018;105:299–311. doi:10.1016/j.biopha.2018.05.098
228. Frangioudakis G, Burchfield JG, Narasimhan S, et al. Diverse roles for protein kinase C delta and protein kinase C epsilon in the generation of high-fat-diet-induced glucose intolerance in mice: regulation of lipogenesis by protein kinase C delta. Diabetologia. 2009;52(12):2616–2620. doi:10.1007/s00125-009-1543-0
229. Zhang J, Burrington CM, Davenport SK, et al. PKCδ regulates hepatic triglyceride accumulation and insulin signaling in Lepr(db/db) mice. Biochem Biophys Res Commun. 2014;450(4):1619–1625. doi:10.1016/j.bbrc.2014.07.048
230. Samuel VT, Liu ZX, Wang A, et al. Inhibition of protein kinase Cepsilon prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J Clin Invest. 2007;117(3):739–745. doi:10.1172/JCI30400
231. Greene MW, Burrington CM, Lynch DT, et al. Lipid metabolism, oxidative stress and cell death are regulated by PKC delta in a dietary model of nonalcoholic steatohepatitis. PLoS One. 2014;9(1):e85848. doi:10.1371/journal.pone.0085848
232. Klymenko K, Novokhatska T, Kizub I, et al.. PKC-δ isozyme gene silencing restores vascular function in diabetic rat. J Basic Clin Physiol Pharmacol. 2014:1–9. doi:10.1515/jbcpp-2013-0147
233. Pereira S, Park E, Mori Y, et al. FFA-induced hepatic insulin resistance in vivo is mediated by PKCδ, NADPH oxidase, and oxidative stress. Am J Physiol Endocrinol Metab. 2014;307(1):E34–E46. doi:10.1152/ajpendo.00436.2013
234. De Mattia G, Bravi MC, Laurenti O, et al. Influence of reduced glutathione infusion on glucose metabolism in patients with non-insulin-dependent diabetes mellitus. Metabolism. 1998;47(8):993–997. doi:10.1016/s0026-0495(98)90357-2
235. Zhang XQ, Xu CF, Yu CH, et al. Role of endoplasmic reticulum stress in the pathogenesis of nonalcoholic fatty liver disease. World J Gastroenterol. 2014;20(7):1768–1776. doi:10.3748/wjg.v20.i7.1768
236. Yang M, Chen Z, Xiang S, et al. Hugan Qingzhi medication ameliorates free fatty acid-induced L02 hepatocyte endoplasmic reticulum stress by regulating the activation of PKC-δ. BMC Complement Med Ther. 2020;20(1):377. doi:10.1186/s12906-020-03164-3
237. Greene MW, Burrington CM, Ruhoff MS, et al. PKC{delta} is activated in a dietary model of steatohepatitis and regulates endoplasmic reticulum stress and cell death. J Biol Chem. 2010;285(53):42115–42129. doi:10.1074/jbc.M110.168575
238. Wang H, Chakrabarty S. Platelet-activating factor activates mitogen-activated protein kinases, inhibits proliferation, induces differentiation and suppresses the malignant phenotype of human colon carcinoma cells. Oncogene. 2003;22(14):2186–2191. doi:10.1038/sj.onc.1206348
239. Yu SM, Tsai SY, Kuo SC, et al. Inhibition of platelet function by A02131-1, a novel inhibitor of cGMP-specific phosphodiesterase, in vitro and in vivo. Blood. 1996;87(9):3758–3767.
240. Richardson L, Dixon CL, Aguilera-Aguirre L, et al. Oxidative stress-induced TGF-beta/TAB1-mediated p38MAPK activation in human amnion epithelial cells. Biol Reprod. 2018;99(5):1100–1112. doi:10.1093/biolre/ioy135
241. Liu YM, Wang X, Nawaz A, et al. Wogonin ameliorates lipotoxicity-induced apoptosis of cultured vascular smooth muscle cells via interfering with DAG-PKC pathway. Acta Pharmacol Sin. 2011;32(12):1475–1482. doi:10.1038/aps.2011.120
242. Inoguchi T, Li P, Umeda F, et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C–dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000;49(11):1939–1945. doi:10.2337/diabetes.49.11.1939
243. Niu W, Huang C, Nawaz Z, et al. Maturation of the regulation of GLUT4 activity by p38 MAPK during L6 cell myogenesis. J Biol Chem. 2003;278(20):17953–17962. doi:10.1074/jbc.M211136200
244. Hong F, Wu N, Ge Y, et al. Nanosized titanium dioxide resulted in the activation of TGF-β/Smads/p38MAPK pathway in renal inflammation and fibration of mice. J Biomed Mater Res A. 2016;104(6):1452–1461. doi:10.1002/jbm.a.35678
245. Leelahavanichkul K, Amornphimoltham P, Molinolo AA, et al. A role for p38 MAPK in head and neck cancer cell growth and tumor-induced angiogenesis and lymphangiogenesis. Mol Oncol. 2014;8(1):105–118. doi:10.1016/j.molonc.2013.10.003
246. Gao W, Du X, Lei L, et al. NEFA-induced ROS impaired insulin signalling through the JNK and p38MAPK pathways in non-alcoholic steatohepatitis. J Cell Mol Med. 2018;22(7):3408–3422. doi:10.1111/jcmm.13617
247. Wang W, Li X, Xu J. Magnesium isoglycyrrhizinate attenuates D-galactosamine/lipopolysaccharides induced acute liver injury of rat via regulation of the p38-MAPK and NF-κB signaling pathways. Immunopharmacol Immunotoxicol. 2018;40(3):262–267. doi:10.1080/08923973.2018.1441300
248. Tang Z, Xia N, Yuan X, et al. PRDX1 is involved in palmitate induced insulin resistance via regulating the activity of p38MAPK in HepG2 cells. Biochem Biophys Res Commun. 2015;465(4):670–677. doi:10.1016/j.bbrc.2015.08.008
249. Liu Y, Song A, Zang S, et al. Jinlida reduces insulin resistance and ameliorates liver oxidative stress in high-fat fed rats. J Ethnopharmacol. 2015;162:244–252. doi:10.1016/j.jep.2014.12.040
250. Zhu Y, Zhang H, Wei Y, et al. Pea-derived peptides, VLP, LLP, VA, and LL, improve insulin resistance in HepG2 cells via activating IRS-1/PI3K/AKT and blocking ROS-mediated p38MAPK signaling. J Food Biochem. 2020;44(11):e13454. doi:10.1111/jfbc.13454
251. Gong XW, Xu YJ, Yang QH, et al. Effect of soothing gan (liver) and invigorating Pi (spleen) recipes on TLR4-p38 MAPK pathway in kupffer cells of non-alcoholic steatohepatitis rats. Chin J Integr Med. 2019;25(3):216–224. doi:10.1007/s11655-018-2829-6
252. Zhang HA, Yang XY, Xiao YF. AMPKα1 overexpression alleviates the hepatocyte model of nonalcoholic fatty liver disease via inactivating p38MAPK pathway. Biochem Biophys Res Commun. 2016;474(2):364–370. doi:10.1016/j.bbrc.2016.04.111
253. Böhm T, Berger H, Nejabat M, et al. Food-derived peroxidized fatty acids may trigger hepatic inflammation: a novel hypothesis to explain steatohepatitis. J Hepatol. 2013;59(3):563–570. doi:10.1016/j.jhep.2013.04.025
254. Luo Y, Tian G, Zhuang Z, et al. Berberine prevents non-alcoholic steatohepatitis-derived hepatocellular carcinoma by inhibiting inflammation and angiogenesis in mice. Am J Transl Res. 2019;11(5):2668–2682.
255. Ceccarelli S, Panera N, Mina M, et al. LPS-induced TNF-α factor mediates pro-inflammatory and pro-fibrogenic pattern in non-alcoholic fatty liver disease. Oncotarget. 2015;6(39):41434–41452. doi:10.18632/oncotarget.5163
256. Zhao ZB, Ji K, Shen XY, et al. Di(2-ethylhexyl) phthalate promotes hepatic fibrosis by regulation of oxidative stress and inflammation responses in rats. Environ Toxicol Pharmacol. 2019;68:109–119. doi:10.1016/j.etap.2019.03.008
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
The Effects and Pathogenesis of PM2.5 and Its Components on Chronic Obstructive Pulmonary Disease

Wang Q, Liu S
International Journal of Chronic Obstructive Pulmonary Disease 2023, 18:493-506
Published Date: 6 April 2023
The Association Between Sarcopenia and Diabetes: From Pathophysiology Mechanism to Therapeutic Strategy
Chen H, Huang X, Dong M, Wen S, Zhou L, Yuan X
Diabetes, Metabolic Syndrome and Obesity 2023, 16:1541-1554
Published Date: 30 May 2023
Therapeutic Approaches for Nonalcoholic Fatty Liver Disease: Established Targets and Drugs
Huang X, Chen H, Wen S, Dong M, Zhou L, Yuan X
Diabetes, Metabolic Syndrome and Obesity 2023, 16:1809-1819
Published Date: 21 June 2023
Quercetin: A Flavonoid with Potential for Treating Acute Lung Injury

Huang M, Liu X, Ren Y, Huang Q, Shi Y, Yuan P, Chen M
Drug Design, Development and Therapy 2024, 18:5709-5728
Published Date: 6 December 2024
Exercise Prescription Training in Chronic Obstructive Pulmonary Disease: Benefits and Mechanisms
Liu S, Yang A, Yu Y, Xu B, Yu G, Wang H
International Journal of Chronic Obstructive Pulmonary Disease 2025, 20:1071-1082
Published Date: 15 April 2025