Back to Journals » Drug Design, Development and Therapy » Volume 19
Current Perspectives on the NF-κB Signaling Axis as a Potential Pharmacological Target in Cardiorenal Syndrome
Authors Liu Q, Wang X ![]() , Cheng P, Yang T, Wang C, Zhou H
, Cheng P, Yang T, Wang C, Zhou H
Received 10 August 2025
Accepted for publication 11 December 2025
Published 23 December 2025 Volume 2025:19 Pages 11557—11583
DOI https://doi.org/10.2147/DDDT.S559816
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Anastasios Lymperopoulos
Qian Liu,1 Xinting Wang,1 Peipei Cheng,1 Tianshu Yang,2 Chen Wang,3,4 Hua Zhou1
1Institute of Cardiovascular Disease, Shuguang Hospital Affiliated to Shanghai University of Traditional Chinese Medicine, Shanghai, 201203, People’s Republic of China; 2Department of Cardiology, Shanghai Municipal Hospital of Traditional Chinese Medicine, Shanghai, 200071, People’s Republic of China; 3Institute of Nephrology, Shuguang Hospital Affiliated to Shanghai University of Traditional Chinese Medicine, Shanghai, 201203, People’s Republic of China; 4TCM Institute of Kidney Disease, Shanghai University of Traditional Chinese Medicine, Shanghai, 201203, People’s Republic of China
Correspondence: Hua Zhou, Email [email protected] Chen Wang, Email [email protected]
Abstract: Cardiorenal syndrome (CRS) is a disease involving two vital organs, the heart and the kidney, which has been increasingly recognized in recent years. The treatment of CRS is highly challenging due to its complex nature, rapid progression, poor prognosis, and high mortality rate. As a protein complex, nuclear factor kappa-B (NF-κB) regulates the transcription of target genes by entering the nucleus and affects cardiac and renal functions through its involvement in inflammatory reactions and oxidative stress. By evaluating established preclinical and clinical research on CRS to date, we explored the potential of NF-κB inhibition to exert unique cardiorenal protective effects as a novel treatment for CRS. In this review, we have synthesized recent advances in the structure and function of NF-κB within the cardiovascular and renal systems, and explored the mechanistic involvement of NF-κB in CRS. Innovatively, we have identified natural compounds that dually inhibit NF-κB activity in both cardiac and renal tissues, thereby conferring concurrent protection to both organs. Furthermore, we discuss the translational potential and clinical applicability of NF-κB-targeted pharmacology, which may provide critical insights for developing novel therapeutics against CRS.
Keywords: cardiorenal syndrome, NF-κB, natural compounds, inflammation, oxidative stress
Introduction
CRS refers to a clinical syndrome characterized by acute or chronic dysfunction in either the heart or the kidney, which may induce acute or chronic dysfunction in the other organ, involving lesions in both the heart and the kidney.1 More than one-fourth patients with heart failure (HF) suffer from chronic kidney disease (CKD).2 The incidence of HF in patients with CKD is 30.7%.3 With the global population aging, the incidence of HF and renal failure is increasing, leading to a high incidence rate of CRS. Current management of CRS largely depends on symptomatic and supportive treatments for individual cardiac or renal dysfunction (eg, diuretics, positive inotropic agents, and vasodilators), while lacking targeted drugs capable of directly blocking or reversing its core pathological processes. Furthermore, existing therapies often focus on a single organ—either the heart or the kidneys—and may adversely affect the other. For instance, the use of diuretics to reduce cardiac load or the administration of positive inotropic agents to enhance myocardial contractility may both lead to deterioration of renal function. Given the extremely poor prognosis and numerous limitations of clinical therapies, the search for effective therapeutic targets is urgently needed.4 According to the 2010 Acute Dialysis Quality Initiative (ADQI) consensus, CRS is divided into two major groups − cardiorenal and renocardiac syndromes − which are further classified into five subtypes.5 In the setting of acute/chronic HF as the primum movens leading to acute/chronic kidney injury in Type 1 (acute cardio-renal syndrome) and Type 2 (chronic cardio-renal syndrome), the focus is on the hemodynamic issues and neurohormonal system activation caused by HF. Due to the inability of the failing heart to pump blood effectively, arterial filling volume decreases while venous congestion increases. Reduced cardiac output (CO) and low blood pressure (BP) result in decreased renal blood flow and renal hypoperfusion. Elevated central venous pressure (CVP), high intra-abdominal pressure (IAP), and increased right atrial pressure (RAP) contribute to renal congestion and impaired intrarenal blood flow. On the other hand, activation of neurohormonal system, including renin-angiotensin-aldosterone system (RAAS), sympathetic nervous system (SNS), and increased secretion of vasoconstrictor mediators, leads to renal vasoconstriction and elevated renal blood flow resistance. All of them reduce glomerular filtration rate (GFR).6 In Type 3 (acute reno-cardiac syndrome) and Type 4 (chronic reno-cardiac syndrome), neurohumoral activation caused by RAAS and renal vasoconstriction caused by SNS are the main mechanisms resulting in HF. The activation of RAAS results in increased proximal tubular sodium and water reabsorption. Water-sodium retention increases cardiac preload and worsens cardiac pumping function. Epinephrine and norepinephrine released from SNS activation drive preglomerular vasoconstriction, which increases cardiac afterload and promotes left ventricular hypertrophy (LVH). All of these factors lead to cardiac systolic and diastolic dysfunction.7 Type 5 (secondary cardio-renal syndrome) is characterized by systemic diseases, such as sepsis, septic shock, and diabetes, that lead to simultaneous cardiac and renal dysfunction. Although CRS is classified into five subtypes, we propose that inflammation and oxidative stress represent the common underlying pathophysiological mechanism across all subtypes (Figure 1). These processes play a crucial role in causing tissue damage in both cardiac and renal tissues, leading to cell apoptosis, necrosis, and fibrosis.8,9 Recent studies have revealed that NF-κB acts as a molecular bridge connecting cardiac and renal injury, functioning as a convergence point in cardiorenal crosstalk and thereby playing a pivotal role in CRS. It is involved in various pathophysiological aspects, including myocardial and renal tissue oxidative stress,10,11 myocardial and renal tissue inflammatory damages,12,13 and myocardial and renal fibrosis.14,15 Specifically, NF-κB expression is upregulated in both animal models and patients with CRS. Inhibiting NF-κB alleviates the condition by reducing inflammatory responses, oxidative stress, and damage to cardiomyocytes and renal cells, thereby addressing a key limitation of current therapies: their inability to provide simultaneous protection for both organs.16 In summary, this review will provide a comprehensive overview of the structure of NF-κB, review its research history in CRS, elucidate its mechanisms of action, and explore the impact or potential of natural compound-derived NF-κB inhibitors on cardiorenal diseases. This review aims to establish a multidimensional association among “natural compounds, NF-κB targets, and cardiorenal protective effects”, thereby providing a reference for preclinical drug development.
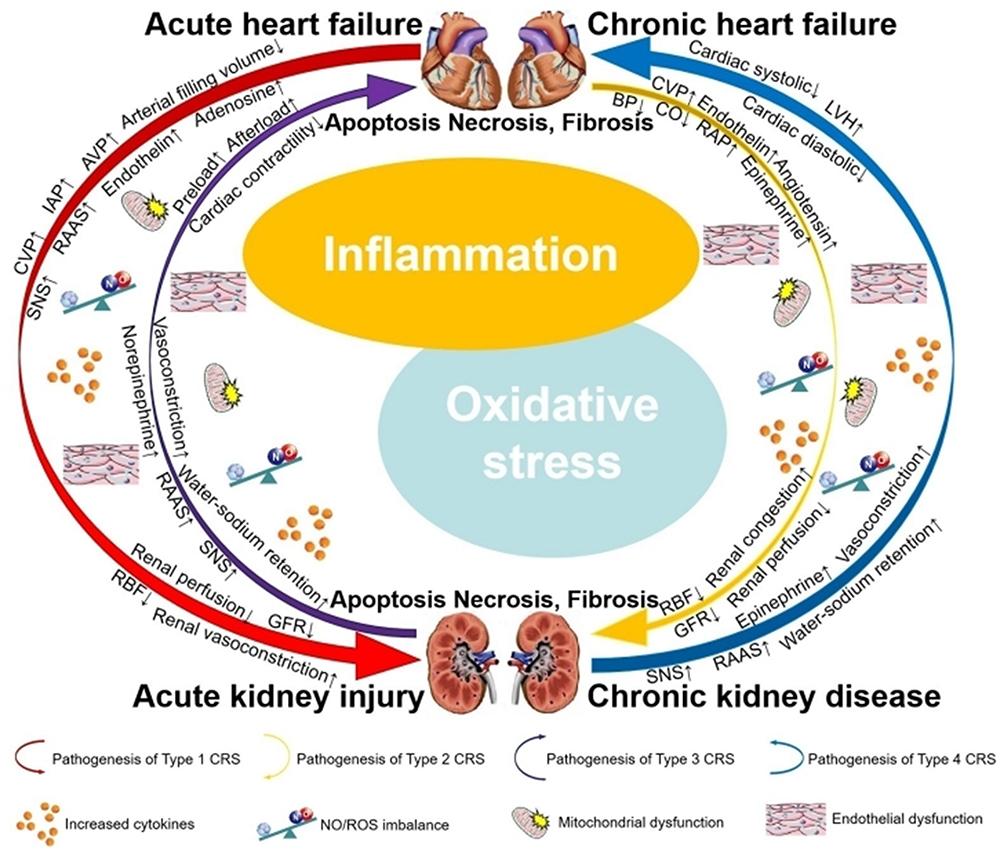 |
Figure 1 Pathophysiologic interaction in CRS. Notes: ↑ indicates promotion or upregulation; ↓ indicates inhibition or downregulation. |
The Basic Region of NF-κB
The N-terminus of NF-κB contains a Rel Homology Domain (RHD), which consists of an approximately 300 amino acid N-terminal domain (NTD) and a C-terminal domain (CTD).17 The RHD includes dimerization domains (DD), DNA-binding domains, nuclear localization sequence (NLS), and binding sites for the inhibitor of NF-κB (IκB).18 The NLS is responsible for transferring NF-κB from the cytoplasm to the nucleus, thereby regulating gene expression and participating in various biological processes. According to the different sequences C-terminal to the RHD, NF-κB proteins are divided into two classes. The first class is NF-κB proteins such as NF-κB1 (p50/p105) and NF-κB2 (p52/p100). Both of them contain long CTD called inhibitory ankyrin repeat domains (ARD), which act to inhibit multiple copies of ankyrin repeats.19 Generally speaking, nuclear import is inhibited through the masking of the NLS by the C-terminal ARD. Therefore, p100 and p105 are considered as cytoplasmic proteins. As the processed products of p100 and p105, p52 and p50 are liberated through the proteolytic cleavage of the C-terminal ARD of p100 and p105, respectively, enabling them to enter the nucleus.20 The second class is Rel proteins such as c-Rel, RelA/p65 and RelB. Their C-terminal transcriptional activation domains (TAD) can activate transcription in many species. NF-κB transcription factors typically exist in the form of homodimers or heterodimers. NF-κB1, formed by the proteolyzed p105 precursor (p50), can bind to RelA/p65, RelB, and c-Rel separately, resulting in the formation of p50-RelA, p50-RelB, and p50/c-Rel heterodimers.21 The p100 precursor (p52) initially binds to RelB and subsequently undergoes processing to form the mature NF-κB2-RelB (Figure 2). As a pivotal regulator linking inflammation to CRS and simultaneously serving as a convergence point for CRS and oxidative stress, elucidating the structure of NF-κB is of paramount importance for mechanistic insights.
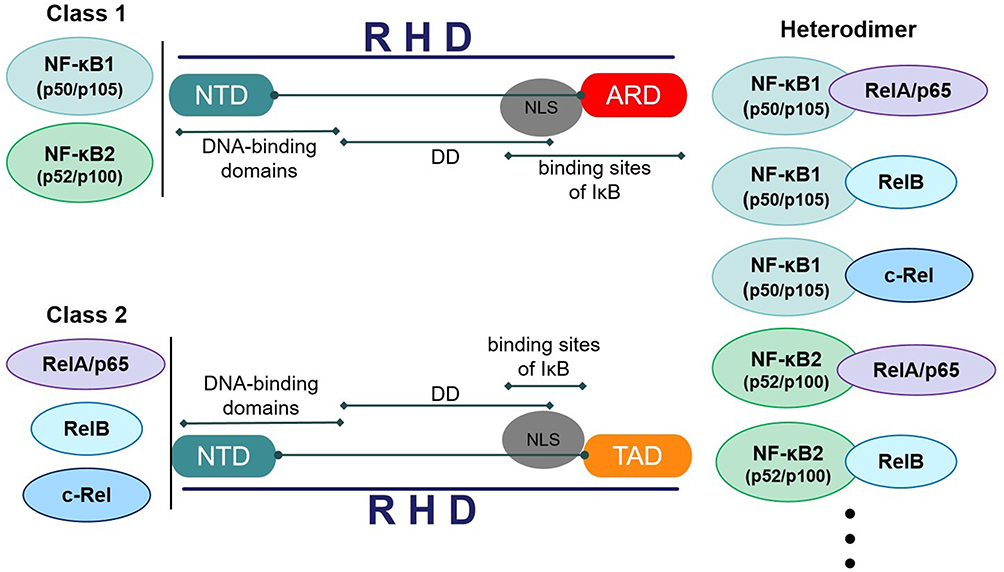 |
Figure 2 Structure and classification of NF-κB. |
The Mechanisms of NF-κB Activation
The NF-κB signaling pathway consists of receptors, receptor-proximal signaling adaptor proteins, IKK complex, IκB proteins, and NF-κB dimers. When cells are subjected to various intracellular and extracellular stimuli, IKK is activated, leading to the phosphorylation and ubiquitination of IκB, which is followed by the degradation of IκB and the release of NF-κB dimers. NF-κB dimers are further activated through various post-translational modifications and then translocated to the nucleus. In the nucleus, they bind to target genes to promote their transcription.22 The signaling pathways leading to NF-κB activation are classified into two types: the canonical and the non-canonical pathways (Figure 3). The canonical pathway, also called the classical pathway, activates the p50/p65 NF-κB dimer. In the stationary state, the p50/p65 heterodimer in the cytoplasm binds to IκB to form a trimer, which inhibits NF-κB activity. The activation of the canonical pathway begins with the initiation of adaptor protein and signal kinase reactions, which leads to the activation of the IKK complex (IKKα, IKKβ, and IKKγ). The activated IKK complex then phosphorylates IκB on the serine residues S32 and S36. With IκB ubiquitination and proteasomal degradation, the p50/p65 heterodimer is released into the nucleus, where it binds to target genes and induces gene expression.23 The non-canonical pathway, also called the alternative pathway, activates the RelB/p52 NF-κB dimer. In this pathway, IKKα and NF-κB-inducing kinase (NIK) are essential. IKKα can be activated by the upstream kinase NIK. IKKα on the serine residues S866 and S870 triggers p100 phosphorylation, ubiquitination and processing. p100 serves as both a precursor to p52 and an IκB-like molecule, specifically inhibiting the nuclear translocation of RelB. As the primary product of NF-κB2, p100 precursor lacks active processing. The processing of p100 induces the nuclear translocation of the RelB/p52 heterodimer while producing p52.24 Based on the two classic pathways mentioned above, we propose that future research could integrate single-cell sequencing with NF-κB signaling pathways to construct a dynamic model of NF-κB signal intensity during CRS progression. This approach would reveal spatiotemporal heterogeneity in NF-κB activation across distinct cardiac and renal cell subpopulations (such as cardiomyocytes versus renal interstitial fibroblasts), which holds critical importance for precise therapeutic targeting in CRS.
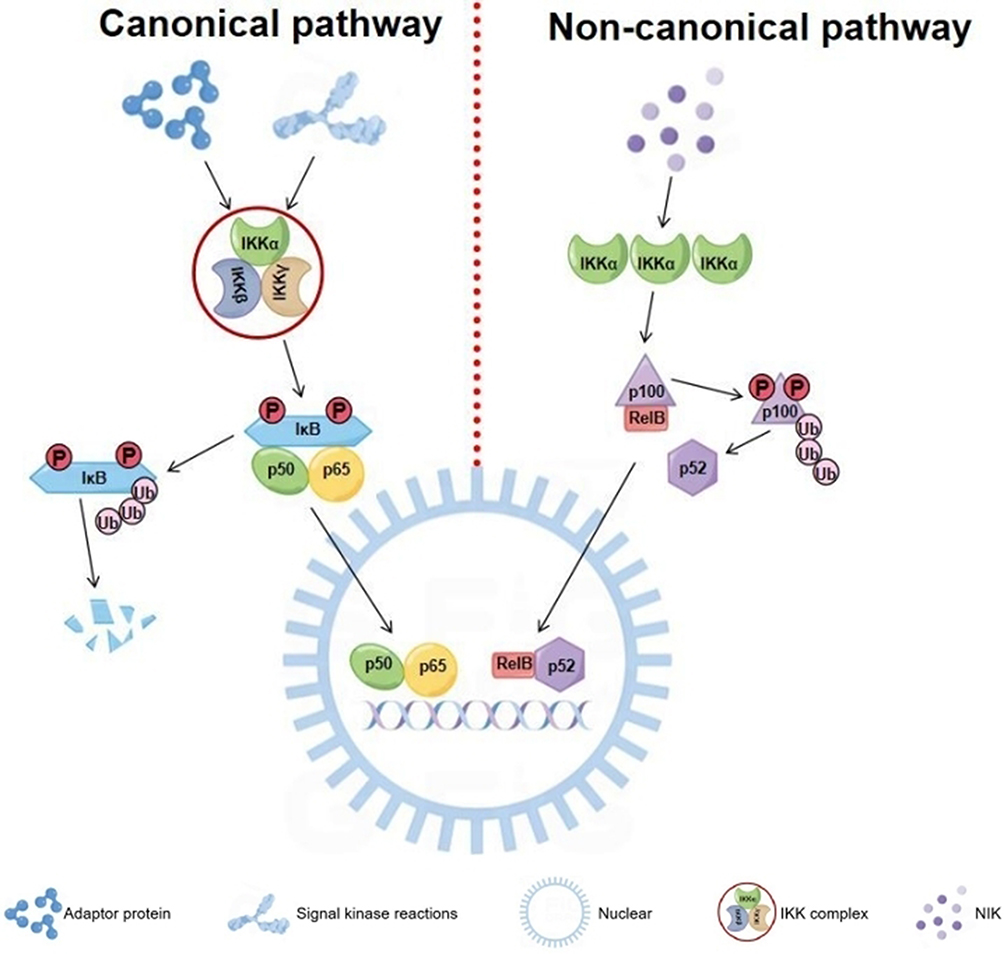 |
Figure 3 Canonical and non-canonical pathways of NF-κB activation. |
The Development of NF-κB
The research field of NF-κB has always been a hot topic. Most critical discoveries have occured over the past 35 years, with intensive research uncovering many functions of NF-κB (Figure 4). As early as 1986, NF-κB was discovered by Ranjan Sen in the laboratory of Nobel laureate David Baltimore through its interaction with the 11 nucleotides (GGGGACTTTCC) located in the immunoglobulin κ light chain gene enhancer in B cells.25 Two years later, NF-κB was found in the cytoplasm of other cells, where its DNA-binding activity was masked by IκB.26 From the 1990s, a large number of studies have focused on the structure and function of NF-κB. Many researchers have gradually discovered the structural domains of NF-κB1, NF-κB2, RelA, RelB, and c-Rel, which encode the entire family of NF-κB transcription factors.27–30 These proteins combine in different pairs to produce various functioning NF-κB dimers.31–33 From 1995 to 1998, the first nfkb1-/- mice, c-rel-/- mice, relb-/- mice, rela-/- mice and nfkb2-/- mice were gradually generated.34–38 Afterwards, multiple mutations of gene knockout mice, such as nfkb1-/- nfkb2-/- mice, rela-/- c-rel-/- mice, nfkb1-/- c-rel-/- mice, and so on, were generated.39–41 The emergence of gene knockout mice has provided a basis for studying the physiological roles of NF-κB signaling components. Soon after the discovery of IκB as an inhibitory protein for NF-κB, how to release NF-κB from its inhibitory IκB partner became an object of intense research. IκBα was purified in 1989.42 The following year, IκBβ was purified, and it was demonstrated that IκB phosphorylation could release NF-κB.43 In 1992, B-cell lymphoma 3 (Bcl-3) was considered as an inhibitor of NF-κB.44,45 The IKK complex, which mediated IκB phosphorylation, was first proposed in 1997.46,47 In the same year, IκBε was discovered.48 After the turn of the millennium, IκBζ/MAIL, IκBNS, and IκBη were discovered as members of mediating NF-κB-dependent regulation.49–51 The canonical pathway was proposed as one of the activation ways of NF-κB in 1999, predominantly targeting the activation of the IKKβ complex.52 Immediately afterwards, the non-canonical pathway, as another important arm of NF-κB signaling, was discovered. This pathway depends on the activation of the p52/RelB NF-κB complex.53
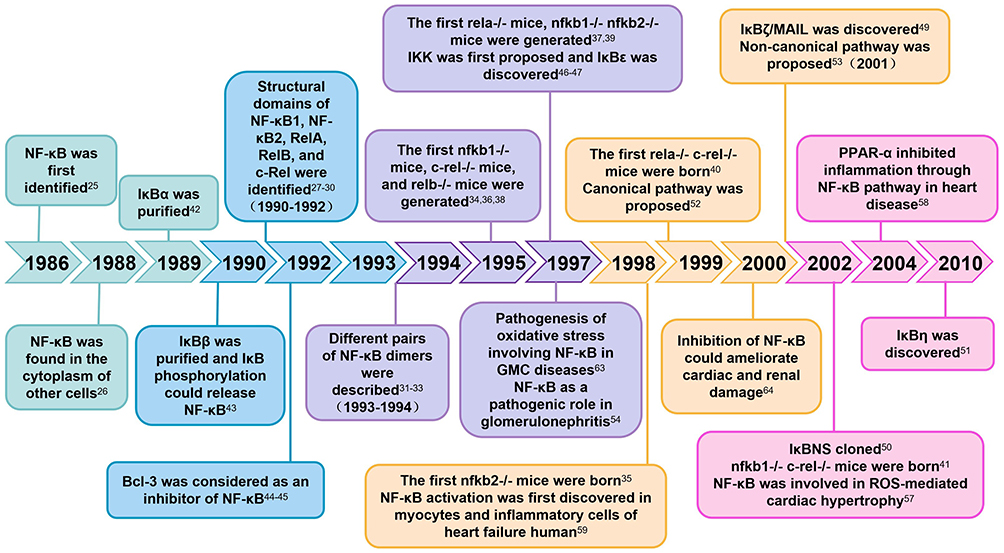 |
Figure 4 Milestones in NF-κB research. |
After clarifying the structure and function of NF-κB, researchers conducted extensive research on its role in disease. Activated NF-κB plays an important pathogenic role in glomerulonephritis by inducing pro-inflammatory factors such as cytokines and cell adhesion molecules.54 Mice with tumor necrosis factor-alpha (TNF-α) receptors TNFR1 or TNFR2 knocked out exhibit reduced renal fibrosis through the inhibition of NF-κB activation.55 In 2001, investigators demonstrated that NF-κB plays a critical role in cardiac hypertrophy. They showed that NF-κB activity was stimulated by hypertrophic agonists in an IKK-dependent manner, and overexpression of NF-κB induced cardiomyocyte hypertrophy.56 Next year, Japanese researchers first reported that apoptosis signal-regulating kinase 1 (ASK1) activated NF-κB, which was involved in reactive oxygen species (ROS)-mediated cardiac hypertrophy.57 Subsequently, investigator clarified that peroxisome proliferator-activated receptor-alpha (PPAR-α) inhibited inflammation through the NF-κB pathway, thereby attenuating myocardial fibrosis in hypertensive heart disease.58 The investigators discovered that the induction of cyclooxygenase (COX) was activated by NF-κB in human HF. They first demonstrated abundant expression of COX-2 and activation of NF-κB in human myocytes and inflammatory cells.59 In 1997, Medzhitov et al first described that a constitutively active mutant of human Toll could induce the activation of NF-κB and the expression of NF-κB-controlled genes.60 Two years later, Chow et al determined that toll-like receptor 4 (TLR4) was involved in lipopolysaccharide (LPS)-induced activation of the NF-κB pathway.61 Subsequently, researchers have proposed that NF-κB, as a downstream transcription factor of TLR4, could be activated by LPS and mediate myocardial ischemia/reperfusion (I/R) injury.62 LPS also induces the expression of vascular cellular adhesion molecule-1 (VCAM-1) on the surface of glomerular mesangial cells (GMCs). The VCAM-1 promoter forms a redox sensitive transcriptional link with oligonucleotides in the NF-κB binding region. Oxidative stress involving NF-κB is an important regulatory signal in the pathogenesis of GMC diseases.63 Until 2000, Dominik et al first reported that inhibition of NF-κB could ameliorate cardiac and renal damage in the double-transgenic rats (dTGR) model, which is suitable for studying the effects of angiotensin II (Ang II) on both cardiac and renal damage.64 Research on NF-κB began quite early, but it was not until after the millennium that extensive research was conducted on the role of NF-κB in the development of CRS.
NF-κB Mediated Signaling Axis in CRS
ASK1/MAPKs/NF-κB
Reactive oxygen species (ROS) have emerged as important intracellular signaling molecules in the oxidative stress response within CRS. ASK1, an ROS-sensitive mitogen-activated kinase, belongs to the MAP3K family. ASK1 is commonly held in an inactive state by binding to thioredoxin (Trx), an antioxidant protein. When CRS occurs, it causes oxidative stress, leading to the liberation of ASK1 from Trx.65 The autophosphorylation of ASK1 triggers a series of events. ASK1 is upstream of extracellular signal-regulated kinases1/2 (ERK1/2), c-Jun N-terminal kinase (JNK), and p38 mitogen activated protein kinase (p38 MAPK), and activates them by phosphorylating their intermediate kinases such as MKK4/7 and MKK3/6.66–68 ROS activate the downstream ASK1 signaling pathway, mediating cardiomyocyte apoptosis, fibrosis, and hypertrophy in myocardial infarction (MI) and HF, as well as mediating kidney cell apoptosis, oxidative stress, and immune activity in renal injury.69,70 As a family of signal transduction proteins and important mediators of inflammation-induced tissue injury, MAPKs convert extracellular signals to the activation of intracellular pathways through a three-tiered cascade in the pathogenesis of cardiorenal-related diseases.71 The phosphorylation of MAPK, which leads to NF-κB activation, has been shown to increase pro-inflammatory cytokine activation in CRS. On the contrary, inhibition of the MAPK/NF-κB pathway could alleviate inflammatory injury in both the heart and kidney.16 The activated ASK1/MAPKs (p38MAPK, ERK1/2)/NF-κB pathway upregulates pro-fibrotic genes, such as transforming growth factor-β1 (TGF-β1) and connective tissue growth factor (CTGF), leading to cardiac fibroblast collagen synthesis and renal cell collagen synthesis in the remodeling of CRS.72 Inhibition of the ASK1/MAPKs/NF-κB pathway could decrease the expression of α-smooth muscle actin (α-SMA), collagen type I (COL-I), brain natriuretic peptide (BNP), serum creatinine (Scr), and blood urea nitrogen (BUN) levels, while also improving myocardial systolic function in rats with CRS.73 ASK1/MAPKs/NF-κB signal pathway in CRS is shown in Figure 5. Based on the aforementioned signaling pathways, we propose a spatiotemporally sequential therapeutic strategy for CRS: acute phase (targeting ASK1 for CRS types 1 and 2), subacute phase (inhibiting p38/JNK for CRS type 5), and chronic phase (blocking NF-κB nuclear translocation for CRS types 2 and 4).
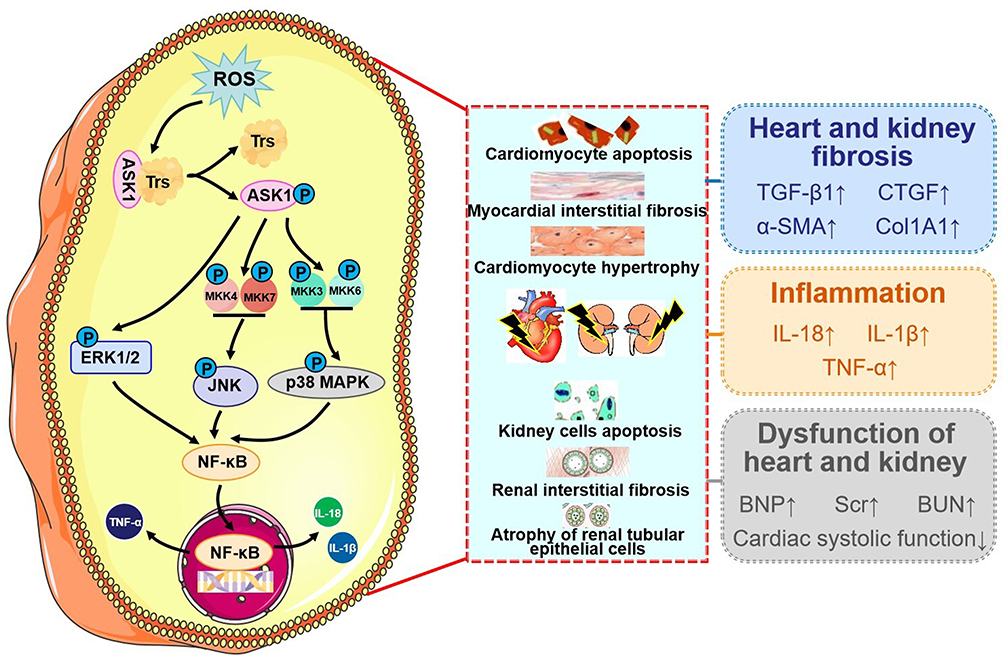 |
Figure 5 Mechanism of NF-κB activation induced by oxidative stress via the ASK1-MAPK axis in CRS. ROS activation triggers oxidative stress and releases ASK1 from the ASK1-Trx complex. The autophosphorylation of ASK1 activates ERK1/2, JNK, and p38 MAPK, contributing to NF-κB translocation from the cytoplasm to the nucleus. This promotes fibrosis and inflammation in the heart and kidney, ultimately leading to the deterioration of cardiac and renal function. Notes: ↑ indicates upregulation; ↓ indicates downregulation. |
PI3K/AKT/NF-κB
After CRS occurs, inflammation and oxidative stress are initiated, leading to increased levels of inflammatory factors and ROS in the body. These changes activate the activity of receptor tyrosine kinases (RTKs) and G protein-coupled receptors (GPCRs).74–76 As transmembrane proteins, RTKs and GPCRs transfer extracellular information to the cell interior. Phosphatidylinositol 3-kinase (PI3K) consists of a p110 (catalytic domain) and a p85 (regulatory domain), and can be activated by both RTKs and GPCRs. PI3K catalyzes the phosphorylation of its substrate, phosphatidylinositol-4,5-bisphosphate (PIP2), to produce the second messenger phosphatidylinositol-3,4,5-trisphosphate (PIP3).77 PIP3 activates 3-phosphinositide-dependent protein kinase 1(PDK1), which phosphorylates AKT at the Thr308 site. AKT promotes the translocation of NF-κB from the cytoplasm to the nucleus through the canonical pathway. NF-κB induces the expression of interleukin-6 (IL-6), an important inflammatory mediator involved in the remodeling of the heart and kidney. In tubular epithelial cells and cardiac fibroblasts, IL-6 trans-signaling plays a key role in the development of renal and cardiac fibrosis.78,79 The activated PI3K/AKT/NF-κB pathway upregulates the expression of TGF-β1, COL-I, and β-MyHC at both the transcriptional and protein levels, leading to cardiac fibrosis and remodeling. The inflammatory genes, such as TNF, interleukin-1 beta (IL-1β), and interleukin-6 (IL-6), were also increased in ventricular myocytes.80 The activities of antioxidant stress factors in the myocardium, such as superoxide dismutase (SOD), NAD(P)H quinone dehydrogenase-1 (NQO1), and heme oxygenase-1 (HO-1), were decreased.81 The activation of the PI3K/AKT/NF-κB pathway also causes kidney injury. This pathway is involved in the inflammatory response and fibroblast proliferation, which leads to an imbalance between the degradation and synthesis of the extracellular matrix (ECM). The specific mechanism includes increasing the expression of IL-18, IL-1β, TGF-β1, fibroblast growth factor 2 (FGF2), alpha-smooth muscle actin (α-SMA), COL-I, COL-IV and fibronectin.82–84 In CRS rats, inhibition of the PI3K/AKT/NF-κB signaling pathway improved cardiac remodeling by increasing ejection fraction (EF), fractional shortening (FS), left ventricular posterior wall dimension in systole and diastole, and interventricular septal thickness in systole and diastole, as well as by reducing left ventricular internal diameter at end-systole and end-diastole (LVIDs, LVIDd). In addition, intervention in the PI3K/AKT/NF-κB pathway can significantly alleviate cardiac and renal damage by reducing serum levels of creatine kinase-MB (CK-MB), lactate dehydrogenase (LDH), Scr, BUN, uric acid (UA), and aspartate aminotransferase (AST). In rats with CRS, the levels of TNF-α and IL-6 were increased in the serum, heart, and kidney.85 PI3K/AKT/NF-κB signal pathway in CRS is shown in Figure 6. We propose that the PI3K/AKT/NF-κB axis functions as the “inflammation igniter” in acute CRS (Types 1 and 3) and, equally, as the “fibrosis switch” in chronic CRS (Types 2 and 4), thereby constituting a unified biological framework spanning all five CRS subtypes.
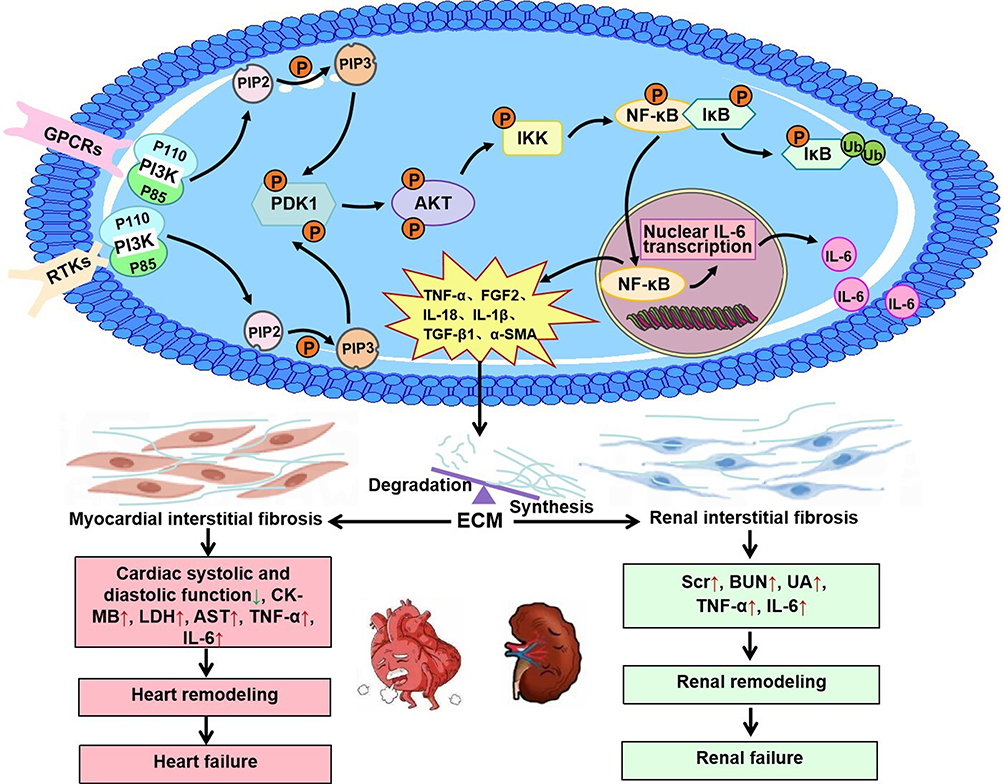 |
Figure 6 Mechanism of NF-κB-mediated fibrosis activated by the PI3K/Akt pathway in CRS. Through the activation of PI3K, RTKs and GPCRs relay extracellular signals to the cell interior. PI3K catalyzes its substrate PIP2 into PIP3. Subsequently, PIP3 facilitates the phosphorylation of AKT through the activation of PDK1. AKT translocates NF-κB from the cytoplasm into the nucleus via the canonical pathway. This process contributes to myocardial and renal interstitial fibrosis and remodeling, ultimately leading to heart and renal failure. Notes: ↑ indicates upregulation; ↓ indicates downregulation. |
TLR4/NF-κB
An increasing number of studies have indicated that LPS plays an important role in the pathogenesis of CRS.86,87 In patients with CRS, elevated central venous pressure and systemic congestion lead to the translocation of Gram-negative bacteria throughout the intestinal villi. These bacteria release large amounts of LPS into the bloodstream, ultimately leading to the activation of oxidative stress and inflammatory pathways.88,89 As the main LPS receptor, TLR4 is expressed on the surface of both myocardial cells and renal tubular epithelial cells. TLR4 contains a Toll/IL-1 receptor (TIR) domain that is responsible for signal transduction.90 The C-terminal portion of myeloid differentiation primary response gene 88 (MyD88) possesses a TIR domain, which can be recruited by TLR4.91 The N-terminal portion of MyD88 is a death domain (DD), which recruits IL-1 receptor-associated kinase 4 (IRAK4). Subsequently, IRAK1/2, as substrates of IRAK4, are phosphorylated.92 Activated IRAK1/2 associate with TNF receptor-associated factor 6 (TRAF6), leading to the activation of the canonical NF-κB signaling pathway.93 The activation of the transcription factor NF-κB induces its translocation into the nucleus, where it promotes the transcription of hypoxia-inducible factor-1α (HIF-1α).94 Under hypoxic conditions, the accumulation of HIF-1α requires activation mediated by NF-κB. Vascular endothelial growth factor (VEGF), as a target gene of HIF-1α, stimulates endothelial cell proliferation, increases vascular permeability, and leads to endothelial dysfunction.95 Endothelial dysfunction induces alterations in glomerular vascular permeability and damages the coronary artery and intramyocardial microvascular architecture in CRS.96 Inhibition of the TLR4/NF-κB/HIF-1α pathway reduces levels of NT-proBNP, Scr, BUN, and 24-hour quantitative urine protein, while improving cardiac function indicators such as EF, FS, and other indicators of cardiac ejection function. Additionally, this inhibition shrinks the heart by decreasing LVIDs and LVIDd. Renal tissue staining showed that activation of the TLR4/NF-κB/HIF-1α pathway caused glomerular enlargement, tubular swelling, basement membrane thickening, and fibrous deposits in the renal interstitium. Additionally, inflammation-related proteins such as monocyte chemoattractant protein-1 (MCP-1), intracellular adhesion molecule-1 (ICAM-1), IL-1β, and IL-6, as well as fibrosis-related genes such as TGF-β, α-SMA, fibronectin, Smad2, Smad3, and E-cadherin, were all significantly increased in CRS rats.97–99
The nucleotide-binding domain, leucine-rich repeat, and pyrin domain (PYD)-containing protein 3 (NLRP3) inflammasome induces the maturation of pro-inflammatory cytokines in cardiac and renal cells. The activation of NLRP3 occurs in two steps: the initiation of TLR4, followed by the triggering of NF-κB-induced transcription of NLRP3.100 NLRP3 transforms from an inactive oligomer to an active multimeric inflammasome. A spatial association and feedback loop exist between NLRP3 and mitochondria. As a mitochondrial-specific phospholipid, cardiolipin serves as the docking site for NLRP3. When the NLRP3 inflammasome signals from the outer mitochondrial membrane, cardiolipin translocates to the outer membrane and recruits NLRP3.101,102 The activation of NLRP3 induces the fragmented release of mitochondrial DNA (mtDNA) and increases ROS generation, converting mtDNA into its oxidized form (ox-mtDNA), which serves as the ultimate ligand for NLRP3 and leads to mitochondrial damage.103 Excessive production of ROS leads to the development of intracellular responses associated with CRS, such as apoptosis, pyroptosis, and cell migration.104 In CRS, dysfunction of the heart and kidney can be caused by the activation of the TLR4/NF-κB/NLRP3 signaling pathway, which triggers inflammatory responses and mitochondrial damage in both organs. As the main innate immune response in cardiorenal associations, the TLR4/NF-κB/NLRP3 pathway produces cytokines and chemokines and recruits leukocytes to myocardial and renal tissues.105 Inhibition of the TLR4/NF-κB/NLRP3 pathway alleviates renal and cardiac collagen deposition, fibrosis, inflammation, oxidative stress, apoptosis, and pyroptosis, ultimately reducing heart and kidney damage.106–108 TLR4/NF-κB signal pathway in CRS is shown in Figure 7. We propose that the TLR4/NF-κB axis serves as the common pathway by which damage-associated molecular patterns (DAMPs) ignite the cardiorenal inflammatory storm in CRS, and as the central hub where inflammation-driven energy imbalance is exacerbated. We recommend that future CRS therapy shift from a “single-track anti-inflammatory” approach to a dual-track strategy of “inflammation blockade coupled with metabolic reprogramming”.
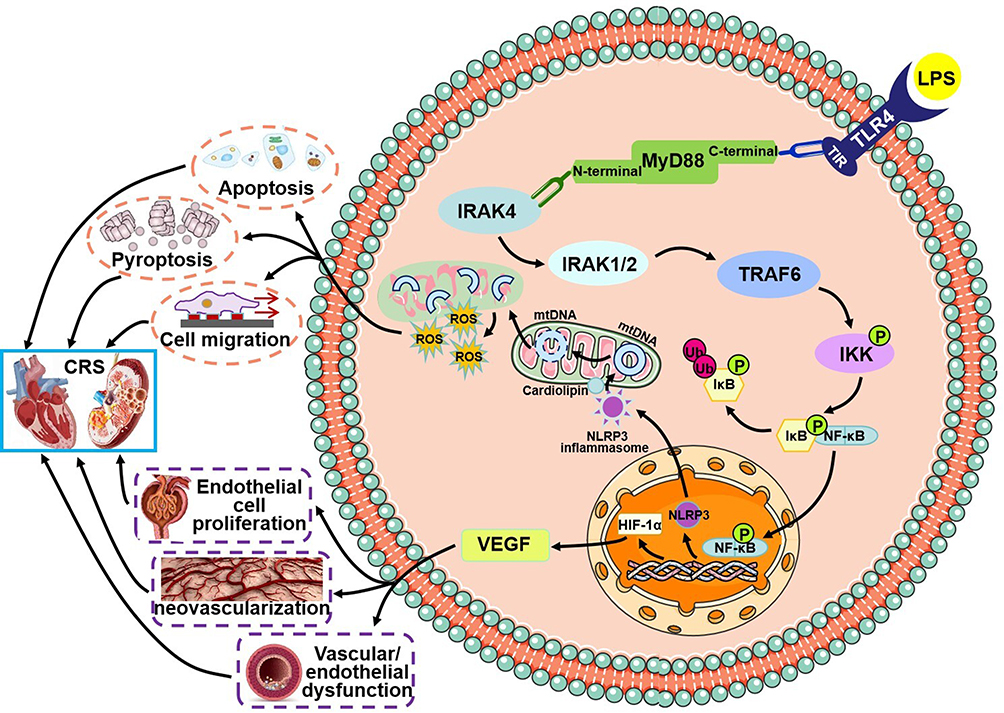 |
Figure 7 Mechanism of NF-κB inflammatory signaling initiation following TLR4 ligand binding in CRS. LPS binds to its receptor TLR4, which recruits MyD88. MyD88, in turn, activates downstream signaling molecules, including IRAK4, IRAK1/2, TRAF6, and the canonical NF-κB signaling pathway. In the TLR4/NF-κB/HIF-1α pathway, NF-κB translocates into the nucleus and promotes the transcription of HIF-1α. HIF-1α then activates its target gene VEGF, which contributes to endothelial dysfunction. In the TLR4/NF-κB/NLRP3 pathway, the transcription of NLRP3 is induced by NF-κB translocating into the nucleus. The activation of NLRP3 induces the fragmented release of mtDNA and increases ROS generation. The activation of these two pathways ultimately leads to damage in both the heart and kidney. |
NF-κB Inhibitors Among Natural Compounds
Terpenoid Compounds
According to their structure, terpenoids can be classified into monoterpenes, sesquiterpenes, diterpenes, triterpenes, and other terpenoid derivatives. Researchers have found that terpenoid compounds can inhibit NF-κB activity, regulate its downstream anti-inflammatory and antioxidant effects, and protect heart and kidney function. Ginsenoside Rg1, an extract of ginseng, attenuates LPS-induced inflammation, oxidative stress, and apoptosis in the heart and kidney by blocking the NF-κB pathway.109,110 Sweroside, a natural iridoid extracted from Swertia pseudochinensis Hara, reduces ROS generation in cardiomyocytes and renal tubular epithelial cells by inhibiting NF-κB p65 nuclear translocation. It demonstrates potent anti-inflammatory and antioxidant activity in cardiac and renal diseases.111,112 As a sesquiterpene alcohol, α-Bisabolol possesses high lipophilicity. It can be used to prevent doxorubicin-induced cardiotoxicity and nephrotoxicity by inhibiting the NF-κB pathway cascade reaction, reducing lipid peroxidation products, and maintaining membrane-stabilizing properties.113,114 Tanshinone IIA, a diterpene quinone and one of the major active components isolated from Danshen, has been shown to inhibit MCP-1 overexpression, reduce macrophage infiltration, and attenuate cardiac and renal inflammatory status by inhibiting NF-κB translocation from the cytoplasm to the nucleus, both in vivo and in vitro.115,116 Oridonin, a diterpenoid natural product isolated from Rabdosia rubescens, inhibits p65 phosphorylation and reduces the levels of pro-inflammatory cytokines, ultimately alleviating myocardial tissue damage, glomerular damage, and tubulointerstitial injury.117,118 Glycyrrhizic acid, a triterpenoid acid and major active compound in licorice, protects against inflammatory responses and oxidative stress damage in the heart and kidney by reducing TNF-α, IL-1β, and IL-6 levels and inhibiting the phosphorylation of NF-κB p65.119,120 Andrographolide, a bicyclic diterpenoid lactone and novel NF-κB inhibitor, is isolated from the leaves of Andrographis paniculata. It impedes cardiac apoptosis, renal hypertrophy, and endothelial injury.121,122 Monotropein, an active iridoid glycoside isolated from Morinda officinalis roots, alleviates vascular endothelial cell and renal tubular cell injuries by inhibiting NF-κB signaling. This mechanism involves reducing oxidative stress and apoptosis, including decreasing malondialdehyde (MDA), BCL2-Associated X (Bax), and cleaved-caspase 3 levels, while increasing SOD and reduced glutathione (GSH) activities and Bcl-2 expression.123,124 Anethole, a terpenoid compound, inhibits the TLR4/MyD88/NF-κB pathway and alleviates inflammation, oxidative stress, lipid peroxidation, and apoptosis, thereby protecting against doxorubicin-induced cardiotoxicity and nephrotoxicity.125
Flavonoid Compounds
Flavonoids refer to a series of compounds consisting of two benzene rings connected to each other through three carbon atoms. They exhibit multiple biological and pharmacological activities, making them valuable in the treatment of heart and kidney diseases. Apigenin, a plant flavone, has been reported to ameliorate cardiac and renal damage through its antioxidant and anti-inflammatory properties by blocking the activation of the MAPK (ERK1/2, JNK, and p38)/NF-κB pathway.126,127 Quercetin, a naturally occurring flavonol, can achieve therapeutic effects through multiple NF-κB-related pathways. Specifically, it inhibits LPS binding to TLR4, blocks MyD88, TRAF6, and NF-κB, thereby alleviating cardiac and renal inflammatory cell infiltration and programmed cell death.128,129 Studies suggest that quercetin remarkably relieves renal toxicity and oxidative stress, promotes cardiomyocyte proliferation, and recovers mitochondrial function by regulating ROS-induced NF-κB activation.130,131 Artesunate, a flavonoid compound and a semi-synthetic derivative of Artemisia, has been shown to have anti-inflammatory and antioxidant functions in the prevention of nephritis and cardiovascular complications by suppressing TLR4/NF-κB pathway activity.132,133 Naringin, a natural flavonoid, plays important roles in repairing mitochondrial dysfunction in heart and kidney diseases. It restores mitochondrial function, increases the cardiac and renal endogenous antioxidant status, and decreases ROS production by inhibiting the activation of NF-κB.134,135 As a major flavonoid and an active ingredient found in Epimedium, Icariin exerts anti-oxidative stress effects in cardiovascular and renal diseases by inhibiting the TLR4/NF-κB pathway. Specifically, it reduces ROS and TGF-β1 levels, increases the expression of antioxidant elements such as SOD and catalase (CAT), thereby increasing the glomerular filtration rate, inhibiting myocardial hypertrophy, and improving myocardial and renal fibrosis.136–139 Puerarin, a known isoflavone extracted from kudzu root, relieves LPS-induced heart and kidney dysfunction by hindering TLR4 binding with MyD88. This results in downstream NF-κB inactivation, thereby reducing inflammation-related damage to the heart and kidney.140,141 In addition, puerarin has been found to alleviate interstitial edema and cell apoptosis in the heart and kidney by modulating the sirtuin 1 (SIRT1)/NF-κB signaling pathway.142,143 With estrogen-like activity, genistein is a nonsteroidal isoflavone that suppresses LPS-induced inflammation in heart and kidney arterial endothelium by inhibiting NF-κB-mediated injury.144 Genistein also improves mitochondrial function in the heart and kidney by inhibiting the expression of NF-κB.145,146 Hydroxysafflor yellow A, a primary water-soluble single-chalcone obtained from the flowers of Carthamus tinctorius L., alleviates inflammatory injuries in ventricular myocytes and renal tubular cells by suppressing the TLR4/NF-κB pathway.147,148 Baicalin, a flavonoid compound, is derived from Radix Scutellariae. It alleviates myocardial apoptosis and renal fibrosis by inhibiting the TLR4/NF-κB signaling pathway.149,150
Glycoside Compounds
A number of biologically active compounds are glycosides. Glycoside compounds regulate various upstream molecules of NF-κB, which affects NF-κB activation and reduces heart and kidney damage through anti-inflammatory and antioxidant effects. As a polysaccharide compound and the main bioactive component extracted from Astragalus membranaceus, astragalus polysaccharide reduces the phosphorylation of TLR4/NF-κB both in vitro and in vivo, thereby alleviating cardiac and renal pathological damage by reducing the expression of inflammatory cytokines IL-1β, IL-6, and MCP-1.151,152 Astragaloside IV, a bioactive saponin and one of the main active ingredients of Radix Astragali, possesses anti-inflammatory and antioxidant stress effects in both heart and kidney diseases.153 Numerous studies have shown that Astragaloside IV inhibits NF-κB-related pathways, including TLR4/NF-κB, ROS/NF-κB, and MAPK (ERK1/2, JNK, p38 MAPK)/NF-κB pathways, to protect GMCs and microvascular endothelial cells, as well as alleviate myocardial and renal interstitial fibrosis.154–159 Salidroside, a phenylpropanoid glycoside and an active component in Rhodiola rosea, decreases levels of ROS, MDA, and NF-κB p65 nuclear translocation. As a result, it increases SOD activity, reduces the release of inflammatory cytokines and levels of biomarkers associated with heart and kidney tissue injury (eg, CK-MB, LDH, Scr, BUN, and UA), ultimately improving heart and kidney dysfunction both in vivo and in vitro.160–162 Notoginsenoside R1, a phytoestrogen isolated from Panax notoginseng, targets estrogen receptors in the heart and kidney, thereby inhibiting the activation of NF-κB and the subsequent myocardial and renal inflammatory and apoptotic responses.163,164 Diosgenin, a natural steroid sapogenin extracted from fenugreek seeds, antagonizes the activation of the TLR4/NF-κB pathway in myocardial and renal tissue damage, thereby reducing the production of inflammatory cytokines.165,166 Mangiferin, a major xanthone glucoside in Rhizoma Anemarrhenae, inhibits ROS accumulation, NF-κB nuclear translocation, and the release of inflammatory cytokines in the heart and kidney.167,168 Polydatin, an active glucoside compound derived from Polygonum cuspidatum, markedly suppresses the activation of NF-κB-related inflammatory cascade in myocardial and kidney injuries, including decreasing the levels of TNF-α, IL-1β, IL-6, and MDA.169,170
Phenolic Compounds
Phenolic compounds, as natural metabolites widely present in plants, contain a large number of phenolic hydroxyl structural units. Multiple studies suggest that these compounds can improve myocardial and renal tissue damage by regulating NF-κB nuclear transcription and its related signaling pathways. Resveratrol, a non-flavonoid phenolic compound, confers protection against oxidative stress and inflammation in both the heart and kidney by intervening NF-κB activity.171 SIRT1 is a nicotinamide adenine dinucleotide (NAD+)-dependent histone deacetylase that regulates the transcriptional activity of NF-κB. Research has demonstrated that resveratrol, as a SIRT1 agonist, activates SIRT1-triggered NF-κB p65 deacetylation and reduces ROS content, thereby attenuating cardiac hypertrophy and protecting renal tubular epithelial cells.172,173 Meanwhile, resveratrol downregulates NAD(P)H oxidase in endothelial cells and mesangial cells by inhibiting NF-κB activity, thereby exerting a protective effect on the coronary artery endothelium and reducing glomerular mesangial cell proliferation.174,175 Sinapic acid is a natural phenolic compound derived from hydroxycinnamic acid. The treatment with sinapic acid augments antioxidant responses and ameliorates cardiac and renal dysfunction related to oxidative/nitrosative stress and apoptosis. The NF-κB and nuclear factor erythroid 2-related factor 2 (Nrf2)/HO-1 signaling pathways may be its primary targets.176,177 Paeonol, an active ingredient extracted from Moutan Cortex, is a natural phenol. The mechanisms of its protective effects on heart and kidney injuries are associated with its ability to attenuate inflammation and suppress the TLR4/NF-κB pathway.178,179 Curcumin, a lipophilic polyphenol and NF-κB inhibitor derived from the rhizomes of Curcuma, protects cardiac and renal tissues against oxidative stress, inflammation, and apoptosis by upregulating Nrf2 and Bcl-2, as well as downregulating NF-κB, IL-1β, Keap1, heme oxygenase 1 (HMOX1), and Bax.180 Curcumin also protects mice from myocarditis and nephritis by inhibiting the PI3K/Akt/NF-κB pathway.181,182 Gallic acid is a polyphenol and one of the strongest naturally occurring antioxidants, derived from Cornus officinalis. Treatment with gallic acid produces significant reversal of oxidative stress parameters, with suppression of the NF-κB/Kim-1 signaling pathway in cardiac and renal toxicity in rats.183 As a phenolic carboxylic acid extracted from Salvia miltiorrhiza, Salvianolic acid A inhibits the expression of p38 MAPK and its downstream NF-κB in renal proximal tubule epithelial cells and myocardium. This alleviates fibrosis and remodeling of the heart and kidney, ultimately attenuating the progression of HF and CKD.184,185
Alkaloid Compounds
Alkaloids are naturally occurring nitrogenous compounds found in plants that have been demonstrated to be effective against heart and kidney injuries. Berberine, a quaternary ammonium alkaloid isolated from Coptis chinensis, can stabilize cardiac hemodynamics, improve systolic and diastolic function of the heart, and attenuate renal cortex inflammation by inhibiting the activation of the TLR4/NF-κB signaling pathway.186,187 Sinomenine, a purified alkaloid from Sinomenium acutum, inhibits the activation of NF-κB, lowers the levels of TNF-α, and decreases LVAWd, LVPWd, Cr, and BUN. These actions demonstrate its efficacy in preventing myocardial cell hypertrophy and tubular cell apoptosis.188,189 Oxymatrine, a quinoline alkaloid extracted from Sophora flavescens, has been reported to attenuate myocardial and renal interstitial edema and fibrosis due to its inactivation of the NF-κB p65 pathway.190,191 Betaine, a major water-soluble component of Lycium chinensis, belongs to the alkaloids. It attenuates right ventricular hypertrophy and proximal tubule damage by decreasing NF-κB expression and increasing anti-inflammatory effects.192,193
Others
In addition to the types of compounds mentioned above, other kinds of compounds also exert both cardiac and renal protective effects. Crocin, one of the most bioactive components of saffron, is an apo-carotenoid that significantly increases cardiac and renal antioxidants such as SOD and GSH while reducing the oxidative load biomarker MDA. The TLR4/NF-κB signaling pathway may play a crucial role in this process.194,195 Lycopene, a non-provitamin phytochemical and another carotenoid component found in tomatoes, abolishes myocardial and renal inflammation by modulating the NF-κB/TNF-α signaling pathway.196,197 Emodin, the main effective monomer of rhubarb, belongs to anthraquinone derivatives. The cardiac and renal protective effects of emodin can be attributed to its suppression of pro-inflammatory cytokines such as TNF-α and IL-1β, which results from its inhibition of NF-κB activity.198,199 Allicin is an organic sulfur compound and the active component of garlic. Some studies have demonstrated that allicin protects cardiac and renal function, prevents cardiac hypertrophy, and reduces urinary protein excretion by inhibiting ROS-dependent MAPK/NF-κB signaling pathways.200–202 Sulforaphane, a member of the isothiocyanate family found in cruciferous vegetables, acts as an Nrf2 agonist. It successfully augments the protective effects of Nrf2 by inhibiting NF-κB activation, suppressing endothelial inflammation and vascular smooth muscle cell proliferation, and improving the antioxidant system in CKD patients by neutralizing ROS.203,204 Osthole, a bioactive coumarin compound extracted from Cnidium monnieri, has been shown to attenuate myocardial infarct size and focal segmental glomerulosclerosis by inhibiting NF-κB-mediated COX-2 expression.205,206
Currently, studies utilizing natural compound monomers to treat CRS in animal models are scarce, and research focusing on cross-talk between cardiac and renal cells in vitro is even more limited. Thus, identifying natural compound monomers capable of simultaneously targeting cardiorenal injury holds significant implications for informing future CRS investigations. All natural NF-κB inhibitors are listed in Table 1.
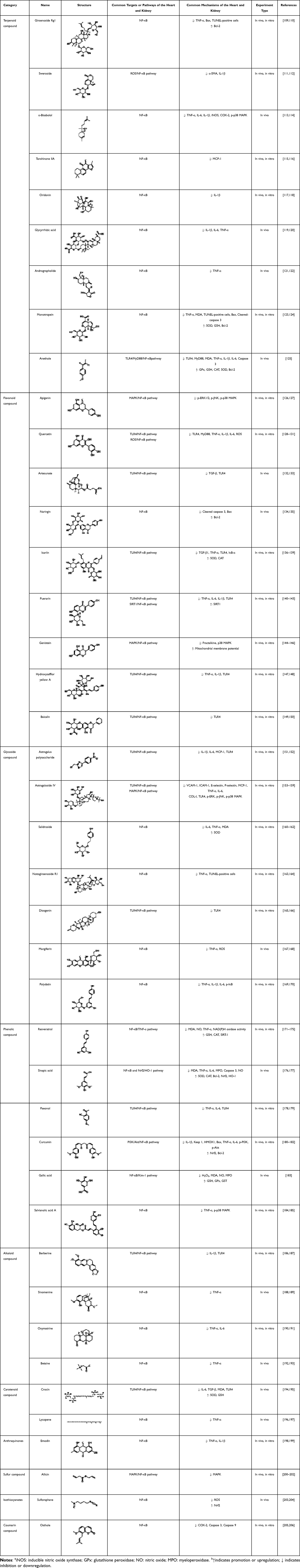 |
Table 1 Inhibitors of NF-κB Natural Compounds |
Limitation and Future Perspectives
Natural plant-derived compounds typically contain multiple active ingredients and exhibit a wide range of biological activities, such as anti-inflammatory, antioxidant, anti-apoptotic, and antifibrotic effects.207,208 They exert protective effects on the heart and kidney by inhibiting NF-κB and its mediated signaling pathways, providing candidate drugs for the treatment of CRS. However, certain limitations remain. ① Given that the majority of foundational research on NF-κB has been conducted in rats, mice, and cells, there is a scarcity of extensive clinical data to substantiate these findings. Basic research serves to inform clinical practice, and future clinical studies are imperative for validation. ② Currently, research on NF-κB inhibitors predominantly focuses on heart disease or kidney disease, with relatively limited exploration into their application for CRS. This scarcity of research renders the study of natural plant extracts for cardiorenal comorbid conditions somewhat constrained, but it also reveals a field with considerable potential for further exploration. This field of study is still in its infancy and necessitates further in-depth exploration to generate novel ideas and effective strategies for the treatment of CRS. ③ In the treatment of heart or kidney diseases, there is a significant lack of safety research on natural plant extracts aimed at systemically or locally inhibiting NF-κB activity. Therefore, it is crucial to conduct more research in this field to ensure safety on the basis of therapeutic efficacy and to establish a solid foundation for clinical applications. ④ CRS involves various modes of programmed cell death in cardiomyocytes and renal cells, including apoptosis, pyroptosis, autophagy, ferroptosis, and cuproptosis. Therefore, an in-depth investigation into the mechanisms by which NF-κB participates in programmed cell death can help elucidate the molecular processes underlying the death of cardiac and renal cells, and furnish a theoretical foundation for the prevention and treatment of CRS. To address the aforementioned limitations, we propose that a comprehensive evaluation of the bioavailability and safety profiles of these natural compounds should first be conducted in preclinical models before progressing to clinical research. The clinical translation could be initiated with preliminary assessments of the compounds’ effects on markers of inflammation and oxidative stress in small patient cohorts, subsequently advancing to large-scale clinical trials designed to evaluate their broader clinical efficacy and safety. Ultimately, the most promising compounds could serve as lead structures for the development of novel dual-organ protective drugs with improved potency and pharmacokinetic properties.
As a rapidly inducible transcription factor, NF-κB nuclear translocation plays a broad role in gene induction across diverse cellular responses. Therefore, most NF-κB inhibitors prevent the nuclear translocation of NF-κB and its binding to κB sites within DNA regulatory sequences. These inhibitors include IKK complex inhibitors, proteasomal inhibitors, IκB degradation inhibitors, and NF-κB translocation inhibitors.209 Since NF-κB is ubiquitously present in cardiomyocytes and renal cells, its downstream mediators cover a wide range of processes, from inflammation to oxidative stress. This implies that NF-κB inhibitors are considered potential therapeutic agents for CRS. There are numerous genes activated by NF-κB, as well as diverse signaling pathways that affect NF-κB. Thus, therapeutic interventions for CRS should not only target individual proteins but also comprehensively regulate the NF-κB-mediated comprehensive network signaling pathway. Preclinical pharmacokinetic and toxicological information provides a foundation for future clinical trials in CRS and holds promise for improving therapeutic outcomes related to cardiorenal injury. In recent years, it has become a popular trend to generate data on the roles of NF-κB in human biology by utilizing the significant phenotypes of human mutants in NF-κB and its related proteins, and comparing them with mouse data. This provides a rich set of tools for studying the signaling and transcription of proteins related to inflammation and oxidative stress in humans.210
The NF-κB inhibitors (eg, sorafenib, bortezomib, curcumin, and naringin) prevent cell proliferation, angiogenesis, invasion, and metastasis, and have therefore been applied in the clinical treatment of cancer, autoimmune diseases, and metabolic syndrome.211–214, Although clinical trials associated with CRS have not been reported, numerous preclinical studies on NF-κB have demonstrated that pharmacological inhibition of NF-κB is beneficial for improving cardiac and renal function in CRS. The activation of NF-κB in animal models exacerbates inflammation and oxidative stress in heart and kidney tissues. However, some NF-κB inhibitors may have adverse effects on the cardiovascular system and cause nephrotoxicity side effects.215,216 Therefore, NF-κB inhibitors without cardiotoxicity and nephrotoxicity should be sought as potential targets for the treatment of CRS. There are not many drugs used clinically to treat CRS, and published studies mainly focus on RAS inhibitors, SGLT2 inhibitors, nonsteroidal mineralocorticoid receptor antagonists, and traditional Chinese medicine formulas. However, the use of some drugs is limited by eGFR, and their targets are unclear. Therefore, the development of monomers with clear composition and chemical structure for CRS treatment is of great significance.
Future research directions could focus on integrating high-throughput multi-omics technologies to construct a comprehensive atlas of the NF-κB signaling network in CRS. Recently, a team developed a novel nitrogen-containing chalcone derivative with enhanced Nrf2 agonist activity through structural optimization after conducting high-throughput luminescence screening of their internal compound library.217 The rise of artificial intelligence has provided a powerful tool for drug screening platforms, enabling the prediction and identification of new NF-κB modulators from vast chemical libraries with unprecedented speed and accuracy. A recently reported novel anti-inflammatory agent was discovered through the screening of a compound library.218 Advanced machine learning has transformed the conventional absorption, distribution, metabolism, excretion, and toxicity (ADMET) assessment of clinical candidate success by deciphering complex structure-property relationships, demonstrating the transformative role of AI in reshaping modern drug discovery and development.219 Furthermore, the application of network pharmacology approaches is essential for deciphering the complex mechanisms of multi-target drugs and systematically understanding the role of the NF-κB pathway. Modern research frequently identifies novel therapeutics for diseases by integrating interdisciplinary strategies, such as biochemical, phytochemical, computational, and pharmacological approaches.220 Such integrated strategies will undoubtedly pave the way for next-generation CRS therapies.
Conclusion
Despite the pivotal role of NF-κB in pathophysiology yielding exciting results, research on NF-κB-mediated cellular functions and the exploration of its clinical applications in CRS are still in the initial stages. In this review, we first outline the pathophysiological interactions underlying CRS. Second, we delineate the structure and activation pathways of NF-κB while updating its evolving research history in cardiorenal diseases. Third, we discuss the mechanistic roles of NF-κB in CRS—specifically through the ASK1/MAPKs/NF-κB, PI3K/AKT/NF-κB, and TLR4/NF-κB signaling axes—and provide critical perspectives. Finally, we identify natural compounds that target NF-κB activation in cardiorenal pathologies. By establishing a multidimensional association among “natural compounds, NF-κB targets, and cardiorenal protective effects”, this review offers cutting-edge references for identifying novel therapeutic targets and guiding future strategies against CRS.
Literature Search Strategy and Selection Criteria
Database sources: A comprehensive literature search was performed using the following electronic databases: PubMed, Web of Science, and Scopus.
Search strategy: The specific keywords, Boolean operators (AND, OR), and search strings used for each database are described as follows. For example: (“NF-κB” OR “nuclear factor kappa B”) AND (“cardiorenal syndrome”).
Selection criteria: Predefined inclusion and exclusion criteria were established to screen the literature. These criteria encompassed publication year, study types (eg, randomized controlled trials, animal studies, and cellular experiments), and language restrictions.
Acknowledgments
This article was supported by the National Natural Science Foundation of China (82274306, 82204859), Three-year Action Plan of the Major Clinical Research Program of Shanghai Shenkang Medical Development Center (SHDC2020CR1053B).
Disclosure
The authors have no conflicts of interest to disclose.
References
1. Rangaswami J, Bhalla V, Blair JEA, et al. Cardiorenal syndrome: classification, pathophysiology, diagnosis, and treatment strategies: a scientific statement from the American Heart Association. Circulation. 2019;139(16):e840–e878. doi:10.1161/CIR.0000000000000664
2. Uduman J. Epidemiology of cardiorenal syndrome. Adv Chronic Kidney Dis. 2018;25(5):391–399. doi:10.1053/j.ackd.2018.08.009
3. Foley RN, Murray AM, Li S, et al. Chronic kidney disease and the risk for cardiovascular disease, renal replacement, and death in the United States Medicare population, 1998 to 1999. J Am Soc Nephrol. 2005;16(2):489–495. doi:10.1681/ASN.2004030203
4. Longhini C, Molino C, Fabbian F. Cardiorenal syndrome: still not a defined entity. Clin Exp Nephrol. 2010;14(1):12–21. doi:10.1007/s10157-009-0257-4
5. Ronco C, McCullough P, Anker SD, et al. Cardio-renal syndromes: report from the consensus conference of the acute dialysis quality initiative. Eur Heart J. 2010;31(6):703–711. doi:10.1093/eurheartj/ehp507
6. Ricci Z, Romagnoli S, Ronco C. Cardiorenal syndrome. Crit Care Clin. 2021;37(2):335–347. doi:10.1016/j.ccc.2020.11.003
7. McCallum W, Testani JM. Updates in cardiorenal syndrome. Med Clin North Am. 2023;107(4):763–780. doi:10.1016/j.mcna.2023.03.011
8. Kumar U, Wettersten N, Garimella PS. Cardiorenal syndrome: pathophysiology. Cardiol Clin. 2019;37(3):251–265. doi:10.1016/j.ccl.2019.04.001
9. Gallo G, Lanza O, Savoia C. New insight in cardiorenal syndrome: from biomarkers to therapy. Int J Mol Sci. 2023;24(6):5089. doi:10.3390/ijms24065089
10. Al-Salam S, Kandhan K, Sudhadevi M, Tariq S. Nootkatone ameliorates doxorubicin induced myocardial injury through modulation of NF-κB signals and oxidative stress. Cell Physiol Biochem. 2022;56(4):401–417. doi:10.33594/000000559
11. Zaaba NE, Al-Salam S, Beegam S, Elzaki O, Yasin J, Nemmar A. Catalpol attenuates oxidative stress and inflammation via mechanisms involving sirtuin-1 activation and NF-κB inhibition in experimentally-induced chronic kidney disease. Nutrients. 2023;15(1):237. doi:10.3390/nu15010237
12. Ning H, Chen H, Deng J, et al. Exosomes secreted by FNDC5-BMMSCs protect myocardial infarction by anti-inflammation and macrophage polarization via NF-κB signaling pathway and Nrf2/HO-1 axis. Stem Cell Res Ther. 2021;12(1):519. doi:10.1186/s13287-021-02591-4
13. Feng Y, Li Z, Wang H, Liu BC, Lee K, He JC. HIPK2 C-terminal domain inhibits NF-κB signaling and renal inflammation in kidney injury. JCI Insight. 2024;9(8):e175153. doi:10.1172/jci.insight.175153
14. Yin C, Ye Z, Wu J, et al. Elevated Wnt2 and Wnt4 activate NF-κB signaling to promote cardiac fibrosis by cooperation of Fzd4/2 and LRP6 following myocardial infarction. EBioMedicine. 2021;74:103745. doi:10.1016/j.ebiom.2021.103745
15. Li H, Duann P, Li Z, et al. The cell membrane repair protein MG53 modulates transcription factor NF-κB signaling to control kidney fibrosis. Kidney Int. 2022;101(1):119–130. doi:10.1016/j.kint.2021.09.027
16. Jiang FW, Guo JY, Lin J, et al. MAPK/NF-κB signaling mediates atrazine-induced cardiorenal syndrome and antagonism of lycopene. Sci Total Environ. 2024;922:171015. doi:10.1016/j.scitotenv.2024.171015
17. Williams LM, Gilmore TD. Looking down on NF-κB. Mol Cell Biol. 2020;40(15):e00104–e00120. doi:10.1128/MCB.00104-20
18. Annemann M, Plaza-Sirvent C, Schuster M, et al. Atypical IκB proteins in immune cell differentiation and function. Immunol Lett. 2016;171:26–35. doi:10.1016/j.imlet.2016.01.006
19. Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132(3):344–362. doi:10.1016/j.cell.2008.01.020
20. Mirzaei S, Saghari S, Bassiri F, et al. NF-κB as a regulator of cancer metastasis and therapy response: a focus on epithelial-mesenchymal transition. J Cell Physiol. 2022;237(7):2770–2795. doi:10.1002/jcp.30759
21. Mussbacher M, Derler M, Basílio J, Schmid JA. NF-κB in monocytes and macrophages - an inflammatory master regulator in multitalented immune cells. Front Immunol. 2023;14:1134661. doi:10.3389/fimmu.2023.1134661
22. Yu H, Lin L, Zhang Z, Zhang H, Hu H. Targeting NF-κB pathway for the therapy of diseases: mechanism and clinical study. Signal Transduct Target Ther. 2020;5(1):209. doi:10.1038/s41392-020-00312-6
23. Dolcet X, Llobet D, Pallares J, Matias-Guiu X. NF-kB in development and progression of human cancer. Virchows Arch. 2005;446(5):475–482. doi:10.1007/s00428-005-1264-9
24. Sun SC. The non-canonical NF-κB pathway in immunity and inflammation. Nat Rev Immunol. 2017;17(9):545–558. doi:10.1038/nri.2017.52
25. Sen R, Baltimore D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell. 1986;46(5):705–716. doi:10.1016/0092-8674(86)90346-6
26. Baeuerle PA, Baltimore D. I kappa B: a specific inhibitor of the NF-kappa B transcription factor. Science. 1988;242(4878):540–546. doi:10.1126/science.3140380
27. Bours V, Villalobos J, Burd PR, Kelly K, Siebenlist U. Cloning of a mitogen-inducible gene encoding a kappa B DNA-binding protein with homology to the rel oncogene and to cell-cycle motifs. Nature. 1990;348(6296):76–80. doi:10.1038/348076a0
28. Ghosh S, Gifford AM, Riviere LR, Tempst P, Nolan GP, Baltimore D. Cloning of the p50 DNA binding subunit of NF-kappa B: homology to rel and dorsal. Cell. 1990;62(5):1019–1029. doi:10.1016/0092-8674(90)90276-k
29. Ruben SM, Dillon PJ, Schreck R, et al. Isolation of a rel-related human cDNA that potentially encodes the 65-kD subunit of NF-kappa B. Science. 1991;254(5028):11. doi:10.1126/science.1925549
30. Ryseck RP, Bull P, Takamiya M, et al. RelB, a new Rel family transcription activator that can interact with p50-NF-kappa B. Mol Cell Biol. 1992;12(2):674–684. doi:10.1128/mcb.12.2.674-684.1992
31. Lernbecher T, Müller U, Wirth T. Distinct NF-kappa B/Rel transcription factors are responsible for tissue-specific and inducible gene activation. Nature. 1993;365(6448):767–770. doi:10.1038/365767a0
32. Brown AM, Linhoff MW, Stein B, et al. Function of NF-kappa B/Rel binding sites in the major histocompatibility complex class II invariant chain promoter is dependent on cell-specific binding of different NF-kappa B/Rel subunits. Mol Cell Biol. 1994;14(5):2926–2935. doi:10.1128/mcb.14.5.2926-2935.1994
33. Hansen SK, Baeuerle PA, Blasi F. Purification, reconstitution, and I kappa B association of the c-Rel-p65 (RelA) complex, a strong activator of transcription. Mol Cell Biol. 1994;14(4):2593–2603. doi:10.1128/mcb.14.4.2593-2603.1994
34. Sha WC, Liou HC, Tuomanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell. 1995;80(2):321–330. doi:10.1016/0092-8674(95)90415-8
35. Franzoso G, Carlson L, Poljak L, et al. Mice deficient in nuclear factor (NF)-kappa B/p52 present with defects in humoral responses, germinal center reactions, and splenic microarchitecture. J Exp Med. 1998;187(2):147–159. doi:10.1084/jem.187.2.147
36. Köntgen F, Grumont RJ, Strasser A, et al. Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev. 1995;9(16):1965–1977. doi:10.1101/gad.9.16.1965
37. Doi TS, Takahashi T, Taguchi O, Azuma T, Obata Y. NF-kappa B RelA-deficient lymphocytes: normal development of T cells and B cells, impaired production of IgA and IgG1 and reduced proliferative responses. J Exp Med. 1997;185(5):953–961. doi:10.1084/jem.185.5.953
38. Weih F, Carrasco D, Durham SK, et al. Multiorgan inflammation and hematopoietic abnormalities in mice with a targeted disruption of RelB, a member of the NF-kappa B/Rel family. Cell. 1995;80(2):331–340. doi:10.1016/0092-8674(95)90416-6
39. Franzoso G, Carlson L, Xing L, et al. Requirement for NF-kappaB in osteoclast and B-cell development. Genes Dev. 1997;11(24):3482–3496. doi:10.1101/gad.11.24.3482
40. Grossmann M, Metcalf D, Merryfull J, Beg A, Baltimore D, Gerondakis S. The combined absence of the transcription factors Rel and RelA leads to multiple hemopoietic cell defects. Proc Natl Acad Sci U S A. 1999;96(21):11848–11853. doi:10.1073/pnas.96.21.11848
41. Pohl T, Gugasyan R, Grumont RJ, et al. The combined absence of NF-kappa B1 and c-Rel reveals that overlapping roles for these transcription factors in the B cell lineage are restricted to the activation and function of mature cells. Proc Natl Acad Sci U S A. 2002;99(7):4514–4519. doi:10.1073/pnas.072071599
42. Baeuerle PA, Baltimore D. A 65-kappaD subunit of active NF-kappaB is required for inhibition of NF-kappaB by I kappaB. Genes Dev. 1989;3(11):1689–1698. doi:10.1101/gad.3.11.1689
43. Ghosh S, Baltimore D. Activation in vitro of NF-kappa B by phosphorylation of its inhibitor I kappa B. Nature. 1990;344(6267):678–682. doi:10.1038/344678a0
44. Hatada EN, Nieters A, Wulczyn FG, et al. The ankyrin repeat domains of the NF-kappa B precursor p105 and the protooncogene bcl-3 act as specific inhibitors of NF-kappa B DNA binding. Proc Natl Acad Sci U S A. 1992;89(6):2489–2493. doi:10.1073/pnas.89.6.2489
45. Franzoso G, Bours V, Park S, Tomita-Yamaguchi M, Kelly K, Siebenlist U. The candidate oncoprotein Bcl-3 is an antagonist of p50/NF-kappa B-mediated inhibition. Nature. 1992;359(6393):339–342. doi:10.1038/359339a0
46. Mercurio F, Zhu H, Murray BW, et al. IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science. 1997;278(5339):860–866. doi:10.1126/science.278.5339.860
47. Zandi E, Rothwarf DM, Delhase M, Hayakawa M, Karin M. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell. 1997;91(2):243–252. doi:10.1016/s0092-8674(00)80406-7
48. Li Z, Nabel GJ. A new member of the I kappaB protein family, I kappaB epsilon, inhibits RelA (p65)-mediated NF-kappaB transcription. Mol Cell Biol. 1997;17(10):6184–6190. doi:10.1128/MCB.17.10.6184
49. Yamazaki S, Muta T, Takeshige K. A novel IkappaB protein, IkappaB-zeta, induced by proinflammatory stimuli, negatively regulates nuclear factor-kappaB in the nuclei. J Biol Chem. 2001;276(29):27657–27662. doi:10.1074/jbc.M103426200
50. Fiorini E, Schmitz I, Marissen WE, et al. Peptide-induced negative selection of thymocytes activates transcription of an NF-kappa B inhibitor. Mol Cell. 2002;9(3):637–648. doi:10.1016/s1097-2765(02)00469-0
51. Yamauchi S, Ito H, Miyajima A. IkappaBeta, a nuclear IkappaB protein, positively regulates the NF-kappaB-mediated expression of proinflammatory cytokines. Proc Natl Acad Sci U S A. 2010;107(26):11924–11929. doi:10.1073/pnas.0913179107
52. Rothwarf DM, Karin M. The NF-kappa B activation pathway: a paradigm in information transfer from membrane to nucleus. Sci STKE. 1999;5:RE1. doi:10.1126/stke.1999.5.re1
53. Senftleben U, Cao Y, Xiao G, et al. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science. 2001;293(5534):1495–1499. doi:10.1126/science.1062677
54. Sakurai H, Shigemori N, Hisada Y, Ishizuka T, Kawashima K, Sugita T. Suppression of NF-kappa B and AP-1 activation by glucocorticoids in experimental glomerulonephritis in rats: molecular mechanisms of anti-nephritic action. Biochim Biophys Acta. 1997;1362(2–3):252–262. doi:10.1016/s0925-4439(97)00068-9
55. Guo G, Morrissey J, McCracken R, Tolley T, Klahr S. Role of TNFR1 and TNFR2 receptors in tubulointerstitial fibrosis of obstructive nephropathy. Am J Physiol. 1999;277(5):F766–772. doi:10.1152/ajprenal.1999.277.5.F766
56. Purcell NH, Tang G, Yu C, Mercurio F, DiDonato JA, Lin A. Activation of NF-kappa B is required for hypertrophic growth of primary rat neonatal ventricular cardiomyocytes. Proc Natl Acad Sci U S A. 2001;98(12):6668–6673. doi:10.1073/pnas.111155798
57. Hirotani S, Otsu K, Nishida K, et al. Involvement of nuclear factor-kappaB and apoptosis signal-regulating kinase 1 in G-protein-coupled receptor agonist-induced cardiomyocyte hypertrophy. Circulation. 2002;105(4):509–515. doi:10.1161/hc0402.102863
58. Ogata T, Miyauchi T, Sakai S, Takanashi M, Irukayama-Tomobe Y, Yamaguchi I. Myocardial fibrosis and diastolic dysfunction in deoxycorticosterone acetate-salt hypertensive rats is ameliorated by the peroxisome proliferator-activated receptor-alpha activator fenofibrate, partly by suppressing inflammatory responses associated with the nuclear factor-kappa-B pathway. J Am Coll Cardiol. 2004;43(8):1481–1488. doi:10.1016/j.jacc.2003.11.043
59. Wong SC, Fukuchi M, Melnyk P, Rodger I, Giaid A. Induction of cyclooxygenase-2 and activation of nuclear factor-kappaB in myocardium of patients with congestive heart failure. Circulation. 1998;98(2):100–103. doi:10.1161/01.cir.98.2.100
60. Medzhitov R, Preston-Hurlburt P, Janeway CA. A human homologue of the Drosophila toll protein signals activation of adaptive immunity. Nature. 1997;388(6640):394–397. doi:10.1038/41131
61. Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J Biol Chem. 1999;274(16):10689–10692. doi:10.1074/jbc.274.16.10689
62. Yang J, Jiang H, Yang J, et al. Valsartan preconditioning protects against myocardial ischemia-reperfusion injury through TLR4/NF-kappaB signaling pathway. Mol Cell Biochem. 2009;330(1–2):39–46. doi:10.1007/s11010-009-0098-1
63. Khachigian LM, Collins T, Fries JW. N-acetyl cysteine blocks mesangial VCAM-1 and NF-kappa B expression in vivo. Am J Pathol. 1997;151(5):1225–1229.
64. Muller DN, Dechend R, Mervaala EM, et al. NF-kappaB inhibition ameliorates angiotensin II-induced inflammatory damage in rats. Hypertension. 2000;35(1 Pt 2):193–201. doi:10.1161/01.hyp.35.1.193
65. Ma FY, Tesch GH, Nikolic-Paterson DJ. ASK1/p38 signaling in renal tubular epithelial cells promotes renal fibrosis in the mouse obstructed kidney. Am J Physiol Renal Physiol. 2014;307(11):F1263–1273. doi:10.1152/ajprenal.00211.2014
66. Mochida Y, Takeda K, Saitoh M, et al. ASK1 inhibits interleukin-1-induced NF-kappa B activity through disruption of TRAF6-TAK1 interaction. J Biol Chem. 2000;275(42):32747–32752. doi:10.1074/jbc.M003042200
67. Valenca SS, Dong BE, Gordon EM, Sun RC, Waters CM. ASK1 regulates bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 2022;66(5):484–496. doi:10.1165/rcmb.2021-0465OC
68. Gerczuk PZ, Breckenridge DG, Liles JT, et al. An apoptosis signal-regulating kinase 1 inhibitor reduces cardiomyocyte apoptosis and infarct size in a rat ischemia-reperfusion model. J Cardiovasc Pharmacol. 2012;60(3):276–282. doi:10.1097/FJC.0b013e31825ea0fa
69. Lai J, Li A, Yue L, Zhong H, Xu S, Liu X. Participation of ASK-1 in the cardiomyocyte-protective role of mechanical ventilation in a rat model of myocardial infarction. Exp Biol Med. 2023;248(18):1579–1587. doi:10.1177/15353702231191205
70. Liu D, Shang H, Liu Y. Stanniocalcin-1 protects a mouse model from renal ischemia-reperfusion injury by affecting ROS-mediated multiple signaling pathways. Int J Mol Sci. 2016;17(7):1051. doi:10.3390/ijms17071051
71. Chen X, Yu W, Li W, et al. An anti-inflammatory chalcone derivative prevents heart and kidney from hyperlipidemia-induced injuries by attenuating inflammation. Toxicol Appl Pharmacol. 2018;338:43–53. doi:10.1016/j.taap.2017.11.003
72. Savira F, Cao L, Wang I, et al. Apoptosis signal-regulating kinase 1 inhibition attenuates cardiac hypertrophy and cardiorenal fibrosis induced by uremic toxins: implications for cardiorenal syndrome. PLoS One. 2017;12(11):e0187459. doi:10.1371/journal.pone.0187459
73. Deng T, Wei Z, Gael A, et al. Higenamine improves cardiac and renal fibrosis in rats with cardiorenal syndrome via ASK1 signaling pathway. J Cardiovasc Pharmacol. 2020;75(6):535–544. doi:10.1097/FJC.0000000000000822
74. Porębska N, Poźniak M, Matynia A, et al. Galectins as modulators of receptor tyrosine kinases signaling in health and disease. Cytokine Growth Factor Rev. 2021;60:89–106. doi:10.1016/j.cytogfr.2021.03.004
75. Caso VM, Manzo V, Cimmino TP, et al. Regulation of inflammation and oxidative stress by formyl peptide receptors in cardiovascular disease progression. Life. 2021;11(3):243. doi:10.3390/life11030243
76. Rudomanova V, Blaxall BC. Targeting GPCR-Gβγ-GRK2 signaling as a novel strategy for treating cardiorenal pathologies. Biochim Biophys Acta Mol Basis Dis. 2017;1863(8):1883–1892. doi:10.1016/j.bbadis.2017.01.020
77. Rodon J, Dienstmann R, Serra V, Tabernero J. Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol. 2013;10(3):143–153. doi:10.1038/nrclinonc.2013.10
78. Chen W, Yuan H, Cao W, et al. Blocking interleukin-6 trans-signaling protects against renal fibrosis by suppressing STAT3 activation. Theranostics. 2019;9(14):3980–3991. doi:10.7150/thno.32352
79. Chou CH, Hung CS, Liao CW, et al. IL-6 trans-signalling contributes to aldosterone-induced cardiac fibrosis. Cardiovasc Res. 2018;114(5):690–702. doi:10.1093/cvr/cvy013
80. Tuo P, Zhao R, Li N, et al. Lycorine inhibits Ang II-induced heart remodeling and inflammation by suppressing the PI3K-AKT/NF-κB pathway. Phytomedicine. 2024;128:155464. doi:10.1016/j.phymed.2024.155464
81. Kalantary-Charvadeh A, Sanajou D, Hemmati-Dinarvand M, et al. Micheliolide protects against doxorubicin-induced cardiotoxicity in mice by regulating PI3K/Akt/NF-kB signaling pathway. Cardiovasc Toxicol. 2019;19(4):297–305. doi:10.1007/s12012-019-09511-2
82. Li J, Zhang K, Xu M, et al. Baicalin - 2- ethoxyethyl ester alleviates renal fibrosis by inhibiting PI3K/AKT/NF-κB signaling pathway. Toxicol Appl Pharmacol. 2024;483:116827. doi:10.1016/j.taap.2024.116827
83. Song Z, Zhu M, Wu J, et al. Fucoidans from Cucumaria frondosa ameliorate renal interstitial fibrosis via inhibition of the PI3K/Akt/NF-κB signaling pathway. Food Funct. 2022;13(3):1168–1179. doi:10.1039/d1fo03067a
84. Liu Z, Xiang H, Deng Q, et al. Baicalin and baicalein attenuate hyperuricemic nephropathy via inhibiting PI3K/AKT/NF-κB signalling pathway. Nephrology. 2023;28(6):315–327. doi:10.1111/nep.14159
85. Wang YY, Liu YY, Li J, Zhang YY, Ding YF, Peng YR. Gualou xiebai decoction ameliorates cardiorenal syndrome type II by regulation of PI3K/AKT/NF-κB signalling pathway. Phytomedicine. 2024;123:155172. doi:10.1016/j.phymed.2023.155172
86. Chen Y, Cao W, Li B, et al. The potential role of hydrogen sulfide in regulating macrophage phenotypic changes via PINK1/parkin-mediated mitophagy in sepsis-related cardiorenal syndrome. Immunopharmacol Immunotoxicol. 2024;46(2):139–151. doi:10.1080/08923973.2023.2281901
87. Liu X, Niu Y, Zhang X, et al. Recombinant α-Klotho protein alleviated acute cardiorenal injury in a mouse model of lipopolysaccharide-induced septic cardiorenal syndrome type 5. Anal Cell Pathol. 2019;2019:5853426. doi:10.1155/2019/5853426
88. Virzì GM, Breglia A, Ankawi G, et al. Plasma lipopolysaccharide concentrations in cardiorenal syndrome type 1. Cardiorenal Med. 2019;9(5):308–315. doi:10.1159/000500480
89. Virzì GM, Breglia A, Castellani C, et al. Lipopolysaccharide in systemic circulation induces activation of inflammatory response and oxidative stress in cardiorenal syndrome type 1. J Nephrol. 2019;32(5):803–810. doi:10.1007/s40620-019-00613-2
90. Shimamoto A, Chong AJ, Yada M, et al. Inhibition of Toll-like receptor 4 with eritoran attenuates myocardial ischemia-reperfusion injury. Circulation. 2006;114(1 Suppl):I270–274. doi:10.1161/CIRCULATIONAHA.105.000901
91. Fitzgerald KA, Palsson-McDermott EM, Bowie AG, et al. Mal (MyD88-adapter-like) is required for Toll-like receptor-4 signal transduction. Nature. 2001;413(6851):78–83. doi:10.1038/35092578
92. Lin SC, Lo YC, Wu H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature. 2010;465(7300):885–890. doi:10.1038/nature09121
93. Lampson BL, Ramίrez AS, Baro M, et al. Positive selection CRISPR screens reveal a druggable pocket in an oligosaccharyltransferase required for inflammatory signaling to NF-κB. Cell. 2024;187(9):2209–2223. doi:10.1016/j.cell.2024.03.022
94. Belaiba RS, Bonello S, Zähringer C, et al. Hypoxia up-regulates hypoxia-inducible factor-1alpha transcription by involving phosphatidylinositol 3-kinase and nuclear factor kappaB in pulmonary artery smooth muscle cell. Mol Biol Cell. 2007;18(12):4691–4697. doi:10.1091/mbc.e07-04-0391
95. Rius J, Guma M, Schachtrup C, et al. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008;453(7196):807–811. doi:10.1038/nature06905
96. Zhang J, Bottiglieri T, McCullough PA. The central role of endothelial dysfunction in cardiorenal syndrome. Cardiorenal Med. 2017;7(2):104–117. doi:10.1159/000452283
97. Xu X, Wang Y, Song Q, et al. Mechanism of Zhenwu Decoction modulating TLR4/NF-κB/HIF-1α loop through miR-451 to delay renal fibrosis in type 2 CRS. Phytomedicine. 2024;132:155632. doi:10.1016/j.phymed.2024.155632
98. Zhu J, Zhang Y, Shi L, et al. RP105 protects against ischemic and septic acute kidney injury via suppressing TLR4/NF-κB signaling pathways. Int Immunopharmacol. 2022;109:108904. doi:10.1016/j.intimp.2022.108904
99. Guo L, Zhang X, Lv N, et al. Therapeutic role and potential mechanism of resveratrol in atherosclerosis: TLR4/NF-κB/HIF-1α. Mediators Inflamm. 2023;2023:1097706. doi:10.1155/2023/1097706
100. McKee CM, Coll RC. NLRP3 inflammasome priming: a riddle wrapped in a mystery inside an enigma. J Leukoc Biol. 2020;108(3):937–952. doi:10.1002/JLB.3MR0720-513R
101. Akbal A, Dernst A, Lovotti M, Mangan MSJ, McManus RM, Latz E. How location and cellular signaling combine to activate the NLRP3 inflammasome. Cell Mol Immunol. 2022;19(11):1201–1214. doi:10.1038/s41423-022-00922-w
102. Mills EL, Kelly B, O’Neill LAJ. Mitochondria are the powerhouses of immunity. Nat Immunol. 2017;18(5):488–498. doi:10.1038/ni.3704
103. Hong Q, Zhu S, Yu Y, et al. The emerging role of mtDNA release in sepsis: current evidence and potential therapeutic targets. J Cell Physiol. 2024;239(11):e31331. doi:10.1002/jcp.31331
104. Tirapelli CR, Padovan JC. Oxidative stress in cardiorenal system. Antioxidants. 2024;13(9):1126. doi:10.3390/antiox13091126
105. Amador-Martínez I, Aparicio-Trejo OE, Bernabe-Yepes B, et al. Mitochondrial impairment: a link for inflammatory responses activation in the cardiorenal syndrome type 4. Int J Mol Sci. 2023;24(21):15875. doi:10.3390/ijms242115875
106. Arnaud C, Billoir E, de Melo Junior AF, Pereira SA, O’Halloran KD, Monteiro EC. Chronic intermittent hypoxia-induced cardiovascular and renal dysfunction: from adaptation to maladaptation. J Physiol. 2023;601(24):5553–5577. doi:10.1113/JP284166
107. Li M, Tan H, Gao T, et al. Gypensapogenin I ameliorates isoproterenol (ISO)-induced myocardial damage through regulating the TLR4/NF-κB/NLRP3 pathway. Molecules. 2022;27(16):5298. doi:10.3390/molecules27165298
108. Niu X, Yao Q, Li W, et al. Harmine mitigates LPS-induced acute kidney injury through inhibition of the TLR4-NF-κB/NLRP3 inflammasome signalling pathway in mice. Eur J Pharmacol. 2019;849:160–169. doi:10.1016/j.ejphar.2019.01.062
109. Luo M, Yan D, Sun Q, et al. Ginsenoside Rg1 attenuates cardiomyocyte apoptosis and inflammation via the TLR4/NF-κB/NLRP3 pathway. J Cell Biochem. 2020;121(4):2994–3004. doi:10.1002/jcb.29556
110. Hu Y, Xiang C, Zhang D, Zhou F, Zhang D. Nephroprotective effect of Ginsenoside Rg1 in lipopolysaccharide-induced sepsis in mice through the SIRT1/NF-κB signaling. Folia Histochem Cytobiol. 2024;62(1):13–24. doi:10.5603/fhc.97140
111. Wang D, Yu X, Gao K, et al. Sweroside alleviates pressure overload-induced heart failure through targeting CaMKIIδ to inhibit ROS-mediated NF-κB/NLRP3 in cardiomyocytes. Redox Biol. 2024;74:103223. doi:10.1016/j.redox.2024.103223
112. Ma X, Guo Z, Zhao W, Chen L. Sweroside plays a role in mitigating high glucose-induced damage in human renal tubular epithelial HK-2 cells by regulating the SIRT1/NF-κB signaling pathway. Korean J Physiol Pharmacol. 2023;27(6):533–540. doi:10.4196/kjpp.2023.27.6.533
113. Meeran MFN, Arunachalam S, Azimullah S, et al. α-Bisabolol, a dietary sesquiterpene, attenuates doxorubicin-induced acute cardiotoxicity in rats by inhibiting cellular signaling pathways, Nrf2/Keap-1/HO-1, Akt/mTOR/GSK-3β, NF-κB/p38/MAPK, and NLRP3 inflammasomes regulating oxidative stress and inflammatory cascades. Int J Mol Sci. 2023;24(18):14013. doi:10.3390/ijms241814013
114. Arunachalam S, Meeran MFN, Azimullah S, et al. α-Bisabolol attenuates doxorubicin induced renal toxicity by modulating NF-κB/MAPK signaling and ccaspase-dependent apoptosis in rats. Int J Mol Sci. 2022;23(18):10528. doi:10.3390/ijms231810528
115. Ren ZH, Tong YH, Xu W, Ma J, Chen Y. Tanshinone II A attenuates inflammatory responses of rats with myocardial infarction by reducing MCP-1 expression. Phytomedicine. 2010;17(3–4):212–218. doi:10.1016/j.phymed.2009.08.010
116. Wu X, Liu L, Xie H, et al. Tanshinone IIA prevents uric acid nephropathy in rats through NF-κB inhibition. Planta Med. 2012;78(9):866–873. doi:10.1055/s-0031-1298487
117. Lin J, Lai X, Fan X, et al. Oridonin protects against myocardial ischemia-reperfusion injury by inhibiting GSDMD-mediated pyroptosis. Genes. 2022;13(11):2133. doi:10.3390/genes13112133
118. Li J, Bao L, Zha D, et al. Oridonin protects against the inflammatory response in diabetic nephropathy by inhibiting the TLR4/p38-MAPK and TLR4/NF-κB signaling pathways. Int Immunopharmacol. 2018;55:9–19. doi:10.1016/j.intimp.2017.11.040
119. Yang J, Shi Y, Chen H, Wang X, Chen Y, Yang B. Glycyrrhizic acid attenuates myocardial injury: involvement of RIP140/NF-kB pathway. Biomed Pharmacother. 2017;95:62–67. doi:10.1016/j.biopha.2017.08.031
120. Zhang H, Zhang R, Chen J, Shi M, Li W, Zhang X. High mobility group box1 inhibitor glycyrrhizic acid attenuates kidney injury in streptozotocin-induced diabetic rats. Kidney Blood Press Res. 2017;42(5):894–904. doi:10.1159/000485045
121. Shu J, Huang R, Tian Y, Liu Y, Zhu R, Shi G. Andrographolide protects against endothelial dysfunction and inflammatory response in rats with coronary heart disease by regulating PPAR and NF-κB signaling pathways. Ann Palliat Med. 2020;9(4):1965–1975. doi:10.21037/apm-20-960
122. Ji X, Li C, Ou Y, et al. Andrographolide ameliorates diabetic nephropathy by attenuating hyperglycemia-mediated renal oxidative stress and inflammation via Akt/NF-κB pathway. Mol Cell Endocrinol. 2016;437:268–279. doi:10.1016/j.mce.2016.06.029
123. Jiang F, Xu XR, Li WM, Xia K, Wang LF, Yang XC. Monotropein alleviates H2O2‑induced inflammation, oxidative stress and apoptosis via NF‑κB/AP‑1 signaling. Mol Med Rep. 2020;22(6):4828–4836. doi:10.3892/mmr.2020.11548
124. Zhang Y, Chen Y, Li B, et al. The effect of monotropein on alleviating cisplatin-induced acute kidney injury by inhibiting oxidative damage, inflammation and apoptosis. Biomed Pharmacother. 2020;129:110408. doi:10.1016/j.biopha.2020.110408
125. Al-Ali MA, Younis NS, Aldhubiab B, Alatawi AS, Mohamed ME, Dayem MSAE. Anethole alleviates Doxorubicin-induced cardiac and renal toxicities: insights from network pharmacology and animal studies. Chem Biol Interact. 2024;401:111155. doi:10.1016/j.cbi.2024.111155
126. Gutiérrez-Venegas G, González-Rosas Z. Apigenin reduce lipoteichoic acid-induced inflammatory response in rat cardiomyoblast cells. Arch Pharm Res. 2017;40(2):240–249. doi:10.1007/s12272-016-0756-2
127. Malik S, Suchal K, Khan SI, et al. Apigenin ameliorates streptozotocin-induced diabetic nephropathy in rats via MAPK-NF-κB-TNF-α and TGF-β1-MAPK-fibronectin pathways. Comparative Study. 2017;313(2):F414–F422. doi:10.1152/ajprenal.00393.2016
128. Yu JH, Hu GL, Guo XQ, Cao HB, Xia ZF, Amin B. Quercetin alleviates lipopolysaccharide-induced cardiac inflammation via inhibiting autophagy and programmed cell death. Biomed Environ Sci. 2024;37(1):54–70. doi:10.3967/bes2024.006
129. Tan J, He J, Qin W, Zhao L. Quercetin alleviates lipopolysaccharide-induced acute kidney injury in mice by suppressing TLR4/NF-κB pathway. Nan Fang Yi Ke Da Xue Bao. 2019;39(5):598–602. doi:10.12122/j.issn.1673-4254.2019.05.16
130. Li F, Liu J, Tang S, et al. Quercetin regulates inflammation, oxidative stress, apoptosis, and mitochondrial structure and function in H9C2 cells by promoting PVT1 expression. Acta Histochem. 2021;123(8):151819. doi:10.1016/j.acthis.2021.151819
131. Liu CM, Sun YZ, Sun JM, Ma JQ, Cheng C. Protective role of quercetin against lead-induced inflammatory response in rat kidney through the ROS-mediated MAPKs and NF-κB pathway. Biochim Biophys Acta. 2012;1820(10):1693–1703. doi:10.1016/j.bbagen.2012.06.011
132. Chen Y, Liang C, Li J, et al. Effect of artesunate on cardiovascular complications in periodontitis in a type I diabetes rat model and related mechanisms. J Endocrinol Invest. 2023;46(10):2031–2053. doi:10.1007/s40618-023-02052-0
133. Wan RJ, Li YH. Effects of Artesunate prevent nephritis via the Toll-like receptor 4/nuclear factor‑κB signaling pathway in rats. Mol Med Rep. 2017;16(5):6389–6395. doi:10.3892/mmr.2017.7362
134. Xu Z, Li J, Su B, et al. A role of ROS-dependent defects in mitochondrial dynamic and autophagy in carbon black nanoparticle-mediated myocardial cell damage. Free Radic Biol Med. 2024;220:249–261. doi:10.1016/j.freeradbiomed.2024.04.241
135. Sahu BD, Tatireddy S, Koneru M, et al. Naringin ameliorates gentamicin-induced nephrotoxicity and associated mitochondrial dysfunction, apoptosis and inflammation in rats: possible mechanism of nephroprotection. Toxicol Appl Pharmacol. 2014;227(1):8–20. doi:10.1016/j.taap.2014.02.022
136. Qi C, Shao Y, Liu X, Wang D, Li X. The cardioprotective effects of icariin on the isoprenaline-induced takotsubo-like rat model: involvement of reactive oxygen species and the TLR4/NF-κB signaling pathway. Int Immunopharmacol. 2019;74:105733. doi:10.1016/j.intimp.2019.105733
137. Qi MY, He YH, Cheng Y, et al. Icariin ameliorates streptozocin-induced diabetic nephropathy through suppressing the TLR4/NF-κB signal pathway. Food Funct. 2021;12(3):1241–1251. doi:10.1039/d0fo02335c
138. Fu S, Li YL, Wu YT, Yue Y, Qian ZQ, Yang DL. Icariside II attenuates myocardial fibrosis by inhibiting nuclear factor-κB and the TGF-β1/Smad2 signalling pathway in spontaneously hypertensive rats. Biomed Pharmacother. 2018;100:64–71. doi:10.1016/j.biopha.2018.01.138
139. Hou L, Lin Z, Xu A, et al. Combined protective effects of icariin and selenomethionine on novel chronic tubulointerstitial nephropathy models in vivo and in vitro. Br J Nutr. 2022;127(1):12–22. doi:10.1017/S0007114521000787
140. Ni SY, Zhong XL, Li ZH, et al. Puerarin alleviates lipopolysaccharide-induced myocardial fibrosis by inhibiting PARP-1 to prevent HMGB1-mediated TLR4-NF-κB signaling pathway. Cardiovasc Toxicol. 2020;20(5):482–491. doi:10.1007/s12012-020-09571-9
141. Hu Z, Chen D, Yan P, et al. Puerarin suppresses macrophage M1 polarization to alleviate renal inflammatory injury through antagonizing TLR4/MyD88-mediated NF-κB p65 and JNK/FoxO1 activation. Phytomedicine. 2024;132:155813. doi:10.1016/j.phymed.2024.155813
142. Wang ZK, Chen RR, Li JH, et al. Puerarin protects against myocardial ischemia/reperfusion injury by inhibiting inflammation and the NLRP3 inflammasome: the role of the SIRT1/NF-κB pathway. Int Immunopharmacol. 2020;89(Pt B):107086. doi:10.1016/j.intimp.2020.107086
143. Guo J, Zhang W, Liang P, et al. Puerarin alleviates lipopolysaccharide-induced acute kidney injury in mice by modulating the SIRT1/NF-κB pathway. Nan Fang Yi Ke Da Xue Xue Bao. 2023;43(7):1248–1253. doi:10.12122/j.issn.1673-4254.2023.07.22
144. Sung MJ, Kim DH, Davaatseren M, et al. Genistein suppression of TNF-alpha-induced fractalkine expression in endothelial cells. Cell Physiol Biochem. 2010;26(3):431–440. doi:10.1159/000320566
145. Hu WS, Lin YM, Ho TJ, et al. Genistein suppresses the isoproterenol-treated H9c2 cardiomyoblast cell apoptosis associated with P-38, Erk1/2, JNK, and NFκB signaling protein activation. Am J Chin Med. 2013;41(5):1125–1136. doi:10.1142/S0192415X13500766
146. Li Y, Ou S, Liu Q, et al. Genistein improves mitochondrial function and inflammatory in rats with diabetic nephropathy via inhibiting MAPK/NF-κB pathway. Acta Cir Bras. 2022;37(6):e370601. doi:10.1590/acb370601
147. Han D, Wei J, Zhang R, et al. Hydroxysafflor yellow A alleviates myocardial ischemia/reperfusion in hyperlipidemic animals through the suppression of TLR4 signaling. Sci Rep. 2016;6:35319. doi:10.1038/srep35319
148. Bai J, Zhao J, Cui D, et al. Protective effect of hydroxysafflor yellow A against acute kidney injury via the TLR4/NF-κB signaling pathway. Sci Rep. 2018;8(1):9173. doi:10.1038/s41598-018-27217-3
149. Sun X, Wang X, He Q, et al. Investigation of the ameliorative effects of baicalin against arsenic trioxide-induced cardiac toxicity in mice. Int Immunopharmacol. 2021;99:108024. doi:10.1016/j.intimp.2021.108024
150. Zhang S, Xu L, Liang R, Yang C, Wang P. Baicalin suppresses renal fibrosis through microRNA-124/TLR4/NF-κB axis in streptozotocin-induced diabetic nephropathy mice and high glucose-treated human proximal tubule epithelial cells. J Physiol Biochem. 2020;76(3):407–416. doi:10.1007/s13105-020-00747-z
151. Xu X, Rui S, Chen C, et al. Protective effects of astragalus polysaccharide nanoparticles on septic cardiac dysfunction through inhibition of TLR4/NF-κB signaling pathway. Int J Biol Macromol. 2020;153:977–985. doi:10.1016/j.ijbiomac.2019.10.227
152. Guo M, Gao J, Jiang L, Dai Y. Astragalus polysaccharide ameliorates renal inflammatory responses in a diabetic nephropathy by suppressing the TLR4/NF-κB pathway. Drug Des Devel Ther. 2023;17:2107–2118. doi:10.2147/DDDT.S411211
153. Zhang WJ, Frei B. Astragaloside IV inhibits NF-κB activation and inflammatory gene expression in LPS-treated mice. Mediators Inflamm. 2015;2015:274314. doi:10.1155/2015/274314
154. Shi H, Zhou P, Gao G, et al. Astragaloside IV prevents acute myocardial infarction by inhibiting the TLR4/MyD88/NF-κB signaling pathway. J Food Biochem. 2021;45(7):e13757. doi:10.1111/jfbc.13757
155. Zhou X, Sun X, Gong X, et al. Astragaloside IV from Astragalus membranaceus ameliorates renal interstitial fibrosis by inhibiting inflammation via TLR4/NF-кB in vivo and in vitro. Int Immunopharmacol. 2017;42:18–24. doi:10.1016/j.intimp.2016.11.006
156. Xu C, Tang F, Lu M, et al. Astragaloside IV improves the isoproterenol-induced vascular dysfunction via attenuating eNOS uncoupling-mediated oxidative stress and inhibiting ROS-NF-κB pathways. Int Immunopharmacol. 2016;33:119–127. doi:10.1016/j.intimp.2016.02.009
157. Su X, Guo H, Zhou Y, et al. Astragaloside IV attenuates high glucose-induced NF-κB-mediated inflammation through activation of PI3K/AKT-ERK-dependent Nrf2/ARE signaling pathway in glomerular mesangial cells. Phytother Res. 2023;37(9):4133–4148. doi:10.1002/ptr.7875
158. He CL, Yi PF, Fan QJ, et al. Xiang-Qi-Tang and its active components exhibit anti-inflammatory and anticoagulant properties by inhibiting MAPK and NF-κB signaling pathways in LPS-treated rat cardiac microvascular endothelial cells. Immunopharmacol Immunotoxicol. 2013;35(2):215–224. doi:10.3109/08923973.2012.744034
159. Che X, Wang Q, Xie Y, et al. Astragaloside IV suppresses transforming growth factor-β1 induced fibrosis of cultured mouse renal fibroblasts via inhibition of the MAPK and NF-κB signaling pathways. Biochem Biophys Res Commun. 2015;464(4):1260–1266. doi:10.1016/j.bbrc.2015.07.116
160. Zhu L, Wei T, Chang X, et al. Effects of salidroside on myocardial injury in vivo in vitro via regulation of Nox/NF-κB/AP1 pathway. Inflammation. 2015;38(4):1589–1598. doi:10.1007/s10753-015-0134-0
161. Li R, Guo Y, Zhang Y, Zhang X, Zhu L, Yan T. Salidroside ameliorates renal interstitial fibrosis by inhibiting the TLR4/NF-κB and MAPK signaling pathways. Int J Mol Sci. 2019;20(5):1103. doi:10.3390/ijms20051103
162. Sun Y, Xun L, Jin G, Shi L. Salidroside protects renal tubular epithelial cells from hypoxia/reoxygenation injury in vitro. J Pharmacol Sci. 2018;137(2):170–176. doi:10.1016/j.jphs.2018.05.011
163. Zhong L, Zhou XL, Liu YS, et al. Estrogen receptor α mediates the effects of notoginsenoside R1 on endotoxin-induced inflammatory and apoptotic responses in H9c2 cardiomyocytes. Mol Med Rep. 2015;12(1):119–126. doi:10.3892/mmr.2015.3394
164. Liu WJ, Tang HT, Jia YT, et al. Notoginsenoside R1 attenuates renal ischemia-reperfusion injury in rats. Shock. 2010;34(3):314–320. doi:10.1097/SHK.0b013e3181ceede4
165. Zhang M, Zhi D, Liu P, Wang Y, Duan M. Protective effects of Dioscin against sepsis-induced cardiomyopathy via regulation of toll-like receptor 4/MyD88/p65 signal pathway. Immun Inflamm Dis. 2024;12(5):e1229. doi:10.1002/iid3.1229
166. Cai S, Chen J, Li Y. Dioscin protects against diabetic nephropathy by inhibiting renal inflammation through TLR4/NF-κB pathway in mice. Immunobiology. 2020;225(3):151941. doi:10.1016/j.imbio.2020.151941
167. Hou J, Zheng D, Fung G, et al. Mangiferin suppressed advanced glycation end products (AGEs) through NF-κB deactivation and displayed anti-inflammatory effects in streptozotocin and high fat diet-diabetic cardiomyopathy rats. Can J Physiol Pharmacol. 2016;94(3):332–340. doi:10.1139/cjpp-2015-0073
168. Pal PB, Sinha K, Sil PC. Mangiferin attenuates diabetic nephropathy by inhibiting oxidative stress mediated signaling cascade, TNFα related and mitochondrial dependent apoptotic pathways in streptozotocin-induced diabetic rats. PLoS One. 2014;9(9):e107220. doi:10.1371/journal.pone.0107220
169. Tan YY, Chen LX, Fang L, Zhang Q. Cardioprotective effects of polydatin against myocardial injury in diabetic rats via inhibition of NADPH oxidase and NF-κB activities. BMC Complement Med Ther. 2020;20(1):378. doi:10.1186/s12906-020-03177-y
170. Gu L, Liu J, Xu D, Lu Y. Polydatin prevents LPS-induced acute kidney injury through inhibiting inflammatory and oxidative responses. Microb Pathog. 2019;137:103688. doi:10.1016/j.micpath.2019.103688
171. El-Sheikh AA, Morsy MA, Okasha AM. Inhibition of NF-κB/TNF-α pathway may be involved in the protective effect of resveratrol against cyclophosphamide-induced multi-organ toxicity. Immunopharmacol Immunotoxicol. 2017;39(4):180–187. doi:10.1080/08923973.2017.1318913
172. Bagul PK, Deepthi N, Sultana R, Banerjee SK. Resveratrol ameliorates cardiac oxidative stress in diabetes through deacetylation of NFkB-p65 and histone 3. J Nutr Biochem. 2015;26(11):1298–1307. doi:10.1016/j.jnutbio.2015.06.006
173. Dong W, Zhang K, Wang X, et al. SIRT1 alleviates Cd nephrotoxicity through NF-κB/p65 deacetylation-mediated pyroptosis in rat renal tubular epithelial cells. Sci Total Environ. 2024;929:172392. doi:10.1016/j.scitotenv.2024.172392
174. Csiszar A, Smith K, Labinskyy N, Orosz Z, Rivera A, Ungvari Z. Resveratrol attenuates TNF-alpha-induced activation of coronary arterial endothelial cells: role of NF-kappaB inhibition. Am J Physiol Heart Circ Physiol. 2006;291(4):H1694–1699. doi:10.1152/ajpheart.00340.2006
175. Zhang L, Pang S, Deng B, et al. High glucose induces renal mesangial cell proliferation and fibronectin expression through JNK/NF-κB/NADPH oxidase/ROS pathway, which is inhibited by resveratrol. Int J Biochem Cell Biol. 2012;44(4):629–638. doi:10.1016/j.biocel.2012.01.001
176. Raish M, Ahmad A, Jardan YAB, et al. Sinapic acid ameliorates cardiac dysfunction and cardiomyopathy by modulating NF-κB and Nrf2/HO-1 signaling pathways in streptozocin induced diabetic rats. Biomed Pharmacother. 2022;145:112412. doi:10.1016/j.biopha.2021.112412
177. Ansari MA. Sinapic acid modulates Nrf2/HO-1 signaling pathway in cisplatin-induced nephrotoxicity in rats. Biomed Pharmacother. 2017;93:646–653. doi:10.1016/j.biopha.2017.06.085
178. Al-Taher AY, Morsy MA, Rifaai RA, Zenhom NM, Abdel-Gaber SA. Paeonol attenuates methotrexate-induced cardiac toxicity in rats by inhibiting oxidative stress and suppressing TLR4-induced NF-κB inflammatory pathway. Mediators Inflamm. 2020;2020:8641026. doi:10.1155/2020/8641026
179. Fan HY, Qi D, Yu C, et al. Paeonol protects endotoxin-induced acute kidney injury: potential mechanism of inhibiting TLR4-NF-κB signal pathway. Oncotarget. 2016;7(26):39497–39510. doi:10.18632/oncotarget.8347
180. Hamdy S, Elshopakey GE, Risha EF, Rezk S, Ateya AI, Abdelhamid FM. Curcumin mitigates gentamicin induced-renal and cardiac toxicity via modulation of Keap1/Nrf2, NF-κB/iNOS and Bcl-2/BAX pathways. Food Chem Toxicol. 2024;183:114323. doi:10.1016/j.fct.2023.114323
181. Song Y, Ge W, Cai H, Zhang H. Curcumin protects mice from coxsackievirus B3-induced myocarditis by inhibiting the phosphatidylinositol 3 kinase/Akt/nuclear factor-κB pathway. J Cardiovasc Pharmacol Ther. 2013;18(6):560–569. doi:10.1177/1074248413503044
182. Yang H, Zhang H, Tian L, et al. Curcumin attenuates lupus nephritis by inhibiting neutrophil migration via PI3K/AKT/NF-κB signalling pathway. Lupus Sci Med. 2024;11(2):e001220. doi:10.1136/lupus-2024-001220
183. Oyagbemi AA, Akinrinde AS, Adebiyi OE, et al. Luteolin supplementation ameliorates cobalt-induced oxidative stress and inflammation by suppressing NF-кB/Kim-1 signaling in the heart and kidney of rats. Environ Toxicol Pharmacol. 2020;80:103488. doi:10.1016/j.etap.2020.103488
184. Dawuti A, Sun S, Wang R, et al. Salvianolic acid A alleviates heart failure with preserved ejection fraction via regulating TLR/Myd88/TRAF/NF-κB and p38MAPK/CREB signaling pathways. Biomed Pharmacother. 2023;168:115837. doi:10.1016/j.biopha.2023.115837
185. Zhang HF, Wang YL, Gao C, et al. Salvianolic acid A attenuates kidney injury and inflammation by inhibiting NF-κB and p38 MAPK signaling pathways in 5/6 nephrectomized rats. Acta Pharmacol Sin. 2018;39(12):1855–1864. doi:10.1038/s41401-018-0026-6
186. Jin JL, Zhang H, Liu Q, Jiang HH, Liu XX, Tang XZ. Berberine protects myocardial injury and cardiac dysfunction in a septic rat model. Zhonghua Yi Xue Za Zhi. 2020;100(35):2779–2784. doi:10.3760/cma.j.cn112137-20200227-00477
187. Zhu L, Han J, Yuan R, Xue L, Pang W. Berberine ameliorates diabetic nephropathy by inhibiting TLR4/NF-κB pathway. Biol Res. 2018;51(1):9. doi:10.1186/s40659-018-0157-8
188. Li L, Fang P, Chen J, Zhang C, Tao H. Protective effect of sinomenine on isoproterenol-induced cardiac hypertrophy in mice. J Appl Biomed. 2021;19(3):142–148. doi:10.32725/jab.2021.014
189. Zhao Z, Guan R, Song S, et al. Sinomenine protects mice against ischemia reperfusion induced renal injury by attenuating inflammatory response and tubular cell apoptosis. Int J Clin Exp Pathol. 2013;6(9):1702–1712.
190. Sun H, Bai J, Sun Y, et al. Oxymatrine attenuated isoproterenol-induced heart failure via the TLR4/NF-κB and MAPK pathways in vivo and in vitro. Eur J Pharmacol. 2023;941:175500. doi:10.1016/j.ejphar.2023.175500
191. Wang HW, Shi L, Xu YP, Qin XY, Wang QZ. Oxymatrine inhibits renal fibrosis of obstructive nephropathy by downregulating the TGF-β1-Smad3 pathway. Ren Fail. 2016;38(6):945–951. doi:10.3109/0886022X.2016.1164185
192. Yang JM, Zhou R, Zhang M, Tan HR, Yu JQ. Betaine attenuates monocrotaline-induced pulmonary arterial hypertension in rats via inhibiting inflammatory response. Molecules. 2018;23(6):1274. doi:10.3390/molecules23061274
193. Norouzzadeh M, Kalantar H, Khorsandi L, Mohtadi S, Khodayar MJ. Betaine ameliorates arsenic-induced kidney injury in mice by mitigating oxidative stress-mediated inflammation. Arch Biochem Biophys. 2024;758:110076. doi:10.1016/j.abb.2024.110076
194. Jin W, Zhang Y, Xue Y, et al. Crocin attenuates isoprenaline-induced myocardial fibrosis by targeting TLR4/NF-κB signaling: connecting oxidative stress, inflammation, and apoptosis. Naunyn Schmiedebergs Arch Pharmacol. 2020;393(1):13–23. doi:10.1007/s00210-019-01704-4
195. Abou-Hany HO, Atef H, Said E, Elkashef HA, Salem HA. Crocin mediated amelioration of oxidative burden and inflammatory cascade suppresses diabetic nephropathy progression in diabetic rats. Chem Biol Interact. 2018;284:90–100. doi:10.1016/j.cbi.2018.02.001
196. He Q, Zhou W, Xiong C, Tan G, Chen M. Lycopene attenuates inflammation and apoptosis in post-myocardial infarction remodeling by inhibiting the nuclear factor-κB signaling pathway. Mol Med Rep. 2015;11(1):374–378. doi:10.3892/mmr.2014.2676
197. Albadrani GM, Altyar AE, Kensara OA, et al. Lycopene alleviates 5-fluorouracil-induced nephrotoxicity by modulating PPAR-γ, Nrf2/HO-1, and NF-κB/TNF-α/IL-6 signals. Ren Fail. 2024;46(2):2423843. doi:10.1080/0886022X.2024.2423843
198. Song ZC, Wang ZS, Bai JH, Li Z, Hu J. Emodin, a naturally occurring anthraquinone, ameliorates experimental autoimmune myocarditis in rats. Tohoku J Exp Med. 2012;227(3):225–230. doi:10.1620/tjem.227.225
199. Li Y, Xiong W, Yang J, et al. Attenuation of inflammation by emodin in lipopolysaccharide-induced acute kidney injury via inhibition of toll-like receptor 2 signal pathway. Iran J Kidney Dis. 2015;9(3):202–208.
200. Kong L, Ji X, Liu Y, Du Y. Effect of artemisinin combined with allicin on improving cardiac function, fibrosis and NF-κB signaling pathway in rats with diabetic cardiomyopathy. Acta Biochim Pol. 2023;70(2):401–405. doi:10.18388/abp.2020_6692
201. Liu C, Cao F, Tang QZ, et al. Allicin protects against cardiac hypertrophy and fibrosis via attenuating reactive oxygen species-dependent signaling pathways. J Nutr Biochem. 2010;21(12):1238–1250. doi:10.1016/j.jnutbio.2009.11.001
202. Xu N, Han W, Yun G, Shi L. Allicin protects renal function, improves oxidative stress and lipid peroxidation in rats with chronic renal failure. Iran J Kidney Dis. 2023;17(3):135–140.
203. Fukunaga N, Kawajiri H, Badiwala MV, et al. Protective role of Nrf2 against ischemia reperfusion injury and cardiac allograft vasculopathy. Am J Transplant. 2020;20(5):1262–1271. doi:10.1111/ajt.15724
204. Ribeiro M, Alvarenga L, Coutinho-Wolino KS, Nakao LS, Cardozo LF, Mafra D. Sulforaphane upregulates the mRNA expression of NRF2 and NQO1 in non-dialysis patients with chronic kidney disease. Free Radic Biol Med. 2024;221:181–187. doi:10.1016/j.freeradbiomed.2024.05.034
205. Duan J, Yang Y, Liu H, Dou PC, Tan SY. Osthole ameliorates acute myocardial infarction in rats by decreasing the expression of inflammatory-related cytokines, diminishing MMP-2 expression and activating p-ERK. Int J Mol Med. 2016;37(1):207–216. doi:10.3892/ijmm.2015.2402
206. Yang SM, Chan YL, Hua KF, et al. Osthole improves an accelerated focal segmental glomerulosclerosis model in the early stage by activating the Nrf2 antioxidant pathway and subsequently inhibiting NF-κB-mediated COX-2 expression and apoptosis. Free Radic Biol Med. 2014;73:260–269. doi:10.1016/j.freeradbiomed.2014.05.009
207. Mohany M, Ahmed MM, Al-Rejaie SS. Molecular mechanistic pathways targeted by natural antioxidants in the prevention and treatment of chronic kidney disease. Antioxidants. 2021;11(1):15. doi:10.3390/antiox11010015
208. Zhang Z, Yang Z, Wang S, Wang X, Mao J. Overview of pyroptosis mechanism and in-depth analysis of cardiomyocyte pyroptosis mediated by NF-κB pathway in heart failure. Biomed Pharmacother. 2024;179:117367. doi:10.1016/j.biopha.2024.117367
209. Pordanjani SM, Hosseinimehr SJ. The role of NF-kB inhibitors in cell response to radiation. Curr Med Chem. 2016;23(34):3951–3963. doi:10.2174/0929867323666160824162718
210. Zhang Q, Lenardo MJ, Baltimore D. 30 years of NF-κB: a blossoming of relevance to human pathobiology. Cell. 2017;168(1–2):37–57. doi:10.1016/j.cell.2016.12.012
211. Kong FH, Ye QF, Miao XY, et al. Current status of sorafenib nanoparticle delivery systems in the treatment of hepatocellular carcinoma. Theranostics. 2021;11(11):5464–5490. doi:10.7150/thno.54822
212. Rajkumar SV. Multiple myeloma: 2022 update on diagnosis, risk stratification, and management. Am J Hematol. 2022;97(8):1086–1107. doi:10.1002/ajh.26590
213. Panknin TM, Howe CL, Hauer M, Bucchireddigari B, Rossi AM, Funk JL. Curcumin supplementation and human disease: a scoping review of clinical trials. Int J Mol Sci. 2023;24(5):4476. doi:10.3390/ijms24054476
214. Chen J, Qin X, Chen M, Chen T, Chen Z, He B. Biological activities, molecular mechanisms, and clinical application of naringin in metabolic syndrome. Pharmacol Res. 2024;202:107124. doi:10.1016/j.phrs.2024.107124
215. Mina R, Cerrato C, Bernardini A, Aghemo E, Palumbo A. New pharmacotherapy options for multiple myeloma. Expert Opin Pharmacother. 2016;17(2):181–192. doi:10.1517/14656566.2016.1115016
216. Ding K, Jiang W, Jia H, Lei M. Synergistically anti-multiple myeloma effects: flavonoid, non-flavonoid polyphenols, and bortezomib. Biomolecules. 2022;12(11):1647. doi:10.3390/biom12111647
217. Deng Y, Xu L, Jiang Z, et al. Discovery of a novel chalcone-derived covalent Keap1 binder for mitigating cisplatin-induced mitochondrial dysfunction and nephrotoxicity. Redox Biol. 2025;85:103737. doi:10.1016/j.redox.2025.103737
218. Zhang F, Zhao B, Fan Y, et al. Discovery of a novel AhR-CYP1A1 axis activator for mitigating inflammatory diseases using an in situ functional imaging assay. Acta Pharm Sin B. 2025;15(1):508–525. doi:10.1016/j.apsb.2024.09.014
219. Fan N, Chen J, Wang J, Chen ZS, Yang Y. Bridging data and drug development: machine learning approaches for next-generation ADMET prediction. Drug Discov Today. 2025;30(11):104487. doi:10.1016/j.drudis.2025.104487
220. Song Y, Zhang F, Guo J, et al. High-efficient discovering the potent anti-Notum agents from herbal medicines for combating glucocorticoid-induced osteoporosis. Acta Pharm Sin B. 2025;15(8):4174–4192. doi:10.1016/j.apsb.2025.06.004
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Platelet-Activating Factor Promotes the Development of Non-Alcoholic Fatty Liver Disease
Yin H, Shi A, Wu J
Diabetes, Metabolic Syndrome and Obesity 2022, 15:2003-2030
Published Date: 8 July 2022
The Effects and Pathogenesis of PM2.5 and Its Components on Chronic Obstructive Pulmonary Disease

Wang Q, Liu S
International Journal of Chronic Obstructive Pulmonary Disease 2023, 18:493-506
Published Date: 6 April 2023
Quercetin: A Flavonoid with Potential for Treating Acute Lung Injury

Huang M, Liu X, Ren Y, Huang Q, Shi Y, Yuan P, Chen M
Drug Design, Development and Therapy 2024, 18:5709-5728
Published Date: 6 December 2024
Exercise Prescription Training in Chronic Obstructive Pulmonary Disease: Benefits and Mechanisms
Liu S, Yang A, Yu Y, Xu B, Yu G, Wang H
International Journal of Chronic Obstructive Pulmonary Disease 2025, 20:1071-1082
Published Date: 15 April 2025
Natural Compounds Targeting Coronary Microvascular Dysfunction: Mechanisms and Beyond
Chen M, Xiang X, Yang X, Fu Y, Wan Z, Yang S, Liu Q, Luo G, Liu M
Drug Design, Development and Therapy 2026, 20:591542
Published Date: 27 March 2026