Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 18
Mettl3 Mediated m6A Methylation Involved in Epithelial-Mesenchymal Transition by Targeting SOCS3/STAT3/SNAI1 in Cigarette Smoking-Induced COPD
Authors Zhang Y, Wang L ![]() , Yan F, Yang M, Gao H, Zeng Y
, Yan F, Yang M, Gao H, Zeng Y ![]()
Received 24 November 2022
Accepted for publication 22 May 2023
Published 30 May 2023 Volume 2023:18 Pages 1007—1017
DOI https://doi.org/10.2147/COPD.S398289
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Yaping Zhang,1,* Lixing Wang,1,* Furong Yan,1 Meili Yang,2 Hongzhi Gao,1 Yiming Zeng3
1Clinical Center for Molecular Diagnosis and Therapy, The Second Affiliated Hospital of Fujian Medical University, Quanzhou, Fujian, People’s Republic of China; 2Department of Neurology, The Second Affiliated Hospital of Fujian Medical University, Quanzhou, Fujian, People’s Republic of China; 3Department of Pulmonary and Critical Care Medicine, The Second Affiliated Hospital of Fujian Medical University, Respirology Medicine Centre of Fujian Province, Quanzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yaping Zhang, Clinical Center for Molecular Diagnosis and Therapy, The Second Affiliated Hospital of Fujian Medical University, No. 34 Zhongshan North Road, Licheng District, Quanzhou, Fujian, People’s Republic of China, Email [email protected]
Purpose: Persistent inflammation and epithelial-mesenchymal transition are essential pathophysiological processes in chronic obstructive pulmonary disease (COPD) and involve airway remodeling. m6A methylation modification was discovered to play an important role in various diseases. Nevertheless, the regulatory role of m6A methylation has not yet been investigated in cigarette smoking-induced COPD. The study aims to explore the regulatory role of m6A methylation in cigarette smoking-induced COPD.
Patients and Methods: In this study, two Gene Expression Omnibus (GEO) datasets were first utilized to analyze the expression profiles of m6A RNA methylation regulators in COPD. We then established a cell model of COPD by exposing human bronchial epithelial cells (HBECs) to cigarette smoke extract (CSE) in vitro and detected the expression of m6A writer Mettl3 and EMT phenotype markers. RNA interference, cycloleucine, RT-qPCR, western blot, MeRIP-sequencing, and cell migration assay were performed to investigate the potential effect of Mettl3 on the EMT process in CSE-induced HBECs.
Results: Our results showed that Mettl3 expression was significantly elevated in cigarette smoking-induced COPD patients and in a cellular model of COPD. Furthermore, Mettl3 silence and cycloleucine treatment inhibited the EMT process of HBECs caused by CSE. Mechanically, Mettl3 silence weakens the m6A methylation of SOCS3 mRNA to enhance the protein expression of SOCS3, inhibiting CSE-induced SOCS3/STAT3/SNAI1 signaling and EMT processes in HBECs.
Conclusion: Our study inferred that Mettl3-mediated m6A RNA methylation modification modulates CSE-induced EMT by targeting SOCS3 mRNA and ultimately serves as a crucial regulator in the emergence of COPD. This conclusion reinforces the regulatory role of m6A methylation in COPD.
Keywords: inflammation, COPD, MeRIP–sequencing, cycloleucine
Introduction
Chronic obstructive pulmonary disease (COPD) is commonly considered a disorder of progressive airflow obstruction and chronic airway inflammation associated with cigarette smoking. Airway remodeling underlies the etiology of COPD, grounded in the inflammation, repair, or remodeling process of airway epithelium.1 IL-6/STAT3 signaling is mainly triggered by IL-6 and involves the persistent airway inflammatory response in COPD,2,3 which is suppressed by SOCS3.4 Moreover, several studies have confirmed the critical role that airway epithelial-mesenchymal transition (EMT) plays in airway remodeling. EMT is defined as the biological process that gradually converts epithelial into mesenchymal cells and involves chronic inflammation, tissue remodeling, and cancer metastasis.5 The airway epithelium of COPD patients displays features of EMT, which is associated with peribronchial fibrosis and airflow limitation.6 Research has shown that Roflumilast N-oxide can achieve therapeutic effects by inhibiting bronchial EMT induced by cigarette smoking in COPD.7 The signaling pathway of IL-6/STAT3 has been identified as a contributing factor in the initiation of EMT in diverse forms of cancer. In prior research efforts, Cong et al have presented evidence to suggest that this particular pathway appears to effectively promote SNAI1 expression and thereby stimulate the manifestation of the EMT phenotype.8
The m6A RNA methylation modification has gained significant attention in biological systems. It plays a crucial role in post-transcriptional gene regulation by impacting mRNA stability, translation efficiency, splicing of methylated transcripts, and localization.9 The process is regulated jointly by writers (Mettl3/14), erasers (FTO and ALKHB5), and m6A-binding proteins (YTHDF/IGF2BP). Of note, growing evidence showed that Mettl3-mediated regulation has a significant role in m6A modification. For instance, Mettl3 enhances the stability of ZMYM1 mRNA through m6A modification, supporting the EMT program.10 Likewise, m6A modification by Mettl3 on TLR4 mRNA shows increased translation and slower degradation, resulting in encouraging neutrophil activation through TLR4 signaling.11 Additionally, Mettl3 can modify SOCS3 mRNA by m6A during T-cell differentiation.12 Mettl3 also promotes the spread of ovarian cancer by controlling the translation of AXL and EMT.13
Insufficient evidence currently exists to substantiate the involvement of m6A RNA methylation in the occurrence of cigarette smoking-induced COPD. Therefore, the objective of this study was to investigate the regulatory impact of m6A RNA methylation in human bronchial epithelial cells stimulated by CSE, with the intent of discovering new insights into the management of COPD.
Materials and Methods
Public Database Acquisition
We acquired data regarding the abundance of four m6A regulators (Mettl3, Mettl14, ALKBH5, and FTO) in the alveolar macrophages gathered from the bronchoalveolar lavage fluid (BALF) of individuals with COPD, smokers, and non-smokers in the GSE130928 database,14 as well as Mettl3 gene expression in the small airway epithelium from 12 non-smokers, 9 early COPD smokers, and 6 COPD-smokers from the GSE5058 database.15
Preparation of Cigarette Smoke Extract
A trusted technique for studying the effects of cigarette smoke exposure in vitro is the use of cigarette smoke extract (CSE). As previously mentioned,16 the procedure involved bubbling 20 mL of medium with the mainstream smoke from two cigarettes (Shishi). The resulting CSE was subjected to sterile filtering using a 0.22 μm filter. The standardization of CSE is achieved by measuring its absorbance at 320 nm in a freshly prepared solution for each experiment. This solution is deemed to be 100% CSE and is subsequently diluted with a culture medium.
Cell Culture
16-Human bronchial epithelial cells (HBECs) were acquired from Shanghai Institute for Biological Sciences and grown in DMEM medium (Gibco) with 10% fetal bovine serum (Gibco) at 37 °C in a humid environment with 5% CO2.
Cycloleucine Treatment
HBECs were cultured as previously described. Cycloleucine (#52-52-8, MCE) was prepared with sterile distilled water. The plated cells were pretreated with 40 mM cycloleucine for 1 h. After 1 hour, HBECs were treated with 5% CSE in CSE groups.
RNA Interference
Double-stranded siRNAs targeting human Mettl3 and scrambled siRNA were purchased from GenePharma (China). The siRNAs were introduced into HBECs using Lipofectamine 3000 (Thermo Fisher) as per the manufacturer’s instructions. Following a 24 h transfection period, the HBECs were exposed to CSE for an additional 24 h, after which total RNA and protein were recovered. The siRNA sequences used were si-Mettl3: 5′ - CCUGCAAGUAUGUUCACUATT −3′, 5′ - UAGUGAACAUACUUGCAGGTT-3′ and scrambled-siRNA: 5′ - UUCUCCGAACGUGUCACGUTT-3′’, 5′ -ACGUGACACGUUCGGAGAATT-3′.
Cell Migration Assay
To evaluate HBEC migration, wound-healing assays were conducted. siRNA-transfected HBECs were cultured in 12-well plates with 90% confluence, after which they were scraped with a 10 µL pipette tip. Following this, the cells were treated with a serum-free medium containing 5% CSE for a period of 24 h. After the removal of CSE, the cells were exposed to a serum-free medium for another 24 h. Microscopic observations were made at 0 h and 48 h, respectively. ImageJ measured the scratch area of migrating cells.
RT-qPCR Analysis
Cellular RNA was isolated from cells with Trizol (Invitrogen) and then subjected to cDNA synthesis via the Primescript RT reagent (Takara). PCR amplification was implemented with the assistance of the SYBR Green PCR kit (Takara) with primers (Shenggong) in the PCR system. For normalization purposes, the GAPDH control was utilized. The relative quantification analysis was conducted utilizing the comparative CT approach. Reference to the employed primer sequences may be found in Supplementary Table 1.
MeRIP–Sequencing
Cloudseq (Shanghai) offered a service referred to as MeRIP-Seq (m6A RNA-Seq).
For the siRNA-mettl3+CSE (HBE/si-Mettl3) and CSE (HBE/si-control) groups, three biological replicates were pooled together. Trizol Reagent was utilized to collect RNA from the two cell samples. Using NanoDrop ND-1000 (Thermo Fisher), the quality and quantity of RNA were evaluated while the integrity of the RNA was determined by performing electrophoresis on a denaturing Agarose gel. Next, depurating mRNA from RNA was accomplished using the Seq-StarTM poly (A) mRNA Isolation Kit. m6A RNA immunoprecipitation was carried out using m6A-MeRIP Kit from GenSeq. NEBNext® Ultra II Directional RNA Library Prep Kit was employed to get an RNA-seq library from the IP samples with m6A immunoprecipitation and the input samples without immunoprecipitation. Lastly, the library’s quality was assessed through the BioAnalyzer 2100 device, and the library was sequenced on an Illumina Hiseq 4000 with 150 bp paired-end reads.
Western Blot
Total cellular protein was extracted using RIPA (#P0013B, Beyotime) and the protein concentration was determined by BCA assay (#BL521A, Biosharp). Subsequently, proteins present in the cell lysate were resolved using 10% SDS-PAGE gels and then transferred to PVDF membranes. To prevent non-specific interactions, the membranes were blocked with 5% milk for 1 h and then were stripped and incubated overnight at 4 °C with different primary antibodies: Mettl3 antibody (#ab66660 or #ab195352, Abcam), SOCS3 antibody (#ab280884, Abcam), SNAI1 antibody (#ab216347, Abcam), STAT3 antibody (#9139, CST), p-STAT3 antibody (#9145, CST), E-cadherin antibody (#ab133597, Abcam), Vimentin antibody (#sc-6260, Santa Cruz), MMP-9 antibody (#sc-21733, Santa Cruz), β-actin antibody (#AF5003, Beyotime, China) and GAPDH antibody (#AF1186, Beyotime, China). Afterward, the signals were visualized with ECL reagents (#P0018S, Beyotime) using ImageQuant LAS 4000 after incubating with the second antibody for 1 h. The grayscale of protein bands was determined by Image J.
mRNA Stability Analysis
HBECs were subjected to exposure to actinomycin D (Sigma) at a concentration of 5 μg/mL for 0, 3, or 6 h to ascertain the stability of mRNA. Following this, the cells were harvested and RNA samples were detected by RT- qPCR.17
Statistical Analysis
The statistical analysis of m6A RNA sequencing involved obtaining paired-end reads from the sequencer and passed to quality control by Q30.18 Using Hisat2, clean reads from all libraries were mapped to HG19. Methylated peaks on RNAs from samples were discovered with the help of MACS software. The result of matching the reads to the genome is visualized using IGV software19 to visually observe the abundance of specific locations in the genome.
The mean ± standard deviation (SD) from three distinct experiments was used to express data. The statistical analysis was undertaken with the employment of GraphPad Prism 9 software and ImageJ. The two-way ANOVA or one-way ANOVA was utilized for statistical analysis. Statistical significance was determined by a P-value lower than 0.05.
Results
Mettl3 Expression Was Increased in Smoker COPD Patients and CSE-Induced HBECs
To explore m6A methylation regulators involved in cigarette smoking-induced COPD, we examined the mRNA expression of m6A “writers” (Mettl3 and Mettl14) and m6A “erasers” (ALKBH5 and FTO) in COPD-smokers in a public database. Analysis of the GSE130928 database revealed that, by comparing the non-smokers and smokers groups, the expression level of Mettl3 in the COPD group was considerably greater (Figure 1A). Conversely, the expression level of m6A eraser ALKBH5 from the COPD group was significantly decreased. While there was no difference in the expression of Mettl14 and FTO in all three groups. In addition, the GSE5058 database analysis revealed a substantial rise in the expression levels of Mettl3 within the small airway epithelium of subjects from the early COPD and COPD groups, as compared to those from the non-smokers group. (Figure 1B).
 |
Figure 1 Mettl3 is significantly increased in COPD caused by cigarette smoking. (A) The expression of m6A RNA methylation regulators (Mettl3, Mettl14, ALKBH5, and FTO) in the alveolar macrophages obtained from the bronchoalveolar lavage fluid of subjects with COPD, smokers, and non-smokers in the GSE130928 database. (B) Mettl3 expression in the airway epithelium of 12 non-smokers, 9 early-COPD, and 6 COPD in the GSE5058 database. (C) The mRNA levels of m6A regulators (Mettl3, Mettl14, ALKBH5, and FTO) in HBECs exposed to CSE for 24 h. (D) The protein level of Mettl3 in HBEC exposed to CSE for 24 h was determined by western blot and quantitatively analyzed by Image J. (E) The mRNA level of Mettl3 in HBEC exposed to CSE for 24 h was determined by RT-qPCR. ****P<0.0005, ***P<0.0005, **P<0.005, *P<0.05. Abbreviation: ns, not significant. |
Next, we evaluated the mRNA expression profiles of m6A writers and erasers in CSE-induced HBECs in vitro. Surprisingly, the results showed a notable elevation of mRNA levels of Mettl3, Mettl14, ALKBH5, and FTO in HBECs upon exposure to 5% CSE, with Mettl3 exhibiting the most significant increase (Figure 1C). Therefore, we focused on studying Mettl3-mediated m6A methylation in CSE-induced HBEC.
Furthermore, we detected the protein and mRNA level of Mettl3 in HBECs exposed to an escalating dose of CSE for 24 h. Our findings revealed a considerable dose-dependent elevation of Mettl3 protein and mRNA levels in HBECs exposed to CSE, as demonstrated by Figure 1D–E.
Mettl3 Knockdown Was Mediated by siRNA in HBECs
To quest the contribution of Mettl3 in cigarette smoking-induced COPD, we performed Mettl3 targeted small interfering RNA to down-regulate Mettl3 expression in HBECs. The efficiency of Mettl3 silence was verified by both western blot and RT- qPCR. Consequently, compared to siRNA control transfected HBECs, the protein and mRNA expression of Mettl3 were suppressed in si-Mettl3 HBECs (Figure 2A and B).
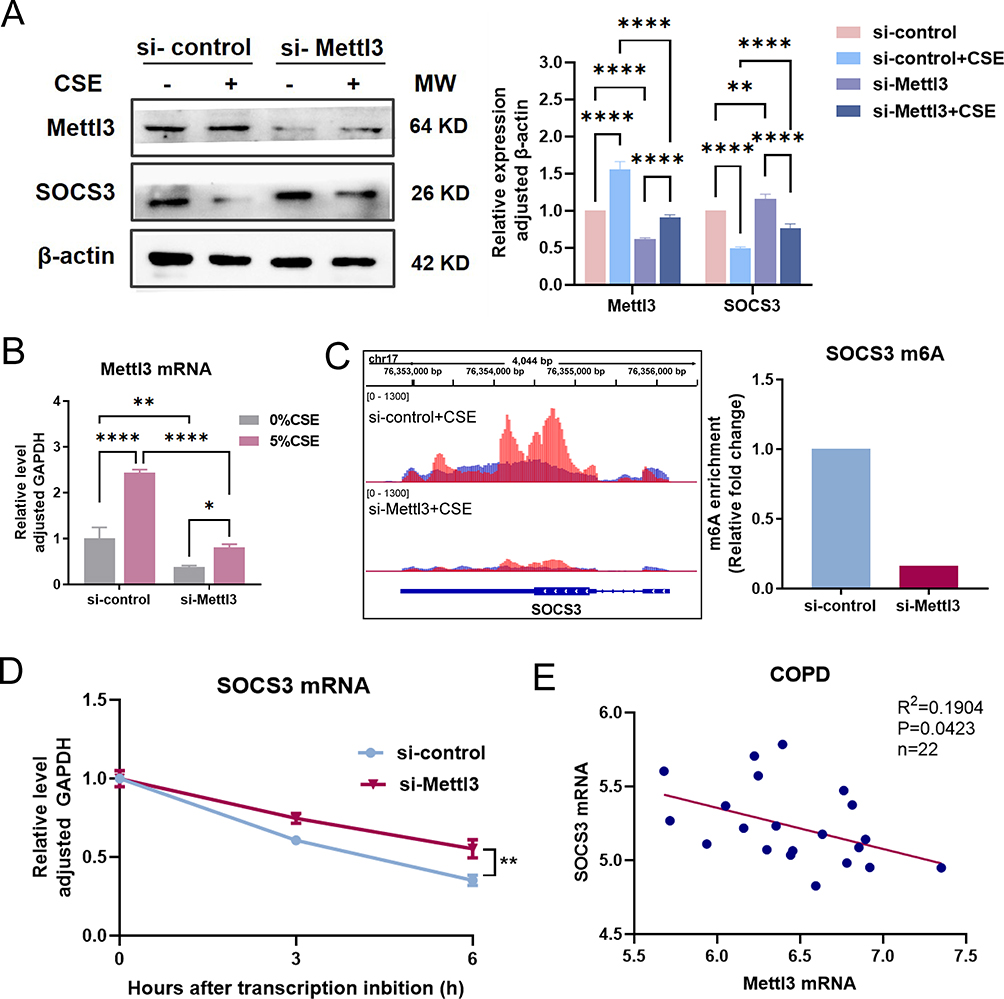 |
Figure 2 The protein expression of SOCS3 is upregulated by Mettl3 silence in HBEC. (A) The protein expression of Mettl3 and SOCS3 were detected in HBEC transfected with siRNA Mettl3 by western blot. (B) The mRNA level of Mettl3 in HBEC transfected with siRNA Mettl3 was determined by RT-qPCR. (C) The m6A peak abundance of SOCS3 mRNA transcripts in HBEC was detected by MeRIP-Seq and shown in IGV (Left). The red peaks were IP, and the blue peaks were Input. And relative m6A enrichment (IP vs Input) of SOCS3 mRNA was analyzed by GraphPad Prism (Right). (D) mRNA stability analysis of SOCS3 mRNA in HBEC transfected with siRNA-Mettl3 or siRNA-control treated with actinomycin D for 3 h and 6 h. (E) Correlation between Mettl3 and SOCS3 mRNA in COPD in the GSE130928 database. ****P<0.0005, **P<0.005, *P<0.05. Abbreviation: ns, not significant. |
SOCS3 Protein Expression Was Upregulated by Mettl3 Silence
The data obtained indicate that the upsurge in SOCS3 activity is regulated by m6A modification, resulting in the hindrance of IL-7-induced STAT5 activation, hence affecting T cell steady-state differentiation.12 This inspired us to explore whether the expression of SOCS3 in HBEC is regulated by Mettl3. As expected, the amount of SOCS3 protein in si-Mettl3 transfected HBEC considerably increased in comparison to the si-control group with or without CSE exposure (Figure 2A).
MeRIP-Seq was performed to investigate the m6A peak change of SOCS3 mRNA in Mettl3 silence HBECs. Notably, we observed a major reduction in the m6A peak of the CDS region of SOCS3 mRNA in Mettl3 silencing cells compared to the si-control group (Figure 2C), which strongly suggested that SOCS3 is most likely regulated via m6A methylation mediated by Mettl3. Furthermore, actinomycin D was used to block the synthesis of new RNA, and the SOCS3 mRNA decay was measured. The result showed that Mettl3 silence massively improved SOCS3 mRNA stability (Figure 2D), implying that m6A contributes to the destability of SOCS3 mRNA. Based on these verifications, we inferred that there is possibly a negative correlation between Mettl3 and SOCS3 expression. In line with this, we checked their mRNA expression in 22 cases with COPD in the GSE130928 database. The result confirmed that the expression of SOCS3 was significantly negatively associated with the expression of Mettl3 in COPD (Figure 2E).
Mettl3 Silence Inhibited IL-6/STAT3/SNAI1 Signaling and EMT in CSE-Induced HBECs
IL-6/STAT3 signaling is widely recognized as an important inflammatory pathway, which is negatively regulated by SOCS3. STAT3 was shown to act directly with SNAI1,8 and IL-6/STAT3/SNAI1 pathway is involved in the process of EMT.20 CSE causes inflammation of HBEC and increases the secretion of IL-6 in HBEC, which has been confirmed in our previous study.21 We previously confirmed that IL-6/STAT3 signaling positively controls the expression of SOCS3 and SNAI1, without concomitant enhancement of Mettl3 (Supplementary Figure 1A). Moreover, SOCS3 silence leads to a rise in STAT3 phosphorylation (Supplementary Figure 1B), which indicates that IL-6/STAT3 is negatively regulated by SOCS3 and aligns with existing theories.
We further observed the response of STAT3/SNAI1 signaling to Mettl3 silence. As shown in Figure 3A, according to the ratio of p-STAT3/STAT3 and the protein level of SNAI1, CSE persistently led to the activation of the IL-6/STAT3/SNAI1 signaling. And compared to the si-control group, Mettl3 silence significantly reduced the response caused by CSE.
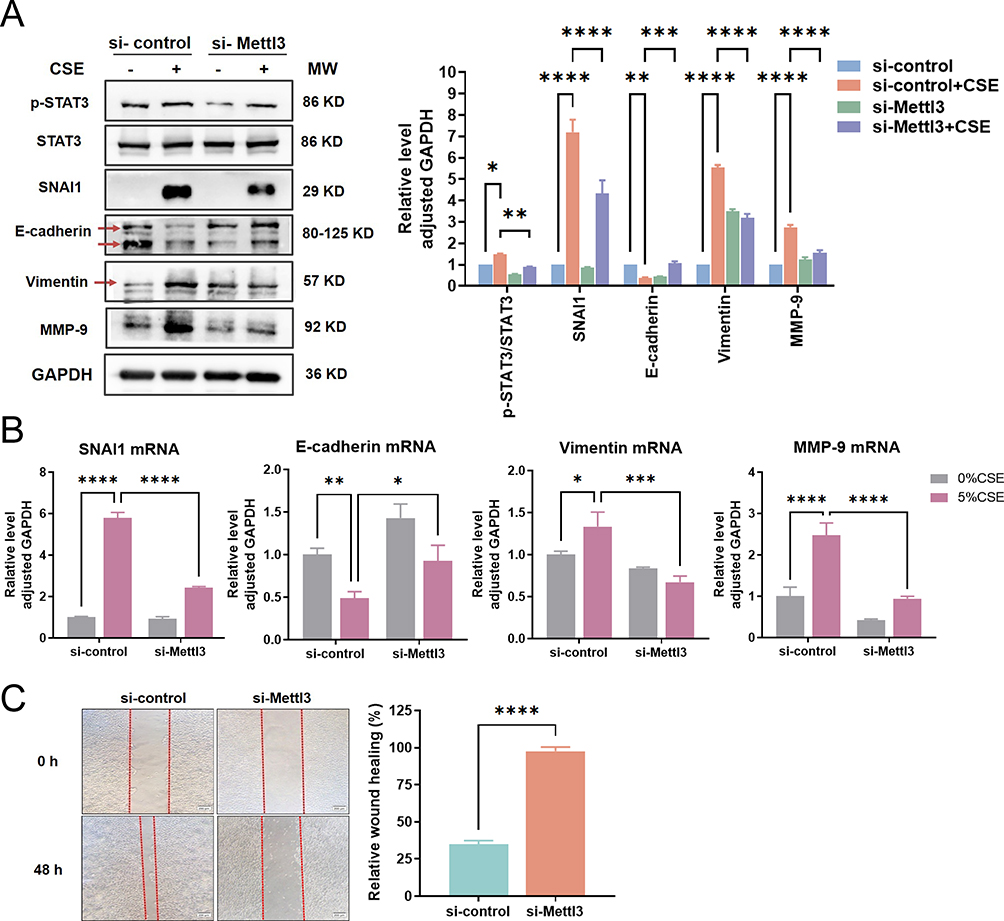 |
Figure 3 IL-6/STAT3 signaling and EMT phenotype were weakened by Mettl3 silence. (A) Western blot analysis of phosphorylation of STAT3 and protein expression levels of EMT phenotype makers in si-Mettl3 HBEC exposed to CSE. (B) RT-qPCR analysis of mRNA levels of EMT phenotype markers in si-Mettl3 HBEC exposed to CSE. (C) The wound healing of si-Mettl3 HBECs treated with 5%CSE for 48 h was recorded (Left) and quantitatively analyzed (Right). ****P<0.0005, ***P<0.0005, **P<0.005, *P<0.05. Abbreviation: ns, not significant. |
Then we conducted an observation of EMT in relation to CSE exposure where we evaluated the EMT phenotype markers through RT-qPCR and western blot. As seen in Figure 3A and B, CSE considerably reduced the protein and mRNA expression of E-cadherin in comparison to the control group, while significantly raising the protein and mRNA levels of SNAI1, Vimentin, and MMP-9. MMPs can degrade the protein components of the alveolar wall extracellular matrix and participate in airway remodeling, which is one of the powerful partners of EMT. MMP-9 has been proven to be persistent in being elevated during repair in the airways of COPD patients.5 All these data indicated that HBECs exposed to CSE presented an EMT-like phenotype.
We further explored the impact of Mettl3 silence on EMT. Vimentin, SNAI1, and MMP-9 protein expression were all dramatically decreased whereas E-cadherin protein expression was increased when Mettl3 was silenced (Figure 3A). In addition, the mRNA levels of the above four indicators were also verified to have a trend consistent with the protein (Figure 3B). Our result confirmed that Mettl3 silence in HBECs weakened the CSE-caused EMT phenotype. Moreover, a wound-healing assay identified the migratory capability of CSE-induced HBEC. The result is shown in Figure 3C, the si-control HBECs significantly repaired the wound healing after 48 h, while Mettl3 silence weakened the migration ability of HBECs. Taken together, our data suggested that Mettl3 contributes to the CSE-induced EMT process.
Cycloleucine Inhibited the EMT Phenotype of HBECs Through SOCS3 Modification
Subsequently, to further verify the actor of m6A methylation in HBECs, a methylation inhibitor cycloleucine (Cyc)22,23 was used to block m6A methylation in HBECs. It was observed that HBECs exposed to CSE exhibited a significant reduction in protein abundance of Mettl3, SNAI1, and Vimentin, while an increase in protein levels of SOCS3 and E-cadherin was noted following a 40 mM cycloleucine treatment (Figure 4), showing a similar result to Mettl3 silence. These findings indicate the critical role played by m6A RNA modification in governing the signaling of SOCS3/STAT3/SNAI1, as well as the EMT process.
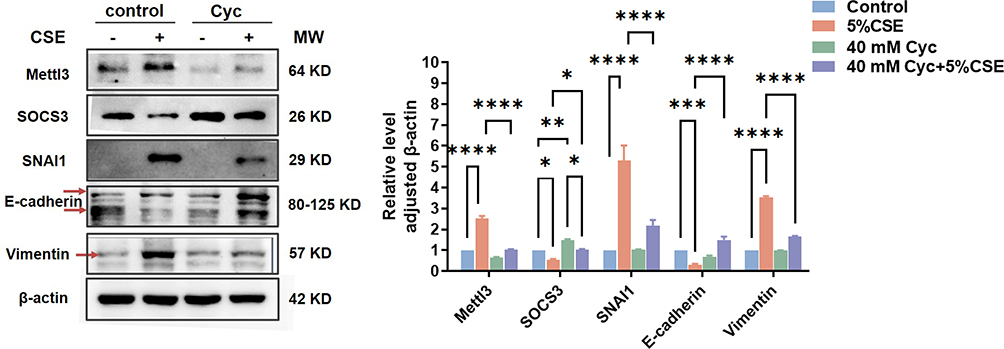 |
Figure 4 Effects of Cycloleucine on the expression levels of Mettl3, SOCS3, and EMT phenotype markers in HBEC induced by CSE. Before CSE treatment, HBEC was pretreated with 40 mM Cycloleucine for 1 h. Protein expression levels were detected by western blot. The grayscale of protein bands was determined by Image J. Two-way ANOVA was used for significance analysis. ****P<0.0005, ***P<0.0005, **P<0.005, *P<0.05. |
Discussion
Chronic obstructive pulmonary disease remains a prevailing contributor to morbidity and mortality on a global scale. Chronic airway inflammation and irreversible airflow restriction are the hallmarks of COPD. Airway remodeling and airflow restriction in COPD is significantly influenced by EMT, which can result in airway wall fibrosis and thickening.24,25 Improving the EMT process and airway remodeling is a potential new direction in the treatment of COPD.26 In this study, our findings also confirmed that cigarette smoke extract induced an EMT phenotype in 16-HBE cells in vitro. Inflammation is another pathologic feature of COPD and aids in airway remodeling. The IL-6/STAT3/SOCS3 signaling plays a crucial role in regulating inflammation and EMT,8,20 with SOCS3 acting as a negative regulator.4 Additionally, STAT3 serves a universal role in determining the expression of SNAI1/2,27 and SNAI1 is a transcriptional repressor of E-cadherin and plays an essential promoting role in the regulation of EMT.28 A study revealed that SOCS3’s transcriptional activity was markedly downregulated in COPD.29 Sabina et al demonstrated that IL-6 levels and p-STAT3 were elevated in mouse lung tissue, while SOCS3 protein was slightly decreased after tobacco smoke exposure3.30 In our study, we also demonstrated that cigarette smoke extract triggered IL-6/STAT3/SNAI1 signaling in HBE cells, which also leads to an EMT phenotype.
The process of m6A methylation serves as a regulatory mechanism for gene expression at the RNA level, occurring after transcription. Research has shown that the relative abundance of m6A in mRNA transcripts can impact RNA splicing, stability, nuclear export, and transcription. m6A can both facilitate RNA translation and promote mRNA degradation. In our investigation, we first explored that the expression of Mettl3 in COPD smokers was significantly increased, which is a validated key m6A methylation writer, implying a potential involvement of m6A modification in the emergence of COPD induced by cigarette smoking.
Currently, only a few studies have shown a connection between Mettl3-mediated m6A methylation and the developmental process of COPD. And our finding referred that m6A inhibition by Mettl3 silence or cycloleucine treatment suppressed CSE-caused STAT3/SNAI1 signaling and EMT phenotype. Finally, we uncovered that Mettl3 silence can promote the stability of SOCS3 mRNA, and cause a decrease in m6A peak abundance of SOCS3 mRNA, indicating that the presence of Mettl3 will interfere with SOCS3 mRNA stability and SOCS3 protein expression during CSE exposure in HBECs. Therefore, this study provides the first mechanistic confirmation of the role of m6A methylation in the onset of COPD.
The previous study indicated that m6A methylation directly affects RNA translation efficiency without altering transcription. A decrease in the abundance of the m6A peak of SNAI1 mRNA also was observed in our finding from MeRIP-seq (Supplementary Figure 2A), confirming that SNAI1 was regulated by m6A methylation. In contrast to the previous study where the mRNA level of SNAI1 did not decrease with decreasing m6A level, our results revealed that the mRNA level of SNAI1 was significantly weakened caused by Mettl3 silence (Supplementary Figure 2B), suggesting that the transcription of SNAI1 was regulated by Mettl3. Summarily, the regulation of SNAI1 protein expression may come from the impairment of IL-6/STAT3 signaling and the direct effects of SNAI1 m6A methylation mediated by Mettl3. As a result, Mettl3 silence increased the protein expression of SOCS3 and decreased SNAI1 expression, which attenuated the STAT3/SNAI1 signaling, and EMT phenotype of CSE-induced HBECs.
Due to the accumulation of previous studies, the actor of the SOCS3/STAT3/SNAI1 pathway in COPD has not been further elaborated in our study. However, we believe that the pathway holds promising potential for the treatment of COPD. By inducing SOCS3 abundance in airway epithelial cells, fluticasone propionate and salmeterol utilized for combination therapy were observed to reduce airway inflammation in COPD patients.31 Evidence suggests that EMT activity and airway blockage in COPD are directly connected to SNAI1.32 We also believe that m6A modification of SOCS3 is a potential and influential therapeutic target in COPD. Of course, it may be a concern that this will inevitably have additional impacts, as Mettl3-mediated m6A methylation also involves in the regulation of other gene transcripts. Anyway, our study provides evidence that the regulation of m6A methylation is a unique molecular mechanism in the progression of COPD induced by cigarette smoking.
Conclusion
In conclusion, inhaled cigarette smoking primarily targets airway epithelial cells. The activation of IL-6/STAT3/SNAI1 signaling triggers EMT processes in airway epithelial cells. Mettl3 induced by cigarette smoking enhances the IL-6/STAT3/SNAI1 signaling, as it reduces the protein expression of SOCS3 through m6A methylation, which contributes to airway remodeling in COPD (Figure 5). This finding reinforces the regulatory function of m6A methylation in COPD.
 |
Figure 5 Schematic illustration of m6A modification involvement in IL-6/STAT3/SNAI1 pathway and EMT in HBEC exposed to CSE. Inhaled cigarette smoking is primarily targeted at the airway epithelial cells. The activation of IL-6/STAT3/SNAI1 signaling triggers EMT processes in airway epithelial cells. Mettl3 induced by cigarette smoking inhibits the negative regulatory effect of SOCS3 on IL-6/STAT3/SNAI1 signaling, as it reduces the protein expression of SOCS3 through m6A methylation, which contributes to airway remodeling in COPD. |
Data Sharing Statement
The dataset used in this study can be obtained from public repositories:
GSE130928 dataset (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE130928);
GSE5058 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE5058);
Our m6A RNA-sequencing dataset is available in the GEO dataset (GSE209802: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE209802).
Ethical/Institutional Review Board and Ethics Approval Number
The ethical/institutional review board: Medical Ethics Committee of the Second Affiliated Hospital of Fujian Medical University.
Ethics approval number: No. 46 [2016].
Acknowledgments
We thank the funding offered by the Science and Technology Department of Fujian Province (China).
Funding
Funding for this study came from grants from “the Natural Science Foundation of Fujian, China, grant number 2019J01171”, “Science and Technology Project of Quanzhou, China, grant number 2018Z122” and “Youth Research Project of Fujian Health Commission, grant number 2018-2-25”.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Hogg JC, Chu F, Utokaparch S, Woods R, Paré P. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(26):2645–2653. doi:10.1056/NEJMoa032158
2. O Neill LAJ. Targeting signal transduction as a strategy to treat inflammatory diseases. Nat Rev Drug Discov. 2006;5(7):549. doi:10.1038/nrd2070
3. Yew-Booth L, Birrell MA, Lau MS, et al. JAK–STAT pathway activation in COPD. Eur Respir J. 2015;46(3):843. doi:10.1183/09031936.00228414
4. Gao H, Ward PA. STAT3 and suppressor of cytokine signaling 3: potential targets in lung inflammatory responses. Expert Opin Ther Targets. 2007;11(7):869. doi:10.1517/14728222.11.7.869
5. Nowrin K, Sohal SS, Patel R, et al. Epithelial-mesenchymal transition as a fundamental underlying pathogenic process in COPD airways: fibrosis, remodeling and cancer: expert Review of Respiratory Medicine. Expert Rev Respir Med. 2014;8(5):547–559. doi:10.1586/17476348.2014.948853
6. Gohy ST, Hupin C, Fregimilicka C, et al. Imprinting of the COPD airway epithelium for dedifferentiation and mesenchymal transition. Eur Respir J. 2015;45(5):1258–1272. doi:10.1183/09031936.00135814
7. Milara J, Peiró T, Serrano A, et al. Roflumilast N-oxide inhibits bronchial epithelial to mesenchymal transition induced by cigarette smoke in smokers with COPD. Pulm Pharmacol Ther. 2014;28(2):138–148. doi:10.1016/j.pupt.2014.02.001
8. Wang C, Dou C, Wang Y, Liu Z, Roberts L, Zheng X. TLX3 repressed SNAI1-induced epithelial-mesenchymal transition by directly constraining STAT3 phosphorylation and functionally sensitized 5-FU chemotherapy in hepatocellular carcinoma. Int J Biol Sci. 2019;15(8):1696–1711. doi:10.7150/ijbs.33844
9. He PC, He C. m6A RNA methylation: from mechanisms to therapeutic potential. EMBO J. 2021;40(3). doi:10.15252/embj.2020105977
10. Yue B, Song C, Yang L, Cui R, Zhao G. METTL3-mediated N6-methyladenosine modification is critical for epithelial-mesenchymal transition and metastasis of gastric cancer. Mol Cancer. 2019;18(1). doi:10.1186/s12943-019-1065-4
11. Luo S, Liao C, Zhang L, et al. METTL3-mediated m6A mRNA methylation regulates neutrophil activation through targeting TLR4 signaling. Cell Rep. 2023;42(3):112259. doi:10.1016/j.celrep.2023.112259
12. H-B L, Tong J, Zhu S, et al. m6A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathways. Nature. 2017;548(7667):338–342. doi:10.1038/nature23450
13. Hua W, Zhao Y, Jin X, et al. METTL3 promotes ovarian carcinoma growth and invasion through the regulation of AXL translation and epithelial to mesenchymal transition. Gynecol Oncol. 2018;151(2):356–365. doi:10.1016/j.ygyno.2018.09.015
14. O’Beirne SL, Kikkers SA, Oromendia C, Salit J, Cloonan SM. Alveolar macrophage immunometabolism and lung function impairment in smoking and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2019;201(6)735–739. doi:10.1164/rccm.201908-1683LE
15. Carolan BJ, Heguy A, Harvey BG, Leopold PL, Ferris B, Crystal RG. Up-regulation of expression of the ubiquitin carboxyl-terminal hydrolase L1 gene in human airway epithelium of cigarette smokers. Cancer Res. 2006;66(22):10729–10740. doi:10.1158/0008-5472.CAN-06-2224
16. Schamberger AC, Mise N, Jia J, et al. Cigarette smoke–induced disruption of bronchial epithelial tight junctions is prevented by transforming growth factor-β. Am J Respir Cell Mol Biol. 2014;50(6):1040–1052. doi:10.1165/rcmb.2013-0090OC
17. Wang X, Lu Z, Gomez A, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505(7481):117–120. doi:10.1038/nature12730
18. Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet J. 2011;17(1). doi:10.14806/ej.17.1.200
19. Helga T, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;2:178–192. doi:10.1093/bib/bbs017
20. Kim SH, Yoo HS, Joo MK, et al. Arsenic trioxide attenuates STAT-3 activity and epithelial-mesenchymal transition through induction of SHP-1 in gastric cancer cells. BMC Cancer. 2018;18(1):150. doi:10.1186/s12885-018-4071-9
21. Zhang Y, Wang L, Liu Y, Yan F. Mettl3-mediated transcriptome-wide m6A methylation induced by cigarette smoking in human bronchial epithelial cells. Toxicol In Vitro. 2023;89:105584. doi:10.1016/j.tiv.2023.105584
22. Kang H, Zhang Z, Yu L, Li Y, Liang M, Zhou L. FTO reduces mitochondria and promotes hepatic fat accumulation through RNA demethylation. J Cell Biochem. 2018;119(7):5676–5685. doi:10.1002/jcb.26746
23. Luo G, Xu W, Zhao Y, et al. RNA m(6) A methylation regulates uveal melanoma cell proliferation, migration, and invasion by targeting c-Met. J Cell Physiol. 2020;235(10):7107–7119. doi:10.1002/jcp.29608
24. Milara J, Peiró T, Serrano A, Cortijo J. Epithelial to mesenchymal transition is increased in patients with COPD and induced by cigarette smoke. Thorax. 2013;68(5):410–420. doi:10.1136/thoraxjnl-2012-201761
25. Sohal SS, Walters EH, Yamaji-Kegan K. Role of epithelial mesenchymal transition (EMT) in chronic obstructive pulmonary disease (COPD). Respir Res. 2013;14(1):1–3. doi:10.1186/1465-9921-14-1
26. Wang Q, Wang Y, Zhang Y, Zhang Y, Xiao W. The role of uPAR in epithelial-mesenchymal transition in small airway epithelium of patients with chronic obstructive pulmonary disease. Respir Res. 2013;14(1):67. doi:10.1186/1465-9921-14-67
27. Kielbik M, Szulckielbik I, Klink M. Impact of selected signaling proteins on SNAIL 1 and SNAIL 2 expression in ovarian cancer cell lines in relation to cells‘cisplatin resistance and EMT markers level. Int J Mol Sci. 2021;22(2):980. doi:10.3390/ijms22020980
28. Puisieux A, Brabletz T, Caramel J. Oncogenic roles of EMT-inducing transcription factors. Nat Cell Biol. 2014;16(6):488–494. doi:10.1038/ncb2976
29. Springer J, Scholz FR, Peiser C, et al. Transcriptional down-regulation of suppressor of cytokine signaling (SOCS)-3 in chronic obstructive pulmonary disease. J Occup Med Toxicol. 2013;8(1):29. doi:10.1186/1745-6673-8-29
30. Lee JH, Kim C, Sethi G, Ahn KS. Brassinin inhibits STAT3 signaling pathway through modulation of PIAS-3 and SOCS-3 expression and sensitizes human lung cancer xenograft in nude mice to paclitaxel. Oncotarget. 2015;6(8):6386. doi:10.18632/oncotarget.3443
31. Nasreen N, Khodayari N, Sukka-Ganesh B, Peruvemba S, Mohammed KA. Fluticasone propionate and Salmeterol combination induces SOCS-3 expression in airway epithelial cells. Int Immunopharmacol. 2012;12(1):217–225. doi:10.1016/j.intimp.2011.11.014
32. Mahmood MQ, Walters EH, Shukla SD, et al. β-catenin, Twist and Snail: transcriptional regulation of EMT in smokers and COPD, and relation to airflow obstruction. Sci Rep. 2017;7(1):10832. doi:10.1038/s41598-017-11375-x
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Severity of Lung Function Impairment Drives Transcriptional Phenotypes of COPD and Relates to Immune and Metabolic Processes
Negewo NA, Gibson PG, Simpson JL, McDonald VM, Baines KJ
International Journal of Chronic Obstructive Pulmonary Disease 2023, 18:273-287
Published Date: 14 March 2023
Analysis of Airway Thickening and Serum Cytokines in COPD Patients with Frequent Exacerbations: A Heart of the Matter
Lin Y, Sang L, Wang J, Chen Y, Lai J, Zhu X, Yang Y, Zhang Z, Liu Y, Wen S, Zhang N, Zhao D
International Journal of Chronic Obstructive Pulmonary Disease 2023, 18:2353-2364
Published Date: 30 October 2023
A miRNA-21-Mediated PTEN/Akt/NF-κB Axis Promotes Chronic Obstructive Pulmonary Disease Pathogenesis
Sai X, Qin C, Zhang Z, Yu H, Bian T
International Journal of Chronic Obstructive Pulmonary Disease 2024, 19:1141-1151
Published Date: 25 May 2024
Potential of the Advanced Lung Cancer Inflammation Index as a Risk Marker for Asthma-Chronic Obstructive Pulmonary Disease Overlap Syndrome and COPD: Evidence from NHANES 2007-2018
Chen N, Liu Y, Wang Q, Wang M, Wang J, Chen W
International Journal of Chronic Obstructive Pulmonary Disease 2025, 20:905-917
Published Date: 31 March 2025
The Inflammatory Roles of n-3 and n-6 Polyunsaturated Fatty Acids in COPD: Clinical Implications and Underlying Mechanisms
Wang Y, Bao Y, Liu B, Li H, Duan H, Wang Y, Zhang J, Wu W, Li P, Liu X
Journal of Inflammation Research 2026, 19:556221
Published Date: 15 January 2026