Back to Journals » International Journal of Nanomedicine » Volume 20
Local M1 Macrophage Reprogramming with Gluconic Acid-Coated Selenium Nanoparticles
Authors Liao ZX ![]() , Ou DL
, Ou DL ![]() , Hsu CL
, Hsu CL ![]() , Lu LN, Wen CH, Lu L
, Lu LN, Wen CH, Lu L ![]() , Chiu CL, Yang PC, Tseng SJ
, Chiu CL, Yang PC, Tseng SJ ![]()
Received 25 July 2025
Accepted for publication 26 November 2025
Published 3 December 2025 Volume 2025:20 Pages 14439—14455
DOI https://doi.org/10.2147/IJN.S556099
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lijie Grace Zhang
Zi-Xian Liao,1 Da-Liang Ou,2 Chia-Lang Hsu,2,3 Lin-Ni Lu,1 Cheng-Han Wen,4 Lin Lu,4 Chun-Lun Chiu,4 Pan-Chyr Yang,5 S.-Ja Tseng6,7
1Institute of Medical Science and Technology, National Sun Yat-sen University, Kaohsiung, Taiwan; 2Graduate Institute of Oncology, National Taiwan University College of Medicine, Taipei, Taiwan; 3Department of Medical Research, National Taiwan University Hospital, Taipei, Taiwan; 4Department of Research and Development, True Diamond Health Co., Ltd, Taoyuan, Taiwan; 5Department of Internal Medicine, National Taiwan University College of Medicine, Taipei, Taiwan; 6Department of Pharmacology, College of Medicine, National Cheng Kung University, Tainan, Taiwan; 7NCKU Center of Applied Nanomedicine, Tainan, Taiwan
Correspondence: Pan-Chyr Yang, Department of Internal Medicine, National Taiwan University College of Medicine, Taipei, Taiwan, Email [email protected] S.-Ja Tseng, Department of Pharmacology, College of Medicine, National Cheng Kung University, Tainan, Taiwan, Email [email protected]
Purpose: While reprogramming tumor-associated macrophages (TAMs) using cytokines shows promise for cancer therapy, its clinical translation is limited by poor bioavailability. Essential mineral selenium (Se), via selenoproteins, is crucial for innate immunity and adaptive immunity regulation.
Methods: Addressing the need for safer, more effective methods to enhance macrophage function, we leveraged the essential mineral Se to create gluconic acid-coated Se nanoparticles (GA-SeNPs). The in vivo efficacy of GA-SeNPs was assessed via intratumoral injection in a B16-F10 melanoma BALB/c mouse model, mirroring the administration route of the first virotherapy for advanced melanoma.
Results: These nanoparticles successfully induced M2-to-M1 macrophage repolarization and inhibited cancer cell growth through reactive oxygen species (ROS) generation. We confirmed through transcriptomic analysis that GA-SeNPs influence the genes of key components in the biosynthesis of selenoproteins. Additionally, GA-SeNPs influence oxidative phosphorylation, inflammatory, and ribosome pathways by promoting the shift of M2 macrophages to an M1 phenotype. Crucially, in a melanoma mouse model, GA-SeNPs treatment yielded a > 4-fold tumor weight reduction and effectively repolarized TAMs to an M1 phenotype while maintaining TAMs levels. GA-SeNPs inhibit cancer growth in vivo by disrupting the immunosuppressive tumor microenvironment. They maintain total TAM counts while strongly promoting M2-to-M1 repolarization.
Conclusion: Their dual localization within both TAMs and cancer cells further highlights their therapeutic potential, presenting a promising strategy to advance TAM-based cancer therapies and improve clinical outcomes.
Keywords: tumor-associated macrophages, selenium nanoparticles, repolarization, reactive oxygen species, apoptosis
Introduction
Tumor-associated macrophages (TAMs)-based cell therapies are crucial for targeting cancer cells or pathogens.1–3 During cancer progression, it is probable that TAMs populations will shift their phenotype, and this shift can contribute to cancer resolution or progression.4 Notably, TAMs-reprogramming therapeutics have seen a marked increase in clinical trials over the past decade.2 TAMs are highly diverse cells that can quickly adapt their function in response to local microenvironmental signals.5,6 Macrophage colony-stimulating factor (M-CSF) and granulocyte-macrophage colony-stimulating factor (GM-CSF) are crucial cytokines for macrophage differentiation in vivo.7 The US Food and Drug Administration (FDA)-approved oncolytic virus Talimogene laherparepvec (T-VEC) exploits this, expressing GM-CSF to enhance anti-tumor immunity in melanoma treatment.8 However, improving their bioavailability is crucial,2,9–13 as the main obstacle in translating preclinical studies into successful clinical TAMs-reprogramming therapies has been their insufficient efficacy.2
Alternatively, cancer cells modify their metabolic pathways, resulting in increased lactate production within the tumor microenvironment.14 Tumor-derived lactate enhances M2 TAM immunosuppression and promotes tumor growth by stimulating their favored metabolic pathway, oxidative phosphorylation, through mitochondrial signaling.6,15 Lactate is crucial for cellular energy and normal physiology, but its role is subverted in cancer. In tumors, accelerated glycolysis dramatically increases lactate production, turning this metabolite into a key driver of tumor cell proliferation.16 Crucially, the tumor microenvironment exerts a profound influence on cancer progression by actively promoting cell survival, facilitating metastasis, and driving overall tumor growth.17 Our previous findings suggest that delivery systems capable of sustained and controlled release of lactate oxidase (LOX), which facilitates the conversion of lactate into pyruvate, could lead the way in pioneering lactate depletion therapy within the tumor microenvironment.6,10–12,15 Therefore, there is a pressing need to develop a safe and effective agent or strategy to address these critical issues and enhance the functionality of TAMs.
Selenium (Se), an essential mineral, is particularly notable for its role in human health.18 When incorporated into proteins as selenoproteins, selenium is crucial for regulating innate immunity and adaptive immunity.19,20 Due to their unique physicochemical properties and excellent biocompatibility, Se-based nanomaterials have shown significant potential as therapeutic agents or drug carriers.21,22 Se plays a multifaceted role in cancer, from reducing incidence and suppressing tumor progression to potentially mitigating treatment side effects.23–25 Its efficacy as a chemopreventive agent is linked to its ability to modulate cell behavior, though clinical outcomes vary depending on the Se compound and dosage used.26
To address the poor therapeutic bioavailability and lactate-driven immunosuppression, we utilized the Se, which is crucial for immune regulation via selenoproteins. We designed gluconic acid-coated Se nanoparticles (GA-SeNPs) to overcome the limitations of traditional cytokine therapy. We designed a one-pot synthesis method to create gluconic acid (GA)-coated SeNPs (GA-SeNPs). This process leverages the chemical transformation of the reductant (glucose, Glu) to simultaneously form and coat the SeNPs. Furthermore, The GA coating facilitates enhanced cellular uptake via its interaction with the cell membrane.27 Taken together, this presents a strong justification for developing GA-SeNPs to optimize macrophage-based therapy (Figure 1A). In acidic condition adjusted by lactate, GA-SeNPs treatment not only repolarized M2-like macrophages to an M1-like phenotype in the tumor microenvironment through upregulation of selenoprotein biosynthetic and M1-associated genes, and downregulation of M2-associated genes but also directly suppressed cancer cell proliferation and migration via reactive oxygen species (ROS) generation. Alternatively, the combination of GA-SeNPs with GM-CSF and LOX effectively enhanced the in vitro conversion ratio of M2- to M1-like macrophages. The in vivo efficacy of GA-SeNPs was tested in a B16-F10 melanoma xenograft mouse model via intratumoral injection, reflecting the route of first T-VEC virotherapy administration for advanced melanoma patients.8 Treatment with GA-SeNPs resulted in effective immunity remodeling and tumor inhibition, significantly improving the outcomes in a mouse model.
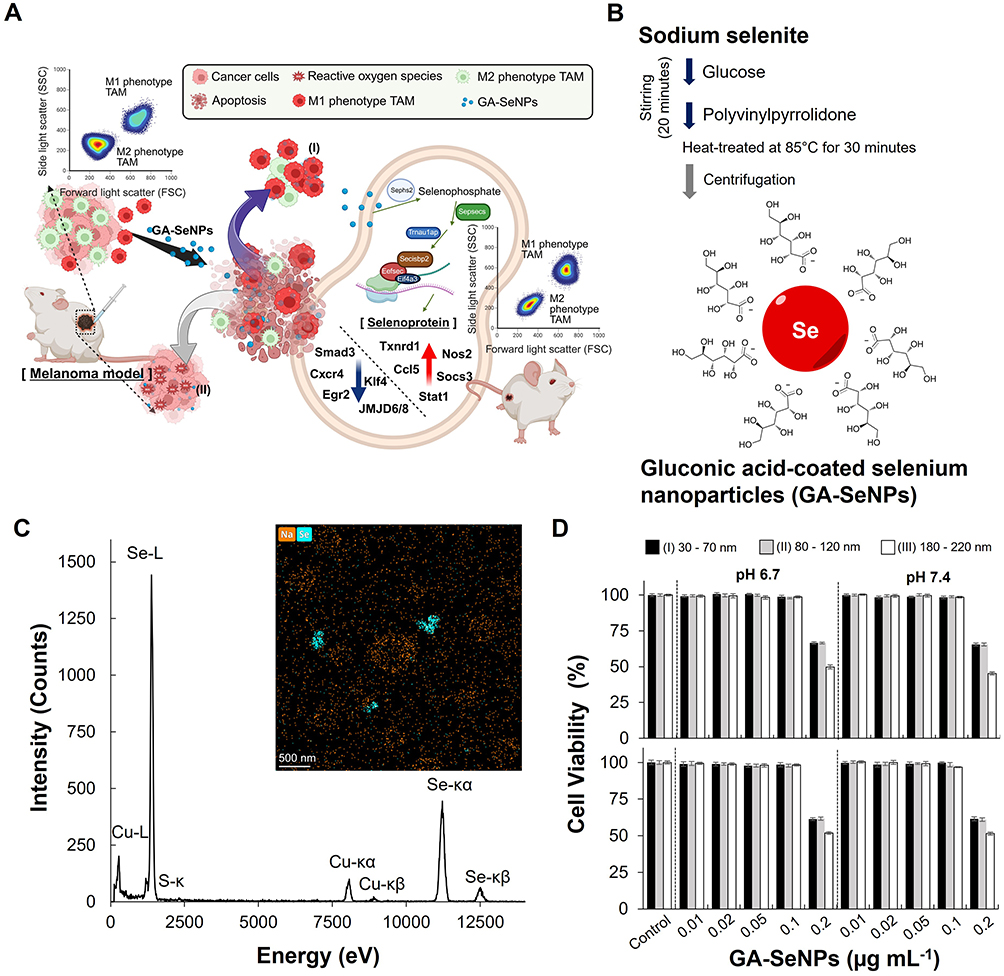 |
Figure 1 Characterization and functional analysis of gluconic acid-coated selenium nanoparticles (GA-SeNPs). (A) Schematic illustrating GA-SeNPs-mediated M2-like to M1-like macrophage repolarization (I) and subsequent reactive oxygen species (ROS)-dependent inhibition of cancer cell proliferation (II). GA-SeNPs treatment converted M2-like macrophages back to an M1-like phenotype in the tumor microenvironment by adjusting gene expression: it upregulated genes for selenoprotein biosynthesis and M1 function, while downregulating M2-associated genes. Blue colored arrow: lower expression; red colored arrow: higher expression. (B) Synthesis scheme and structure of GA-SeNPs. (C) Energy-dispersive X-ray spectroscopy (EDS) spectrum and elemental mapping of GA-SeNPs, confirming selenium (Se) presence. Labelled peaks indicate Se X-ray emissions. (D) Cytotoxicity of GA-SeNPs (varying diameters and concentrations) on M2-like macrophages (upper panel) and B16-F10 cells (lower panel) after 24h incubation. Data represent the mean of triplicate measurements ± s.d. The particle size of the GA-SeNPs was categorized into three distinct ranges: 30 to 70 nm (I), 80 to 120 nm (II), and 180 to 220 nm (III). |
Materials and Methods
Materials
Sodium selenite, polyvinylpyrrolidone, glucose, phosphate-buffered saline (PBS, pH 7.4), 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI), lactate oxidase (LOX) from Aerococcus viridians, and the Nitrite/Nitrate Assay Kit (colorimetric) were obtained from Sigma-Aldrich Co. (St. Louis, MO). The CellTiter 96 AQueous One Solution Cell Proliferation Assay kit for the MTS assay (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) was purchased from Promega (Madison, WI). The anti-iNOS antibody was purchased from Abcam (Cambridge, MA), and the mouse MMR/CD206 antibody was supplied by R&D Systems (Minneapolis, MN). Goat anti-rabbit IgG (H+L)-TAMRA was obtained from Leadgene Biomedical, Inc. (Taiwan). Mouse recombinant interleukin-4 (IL-4) was purchased from GeneTex (Irvine, USA). Mouse granulocyte-macrophage colony-stimulating factor (GM-CSF) was purchased from abcam (Cambridge, UK). Mouse IL-10 ELISA Kit and TNF-α ELISA Kit were sourced from BioLegend (Hsinchu, Taiwan). The Click-iT Plus TUNEL Assay Kits and CellROX Green Reagent were purchased from Thermo Fisher Scientific (Waltham, USA). IRF5 antibody, arginase 1 antibody, and goat anti-rabbit IgG antibody (HRP) for Western blotting were obtained from GeneTex (Irvine, USA).
In vitro Culture of Mouse Macrophages and Cancer Cells
The mouse macrophage RAW 264.7 cell line (ATCC TIB-71), representing M0 macrophages, was cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U mL−1 penicillin, 100 µg mL−1 streptomycin, and 3.7 g L−1 sodium bicarbonate. The cells were maintained in a 37°C incubator with 5% CO2. To induce the polarization of M2-like macrophages from M0 macrophages, the M0 macrophages were incubated in a culture medium containing 20 ng mL−1 interleukin-4 (IL-4) for 24 hours to generate M2-like macrophages.6 Following incubation, the treated macrophages were washed three times with PBS and then used as M2-like macrophages in subsequent studies.
The B16-F10 murine melanoma cell line (ATCC CRL-6475) was cultured at 37°C with 5% CO2 in complete medium composed of DMEM supplemented with 10% FBS, 100 U mL−1 penicillin, 100 µg mL−1 streptomycin, and 3.7 g L−1 sodium bicarbonate.
Synthesis and Characterization of Gluconic Acid-Coated Selenium Nanoparticles (GA-SeNPs)
0.5 g of sodium selenite was dissolved in 5 mL of deionized water. 1.1 g of glucose (Glu) as a reductant was gradually added into the precursor solution, followed by 0.7 mL of polyvinylpyrrolidone (1 mg mL−1). The precursor mixture was continuously stirring for 20 minutes until a homogeneous solution is formed. Subsequently, the mixed solution was heated at 85°C for 30 minutes, allowing it to develop a dark reddish-brown color. The reacted solution was rapidly cooled and centrifuged at 12,000 rpm to obtain gluconic acid (GA)-coated selenium nanoparticles (GA-SeNPs). The resulting particles are spherical, with a diameter of 30–70 nm. Additionally, varying the concentration of polyvinylpyrrolidone (1.0 mL or 1.5 mL) and concentration of Glu (1.0 g), and centrifugation conditions (12,000 rpm or 10,000 rpm) produced GA-SeNPs with a diameter of 80–120 nm or 180–220 nm. The GA-SeNPs collected after the various centrifugation steps were stored for up to seven months at 25 °C until use. X-ray diffraction (XRD) was performed to determine the atomic and molecular structure of GA–SeNPs. Fourier Transform Infrared (FT-IR) spectroscopy was conducted in the 4000–400 cm−1 range using a Bruker FTIR spectrophotometer to characterize the GA-SeNPs. The physical and chemical properties of the samples were verified using transmission electron microscopy (TEM) and energy-dispersive X-ray spectroscopy (EDS).
Cytotoxicity of GA-SeNPs
To thoroughly evaluate the impact of GA-SeNPs on the viability of M2-like macrophages or B16-F10 cells, M2-like macrophages or B16-F10 cells were exposed to GA-SeNPs of varying diameters at different concentrations (0.01, 0.02, 0.05, 0.1, or 0.2 μg mL−1). M2-like macrophages (1 x 104 cells) were seeded into each well of a 96-well plate and allowed to adhere overnight in culture medium. Various concentrations of GA-SeNPs were added daily, and the cells were incubated with the GA-SeNPs for a total of 24 hours to ensure sufficient interaction time. Following the treatment, cell viability was assessed using the MTS assay.
In vitro Analysis of Macrophages Treated with GA-SeNPs
Each well of a 24-well plate was seeded with 7×104 M2-like macrophages and cultured overnight. To mimic different physiological environments, the culture medium was carefully adjusted to either a neutral pH of 7.4 or a more acidic pH of 6.7 using L-lactate at a concentration of 1.0 M. This pH adjustment was crucial for investigating the potential influence of the microenvironment on the macrophages’ response to the treatments. Macrophages were exposed to GA-SeNPs at varying diameters and concentrations (0.01, 0.02, 0.05, or 0.1 μg mL−1) in pH-adjusted culture medium (7.4 or 6.7) for 72 hours, with the culture medium replaced daily.
To confirm the specificity of M1-like or M2-like macrophage induction, markers such as iNOS and CD206 were assessed in all macrophages,6 including those induced to the M1 or M2 phenotypes. The treated macrophages were fixed with 4% paraformaldehyde (PFA) and immune-stained using Anti-iNOS antibody (Abcam, Cambridge, MA) and Anti-CD206 antibody (R&D system, Minneapolis, MN), which are specific to the iNOS and CD206 markers, respectively, to observe the distribution of M1-like (iNOS+CD206−) or M2-like macrophages (iNOS−CD206+). Signal amplification was achieved using Rabbit Anti-Goat IgG antibody-FITC (Genetex, Irvine, USA) for CD206 and Goat anti-Rabbit IgG (H+L)-TAMRA (Leadgene, Taiwan) for iNOS. The stained cells were then observed under a confocal microscope. Additionally, the cell nuclei were stained with DAPI. Alternatively, immune-stained cells were quantitatively analyzed by flow cytometry (Beckman Coulter, Fullerton, CA, USA), where cells were appropriately gated using forward and side scatter, and 10,000 events per sample were collected. Untreated cells served as the negative control.
GA-SeNPs in the Treatment of B16-F10 Melanoma Cells
To more effectively observe the antitumor effects in our experimental setup, we utilized a strategy that involved infecting mouse melanoma B16-F10 cells with a lentivirus encoding green fluorescent protein (GFP). This approach enabled us to create a B16-F10 cell line (B16-F10-GFP) that stably expresses GFP, providing a reliable marker for tracking and visualizing the cancer cells during co-culture with GA-SeNPs or macrophages. After transducing the B16-F10 cells with the GFP-encoding lentivirus, the B16-F10-GFP cells were cultured under specific conditions to maintain their viability and promote stable GFP expression. The transduced cells were grown in DMEM, supplemented with 10% FBS to provide essential growth factors, 100 U mL−1 penicillin and 100 µg mL−1 streptomycin to prevent bacterial contamination, 3.7 g L−1 sodium bicarbonate to maintain pH balance, and 2 µg mL−1 puromycin to selectively maintain only those cells that had successfully integrated the GFP gene. This selective pressure ensured that the resulting B16-F10-GFP cell line consistently expressed GFP, making it an ideal model for subsequent antitumor studies.
To assess the inhibitory effect of GA-SeNPs on cancer cell migration, a wound healing assay was conducted using B16-F10 cells. These cells were seeded into 24-well culture plates at a density of 1×106 cells per well and allowed to culture overnight. The following day, the cells were treated daily with GA-SeNPs of varying diameters in fresh culture medium. A vertical scratch was then made across the center of each well using a pipette tip to create a cell-free zone. After washing the wells twice with PBS to remove any non-adherent cells, fresh culture medium was added, and the cells were incubated for 3 days. The plates were imaged using a confocal microscope.
Novelty, SeNPs induce the ROS-mediated cell death in cancer cells.22,28 ROS levels in B16-F10 cells treated daily with GA-SeNPs of varying diameters for 72 hours were stained by using the CellROX green reagent (Invitrogen, Camarillo, CA) and observed with a confocal microscope, as previously described.11 Alternatively, B16-F10 cells treated with GA-SeNPs of varying diameters were stained with fluorescein using the Click-iT Plus TUNEL Assay Kits for In Situ Apoptosis Detection (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. Apoptotic cells were qualitatively analyzed using a fluorescence microscope, with untreated cells as the negative control. Additionally, cell nuclei were stained with DAPI.
Modulating M2-Like Macrophage and Cancer Cell Dynamics in Co-Culture with GA-SeNPs
We next investigated the interactions between GA-SeNPs, M2-like macrophages, and B16-F10-GFP cells to mimic the systemic circulation of GA-SeNPs in the tumor microenvironment. M2-like macrophages were seeded in each well of a 24-well plate at densities of 1 x 104, 3 x 104, and 5×104 cells, along with a fixed number of B16-F10-GFP cells (1 x 104 cells). After overnight incubation, the cells were treated daily with GA-SeNPs of different diameters in the culture medium and incubated for three days. After three days treatment, treated cells were fixed with 4% PFA. Furthermore, the fixed cell nuclei were stained with DAPI. Confocal microscopy was used to image the plates. The GFP-expressing cells were analyzed using quantitative flow cytometry (Beckman Coulter, Fullerton, CA, USA), acquiring a minimum of 10,000 events. Untreated cells served as the negative control.
Treatment of GA-SeNPs Combined with GM-CSF, LOX, and GM-CSF/LOX on M2-Like Macrophages
Each well of a 24-well plate was seeded with a specified number (7 x 104) of M2-like macrophages. The M2-like macrophages were allowed to adhere and stabilize by culturing them overnight under standard conditions. Following this initial culture period, the M2-like macrophages were subjected to treatment with GA-SeNPs in combination with GM-CSF (25ng mL−1)29 or LOX (0.025 U mL−1),6 or a dual combination of GM-CSF/LOX at pH 7.4 or pH 6.7, with daily culture medium replacement. The macrophages were then incubated with the treatment combinations for a period of 72 hours, allowing sufficient time for the cellular responses to develop before further analysis. M2-like macrophages were subjected to a 72-hour treatment with GA-SeNPs in combination with GM-CSF, LOX, or a combination of GM-CSF/LOX to assess the impact on cytokine production. Following the treatment period, the supernatants from the culture medium were carefully collected for analysis. To quantify the levels of key cytokines, including pro-inflammatory tumor necrosis factor-α (TNF-α)30 and anti-inflammatory interleukin 10 (IL-10),31 an enzyme-linked immunosorbent assay (ELISA) was employed.
Mice Studies
The animal experiments (IACUC-20160366) were approved by the Institutional Animal Care and Use Committee of the College of Medicine, National Taiwan University, and conducted in compliance with the guidelines outlined in the Guide for the Care and Use of Laboratory Animals. Six-week-old male BALB/c mice, sourced from the National Laboratory Animal Center in Taiwan, were housed under controlled conditions with a 12-hour light/dark cycle. Up to five mice were housed per cage, with unrestricted access to food and water.
To assess the effects of GA-SeNPs, 2×106 B16-F10 cells were subcutaneously injected into the right flank of mice to establish xenograft tumors. The design of experimental timeline and protocol followed the schematic. Once tumor volumes reached approximately 200 mm3, mice were randomly assigned to three experimental groups (n = 5 per group). GA-SeNPs (30–70 nm; selenium concentration: 0.1 mg kg−1 day−1) were administered directly into the tumors at varying frequencies (six or nine injections). The control group received PBS injections as a placebo treatment. Tumor volumes were measured with calipers post-excision or after tumor fixation using the formula: 0.5L2W (where L is the tumor length and W is the tumor width).
In vivo Assay
Xenograft tumors were harvested for comprehensive analysis. Tumors or organs (liver, heart, kidney, lung, and spleen) were formalin-fixed, embedded in paraffin (FFPE), sectioned, and subjected to standard histopathological imaging and analysis. To assess apoptosis, xenograft tumor sections were performed on 5-μm sections and stained using the Fluorescein In Situ Cell Death Detection Kit. To analyze immune cell distribution within the melanoma microenvironment, tumors were processed into FFPE sections and evaluated for lymphocyte infiltration. The Polaris system (Akoya Biosciences) was used in conjunction with the Opal 7-Color Manual IHC Kit and the anti-Rabbit Manual IHC Kit (Akoya Biosciences, NEL810001KT and NEL840001KT) to detect specific immune cell markers in FFPE sections. Opal dyes were assigned as follows: Opal620 for rabbit anti-CD4 (Abcam), Opal570 for rabbit anti-CD8 (Bioss), Opal650 for rabbit anti-MHC II (Bioss), Opal520 for rabbit anti-CD19 (HistoSure), Opal520 for rabbit anti-CD206 (Bioss), and Opal690 for rabbit anti-F4/80 (HistoSure). Multispectral imaging of tumor sections was performed using the Phenochart and inForm software (Akoya Biosciences). The inForm software’s machine-learning mode was applied, with ten representative tumor images selected as the training set. These images were manually annotated to identify various cell types based on marker expression: B cells (CD19⁺), CD4⁺ T cells (CD4⁺), CD8⁺ T cells (CD8⁺), M1 phenotype TAMs (F4/80⁺MHC II⁺CD206−), and M2 phenotype TAMs (F4/80⁺MHC II−CD206⁺). Cell segmentation was performed using nuclear staining with DAPI.
RNA Sequencing and Data Analysis
Total RNA was extracted using the TRIzol reagent (Thermo Fisher Scientific, CA, USA). RNA sequencing libraries were prepared with the Illumina Stranded mRNA Prep Kit (Illumina, CA, USA) and sequenced on an Illumina NovaSeq™ 6000 platform. Raw sequencing reads in FASTQ format underwent quality control (QC) using FastQC (v0.11.9), followed by adaptor sequence trimming with cutadapt (v3.5). High-quality reads were then aligned to the mouse reference genome (GRCm38) using STAR aligner (v2.7.8a)32 with the --quantMode parameter enabled to generate gene-level counts based on the GENCODE vM25 annotation. Between-sample normalization was performed using the trimmed mean of M-values (TMM) method, and transcripts per million (TPM) values were calculated for downstream analysis. Differential expression analysis was conducted using NOISeq, with genes exhibiting a probability > 0.8 considered differentially expressed. Gene set enrichment analysis (GSEA) of gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) were performed using the R package clusterProfiler,33 incorporating gene sets from MSigDB (v7.4). All analyses were conducted in R (v4.4.1).
Statistical Analysis
All in vitro experiments were performed in triplicate, and the results are presented as mean ± standard deviation (s.d). Differences between the control and experimental groups were evaluated using a two-tailed Student’s t-test. Statistical significance was defined as P < 0.05 (*) or P > 0.4 (#).
Results
Properties of Synthesized GA-SeNPs
To synthesize GA-SeNPs, Glu and polyvinylpyrrolidone were introduced into a sodium selenite solution, driven by the oxidation of Glu. Following the addition of these components, the reaction mixture was subjected to centrifugation and facilitated the formation and separation of the GA-SeNPs from the solution (Figure 1B), which exhibited the average zeta potential of −16.25 mV (Figure S1). Subsequently, The FT-IR spectra of GA-SeNPs’ carboxylic acids exhibited a strong C=O band at 1645 cm−1 and a broad O-H stretch band ranging from 3700 to 2500 cm−1 (Figure S2). The combined presence of these features strongly confirmed the presence of a carboxylic acid of GA. The XRD characterization shown in Figure S3A revealed a peak in curve A, indicating the crystalline growth of SeNPs. The EDS spectra and mapping in Figure 1C confirmed the presence of Se and copper (Cu), validating the sample’s purity and absence of contamination from other elements. The detected Cu was attributed to the copper grid substrate used in the analysis. Additionally, the results confirmed that the GA-SeNPs consisted of spherical Se. The GA-SeNPs were studied at three particle size increments: 30–70 nm, 80–120 nm, and 180–220 nm, as observed through TEM analysis (Figure S3B).
To comprehensively assess the effects of GA-SeNPs on the viability of M2-like macrophages and B16-F10 melanoma cells, we conducted a series of experiments where these cells were exposed to GA-SeNPs of varying diameters and concentrations. The aim was to determine the optimal concentration that would have a minimal cytotoxic impact on the cells while still allowing us to study the nanoparticles’ biological effects. Cells were treated with GA-SeNPs at different concentrations, ranging from 0.01 μg mL−1 to 0.2 μg mL−1, to observe any potential changes in cell viability (Figure 1D). The results indicated that concentrations below 0.1 μg mL−1 had no significant impact on the viability of either M2-like macrophages or B16-F10 cells. This finding was consistent across all tested diameters of the nanoparticles, suggesting that GA-SeNPs at these lower concentrations are biocompatible and do not induce cytotoxicity under the conditions tested. Based on these observations, we determined that a concentration of 0.1 μg mL−1 is appropriate for use in subsequent experiments. This concentration was selected as it provides a balance between maintaining cell viability and allowing for effective investigation the biological activities of GA-SeNPs.
GA-SeNPs Induced Macrophage Repolarization Under Acidic Conditions
Tumor-secreted lactate could drive M2 phenotype TAMs polarization, with M2-like macrophages being more prevalent in acidic tumor microenvironments.6,14,29,34 To assess the effect of GA-SeNPs on macrophage repolarization, we analyzed the distribution of M1-like and M2-like macrophages, starting with M2-like macrophages and treating them with various concentrations of GA-SeNPs of varying diameters at pH 7.4 or pH 6.7. At both pH 7.4 and pH 6.7, an increase in M1-like macrophage intensity was observed following stimulation with GA-SeNPs of varying diameters (Figure 2A). Exposure to the culture medium containing GA-SeNPs at pH 6.7 led to a significant shift in M1-like macrophage populations over a 3-day period. Specifically, the proportion of M2-like macrophages decreased from 94 ± 0.4% to 43 ± 0.39%, indicating that this dosage ranged from 0.01 μg mL−1 to 0.1 μg mL−1 of GA-SeNPs with diameter of 30–70 nm effectively increased the conversion percentage (p < 0.05). In contrast, the percentage of M1-like macrophages increased from 5 ± 0.3% to 45 ± 0.7%. These data indicated that the particle size of GA-SeNPs at the concentration of 0.1 μg mL−1 had a small effect on the efficiency of converting M2-like macrophages to M1-like macrophages, with conversion percentage of 45 ± 0.7% for 30–70 nm, 38.9 ± 0.8% for 80–120 nm, and 36.9 ± 2% for 180–220 nm. Taken together, the 30–70 nm group demonstrated the higher performance, with a conversion rate that is about 1.16 (80–120 nm) to 1.22-fold (180–220 nm) greater than the larger particle diameter groups. Likewise, treatments at pH 7.4 showed similar profiles for the distribution of M1-like and M2-like macrophages after treating M2-like macrophages with GA-SeNPs of different diameters. Consistent with those findings, the fluorescence images revealed similar profiles for the distribution of M1-like and M2-like macrophages after treating M2-like macrophages with GA-SeNPs of varying diameters. (Figure 2B). Notably, GA-SeNPs treatment led to a significant decrease in the expression of CD206, a marker associated with M2-like macrophages (iNOS−CD206+),6,29 and a marked increase in the expression of iNOS, a marker associated with M1-like macrophages (iNOS+CD206−).6,29
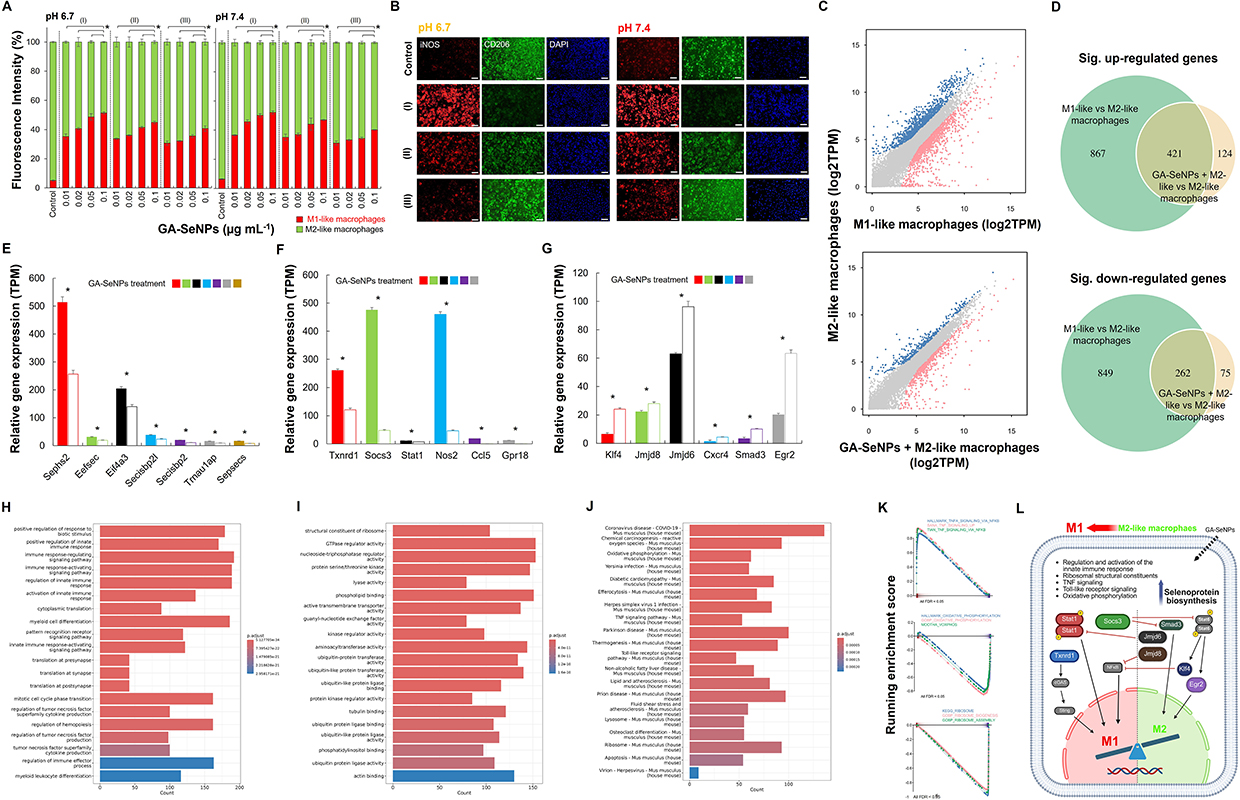 |
Figure 2 The effect of GA-SeNPs on macrophage repolarization. (A) Flow cytometry analysis of M2-like macrophage repolarization after 3 days of treatment with GA-SeNPs (varying diameters) at pH 7.4 and 6.7 (daily medium change) (*p < 0.05, based on a two-tailed t-test assuming unequal variances). The GA-SeNPs had three distinct particle size ranges: 30 to 70 nm (I), 80 to 120 nm (II), and 180 to 220 nm (III). Data represent the mean of triplicate measurements ± s.d. (B) Fluorescence microscopy images showing M1/M2 marker distribution in macrophages after 3-day treatment with GA-SeNPs (varying diameters at the concentration of 0.1 μg mL−1). The GA-SeNPs’ particle sizes fell into three categories: (I) 30–70 nm, (II) 80–120 nm, and (III) 180–220 nm. Scale bars = 200 µm. (C) Scatter plots showing gene expression distribution between M1 vs M2 macrophages, and GA-SeNPs-treated M2 vs M2 macrophages (grey: not significant, red: downregulated, blue: upregulated). (D) Venn diagram illustrating overlap of gene expression distribution. Normalized gene expressed levels (TPM) from RNA-seq data for 3 different gene copy groups for genes involved in (E) selenoprotein biosynthesis (Sephs2, Eefsec, Eif4a3, Secisbp2l, Secisbp2, Trnau1ap, and Sepsecs), driving M1 phenotype (Txnrd1, Socs3, Stat1, Nos2, Ccl5, and Gpr18) (F) or M2 phenotype (Klf4, Jmjd8, Jmjd6, Cxcr4, Smad3, and Egr2) (G). Whiskers represent standard errors of the mean (SEM). Asterisks show the level of significance based on the Student’s t-test (*p < 0.05). The biological processes (BP) (H) and molecular functions (MF) (I) of gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) (J) pathways comparing M1-like vs M2-like macrophages and GA-SeNPs-treated M2-like vs M2-like macrophages. (K) GSEA plots of TNF, oxidative phosphorylation, and ribosome signatures. (L) Schematic summarizing GA-SeNPs impact on macrophage inflammatory signaling, selenoprotein synthesis (blue gradient arrow: higher expression), metabolism, and mitochondrial activity pathways, leading to the repolarization and formation of M1-like macrophages (black-colored arrows indicate specific gene expression, while red-colored arrows indicate inhibition). |
Analysis of the Transcriptome of M2-Like Macrophages Following GA-SeNPs Treatment
To further elucidate the repolarization of M2-like macrophages induced by GA-SeNPs, transcriptomic analysis was performed. Scatter plots illustrated the gene expression distribution in M1-like, M2-like macrophages, and GA-SeNPs-treated M2-like macrophages (Figure 2C). Differential expression analysis using NOISeq identified 1,288 up-regulated and 1,111 down-regulated genes in M1-like macrophage compared to M2-like macrophage, while 545 gene were up-regulated and 337 genes were down-regulated in GA-SeNPs-treated M2-like macrophage compared to untreated M2-like macrophage (Figure 2D), indicating that GA-SeNPs played a crucial role in regulating M2-like macrophage behavior. Furthermore, the Venn diagram revealed a significantly overlap (p < 0.001, Fisher’s exact test) between GA-SeNPs-responsive genes in M2-like macrophages (GA-SeNPs-treated vs untreated) and M1-specific genes (M1-like vs M2-like macrophages) (Figure 2D). The heatmap clustering visualized gene expression similarities among M1-like, M2-like, and GA-SeNPs-treated M2-like macrophages (Figure S4). M2-like macrophages treated with GA-SeNPs exhibited greater similarity to M1-like macrophages than to untreated M2-like macrophages. Otherwise, heatmap clustering revealed differential gene expression (Figure S5), showing a shift in M2-like macrophages toward an M1-like profile after GA-SeNPs treatment. Overall, this strongly suggests that the GA-SeNPs are not merely causing random gene expression changes, but are specifically driving M2 macrophages towards an M1 transcriptional program. Heatmap clustering further corroborated this, visually demonstrating that GA-SeNPs-treated M2 macrophages aligned more closely with M1-like macrophages in their transcriptional profiles than with untreated M2-like cells, signifying a clear phenotypic shift.
Figure 2E–2G presented RNA sequencing data showing normalized gene expression levels (TPM) in M2-like macrophages following GA-SeNPs treatment. First, we examined the expression of genes involved in selenoprotein biosynthesis, including Sephs2 (Selenophosphate synthetase 2), Eefsec (Eukaryotic elongation factor, selenocysteine-tRNA-specific), Eif4a3 (Eukaryotic initiation factor 4A3), Secisbp2l (SECIS binding protein 2-like), Secisbp2 (SECIS binding protein 2), Trnau1ap (tRNA selenocysteine 1 associated protein 1), and Sepsecs (O-phosphoseryl-tRNA:selenocysteinyl-tRNA synthase). Following treatment with GA-SeNPs, M2-like macrophages exhibited a significant upregulation in the expression of all these biosynthetic genes of selenoprotein compared to untreated M2-like macrophage controls (Figure 2E). Next, given that M1-like macrophages are typically pro-inflammatory and play a role in fighting infections and cancer,1–3 we investigated genes promoting the M1 phenotype. Specifically, the expression of Txnrd1 (Thioredoxin reductase 1), Socs3 (Suppressor of cytokine signaling 3), Stat1 (Signal transducer and activator of transcription 1), Nos2 (Nitric oxide synthase 2, inducible), Ccl5 (C-C motif chemokine ligand 5), and Gpr18 (G protein-coupled receptor 18) was assessed. Treatment with GA-SeNPs significantly upregulated these M1-polarizing genes in M2-like macrophages, suggesting a phenotypic shift towards an M1 state relative to untreated controls (Figure 2F). Conversely, as M2 macrophages are generally associated with anti-inflammatory responses,1–3 key M2-polarizing genes were examined. These included Klf4 (Kruppel-like factor 4), Jmjd8 (Jumonji domain containing 8), Jmjd6 (Jumonji domain containing 6), Cxcr4 (C-X-C motif chemokine receptor 4), Smad3 (SMAD family member 3), and Egr2 (Early growth response 2). Treatment with GA-SeNPs led to a significant downregulation of these M2-associated genes in M2-like macrophages, indicating a shift away from the M2 phenotype and towards an M1-like profile when compared to untreated M2-like macrophage controls (Figure 2G).
Gene Ontology (GO) enrichment analysis of differentially expressed genes (DEGs) between GA-SeNPs-treated and untreated M2-like macrophages revealed significant enrichment in several biological processes (BP), primarily related to the regulation and activation of the innate immune response and associated signaling pathways (Figure 2H). Enriched molecular functions (MF) included ribosomal structural constituents and protein serine/threonine kinase activity, which are associated with selenoprotein biosynthesis (Figure 2I). This MF finding is notable, as selenium incorporation into selenoproteins has established links to ribosomal function,35 suggesting GA-SeNPs may similarly influence ribosomal components involved in macrophage activity. Further pathway analysis using KEGG indicated that these DEGs were enriched in pathways such as Coronavirus disease - COVID-19, TNF signaling, Ribosome, Toll-like receptor signaling, and oxidative phosphorylation (Figure 2J). Consistent with this, Gene Set Enrichment Analysis (GSEA) plots were also found to regulate signaling pathways associated with TNF-α inflammatory responses, oxidative phosphorylation, and the ribosome in between GA-SeNPs-treated M2-like macrophages and M2 macrophages (Figure 2K).
To further explore these alterations, GSEA of GO and KEGG pathways was performed. Network mapping visualized these enrichments, comparing M1-like versus M2-like macrophages (Figure S6) and GA-SeNPs-treated versus untreated M2-like macrophages (Figure S7). These GSEA results suggest that GA-SeNPs treatment impacts inflammatory signaling, ribosome, and cellular metabolism, potentially leading to altered macrophage function and mitochondrial activity. Collectively, these comprehensive analyses indicate that GA-SeNPs directly influence macrophage behavior, including their phenotype and polarization state, even in the presence of lactate. Collectively, these multi-faceted transcriptomic data strongly suggest that GA-SeNPs are not just causing random gene expression changes but are specifically driving M2 macrophages towards an M1-like transcriptional program and functional state (Figure 2L), even under conditions like the presence of lactate. This indicates a direct and profound influence of GA-SeNPs on macrophage behavior and polarization.
GA-SeNPs Triggered Cancer Cell Apoptosis via ROS Production
Cell migration plays a crucial role throughout cancer development, particularly during invasion, which is the initial stage of metastasis.36 After chemotactic migration, cancer cells penetrate surrounding tissues and local blood vessels. To assess the effect of GA-SeNPs on cancer cell migration, we conducted a wound healing assay using B16-F10-GFP cells. We compared the migration of B16-F10-GFP cells treated with GA-SeNPs to that of untreated control cells. As depicted in Figure 3A, the untreated Control group exhibited substantial wound closure over three days, with a migration rate of approximately 75%, establishing the maximal migration baseline. In contrast, B16-F10-GFP cells treated with GA-SeNPs (at 0.1 μg mL−1) across all diameters demonstrated significant inhibition of migration relative to the Control. Specifically, GA-SeNPs (30–70 nm) was the most effective, limiting migration to approximately 48.47 ± 8.24% closure, confirming the therapeutic potential of the GA-SeNPs in suppressing cancer cell mobility. At a concentration of 0.1 μg mL−1, GA-SeNPs with diameters of 80–120 nm or 180–220 nm achieved migration rates of 59.24 ± 3.53% or 64.12 ± 4.71%, respectively, after 72 hours (Figure 3B). Overall, the 30–70 nm GA-SeNPs provided a substantially better therapeutic effect against cancer cell migration, demonstrating 1.68 to 2.44 times greater inhibition compared to the groups of 80 nm-120 nm and 180 nm-220 nm.
 |
Figure 3 Impact of GA-SeNPs on cancer cell proliferation through ROS generation. Representative images (A) and quantification (B) of wound healing assays showing the migration of B16-F10-GFP cells treated daily with GA-SeNPs of different diameters across various time points (*p < 0.05, based on a two-tailed t-test assuming unequal variances). The particle size of GA-SeNPs was distributed across three distinct ranges: 30 to 70 nm (I), 80 to 120 nm (II), and 180 to 220 nm (III). The cell-free area, which represents the initial scratch distance, is demarcated by a white dotted line. The data represent the mean of triplicate measurements ± s.d. Scale bars = 200 µm. (C) Representative photographs depicting ROS production (Upper panel) and TUNEL assay identifying apoptotic cells (Lower panel) induced by GA-SeNPs of different diameters in B16-F10 cells. Scale bars = 200 µm. The GA-SeNPs exhibited three particle size ranges: 30 to 70 nm (I), 80 to 120 nm (II), and 180 to 220 nm (III). (D) Percentages of GFP-expressing B16-F10 cells co-cultured with M2-like macrophages at various ratios of M2-like macrophages to B16-F10 cells, 3 days post-treatment, following the daily addition of GA-SeNPs with different diameters at pH 6.7, analyzed by flow cytometry. Data represent the mean of triplicate measurements ± s.d. (E) In vitro modulation of M2-like macrophage repolarization after 3 days of treatment with or without GA-SeNPs, GM-CSF, or LOX at pH 7.4 and pH 6.7, with daily culture medium replacement. Data are presented as the mean of triplicate measurements ± s.d. (F) Quantitative analysis of co-treatment effects on pro-inflammatory cytokine TNF-α (upper panel) and anti-inflammatory cytokine IL-10 (lower panel), assessing M2-like macrophage repolarization after 3 days of treatment with or without GA-SeNPs, GM-CSF, or LOX at pH 7.4 and pH 6.7 (*p < 0.05, based on a two-tailed t-test assuming unequal variances). Data are presented as the mean of triplicate measurements ± s.d. |
SeNPs have demonstrated enhanced anticancer activity against cancer cells, primarily through the ROS generation.28 These ROS play a crucial role in inducing oxidative stress within the cancer cells, leading to cellular damage and apoptosis. We assessed ROS levels in B16-F10 cells under different treatments using the CellROX green reagent. As illustrated in Figure 2C and Figure S8, GA-SeNPs treatment significantly increased ROS generation in cancer cells compared to the untreated control, highlighting ROS role in inducing apoptosis.28 Based on this evidence, it could be concluded that GA-SeNPs are metabolized into selenoprotein. This metabolite regulates selenoprotein expression in cancer cells, alleviates endoplasmic reticulum (ER) stress, and promotes robust ROS production, leading to cancer cell apoptosis.37 The TUNEL assay further confirmed the high therapeutic efficacy of the GA-SeNPs treatment strategy (Figure 3C and Figure S9). By generating ROS, GA-SeNPs disrupt normal cellular processes in cancer cells, resulting in reduced proliferation and survival. This mechanism emphasized the potential of GA-SeNPs as effective therapeutic agents in cancer treatment.
GA-SeNPs Disrupted Pro-Tumor Interactions Between M2-Like Macrophages and Cancer Cells in Co-Culture
Given the pivotal role of GA-SeNPs in both repolarizing M2-like macrophages to a pro-inflammatory M1-like phenotype (Figure 2) and inducing ROS generation within cancer cells (Figure 3C), we aimed to further explore their dual functionality in a more complex biological context. Specifically, we investigated how GA-SeNPs of varying diameters concurrently interact with M2-like macrophages and B16-F10-GFP cells to simulate the systemic delivery and circulation of GA-SeNPs within the tumor microenvironment. It was confirmed that GA-SeNPs of varying diameters significantly enhance the ability of reconverted M1-like macrophages to eliminate B16-F10-GFP cells, with percentages of GFP-positive cells (%) of 24 ± 0.94% for 30–70 nm, 27 ± 1.89% for 80–120 nm, and 36 ± 2.55% for 180–220 nm, especially at highest ratio (5:1) of M2-like macrophages to cancer cells (Figure 3D). Furthermore, the 30–70 nm GA-SeNPs was significantly superior in mediating cancer cell elimination, resulting in 1.125 to 1.5-fold fewer remaining cancer cells compared to the groups of 80 nm-120 nm and 180 nm-220 nm. This highlights a clear, size-dependent relationship where smaller nanoparticles enhance the therapeutic function of the resulting M1-like macrophages. This approach allows us to better understand the potential of GA-SeNPs to not only modulate the immune landscape by converting tumor-promoting macrophages into tumor-fighting ones but also to directly impact cancer cells by triggering oxidative stress, thereby providing a multifaceted therapeutic strategy. Based on these findings, we concluded that GA-SeNPs with a diameter range of 30–70 nm are appropriate for use in future experiments.
GA-SeNPs Combination Therapy Enhanced Macrophage Repolarization
Tumor-derived lactate leverages mitochondrial signaling to activate oxidative phosphorylation in M2 TAMs, enhancing their immunosuppressive activity.6,14 TAMs differentiation in vivo is driven by GM-CSF.7,20 Tumor cells alter their metabolic pathways, increasing lactate production, which enhances the immunosuppressive functions of M2-like TAMs and promotes tumor growth.6,14,29 Our previous research indicates that employing delivery systems for the sustained release of LOX, which converts lactate into pyruvate, may offer a promising approach for lactate depletion therapy in tumors.6,10,11,14 As anticipated, co-treatment with GA-SeNPs (30–70 nm), GM-CSF (25 ng mL−1),29 and LOX (0.025 U mL−1)6 was highly effective in converting M2-like macrophages to an M1-like phenotype, with conversion of 65 ± 0.7% at pH 7.4 and 83 ± 0.54% at pH 6.7. These results were comparable to those observed with each individual treatment group (Figure 3E). This outcome underscored the robust ability of the combination therapy to repolarize macrophages towards a pro-inflammatory state, which was crucial for effective anti-tumor immune responses. Further analysis revealed that the co-treatment significantly enhanced the production of pro-inflammatory cytokines, particularly TNF-α, a key mediator in the immune response against cancer cells. Simultaneously, there was a marked downregulation in the expression of anti-inflammatory cytokines, such as IL-10, which is typically associated with immune suppression and tumor progression (Figure 3F). As anticipated, co-treatment with GA-SeNPs, GM-CSF, and LOX was highly effective in inducing TNF-α expression, with levels of 4.8 ± 0.2 ng mL−1 at pH 6.7 (compared to control, p = 7.05E-16) and 5.1 ± 0.2 ng mL−1 at pH 7.4 (p = 1.56E-15, compared to control). In contrast, this co-treatment was less effective at inducing IL-10, with levels of 0.9 ± 0.13 ng mL−1 at pH 6.7 (p = 3.36E-15, compared to control) and 0.7 ± 0.13 ng mL−1 at pH 7.4 (p = 8.46E-15, compared to control). These findings highlighted the therapeutic potential of the GA-SeNPs, GM-CSF, and LOX combination in not only promoting an anti-tumor immune environment but also in potentially overcoming the immunosuppressive barriers commonly present in the tumor microenvironment.
GA-SeNPs Reduced B16-F10 Tumor Burden in Mice
To advance the proof-of-principle findings (Figures 1–3) toward preclinical application, we administered GA-SeNPs to BALB/c mice bearing B16-F10 melanoma xenograft tumors through intratumoral injection, mirroring the pioneering virotherapy administered route for advanced melanoma patients.8 Additionally, TAMs can constitute up to 50% of the tumor mass, with the majority exhibiting the M2 phenotype, as widely recognized.38 To ensure M2 macrophages occupied the entire tumor and tumor microenvironment, tumors were grown to ~200 mm3 before treatment. The 30–70 nm diameter, being the most effective at M2-to-M1 repolarization and cancer cell migration suppression, was consequently chosen for the in vivo anti-melanoma studies. Experimental groups then received GA-SeNPs at varying frequencies or PBS via intratumoral injection (Figure 4A). Remarkably, GA-SeNPs treatment demonstrated significant tumor growth suppression (Figure 4B). Six consecutive injections of GA-SeNPs led to a significant > 3.4-fold or > 5-fold reduction in tumor volume (573.8 ± 168.2 mm3) or tumor weight (495.0 ± 123.2 mg) compared to the PBS group (tumor volume: 2068.8 ± 428.2 mm3, p = 0.00278; tumor weight: 2880.8 ± 702.9 mg, p = 0.00176) (Figure 4C and 4D). As expected, nine consecutive injections of GA-SeNPs achieved similar tumor suppression, reducing tumor weight and volume to 426.7% and 562.4% of the PBS group values, respectively. Overall, GA-SeNPs significantly reduced the tumor burden, resulting in a greater than 4-fold reduction in tumor weight when compared to the PBS control groups.
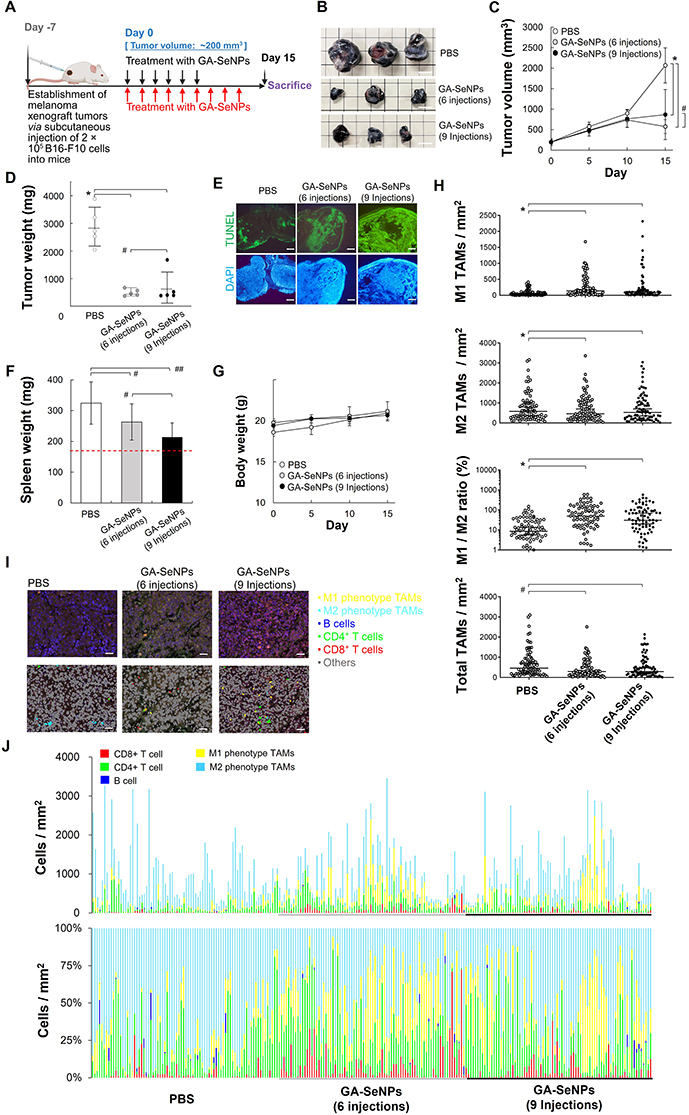 |
Figure 4 Animal studies evaluating the effects of various formulations on B16-F10 xenografts. (A) Treatment regimen for mice with B16-F10 xenografts receiving various treatments. (B) Representative photographs of B16-F10 xenograft tumors following various treatments, alongside excised tumors collected on Day 15 (scale bars = 1 cm). (C) Tumor volume comparison across treatment groups, with statistical significance indicated (*p < 0.05; #p > 0.10, based on a two-tailed t-test assuming unequal variances). Data represent the mean of sextuplicate measurements ± s.d. (D) Tumor weight comparison across groups, analyzed with the same statistical parameters as in (C). (E) Histological analysis of tumor sections from PBS-treated and GA-SeNPs-treated mice (6 or 9 injections) at Day 15. Sections were stained using the In Situ Cell Death Detection Kit with fluorescein (green fluorescence) and DAPI (blue fluorescence). Scale bars = 500 μm. (F) Spleen weight measurements for mice under different treatment regimens (#p > 0.10; ##p > 0.05, based on a two-tailed t-test assuming unequal variances). The spleen weight of healthy mice, represented by the red dotted line, was 169 ± 3.0 mg. (G) Body weight of mice measured at specified time points across treatment groups. Data represent the mean of quintuplicate measurements ± s.d. (H) Measurement of tumor-infiltrating M1 phenotype TAMs (F4/80⁺MHC II⁺CD206−) and M2 phenotype TAMs (F4/80⁺MHC II−CD206⁺) in xenograft tumors (*p < 0.05; #p > 0.10, based on a two-tailed t-test assuming unequal variances). (I) Representative spectrally unmixed composite images of tumor-infiltrating M1 and M2 phenotype TAMs, CD8⁺ T cells, CD4⁺ T cells, and B cells in formalin-fixed, paraffin-embedded tumor tissues, quantified using multiplex immunofluorescence staining after various treatments following color coding: yellow for M1 phenotype TAMs, light blue for M2 phenotype TAMs, green for CD4⁺ T cells, red for CD8⁺ T cells, and dark blue for B cells. Scale bar = 50 μm. (J) The corresponding quantitative results are presented with the following color coding: yellow for M1 phenotype TAMs, light blue for M2 phenotype TAMs, green for CD4⁺ T cells, red for CD8⁺ T cells, and dark blue for B cells. |
Histological analysis of melanoma xenograft tumors demonstrated a statistically significant increase in apoptotic cell count following GA-SeNPs treatment (Figure 4E), consistent with the observed tumor shrinkage. The spleen plays a crucial role in extramedullary hematopoiesis and promoting tumor immunotolerance.39 All tumor-bearing conditions have exhibited an enlarged spleen compared to healthy mice, indicative of a robust immune response characterized by leukocytosis, lymphocytosis, or granulocytosis in reaction to exogenous tumor cells.39,40 In comparison to PBS, the results in Figure 4F suggested a localized repolarization or activation of the immune system after mice received GA-SeNPs (p = 0.169 for six injections of GA-SeNPs; p = 0.065 for nine injections of GA-SeNPs). The GA-SeNPs formulation was well tolerated, as indicated by stable weight measurements (Figure 4G). Histopathological examination, using hematoxylin and eosin (H&E) stained micrographs of major organs, revealed no significant injuries or pathological changes after the various treatments (Figure S10).
GA-SeNPs Effectively Repolarize TAMs While Maintaining Their Population
In vivo experiments (Figure 4B–E) explored the hypothesis that the antitumor effects of GA-SeNPs stem, in part, from their ability to modulate the immunosuppressive tumor microenvironment. To test this, we analyzed tumor-infiltrating immune cells following PBS or GA-SeNPs treatment (6 or 9 injections). TAMs, the predominant immune cells in the tumor microenvironment, are largely skewed toward the immunosuppressive M2 phenotype TAMs.13,39,41 GA-SeNPs treatment (6 or 9 injections) significantly increased the population of M1 phenotype TAMs (F4/80⁺MHC II⁺CD206−) compared to PBS (75.4 cells/mm3), with increases of approximately 264.3 cells/mm3 (6 injections) and 239.0 cells/mm3 (9 injections) (Figure 4H). In contrast, M2 phenotype TAMs (F4/80⁺MHC II−CD206⁺) were notably reduced in both the 6-injection (431.2 cells/mm3) and 9-injection (510.5 cells/mm3) GA-SeNPs groups compared to PBS (701.8 cells/mm3). This resulted in a significant increase (> 4.0-fold) in the M1/M2 TAMs ratio compared to PBS (p = 4.57E-7 for 6 injections of GA-SeNPs; p = 5.23E-6 for 9 injections of GA-SeNPs), in line with findings in Figures 4I and J. Furthermore, our observations indicated that the overall macrophage count showed no significant difference in response to GA-SeNPs compared to PBS (p = 0.47 for six injections of GA-SeNPs; p = 0.79 for nine injections of GA-SeNPs). These results strongly indicate that GA-SeNPs exert immunomodulatory effects, effectively repolarizing M2 phenotype into the M1 phenotype TAMs within the tumor microenvironment. This shift correlates with enhanced tumor control when GA-SeNPs are administered intratumorally, as demonstrated in Figure 4B–E.
Notably, M1 phenotype TAMs play a crucial role in promoting antitumor inflammation, including enhancing IFN-γ production by T cells.3 They mediate cytotoxic tumor killing, facilitate phagocytosis of cancer cells, and modulate immune responses via CD8⁺ T lymphocytes and interferons.3,13 Given their role in T cell recruitment, we anticipated an increase in tumor-infiltrating CD8⁺ T cells (CD8⁺ stained) in the GA-SeNPs-treated group (Figure S11), suggesting a localized inflamed tumor immune phenotype. Interestingly, none of the treatments significantly affected the recruitment of CD4⁺ T cells (CD4⁺ stained) or B cells (CD19⁺ stained). Collectively, these in vivo findings suggest that GA-SeNPs enhance CD8⁺ T cell responses, likely through the recruitment and activation of M1 phenotype TAMs (Figure 4I and J).
Discussion
The repolarization of TAMs is crucial, as these cells are abundant in the tumor microenvironment and predominantly exhibit an M2 phenotype that fosters tumor growth, angiogenesis, and immunosuppression.13,39,41 Therefore, strategies to re-educate these TAMs towards an M1 anti-tumor phenotype are highly sought after in cancer immunotherapy.42 Furthermore, TAMs adapt to local signals,5,6 with cytokines like M-CSF and GM-CSF crucial for their differentiation.7 In this study, the GA-SeNPs demonstrated a potent dual anti-cancer action by robustly repolarizing pro-tumor M2 TAMs towards an anti-tumor M1 phenotype and directly inducing cancer cell apoptosis, leading to significant in vivo tumor suppression.1–4
Notably, SeNPs trigger apoptosis in different cancer cells, likely through mechanisms that involve altering the intracellular redox balance and, consequently, increasing ROS generation.23–25 Delving into the molecular mechanisms of M2 macrophages treated with GA-SeNPs, the transcriptomic analysis provided compelling evidence that GA-SeNPs orchestrate the reprogramming of M2 macrophages. The significant overlap between GA-SeNPs-responsive genes and M1-specific genes confirms a directed phenotypic switch rather than non-specific cellular stress. Critically, a highly relevant finding was the significant upregulation of the entire suite of genes (eg, Sephs2, Eefsec, and Secisbp2) involved in selenoprotein biosynthesis. This strongly suggests that the immunomodulatory effects of GA-SeNPs are, at least in part, mediated by their metabolic incorporation into functional selenoproteins. Selenoproteins, such as Txnrd1, are increasingly recognized for their crucial roles in redox regulation, immune cell function, and inflammatory responses.43 The observed upregulation of canonical M1-polarizing genes (Stat1, Nos2, and Ccl5) and downregulation of M2-associated genes (Klf4, Cxcr4, and Egr2), alongside enrichment in pathways like TNF signaling and Toll-like receptor signaling, further solidifies the M1-skewing effect. GO and KEGG analyses further supported this, highlighting enrichment in oxidative phosphorylation (the favored metabolic pathway of M2 phenotype), TNF signaling, and ribosomal functions, consistent with the known roles of selenoproteins in immunity and protein synthesis.44
Beyond immunomodulation, GA-SeNPs demonstrated direct anti-cancer properties by inhibiting B16-F10 melanoma cell migration and inducing apoptosis via significant ROS generation. The ROS-mediated cytotoxicity is a well-established anti-cancer mechanism for various selenium compounds and SeNPs, often exploiting the higher basal oxidative stress in cancer cells.45 The hypothesis that GA-SeNPs are metabolized into selenoproteins that then modulate ROS levels is consistent with the known bioactivity of selenium. This highlights a synergistic interplay where GA-SeNPs not only directly attack cancer cells but also create a hostile immune environment by re-educating TAMs. This multi-pronged attack is often more effective than single-target therapies.13
Intratumoral administration of GA-SeNPs significantly suppressed B16-F10 tumor growth in mice, which correlated with increased tumor cell apoptosis. Evidence for the therapeutic effect came from the successful repolarization of TAMs within the tumor: the M1/M2 ratio significantly increased, yet the total TAM population was maintained. This stability is crucial, indicating true phenotypic reprogramming rather than selective depletion of M2 cells. Perhaps most importantly, GA-SeNPs treatment led to an increase in tumor-infiltrating CD8⁺ T cells. CD8⁺ T cells are the primary effectors of anti-tumor immunity, and their presence is often correlated with improved prognosis and response to immunotherapy.13,39 M1 TAMs, known to secrete chemokines like Ccl5 and improve antigen presentation, likely facilitated this CD8⁺ T cell recruitment,46 consistent with our in vitro and in vivo observations. This study contributes significantly to the burgeoning field of cancer nanomedicine and immunotherapy. The demonstration that GA-SeNPs can effectively repolarize TAMs by engaging selenium metabolic pathways and upregulating selenoprotein biosynthesis offers a new mechanistic insight beyond generic nanoparticle-induced inflammation. This is particularly important as selenium status itself has been linked to immune competence and cancer risk.47
Despite demonstrating that GA-SeNPs effectively repolarize TAMs and inhibit melanoma growth, our study has limitations that guide future work. Our in vivo efficacy was exclusively validated in the B16-F10 melanoma model, a constraint on the generalizability of the M2-to-M1 repolarization strategy. Therefore, future studies will expand testing to a broader range of solid tumors. Additionally, the intratumoral injection route must be directly compared against systemic administration to establish the clinical delivery method. Translational development faces two primary challenges: delivery and manufacturing. The localized injection strategy is unsuitable for treating metastasis, making it crucial to develop systemic delivery systems that achieve targeted tumor accumulation. Simultaneously, the scalability and cost-effectiveness of GA-SeNPs synthesis require optimization to move the therapy closer to clinical application.
Conclusion
GA-SeNPs represent a promising multi-functional therapeutic agent that effectively combats cancer by directly inducing tumor cell apoptosis and, importantly, by reprogramming the tumor immune microenvironment through TAM polarization towards an M1 phenotype. These findings highlight the potential of GA-SeNPs as a standalone or combination therapy to enhance anti-tumor immunity and improve cancer treatment outcomes.
Acknowledgments
This work was supported by the National Science and Technology Council (111-2628-E-110-007-MY3, 112-2221-E-006-196, 112-2314-B-002-148, 112-2628-B-110-004-MY3, 113-2622-B-006-009, 113-2314-B-002-115-MY3, 114-2223-E-110-003-MY3, 114-2314-B-006-109-MY3, and 114-2622-B-006-013), Excellent Translational Medicine Research Projects of National Taiwan University College of Medicine and National Taiwan University Hospital (NSCCMOH-114C101-72-74), and YongLin Chair Grant (S-01). Da-Liang Ou belongs National Taiwan University YongLin Institute of Health Scholar.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Li C, Xu X, Wei S, Jiang P, Xue L, Wang J. Tumor-associated macrophages: potential therapeutic strategies and future prospects in cancer. J Immunother Cancer. 2021;
2. Rannikko JH, Hollmén M. Clinical landscape of macrophage-reprogramming cancer immunotherapies. Br J Cancer. 2024;131:627–640. doi:10.1038/s41416-024-02715-6
3. Chen S, Saeed AFUH, Liu Q, et al. Macrophages in immunoregulation and therapeutics. Signal Transduct Target Ther. 2023;8:207. doi:10.1038/s41392-023-01452-1
4. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi:10.1038/nri2448
5. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–737. doi:10.1038/nri3073
6. Liao ZX, Fa YC, Kempson IM, Tseng SJ. Repolarization of M2 to M1 macrophages triggered by lactate oxidase released from methylcellulose hydrogel. Bioconjug Chem. 2019;30:2697–2702. doi:10.1021/acs.bioconjchem.9b00618
7. Lee KMC, Achuthan AA, Hamilton JA. GM-CSF: a promising target in inflammation and autoimmunity. Immunotargets Ther. 2020;9:225–240. doi:10.2147/ITT.S262566
8. Pol J, Kroemer G, Galluzzi L. First oncolytic virus approved for melanoma immunotherapy. OncoImmunology. 2016;
9. Tseng SJ, Huang KY, Kempson IM, et al. Remote control of light-triggered virotherapy. ACS Nano. 2016;10:10339–10346. doi:10.1021/acsnano.6b06051
10. Tseng SJ, Kempson IM, Huang KY, et al. Targeting tumor microenvironment by bioreduction-activated nanoparticles for light-triggered virotherapy. ACS Nano. 2018;12:9894–9902. doi:10.1021/acsnano.8b02813
11. Tseng SJ, Kempson IM, Liao Z-X, Ho YC, Yang PC. An acid degradable, lactate oxidizing nanoparticle formulation for non-small cell lung cancer virotherapy. Nano Today. 2022;46:101582. doi:10.1016/j.nantod.2022.101582
12. Liao ZX, Hsu SH, Tang SC, Kempson I, Yang PC, Tseng SJ. Potential targeting of the tumor microenvironment to improve cancer virotherapy. Pharmacol Ther. 2023;250:108521. doi:10.1016/j.pharmthera.2023.108521
13. Ou DL, Liao ZX, Kempson IM, Li L, Yang PC, Tseng SJ. Nano-modified viruses prime the tumor microenvironment and promote the photodynamic virotherapy in liver cancer. J Biomed Sci. 2024;31:1. doi:10.1186/s12929-023-00989-1
14. Kostiuchenko O, Lushnikova I, Skibo G. The role of gut microbiota metabolites in the regeneration and protection of nervous tissue: a narrative review. Regen Med Rep. 2024;1:12–30.
15. Liao ZX, Kempson IM, Hsieh CC, Tseng SJ, Yang PC. Potential therapeutics using tumor-secreted lactate in nonsmall cell lung cancer. Drug Discov Today. 2021;26:2508–2514. doi:10.1016/j.drudis.2021.07.014
16. Kurwe MS, Karim HMR, Singha SK, Neema PK, Panda CK. Effect of real-time oxygen consumption versus fixed flow-based low flow anesthesia on oxygenation and perfusion: a randomized, single-blind study. Med Gas Res. 2024;14:108–114. doi:10.4103/2045-9912.385938
17. Deng Q, Yang X, Li Z. Hyperbaric oxygen: a multifaceted approach in cancer therapy. Med Gas Res. 2024;14:130–132. doi:10.4103/mgr.MEDGASRES-D-23-00028
18. Kieliszek M, Serrano Sandoval SN. The importance of selenium in food enrichment processes. A comprehensive review. J Trace Elem Med Biol. 2023;79:127260. doi:10.1016/j.jtemb.2023.127260
19. Lai H, Xu L, Liu C, et al. Universal selenium nanoadjuvant with immunopotentiating and redox-shaping activities inducing high-quality immunity for SARS-CoV-2 vaccine. Sig Transduct Target Ther. 2023;8:88. doi:10.1038/s41392-023-01371-1
20. Kang D, Lee J, Wu C, et al. The role of selenium metabolism and selenoproteins in cartilage homeostasis and arthropathies. Exp Mol Med. 2020;52:1198–1208. doi:10.1038/s12276-020-0408-y
21. Jin K, Shi S, Huang D, et al. Maintaining cardiac homeostasis by translational selenium nanoparticles with rapid selenoproteins regulation to achieve radiation-induced heart prevention. Chem Eng J. 2025;506:160005. doi:10.1016/j.cej.2025.160005
22. Liu S, Li X, Wei W, et al. Translational selenium nanoparticles enhance NKG2D-mediated cytotoxicity of NK cells against malignant pleural mesothelioma cells through the TrxR1-pSTAT3 pathway. Nano Today. 2025;62:102720. doi:10.1016/j.nantod.2025.102720
23. Ferro C, Florindo HF, Santos HA. Selenium nanoparticles for biomedical applications: from development and characterization to therapeutics. Adv Healthc Mater. 2021;10:2100598. doi:10.1002/adhm.202100598
24. Ferro C, Matos AI, Serpico L, et al. Selenium nanoparticles synergize with a KRAS nanovaccine against breast cancer. Adv Healthc Mater. 2025;14:2401523. doi:10.1002/adhm.202401523
25. Hosnedlova B, Kepinska M, Skalickova S, et al. Nano-selenium and its nanomedicine applications: a critical review. Int J Nanomed. 2018;13:2107–2128. doi:10.2147/IJN.S157541
26. Ichikawa Y, Sato B, Hirano SI, Takefuji Y, Satoh F. Hydrogen inhalation therapy may ameliorate amyotrophic lateral sclerosis. Med Gas Res. 2024;14:149–150. doi:10.4103/2045-9912.390249
27. Pan XW, Huang JS, Liu SR, et al. Evaluation of the liver targeting and anti‑liver cancer activity of artesunate‑loaded and glycyrrhetinic acid‑coated nanoparticles. Exp Ther Med. 2023;26:516. doi:10.3892/etm.2023.12215
28. Zhuang Y, Li L, Feng L, et al. Mitochondrion-targeted selenium nanoparticles enhance reactive oxygen species-mediated cell death. Nanoscale. 2020;12:1389–1396. doi:10.1039/C9NR09039H
29. Hsieh CC, Hsieh MJ, Wang YH, Liao ZX. Macrophage distribution affected by virus-encoded granulocyte macrophage colony stimulating factor combined with lactate oxidase. ACS Omega. 2022;7:24020–24026. doi:10.1021/acsomega.2c03213
30. Yamauchi T, Makinodan M, Toritsuka M, et al. Tumor necrosis factor-α expression aberration of M1/M2 macrophages in adult high-functioning autism spectrum disorder. Autism Res. 2021;14:2330–2341. doi:10.1002/aur.2585
31. Chuang Y, Hung ME, Cangelose BK, Leonard JN. Regulation of the IL-10-driven macrophage phenotype under incoherent stimuli. Innate Immun. 2016;22:647–657. doi:10.1177/1753425916668243
32. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi:10.1093/bioinformatics/bts635
33. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16:284–287. doi:10.1089/omi.2011.0118
34. Mu X, Shi W, Xu Y, et al. Tumor-derived lactate induces M2 macrophage polarization via the activation of the ERK/STAT3 signaling pathway in breast cancer. Cell Cycle. 2018;17:428–438. doi:10.1080/15384101.2018.1444305
35. Li Z, Ferguson L, Deol KK, et al. Ribosome stalling during selenoprotein translation exposes a ferroptosis vulnerability. Nat Chem Biol. 2022;18:751–761. doi:10.1038/s41589-022-01033-3
36. Novikov NM, Zolotaryova SY, Gautreau AM, Denisov EV. Mutational drivers of cancer cell migration and invasion. Br J Cancer. 2021;124:102–114. doi:10.1038/s41416-020-01149-0
37. Liu T, Xu L, He L, et al. Selenium nanoparticles regulates selenoprotein to boost cytokine-induced killer cells-based cancer immunotherapy. Nano Today. 2020;35:100975. doi:10.1016/j.nantod.2020.100975
38. Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol. 2009;86:1065–1073. doi:10.1189/jlb.0609385
39. Ou DL, Tseng SJ, Kempson IM, Hsu CL, Yang PC, Liao ZX. Enhanced targeting and immune activation of tumor microenvironment by nanomodified anti-PD1 in liver cancer. Adv Therap. 2021;4:2100048. doi:10.1002/adtp.202100048
40. Richards J, McNally B, Fang X, Caligiuri MA, Zheng P, Liu Y. Tumor growth decreases NK and B cells as well as common lymphoid progenitor. PLoS One. 2008;
41. Guo N, Zhou W, Zhang Z, et al. Macrophage and mitochondria targeted nanoplatform to deplete and polarize M1-like macrophages for rheumatoid arthritis treatment. Chem Eng J. 2025;503:158468. doi:10.1016/j.cej.2024.158468
42. Zhang S, Yu B, Sheng C, et al. SHISA3 reprograms tumor-associated macrophages toward an antitumoral phenotype and enhances cancer immunotherapy. Adv Sci. 2024;11:2403019. doi:10.1002/advs.202403019
43. Scholzen KC, Arnér ESJ. Cellular activity of the cytosolic selenoprotein thioredoxin reductase 1 (TXNRD1) is modulated by copper and zinc levels in the cell culture medium. J Trace Elem Med Biol. 2025;88:127624. doi:10.1016/j.jtemb.2025.127624
44. Hatfield DL, Tsuji PA, Carlson BA, Gladyshev VN. Selenium and selenocysteine: roles in cancer, health, and development. Trends Biochem Sci. 2014;39:112–120. doi:10.1016/j.tibs.2013.12.007
45. Chirakara D, Lotlikar S, Nannan M, Dunna NR, Venkatabalasubramanian S. Exploring the liposomal encapsulation and enhanced cytotoxicity of selenium nanoparticles against lung cancer cells. Next Nanotechnol. 2025;7:100121. doi:10.1016/j.nxnano.2024.100121
46. Schol P, van Elsas MJ, Middelburg J, et al. Myeloid effector cells in cancer. Cancer Cell. 2024;42:1997–2014. doi:10.1016/j.ccell.2024.11.002
47. Rayman MP. Selenium and human health. Lancet. 2012;379:1256–1268. doi:10.1016/S0140-6736(11)61452-9
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Andrographolide Inhibits Static Mechanical Pressure-Induced Intervertebral Disc Degeneration via the MAPK/Nrf2/HO-1 Pathway
Zhang C, Lu Z, Lyu C, Zhang S, Wang D
Drug Design, Development and Therapy 2023, 17:535-550
Published Date: 20 February 2023
Antitumor Activity of Berberine by Activating Autophagy and Apoptosis in CAL-62 and BHT-101 Anaplastic Thyroid Carcinoma Cell Lines
Shi XZ, Zhao S, Wang Y, Wang MY, Su SW, Wu YZ, Xiong C
Drug Design, Development and Therapy 2023, 17:1889-1906
Published Date: 26 June 2023
Design and Mechanism Study of 6c, a Novel Artesunate Derivatives, for Anti-Hepatocellular Carcinoma

Xiong SS
Journal of Hepatocellular Carcinoma 2025, 12:149-167
Published Date: 25 January 2025
PEDF Overexpression Ameliorates Cardiac Lipotoxicity in Diabetic Cardiomyopathy via Regulation of Energy Metabolism
Mao T, Wang Y
Diabetes, Metabolic Syndrome and Obesity 2025, 18:217-231
Published Date: 27 January 2025
Chitosan Oligosaccharide-Functionalized Ruthenium-Curcumin Nanodots for Targeted Therapy of Acute Kidney Injury
Yu Q, Tan XY, Liu X, Mao HB, Chen ZG
International Journal of Nanomedicine 2026, 21:601940
Published Date: 30 June 2026