Back to Journals » Journal of Hepatocellular Carcinoma » Volume 11
Integration of Single-Cell RNA Sequencing and Bulk RNA Sequencing to Identify an Immunogenic Cell Death-Related 5-Gene Prognostic Signature in Hepatocellular Carcinoma
Received 11 November 2023
Accepted for publication 3 May 2024
Published 16 May 2024 Volume 2024:11 Pages 879—900
DOI https://doi.org/10.2147/JHC.S449419
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Mohamed Shaker
Liqun Peng,1,2 Shaohua Xu,3 Jian-Liang Xu4
1Department of Hepatobiliary and Pancreatic Surgery, Zhongnan Hospital of Wuhan University, Wuhan, People’s Republic of China; 2Clinical Medicine Research Center for Minimally Invasive Procedure of Hepatobiliary & Pancreatic Diseases of Hubei Province, Wuhan, People’s Republic of China; 3Department of Clinical Laboratory, Center for Gene Diagnosis & Program of Clinical Laboratory, Zhongnan Hospital of Wuhan University, Wuhan, People’s Republic of China; 4Department of Hepatobiliary Surgery, the Third Affiliated Hospital of Sun Yat-sen University, Guangzhou, People’s Republic of China
Correspondence: Jian-Liang Xu, Department of Hepatobiliary Surgery, the Third Affiliated Hospital of Sun Yat-sen University, 600 Tianhe Road, Guangzhou, 510630, People’s Republic of China, Tel +86-13580575810, Fax +8620-85253178, Email [email protected] Shaohua Xu, Department of Clinical Laboratory, Center for Gene Diagnosis & Program of Clinical Laboratory, Zhongnan Hospital of Wuhan University, 169 Donghu Road, Wuhan, 430071, People’s Republic of China, Email [email protected]
Introduction: Immunogenic cell death (ICD) can enhance the potency of immunotherapy in cancer treatment. Nevertheless, it is ambiguous how ICD-related genes (ICDRGs) contribute to hepatocellular carcinoma (HCC).
Methods: Single-cell RNA sequencing (scRNA-seq) data were used to distinguish malignant cells from normal cells in the HCC tumor microenvironment(TME). Bulk RNA sequencing data was employed to acquire the landscape of the 33 ICDRGs. Unsupervised clustering identified two ICD molecular subtypes. The cellular infiltration characteristics and biological behavior in different subtypes were analyzed by ssGSEA. Subsequently, differentially expressed genes (DEGs) between the two subtypes were determined, based on which patients were classified into three gene clusters. Then, the prognostic model was constructed by Lasso-Cox analysis. Finally, we investigated the expression of risk genes in cancer cell line encyclopedia (CCLE) and validated the function of NKX3-2 in vitro experiments.
Results: ICD scores and ICDRGs expression in malignant cells were significantly lower than in normal cells by scRNA-seq analysis. ICD-high subtype was characterized by ICD-related gene overexpression and high levels of immune infiltration abundance and immune checkpoints; Three DEGs-related gene clusters were likewise strongly linked to stromal and immunological activation. In the ICD-related prognostic model consisting of NKX3-2, CHODL, MMP1, NR0B1, and CTSV, the low-risk group patients had a better endpoint and displayed increased susceptibility to immunotherapy and chemotherapeutic drugs like 5-Fluorouracil, afatinib, bortezomib, cediratinib, lapatinib, dasatinib, gefitinib and crizotinib. Moreover, NKX3-2 amplification in HCC samples has been verified by experiments, and its disruption suppressed the proliferation and invasion of tumor cells.
Conclusion: Our study highlighted the potential of the ICDRGs risk score as a prognostic indicator to aid in the accurate diagnosis and immunotherapy sensitivity of HCC.
Keywords: Hepatocellular carcinoma, prognostic model, immunotherapy, immunogenic cell death
Introduction
Hepatocellular carcinoma (HCC) is the sixth most prevalent cancer in the world, and its morbidity and mortality rates continue to rise.1 Recently, despite the development of new early screening and treatment methods for HCC,2–4 the prognosis of HCC patients is still dismal.5,6 In addition, the selection of treatment criteria and identification of high-risk patients remains ambitious owing to individual heterogeneity. Consequently, there is a necessity to discover novel prognostic biomarkers for HCC and to guide individualized treatment. Clinical trials for advanced HCC employing immunotherapy represented by immune checkpoint inhibitors (ICIs) have recently shown encouraging results.7 However, the highly heterogeneous and suppressive characteristics of the immune TME of HCC substantially restrict the therapeutic efficacy.8,9 To enhance the effectiveness of immunotherapy and the prognosis of patients, it is crucial to reshape the tumor microenvironment of HCC.
Immunogenic cell death (ICD) is a novel model of cell death that increases the immunogenicity of cancer cells and activates the body’s anti-tumor immunity.10 A variety of triggers and anti-tumor treatments can induce ICD, including chemotherapy,11 radiotherapy,12 extracorporeal phototherapy,13 photodynamic therapy,14 lysoviral therapy,15 and targeted therapy.16 ICD is manifested by the induced stimulation of tumor cells after upregulation of certain characteristic protein molecules (calcium reticulum protein CRT, adenosine triphosphate ATP, and HMGB1 expressed on the cell membrane surface or released into the extracellular compartment.17 These protein molecules are known as damage-associated molecular patterns (DAMPs).18 DAMPs enhance the immunogenicity of tumor cells, promote the recognition and antigen-presenting ability of DCs to cancer cells, stimulate DCs maturation, and mature DCs activate specific T cells to attack cancer cells. Currently, merely a few clinically relevant drugs have been demonstrated to trigger true ICD, such as adriamycin and onioncyclines, oxaliplatin,19 lurbinectedin,20 and belantamab mafodotin,21,22 which have been employed in the treatment of several malignancies. Previous studies demostrated that Oxaliplatin causes ICD in HCC and has a synergistic impact when combined with anti-PD-1 antibodies.23 The angiogenesis inhibitor lenvatinib induces ICD in HCC cells, and ICD-associated DAMPs further activate TLR3 and TLR4 to exert antitumor immune responses.24 The STAT3 inhibitor napabucasin upregulates translocation of calreticulin, ERp57, and HSPs to the surface of HCC cells, and downregulates the “don’t eat me” molecule CD47, which promotes ICD and anti-tumor immunomemory in HCC.25 Regarding the therapeutic application of ICD-inducing medications and the investigation of ICD-related indicators, insufficient evidence was utilized in HCC. Therefore, exploring ICD-related biomarkers might be crucial for comprehending the pathophysiology of HCC and improving the clinical outcomes of chemotherapeutic medications.
In this study, we used TCGA-LIHC and GEO data to develop molecular subtypes combined with ICD-related prognostic risk models to evaluate the TME, clinical outcomes, and response to chemotherapy and immunotherapy of HCC patients. The expression of key molecular genes was confirmed in a series of HCC patients and HCC cell lines.
Materials and Methods
HCC Datasets and Processing
In our study, we integrated HCC samples from two databases: 374 samples of TCGA-LIHC and 115 samples of GEO (GSE76427). Additional information includes survival data, somatic mutation information, clinical information, and copy number variation (CNV). We excluded HCC patients with incomplete survival information in subsequent analyses. After screening, 481 HCC patients were studied, including 370 TCGA patients and 111 GEO patients. RNA sequencing data (FPKM values) from the GEO and TCGA were obtained as normalized matrix files. For combinatorial analysis, the FPKM data were then transformed into transcripts with per kilobase million (TPM) values.26
Single Cell Analysis
The single-cell dataset GSE189903 was downloaded from the GEO database, which contains multi-regional paired samples of tumor margins, tumor cores, and paracancerous normal tissues from four HCC and three iCCA patients.27 We selected only core tumor samples from HCC patients for analysis, and these samples included a total of 27,819 cells. The R package “Seurat”28 was subsequently used for single-cell data analysis. The specific process is as follows: first, we filtered low-quality cells by two metrics: the number of expressed genes (nFeature_RNA) and the percentage of mitochondrial gene expression (percent.mt). Only cells with 500 < nFeature_RNA < 6000 and percent.mt < 10 were retained, resulting in 26,090 cells. After normalizing the data using the “NormalizeData” and “ScaleData” functions, 2000 highly differentially expressed genes were found by the “FindVariableFeatures” function. These genes were normalized and then subjected to PCA downscaling, and the cells were clustered and grouped using the first 20 dimensions by the “FindNeighbors” and “FindClusters” functions. The resolution parameter is set to 0.15. Then UMAP visualization is performed. We defined cell subpopulations using classical marker genes collected from the literature.29,30 And the “CellCycleScoring” function was used to calculate the cell cycle score. We downloaded 50 classical pathways from the MSigDB (https://www.gsea-msigdb.org/gsea/msigdb/) database and then used the “AddModuleScore” function to calculate the scores of these pathways in all single cells. This function calculates a score for every single cell on the basis of the average expression level of genes within the pathway. For the ICD signature collected from the literature, we used the “AddModuleScore” function to calculate the signature score. All other visualizations were done using the R package “ggplot2”.31
Unsupervised Clustering Based on ICDRGs
33 ICDRGs (Table S1) were acquired from previous literature and available data to investigate various ICD-associated patterns in HCC. Hierarchical aggregation clustering was employed to cluster the samples via the R package “ConsensusClusterPlus”.32 Stability evidence was then used for unsupervised analysis to evaluate cluster counts and membership.
TME and Immune Correlation Analysis
Single-sample gene set enrichment analysis (ssGSEA) was utilized to calculate the infiltration level of 23 immune cell types and evaluate their immune function and association. Furthermore, the “estimate” package33 was used to compute the immune and stromal scores of each HCC patient. Moreover, to assess the association between immune cell infiltration and risk score, several methods were employed, including XCELL, QUANTISEQ, TIMER, EPIC, MCPcounter, and CIBERSORT.
Enrichment Functional Analysis
The cut-off criterion of log|FC| ≥ 1 and adjusted P< 0.05 were used to find DEGs between the various ICD subtypes using the R program “limma”. The R package “clusterProfiler”34 was utilized to carry out GO and KEGG analysis of all DEGs. The c2.cp.kegg.v7.4.symbols.gmt gene set annotation file was used by GSEA, and pathways with p values<0.05 were deemed significant.
Construction and Validation of an ICD-Related Risk Score
112 prognostic DEGs out of 519 overlapping DEGs in two subtypes were identified using univariate Cox analysis. Prognostic DEGs were successively subjected to Lasso Cox regression analysis and multivariate Cox regression analysis. Finally, five genes were screened out to construct the risk model, including NKX3-2, CHODL, MMP1, NR0B1, and CTSV. We computed the risk score according to the formula: Risk score=∑(Expi*coefi). This prognostic model divided HCC patients into two different groups by the median value of the risk scores. In addition, the area under the curve (AUC) corresponding to various years and clinicopathological features was plotted utilizing the receiver operating characteristic (ROC) curve analysis. We generated nomograms from clinicopathological data using the “regplot” R tool. Nomograms can be used to examine the 1-, 3-, and 5-year overall survival(OS) probability for HCC. Additionally, we generated heatmaps to analyze the distribution of various clinicopathological data in various groups.
Response of Immunotherapy and Drug Susceptibility Analysis
From the Cancer Immunome Database (TCIA, https://tcia.at/home), we were able to acquire the Immunophenoscores (IPS) of patients with HCC. The Wilcoxon rank-sum test was employed to examine the connection between IPS and the risk signature. Furthermore, the R package “pRRophetic” was used to examine the half inhibitory concentration (IC50) of common anti-tumor drugs.
Cell Culture, Transfection and Real-Time PCR
THLE-2 cells were obtained from Meisen Cell Technology Co., Ltd (Zhejiang, China). Three human HCC cells, HepG2, Hep3B, and Huh7, were originally derived from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). HCCLM3 cell lines were originally purchased from iCell Bioscience Inc. (Shanghai, China). At 37°C and 5% CO2, all cell lines were grown in DMEM medium with 10% fetal bovine serum (FBS). Three different small interfering RNAs were developed and manufactured by GenePharma (Suzhou, China) to target NKX3-2. The target sequences were: 5’-CCAACACCUUGACGUCCUUTT-3’(siNKX3-2#1);5’-GCUUUAACCACCAGCGCUATT-3’(siNKX3-2#2); 5’-GCGACGACCAGAGACAAUATT-3’(siNKX3-2#3) and 5’-UUCUCCGAACGUGUCACGUTT-3’(siCtrl). All transfections were performed using JetPRIME® (Polyplus-transfection S.A, Illkirch, France) based on the manufacturer’s instructions. The total RNA of the cell lines was extracted using the TRIzol reagent (Invitrogen, CA, USA). The PrimeScript RT kit was then used to perform reverse transcription to create cDNA (Nazyme, Nanjing, China). Then, qRT-PCR was performed using a Universal SYBR qPCR kit (Vazyme, Nanjing, China) in the CFX96TM Real-Time System (Bio-Rad, California, USA). Primers used in this study were listed as follows: NKX3-2 (Forward): CCTTAAACAGGTGATCCCAC; NKX3-2 (Reverse):ATACAGCGAATAGAGCTTCC.GAPDH (Forward):GTCTCCTCTGACTTCAACAGCG;GAPDH(Reverse):ACCACCCTGTTGCTGTAGCCAA. Relative quantification was determined using the 2-ΔΔCt method.
Immunohistochemistry
15 freshly harvested human HCC tumor samples and matched normal samples were procured from the Third Affiliated Hospital of Sun Yat-sen University (SYSU), Guangzhou, China, with informed patient consent. Between June 2019 and September 2022, these patients underwent treatment for HCC at the Third Affiliated Hospital of SYSU. HCC diagnosis was confirmed based on pathological findings or the American Association for the Study of Liver Disease radiological criteria using either computed tomography or magnetic imaging resonance results. All patients and healthy controls were screened for serum human immunodeficiency virus antibodies, hepatitis B surface antigen, hepatitis C virus antibodies, hepatitis D virus antigens, and hepatitis D virus antibodies. Patients who were positive for human immunodeficiency virus; were pregnant; had received systemic corticosteroids, immunosuppressive agents, or anti-cancer therapies were excluded.
After being deparaffinized with xylene, the tissue sections from HCC patients were rehydrated with an alcohol gradient. To prevent any potential influence from endogenous peroxidase, a treatment with 3% H2O2 was carried out. NKX3-2 antibody (1:500; ab196020, Abcam) was incubated on the samples for one night at 4 °C, and then the samples were treated with Goat Anti-Rabbit IgG H&L (HRP) (1:200; G1213, Servicebio) secondary antibody for 2 hours at room temperature. After applying the DAB chromogenic kit (AFIHC004, Aifang) to the samples, the nuclei were counterstained with hematoxylin.
Cell Counting Kit (CCK)-8 Assay
Transfected HCC cells were inoculated into 96-well plates. At the same time each day for the following 1 to 4 days, 10 µL of CCK-8 reagent (Biosharp, Anhui) was added to each well and incubated at 37 °C for 2 hours away from light. Absorbance was measured at 450 nm.
Colony Formation Assay
500 transfected HCC cells were inoculated in a 6-well plate and maintained for 2 weeks. Cells were then fixed with 4% paraformaldehyde for 15 minutes, stained with crystal violet (Biosharp) for 15 minutes, and rinsed with PBS, and colonies were counted.
Wound Healing and Transwell Assays
In the wound healing assay, transfected cells were grown in a 6-well plate for 24 hours, and then the wounds were scratched with a 20-microliter sterile pipette tip. Cells were rinsed with PBS and photographs of each wound were taken at 0 h and 24 h under an inverted microscope (Olympus, Japan). For the Transwell assay, 200 µL of serum-free medium containing 2×104 cells was added to the upper chamber (10 µm) and 500 µL of complete medium with serum was added to the lower chamber. The ImageJ software was used to quantify the statistical cell migration capacity.
Statistical Analyses
The Student’s t-test was used to compare the gene expression levels in HCC and normal tissues. The OS of patients in different risk groups was compared using the K-M analysis and Log rank test. A Chi-square test was utilized for comparisons among the clinical variances within each cohort. The Mann–Whitney test and Benjamini-Hochberg (BH) technique were used to compare the ssGSEA scores of immune cells and pathways between the two groups. R software (version 4.2.2), GraphPad Prism software (version 8.0), and SPSS (Version 23.0) were used for the statistical analyses in this study. The definition of statistical significance was as follows: ns, not significant; *p <0.05; **p ≤0.01; ***p ≤0.001; ****p < 0.0001.
Results
The Landscape of the ICDRGs in TCGA-LIHC
The framework of the overall study is demonstrated in Figure 1A. This study comprised a total of 33 ICDRGs, which were chosen based on existing literature publications.10 Firstly, we investigated the incidence of somatic mutations and the copy number variation(CNV) of ICDRGs in the TCGA-LIHC cohort. 74 of 371 HCC samples carried ICDRGs mutations, exhibiting a 19.95% mutation frequency. The most frequently altered gene among them was PIK3CA, which was promptly followed by NLRP3, IL1R1, HSP90AA1, CXCR3, and CALR (Figure 1B). Figure 1C showed that CNV was frequently observed and that deletions or amplifications of certain ICDRGs had distinctive features. The genes with higher frequency of copy number amplification included NLRP3, IL10, PIK3CA, and TNF. The genes with higher deletion frequency included HMGB1, HSP90AA1, IFNGR1, and ATG5. The Spearman correlation heatmap provides a comprehensive overview of the interactions of the 33 ICDRGs (Figure 1D). The boxplot showed that the expression patterns of 24 of the 33 ICDRGs differed significantly between normal and HCC samples (Figure 1E). Numerous ICDRGs with CNV gain had excessive expression in HCC samples, including BAX, CASP8, CXCR3, EIF2AK3, LY96, and PIK3CA. While ICDRGs with CNV loss, including CD4, NT5E, and TLR4, showed decreased expression levels in HCC samples. These findings indicated a possible correlation between the expression of these ICDRGs and CNV changes in HCC. Collectively, the genetic alterations and expression features of ICDRGs in normal and HCC samples were highly heterogeneous, suggesting that they occupy important roles in the advancement of HCC.
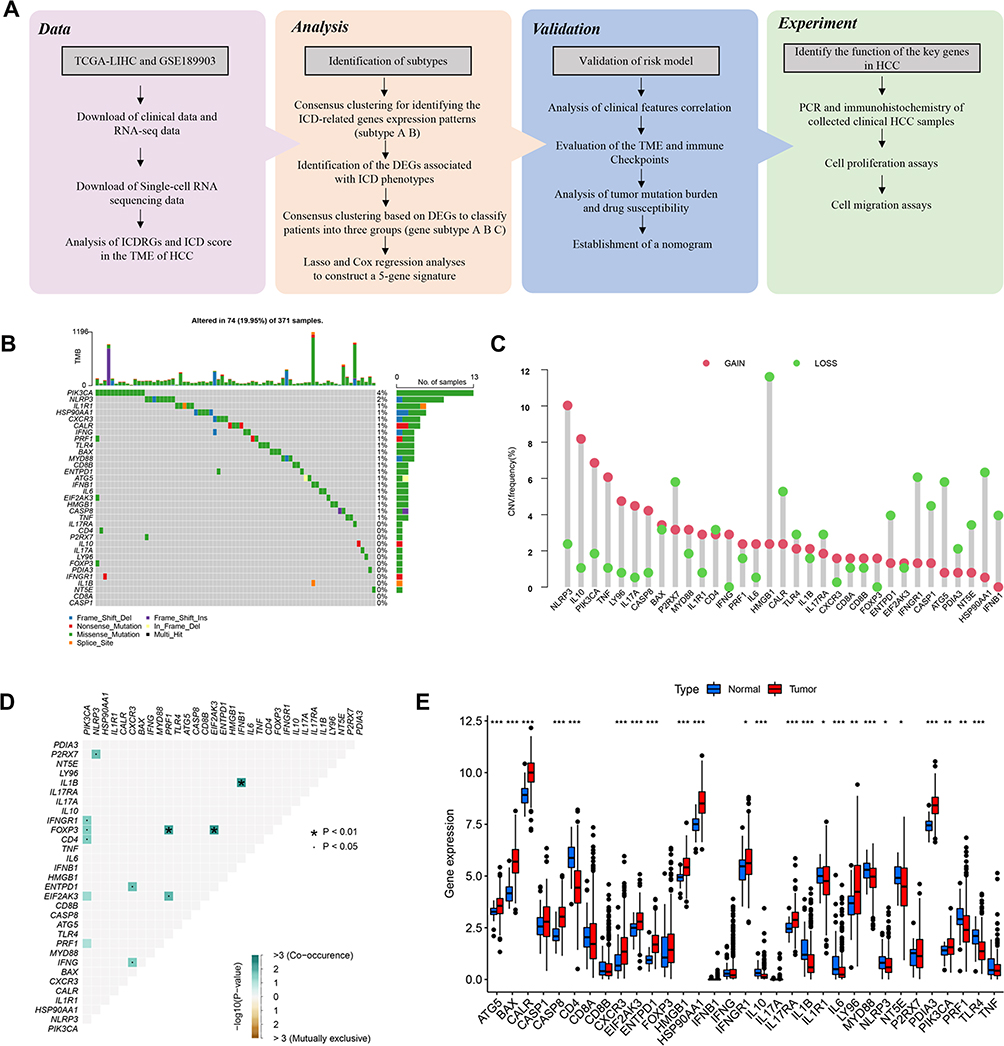 |
Figure 1 Genetic alterations and expression features of ICDRGs in TCGA-LIHC. (A)The flowchart of the entire study. (B) The mutation frequency of 33 ICDRGs in 371 HCC patients from TCGA. (C) CNV frequency of ICDRGs in TCGA-LIHC cohort. The height of the column represents the alteration frequency. The green dot represents deletion frequency. The red dot represents the amplification frequency. (D) Spearman correlations between ICDRGs. (E) Differences in mRNA expression levels of ICDRGs in normal and tumor samples.*P < 0.05; **P < 0.01; ***P < 0.001; ns, no significance. |
ICD-Related Molecular Subtypes and Immune Landscape in HCC Based on Bulk RNA Data
To discover ICD-based molecular subtypes for HCC, unsupervised clustering analysis was performed using 33 ICDRGs in TCGA-LIHC cohort. At a consensus matrix k value of 2, there were the fewest crossovers among the HCC samples (Figure 2A). 370 patients were eventually categorized into two subgroups, subtype A (203 samples) and subtype B (167 samples), based on ICDRGs expression. The patients in subtype A showed better OS compared to subtype B (P<0.001) (Figure 2B). The heatmap indicated the variations in clinicopathological information between the two subtypes, including age, gender, TNM stage, and tumor grade (Figure 2C).
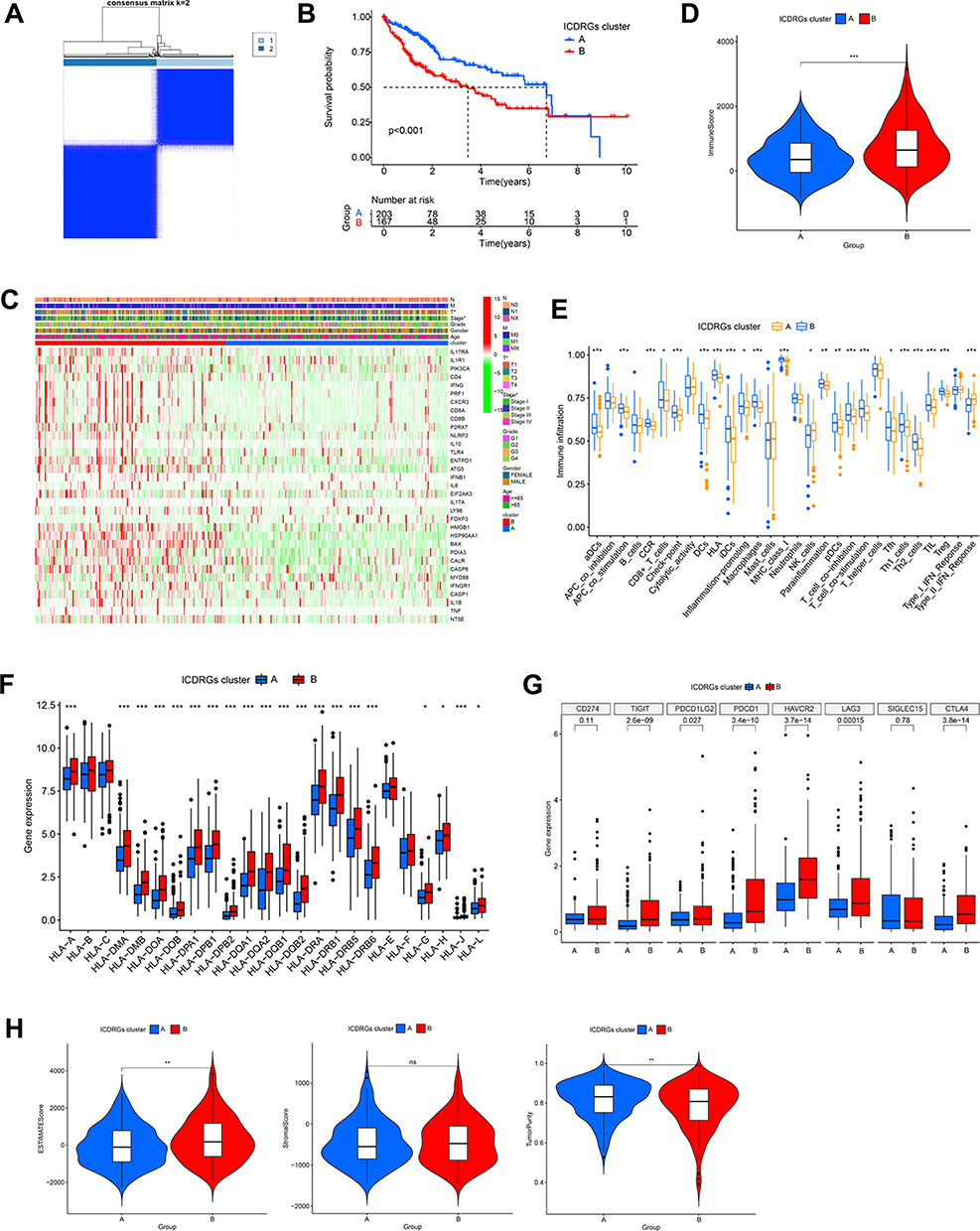 |
Figure 2 Identification of ICD-related gene subtypes in HCC. (A) Consensus clustering of HCC patients with k=2. (B) Kaplan-Meier curve showing the survival advantage of the ICD-low subtype was considerably better. (C) Heatmap displaying the association between the two subtypes and clinicopathological factors. (D) The immune score between two subtypes. (E) The landscape of tumor-infiltrating immune cells was analyzed by ssGSEA. Box plots revealing differential expression of HLA genes (F) and multiple ICGs (G) between two subtypes. (H) Violin plots revealing the ESTIMATE score, stroma score, and tumor purity in two subtypes. *P < 0.05; **P < 0.01; ***P < 0.001; ns, no significance. |
Then we explored the infiltrating level of immune cells and immune-related functions across different subtypes using ssGSEA. Notably, the infiltrating level of several immune cells elevated in subtype B, such as various types of DCs (aDCs, iDCs, pDCs, and DCs), CD8+ T cells, macrophages, Th1 cells, Th2 cells, and TILs (Figure 2E). Simultaneously, APC co-stimulation, checkpoint, inflammation-promoting, and T-cell co-stimulation showed similar trends in subtype B indicating antitumor immunity and highly active antigen presentation. Besides, the subtype B displayed increased immune scores, ESTIMATE scores, and reduced tumor purity than the subtype A (Figure 2D–H). This suggested that subtype B was characterized by increased stromal and immune cell infiltration and activity in the tumor microenvironment. Subtype B was also upregulated in most of the HLA genes and common immune checkpoint genes(ICGs)including PD1 (PDCD1), PDL2 (PDCD1LG2), LAG3, HAVCR2, TIGIT and CTLA4, and the opposite for the subtype A (Figure 2F and G). In a word, these findings revealed that ICDRGs were possibly related to immunotherapy, and that subtype B exhibited specific TME characteristics with higher stromal and immune cell infiltration and activity, suggesting that this subtype might be more immunogenic and dynamic tumor microenvironment.
Identification of Gene Subtypes Based on 519 Overlapping DEGs
To comprehend the biological mechanisms contributing to different prognoses in both subtypes, we identified differential expression genes and related signaling pathways. Differential gene expression analysis was conducted for the subtype A and subtype B with the screening criterion of |log2(Fold Change)|>2 and p<0.05. There were 519 DEGs, of which 44 were decreased and 475 were evaluated (Figure S1A, B). GO analysis demonstrated that the most abundant processes of DEGs were immune-related biological processes, such as the B-cell receptor signaling pathway, positive regulation of lymphocyte activation, activation of the immune response, antigen receptor-mediated signaling pathway, antigen binding, T-cell receptor complex, immunoglobulin complex, and cytokine activity (Figure S1C). The KEGG analysis displayed that the most abundant signaling pathways of DEGs were cancer-related pathways, including retinol metabolism, drug metabolism-cytochrome P450, cell adhesion molecules, ECM-receptor interaction, glycolysis, and NF-κB signaling pathway (Figure S1D). Furthermore, we utilized GSEA to compare the biological functions and signaling pathways between the two subtypes and found that several cancer metastasis pathways were enriched in the subtype B, including cell adhesion molecules and ECM receptor interaction (Figure S1E). While several metabolic pathways including fatty acid metabolism, primary bile acid biosynthesis, glycine serine and threonine metabolism, and drug metabolism, were concentrated in the subtype A (Figure S1F).
We carried out a univariate Cox regression algorithm of DEGs and discovered 112 prognostic-related DEGs for further analysis (Figure S2). Unsupervised cluster analysis was used to separate the TCGA-LIHC cohort of patients into 3 gene clusters based on the 112 DEGs (Figure 3A). Compared with gene clusters A and C, gene cluster B exhibited a significant survival disadvantage (Figure 3B). The DEGs between several gene clusters were statistically varied, as shown by a heatmap. While the majority of DEGs are upregulated in gene cluster A (Figure 3D). The three gene clusters were also examined for clinical pathological variables, including sex, age, TNM staging, and survival status while there were no statistical differences. Then, we compared the expression of 33 ICDRGs in three gene clusters, with gene cluster A having the highest levels (Figure 3E).
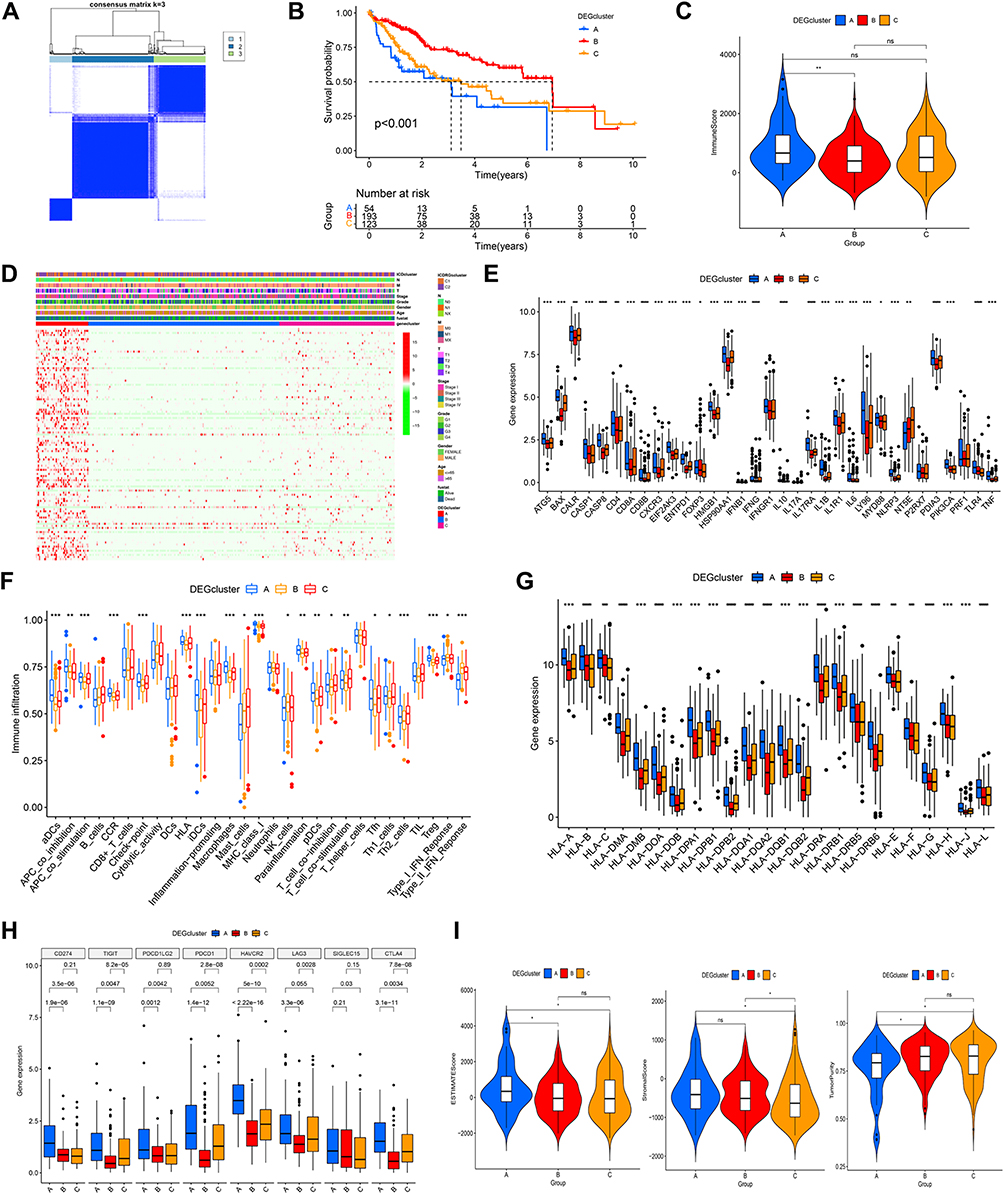 |
Figure 3 Associations between the DEG-based genotypes and tumor immune cell infiltration (A) Consensus clustering of HCC patients with k = 3. (B) The survival curve showed patients in gene cluster B exhibited higher OS(p < 0.001). (C) The immune score were analyzed by ssGSEA in three gene clusters. (D) Variations in clinical factors among patients of the three gene clusters, with fustat, age, gender, tumor grade, and stage as reference indicators. (E) Expression profile of 33 ICDRGs in three gene clusters. (F) The population of tumor-infiltrating immune cells was analyzed by ssGSEA in three gene clusters. Box plots indicating the difference of HLA genes (G) and multiple ICGs (H) between three gene clusters. (I) Violin plots showing the differences in ESTIMATE score, stromal score, and tumor purity between three gene clusters. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001;ns, no significance. |
Interestingly, in contrast to the survival disadvantage, gene clusters A and C displayed a higher abundance of most infiltrating immune cells correlated with antigen presentation, tumor killing, processing, and stronger immune functions (Figure 3F). The immune score in the three clusters was consistent with the prior findings (Figure 3C). Variations of the HLA genes and ICGs were also found within the different gene clusters (Figures 3G and H). GeneCluster A exhibited the most enhanced expression of these genes, suggesting that it may be more responsive to immunotherapy. In summary, the above outcomes indicated gene Cluster A represented an immunological “hot” phenotype. The ESTIMATE score and stroma score of gene cluster A were also higher than that in gene clusters B and C (p < 0.001), while the tumor purity was the opposite (Figure 3I).
Construction and Verification of the Risk Model
According to the 112 prognosis-related genes obtained previously, we conducted multivariate Cox analysis and LASSO algorithm to screen out 5 genes to construct the prognosis model (Figure 4A and B). The model formula is as follows:Risk Score=0.446301544408387*NKX3-2+0.214878258114391*CHODL+0.290746555855764*MMP1+0.236487283993298*NR0B1+0.138635202753353*CTSV. Forest plot showed all of 5 genes predicting undesirable prognosis of HCC (HR>1, p<0.01) (Figure 4C). Then, the TCGA dataset was 7:3 divided into training and testing groups, and the GSE76427 dataset was employed for the external testing set. HCC samples were classified into high-risk and low-risk groups by employing the median risk score as the cut-off point. Kaplan-Meier curves revealed that the low-risk group presented with a considerably higher survival rate than the high-risk group in both the training set and the validation set (Figure 4D–G).
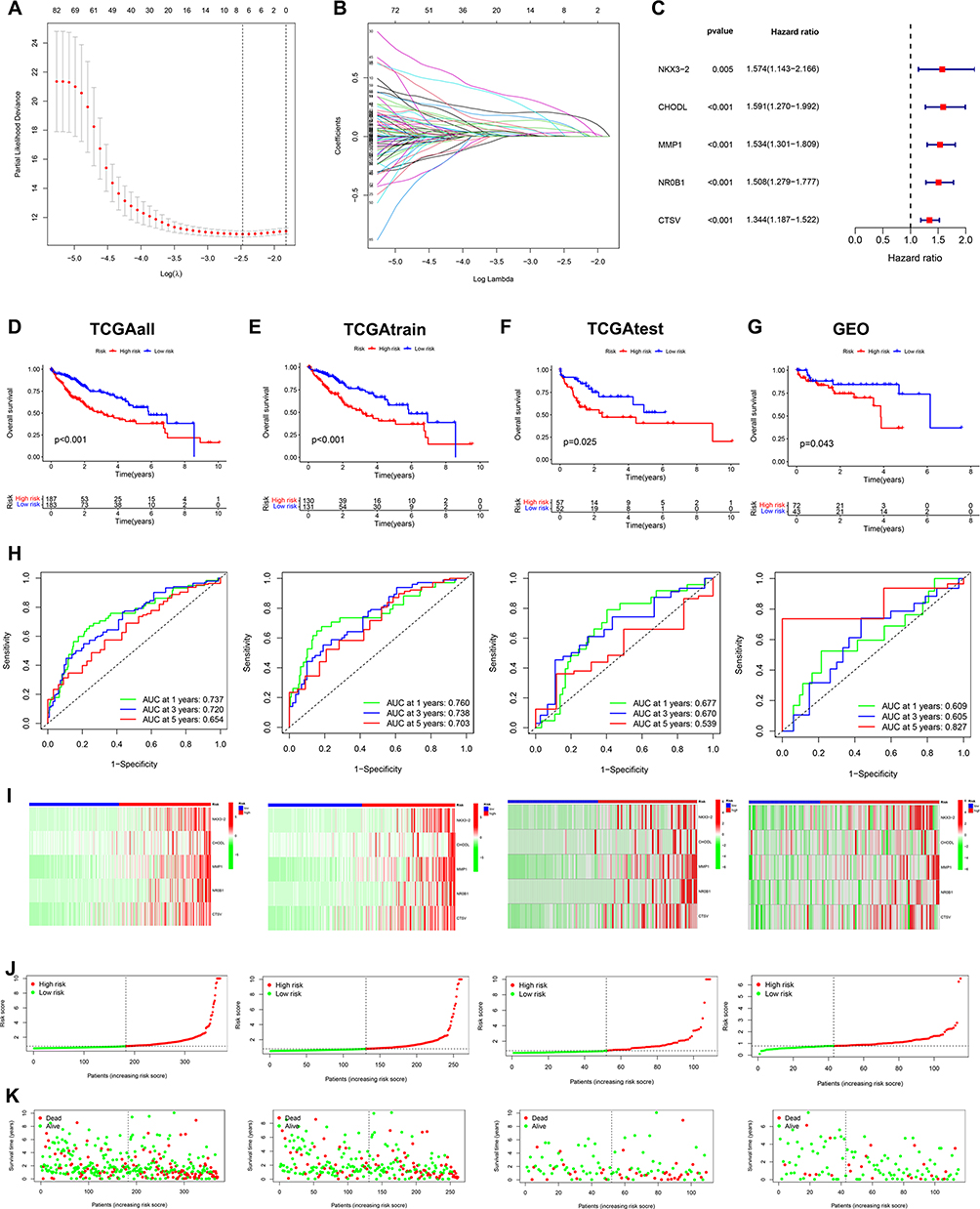 |
Figure 4 ICD-related prognostic signature generation and validation. (A-C) Lasso-Cox analysis of survival-related DEGs to identify five risk genes and construct the ICD-related prognostic model. Survival curves of different risk groups in TCGA cohort (D), TCGA training cohort (E), TCGA-testing cohort (F), and GEO validating cohort GSE76427 (G). (H) ROC curves in four datasets to estimate 1-, 3-, and 5-year survival. (I) Heatmaps of the 5 ICD-related gene signatures in the four datasets. (J, K) Distribution and scatterplot of risk scores in the four datasets. |
Then we utilized ROC analysis to determine the diagnostic effectiveness of the risk scores. The AUC values at 1, 3, and 5 years were 0.760, 0.738, and 0.703 in the TCGA training group, 0.677, 0.670, and 0.539 in the TCGA testing group, and 0.609, 0.605 and 0.827 in GEO validating group (Figure 4H). The high-risk group demonstrated higher gene expression distribution and higher mortality, as can be observed from the patient survival status, risk score distribution, and gene expression profile (Figure 4I–K).
Discovery of Molecular Functions and Pathways in Different Risk Groups
To discover the molecular variances and signaling pathways that underlie the various groups identified by the 5-gene risk model, GSEA was conducted. As shown in Figure S3A, multiple cell proliferation pathways were enriched in the high-risk group, such as base excision repair, cell cycle, DNA replication, neurotrophin signaling pathway, RNA degradation, and so on. While numerous metabolic pathways were encompassed in the low-risk group, such as butanoate metabolism, fatty acid metabolism, drug metabolism, primary bile acid biosynthesis, and so on (Figure S3B). GO and KEGG analysis were further conducted on the DEGs between the two risk groups (p<0.05). GO analysis suggested that cellular replication and transcriptional events were notably enriched, such as DNA replication, RNA splicing, and nuclear division (Figure S3C). The KEGG analysis revealed that many immune-related pathways were remarkably concentrated, including Fc gamma R mediated infection, Th1, and Th2 cell differentiation, and TNF signaling pathway (Figure S3D).
Relationship of ICD-Related Risk Model with the TME in HCC
The association between the ICD-related risk model and TME was also examined. In terms of immune infiltration abundance, higher concentrations of B cells, mast cells, neutrophils, and NK cells were observed in the low-risk group (Figure 5A). While the high-risk group displayed elevated levels of aDCs, iDCs, macrophages, and Tregs. In addition, the low-risk group also exhibited enhanced antitumor effects, such as type I IFN response, type II IFN response, and cytolytic activity (Figure 5B). However, we found no significant differences between the ESTIMATE score and immune score in the two risk groups (Figure 5C). Heatmap showing the correlation between the 5 ICD genes, risk score, and immune cells and immune function (Figure 5D), in which NKX3-2, MMP1, and CTSV showed the most significant correlation. Furthermore, we addressed the association between the risk scores and common ICIs in this model, respectively, revealing risk scores were correlated with the expression of ICGs such as CD276, CD274, CTLA4, and so on (Figure 5E). Finally, we examined the relevance between risk scores and the infiltration levels of immune cells obtained by various algorithms including CIBERSORT, TIMER, XCELL, CIBERSORT, MCPCOUNTER, QUANTISEQ, and EPIC (Figure 5F, Figure S4).
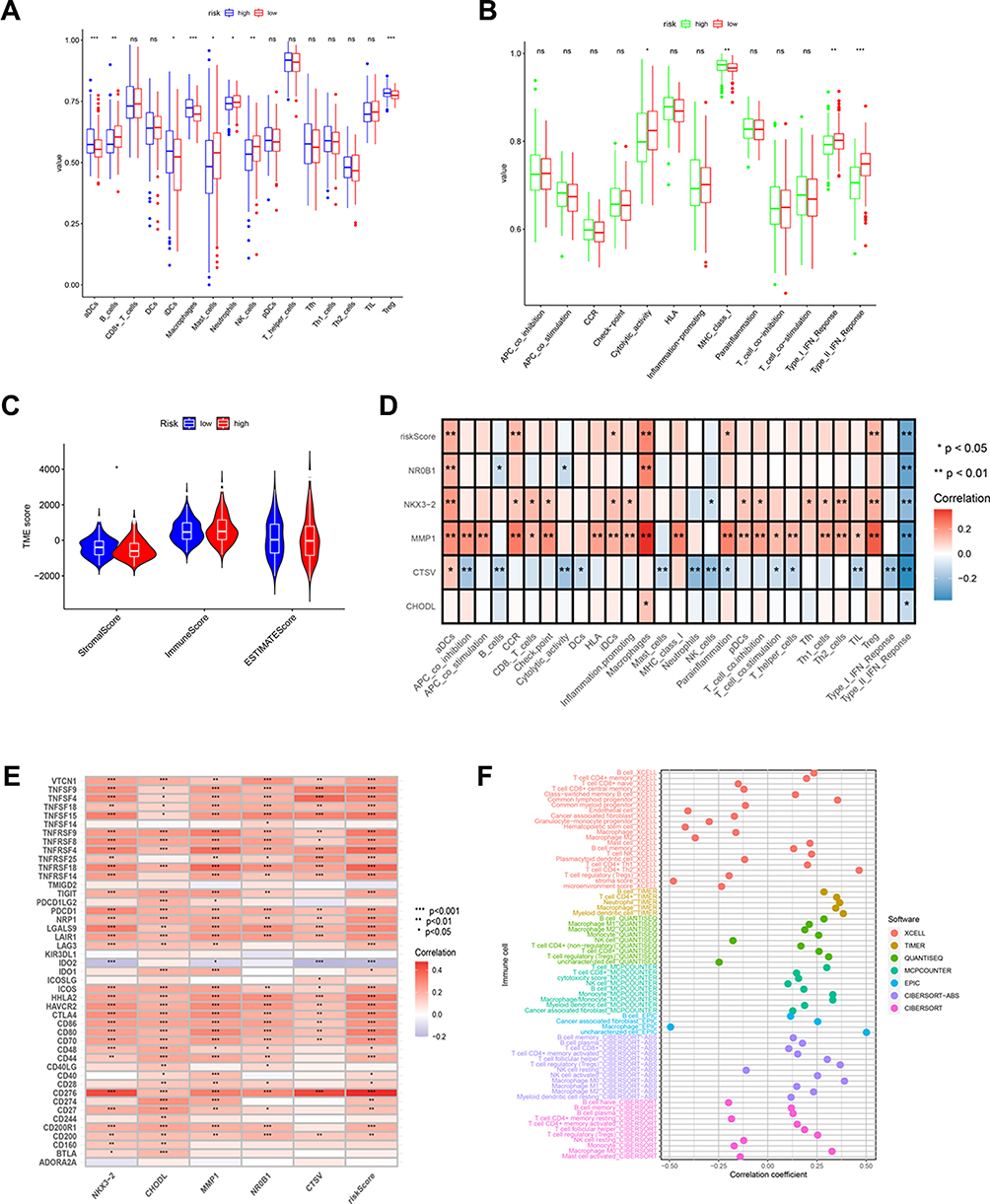 |
Figure 5 Relationship of ICD-related gene signature with the TME. (A) The infiltration abundance of immune cells in two risk groups. (B) The difference in immune functions between the two risk groups. (C) The stromal score, immune score, and ESTIMATE score between the two risk groups. (D) Heat map revealing the relationship between the 5 ICD genes, risk score, and immune cells and immune function. (E)Heatmap indicating the relationship between ICGs and 5 ICD genes, and risk scores. (F)The connection between stromal and immune cells and risk score was determined by seven different algorithms. *P < 0.05; **P < 0.01; ***P < 0.001; ns, no significance. |
Analysis of Tumor Mutation Burden and Drug Susceptibility
There is growing evidence that tumor mutational load (TMB) is a reliable indicator of tumor immunotherapy. Thus, we assessed variations in the distribution of somatic mutations between high and low-risk groups. TP53, CTNNB1, TTN, MUC16, and PCLO had the highest mutation rates in different risk groups, all with mutation rates above 15%. However, TMB was not statistically significant between the two risk groups (Figure 6A–C). Spearman correlation analysis also suggested no significant correlation between risk score and TMB (Figure 6D). Interestingly, the prognosis of patients with low TMB was remarkably better than that of patients with high TMB (Figure 6E). And the low-risk low-TMB group displayed the highest rates of survival than other subgroups (Figure 6F).
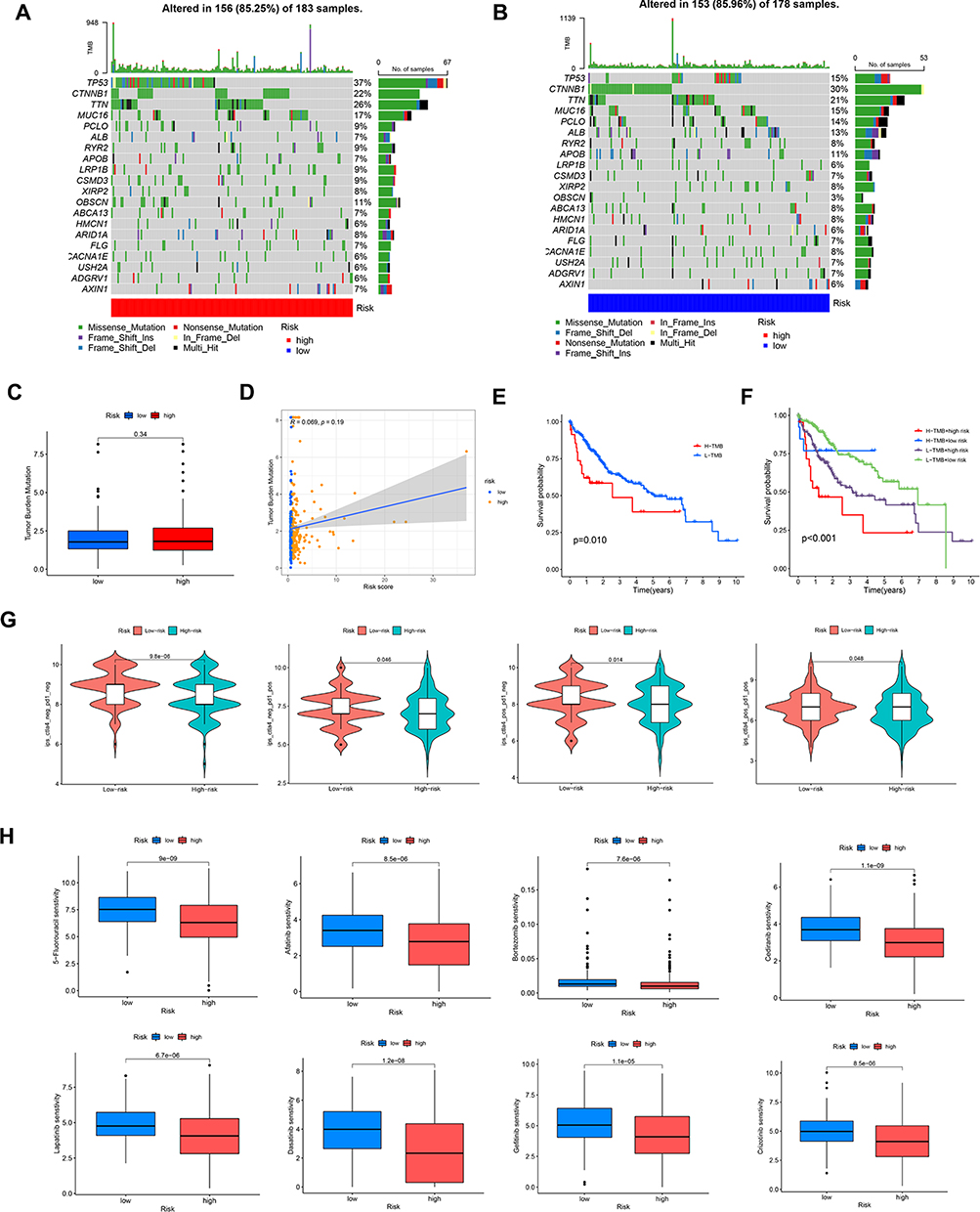 |
Figure 6 Analysis of TMB and drug susceptibility. (A, B) Mutations in high- and low-risk populations are summarized via a waterfall plot. (C)TMB variation between the two risk groups. (D) Relationship between the risk score and TMB. (E and F) The KM curve of the tumor mutation burden versus the OS. (G)Difference of immunotherapy response with CTLA4- and PD-1-, CTLA4- and PD-1+, CTLA4+ and PD-1, and CTLA4+ and PD-1+ between two risk groups. (H)Prediction of the drug sensitivity (IC50) of 5-Fluorouracil, Afatinib, Bortezomib, Cediranib, Lapatinib, Dasatinib, lapatinib, and Crizotinib. |
The sensitivity to immunotherapy and chemotherapy medications of patients in two separate risk groups was then evaluated. Notably, HCC patients in the low-risk group were more responsive to CTLA-4 inhibitors alone, PD-1 inhibitors alone, and the combination of the two drugs (p < 0.05) (Figure 6G). Besides, patients in the low-risk group exhibited greater susceptibility to 5-Fluorouracil, afatinib, bortezomib, cediratinib, lapatinib, dasatinib, gefitinib, and crizotinib than the high-risk group (Figure 6H). These information implied that the patients in the low-risk group might have a better prognosis and lower chemotherapeutic treatment resistance.
Analysis of Clinical Correlation and Construction of a Nomogram
Then, the association between risk scores and clinical characteristics was assessed. The risk score was p < 0.001, HR = 1.112, CI = 1.078–1.148 in univariate Cox analysis (Figure 7A); and the risk score was p < 0.001, HR = 1.113, CI = 1.075–1.152 in multivariate Cox analysis (Figure 7B). TNM stage was another important predictive factor for OS (p 0.001, HR = 1.679). Significant differences in 5 risk gene expression, age, gender, tumor grade, and TNM stage between the two risk groups could be seen in the heatmap (Figure 7C). Correlation analysis with clinical features displayed that risk scores were positively related to advanced tumor status. HCC patients in grades 1–2, stage I–II, and stage T1-2 presented lower risk scores than those of the other groups, with no variation across age, N, or M stages (Figure S5). Then, a nomogram was created combined with the risk score, stage, and clinical factors (Figure 7E). The calibration plots showed the nomogram was comparable to an ideal model with good effectiveness and accuracy (Figure 7G). The nomogram represented the best performance followed by risk and stage. The AUC values of the nomogram were 0.746, 0.720, and 0.695 in predicting 1,3, and 5-year OS of HCC patients (Figure 7D, F and H).
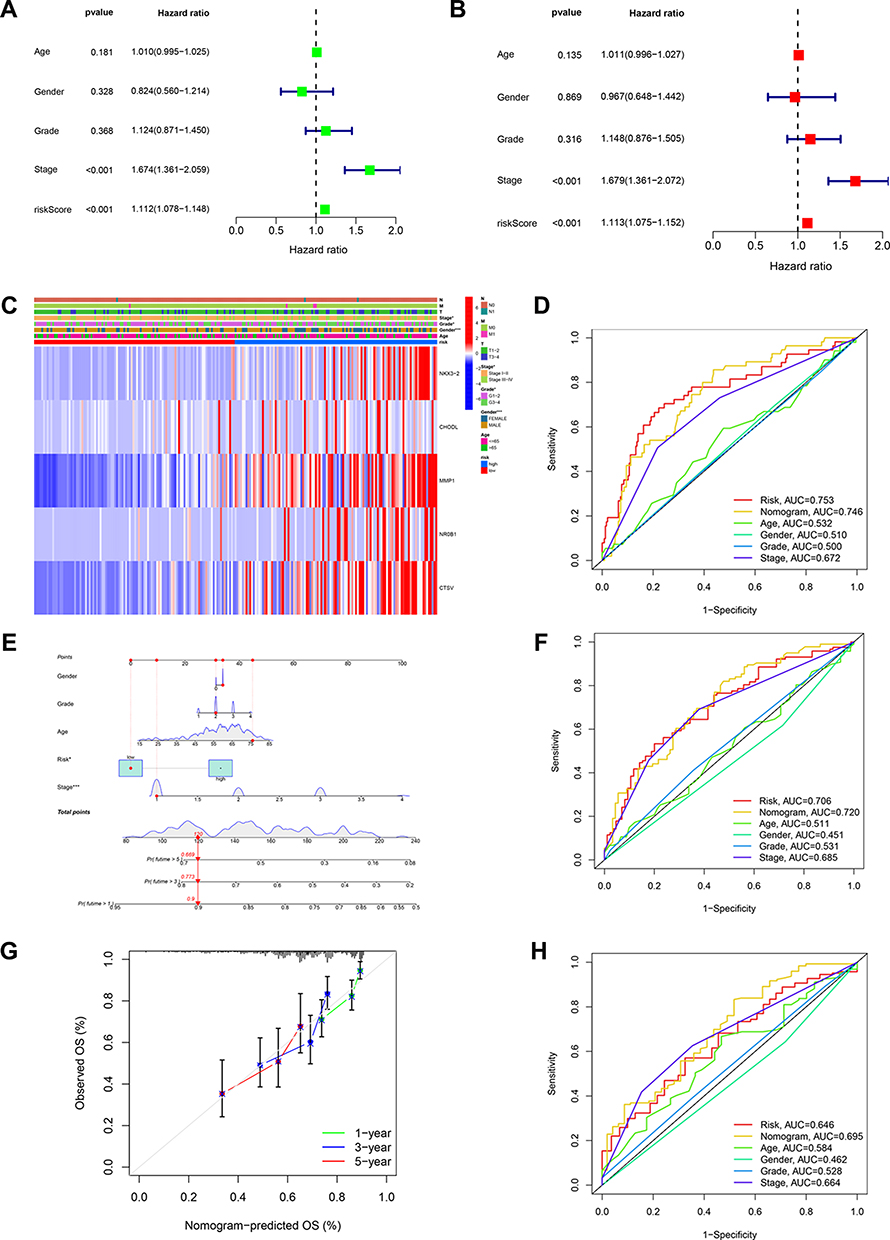 |
Figure 7 Analysis of clinical correlation and development of a nomogram. (A) The univariate Cox regression analysis and(B) multivariate Cox regression analysis of prognostic factors. (C) Heatmap for differential clinicopathological features of two risk groups. (D) ROC curves of the nomogram, risk, stage, and age for estimating 1-year OS of HCC. (E) Development of a nomogram based on risk, gender, age grade, and stage. (F) ROC curves of the nomogram, risk, stage, and age in predicting 3-year OS of HCC. (G) Calibration curves. (H) ROC curves of the age, risk, stage, and nomogram for estimating 5-year OS of HCC. |
Validation of the Effect of NKX3-2 on the Biological Behavior of HCC Cells in vitro
To verify the 5 risk ICD genes, we obtained the expression data from HCC patients and cell lines. TCGA data showed HCC samples exhibited higher levels of NKX3-2, CHODL, MMP1, NR0B1, and CTSV expression than normal samples (Figure S6A). Figure S6B indicated that CTSV, MMP1, and NR0B1 were expressed at high levels in most HCC cell lines according to the CCLE data. Survival analyses revealed that increased expression of the 5 risk ICD genes in HCC patients was linked to poor outcomes (Figure S6C). Since NKX3-2 had a relatively high hazard ratio and its correlation coefficient was the largest in the established risk-prognosis model, it was selected to be functionally validated. Firstly, NKX3-2 mRNA levels were examined in 15 HCC and 15 adjacent normal tissues by qPCR, demonstrating a substantial upregulation in HCC samples (Figure 8A). Additionally, immunohistochemistry revealed HCC tumor tissues exhibited higher levels of NKX3-2 protein levels than the nearby normal tissues. (Figure 8B). Then, qRT-PCR detected the mRNA expression levels of NKX3-2 in HCC cell lines (Figure 8C). HepG2 and HCCLM3 cells with comparatively high expression of NKX3-2 were performed for knockdown experiments. Knockdown efficiencies of siNKX3-2 were confirmed (Figure 8D and E). The clone formation experiments and CCK-8 assays showed the proliferation of HepG2 and HCCLM3 cells decreased after NKX3-2 silencing (Figure 8F, H and I). Similarly, the migratory potential of the HepG2 and HCCLM3 cells was greatly reduced after NKX3-2 knockdown by transwell and wound healing assays (Figure 8G–J).
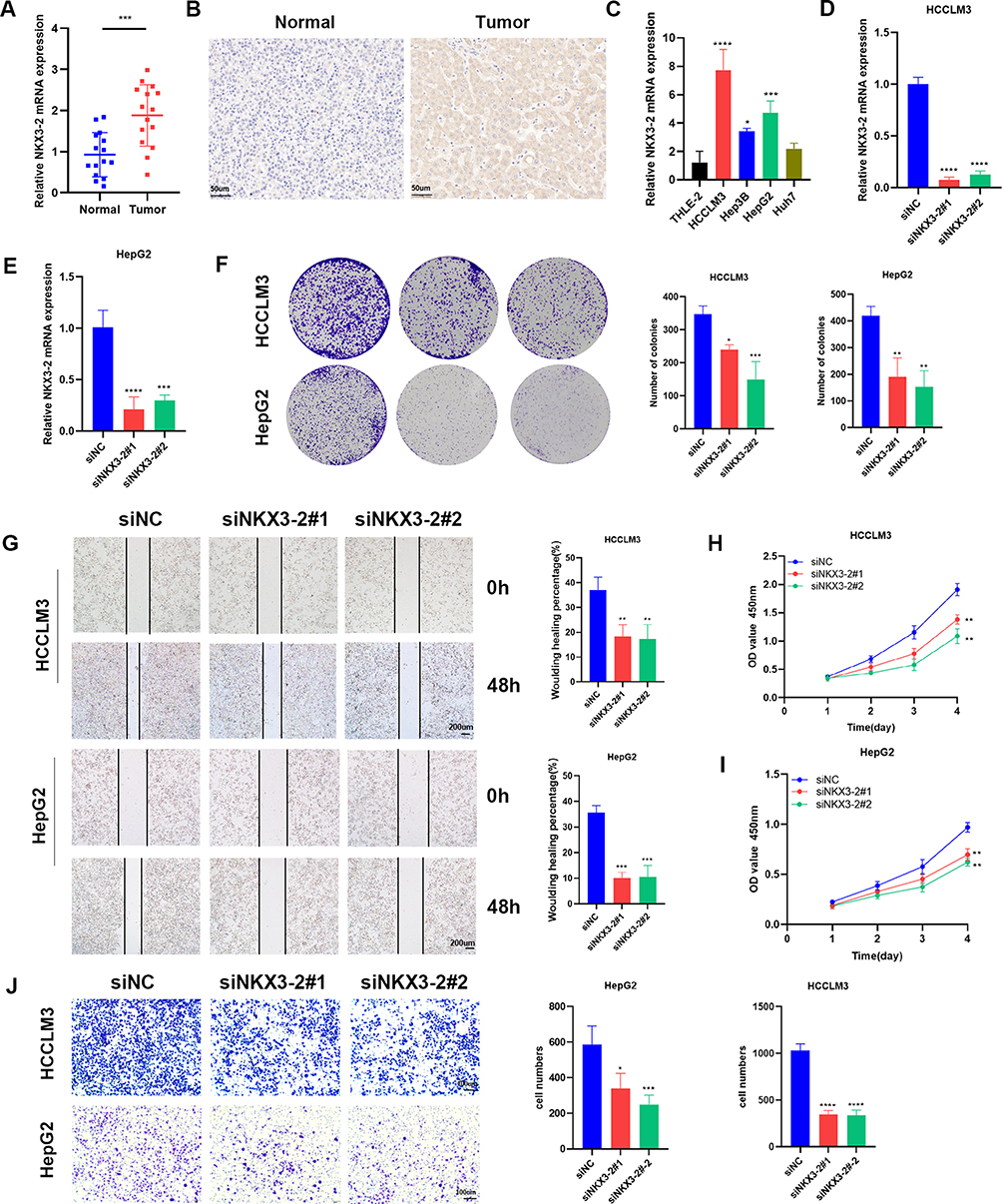 |
Figure 8 NKX3-2 exhibited amplification in HCC specimens and silencing NKX3-2 decreased the proliferation and migration of HCC cells. (A) NKX3-2 mRNA expression in tumor and adjacent normal tissues. (B) Representative images and staining scores of the IHC assay (scale bars: 50μm). (C) qRT-PCR showed NKX3-2 was up-regulated in HepG2, HCCLM3 and Hep3B HCC cells than in THLE-2 cells. (D) HCCLM3 cells and (E) HepG2 cells were transfected with siNKX3-2 for 48h and qRT-PCR detected the knockdown efficiency. (F) After NKX3-2 knockdown, the cloning ability of HCCLM3 and HepG2 cells decreased. (G) Wound healing experiments showing the migration ability of HCCLM3 and HepG2 cells was inhibited after NKX3-2 knockdown (scale bars: 200 μm;). CCK8 assays showing the proliferation of HCCLM3 (H) and HepG2 (I) cells was inhibited after NKX3-2 knockdown. (J) Transwell assays showing the invasion capability of HCCLM3 and HepG2 cells were suppressed after NKX3-2 silencing (scale bars: 100 μm;). **P < 0.01; ***P < 0.001; ****P < 0.0001; ns, no significance. |
ICDRGs and ICD Score in the TME of HCC Based on scRNA-Seq Analysis
The immune cells in TME might also be influenced by ICD. Thus, we investigated the ICD gene signature in multiple stroma cells based on the single-cell dataset GSE189903, which contains multi-regional paired samples of tumor margins, tumor cores, and paracancerous normal tissue from four HCC and three iCCA patients. To comprehend the precise function of ICDRGs in the immune TME of HCC, the single-cell dataset GSE189903 was analyzed, which contains multi-regional paired samples of tumor margins, tumor cores, and paracancerous normal tissue from four HCC and three iCCA patients.27 We selected only core tumor samples from HCC patients for analysis, and these samples included a total of 27,819 cells. After quality control and normalization, the final 26,090 cells were retained. UMAP downscaling visualization was performed, and 12 cell clusters were obtained (Figure 9A–C). We defined cell subpopulations manually by using classical marker genes and obtained seven cell types: malignant cells (EPCAM, KRT19), hepatic parenchymal cells (TTR, ALB), fibroblasts (FN1, COL1A1), endothelial cells (PECAM1, VWF), myeloid cells (LYZ, CD68), B cells (MS4A1, CD79A), T cells (CD3E, CD3D) (Figure 9D and E). DEGs of different cell types were discovered (Figure 9F; Table S2), and GVSA enrichment analysis was performed to show the relevant biological pathways between malignant cells and other cells (Figure S7). Results revealed that metabolism-related BPs, including xenobiotic metabolism, fatty acid metabolism, adipogenesis, and glycolysis, were highly expressed in malignant cells, compared with normal cells (Figure S7). We also examined the heterogeneity of cell composition in these samples (Figure 9G and H). The results showed that similar TME cell patterns were observed in multiple tumor regions or samples, but with minor differences. And the T-cell transcriptional profile is stable across different sampling regions.
 |
Figure 9 Correlation of ICDRGs with immune TME in single-cell RNA data of HCC. (A–C) View UMAP plots by sample_id, seurat_clusters, and celltype. (D) View marker gene expression by defined cell type. (E) View marker genes expression in each cluster. (F) DEGs of different cell types. (G) The overall percentage of different cell types. (H) The relative percentage of content of each cell type in each HCC patient. (I) Dot plot plots showing ICDRGs expression in each cell type. (J) Heatmap showing ICD pathway scores for each cell type in 4 HCC samples. (K) View of UMAP plots by cell cycle (phase). (L) The relative proportion of cells at each cell cycle stage. |
Next, we used the UMAP to investigate the expression profile of ICDRGs in HCC samples at the single-cell level (Figure S8). 32 genes were distributed in seven cell types. The ICDRGs showed relatively higher expression proportions and levels in the myeloid cells and endothelial cells (Figure 9I). It suggested that these two cell types might occupy more vital roles in the ICD mechanisms correlated with the 33 ICDRGs. Moreover, expression of genes HMGB1, HSP90AA1, BAX, PDIA3, and CALR were ubiquitous in the TME of HCC patients, indicating their potentially critical regulatory function roles in TME, while IFNB1 was not detected. We then used the AddModuleScore algorithm to calculate the ICD score for each cell. The mean of ICD pathway scores in each patient and each cell type was visualized with a heat map (Figure 9J). ICD scores were lowest in hepatic malignant cells, suggesting that ICDRGs may exhibit a protective role in HCC development. Additionally, the heterogeneity of cell cycle distribution and ICDRGs expression in each phase were also compared (Figure 9K and L). As indicated in Figure 9L, the majority of cells exhibited the largest number and proportion in G1 phases except for B cells and T cells, whose G1 phase was almost identical to the S phase.
Discussion
Cancer therapy-triggered ICD reshapes the tumor immune microenvironment mainly by stimulation of dead tumor cells displaying or releasing molecular patterns associated with injury, resulting in T-cell activation and the suppression of immune responses.10,35 It is anticipated that the induction of ICD will address the present low response rates and numerous negative effects of tumor immunotherapy.36 Presently, with the rapid development of sequencing technologies, high-throughput genomics allows us to carry out an in-depth study of the cellular mechanisms of tumorigenesis and development. To examine the crucial role of ICDRGs in HCC and its potential connection with the survival prognosis and immunophenotypic characteristics, a combined analysis of scRNA-seq and bulk RNA-seq data was carried out in our study. The ICD-related gene signature was associated with OS, TME, gene alternation profiles, potential response to ICIs and chemotherapies, which indicated that ICD may serve as an independent prognostic factor for the overall survival of HCC.
The TME of HCC consists of stromal cells, various immune cells, and tumor cells with different degrees of differentiation and possesses strong heterogeneity. This characteristic determines the limited benefit that HCC patients derive from clinical treatment.27 ScRNA-seq is capable of identifying subpopulations of cells inside the TME and determining the geographic heterogeneity of various tumor cells. It may offer novel insights into the mechanisms underlying the interactions between stromal, immunological, and tumor cells in the advancement and treatment of HCC. Therefore, we investigated for the first time the features of ICDRGs in the HCC tumor microenvironment at the single-cell level. The cell cycle distribution and gene expression heterogeneity, potential biological pathways, and ICD scores of these cell types were subsequently evaluated. Results showed the majority of ICDRGs were ubiquitously expressed in the TME of HCC, especially HMGB1, HSP90AA1, BAX, PDIA3, and CALR, indicating their potentially critical regulatory function roles. It is well known that antigenicity, adjuvant and favorable microenvironment are three factors that need to be fulfilled for the development of ICD.37 HMGB1 and CALR are major determinants of cellular adjuvants, ie, the ability of stressed and dying cells to transmit co-stimulatory signals to immune cells.38 PDIA3, also known as ERp57, can co-translocate to the cell surface with CALR to enhance immunogenicity, and CRT will maintain ER localization in the absence of ERp57.39,40 Similarly, BAX is essential for CRT exposure. Translocation of CRT from the ER lumen to the plasma membrane surface occurs when the ER stress response initiates the “apoptotic module” of the ER exposure pathway, which includes cleavage of Bap31, activation of cysteine asparagine-8 and conformational activation of Bak and Bax.41 Exposure of the heat shock protein HSP90AA1 in response to cell death increases tumor cell immunogenicity, and binding of HSP90-CD91 to immune cells promotes DC maturation and Th1/17 initiation.42 In addition, the two cell types with the highest ICD scores were T cells and myeloid cells, while tumor cells had the lowest ICD scores overall. These findings indicated that ICD might be a protective factor in HCC and that tumor cells might protect themselves by downregulating ICD activity. However, a series of studies investigating the immune cell compositions and functions in TME, and their association with ICD and the efficacy of ICIs, are needed. The induction of ICD based on multiple anti-cancer treatments might be of clinical usage.
Immunotherapy in conjunction with chemotherapy is the first choice of treatment for advanced or surgically incurable HCC. However, identifying individuals who could benefit from immunotherapy remains a difficult task. In the bulk RNA-seq data, most ICD genes were substantially overexpressed in HCC patients and strongly linked to prognosis. HCC patients were separated into the ICD-high and ICD-low subgroups based on unsupervised clustering analysis. Results showed that both subgroups were effective in differentiating the survival and prognosis of HCC patients. ssGSEA analysis indicated the ICD-high subgroup had a significantly higher proportion of immune cell infiltration (eg, DC cells, CD8+ T cells, macrophages, Th1 cells, Th2 cells, and TILs) and higher levels of ICGs expression. In contrast, the ICD-low subgroup displayed low immune scores and inactivated immune function against tumor cells. To elucidate the underlying mechanisms, functional enrichment analysis suggested that DEGs between the two subgroups were especially concentrated in adaptive immune response pathways, regulatory immune processes, and immune-related cytokine signaling. Three gene clusters were defined depending on the DEGs to further categorize HCC patients. Gene cluster B, which displayed an activated TME landscape consistent with the earlier findings, had the best survival benefit over the other two groups. The distinct characterization of ICDRGs described above in HCC samples motivated us to develop an ICD-related prognostic signature. Kaplan-Meier survival analysis showed that risk scores can clearly distinguish HCC patients with good or unfavorable prognoses. ROC curves suggested that risk score was highly accurate in predicting OS clinical outcomes, both in internal and external validation cohorts. Moreover, different risk groups exhibited different clinical features, mutation patterns, immune cell infiltration, immune checkpoint features, and drug treatment responses. The low-risk group with a favorable prognosis had altered immune cell infiltration (eg, B cells, mast cells, neutrophils, and NK cells) and ICGs expression (eg, PD-L1, CTLA-4, TIGIT), and a potentially better response to ICB and chemotherapy.
The signature was composed of 5 ICDRGs with prognostic capability, all correlated with an increased risk of HCC. The Nirenberg and Kim homeobox gene Bapx1 (NKX3-2), a key gene in embryonic development,43 has received little attention in regulating tumor development and progression. NKX3-2 expression has reportedly been remarkably increased in gastric cancer and correlated with TNM staging, metastasis, and poor OS.44 Nevertheless, its potential role in HCC has not been previously reported. Chondrolectin (CHODL) might act as a biomarker for multiple malignancies, including colorectal cancer45 and lung cancer.46 Additionally, overexpression of CHODL facilitated the invasion and migration of HCC cells in vitro experiments.47 Matrix metalloproteinase 1 (MMP1), a commonly recognized chemokine, stimulated the epithelial-mesenchymal transition (EMT), which facilitated cancer cell metastasis48 and was reported to be a negative independent prognostic factor for HCC.49 The nuclear receptor transcription factor DAX-1 (NR0B1) may function as an oncogene in various tumors, such as prostate and breast cancer,50 cervical cancer,51 Ewing sarcoma,52 and lung cancer,53 while its function in HCC has rarely been investigated. Cathepsin V (CTSV) has been reported to be upgraded and linked to worse prognosis in breast ductal adenocarcinoma54 and lung cancer.55 Besides, CTSV facilitated tumor cell invasion and proliferation in bladder cancer56 and colorectal cancer,57 but its involvement in HCC was not apparent. In conclusion, our current study is the first to report the combined effect of these ICDRGs on HCC.
It should be acknowledged that this study has some limitations. Firstly, since the data of all HCC samples utilized in our study were collected from public databases, further observational research is necessary to establish the prognostic value of the ICDRGs. Secondly, although we validated the expression and function of NKX3-2 in vitro experiments, in-depth experiments might provide more convincing results. And the molecular mechanisms promoting the malignant progression of HCC still need to be investigated more thoroughly.
Conclusion
In the present study, the ICD signature of HCC was investigated with its association with T cell and myeloid cell infiltration identified. HCC patients in different ICD clusters presented distinct prognoses. The risk model based on the ICDRGs we built classified HCC patients into high-risk and low-risk subgroups, with significantly different OS, TME, gene alternation profiles, and potential responses to ICIs and chemotherapies. Nomogram prognostic models and the effect of NKX3-2 on the biological behavior of HCC cells in vitro supported the conclusion that ICD might be of clinical usage for the treatment of HCC.
Abbreviations
HCC, Hepatocellular carcinoma; ICD, Immunogenic cell death; ICDRGs, ICD-related genes; TME, tumor microenvironment; DCs, dendritic cells; DAMPs, damage-associated molecular patterns;scRNA-seq, Single-cell RNA sequencing; RNA-seq, RNA sequencing; TCGA, The Cancer Genome Atlas; GEO, Gene Expression Omnibus; ICGC, International Cancer Genome Consortium; CCLE, Cancer Cell Line Encyclopedia;GSVA, Gene set variation analysis; ICB, Immune checkpoint blockade; KEGG, Kyoko Gene and Genome Encyclopedia; GO, Gene Ontology; TMB, Tumor mutational burden; LASSO, Least absolute shrinkage, and selection operator; DEGs, differentially expressed genes; TLR3, Toll-Like Receptor 3; TLR4, Toll-Like Receptor 4; STAT3, Signal Transducer And Activator Of Transcription 3; HSPs, Heat-shock proteins; CD47, Antigenic Surface Determinant Protein OA3; HMGB1, High Mobility Group Box 1; HSP90AA1, Heat Shock Protein 90 Alpha Family Class A Member 1; BAX, Bcl-2-associated X protein; PDIA3/ERp57, Protein Disulfide Isomerase A3; CALR, Calreticulin.
Data Sharing Statement
Full data will be available from the corresponding author upon reasonable request.
Acknowledgments
We would like to acknowledge the TCGA, GEO, and CCLE for providing relevant data.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Ethics Approval and Informed Consent
This study conformed to the Declaration of Helsinki. The tissue samples were collected with the approval of the ethics committee of the Third Affiliated Hospital of Sun Yat-sen University (Guangzhou, China; no. RG2023-106-02).
Funding
This study was supported by the Guangdong Basic and Applied Basic Research Foundation (No. 2023A1515012544 and 2022A1515012659), Tip-top Scientific and Technical Innovative Youth Talents of the Third Affiliated Hospital of Sun Yat-sen University, and Guangzhou Science and Technology Plan Project (2024A03J0097).
Disclosure
The authors declare that they have no conflicts of interest in this work.
References
1. Vogel A, Meyer T, Sapisochin G, Salem R, Saborowski A. Hepatocellular carcinoma. Lancet. 2022;400:1345–1362. doi:10.1016/s0140-6736(22)01200-4
2. Jiang Y, Sun A, Zhao Y, et al. Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature. 2019;567:257–261. doi:10.1038/s41586-019-0987-8
3. Llovet JM, Montal R, Sia D, Finn RS. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat Rev Clin Oncol. 2018;15:599–616. doi:10.1038/s41571-018-0073-4
4. Su GL, Altayar O, O’Shea R, et al. AGA Clinical practice guideline on systemic therapy for hepatocellular carcinoma. Gastroenterology. 2022;162:920–934. doi:10.1053/j.gastro.2021.12.276
5. Peters NA, Javed AA, He J, Wolfgang CL, Weiss MJ. Association of socioeconomics, surgical therapy, and survival of early stage hepatocellular carcinoma. J Surg Res. 2017;210:253–260. doi:10.1016/j.jss.2016.11.042
6. Zhang G, Li R, Deng Y, Zhao L. Conditional survival of patients with hepatocellular carcinoma: results from the surveillance, epidemiology, and end results registry. Expert Rev Gastroenterol Hepatol. 2018;12:515–523. doi:10.1080/17474124.2018.1453806
7. Sangro B, Sarobe P, Hervás-Stubbs S, Melero I. Advances in immunotherapy for hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2021;18:525–543. doi:10.1038/s41575-021-00438-0
8. Oura K, Morishita A, Tani J, Masaki T. Tumor immune microenvironment and immunosuppressive therapy in hepatocellular carcinoma: a review. Int J Mol Sci. 2021;22. doi:10.3390/ijms22115801
9. Zhou Z, Hu Y, Wu Y, et al. The immunosuppressive tumor microenvironment in hepatocellular carcinoma-current situation and outlook. Mol Immunol. 2022;151:218–230. doi:10.1016/j.molimm.2022.09.010
10. Galluzzi L, Vitale I, Warren S, et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J Immunother Cancer. 2020;8. doi:10.1136/jitc-2019-000337
11. Tesniere A, Schlemmer F, Boige V, et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene. 2010;29:482–491. doi:10.1038/onc.2009.356
12. Zhu M, Yang M, Zhang J, et al. Immunogenic cell death induction by ionizing radiation. Front Immunol. 2021;12:705361. doi:10.3389/fimmu.2021.705361
13. Ventura A, Vassall A, Robinson E, et al. Extracorporeal photochemotherapy drives monocyte-to-dendritic cell maturation to induce anticancer immunity. Cancer Res. 2018;78:4045–4058. doi:10.1158/0008-5472.Can-18-0171
14. Turubanova VD, Balalaeva IV, Mishchenko TA, et al. Immunogenic cell death induced by a new photodynamic therapy based on photosens and photodithazine. J Immunother Cancer. 2019;7:350. doi:10.1186/s40425-019-0826-3
15. Shekarian T, Sivado E, Jallas AC, et al. Repurposing rotavirus vaccines for intratumoral immunotherapy can overcome resistance to immune checkpoint blockade. Sci Transl Med. 2019:11. doi:10.1126/scitranslmed.aat5025
16. Tong J, Tan X, Song X, et al. CDK4/6 inhibition suppresses p73 phosphorylation and activates DR5 to potentiate chemotherapy and immune checkpoint blockade. Cancer Res. 2022;82:1340–1352. doi:10.1158/0008-5472.Can-21-3062
17. Wang Q, Ju X, Wang J, Fan Y, Ren M, Zhang H. Immunogenic cell death in anticancer chemotherapy and its impact on clinical studies. Cancer Lett. 2018;438:17–23. doi:10.1016/j.canlet.2018.08.028
18. Krysko DV, Garg AD, Kaczmarek A, Krysko O, Agostinis P, Vandenabeele P. Immunogenic cell death and DAMPs in cancer therapy. Nat Rev Cancer. 2012;12:860–875. doi:10.1038/nrc3380
19. Maharjan R, Choi JU, Kweon S, et al. A novel oral metronomic chemotherapy provokes tumor specific immunity resulting in colon cancer eradication in combination with anti-PD-1 therapy. Biomaterials. 2022;281:121334. doi:10.1016/j.biomaterials.2021.121334
20. Kepp O, Zitvogel L, Kroemer G. Lurbinectedin: an FDA-approved inducer of immunogenic cell death for the treatment of small-cell lung cancer. Oncoimmunology. 2020;9:1795995. doi:10.1080/2162402x.2020.1795995
21. Montes de Oca R, Alavi AS, Vitali N, et al. Belantamab Mafodotin (GSK2857916) drives immunogenic cell death and immune-mediated antitumor responses in vivo. Mol Cancer Ther. 2021;20:1941–1955. doi:10.1158/1535-7163.Mct-21-0035
22. Kostopoulos IV, Kakalis A, Birmpilis A, et al. Belantamab mafodotin induces immunogenic cell death within 24 h post-administration in newly diagnosed multiple myeloma patients. Am J Hematol. 2022. doi:10.1002/ajh.26823
23. Zhu H, Shan Y, Ge K, Lu J, Kong W, Jia C. Oxaliplatin induces immunogenic cell death in hepatocellular carcinoma cells and synergizes with immune checkpoint blockade therapy. Cell Oncol. 2020;43:1203–1214. doi:10.1007/s13402-020-00552-2
24. Zhou C, Yang ZF, Sun BY, et al. Lenvatinib induces immunogenic cell death and triggers toll-like receptor-3/4 ligands in hepatocellular carcinoma. J Hepatocell Carcinoma. 2023;10:697–712. doi:10.2147/jhc.S401639
25. Li Y, Song Z, Han Q, et al. Targeted inhibition of STAT3 induces immunogenic cell death of hepatocellular carcinoma cells via glycolysis. Mol Oncol. 2022;16:2861–2880. doi:10.1002/1878-0261.13263
26. Zhao Y, Li MC, Konaté MM, et al. TPM, FPKM, or normalized counts? A comparative study of quantification measures for the analysis of RNA-seq Data from the NCI patient-derived models repository. J Transl Med. 2021;19:269. doi:10.1186/s12967-021-02936-w
27. Ma L, Heinrich S, Wang L, et al. Multiregional single-cell dissection of tumor and immune cells reveals stable lock-and-key features in liver cancer. Nat Commun. 2022;13:7533. doi:10.1038/s41467-022-35291-5
28. Mangiola S, Doyle MA, Papenfuss AT. Interfacing Seurat with the R tidy universe. Bioinformatics. 2021;37:4100–4107. doi:10.1093/bioinformatics/btab404
29. Zhang Q, He Y, Luo N, et al. Landscape and dynamics of single immune cells in hepatocellular carcinoma. Cell. 2019;179:829–845.e820. doi:10.1016/j.cell.2019.10.003
30. Sun Y, Wu L, Zhong Y, et al. Single-cell landscape of the ecosystem in early-relapse hepatocellular carcinoma. Cell. 2021;184:404–421.e416. doi:10.1016/j.cell.2020.11.041
31. Gustavsson EK, Zhang D, Reynolds RH, Garcia-Ruiz S, Ryten M. ggtranscript: an R package for the visualization and interpretation of transcript isoforms using ggplot2. Bioinformatics. 2022;38:3844–3846. doi:10.1093/bioinformatics/btac409
32. Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010;26:1572–1573. doi:10.1093/bioinformatics/btq170
33. Scire J, Huisman JS, Grosu A, et al. estimateR: an R package to estimate and monitor the effective reproductive number. BMC Bioinf. 2023;24:310. doi:10.1186/s12859-023-05428-4
34. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16:284–287. doi:10.1089/omi.2011.0118
35. Rodrigues MC, Morais JAV, Ganassin R, et al. An overview on immunogenic cell death in cancer biology and therapy. Pharmaceutics. 2022;14. doi:10.3390/pharmaceutics14081564
36. Li Z, Lai X, Fu S, et al. Immunogenic cell death activates the tumor immune microenvironment to boost the immunotherapy efficiency. Adv Sci. 2022;9:e2201734. doi:10.1002/advs.202201734
37. Kepp O, Senovilla L, Vitale I, et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology. 2014:
38. Fucikova J, Spisek R, Kroemer G, Galluzzi L. Calreticulin and cancer. Cell Res. 2021;31:5–16. doi:10.1038/s41422-020-0383-9
39. Pol JG, Plantureux C, Pérez-Lanzón M, Kroemer G. PDIA3 as a potential bridge between immunogenic cell death and autoreactivity. Oncoimmunology. 2022;11:2130558. doi:10.1080/2162402x.2022.2130558
40. Panaretakis T, Joza N, Modjtahedi N, et al. The co-translocation of ERp57 and calreticulin determines the immunogenicity of cell death. Cell Death Differ. 2008;15:1499–1509. doi:10.1038/cdd.2008.67
41. Zitvogel L, Kepp O, Senovilla L, Menger L, Chaput N, Kroemer G. Immunogenic tumor cell death for optimal anticancer therapy: the calreticulin exposure pathway. Clin Cancer Res. 2010;16:3100–3104. doi:10.1158/1078-0432.Ccr-09-2891
42. Garg AD, Romano E, Rufo N, Agostinis P. Immunogenic versus tolerogenic phagocytosis during anticancer therapy: mechanisms and clinical translation. Cell Death Differ. 2016;23:938–951. doi:10.1038/cdd.2016.5
43. Verzi MP, Stanfel MN, Moses KA, et al. Role of the homeodomain transcription factor Bapx1 in mouse distal stomach development. Gastroenterology. 2009;136:1701–1710. doi:10.1053/j.gastro.2009.01.009
44. Ouyang S, Zhu G, Ouyang L, et al. Bapx1 mediates transforming growth factor-β- induced epithelial-mesenchymal transition and promotes a malignancy phenotype of gastric cancer cells. Biochem Biophys Res Commun. 2017;486:285–292. doi:10.1016/j.bbrc.2017.03.029
45. Zhang X, Wu K, Huang Y, Xu L, Li X, Zhang N. Promoter hypermethylation of CHODL contributes to carcinogenesis and indicates poor survival in patients with early-stage colorectal cancer. J Cancer. 2020;11:2874–2886. doi:10.7150/jca.38815
46. Masuda K, Takano A, Oshita H, et al. Chondrolectin is a novel diagnostic biomarker and a therapeutic target for lung cancer. Clin Cancer Res. 2011;17:7712–7722. doi:10.1158/1078-0432.Ccr-11-0619
47. Huang Z, Zhang N, Li W, Cao J, Zhang L, Chen Y. Expression of CHODL in hepatocellular carcinoma affects invasion and migration of liver cancer cells. Oncol Lett. 2017;13:715–721. doi:10.3892/ol.2016.5466
48. Bassiouni W, Ali MAM, Schulz R. Multifunctional intracellular matrix metalloproteinases: implications in disease. Febs j. 2021;288:7162–7182. doi:10.1111/febs.15701
49. Xu L, Yang H, Yan M, Li W. Matrix metalloproteinase 1 is a poor prognostic biomarker for patients with hepatocellular carcinoma. Clin Exp Med. 2022. doi:10.1007/s10238-022-00897-y
50. Roshan-Moniri M, Hsing M, Butler MS, Cherkasov A, Rennie PS. Orphan nuclear receptors as drug targets for the treatment of prostate and breast cancers. Cancer Treat Rev. 2014;40:1137–1152. doi:10.1016/j.ctrv.2014.10.005
51. Liu XF, Li XY, Zheng PS, Yang WT. DAX1 promotes cervical cancer cell growth and tumorigenicity through activation of Wnt/β-catenin pathway via GSK3β. Cell Death Dis. 2018;9:339. doi:10.1038/s41419-018-0359-6
52. Kinsey M, Smith R, Lessnick SL. NR0B1 is required for the oncogenic phenotype mediated by EWS/FLI in Ewing’s sarcoma. Mol Cancer Res. 2006;4:851–859. doi:10.1158/1541-7786.Mcr-06-0090
53. Susaki Y, Inoue M, Minami M, et al. Inhibitory effect of PPARγ on NR0B1 in tumorigenesis of lung adenocarcinoma. Int J Oncol. 2012;41:1278–1284. doi:10.3892/ijo.2012.1571
54. Toss M, Miligy I, Gorringe K, et al. Prognostic significance of cathepsin V (CTSV/CTSL2) in breast ductal carcinoma in situ. J Clin Pathol. 2020;73:76–82. doi:10.1136/jclinpath-2019-205939
55. Yang L, Zeng Q, Deng Y, Qiu Y, Yao W, Liao Y. Glycosylated cathepsin V serves as a prognostic marker in lung cancer. Front Oncol. 2022;12:876245. doi:10.3389/fonc.2022.876245
56. Xia Y, Ge M, Xia L, Shan G, Qian H. CTSV (cathepsin V) promotes bladder cancer progression by increasing NF-κB activity. Bioengineered. 2022;13:10180–10190. doi:10.1080/21655979.2022.2061278
57. Wang CH, Wang LK, Wu CC, et al. Cathepsin V mediates the tazarotene-induced gene 1-induced reduction in invasion in colorectal cancer cells. Cell Biochem Biophys. 2020;78:483–494. doi:10.1007/s12013-020-00940-3
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Trial Designs for Integrating Novel Therapeutics into the Management of Intermediate-Stage Hepatocellular Carcinoma
Su YY, Liu YS, Hsiao CF, Hsu C, Chen LT
Journal of Hepatocellular Carcinoma 2022, 9:517-536
Published Date: 2 June 2022
Macrotrabecular-Massive Hepatocellular Carcinoma: Light and Shadow in Current Knowledge
Sessa A, Mulé S, Brustia R, Regnault H, Galletto Pregliasco A, Rhaiem R, Leroy V, Sommacale D, Luciani A, Calderaro J, Amaddeo G
Journal of Hepatocellular Carcinoma 2022, 9:661-670
Published Date: 27 July 2022
Lenvatinib Induces Immunogenic Cell Death and Triggers Toll-Like Receptor-3/4 Ligands in Hepatocellular Carcinoma

Zhou C, Yang ZF, Sun BY, Yi Y, Wang Z, Zhou J, Fan J, Gan W, Ren N, Qiu SJ
Journal of Hepatocellular Carcinoma 2023, 10:697-712
Published Date: 24 April 2023
Development and Validation of a Novel Nomogram Integrated with Hypoxic and Lactate Metabolic Characteristics for Prognosis Prediction in Hepatocellular Carcinoma
Qiu X, Dong L, Wang K, Zhong X, Xu H, Xu S, Guo H, Wei X, Chen W, Xu X
Journal of Hepatocellular Carcinoma 2024, 11:241-255
Published Date: 2 February 2024
Immunotherapy Efficacy in Advanced Hepatocellular Carcinoma in a Diverse and Underserved Population in the United States
Bteich F, Desai K, Zhang C, Kaur A, Levy RA, Bioh L, Wang A, Sultana S, Kaubisch A, Kinkhabwala M, Bellemare S, Fidvi S, Kanmaniraja D, Berkenblit R, Moon JY, Adedimeji A, Tow CY, Saenger Y
Journal of Hepatocellular Carcinoma 2024, 11:257-269
Published Date: 3 February 2024