Back to Journals » OncoTargets and Therapy » Volume 18
Immunotherapy Resistance and Therapeutic Strategies in PD-L1 High Expression Non-Small Cell Lung Cancer
Authors Liu J, Cai Y, Liu J ![]() , Chen D, Wu X
, Chen D, Wu X
Received 13 May 2025
Accepted for publication 8 August 2025
Published 29 August 2025 Volume 2025:18 Pages 953—966
DOI https://doi.org/10.2147/OTT.S539978
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjay Singh
Jianhua Liu,1,* Yin Cai,2,* Jiang Liu,2 Dadong Chen,2 Xiang Wu2
1Department of Otorhinolaryngology, Xinghua People’s Hospital Affiliated to Yangzhou University, Xinghua, Jiangsu, People’s Republic of China; 2Department of Oncology, Xinghua People’s Hospital Affiliated to Yangzhou University, Xinghua, Jiangsu, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jiang Liu, Department of Oncology, Xinghua People’s Hospital Affiliated to Yangzhou University, 419 Ying Wu Nan Road, Xinghua, Jiangsu, 225700, People’s Republic of China, Email [email protected] Xiang Wu, Department of Oncology, Xinghua People’s Hospital Affiliated to Yangzhou University, 419 Ying Wu Nan Road, Xinghua, Jiangsu, 225700, People’s Republic of China, Email [email protected]
Abstract: Non-small cell lung cancer (NSCLC) is the most common subtype of lung cancer, and high programmed death-ligand 1 (PD-L1) expression (≥ 50%) is a key biomarker for predicting clinical benefit from immune checkpoint inhibitors (ICIs). This therapy has substantially improved long-term survival rates, with a five-year survival rate exceeding 25%. Nevertheless, primary or acquired resistance occurs in 30– 40% of PD-L1-high patients. This resistance arises from multifactorial mechanisms involving tumor-intrinsic adaptations, immune microenvironment reprogramming, and extrinsic immunosuppressive signals. In this review, we systematically dissect the biological and clinical drivers of ICIs resistance in PD-L1-high NSCLC and explore emerging strategies to overcome these barriers, including novel combinatorial approaches and biomarker-guided therapies.
Keywords: PD-L1, non-small cell lung cancer, immunotherapy, immunotherapy resistance, biomarkers
Introduction
The rising incidence of NSCLC constitutes a significant challenge within the field of oncology, as it is the leading cause of cancer-related deaths worldwide. Immunotherapy has emerged as a crucial strategy in the management of this malignancy. The PD-L1 molecule, an essential immune checkpoint, plays a significant role in the tumor microenvironment by mediating immune evasion. Elevated PD-L1 expression is frequently associated with enhanced responses to ICIs, making it a key biomarker for patient stratification and treatment planning.1 Despite the favorable outcomes observed in a subset of patients treated with programmed death-1 (PD-1) /PD-L1 inhibitors, a considerable proportion continues to experience resistance to these therapies. The mechanisms underlying resistance in PD-L1 high NSCLC may differ fundamentally from those in PD-L1 low or negative tumors. While all NSCLC patients may develop resistance through tumor-intrinsic factors or microenvironmental influences, PD-L1 high tumors present unique challenges. First, their strong PD-L1 expression suggests active immune evasion pathways that may be more dependent on alternative checkpoints (eg, LAG-3, TIM-3) when PD-1/PD-L1 blockade is applied. Second, the inflammatory microenvironment in these tumors may foster more rapid adaptive resistance through T-cell exhaustion or the recruitment of regulatory T cells. Third, spatial heterogeneity of PD-L1 expression is particularly problematic in high expressers, where biopsy sampling errors could lead to inappropriate treatment decisions.
Resistance can be categorized as either primary or acquired, underscoring the complexity of immune evasion mechanisms.2 Primary resistance denotes a lack of response upon initial treatment, while acquired resistance manifests an initial positive response, ultimately leading to disease progression (Figure 1). Mechanisms of resistance to immune checkpoint blockade are multifaceted and can arise from tumor-intrinsic factors and elements from the tumor microenvironment. Tumor cells may develop genetic alterations that promote immune evasion, such as mutations in key oncogenic pathways or upregulation of alternative immune checkpoints. Research has indicated that the epidermal growth factor receptor (EGFR) mutations may influence PD-L1 expression and contribute to resistance against PD-1/PD-L1 inhibitors.3–6 Additionally, the tumor microenvironment, often characterized by immunosuppressive cells and cytokines, can exacerbate resistance mechanisms. Tumor-associated macrophages (TAMs) and regulatory T cells (Tregs) may express PD-L1, establishing a feedback loop that diminishes T-cell activation and fosters tumor survival.7 However, these findings have not been systematically integrated for PD-L1-high subgroups. Despite PD-L1’s predictive value, its utility is limited by spatial and temporal heterogeneity. The heterogeneity of PD-L1 expression within tumors presents further complications in accurately predicting treatment responses. This variability can occur not only among different patients but within the same tumor, resulting in inconsistencies in therapeutic effectiveness.8 Recent research suggests that combining PD-L1 expression profiling with other biomarkers, such as tumor mutational burden (TMB) and microsatellite instability (MSI), may improve predictive accuracy.9 Current literatures10,11 lack a unified discussion of why PD-L1-high NSCLC fails ICIs despite biomarker positivity. Key gaps are as follows: (a) The impact of co-occurring genomic alterations on PD-L1 regulation. (b) Dynamic changes in PD-L1 and alternative checkpoints (eg, LAG-3) post-ICI exposure. A comprehensive understanding of mechanisms underlying these resistance patterns is essential for augmenting therapeutic efficacy and improving patient outcomes.12
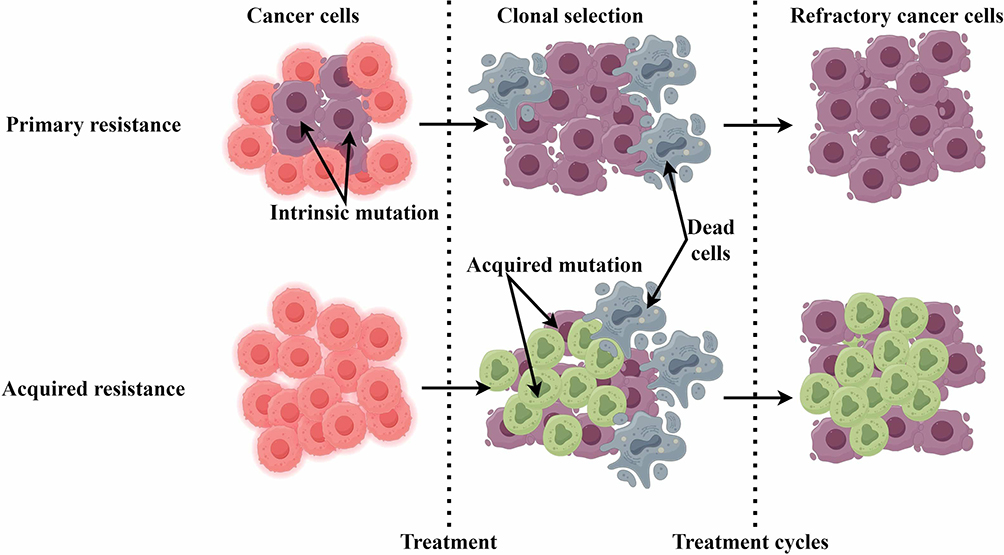 |
Figure 1 A general diagram illustrating how cancer resistance develops following immunotherapy treatment. Intrinsic mutations in cancer cells primarily render them resistant to immunotherapy, resulting in refractory cancer cells. Acquired resistance emerges in surviving cancer cells after immunotherapy, leading to further refractory cancer cells. |
Investigating the signaling pathways and their regulation within the tumor microenvironment is essential for developing novel therapeutic strategies to overcome resistance. Targeting pathways involved in PD-L1 regulation, such as the PI3K/AKT/mTOR signaling pathway, represents a promising approach to enhancing the efficacy of immunotherapies.13 Additionally, the innovative combination therapies that integrate ICIs with other treatment modalities is underway, to improve clinical responses and mitigate resistance.14 A deepen understanding of the mechanisms that contribute to primary and acquired resistance is critical for optimizing immunotherapy strategies and enhancing patient outcomes. Future research should focus on identifying reliable predictive biomarkers and developing combination therapies that can effectively address the complex nature of immunotherapy resistance.15 This review will focus specifically on resistance mechanisms in PD-L1 high NSCLC, analyzing both tumor-intrinsic factors and microenvironmental influences that contribute to immunotherapy failure. We will highlight emerging predictive biomarkers beyond PD-L1 and discuss novel therapeutic approaches targeting the unique biology of these tumors. By focusing on this subgroup, we aim to provide insights that can directly inform treatment strategies for patients most likely to benefit from immunotherapy.
Tumor-Intrinsic Resistance to Immunotherapy
Defects in Antigen Presentation
Defects in antigen presentation represent critical factors facilitating immune evasion by tumor cells. Antigen presentation is a fundamental biological process in which major histocompatibility complex (MHC) molecules display peptide fragments derived from intracellular proteins to T cells, initiating an immune response. However, in various cancers, including NSCLC, this process is significantly impaired, which diminishes the capacity of the immune system to recognize and attack tumor cells. One primary mechanism through which tumors evade immune detection is the down-regulation of MHC class I molecules on their surface. This down-regulation is not merely a passive occurrence; it can be actively modulated by various signaling pathways. For instance, the phosphoinositide 3-kinase (PI3K) pathway has been demonstrated to inhibit the expression of MHC class I molecules, facilitating immune escape.16 Additionally, the tumor microenvironment (TME), characterized by hypoxia and immunosuppressive cytokines, can exacerbate the down-regulation of MHC class I expression.17 Research has demonstrated that various tumor cells can lose MHC class I expression due to genetic alterations or epigenetic modifications. This loss of expression hinders the presentation of tumor antigens to CD8+ cytotoxic T lymphocytes.18 The down-regulation of MHC class I is frequently accompanied by deficiencies in the antigen processing machinery (APM), which encompasses proteins essential for generating and transporting peptide fragments to MHC molecules. Such impairments prevent the effective presentation of tumor-specific antigens, enabling tumors to evade immune surveillance, as T cells are rendered unable to recognize and respond to the tumor cells.19 The TME also plays a significant role in contributing to these defects. Factors secreted by tumor cells, such as immunosuppressive cytokines, can disrupt the functionality of antigen-presenting cells (APCs), further compromising their ability to present antigens effectively.18
Another significant aspect of antigen presentation defects in tumors is the phenomenon known as “antigenic modulation.” Tumor cells may alter the expression of target antigens in response to immune pressure, a mechanism observed across various cancer types. This modulation may result in the loss of specific antigens recognized by T cells, thereby facilitating the evasion of immune detection.20 The implications of these defects in antigen presentation are substantial, contributing to immune evasion, particularly ICIs that depend on the presence of functional T cells to exert their effects. Tumors that exhibit low levels of MHC class I expression often demonstrate poor responses to ICIs, as the absence of effective antigen presentation restricts the activation of T cells capable of targeting the tumor.21 Therapeutic strategies aimed at restoring the expression of MHC class I, including small molecules to inhibit the PI3K pathway or enhance transporter associated with antigen processing (TAP) function, have demonstrated promise in preclinical models.22 Combination therapies that integrate ICIs could counteract the impact of MHC class I down-regulation by revitalizing exhausted T cells, thus enhancing anti-tumor immunity.23
Spatiotemporal Heterogeneity of PD-L1 Expression
PD-L1 expression is a critical determinant influencing the efficacy of ICIs. However, PD-L1 expression possesses significant spatiotemporal heterogeneity, making it challenging a predictive biomarker for immunotherapy responses. This heterogeneity can be observed within a single tumor, between different metastatic sites within the same patient, and even over time as the tumor evolves in response to treatment.23 The heterogeneity in PD-L1 expression is affected by several factors, including the TME, genetic alterations, and the patient’s treatment history. A study has demonstrated that PD-L1 expression can vary markedly between primary and metastatic tumors, with some metastatic sites displaying higher PD-L1 levels than the primary tumor.8 The spatial heterogeneity of PD-L1 expression introduces additional complexity. Tumors can upregulate PD-L1 as a response to immune pressure from therapies such as chemotherapy or radiation, and immune cells in the TME.24 The regulation of PD-L1 expression is significantly influenced by epigenetic factors. DNA methylation, mediated by enzymes such as EZH2, plays a key role in modulating PD-L1 levels. A report has shown that the upregulation of EZH2 in lung cancer tissues correlates with increased PD-L1 expression, leading to an immunosuppressive TME.25 This epigenetic modification can occur in response to various stimuli, including hypoxia and inflammatory cytokines, complicating the therapeutic landscape. In addition, the PI3K/AKT signaling pathway plays a significant role in modifying PD-L1 expression. Persistent activation of this pathway has been associated with increased PD-L1 levels, facilitating immune evasion and resistance to ICI therapies.
To mitigate the challenges presented by spatiotemporal heterogeneity of PD-L1 expression, there is an increasing interest in developing non-invasive imaging techniques capable of dynamically assessing PD-L1 expression in real-time. The implementation of radiolabeled antibodies targeting PD-L1 for positron emission tomography (PET) imaging has shown promise in providing a comprehensive overview of PD-L1 expression across multiple tumor sites.26 The spatiotemporal heterogeneity of PD-L1 expression poses significant challenges in cancer immunotherapy. A thorough understanding of the factors driving spatiotemporal heterogeneity of PD-L1 expression, coupled with the advancement of innovative imaging techniques, will be essential in optimizing the application of immune checkpoint inhibitors and enhancing patient outcomes in cancer treatment.
Upregulation of Immunosuppressive Molecules
The upregulation of immunosuppressive molecules in the TME is a key mechanism through which tumors evade the immune system surveillance. This phenomenon is evident in NSCLC, where the upregulation of PD-L1 expression on both tumor cells and immune cells significantly contributes to the suppression of T-cell responses. Immunosuppressive factors, including various cytokines and immune checkpoint molecules, foster an environment that promotes tumor growth while inhibiting effective anti-tumor immunity. The level of PD-L1 expression is significantly higher in NSCLC tumors compared to adjacent normal tissues, and this increased expression is associated with poor clinical outcomes.24 The upregulation of PD-L1 and other immunosuppressive molecules, such as transforming growth factor-beta (TGF-β), has been associated with the recruitment and activation of regulatory T cells (Tregs), which further undermine the anti-tumor immune response.27 Tumor cells upregulating various immune checkpoints, including cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), lymphocyte-activation gene 3 (LAG-3), and T-cell immunoglobulin and mucin-domain containing-3 (TIM-3), is increasingly recognized as a critical mechanism by which tumors evade immune surveillance. The upregulation of these checkpoints can establish a complex network of immune inhibitory signals that not only attenuate T-cell activation but also contribute to establishing an immunosuppressive environment. Specifically, the interplay between PD-1/PD-L1 and other checkpoints such as CTLA-4 and LAG-3 may result in T-cell exhaustion, ultimately culminating in impaired anti-tumor immunity.8
Tumor Heterogeneity
Tumor heterogeneity denotes the presence of various cell populations within a single tumor, which can differ in genetic, phenotypic, and functional characteristics. This heterogeneity is a significant factor contributing to treatment resistance and disease progression. Single-cell RNA sequencing (scRNA-seq) has emerged as an effective tool for examining this heterogeneity, facilitating the analysis of gene expression profiles at the cell level. Distinct subpopulations of cancer cells may demonstrate varying responses to therapeutic interventions, which lead to the survival of resistant clones capable of repopulating the tumor after treatment.28 Additionally, spatial transcriptomics can mirror these heterogeneous populations within the tumor, elucidating how TME influences tumor cell behavior and therapeutic responses.29 Investigating the mechanisms driving this heterogeneity is essential for developing more effective therapies and for enhancing patient prognoses.
Immunosuppressive Tumor Microenvironment
TME is a complex and dynamic ecosystem that plays a critical role in tumor progression and immune evasion. It consists of diverse cell types, including tumor cells, immune cells, fibroblasts, and endothelial cells, which interact to form a supportive niche for tumor growth. A key feature of the TME is its immune suppressive nature, which hinders effective anti-tumor immune responses.30–33 Immunosuppressive cells, such as regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), and tumor-associated macrophages (TAMs), are frequently abundant in the TME. These cells can secrete immunosuppressive cytokines, including TGF-β and interleukin-10 (IL-10), which inhibit the functions of effector T cells and foster an environment conducive to tumor growth.34,35 TAMs and MDSCs often exhibit high levels of PD-L1 expression, which not only diminishes T cell activation but also promotes the survival of tumor cells.8,36 Consequently, these immune suppressive cells can induce a state of immune tolerance, where the immune system fails to recognize and effectively attack tumor cells.
Immunosuppressive Cell Infiltration
Immunosuppressive cell infiltration represents a defining characteristic of the TME and significantly impacts the efficacy of cancer therapies, particularly immunotherapies. Tregs can directly suppress effector T cell functions by secreting immunosuppressive cytokines such as TGF-β and IL-10, as well as by engaging inhibitory receptors on T cells. TGF-β is a multifunctional cytokine that inhibits both the proliferation and activation of effector T cells while promoting the differentiation of naïve T cells into Tregs, thus perpetuating a cycle of immunosuppression within the tumor microenvironment.37 In contrast, IL-10 is essential for preserving an anti-inflammatory environment that facilitates tumor growth by inhibiting the activity of pro-inflammatory immune cells.8,38 Their presence within the TME is frequently correlated with poor prognosis and reduced efficacy of immune checkpoint inhibitors.8 MDSCs are another critical component of the immune-suppressive milieu within tumors. They exert their effects by producing elevated levels of arginase, nitric oxide, and reactive oxygen species, which can induce T-cell dysfunction and apoptosis.39 MDSCs are often associated with unfavorable prognostic outcomes in various cancers, as they contribute to the establishment of an immunosuppressive niche that enables tumors to evade immune surveillance.40,41 The accumulation of MDSCs in the TME has also been linked to resistance to immunotherapy, making them prime targets for novel therapeutic strategies.42,43 TAMs also significantly influence the immune landscape of tumors, exhibiting both pro-inflammatory and anti-inflammatory phenotypes.44,45 The polarization of TAMs towards an M2-like phenotype is typically associated with immune suppression and tumor progression.46 The dynamic interplay among these immune cells creates a feedback loop that not only inhibits effector T cell function but also fosters tumor progression, rendering them attractive targets for therapeutic intervention in cancer treatment. A recent study47 revealed that in PD-L1-high lung cancer, cell-intrinsic PD-L1 signaling drives immunosuppression by myeloid-derived suppressor cells through the IL-6/Jak/Stat3 pathway. Overall, the infiltration of immune suppressive cells into the TME is a critical factor contributing to the immune evasion of tumor cells.
Metabolic Microenvironment Dysregulation
The metabolic microenvironment of tumors is frequently dysregulated, significantly affecting tumor cell behavior and the immune response. Tumor cells often undergo abnormal metabolic pathways that allow them to thrive in hypoxic and nutrient-deprived environments. This metabolic reprogramming is defined by an increase in glycolysis, even in the presence of oxygen—a phenomenon referred to as the Warburg effect.48,49 This shift in metabolism not only facilitates rapid cell proliferation but also results in the accumulation of metabolic by-products, such as lactate, which can further inhibit immune cell function.50 Lactate generated in the TME may impede T cell activation and promote the differentiation of Tregs, contributing to immune evasion.51 The dysregulated metabolism within the TME adversely affects the functionality of Tregs, MDSCs, and TAMs. The interactions between tumor cells and immune cells are mediated by various metabolites that can influence immune cell differentiation and function. Elevated levels of TGF-β and IL-10 in the TME may induce M2 polarization in macrophages, which promotes tumor growth and suppresses effective anti-tumor immunity.52 The metabolic requirements of tumor cells can lead to competition for nutrients, exacerbating the immunosuppressive environment. Overall, the dysregulation of metabolic pathways within the TME not only supports the survival of tumor cells but modulates the functions of immune cells, perpetuating a cycle of immunosuppression. Targeting these pathways and the interactions among immune cells may offer promising avenues for therapeutic intervention in cancer treatment.53
Formation of Physical Barriers
The physical barriers within the TME influence T cell infiltration and activity. The TME consists of a dense extracellular matrix (ECM) and abnormal vasculature, which can obstruct the effective migration of T cells to tumor sites. These physical barriers not only hinder T-cell access but contribute to an overall immunosuppressive environment by promoting fibrotic reactions that further isolate tumor cells from the immune system. Research54 has indicated that the remodeling of the ECM, through the activity of cancer-associated fibroblasts (CAFs), can significantly impact T cell behavior. CAFs secrete various cytokines and growth factors that alter the composition of the ECM, making it stiffer and less permeable to immune cells. The mechanical properties of the ECM can directly affect T cell behavior.55 This indicates that the physical characteristics of the ECM are not just passive barriers; they actively modulate T-cell responses. The spatial organization of CAFs and ECM components can create niches that exclude T cells, a phenomenon referred to as immune exclusion.56,57 In such cases, T cells may be present within the TME but remain unable to infiltrate the tumor mass. This remodeling creates a physical barrier that prevents T cell access to tumor cells, facilitating tumor immune evasion.58,59
The Dysfunction of Host Immune System
The dysfunction of the host immune system presents a significant challenge to effective cancer immunotherapy, often manifesting as T-cell exhaustion. This phenomenon occurs when T-cells lose their effectiveness in responding to antigens due to prolonged exposure to their targets, resulting in a state where they fail to proliferate or produce cytokines efficiently.60 T-cell exhaustion mainly refers to the upregulation of inhibitory receptors, including PD-1, CTLA-4, and TIM-3, which dampen T-cell activity and promote immune tolerance.61 Within the TME, the persistent presence of tumor antigens can lead to chronic stimulation of T cells, ultimately causing their functional decline and exhaustion. The immunosuppressive cytokines may aggravate T cell dysfunction.62 The metabolic status of T-cells can significantly impact their functionality. In nutrient-deprived environments, T-cells frequently exhibit impaired proliferation and compromised effector functions, which is exacerbated by the higher metabolic demands of tumor cells.63 Patients exhibiting elevated levels of exhausted T cells tend to have poorer prognoses and diminished responses to immunotherapies.64,65 Additionally, studies have demonstrated that PD-L1 overexpression in breast cancer induces exhaustion of persistently activated CD4+ T cells, thereby compromising immunotherapy efficacy.66 T cell exhaustion represents a critical barrier to effective cancer immunotherapy, underscoring the need for a multifaceted approach to restore T cell functionality and enhance anti-tumor immunity. Therefore, it is reasonable to speculate that in non-small cell lung cancer with high PD-L1 expression, the exhaustion of T cells may be one of the reasons for its resistance.
Genomic and Epigenetic Regulatory Abnormalities
Genomic Instability
Genomic instability significantly impacts tumor progression and therapeutic response, particularly in elderly patients aged 75 and above. The interaction between age-related immunosenescence and genomic instability is essential for evaluating treatment outcomes in this population.67 The aging process is associated with a decline in immune function, diminished T-cell responses, and increased chronic inflammation. These changes can exacerbate the effects of genomic instability. Compromised immune surveillance allows for the increased genetic alterations that may lead to tumorigenesis. This is especially pertinent in NSCLC, where mutations in the TP53 tumor suppressor gene are prevalent.68 Such mutations not only contribute to the malignant phenotype but impede the immune system’s capacity to recognize and eliminate tumor cells. Although genomic instability can result in an increased mutation burden that may enhance the immunogenicity of tumors, making them more susceptible to immunotherapy, the overall efficacy of immunotherapy in elderly patients may be diminished.69,70 This reduction in effectiveness is due to the combined effects of immunosenescence and the altered TME, which is affected by genomic instability.
Chromosomal instability (CIN) mainly refers to an increased rate of chromosomal alterations, which include gains, losses, and structural rearrangements.71 This instability may result in mutations or deletions within critical genes associated with drug resistance, particularly those involved in the JAK/STAT signaling pathway.72,73 When chromosomal instability induces mutations or deletions in the genes of the JAK/STAT pathway, it can severely disrupt the signaling cascade mediated by Upd3-a cytokine with homology to interleukin-6.74 This disruption often results in reduced expression of PD-L1. The downregulation of PD-L1 expression not only undermines the tumor’s ability to escape immune surveillance but diminishes the overall efficacy of immunotherapies targeting the PD-1/PD-L1 axis. Tumors exhibiting high levels of CIN tend to display a more aggressive phenotype and correlate with a poorer prognosis, chiefly due to their capacity to adapt to therapeutic pressures and evade immune detection.75,76
The role of long non-coding RNAs (lncRNAs) in mediating genomic instability has become increasingly prominent in recent years.69,77 These lncRNAs can regulate the expression of genes associated with DNA repair and cell cycle regulation, thus impacting the stability of the genome. In lung cancer, specific lncRNA signatures linked to genomic instability have been indicated to correlate with the prognosis and treatment response.78 Genomic instability plays a pivotal role in cancer progression and the efficacy of treatment, particularly in elderly patients. The interaction between age-related immune decline and genomic alterations necessitates a thorough understanding of these elements to refine therapeutic strategies and improve survival outcomes in this at-risk population. Future research should focus on elucidating the molecular mechanisms that link genomic instability to immune dysfunction and exploring targeted therapies.
Epigenetic Reprogramming
Epigenetic reprogramming refers to the heritable changes in gene expression that do not involve alterations to the underlying DNA sequence.79 Epigenetic modifications, including DNA methylation, histone modifications, and non-coding RNA expression, can markedly affect the expression of genes involved in immune regulation. The enhancer of zeste homolog 2 (EZH2), an epigenetic regulator, has been demonstrated to promote the formation of an immunosuppressive TME by upregulating PD-L1 expression through HIF-1α.25 This epigenetic control can lead to the silencing of genes that would typically promote anti-tumor immunity, thereby facilitating immune evasion. The interplay between epigenetic modifications and chromosomal instability can establish a feedback loop that exacerbates tumor aggressiveness and therapeutic resistance.80 DNA methylation and histone modifications are critical epigenetic mechanisms that regulate gene expression in various biological processes, including immune responses. In the cancer, these modifications can lead to the silencing of immune activation genes, such as MHC-I, which is essential for presenting antigens to T cells and initiating immune responses. The down-regulation of MHC-I expression in tumors allows cancer cells to evade detection and elimination by the immune system, contributing to tumor progression and metastasis.81 Moreover, the interplay between DNA methylation and histone modifications is intricate, and can lead to a feedback loop that exacerbates immune suppression.82 This dynamic fosters a TME that not only supports cancer cell growth but actively undermines anti-tumor immune responses, making it challenging to achieve effective immunotherapy outcomes. Consequently, targeting the epigenetic machinery presents a promising avenue for therapeutic intervention, potentially restoring sensitivity to ICIs and improving outcomes in cancers with high levels of epigenetic reprogramming.
Strategies for Overcoming Drug Resistance
Multiple mechanisms are involved in the process of immunotherapy resistance (Figure 2). Overcoming resistance in cancer immunotherapy represents a complex challenge that necessitates innovative strategies.83,84 One promising method involves the optimization of combination therapies, which can enhance efficacy by targeting multiple pathways concurrently. The application of epigenetic modulators like DNA methyltransferase inhibitors can reverse silencing in immune activation genes and restore the expression of MHC-I in tumor cells. In addition, directly targeting signaling pathways of immune suppression can significantly enhance the effectiveness of immunotherapies. Another critical aspect of addressing resistance is to select biomarkers capable of predicting patient responses to combination therapies. By stratifying patients according to their molecular profiles, clinicians can develop tailored treatment regimens likely to yield positive outcomes for patients.
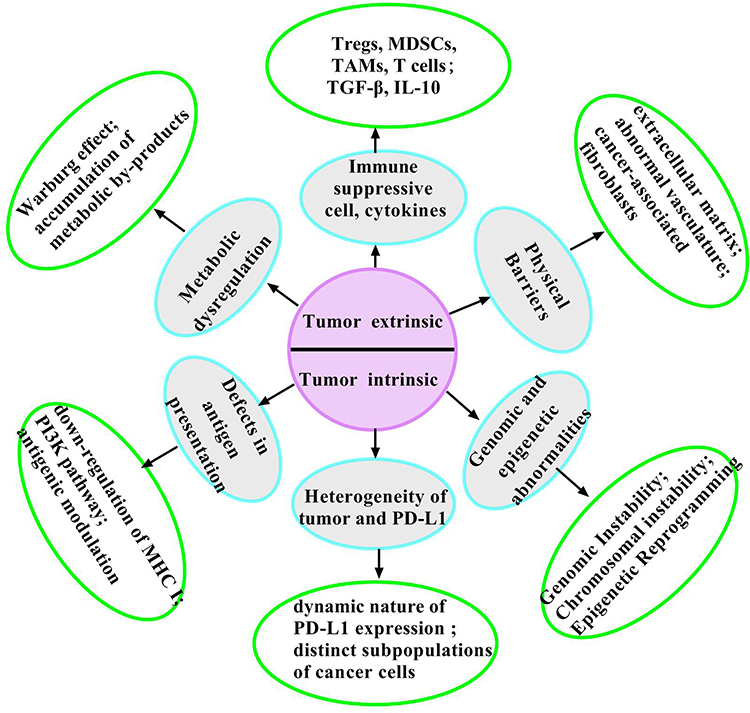 |
Figure 2 Mechanisms related to immunotherapy resistance. The limited effectiveness of immunotherapy is primarily due to various mechanisms of immunotherapy resistance. Tregs, MDSCs, and TAMs, combined with tumor-released immunosuppressive cytokines, contribute to the exhaustion of tumor-infiltrating lymphocytes (TILs). In the TME, Tumor cells often undergo abnormal metabolic pathways that allow them to thrive in hypoxic and nutrient-deprived environments. This metabolic reprogramming is defined by an increase in glycolysis, even in the presence of oxygen—a phenomenon referred to as the Warburg effect. The remodeling of the ECM and abnormal vasculature can significantly impact T cell behavior. The down-regulation of MHC class I molecules on the tumor surface, some signaling pathways, and antigenic modulation impair the process of antigen presentation. PD-L1 expression possesses significant spatiotemporal heterogeneity. Tumor heterogeneity denotes the presence of various cell populations within a single tumor, which can differ in genetic, phenotypic, and functional characteristics. Genomic instability and chromosomal instability significantly impact tumor progression and therapeutic response. Epigenetic reprogramming supports tumor cell growth but actively undermines anti-tumor immune responses. |
Optimization of Combination Therapy Strategies
The combination therapy strategies are crucial for enhancing the efficacy of cancer treatments, particularly in overcoming resistance. A promising approach is the integration of ICIs with various therapeutic modalities, including chemotherapy, targeted therapy, and radiotherapy.85 These multifaceted strategies seek to harness the synergistic effects of different treatments to improve patient outcomes.
Combining ICIs with chemotherapy can significantly enhance T cell activation, resulting in improved anti-tumor responses.86 The timing and sequencing of these combination therapies are critical factors that can substantially influence their effectiveness. Administering chemotherapy before ICIs may help reduce tumor burden and enhance the infiltration of immune cells into the TME. Conversely, immunotherapy as a neoadjuvant treatment can start the immune system, augmenting the response to subsequent therapies.87
One of the most promising strategies in cancer immunotherapy is dual ICIs targeting PD-1 in conjunction with cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) or lymphocyte activation gene 3 (LAG-3).88 These combinations have demonstrated the ability to activate T cells synergistically. However, the balance between efficacy and toxicity remains a critical consideration in the clinical application of these therapies. The PD-1/PD-L1 pathway is crucial in tumor-induced immune evasion. Elevated levels of PD-L1 expression are often associated with preferable responses to PD-1 inhibitors, such as pembrolizumab and nivolumab, which have become standard treatments for patients with advanced NSCLC exhibiting PD-L1 expression levels of 1% or greater.8 The combination of PD-1 and CTLA-4 inhibitors has yielded promising results, demonstrating increased overall survival rates in various cancers, including lung cancer.89 This dual blockade enhances T cell activation and proliferation, culminating in a more robust immune response against tumor cells. LAG-3 serves as another checkpoint that inhibits T cell activation and proliferation; its blockade may enhance the efficacy of PD-1 inhibition. Combining PD-1 with LAG-3 inhibitors may improve therapeutic outcomes.90 Preclinical investigations have indicated that dual blockade targeting PD-1 and LAG-3 can result in significant tumor regression and improved survival rates in murine tumor models, indicating potential benefits in clinical settings.91 Nevertheless, the increased efficacy of these combination therapies often corresponds with heightened toxicity. Immune-related adverse events (irAEs) are a prominent concern associated with dual checkpoint inhibitors, as they can lead to serious complications affecting multiple organ systems, including the skin, gastrointestinal tract, and endocrine organs.92 The incidence of irAEs tends to be substantially higher with combination therapies compared to monotherapy, necessitating careful patient selection and monitoring. By harnessing the synergistic effects of various therapeutic modalities and tailoring treatment regimens according to individual patient profiles, clinicians can enhance efficacy and overcome drug resistance.
Biomarker-Guided Precision Therapy
Biomarkers play a vital role in the precise treatment of tumors, as they facilitate therapeutic decision-making and enable the stratification of patients for immunotherapy. Among these biomarkers, PD-L1 has emerged as a critical factor influencing the efficacy of ICIs in NSCLC. The heterogeneity of PD-L1 expression and the variability in response rates to ICIs underscore the necessity for additional biomarkers that may stratify patients. Bone Marrow Stromal Antigen 1 (BST1) has garnered significant attention due to its role in immune response and potential association with PD-L1 expression. A recent study indicates that BST1 may be a promising biomarker for predicting response to chemoimmunotherapy.93 TMB has been recognized as a substantial predictor of response to ICIs across diverse tumor types, including NSCLC.94–96 TMB quantifies the total number of mutations within a tumor’s genome and has been correlated with an increased neoantigen load, which enhances the likelihood of immune recognition and results in a more favorable therapeutic response. Patients with high TMB levels generally have improved responses to PD-1/PD-L1 inhibitors compared with those with lower TMB levels.97–99 Additionally, in patients with advanced non-small cell lung cancer (NSCLC) and high tumor mutational burden, irrespective of PD-L1 expression status, first-line treatment with the combination of nivolumab and ipilimumab demonstrated significantly prolonged PFS compared to platinum-based chemotherapy.100 The findings establish TMB as an independent predictive biomarker for patient selection in dual immune checkpoint inhibitor therapy, highlighting its potential to guide treatment decisions beyond PD-L1 stratification. Integration of PD-L1, BST1, and TMB provides a comprehensive approach to patient stratification in NSCLC. While PD-L1 serves as a well-established biomarker for predicting responses to ICIs, the inclusion of BST1 and TMB enhances the precision of patient selection. This multifaceted biomarker strategy aligns with the principles of precision medicine, which endeavors to tailor treatments based on the individual characteristics of patients.
Future Directions
Application of Circulating Tumor DNA (ctDNA)
ctDNA has emerged as a non-invasive biomarker that provides real-time insights into tumor dynamics, including mutation status, treatment response, and the detection of minimal residual disease (MRD). Recent advancements in high-throughput sequencing technologies have significantly enhanced the sensitivity and specificity of circulating tumor DNA (ctDNA) detection, enabling its application in early cancer diagnosis, monitoring treatment efficacy, and predicting recurrence.101,102 In NSCLC, ctDNA can predict patient outcomes, making it a valuable tool for guiding treatment decisions.103 The technology has evolved to include next-generation sequencing and digital droplet PCR, which enhance the sensitivity of ctDNA detection, allowing for the identification of low-frequency mutations that may not be captured in traditional tissue biopsies. Moreover, ctDNA can provide insights into the clonal evolution of tumors, helping to identify emerging resistance mechanisms during therapy.104
Multi-Omics Strategies for Optimizing Immunotherapy Benefit Population Selection
The integration of multi-omics strategies, encompassing genomics, transcriptomics, proteomics, and epigenomics, is revolutionizing the landscape of cancer treatment, particularly in the context of immunotherapy.105 By analyzing the tumor microenvironment and the molecular characteristics of tumors at a comprehensive level, researchers can identify distinct patient populations that are more likely to benefit from immunotherapy. This approach enables the stratification of patients based on their unique tumor biology, resulting in more tailored and effective treatment regimens. Furthermore, multi-omics analyses can reveal insights into the mechanisms of resistance that tumors may develop against immunotherapies, enabling the design of combination therapies that can overcome these barriers.105 The advent of spatial transcriptomics has revolutionized our understanding of tumor heterogeneity and the mechanisms underlying resistance to therapies in cancers such as lung cancer.106 This innovative technique allows researchers to map gene expression profiles within the context of the tumor microenvironment, providing insights into the spatial distribution of resistant clones.107 By examining how different regions of a tumor respond to treatment, spatial transcriptomics can reveal the presence of subclonal populations that exhibit distinct biological behaviors and therapeutic responses. As the technology continues to advance, the ability to integrate multi-omics data will enhance our understanding of tumor heterogeneity and the immune landscape, ultimately guiding more precise therapeutic strategies and improving patient outcomes in the realm of cancer immunotherapy.
Conclusion
The intricate landscape of immunotherapy resistance in PD-L1 high-expressing NSCLC underscores the intricacies of cancer treatment and emphasizes the necessity for a comprehensive approach in research and clinical practice. Mechanisms of resistance are not isolated; instead, they represent interconnected processes influenced by intrinsic cellular characteristics and the TME. This review elucidates various factors contributing to immunotherapy resistance, including genetic alterations, epigenetic reprogramming, and the dynamic nature of the TME. The complexity of immunotherapy resistance necessitates a personalized approach to treatment. Current biomarkers, such as PD-L1 and TMB, exhibit limitations in their predictive accuracy. These highlight the need to identify novel biomarkers that more accurately predict a patient’s likelihood of benefiting from ICIs. Combination therapies represent a promising approach for overcoming resistance. The combination of targeted therapies, chemotherapy, and innovative immunotherapeutic agents may yield a synergistic effect. Nevertheless, achieving optimal integration of these modalities requires a profound understanding of the mechanisms underlying resistance. Insights derived from investigations into the tumor microenvironment hold particular promise. Immunosuppressive cells, cytokines and extracellular matrix components have a certain role in inhibiting immune response. Targeting these elements may be possible to reverse resistance and improve patient outcomes.
In summary, addressing immunotherapy resistance in PD-L1 high-expressing NSCLC demands urgent, coordinated efforts to translate mechanistic insights into clinical breakthroughs. The interconnected resistance drivers—including genetic alterations, epigenetic reprogramming, and dynamic tumor microenvironment (TME) interactions—while advancing beyond current biomarker limitations. The complexity of these resistance pathways underscores the necessity for personalized therapeutic strategies and innovative combination approaches targeting immunosuppressive TME elements. Looking forward, emerging therapies leveraging multi-omics profiling and novel TME modulators hold significant promise for overcoming resistance and optimizing precision medicine in NSCLC immunotherapy.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Disclosure
All authors have declared that they have no conflicts of interest that might be relevant to the contents of this manuscript.
References
1. Zhan QY, Xie LX, Wang C. Promoting critical care system and capacity building in pulmonary and critical care medicine subspecialties. Zhonghua Yi Xue Za Zhi. 2023;103(40):3149–3151. doi:10.3760/cma.j.cn112137-20230602-00919
2. Wang F, Wang S, Zhou Q. the resistance mechanisms of lung cancer immunotherapy. Front Oncol. 2020;10(568059).
3. Hayashi H, Mitsudomi T. Immune-checkpoint inhibitors for lung cancer with EGFR mutation. Gan to Kagaku Ryoho Cancer Chemother. 2017;44(9):722–726.
4. Mazieres J, Drilon A, Lusque A, et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: results from the IMMUNOTARGET registry. Annals Oncology. 2019;30(8):1321–1328. doi:10.1093/annonc/mdz167
5. Chen X, Gao A, Zhang F, et al. ILT4 inhibition prevents TAM- and dysfunctional T cell-mediated immunosuppression and enhances the efficacy of anti-PD-L1 therapy in NSCLC with EGFR activation. Theranostics. 2021;11(7):3392–3416. doi:10.7150/thno.52435
6. Ito A, Tarukawa T, Suzuki Y, et al. Clinicopathological and molecular characteristics promoting PD-L1 expression in early-stage lung adenocarcinoma and squamous cell carcinoma. Anticancer Res. 2023;43(11):5197–5204. doi:10.21873/anticanres.16721
7. Shinchi Y, Ishizuka S, Komohara Y, et al. The expression of PD-1 ligand 1 on macrophages and its clinical impacts and mechanisms in lung adenocarcinoma. Cancer Immunol Immunother. 2022;71(11):2645–2661. doi:10.1007/s00262-022-03187-4
8. Ullah A, Pulliam S, Karki NR, et al. PD-L1 over-expression varies in different subtypes of lung cancer: will this affect future therapies? Clinics and Practice. 2022;12(5):653–671. doi:10.3390/clinpract12050068
9. Sánchez-Magraner L, Gumuzio J, Miles J, et al. Functional engagement of the PD-1/PD-L1 COMPLEX BUT Not PD-l1 expression is highly predictive of patient response to immunotherapy in non-small-cell lung cancer. J Clin Oncology. 2023;41(14):2561–2570. doi:10.1200/JCO.22.01748
10. Lamberti G, Sisi M, Andrini E, et al. The mechanisms of PD-L1 Regulation in Non-Small-Cell Lung Cancer (NSCLC): which are the involved players? Cancers. 2020;12(11):3129. doi:10.3390/cancers12113129
11. Cui JW, Li Y, Yang Y, et al. Tumor immunotherapy resistance: revealing the mechanism of PD-1 / PD-L1-mediated tumor immune escape. Biomed Pharmacothe. 2024;171(116203).
12. Pathak R, Pharaon RR, Mohanty A, et al. Acquired resistance to PD-1/PD-L1 blockade in lung cancer: mechanisms and patterns of failure. Cancers. 2020;12(12):3851. doi:10.3390/cancers12123851
13. Huang C, Ren S, Chen Y, et al. PD-L1 methylation restricts PD-L1/PD-1 interactions to control cancer immune surveillance. Sci Adv. 2023;9(21):eade4186. doi:10.1126/sciadv.ade4186
14. Franzi S, Mattioni G, Rijavec E, et al. Neoadjuvant chemo-immunotherapy for locally advanced non-small-cell lung cancer: a review of the literature. J Clin Med. 2022;11(9):2629. doi:10.3390/jcm11092629
15. Zheng X, Song X, Zhu G, et al. Nanomedicine combats drug resistance in lung cancer. Adv Materials. 2024;36(3):e2308977.
16. Erhart CC, Cefalì M, Mangan D, et al. Prognostic value of KRAS G12C in advanced non-small cell lung cancer with high PD-L1 expression treated with upfront immunotherapy: a systematic review and meta-analysis. Swiss Med Weekly. 2024;154(3695):3695. doi:10.57187/s.3695
17. Pramanik A, Patibandla S, Gao Y, et al. Bio-conjugated magnetic-fluorescence nanoarchitectures for the capture and identification of lung-tumor-derived programmed cell death lighand 1-positive exosomes. ACS Omega. 2022;7(18):16035–16042. doi:10.1021/acsomega.2c01210
18. Kim S, Cho S, Kim JH. CD1-mediated immune responses in mucosal tissues: molecular mechanisms underlying lipid antigen presentation system. Experimental & Molecular Medicine. 2023;55(9):1858–1871. doi:10.1038/s12276-023-01053-6
19. Pradhan S, Ghosh S, Hussain S, et al. Linking membrane fluidity with defective antigen presentation in leishmaniasis. Parasite Immunol. 2021;43(7):e12835. doi:10.1111/pim.12835
20. Schmitz K, Wilken-Schmitz A, Vasic V, et al. Progranulin deficiency confers resistance to autoimmune encephalomyelitis in mice. Cell. Mol. Immunol. 2020;17(10):1077–1091. doi:10.1038/s41423-019-0274-5
21. Kwantwi LB. Genetic alterations shape innate immune cells to foster immunosuppression and cancer immunotherapy resistance. Clin Exp Med. 2023;23(8):4289–4296. doi:10.1007/s10238-023-01240-9
22. Zhang X, Zhang J, Liu P, et al. Immunotherapy progress and clinical strategy of unresectable locally advanced non-small cell lung cancer. Front Oncol. 2023;13(1022042).
23. Kumar S, Pandey M, Mir IA, et al. Evaluation of the programmed death-ligand 1 mRNA expression and immunopositivity and their correlation with survival outcomes in Indian lung cancer patients. Human Cell. 2022;35(1):286–298. doi:10.1007/s13577-021-00647-4
24. Katakura S, Kobayashi N, Hashimoto H, et al. MicroRNA-200b is a potential biomarker of the expression of PD-L1 in patients with lung cancer. Thoracic Cancer. 2020;11(10):2975–2982. doi:10.1111/1759-7714.13653
25. Zhao Y, Wang XX, Wu W, et al. EZH2 regulates PD-L1 expression via HIF-1α in non-small cell lung cancer cells. Biochem Biophys Res Commun. 2019;517(2):201–209. doi:10.1016/j.bbrc.2019.07.039
26. Brown EL, DeWeerd RA, Zidel A, et al. Preclinical antibody-PET imaging of PD-L1. Front Nuclear Med. 2022;2(953202). doi:10.3389/fnume.2022.953202.
27. Li Y, Cao L, Qian Z, et al. Mifepristone regulates Tregs function mediated by dendritic cells through suppressing the expression of TGF-β. Immunopharmacology and Immunotoxicology. 2021;43(1):85–93. doi:10.1080/08923973.2020.1867998
28. Petti AA, Williams SR, Miller CA, et al. A general approach for detecting expressed mutations in AML cells using single cell RNA-sequencing. Nat Commun. 2019;10(1):3660. doi:10.1038/s41467-019-11591-1
29. Chen A, Hu S, Qf W. Tumor heterogeneity of acute myeloid leukemia: insights from single-cell sequencing. Blood Sci. 2019;1(1):73–76. doi:10.1097/BS9.0000000000000015
30. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14(10):1014–1022. doi:10.1038/ni.2703
31. Fridman WH, Zitvogel L, Sautès-Fridman C, et al. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol. 2017;14(12):717–734. doi:10.1038/nrclinonc.2017.101
32. Sakaguchi S, Yamaguchi T, Nomura T, et al. Regulatory T cells and immune tolerance. Cell. 2008;133(5):775–787. doi:10.1016/j.cell.2008.05.009
33. Oida T, Zhang X, Goto M, et al. CD4+CD25- T cells that express latency-associated peptide on the surface suppress CD4+CD45RBhigh-induced colitis by a TGF-beta-dependent mechanism. J Immunol. 2003;170(5):2516–2522. doi:10.4049/jimmunol.170.5.2516
34. Mirlekar B. Tumor promoting roles of IL-10, TGF-β, IL-4, and IL-35: its implications in cancer immunotherapy. SAGE Open Med. 2022;10(20503121211069012). doi:10.1177/20503121211069012
35. Wang H, Yung MMH, Ngan HYS, et al. The impact of the tumor microenvironment on macrophage polarization in cancer metastatic progression. Int J Mol Sci. 2021;22(12).
36. Xu X, Xie K, Li B, et al. Adaptive resistance in tumors to anti-PD-1 therapy through re-immunosuppression by upregulation of GPNMB expression. Int Immunopharmacol. 2021;101(Pt B):108199. doi:10.1016/j.intimp.2021.108199
37. Monjaras-Avila CU, Lorenzo-Leal AC, Luque-Badillo AC, et al. The tumor immune microenvironment in clear cell renal cell carcinoma. Int J Mol Sci. 2023;24(9):7946. doi:10.3390/ijms24097946
38. Sundstedt A, O’Neill EJ, Nicolson KS, et al. Role for IL-10 in suppression mediated by peptide-induced regulatory T cells in vivo. J Immunol. 2003;170(3):1240–1248. doi:10.4049/jimmunol.170.3.1240
39. Bhardwaj V, Sm A. Modulation of T-cell function by myeloid-derived suppressor cells in hematological malignancies. Front Cell Develop Biol. 2023;11(1129343). doi:10.3389/fcell.2023.1129343
40. Meyer C, Cagnon L, Costa-Nunes CM, et al. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother. 2014;63(3):247–257. doi:10.1007/s00262-013-1508-5
41. Kodumudi KN, Weber A, Sarnaik AA, et al. Blockade of myeloid-derived suppressor cells after induction of lymphopenia improves adoptive T cell therapy in a murine model of melanoma. J Immunol. 2012;189(11):5147–5154. doi:10.4049/jimmunol.1200274
42. Lasser SA, Ozbay KFG, Arkhypov I, et al. Myeloid-derived suppressor cells in cancer and cancer therapy. Nat Rev Clin Oncol. 2024;21(2):147–164. doi:10.1038/s41571-023-00846-y
43. He S, Zheng L, Qi C. Myeloid-derived suppressor cells (MDSCs) in the tumor microenvironment and their targeting in cancer therapy. Mol Cancer. 2025;24(1):5. doi:10.1186/s12943-024-02208-3
44. De PM, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell. 2013;23(3):277–286. doi:10.1016/j.ccr.2013.02.013
45. Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell. 2015;27(4):462–472. doi:10.1016/j.ccell.2015.02.015
46. Bui I, Bonavida B. Polarization of M2 Tumor-Associated Macrophages (TAMs) in Cancer Immunotherapy. Critical Rev Oncogenesis. 2024;29(4):75–95. doi:10.1615/CritRevOncog.2024053830
47. Jeong H, Koh J, Kim S, et al. Cell-intrinsic PD-L1 signaling drives immunosuppression by myeloid-derived suppressor cells through IL-6/Jak/Stat3 in PD-L1-high lung cancer. J ImmunoTherapy Cancer. 2025;13(3):e010612. doi:10.1136/jitc-2024-010612
48. Fukushi A, Kim HD, Chang YC, et al. Revisited metabolic control and reprogramming cancers by means of the warburg effect in tumor cells. Int J Mol Sci. 2022;23(17):10037. doi:10.3390/ijms231710037
49. Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11(5):325–337. doi:10.1038/nrc3038
50. Malhotra D, Gabrani R. Metabolic shifts in glioblastoma: unraveling altered pathways and exploring novel therapeutic avenues. Mol Bio Reports. 2025;52(1):146. doi:10.1007/s11033-025-10242-7
51. Apostolova P, Pearce EL. Lactic acid and lactate: revisiting the physiological roles in the tumor microenvironment. Trends Immunol. 2022;43(12):969–977. doi:10.1016/j.it.2022.10.005
52. Shen T, Miao S, Zhou Y, et al. Exosomal AP000439.2 from clear cell renal cell carcinoma induces M2 macrophage polarization to promote tumor progression through activation of STAT3. Cell Comm Signaling. 2022;20(1):152. doi:10.1186/s12964-022-00957-6
53. Bader JE, Voss K, Rathmell JC. Targeting Metabolism to Improve the Tumor Microenvironment for Cancer Immunotherapy. Molecular Cell. 2020;78(6):1019–1033. doi:10.1016/j.molcel.2020.05.034
54. Yamauchi M, Gibbons DL, Zong C, et al. Fibroblast heterogeneity and its impact on extracellular matrix and immune landscape remodeling in cancer. Matrix Biology: Journal of the International Society for Matrix Biology. 2020;92(8).
55. Zheng M, Zhang H, Dai M, et al. A PTT-induced feed-back carbon nanosystem for enhanced breast cancer therapy by extracellular matrix remodeling. Nano Lett. 2025;25(8):3180–3190. doi:10.1021/acs.nanolett.4c05625
56. Yu KX, Yuan WJ, Wang HZ, et al. Extracellular matrix stiffness and tumor-associated macrophage polarization: new fields affecting immune exclusion. Cancer Immunol Immunother. 2024;73(6):115. doi:10.1007/s00262-024-03675-9
57. Lu X, Gou Z, Chen H, et al. Extracellular matrix cancer-associated fibroblasts promote stromal fibrosis and immune exclusion in triple-negative breast cancer. J Pathol. 2025;265(3):385–399. doi:10.1002/path.6395
58. Min KW, Kim DH, Noh YK, et al. Cancer-associated fibroblasts are associated with poor prognosis in solid type of lung adenocarcinoma in a machine learning analysis. Sci Rep. 2021;11(1):16779. doi:10.1038/s41598-021-96344-1
59. Zhang H, Yue X, Chen Z, et al. Define cancer-associated fibroblasts (CAFs) in the tumor microenvironment: new opportunities in cancer immunotherapy and advances in clinical trials. Mol Cancer. 2023;22(1):159. doi:10.1186/s12943-023-01860-5
60. Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015;36(4):265–276. doi:10.1016/j.it.2015.02.008
61. Ej W. T cell exhaustion. Nat Immunol. 2011;12(6):492–499. doi:10.1038/ni.2035
62. Jiang W, He Y, He W, et al. Exhausted CD8+T cells in the tumor immune microenvironment: new pathways to therapy. Front Immunol. 2020;11(622509).
63. Pearce EL, Poffenberger MC, Chang CH, et al. Fueling immunity: insights into metabolism and lymphocyte function. Science. 2013;342(6155):1242454. doi:10.1126/science.1242454
64. Miller BC, Sen DR, Al AR, et al. Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol. 2019;20(3):326–336. doi:10.1038/s41590-019-0312-6
65. Kurachi M. CD8(+) T cell exhaustion. Semin Immunopathol. 2019;41(3):327–337. doi:10.1007/s00281-019-00744-5
66. Jancewicz I, Szarkowska J, Konopinski R, et al. PD-L1 overexpression, SWI/SNF complex deregulation, and profound transcriptomic changes characterize cancer-dependent exhaustion of persistently activated CD4(+) T cells. Cancers. 2021;13(16):4148. doi:10.3390/cancers13164148
67. Islam MA, Sehar U, Sultana OF, et al. SuperAgers and centenarians, dynamics of healthy ageing with cognitive resilience. Mechanisms Ageing Development. 2024;219(111936). doi:10.1016/j.mad.2024.111936.
68. Levine AJ. p53: 800 million years of evolution and 40 years of discovery. Nat Rev Cancer. 2020;20(8):471–480. doi:10.1038/s41568-020-0262-1
69. Yang K, Liang X, Wen K. Long non‑coding RNAs interact with RNA‑binding proteins to regulate genomic instability in cancer cells (Review). Oncol Rep. 2022;48(4). doi:10.3892/or.2022.8390
70. Chen M, Linstra R, van Vugt M. Genomic instability, inflammatory signaling and response to cancer immunotherapy. Biochim. Biophys. Acta, Rev. Cancer. 2022;1877(1):188661. doi:10.1016/j.bbcan.2021.188661
71. Chen X, Agustinus AS, Li J, et al. Chromosomal instability as a driver of cancer progression. Nat Rev Genet. 2025;26(1):31–46. doi:10.1038/s41576-024-00761-7
72. Gravina AG, Pellegrino R, Esposito A, et al. The JAK-STAT pathway as a therapeutic strategy in cancer patients with immune checkpoint inhibitor-induced colitis: a narrative review. Cancers. 2024;16(3):611. doi:10.3390/cancers16030611
73. Zhou T, Yang P, Tang S, et al. Classification of lung adenocarcinoma based on immune checkpoint and screening of related genes. J Oncol. 2021;2021(5512325):1–12. doi:10.1155/2021/5512325
74. Barrio L, Gaspar AE, Muzzopappa M, et al. Chromosomal instability-induced cell invasion through caspase-driven DNA damage. Curr Bio. 2023;33(20):4446–4457e4445. doi:10.1016/j.cub.2023.09.004
75. Carloni V, Morganti E, Galli A, et al. The adaptability of chromosomal instability in cancer therapy and resistance. Int J Mol Sci. 2022;24(1):245. doi:10.3390/ijms24010245
76. Requesens M, Foijer F, Nijman HW, et al. Genomic instability as a driver and suppressor of anti-tumor immunity. Front Immunol. 2024;15(1462496). doi:10.3389/fimmu.2024.1462496.
77. Mohapatra S, Winkle M, Ton AN, et al. The role of non-coding RNAs in chromosomal instability in cancer. J Pharmacol Exp Ther. 2023;384(1):10–19. doi:10.1124/jpet.122.001357
78. Yang L, Guo G, Yu X, et al. Mutation-derived long noncoding rna signature predicts survival in lung adenocarcinoma. Front Oncol. 2022;12(780631).
79. Goel S, Bhatia V, Biswas T, et al. Epigenetic reprogramming during prostate cancer progression: a perspective from development. Semi Cancer Biol. 2022;83(136):136–151. doi:10.1016/j.semcancer.2021.01.009
80. Gallagher SJ, Shklovskaya E, Hersey P. Epigenetic modulation in cancer immunotherapy. Curr. Opin. Pharmacol. 2017;35(48):48–56. doi:10.1016/j.coph.2017.05.006
81. Morrison BJ, Steel JC, Morris JC. Reduction of MHC-I expression limits T-lymphocyte-mediated killing of Cancer-initiating cells. BMC Cancer. 2018;18(1):469. doi:10.1186/s12885-018-4389-3
82. Amirkhah R, Naderi-Meshkin H, Shah JS, et al. The intricate interplay between epigenetic events, alternative splicing and noncoding RNA deregulation in colorectal cancer. Cells. 2019;8(8):929. doi:10.3390/cells8080929
83. Zheng DX, Bozym DJ, Tarantino G, et al. Overcoming resistance mechanisms to melanoma immunotherapy. American Journal of Clinical Dermatology. 2025;26(1):77–96. doi:10.1007/s40257-024-00907-7
84. Horvath L, Thienpont B, Zhao L, et al. Overcoming immunotherapy resistance in non-small cell lung cancer (NSCLC) - novel approaches and future outlook. Mol Cancer. 2020;19(1):141. doi:10.1186/s12943-020-01260-z
85. Passaro A, Brahmer J, Antonia S, et al. Managing resistance to immune checkpoint inhibitors in lung cancer: treatment and novel strategies. Journal of Clinical Oncology. 2022;40(6):598–610. doi:10.1200/JCO.21.01845
86. Zouein J, Haddad FG, Eid R, et al. The combination of immune checkpoint inhibitors and chemotherapy in advanced non-small-cell lung cancer: the rational choice. Immunotherapy. 2022;14(2):155–167. doi:10.2217/imt-2021-0014
87. Sorin M, Prosty C, Ghaleb L, et al. Neoadjuvant Chemoimmunotherapy for NSCLC: a systematic review and meta-analysis. JAMA Oncology. 2024;10(5):621–633. doi:10.1001/jamaoncol.2024.0057
88. Kalinka E, Wojas-Krawczyk K, Krawczyk P. Double guard efficiency and safety-overcoming resistance to immunotherapy by blocking or stimulating several immune checkpoints in non-small cell lung cancer patients. Cancers. 2023;15(13):3499. doi:10.3390/cancers15133499
89. Cheng W, Kang K, Zhao A, et al. Dual blockade immunotherapy targeting PD-1/PD-L1 and CTLA-4 in lung cancer. J Hematol Oncol. 2024;17(1):54. doi:10.1186/s13045-024-01581-2
90. Lichtenegger FS, Rothe M, Schnorfeil FM, et al. Targeting LAG-3 and PD-1 to enhance t cell activation by antigen-presenting cells. Front Immunol. 2018;9(385). doi:10.3389/fimmu.2018.00385.
91. Foy SP, Sennino B, Dela Cruz T, et al. Poxvirus-based active immunotherapy with PD-1 and LAG-3 dual immune checkpoint inhibition overcomes compensatory immune regulation, yielding complete tumor regression in mice. PLoS One. 2016;11(2):e0150084. doi:10.1371/journal.pone.0150084
92. Li Y, Pond G, McWhirter E. Multisystem immune-related adverse events from dual-agent immunotherapy use. Current Oncol. 2024;31(1):425–435. doi:10.3390/curroncol31010028
93. Yi C, Bian D, Wang J, et al. Anti-PD1 based precision induction therapy in unresectable stage III non-small cell lung cancer: a Phase II umbrella clinical trial. Nat Commun. 2025;16(1):1932. doi:10.1038/s41467-025-57184-z
94. Boumber Y. Tumor mutational burden (TMB) as a biomarker of response to immunotherapy in small cell lung cancer. J Thoracic Dis. 2018;10(8):4689–4693. doi:10.21037/jtd.2018.07.120
95. Deng H, Zhao Y, Cai X, et al. PD-L1 expression and Tumor mutation burden as Pathological response biomarkers of Neoadjuvant immunotherapy for Early-stage Non-small cell lung cancer: a systematic review and meta-analysis. Critical Rev Oncol/Hematol. 2022;170(103582):103582. doi:10.1016/j.critrevonc.2022.103582
96. Marabelle A, Fakih M, Lopez J, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, Phase 2 KEYNOTE-158 study. Lancet Oncol. 2020;21(10):1353–1365. doi:10.1016/S1470-2045(20)30445-9
97. Yoh K, Matsumoto S, Furuya N, et al. Comprehensive assessment of PD-L1 expression, tumor mutational burden and oncogenic driver alterations in non-small cell lung cancer patients treated with immune checkpoint inhibitors. Lung Cancer. 2021;159(128):128–134. doi:10.1016/j.lungcan.2021.07.015
98. Samstein RM, Lee CH, Shoushtari AN, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nature Genet. 2019;51(2):202–206. doi:10.1038/s41588-018-0312-8
99. Palmeri M, Mehnert J, Silk AW, et al. Real-world application of tumor mutational burden-high (TMB-high) and microsatellite instability (MSI) confirms their utility as immunotherapy biomarkers. ESMO Open. 2022;7(1):100336. doi:10.1016/j.esmoop.2021.100336
100. Hellmann MD, Ciuleanu TE, Pluzanski A, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. New Engl J Med. 2018;378(22):2093–2104.
101. Pellini B, Szymanski J, Chin RI, et al. liquid biopsies using circulating tumor DNA in non-small cell lung cancer. Thoracic Surg Clin. 2020;30(2):165–177. doi:10.1016/j.thorsurg.2020.01.005
102. Desai A, Pasquina LW, Nulsen C, et al. Putting comprehensive genomic profiling of ctDNA to work: 10 proposed use cases. J Liquid Biopsy. 2024;4(100140):100140. doi:10.1016/j.jlb.2024.100140
103. Anagnostou V, Ho C, Nicholas G, et al. ctDNA response after pembrolizumab in non-small cell lung cancer: phase 2 adaptive trial results. Nature Med. 2023;29(10):2559–2569. doi:10.1038/s41591-023-02598-9
104. Ravi P, Ravi A, Riaz IB, et al. Longitudinal evaluation of circulating tumor DNA using sensitive amplicon-based next-generation sequencing to identify resistance mechanisms to immune checkpoint inhibitors for advanced urothelial carcinoma. Oncologist. 2022;27(5):e406–e409. doi:10.1093/oncolo/oyac037
105. Guan A, Quek C. Single-cell multi-omics: insights into therapeutic innovations to advance treatment in cancer. Int J Mol Sci. 2025;26(6):2447. doi:10.3390/ijms26062447
106. Kang DH, Kim Y, Lee JH, et al. Spatial Transcriptomics in lung cancer and pulmonary diseases: a comprehensive review. Cancers. 2025;17(12):1912. doi:10.3390/cancers17121912
107. Liu W, Puri A, Fu D, et al. Dissecting the tumor microenvironment in response to immune checkpoint inhibitors via single-cell and spatial transcriptomics. Clin Exp Metastasis. 2024;41(4):313–332. doi:10.1007/s10585-023-10246-2
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
The DPY30-H3K4me3 Axis-Mediated PD-L1 Expression in Melanoma
Zhang Z, Han Y, Sun Q, Wang Y, Sun L
Journal of Inflammation Research 2022, 15:5595-5609
Published Date: 26 September 2022
From COPD to Lung Cancer: Mechanisms Linking, Diagnosis, Treatment, and Prognosis

Qi C, Sun SW, Xiong XZ
International Journal of Chronic Obstructive Pulmonary Disease 2022, 17:2603-2621
Published Date: 17 October 2022
The Path to Personalized Treatment in KRAS-Mutant Non-Small Cell Lung Cancer: A Review of Targeted Therapies and Immunotherapy
Shu CL, Liu YL
Cancer Management and Research 2022, 14:3485-3492
Published Date: 16 December 2022
Role of PD-1 Inhibitors in the Treatment of Esophagogastric Adenocarcinoma: Patient Selection and Reported Outcomes
Epistola R, Sperandio R, Wainberg Z, Iqbal S, Chao J
Cancer Management and Research 2023, 15:265-275
Published Date: 18 March 2023
Ability of Blood Cell Parameters to Predict Clinical Outcomes of Nivolumab Monotherapy in Advanced Esophageal Squamous Cell Carcinoma
Hamai Y, Emi M, Ibuki Y, Kurokawa T, Yoshikawa T, Ohsawa M, Hirohata R, Kitasaki N, Okada M
OncoTargets and Therapy 2023, 16:263-273
Published Date: 10 April 2023