Back to Journals » Journal of Inflammation Research » Volume 19
IL-27 Aggravates Sepsis-Induced ARDS by Driving Macrophage Ferroptosis Through Activation of NCOA4-Mediated Ferritinophagy
Authors Guo M ![]() , Xiong M, Shi J, Peng Q, Lin S, Xu F
, Xiong M, Shi J, Peng Q, Lin S, Xu F ![]()
Received 18 November 2025
Accepted for publication 21 February 2026
Published 3 March 2026 Volume 2026:19 575933
DOI https://doi.org/10.2147/JIR.S575933
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Xin Du
Minkang Guo,1,2 Meng Xiong,1 Jindian Shi,1 Qiaozhi Peng,1 Shihui Lin,1 Fang Xu1
1Department of Critical Care Medicine, the First Affiliated Hospital of Chongqing Medical University, Chongqing, 400016, People’s Republic of China; 2The Chongqing Key Laboratory of Translational Medicine in Major Metabolic Diseases, Chongqing, 400016, People’s Republic of China
Correspondence: Fang Xu, Department of Critical Care Medicine, the First Affiliated Hospital of Chongqing Medical University, Chongqing, 400016, People’s Republic of China, Email [email protected] Shihui Lin, Department of Critical Care Medicine, the First Affiliated Hospital of Chongqing Medical University, Chongqing, 400016, People’s Republic of China, Email [email protected]
Background: Acute respiratory distress syndrome (ARDS) induced by sepsis is a clinical syndrome characterized by high morbidity and mortality rates. This study aims to clarify the effects of recombinant mouse IL-27 protein on macrophage ferritinophagy, macrophage polarization, and its interventional role in sepsis-induced ARDS.
Methods: This study utilized wild-type (WT) and IL-27 receptor knockout (IL-27R−/−) mice to establish a cecal ligation and puncture (CLP)-induced sepsis model. The post-modeling mice were randomly divided into four groups: control group, CLP model group, IL-27 combined with CLP treatment group, and IL-27+CLP+PROTAC NCOA4 degrader-1 (Compound V3, CV3) combined treatment group. For the in vitro experiments using RAW 264.7 cells and Bone Marrow-Derived Macrophages (BMDMs), the cells were divided into four main groups as follows: the control group, the lipopolysaccharide (LPS) model group, the LPS+IL-27 treatment group, and the LPS+IL-27+CV3 combination treatment group.
Results: This study investigates the role of IL-27 in exacerbating ferritinophagy and ferroptosis in macrophages and septic lung injury, and explores the therapeutic potential of the NCOA4 degrader CV3. We found that IL-27 synergizes with LPS to enhance NCOA4-mediated ferritinophagy, leading to increased degradation of FTH1, upregulation of LC3A/B, and promotion of ferroptosis. Ferritinophagy amplification drove M1 macrophage polarization and inflammatory cytokine release. CV3, a PROTAC-based NCOA4 degrader, effectively disrupted the NCOA4-FTH1 interaction, inhibited ferritinophagy, and mitigated ferroptosis and inflammation. In murine models of sepsis-induced ARDS, CV3 alleviated lung injury, restored antioxidant defenses, and reduced ferroptosis. Notably, IL-27R−/− mice were resistant to IL-27-induced exacerbation of ferritinophagy and lung injury, confirming the specificity of IL-27 signaling.
Conclusion: These findings reveal a potential mechanistic link between NCOA4-mediated ferritinophagy and sepsis-associated ARDS pathogenesis. Targeting this pathway with CV3 may offer a novel therapeutic strategy, which warrants further investigation.
Keywords: sepsis, ARDS, macrophage polarization, IL-27, ferritinophagy, NCOA4, FTH1
Introduction
Sepsis is a deadly medical emergency triggered by a dysfunctional overreaction to infection, causing the body to harm its own tissues and organs.1 The excessive inflammatory response it triggers can induce multiple organ dysfunction, and approximately 45% of sepsis patients develop acute respiratory distress syndrome (ARDS).2 ARDS is the leading cause of both short-term mortality and long-term decline in quality of life in sepsis patients.3,4 Despite advances in understanding the pathogenesis of ARDS, the mortality rate remains as high as 45% due to the lack of specific and effective drug therapies.5–7 Current therapeutic approaches remain inadequate in fundamentally interrupting disease progression at the pathogenetic level to achieve significant improvement in clinical outcomes. Therefore, in-depth exploration of novel intervention strategies capable of precisely modulating the inflammatory cascade is of paramount clinical importance.
IL-27 is a proinflammatory cytokine composed of two proteins, EBI3 and p28. Its biological effects are mainly mediated by the IL-27 receptors highly expressed on the surface of macrophages.8 Existing studies have confirmed that the level of IL-27 is significantly elevated in both the CLP septic mouse model and the plasma of septic patients, suggesting its potential value as a diagnostic biomarker for sepsis.9,10 Accumulating evidence has confirmed that IL-27 can regulate the autophagic activity of fibroblast-like synoviocytes in rheumatoid arthritis via the STAT3 signaling pathway, thereby inhibiting the proliferative capacity of these cells.11 Further mechanistic studies have shown that the increased level of IL-27 can participate in the occurrence and development of sepsis-induced myocardial dysfunction by regulating the inflammatory response;12 meanwhile, other research indicates that suppressing IL-27 secretion can effectively alleviate sepsis-induced ARDS.13
Ferroptosis is an iron-dependent form of regulated cell death with a unique mechanism distinct from other programmed cell death pathways.14,15 Ferritinophagy, the selective autophagic degradation of ferritin, plays a crucial role in regulating iron homeostasis and is closely associated with ferroptosis. Nuclear receptor coactivator 4 (NCOA4) is a key mediator of ferritinophagy. Through quantitative proteomics, it has been identified as the cargo receptor responsible for transporting ferritin in this process.16 During ferritinophagy, NCOA4 directly recognizes and binds to ferritin heavy chain 1 (FTH1), which culminates in the lysosomal degradation of the complex and the liberation of iron.17 This process results in the release of large amounts of chelated iron from the ferritin complex.16,18,19 There is increasing evidence that dysregulated ferroptosis is implicated in the pathogenesis of numerous human diseases,14 and targeting the modulation of ferroptosis holds significant promise for the design of cancer therapies.20
The pathological features of sepsis-related ARDS are primarily characterized by significant inflammatory cell infiltration and an excessive cytokine release response.21 Alveolar macrophages (AMs) are key effector cells in pulmonary immune defense, responsible for recognizing and phagocytizing exogenous pathogens and endogenous damage-associated molecular patterns (DAMPs), playing a crucial role in maintaining pulmonary immune homeostasis.22 Studies have shown that lipopolysaccharide (LPS) can activate AMs, prompting them to release large amounts of pro-inflammatory factors, triggering a cascade of inflammatory responses and thereby exacerbating lung tissue damage.23 Research indicates that melatonin inhibits ferroptosis in macrophages by mediating ferritinophagy via NCOA4, thereby improving sepsis-induced ARDS.24 Under hypoxic conditions, the expression of NCOA4 decreases, inhibiting ferritinophagy and subsequently reducing the inflammatory response in macrophages.25 Therefore, early identification and regulation of abnormal inflammatory responses in macrophages may provide a potential strategy for the prevention and treatment of sepsis-induced ARDS.1
Materials and Methods
Cell Culture
The RAW 264.7 cells were acquired from the Cell Bank of Chinese Academy of Sciences. RAW 264.7 cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) enriched with 10% fetal bovine serum, 100 U/mL penicillin, and 100 µg/mL streptomycin, under a humidified atmosphere of 5% CO2 at 37°C.In vitro treatment, cells were pretreated with either IL-27 (50 ng/mL) or CV3 (10 μM) for 18 hours. Following this pretreatment, the cells were stimulated with LPS at a concentration of 100 ng/mL for 6 hours.
Construction of CLP Model
Male C57BL/6 mice (approximately 8 weeks old, 18–22 g) were supplied by the Animal Experiment Center of Chongqing Medical University. The mice were housed in a specific pathogen-free (SPF) barrier facility. The animal study protocol received approval from the Research Ethics Committee of the First Affiliated Hospital of Chongqing Medical University (No. IACUC-CQMU-2023-0466).
The experiment utilized a CLP procedure to construct the model. Anesthesia was induced and maintained using isoflurane. After shaving the abdominal hair, a 1 cm midline abdominal incision was made. The cecum was ligated at a point 1 cm from its distal tip and subsequently punctured at the site of ligation with a 21-gauge needle. Control group mice did not undergo CLP. This experiment induced a sepsis-induced ARDS model in wild-type (WT) and IL-27 receptor gene knockout (IL-27R−/−) mice through CLP. Some mice were intraperitoneally injected with recombinant mouse IL-27 protein (at a dose of 50 μg/kg) 2 hours before the CLP procedure. CV3 was administered intraperitoneally at a dose of 10 mg/kg. The compound was dissolved in a vehicle solution consisting of 5% DMSO, 45% PEG 300, and 50% saline. The administration was performed 2 hours before the CLP procedure. 24 h after the intervention, the experimental animals were humanely euthanized using isoflurane, and serum and lung tissue samples were collected for subsequent analysis.
Lung Wet to Dry Ratio
The freshly isolated mouse lungs tissues were excised, weighed, and then placed in a constant-temperature metal bath at 45 °C for 24 h.
Western Blot Assay
After collecting the samples, pre-cooled RIPA lysis buffer was added, followed by lysis on ice for 30 minutes. The lysates were then centrifuged at 4°C and 12,000 rpm for 30 minutes to collect the supernatant. Protein concentration was determined using the BCA protein assay, and the samples were subsequently used for SDS-PAGE electrophoresis. Following transfer to a polyvinylidene fluoride (PVDF) membrane, the membrane was blocked in 5% skim milk for 1 hour at room temperature. Following blocking, the membrane was treated with the specific primary antibody. Subsequently, it was incubated with a corresponding secondary antibody. (primary antibodies: anti-NCOA4, Abclonal, A25307, 1:1000; anti-FTH1, Proteintech, 11682-1-AP, 1:1000; anti-LC3A/B, Abclonal, A27200PM, 1:10000; anti-HO-1, Abclonal, A27713, 1:2000; anti-GPX4, Abclonal, A11243, 1:1000; anti-Ptgs2, Abclonal, A3560, 1:2000; anti-iNOS, abcam, ab202417, 1:1000; anti-IL-1β, abcam, ab234437, 1:1000; anti-Arg1, Abclonal, A25808, 1:10000; anti-IL-10, Abclonal, A2171, 1:1000; anti-ACTB, Abclonal, AC026, 1:20,000;)
Co-Immunoprecipitation (Co-IP)
For the experiment using BeaverBeads® Protein A Immunoprecipitation Kit, briefly, lyse the cells in binding buffer for 30 minutes, then centrifuge at 14,000 g for 10 minutes at 4°C and retain the supernatant. Incubate the specific antibody with the beads on a rotatory mixer at room temperature for 15 minutes. Then incubate the beads with the supernatant at room temperature for 60 minutes to allow the target antigen to specifically bind to the antibody-bead complex.
Quantitative PCR (qPCR)
Total RNA was isolated using RNAiso Plus (TAKARA, Japan); subsequently, cDNA was synthesized from the RNA using a commercial reverse transcription kit. qPCR was performed using the cDNA template, and gene expression was analyzed via the 2−ΔΔCT method. Primer sequences are listed in Table 1.
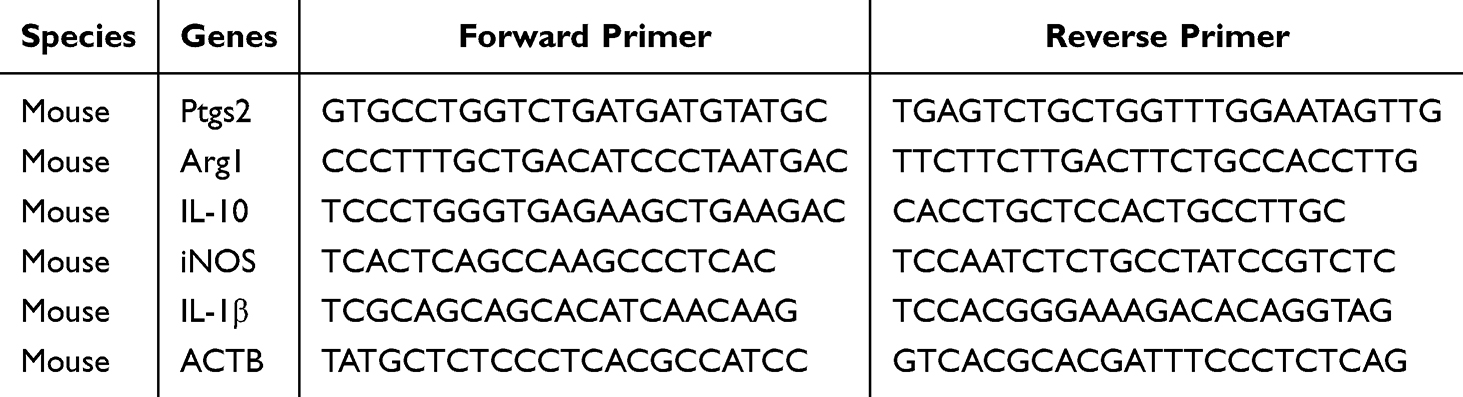 |
Table 1 Primer Sequences |
Histopathological Analysis
Following fixation in 4% paraformaldehyde for 24 hours, the lung tissues were processed, embedded in paraffin, and sectioned. Subsequently, the sections were stained with hematoxylin and eosin (H&E), and lung tissue injury was assessed using the MIKAWA score.26
Immunohistochemistry (IHC)
Paraffin-embedded tissue sections (4–5 μm) were deparaffinized and subjected to antigen retrieval with citrate buffer. Endogenous peroxidase was blocked with 3% H2O2 (10 min, RT), followed by serum blocking (30 min, RT). Sections were incubated with primary antibody overnight at 4°C, then with HRP-conjugated secondary antibody (1 h, RT). Signals were developed with DAB, and nuclei were counterstained with hematoxylin. Images were acquired under a microscope for analysis.
Immunofluorescence Staining
Paraffin sections (4–5 μm) were deparaffinized, rehydrated, and underwent citrate-based antigen retrieval. After blocking with 5% BSA for 1 h at room temperature, sections were incubated with primary antibody overnight at 4°C, followed by Alexa Fluor-conjugated secondary antibody for 1 h in the dark. Nuclei were stained with DAPI and slides were mounted with anti-fade medium. Images were captured using a fluorescence microscope.
Isolation and Culture of BMDMs
The femur and tibia were flushed with PBS to harvest bone marrow, which was then passed through a 70 μm cell strainer to prepare cells suspension. After culturing in complete medium for 7 days, BMDMs were differentiated from progenitor cells by stimulation with Macrophage colony-stimulating factor (M-CSF).
Confocal Laser Scanning Microscope
The cells were cultured in confocal dishes. After treatment according to the experimental groups, following fixation with 4% paraformaldehyde, the cells were permeabilized with 0.5% Triton X-100. Subsequently, the cells were co-incubated with primary antibodies against iNOS and CD206.
ELISA
Mouse plasma and cell culture medium were collected, and the supernatants were obtained after centrifugation. The levels of IL-1β and TNF-α were detected using mouse ELISA kits.
Detection of Glutathione (GSH), Malondialdehyde (MDA), Superoxide Dismutase (SOD), and Iron Content
The lung tissue was homogenized using sample preparation buffer, and the homogenate was then centrifuged at 12,000 g for 10 mins. Following collection, the supernatant was utilized for further analysis in strict adherence to the manufacturer’s specifications.
Reactive Oxygen Species (ROS)
BMDMs were seeded into confocal dishes and 96-well plates, treated as required, and then incubated with DCFH-DA. Cellular ROS levels were observed using a confocal laser scanning microscope, while fluorescence intensity was quantified using a fluorescence microplate reader at 488 nm excitation and 525 nm emission.
Transmission Electron Microscopy (TEM)
BMDMs were immersed in glutaraldehyde-based EM fixative, trimmed into 1 mm3 pieces, and fixed overnight at 4 °C in the fixative solution. After staining with uranyl acetate, the ultrastructural morphology of macrophages was examined using a Hitachi H-7650 TEM.
Statistical Analysis
The data are presented as the mean ± standard deviation (SD). All experiments were independently repeated at least three times to ensure reproducibility. Statistical analyses were performed using GraphPad Prism 10 software. For normally distributed data with equal variances, parametric tests were applied: unpaired two-tailed Student’s t-test was used for comparisons between two groups, and one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test was used for comparisons among three or more groups. To enhance statistical rigor, non-parametric tests were employed for data that did not meet the assumptions of normality or homogeneity of variance. Specifically, the Mann–Whitney U-test was used for comparisons between two groups, and the Kruskal–Wallis test was applied for comparisons involving three or more groups. P< 0.05 was considered statistically significant.
Results
IL-27 Promotes Ferritinophagy in Macrophages and Lung Tissues
In our previous research, we found that IL-27 combined with LPS can further exacerbate macrophage ferroptosis, aggravating sepsis-induced ARDS.27 Evidence suggests a link between the activation of NCOA4-mediated ferritinophagy and the induction of apoptosis. In this process, NCOA4 can bind to both ferritin and autophagosome proteins, promoting the degradation of ferritin in lysosomes.28,29 In this study, LPS increases the expression of NCOA4, suppresses FTH1, and promotes ferritinophagy. When LPS is combined with IL-27, IL-27 further increases the expression of NCOA4, severely suppresses FTH1, and exacerbates ferritinophagy compared to the LPS-only treatment group, thereby promoting ferroptosis. Additionally, the combination of LPS and IL-27 significantly increases the expression of the autophagy-related protein LC3A/B (Figure 1A and B). The protein-protein interaction (PPI) between NCOA4 and FTH1 constitutes a central regulatory node controlling the ferritinophagy pathway. Results demonstrated that LPS enhances the interaction between NCOA4 and FTH1. Treatment with IL-27 in combination with LPS significantly strengthened the interaction between NCOA4 and FTH1, thereby promoting ferritinophagy (Figure 1C). IHC results of lung tissues from WT mice showed that after CLP treatment, the FTH1 content in lung tissue decreased, indicating that ferritinophagy was promoted. FTH1 content was further reduced in the CLP+IL-27 group versus the CLP group, indicating an exacerbation of CLP-induced ferritinophagy by IL-27. (Figure 1D).
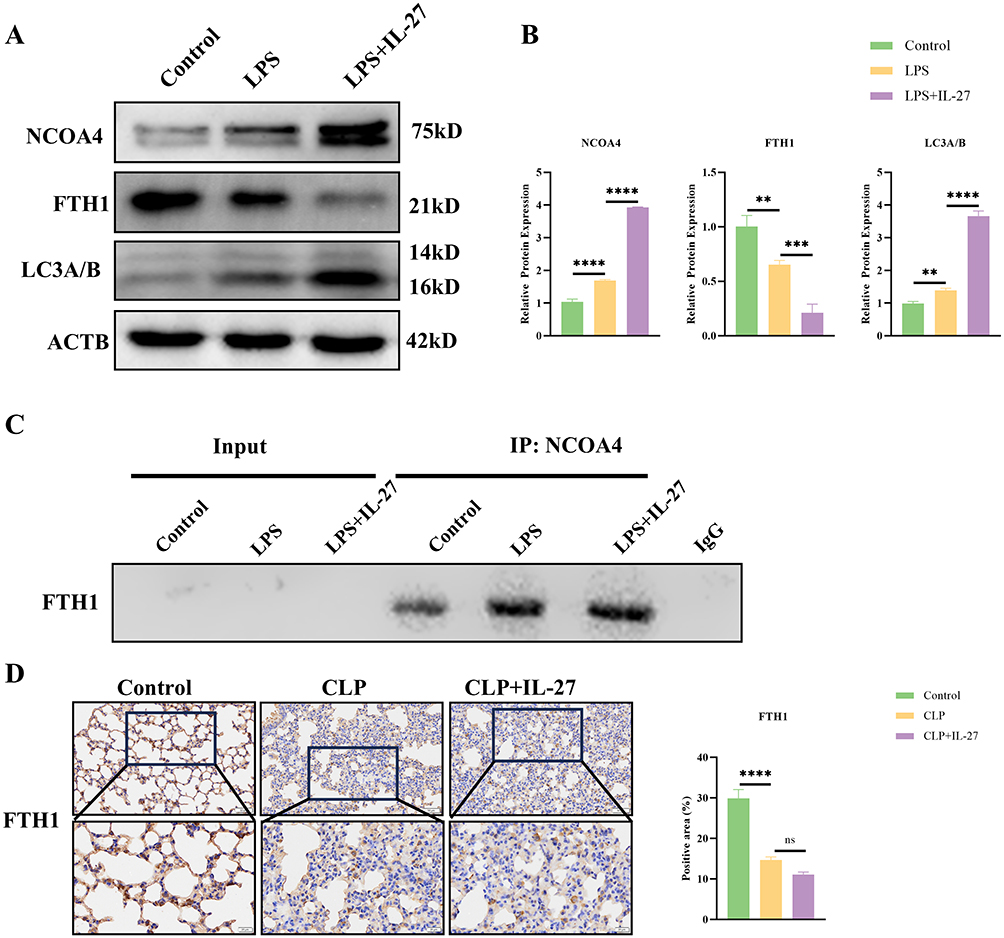 |
Figure 1 IL-27 promotes ferritinophagy in macrophages and lung tissues. (A and B) The expression levels of NCOA4, FTH1, and LC3A/B in RAW 264.7 cells and the statistical analysis. (C) Western blot analysis of CoIP results for NCOA4-FTH1. (D) Representative images of FTH1 expression and the immunohistochemical staining score for FTH1. **p<0.01, ***p<0.001, ****p<0.0001.Scale bar: 20 μm and 50 μm. Abbreviation: ns, not significant. |
Promote NCOA4 Degradation to Inhibit Macrophage Ferroptosis
Compound V3 (CV3) is a PROTAC-based NCOA4 degrader that reduces NCOA4 levels. CV3 treatment promotes the degradation of NCOA4, thereby reducing FTH1 degradation and increasing total FTH1 levels. Simultaneously, CV3 downregulates LC3A/B expression (Figure 2A and B). This study investigated whether CV3 inhibits ferritinophagy by disrupting this interaction using CoIP assays. The results demonstrated that IL-27 combined with LPS treatment significantly enhances the interaction between NCOA4 and FTH1, thereby promoting ferritinophagy. In contrast, CV3 treatment effectively inhibits the NCOA4-FTH1 interaction, ultimately blocking the ferritinophagy process (Figure 2C). Subsequently, we examined key antioxidant and stress-response enzymes GPX4 and HO-1 in vivo. Western blot analysis demonstrated that LPS treatment significantly suppressed GPX4 and HO-1 expression while upregulating the ferroptosis marker Ptgs2, indicating marked exacerbation of macrophage ferroptosis. The pro-ferroptosis effect was further enhanced when IL-27 was co-administered with LPS. In contrast, CV3 treatment partially restored GPX4 and HO-1 expression in macrophages while suppressing Ptgs2 levels, thereby attenuating the ferroptosis progression in macrophages (Figure 2D and E). The PCR results further confirmed that CV3 could suppress Ptgs2 expression, thereby inhibiting macrophage ferroptosis (Figure 2F). Additionally, we examined the regulatory effects of CV3 on Fe2+, MDA, GSH, and SOD. The data demonstrated that combined IL-27 and LPS treatment significantly increased intracellular iron levels and MDA content (a terminal product of lipid peroxidation), while reducing both GSH levels and SOD activity (Figure 2G–J). These findings suggest that this combined treatment exacerbates oxidative damage in macrophages and promotes ferroptosis. However, CV3 treatment effectively reversed these effects and attenuated macrophage ferroptosis. We quantified intracellular ROS levels in BMDMs using both a fluorescence microplate reader and confocal microscopy. A significant enhancement in ROS generation was observed in macrophages treated with IL-27 and LPS, while CV3 treatment reduced this accumulation, suggesting that CV3 may counteract the pro-oxidant effects of IL-27 and LPS (Figure 2K and L). Finally, we observed the mitochondrial state of BMDMs using transmission electron microscopy. The results showed that in LPS-treated macrophages, the mitochondria exhibited features such as reduced volume, increased membrane density, and decreased mitochondrial cristae. In the LPS combined with IL-27 treatment group, mitochondrial damage was further exacerbated, with many mitochondria entering lysosomes and more pronounced ferritinophagy. In stark contrast, CV3 effectively alleviated ferritinophagy in macrophages, inhibited the ferroptosis process, and promoted the recovery of mitochondrial function (Figure 2M).
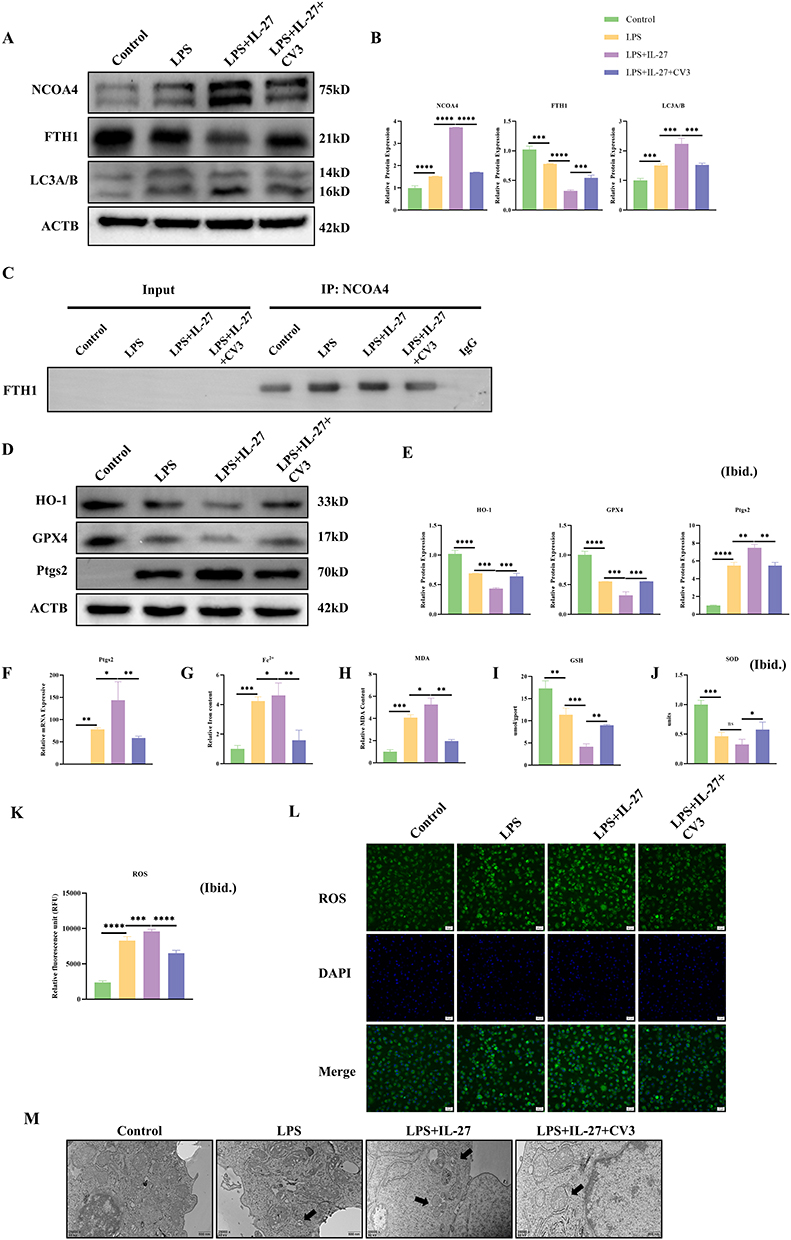 |
Figure 2 CV3 Promote NCOA4 degradation to inhibit macrophage ferroptosis. (A and B) Western blotting and statistical analysis of key ferritinophagy-related proteins such as NCOA4, FTH1, and LC3-A/B. (C) Western blot analysis of CoIP results for NCOA4-FTH1. (D and E) Western blotting and statistical analysis of key ferroptosis-associated proteins. (F) qPCR results of the ferroptosis marker Ptgs2. (G–J) Detection of core ferroptosis markers in macrophages: Fe2⁺, MDA, GSH, and SOD. (K) Detection of ROS levels in BMDMs using a fluorescence microplate reader. (L) ROS levels in BMDMs were determined using DCFH-DA. (M) Representative images showing mitochondria in lung tissue macrophages. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Scale bar: 20 μm and 500 nm. Abbreviation: ns, not significant. |
Inhibiting Ferritinophagy Effectively Alleviates the Inflammatory Response in Macrophages
Consistent with previous findings, LPS promotes macrophage polarization toward the M1 phenotype while suppressing their polarization toward the M2 phenotype. Western blot results showed that, compared to the LPS-only treatment group, the combined treatment of IL-27 and LPS further upregulated the expression of M1 polarization markers (IL-1β and iNOS) and further suppressed the expression of M2 polarization markers (IL-10 and Arg1), indicating an exacerbation of the inflammatory response in macrophages (Figure 3A and B). Additionally, PCR results were consistent with the Western blot findings (Figure 3C). After co-incubation of BMDMs with iNOS and CD206 antibodies, laser confocal microscopy observations revealed that in the IL-27 and LPS co-treatment group, iNOS expression was significantly upregulated, while CD206 expression was markedly downregulated. In contrast, CV3 treatment significantly suppressed iNOS expression while upregulated CD206 expression, suggesting its potential to effectively inhibit the inflammatory response in macrophages (Figure 3D). ELISA analysis of cell supernatants revealed that IL-27 and LPS co-treatment further increased inflammatory cytokine levels compared to LPS alone, aggravating macrophage inflammation. Conversely, CV3 treatment suppressed inflammatory cytokine release by inhibiting NCOA4, demonstrating its anti-inflammatory effect (Figure 3E).
 |
Figure 3 Inhibition of ferritinophagy to suppress the inflammatory response in macrophages. (A and B) Western blotting and statistical analysis of the polarization-related indicators iNOS, IL-1β, Arg1, and IL-10. (C) qPCR results of macrophage polarization markers. (D) Representative confocal microscopy images from each experimental group. (E) Detection of inflammatory cytokines in macrophage culture supernatant by ELISA. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Scale bar: 20 μm. |
CV3 Ameliorates CLP-Induced ARDS in WT Mice via Attenuation of Ferroptosis in the Lung
To determine the role of CV3 in CLP-induced ARDS, we performed a series of in vivo. Histological analysis of lung tissue by HE staining demonstrated that CLP induced lung injury, including features such as alveolar structural damage, interstitial edema, and abundant inflammatory cell infiltration. Moreover, lung injury was more severe in the CLP+IL-27 group, notably featuring exacerbated interstitial edema and a heightened inflammatory cell infiltrate. (Figure 4A). In IL-27R−/− mice, although CLP still induced lung injury, the degree of interstitial edema and alveolar structural destruction was milder compared to the WT mouse CLP group. Additionally, there was no difference in lung injury severity between the CLP+IL-27 group and the CLP group in these mice, and recombinant mouse IL-27 did not produce any significant exacerbating effects on lung injury following CLP, suggesting that IL-27 cannot participate in septic lung injury in the absence of the IL-27 receptor (Figure 4B). Sepsis can trigger an inflammatory cytokine storm, leading to an excessive systemic inflammatory response. This study utilized ELISA to measure the expression levels of key inflammatory factors in mouse plasma to investigate the effects of related interventions. In WT mice, the plasma levels of two core inflammatory factors, TNF-α and IL-1β, were significantly higher in the CLP-induced sepsis model group compared to the normal control group. In the CLP + IL-27 group, the levels of these inflammatory factors were further elevated compared to the CLP group, suggesting that IL-27 may exacerbate sepsis-induced inflammatory responses. In contrast, CV3 effectively downregulated the high expression levels of TNF-α and IL-1β in the plasma of CLP model mice, significantly alleviating the CLP-induced hyperinflammatory state (Figure 4C). The lung W/D was increased in CLP model mice, indicating the presence of alveolar edema. Treatment with IL-27 further elevated the W/D ratio, whereas CV3 reduced it, suggesting that CV3 alleviates ARDS injury in mice by mitigating pulmonary edema (Figure 4D). CV3 treatment significantly alleviated lung injury and reduced pathological scores of lung injury. Meanwhile, IHC results showed that CLP induced a reduction in FTH1 in lung tissue, and the CLP+IL-27 group exhibited a further decrease in FTH1, indicating aggravated ferritinophagy. CV3 significantly inhibited ferritinophagy and increased FTH1 levels (Figure 4E). Compared to the control group, the expression level of GPX4 was decreased in the CLP group and significantly lower in the CLP+IL-27 group. The CV3+CLP+IL-27 group showed higher GPX4 expression compared to the CLP+IL-27 group (Figure 4F). The CLP+IL-27 group also exhibited significantly higher Ptgs2 expression, suggesting exacerbated ferroptosis, which could be alleviated by CV3 (Figure 4G). Lung tissue homogenate tests further revealed that SOD and GSH levels were reduced in the CLP group and further decreased in the CLP+IL-27 group (indicating aggravated ferroptosis). CV3 increased SOD and GSH levels to mitigate ferroptosis, thereby protecting lung tissue from injury (Figure 4H and I).
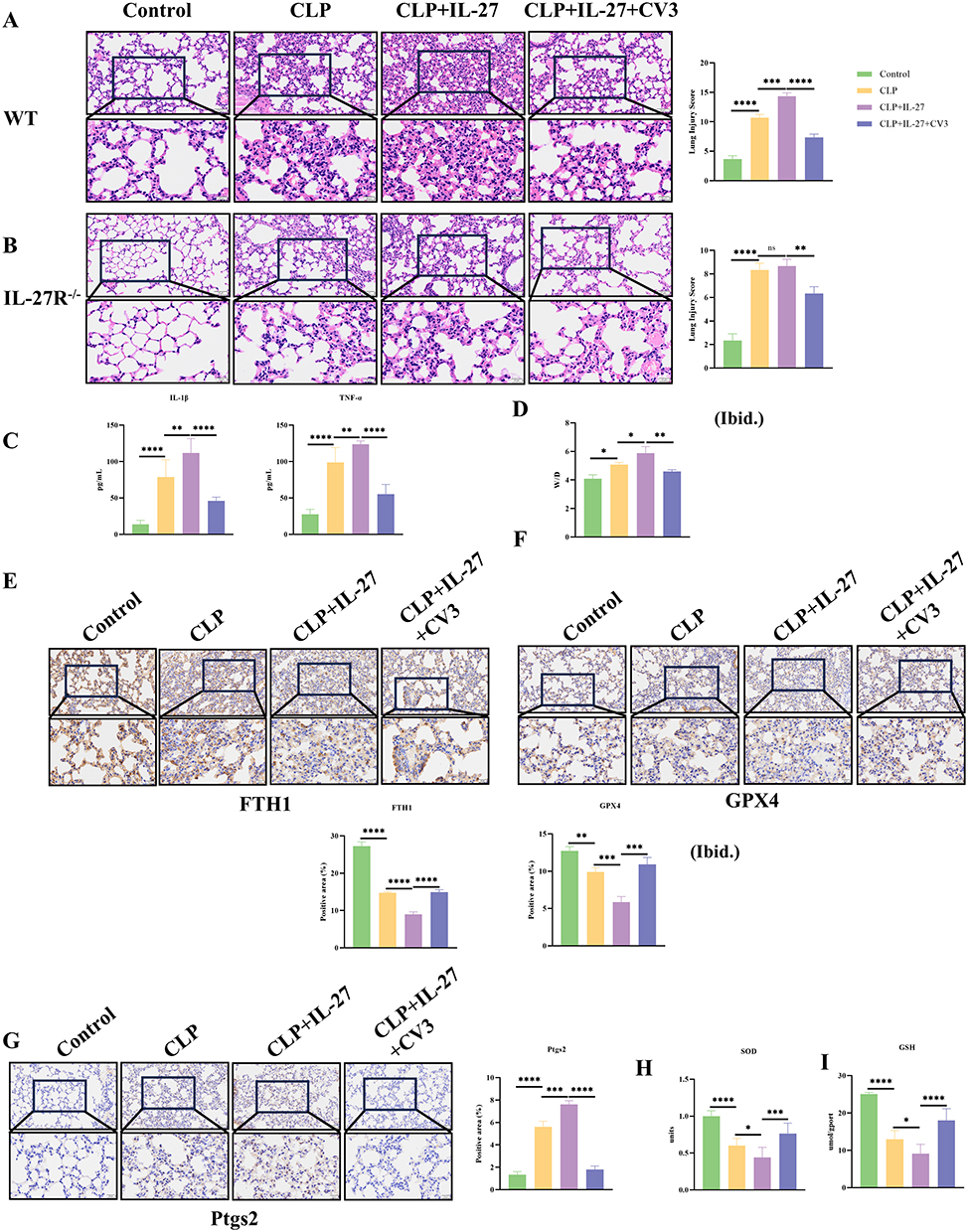 |
Figure 4 CV3 ameliorates ARDS in WT mice via attenuation of ferritinophagy in lung tissue. (A) Histological analysis of lung injury: Representative H&E-stained sections and injury scores in WT mice. (B) Histological analysis of lung injury: Representative H&E-stained sections and injury scores in IL-27R−/− mice. (C) ELISA of plasma inflammatory factors in WT mice. (D) lung W/D. (E) Representative images and quantitative analysis of FTH1 expression levels by immunohistochemical staining. (F) Representative images and quantitative analysis of GPX4 expression levels by immunohistochemical staining. (G) Representative images and quantitative analysis of Ptgs2 expression levels by immunohistochemical staining. (H and I) SOD and GSH levels were measured in lung tissue homogenates. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Scale bar: 20 μm and 50 μm. Abbreviation: ns, not significant. |
CV3 Inhibits CLP-Induced ARDS in IL-27R−/− Mice by Mitigating Ferroptosis in Lung Tissue
In IL-27R−/− mice, the expression levels of the inflammatory cytokines TNF-α and IL-1β in plasma were higher than those in normal control mice. In the CLP+IL-27 group, the levels of these inflammatory factors were largely consistent with those in the CLP group. CV3 effectively alleviated the CLP-induced hyperinflammatory state (Figure 5A). The lung W/D was increased in CLP model mice, and no significant difference in W/D was observed after IL-27 treatment. In contrast, CV3 reduced the W/D ratio, indicating that CV3 mitigates lung injury in mice by attenuating pulmonary edema (Figure 5B). In IL-27R−/− mice, CLP treatment still promoted the degradation of FTH1. However, in the CLP+IL-27 group, due to the lack of the IL-27 receptor, IL-27 lost its ability to exacerbate FTH1 degradation, and no difference was observed between the CLP+IL-27 group and the CLP group. The ferritinophagy inhibitor CV3 suppressed FTH1 degradation and inhibited CLP+IL-27-induced ferritinophagy (Figure 5C). In the GPX4 IHC experiment, the expression of GPX4 in lung tissue increased in the CLP group, which may be attributed to the lack of the IL-27 receptor, resulting in attenuated CLP-induced inflammation and ferroptosis, thereby showing a protective increase. The CLP+IL-27 group exhibited similar results to the CLP group (Figure 5D). The results of Ptgs2 immunohistochemical staining demonstrated that CLP induced ferroptosis in lung tissue; however, no statistically significant difference was observed between the CLP+IL-27 and CLP groups. The application of CV3 ameliorated ferroptosis and alleviated CLP-induced lung injury (Figure 5E). Further analysis of lung tissue homogenates revealed decreased levels of SOD and GSH in the CLP group, and the CLP+IL-27 group did not differ significantly from the CLP group. CV3 increased SOD and GSH levels to mitigate ferroptosis, thereby protecting lung tissue from damage (Figure 5F and G).
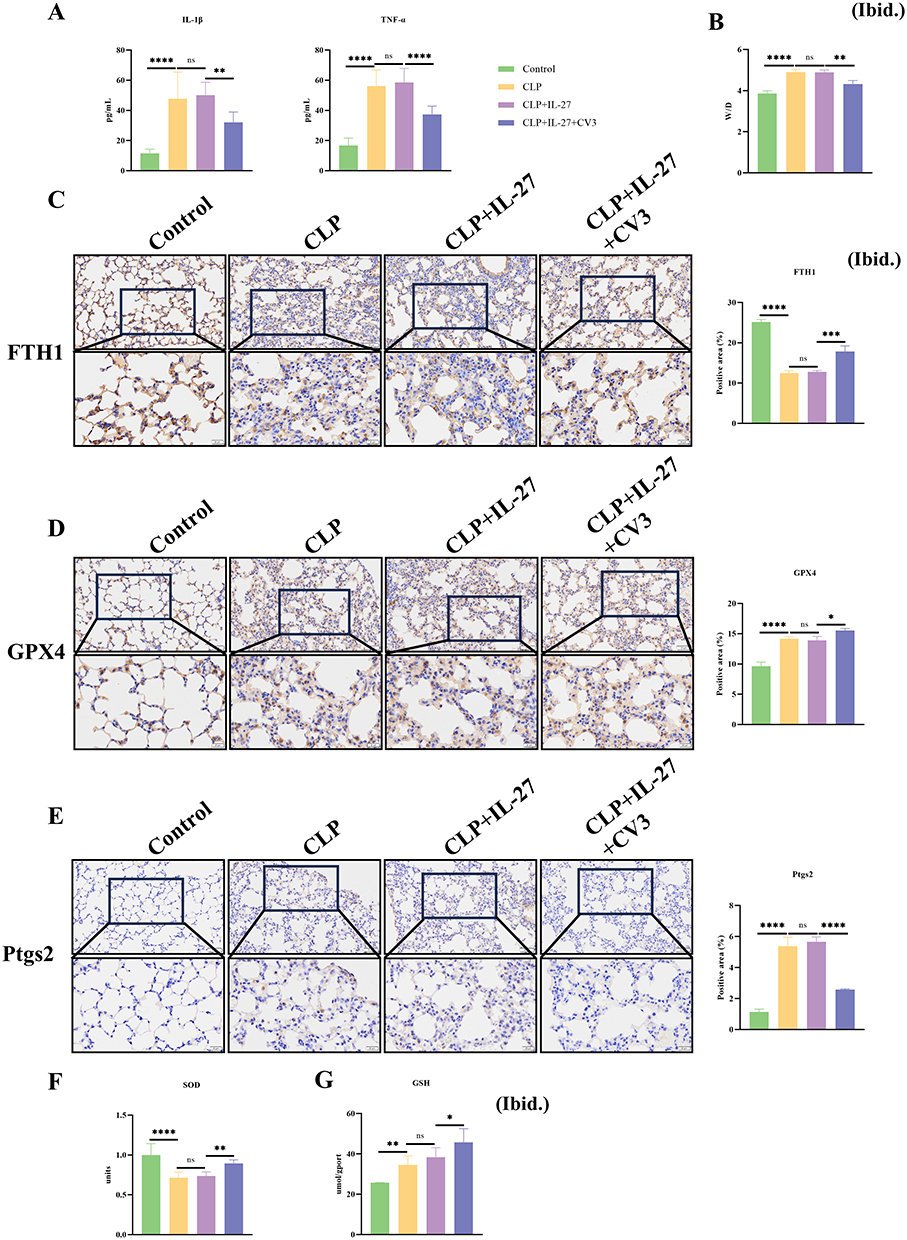 |
Figure 5 CV3 alleviated ARDS in IL-27R−/− mice by attenuating ferritinophagy levels in lung tissue. (A) ELISA of plasma inflammatory factors in IL-27R−/− mice. (B) lung W/D. (C) Representative images and quantitative analysis of FTH1 expression levels by immunohistochemical staining. (D) Representative images and quantitative analysis of GPX4 expression levels by immunohistochemical staining. (E) Representative images and quantitative analysis of Ptgs2 expression levels by immunohistochemical staining. (F and G) The levels of SOD and GSH in lung tissue homogenates were quantified to assess the oxidative stress status. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Scale bar: 20 μm and 50 μm. Abbreviation: n, not significant. |
Discussion
Sepsis-induced ARDS is the primary clinical subtype of ARDS, with significantly higher severity and harm compared to other subtypes.30 Despite considerable advancements in clinical treatment strategies for sepsis-induced ARDS, a subset of patients still exhibits suboptimal therapeutic responses. Therefore, identifying novel therapeutic targets to improve the clinical prognosis of sepsis-induced ARDS has become an urgent issue to be addressed. This study explored the regulatory mechanism of IL-27 in sepsis-induced ARDS, focusing on its role in regulating macrophage ferritinophagy via the NCOA4-FTH1 pathway. The findings of this study indicate that IL-27 can induce macrophage ferritinophagy, which may further exacerbate ferroptosis, amplify the inflammatory response, and ultimately aggravate the pathological damage in sepsis-induced ARDS. These results suggest that IL-27 may play an important role in regulating the pathological progression of sepsis-induced ARDS, providing a novel potential direction for exploring targeted therapeutic strategies for this disease.
Macrophages are central drivers of the pathological progression in sepsis-induced ARDS. Within the pathological microenvironment of sepsis-induced ARDS, macrophages can sense and respond to external stress signals, triggering inflammatory responses that exacerbate lung tissue damage.31,32 Notably, macrophages exhibit functional duality in ARDS: upon exposure of lung tissue to LPS, the total number of macrophages increases significantly, predominantly driven by the elevation of pro-inflammatory M1-type macrophages. These inflammatory macrophages not only serve as the primary source of inflammatory factors (eg, IL-6, IL-1β, TNF-α) but also their released inflammatory factors can regulate the activity of enzymes related to iron metabolism and lipid metabolism, ultimately amplifying the pulmonary inflammatory cascade and alveolar structural damage.33 In contrast, anti-inflammatory M2-type macrophages exert a protective effect in alleviating ARDS-related lung injury by promoting alveolar epithelial repair and tissue regeneration. In this study, Western blot and PCR experiments were performed to verify the regulatory effect of IL-27 on the inflammatory phenotype of macrophages. The combined IL-27 and LPS treatment significantly enhanced the expression of IL-1β and iNOS in macrophages and augmented the secretion of pro-inflammatory cytokines, including IL-1β and TNF-α. Conversely, treatment with CV3 significantly inhibited the inflammatory activation of macrophages. Further mechanistic analysis revealed that combined treatment with IL-27 and LPS induced an imbalance in macrophage phenotypes, particularly promoting polarization toward a pro-inflammatory phenotype and triggering inflammatory responses. This imbalance ultimately amplifies macrophage-mediated inflammatory responses, which may represent one of the crucial mechanisms by which IL-27 exacerbates lung injury in sepsis-induced ARDS.
Ferroptosis is a regulated form of cell death first identified in 2012, characterized by iron-dependent non-apoptotic cell death, which distinguishes it from traditional cell death types such as apoptosis and necrosis.15 Iron overload is one of the most typical pathological features of ferroptosis: abnormal accumulation of intracellular free iron triggers the generation of large amounts of ROS, which can cause oxidative damage to intracellular DNA, proteins, and organelles such as mitochondria, ultimately leading to cell death.34 Previous studies have confirmed that exogenous supplementation of uridine can alleviate sepsis-induced lung injury by inhibiting macrophage ferroptosis. This mechanism is closely related to uridine’s upregulation of Nrf2 expression, thereby activating the Nrf2-dependent antioxidant pathway (eg, inducing the expression of antioxidant genes such as GPX4 and HO1).35,36 Cell experiments showed that combined treatment with IL-27 and LPS further promotes macrophage ferroptosis. Animal experiments similarly revealed that in a sepsis-induced WT mouse model, IL-27 significantly upregulates the expression of Ptgs2 protein (a key marker of ferroptosis) in lung tissue, while reducing the levels of SOD and GSH and downregulating GPX4 protein expression, ultimately significantly activating ferroptosis in lung tissue cells.27 In IL-27R−/− mice, due to the lack of IL-27 receptor, lung injury in the CLP model was alleviated, and GPX expression was higher compared to the control group, which may be attributed to a protective increase in response to mild injury.
It is known that ferritinophagy is a core mechanism regulating macrophage ferroptosis and plays a key regulatory role in the pathological processes of various diseases. However, whether IL-27 participates in the ferroptosis process of sepsis-induced ARDS by regulating ferritinophagy remains unclear. Based on this, this study further investigated the regulatory effects of IL-27 on a sepsis ARDS animal model and an LPS-induced macrophage inflammation model. The results showed that in vivo experiments, IL-27 significantly exacerbated pulmonary pathological damage in septic mice, specifically manifested as enhanced pulmonary inflammatory response and aggravated lung tissue edema. At the mechanistic level, IL-27 promoted ferritinophagy activity in lung tissue cells, accompanied by decreased levels of SOD and GSH, increased intracellular Fe2⁺ concentration, and ultimately led to a significant increase in lung tissue ferroptosis. However, when the ferritinophagy-specific inhibitor CV3 was applied, the activation of ferroptosis in lung tissue was significantly alleviated, and the mediated lung tissue damage was also markedly mitigated. In the LPS-induced macrophage inflammation model, it was further confirmed that IL-27 exacerbates macrophage ferroptosis by promoting ferritinophagy, ultimately inducing macrophage polarization toward the M1 phenotype. CoIP experimental results revealed that, compared to the control group, the IL-27 combined with LPS treatment group exhibited the following characteristics: total FTH1 protein levels were significantly reduced, and the binding level between NCOA4 and FTH1 was markedly increased. These findings indicate enhanced ferritinophagy activity in macrophages of this treatment group, with more NCOA4-FTH1 complexes being transported to lysosomes for degradation, leading to increased intracellular Fe2⁺ concentration and subsequently exacerbating the ferroptosis process. Further mechanistic validation showed that after adding the ferritinophagy inhibitor CV3, the binding between NCOA4 and FTH1 and the subsequent complex transport process were blocked, ferroptosis was effectively inhibited, and the inflammatory state of macrophages was significantly alleviated.
In sepsis-induced ARDS, in addition to immunological mechanisms such as macrophage ferroptosis, the management of endothelial injury and microvascular complications is also critical for clinical outcomes. For example, clinical studies have demonstrated that agents such as iloprost, which possess endothelial-protective and anti-inflammatory properties, can improve oxygenation in ARDS.37
The present study has several noteworthy limitations. First, the 24-hour experimental timeframe, while suitable for assessing acute inflammatory and ferroptotic responses, does not capture longer-term processes such as tissue remodeling or late-stage resolution; future studies should incorporate multiple time points to delineate the dynamics of these pathways throughout the progression of sepsis-associated ARDS. Second, the small sample size per group (n=5) may reduce statistical power, particularly in detecting subtle between-group differences. This work is primarily positioned as a mechanistic exploration and proof-of-concept investigation, aiming to preliminarily elucidate relevant phenotypes and pathways. Furthermore, although CV3 was utilized for targeted pathway modulation with dose optimization and negative controls to minimize non-specific effects, its potential off-target actions cannot be entirely excluded. CV3 may influence other pathways related to macrophage iron metabolism or autophagy, thereby exerting superimposed effects on the observed phenotypes. Thus, the specificity of the related conclusions requires further verification through approaches such as target-specific genetic knockout or overexpression, or the use of more selective pharmacological tools. Finally, the role of NCOA4 demonstrated here is supported mainly by pharmacological and biochemical evidence. While these results provide robust supportive data, the establishment of causality remains confined to the experimental system employed. Further validation using genetic methods (eg, knockout or knockdown models) in future studies will help strengthen this inference and offer deeper mechanistic insights into the function of NCOA4.
Conclusion
Our findings suggest that IL-27 may promote macrophage ferritinophagy through enhancing the NCOA4-FTH1 interaction. This process appears to facilitate the release of intracellular stored iron (Fe2⁺), which could subsequently contribute to elevated ROS generation. The resulting oxidative stress may disturb mitochondrial homeostasis, potentially leading to macrophage ferroptosis via the described cascade. Furthermore, this study supports the possibility that IL-27 exacerbates sepsis-induced ARDS lung injury by modulating the macrophage ferritinophagy-ferroptosis axis. These observations provide preliminary experimental evidence for understanding the pathological mechanisms underlying sepsis-induced ARDS and may offer theoretical insights for future targeted intervention strategies.
Abbreviations
Ams, Alveolar macrophages; ARDS, Acute Respiratory Distress Syndrome; BALF, Bronchoalveolar lavage fluid; BMDMs, bone marrow-derived macrophages; CLP, cecal ligation and puncture; Co-IP, Co-immunoprecipitation; CV3, Compound V3; DAMPs, damage-associated molecular patterns; DMEM, Dulbecco’s Modified Eagle Medium; ELISA, Enzyme-linked immunosorbent assay; FTH1, ferritin heavy chain; GSH Detection of Glutathione; H&E, hematoxylin and eosin; IHC, Immunohistochemistry; IL-27R−/−, IL-27 receptor knockout; LPS, lipopolysaccharide; M-CSF, Macrophage colony-stimulating factor; MDA, Malondialdehyde; NCOA4, Nuclear receptor coactivator 4; PVDF, polyvinylidene fluoride; qPCR, Quantitative PCR; ROS, Reactive oxygen species; SOD, Superoxide Dismutase; TEM, Transmission electron microscopy; WT, wild-type.
Data Sharing Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study protocol received approval from the Research Ethics Committee of the First Affiliated Hospital of Chongqing Medical University (No. IACUC-CQMU-2023-0466). The study was conducted in strict accordance with the Guide for the Care and Use of Laboratory Animals (8th edition, National Research Council, 2011). And this study adhered to the ARRIVE guidelines.
Author Contributions
Minkang Guo conducted the investigation, performed formal analysis, and wrote the original draft. Shihui Lin and Fang Xu provided conceptualization, supervision, and reviewed and edited the manuscript. Meng Xiong, Jindian Shi, and Qiaozhi Peng participated in methodology development, data curation, and validation. All authors reviewed the manuscript. All authors took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
General Program of the National Natural Science Foundation of China [82172161], The Doctoral Program of the First Affiliated Hospital of Chongqing Medical University [CYYY-BSYJSCXXM-202340], General Program of the Natural Science Foundation of Chongqing Municipality [CSTB2024NSCQ-MSX0956] supported this study, and Science and Technology Research Program of Chongqing Municipal Education Commission [KJQN202400101].
Disclosure
The authors declare no competing interests.
References
1. Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA. 2016;315(8):801–16. doi:10.1001/jama.2016.0287
2. Shankar-Hari M, Phillips GS, Levy ML, et al. Developing a new definition and assessing new clinical criteria for septic shock: for the third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA. 2016;315(8):775–787. doi:10.1001/jama.2016.0289
3. Bos LDJ, Ware LB. Acute respiratory distress syndrome: causes, pathophysiology, and phenotypes. Lancet. 2022;400(10358):1145–1156. doi:10.1016/S0140-6736(22)01485-4
4. Ranieri VM, Ranieri VM, Rubenfeld GD, et al. Acute respiratory distress syndrome: the Berlin definition. JAMA. 2012;307(23):2526–2533. doi:10.1001/jama.2012.5669
5. Bellani G, Laffey JG, Pham T, et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA. 2016;315(8):788–800. doi:10.1001/jama.2016.0291
6. Fan E, Brodie D, Slutsky AS. Acute respiratory distress syndrome: advances in diagnosis and treatment. JAMA. 2018;319(7):698–710. doi:10.1001/jama.2017.21907
7. Rubenfeld GD, Caldwell E, Peabody E, et al. Incidence and outcomes of acute lung injury. New Engl J Med. 2005;353(16):1685–1693. doi:10.1056/NEJMoa050333
8. Pflanz S, Timans JC, Cheung J, et al. IL-27, a heterodimeric cytokine composed of EBI3 and p28 protein, induces proliferation of naive CD4+ T cells. Immunity. 2002;16(6):779–790. doi:10.1016/S1074-7613(02)00324-2
9. Wirtz S, Tubbe I, Galle PR, et al. Protection from lethal septic peritonitis by neutralizing the biological function of interleukin 27. J Exp Med. 2006;203(8):1875–1881. doi:10.1084/jem.20060471
10. O’dwyer MJ, Mankan AK, White M, et al. The human response to infection is associated with distinct patterns of interleukin 23 and interleukin 27 expression. Intens Care Med. 2008;34(4):683–691. doi:10.1007/s00134-007-0968-5
11. Hou JH, Gao W. IL-27 regulates autophagy in rheumatoid arthritis fibroblast-like synoviocytes via STAT3 signaling. Immunobiology. 2022;227(4):152241. doi:10.1016/j.imbio.2022.152241
12. Gao F, Yang Y-Z, Feng X-Y, et al. Interleukin-27 is elevated in sepsis-induced myocardial dysfunction and mediates inflammation. Cytokine. 2016;88:1–11. doi:10.1016/j.cyto.2016.08.006
13. Wang X, Liu D, Zhang X, et al. Exosomes from adipose-derived mesenchymal stem cells alleviate sepsis-induced lung injury in mice by inhibiting the secretion of IL-27 in macrophages. Cell Death Discovery. 2022;8(1):18. doi:10.1038/s41420-021-00785-6
14. Stockwell BR, Friedmann Angeli JP, Bayir H, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273–285. doi:10.1016/j.cell.2017.09.021
15. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi:10.1016/j.cell.2012.03.042
16. Mancias JD, Wang X, Gygi SP, et al. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509(7498):105–109. doi:10.1038/nature13148
17. Fang Y, Chen X, Tan Q, et al. Inhibiting ferroptosis through disrupting the NCOA4-FTH1 interaction: a new mechanism of action. ACS Cent Sci. 2021;7(6):980–989. doi:10.1021/acscentsci.0c01592
18. Zhou B, Liu J, Kang R, et al. Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol. 2020;66:89–100. doi:10.1016/j.semcancer.2019.03.002
19. Dowdle WE, Nyfeler B, Nagel J, et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat Cell Biol. 2014;16(11):1069–1079. doi:10.1038/ncb3053
20. Chen X, Kang R, Kroemer G, et al. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol. 2021;18(5):280–296. doi:10.1038/s41571-020-00462-0
21. Matthay MA, Zemans RL, Zimmerman GA, et al. Acute respiratory distress syndrome. Nat Rev Dis Primers. 2019;5(1):18. doi:10.1038/s41572-019-0069-0
22. Evren E, Ringqvist E, Willinger T. Origin and ontogeny of lung macrophages: from mice to humans. Immunology. 2020;160(2):126–138. doi:10.1111/imm.13154
23. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122(3):787–795. doi:10.1172/JCI59643
24. Xu W, Wu Y, Wang S, et al. Melatonin alleviates septic ARDS by inhibiting NCOA4-mediated ferritinophagy in alveolar macrophages [J]. Cell Death Discovery. 2024;10(1):253. doi:10.1038/s41420-024-01991-8
25. Fuhrmann DC, Mondorf A, Beifuß J, et al. Hypoxia inhibits ferritinophagy, increases mitochondrial ferritin, and protects from ferroptosis. Redox Biol. 2020;36:101670. doi:10.1016/j.redox.2020.101670
26. Mikawa K, Nishina K, Takao Y, et al. ONO-1714, a nitric oxide synthase inhibitor, attenuates endotoxin-induced acute lung injury in rabbits. Anesthesia Analg. 2003;97(6):1751–1755. doi:10.1213/01.ANE.0000086896.90343.13
27. Xiong M, Luo R, Zhang Z, et al. IL-27 regulates macrophage ferroptosis by inhibiting the Nrf2/HO1 signaling pathway in sepsis-induced ARDS. Inflamm Res. 2025;74(1):39. doi:10.1007/s00011-024-01986-2
28. Hou W, Xie Y, Song X, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12(8):1425–1428. doi:10.1080/15548627.2016.1187366
29. Wu H, Liu Q, Shan X, et al. ATM orchestrates ferritinophagy and ferroptosis by phosphorylating NCOA4. Autophagy. 2023;19(7):2062–2077. doi:10.1080/15548627.2023.2170960
30. Grange C, Bussolati B. Extracellular vesicles in kidney disease. Nat Rev Nephrol. 2022;18(8):499–513. doi:10.1038/s41581-022-00586-9
31. Li R, Li X, Zhao J, et al. Mitochondrial STAT3 exacerbates LPS-induced sepsis by driving CPT1a-mediated fatty acid oxidation. Theranostics. 2022;12(2):976–998. doi:10.7150/thno.63751
32. Yang L, Xie M, Yang M, et al. PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis. Nat Commun. 2014;5(1):4436. doi:10.1038/ncomms5436
33. Murray PJ, Allen JE, Biswas SK, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41(1):14–20. doi:10.1016/j.immuni.2014.06.008
34. Li S, Zheng L, Zhang J, et al. Inhibition of ferroptosis by up-regulating Nrf2 delayed the progression of diabetic nephropathy [J]. Free Radic Biol Med. 2021;162:435–449. doi:10.1016/j.freeradbiomed.2020.10.323
35. Zheng J, Conrad M. The metabolic underpinnings of ferroptosis. Cell Metab. 2020;32(6):920–937. doi:10.1016/j.cmet.2020.10.011
36. Lai K, Song C, Gao M, et al. Uridine alleviates sepsis-induced acute lung injury by inhibiting ferroptosis of macrophage. Int J Mol Sci. 2023;24(6):5093. doi:10.3390/ijms24065093
37. Küçük RS, Uluç K, Çolakoğlu ŞM, et al. The effect of using iloprost on prognosis in COVID-19 patients with ARDS: a retrospective clinical study. Eur Rev Med Pharmacol Sci. 2023;27(9):4269–4279. doi:10.26355/eurrev_202305_32337
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Modified Xi-Jiao-Di-Huang Decoction Alleviates Sepsis via Regulating Macrophage Polarization by Inhibiting the PIM2/NF-κB Pathway
Ge F, Tian F, Zhu Y, Yan Q, Sun Q, Lu J
Journal of Inflammation Research 2025, 18:5017-5030
Published Date: 13 April 2025
Association Between Ferritinophagy and M1 Macrophage Polarization During the Acute Phase of Childhood Allergic Asthma
Li J, Wang X, Shi X, Jiang Y, Shen R
Journal of Inflammation Research 2026, 19:569956
Published Date: 18 February 2026
Circulating and Respiratory Biomarkers in Sepsis-Induced ARDS: Diagnostic and Prognostic Insights – A Narrative Review
Muhoza BG, Liu S, Niyonkuru E, Sun T
Journal of Inflammation Research 2026, 19:571504
Published Date: 25 March 2026
Exosomal miR-148a-3p from LPS-Activated Macrophages Promotes M1 Polarization and Ferroptosis-Related Characteristics of Recipient Macrophages by Reducing SLC7A11
Liang J, Cai Y, Shao Y
Infection and Drug Resistance 2026, 19:590998
Published Date: 20 May 2026