Back to Journals » Infection and Drug Resistance » Volume 19
Exosomal miR-148a-3p from LPS-Activated Macrophages Promotes M1 Polarization and Ferroptosis-Related Characteristics of Recipient Macrophages by Reducing SLC7A11
Authors Liang J ![]() , Cai Y, Shao Y
, Cai Y, Shao Y ![]()
Received 22 December 2025
Accepted for publication 13 May 2026
Published 20 May 2026 Volume 2026:19 590998
DOI https://doi.org/10.2147/IDR.S590998
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Oliver Planz
Jun Liang,1,2 Yingli Cai,1,2 Yiming Shao3
1Clinical Medical College, Jinan University, Guangzhou, Guangdong, 510632, People’s Republic of China; 2Department of Emergency, The First People’s Hospital of Zhaoqing, Zhaoqing, Guangdong, 526060, People’s Republic of China; 3Dongguan Key Laboratory of Sepsis Translational Medicine, The Intensive Care Unit, The First Dongguan Affiliated Hospital, Guangdong Medical University, Dongguan, Guangdong, 523710, People’s Republic of China
Correspondence: Yiming Shao, Dongguan Key Laboratory of Sepsis Translational Medicine, The Intensive Care Unit, The First Dongguan Affiliated Hospital, Guangdong Medical University, Jiaoping Road 42, Tangxia Town, Dongguan, 523710, Guangdong, People’s Republic of China, Email [email protected]
Background: Gram-negative bacterial infection is a major cause of severe inflammatory disorders and cellular damage, with lipopolysaccharide (LPS) as a key pathogenic trigger. Macrophages are central players in LPS-induced inflammatory cascades, yet the mechanisms by which macrophage-derived exosomes propagate inflammatory signals and cellular injury remain elusive. This study aimed to investigate whether LPS-activated macrophage exosomes drive M1 polarization and ferroptosis-related characteristics in recipient macrophages.
Methods: RAW264.7 macrophages were treated with LPS. Exosomes were extracted through ultracentrifugation and their characteristics were determined utilizing Western blot, transmission electron microscopy, and nanoparticle tracking analysis. MicroRNA levels were quantified by droplet digital PCR. The functional effects of exosomes and miR-148a-3p inhibitor on recipient macrophages were evaluated via quantitative real-time PCR, Western blot, enzyme-linked immunosorbent assay, malondialdehyde (MDA) detection and ferrostatin-1 (Fer-1) rescue assay. Dual-luciferase reporter assay validated direct targeting relationships.
Results: LPS directly induced M1 polarization and pro-inflammatory cytokine secretion in RAW264.7 cells. Exosomes from LPS-stimulated macrophages were enriched with miR-148a-3p and promoted M1 polarization and ferroptosis-related characteristics in recipient cells. Inhibition of miR-148a-3p and Fer-1 treatment effectively negated the LPS-exosome-mediated effects. Mechanistically, miR-148a-3p directly interacted with and inhibited SLC7A11.
Conclusion: Exosomal miR-148a-3p derived from LPS-activated macrophages promotes M1 polarization and ferroptosis-related characteristics in recipient macrophages by reducing SLC7A11. This finding uncovers a novel LPS-induced inflammatory signaling pathway in macrophage–macrophage communication and suggests a potential candidate target for intervention in Gram-negative bacteria–related inflammatory injury. The diagram illustrates the interaction between LPS-activated macrophages and recipient macrophages. On the left, LPS-activated macrophages release exosomes, which are depicted as small particles moving towards the recipient macrophages on the right. The exosomes contain exosomal miR-148a-3p, which binds to SLC7A11 in the recipient macrophages. This binding promotes ferroptosis and M1 polarization, leading to the upregulation of iNOS, TNF-alpha, IL-1beta and IL-6. The diagram shows the processes of upregulated and downregulated expression, indicating the influence of exosomes on gene expression in recipient macrophages.LPS-activated macrophages release exosomes, inducing ferroptosis and M1 polarization in recipients.
Keywords: exosome, miR-148a-3p, macrophage polarization, ferroptosis-related characteristics, LPS
Introduction
Gram-negative bacterial infection triggers robust host inflammatory responses mediated by pattern recognition receptors sensing lipopolysaccharide (LPS), leading to excessive immune activation and cellular dysfunction.1–3 Macrophages, as pivotal innate immune effector cells, undergo phenotypic polarization into pro-inflammatory M1 macrophages upon LPS stimulation, and secrete large amounts of inflammatory cytokines, amplifying local and systemic inflammatory reactions.4–6
Ferroptosis, characterized as a regulated mode of cellular demise reliant on iron and propelled by the buildup of lipid peroxides, is progressively implicated in sepsis and related conditions.7–9 The cystine/glutathione (GSH) axis is central to ferroptosis regulation, with SLC7A11, which serves as a subunit of the system Xc−, being critical for the uptake of cystine and subsequent GSH synthesis. Meanwhile, GPX4 dependents on GSH to eliminate lipid peroxides.10,11 LPS stimulation has been shown to induce M1 polarization accompanied by ferroptotic features, forming a vicious cycle that exacerbates sepsis progression.12–15
Exosomes (30–150 nm extracellular vesicles) mediate intercellular communication by transporting proteins, lipids, and RNAs, and are key regulators of sepsis pathology.16,17 Previous studies have shown that the extracellular vesicles from the plasma of patients suffering from sepsis-induced acute lung injury regulate macrophage polarization and ferroptosis.18 Additionally, exosomes derived from LPS-activated macrophages can trigger ferroptosis in alveolar epithelial cells.19,20 However, the mechanisms by which they disseminate “M1 polarization–ferroptosis” signals among homologous recipient macrophages remain unclear.
Accumulating evidence has highlighted the pivotal role of exosomal miRNAs in orchestrating immune cell function during sepsis, a severe systemic inflammatory syndrome often initiated by Gram-negative bacterial infection.21,22 Plasma exosomal miR-17-5p has been identified as a modulator of macrophage polarization in sepsis-induced lung injury,23 while neutrophil-derived exosomal miR-30d-5p drives M1 macrophage polarization and primes pyroptosis in the context of sepsis-related acute lung injury.24 Beyond immune cell sources, endothelial cell-derived exosomal miR-184-3p, under the modulation of heme oxygenase-1, fine-tunes macrophage polarization and alleviates sepsis-induced lung injury,25 and LPS-preconditioned mesenchymal stem cell-secreted exosomal miR-150-5p promotes M2 macrophage polarization to exert protective effects against sepsis.26 Despite these insightful findings, the majority of current research has focused on the cross-cell-type regulatory effects of exosomal miRNAs, with scarce attention paid to the biological significance of homologous cell communication mediated by exosomal cargos. This knowledge gap is particularly prominent in Gram-negative bacterial infection, where the paracrine crosstalk between LPS-activated macrophages and naive homologous macrophages via exosomal miRNAs remains largely uncharacterized.
miR-148a-3p has recently emerged as a potential regulatory factor in sepsis and ferroptosis: it modulates macrophage polarization in murine sepsis models and promotes ferroptosis by targeting SLC7A11 in other cell types.27,28 However, whether miR-148a-3p is packaged into exosomes of LPS-activated macrophages, and if this exosomal miRNA acts as a functional messenger to drive concurrent M1 polarization and ferroptosis in recipient homologous macrophages during Gram-negative bacterial infection, has not been elucidated. This dual process is critical for the amplification of inflammatory injury in Gram-negative bacterial infection.
In this study, our objective was to tackle these unaddressed research gaps by isolating exosomes derived from LPS-activated murine macrophages (LPS-exo) and exploring their functional impacts on M1 polarization and ferroptosis-related characteristics in recipient macrophages. Additionally, we aimed to elucidate the role of exosomal miR-148a-3p in mediating these effects and to validate its direct targeting of SLC7A11. Our research centers on homologous macrophage-to-macrophage communication, a regulatory axis that has been overlooked in the context of Gram-negative bacterial infections, and seeks to unveil a novel exosomal miRNA-mediated signaling pathway that orchestrates M1 polarization and ferroptosis-related characteristics. This investigation not only bridges the knowledge gap concerning the function of exosomal miRNAs during homologous immune cell interactions in vitro but also offers a potential molecular target for intervening in inflammatory injury triggered by Gram-negative bacterial infections at the cellular level, setting it apart from prior sepsis studies that primarily focus on cross-cell-type regulation and modulation of single biological processes.
Materials and Methods
Cell Lines and Their Culture Conditions
RAW264.7 and HEK293T were procured from the ATCC (Manassas, VA, USA) and maintained in DMEM (Gibco, Thermo Fisher Scientific, Waltham, MA, USA). The culture medium was fortified with 10% FBS (Sigma-Aldrich, St. Louis, MO, USA), along with 1% penicillin-streptomycin (HyClone, Logan, UT, USA). The cell cultures were sustained at a temperature of 37°C in a humidified environment enriched with 5% carbon dioxide.
Isolation and Culture of Mouse Bone Marrow-Derived Macrophages (BMDMs)
Primary BMDMs were isolated from 6–8-week-old C57BL/6 mice. Briefly, femurs and tibias were dissected, and bone marrow was flushed with sterile PBS. After red blood cell lysis, cells were filtered and cultured in DMEM containing 10% FBS and 20 ng/mL M-CSF for 7 days to induce macrophage differentiation. BMDMs were used for exosome extraction and miRNA detection.
Cell Viability Assessment via CCK-8 Method
RAW264.7 cells were evenly distributed into 96-well culture plates at a seeding density of 5 × 103 cells per individual well, followed by an overnight incubation period to allow for cell adherence and initial growth. Subsequently, the cells were subjected to treatment with LPS at final concentrations of 0, 0.5, 1, or 10 µg/mL for 24 hours. Following the treatment phase, 10 µL of CCK-8 reagent was introduced into each sample, then the plates were placed back into the incubator at 37°C for an additional 2-hour incubation. Finally, a microplate reader from BioTek (Winooski, VT, USA) was employed to measure each sample’s absorbance at 450 nm wavelength.
RNA Extraction and Complementary DNA (cDNA) Generation
Total RNA was isolated from RAW264.7 cells utilizing TRIzol (Invitrogen, Carlsbad, CA), following the protocol provided by the manufacturer. The assessment of purity and concentration of RNA was conducted with a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific). The extraction of exosomal RNA was performed utilizing the ExoRNeasy Serum/Plasma Kit (Qiagen, Hilden, Germany). To ensure the removal of any remaining genomic DNA contamination, the samples were subsequently treated with DNase I (Qiagen). To synthesize cDNA from cellular RNA, 1 µg of cellular RNA was subjected to reverse transcription utilizing the PrimeScript RT Reagent Kit (Takara, Tokyo, Japan) with random primers for mRNA analysis. In the case of miRNA analysis, 500 ng of either exosomal or cellular RNA underwent reverse-transcribed using the miScript Reverse Transcription Kit (Qiagen), which incorporates miRNA-specific stem-loop primers for targeted amplification.
Quantitative Real-Time PCR (RT-qPCR)
RT-qPCR was performed using Platinum ® SYBR ® Green qPCR SuperMix-UDG (Invitrogen). The thermal cycling parameters followed those described previously.29 The expression of mRNA and miRNA was normalized to GAPDH and U6 small nuclear RNA, respectively. The relative expression levels of the target genes were then calculated employing the 2^(-ΔΔCt) method. The specific primer sequences utilized in the RT-qPCR experiments are detailed in Table 1.
 |
Table 1 The Primer Sequences Used for RT-qPCR |
Droplet Digital PCR (ddPCR)
The absolute quantification of candidate miRNAs by ddPCR used Forevergen Co., Ltd.’s (Guangzhou, China) instruments. Each reaction mixture contained 10 μL of ultra-sensitive ddPCR premix (Forevergen), 10 μM of forward and reverse primer, 6 µL RNase-free water and 2 μL cDNA, with a total volume of 20 μL. Droplets were generated with the MicroDrop-100A and transferred to a 96-well PCR plate. The ddPCR program as previously reported.30 Droplet analysis was done using the MicroDrop-100B Biochip Reader, and data were processed with QuantDrop V1 software for accurate quantification.
Western Blotting
On ice, purified exosomes and RAW264.7 cell pellets were subjected to lysis in RIPA buffer that had been fortified with 1% phenylmethylsulfonyl fluoride, with both reagents sourced from Beyotime (Shanghai, China). The cell lysates underwent centrifuged at 12,000 × g for 15 minutes at 4°C to gather the supernatants. Protein concentrations were measured by the BCA Protein Assay Kit (Beyotime). Equal quantities of protein (30 µg per lane for cells, 40 µg per lane for exosomes) were subjected to separation using either 10% or 12% SDS-PAGE, followed by transfer onto polyvinylidene difluoride (PVDF) membranes (Millipore, Burlington, MA, USA). The membranes underwent blocking in 5% non-fat milk dissolved in TBST for one hour at ambient temperature. This was succeeded by an overnight incubation with primary antibodies, with the specific dilutions detailed in Table 2. After three Subsequent to three rounds of washing with TBST, the membranes were treated with secondary antibodies for one hour at ambient temperature. The visualization of protein bands was accomplished using an Enhanced Chemiluminescence (ECL) Detection Kit (Millipore), and quantification was carried out utilizing ImageJ software (NIH, Bethesda, MD, USA). GAPDH was utilized as the loading control for cellular proteins, while the analysis of exosomal proteins was conducted to determine the presence or absence of specific markers without utilizing an internal control.
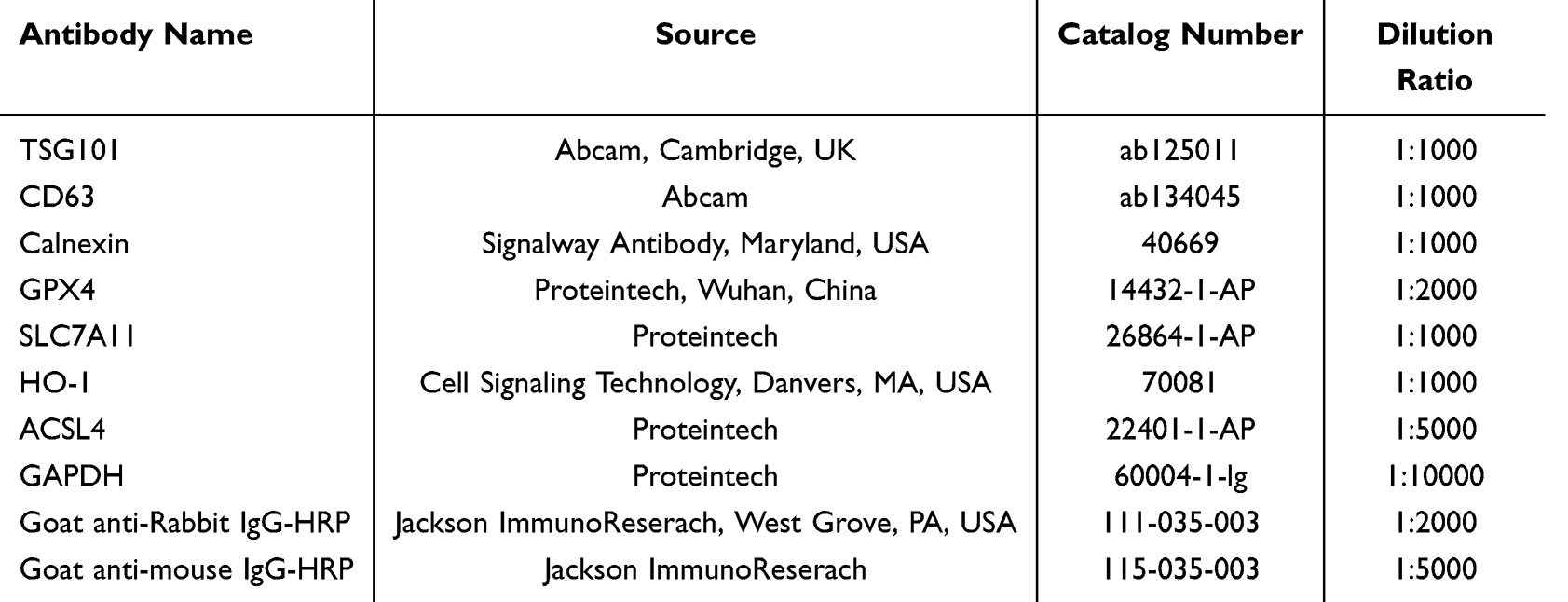 |
Table 2 Primary and Secondary Antibodies Used in Western Blotting |
Enzyme-Linked Immunosorbent Assay (ELISA)
The concentrations of pro-inflammatory cytokines were quantified utilizing commercially available ELISA kits (Cusabio Biotech, Wuhan, China) according to the protocols provided by companies. The absorbance values were recorded at a wavelength of 450 nm.
Isolation and Characterization of Exosomes
RAW264.7 cells were cultured until they reached approximately 80% confluence. The culture medium was then substituted with 10 mL of serum-free DMEM, supplemented with 10 µg/mL LPS for the experimental group, while the control group received an equivalent volume of phosphate-buffered saline (PBS). Following incubation for 16 hours, the supernatants underwent a series of ultracentrifugation steps to isolate exosomes as previously described.31 For transmission electron microscopy (TEM) analysis, exosomes were diluted 1:10 with PBS, deposited onto carbon-coated copper grids, air-dried for 5 minutes, and stained with 2% phosphotungstic acid (pH 7.0) for 3 minutes before being examined under a TEM at 80 kV. For nanoparticle tracking analysis (NTA), exosome suspensions were diluted with PBS, and the size distribution and concentration were assessed using the ZetaView PMX 110 system (ZetaView 8.04.02 software). Western blotting was conducted to identify exosomal markers (TSG101, CD63) alongside a negative control marker (Calnexin), as described above.
Exosome Uptake Assay and Effects of LPS-Exo on RAW264.7 Cells
Exosomes (20 µg) were labeled with 10 µM PKH67 dye (Sigma-Aldrich) for a period of 5 minutes. The labeling reaction was halted by incorporating 2 mL of exosome-depleted FBS, followed by the elimination of any unbound dye through ultracentrifugation. The labeled exosomes (20 µg) were then introduced to RAW264.7 cells and instigated for a period of 24 hours. After the incubation period, the cells were fixed using 4% paraformaldehyde and subsequently stained with DAPI (1 µg/mL; Sigma-Aldrich) to enable visualization the nuclei, after which observations were conducted using a fluorescence microscope. To assess the impact of LPS-exo, 2 × 106 RAW264.7 cells were co-cultured with 100 µg/mL exosomes for a period of 24 hours. Post co-culture, both cells and supernatants were harvested for further experimental analyses.
Cell Transfection
2 × 106 RAW264.7 cells were cultured into 6-well plates until 70–80% confluency. Transient transfection was conducted utilizing Lipofectamine 3000 reagent (Thermo Fisher Scientific), incorporating 150 pmol of either mmu-miR-148a-3p inhibitor or a negative control (NC) inhibitor (Ambion, Life Technologies, USA) for each well. After transfection for 24 hours, cells were harvested for subsequent assay.
Rescue Experiments
For ferroptosis-related functional rescue experiments, cells were pretreated with ferrostatin-1 (Fer-1, 1 μM, Sigma-Aldrich) for 2 hours, followed by co-incubation with 100 μg/mL LPS-exo for an additional 24 hours prior to sample collection for related index detection.
Malondialdehyde (MDA) Detection
Intracellular MDA levels, a marker of lipid peroxidation, were determined using a commercial MDA assay kit (Beyotime). Briefly, cells were collected and lysed, and supernatants were incubated with the working solution. The absorbance at 532 nm was measured, and MDA concentrations were calculated based on a standard curve.
Dual-Luciferase Reporter Assay
The potential interaction sites for mmu-miR-148a-3p within the 3′ untranslated region (3′UTR) of SLC7A11 were identified utilizing TargetScanHuman (https://www.targetscan.org/). The wild-type (WT) binding sequence “UGCACUG” and a corresponding mutated (MUT) sequence “ACGUGAC” were subsequently cloned into the pmirGLO vector (Promega, Madison, WI, USA) (IGE Biotechnology Co., Ltd., Guangzhou, China). Following this, HEK293T cells were co-transfected with 0.5 µg of either the WT or MUT plasmid alongside 50 nM of mmu-miR-148a-3p mimics or negative control (NC) mimics (Ambion) using Lipofectamine 3000 as the transfection reagent. After a 48-hour incubation period, the cells were lysed using Passive Lysis Buffer (Promega), and the activities of firefly and renilla luciferases were quantified employing the Dual-Luciferase Reporter Assay System (Promega). Firefly luciferase activity was normalized to renilla luciferase activity, and the relative luciferase ratio was calculated.
Statistical Analysis
All data were first tested for normality of distribution using Shapiro–Wilk test. Data are presented as the mean ± standard error of the mean (SEM). Statistical evaluations were performed utilizing GraphPad Prism 7.0 (GraphPad Software, San Diego, CA, USA). For comparisons among three or more groups, one-way ANOVA followed by Bonferroni’s post-hoc test was used only when data conformed to normal distribution. A p-value of less than 0.05 was deemed statistically significant.
All experiments were performed with at least 3 independent biological replicates (n=3).
Results
LPS Induces M1 Polarization and Inflammatory Response in RAW264.7 Macrophages
To investigate the direct activating effects of LPS on macrophages, RAW264.7 cells were treated with varying LPS concentrations. Our findings revealed that LPS not only enhanced cell proliferation (Figure 1A) and triggered a morphological shift towards a spindle-shaped phenotype (Figure 1B), but also markedly elevated the mRNA expression (Figure 1C–E) and protein release (Figure 1F-H) of TNF-α, IL-1β, and IL-6. Furthermore, LPS challenge resulted in the upregulation of iNOS and concurrent downregulation of Arg-1 (Figure 1I and J). These collective observations indicated that LPS effectively provoked macrophage activation, instigating a robust inflammatory response and driving polarization towards the M1 phenotype.
 |
Figure 1 LPS induces M1 polarization and inflammatory response in RAW264.7 macrophages. (A) Cell viability was evaluated using the CCK-8 assay (n = 3) in RAW264.7 cells following 24-hour exposure to LPS at concentrations from 0 to 10 μg/mL. One-way ANOVA was applied, and significance levels compared to 0 μg/mL LPS are indicated as ***p < 0.001, ****p < 0.0001. (B) Morphological changes observed in RAW264.7 cells following 24-hour LPS exposure (scale bar = 200 μm). (C–E) The mRNA levels of pro-inflammatory cytokines in RAW264.7 cells treated with LPS for 6 hours and 16 hours were quantified through qRT-PCR (n = 3). (F–H) ELISA (n = 3) was employed to measure cytokine secretion levels in the culture supernatants following LPS treatment for 6 and 16 hours. (I and J) The expression levels of iNOS and Arg-1 in RAW264.7 cells subjected to LPS for 6 hours and 16 hours were analyzed through qRT-PCR (n = 3). Statistical analysis for (C–J) was performed using one-way ANOVA. Significance levels compared to 0 μg/mL LPS for 6 hours are represented as follows: *p < 0.05; **p < 0.01; *** p < 0.001; **** p < 0.0001; while “ns” indicates not significant. Significance levels compared to 0 μg/mL LPS for 16 hours are represented as follows: #p < 0.05; ##p < 0.01; ###p < 0.001; ####p < 0.0001. |
Macrophages Stimulated by LPS Secrete Exosomes Enriched with miR-148a-3p
We subsequently isolated and characterized exosomes from cell culture supernatants. TEM revealed that the isolated vesicles exhibited the classic cup-shaped morphology (Figure 2A). NTA analysis defined their size distribution and concentration profiles (Figure 2B), while Western blotting confirmed the presence of exosomal marker proteins, specifically TSG101 and CD63, alongside the absence of the negative marker Calnexin (Figure 2C), verifying the acquisition of high-purity exosomes. Internalization assays using PKH67 labeling demonstrated that these exosomes were efficiently taken up by recipient RAW264.7 cells (Figure 2D). ddPCR revealed that miR-148a-3p was enriched by 4.3-fold and miR-128-3p by 3.5-fold in exosomes derived from LPS-stimulated RAW264.7 cells (LPS-exo) compared with PBS-exo (Figure 2E). Subsequent qRT-PCR further confirmed that miR-148a-3p was effectively delivered into and upregulated in recipient macrophages, whereas miR-128-3p exhibited no significant alteration in recipient cells (Figure 2F). Consistently, miR-148a-3p was also significantly enriched by 3.0-fold in LPS-exo isolated from primary mouse BMDMs (Figure 2G), in line with observations in RAW264.7 cells. Collectively, these results demonstrate that LPS stimulation remodels the miRNA cargo of macrophage-derived exosomes and identifies miR-148a-3p as a key transferable effector molecule within LPS-exo.
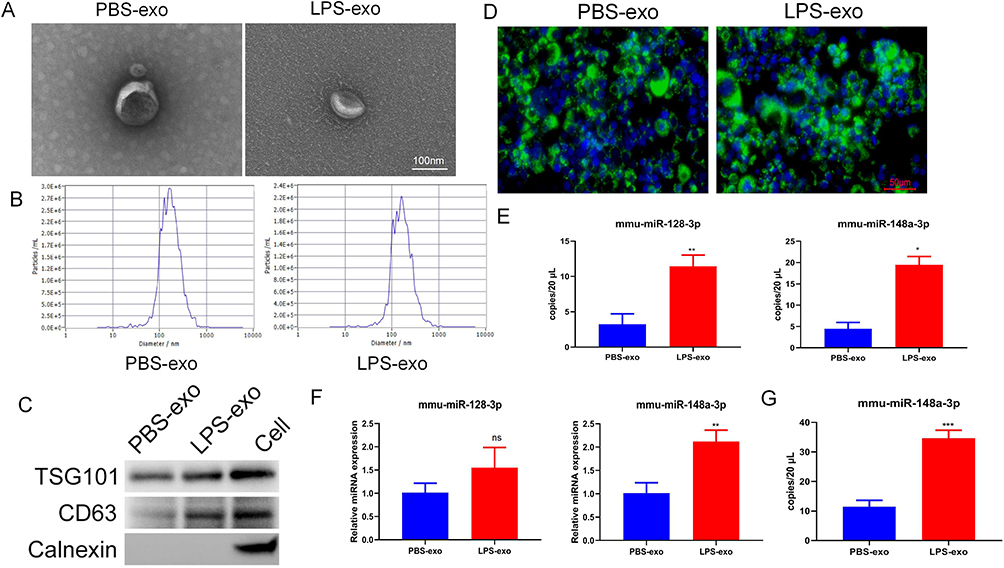 |
Figure 2 Macrophages stimulated by LPS secrete exosomes enriched with miR-148a-3p. (A) A representative TEM image showing the morphology of isolated exosomes (scale bar = 100 nm). (B) Exosome size distribution and concentration were analyzed by NTA. (C) Exosomal markers, specifically TSG101 and CD63, along with the negative control marker Calnexin were detected by Western blotting. (D) Fluorescence imaging of RAW264.7 cells after a 24-hour incubation with 20 μg of PKH67-labeled exosomes (green: PKH67-labeled exosomes; blue: DAPI-stained nuclei; scale bar = 50 μm). (E) The absolute quantification of candidate miRNAs in exosomes isolated from RAW264.7 cells treated with PBS or LPS was performed using ddPCR (n = 3). (F) The relative expression levels of candidate miRNAs in recipient RAW264.7 cells post-exosome uptake were determined via qRT-PCR (n = 3). (G) The absolute quantification of candidate miRNAs in exosomes isolated from BMDMs treated with PBS or LPS was performed using ddPCR (n = 3). For (E and G), statistical analysis was performed using Student’s t-test. Significance levels, in comparison to the PBS-exo group, are indicated as follows: *p < 0.05; **p < 0.01; ***p < 0.001; ns, not significant. |
Macrophage-Derived LPS-Exo Instigates M1 Polarization and Ferroptosis-Related Characteristics in Recipient Cells
To delineate the biological functions of LPS-exo, RAW264.7 cells were subjected to treatment with the purified vesicles. As illustrated in Figure 3, compared to PBS-exo treatment, exposure to LPS-exo significantly upregulated iNOS and downregulated Arg1 at the mRNA level (Figure 3A). At the same time, it enhanced the production of TNF-α, IL-1β, and IL-6, with their expression being elevated at both transcriptional (Figure 3B) and translational (Figure 3C) levels. More importantly, LPS-exo significantly upregulated ACSL4/HO-1 and downregulated SLC7A11/GPX4 at mRNA and protein levels (Figure 3D and E), and increased intracellular MDA content (Figure 3F). These findings demonstrated that LPS-exo alone is sufficient to function as an effector, inducing concurrent M1 polarization and ferroptosis-related characteristics in recipient macrophages.
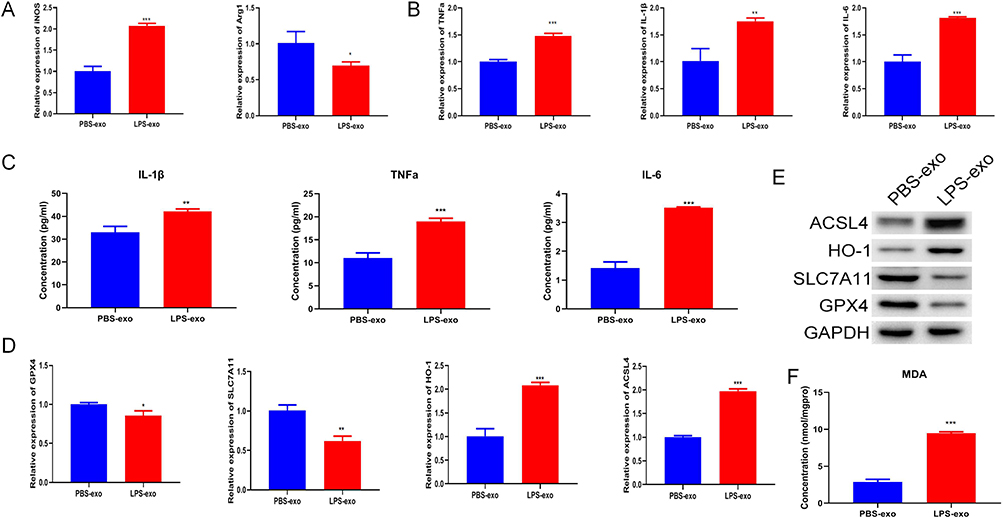 |
Figure 3 Macrophage-derived LPS-exo instigates M1 polarization and ferroptosis-related characteristics in recipient cells. (A) M1/M2 polarization markers in RAW264.7 cells treated with 100 μg/mL exosomes for 24 hours were quantified using qRT-PCR (n = 3). (B) The mRNA levels of inflammatory cytokines in RAW264.7 cells following exosome treatment were analyzed using qRT-PCR (n = 3). (C) Levels of secreted cytokines in culture supernatants after exosome treatment were measured by ELISA (n = 3). (D) mRNA expression of ferroptosis-related genes (GPX4, SLC7A11, HO-1, ACSL4) in RAW264.7 cells treated with exosomes was evaluated via qRT-PCR (n = 3). (E) Protein levels of ferroptosis-related markers in RAW264.7 cells post-exosome treatment were detected by Western blotting. (F) The MDA concentration in RAW264.7 cells following exosome treatment was assessed (n = 3). Statistical analysis for (A–D and F) was performed using Student’s t-test. Significance levels relative to the PBS-exo group are marked as: *p < 0.05; **p < 0.01; ***p < 0.001. |
Fer-1 Inhibits LPS-Exo-Induced M1 Polarization and Ferroptosis-Related Characteristics
To further determine whether ferroptosis-related processes are involved in LPS-exo-mediated macrophage activation, recipient RAW264.7 cells were co-treated with Fer-1, a classical ferroptosis inhibitor. Our results showed that Fer-1 treatment significantly reversed the LPS-exo-mediated dysregulation of M1/M2 polarization markers (Figure 4A). Meanwhile, Fer-1 markedly suppressed the elevated mRNA expression of pro-inflammatory cytokines induced by LPS-exo (Figure 4B). Furthermore, Fer-1 restored the mRNA expression levels of ferroptosis-related genes (Figure 4C), reduced intracellular MDA content (Figure 4D), and reversed the abnormal protein expression of ferroptosis-associated molecules (Figure 4E). Collectively, these data demonstrate that Fer-1 effectively rescues LPS-exo-induced M1 polarization and ferroptosis-related characteristics in recipient macrophages.
 |
Figure 4 Fer-1 inhibits LPS-exo-induced M1 polarization and ferroptosis-related characteristics. (A) The expression levels of M1/M2 macrophage markers in RAW264.7 cells treated with PBS-exo, LPS-exo or LPS-exo + Fer-1 were analyzed by qRT-PCR (n = 3). (B) The mRNA expression of inflammatory cytokines in the above-treated cells was determined by qRT-PCR (n = 3). (C) The transcript levels of ferroptosis-related genes were measured by qRT-PCR (n = 3). (D) The MDA concentration in RAW264.7 cells under the above treatments was detected (n = 3). (E) The protein expression levels of ferroptosis-related markers were examined by Western blotting. Statistical analysis was performed by one-way ANOVA. Compared with the LPS-exo group, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. |
Inhibition of miR-148a-3p Reverses LPS-Exo-Induced M1 Polarization and Ferroptosis-Related Characteristics
To ascertain the pivotal effect of miR-148a-3p in mediating the influences of LPS-exo, we utilized a miR-148a-3p inhibitor. The efficiency of the knockdown was validated through qRT-PCR (Figure 5A). Subsequent functional rescue experiments demonstrated that suppressing miR-148a-3p effectively counteracted the LPS-exo-driven alterations, including M1/M2 polarization balance (Figure 5B), inflammatory cytokines production and secretion (Figure 5C and D), aberrant mRNA and protein expression of ferroptosis-associated molecules (GPX4, SLC7A11, HO-1, ACSL4) (Figure 5E and F) levels, as well as intracellular MDA levels (Figure 5G). In summary, miR-148a-3p served as a crucial effector molecule responsible for the pro-M1 polarization and pro-ferroptosis-related effects elicited by LPS-exo.
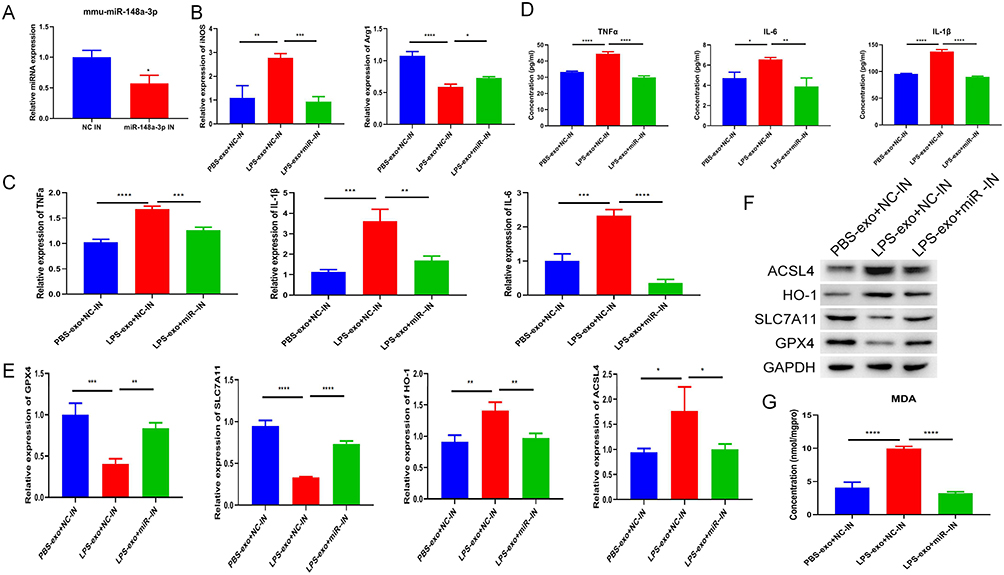 |
Figure 5 Inhibition of miR-148a-3p reverses LPS-exo-induced M1 polarization and ferroptosis-related characteristics. (A) The knockdown efficiency of miR-148a-3p in RAW264.7 cells transfected with the inhibitor for 48 hours was confirmed by qRT-PCR (n = 3). (B) The expression levels of M1/M2 macrophage markers in cells that underwent transfection with the inhibitor and were subsequently treated with exosomes were analyzed via qRT-PCR (n = 3). (C) The mRNA expression of inflammatory cytokines in cells subjected to inhibitor transfection and exosome treatment was quantified using qRT-PCR (n = 3). (D) The production levels of cytokines in culture supernatants under inhibitor transfection and exosome treatment conditions were assessed through ELISA (n = 3). (E) Following inhibitor transfection and exosome treatment, the transcript levels of ferroptosis-related genes were evaluated using qRT-PCR (n = 3). (F) The protein expression levels of markers indicative of ferroptosis-related characteristics were analyzed by Western blotting following inhibitor transfection and exosome treatment. (G) The MDA concentration in RAW264.7 cells following inhibitor transfection and exosome treatment was measured (n = 3). Statistical analysis for all above experiments was performed as follows: For (A), Student’s t-test was used, with statistical significance denoted as *p < 0.05 compared to the NC inhibitor group. For (B–F), one-way ANOVA was applied, and significance levels compared to the LPS-exo + NC inhibitor group are indicated as *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. |
miR-148a-3p Directly Targets the Ferroptosis-Related Regulator SLC7A11
To clarify the direct molecular mechanismby which miR-148a-3p influences ferroptosis-related processes, we sought its downstream target. Bioinformatic analysis indicated a potential binding site for miR-148a-3p within the 3’UTR of SLC7A11, a key gene in ferroptosis-related regulation (Figure 6A). A dual-luciferase reporter assay substantiated this prediction, showing that miR-148a-3p mimics reduced the luciferase activity of WT SLC7A11 3’UTR reporter by 1.8-fold, while no significant change was observed in MUT group (Figure 6B). These data provide compelling evidence that SLC7A11 is indeed a genuine functional target of miR-148a-3p.
 |
Figure 6 miR-148a-3p directly targets the ferroptosis regulator SLC7A11. (A) Predicted binding of miR-148a-3p to SLC7A11 (red letters indicate binding bases). (B) Luciferase activity was measured in 293T cells that were co-transfected with either miR-148a-3p mimics or negative control mimics and WT or MUT reporters of the SLC7A11 3’ untranslated region (UTR) (n = 3, one-way ANOVA; ****p < 0.0001 vs. mimics NC + WT reporter; ns, not significant vs. mimics NC + MUT reporter). |
Discussion
In this study, we demonstrate that LPS-stimulated Raw264.7 cells and primary BMDMs secrete exosomes highly enriched with miR-148a-3p. By directly targeting and suppressing SLC7A11, this exosomal miRNA concurrently induces M1 polarization and ferroptosis-related characteristics in recipient homologous macrophages in vitro, forming a paracrine amplification loop for inflammatory activation at the cellular level. These findings reveal a novel mechanism of homologous macrophage communication in response to LPS stimulation.
Although exosomal miRNAs are known to regulate macrophage function during bacterial infection, most studies have focused on exosomes from plasma or non-macrophage sources.23–26 In contrast, our findings highlight the critical role of homologous macrophage communication. We confirm that exosomes from LPS-activated macrophages are sufficient to drive M1 polarization independently of direct LPS exposure. Notably, we identify miR-148a-3p as the key functional mediator within these exosomes, while miR-128-3p fails to be effectively transferred and exert regulatory effects, possibly due to specific sorting and loading mechanisms of exosomal miRNAs.32–34 This shifts the paradigm, positioning exosomal miRNAs as active mediators rather than passive bystanders in immune regulation.
Beyond regulating polarization, we further show that exosomal miR-148a-3p induces ferroptosis-related characteristics in macrophages by downregulating SLC7A11/GPX4 and upregulating ACSL4/HO-1, leading to elevated MDA levels. This dual mechanism promoting both inflammation and ferroptosis-related characteristics creates a positive feedback loop that exacerbates cellular dysfunction in vitro. While previous models proposed a “vicious cycle” between these processes,35–37 we identify exosomal miR-148a-3p as a critical coordinator of this crosstalk in homologous macrophage communication in vitro.
Our findings identify exosomal miR-148a-3p as potential cellular targets for ameliorating Gram-negative bacterial infection-induced inflammatory macrophage activation in vitro. Inhibiting miR-148a-3p or restoring SLC7A11 expression may simultaneously mitigate excessive inflammation and ferroptosis-related dysfunction in macrophages, providing a new dual-target research direction for in vitro anti-inflammatory injury strategies. However, several limitations and unanswered questions merit consideration. First, this study is confined to murine macrophage cell lines and primary BMDMs. Consequently, the findings cannot be directly generalized to human macrophages or clinical scenarios. Second, all experimental procedures were conducted in vitro, and the in vivo relevance of these findings still requires further validation. Third, our investigation solely focused on detecting ferroptosis-related characteristics, such as MDA levels and the rescue effect of Fer-1, while lacking direct lipid peroxidation assays (eg, BODIPY-C11 staining) and definitive measurements of ferroptosis-induced cell death. Therefore, we cannot definitively conclude that genuine ferroptosis has occurred. Fourth, the upstream mechanism governing the sorting of miR-148a-3p into exosomes remains elusive. Future research endeavors should encompass studies involving human macrophages, in vivo models, direct ferroptosis detection methods, and in-depth mechanistic exploration of exosomal miRNA sorting processes.
Conclusion
Collectively, this study demonstrates that exosomal miR-148a-3p derived from LPS-activated murine macrophages mediates M1 polarization and ferroptosis-related characteristics in recipient macrophages by targeting SLC7A11 in vitro. These findings reveal a new paracrine signaling pathway in macrophage inflammatory amplification induced by Gram-negative bacterial stimulation. Due to the in vitro nature and species limitation of this study, conclusions are restricted to cellular-level mechanisms and do not support direct generalization to systemic inflammation, sepsis progression, or clinical therapy. Further validation in human cells and in vivo models is required before translational applications can be considered.
Data Sharing Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Funding
There is no funding to report.
Disclosure
The authors declare no conflicts of interest.
References
1. Armina-Rodriguez A, Dorta-Estremera SM, Mendez LB, et al. Protection of mice against septic shock induced by lethal intraperitoneal dose of LPS by a recombinant Fasciola hepatica fatty acid binding protein. Microbiol Spectr. 2026;14(4):e0275625. doi:10.1128/spectrum.02756-25
2. Zhao X, Shao R, Yu Y, et al. TA-Zr/Ce nanozyme-mediated lipopolysaccharide dephosphorylation: a targeted strategy for sepsis treatment with hemoperfusion applications. ACS Nano. 2026;20(2):2523–13. doi:10.1021/acsnano.5c21151
3. Zhang S, Zhang A, Yu S, et al. Construction and activity evaluation of biphenyl hydroxamic acid dual-target antibacterial inhibitor and nanocarrier. ACS Infect Dis. 2026;12(2):827–840. doi:10.1021/acsinfecdis.5c00928
4. Yang X, Lu L, Wu C, et al. ATP2B1-AS1 exacerbates sepsis-induced cell apoptosis and inflammation by regulating miR-23a-3p/TLR4 axis. Allergol Immunopathol. 2023;51(2):17–26. doi:10.15586/aei.v51i2.782
5. Li H, Qiu D, Yuan Y, et al. Trichinella spiralis cystatin alleviates polymicrobial sepsis through activating regulatory macrophages. Int Immunopharmacol. 2022;109:108907. doi:10.1016/j.intimp.2022.108907
6. Arai Y, Asano K, Mandai S, et al. WNK1-TAK1 signaling suppresses lipopolysaccharide-induced cytokine production and classical activation in macrophages. Biochem Biophys Res Commun. 2020;533(4):1290–1297. doi:10.1016/j.bbrc.2020.10.007
7. Li H, Wang X, Liang X, et al. Verapamil inhibits ferroptosis in septic acute lung injury by blocking L-type calcium channels. Biochem Biophys Res Commun. 2025;744:151202. doi:10.1016/j.bbrc.2024.151202
8. Lin S, Yan J, Wang W, et al. STAT3-mediated ferroptosis is involved in sepsis-associated acute respiratory distress syndrome. Inflammation. 2024;47(4):1204–1219. doi:10.1007/s10753-024-01970-2
9. Sun B, Wang L, Zhang T. Role of ferroptosis in alveolar epithelial cells in acute respiratory distress syndrome. J Inflamm Res. 2025;18:9679–9698. doi:10.2147/JIR.S528482
10. Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi:10.1016/j.cell.2012.03.042
11. Lee J, Roh JL. SLC7A11 as a gateway of metabolic perturbation and ferroptosis vulnerability in cancer. Antioxidants. 2022;11(12):2444. doi:10.3390/antiox11122444
12. Lv Y, Zhang L. IRF7 activates LCN2 transcription to enhance LPS-induced acute lung injury by inducing macrophage ferroptosis and M1 polarization. Cell Biochem Biophys. 2025;83(2):2415–2430. doi:10.1007/s12013-024-01651-9
13. Ju M, Hao M, Lin D, et al. Cefiderocol has immunoregulative effects in LPS-induced vitro experimental model via inhibiting inflammation and ferroptosis. Int J Antimicrob Agents. 2024;64(6):107374. doi:10.1016/j.ijantimicag.2024.107374
14. Wang Z, Zhang W, Zhu X, et al. Purine-rich element-binding Protein B mediates ferroptosis in lipopolysaccharide-induced Raw264.7 macrophage inflammation. J Physiol Investig. 2024;67(4):187–197. doi:10.4103/ejpi.EJPI-D-24-00008
15. Mo G, Guo J, Zhang L, et al. miR-129-2-3p mediates LPS-induced macrophage polarization and ferroptosis by targeting the SMAD3-GPX4 axis. Gene. 2024;894:147962. doi:10.1016/j.gene.2023.147962
16. Kalluri R, Lebleu VS. The biology, function, and biomedical applications of exosomes. Science. 2020;367(6478). doi:10.1126/science.aau6977
17. Shi ZY, Yang XX, Malichewe C, et al. Exosomal microRNAs-mediated intercellular communication and exosome-based cancer treatment. Int J Biol Macromol. 2020;158:530–541. doi:10.1016/j.ijbiomac.2020.04.228
18. Ye R, Wei Y, Li J, et al. Plasma-derived extracellular vesicles prime alveolar macrophages for autophagy and ferroptosis in sepsis-induced acute lung injury. Mol Med. 2025;31(1):40. doi:10.1186/s10020-025-01111-x
19. Wang W, Zhu L, Li H, et al. Alveolar macrophage-derived exosomal tRF-22-8BWS7K092 activates hippo signaling pathway to induce ferroptosis in acute lung injury. Int Immunopharmacol. 2022;107:108690. doi:10.1016/j.intimp.2022.108690
20. Rao H, Ding Q, Liu A, et al. The synergistic role of P2rx7 and Panx1 in regulating alveolar macrophage pyroptosis and exosome-mediated ferroptosis of alveolar epithelial cells in lipopolysaccharide-induced acute respiratory distress syndrome. FASEB J. 2025;39(16):e70904. doi:10.1096/fj.202403385RR
21. Saadh MJ, Saeed TN, Alfarttoosi KH, et al. Exosomes and MicroRNAs: key modulators of macrophage polarization in sepsis pathophysiology. Eur J Med Res. 2025;30(1):298. doi:10.1186/s40001-025-02561-z
22. Xiao Y, Yuan Y, Hu D, et al. Exosome-derived microRNA: potential target for diagnosis and treatment of sepsis. J Immunol Res. 2024;2024(1):4481452. doi:10.1155/2024/4481452
23. Xian Y, Sun Y, Wang L, et al. Plasma exosomal miR-17-5p regulates macrophage polarization by targeting Bcl11b in sepsis-induced lung injury. J Inflamm Res. 2025;18:10651–10668. doi:10.2147/JIR.S524742
24. Jiao Y, Zhang T, Zhang C, et al. Exosomal miR-30d-5p of neutrophils induces M1 macrophage polarization and primes macrophage pyroptosis in sepsis-related acute lung injury. Crit Care. 2021;25(1):356. doi:10.1186/s13054-021-03775-3
25. Chen W, Zhang Y, Chen J, et al. Heme oxygenase-1 modulates macrophage polarization through endothelial exosomal miR-184-3p and reduces sepsis-induce lung injury. Int J Nanomed. 2025;20:5039–5057. doi:10.2147/IJN.S506830
26. Zheng T, Li S, Zhang T, et al. Exosome-shuttled miR-150-5p from LPS-preconditioned mesenchymal stem cells down-regulate PI3K/Akt/mTOR pathway via Irs1 to enhance M2 macrophage polarization and confer protection against sepsis. Front Immunol. 2024;15:1397722. doi:10.3389/fimmu.2024.1397722
27. Bai X, Li J, Li L, et al. Extracellular vesicles from adipose tissue-derived stem cells affect Notch-miR148a-3p axis to regulate polarization of macrophages and alleviate sepsis in mice. Front Immunol. 2020;11:1391. doi:10.3389/fimmu.2020.01391
28. Martino E, Balestrieri A, Aragona F, et al. MiR-148a-3p promotes colorectal cancer cell ferroptosis by targeting SLC7A11. Cancers. 2023;15(17):4342. doi:10.3390/cancers15174342
29. Li J, Chen Y, Liu J, et al. Elevated expression and activity of sodium leak channel contributes to neuronal sensitization of inflammatory pain in rats. Front Mol Neurosci. 2021;14:723395. doi:10.3389/fnmol.2021.723395
30. Xu W, Shen P, Li R, et al. Development of an event-specific droplet digital pcr assay for quantification and evaluation of the transgene DNAs in trace samples of GM PRNP-Knockout goat. Foods. 2022;11(6):1.
31. Shang A, Gu C, Wang W, et al. Exosomal circPACRGL promotes progression of colorectal cancer via the miR-142-3p/miR-506-3p- TGF-beta1 axis. Mol Cancer. 2020;19(1):117. doi:10.1186/s12943-020-01235-0
32. Rutecki S, Ksiazek K, Mikula-Pietrasik J. Exosomal cargo-derived mediators of ovarian cancer chemoresistance. Mol Diagn Ther. 2026;30(3):463–474. doi:10.1007/s40291-026-00844-7
33. Zhang D, Xu G, Jiang W, et al. Differentially significant miRNA target gene analysis in exosomes extracted from seneca valley virus infected IBRS-2 cells. Microb Pathogenesis. 2026;212:108300. doi:10.1016/j.micpath.2026.108300
34. Yuan HY, Xu J, Zhang YY, et al. The role of exosomes in ankylosing spondylitis: from biological features and functional cargo to clinical applications. Front Mol Biosci. 2026;13:1744396. doi:10.3389/fmolb.2026.1744396
35. Lai X, Wu A, Liu Y, et al. Ferritinophagy activation states determine the susceptibility to ferroptosis of macrophages in bone marrow and spleen. Int J Bio Sci. 2025;21(10):4567–4585. doi:10.7150/ijbs.114545
36. Chen X, Kang R, Kroemer G, et al. Ferroptosis in infection, inflammation, and immunity. J Exp Med. 2021;218(6). doi:10.1084/jem.20210518.
37. Dai Y, Cui C, Jiao D, et al. JAK/STAT signaling as a key regulator of ferroptosis: mechanisms and therapeutic potentials in cancer and diseases. Can Cell Inter. 2025;25(1):83. doi:10.1186/s12935-025-03681-6
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
The Emerging Role of Ferroptosis in Sepsis, Opportunity or Challenge?
Huang Q, Ding Y, Fang C, Wang H, Kong L
Infection and Drug Resistance 2023, 16:5551-5562
Published Date: 23 August 2023
The Emerging Roles of Ferroptosis in Pathophysiology and Treatment of Acute Lung Injury
Wang Y, Zhao Z, Xiao Z
Journal of Inflammation Research 2023, 16:4073-4085
Published Date: 14 September 2023
Moderate Hypothermia Alleviates Sepsis-Associated Acute Lung Injury by Suppressing Ferroptosis Induced by Excessive Inflammation and Oxidative Stress via the Keap1/GSK3β/Nrf2/GPX4 Signaling Pathway
Xu J, Tao L, Jiang L, Lai J, Hu J, Tang Z
Journal of Inflammation Research 2024, 17:7687-7704
Published Date: 25 October 2024
Ferroptosisand Its Role in the Treatment of Sepsis-Related Organ Injury: Mechanisms and Potential Therapeutic Approaches
Zhang P, Liu W, Wang S, Wang Y, Han H
Infection and Drug Resistance 2024, 17:5715-5727
Published Date: 20 December 2024
Serum Exosomes miR-122-5P Induces Hepatic and Renal Injury in Septic Rats by Regulating TAK1/SIRT1 Pathway
Wang J, Jiang Y, Yuan Y, Ma X, Li T, Lv Y, Zhang J, Chen L, Zhou J, Meng Y, Zhang B, Dong X, Ma L
Infection and Drug Resistance 2025, 18:185-197
Published Date: 9 January 2025