Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 20
Identifying Common Diagnostic Biomarkers and Therapeutic Targets between COPD and Sepsis: A Bioinformatics and Machine Learning Approach
Authors Li X, Xiao Y ![]() , Yang M
, Yang M ![]() , Zhang X, Yuan Z, Zhang Z, Zhang H, Liu L
, Zhang X, Yuan Z, Zhang Z, Zhang H, Liu L ![]() , Zhao M
, Zhao M ![]()
Received 7 December 2024
Accepted for publication 20 May 2025
Published 28 May 2025 Volume 2025:20 Pages 1761—1786
DOI https://doi.org/10.2147/COPD.S510846
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Xinyi Li,1,* Yuyang Xiao,1,* Meng Yang,1 Xupeng Zhang,1 Zhangchi Yuan,1 Zaiqiu Zhang,1 Hanyong Zhang,2 Lin Liu,1 Mingyi Zhao1
1Department of Pediatrics, The Third Xiangya Hospital of Central South University, Changsha, Hunan, 410013, People’s Republic of China; 2Development of Novel Pharmaceutical Preparations, Changsha Medical University, Changsha, 410219, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Mingyi Zhao, Department of Pediatrics, Third Xiangya Hospital of Central South University, Changsha, Hunan, 410013, People’s Republic of China, Email [email protected] Lin Liu, Department of Pediatrics, Third Xiangya Hospital of Central South University, Changsha, Hunan, 410013, People’s Republic of China, Email [email protected]
Background: Evidence suggests a bidirectional association between chronic obstructive pulmonary disease (COPD) and sepsis, but the underlying mechanisms remain unclear. This study aimed to explore shared diagnostic genes, potential mechanisms, and the role of immune cells in the COPD-sepsis relationship using Mendelian randomization (MR) and bioinformatics approaches, while also identifying potential therapeutic drugs.
Methods: Two-sample MR analysis was performed using genome-wide association data to assess genetically predicted COPD and sepsis. Immune cell-mediated effects were quantified using a two-way two-sample MR analysis. Differential expression gene (DEG) analysis and weighted gene co-expression network analysis (WGCNA) were used to identify common genes. Functional enrichment analyses were conducted to explore the biological roles of these genes. LASSO and SVM-RFE algorithms identified shared diagnostic genes, which were evaluated using receiver operating characteristic (ROC) curves. Immune cell infiltration was analyzed with CIBERSORT, while transcription factor (TF) and miRNA networks were constructed using NetworkAnalyst. Drug predictions were made using DSigDB, and molecular docking validated potential drugs.
Results: Three immune cell types were identified as mediators between COPD and sepsis, with genetically predicted effects mediated by these cells at rates of 6.5%, 12.8%, and 3.9%. A total of 33 overlapping genes were identified, and AIM2 and RNF125 were highlighted as key diagnostic genes. Immune infiltration analysis revealed dysregulated monocyte, macrophage, plasma, and dendritic cells. Regulatory network analysis identified nine key co-regulators. Ten potential drug targets were identified, with seven validated via molecular docking.
Conclusion: AIM2 and RNF125 may serve as diagnostic biomarkers, and identified immune cell subsets could mediate the COPD-sepsis connection, offering insights into potential therapeutic targets.
Keywords: Mendelian randomization, comprehensive bioinformatics analysis, machine learning, molecular docking, chronic obstructive pulmonary disease, sepsis, immune cells, co-diagnostic genes, predictive drugs
Introduction
Chronic obstructive pulmonary disease (COPD) is a heterogeneous condition1 characterized by persistent respiratory symptoms and progressive airflow obstruction.2 In 2017, approximately 544.9 million individuals worldwide were diagnosed with chronic respiratory diseases, with COPD accounting for about 55%, making it the third leading cause of death globally.3,4 Data statistics show that in 2020, approximately 384 million people worldwide had COPD, resulting in about 3.2 million deaths, which accounted for 6% of the total global mortality.5,6 Patients with COPD frequently experience a range of complications, including cardiovascular disease, depression, and anxiety,7,8 which significantly threaten their health and quality of life. Therefore, there is an urgent need to explore novel treatment strategies for COPD.
Sepsis is a clinical syndrome resulting from an abnormal immune response to infection9 and is characterized by life-threatening multiple organ dysfunction.10 The disease course can be divided into two stages: (i) the initial stage of systemic inflammatory response syndrome (SIRS), which may lead to multiple organ failure (MOF) and septic shock; and (ii) the subsequent stage of compensatory anti-inflammatory response syndrome (CARS), which results in sepsis-induced immunosuppression, increasing the risk of late infection and mortality.11 Approximately 48.9 million individuals are diagnosed with sepsis worldwide each year, with about 11 million fatalities resulting, accounting for 19.7% of total global deaths.12 Sepsis is heterogeneous and can result in a variety of infections caused by different pathogens, leading to various types of organ damage.13 Therefore, despite the gradual standardization of sepsis treatment methods, significant challenges and opportunities for improvement remain,14 necessitating further research.
Studies have indicated that the prevalence and severity of COPD are elevated in patients with sepsis, resulting in more severe comorbidities, including increased complications, significant inflammation, and metabolic abnormalities.15 In summary, although significant differences exist in the pathogenesis and clinical manifestations of COPD and sepsis, an intrinsic relationship exists between the two, both involving abnormalities in the immune system and inflammatory responses. Understanding the relationship between COPD and sepsis, along with their common influencing factors, will elucidate their pathogenic mechanisms and provide new insights for diagnosis and treatment. In this study, we employed Mendelian randomization analysis, comprehensive bioinformatics analysis, and machine learning to elucidate the pathogenesis of comorbidity between COPD and sepsis. This research aims to enhance our understanding of the pathogenesis of COPD and sepsis while providing new insights and directions for potential therapeutic targets, which holds significant importance and value.
Materials and Methods
Research Design
In this study, we investigated the causal relationship between COPD and sepsis through a two-way two-sample Mendelian randomization (MR) and examined the mediating role of immune cells. In our MR study, single nucleotide polymorphisms (SNPs) were designated as instrumental variables (IVs).16 Subsequently, comprehensive bioinformatics analysis methods and two machine learning approaches were employed to identify co-diagnostic genes for COPD and sepsis, facilitating immune infiltration analysis, identification of transcription factors and miRNAs, and evaluation of candidate drugs. Figure 1 presents the workflow of this study.
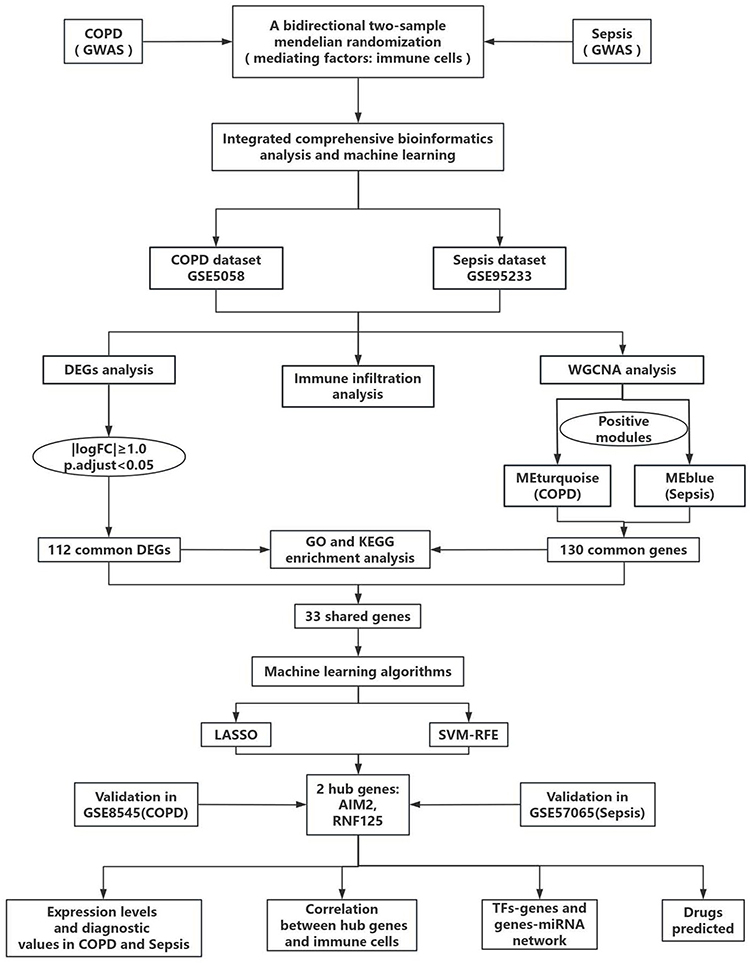 |
Figure 1 The work flow chart of this study. Abbreviations: COPD, chronic obstructive pulmonary disease; GWAS, genome-wide association study; logFC, log fold change; GSE, Gene Expression Omnibus Series; DGEs, differentially expressed genes; WGCNA, weighted gene co-expression network analysis; GO, gene ontology; KEGG, kyoto encyclopedia of genes and genomes; LASSO, Logical Regression of Selection Operators; SVM-RFE, support vector machine recursive feature elimination; TFs, transcription factors. |
Data Sources
In the Mendelian randomization (MR) analysis, we utilized summary-level data obtained from the largest publicly accessible genome-wide association study (GWAS) database (https://gwas.mrcieu.ac.uk/) for each trait (Table 1). Exposure (COPD) and outcome (sepsis) data were extracted from the FinnGen Biobank (FREEZE 11; https://r11.finngen.fi/) and the UK Biobank. All participants included in the analysis were of European ancestry to minimize potential race-related influences on the outcomes.
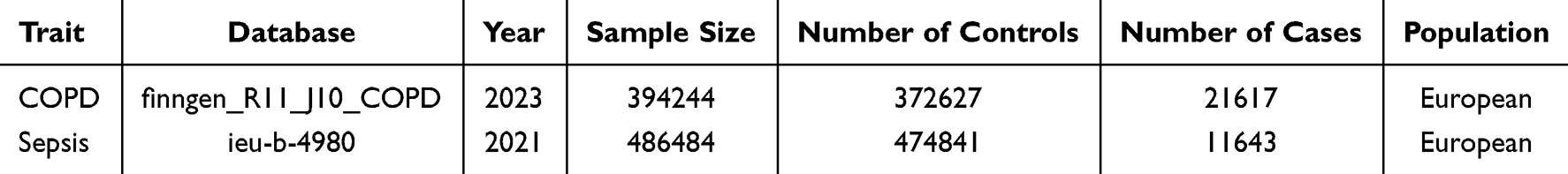 |
Table 1 Research Details of COPD and Sepsis Data Sets in MR Analysis |
GWAS summary statistics for each immune trait are publicly available from the GWAS Catalog (accession numbers GCST90001391 to GCST90002121).17 A total of 731 immunophenotypes were included, encompassing absolute cell (AC) counts (n=118), median fluorescence intensities (MFI) reflecting surface antigen levels (n=389), morphological parameters (MP) (n=32), and relative cell (RC) counts (n=192). Specifically, the MFI, AC, and RC features include B cells, CDCs, mature stages of T cells, monocytes, myeloid cells, TBNK (T cells, B cells, natural killer cells), and Treg panels, while the MP feature comprises CDC and TBNK panels. The original GWAS on immune traits was conducted using data from 3757 European individuals, with no overlapping cohorts. Approximately 22 million single nucleotide polymorphisms (SNPs) genotyped with high-density arrays were imputed using the Sardinian sequence-based reference panel,18 and associations were tested after adjusting for covariates (ie, sex, age, and age²).
The gene expression datasets for COPD and sepsis were obtained from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/).19 The following criteria guided the search: (1) the samples were from Homo sapiens, (2) the experimental data type was microarray, (3) the dataset included both the control and disease groups, and (4) the total number of samples in each cohort was at least 10. Finally, four GEO datasets—GSE5058, GSE95233, GSE8545, and GSE57065—were selected as the datasets for COPD and sepsis. Among them, GSE5058 (COPD) and GSE95233 (sepsis) were designated as the discovery cohorts. GSE8545 and GSE57065 were used to validate the hub genes of COPD and sepsis, respectively. The detailed information regarding the aforementioned datasets is presented in Table 2.
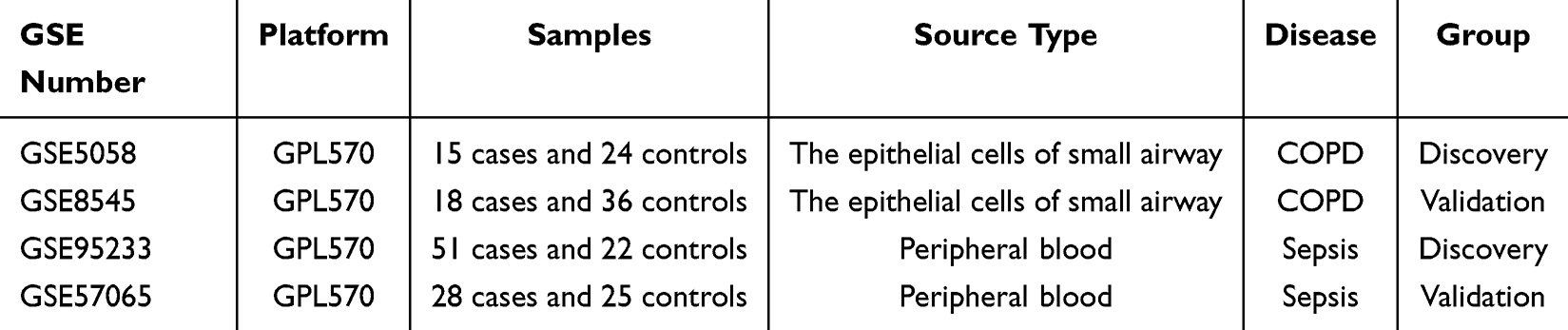 |
Table 2 The Basic Information of GEO Dataset Used in This Study |
Instrumental Variable Selection and Data Reconciliation in MR Analysis
We included genome-wide significant single nucleotide polymorphisms (SNPs) (P < 5 × 10−8). If no genome-wide significant SNPs were available as instrumental variables (IVs), SNPs below the genome-wide significance level (P < 5 × 10−6) were utilized as candidate IVs. If no available SNPs are screened out within these two ranges, then SNPs below the genome-wide significance level (P < 1 × 10−5) were utilized as candidate IVs. SNP selection was based on linkage disequilibrium clustering (window size = 10,000 KB, r² < 0.001). Linkage disequilibrium levels were estimated from the 1000 Genomes Project based on European samples.20 If no specifically exposed SNPs are identified in the resultant dataset, the linkage disequilibrium (LD) marker employs proxy SNPs. The Mendelian randomization analysis excluded palindromic and ambiguous SNPs from the IVs.21 The F statistic is calculated based on the variance explained by the SNP for each exposure, specifically as (N−K−1)K/R2(1−R2)\frac{(N-K-1)}{K}\bigg/\frac{R²}{(1-R²)}K(N−K−1)/(1−R2)R2, where K represents the number of genetic variations and N denotes the sample size. Weak instrumental variables (F statistic < 10) were excluded in this study.22,23 The minor allele frequency (MAF) was set to > 1%.
Main MR Analysis
Figure 2 presents a summary of the analysis. We conducted a two-sample two-way Mendelian randomization (MR) analysis to assess the causal relationship between chronic obstructive pulmonary disease (COPD) and sepsis (Figure 2A), designating it as the total effect. Inverse variance weighting (IVW) was employed to perform a meta-analysis that combines the Wald ratio of each SNP’s causal effect.21,24 Subsequently, the MR-Egger25 and weighted median26 methods were utilized as supplementary analyses to IVW. Different methods are employed to obtain MR estimates based on varying validity assumptions. The application of IVW relies on the premise that all SNPs are valid instrumental variables. Consequently, this method can yield accurate estimation results. The MR-Egger method assesses the directional pleiotropy of instrumental variables, where the intercept can be interpreted as an estimate of the average pleiotropy of genetic variations. In comparison to the MR-Egger analysis, the weighted median method has the advantage of achieving higher accuracy (ie, smaller standard deviation). In the presence of horizontal pleiotropy, even if 50% of the genetic variations are invalid instrumental variables (IVs), the weighted median method can provide consistent estimates.27
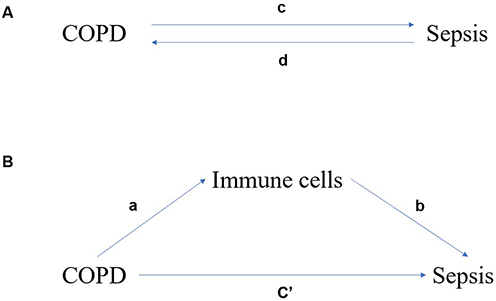 |
Figure 2 The associated charts studied in this study. (A) Bidirectional MR analysis of chronic obstructive pulmonary disease (COPD) and sepsis. c is the total effect of exposure and sepsis as a result of gene-predicted COPD. d is the total effect of genetic prediction of sepsis as a result of exposure and COPD as a result. (B) The total effect was decomposed into: (i) indirect effect using two-step method (a is the effect of COPD on immune cells, b is the effect of immune cells on sepsis) and product method (a×b); (ii) Direct effect (c′ = c−a×b). The proportion mediated by immune cells is the indirect effect divided by the total effect. |
Mediation MR Analysis
We additionally conducted a mediation analysis employing a two-step Mendelian randomization (MR) design to investigate whether immune cells mediate the causal pathway from chronic obstructive pulmonary disease (COPD) to sepsis outcomes (Figure 2). The overall effect can be decomposed into an indirect effect (mediated by immune cells) and a direct effect (without mediation).28 The total effect of COPD on sepsis was divided into (1) the direct effect of COPD on sepsis (c′ in Figure 2B) and (2) the indirect effect of COPD mediated by immune cells (a × b in Figure 2B). We calculated the percentage mediated by the mediating effect by dividing the indirect effect by the total effect. Simultaneously, the 95% confidence interval was calculated using the delta method.29
Sensitivity Analysis
The causal direction of each extracted SNP concerning exposure and outcomes was assessed using MR Steiger filtering.30 This method calculates the variance explained by the exposure and instrumental SNPs, testing whether the variance in the outcomes is less than that in the exposure. The “TRUE” MR Steiger results indicate that the causal relationship is in the expected direction, whereas the “FALSE” results suggest that the causal relationship is in the opposite direction. SNPs resulting in “FALSE” were excluded, indicating that these SNPs exerted a primary effect on the outcomes rather than serving as evidence of exposure.
Heterogeneity among SNPs was assessed using Cochran’s Q statistics and funnel plots.31,32 The MR-Egger intercept method25 and the MRPRESSO method33 were employed to detect horizontal pleiotropy. If an outlier is detected, it is removed, and the MR causal estimation is re-evaluated. If heterogeneity remains high after removal, the stability of the results is evaluated using a random effects model, which is less susceptible to weak SNP exposure associations. Finally, omission analysis was conducted to verify the impact of each SNP on the overall causal estimation.
Identification of Differentially Expressed Genes (DEGs)
Preprocessing of all original datasets, including background adjustment and normalization, was conducted using the “affy” package of R software (version 4.3.0). Utilizing the GPL570 platform (Affymetrix Human Genome U133 Plus 2.0 Array), probes were transformed into gene symbols. The average value was chosen as the gene expression level for genes matching multiple probes. Subsequently, differentially expressed genes (DEGs) in the COPD and sepsis datasets were identified using the “Limma” R software package. The threshold criteria for DEGs were set at an adjusted p-value < 0.05 and |log2 fold change (FC)| ≥ 1.0. The “ggplot2” and “pheatmap” R packages were employed to generate the differential gene volcano plot and cluster heatmap. The “Venn” package was utilized to create a Venn diagram to identify the common DEGs between COPD and sepsis.
Weighted Gene Co-Expression Network Analysis (WGCNA) and Module Gene Identification
To explore gene interactions, the systems biology method Weighted Gene Co-expression Network Analysis (WGCNA) was employed to construct a gene co-expression network. First, genes exhibiting more than 25% variation in the samples were integrated, and the integrated dataset was imported into WGCNA. Second, to ensure the reliability of the network construction results, outlier samples were removed. Third, the adjacency degree was calculated from the soft threshold power β derived from the co-expression similarity using the pickSoftThreshold function. Subsequently, the adjacency relationship was transformed into a topological overlap matrix (TOM), and the corresponding dissimilarity (1-TOM) was calculated. Fourth, modules were detected through hierarchical clustering and the dynamic tree cut function. To classify genes with similar expression profiles into gene modules, average linkage hierarchical clustering was performed based on the TOM-based dissimilarity measure. The minimum size of the gene tree was set to 50.34 Fifth, module membership (MM) and gene significance (GS) were calculated for modules related to clinical attributes. Finally, the characteristic gene network was visualized. The differentially expressed genes (DEGs) screened from the integrated dataset intersected with the genes in the significant module to identify common genes (cg).
Gene Ontology (GO) Annotation and Kyoto Encyclopedia of Genes and Genomes (KEGG) Cg Pathway Enrichment Analysis
DAVID (Database for Annotation, Visualization and Integrated Discovery) 6.8 (https://david.ncifcrf.gov/tools.jsp) is a gene function classification tool, which is used to perform gene ontology (GO) by enrichment analysis of common genes (cg).35 The enriched GO terms were divided into biological process (BP), cellular component (CC) and molecular function (MF) ontology. In order to understanding the biological function of common genes (cg), we uploaded the common gene (cg) to the DAVID database (Gene Function Classification Tool, https://david.ncifcrf.gov/tools.jsp).36 The significant enrichment threshold of GO and KEGG analysis was p<0.05, and the count was≥2.
Machine Learning for Identifying Candidate Biomarkers
Based on potential diagnostic factors, two machine learning algorithms were employed to predict the status of COPD and sepsis. LASSO is a regression analysis algorithm that employs regularization techniques to enhance prediction accuracy. The LASSO regression analysis was conducted using the “glmnet” software package in R to identify genes significantly associated with the diagnostic capability for COPD, sepsis, and healthy specimens. Support Vector Machine (SVM) is a widely utilized machine learning technique for classification and regression analysis. To prevent overfitting, the Recursive Feature Elimination (RFE) algorithm is employed to select the optimal genes from the metadata queue. Consequently, SVM Recursive Feature Elimination (SVM-RFE) is employed to identify the gene set with the highest recognition capability.
Expression Analysis and Diagnostic Evaluation of Candidate Biomarkers
The expression levels of candidate biomarkers in the control group and the disease group were visualized using the “ggplot2” package (p<0.05). The area under the curve (AUC) score of the receiver operating characteristic (ROC) curve was calculated, and the diagnostic accuracy of the candidate biomarkers was evaluated using the “pROC” package.
Immune Infiltration Analysis
CIBERSORT is a deconvolution algorithm that utilizes gene expression data to identify 22 different types of immune cells. The “CIBERSORT” software package is employed to analyze immune cell infiltration. The results are visualized using the “ggplot2”, “corrplot”, and “vioplot” packages. Spearman correlation analysis was employed to assess the correlation between immune cells and candidate biomarkers.
Identification of Transcription Factors and miRNAs
Utilizing the ENCODE and TarBase databases, transcription factor-gene and gene-miRNA regulatory networks were constructed using the NetworkAnalyst 3.0 platform (https://www.networkanalyst.ca/). ENCODE is a project that offers comprehensive and integrated data regarding the functional genomic characteristics of human and mouse cells and tissues. TarBase is the primary experimental validation database for miRNA-mRNA interactions. Finally, Cytoscape was employed to visualize these interactions.
Evaluation of Candidate Drugs
The enrichment platform was used to identify the relationship between drug molecules and hub genes, and the data were from the drug feature database (DSigDB, https://tanlab.ucdenver.edu/DSigDB). The database currently has 22,527 gene sets, including 17,389 drugs, covering 19,531 genes.37 The three-dimensional structure of drug target molecules was obtained from PubChem (https://pubchem.ncbi.nlm.nih.gov/)38 and saved in SDF file format. In Protein Data Bank (PDB; the https://www.rcsb.org/) database39 obtains the 3D structure of key gene targets and saves them as files in PDB format. Molecular docking and visual analysis were performed using AutoDock Vina40 and PyMol 1.8.1 (Schrödinger, http://www.pymol.org/) to show the interaction between drug molecules and gene targets.
Statistical Analysis
Statistical analysis was conducted using R software (version 4.3.0), with p<0.05 regarded as statistically significant. Mendelian randomization (MR) analysis was conducted using the “TwoSampleMR” package.41 MR-pleiotropic residuals and outliers (MR-PRESSO) and robustly adjusted profile scores (MR.RAPS) were calculated using the R software packages “MRPRESSO” and “MR.RAPS”, respectively. Additionally, the PhenoScanner search was utilized to evaluate all known phenotypes associated with the genetic tools included in our analysis. In the screening of differentially expressed genes (DEGs), module genes, enrichment analysis, transcription factor (TF)-gene networks, gene-miRNA networks, and immune infiltration analysis, p<0.05 was regarded as significant.
Result
Relationship Between COPD and Sepsis
Following the exclusion of palindromic and ambiguous SNPs identified through MR Steiger filtering, as well as non-proxy SNPs and those with incorrect causal directions, the remaining SNPs were utilized as instrumental variables. In the context of COPD, 17 SNPs were identified, while 13 SNPs were identified in the context of sepsis (Supplementary Tables S1 and S2).
The IVW, MR-Egger, and weighted median methods were employed to estimate the causal relationship between genetically predicted COPD and sepsis (Figure 3A). All three MR methods indicated a positive correlation between COPD and sepsis, with the IVW odds ratio (OR) for each standard deviation (SD) increase in COPD being 1.1428 (95% CI, 1.0476–1.2466; P = 0.0026); the MR-Egger OR being 1.3435 (95% CI, 1.1176–1.6151; P = 0.0067); and the weighted median OR being 1.2204 (95% CI, 1.0735–1.3873; P = 0.0023). However, our MR analysis results indicated no evidence of a reverse causal relationship, meaning that genetically predicted sepsis does not exhibit a causal effect on COPD. Specifically, the IVW OR for each SD increase in sepsis was 1.0779 (95% CI, 0.9688–1.1992; P = 0.1682); the MR-Egger OR was 0.9937 (95% CI, 0.7936–1.2443; P = 0.9572); and the weighted median OR was 0.9917 (95% CI, 0.8705–1.1300; P = 0.9009). The results are illustrated in Figure 3B.
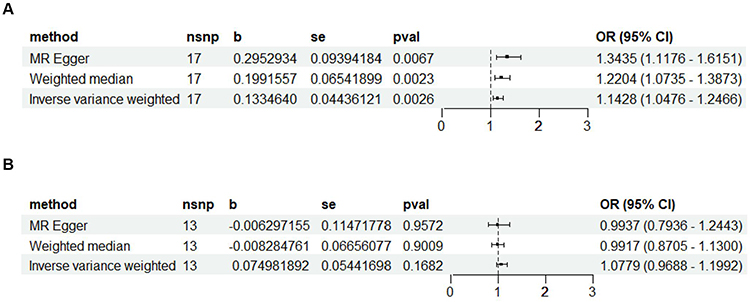 |
Figure 3 Forest plot illustrating the causal relationship of COPD with sepsis. (A) With COPD as the exposure factor and sepsis as the outcome factor, using the IVW, MR Egger, and weighted median methods to study the causal relationship between the two. (B) With sepsis as the exposure factor and COPD as the outcome factor, using the IVW, MR Egger, and weighted median methods to study the causal relationship between the two. |
Screening of Immune Cells Mediating the Relationship Between COPD and Sepsis
Initially, we treated COPD as the exposure factor and immune cell characteristics as the outcome factors. After removing palindromic and ambiguous SNPs, non-proxy SNPs, and SNPs identified with incorrect causal directions by MR Steiger filtering, we extracted a total of 19 genome-wide significant SNPs as instrumental variables (Supplementary Table S3). Using the IVW method for batch screening, we identified causal relationships between immune cell characteristics and COPD, resulting in three findings (Supplementary Table S4).
Subsequently, we extracted genome-wide significant SNPs for immune cell characteristics using the same methodology. SNPs identified with COPD as the exposure factor were removed, and the remaining SNPs were utilized as instrumental variables (Supplementary Tables S5–S7). Using sepsis as the outcome factor, we employed the IVW method to screen for immune cell characteristics with causal relationships to sepsis, resulting in three identified findings: CD33bright HLA DR+ CD14- %CD33bright HLA DR+, BAFF-R on IgD- CD38bright, and CD45 on CD33- HLA DR+ (Supplementary Table S8).
Proportion of Association Between COPD and Sepsis Mediated by Immune Cells
We analyzed these three immune cell characteristics as mediators in the pathway from COPD to sepsis. We found that the risk of sepsis was positively correlated with CD33bright HLA DR+ CD14- %CD33bright HLA DR+, which, in turn, was positively correlated with COPD risk; the risk of sepsis was negatively correlated with BAFF-R on IgD- CD38bright and CD45 on CD33- HLA DR+, which were also negatively correlated with COPD risk. As shown in Figure 4A, our study indicates that CD33bright HLA DR+ CD14- %CD33bright HLA DR+ accounts for 6.5% of the increased risk of sepsis associated with COPD (Indirect effect: 0.0651). As depicted in Figure 4B, BAFF-R on IgD- CD38bright accounts for 12.8% of the increased risk of sepsis associated with COPD (Indirect effect: 0.1279). As illustrated in Figure 4C, CD45 on CD33- HLA DR+ accounts for 3.9% of the increased risk of sepsis associated with COPD (Indirect effect: 0.0389). Detailed data are presented in Supplementary Table S9.
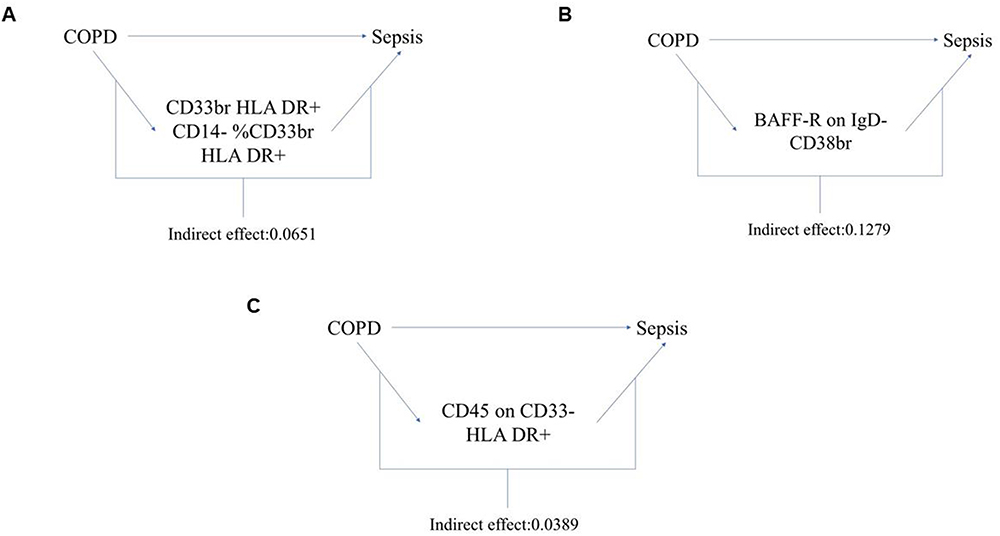 |
Figure 4 Schematic illustration of the mediating role of immune cell characteristics. (A) The immune cell CD33bright HLA DR+ CD14- %CD33bright HLA DR+ play a mediating role between COPD and sepsis and its indirect effect values. (B) The immune cell BAFF-R on IgD- CD38bright play a mediating role between COPD and sepsis and its indirect effect values. (C) The immune cell CD45 on CD33- HLA DR+ play a mediating role between COPD and sepsis and its indirect effect values. |
Sensitivity Analysis
Several sensitivity analyses were performed to assess and address the presence of pleiotropy in causal estimates. The Cochran’s Q test and funnel plot revealed no evidence of heterogeneity or asymmetry in the causal relationships among these SNPs (Supplementary Table S10 and Supplementary Figures S1–S8). The impact of each SNP on the overall causal estimate was validated through a leave-one-out analysis (Supplementary Figures S9–S16). After systematically removing each SNP and reanalyzing the remaining SNPs using MR, the results remained consistent, indicating that the inclusion of all SNPs rendered the causal relationship significant.
Identification of Differentially Expressed Genes
In the COPD dataset (GSE5058), a total of 2998 differentially expressed genes (DEGs) were identified, including 1363 upregulated and 1635 downregulated genes (Figure 5A). In the sepsis dataset (GSE95233), a total of 1101 DEGs were identified, with 608 upregulated and 493 downregulated genes (Figure 5B). Cluster heatmaps based on all differentially expressed genes for both COPD and sepsis are presented (Figure 5C and D). Using a Venn diagram, a total of 112 common DEGs were identified between COPD and sepsis (Figure 5E).
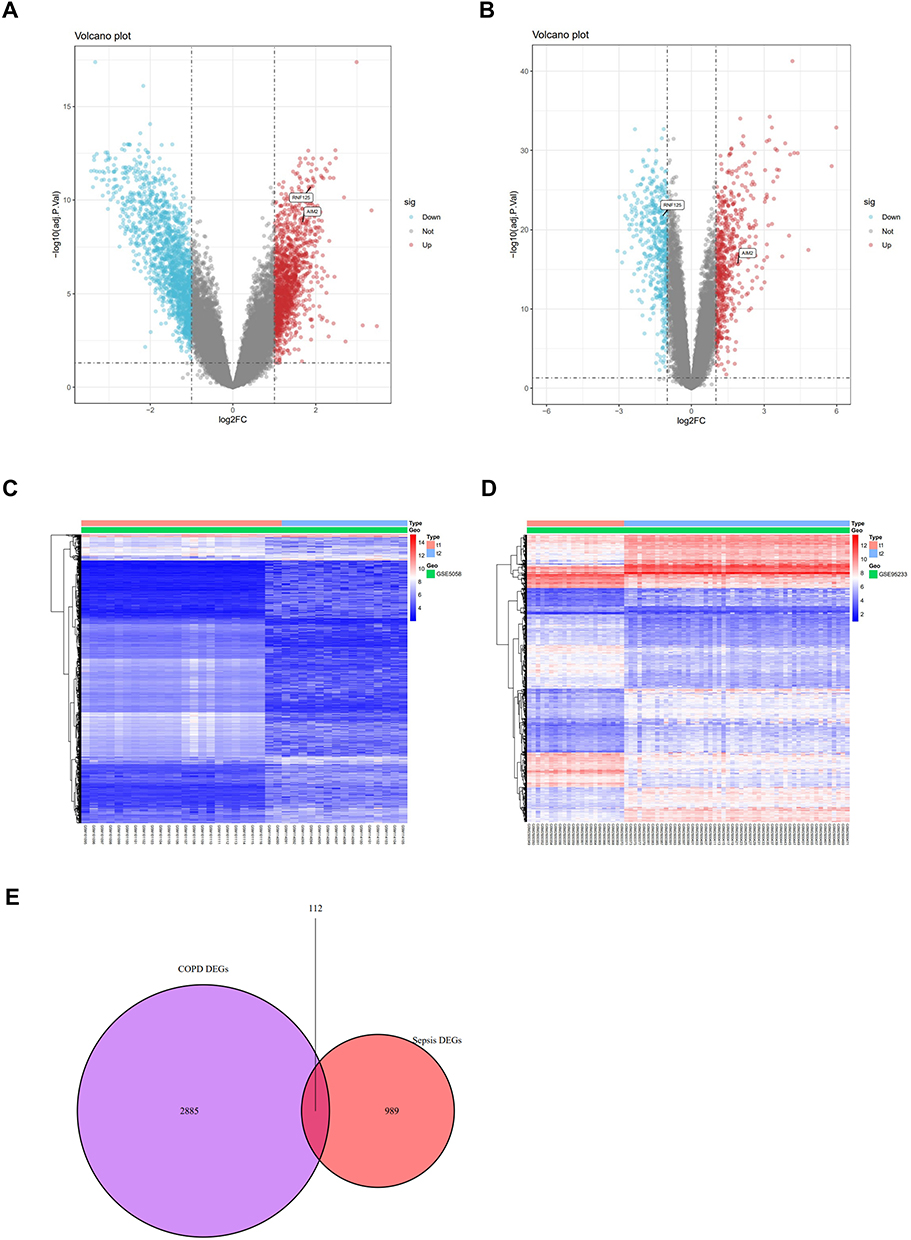 |
Figure 5 Identification of differentially expressed genes (DEGs). (A and B) Volcano plots of all DEGs in COPD (GSE5058) and sepsis (GSE95233), respectively. (C and D) Heatmaps of all DEGs in COPD (GSE5058) and sepsis (GSE95233), respectively. (E) Venn diagram of overlapping DEGs between COPD and sepsis. |
Functional Enrichment Analysis of Common Differentially Expressed Genes
Functional enrichment analyses employing Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were conducted to explore the biological characteristics and pathways associated with the 112 common differentially expressed genes (DEGs). The GO analysis identified a total of 47 terms, comprising 29 biological processes (BP), 13 cellular components (CC), and 5 molecular functions (MF). The most significant 15 terms are presented here, encompassing 5 biological processes (BP), 5 cellular components (CC), and 5 molecular functions (MF). The biological processes (BP) were primarily enriched in the innate immune response, positive regulation of natural killer cell-mediated cytotoxicity, immune response, pyroptotic inflammatory response, and inflammatory response. The cellular components (CC) were enriched in tertiary granule membrane, specific granule membrane, tertiary granule lumen, specific granule lumen, and plasma membrane. The molecular functions (MF) were enriched in carbohydrate binding, pattern recognition receptor activity, lipopolysaccharide binding, beta-glucuronidase activity, and G protein-coupled purinergic nucleotide receptor activity. The KEGG pathway enrichment analysis focused on two primary pathways, namely neutrophil extracellular trap formation and transcriptional misregulation in cancer. The results of the GO and KEGG enrichment analyses are presented in Figure 6, with complete findings listed in Supplementary Table S11. These findings indicate a significant association between chronic obstructive pulmonary disease (COPD), sepsis, and inflammatory responses, as well as immune processes.
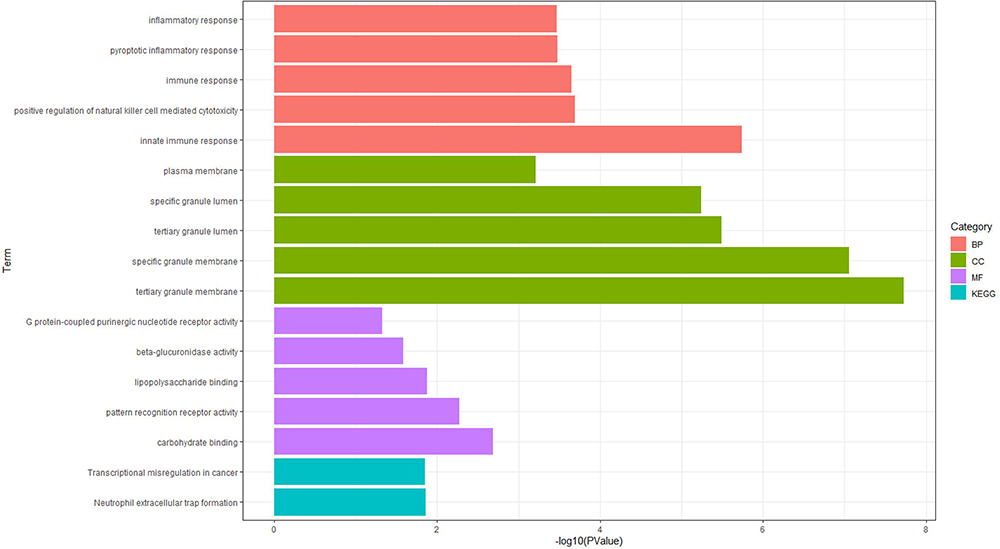 |
Figure 6 Functional enrichment analysis of common DEGs between COPD and sepsis. GO analysis results of the common DEGs: displaying the top 5 biological process (BP) terms, top 5 cellular component (CC) terms, and top 5 molecular function (MF) terms. Two KEGG pathways associated with the common DEGs. |
Weighted Gene Co-Expression Network Analysis (WGCNA) and Module Gene Identification
We conducted Weighted Gene Co-expression Network Analysis (WGCNA) to identify the most pertinent gene modules in chronic obstructive pulmonary disease (COPD) and sepsis. The optimal soft-thresholding power (β) was set to 5 for the COPD dataset (GSE5058) and 20 for the sepsis dataset (GSE95233) (Figure 7A and B). In the COPD dataset, five modules were identified, with the turquoise module exhibiting the strongest negative correlation with COPD (correlation coefficient = −0.88, p = 2e-13) and comprising 4256 genes (Figure 7C and E). In the sepsis dataset, seven modules were identified, with the blue module demonstrating the strongest negative correlation (correlation coefficient = −0.91, p = 2e-29) and comprising 1009 genes (Figure 7D and F). Scatter plots illustrate the correlation between module membership and gene significance in the turquoise module for COPD and in the blue module for sepsis (Figure 7G and H). Subsequently, a Venn diagram was employed to identify 130 overlapping genes within the modules associated with both COPD and sepsis (Figure 7I).
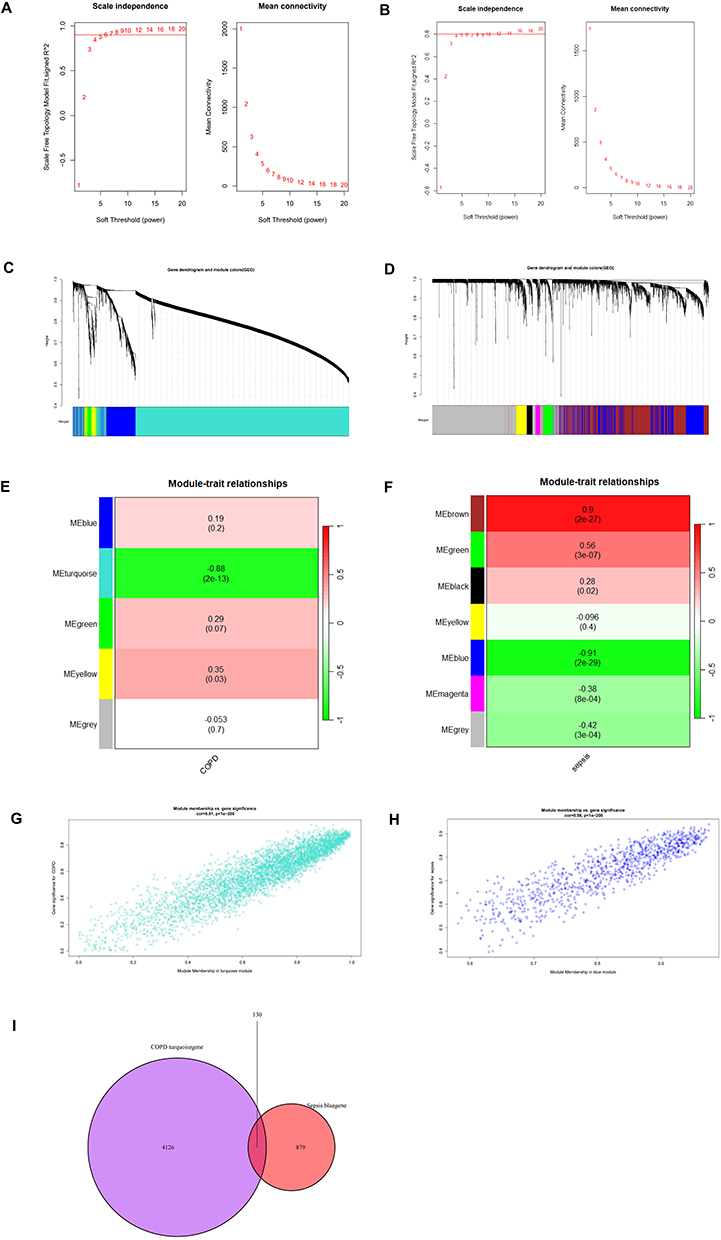 |
Figure 7 Construction of WGCNA networks and identification of key modules. (A and B) Selection of soft thresholds for COPD (GSE5058) and sepsis (GSE95233), respectively. (C and D) Hierarchical clustering trees of co-expression gene clusters for COPD (GSE5058) and sepsis (GSE95233), respectively. (E and F) Heatmaps showing the correlation of each module with clinical characteristics in COPD (GSE5058) and sepsis (GSE95233), respectively. (G) Scatter plot of module membership versus gene significance for the Turquoise module in COPD (GSE5058). (H) Scatter plot of module membership versus gene significance for the Blue module in sepsis (GSE95233). (I) Venn diagram of overlapping genes between COPD and sepsis identified through WGCNA analysis. |
Functional Enrichment Analysis of Disease-Related Common Genes
Subsequently, we conducted Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses to elucidate the biological functions and pathways associated with disease-related common genes. A total of 48 GO terms were identified, comprising 31 biological processes (BP), 9 cellular components (CC), and 8 molecular functions (MF). The 15 most significant GO terms are presented here, encompassing 5 BP, 5 CC, and 5 MF. Additionally, nine KEGG pathways were identified, with the top five pathways highlighted (Figure 8). Detailed results are available in Supplementary Table S12. In the BP category, most genes were primarily involved in the adaptive immune response, positive regulation of natural killer cell-mediated cytotoxicity, positive regulation of type II interferon production, cell surface receptor signaling pathways, and immune response. Within the CC category, the majority of genes participated in the external side of the plasma membrane, the plasma membrane, the immunological synapse, the cell surface, and membrane rafts. Regarding the MF category, most genes were involved in transmembrane signaling receptor activity, signaling receptor binding, phosphatidylcholine binding, cytokine activity, and non-membrane-spanning protein tyrosine kinase activity. The KEGG pathway enrichment analysis revealed that most genes were significantly enriched in the T cell receptor signaling pathway, Th1 and Th2 cell differentiation, viral protein interactions with cytokines and cytokine receptors, Th17 cell differentiation, and the PD-L1 expression and PD-1 checkpoint pathway in cancer. These findings suggest that COPD and sepsis are primarily associated with inflammatory and immune responses, aligning with the enrichment analysis results for the common differentially expressed genes (DEGs).
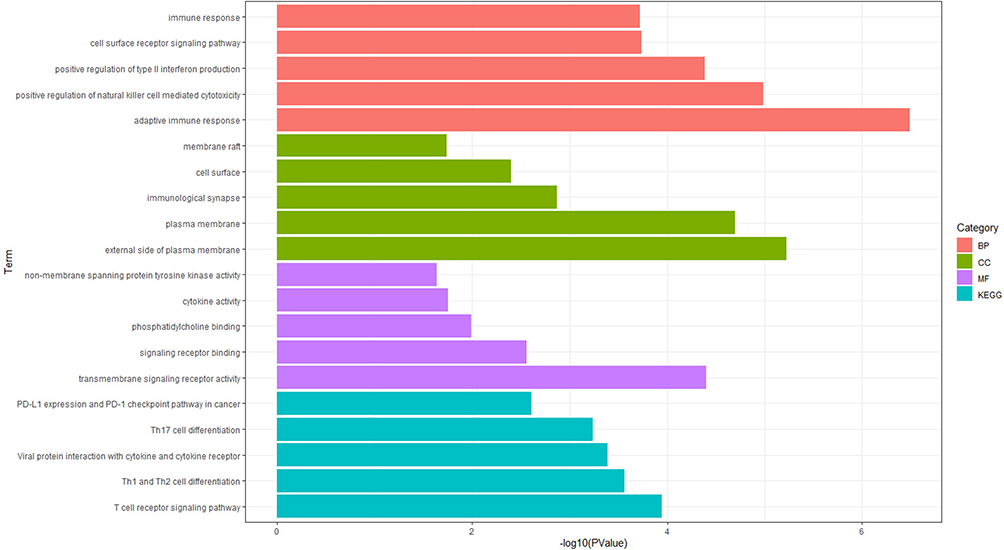 |
Figure 8 Functional enrichment analysis based on disease-related common genes. GO classifications obtained from WGCNA analysis between COPD and sepsis, displaying the top 5 terms for biological process (BP), molecular function (MF), and cellular component (CC). KEGG enrichment analysis results obtained from WGCNA analysis between COPD and sepsis, showing the top 5 pathways. |
Identification of Candidate Biomarkers Through Machine Learning
To further investigate the shared pathogenic mechanisms between COPD and sepsis, we identified 33 genes by intersecting the 112 DEGs and the 130 genes detected through WGCNA (Figure 9A). Subsequently, two machine learning algorithms were applied to screen for potential candidate biomarkers among these 33 shared genes. In the COPD dataset (GSE5058), LASSO regression identified five genes (Figure 9B and C), while the SVM-RFE algorithm identified 26 genes with the lowest root mean square error (RMSE) (Figure 9D). Ultimately, five overlapping genes were identified in the COPD group through the intersection of these two algorithms (Figure 9E). Similarly, in the sepsis dataset (GSE95233), LASSO regression identified 11 genes, and the SVM-RFE algorithm selected 21 genes (Figure 9F–H). Ten overlapping genes were subsequently identified in the sepsis group (Figure 9I). Finally, AIM2 and RNF125 were identified as candidate biomarkers through a Venn diagram (Figure 9J).
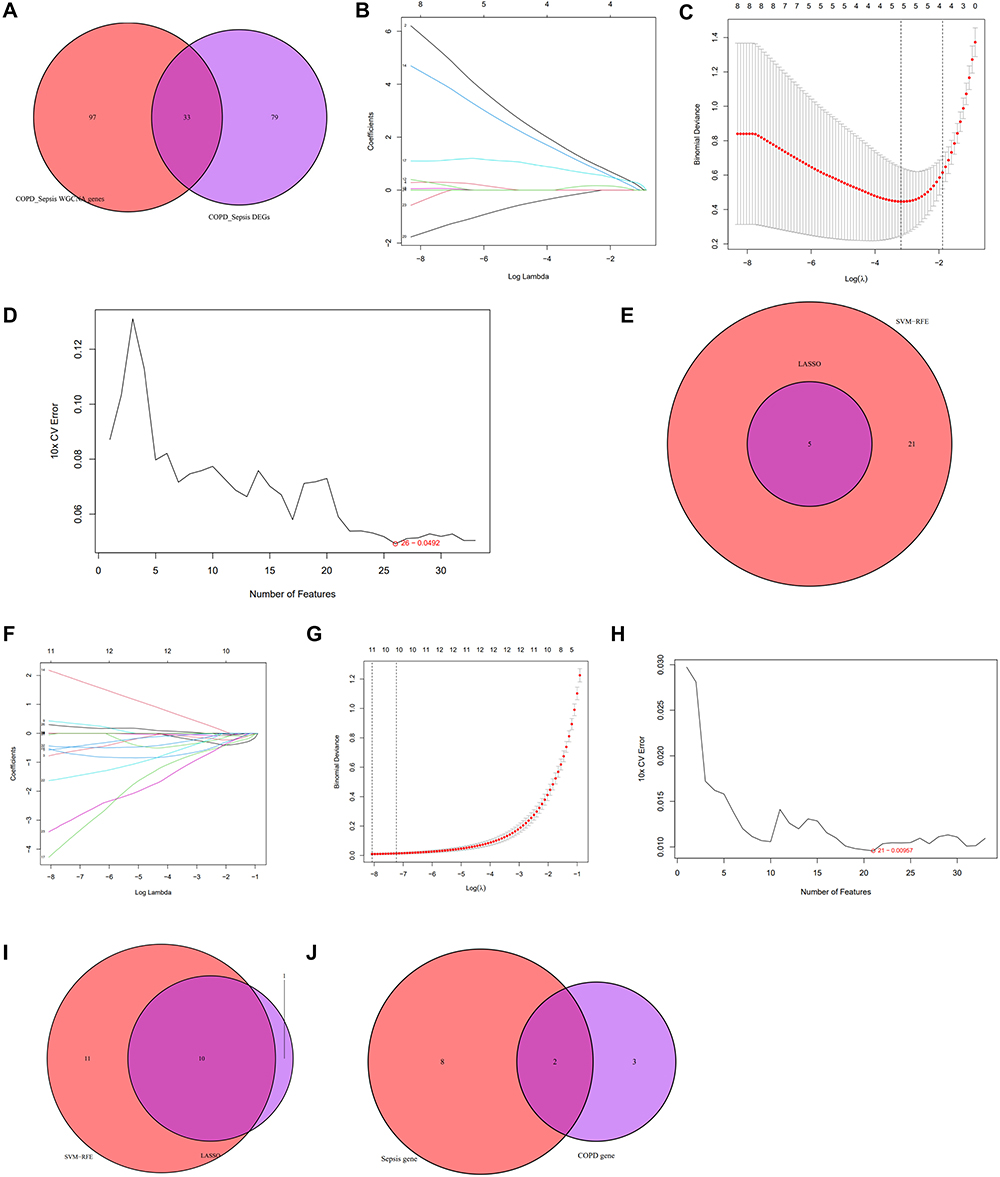 |
Figure 9 Machine learning for screening candidate biomarkers. (A) Venn diagram showing the intersection of common genes obtained from WGCNA and DEGs. (B and C) Five genes identified as the most suitable for COPD diagnosis based on the Lasso regression algorithm at the minimum binomial deviance. (D) The top 26 genes with the minimum error and highest accuracy in COPD selected based on SVM-RFE. (E) Venn diagram of intersecting genes obtained from two machine learning algorithms in COPD. (F and G) Eleven genes identified as the most suitable for diagnosing sepsis based on the Lasso regression algorithm at the minimum binomial deviance. (H) The top 21 genes with the minimum error and highest accuracy in sepsis selected based on SVM-RFE. (I) Venn diagram of intersecting genes obtained from two machine learning algorithms in sepsis. (J) Venn diagram showing the overlap of final candidate biomarkers between COPD and sepsis. |
Diag Nostic Value and Verification of Candidate Biomarkers
To verify whether AIM2 and RNF125 serve as diagnostic markers for COPD and sepsis, we further analyzed their expression levels and diagnostic efficacy. As shown in Figure 10A and B, for the COPD dataset, the expression levels of these two genes were significantly higher in the disease group compared to the control group. In Figure 10C and D, for the sepsis dataset, the expression level of AIM2 was significantly higher in the disease group compared to the control group, while the expression level of RNF125 was significantly lower in the disease group. The ROC curve analysis indicated that each biomarker had good predictive performance (Figure 10E–H): for the COPD dataset (GSE5058), AIM2 (AUC: 0.967, 95% CI: 0.911–1.000) and RNF125 (AUC: 0.986, 95% CI: 0.950–1.000) (Figure 10E and F); for the sepsis dataset (GSE95233), AIM2 (AUC: 0.983, 95% CI: 0.949–1.000) and RNF125 (AUC: 0.999, 95% CI: 0.995–1.000) (Figure 10G and H).
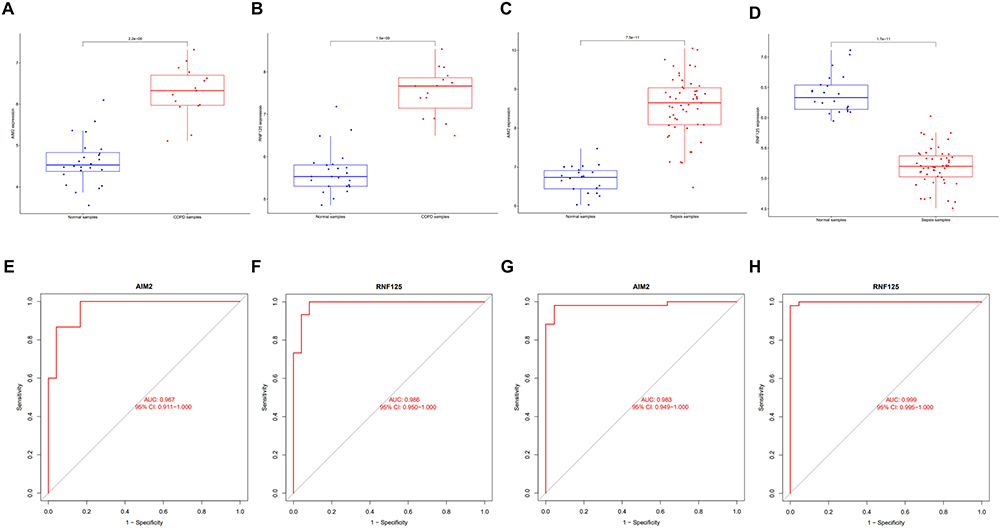 |
Figure 10 Assessment of the diagnostic value of candidate biomarkers in the discovery dataset. (A and B) Expression differences of two shared genes in the COPD (GSE5058) discovery dataset. (C and D) Expression differences of two shared genes in the sepsis (GSE95233) discovery dataset. (E and F) ROC curves for the two shared genes in the COPD (GSE5058) discovery dataset. (G and H) ROC curves for the two shared genes in the sepsis (GSE95233) discovery dataset. |
To further assess the accuracy of the candidate biomarkers, we validated them in two external datasets (GSE8545 for COPD and GSE57065 for sepsis). Consistent with previous results, the expression of both biomarkers was significantly upregulated in the disease group of the validation COPD dataset (Figure 11A and B), and the ROC curve analysis confirmed that all candidate genes had good diagnostic value (Figure 11E and F). In the validation sepsis dataset, the expression of AIM2 was significantly upregulated, while the expression of RNF125 was significantly downregulated in the disease group (Figure 11C and D), and the ROC curve analysis confirmed that all candidate genes had good diagnostic value (Figure 11G and H). In summary, the above research results indicate that AIM2 and RNF125 can serve as promising diagnostic biomarkers for COPD and sepsis.
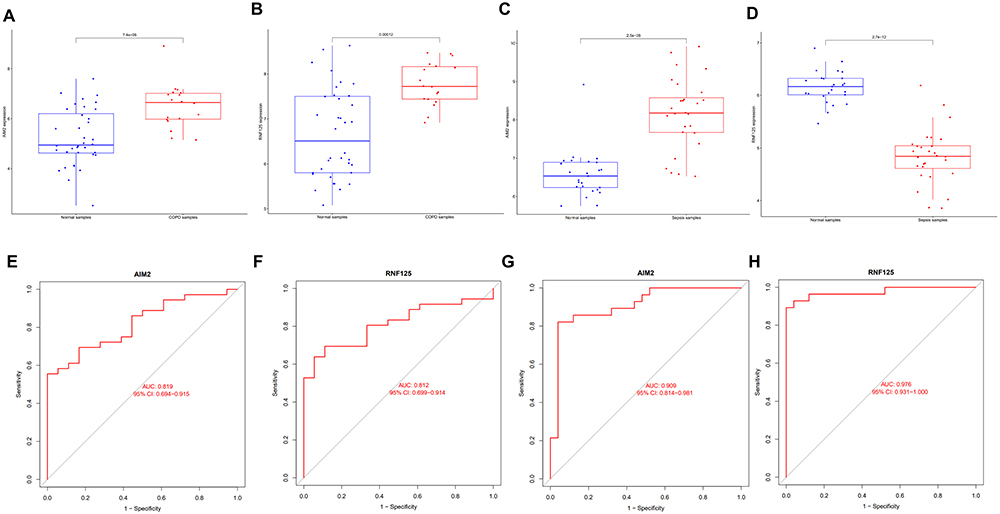 |
Figure 11 Assessment of the diagnostic value of candidate biomarkers in the validation dataset. (A and B) Expression differences of two shared genes in the COPD (GSE8545) validation dataset. (C and D) Expression differences of two shared genes in the sepsis (GSE57065) validation dataset. (E and F) ROC curves for the two shared genes in the COPD (GSE8545) validation dataset. (G and H) ROC curves for the two shared genes in the sepsis (GSE57065) validation dataset. |
Analysis of Immune Cell Infiltration
Both COPD and sepsis are infection-related diseases characterized by strong immune responses, which prompted us to use CIBERSORT to analyze the proportions of immune-infiltrating cells. Figure 12A and B show the distribution of 22 immune cell types in COPD and sepsis samples, respectively. Correlation heatmaps of immune cell interactions are presented in Figure 12C and D (Figure 12C for COPD and Figure 12D for sepsis).
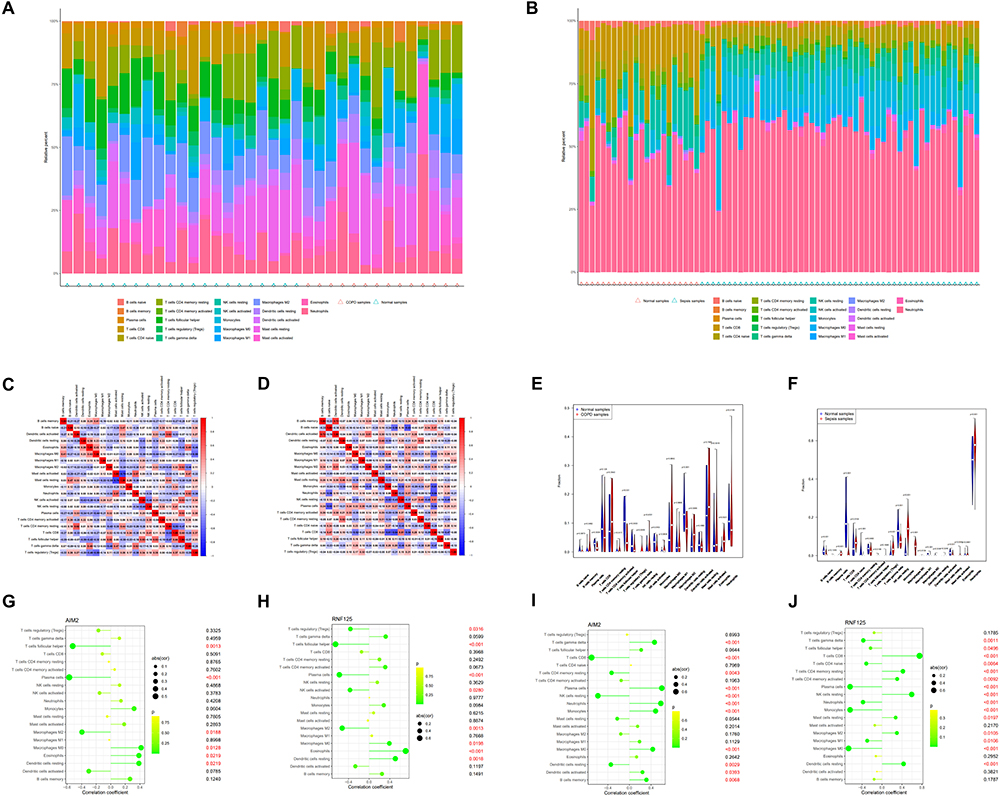 |
Figure 12 Analysis of immune cell infiltration. (A–D) Percentages of 22 immune cells identified by the CIBERSORT algorithm in the COPD dataset (GSE5058) and sepsis dataset (GSE95233). (E and F) Box plots showing the proportions of immune cells in COPD and sepsis patients versus controls. (G and H) Correlation of AIM2 (G) and RNF125 (H) with infiltrating immune cells in COPD and normal samples. (I and J) Correlation of AIM2 (I) and RNF125 (J) with infiltrating immune cells in sepsis and normal samples. |
In COPD samples, we observed dysregulation in B cells memory, plasma cells, activated CD4 memory T cells, follicular helper T cells, gamma delta T cells, activated NK cells, M2 macrophages, resting dendritic cells, and eosinophils compared to normal samples (Figure 12E). In sepsis samples, dysregulated levels were observed in naïve B cells, plasma cells, CD8 T cells, naïve CD4 T cells, resting and activated CD4 memory T cells, follicular helper T cells, gamma delta T cells, resting NK cells, monocytes, M0, M1, and M2 macrophages, resting dendritic cells, resting and activated mast cells, eosinophils, and neutrophils compared to normal samples (Figure 12F).
Next, we investigated the correlation between the shared biomarkers and immune cells. In COPD samples, AIM2 was negatively correlated with follicular helper T cells, plasma cells, and M2 macrophages, while it was positively correlated with M0 macrophages, eosinophils, and resting dendritic cells (Figure 12G). RNF125 exhibited a negative correlation with regulatory T cells (Tregs), follicular helper T cells, plasma cells, activated NK cells, and M2 macrophages, and a positive correlation with M0 macrophages, eosinophils, and resting dendritic cells (Figure 12H). In sepsis samples, AIM2 was negatively correlated with CD8 T cells, resting CD4 memory T cells, resting NK cells, and resting dendritic cells, but positively correlated with gamma delta T cells, plasma cells, neutrophils, monocytes, M0 macrophages, activated dendritic cells, and memory B cells (Figure 12I). RNF125 was negatively correlated with gamma delta T cells, follicular helper T cells, naïve CD4 T cells, plasma cells, neutrophils, monocytes, M1 and M0 macrophages, while it was positively correlated with CD8 T cells, resting CD4 memory T cells, activated CD4 memory T cells, resting NK cells, resting mast cells, M2 macrophages, and resting dendritic cells (Figure 12J).
These findings suggest that AIM2 and RNF125 are closely linked to immune cell types, particularly macrophages, dendritic cells, and regulatory T cells, underscoring their potential role in the immune landscape of both COPD and sepsis.
Construction of TF (Transcription Factors)-Gene and Gene-miRNA Regulatory Networks
Transcription factors (TFs) and microRNAs (miRNAs) play crucial roles in understanding disease progression. To identify the key transcriptional and post-transcriptional regulatory elements for the shared biomarkers, we constructed TF-gene and gene-miRNA networks, thereby deepening our insight into the regulatory mechanisms of the disease. The TF-gene network consists of 15 nodes and 17 edges (Figure 13A), while the gene-miRNA network includes 83 nodes and 86 edges (Figure 13B). In the TF-gene network, RELA, NFKB1, PPARG, and FOXC1 were found to interact with both hub genes, suggesting their potential as key transcriptional regulators. Similarly, in the gene-miRNA network, hsa-miR-196a-5p, hsa-miR-34a-5p, hsa-miR-124-3p, hsa-miR-21-5p, and hsa-miR-203a-3p were identified as interacting with the two hub genes, highlighting their possible role as post-transcriptional regulators. These findings suggest that these transcription factors and miRNAs may serve as common regulatory elements for the hub genes, though further validation is required to confirm their functional roles.
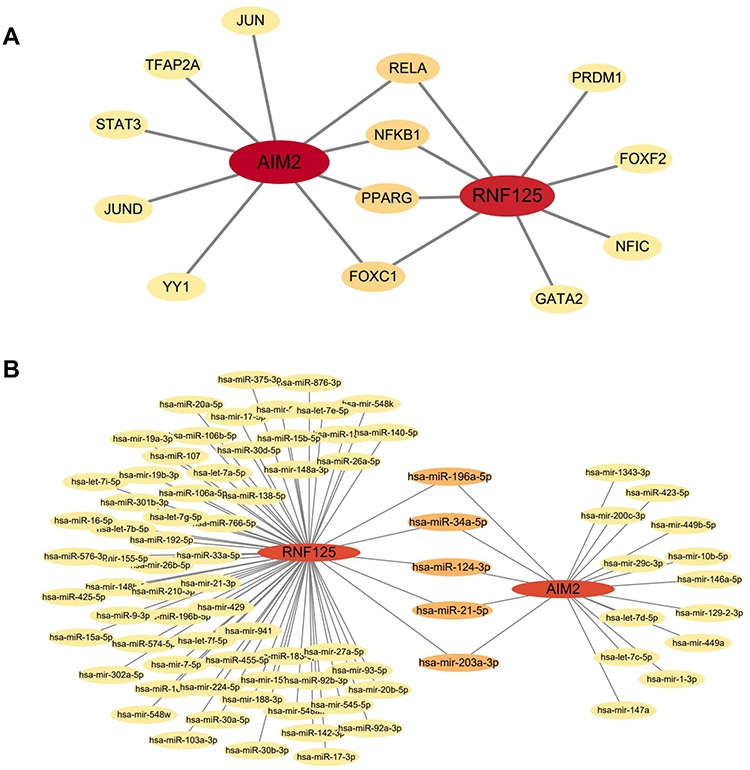 |
Figure 13 Gene-miRNA network of Tfs genes and two shared genes. (A) Regulatory network of Tfs genes for the two shared genes. (B) Gene-miRNA regulatory network for the two shared genes. |
Identification of Candidate Drugs
We utilized the DSigDB database on the Enrichr platform to search for potential drugs targeting the hub genes, resulting in the identification of 32 gene-drug associations, with detailed results provided in Supplementary Table S13. We extracted the top 10 drugs sorted by p-value, which include carbamazepine (HL60 DOWN), Gadodiamide hydrate (CTD 00002623), Melittin (CTD 00006261), metoclopramide (HL60 DOWN), benfotiamine (PC3 DOWN), isotretinoin (HL60 UP), MeIQx (CTD 00001739), iohexol (HL60 DOWN), suloctidil (HL60 UP), and Aizen uranine (BOSS) (Table 3).
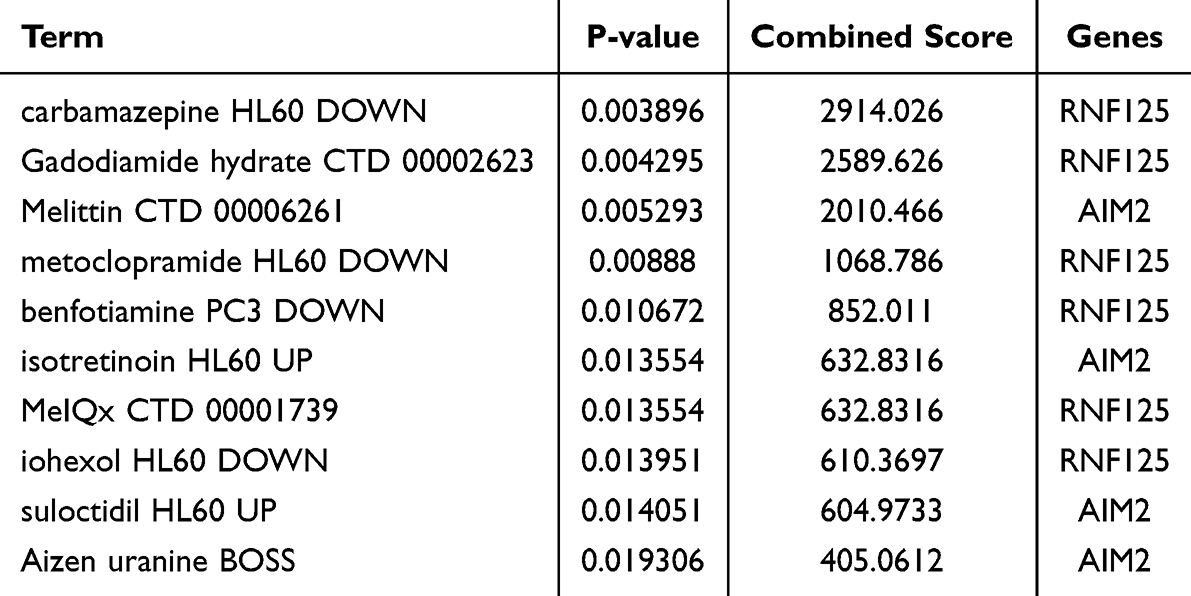 |
Table 3 Predicted Top 10 Drug Compounds |
Molecular docking was performed between the drug molecular targets identified and the corresponding key genes. Protein structures of the gene targets were downloaded from the PDB database, and molecular ligands of the drugs were retrieved from PubChem for docking analysis. The docking results indicated that the binding energies of the drug ligands with the potential gene targets were all below −4.5 kJ/mol, suggesting that the conformational energies of the drug ligands with the gene targets are low, indicating strong binding activity (Table 4). Notably, the three-dimensional structures of Gadodiamide hydrate (CTD 00002623) and Melittin (CTD 00006261) were not found in PubChem. Additionally, the docking results for suloctidil (HL60 UP) showed that no hydrogen bonds were formed between the drug molecule and the corresponding gene. Therefore, only seven drug targets were validated through molecular docking, as shown in Figure 14.
 |
Table 4 Corresponding IDs and Docking Binding Energies of Each Drug Ligand with Potential Gene Targets |
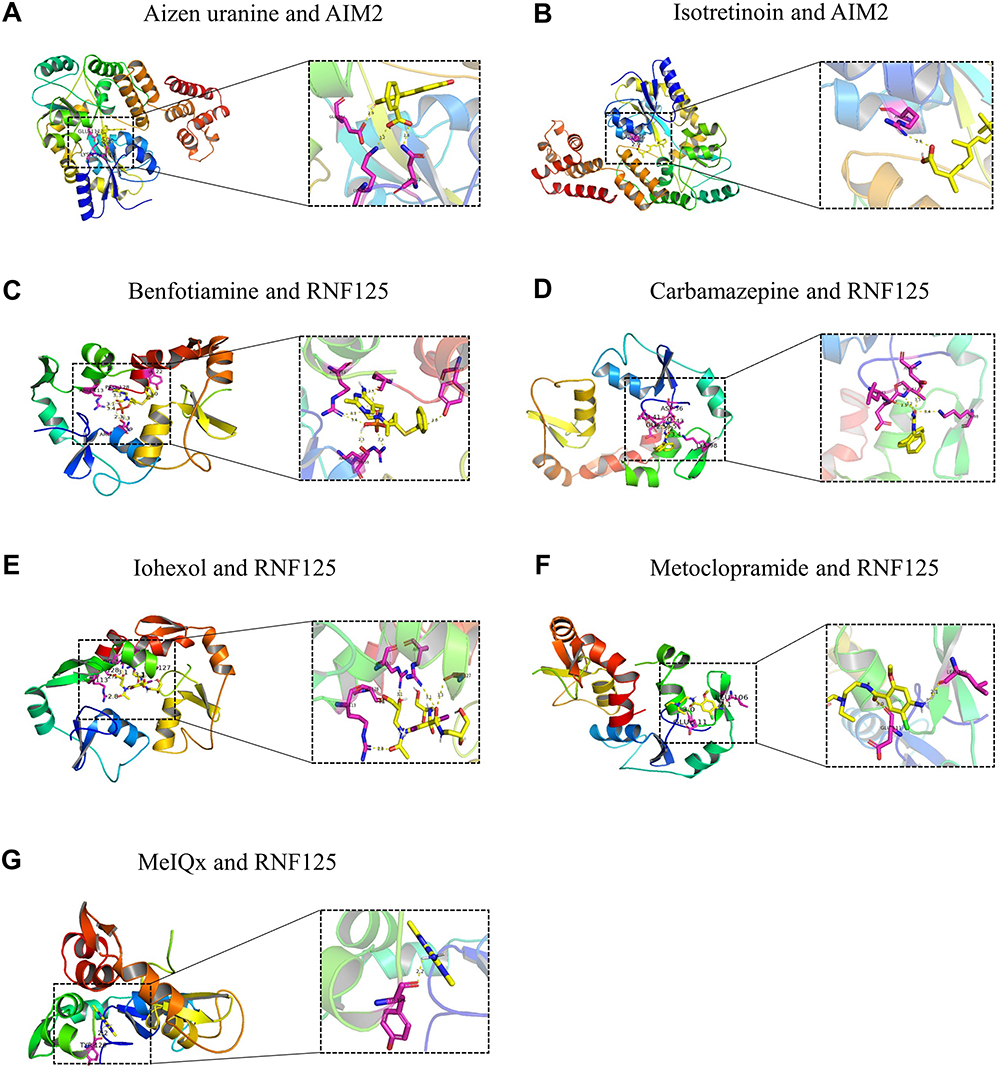 |
Figure 14 Molecular docking diagrams of seven drugs. (A) The molecular docking results of drug Aizen uranine and gene AIM2. (B) The molecular docking results of drug Isotretinoin and gene AlM2. (C) The molecular docking results of drug Benfotiamine and gene RNF125. (D) The molecular docking results of drug Carbamazepine and gene RNF125. (E) The molecular docking results of drug lohexol and gene RNF125. (F) The molecular docking results of drug Metoclopramide and gene RNF125. (G) The molecular docking results of drug MelOx and gene RNF125. |
Discussion
Chronic obstructive pulmonary disease (COPD) is a long-term respiratory condition characterized by persistent respiratory symptoms and airflow limitation, often accompanied by multiple complications.42,43 It is the third leading cause of death globally.44 Sepsis, a severe infection associated with high mortality, is closely linked to septic shock and multi-organ dysfunction.45 In recent years, the incidence of sepsis has risen, placing significant strain on healthcare systems.46 While several systematic reviews and meta-analyses have indicated a bidirectional relationship between COPD and sepsis, the underlying pathophysiology remains complex and not fully understood. Exploring the causal link between COPD and sepsis through Mendelian randomization (MR) analysis can help reduce the confounding effects of potential biases.
Bioinformatics analysis is a valuable tool for comprehensively understanding the genetic-level pathophysiological mechanisms of diseases. Although prior studies have investigated the mechanisms of COPD and sepsis independently, no studies have focused on their common mechanisms. This study integrates MR analysis with bioinformatics and machine learning methods to explore the shared transcriptional features between COPD and sepsis, with a particular focus on the mediating role of immune cells.
Initially, we used MR analysis to assess the role of immune cells as mediators in the increased risk of sepsis among COPD patients. Our goal was to explore the causal relationship between COPD and sepsis using existing genome-wide association study (GWAS) data, and to determine whether this relationship is mediated by immune cells. The results indicated that genetically predicted COPD is associated with a 14.3% increased risk of sepsis for each standard deviation increase in COPD severity. The mediation effects of immune cells were quantified as follows: CD33br HLA DR+ CD14- %CD33br HLA DR+ accounted for 6.5%, BAFF-R on IgD-CD38br for 12.8%, and CD45 on CD33- HLA DR+ for 3.9%. To our knowledge, this is the first study to use MR to investigate the role of immune cells in mediating the increased risk of sepsis in COPD. These findings align with previous observational studies that have shown a strong association between postoperative sepsis and COPD, particularly in surgeries such as craniotomy, left ventricular surgery, and hip fracture procedures.47–49 However, unlike observational studies, which are prone to reverse causality and confounding factors, our MR approach provides stronger causal inference.
Nevertheless, our study has several limitations. First, the analysis was conducted on a European population, which limits its generalizability to other populations. Second, the GWAS dataset for sepsis included a relatively small number of sepsis cases, and future studies would benefit from larger datasets for validation. Third, although we took measures to identify and remove pleiotropic effects, the possibility that pleiotropy influenced our results cannot be entirely ruled out. Fourth, the use of aggregate-level statistics instead of individual-level data precluded subgroup analyses, such as sex-specific causal relationships. Lastly, while we identified genetic prediction rates for immune cell-mediated COPD at 6.5%, 12.8%, and 3.9%, these rates are relatively low, suggesting the need for further research to identify additional mediators.
We identified 112 differentially expressed genes (DEGs) and 130 disease-associated module genes shared between COPD and sepsis. Functional enrichment analysis revealed that these genes were predominantly involved in innate and adaptive immune responses, as well as inflammatory processes. Collectively, these findings suggest that the regulation of immune response and cytokine secretion may be a crucial link between COPD and sepsis. By applying two machine learning algorithms (LASSO and SVM-RFE), we identified two key diagnostic genes, AIM2 and RNF125, for both diseases. The expression of these genes was significantly higher in COPD patients compared to the control group. In sepsis patients, AIM2 expression was elevated, while RNF125 expression was reduced relative to the control group. Furthermore, receiver operating characteristic (ROC) analysis demonstrated their potential as diagnostic biomarkers for COPD and sepsis, with area under the curve (AUC) values exceeding 0.80, underscoring their vital role in the diagnosis and progression of these conditions.
AIM2 (Absent in Melanoma 2), located on the 1q23 region of the human chromosome, consists of an N-terminal pyrin domain (PYD) and a C-terminal hematopoietic interferon-induced nucleoprotein domain (HIN200), which is a cytosolic DNA receptor belonging to the HIN-200 protein family. It is primarily localized in the cytoplasm, where the HIN domain recognizes double-stranded DNA (dsDNA). The PYD domain recruits and binds to apoptosis-associated speck-like proteins (ASC) to assemble inflammasomes.50–53 When AIM2 binds dsDNA, it recruits ASC and pro-Caspase-1, forming inflammatory complexes that lead to the secretion of pro-inflammatory cytokines IL-1β and IL-18, thus triggering pyroptosis.54–56 AIM2 plays a role in cancer,57 autoimmune diseases,58 infectious diseases,59 and cardiovascular conditions.60,61 It can induce antigen-specific antibody responses via CD8+ T cells, enhancing tumor treatment.62 Elevated AIM2 expression in B cells has been observed in systemic lupus erythematosus (SLE), where its regulation of the Bcl6-Blimp-1 signaling axis promotes B cell differentiation, positioning AIM2 as a therapeutic target for SLE.63 In atherosclerotic stroke, AIM2 activation by circulating free dsDNA contributes to plaque instability and thrombosis, exacerbating post-stroke inflammation.60,61
AIM2 also plays a pivotal role in immune cell function. In macrophages, it detects cytoplasmic dsDNA, activates inflammasomes, and promotes IL-1β and IL-18 secretion, inducing pyroptosis and contributing to innate immunity.58 In dendritic cells, AIM2 enhances pro-inflammatory effects, promotes T cell activation and differentiation, and influences adaptive immunity.64 Additionally, AIM2 helps maintain the function and stability of Treg cells, potentially alleviating autoimmune diseases.65,66 Our findings similarly show that AIM2 is closely associated with macrophages, dendritic cells, and Treg cells in both COPD and sepsis, and its regulation of these immune cells may offer a therapeutic strategy for these diseases.
RNF125 (Ring Finger Protein 125), located on chromosome 17, is an E3 ubiquitin ligase that regulates protein stability and function through ubiquitination.67 It modifies the TRIM14 protein via ubiquitination, affecting its role in antiviral immunity.68 RNF125 regulates various signaling pathways, including Wnt/β-catenin, and plays a significant role in osteoarthritis development.69 As a tumor suppressor, RNF125 is downregulated in some cancers and is involved in cell cycle control and tumor inhibition by ubiquitinating p53 and other proteins.70 Moreover, RNF125 promotes PD-L1 degradation through ubiquitination, enhancing the tumor immune response.71 In the tumor microenvironment, high RNF125 expression correlates with increased infiltration of T cells and macrophages, suggesting that RNF125 regulates immune cell activity and tumor immune escape by modulating PD-L1 expression.71 Our results indicate that RNF125 is closely linked to macrophages and T cells in both COPD and sepsis, and its regulation of these cells may provide a promising therapeutic approach for both diseases.
Both COPD and sepsis are strongly associated with pathogen infections. Infections are a major cause of COPD exacerbations,72,73 and they represent one of the leading causes of sepsis.74,75 Studies have shown that the systemic immune inflammatory index (SII) is closely related to COPD, with higher SII levels linked to increased COPD prevalence and a heightened risk of all-cause mortality in COPD patients.76 Infections trigger immune responses, which are closely associated with the function of various immune cells. Although direct research on the link between COPD and sepsis is limited, the pathophysiological connection between the two diseases is well established and is heavily influenced by immune cells. Long-term use of oral or inhaled corticosteroids has been shown to increase the risk of sepsis in COPD patients, potentially due to immunosuppression.77,78 Moreover, research suggests that macrophages play a significant role in both COPD and sepsis.79 Our study further supports that the monocyte-macrophage system acts as an essential bridge between COPD and sepsis. Immune infiltration analysis also revealed that the monocyte-macrophage system is strongly correlated with the expression levels of the candidate biomarkers AIM2 and RNF125.
The results demonstrated that RELA, NFKB1, PPARG, FOXC1, has-miR-196a-5p, has-miR-34a-5p, has-miR-124-3p, has-miR-21-5p, and has-miR-203a-3p are key regulators of the two shared genes. RELA (also known as p65), a vital member of the NF-κB family,80 plays a crucial role in regulating inflammatory responses, cell proliferation, and differentiation,81 and is closely associated with tumors,82 metabolic diseases,83 and immune-related disorders.84 NFKB1 (also known as p50), another member of the NF-κB family, is integral to cytokine signaling, inflammatory responses, and cell survival. It can form dimers that bind to NF-κB response elements, modulating target gene expression, influencing the cell cycle, and inhibiting apoptosis.85 PPARG encodes peroxisome proliferator-activated receptor γ (PPARγ), a nuclear receptor involved in adipocyte differentiation, glucose homeostasis, and inflammatory responses.86–88 It promotes fatty acid oxidation and inhibits NF-κB-mediated pro-inflammatory responses to maintain intestinal homeostasis.89 FOXC1 encodes forkhead frame C1 (FoxC1), a transcription factor involved in cell migration, angiogenesis, and tumor development, with abnormalities linked to ocular diseases and cancers.90–93
The identified miRNAs also play significant roles: miR-196a-5p regulates gene expression by binding to the 3′ untranslated region of target mRNA and promotes tumor growth by targeting HOXA5, driving proliferation and invasion of non-small cell lung cancer cells.94–96 miR-34a-5p is involved in regulating the cell cycle, differentiation, and apoptosis, acting as a tumor suppressor by inhibiting pro-tumor genes.97,98 miR-124-3p plays a critical role in the development and maintenance of the nervous system, influencing neuronal cell survival, differentiation, and synaptic plasticity through the regulation of various target genes.99,100 miR-21-5p affects cell proliferation, apoptosis, migration, and angiogenesis, and is strongly linked to cancer, cardiovascular diseases, and neurodegenerative disorders.101–104 miR-203a-3p regulates cell proliferation, differentiation, and apoptosis, and its dysregulated expression across various cancers affects tumor development and treatment outcomes by targeting different genes.105–107
Currently, research on these regulators in the context of COPD and sepsis is limited. However, based on our analysis of the co-regulators, we hypothesize that the connection between these diseases may be mediated through the NF-κB pathway, apoptosis, and processes of cell proliferation and differentiation. Additionally, using DSigDB, we predicted 10 potential drug compounds based on the two hub genes. Molecular docking showed stable binding for seven of these compounds, further suggesting their therapeutic potential for COPD and sepsis. Nevertheless, the efficacy and pathways of these drugs need further validation through cellular experiments and clinical trials.
There are some limitations to our study. First, the dataset used was relatively small. Additionally, this study lacked data on patients with both COPD and sepsis. Future studies should incorporate larger sample sizes and consider comorbidity datasets. Finally, these results require further validation through in vivo and in vitro experiments. Despite these limitations, our preliminary identification of the two biomarkers provides a foundation for future research and the generation of new hypotheses.
This study is the first to explore the relationship between COPD and sepsis using bioinformatics methods, with immune cells serving as an intermediate factor. The results are supported by robust statistical analyses and align with current observational evidence, highlighting the innovative nature of our approach. By employing a variety of analytical methods, we thoroughly explored the genes and regulatory factors related to COPD, sepsis, and immune cells. We also predicted relevant cell signaling pathways and potential therapeutic drug targets, offering new directions for basic and clinical research. Our findings provide valuable bioinformatics evidence for further investigation into COPD and sepsis, contributing novel insights for future research and development in this field.
Conclusion
In MR studies, we identified a causal relationship between COPD and sepsis, as well as the proportion of immune cell-mediated effects. However, further investigation is required to explore other risk factors that may serve as potential mediators. Clinically, more attention should be given to peripheral blood markers of sepsis in patients with COPD. Through GEO analysis, we identified two diagnostic genes (AIM2 and RNF125) that are shared between COPD and sepsis, along with co-regulatory pathways and common immune characteristics. Furthermore, we screened regulatory molecules and targeted drugs based on the two hub genes, offering a novel strategy for the pharmacological treatment of both COPD and sepsis. These findings provide a theoretical foundation for understanding the molecular mechanisms underlying the comorbidity of COPD and sepsis from multiple perspectives, including genetics, signaling pathways, and immune infiltration.
Abbreviations
AC, Absolute cell; AIM2, Absent in Melanoma 2; ASC, Apoptosis-associated speck-like proteins; AUC, Area under the curve; BP, Biological process; CARS, Compensatory anti-inflammatory response syndrome; CC, Cellular component; CDCs, Classic dendritic cells; COPD, Chronic obstructive pulmonary disease; DEGs, Differentially expressed genes; FC, Fold change; FoxC1, forkhead frame C1; GO, Gene ontology; GS, Gene significance; dsDNA, Double-stranded DNA; GSE, Gene Expression Omnibus Series; GWAS, Genome-wide association study; IVs, Instrumental variables; KEGG, Kyoto encyclopedia of genes and genomes; LASSO, Logical Regression of Selection Operators; LD, Linkage disequilibrium; logFC, log fold change; MAF, Minor allele frequency; MF, Molecular function; MFI, Median fluorescence intensities; miRNA, microRNAs; MM, Module membership; MOF, Multiple organ failure; MP, Morphological parameters; MR, Mendelian randomization; MR-PRESSO, MR-pleiotropic residuals and outliers; MR.RAPS, MR robustly adjusted profile scores; OR, Odds ratio; PPARγ, peroxisome proliferator-activated receptor γ; PYD, Pyrin domain; RC, Relative cell; SD, Standard deviation; RFE, Recursive Feature Elimination; SLE, Systemic lupus erythematosus; RNF125, Ring Finger Protein 125; ROC, Receiver operating characteristic; SII, Systemic immune inflammatory index; SIRS, Systemic inflammatory response syndrome; SNPs, Single nucleotide polymorphisms; SVM, Support Vector Machine; SVM-RFE, Support vector machine recursive feature elimination; TFs, Transcription factors; TOM, Topological overlap matrix; WGCNA, Weighted gene co-expression network analysis.
Data Sharing Statement
The datasets generated and/or analysed during the current study are available in the GWAS database (https://gwas.mrcieu.ac.uk/), FinnGen Biobank (FREEZE 11; https://r11.finngen.fi/), Gene Expression Omnibus (GEO) repository (https://www.ncbi.nlm.nih.gov/gds/?term=), PubChem (https://pubchem.ncbi.nlm.nih.gov/) and Protein Data Bank (PDB; https://www.rcsb.org/).
Ethics Approval and Consent to Participate
This study was conducted according to the guidelines laid down in the Declaration of Helsinki, and it has been reviewed and approved by the Ethics Committee of the Third Xiangya Hospital of Central South University (25225).
Author Contributions
XL, YX and MZ designed this study. XL, YX, MY, XZ, ZY, ZZ and MZ reviewed and revised the manuscript. XL and YX drafted the original manuscript. XL and MY designed and completed the figures and tables. All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research was supported by the Wisdom Accumulation and Talent Cultivation Project of the Third Xiangya Hospital of Central South University (No. YX202212), National Natural Science Foundation of China (No.82102280), Natural Scientific Foundation of Hunan Province (No.2022JJ30893), National College Students’ Innovation and Entrepreneurship Training Program (No. X202410533614).
Disclosure
Xinyi Li and Yuyang Xiao are co-first authors for this study. The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. Segal LN, Martinez FJ. Chronic obstructive pulmonary disease subpopulations and phenotyping. J Allergy Clin Immunol. 2018;141(6):1961–1971. doi:10.1016/j.jaci.2018.02.035
2. Labaki WW, Rosenberg SR. Chronic obstructive pulmonary disease. Ann Intern Med. 2020;173(3):Itc17–Itc32. doi:10.7326/AITC202008040
3. Christenson SA, Smith BM, Bafadhel M, Putcha N. Chronic obstructive pulmonary disease. Lancet. 2022;399(10342):2227–2242. doi:10.1016/S0140-6736(22)00470-6
4. Sandelowsky H, Weinreich UM, Aarli BB, et al. COPD - do the right thing. BMC Fam Pract. 2021;22(1):244. doi:10.1186/s12875-021-01583-w
5. Agusti A, Böhm M, Celli B, et al. GOLD COPD DOCUMENT 2023: a brief update for practicing cardiologists. Clin Res Cardiol. 2024;113(2):195–204. doi:10.1007/s00392-023-02217-0
6. Venkatesan P. GOLD COPD report: 2025 update. Lancet Respir Med. 2025;13(1):e7–e8. doi:10.1016/S2213-2600(24)00413-2
7. Kunisaki KM, Dransfield MT, Anderson JA, et al. Exacerbations of chronic obstructive pulmonary disease and cardiac events. A post hoc cohort analysis from the SUMMIT randomized clinical trial. Am J Respir Crit Care Med. 2018;198(1):51–57. doi:10.1164/rccm.201711-2239OC
8. Wang M, Lin EP-Y, Huang L-C, Li C-Y, Shyr Y, Lai C-H. Mortality of cardiovascular events in patients with COPD and preceding hospitalization for acute exacerbation. Chest. 2020;158(3):973–985. doi:10.1016/j.chest.2020.02.046
9. Liu AC, Patel K, Vunikili RD, et al. Sepsis in the era of data-driven medicine: personalizing risks, diagnoses, treatments and prognoses. Brief Bioinform. 2020;21(4):1182–1195. doi:10.1093/bib/bbz059
10. Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. 2016;315(8):801–810. doi:10.1001/jama.2016.0287
11. Clere-Jehl R, Mariotte A, Meziani F, Bahram S, Georgel P, Helms J. JAK-STAT targeting offers novel therapeutic opportunities in sepsis. Trends Mol Med. 2020;26(11):987–1002. doi:10.1016/j.molmed.2020.06.007
12. Rudd KE, Johnson SC, Agesa KM, et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the Global Burden of Disease Study. Lancet. 2020;395(10219):200–211. doi:10.1016/S0140-6736(19)32989-7
13. Maslove DM, Tang B, Shankar-Hari M, et al. Redefining critical illness. Nat Med. 2022;28(6):1141–1148. doi:10.1038/s41591-022-01843-x
14. Laupland KB, Kirkpatrick AW, Delaney A. Polyclonal intravenous immunoglobulin for the treatment of severe sepsis and septic shock in critically ill adults: a systematic review and meta-analysis. Crit Care Med. 2007;35(12):2686–2692.
15. Chen Y, Lu L, Li X, et al. Association between chronic obstructive pulmonary disease and 28-day mortality in patients with sepsis: a retrospective study based on the MIMIC-III database. BMC Pulm Med. 2023;23(1):435. doi:10.1186/s12890-023-02729-5
16. Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23(R1):R89–98. doi:10.1093/hmg/ddu328
17. Orrù V, Steri M, Sidore C, et al. Complex genetic signatures in immune cells underlie autoimmunity and inform therapy. Nat Genet. 2020;52(10):1036–1045. doi:10.1038/s41588-020-0684-4
18. Sidore C, Busonero F, Maschio A, et al. Genome sequencing elucidates Sardinian genetic architecture and augments association analyses for lipid and blood inflammatory markers. Nat Genet. 2015;47(11):1272–1281. doi:10.1038/ng.3368
19. Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–210. doi:10.1093/nar/30.1.207
20. Abecasis GR, Altshuler D, Auton A, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467(7319):1061–1073.
21. Hemani G, Zheng J, Elsworth B, et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife. 2018;7. doi:10.7554/eLife.34408
22. Li BB, Martin EB. An approximation to the F distribution using the chi-square distribution. Comput Stat Data Anal. 2002;40(1):21–26. doi:10.1016/S0167-9473(01)00097-4
23. Burgess S, Thompson SG. Avoiding bias from weak instruments in Mendelian randomization studies. Int J Epidemiol. 2011;40(3):755–764. doi:10.1093/ije/dyr036
24. Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37(7):658–665. doi:10.1002/gepi.21758
25. Burgess S, Thompson SG. Interpreting findings from Mendelian randomization using the MR-Egger method. Eur J Epidemiol. 2017;32(5):377–389. doi:10.1007/s10654-017-0255-x
26. Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40(4):304–314. doi:10.1002/gepi.21965
27. Zhang Y, Liu Z, Choudhury T, Cornelis MC, Liu W. Habitual coffee intake and risk for nonalcoholic fatty liver disease: a two-sample Mendelian randomization study. Eur J Nutr. 2021;60(4):1761–1767. doi:10.1007/s00394-020-02369-z
28. Carter AR, Sanderson E, Hammerton G, et al. Mendelian randomisation for mediation analysis: current methods and challenges for implementation. Eur J Epidemiol. 2021;36(5):465–478. doi:10.1007/s10654-021-00757-1
29. Lynch M, Walsh B. Genetics and Analysis of Quantitative Traits. Sunderland, MA: Sinauer; 1998.
30. Hemani G, Tilling K, Davey Smith G. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet. 2017;13(11):e1007081. doi:10.1371/journal.pgen.1007081
31. Tan JS, Liu NN, Guo TT, Hu S, Hua L. Genetically predicted obesity and risk of deep vein thrombosis. Thromb Res. 2021;207:16–24. doi:10.1016/j.thromres.2021.08.026
32. Tan JS, Ren JM, Fan L, et al. Genetic Predisposition Of Anti-Cytomegalovirus Immunoglobulin G levels and the risk of 9 cardiovascular diseases. Front Cell Infect Microbiol. 2022;12:884298. doi:10.3389/fcimb.2022.884298
33. Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50(5):693–698. doi:10.1038/s41588-018-0099-7
34. Ravasz E, Somera AL, Mongru DA, Oltvai ZN, Barabási AL. Hierarchical organization of modularity in metabolic networks. Science. 2002;297(5586):1551–1555. doi:10.1126/science.1073374
35. Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13. doi:10.1093/nar/gkn923
36. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi:10.1038/nprot.2008.211
37. Yoo M, Shin J, Kim J, et al. DSigDB: drug signatures database for gene set analysis. Bioinformatics. 2015;31(18):3069–3071. doi:10.1093/bioinformatics/btv313
38. Wang Y, Suzek T, Zhang J, et al. PubChem BioAssay: 2014 update. Nucleic Acids Res. 2014;42(Database issue):D1075–82. doi:10.1093/nar/gkt978
39. Berman HM, Westbrook J, Feng Z, et al. The protein data bank. Nucleic Acids Res. 2000;28(1):235–242. doi:10.1093/nar/28.1.235
40. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. doi:10.1002/jcc.21334
41. Broadbent JR, Foley CN, Grant AJ, Mason AM, Staley JR, Burgess S. MendelianRandomization v0.5.0: updates to an R package for performing Mendelian randomization analyses using summarized data. Wellcome Open Res. 2020;5:252. doi:10.12688/wellcomeopenres.16374.2
42. Kahnert K, Jörres RA, Behr J, Welte T. The diagnosis and treatment of COPD and its comorbidities. Dtsch Arztebl Int. 2023;120(25):434–444. doi:10.3238/arztebl.m2023.027
43. Negewo NA, Gibson PG, McDonald VM. COPD and its comorbidities: impact, measurement and mechanisms. Respirology. 2015;20(8):1160–1171. doi:10.1111/resp.12642
44. Lozano R, Naghavi M, Foreman K, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380(9859):2095–2128. doi:10.1016/S0140-6736(12)61728-0
45. Esposito S, De Simone G, Boccia G, De Caro F, Pagliano P. Sepsis and septic shock: new definitions, new diagnostic and therapeutic approaches. J Glob Antimicrob Resist. 2017;10:204–212. doi:10.1016/j.jgar.2017.06.013
46. Srzić I, Nesek Adam V, Tunjić Pejak D. Sepsis definition: what’s new in the treatment guidelines. Acta Clin Croat. 2022;61(Suppl 1):67–72. doi:10.20471/acc.2022.61.s1.11
47. Zhang J, Li YI, Pieters TA, et al. Sepsis and septic shock after craniotomy: predicting a significant patient safety and quality outcome measure. PLoS One. 2020;15(9):e0235273. doi:10.1371/journal.pone.0235273
48. Gonuguntla K, Patil S, Gadela NV, et al. Trends, Outcomes, and Predictors of Sepsis and Severe Sepsis in Patients with Left Ventricular Assist Devices. Cureus. 2020;12(4):e7523. doi:10.7759/cureus.7523
49. Gonzalez CA, O’Mara A, Cruz JP, Roth D, Van Rysselberghe NL, Gardner MJ. Postoperative sepsis and septic shock after Hip fracture surgery. Injury. 2023;54(8):110833. doi:10.1016/j.injury.2023.05.064
50. Jin T, Perry A, Jiang J, et al. Structures of the HIN domain: DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity. 2012;36(4):561–571. doi:10.1016/j.immuni.2012.02.014
51. Wang B, Yin Q. AIM2 inflammasome activation and regulation: a structural perspective. J Struct Biol. 2017;200(3):279–282. doi:10.1016/j.jsb.2017.08.001
52. Wang B, Tian Y, Yin Q. AIM2 inflammasome assembly and signaling. Adv Exp Med Biol. 2019;1172:143–155.
53. Wang B, Bhattacharya M, Roy S, Tian Y, Yin Q. Immunobiology and structural biology of AIM2 inflammasome. Mol Aspects Med. 2020;76:100869. doi:10.1016/j.mam.2020.100869
54. Hornung V, Ablasser A, Charrel-Dennis M, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458(7237):514–518. doi:10.1038/nature07725
55. Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458(7237):509–513. doi:10.1038/nature07710
56. Roberts TL, Idris A, Dunn JA, et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science. 2009;323(5917):1057–1060. doi:10.1126/science.1169841
57. Choubey D. Absent in melanoma 2 proteins in the development of cancer. Cell Mol Life Sci. 2016;73(23):4383–4395. doi:10.1007/s00018-016-2296-9
58. Zhu H, Zhao M, Chang C, Chan V, Lu Q, Wu H. The complex role of AIM2 in autoimmune diseases and cancers. Immun Inflamm Dis. 2021;9(3):649–665. doi:10.1002/iid3.443
59. Zhu W, Zu X, Liu S, Zhang H. The absent in melanoma 2 (AIM2) inflammasome in microbial infection. Clin Chim Acta. 2019;495:100–108. doi:10.1016/j.cca.2019.04.052
60. Du L, Wang X, Chen S, Guo X. The AIM2 inflammasome: a novel biomarker and target in cardiovascular disease. Pharmacol Res. 2022;186:106533. doi:10.1016/j.phrs.2022.106533
61. Zhao ZZ, Zheng XL, Jiang ZS. Emerging roles of absent in melanoma 2 in cardiovascular diseases. Clin Chim Acta. 2020;511:14–23. doi:10.1016/j.cca.2020.08.031
62. Fukuda K, Okamura K, Riding RL, et al. AIM2 regulates anti-tumor immunity and is a viable therapeutic target for melanoma. J Exp Med. 2021;218(9). doi:10.1084/jem.20200962
63. Yang M, Long D, Hu L, et al. AIM2 deficiency in B cells ameliorates systemic lupus erythematosus by regulating Blimp-1-Bcl-6 axis-mediated B-cell differentiation. Signal Transduct Target Ther. 2021;6(1):341. doi:10.1038/s41392-021-00725-x
64. Corrales L, Woo SR, Williams JB, McWhirter SM, Dubensky TW, Gajewski TF. Antagonism of the STING Pathway via Activation of the AIM2 Inflammasome by Intracellular DNA. J Immunol. 2016;196(7):3191–3198. doi:10.4049/jimmunol.1502538
65. Lozano-Ruiz B, Tzoumpa A, Martínez-Cardona C, et al. Absent in Melanoma 2 (AIM2) regulates the stability of regulatory T cells. Int J Mol Sci. 2022;23(4):2230. doi:10.3390/ijms23042230
66. Chou WC, Guo Z, Guo H, et al. AIM2 in regulatory T cells restrains autoimmune diseases. Nature. 2021;591(7849):300–305. doi:10.1038/s41586-021-03231-w
67. Zhao H, Li CC, Pardo J, et al. A novel E3 ubiquitin ligase TRAC-1 positively regulates T cell activation. J Immunol. 2005;174(9):5288–5297. doi:10.4049/jimmunol.174.9.5288
68. Jia X, Zhou H, Wu C, et al. The ubiquitin ligase RNF125 targets innate immune adaptor protein TRIM14 for ubiquitination and degradation. J Immunol. 2017;198(12):4652–4658. doi:10.4049/jimmunol.1601322
69. Lv R, Du L, Bai L. RNF125, transcriptionally regulated by NFATC2, alleviates osteoarthritis via inhibiting the Wnt/β-catenin signaling pathway through degrading TRIM14. Int Immunopharmacol. 2023;125(Pt B):111191. doi:10.1016/j.intimp.2023.111191
70. Yang L, Zhou B, Li X, et al. RNF125 is a ubiquitin-protein ligase that promotes p53 degradation. Cell Physiol Biochem. 2015;35(1):237–245. doi:10.1159/000369691
71. Wei M, Mo Y, Liu J, et al. Ubiquitin ligase RNF125 targets PD-L1 for ubiquitination and degradation. Front Oncol. 2022;12:835603. doi:10.3389/fonc.2022.835603
72. Wedzicha JA, Seemungal TA. COPD exacerbations: defining their cause and prevention. Lancet. 2007;370(9589):786–796. doi:10.1016/S0140-6736(07)61382-8
73. Cafferkey J, Coultas JA, Mallia P. Human rhinovirus infection and COPD: role in exacerbations and potential for therapeutic targets. Expert Rev Respir Med. 2020;14(8):777–789. doi:10.1080/17476348.2020.1764354
74. Rowe TA, McKoy JM. Sepsis in older adults. Infect Dis Clin North Am. 2017;31(4):731–742. doi:10.1016/j.idc.2017.07.010
75. Lin HY. The severe COVID-19: a sepsis induced by viral infection? And its immunomodulatory therapy. Chin J Traumatol. 2020;23(4):190–195. doi:10.1016/j.cjtee.2020.06.002
76. Ye C, Yuan L, Wu K, Shen B, Zhu C. Association between systemic immune-inflammation index and chronic obstructive pulmonary disease: a population-based study. BMC Pulm Med. 2023;23(1):295. doi:10.1186/s12890-023-02583-5
77. Ernst P, Coulombe J, Brassard P, Suissa S. The risk of sepsis with inhaled and oral corticosteroids in patients with COPD. COPD. 2017;14(2):137–142. doi:10.1080/15412555.2016.1238450
78. Wang CY, Lin YS, Wang YH, et al. Risk of sepsis among patients with COPD treated with fixed combinations of inhaled corticosteroids and long-acting Beta2 agonists. Aging. 2019;11(17):6863–6871. doi:10.18632/aging.102217
79. Arora S, Dev K, Agarwal B, Das P, Syed MA. Macrophages: their role, activation and polarization in pulmonary diseases. Immunobiology. 2018;223(4–5):383–396. doi:10.1016/j.imbio.2017.11.001
80. Schmitz ML, Baeuerle PA. The p65 subunit is responsible for the strong transcription activating potential of NF-kappa B. EMBO J. 1991;10(12):3805–3817. doi:10.1002/j.1460-2075.1991.tb04950.x
81. Chen S, Jiang S, Zheng W, et al. RelA/p65 inhibition prevents tendon adhesion by modulating inflammation, cell proliferation, and apoptosis. Cell Death Dis. 2017;8(3):e2710. doi:10.1038/cddis.2017.135
82. Lomert E, Turoverova L, Kriger D, et al. Co-expression of RelA/p65 and ACTN4 induces apoptosis in non-small lung carcinoma cells. Cell Cycle. 2018;17(5):616–626. doi:10.1080/15384101.2017.1417709
83. Sun HJ, Xiong SP, Cao X, et al. Polysulfide-mediated sulfhydration of SIRT1 prevents diabetic nephropathy by suppressing phosphorylation and acetylation of p65 NF-κB and STAT3. Redox Biol. 2021;38:101813. doi:10.1016/j.redox.2020.101813
84. Zhang S, Xu P, Zhu Z, et al. Acetylation of p65(Lys310) by p300 in macrophages mediates anti-inflammatory property of berberine. Redox Biol. 2023;62:102704. doi:10.1016/j.redox.2023.102704
85. Concetti J, Wilson CL. NFKB1 and Cancer: friend or Foe? Cells. 2018;7(9):133. doi:10.3390/cells7090133
86. Debril MB, Renaud JP, Fajas L, Auwerx J. The pleiotropic functions of peroxisome proliferator-activated receptor gamma. J Mol Med. 2001;79(1):30–47. doi:10.1007/s001090000145
87. Jabbari P, Sadeghalvad M, Rezaei N. An inflammatory triangle in Sarcoidosis: PPAR-γ, immune microenvironment, and inflammation. Expert Opin Biol Ther. 2021;21(11):1451–1459. doi:10.1080/14712598.2021.1913118
88. Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem. 2008;77:289–312. doi:10.1146/annurev.biochem.77.061307.091829
89. Le Loupp AG, Bach-Ngohou K, Bourreille A, et al. Activation of the prostaglandin D2 metabolic pathway in Crohn’s disease: involvement of the enteric nervous system. BMC Gastroenterol. 2015;15:112. doi:10.1186/s12876-015-0338-7
90. Elian FA, Yan E, Walter MA. FOXC1, the new player in the cancer sandbox. Oncotarget. 2018;9(8):8165–8178. doi:10.18632/oncotarget.22742
91. Yang Z, Jiang S, Cheng Y, et al. FOXC1 in cancer development and therapy: deciphering its emerging and divergent roles. Ther Adv Med Oncol. 2017;9(12):797–816. doi:10.1177/1758834017742576
92. Wiggs JL, Pasquale LR. Genetics of glaucoma. Hum Mol Genet. 2017;26(R1):R21–r7. doi:10.1093/hmg/ddx184
93. Kume T. The cooperative roles of Foxc1 and Foxc2 in cardiovascular development. Adv Exp Med Biol. 2009;665:63–77.
94. Zhu Y, Tang Y, Fan Y, Wu D. MiR-196a-5p facilitates progression of estrogen-dependent endometrial cancer by regulating FOXO1. Histol Histopathol. 2023;38(10):1157–1168. doi:10.14670/HH-18-572
95. Yao F, Shi W, Fang F, et al. Exosomal miR-196a-5p enhances radioresistance in lung cancer cells by downregulating NFKBIA. Kaohsiung J Med Sci. 2023;39(6):554–564. doi:10.1002/kjm2.12673
96. Bao M, Pan S, Yang W, Chen S, Shan Y, Shi H. Serum miR-10a-5p and miR-196a-5p as non-invasive biomarkers in non-small cell lung cancer. Int J Clin Exp Pathol. 2018;11(2):773–780.
97. Chen S, Yuan M, Chen H, et al. MiR-34a-5p suppresses cutaneous squamous cell carcinoma progression by targeting SIRT6. Arch Dermatol Res. 2024;316(6):299. doi:10.1007/s00403-024-03106-w
98. Li W, Pan T, Jiang W, Zhao H. HCG18/miR-34a-5p/HMMR axis accelerates the progression of lung adenocarcinoma. Biomed Pharmacother. 2020;129:110217. doi:10.1016/j.biopha.2020.110217
99. Yelick J, Men Y, Jin S, Seo S, Espejo-Porras F, Yang Y. Elevated exosomal secretion of miR-124-3p from spinal neurons positively associates with disease severity in ALS. Exp Neurol. 2020;333:113414. doi:10.1016/j.expneurol.2020.113414
100. Jiang M, Zhang X, Wang X, et al. MicroRNA-124-3p attenuates the development of nerve injury-induced neuropathic pain by targeting early growth response 1 in the dorsal root ganglia and spinal dorsal horn. J Neurochem. 2021;158(4):928–942. doi:10.1111/jnc.15433
101. Ma S, Zhang A, Li X, et al. MiR-21-5p regulates extracellular matrix degradation and angiogenesis in TMJOA by targeting Spry1. Arthritis Res Ther. 2020;22(1):99. doi:10.1186/s13075-020-2145-y
102. Zhan L, Mu Z, Jiang H, et al. MiR-21-5p protects against ischemic stroke by targeting IL-6R. Ann Transl Med. 2023;11(2):101. doi:10.21037/atm-22-6451
103. Rodrigues PM, Afonso MB, Simão AL, et al. miR-21-5p promotes NASH-related hepatocarcinogenesis. Liver Int. 2023;43(10):2256–2274. doi:10.1111/liv.15682
104. Garcia G, Pinto S, Ferreira S, et al. Emerging role of miR-21-5p in neuron-glia dysregulation and exosome transfer using multiple models of Alzheimer’s disease. Cells. 2022;11(21):3377. doi:10.3390/cells11213377
105. An N, Zheng B. MiR-203a-3p Inhibits Pancreatic Cancer Cell Proliferation, EMT, and Apoptosis by Regulating SLUG. Technol Cancer Res Treat. 2020;19:1533033819898729. doi:10.1177/1533033819898729
106. Luo Z, Dou J, Xie F, et al. miR-203a-3p promotes loureirin A-induced hair follicle stem cells differentiation by targeting Smad1. Anat Rec. 2021;304(3):531–540. doi:10.1002/ar.24480
107. Yang P, Zhang D, Zhou F, et al. miR-203a-3p-DNMT3B feedback loop facilitates non-small cell lung cancer progression. Hum Cell. 2022;35(4):1219–1233. doi:10.1007/s13577-022-00728-y
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
The Causal Relationship Between Gastroesophageal Reflux Disease and Chronic Obstructive Pulmonary Disease: A Bidirectional Two-Sample Mendelian Randomization Study
Liu B, Chen M, You J, Zheng S, Huang M
International Journal of Chronic Obstructive Pulmonary Disease 2024, 19:87-95
Published Date: 10 January 2024
Association of Chronic Obstructive Pulmonary Disease with Risk of Psychiatric Disorders: A Two-Sample Mendelian Randomization Study
Zhang Q, Zhang H, Xu Q
International Journal of Chronic Obstructive Pulmonary Disease 2024, 19:343-351
Published Date: 1 February 2024
Identification and Analysis of PANoptosis-Related Genes in Sepsis-Induced Lung Injury by Bioinformatics and Experimental Verification
Yang Z, Kao X, Huang N, Yuan K, Chen J, He M
Journal of Inflammation Research 2024, 17:1941-1956
Published Date: 28 March 2024
ER Stress-Related Biomarkers in Chronic Obstructive Pulmonary Disease: A Comprehensive Transcriptome, Mendelian Randomization, and Machine-Learning Analysis
Li J, Li G, Liu J, Li L, Zhou H, Fei X, Wen Y, Zhao D
International Journal of Chronic Obstructive Pulmonary Disease 2025, 20:3803-3818
Published Date: 26 November 2025
Mitochondrial Hub Genes in Rheumatoid Arthritis Identified by Machine Learning and Mendelian Randomization with Experimental Validation
Luo X, Dong Z, Song Y, Hu J, Liao S, Ding X
Journal of Inflammation Research 2026, 19:576901
Published Date: 27 March 2026