Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 20
ER Stress-Related Biomarkers in Chronic Obstructive Pulmonary Disease: A Comprehensive Transcriptome, Mendelian Randomization, and Machine-Learning Analysis
Authors Li J, Li G, Liu J ![]() , Li L, Zhou H, Fei X, Wen Y, Zhao D
, Li L, Zhou H, Fei X, Wen Y, Zhao D
Received 19 June 2025
Accepted for publication 10 November 2025
Published 26 November 2025 Volume 2025:20 Pages 3803—3818
DOI https://doi.org/10.2147/COPD.S548160
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Jiajia Li,1 Guofeng Li,1 Junnan Liu,2 Lijie Li,2 Huiling Zhou,1 Xinru Fei,3 Yuhua Wen,3 Dongkai Zhao2
1School of Traditional Chinese Medicine, Changchun University of Chinese Medicine, Changchun, Jilin, 130117, People’s Republic of China; 2Department of Respiration, Third Affiliated Hospital to Changchun University of Chinese Medicine, Changchun, Jilin, 130022, People’s Republic of China; 3School of Rehabilitation Medicine, Changchun University of Chinese Medicine, Changchun, Jilin, 130117, People’s Republic of China
Correspondence: Dongkai Zhao, Department of Respiration, Third Affiliated Hospital to Changchun University of Chinese Medicine, Changchun, Jilin, 130022, People’s Republic of China, Email [email protected]
Background: Chronic obstructive pulmonary disease (COPD) is a common respiratory disease; however, measures for preventing COPD and delaying disease progression are limited. Therefore, identifying genetic variations and novel biomarkers related to COPD incidence and progression is crucial for improving clinical outcomes. Here, we investigated the potential of the endoplasmic reticulum stress-related gene DNAJB1 as a risk gene in COPD and its clinical value via bioinformatics and Mendelian randomization.
Methods: We first performed differential gene analysis on single-cell sequencing datasets then identified candidate genes and genetic loci using Mendelian randomization analysis and co-localization analysis, respectively. Machine-learning analysis of microarray data was used to identify potential biomarkers. Subsequently, we explored the biological role of DNAJB1 through cellular communication, functional enrichment, and correlation analyses with inflammatory factors.
Results: DNAJB1 was identified as a risk gene for COPD that shares genetic variants with COPD. Nine key biological genes, including DNAJB1, were identified as potential diagnostic biomarkers. High DNAJB1 expression and high scores for the endoplasmic reticulum stress gene set were validated using the microarray dataset.
Conclusion: Our finding reveals DNAJB1 as a COPD risk gene and identifies a diagnostic genetic marker panel, providing useful perspectives for early diagnosis and the development of potential therapeutic targets.
Keywords: chronic obstructive pulmonary disease, endoplasmic reticulum stress, biomarker gene, Mendelian randomization, machine learning
Introduction
Chronic obstructive pulmonary disease (COPD) refers to a heterogeneous group of pulmonary disorders characterized by deteriorating lung function and persistent inflammatory processes, whose primary risk factors include tobacco smoke, toxic gases/particles, genetic variants, and age.1 Current treatments for COPD focus on improving clinical symptoms and preventing deterioration but do not address disease progression or reverse patient outcomes. Accordingly, novel therapeutic methods are required that target the potential mechanisms involved in COPD progression, with a focus on treatments targeting specific molecular pathways rather than disease symptoms. The pathogenesis of COPD is related to endoplasmic reticulum (ER) stress. CD4+ T cells, as key immune cells, play an important role in the progression of COPD. However, current research on the mechanism by which endoplasmic reticulum stress in CD4+ T cells regulates the progression of COPD is still relatively limited, especially the related biomarkers have not been fully clarified. This knowledge gap restricts the development of COPD regulatory targets and diagnostic markers.
COPD pathogenesis is commonly associated with ER stress. Specifically, ER stress is closely associated with COPD risk factors. The presence of reactive oxygen and nitrogen species and reactive intermediates such as acrolein in cigarette smoke promotes oxidative damage and protein misfolding in pulmonary cells, thereby initiating ER stress.2 Several studies have demonstrated that the unfolded protein response (UPR) leads to apoptosis and inflammatory responses in the lung tissues of some patients with COPD.3 Alpha-1 antitrypsin deficiency is a known genetic disorder associated with ER stress in patients with COPD. UPR activation, increased expression of BiP/GRP78, ATF4, GRP94, and increased production of cytokines (IL-10, IL-6, and IL-8) have been observed in cells isolated from patients with alpha-1 antitrypsin deficiency.4 However, our current understanding of how the UPR contributes to COPD pathogenesis is limited, and its precise molecular mechanisms remain unclear. Activation of the UPR may induce and prevent the development of COPD. Although initially designed to adapt to the environment and promote survival, the UPR switches to an anti-survival mode when ER stress is severe or prolonged. Elevated UPR activity may result in apoptosis in lung tissue, whereas decreased UPR activity may cause misfolded protein accumulation and impaired antioxidant defense in COPD.3,5 Furthermore, the expression of UPR-related genes is highly heterogeneous among different individuals; these genes impact the ER stress response not only genetically but also through germline genetic polymorphisms.6
The immune responses in COPD include both innate and adaptive immune responses, with disorders of both immune systems likely contributing to some of the clinical features of COPD heterogeneity, including acute exacerbations, emphysema, and excessive sputum production involving cells such as neutrophils, T lymphocytes, and macrophages.7 During the inflammatory response in COPD, T cells are the main effector cells in the airways and lung parenchyma and are correlated with the degree of alveolar damage in patients.8 Both CD4+ and CD8+ T cells play essential roles in the airway inflammatory responses in COPD. Many studies have investigated pulmonary CD8+ T cells, which increase the production of IFN-γ and the expression of cytotoxic molecules.9 Although few studies have analyzed lung CD4+ T cells, several recent findings have suggested that these cells also contribute to COPD progression. For example, an analysis of T lymphocytes in the lung tissue of patients with moderate-to-severe COPD highlighted the critical role of CD4+ T cells and the contribution of the systemic immune response to smoke-induced inflammation.7,10 Chronic airway inflammation promoted by CD4+ T cells and alterations in the cytokine microenvironment are critical for the immunopathogenesis of chronic COPD injury. CD4+ T cells require large amounts of proteins during their developmental and differentiation stages, which may cause unfolded and misfolded proteins to accumulate in the ER, thereby inducing ER stress.11 ER stress is an important pathological mechanism involved in the development of various autoimmune diseases, including rheumatoid arthritis, multiple sclerosis pathology, and type 1 diabetes mellitus, through the regulation of T-cell homeostasis.12–14 Accordingly, we hypothesize that ER stress may also contribute to the occurrence and progression of COPD by affecting T-cell homeostasis during the course of the disease.
In recent years, advances in single-cell RNA sequencing (scRNA-seq) technology have led to rapid progress in the fields of biology, immunology, and oncology through the high-throughput sequencing of genomes, transcriptomes, and even spatial transcriptomes at the single-cell level. This has provided groundbreaking insights into the cellular heterogeneity of tissues, revealing gene expression in individual cells and identifying new cellular subtypes associated with diseases.15 Furthermore, Mendelian randomization uses genetic variants as instrumental variables to infer causal relationships between exposures and outcomes, effectively reducing interference from confounding biases. The increasing availability of large-scale genome-wide association study (GWAS) data and quantitative trait loci data has allowed researchers to link genes with phenotypes and use expression quantitative trait loci (eQTL) to reveal gene–phenotype relationships by understanding how specific genetic variations affect gene expression.16 The machine learning algorithms facilitated the transition from gene discovery to practical diagnostic applications. This approach effectively identified crucial biomarkers from genetic data and captured the complex gene-disease associations through algorithmic modeling, ultimately constructing high-performance diagnostic models with clinical utility.
In this study, we hypothesize that endoplasmic reticulum stress-related differentially expressed genes (DEGs) in CD4+ T cells contribute to the pathogenesis of COPD and may serve as potential biomarkers. To test this hypothesis, we utilized scRNA-seq data to identify differentially expressed genes, applied Mendelian randomization analysis to validate the causal relationship between key genes and COPD, and finally employed a machine learning approach using microarray data to construct a diagnostic model for COPD based on these genes.
Materials and Methods
Data Sources
scRNA-seq and microarray data for COPD were obtained from the GEO database, including datasets GSE196638 (with 3 COPD cases and 3 controls), GSE47460 (with 220 COPD cases and 108 controls), and GSE38974 (with 23 COPD cases and 9 controls). GWAS and eQTL data were sourced from the IEU database, with the numbers “finn-b-J10_COPD” (including 6,915 cases and 186,723 controls) and “eqtl-a-ENSG00000132002” (including 14,263 participants). Endoplasmic reticulum stress-related genes obtained from GeneCards database and previous studies,17 as shown in Supplementary Table S1.
Single-Cell Data Analysis
The scRNA-seq analysis was based on the Seurat package (version 4.4.0). Quality control of single-cell data was based on the criteria of nFeature_RNA > 200, nFeature_RNA < 7,000, and percentage of mitochondrial genes < 15. Subsequently, we normalized the data, screened for highly variable genes, and performed principal component analysis. We employed the Harmony package for batch-effect correction, followed by cell clustering using the FindNeighbors and FindClusters functions. Cell clusters were manually annotated according to the reported marker genes for each cell type.18,19 The FindMarkers function was used to identify upregulated differential genes in CD4+ T cells, and the difference threshold was set to logFC > 0.5. Functional enrichment analyses were conducted using the ClusterProfiler package, with screening criteria of P < 0.05 and Q < 0.05. Intercellular communication networks were investigated using CellChat, which enabled the systematic analysis of ligand–receptor interactions and identification of key signaling pathways in T-cell populations. Subsequently, the metabolic activity of different cell types was quantified using the scMetabolism package.
Mendelian Randomization and Co-Localization Analysis
SNP information was extracted based on differential genes using the following screening criteria: P value of the exposed instrumental variable (P < 5×10−8), threshold r2 < 0.001 for removal of linkage disequilibrium, and F-statistic >10. Mendelian randomization analysis was conducted using the TwoSampleMR package, where genetic instruments represented the exposure and disease traits represented the outcome. The choice of Mendelian randomization methods depends on the availability of instrumental SNPs. The inverse variance weighted (IVW) method is the primary method for exposures with multiple SNPs, while the Wald ratio is the estimator for genes with a single SNP. Sensitivity analyses, including MR-Egger, weighted median, weighted mode, and simple mode, were conducted where applicable. The P value determined by the Bonferroni method after multiplicity correction was 0.05/185=0.00027. Additionally, heterogeneity of the associations between SNPs and the outcome was assessed using Cochran’s Q test. While the limited number of instrumental variables (IVs) restricted the application of methods such as MR-Egger, we have provided a certain degree of support for the robustness of our conclusions through rigorous screening of strong IVs and assessment of heterogeneity. Co-localization analysis was performed with the “coloc” package to explore shared causal variants between the target genes and COPD. Eqtl-a-ENSG00000132002 was used as the exposure and finn-b-J10_COPD was used as the outcome, with the co-localization window selected as 1 MB upstream and downstream of the SNP with the highest correlation (lowest P value). Five different posterior probabilities were reported in the co-localization analysis results, but we focused on two traits: H3, which is associated with COPD risk and gene expression, with different causal variations; and H4, which is associated with COPD risk and gene expression and shares a common causal variation. The prior probabilities required to consider the causal variations associated only with trait 1, only with trait 2, and with both were set to 1 × 10−4, 1 × 10−4, and 1 × 10−5, respectively. The posterior probability of hypothesis 4 exceeding 0.70 was regarded as significant evidence supporting co-localization.20
Machine Learning
We intersected the differential genes with a set of ER stress-related genes to obtain intersecting genes and screened key genes by LASSO regression analysis. We then used the key genes as variables to construct a COPD diagnostic model with GSE47460 and GSE38974 as the training and validation sets, respectively, using the R package “mlr3verse.” The performance of five classical algorithms, including Logistic Regression (LR), Linear Discriminant Analysis (LDA), Support Vector Machine (SVM), Naive Bayes (NB), and Random Forest (RF), was evaluated and compared using 10 repeats of 5-fold cross-validation. Based on its higher specificity and stable AUC performance, the SVM algorithm was selected to construct the final diagnostic model. Evaluation on an independent validation set confirmed the model’s robust performance stability, with an achieved AUC of 0.705. The robustness of the results was strengthened through a combination of measures such as ComBat-based batch effect correction, LASSO-based gene selection, repeated cross-validation, and independent dataset validation.
Microarray Data Analysis
We compared the expression levels of ER stress-associated gene set as well as the specific gene DNAJB1 in the GSE38974 dataset and created heat maps to explore intergroup differences between the disease and normal groups. Correlations between intersecting genes and inflammatory factor-related genes were calculated using a correlation test in the psych package.
Results
Identification of Cell Types
We first classified the cells into four categories: mesenchymal, epithelial, immune, and endothelial cells. Immune cells were extracted and further categorized into B, mast, natural killer, dendritic, CD8+ T, plasma, CD4+ T, proliferative, and myeloid cells. The annotation results of single-cell clustering were visualized using t-distributed stochastic neighbor embedding (Figure 1A and B). Of these, cell clusters unrelated to the marker genes and with a small number of cells were excluded. The final classification results and marker genes are shown in Figure 1C and D and the proportions of cell types are shown in Figure 1E. The proportion of CD4+ T cells was higher in the COPD group than in the normal group, and the abundance of CD4+ T cells in the immune microenvironment of the COPD group was higher than that of other immune cell types. We obtained 185 upregulated DEGs(Supplementary Table S2) and 13 hub genes after intersecting with ER stress-related genes (Figure 2A). According to functional enrichment analysis (Figure 2B and C), DEGs showed enrichment in biological processes related to T-cell activation and differentiation, molecular functions related to mitogen-activated protein kinase (MAPK) and tumor necrosis factor (TNF) pathways, and cellular components including ribosomes, plasma membranes, and T-cell receptor complexes. Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis revealed enrichment in T-cell differentiation, cytokine interactions, and the TNF and MAPK signaling pathways.
 |
Figure 1 Identification of cell types in lung tissue from 3 COPD patients and 3 healthy controls. (A) The tSNE plot depicts the distribution of cell types in the lung tissues. (B) The tSNE plot depicts the distribution of subpopulations in the immune cells. (C) Marker genes for cell subpopulations initially classified in the lung tissues. (D) Marker genes for immune cell subpopulations in the lung tissues. (E) Proportion of cell types in the COPD and normal groups. |
 |
Figure 2 Differential gene analysis of CD4 T cells. (A) Intersection of differential genes with endoplasmic reticulum stress-related genes. (B and C) GO and KEGG enrichment analysis of differential genes. |
Identification of Risk Genes
Mendelian randomization analysis revealed 15 DEGs that were causally associated with COPD (Figure 3). Significant risk genes (P < 0.05) are shown in the volcano plot (Figure 4A). DNAJB1 was identified as a potential risk factor for COPD, whereby increased DNAJB1 expression may lead to an increased risk of COPD (OR 1.3947, 95% CI 1.1711–1.6610, P = 0.000190). HSPA8 was also identified as an ER-associated DEG that was positively associated with COPD risk (OR 1.4094, 95% CI 1.0646–1.8657, P = 0.016507). Furthermore, Cochran’s Q test found no significant heterogeneity in the causal estimates from the instrumental variables (SNPs) for these two genes (P > 0.05). In the single-cell dataset, DNAJB1 and HSPA8 showed high expression in CD4+ T-cell subsets (Figure 4B). The results of co-localization analysis (Figure 4C) showed that PPH4 = 0.70, suggesting that causal variants are shared between DNAJB1 and COPD. The lead SNP in the association region was rs7003. Reverse causality analyses with COPD and DNAJB1 as the exposure and outcome, respectively, revealed no causal relationship (P > 0.05).
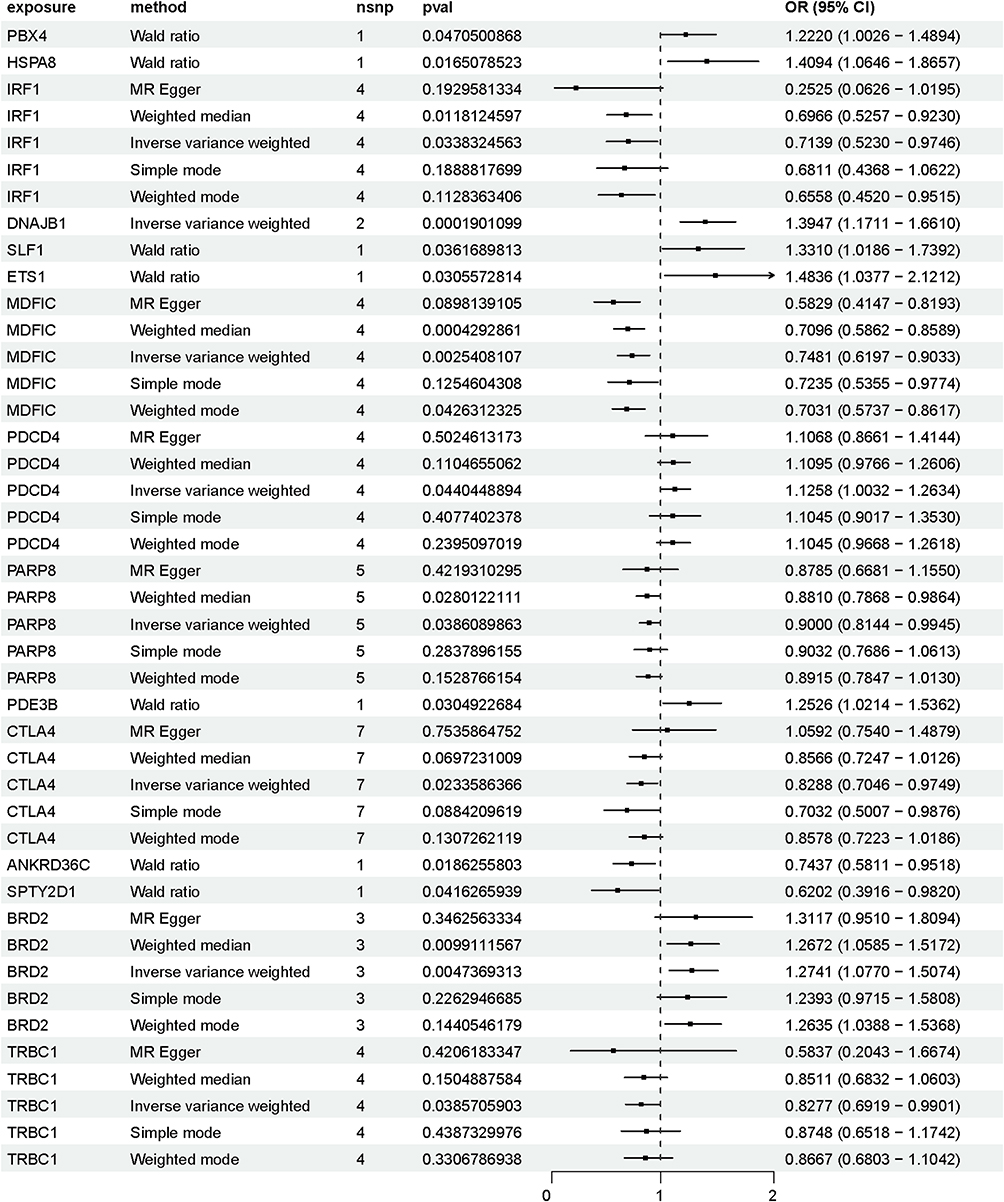 |
Figure 3 Mendelian randomization analysis of differential genes and COPD. |
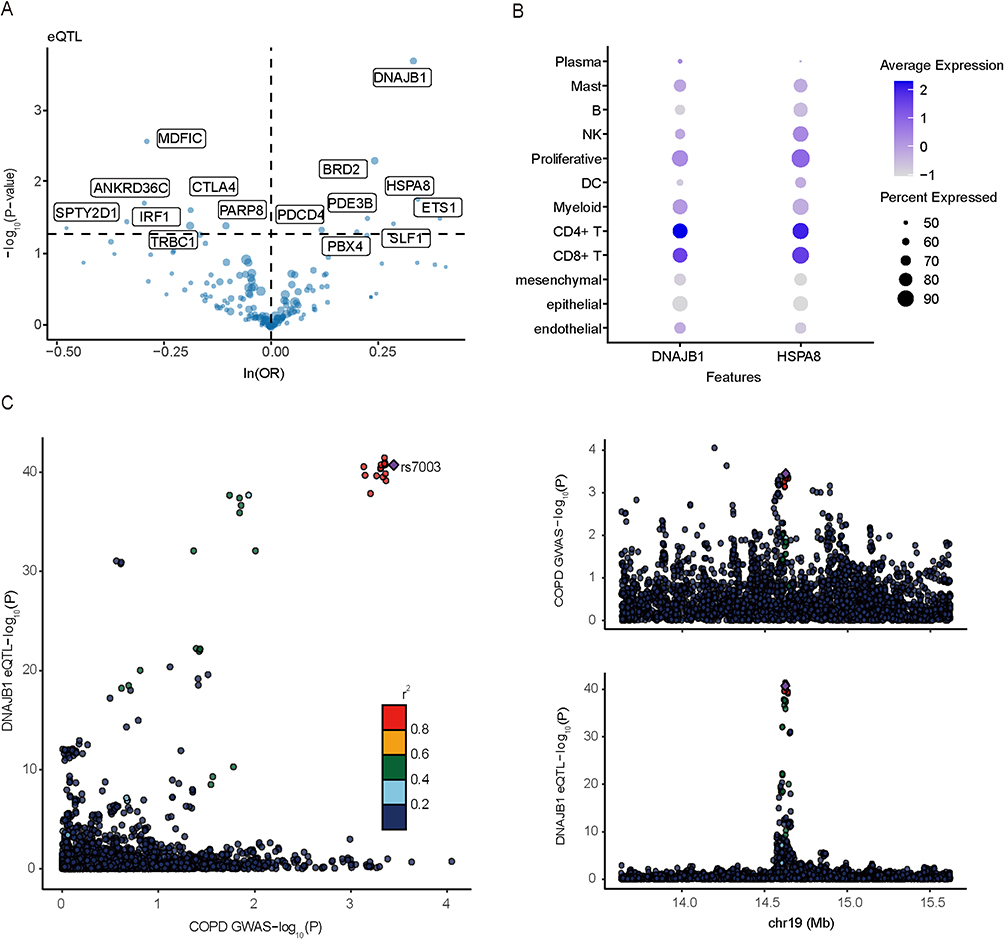 |
Figure 4 Visualization of MR and co-localization analysis results. (A) Volcano plots of MR analysis results for differential genes and COPD. (B) Expression levels of causal genes for COPD in cell subsets. (C) Regional association plots of DNAJB1 and COPD. |
Cell Function Analysis
Based on the expression levels of DNAJB1, we classified CD4+ T cells into DNAJB1-positive and DNAJB1-negative types. The scMetabolism package was used to calculate the scores of the active metabolic pathways. Figure 5A illustrates the metabolic activity levels of the different cell types. We found that CD4+ T cells showed higher glucose and amino acid metabolic activity, with higher glutamine activity in DNAJB1-positive CD4+ T cells than in DNAJB1-negative CD4+ T cells. Myeloid cells showed higher fatty acid activity and CD8+ T cells showed higher steroid metabolism activity. The intercellular communication networks between CD4+ T lymphocytes and other cell types in the COPD microenvironment were systematically investigated using the CellChat package. As shown in Figure 5B, CD4+ T cells communicated with myeloid and CD8+ T cells in greater numbers with stronger interactions, and more pronounced communication was observed for DNAJB1-positive CD4+ T cells. Subsequent analysis of the receptor–ligand pairs of the CD4+ T and myeloid systems and CD8+ T cells (Figure 5C) showed that macrophage migration inhibitory factor (MIF)-(CD74+CXCR4) and MIF-(CD74+CD44) pathways only formed from the communication of DNAJB1-positive T cells. The TNF–TNFRSF1B pathway mediated CD4+ T cells communication with myeloid and CD8+ T cells. The CD99-CD99 pathway was more prominent for communication between DNAJB1-positive CD4+ T and CD8+ T cells, and the CD6-ALCAM pathway for communication with myeloid cells.
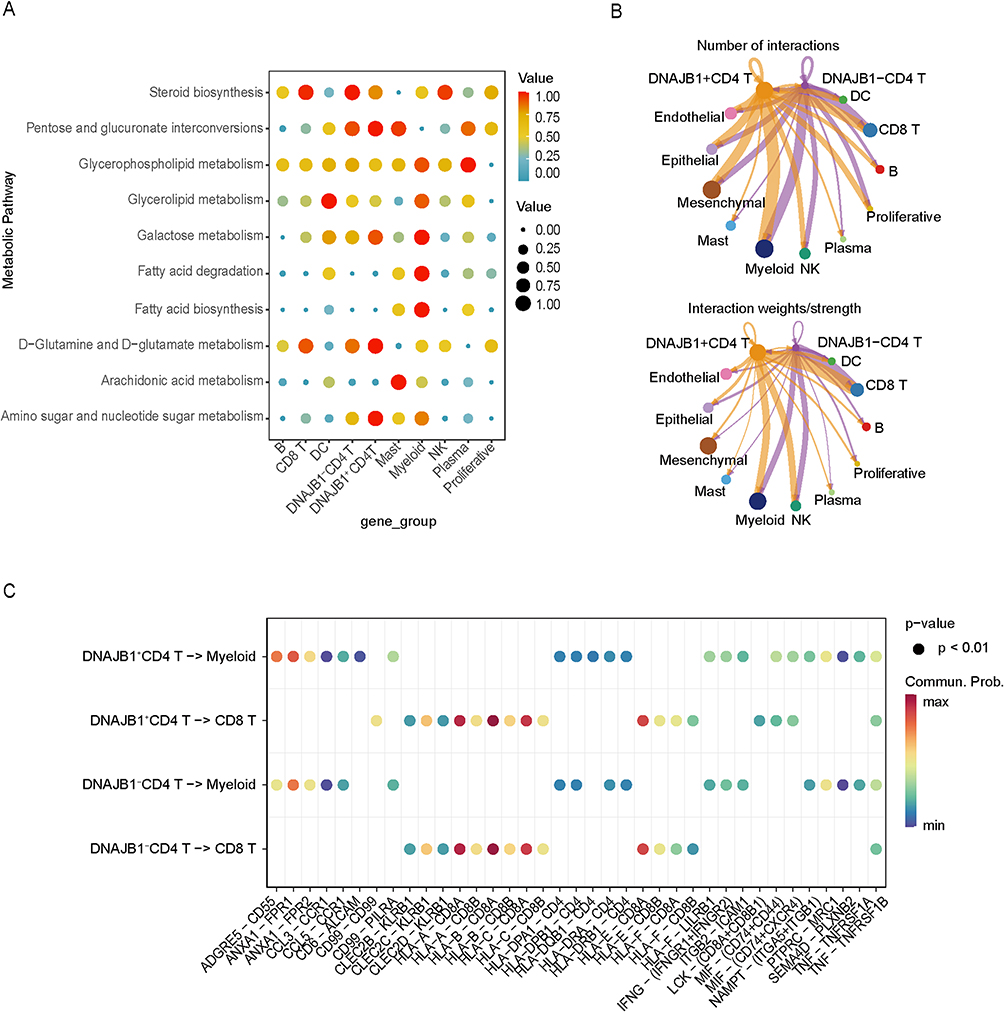 |
Figure 5 Comparison of characteristics between DNAJB1 positive and negative CD4 T cells. (A) Differences in metabolic activity and pathways between cell subpopulations. (B) Number and strength of ligand-receptor pair interactions in DNAJB1-positive and negative CD4 T cells. (C) The ligand-receptor pairs of DNAJB1-positive and DNAJB1-negative CD4 T cells interacting with CD8 T cells and myeloid cells respectively. |
Diagnostic Model Construction and Functional Validation
The LASSO regression algorithm was used to further screen intersecting genes and identify the best candidate for the model. Nine genes (DNAJB1, CXCR4, FUS, IFNG, BCL2, TNF, PPP1R15A, CASP8, and RPS27A) were selected to construct the final diagnostic model. We applied five different machine-learning algorithms and evaluated model performance by 10-fold cross-validation over five iterations (Figure 6A). We used the area under the curve (AUC) value as an indicator of the diagnostic accuracy of the models (Figure 6B). The “SVM” machine-learning algorithm model was selected and validated in the validation dataset; the AUC value of 0.705 (Figure 6C) indicated its potential utility for diagnosing COPD. We then demonstrated the expression levels of the ER stress-related gene set and DNAJB1 in the GSE38974 dataset (Figure 7A and B), and found higher expression levels in the disease group. The expression levels of intersecting genes in GSE38974 (ER stress-related genes and DEGs) were also characterized, as shown in the heat map (Figure 7C). The correlation plot of inflammatory factors and intersecting genes shows that DNAJB1 was correlated with IL-6 and colony-stimulating factor 3 (CSF3) (Figure 7D).
 |
Figure 6 Construction of the diagnostic model for COPD based on key genes. (A) Diagnostic models construction through five machine learning algorithms. (B) The ROC curve compared the performance of the COPD diagnostic model based on genetic characteristics on the training set (220 COPD patients, 108 controls). (C) The ROC curve depicts the diagnostic prediction accuracy in the validation set (23 COPD patients, 9 controls). |
 |
Figure 7 Gene expression analysis of microarray datasets. (A) Expression difference of DNAJB1 between the two groups. (B) Differential expression of endoplasmic reticulum stress-related gene set between the two groups. (C) Expression difference of intersection genes between the two groups. (D) Correlation of intersection genes with inflammatory factors. Values are correlation coefficients, with significance levels indicated as *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001. |
Discussion
COPD represents a global public health challenge through its role in chronic morbidity and mortality. With increasing smoking rates and population aging, the future prevalence of COPD is predicted to continue increasing. According to one study, the global prevalence of COPD is predicted to reach 600 million by 2050, indicating a 23% relative increase in the number of patients with COPD from 2020.21 COPD results from complex and dynamic interactions between genes and the environment during an individual’s life course.1 Thus, exploring the risk genes and potential markers associated with COPD may improve the accuracy of COPD prevention and diagnosis.
In individuals with COPD, CD8+ and CD4+ T cells accumulate in the lung tissue. CD4+ helper T lymphocytes can differentiate into multiple subgroups, with subpopulation Th1 cells producing IFN-γ, which stimulates the inflammatory response.22 Th2 cell subset can promote airway pathological remodeling and contribute to airway obstructive changes, whereas reduced Treg-cell function can lead to immune dysregulation.23,24 These characteristics of CD4+ T cells all exacerbate COPD progression. T-cell-mediated immune responses require many newly synthesized proteins to support their proliferation and differentiation.25 Research has highlighted the UPR as an important mediator of T-cell-mediated diseases and a critical actor regulating T-cell function and the immune response.11,26 As an important mechanism affecting the biological function of T cells, ER stress and its resulting UPR profoundly influence the immune response and related disease processes. Recent studies have emphasized the importance of UPR and ER stress in COPD.27,28
In this study, scRNA-seq analysis verified the increased proportion of CD4+ T cells in COPD and the differential expression of the ER stress-related gene set and DNAJB1 between COPD and normal groups. Gene ontology and KEGG enrichment analyses indicated that the DEGs of CD4+ T cells were mainly enriched in ribosomes, T-cell activation and differentiation, and MAPK pathways. The functions of ribosomes and ER are closely related and mainly reflected in the synthesis and accumulation of misfolded proteins. Secretory and membrane proteins are synthesized in the ribosome and enter the ER, where they become mature proteins through folding and a variety of other modifications. This process is subjected to stringent quality control to maintain intracellular homeostasis. Activation of the UPR after ER stress may further block protein synthesis in the ribosome.29 The MAPK and TNF-α pathways are associated with ER stress, and both pathways act as key effectors in driving the subsequent inflammatory responses.30
Given the critical regulatory role of ER stress in COPD pathogenesis, we screened for causally differentiated genes in COPD using Mendelian randomization analysis and took the intersection with ER stress genes. Finally, we identified two genes: DNAJB1 and HSPA8. The molecular chaperone protein DNAJB1 belongs to the Hsp40 family, whereas HSPA8 (also known as Hsc70) is a representative member of the Hsp70 family. As members of the heat shock protein (HSP) family, these two proteins play a central role in maintaining protein homeostasis in cells by regulating conformational changes in complex peptides and by participating in ER stress response mechanisms.31,32 HSPs are classified according to their molecular weight; each member exhibits a different function and is correlated with a number of diseases, including cancer and infections.33 Thermal stimulation, inflammation, viruses, reactive oxygen species, and other stimulants can affect the production of these molecules. HSPs represent diagnostic biomarkers and potential therapeutic targets for diseases, such as COVID-19, malaria, and cancer.34–36 Chaperone Hsp40 is involved in the correct folding of proteins by assisting Hsp70, and its role in diseases associated with misfolded and aggregated proteins, such as ischemic stroke and COPD, has been well validated. In brain injury, significant induction of HSP70 and its co-chaperone HSP40 has been reported.37,38 In animal models of COPD, certain HSPs enhance activation of the TLR4 and NLRP3 inflammasome pathways, leading to elevated levels of inflammatory mediators and structural damage to the lung tissue.39 Increased DNAJB1 expression has also been detected in the lung tissue of a mouse model of cigarette smoke-induced emphysema.40 In addition, SNP rs1008438 in the HSP gene is associated with genetic susceptibility to COPD, which predisposes smokers to severe COPD.41 In this research, it is suggested that DNAJB1 serves not only as a risk factor for COPD but also as an essential component of the COPD biomarker panel.
Next, the scMetabolism package was used to visualize and quantify the metabolic diversity of cells. We found that CD4+ T cells exhibited higher amino acid metabolic activities. In particular, DNAJB1-positive CD4+ T cells showed higher glutamine activity than DNAJB1-negative CD4+ T cells. Amino acid metabolism plays a key role in the activation, proliferation, and differentiation of CD4+ T cells. In COPD, alterations in the amino acid metabolism of CD4+ T cells affect cell function and inflammatory responses.42 On the one hand, amino acids can affect the level of DNAJB1.43 On the other hand, DNAJB1 influences protein synthesis and degradation by assisting in the correct folding of nascent peptide chains, thereby regulating amino acid recycling. Under ER stress, misfolded proteins are degraded, thereby releasing amino acids to re-engage in metabolism, which in turn leads to an increase in glutamine activity. In addition, fatty acid metabolic activity was lower in T cells, which is consistent with previous research. Because of their rapid proliferation, differentiation, and secretion of abundant cytokines that require large amounts of energy and biosynthetic precursors, T cells shift their metabolic pathway from fatty acids to glutamine, pentose phosphate, and glycolytic metabolic pathways.44 Cell communication analysis revealed that CD4+ T cells communicating with CD8+ T and myeloid cells were enriched in the TNF-TNFRSF1B pathway, whereas MIF-(CD74+CD44) and MIF-(CD74+CXCR4) pathways were enriched only in DNAJB1-positive CD4+ T cells. MIF is a key mediator of the inflammatory response and is closely associated with many autoimmune diseases, tumors, and other diseases.45 ATF6 and MIF exhibit simultaneously elevated expression in inflammatory diseases and interact under ER stress conditions.46 As a promoter of the ATF6 pathway, MIF promotes the differentiation of CD4+ T cells by activating ATF6 signaling. TNF is a key cytokine for the hyperresponsiveness of airway tissues, and its action is mediated by two receptor subtypes, TNFR1 and TNFR2.47 Although both receptors are involved in COPD pathogenesis, TNFR2 plays a more active regulatory role in the inflammatory response and airway remodeling.48 The expression of TNFR2 has cell lineage specificity, and its preferential expression pattern in CD4+Foxp3+ regulatory T cells determines the critical role of this receptor in regulating Treg-cell activation, proliferation, and functional stability.49 Based on the above results, it can be inferred that the specific upregulation of DNAJB1 in CD4+ T cells suggests that ER stress drives T cell dysfunction, and the mechanism may be related to the glutamine metabolic pathway and MIF-mediated cell communication.
To further understand the role of CD4+ T cells in COPD, we used machine-learning algorithms to identify several diagnostic genes, including DNAJB1, and constructed a diagnostic model. Through validation with external dataset, we confirmed the high diagnostic efficacy of these diagnostic genes, which is important for identifying biomarkers closely related to COPD. We then analyzed the correlation between intersecting genes and cytokines and found that DNAJB1 was significantly correlated with IL-6 and CSF3. As a key pro-inflammatory mediator, IL-6 was originally recognized as a B cell growth factor. However, it also plays an important role in regulating CD4+ T-cell effector function, affecting the immune response and leading to inflammation by regulating T-cell differentiation.50,51 This factor plays an important role in pathological progression and tissue destruction in a variety of autoimmune and inflammation-related diseases, such as rheumatoid arthritis and COPD.52,53 A correlation exists between DNAJB1 and IL-6, and the potential connection is related to chronic UPR activation through the NF-κB pathway and AP-1 leading to inflammation.54,55 Granulocyte-CSF deficiency greatly reduces airway inflammation and lung tissue destruction in animal models, and bronchoalveolar lavage fluid from patients with COPD showed elevated granulocyte-CSF levels in human clinical translation studies.56 Genetic variations affect pulmonary function in patients with COPD.57 However, few studies have investigated the relationship between CSF and the ER stress-related gene DNAJB1; thus, further investigation is required.
Conclusion
The main strength of this study lies in the comprehensive application of multiple complementary bioinformatics and genetic methods for cross-validation, which significantly enhances the credibility of DNAJB1 as a risk gene for COPD. Single-cell analysis revealed the specific upregulation of DNAJB1 in CD4+ T cells and elucidated the related mechanisms, while Mendelian randomization indicated causal effects, colocalization analysis supported shared genetic loci, and machine learning confirmed the diagnostic potential. In conclusion, this study further highlights the importance of ER stress, especially in CD4+ T cells, in the pathogenesis of COPD, providing a new perspective for understanding disease heterogeneity. As a master regulator of the ER stress pathway, DNAJB1 represents a compelling therapeutic target. For example, developing small-molecule drugs that target its chaperone function to alleviate ER stress, or formulating strategies to regulate its expression level, may lead to the development of novel COPD treatments for immune dysregulation, which are currently poorly managed by existing therapies. Nevertheless, this study has some limitations that must be acknowledged. First, the findings rely on publicly available datasets, some of which contain incomplete clinical annotations, such as data on age, sex, and smoking history, making it challenging to fully control for the influence of these confounding factors. Additionally, the Mendelian randomization analyses were primarily based on genetic data from European populations, which may limit the generalizability of our results across different ethnic groups. Another key limitation is that the current evidence is mainly derived from computational and genetic analyses. Although our findings suggest a critical role for DNAJB1 in the pathogenesis of COPD, its specific biological functions and mechanistic contributions, particularly in endoplasmic reticulum stress and immune regulation, still lack direct experimental validation. Furthermore, while DNAJB1 shows potential as a biomarker, its clinical applicability requires rigorous evaluation in independent, prospectively designed cohorts. Future research should focus on several key directions. First, prospective studies with comprehensive clinical annotations are needed to verify these findings while adequately controlling for confounders. Second, the generalizability of the genetic evidence requires validation in multi-ethnic populations beyond the European cohorts. Third, future studies should develop a composite diagnostic model that integrates biomarkers identified in this study with other established COPD biomarkers and validate it in independent cohorts. Finally, both mechanistic studies to elucidate DNAJB1’s role in endoplasmic reticulum stress and CD4+ T cell regulation and systematic evaluation of its clinical utility are essential to facilitate translation into COPD management.
Abbreviations
COPD, Chronic obstructive pulmonary disease; scRNA-seq, single-cell RNA sequencing; ER stress, endoplasmic reticulum stress; DEGs, Differentially expressed genes; GWAS, Genome-wide association study; GO, Gene ontology; KEGG, Kyoto encyclopedia of genes and genomes; LASSO, Logical Regression of Selection Operators; logFC, log fold change; MR, Mendelian randomization; OR, Odds ratio; AUC, Area under the curve; ROC, Receiver operating characteristic; SNP, Single nucleotide polymorphism; GEO, Gene Expression Omnibus.
Data Sharing Statement
The scRNA-seq and microarray datasets supporting the findings of this study are available in the GEO database (https://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE196638, GSE47460, and GSE38974. The GWAS and eQTL data were obtained from the IEU OpenGWAS database (https://gwas.mrcieu.ac.uk/) with accession IDs finn-b-J10_COPD (GWAS) and eqtl-a-ENSG00000132002 (eQTL for DNAJB1).
Ethical Approval
This study has been reviewed by the Institutional Review Board of the Third Affiliated Hospital of Changchun University of Chinese Medicine. Since the research involves the analysis of legally publicly available data and does not involve intervention in public behavior, it complies with Articles 32(1) and (2) of the “Ethical Review Measures for Life Sciences and Medical Research Involving Humans” regarding exemption from ethical approval. Therefore, this study does not require additional IRB approval.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was partially supported by the Natural Science Foundation of Jilin Province (Grant No. YDZJ202401121ZYTS).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Agusti A, Melen E, DeMeo DL, Breyer-Kohansal R, Faner R. Pathogenesis of chronic obstructive pulmonary disease: understanding the contributions of gene-environment interactions across the lifespan. Lancet Respir Med. 2022;10(5):512–524. doi:10.1016/S2213-2600(21)00555-5
2. Yu Y, Yang A, Yu G, Wang H. Endoplasmic reticulum stress in chronic obstructive pulmonary disease: mechanisms and future perspectives. Biomolecules. 2022;12(11):1637. doi:10.3390/biom12111637
3. Kelsen SG. The unfolded protein response in chronic obstructive pulmonary disease. Ann Am Thorac Soc. 2016;13(Suppl 2):S138–45. doi:10.1513/AnnalsATS.201506-320KV
4. Carroll TP, Greene CM, O’Connor CA, Nolan AM, O’Neill SJ, McElvaney NG. Evidence for unfolded protein response activation in monocytes from individuals with alpha-1 antitrypsin deficiency. J Immunol. 2010;184(8):4538–4546. doi:10.4049/jimmunol.0802864
5. Min T, Bodas M, Mazur S, Vij N. Critical role of proteostasis-imbalance in pathogenesis of COPD and severe emphysema. J Mol Med. 2011;89(6):577–593. doi:10.1007/s00109-011-0732-8
6. Dombroski BA, Nayak RR, Ewens KG, Ankener W, Cheung VG, Spielman RS. Gene expression and genetic variation in response to endoplasmic reticulum stress in human cells. Am J Hum Genet. 2010;86(5):719–729. doi:10.1016/j.ajhg.2010.03.017
7. Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(26):2645–2653. doi:10.1056/NEJMoa032158
8. Finkelstein R, Fraser RS, Ghezzo H, Cosio MG. Alveolar inflammation and its relation to emphysema in smokers. Am J Respir Crit Care Med. 1995;152(5 Pt 1):1666–1672. doi:10.1164/ajrccm.152.5.7582312
9. Hodge G, Hodge S. Steroid resistant CD8(+)CD28(null) NKT-like pro-inflammatory cytotoxic cells in chronic obstructive pulmonary disease. Front Immunol. 2016;7:617. doi:10.3389/fimmu.2016.00617
10. Grumelli S, Corry DB, Song LZ, et al. An immune basis for lung parenchymal destruction in chronic obstructive pulmonary disease and emphysema. PLoS Med. 2004;1(1):e8. doi:10.1371/journal.pmed.0010008
11. Chen S, Wang Q, Wang H, Xia S. Endoplasmic reticulum stress in T cell-mediated diseases. Scand J Immunol. 2023;98(3):e13307. doi:10.1111/sji.13307
12. Ao Q, Hu H, Huang Y. Ferroptosis and endoplasmic reticulum stress in rheumatoid arthritis. Front Immunol. 2024;15:1438803. doi:10.3389/fimmu.2024.1438803
13. Andhavarapu S, Mubariz F, Arvas M, Bever C, Makar TK. Interplay between ER stress and autophagy: a possible mechanism in multiple sclerosis pathology. Exp Mol Pathol. 2019;108:183–190. doi:10.1016/j.yexmp.2019.04.016
14. Piganelli JD, Mamula MJ, James EA. The role of beta cell stress and neo-epitopes in the immunopathology of type 1 diabetes. Front Endocrinol. 2020;11:624590. doi:10.3389/fendo.2020.624590
15. Luecken MD, Theis FJ. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol Syst Biol. 2019;15(6):e8746. doi:10.15252/msb.20188746
16. Ghaffar A. International headache genetics C, Nyholt DR. Integrating eQTL and GWAS data characterises established and identifies novel migraine risk loci. Hum Genet. 2023;142(8):1113–1137. doi:10.1007/s00439-023-02568-8
17. Mo J, Ruan S, Yang B, et al. A novel defined risk signature of endoplasmic reticulum stress-related genes for predicting the prognosis and immune infiltration status of ovarian cancer. J Zhejiang Univ Sci B. 2023;24(1):64–77. doi:10.1631/jzus.B2200272
18. Habermann AC, Gutierrez AJ, Bui LT, et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci Adv. 2020;6(28):eaba1972. doi:10.1126/sciadv.aba1972
19. Bischoff P, Trinks A, Obermayer B, et al. Single-cell RNA sequencing reveals distinct tumor microenvironmental patterns in lung adenocarcinoma. Oncogene. 2021;40(50):6748–6758. doi:10.1038/s41388-021-02054-3
20. Chen J, Ruan X, Sun Y, et al. Multi-omic insight into the molecular networks of mitochondrial dysfunction in the pathogenesis of inflammatory bowel disease. EBioMedicine. 2024;99:104934. doi:10.1016/j.ebiom.2023.104934
21. Boers E, Barrett M, Su JG, et al. Global burden of chronic obstructive pulmonary disease through 2050. JAMA Network Open. 2023;6(12):e2346598. doi:10.1001/jamanetworkopen.2023.46598
22. Chen J, Xiang X, Nie L, et al. The emerging role of Th1 cells in atherosclerosis and its implications for therapy. Front Immunol. 2022;13:1079668. doi:10.3389/fimmu.2022.1079668
23. Sakaguchi S, Mikami N, Wing JB, et al. Cells and human disease. Annu Rev Immunol. 2020;38:541–566. doi:10.1146/annurev-immunol-042718-041717
24. Qi Y, Yan Y, Tang D, et al. Inflammatory and immune mechanisms in COPD: current status and therapeutic prospects. J Inflamm Res. 2024;17:6603–6618. doi:10.2147/JIR.S478568
25. Shah K, Al-Haidari A, Sun J, Kazi JU. T cell receptor (TCR) signaling in health and disease. Signal Transduct Target Ther. 2021;6(1):412. doi:10.1038/s41392-021-00823-w
26. Zhang W, Cao X. Unfolded protein responses in T cell immunity. Front Immunol. 2024;15:1515715. doi:10.3389/fimmu.2024.1515715
27. Naiel S, Tat V, Padwal M, et al. Protein misfolding and endoplasmic reticulum stress in chronic lung disease: will cell-specific targeting be the key to the cure? Chest. 2020;157(5):1207–1220. doi:10.1016/j.chest.2019.11.009
28. Wei J, Rahman S, Ayaub EA, Dickhout JG, Ask K. Protein misfolding and endoplasmic reticulum stress in chronic lung disease. Chest. 2013;143(4):1098–1105. doi:10.1378/chest.12-2133
29. Kaneko M, Imaizumi K, Saito A, et al. ER stress and disease: toward prevention and treatment. Biol Pharm Bull. 2017;40(9):1337–1343. doi:10.1248/bpb.b17-00342
30. Akhter N, Wilson A, Arefanian H, et al. Endoplasmic reticulum stress promotes the expression of TNF-alpha in THP-1 cells by mechanisms involving ROS/CHOP/HIF-1alpha and MAPK/NF-kappaB pathways. Int J Mol Sci. 2023;24(20):15186. doi:10.3390/ijms242015186
31. Makhoba XH. Two sides of the same coin: heat shock proteins as biomarkers and therapeutic targets for some complex diseases. Front Mol Biosci. 2025;12:1491227. doi:10.3389/fmolb.2025.1491227
32. Donnelly BF, Needham PG, Snyder AC, et al. Hsp70 and Hsp90 multichaperone complexes sequentially regulate thiazide-sensitive cotransporter endoplasmic reticulum-associated degradation and biogenesis. J Biol Chem. 2013;288(18):13124–13135. doi:10.1074/jbc.M113.455394
33. Hu C, Yang J, Qi Z, et al. Heat shock proteins: biological functions, pathological roles, and therapeutic opportunities. MedComm. 2022;3(3):e161. doi:10.1002/mco2.161
34. Kasperkiewicz M, Tukaj S. Targeting heat shock proteins 90 and 70: a promising remedy for both autoimmune bullous diseases and COVID-19. Front Immunol. 2022;13:1080786. doi:10.3389/fimmu.2022.1080786
35. Barth J, Schach T, Przyborski JM. HSP70 and their co-chaperones in the human malaria parasite P. falciparum and their potential as drug targets. Front Mol Biosci. 2022;9:968248. doi:10.3389/fmolb.2022.968248
36. Zhao K, Zhou G, Liu Y, et al. HSP70 family in cancer: signaling mechanisms and therapeutic advances. Biomolecules. 2023;13(4):601. doi:10.3390/biom13040601
37. Konstantinova EV, Chipigina NS, Shurdumova MH, Kovalenko EI, Sapozhnikov AM. Heat shock protein 70 kDa as a target for diagnostics and therapy of cardiovascular and cerebrovascular diseases. Curr Pharm Des. 2019;25(6):710–714. doi:10.2174/1381612825666190329123924
38. Paschen W, Linden T, Doutheil J. Effects of transient cerebral ischemia on hsp40 mRNA levels in rat brain. Brain Res Mol Brain Res. 1998;55(2):341–344. doi:10.1016/s0169-328x(98)00027-8
39. Ou G, Zhu M, Huang Y, et al. HSP60 regulates the cigarette smoke-induced activation of TLR4-NF-kappaB-MyD88 signalling pathway and NLRP3 inflammasome. Int Immunopharmacol. 2022;103:108445. doi:10.1016/j.intimp.2021.108445
40. Li K, Ye X, Xu M, et al. MiR-23a-3p alleviates cigarette smoke extract-induced pulmonary vascular endothelial cell apoptosis by targeting DNAJB1 in emphysema. Clin Respir J. 2023;17(12):1223–1232. doi:10.1111/crj.13707
41. Ambrocio-Ortiz E, Perez-Rubio G, Ramirez-Venegas A, et al. Effect of SNPs in HSP family genes, variation in the mRNA and intracellular Hsp levels in COPD secondary to tobacco smoking and biomass-burning smoke. Front Genet. 2019;10:1307. doi:10.3389/fgene.2019.01307
42. Ma S, Ming Y, Wu J, Cui G. Cellular metabolism regulates the differentiation and function of T-cell subsets. Cell Mol Immunol. 2024;21(5):419–435. doi:10.1038/s41423-024-01148-8
43. Hensen SM, Heldens L, van Enckevort CM, van Genesen ST, Pruijn GJ, Lubsen NH. Heat shock factor 1 is inactivated by amino acid deprivation. Cell Stress Chaperones. 2012;17(6):743–755. doi:10.1007/s12192-012-0347-1
44. Wang R, Dillon CP, Shi LZ, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35(6):871–882. doi:10.1016/j.immuni.2011.09.021
45. Fey RM, Nichols RA, Tran TT, Vandenbark AA, Kulkarni RP. MIF and CD74 as emerging biomarkers for immune checkpoint blockade therapy. Cancers. 2024;16(9):1773. doi:10.3390/cancers16091773
46. Yan G, Song R, Zhang J, et al. MIF promotes Th17 cell differentiation in rheumatoid arthritis through ATF6 signal pathway. Mol Med. 2024;30(1):237. doi:10.1186/s10020-024-01005-4
47. McFarlane SM, Jupp OJ, Cobban HJ, et al. Stimulation of stress-activated but not mitogen-activated protein kinases by tumour necrosis factor receptor subtypes in airway smooth muscle. Biochem Pharmacol. 2001;61(6):749–759. doi:10.1016/s0006-2952(01)00530-5
48. Caram LMO, Ferrari R, Nogueira DL, et al. Tumor necrosis factor receptor 2 as a possible marker of COPD in smokers and ex-smokers. Int J Chron Obstruct Pulmon Dis. 2017;12:2015–2021. doi:10.2147/COPD.S138558
49. Islam MS, Yang Y, Chen X. TNF-TNFR2 signal plays a decisive role in the activation of CD4(+)Foxp3(+) regulatory T cells: implications in the treatment of autoimmune diseases and cancer. Adv Exp Med Biol. 2021;1278:257–272. doi:10.1007/978-981-15-6407-9_13
50. Cheng X, Yu X, Ding YJ, et al. The Th17/Treg imbalance in patients with acute coronary syndrome. Clin Immunol. 2008;127(1):89–97. doi:10.1016/j.clim.2008.01.009
51. Nish SA, Schenten D, Wunderlich FT, et al. T cell-intrinsic role of IL-6 signaling in primary and memory responses. Elife. 2014;3:e01949. doi:10.7554/eLife.01949
52. Pandolfi F, Franza L, Carusi V, Altamura S, Andriollo G, Nucera E. Interleukin-6 in Rheumatoid Arthritis. Int J Mol Sci. 2020;21(15):5238. doi:10.3390/ijms21155238
53. Wu D, Gong Z, Hao X, Liu L. Genetic perturbation of IL-6 receptor signaling pathway and risk of multiple respiratory diseases. J Transl Med. 2024;22(1):581. doi:10.1186/s12967-024-05366-6
54. Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140(6):900–917. doi:10.1016/j.cell.2010.02.034
55. Lenna S, Farina AG, Martyanov V, et al. Increased expression of endoplasmic reticulum stress and unfolded protein response genes in peripheral blood mononuclear cells from patients with limited cutaneous systemic sclerosis and pulmonary arterial hypertension. Arthritis Rheum. 2013;65(5):1357–1366. doi:10.1002/art.37891
56. Tsantikos E, Lau M, Castelino CM, et al. Granulocyte-CSF links destructive inflammation and comorbidities in obstructive lung disease. J Clin Invest. 2018;128(6):2406–2418. doi:10.1172/JCI98224
57. He JQ, Shumansky K, Connett JE, Anthonisen NR, Pare PD, Sandford AJ. Association of genetic variations in the CSF2 and CSF3 genes with lung function in smoking-induced COPD. Eur Respir J. 2008;32(1):25–34. doi:10.1183/09031936.00040307
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
The Causal Relationship Between Gastroesophageal Reflux Disease and Chronic Obstructive Pulmonary Disease: A Bidirectional Two-Sample Mendelian Randomization Study
Liu B, Chen M, You J, Zheng S, Huang M
International Journal of Chronic Obstructive Pulmonary Disease 2024, 19:87-95
Published Date: 10 January 2024
Exploring a Potential Causal Link Between Dietary Intake and Chronic Obstructive Pulmonary Disease: A Two-Sample Mendelian Randomization Study
Zhang C, Yu L, Xiong T, Zhang Y, Liu J, Zhang J, He P, Xi Y, Jiang Y
International Journal of Chronic Obstructive Pulmonary Disease 2024, 19:297-308
Published Date: 26 January 2024
Association of Chronic Obstructive Pulmonary Disease with Risk of Psychiatric Disorders: A Two-Sample Mendelian Randomization Study
Zhang Q, Zhang H, Xu Q
International Journal of Chronic Obstructive Pulmonary Disease 2024, 19:343-351
Published Date: 1 February 2024
A Bidirectional Mendelian Randomization Study Investigating the Causal Relationship Between Ankylosing Spondylitis and Chronic Obstructive Pulmonary Disease
Pan D, Dai X, Li P, Xue L
International Journal of Chronic Obstructive Pulmonary Disease 2025, 20:259-271
Published Date: 8 February 2025
Identifying Common Diagnostic Biomarkers and Therapeutic Targets between COPD and Sepsis: A Bioinformatics and Machine Learning Approach
Li X, Xiao Y, Yang M, Zhang X, Yuan Z, Zhang Z, Zhang H, Liu L, Zhao M
International Journal of Chronic Obstructive Pulmonary Disease 2025, 20:1761-1786
Published Date: 28 May 2025