Back to Journals » Drug Design, Development and Therapy » Volume 20
Cuproptosis in Sepsis: Cell Type-Specific Mechanisms and Clinical Prospects
Authors Fang S, Li W, Chang Z, Tang X, Niu X, Chen Y, Hu X ![]()
Received 2 February 2026
Accepted for publication 16 April 2026
Published 28 April 2026 Volume 2026:20 600729
DOI https://doi.org/10.2147/DDDT.S600729
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Anastasios Lymperopoulos
Shangping Fang,1,* Wanning Li,1,* Zhaorong Chang,1 Xiaoyu Tang,2 Xin Niu,1 Yongquan Chen,3 Xianwen Hu4
1School of Anesthesiology, Wannan Medical University, Wuhu, Anhui, People’s Republic of China; 2School of Clinical Medicine, Wannan Medical University, Wuhu, Anhui, People’s Republic of China; 3Department of Anesthesiology, The First Affiliated Hospital of Wannan Medical University, Wuhu, Anhui, People’s Republic of China; 4Department of Anesthesiology, The Second Affiliated Hospital of Anhui Medical University, Hefei, Anhui, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yongquan Chen, Department of Anesthesiology, The First Affiliated Hospital of Wannan Medical University, No. 2, Zheshan West Road, Wuhu, Anhui, 241001, People’s Republic of China, Email [email protected] Xianwen Hu, Department of Anesthesiology, The Second Affiliated Hospital of Anhui Medical University, NO.678, Furong Road, Hefei, Anhui, 230601, People’s Republic of China, Email [email protected]
Abstract: Sepsis is a life-threatening clinical syndrome caused by a severely dysregulated host response to infection. As a major global health challenge, it continues to exhibit high mortality. Copper, an essential trace element crucial for biological homeostasis, is central to a recently defined form of cell death: cuprotosis. This novel, copper-dependent mitochondrial cell death pathway is mechanistically distinct from classical apoptosis and pyroptosis. In sepsis, cuprotosis contributes significantly to immune dysfunction and organ failure by mediating the death of both immune cells (e.g. macrophages, lymphocytes) and parenchymal cells (e.g. cardiomyocytes, renal tubular cells). Therefore, modulating this regulatory mechanism in a cell type–specific manner may represent a novel potential therapeutic avenue for sepsis, although substantial clinical validation is still required. This review systematically outlines the core mechanisms of cuprotosis, elucidates its pathophysiological role in sepsis, and evaluates the potential and challenges of targeting cuprotosis for sepsis therapy.
Keywords: cuproptosis, sepsis, mechanism, treatment
Introduction
Sepsis is a life-threatening critical illness characterized by infection-triggered systemic inflammatory response syndrome (SIRS).1 Globally, it causes over 48.9 million new cases and 11 million deaths annually, accounting for 19.7% of all fatalities. In China, the in-hospital mortality rate reaches 35.5%. Its incidence is rising annually due to population aging and increased invasive medical procedures.2 During sepsis, excessive inflammatory cytokines disrupt the endothelial barrier, leading to microcirculatory failure, metabolic disturbances, and mitochondrial dysfunction via multiple cell death pathways including pyroptosis, necrosis, and apoptosis.3,4 Uncontrolled infection-induced immune responses, excessive inflammation, and immunosuppression collectively damage host tissues, culminating in multiple organ dysfunction syndrome (MODS) and markedly increased mortality.5
Clinically, sepsis is defined as life-threatening organ dysfunction caused by a dysregulated host response to infection, characterized by systemic inflammation, endothelial injury, microcirculatory disturbance, and metabolic disorders.6 Current standard therapies focus on early antibiotics, fluid resuscitation, hemodynamic support, and organ supportive care.7 As an innovative adjuvant strategy, hemoadsorption has emerged to scavenge inflammatory mediators and toxins, helping alleviate cytokine storm in severe sepsis.8
Cuproptosis, a novel copper-dependent form of programmed cell death discovered in recent years, operates through a mechanism distinct from traditional pathways. This process is highly dependent on intracellular levels of free copper ions. Excess copper induces abnormal oligomerization of lipoylated proteins,9 which disrupts the mitochondrial TCA cycle, impairs respiratory function, and ultimately leads to cell death via toxic protein aggregation.
The initial inflammatory response in sepsis generates excessive ROS, disrupting intracellular copper homeostasis and promoting cuprotosis. Crucially, cuprotosis is not merely a consequence but also a potent amplifier of inflammation, forming a vicious cycle that drives disease progression.10 Given the central role of cell death in the development and progression of sepsis, cuprotosis—by mediating damage to both immune and parenchymal cells—may represent a novel potential target for sepsis intervention. Although current strategies targeting cuprotosis have involved various approaches, ranging from the optimization of copper chelators to cell-specific delivery systems, their clinical translation still faces major challenges: the regulatory network of cuprotosis across different cell types and disease stages in sepsis remains unclear; there is a lack of biomarkers for real-time monitoring and patient stratification; achieving precise, cell-specific delivery while minimizing off-target effects remains a major technical hurdle; and its crosstalk with other cell death pathways such as pyroptosis and ferroptosis awaits systematic elucidation.
This review aims to systematically elaborate the molecular mechanisms of cuprotosis and its role in the pathophysiology of sepsis, evaluate the potential and challenges of corresponding therapeutic strategies, and provide a theoretical foundation and future directions for the development of novel sepsis therapeutics.
Core Molecular Mechanisms of Cuproptosis
Copper Ion Homeostasis Disruption
As a crucial cofactor, copper’s biological functions depend on its redox activity.11,12 OOSTERHEERT et al13 and HUSSAIN et al14 experimentally identified that extracellular Cu2⁺ is reduced to Cu⁺ by the STEAP reductase family upon reaching the cell surface, then transported into the cytoplasm via the high-affinity copper transporter CTR1 located on the cell membrane. Studies by LU et al15 ZHAO et al16 and COBINE et al17 revealed that intracellular copper ions are directed by chaperone proteins such as copper chaperone for superoxide dismutase (CCS), antioxidant 1 (ATOX1), and cytochrome c oxidase assembly factor 17 (COX17) to mitochondria, antioxidant stress pathways, and energy metabolism pathways, respectively, for delivery to different organelles. Specifically, SHAO et al18 demonstrated, using tissue microarray analysis, that the metal ion transporter ATP7A transports copper to copper-containing enzymes in the secretory pathway. Meanwhile, BITTER et al19 and YU et al20 revealed through structural studies that ATP7B transports copper from the liver to ceruloplasmin, thereby mediating copper excretion into bile. When intracellular copper ion concentrations become excessively high, CTR1 is removed from the cell membrane, shutting down the pathway for cellular copper uptake.21,22 ATP7A and ATP7B then depart from the Golgi apparatus and initiate an export transport program, respectively pumping excess copper out of the cell or secreting it into bile, thereby preventing copper toxicity.23–26 Upon restoration of intracellular copper ion concentrations to normal levels, CTR1 re-localizes to the cell membrane, while ATP7A and ATP7B return to the Golgi apparatus, resuming normal function. Thus, CTR1, ATP7A, and ATP7B work synergistically to form the core regulatory network governing cellular and systemic copper homeostasis.27
Exogenous exposure and genetic defects are driving factors of copper overload. ZHANG et al28 pointed out that exogenous exposure involves excessive copper intake from external sources such as drinking water, occupational exposure, and diet, exceeding the body’s processing and excretion capacity, leading to continuous copper accumulation within the body.29,30 Genetic defects refer to congenital failures in the body’s copper metabolic pathways due to genetic mutations. BULL et al31 discovered that Wilson’s disease (hepatolenticular degeneration) results from ATP7B gene mutations, causing low serum ceruloplasmin levels.32,33 Copper accumulates extensively in the liver;34 once the liver’s storage capacity is reached, copper is released into the bloodstream, causing severe damage to organs such as the cornea and kidneys.35,36
ATRIáN-BLASCO et al37 found that metallothioneins (MTs) are key intracellular copper-buffering proteins that regulate metal homeostasis and mitigate heavy metal toxicity, DNA damage, and oxidative stress. However, both exogenous and genetic copper overload can overwhelm MT buffering capacity.38,39 Saturated MT binding sites or persistent copper accumulation lead to a sharp increase in cytoplasmic free copper ions.25 This not only induces severe oxidative stress, resulting in hepatocyte death, hepatitis, cirrhosis, and liver failure, but also disrupts the metabolism of other essential metals.31
FDX1-Mediated Copper Reduction and Abnormal Lipoylation
TSVETKOV et al9 demonstrated that ferredoxin 1 (FDX1), a small iron-sulfur (Fe-S) cluster protein functioning as an electron carrier in the mitochondrial matrix, receives electrons from the mitochondrial electron transport chain during cellular respiration. This process renders FDX1 negatively charged, endowing it with potent reductive activity capable of reducing Cu2⁺ to Cu⁺. Dihydrolipoamide S-acetyltransferase (DLAT) and dihydrolipoamide dehydrogenase (DLD) are core components of the pyruvate dehydrogenase complex, a key enzyme in the TCA cycle that also includes critical subunits such as pyruvate dehydrogenase E1 alpha subunit (PDHA1) and pyruvate dehydrogenase E1 beta subunit (PDHB). These proteins depend on the lipoylation of specific lysine residues by lipoic acid synthase (LIAS) to maintain TCA cycle‑driven cellular energy metabolism.40 Conversely, Cu⁺ reduced by FDX1 specifically induces abnormal oligomerization of lipoylated proteins—particularly DLAT and DLD—promoting the aggregation of multiple protein molecules through noncovalent interactions into oligomers. This oligomerization progressively evolves into insoluble, fibrillar abnormal protein aggregates.40,41 When DLAT and DLD form insoluble aggregates, their enzymatic activities are lost, leading to the dysfunction of the pyruvate dehydrogenase complex. The TCA cycle is disrupted, resulting in a severe deficiency in ATP (cellular energy) production.9
Mitochondrial Dysfunction
The failure of the pyruvate dehydrogenase complex blocks the conversion of pyruvate to acetyl-CoA, preventing glucose from entering the TCA cycle. Once intermediate metabolites in the TCA cycle are depleted, the entire cycle stalls, leading to a sharp decrease in NADH and FADH2 production. This, in turn, deprives the electron transport chain of electron donors, causing respiratory chain complexes to malfunction.42 Following the collapse of both the TCA cycle and electron transport chain, ATP synthesis becomes severely inadequate, causing rapid depletion of intracellular ATP levels and ultimately inducing cell death.43 Concurrently, dysfunction in the electron transport chain allows highly reduced electron carriers to readily transfer electrons to oxygen (O2), generating superoxide anion (·O2−) and other reactive oxygen species (ROS). Furthermore, intracellular Cu⁺ can catalyze the Fenton reaction, converting hydrogen peroxide (H2O2) into highly toxic hydroxyl radicals (·OH), significantly exacerbating oxidative damage. These radicals attack mitochondrial proteins, lipids, and DNA, further disrupting mitochondrial membrane structure, intensifying energy depletion, and creating a vicious cycle that ultimately leads to cellular collapse.44
Schematic diagram of the core molecular mechanism of cuproptosis are illustrated in Figure 1.
 |
Figure 1 Schematic diagram of the core molecular mechanism of cuproptosis. Abbreviations: STEAP, Six-Transmembrane Epithelial Antigen of the Prostate; CTR1, Copper Transporter 1; ATP7A/B, ATPase Copper Transporting Alpha/Beta; CCS, Copper Chaperone for Superoxide Dismutase; ATOX1, Antioxidant Protein 1; SOD1, Superoxide Dismutase 1; COX17, Cytochrome C Oxidase Assembly Factor 17; COX11, Cytochrome C Oxidase Assembly Factor 11; FDX1, Ferredoxin 1; LIAS, Lipoic Acid Synthase; DLAT, Dihydrolipoamide S-Acetyltransferase; Fe-S, Iron-Sulfur Cluster. Notes: Extracellular Cu2⁺ is reduced to Cu⁺ by STEAP and then taken up into cells through CTR1. Intracellular Cu⁺ is transported via distinct routes by copper chaperones such as CCS, ATOX1 and COX17: some is supplied to SOD1 to sustain redox balance, some is shuttled to the Golgi apparatus or effluxed out of cells via ATP7A/B for copper homeostasis regulation, and the rest moves into mitochondria to take part in respiratory chain assembly and support the TCA cycle. When copper overload occurs, Cu⁺ directly impairs mitochondrial Fe-S clusters and activates the FDX1-LIAS pathway to induce DLAT aggregation, eventually triggering cuproptosis. (Arrows represent promotion, transport, or causal effects). |
Interplay Between Cuproptosis and Other Cell Death Pathways
As a novel form of cell death, a key direction in studying cuproptosis mechanisms lies in elucidating its relationship with other known cell death pathways.
Ferroptosis
Similar to ferroptosis, both represent cell death modes regulated by the accumulation of specific metal ions, and both pathways involve bursts of reactive oxygen species and cellular metabolism. However, significant differences exist in their key mechanisms: Cuproptosis relies on abnormal copper ion accumulation within mitochondria, causing abnormal lipoylation of target proteins, which in turn induces proteotoxic stress and TCA cycle collapse. Copper further promotes ROS production through the Fenton reaction, accelerating cellular damage.9,45 Conversely, ferroptosis relies on the accumulation of iron ions, as highlighted by GUO et al46 and YIN et al47 Its critical pathway involves loss of GPX4 activity within the plasma membrane/endoplasmic reticulum or excessive phospholipid peroxidation, leading to the generation of uncontrolled, toxic lipid ROS. This directly causes plasma membrane rupture and cell death, while peroxidation products of polyunsaturated fatty acids further compromise membrane integrity. Furthermore, although both forms of cell death are accompanied by severe mitochondrial dysfunction, their underlying mechanisms and morphological features differ substantially. Copper exerts direct toxic effects: elevated copper levels target mitochondria, impairing the function of electron transport chain (ETC) complexes and stimulating a sharp increase in reactive oxygen species via the Fenton reaction. These changes rapidly lead to mitochondrial swelling, cristae breakdown, and a marked drop in cellular energy metabolism. Ferroptosis, by contrast, is driven primarily by iron-dependent lipid peroxidation. In this scenario, mitochondria are not just passive targets but also actively contribute to and amplify peroxidative injury, thereby accelerating cell death. Morphologically, ferroptotic cells show distinctly shrunken mitochondria with ruptured outer membranes and unusually high electron density in mitochondrial membranes.48,49
Apoptosis
SATTLER et al50 and ELMORE et al51 propose that apoptosis represents a highly programmed form of cell death, primarily driven by the cascade activation of proteases within the Caspase family—specifically the initiator enzymes Caspase-8/9 and the effector enzyme Caspase-3—ultimately leading to the formation of apoptotic bodies that are phagocytosed and cleared. In contrast, cuproptosis is entirely independent of caspase activation. Broad-spectrum caspase inhibitors fail to inhibit their occurrence, and affected cells exhibit non-apoptotic morphology resembling a necrosis-like phenotype.9
Pyroptosis
Beyond this, pyroptosis—another form of programmed cell death—is activated by inflammatory caspases (Caspase-1/4/5/11), as noted by HE et al52 These activated caspases cleave the Gasdermin D (GSDMD) protein, whose N-terminal domain perforates the cell membrane. This leads to cellular swelling, rupture, and the release of pro-inflammatory cytokines (such as IL-1β, IL-18), triggering a robust inflammatory response.53,54 However, although cuproptosis may also exacerbate pyroptosis by releasing inflammatory mediators and damage-associated molecular patterns (DAMPs) due to cellular contents leakage, this release is passive and independent of GSDMD cleavage and pore formation.9
Distinctive features and interconnections among cuproptosis, ferroptosis, apoptosis, and pyroptosis are presented in Table 1.
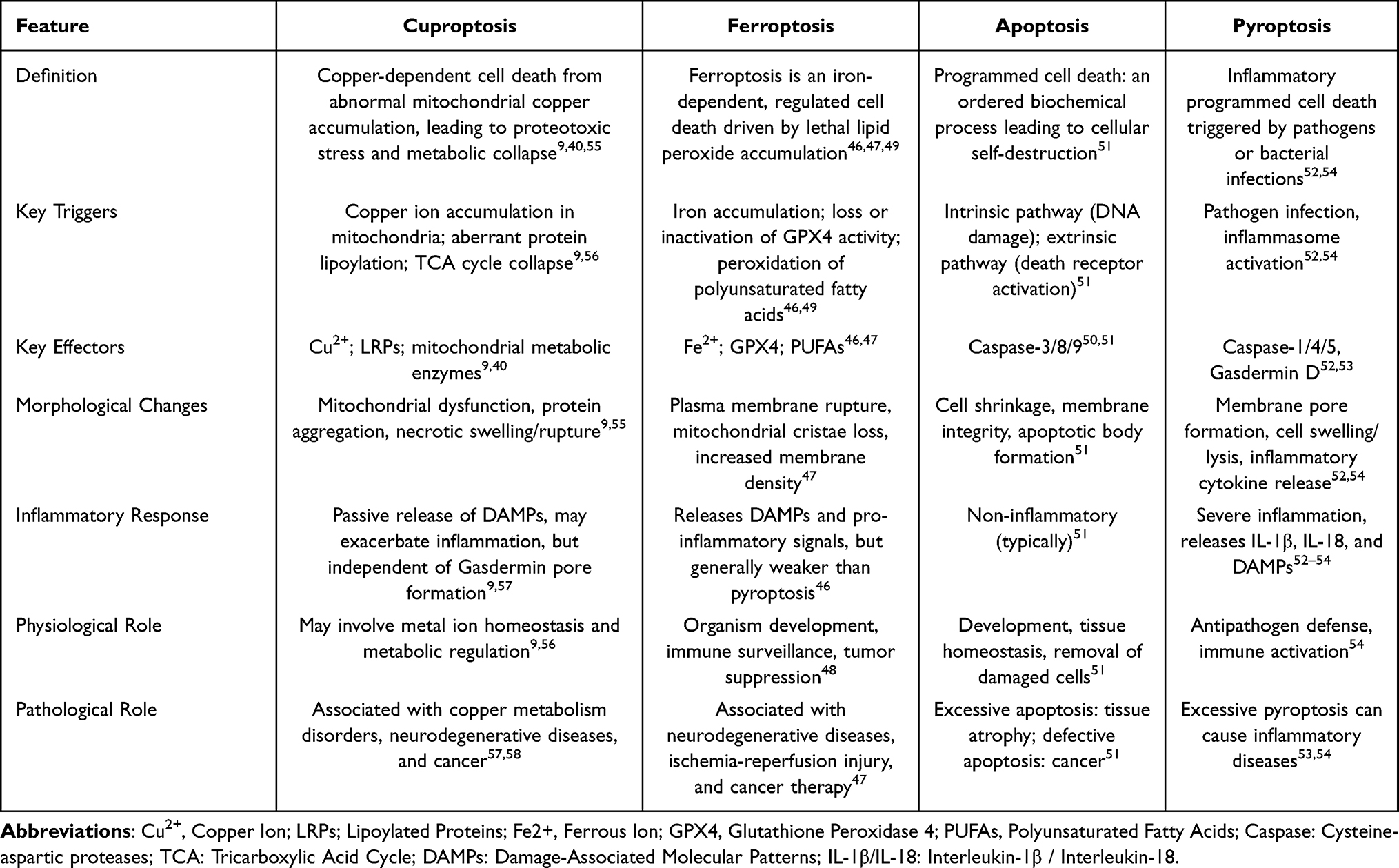 |
Table 1 Distinctive Features and Interconnections Among Cuproptosis, Ferroptosis, Apoptosis and Pyroptosis |
Pathological Role of Cuproptosis in Sepsis
Sepsis Microenvironment Drives Copper Accumulation
Following sepsis onset, the body releases massive pro-inflammatory factors, triggering a cytokine storm. Among these, TNF-α and IL-6 emerge as core mediators with markedly elevated levels.57 LIU et al59 demonstrated that elevated TNF‑α strongly inhibits the activity of the copper transporter ATP7B, impairing copper ion efflux and leading to substantial copper retention in hepatocytes. Concurrently, sepsis frequently complicates into acute liver injury due to ischemia, hypoxia, and inflammatory mediator attack. This hepatic dysfunction directly reduces the synthesis of proteins including ceruloplasmin—a key copper‑transport protein—further decreasing circulating ceruloplasmin and promoting systemic copper accumulation.
Moreover, sepsis‑induced microcirculatory disturbances and metabolic dysfunction cause local tissue hypoxia, which activates hypoxia‑inducible factor‑1α (HIF‑1α). FU et al60 showed that activated HIF‑1α significantly upregulates the expression of the copper importer CTR1 on the cell membrane, driving increased cellular copper uptake. Together, these pathways elevate intracellular copper concentration beyond the threshold that triggers cuproptosis.
Thus, ATP7B inhibition establishes a vicious cycle between cuproptosis and cytokine storm: pro‑inflammatory cytokines (eg., TNF‑α, IL‑6) suppress copper export, leading to accumulation and cuproptosis; in turn, cuproptosis amplifies inflammation through DAMPs release, ROS bursts from mitochondrial dysfunction, and immune cell death, further exacerbating the cytokine storm and driving sepsis progression.10,41,57,61
Interaction Between Sepsis Metabolic Reprogramming and Cuproptosis
Sepsis metabolic reprogramming manifests in two forms: impaired aerobic glycolysis and disrupted fatty acid oxidation (FAO). FAO serves as a crucial source of acetyl-CoA. Research has demonstrated62 that the inflammatory response in sepsis disrupts FAO, leading to decreased acetyl-CoA levels and further impairing the already compromised TCA cycle. Concurrently, impaired FAO induces accumulation of long-chain fatty acids (LCFAs) in the cytoplasm, potentially affecting key enzymes for protein lipoylation. However, due to disrupted TCA cycle and impaired mitochondrial membrane potential, these lipoylated key enzymes cannot be effectively integrated into functional enzyme complexes for efficient catalytic reactions, resulting in unstable lipoylated proteins. This metabolic disorder, characterized by conformationally unstable acylated proteins, significantly heightens cellular sensitivity to copper ion toxicity.9,56 Furthermore, ZHAO et al63 observed that cells in a sepsis environment preferentially utilize aerobic glycolysis for energy production. Reduced pyruvate entry into mitochondria, coupled with impaired TCA cycle function, decreased flux, and diminished energy output, directly induces susceptibility to cuproptosis. The deceleration and disruption of the TCA cycle impede the function and cause conformational instability of associated acyltransferase complexes.9,58 This leads to a shortage of substrates for the mitochondrial respiratory chain, further impairing mitochondrial function. Accumulated intracellular copper ions, mediated by molecules such as FDX1, directly attack and oligomerize unstable acylated proteins. This leads to complete collapse of the TCA cycle and disruption of the electron transport chain, resulting in total ATP production failure and explosive ROS generation. The massive ROS surge further exacerbates copper toxicity through the Fenton reaction, damaging additional metabolic enzymes and mitochondrial components. Collapsed cells release DAMPs, further activating potent inflammatory responses (eg., NF-κB pathway),9 exacerbating the pathological process of sepsis itself and forming a vicious cycle of interaction between sepsis and cuproptosis.
Cell Type-Specific Effects of Cuproptosis in Sepsis
Cuproptosis in Immune Cell Compartments and Immune Dysregulation
Myeloid Cells
Macrophages are pivotal members of the myeloid cell lineage, with their metabolic state directly correlating to functional phenotypes. LIU et al64 proposed that classically activated M1 (pro-inflammatory) macrophages are highly dependent on aerobic glycolysis. Disruption of the TCA cycle leads to succinate accumulation, which in turn promotes the production of inflammatory mediators such as IL-1β by stabilizing HIF-1α. This metabolic state closely resembles the susceptibility metabolic profile observed in cuproptosis. Concurrently, cuproptosis drives copper ion accumulation and FDX1 upregulation, which further enhances glycolysis and the M1 phenotype by stabilizing HIF-1α. Moreover, cuproptosis disrupts the TCA cycle, directly inhibiting the alternative (M2) activation phenotype and thereby indirectly reinforcing the M1 phenotype. Functionally, macrophage phagocytosis and bactericidal activity are energy-intensive processes requiring substantial ATP. Mitochondrial energy collapse induced by cuproptosis renders cells incapable of completing phagocytosis. Furthermore, phagocytosis necessitates cytoskeletal remodeling, and cuproptosis may disrupt actin dynamics via oxidative stress, directly leading to phagocytic dysfunction. Conversely, Fu et al60 further demonstrated that copper-induced lipoylation dysfunction impairs protein function, leading to succinate accumulation. This, in turn, promotes IL-1β production via HIF-1α activation and inhibition of histone demethylation,9 establishing a vicious cycle where inflammation and metabolic dysfunction mutually exacerbate each other.
Lymphocytes
T cells within lymphocytes, particularly effector T cells, undergo extensive depletion and apoptosis during the late stages of sepsis, forming the core of immune suppression in the body. YANG et al65 proposed that upon T-cell activation, increased mitochondrial biogenesis, elevated TCA cycle flux, and heightened lipoylation render them susceptible to cuproptosis. Copper ions accumulated in the septic microenvironment preferentially target and eliminate these effector T cells, which should otherwise mediate immune responses. Even if cuproptosis does not directly kill T cells, sublethal copper stress is sufficient to cause severe mitochondrial dysfunction.9,10,55 Concurrently, WANG et al66 revealed through comprehensive biological sequencing analysis that mitochondrial morphological and functional defects are hallmarks of T cell exhaustion. Thus, cuproptosis selectively eliminates or exhausts functional effector T cells, directly promoting immune paralysis and impairing the body’s ability to clear secondary infections.
Neutrophils
Neutrophils serve as the first responders in the inflammatory response to sepsis, exhibiting dynamic changes in functional status. Their unique bactericidal mechanism—NETosis (neutrophil extracellular trap formation)—requires reactive oxygen species (ROS) production, a process closely linked to mitochondrial function. Copper ions themselves serve as potent inducers of NETosis.67,68 ZHANG et al69 suggested that the high-copper microenvironment formed in sepsis may excessively activate the NETosis pathway. While early NETosis aids in trapping and eliminating pathogens, excessive or uncontrolled NET formation damages vascular endothelium and promotes microthrombus formation, becoming a key mechanism for sepsis-related organ injury. CICHON et al67 propose that the energy crisis and enzymatic dysfunction induced by cuproptosis may prevent neutrophils from forming structurally intact NETs or impair their antimicrobial functions, significantly reducing bactericidal capacity. The metabolic collapse caused by cuproptosis comprehensively weakens chemotaxis, phagocytosis, and oxidative burst capabilities, rendering immune functions ineffective. Ultimately, this exacerbates immune-pathological damage and leads to uncontrolled infection.
In summary, prior work has shown that copper accumulation driven by cuproptosis contributes to immune dysfunction and exaggerated cytokine storms in sepsis.10 A self-amplifying pathological cycle has been proposed: impaired copper export causes intracellular copper overload, which then induces cuproptosis in key immune cells. In macrophages, this pathway stimulates IL‑1β release via the succinate/HIF‑1α axis; in T cells, it leads to cellular exhaustion; and in neutrophils, it triggers irregular NETosis. Collectively, these events intensify inflammatory reactions and further weaken copper transport.9,70,71 This cycle worsens immune imbalance and organ injury.
In addition to sepsis, disrupted copper transport may also contribute to cytokine storm syndromes in surgical settings such as traumatic injury and ischemia‑reperfusion injury. Relevant studies indicate that tissue injury and hypoxia can trigger the release of proinflammatory mediators including TNF‑α and IL‑6, which in turn suppress copper export mediated by ATP7A/B.72 The local copper buildup that follows triggers cuproptosis in parenchymal and immune cells, which amplifies inflammation through DAMP release and mitochondrial damage, ultimately worsening tissue injury.9 Though still exploratory, targeting copper efflux represents a promising strategy for managing inflammation in such settings.
The regulatory mechanisms of cuproptosis on macrophages, T cells, and neutrophils are shown in Figure 2.
 |
Figure 2 Regulatory mechanisms of cuproptosis on macrophages, T cells, and neutrophils. Abbreviations: IL-1β, Interleukin-1 Beta; HIF-1α, Hypoxia-Inducible Factor-1 Alpha; ROS, Reactive Oxygen Species; NETosis, Neutrophil Extracellular Trap; DAMPs, Damage-Associated Molecular Patterns. Notes: TCA cycle impairment causes succinate accumulation, stabilizing HIF-1α to drive M1 activation and IL-1β secretion. Cuproptosis-driven copper accumulation and FDX1 upregulation enhance glycolysis and sustain the M1 phenotype. Meanwhile, ATP depletion and actin damage lead to irreversible macrophage phagocytic exhaustion. T cell: T cell activation boosts mitochondrial biogenesis, TCA cycle flux, and acyl-protein expression, rendering cells highly sensitive to cuproptosis. In sepsis, accumulated copper ions target and eliminate effector T cells, induce mitochondrial dysfunction and apoptosis, resulting in immune paralysis. Neutrophil: Excessive Cu2⁺ and ROS overactivate the NETosis pathway in sepsis; aberrant NETs damage vascular endothelium and promote microthrombosis. Cuproptosis-induced energy crisis and enzymatic dysfunction inhibit NETosis activation and antibacterial activity, significantly reducing neutrophil bactericidal capacity. (T-shaped arrows indicate inhibition; solid arrows indicate promotion/induction; cross symbols indicate blockade;↑, upregulation/increase). |
Cuproptosis and Organ Dysfunction in Substantial Cellular Injury
Endothelial Cells
KLEIN et al73 demonstrated that vascular endothelial dysfunction is the initiating event in acute lung injury (ALI). Endothelial cells sustain primary injury under septic conditions, with their vulnerability highly dependent on mitochondrial energy and intact cytoskeletal integrity. MUHETAER et al74 analyzed the GEO database and indicated that VE-cadherin, a core molecule maintaining endothelial cell-cell junctions and regulating vascular permeability, can be directly modified by copper ions through oxidative stress. Copper ions modify cysteine residues in the extracellular domain of VE-cadherin, disrupting homodimerization and leading to the opening of the endothelial barrier. Under cuproptosis conditions, ATP depletion directly disrupts the polymerization-depolymerization equilibrium of actin, causing stress fiber rupture, cellular collapse, and the formation of intercellular gaps.9 Sublethal copper stress may activate proteases such as caspases, cleaving VE-cadherin and cytoskeletal proteins to further accelerate barrier dysfunction.75–78 Under the influence of cuproptosis, the endothelial barrier is disrupted, leading to a sharp increase in vascular permeability.79 Excessive leakage of fluid and plasma proteins then gives rise to pulmonary edema and hypoxemia, the key pathological features of sepsis-related acute respiratory distress syndrome (ARDS).80,81
Cardiomyocytes
Cardiomyocytes possess the highest mitochondrial density among human cells, with their contractile function highly dependent on ATP. Under cuproptosis conditions, key acylated proteins in the TCA cycle of cardiomyocytes can be directly targeted and oligomerized, inhibiting the entry of acetyl-CoA into the TCA cycle. This leads to a drastic reduction in NADH and FADH2 production, causing ATP yield to collapse.9 YAN et al82 demonstrated that excitation-contraction coupling in cardiomyocytes requires precise calcium ion regulation, an energy-consuming process. ATP depletion leads to dysfunction of calcium pumps on the sarcoplasmic reticulum and sodium-potassium pumps on the cell membrane, causing intracellular calcium overload and diminished contractility. SHEN et al83 noted that ROS bursts triggered by elevated copper ions may directly oxidize cardiac myosin and actin, impairing their contractile activity. This ultimately leads to septic myocardial suppression, reduced cardiac output, and systemic hypoperfusion.84 The combination of energy collapse, calcium dysregulation, and protein injury accounts for the metabolic basis of myocardial suppression and decreased cardiac output in sepsis-induced cardiomyopathy (SICM).85
Renal Tubular Epithelial Cells
Renal tubules, particularly the proximal tubule epithelial cells, serve as the primary injury target in acute kidney injury (AKI). These cells possess a large number of mitochondria and rely on FAO for energy to perform reabsorption functions, making them extremely sensitive to ischemia, hypoxia, and toxic insults. CHEN et al86 ZOU et al87 and SUN et al88 proposed that the kidney, as a vital organ for copper clearance and homeostasis regulation, is susceptible to suppression in the septic environment due to the FAO-dependent metabolic characteristics of renal tubular epithelial cells. Furthermore, owing to their sensitivity, copper ions readily accumulate at high concentrations in these cells. When copper-dependent cell death takes place, extensive loss and shedding of tubular epithelial cells cause tubular blockage and structural damage, allowing filtered fluid to flow backward. This is closely linked to acute tubular necrosis in acute kidney injury (AKI) and is a key mechanism responsible for the rapid deterioration of renal function.89
Hepatocytes and Kupffer Cells
The liver serves as the central organ for metabolic dysregulation and amplified inflammation in sepsis. Hepatocytes, as the primary cells responsible for synthesizing biliverdin and excreting copper, undergo cuproptosis. This triggers a failure in the central regulation of systemic copper homeostasis, exacerbating copper distribution disorders. Necrotic hepatocytes release large quantities of DAMPs signaling molecules, activating Kupffer cells within the liver. Kupffer cells, the primary macrophages within the liver, release substantial inflammatory mediators upon activation. Furthermore, GUO et al90 observed that cuproptosis impairs their phagocytic function, exacerbating local or systemic inflammatory responses and creating a vicious cycle.91 This vicious cycle of liver injury, aggravated inflammation, and systemic homeostasis breakdown greatly speeds up sepsis’s progression toward multiple organ dysfunction.92
Mechanisms of cuproptosis-induced parenchymal cell injury and organ dysfunction are shown in Figure 3. Cascade of key parenchymal cell injuries induced by cuproptosis is shown in Figure 4. Cell-type-specific effects of cuproptosis in sepsis and supporting references are shown in Table 2.
 |
Table 2 Cell-Type-Specific Effects of Cuproptosis in Sepsis and Supporting References |
 |
Figure 3 Mechanisms of cuproptosis-induced parenchymal cell injury and organ dysfunction. Notes: Extracellular Cu2⁺ is reduced to Cu⁺ and transported into cells via CTR1. Excess Cu⁺ triggers cuproptosis by damaging mitochondrial Fe-S clusters and inducing DLAT aggregation. Cuproptosis then drives multi-organ injury: cardiomyocytes suffer reduced contractility from energy depletion and calcium overload; renal tubular epithelial cell sloughing causes acute kidney injury; vascular endothelial damage elevates permeability and leads to tissue edema; and hepatocyte injury releases inflammatory signals to amplify systemic inflammation.(Arrows represent promotion, transport, or causal effects; The intersection node indicates that both upstream factors jointly lead to the downstream effects). |
 |
Figure 4 Cascade of key parenchymal cell injuries induced by cuproptosis. Notes: Arrows indicate changes in expression levels (↑, upregulation/increase; ↓, downregulation/decrease). |
Precision Therapeutic Strategies Targeting the Cuproptosis Network
Reassessment and Optimization of Traditional Intervention Strategies
HUO et al84 noted that copper homeostasis can be regulated by selecting the dosage and timing of copper chelators (TTM). TTM directly reduces intracellular free copper levels.75 This decrease relieves the feedback inhibition of the copper importer CTR1 seen under high-copper conditions, helping maintain its membrane localization and support basic copper uptake. The resulting low-copper signal reshapes intracellular copper distribution, prompting copper-transporting ATPases (ATP7A and ATP7B) to relocate to the Golgi apparatus and facilitate cuproenzyme production. TTM also limits copper entry into mitochondria, thus avoiding copper overload, oxidative injury, and subsequent dysfunction, which helps preserve cellular energy metabolism. The overall reduction in cellular copper burden also aids the normal function of other ion channels. By coordinately regulating copper uptake, transport, and utilization, TTM ultimately restores equilibrium to the disordered copper metabolic network.75 During early sepsis, when copper ion levels surge, TTM administration rapidly controls the copper storm and mitigates damage to cellular mitochondria. Thus, the early sepsis phase represents a critical window for TTM therapy.10 Conversely, copper serves as a cofactor for numerous essential enzymes in the body. Systemic, prolonged, indiscriminate chelation leads to copper deficiency, impairing immune function and tissue repair. Thus, short-term, pulse-based, precision dosing guided by serum copper monitoring is more appropriate than long-term fixed-dose administration. Additionally, rather than directly targeting copper, mitochondrial resilience can be enhanced. Utilizing the targeted antioxidant SS-31 stabilizes mitochondrial inner membrane structure, reduces ROS production, and promotes ATP generation. This approach counters copper-induced mitochondrial oxidative stress and energy collapse in the septic environment, aiding cellular recovery.93,94 Conversely, SAHEBNASAGH et al95 and AGRAWAL et al96 propose addressing FAO impairment and TCA cycle disruption by supplementing with L-carnitine to facilitate fatty acid transport into mitochondria. Supplementing with dicarboxylic acids or ketone bodies bypasses damaged enzymatic pathways, supplying substrates to the TCA cycle.
Precision Targeted Intervention Strategies
Cell-Specific Nanomedicine Delivery Systems
In sepsis, KUNZ et al97 observed that activated endothelial cells highly express adhesion molecules such as ICAM-1 and VCAM-1. Nanoparticles targeting these molecules (eg., functionalized liposomes or polymeric nanoparticles) encapsulating mitochondrial protectants like TTM or SS-31 can be specifically delivered to endothelial cells. This approach mitigates oxidative stress and mitochondrial damage, thereby preserving vascular barrier integrity and reducing plasma leakage and organ injury. Concurrently, microenvironmental characteristics like ROS during disease progression can promote targeted release of nanoscale systems, further enhancing therapeutic precision. Cell-derived nanoparticles (CDNPs) are efficiently internalized by macrophages and enhance their MHCII expression, boosting antigen presentation capacity.97 Furthermore, scavenger receptors on macrophage surfaces recognize specific ligands. Leveraging this pathway, nanoparticles modified with phospholipids or polysaccharides can deliver FDX1 inhibitors or anti-inflammatory drugs, modulating macrophage polarization from pro-inflammatory M1 to anti-inflammatory M2 types and reversing immunosuppression. Furthermore, ZHANG et al98 demonstrated that a DNAzyme nanoplatform targeting ATP7B can regulate copper homeostasis, synergistically enhancing tumor cell death when combined with cuproptosis inducers (eg., Elesclomol). This strategy holds promise for addressing dysregulated immune cells in sepsis. Building on this foundation, nanotechnology can further modulate the sensitivity of immune cells to copper by precisely regulating key cuproptosis-related proteins such as LIAS, PDHA1, and PDHB in these cells.98 For example, nanoparticles can be designed to co-deliver elesclomol and siRNA targeting LIAS or PDHA1. Such a system not only transports copper ions into overactivated immune cells but also specifically inhibits the lipoic acid metabolic pathway, thereby actively inducing a metabolically vulnerable state. This dual strategy renders these cells highly susceptible to copper ion toxicity, allowing more precise and efficient elimination of pathogenic immune cells with minimal impact on normal cell function. Accordingly, this approach provides a novel direction for the precise intervention of immunometabolism in sepsis.99
Key Node Molecular Intervention: FDX1 Inhibition
FDX1 serves as the central molecule in the cuproptosis signaling pathway. TSVETKOV et al9 demonstrated that knocking down FDX1 completely rescues cuproptosis. Consequently, current intervention strategies primarily focus on directly targeting FDX1 or inhibiting it by regulating its upstream modulators. Building on existing research, Huang et al100 activated the MAPK signaling pathway to inhibit FDX1, thereby directly shutting down the cuproptosis signaling pathway without altering systemic copper levels. This approach is particularly crucial for patients requiring preservation of copper’s other physiological functions. SHANG et al101 employed Met (metformin) to directly reduce the expression levels of copper proteins, including FDX1, effectively suppressing cuproptosis and significantly protecting neuronal cells.
Pathophysiological Phase-Based Sequential and Combined Therapies
Differentiated Intervention Strategies for Different Immunometabolic Phases of Sepsis
HOTCHKISS et al102 proposed that the pathological progression of sepsis is not a singular inflammatory process, but rather a dynamic, continuous, and often intertwined immune-metabolic reprogramming process. Its classical paradigm is typically summarized into two dominant phases: the SIRS phase (high inflammation) and the CARS phase (immune suppression). During the SIRS phase of sepsis, copper levels in the body rise significantly, triggering an inflammatory storm. Cells enter a state of hypermetabolism, characterized by widespread apoptosis and dysfunction of immune cells alongside a predominance of anti-inflammatory mediators. At this stage, short-term use of TTM combined with SS-31 should be considered to actively suppress immune cells, mitigate inflammatory responses, and thereby protect organ function.103–105 To prevent excessive inflammatory damage, the SIRS-phase body activates robust endogenous anti-inflammatory mechanisms, transitioning into the CARS phase. Here, immune cell depletion, functional impairment, and copper homeostasis disruption prevail, manifesting as a collapse of defense capabilities dominated by immunosuppression and metabolic exhaustion, making secondary infections likely. Immediate intervention should include energy substrate supplementation, enhanced immune modulation, secondary infection prevention, and protection of parenchymal cells.106,107
Combined Therapies Targeting Multimodal Death Networks
Overcoming the limitations of monotherapy hinges on synergistically targeting the interwoven cell death networks in sepsis. MAO et al108 demonstrated that ferroptosis inhibitors (eg., ferrostatin-1) block ferroptosis by inhibiting lipid peroxidation and glutathione depletion. This approach not only releases DAMPs to activate dendritic cells and T cells but also protects CD8⁺ T cells under specific conditions, thereby preserving immune function. GUO et al99 and IMAM et al109 demonstrated that cuproptosis inhibitors (eg., copper chelators) prevent mitochondrial protein lipidation and metabolic dysfunction by regulating copper homeostasis proteins (eg., ATP7A/B, FDX1). Both pathways share GSH metabolic nodes, and copper ion accumulation further promotes reactive oxygen species (ROS) production, intersecting with ferroptosis pathways. MENG et al110 demonstrated that immunomodulators (eg., anti-PD-1 antibodies) remodel the tumor microenvironment to release T-cell suppression and enhance immune responses. Their combined use creates a synergistic mechanism: copper/iron death inhibitors increase cellular immunogenicity and susceptibility to immune attacks while modulating the immune microenvironment; immunomodulators further amplify immune effects triggered by death signals. Studies confirm that in models like breast cancer, combination therapy significantly elevates tumor-infiltrating lymphocyte levels and prolongs survival, demonstrating multi-target synergistic therapeutic potential.109 TONG et al111 building on prior research, concluded that copper-mediated death engages cross-talk with other cell death pathways, including apoptosis, pyroptosis, and ferroptosis. They summarized that multiple drugs can produce synergistic effects by jointly blocking diverse death modes.
Prospects
Translating cuproptosis research into clinical practice presents a key challenge: how to achieve precise interventions amid rapidly evolving disease states. As a vital micronutrient, copper demands tight therapeutic control. Although excess copper induces toxicity, insufficient levels can lead to severe complications including immunosuppression. This delicate balance is particularly hard to sustain in sepsis, a condition marked by rapid progression and high patient heterogeneity.
At present, real-time in vivo measurement of cuproptosis in specific tissues or cell populations remains impractical due to a shortage of robust biomarkers, severely limiting progress in targeted therapies. Lipoylation-associated proteins LIAS and PDHB have emerged as promising indicators. In patients with sepsis, reduced LIAS expression is linked to more severe organ damage and inflammation, supporting its use in early risk stratification. Elevated serum soluble PDHB (sPDHB) in critically ill individuals is also associated with poorer clinical outcomes. While combined assessment of these markers may enhance diagnostic accuracy and treatment monitoring, standardized protocols and large-cohort validation are still required.
Still, the most daunting obstacle remains the highly dynamic character of sepsis. Without biomarkers that capture the spatial and temporal changes of copper-mediated cell death, delivering tailored treatment to the right targets at optimal times remains challenging.
Future studies should take advantage of the cell-type specificity of cuproptosis. Single-cell sequencing and spatial transcriptomics can be used to generate detailed molecular profiles of injured organs in sepsis, helping identify tissue-specific regulatory hubs to inform geted drug development. Identifying sensitive and dynamic biomarkers for cuproptosis will be essential. Such tools will support more accurate clinical interventions, improve early risk evaluation, and ultimately enhance prognosis for patients with sepsis.
Conclusion
In summary, cuproptosis is a novel copper-dependent programmed cell death pathway that plays a cell type-specific role in sepsis pathology. It mediates the death of immune cells (macrophages, T cells, neutrophils) and parenchymal cells (endothelial cells, cardiomyocytes, renal tubular epithelial cells, hepatocytes), thereby driving immune dysfunction, systemic inflammation, and multi-organ injury. Specifically, cuproptosis promotes M1 polarization and phagocytic dysfunction in macrophages, exhausts effector T cells, and induces abnormal NETosis in neutrophils, collectively amplifying the cytokine storm and forming a vicious cycle with copper accumulation and metabolic dysregulation. In parenchymal cells, it disrupts mitochondrial function and metabolism, contributing to sepsis-related organ injuries such as ALI/ARDS, SICM, and AKI. Cuproptosis also interacts with other cell death pathways—ferroptosis, apoptosis, and pyroptosis—through distinct mechanisms, further contributing to sepsis pathogenesis.
Current evidence on cuproptosis in sepsis remains largely mechanistic, with limited clinical validation. This review summarizes its core mechanisms, cell type-specific roles, and crosstalk with other death pathways, while identifying key knowledge gaps: the regulatory network of cuproptosis across cell types and disease stages in sepsis needs clarification; biomarkers such as LIAS and PDHB require large-scale clinical validation for diagnosis and stratification; the mechanisms by which cuproptosis contributes to organ dysfunction (eg., ALI, SICM) need further exploration; and therapeutic strategies targeting cuproptosis—including copper chelators, nanotechnology, and drug repurposing—face translational challenges. Despite these limitations, targeting cuproptosis and its cell type-specific regulation offers a novel theoretical and potential therapeutic avenue for improving outcomes in sepsis.
Data Sharing Statement
No data was used for the research described in the article.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by funding from Anhui University Research Project (No. 2023AH040252), Natural Science Foundation of Anhui Province (No. 2208085MC55), the Key Project of Anhui Province “Leading the Charge with Open Competition”, and Anhui Province Clinical Key Specialty Construction Project, Anhui Province College Student Innovation and Entrepreneurship Project (S202510368015).
Disclosure
The authors declare that they have no conflicts of interest.
References
1. Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. 2016;315(8):801–17. doi:10.1001/jama.2016.0287
2. Rudd KE, Johnson SC, Agesa KM, et al. Global, regional, and national sepsis incidence and mortality, 1990-2017: analysis for the Global Burden of Disease Study. Lancet. 2020;395(10219):200–211. doi:10.1016/S0140-6736(19)32989-7
3. Xu X, An J, Yang T, Li B, Dou Z. Research progress on the role of mesenchymal stem cells in pyroptosis in sepsis. Stem Cell Res Ther. 2025;17(1):24. doi:10.1186/s13287-025-04837-x
4. Kashif AM, Ouyang Y, Li Y, Pan B. NETosis and pyroptosis of immune cells in sepsis. J Translat Internal Med. 2025;13(4):318–327. doi:10.1515/jtim-2025-0035
5. Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol. 2013;13(12):862–874. doi:10.1038/nri3552
6. Arina P, Hofmaenner DA, Singer M. Definition and epidemiology of sepsis. Seminars Respirat Critic Care Med. 2024;45(4):461–468. doi:10.1055/s-0044-1787990
7. Gotts JE, Matthay MA. Sepsis: pathophysiology and clinical management. BMJ. 2016;353:i1585. doi:10.1136/bmj.i1585
8. Sazonov V, Abylkassov R, Tobylbayeva Z, Saparov A, Mironova O, Poddighe D. Case series: efficacy and safety of hemoadsorption with HA-330 adsorber in septic pediatric patients with cancer. Front Pediatrics. 2021;9:672260. doi:10.3389/fped.2021.672260
9. Tsvetkov P, Coy S, Petrova B, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375(6586):1254–1261. doi:10.1126/science.abf0529
10. Tan R, Wen K, Zhao T, et al. Deciphering cuproptosis in sepsis: mechanisms, consequences, and therapeutic opportunities. J Inflamm Res. 2025;18:9879–9890. doi:10.2147/JIR.S533967
11. Kim BE, Nevitt T, Thiele DJ. Mechanisms for copper acquisition, distribution and regulation. Nature Chem Biol. 2008;4(3):176–185. doi:10.1038/nchembio.72
12. Lutsenko S. Human copper homeostasis: a network of interconnected pathways. Curr Opin Chem Biol. 2010;14(2):211–217. doi:10.1016/j.cbpa.2010.01.003
13. Oosterheert W, van Bezouwen LS, Rodenburg RNP, et al. Cryo-EM structures of human STEAP4 reveal mechanism of iron(III) reduction. Nat Commun. 2018;9(1):4337. doi:10.1038/s41467-018-06817-7
14. Hussain Q, Ye T, Li S, Nkoh JN, Zhou Q, Shang C. Genome-wide identification and expression analysis of the copper transporter (COPT/Ctr) gene family in kandelia obovata, a typical mangrove plant. Int J Mol Sci. 2023;24(21).
15. Lu Y, Chan YT, Wu J, et al. CRISPR/Cas9 screens unravel miR-3689a-3p regulating sorafenib resistance in hepatocellular carcinoma via suppressing CCS/SOD1-dependent mitochondrial oxidative stress. Drug Resist Updates. 2023;71:101015. doi:10.1016/j.drup.2023.101015
16. Zhao P, Shi W, Ye Y, et al. Atox1 protects hippocampal neurons after traumatic brain injury via DJ-1 mediated anti-oxidative stress and mitophagy. Redox Biol. 2024;72:103156. doi:10.1016/j.redox.2024.103156
17. Cobine PA, Moore SA, Leary SC. Getting out what you put in: copper in mitochondria and its impacts on human disease. Biochimica Et Biophysica Acta Mol Cell Res. 2021;1868(1):118867. doi:10.1016/j.bbamcr.2020.118867
18. Shao K, Shen H, Chen X, et al. Copper transporter gene ATP7A: a predictive biomarker for immunotherapy and targeted therapy in hepatocellular carcinoma. Int Immunopharmacol. 2023;114:109518. doi:10.1016/j.intimp.2022.109518
19. Bitter RM, Oh S, Deng Z, Rahman S, Hite RK, Yuan P. Structure of the Wilson disease copper transporter ATP7B. Sci Adv. 2022;8(9):eabl5508. doi:10.1126/sciadv.abl5508
20. Yu CH, Lee W, Nokhrin S, Dmitriev OY. The structure of metal binding domain 1 of the copper transporter ATP7B reveals mechanism of a singular wilson disease mutation. Sci Rep. 2018;8(1):581. doi:10.1038/s41598-017-18951-1
21. Wen MH, Chen H, Yan G, et al. Elevated intracellular copper induces CTR1 monomerization and prevents copper uptake. Nat Commun. 2025;16(1):11500. doi:10.1038/s41467-025-66283-w
22. Chatterjee SK, Kar S, Nikte SV, et al. Noncanonical regulation of the plasma membrane copper transporter CTR1 through modulation of membrane mechanical properties. Mol Biol Cell. 2026;37(2):ar13. doi:10.1091/mbc.E25-04-0159
23. Gao S, Zhang H, Zhang X, Wang J, Bai W, Jiang B. COX19 is a new target of MACC1 and promotes colorectal cancer progression by regulating copper transport in mitochondria. J Nutr. 2024;154(2):381–394. doi:10.1016/j.tjnut.2023.12.032
24. Ruturaj Mishra M, Saha S, Maji S, et al. Regulation of the apico-basolateral trafficking polarity of the homologous copper-ATPases ATP7A and ATP7B. J Cell Sci. 2024;137(5).
25. Guo Z, Chen D, Yao L, et al. The molecular mechanism and therapeutic landscape of copper and cuproptosis in cancer. Signal Transduct Target Ther. 2025;10(1):149. doi:10.1038/s41392-025-02192-0
26. More SJ, Bampidis V, Benford D, et al. Re-evaluation of the existing health-based guidance values for copper and exposure assessment from all sources. EFSA J Eur Food Safety Authority. 2023;21(1):e07728. doi:10.2903/j.efsa.2023.7728
27. Wang Y, Pei P, Yang K, Guo L, Li Y. Copper in colorectal cancer: from copper-related mechanisms to clinical cancer therapies. Clin Translat Med. 2024;14(6):e1724. doi:10.1002/ctm2.1724
28. Zhang K, Nulali J, Zhang C, et al. Association between dietary copper intake and respiratory disease mortality. J Nutr Biochem. 2026;151:110273. doi:10.1016/j.jnutbio.2026.110273
29. Gan X, He P, Zhou C, et al. J-shaped association between dietary copper intake and all-cause mortality: a prospective cohort study in Chinese adults. Br J Nutr. 2023;129(11):1841–1847. doi:10.1017/S0007114522002732
30. Gerhardsson L, Englyst V, Lundström NG, Sandberg S, Nordberg G. Cadmium, copper and zinc in tissues of deceased copper smelter workers. J Trace Elem Med Biol. 2002;16(4):261–266. doi:10.1016/S0946-672X(02)80055-4
31. Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nature Genet. 1993;5(4):327–337. doi:10.1038/ng1293-327
32. Sasula MJ, Held ATJ, Schefczyk S, et al. High copper levels induce oxidative stress and inflammatory processes in a cell culture model of Wilson’s disease. Mol Cell Biochem. 2026;481(3):1487–1499. doi:10.1007/s11010-026-05481-6
33. Muchenditsi A, Talbot CC, Gottlieb A, et al. Systemic deletion of Atp7b modifies the hepatocytes’ response to copper overload in the mouse models of Wilson disease. Sci Rep. 2021;11(1):5659. doi:10.1038/s41598-021-84894-3
34. El Nachef L, Al-Choboq J, Bourguignon M, Foray N. Response of fibroblasts from menkes’ and wilson’s copper metabolism-related disorders to ionizing radiation: influence of the nucleo-shuttling of the ATM protein kinase. Biomolecules. 2023;13(12):1746. doi:10.3390/biom13121746
35. Abbassi N, Bourrahouat A, Bedoya EC, et al. Epidemiology, clinical features, and mortality rate of Wilson disease in Moroccan children: a pediatric case series. Archiv de Pediatrie. 2022;29(6):453–458. doi:10.1016/j.arcped.2022.03.010
36. Lv D, Yu Y. Copper-Induced cell death in renal diseases: molecular mechanisms and therapeutic implications. Drug Des Devel Ther. 2025;19:11849–11861. doi:10.2147/DDDT.S562664
37. Atrián-Blasco E, Santoro A, Pountney DL, Meloni G, Hureau C, Faller P. Chemistry of mammalian metallothioneins and their interaction with amyloidogenic peptides and proteins. Chem Soc Rev. 2017;46(24):7683–7693. doi:10.1039/C7CS00448F
38. Klaassen CD, Liu J, Choudhuri S. Metallothionein: an intracellular protein to protect against cadmium toxicity. Annu Rev Pharmacol Toxicol. 1999;39:267–294. doi:10.1146/annurev.pharmtox.39.1.267
39. Lazo JS, Pitt BR. Metallothioneins and cell death by anticancer drugs. Annu Rev Pharmacol Toxicol. 1995;35:635–653. doi:10.1146/annurev.pa.35.040195.003223
40. Hsiao JC, Warui DM, Kwon JJ, et al. Deep mutational scanning of FDX1 identifies key structural determinants of lipoylation and cuproptosis. Nat Commun. 2025;17. doi:10.1038/s41467-025-67869-0
41. Xie J, Yang Y, Gao Y, He J. Cuproptosis: mechanisms and links with cancers. Mol Cancer. 2023;22(1):46. doi:10.1186/s12943-023-01732-y
42. Zhu S, Niu Y, Zhou W, et al. Mitochondrial copper overload promotes renal fibrosis via inhibiting pyruvate dehydrogenase activity. Cell Mol Life Sci. 2024;81(1):340. doi:10.1007/s00018-024-05358-1
43. Wu J, Zhang W, Zhao S, et al. Research of progress of pyruvate dehydrogenase complex in sepsis metabolism. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. 2021;33(6):765–768. doi:10.3760/cma.j.cn121430-20210127-00133
44. Mao L, Sun M, Chen Z, et al. The pyruvate dehydrogenase complex mitigates LPS-Induced endothelial barrier dysfunction by metabolic regulation. Shock. 2022;57(6):308–317. doi:10.1097/SHK.0000000000001931
45. Jenkins H, MacLean L, McClean S, et al. Structural and solution speciation studies on selected [Cu(NN)(OO)] complexes and an investigation of their biomimetic activity, ROS generation and their cytotoxicity in normoxic, hypoxic and anoxic environments in MCF-7 breast cancer-derived cells. J Inorganic Biochem. 2023;249:112383. doi:10.1016/j.jinorgbio.2023.112383
46. Guo Z, Liu Y, Chen D, et al. Targeting regulated cell death: apoptosis, necroptosis, pyroptosis, ferroptosis, and cuproptosis in anticancer immunity. J Translat Internal Med. 2025;13(1):10–32. doi:10.1515/jtim-2025-0004
47. Yin S, Li Z, Ou WB. Ferroptosis: mechanisms, comparison with cuproptosis and emerging horizons in therapeutics. Oncology Res. 2025;34(1):8. doi:10.32604/or.2025.069049
48. Bock FJ, Tait SWG. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol. 2020;21(2):85–100. doi:10.1038/s41580-019-0173-8
49. Jiang Y, Zhang M, Sun M. ACSL4 at the helm of the lipid peroxidation ship: a deep-sea exploration towards ferroptosis. Front Pharmacol. 2025;16:1594419. doi:10.3389/fphar.2025.1594419
50. Sattler M, Liang H, Nettesheim D, et al. Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science. 1997;275(5302):983–986. doi:10.1126/science.275.5302.983
51. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495–516. doi:10.1080/01926230701320337
52. He WT, Wan H, Hu L, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015;25(12):1285–1298. doi:10.1038/cr.2015.139
53. Rao Z, Zhu Y, Yang P, et al. Pyroptosis in inflammatory diseases and cancer. Theranostics. 2022;12(9):4310–4329. doi:10.7150/thno.71086
54. Yu P, Zhang X, Liu N, Tang L, Peng C, Chen X. Pyroptosis: mechanisms and diseases. Signal Transduct Target Ther. 2021;6(1):128. doi:10.1038/s41392-021-00507-5
55. Duan WJ, He RR. Cuproptosis: copper-induced regulated cell death. Sci China Life Sci. 2022;65(8):1680–1682. doi:10.1007/s11427-022-2106-6
56. Tang D, Chen X, Kroemer G. Cuproptosis: a copper-triggered modality of mitochondrial cell death. Cell Res. 2022;32(5):417–418. doi:10.1038/s41422-022-00653-7
57. Li J, Liu H, Shan Z, Zhong K, Liang Q. Molecular mechanisms and potential implications of ferroptosis, cuproptosis, and disulfidptosis in septic lung injury. Front Med. 2025;12:1615264. doi:10.3389/fmed.2025.1615264
58. Chen P, He P, Rao X, et al. Emerging trends in cardiovascular diseases: the impact of ferroptosis and cuproptosis on cardiomyocyte death. Mol Cell Biochem. 2025;480(10):5323–5344. doi:10.1007/s11010-025-05340-w
59. Liu L, Geng X, McDermott J, et al. Copper deficiency in the lungs of TNF-α transgenic mice. Front Physiol. 2016;7:234. doi:10.3389/fphys.2016.00234
60. Fu DG, He JZ, Mu QC, et al. Inhibition of CTR1 expression improves hypoxia/reoxygenation-induced myoblast injury by blocking cuproptosis. Biochem Biophys Res Commun. 2024;735:150804. doi:10.1016/j.bbrc.2024.150804
61. Roelofsen H, Wolters H, Van Luyn MJ, Miura N, Kuipers F, Vonk RJ. Copper-induced apical trafficking of ATP7B in polarized hepatoma cells provides a mechanism for biliary copper excretion. Gastroenterology. 2000;119(3):782–793. doi:10.1053/gast.2000.17834
62. Sun Z, Zhao Q, Zhang J, et al. Bioinformatics reveals diagnostic potential of cuproptosis-related genes in the pathogenesis of sepsis. Heliyon. 2024;10(1):e22664. doi:10.1016/j.heliyon.2023.e22664
63. Zhao T, Guo Y, Li J. Identification and experimental validation of cuproptosis regulatory program in a sepsis immune microenvironment through a combination of single-cell and bulk RNA sequencing. Front Immunol. 2024;15:1336839. doi:10.3389/fimmu.2024.1336839
64. Liu T, Wen Z, Shao L, et al. ATF4 knockdown in macrophage impairs glycolysis and mediates immune tolerance by targeting HK2 and HIF-1α ubiquitination in sepsis. Clin Immunol. 2023;254:109698. doi:10.1016/j.clim.2023.109698
65. Yang J, Chen N, Zhao P, et al. Diminished expression of GLS in CD4 + T cells serves as a prognostic indicator associated with cuproptosis in septic patients. Shock. 2024;62(1):51–62. doi:10.1097/SHK.0000000000002370
66. Wang T, Fang X, Sheng X, et al. Identification of immune characteristic biomarkers and therapeutic targets in cuproptosis for sepsis by integrated bioinformatics analysis and single-cell RNA sequencing analysis. Heliyon. 2024;10(5):e27379. doi:10.1016/j.heliyon.2024.e27379
67. Cichon I, Ortmann W, Bednarz A, Lenartowicz M, Kolaczkowska E. Reduced Neutrophil Extracellular Trap (NET) formation during systemic inflammation in mice with Menkes disease and Wilson disease: copper requirement for NET release. Front Immunol. 2019;10:3021. doi:10.3389/fimmu.2019.03021
68. Thoraval L, Thiébault E, Siboni R, et al. The acute inflammatory response to copper(II)-doped biphasic calcium phosphates. Mater Today Bio. 2023;23:100814. doi:10.1016/j.mtbio.2023.100814
69. Zhang H, Wang Y, Qu M, et al. Neutrophil, neutrophil extracellular traps and endothelial cell dysfunction in sepsis. Clin Translat Med. 2023;13(1):e1170. doi:10.1002/ctm2.1170
70. Li X, Lin Y, Cheng X, et al. Ovarian ferroptosis induced by androgen is involved in pathogenesis of PCOS. Human Reproduction Open. 2024;2024(2):hoae013. doi:10.1093/hropen/hoae013
71. Vaseruk A, Bila G, Bilyy R. Nanoparticles for stimulation of neutrophil extracellular trap-mediated immunity. Eur J Immunol. 2024;54(4):e2350582. doi:10.1002/eji.202350582
72. Yang S, Li X, Yan J, et al. Disulfiram downregulates ferredoxin 1 to maintain copper homeostasis and inhibit inflammation in cerebral ischemia/reperfusion injury. Sci Rep. 2024;14(1):15175. doi:10.1038/s41598-024-64981-x
73. Klein D. The vascular endothelium as decision maker in lung injury. Front Cell Develop Biol. 2025;13:1564627. doi:10.3389/fcell.2025.1564627
74. Muhetaer M, He T, Zhu H, et al. Analysis of the correlation between cuproptosis and instability of atherosclerotic plaques. Biomedicines. 2025;13(12):2983. doi:10.3390/biomedicines13122983
75. Wei H, Zhang WJ, Leboeuf R, Frei B. Copper induces--and copper chelation by tetrathiomolybdate inhibits--endothelial activation in vitro. Redox Rep. 2014;19(1):40–48. doi:10.1179/1351000213Y.0000000070
76. Herren B, Levkau B, Raines EW, Ross R. Cleavage of beta-catenin and plakoglobin and shedding of VE-cadherin during endothelial apoptosis: evidence for a role for caspases and metalloproteinases. ?mol Biol Cell. 1998;9(6):1589–1601. doi:10.1091/mbc.9.6.1589
77. Chen X, Cai Q, Liang R, et al. Copper homeostasis and copper-induced cell death in the pathogenesis of cardiovascular disease and therapeutic strategies. Cell Death Dis. 2023;14(2):105. doi:10.1038/s41419-023-05639-w
78. Su W, Kowalczyk AP. The VE-cadherin cytoplasmic domain undergoes proteolytic processing during endocytosis Mol Biol Cell. 2017;28(1):76–84. doi:10.1091/mbc.e16-09-0658
79. Wang Z, Wu C, Li R, et al. Cuproptosis and cardiovascular diseases: mechanisms, pathophysiology, and therapeutic Strategies-A narrative review. Rev Cardiovasc Med. 2025;26(9):38833. doi:10.31083/RCM38833
80. Cusack R, Bos LD, Povoa P, Martin-Loeches I. Endothelial dysfunction triggers acute respiratory distress syndrome in patients with sepsis: a narrative review. Front Med. 2023;10:1203827. doi:10.3389/fmed.2023.1203827
81. Sheu CC, Gong MN, Zhai R, et al. Clinical characteristics and outcomes of sepsis-related vs non-sepsis-related ARDS. Chest. 2010;138(3):559–567. doi:10.1378/chest.09-2933
82. Yan J, Li Z, Li Y, Zhang Y. Sepsis induced cardiotoxicity by promoting cardiomyocyte cuproptosis. Biochem Biophys Res Commun. 2024;690:149245. doi:10.1016/j.bbrc.2023.149245
83. Shen Z, Liu Z, Cai S, et al. Copper homeostasis and cuproptosis in myocardial infarction: molecular mechanisms, treatment strategies and potential therapeutic targets. Front Pharmacol. 2025;16:1525585. doi:10.3389/fphar.2025.1525585
84. Huo S, Wang Q, Shi W, et al. ATF3/SPI1/SLC31A1 signaling promotes cuproptosis induced by advanced glycosylation end products in diabetic myocardial injury. Int J Mol Sci. 2023;24(2):1667. doi:10.3390/ijms24021667
85. Kuroshima T, Kawaguchi S, Okada M. Current perspectives of mitochondria in sepsis-induced cardiomyopathy. Int J Mol Sci. 2024;25(9):4710. doi:10.3390/ijms25094710
86. Chen S, Chen T, Xu C, et al. Iron overload exaggerates renal ischemia-reperfusion injury by promoting tubular cuproptosis via interrupting function of LIAS. Redox Biol. 2025;86:103795. doi:10.1016/j.redox.2025.103795
87. Zou Y, Wu S, Xu X, et al. Cope with copper: from molecular mechanisms of cuproptosis to copper-related kidney diseases. Int Immunopharmacol. 2024;133:112075. doi:10.1016/j.intimp.2024.112075
88. Sun DQ, Zhong MY, Zhang JH, et al. Oxidized-LDL aggravates renal injury via tubular cuproptosis. Cell Signalling. 2025;132:111839. doi:10.1016/j.cellsig.2025.111839
89. Deng XJ, Wang YN, Lv CB, et al. Effect of cuproptosis on acute kidney injury after cardiopulmonary bypass in diabetic patients. World J Diabet. 2024;15(10):2123–2134. doi:10.4239/wjd.v15.i10.2123
90. Guo Y, Yang M, Sun S, et al. FDX1-mediated cuproptosis promotes cholestatic liver injury exacerbated by taurocholic acid-enhanced copper accumulation. Cell Death Discovery. 2026;12(1):12. doi:10.1038/s41420-025-02861-7
91. Chen H, Li D, Zhang H, et al. Mechanisms of copper metabolism and cuproptosis: implications for liver diseases. Front Immunol. 2025;16:1633711. doi:10.3389/fimmu.2025.1633711
92. Strnad P, Tacke F, Koch A, Trautwein C. Liver - guardian, modifier and target of sepsis. Nat Rev Gastroenterol Hepatol. 2017;14(1):55–66. doi:10.1038/nrgastro.2016.168
93. Zhong L, Ren X, Ai Y, Liu Z. SS-31 improves cognitive function in sepsis-associated encephalopathy by inhibiting the Drp1-NLRP3 inflammasome activation. Neuromol Med. 2023;25(2):230–241. doi:10.1007/s12017-022-08730-1
94. Sun M, Ma J, Ye J, Fan H, Le J, Zhu J. Protective effect of mitochondria-targeted antioxidant peptide SS-31 in sepsis-induced acute kidney injury. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. 2021;33(12):1418–1422. doi:10.3760/cma.j.cn121430-20210719-01063
95. Sahebnasagh A, Avan R, Monajati M, et al. L-carnitine: searching for new therapeutic strategy for sepsis management. Curr Med Chem. 2022;29(18):3300–3323. doi:10.2174/0929867328666211117092345
96. Agrawal V, Hemnes AR, Shelburne NJ, et al. l-Carnitine therapy improves right heart dysfunction through Cpt1-dependent fatty acid oxidation. Pulmonary Circulat. 2022;12(3):e12107. doi:10.1002/pul2.12107
97. Kunz N, Xia BT, Kalies KU, et al. Cell-Derived nanoparticles are endogenous modulators of sepsis with therapeutic potential. Shock. 2017;48(3):346–354. doi:10.1097/SHK.0000000000000855
98. Zhang H, Ding B, Kou Q. Functional nano-nucleic acid platform promotes copper-induced gastric cancer cell death. RSC Adv. 2025;15(57):49643–49651. doi:10.1039/D5RA07735D
99. Guo B, Yang F, Zhang L, et al. Cuproptosis induced by ROS responsive nanoparticles with elesclomol and copper combined with αPD-L1 for enhanced cancer immunotherapy. Adv Mater. 2023;35(22):e2212267. doi:10.1002/adma.202212267
100. Huang XX, Xie CK, Mo YC, et al. Targeting SERPINB3-MAPK axis-mediated cuproptosis resistance enhances the response to antitumor immunotherapy. Mol Cancer. 2025;25(1). doi:10.1186/s12943-025-02529-x
101. Shang H, Wang Z, Sun Y, et al. Metformin inhibits microglial activation-mediated cuproptosis by modulating the TLR4/Myd88/NF-κB signaling pathway in parkinson’s disease. Mol Neurobiol. 2025;63(1):95. doi:10.1007/s12035-025-05499-9
102. Hotchkiss RS, Coopersmith CM, McDunn JE, Ferguson TA. The sepsis seesaw: tilting toward immunosuppression. Nature Med. 2009;15(5):496–497. doi:10.1038/nm0509-496
103. Moore AR, Zheng H, Ganesan A, et al. Author correction: a consensus immune dysregulation framework for sepsis and critical illnesses. Nature Med. 2025;31(12):4313. doi:10.1038/s41591-025-04066-y
104. Huang Z, Cao L, Yan D. Inflammatory immunity and bacteriological perspectives: a new direction for copper treatment of sepsis. J Trace Elem Med Biol. 2024;84:127456. doi:10.1016/j.jtemb.2024.127456
105. You W. Roles of cytokine storm in sepsis progression: biomarkers, and emerging therapeutic strategies. Front Immunol. 2025;16:1696366. doi:10.3389/fimmu.2025.1696366
106. McBride MA, Owen AM, Stothers CL, et al. The metabolic basis of immune dysfunction following sepsis and trauma. Front Immunol. 2020;11:1043. doi:10.3389/fimmu.2020.01043
107. Chapman NM, Chi H. Metabolic adaptation in CD4(+) T cells promotes immunosuppression in sepsis. Nat Immunol. 2026;27(2):178–180. doi:10.1038/s41590-025-02403-4
108. Mao Z, Hu Y, Zhao Y, et al. The mutual regulatory role of ferroptosis and immunotherapy in anti-tumor therapy. Apoptosis. 2024;29(9–10):1291–1308. doi:10.1007/s10495-024-01988-9
109. Imam M, Ji J, Zhang Z, Yan S. Targeting the initiator to activate both ferroptosis and cuproptosis for breast cancer treatment: progress and possibility for clinical application. Front Pharmacol. 2024;15:1493188. doi:10.3389/fphar.2024.1493188
110. Meng Y, Ye F, Nie P, et al. Immunosuppressive CD10(+)ALPL(+) neutrophils promote resistance to anti-PD-1 therapy in HCC by mediating irreversible exhaustion of T cells. J Hepatol. 2023;79(6):1435–1449. doi:10.1016/j.jhep.2023.08.024
111. Tong X, Tang R, Xiao M, et al. Targeting cell death pathways for cancer therapy: recent developments in necroptosis, pyroptosis, ferroptosis, and cuproptosis research. J hematol Oncol. 2022;15(1):174.
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
The Emerging Role of Ferroptosis in Sepsis, Opportunity or Challenge?
Huang Q, Ding Y, Fang C, Wang H, Kong L
Infection and Drug Resistance 2023, 16:5551-5562
Published Date: 23 August 2023
Cuproptosis-Related Biomarkers and Characterization of Immune Infiltration in Sepsis
Wang Y, Qiu X, Liu J, Liu X, Pan J, Cai J, Liu X, Qu S
Journal of Inflammation Research 2024, 17:2459-2478
Published Date: 22 April 2024
Research Progress on Mechanisms and Treatment of Sepsis-Induced Myocardial Dysfunction
Hao Y, Liu R, Wang H, Rui T, Guo J
International Journal of General Medicine 2024, 17:3387-3393
Published Date: 5 August 2024
Mechanisms and Management of Albumin-Paclitaxel-Induced Peripheral Neuropathy in Breast Cancer
Xu X, Han Q, Qiu S, Gao S, Ren C, Li X
Breast Cancer: Targets and Therapy 2025, 17:693-709
Published Date: 13 August 2025
Copper-Induced Cell Death in Renal Diseases: Molecular Mechanisms and Therapeutic Implications
Lv D, Yu Y
Drug Design, Development and Therapy 2025, 19:11849-11861
Published Date: 31 December 2025