Back to Journals » Journal of Hepatocellular Carcinoma » Volume 11
Construction of a Prognostic Model for Hepatocellular Carcinoma Based on Macrophage Polarization-Related Genes
Authors Chen H, Li J ![]() , Cao D
, Cao D ![]() , Tang H
, Tang H
Received 22 December 2023
Accepted for publication 7 May 2024
Published 11 May 2024 Volume 2024:11 Pages 857—878
DOI https://doi.org/10.2147/JHC.S453080
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Manal Hassan
Han Chen,1,2 Jianhao Li,1,2 Dan Cao,1,2 Hong Tang1,2
1Center of Infectious Diseases, West China Hospital of Sichuan University, Chengdu, 610041, People’s Republic of China; 2Division of Infectious Diseases, State Key Laboratory of Biotherapy and Center of Infectious Diseases, West China Hospital of Sichuan University, Chengdu, 610041, People’s Republic of China
Correspondence: Hong Tang, Email [email protected]
Background: The progression of hepatocellular carcinoma (HCC) is related to macrophage polarization (MP). Our aim was to identify genes associated with MP in HCC patients and develop a prognostic model based on these genes.
Results: We successfully developed a prognostic model consisting of six MP-related genes (SCN4A, EBF3, ADGRB2, HOXD9, CLEC1B, and MSC) to calculate the risk score for each patient. Patients were then classified into high- and low-risk groups based on their median risk score. The performance of the MP-related prognostic model was evaluated using Kaplan-Meier and ROC curves, which yielded favorable results. Additionally, the nomogram demonstrated good clinical effectiveness and displayed consistent survival predictions with actual observations. Gene Set Enrichment Analysis (GSEA) revealed enrichment of pathways related to KRAS signaling downregulation, the G2M checkpoint, and E2F targets in the high-risk group. Conversely, pathways associated with fatty acid metabolism, xenobiotic metabolism, bile acid metabolism, and adipogenesis were enriched in the low-risk group. The risk score positively correlated with the number of invasion-related genes. Immune checkpoint expression differed significantly between the two groups. Patients in the high-risk group exhibited increased sensitivity to mitomycin C, cisplatin, gemcitabine, rapamycin, and paclitaxel, while those in the low-risk group showed heightened sensitivity to doxorubicin. These findings suggest that the high-risk group may have more invasive HCC with greater susceptibility to specific drugs. IHC staining revealed higher expression levels of SCN4A in HCC tissues. Furthermore, experiments conducted on HepG2 cells demonstrated that supernatants from cells with reduced SCN4A expression promoted M2 macrophage polarization marker, CD163 in THP-1 cells. Reduced SCN4A expression induced HCC-related genes, while increased SCN4A expression reduced their expression in HepG2 cells.
Conclusion: The MP-related prognostic model comprising six MPRGs can effectively predict HCC prognosis, infer invasiveness, and guide drug therapy. SCN4A is identified as a suppressor gene in HCC.
Keywords: hepatocellular carcinoma, macrophage polarization-related genes, tumor microenvironment, prognosis, SCN4A
Introduction
Hepatocellular carcinoma (HCC) is a highly aggressive malignancy with a significant global impact, resulting in a substantial number of deaths worldwide.1,2 The limited availability of effective pharmacotherapeutic interventions contributes to the high mortality rate in patients.3 HCC is often diagnosed at an advanced stage because of the absence of noticeable clinical symptoms in the early stages.4 Consequently, the overall survival (OS) rates for HCC treatment remain suboptimal, highlighting the need for improved therapeutic strategies.5,6 To address these challenges, it is crucial to gain a comprehensive understanding of the molecular mechanisms involved in HCC development and to identify novel therapeutic targets. Moreover, the identification of novel prognostic markers and the development of predictive models for HCC prognosis have significant implications for guiding therapeutic interventions in patients with HCC. By identifying reliable prognostic markers and implementing accurate predictive models, clinicians can make informed decisions regarding personalized treatment approaches, ultimately improving patient outcomes.
HCC is a malignancy closely associated with inflammation. The stromal component of HCC primarily consists of fibroblasts, endothelial cells, tumor-infiltrating inflammatory cells, and soluble molecules.7–10 The tumor microenvironment (TME), which encompasses these cellular components and molecular factors, plays a vital role in regulating neoplastic cell behavior, tumorigenesis, and therapeutic responses.11,12 Tumor-associated macrophages (TAMs) are the major contributors.13 TAMs can be classified into two subtypes: M1 (classical activated macrophages) and M2 (alternatively activated macrophages), each with distinct receptors, cytokines, and chemokines.14,15 M1-type TAMs express inflammatory factors such as tumor necrosis factor α (TNF-α), IL-1β, IL-6, IL-12, and IL-23, which promote a Th1 (cytotoxic) type response to inhibit tumor growth.16 Conversely, M2-type TAMs express cytokines such as IL-4, IL-13, IL-8, and IL-10 and growth factors such as epidermal growth factor (EGF), platelet-derived growth factor (PDGF), vascular endothelial growth factor A (VEGFA), and basic fibroblast growth factor (bFGF).17 The polarization of M1/M2 macrophages and the secretion of these cytokines play critical roles in regulating tumor cell proliferation, tumor cell metabolism, and tumor-related angiogenesis.18,19 Therefore, it is essential to identify genes and biomarkers related to tumor-associated macrophage polarization for the effective treatment and prognosis of HCC. Such markers will aid in monitoring HCC immunotherapy responses and furthering our understanding of immune infiltration mechanisms.20 Therefore, in this study, we employed bioinformatic methodologies to identify differentially expressed genes in HCC, establish a prognostic model related to macrophage polarization to predict the prognosis of patients with HCC, and validate the robust performance of our model. The findings from this study provide novel insights into the influence of macrophages on HCC and serve as a theoretical foundation for enhancing prognosis prediction and treatment strategies for patients with HCC.
Materials and Methods
Data Acquiring
HCC-related datasets were obtained from the The Cancer Genome Atlas (TCGA) and International Cancer Genome Consortium (ICGC) databases. The TCGA-HCC dataset consists of 50 normal tissue samples and 374 cancer tissue samples, with survival information available for 368 of these samples. The ICGC-LIHC dataset served as the validation set and included 232 cancer tissue samples with survival time records available for 215 samples. To identify the relevant genes associated with macrophage polarization (MP), we mined 35 MP-related genes (MPRGs) from published papers. These datasets and gene sets formed the basis of our study to investigate the relationship between MPRGs and HCC prognosis.21
Weighted Gene Co-Expression Network Analysis (WGCNA)
MPRGs score was was calculated using the “GSVA” package through ssGSEA analysis.22 To identify gene modules associated with the MPRG score in the TCGA-HCC dataset, we used the “WGCNA” package.23 Initially, clustering analysis was performed on the samples and outliers were excluded from further analysis. Subsequently, an appropriate soft threshold is determined for the data. A scale-free network was then constructed and genes were divided into several modules using a minimum module size of 30, as determined by the dynamic tree-cutting algorithm. Similar modules were subsequently merged, with the merge CutHeight set to 0.45. The correlation between the MPRG score and each module was assessed. Finally, modules showing the strongest association with the MPRG score (based on a significance threshold of p < 0.05 and |correlation| ≥ 0.3) were identified, and key genes within these modules were selected for subsequent analysis.
Recognition and Enrichment Analysis of Sifferentially Expressed MPRGs (DEMPRGs)
To identify differentially expressed genes (DEGs), we used the “DESeq2” package24 with a significance threshold of adjusted p < 0.05 and |Log2FC| > 2 on the TCGA-HCC dataset.24 DEMPRGs were identified by overlapping them with the key module genes related to the MPRGs score. Volcano Plot was plotted by package “ggplot2” (H. Wickham. ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag New York, 2016.). A heat map was drawn using the ‘pheatmap package (Kolde R (2019). Pheatmap: Pretty Heatmaps. R package version 1.0.12). GO and KEGG enrichment analyses of DEMPRGs were carried out using the “clusterProfiler” package (adjusted p < 0.05).25
Construction of Risk Model
A total of 368 HCC patients with survival information in the TCGA-HCC dataset (training set) and 215 HCC patients with survival records from the ICGC-LIHC dataset (validation set) were used to establish and validate the MP-related gene signature. The univariate Cox (survival package) (Terry M. Therneau, Patricia M. Grambsch (2000). Modeling Survival Data: Extending the Cox Model Springer, New York. ISBN 0–387-98784-3.) and LASSO regression (glmnet package)26 analyses were initially applied to screen for MP-related prognostic genes in the TCGA-HCC dataset.
Next, we conducted multivariate Cox analysis to identify prognostic genes. We constructed an MP-related prognostic model using multivariate Cox regression analysis in combination with stepwise regression. To categorize patients with HCC into high- and low-risk groups, we utilized the median risk score as a threshold. To evaluate the performance of our prognostic model, we employed several measures. First, Kaplan-Meier (K-M) survival analysis was performed to assess the differences in overall survival between the high- and low-risk groups. Additionally, receiver operating characteristic (ROC) curve analysis was conducted to determine the predictive accuracy of our model. Finally, risk curves were generated to visualize the relationship between the risk score and patient outcomes. Collectively, these analyses provided insights into the effectiveness of our MP-related prognostic model in predicting patient prognosis.
Correlation and Independent Prognostic Analysis Between Clinical Features and Risk Score
First, the differences in the risk score in clinical information (M, N, T, tumor stage, grade, sex, age, and extent of adjacent hepatic tissue inflammation) were analyzed using the Wilcoxon test. Next, the independent prognostic ability of clinical features and risk scores was explored using univariate and multivariate Cox models. Finally, we developed a nomogram model based on independent prognostic factors.
GSEA and TME Analysis
First, we downloaded ‘h.all.v7.5.1.symbols.gmt gene sets in MSigDB and GSEA were performed. The enriched pathways in the two risk subgroups are presented. The differences in the ESTIMATE, stromal, and immune scores between the two risk subgroups were analyzed using the Wilcoxon test. In addition, the correlation between biomarkers and tumor purity, ESTIMATE, stromal, and immune scores was analyzed.
Immunotherapy and Chemotherapy Analyses
First, the expression of immune checkpoints was analyzed using the Wilcoxon test in high-risk groups. Next, TIDE and immunophenoscore (IPS) scores were used to evaluate sensitivity to immunotherapy. The correlation between the biomarkers and the TIDE and IPS scores was further evaluated.
We downloaded invasion-related genes from the cancer SEA database and epithelial-mesenchymal transition (EMT)-related genes from the dbEMT2 database. We then analyzed the correlation between the risk score and invasion-related and EMT-related genes using the Spearman method. Next, the ssGSEA algorithm was used to calculate invasion and EMT scores based on invasion-related and EMT-related genes. Correlations between biomarkers, invasion, and EMT scores were analyzed using the Spearman method. Finally, the “pRRophetic” package (Geeleher (2014) was used. _pRRophetic: Predicts the clinical chemotherapeutic response from pre-treatment tumor gene expression levels. _. R package version 0.5.) software was used to calculate drug sensitivity of each sample. IC50 values of the two risk groups are presented.
Clinical Samples
A total of 64 para-tumor HCC tissues were obtained after informed consent was obtained from patients who underwent liver resection at the Western China Hospital of Sichuan University from 2018 to 2020. All subjects provided informed consent for inclusion before participating in the study, and the tissue was used for future studies.
IHC Staining
Eight tissue specimens from patients with HCC were subjected for immunohistochemistry (IHC) staining. The primary antibodies used for IHC staining were anti-SCN4A polyclonal antibody and DF4513 (Affbiotech). IHC staining was performed according to the manufacturer’s instructions using the Envision+ kit (Dako, Carpinteria, CA, USA). Immunohistochemical images were captured using an Olympus BX63 microscope (Olympus, Japan), allowing the visualization and analysis of the staining patterns and distribution of the target protein in the HCC tissue samples.
Cell Lines and Reagents
The HCC cell line HepG2 and the monocyte cell line THP-1 were obtained from Procell Life Science and Technology (Wuhan, China). HepG2 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; HyClone, Cytiva, USA) supplemented with 10% fetal bovine serum (FBS; Gibco) at 37 °C. THP-1 monocytes were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS; Gibco) and 0.05nM β-mercaptoethanol. THP-1 monocytes were incubated with 100 ng/mL phorbol 12-myristate 13-acetate (PMA, Sigma, P8139) for 24 h in RPMI 1640 medium to differentiate them into macrophages. Macrophage M2 polarization was achieved by incubating PMA-differentiated macrophages with 20 ng/mL of interleukin 4 (GMP-CD03, Novoprotein, Suzhou, China) and 20 ng/mL of interleukin 13 (CC89, Novoprotein, Suzhou, China) for 24 h in the presence of PMA. This protocol allowed us to generate M2-polarized macrophages from THP-1 monocytes for further experiments.
RNA Interference Transfection
SCN4A-specific and negative control siRNAs were designed and obtained from RiboBio (Guangzhou, China). Prior to siRNA transfection, the cells were cultured in medium without fetal bovine serum (FBS) for 24 h. The siRNA constructs were transfected into the cells using Lipofectamine RNAiMAX (Invitrogen), following the manufacturer’s protocol. At 4–6 hours post-transfection, the cells were washed with phosphate-buffered saline (PBS) to ensure the complete removal of the siRNA constructs from the medium. Subsequently, the cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS, which provides the growth factors and nutrients necessary for cell viability and proliferation.
Real-Time Quantitative PCR
Total RNA was extracted from the samples using a High Pure RNA Isolation Kit (RNeasy Mini Kit, 74104, QIAGEN) following the manufacturer’s instructions. A PrimeScript™ RT Reagent Kit (Perfect Real Time, Takara, Japan) was used to obtain complementary DNA (cDNA). Specific primers were used for the amplification of target genes. The expression levels of the selected genes were measured in triplicate using a LightCycler 96 system (Roche). To ensure accurate normalization, the expression of each sample was normalized to that of the reference gene ACTB.
Western Blotting
Cell lysates were prepared using RIPA buffer for protein extraction. For Western blotting, the following primary antibodies were used: anti-SCN4A polyclonal antibody DF4513 (AffBiotech) and anti-β-actin monoclonal antibody TA-09 (ZSBIO, Beijing, China). To detect immune complexes, enhanced chemiluminescence detection reagents (4A BIOTECH, Beijing, China) were used according to the manufacturer’s instructions. This allowed for the visualization of the protein bands on the Western blot, aiding in the analysis and interpretation of the results.
Results
Identification of MPRGs (Macrophage Polarization Related Genes) Score-Related Module Genes (Scale-Free R2 = 0.9)
A flowchart of the study is presented in Figure 1. The ssGSEA algorithm was used to calculate the MPRGs score for each sample. Subsequently, WGCNA was used to identify module genes associated with the MPRG score. Initially, we removed five outlier samples, resulting in 419 samples that were used for the subsequent analysis (Figure 2A). Next, we created a scale-free network using a soft threshold of β = 8, which achieved a scale-free R2 value of 0.9, indicating an appropriate fit for the data (Figure 2B). We then performed a cluster dendrogram analysis using the dynamic tree cut algorithm, resulting in the division of the network into 40 modules. After merging similar modules, we obtained a final set of 16 modules (Figure 2C). We further investigated the correlation between the modules and MPRGs score. Analysis revealed that the cyan module exhibited a strong correlation with the MPRGs score (Figure 2D). Finally, we selected 3510 genes from the cyan module for subsequent analysis considering their potential relevance to the MPRGs score.
 |
Figure 1 The brief flowchart of this study. |
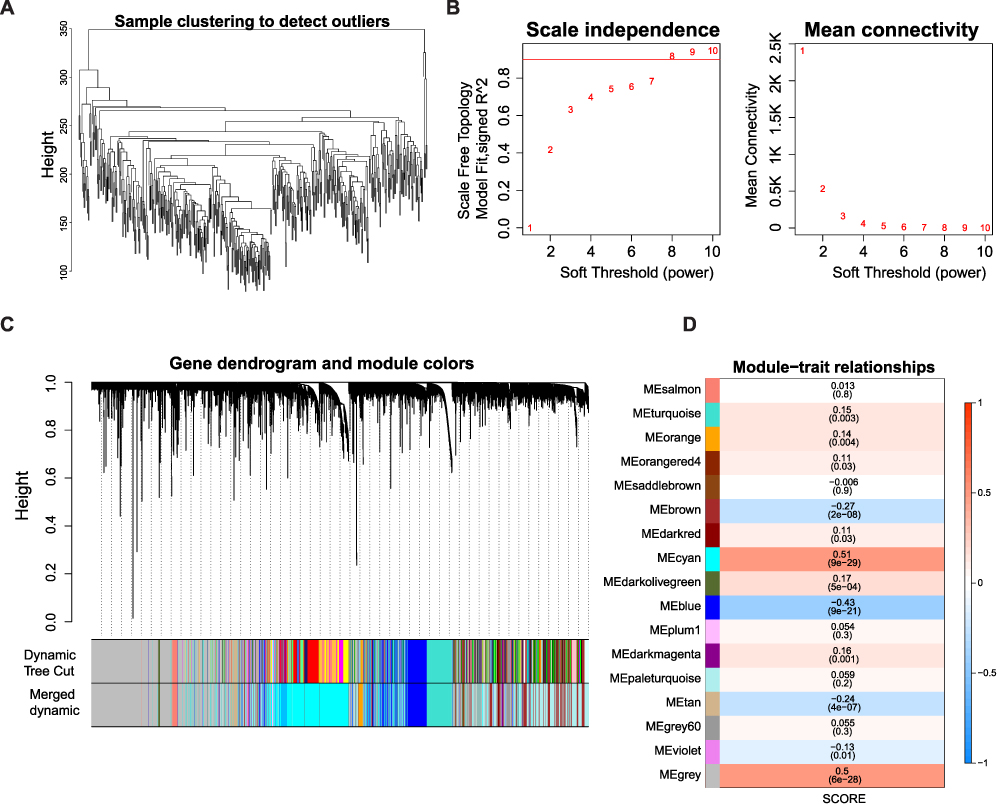 |
Figure 2 Identification of MPRGs score-related module genes (A) Sample clustering to detect the outliers in the TCGA-HCC dataset. The red line represents the height of the screening outliers. (B) Analyses of the appropriate soft threshold power and minimum mean connectivity to construct topological overlap matrix T(OM). (C) Clustering dendrograms of the co-expression network modules. (D) The correlation analysis between the TME score and module, correlation coefficient, and p-values are shown. |
Identification of DEMPRGs and Enrichment Analysis
We identified 2942 differentially expressed genes (DEGs) in the HCC groups of the TCGA-HCC dataset, consisting of 361 downregulated genes and 2581 up-regulated genes (Figure 3A). To further investigate the genes that were differentially expressed and relevant to the MPRG scores, we performed an intersection analysis between the 2942 DEGs and 3510 module genes. This analysis identified 283 differentially expressed MPRGs (DEM-PRGs). These 283 DEM-PRGs represent the overlap between the DEGs and the module genes, signifying their potential significance in relation to both differential expression and MPRG scores. Moreover, we conducted GO and KEGG enrichment analyses to gain insights into the biological functions and signaling pathways associated with the differentially expressed MPRGs (DEMPRGs). These analyses revealed various significant biological processes including epithelial cell proliferation, endothelial cell proliferation, regulation of opsonization, axonogenesis, positive chemotaxis, cell recognition, cell chemotaxis, and sprouting angiogenesis. Additionally, several signaling pathways were found to be related to DEMPRGs, such as cytokine-cytokine receptor interactions, systemic lupus erythematosus, malaria, nicotine addiction, and amphetamine addiction (Supplementary Table 1, Figure 3B and C).
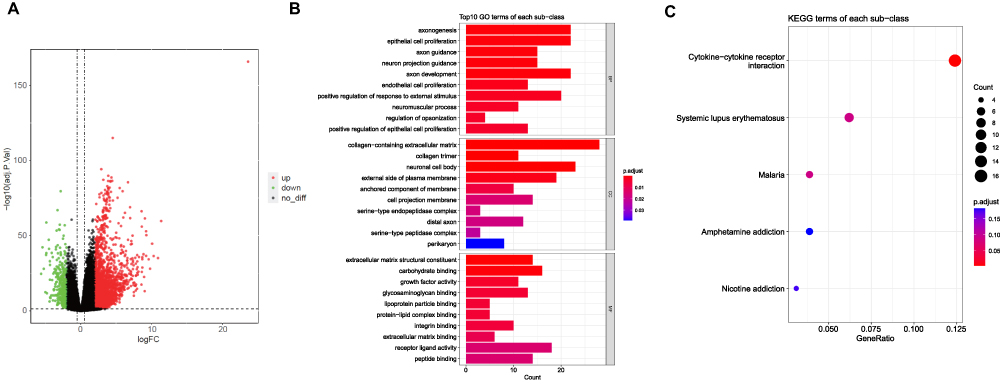 |
Figure 3 Identification of DEMPRGs and enrichment analysis (A) Volcano plot of DEGs between two clusters with different MP-related gene expression. The red and green points represent up- and down-regulated genes with statistical significance, respectively. (B) The bar plot of enriched Gene Ontology (GO) terms. BP indicates biological process, CC indicates cellular component, and MF indicates molecular function. The length of each column represents the count of genes; the shade of color represents the p value. (C) KEGG analysis of DEGs that were upregulated of DEGs. |
Creation and Validation of a MP-Related Risk Model
To screen for genes associated with HCC prognosis in the TCGA-HCC dataset (training set), 49 genes were mined using a univariate Cox model (p<0.05) (Table 1 and Figure 4A). 12 genes (SCN4A, CD5L, EBF3, ADGRB2, HOXD9, HOXD10, KDM8, CTHRC1, CLEC1B, MSC, KCNQ3, and MAPT) were further acquired by LASSO regression analysis (Figure 4B and C). Six prognostic genes (SCN4A, EBF3, ADGRB2, HOXD9, CLEC1B, and MSC) were identified using multivariate COX and stepwise regression analyses to create a risk model (Table 2 and Figure 4D). Meanwhile, a risk score model was developed to calculate a patient’s risk score (Riskscore = h0 (t) * exp ((−0.162 SCN4A expression) + (−0.109 * EBF3 expression) + (0.108 * ADGRB2 expression) + (0.117 * HOXD9 expression) + (−0.129 * CLEC1B expression) + (0.078 * MSC expression))), and according to median of risk score, we classified patients into high and low risk groups. The patient survival time gradually decreased as the risk score increased. The results of the heat map revealed that ADGRB2, HOXD9 and MSC were highly expressed in high-risk patients, while the high expression of CLEC1B, SCN4A and EBF3 was associated with a low risk of HCC (Figure 4E and F). Kaplan-Meier survival analysis revealed that high-risk HCC patients had significantly poorer survival than those with low risk (Figure 4G). Finally, the results of the ROC curves illustrated that the AUC values for OS were 0.737 (1 year), 0.708 (3 years), and 0.742 (5 years) in the TCGA-HCC dataset, indicating the decent performance of the prognostic model (Figure 4H).
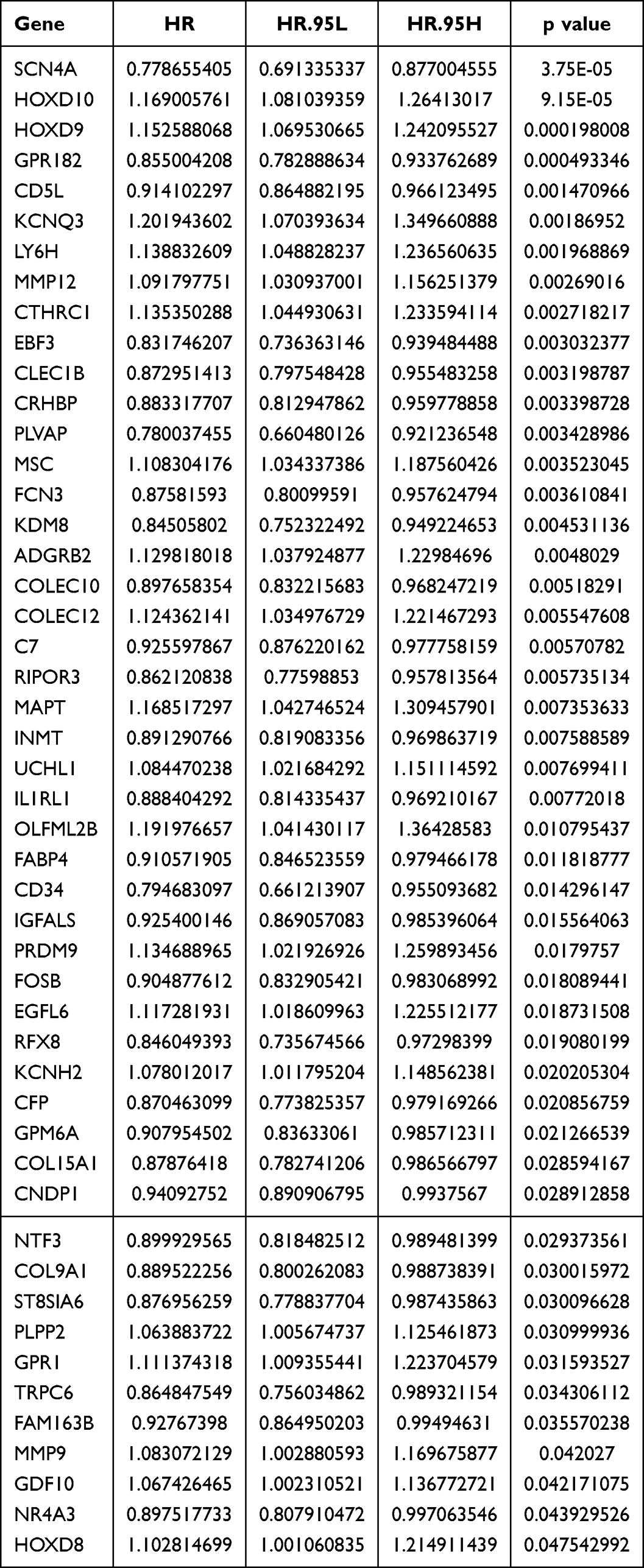 |
Table 1 Result of Univariate Cox Model |
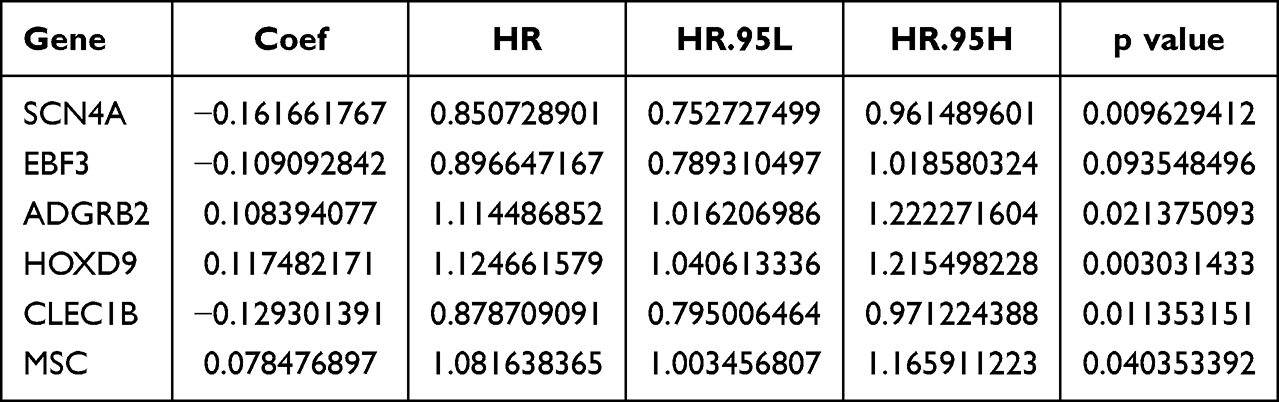 |
Table 2 Prognostic Genes Identified by Multivariate COX |
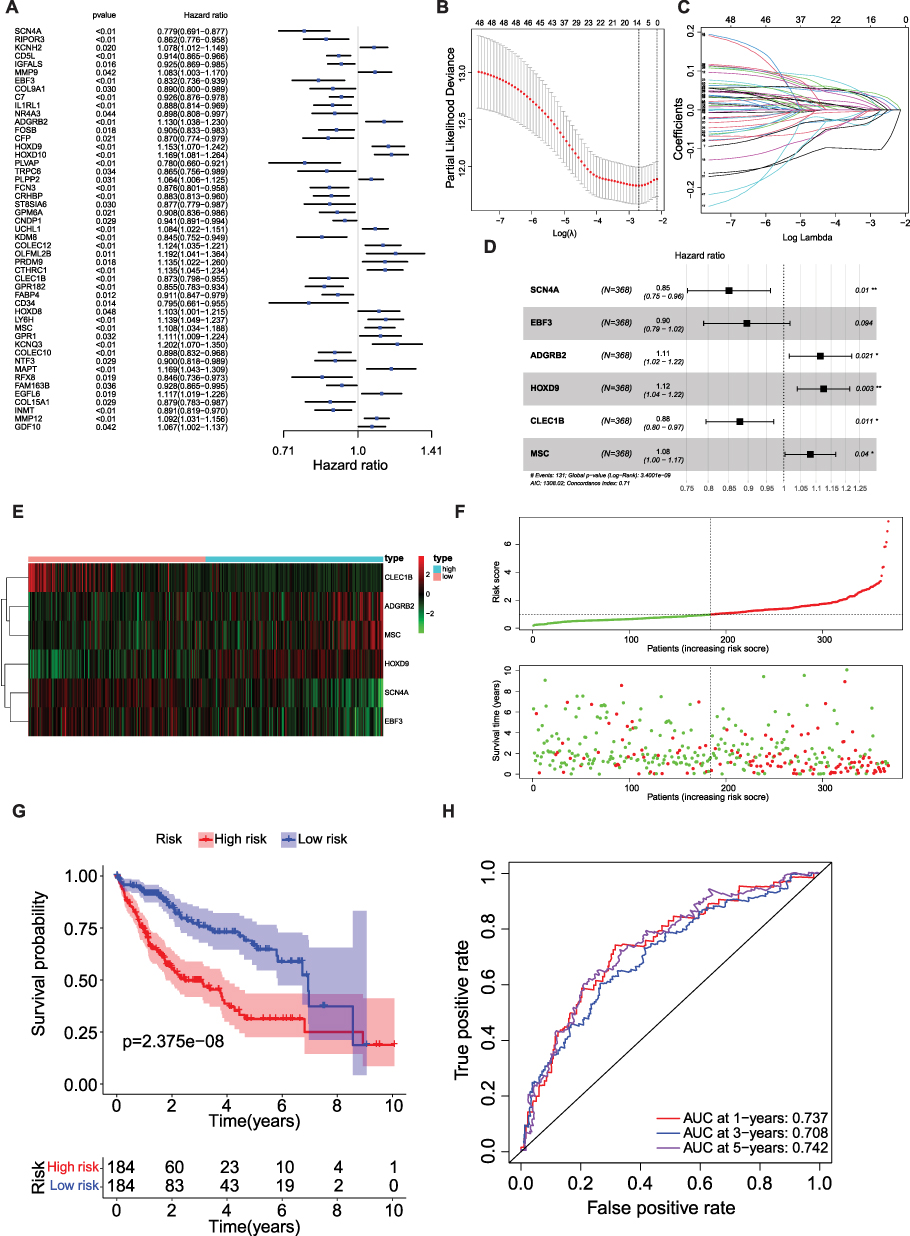 |
Figure 4 Creation and validation of a MP-related risk model (A) Forest plot of the univariate Cox regression analysis showing the prognostic metabolic genes. Genes with HR < 1 have better overall survival outcomes, while genes with HR > 1 have worse overall survival outcomes. (B) Lasso regression. Finally, 12 MP-related genes were obtained. (C) Lasso filters variables. (D) Forest plot of HR of six genes. (E and F) Expression heat map, risk score distribution and survival status. (G) Survival curve of low and high risk subgroup. (H) Time-dependent ROC curves for the efficacy evaluation of the 6-gene MP-related signature. * P < 0.05, ** P ≤ 0.01. |
To further validate the predictive applicability of the risk model, the aforementioned analyses were performed using the ICGC-LIHC dataset (the validation set). Consistent trends were observed in the ICGC-LIHC dataset (Supplementary Figure 1).
Clinical Significance and Independent Prognostic Analysis of the Risk Model
To explore the link between clinical characteristics and risk scores in patients with HCC, the differences in risk scores among different clinical characteristics were evaluated using the Wilcoxon test. The results showed that the risk score was correlated with the HCC grade, HCC stage, and T stage (p<0.05) (Figure 5A).
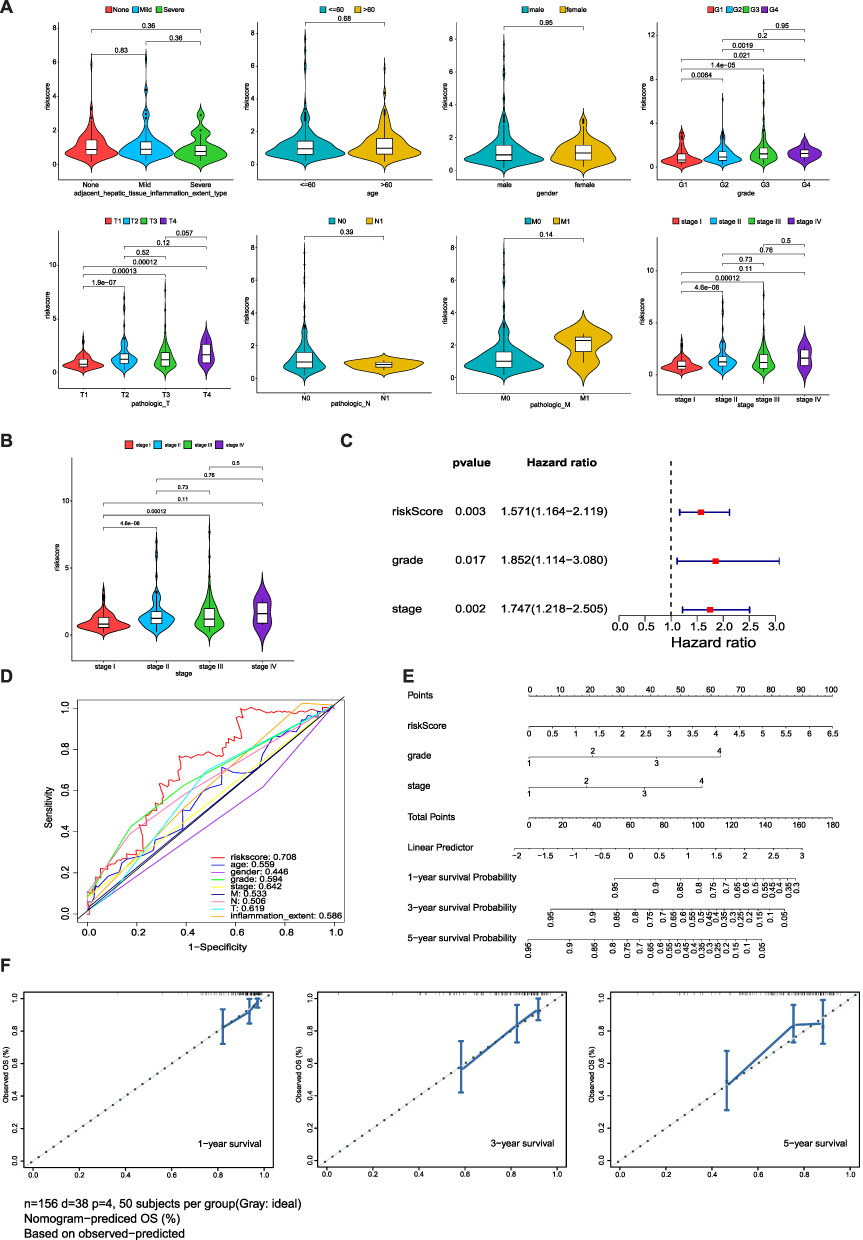 |
Figure 5 Clinical significance and independent prognostic analysis of the risk model (A) The correlation between risk score and patient parameters. (B) Risk score of different stages in HCC. (C) Forest maps of the univariate and multivariate Cox regression analysis, including risk score, grade, and stage. (D) ROC curves presented AUC value of risk score. (E) Nomogram based on risk score and clinical factors, including a prediction of 1-, 3-, and 5-year OS. (F) Calibration curves were predicted and observed 1-, 3-, and 5-year survival nomogram consistency. |
To determine the prognostic ability of clinicopathological features and risk score, univariate and multivariate Cox analyses were performed. We found that risk score, age, and grade could be used as independent prognostic factors (p<0.05) (Tables 3 and 4, Figure 5B and C). The ROC curves presented an AUC value (0.708) for the risk score (Figure 5D). We then established a nomogram model with risk score, stage, and grade to predict the survival rate of patients at different times (Figure 5E). The nomogram model had a significant predictive effect for patients with HCC (Figure 5F).
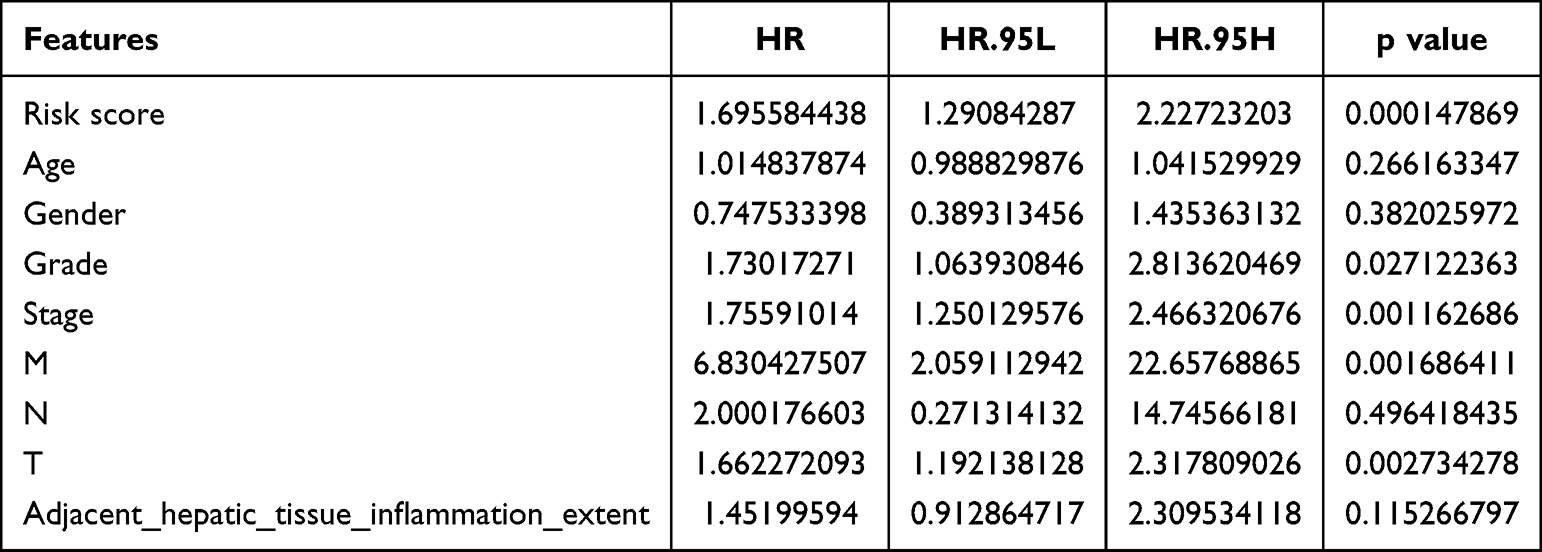 |
Table 3 Result of Independent Prognostic Univariate COX Analysis |
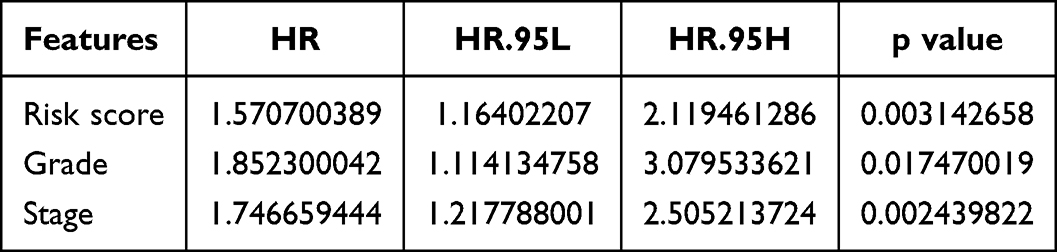 |
Table 4 Result of Independent Prognostic Multivariate COX Analysis |
GSEA and TME Analysis in Two Risk Subgroups
GSEA was performed to analyze the differential GO enrichment pathways in the two risk groups. The results revealed that pathways of the G2M checkpoint, KRAS signaling down regulation (DN), and E2F targets were enriched in the high-risk groups, whereas fatty acid metabolism, xenobiotic metabolism, bile acid metabolism, and adipogenesis were related to the low-risk group (Figure 6A).
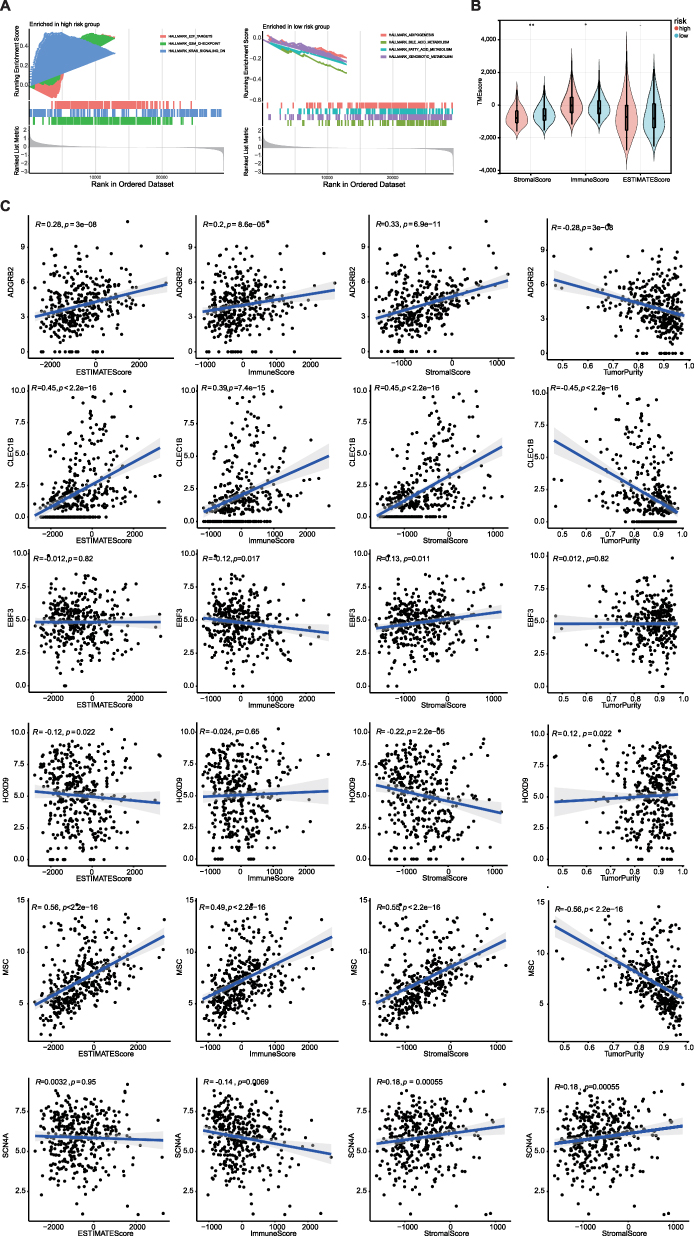 |
Figure 6 GSEA analysis showing the enriched pathways of the high-and low-risk groups. (A) Left chart: Multiple GSEA showing enriched pathways in the high-risk group. Right chart: Multiple GSEA showing enriched pathways in the low-risk group. (B) Violin plots of stromal scores, immune scores and ESTIMATE scores between high and low risk groups. (C) The correlation between MPRGs and stromal scores, immune scores, ESTIMATE scores.* P < 0.05, ** P < 0.01. |
Next, we analyzed the differences in TME between the two risk subgroups. The results revealed that the immune and stromal scores were significantly difference between the two risk subgroups (Figure 6B). The correlation between each biomarker and tumor purity, ESTIMATE, and stromal and immune scores is presented in (Figure 6C). MSC and CLEC1B were positively correlated with ESTIMATE and stromal and immune scores, whereas MSC and CLEC1B were negatively correlated with tumor purity.
Correlation Analysis of Metastasis/Invasion Indicators in HCC
To better assess the effect of MPRGs on HCC development, a correlation between the expression of invasion-related and EMT-related genes was performed. The results demonstrated a positive correlation between the risk score and the majority of invasive-related genes (Figure 7A), while also revealing diverse associations between the risk score and EMT-related genes (Figure 7B). The correlations of biomarkers with invasion and EMT scores are presented in Figure 7C, in which ADGRB2, MSC and HOXD9 were positively correlated with EMT and invasion scores, indicating that the biomarkers were related to tumor invasion.
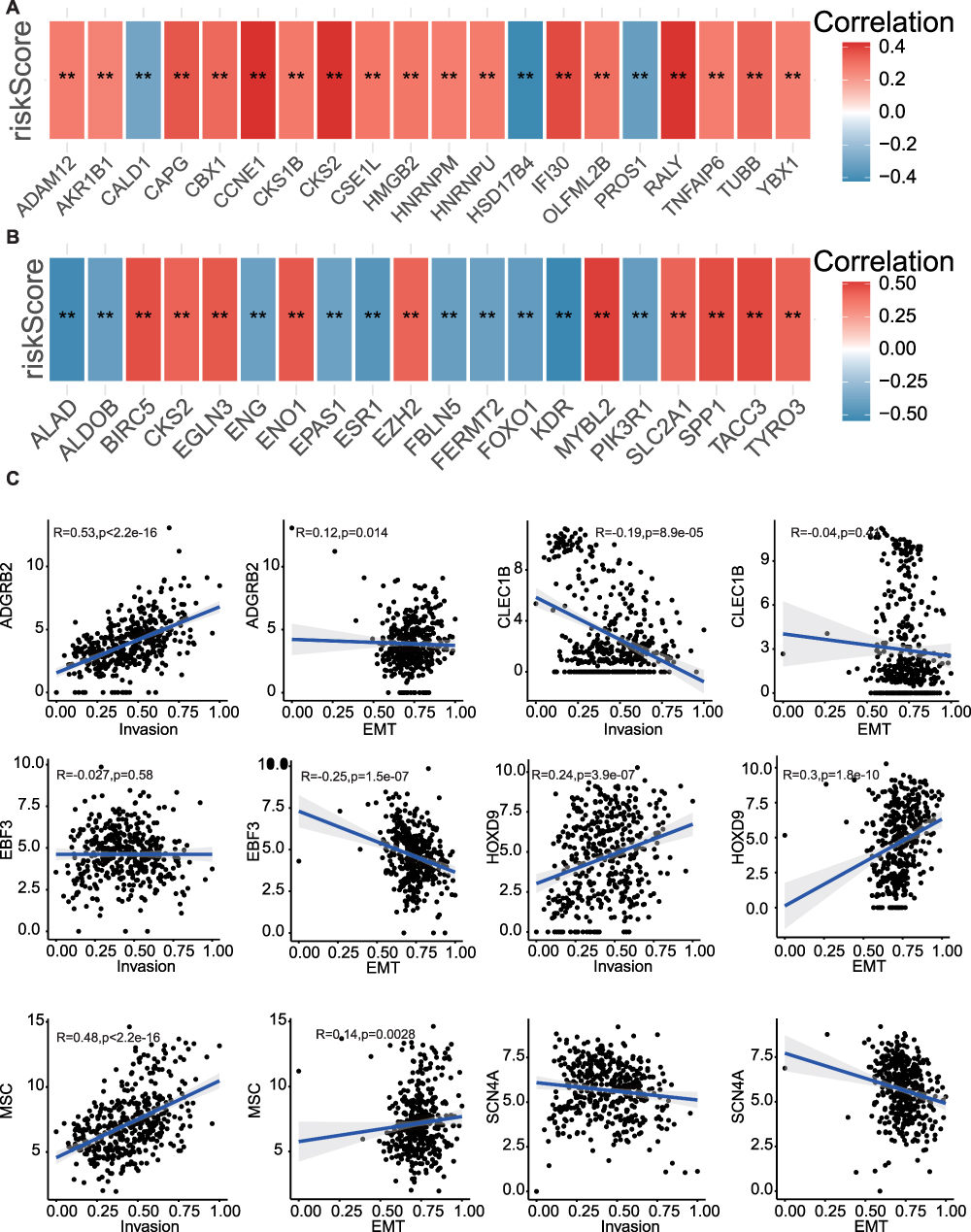 |
Figure 7 Correlation analysis of metastasis/invasion indicators in HCC. (A) The correlation between risk scores and invasion-related related genes. (B) The correlation between risk scores and EMT related genes. (C) The correlation between MPRGs and invasion score, EMT score. ** P < 0.01. |
Immunotherapy and Chemotherapy Response in Two Risk Subgroups
Previous studies have shown that macrophages play a dominant role in immunotherapy and are related to the response to immune checkpoint blockade.27,28 Based on this, we analyzed the expression of many immune checkpoints in the low- and high-risk groups. The results indicated that most of the immune checkpoints were significantly different between the low- and high-risk groups (Figure 8A). Drug sensitivity tests suggested that high-risk patients had lower IC50 values for mitomycin C, cisplatin, gemcitabine, rapamycin, and paclitaxel, indicating higher chemotherapeutic sensitivity to these drugs. Patients in the low-risk group had a lower IC50 value for Doxorubicin, suggesting that they had a higher response to chemotherapy of Doxorubicin (Figure 8B).
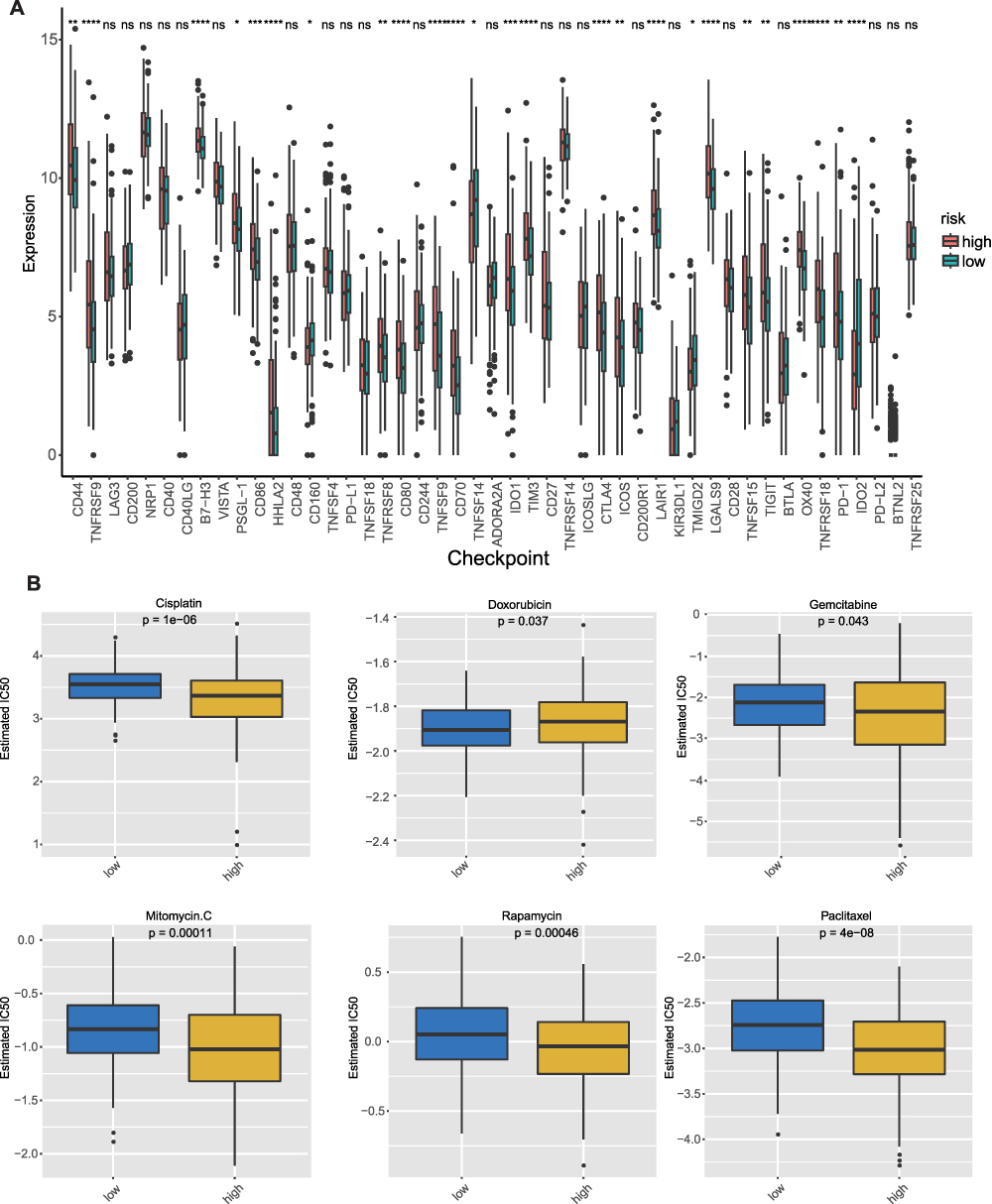 |
Figure 8 Immune checkpoint expressions and chemotherapy response in two risk subgroups (A) Immune checkpoint expressions in high risk group and low risk group. (B) IC50 value of chemotherapeutic drugs in high risk group and low risk group. ns=not significant, * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001. |
SCN4A is Highly Expressed in HCC and Inhibition of SCN4A Promotes Macrophage M2 Polarization in HCC Tumor Micro-Environment
As the MP-related risk model was composed of six genes (SCN4A, EBF3, ADGRB2, HOXD9, CLEC1B, and MSC), their expression levels were extracted from the TCGA-HCC dataset. Combined with the sample grouping information of the data set, the expression of biomarkers in the data set was plotted by the wilcox.test method using the R package “ggplot2” (Figure 9A). In the subsequent analysis, we investigating the expression of SCN4A in 64 HCC tissues using qPCR. These results indicate that SCN4A exhibited higher expression levels in HCC tissues than in para-carcinoma tissues. Of the 64 HCC tissues analyzed, 59 showed elevated expression of SCN4A, while 6 displayed decreased expression (Figure 9B). To further validate these findings, IHC staining for SCN4A was performed on 8 HCC specimens. Among these samples, six patients exhibited higher expression levels of SCN4A in HCC tissues than in para-carcinoma tissues (Figure 9C), whereas two patients demonstrated lower expression levels (data not shown). These findings provided additional support for the upregulation of SCN4A in HCC tissues, as observed by qPCR analysis.
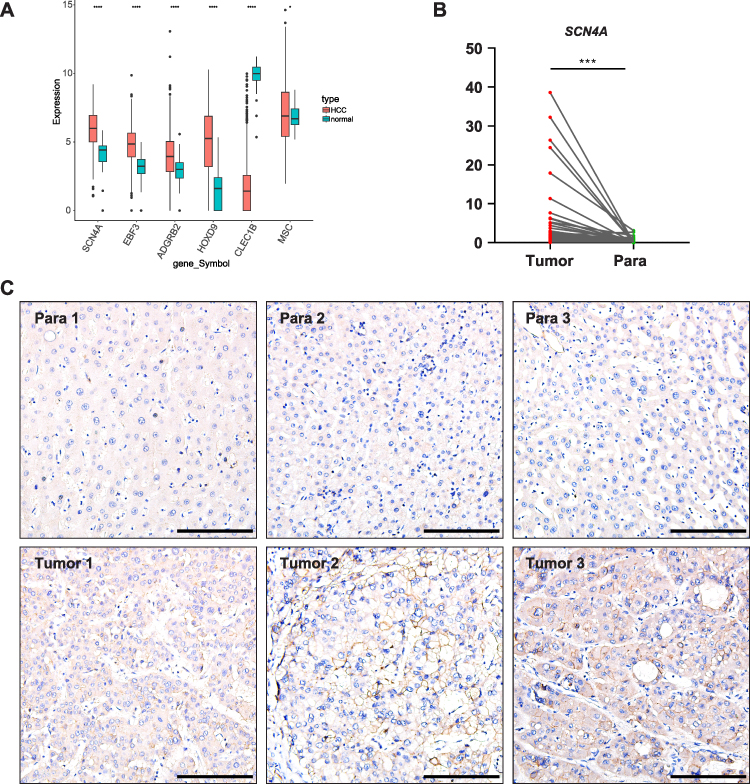 |
Figure 9 SCN4A is highly expressed in HCC tissues (A) MPRGs expressions in HCC of TCGA-HCC dataset. (B) qPCR result of SCN4A expressions in HCC of patients. (C) Representative IHC staining of SCN4A expressions in HCC of patients. Bars=100 μm. * P < 0.05, *** P < 0.001, **** P < 0.0001. |
Given the strong association between M2 polarization and tumor development, we investigated whether SCN4A influenced M2 polarization in the HCC tumor microenvironment. To achieve this, we differentiated THP-1 monocytes into M2-type macrophages as previously described (Figure 10A). Subsequently, we employed siRNA to downregulate the expression of SCN4A in HepG2 cells and utilized a plasmid to overexpress SCN4A in HepG2 cells. To establish an indirect co-culture model, supernatants from HepG2 cells transfected with either siSCN4A/siCtrl or SCN4A plasmid/vector for 48 h were transferred to M2-type THP-1 cells (Figure 10B). After stimulation with HepG2 supernatants for 24 h, M2-type THP-1 cells were harvested for qPCR analysis. qPCR results demonstrated that, in comparison to THP-1 cells cultured in the supernatant of HepG2 cells transfected with siCtrl, the M2-type macrophage biomarker CD163 exhibited upregulation upon exposure to the supernatant of HepG2 cells transfected with siSCN4A. Conversely, when compared to THP-1 cells cultured in the supernatant of HepG2 cells transfected with the vector, the M2-type macrophage biomarker CD163 showed downregulation upon exposure to the supernatant of HepG2 cells transfected with the SCN4A plasmid. These findings imply that SCN4A may play a role in inhibiting the M2 polarization of macrophages within the HCC tumor microenvironment (Figure 10C). Next, in order to explore whether SCN4A acts as an oncogene or a tumor suppressor gene, we further investigated the impact of SCN4A on liver cancer cell proliferation. Results from CCK-8 assays and colony formation experiments indicated that downregulation of SCN4A expression in HepG2 cells promotes their proliferation, while overexpression of SCN4A inhibits their proliferation (Figure 10D and E). These findings suggest that SCN4A may function as a tumor-suppressive gene in liver cancer. To further confirm that SCN4A is a tumor-suppressive gene in liver cancer, we explored the changes in the expression levels of several molecules associated with liver cancer progression in HepG2 cells following knockdown/overexpression of SCN4A. We found that upon knocking down SCN4A, the expression of molecules closely linked to hepatocellular carcinoma progression, such as β-catenin, c-myc, CyclinD1, NOTCH-1, HIF-1α, VEGFA, and N-cadherin, increased, while the expression of the liver cancer inhibitory gene E-cadherin decreased. Conversely, in HepG2 cells overexpressing SCN4A, the expression of β-catenin, c-myc, CyclinD1, NOTCH-1, HIF-1α, VEGFA, and N-cadherin decreased, while the expression of E-cadherin, the liver cancer inhibitory gene, increased (Figure 10F). These research findings indicate that SCN4A is a gene that suppresses the progression of liver cancer.
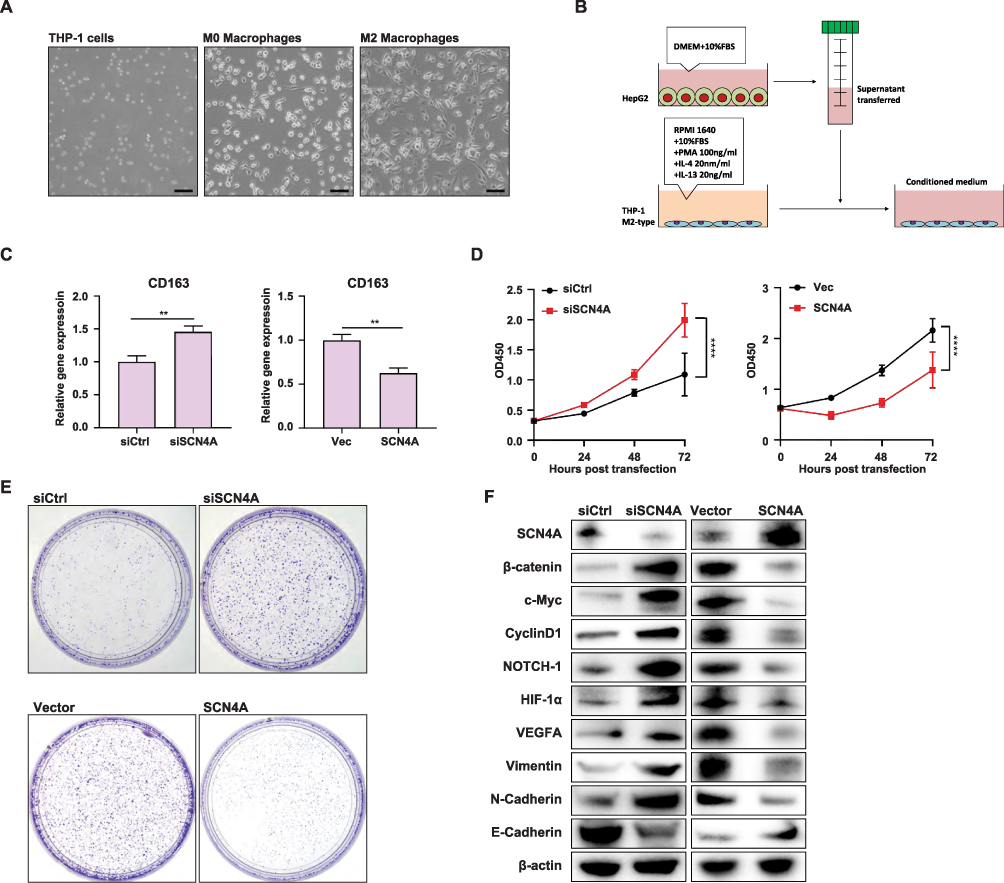 |
Figure 10 SCN4A expression in HCC cells could regulate M2 macrophage polarization in HCC tumor microenvironment and HCC progression (A) THP-1 cells were induced to differentiate into macrophages displaying M2 polarization by treatment with IL-4 (20 ng/mL) and IL-13 (20 ng/mL), in the presence of PMA (100 ng/mL). (B) Schematic of the indirect co-culture model. (C) qPCR result of CD163 expression in MP macrophages of the indirect co-culture model. (D) CCK-8 experiments showed the growth curve of HepG2 cells. (E) Colony formation assay showed the growth of HepG2 cells. (F)Western blot analysis of SCN4A, β-catenin, c-Myc, CyclinD1, NOTCH-1, HIF-1α, VEGFA, Vimentin, N-Cadherin and E-Cadherin in HepG2 cells. **** P < 0.0001. |
Discussion
HCC is a highly malignant tumor with poor prognosis worldwide. TAMs have been found to play a crucial role in all stages of HCC.29 Recent studies have revealed the significance of M1 and M2 macrophage polarization in the development of HCC. M1 TAMs primarily exhibit antitumor functions, whereas M2 TAMs contribute to cancer cell proliferation, aggressiveness, and metastasis through various mechanisms.14,30 Moreover, M2 TAMs are involved in promoting angiogenesis in liver cancers. This effect is achieved by upregulating the expression of tyrosine kinase receptor Tie-2, human macrophage metalloelastase (HME), and VEGF. These factors collectively enhance angiogenesis within the tumor and facilitate HCC.31–34
Based on these findings, we integrated MPRGs with clinical parameters to develop a prognostic model for HCC. GO and KEGG pathway analyses revealed that these MPRGs were primarily associated with axon development, epithelial cell function, and played a significant role in HCC prognosis through growth factor activity, extracellular matrix structural constituents, and binding to extracellular components. These results align with those of previous studies, suggesting that morphological changes, such as axon sprouting, occur during macrophage polarization. Consequently, macrophages may interact with epithelial cells, whereas numerous extracellular components can bind to macrophages. Using univariate Cox regression, LASSO regression, and multivariate Cox regression, we selected six MPRGs, ADGRB2, HOXD9, MSC, CLEC1B, SCN4A, and EBF3, to construct the risk model. Among these genes, ADGRB2, HOXD9, and MSC exhibited higher expression levels in patients at higher risk, whereas CLEC1B, EBF3, and SCN4A were highly expressed in patients at a lower risk of HCC. These findings provide valuable insights into the potential utility of MPRGs as a prognostic marker for HCC.
To gain a deeper understanding of MPRGs included in the model, we conducted a literature review to explore their known functions. The first gene, ADGRB2 (Adhesion G Protein-Coupled Receptor B2), also referred to as BAI2 (brain angiogenesis inhibitor 2), is a p53-target gene that specifically encodes a brain-specific angiogenesis inhibitor.35 To date, no study has specifically explored the role of ADGRB2 in cancer have been published. Therefore, the function of ADGRB2 in cancer remains unclear. Another MPRG, HOXD9 (Homeobox D9), belongs to a superfamily of genes that regulates the development and control of various cellular processes. In HCC, HOXD9 promotes metastasis by regulating ZEB1.36 Furthermore, HOXD9 has been frequently used in risk prediction models for HCC using bioinformatics methods, indicating its potential significance in prognostic evaluation.37–40 The MSC gene encodes musculin, which acts as a transcriptional repressor, impeding myogenesis and activating E-box-dependent muscle genes. Its involvement in the TME suggests that it may play a role in HCC prognosis by influencing the TME dynamics. Further investigations are needed to elucidate the precise mechanisms underlying HCC.41
These results are consistent with our findings that ADGRB2, HOXD9 and MSC are highly expressed in high-risk patients. CLEC1B (C-Type Lectin Domain Family 1 Member B), also known as C-type lectin-like receptor 2 (CLEC-2), is expressed on platelets, Kupffer cells and other immune cells, and binds to various ligands.42 Notably, the expression of CLEC1B, and Programmed death-ligand 1 (PD-L1) has been identified as a predictor of clinical outcomes in HCC cases associated with tumor hemorrhage.43 Furthermore, low expression of CLEC1B has been observed in HCC, highlighting its potential role as a biomarker for suppressing the microenvironment, stemness, and hypoxia.44,45 EBF3 (Early B cell Factor 3) is a member of the early B-cell factor (EBF) family, which comprises DNA binding transcription factors. EBF proteins play critical roles in B cell differentiation, bone development, and neurogenesis. Although there are reports suggesting that EBF3 may promote metastasis in certain tumors,46,47 it is generally considered to function as a tumor suppressor.48–51 SCN4A (Sodium Voltage-Gated Channel Alpha Subunit 4) is predominantly expressed in skeletal muscles, and its mutations have been associated with various myotonia and periodic paralysis disorders.52 Interestingly, a correlation between SCN4A and HCC prognosis was initially identified using bioinformatics.53,54 This highlights the potential significance of SCN4A in predicting the outcomes of previously unknown HCC cases. Our findings are consistent with these results, indicating that CLEC1B, EBF3, and SCN4A are highly expressed in patients at a lower risk.
Following the modeling of these six MPRGs, we proceeded with relevant verification steps. First, we established a nomogram model incorporating the risk score, stage, and grade, which demonstrated a favorable predictive effect for patients with HCC. Subsequently, we analyzed the correlation between tumor purity, ESTIMATE scores, stromal scores, immune scores, and the six MPRGs. Additionally, we examined the relationship between each biomarker and tumor purity, ESTIMATE scores, stromal scores, and immune scores. The results revealed that MSC and CLEC1B were positively correlated with ESTIMATE and stromal and immune scores, while demonstrating a negative correlation with tumor purity. Furthermore, we analyzed the association of each biomarker with invasion scores, which indicated a positive correlation between ADGRB2 and MSC with tumor invasion.
According to previous studies, it was the third time that SCN4A was identified to be strongly correlated with HCC prognosis by our research. However, the nature of this relationship remains unclear. To delve deeper into its role, we conducted qPCR and IHC staining of SCN4A and discovered that it exhibited higher expression levels than adjacent para-tumor tissues. To further investigate the effect of SCN4A, we induced THP-1 cells into an M2-like subtype and established an indirect co-culture model using the supernatants from HepG2 cells. Interestingly, When the SCN4A gene was knocked down in HepG2 cells, the supernatants obtained induced M2 polarization in THP-1 cells. Conversely, when the SCN4A gene was overexpressed in HepG2 cells, the resulting supernatants reduced M2 polarization in THP-1 cells. This observation aligns with our risk model, which implicates SCN4A in patients at lower risk of HCC. Based on these findings, we can infer that SCN4A deficiency promotes polarization of M2 macrophages in HCC. Thus, SCN4A may act as a negative regulator of M2 macrophages in HCC. Furthermore, we confirmed through cellular experiments that SCN4A is a gene that inhibits HCC.
We also analyzed the enrichment pathways of the high and low-risk groups. The E2F targets signaling, G2M checkpoint signaling and KRAS signaling were enriched in high-risk group and signalings related to adipogenesis, bile acid metabolism, fatty acid metabolism were enriched in low risk group. E2F targets are a group of genes that encode transcription factors in higher eukaryotes, participating in the cell cycle regulation and synthesis of DNA in mammalian cells. Activation of E2F targets promotes cell cycle progression. G2M checkpoint signaling is also a signaling pathway involved in the cell cycle. KRAS is one of the important members of the RAS family, and its main downstream signaling pathways include important signaling transduction pathways such as PI3K/AKT. It plays a crucial regulatory role in signaling pathways involved in tumor cell growth and angiogenesis processes. However, the enriched pathways in the low-risk group are primarily associated with metabolism, particularly lipid metabolism. The above research findings indicate that in the future development of anti-tumor drugs, efforts should be focused on targeting the E2F, G2M checkpoint, and KRAS signaling pathways. Additionally, exploring the development of drugs that regulate tumor lipid metabolism could also be beneficial.
In addition, we compared immunotherapy and chemotherapy responses between high- and low-risk subgroups. This analysis revealed that the patients in the high-risk subgroup exhibited greater sensitivity to mitomycin C, cisplatin, gemcitabine, rapamycin, and paclitaxel. Conversely, patients in the low-risk subgroup showed a better response to doxorubicin-based chemotherapy. These findings have crucial implications for guiding clinical decision making in terms of chemotherapy selection for patients with HCC.
This study has several limitations that should be acknowledged. First, our experimental focus was primarily on SCN4A, with only eight sections of IHC staining conducted to examine its effect on M2 polarization. Further investigations are needed to determine whether SCN4A overexpression can reduce CD163 expression in M2-like THP-1 cells, explore the impact of SCN4A on M1 polarization in HCC, and elucidate the precise mechanism by which SCN4A regulates M1/M2 polarization. Additionally, it is important to note that the expression of the other five MPRGs in the HCC samples was not thoroughly examined in this study. Future studies should aim to clarify their expression and role in HCC. Moving forward, we plan to expand our sample size to validate and optimize our model, specifically focusing on elucidating the mechanisms by which these genes modulate M1 or M2 polarization in HCC. By addressing these limitations, we can gain a deeper understanding of the complex interplay between the tumor microenvironment and HCC progression. Despite these limitations, our study provides a TME score-related prognostic model for HCC, contributing valuable insights that may aid the discovery of new treatment strategies for this disease.
Abbreviation
ACTB, beta-actin; ADGRB2, adhesion G protein-coupled receptor B2; BAI2, brain angiogenesis inhibitor 2; bFGF, basic fibroblast growth factor; C7, complement C7; CD163, Cluster of Differentiation 163; CD34, CD34 molecule; CD5L, CD5 molecule like; cDNA, complementary DNA; CFP, complement factor properdin; CLEC1B, C-type lectin domain family 1 member B; CNDP1, carnosine dipeptidase 1; COL15A1, collagen type XV alpha 1 chain; COL9A1, collagen type IX alpha 1 chain; COLEC10, collectin subfamily member 10; COLEC10, collectin subfamily member 12; CRHBP, corticotropin releasing hormone binding protein; CTHRC1, collagen triple helix repeat containing 1; DEG, differentially expressed gene; DEMPRG, differentially expressed MP-related gene; DN, Down regulation; EBF3, early B cell factor 3; EMT, epithelial-mesenchymal transition; EGF, epidermal growth factor; EGFL6, EGF like domain multiple 6; FABP4, fatty acid binding protein 4; FAM163B, family with sequence similarity 163 member B; FCN3, ficolin 3; FOSB, FBJ osteosarcoma oncogene B; G2/M, G2 phase to mitotic phase; GDF10, growth differentiation factor 10; GO, Gene Ontology; GPR1, G protein-coupled receptor 1; GPR182, G protein-coupled receptor 182; GPM6A, glycoprotein M6A; GSEA, Gene Set Enrichment Analysis; GSVA, Gene Set Variation Analysis; HCC, hepatocellular carcinoma; HOXD8, homeobox D8; HOXD9, homeobox D9; HOXD10, homeobox D10; HME, human macrophage metalloelastase; ICGC, International Cancer Genome Consortium; IGFALS, insulin like growth factor binding protein acid labile subunit; IHC, Immunohistochemistry; IL-, Interleukin-; IL1RL1, interleukin 1 receptor like 1; INMT, indolethylamine N-methyltransferase; KCNH2, potassium voltage-gated channel subfamily H member 2; KCNQ3, potassium voltage-gated channel subfamily Q member 3; KDM8, lysine demethylase 8; KEGG, Kyoto Encyclopedia of Genes and Genomes; KRAS, Kirsten rat sarcoma; LY6H, lymphocyte antigen 6 family member H; MAPT, microtubule associated protein tau; MMP9, matrix metallopeptidase 9; MMP12, matrix metallopeptidase 12; MP, macrophage polarization; MSC, musculin; NR4A3, nuclear receptor subfamily 4 group A member 3; NTF3, neurotrophin 3; OLFML2B, olfactomedin like 2B; OS, overall survival; PBS, phosphate-buffered saline; PD-L1, Programmed death-ligand 1; PDGF, platelet-derived growth factor; PLVAP, plasmalemma vesicle associated protein; PLPP2, phospholipid phosphatase 2; PRDM9, PR/SET domain 9; ROC curves, receiver operating characteristic curve; RIPOR3, RIPOR family member 3; RFX8, regulatory factor X8; SCN4A, Sodium Voltage-Gated Channel Alpha Subunit 4; ST8SIA6, ST8 alpha-N-acetyl-neuraminide alpha-2,8-sialyltransferase 6; TAM, Tumor-associated macrophage; TCGA, The Cancer Genome Atlas; TME, tumor microenvironment; TNF-α, tumor necrosis factor α; TRPC6, transient receptor potential cation channel subfamily C member 6; UCHL1, ubiquitin C-terminal hydrolase L1; VEGFA, vascular endothelial growth factor A; WGCNA, Weighted Gene Co-expression Network Analysis.
Data Sharing Statement
Gene expression data described in this study were deposited in TCGA and GTE databases. Additional relevant data supporting the findings of this study are available from the corresponding authors upon request. The datasets used and materials in this study are available upon reasonable request.
Ethics Approval and Consent to Participate
Clinical Samples
The study was conducted in accordance with the guidelines of the Declaration of Helsinki and approved by the ethics committee of the West China Hospital of Sichuan University (No. 2016-91).
Consent for Publication
All the authors agreed to publish the article.
Acknowledgment
We thank Miss Mingming Zhang for preparing HCC specimens.
Author Contributions
All authors made a significant contribution to the work reported, whether in the conception, study design, execution, acquisition of data, analysis, and interpretation, or in all these areas, took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by 1.3.5 project for disciplines of excellence, West China Hospital, Sichuan University (No.ZYGD23030).
Disclosure
The authors declare no potential conflicts of interest regarding the research, authorship, or publication of this article.
References
1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
2. Llovet JM, Kelley RK, Villanueva A, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7(1):6. doi:10.1038/s41572-020-00240-3
3. Huang A, Yang X-R, Chung W-Y, et al. Targeted therapy for hepatocellular carcinoma. Signal Transduct Target Ther. 2020;5(1):146. doi:10.1038/s41392-020-00264-x
4. Bruix J, Reig M, Sherman M. Evidence-based diagnosis, staging, and treatment of patients with hepatocellular carcinoma. Gastroenterology. 2016;150(4):835–853. doi:10.1053/j.gastro.2015.12.041
5. Jacome AA, Castro ACG, Vasconcelos JPS, et al. Efficacy and safety associated with immune checkpoint inhibitors in unresectable hepatocellular carcinoma: a meta-analysis. JAMA Network Open. 2021;4(12):e2136128. doi:10.1001/jamanetworkopen.2021.36128
6. Sonbol MB, Riaz IB, Naqvi SAA, et al. Systemic Therapy and Sequencing Options in Advanced Hepatocellular Carcinoma: a Systematic Review and Network Meta-analysis. JAMA Oncol. 2020;6(12):e204930. doi:10.1001/jamaoncol.2020.4930
7. Peng H, Zhu E, Zhang Y. Advances of cancer-associated fibroblasts in liver cancer. Biomark Res. 2022;10(1):59. doi:10.1186/s40364-022-00406-z
8. Baglieri J, Brenner DA, Kisseleva T. The role of fibrosis and liver-associated fibroblasts in the pathogenesis of hepatocellular carcinoma. Int J Mol Sci. 2019;20(7):1723. doi:10.3390/ijms20071723
9. Pineiro Fernandez J, Luddy KA, Harmon C, et al. Hepatic tumor microenvironments and effects on NK cell phenotype and function. Int J Mol Sci. 2019;20(17):4131. doi:10.3390/ijms20174131
10. Chen H, Zhou X-H, Li J-R, et al. Neutrophils: driving inflammation during the development of hepatocellular carcinoma. Cancer Lett. 2021;522:22–31. doi:10.1016/j.canlet.2021.09.011
11. Wang Z, Wang Y, Gao P, et al. Immune checkpoint inhibitor resistance in hepatocellular carcinoma. Cancer Lett. 2023;555:216038. doi:10.1016/j.canlet.2022.216038
12. Zhang J, Han H, Wang L, et al. Overcoming the therapeutic resistance of hepatomas by targeting the tumor microenvironment. Front Oncol. 2022;12:988956. doi:10.3389/fonc.2022.988956
13. Tian Z, Hou X, Liu W, et al. Macrophages and hepatocellular carcinoma. Cell Biosci. 2019;9:79. doi:10.1186/s13578-019-0342-7
14. Cheng K, Cai N, Zhu J, et al. Tumor-associated macrophages in liver cancer: from mechanisms to therapy. Cancer Commun. 2022;42(11):1112–1140. doi:10.1002/cac2.12345
15. He Z, Zhang S. Tumor-associated macrophages and their functional transformation in the hypoxic tumor microenvironment. Front Immunol. 2021;12:741305. doi:10.3389/fimmu.2021.741305
16. Tamura R, Tanaka T, Yamamoto Y, et al. Dual role of macrophage in tumor immunity. Immunotherapy. 2018;10(10):899–909. doi:10.2217/imt-2018-0006
17. Wu K, Lin K, Li X, et al. Redefining tumor-associated macrophage subpopulations and functions in the tumor microenvironment. Front Immunol. 2020;11:1731. doi:10.3389/fimmu.2020.01731
18. Boutilier AJ, Elsawa SF. Macrophage polarization states in the tumor microenvironment. Int J Mol Sci. 2021;22(13):6995. doi:10.3390/ijms22136995
19. Mehla K, Singh PK. Metabolic regulation of macrophage polarization in cancer. Trends Cancer. 2019;5(12):822–834. doi:10.1016/j.trecan.2019.10.007
20. Li R, Zhao W, Liang R, et al. Identification and validation of a novel tumor microenvironment-related prognostic signature of patients with hepatocellular carcinoma. Front Mol Biosci. 2022;9:917839. doi:10.3389/fmolb.2022.917839
21. Zhao Y, Li M, Yang Y, et al. Identification of macrophage polarization-related genes as biomarkers of chronic obstructive pulmonary disease based on bioinformatics analyses. Biomed Res Int. 2021;2021:9921012. doi:10.1155/2021/9921012
22. Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinf. 2013;14:7. doi:10.1186/1471-2105-14-7
23. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinf. 2008;9:559. doi:10.1186/1471-2105-9-559
24. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi:10.1186/s13059-014-0550-8
25. Yu G, Wang L-G, Han Y, et al. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–287. doi:10.1089/omi.2011.0118
26. Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J Stat Softw. 2010;33(1):1–22. doi:10.18637/jss.v033.i01
27. Mantovani A, Allavena P, Marchesi F, et al. Macrophages as tools and targets in cancer therapy. Nat Rev Drug Discov. 2022;21(11):799–820. doi:10.1038/s41573-022-00520-5
28. Pu Y, Ji Q. Tumor-associated macrophages regulate PD-1/PD-L1 Immunosuppression. Front Immunol. 2022;13:874589. doi:10.3389/fimmu.2022.874589
29. Zhou D, Luan J, Huang C, et al. Tumor-associated macrophages in hepatocellular carcinoma: friend or foe? Gut Liver. 2021;15(4):500–516. doi:10.5009/gnl20223
30. Shirabe K, Mano Y, Muto J, et al. Role of tumor-associated macrophages in the progression of hepatocellular carcinoma. Surg Today. 2012;42(1):1–7. doi:10.1007/s00595-011-0058-8
31. Matsubara T, Kanto T, Kuroda S, et al. TIE2-expressing monocytes as a diagnostic marker for hepatocellular carcinoma correlates with angiogenesis. Hepatology. 2013;57(4):1416–1425. doi:10.1002/hep.25965
32. Gorrin-Rivas MJ, Arii S, Mori A, et al. Implications of human macrophage metalloelastase and vascular endothelial growth factor gene expression in angiogenesis of hepatocellular carcinoma. Ann Surg. 2000;231(1):67–73. doi:10.1097/00000658-200001000-00010
33. Hou ZH, Xu X-W, Fu X-Y, et al. Long non-coding RNA MALAT1 promotes angiogenesis and immunosuppressive properties of HCC cells by sponging miR-140. Am J Physiol Cell Physiol. 2020;318(3):C649–C663. doi:10.1152/ajpcell.00510.2018
34. Hasita H, Komohara Y, Okabe H, et al. Significance of alternatively activated macrophages in patients with intrahepatic cholangiocarcinoma. Cancer Sci. 2010;101(8):1913–1919. doi:10.1111/j.1349-7006.2010.01614.x
35. Watkins LR, Orlandi C. Orphan G Protein coupled receptors in affective disorders. Genes. 2020;11(6):694. doi:10.3390/genes11060694
36. Lv X, Li L, Lv L, et al. HOXD9 promotes epithelial-mesenchymal transition and cancer metastasis by ZEB1 regulation in hepatocellular carcinoma. J Exp Clin Cancer Res. 2015;34:133. doi:10.1186/s13046-015-0245-3
37. Wang J, Ding Z-W, Chen K, et al. A predictive and prognostic model for hepatocellular carcinoma with microvascular invasion based TCGA database genomics. BMC Cancer. 2021;21(1):1337. doi:10.1186/s12885-021-09047-1
38. Long J, Zhang L, Wan X, et al. A four-gene-based prognostic model predicts overall survival in patients with hepatocellular carcinoma. J Cell Mol Med. 2018;22(12):5928–5938. doi:10.1111/jcmm.13863
39. Zhang Z, Li J, He T, et al. Two predictive precision medicine tools for hepatocellular carcinoma. Cancer Cell Int. 2019;19:290. doi:10.1186/s12935-019-1002-z
40. Chen C, Liu YQ, Qiu SX, et al. Five metastasis-related mRNAs signature predicting the survival of patients with liver hepatocellular carcinoma. BMC Cancer. 2021;21(1):693. doi:10.1186/s12885-021-08431-1
41. Zhang F-P, Huang Y-P, Luo W-X, et al. Construction of a risk score prognosis model based on hepatocellular carcinoma microenvironment. World J Gastroenterol. 2020;26(2):134–153. doi:10.3748/wjg.v26.i2.134
42. Meng D, Luo M, Liu B. The Role of CLEC-2 and Its Ligands in Thromboinflammation. Front Immunol. 2021;12:688643. doi:10.3389/fimmu.2021.688643
43. Hu K, Wang Z-M, Li J-N, et al. CLEC1B expression and PD-L1 expression predict clinical outcome in hepatocellular carcinoma with tumor hemorrhage. Transl Oncol. 2018;11(2):552–558. doi:10.1016/j.tranon.2018.02.010
44. Liang X, Song F, Fang W, et al. CLEC1B is a promising prognostic biomarker and correlated with immune infiltration in hepatocellular carcinoma. Int J Gen Med. 2022;15:5661–5672. doi:10.2147/IJGM.S363050
45. Zhang G, Zhang K, Zhao Y, et al. A novel stemness-hypoxia-related signature for prognostic stratification and immunotherapy response in hepatocellular carcinoma. BMC Cancer. 2022;22(1):1103. doi:10.1186/s12885-022-10195-1
46. Ding S, Wang X, Lv D, et al. EBF3 reactivation by inhibiting the EGR1/EZH2/HDAC9 complex promotes metastasis via transcriptionally enhancing vimentin in nasopharyngeal carcinoma. Cancer Lett. 2022;527:49–65. doi:10.1016/j.canlet.2021.12.010
47. Rodger EJ, Chatterjee A, Stockwell PA, et al. Characterisation of DNA methylation changes in EBF3 and TBC1D16 associated with tumour progression and metastasis in multiple cancer types. Clin Clin Epigenet. 2019;11(1):114. doi:10.1186/s13148-019-0710-5
48. Liao D. Emerging roles of the EBF family of transcription factors in tumor suppression. Mol Cancer Res. 2009;7(12):1893–1901. doi:10.1158/1541-7786.MCR-09-0229
49. Kim J, Min SY, Lee HE, et al. Aberrant DNA methylation and tumor suppressive activity of the EBF3 gene in gastric carcinoma. Int, J, Cancer. 2012;130(4):817–826. doi:10.1002/ijc.26038
50. Tao YF, Xu L-X, Lu J, et al. Early B-cell factor 3 (EBF3) is a novel tumor suppressor gene with promoter hypermethylation in pediatric acute myeloid leukemia. J Exp Clin Cancer Res. 2015;34(1):4. doi:10.1186/s13046-014-0118-1
51. Zhao LY, Niu Y, Santiago A, et al. An EBF3-mediated transcriptional program that induces cell cycle arrest and apoptosis. Cancer Res. 2006;66(19):9445–9452. doi:10.1158/0008-5472.CAN-06-1713
52. Hudson AJ, Ebers GC, Bulman DE. The skeletal muscle sodium and chloride channel diseases. Brain. 1995;118:547–563. doi:10.1093/brain/118.2.547
53. Wang Z, Embaye KS, Yang Q, et al. A novel metabolism-related signature as a candidate prognostic biomarker for hepatocellular carcinoma. J Hepatocell Carcinoma. 2021;8:119–132. doi:10.2147/JHC.S294108
54. Yan Y, He W, Chen Y, et al. Comprehensive analysis to identify the encoded gens of sodium channels as a prognostic biomarker in hepatocellular carcinoma. Front Genet. 2021;12:802067. doi:10.3389/fgene.2021.802067
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Decaprenyl Diphosphate Synthase Subunit 1 (PDSS1): A Potential Prognostic Biomarker and Immunotherapy-Target for Hepatocellular Carcinoma
Yang Y, Li J, Tang M, Nie B, Huang W
Cancer Management and Research 2022, 14:1627-1639
Published Date: 3 May 2022
Development and Validation of a Propionate Metabolism-Related Gene Signature for Prognostic Prediction of Hepatocellular Carcinoma
Xiao J, Wang J, Zhou C, Luo J
Journal of Hepatocellular Carcinoma 2023, 10:1673-1687
Published Date: 2 October 2023
Molecular Subtyping of Hepatocellular Carcinoma via Lysosome-Related Genes for Prognosis and Therapy Prediction
Yao Y, Zhu T, Shen X, Ma J, Zhu X, Jiang J
International Journal of General Medicine 2025, 18:3933-3950
Published Date: 16 July 2025
The Predictive Significance of Interleukin-2 Receptor in Patients with Hepatocellular Carcinoma
Tian Z, Guo Y, Yang R, Yang W
Journal of Hepatocellular Carcinoma 2025, 12:1893-1904
Published Date: 22 August 2025
Noninvasive Prediction of High Ki-67 Expression in Hepatocellular Carcinoma Using Multiparametric MRI and Clinical Biomarkers
Zhang F, Chen G, Huang M, Yang Y, Wang Z, Shen Y, Luo Y, Hu X, Li Z
Journal of Hepatocellular Carcinoma 2026, 13:574222
Published Date: 17 February 2026