Back to Journals » Journal of Blood Medicine » Volume 13
Clinical Characteristics and Prognostic Risks of Philadelphia-Negative Myeloproliferative Neoplasms at Cipto Mangunkusumo General Hospital
Authors Sukrisman L ![]()
Received 31 May 2022
Accepted for publication 23 August 2022
Published 12 September 2022 Volume 2022:13 Pages 495—503
DOI https://doi.org/10.2147/JBM.S374636
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Lugyanti Sukrisman
Division of Hematology and Medical Oncology, Department of Internal Medicine, Faculty of Medicine, Universitas Indonesia/Cipto Mangunkusumo General Hospital, Jakarta, Indonesia
Correspondence: Lugyanti Sukrisman, Division of Hematology and Medical Oncology, Department of Internal Medicine, Faculty of Medicine, Universitas Indonesia/Cipto Mangunkusumo General Hospital, Jakarta, Indonesia, Email [email protected]
Background: Philadelphia-negative myeloproliferative neoplasms (Ph-MPNs) are clonal hematopoietic cell malignancy that comprises polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF). They prone to develop thrombosis, bleeding, fibrotic progression, and leukemic transformation. We aimed to study clinical characteristics, thrombosis complications, and prognostic risk in Indonesians with Ph-MPNs.
Methods: This was a single-center retrospective cross-sectional study, including patients with Ph-MPNs who attended Hematology and Medical Oncology outpatient clinic at Cipto Mangunkusumo General Hospital, Jakarta, between 2016 and 2021. Demographic data, clinical characteristics, previous antiplatelet treatment, baseline laboratory data, JAK2V617F mutation, and treatment were reviewed for analysis. The prognostic risk model was assessed for PV (IPSS), ET (IPSET-thrombosis), and PMF (IPSS and DIPPS) patients.
Results: One hundred forty-six patients were classified as PV (31.5%), ET (38.4%), and PMF (30.1%) with median ages of 54, 53.5, and 55 years, respectively. PMF presented with the most diverse clinical presentations. JAK2V617 mutations were detected in 82%, 42.5%, and 76.5% of PV, ET, and PMF patients. PV had the highest thrombosis event pre/at diagnosis (26.1%), with predominantly arterial thrombosis. Bleeding occurred more in PMF (20.5%). Phlebotomy and hydroxyurea are the only treatment available for Ph-MPNs in the government hospital in Indonesia. IPSS intermediate risk in PV (47.8%), IPSET-thrombosis low risk in ET (46.4%), and IPSS and DIPSS intermediate-2 in PMF (38.6% and 50% respectively) were the most common risk groups.
Conclusion: Essential thrombocythemia was the most prevalent Ph-MPNs in Indonesia. Arterial thrombosis was the highest thrombosis event pre/at diagnosis in PV despite IPSET-thrombosis low risk being the most common risk group. In JAK2V617 mutation-positive MPN population, thrombosis event was also the highest in PV.
Keywords: Ph-MPNs, clinical characteristics, thrombosis, prognosis
Introduction
Philadelphia-negative myeloproliferative neoplasms (Ph-MPNs) are a group of hematopoietic malignancies which encompasses polycythemia vera (PV), essential thrombocythemia (ET), and myelofibrosis (MF).1 All entities of Ph-MPNs shared a feature of mutations that is associated with physiologic pathways for hematopoiesis, as the disease arises from single hematopoietic stem cell that underwent somatic mutation.2
A study of Ph-MPNs in 17 countries unveiled the prevalence of Ph-MPNs in 2016 of 64.14 per 100,000 and the incidence cases of 12.05 per 100,000.3 Another study reported the incidence rates of Ph-MPNs per 100,000 population were between 1.0 and 1.5 for PV, 2.0 and 3.0 for ET, and 0.3 and 0.5 for PMF.4
In the initial stage, Ph-MPNs generally appear with no symptoms, thus usually diagnosed accidentally based on routine laboratory findings or when symptoms or complications occurred.5,6 These complications influence the survival and prognosis of MPN patients.
Patients with Ph-MPNs experienced poor quality of life due to symptoms that affect general activity, anxiety, mood disorder, and cancelation of life plan.7,8 On top of that, those with Ph-MPNs have an increased risk of morbidity and mortality compared to the general population. The relative survival ratios (RSR) of 10 years was 0.64, 0.68, and 0.21 for patients with PV, ET, and PMF, respectively.9 Previous prospective study showed that the median life expectancy among patients with Ph-MPNs was 91.7 months (7.5 years).10
Thrombosis and major cardiovascular adverse events are important causes of mortality in Ph-MPNs patients.11,12 Moreover, patients with Ph-MPNs are at risk for developing leukemic and fibrotic progression which contribute further to the morbidity and mortality of the patients.13,14
Given the insufficient data on Ph-MPNs in Indonesia, we conducted this study to find out the clinical characteristics, thrombosis complications, and prognosis in Indonesian with Ph-negative MPN.
Methods
This cross-sectional study was performed at Cipto Mangunkusumo General Hospital, Jakarta. The information of all Ph-negative patients (PV, ET, and PMF) who have attended the Hematology and Medical Oncology outpatient clinic from 2016 until 2021 were retrospectively retrieved from medical records, including demographic data (age, sex), clinical characteristics (comorbidities, signs and symptoms, thrombosis and bleeding events pre/at diagnosis, previous antiplatelet treatment), baseline laboratory data at diagnosis (complete blood count), JAK2V617F mutation status, and treatment administered.
The status of JAK2V617D mutation was determined qualitatively using RNA extraction kit which employs silica-based membrane followed by cDNA synthesis, endpoint/conventional PCR and capillary/Sanger sequencing. This test has up to 20% sensitivity to detect mutation within the samples. It has 100% specificity to detect V617F mutation.
For PV patients, the International Study of PV15 was applied for estimated survival (Supplementary Table 1). For ET patients, the International Prognostic Score for ET-thrombosis (IPSET-thrombosis)16 was evaluated for thrombosis risk (Supplementary Table 2). For PMF patients, the International Prognosis Scoring System (IPSS)17 and Dynamic IPSS (DIPSS)18 were calculated for estimated survival (Supplementary Table 3).
This study was conducted in accordance with Declaration of Helsinki, and the study protocol was approved by the Local Ethics Committee of Faculty of Medicine Universitas Indonesia (No. KET-558/UN.2.F1/ETIK/PPM.00.02/2021). Verbal informed consent was obtained from all subjects before blood sampling. Since our study did not apply any intervention, The Ethical Committee waived the requirement for written informed consent. The data were analyzed by descriptive statistics. The data were expressed in frequency (percentage, %) for categorical variables and median (range) for continuous variables. All analysis was performed using SPSS version 24 for Mac.
Results
We identified 146 patients with Ph-negative Ph-MPNs, including 46 patients with PV (31.5%), 56 patients with ET (38.4%), and 44 patients with PMF (30.1%). Baseline clinical and laboratory characteristics of the subjects are represented in Table 1. The median age of PV, ET, and PMF patients were 54, 53.5, and 55 years, respectively. PV and ET occurred more in males.
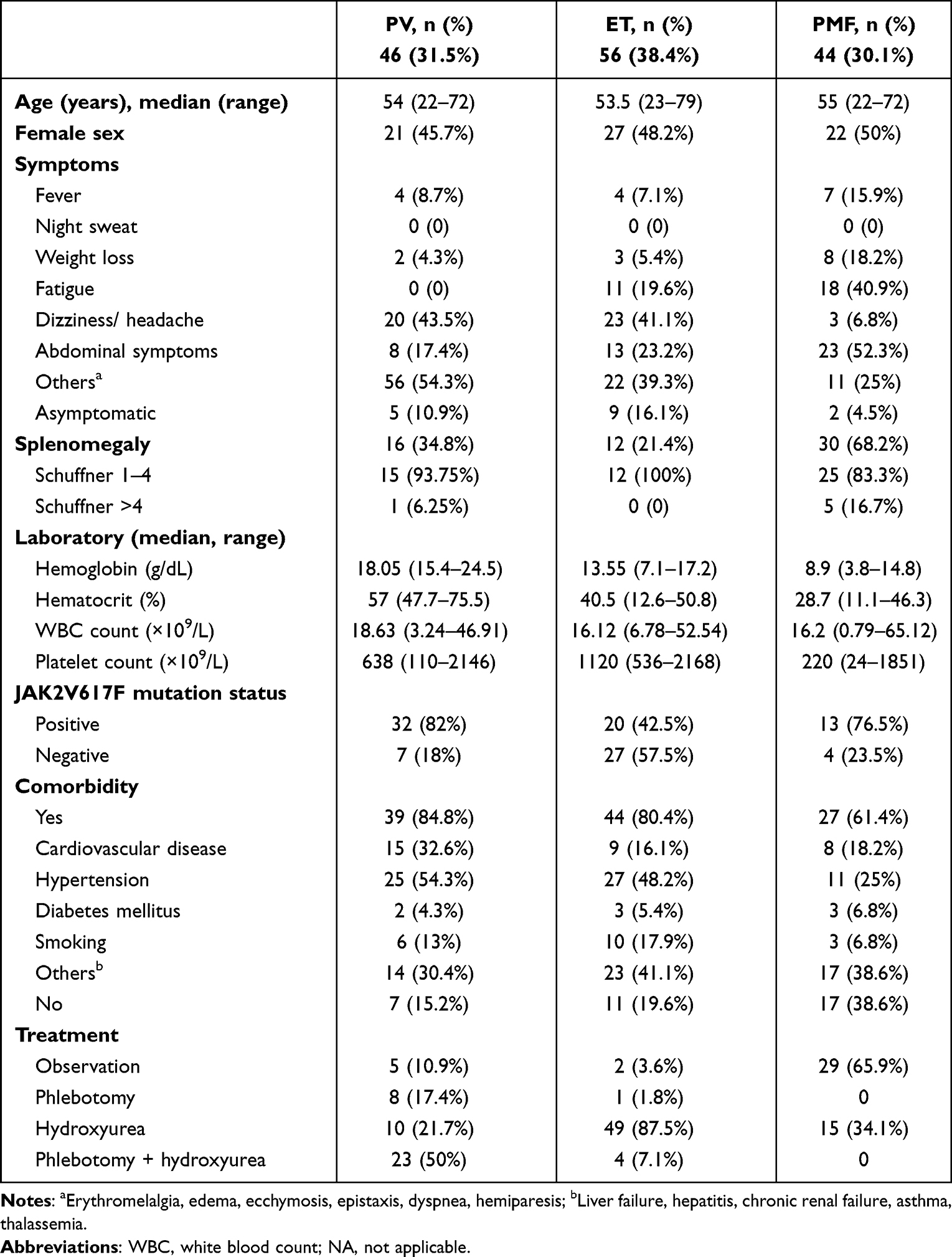 |
Table 1 Baseline Clinical and Laboratory Characteristics of 146 Subjects |
Asymptomatic cases were found in 10.9% of PV, 16.1% of ET, and 4.5% the of PMF patients. The most common signs and symptoms in PV patients were dizziness/headache (43.5%), splenomegaly (34.8%), and abdominal symptoms (17.4%). Similarly, most ET patients had dizziness/headache (41.1%), abdominal symptoms (23.2%), and splenomegaly (21.4%). PMF patients presented with splenomegaly (68.2%), abdominal symptoms (52.3%), fatigue (40.9%), and fever (15.9%).
Regarding complete blood count parameters, the median hemoglobin and hematocrit of PV patients were 18.05 g/dL and 57%. Median platelet counts in ET and PV patients were 1120 × 109/L and 638 × 109/L. Patients with PMF had a wide range of WBC and platelet counts with median WBC of 16.2 × 109/L (range 0.79–65.12 × 109/L) and median platelet count of 220 × 109/L (range 24–1851 × 109/L).
Due to lack of testing, JAK2V617F mutation analyses could not be reported in a total of 43 patients. The majority of PV were positive for JAK2V617 mutation (82%), and only 42.5% of ET and 76.5% of PMF harbored this mutation. About 84.8% of PV, 80.4% of ET, and 61.4% of PMF patients had at least one comorbidity; hypertension and cardiovascular disease were the most prevalent.
Table 2 shows the thrombosis and bleeding events in our patients. A 26.1% of PV patients experienced thrombosis pre/at diagnosis, which was more common than in ET and PMF patients (21.4% and 4.5%, respectively). Arterial thrombosis was more prevalent than venous thrombosis, primarily manifesting with ischemic stroke. Bleeding events pre/at diagnosis occurred more frequently in PMF (20.5%) and ET (10.7%) patients, manifesting commonly as gastrointestinal hemorrhage and skin and/or mucosal bleeding. About 30% of PV and ET patients had been prescribed antiplatelet therapy, either aspirin and/or clopidogrel. Table 3 shows thrombosis and bleeding events on the JAK2-positive MPN subjects. Similarly, thrombosis in patients with positive JAK2 V617F mutation was more common in PV (21.8%), compared to ET (15%) and PMF (7.7%).
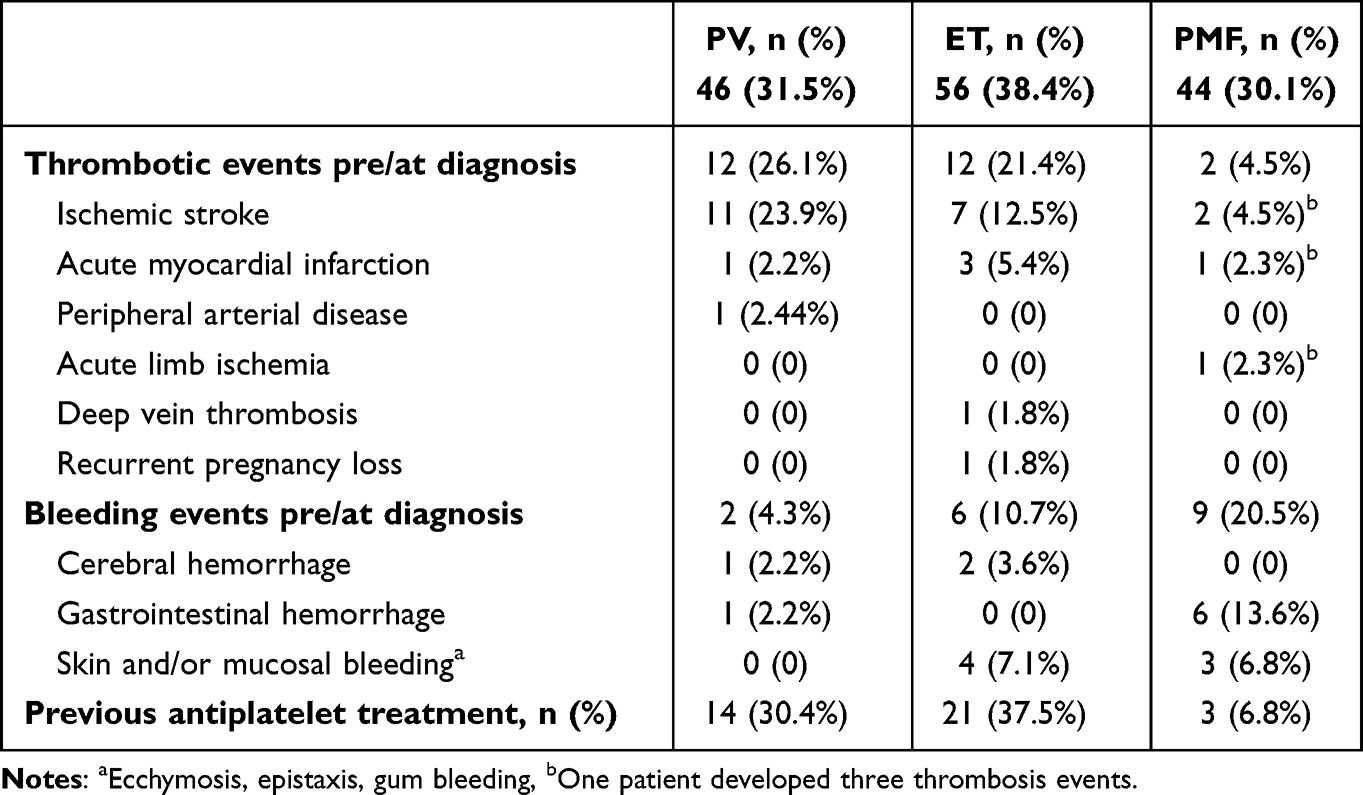 |
Table 2 Thrombotic and Bleeding Events of 146 Subjects |
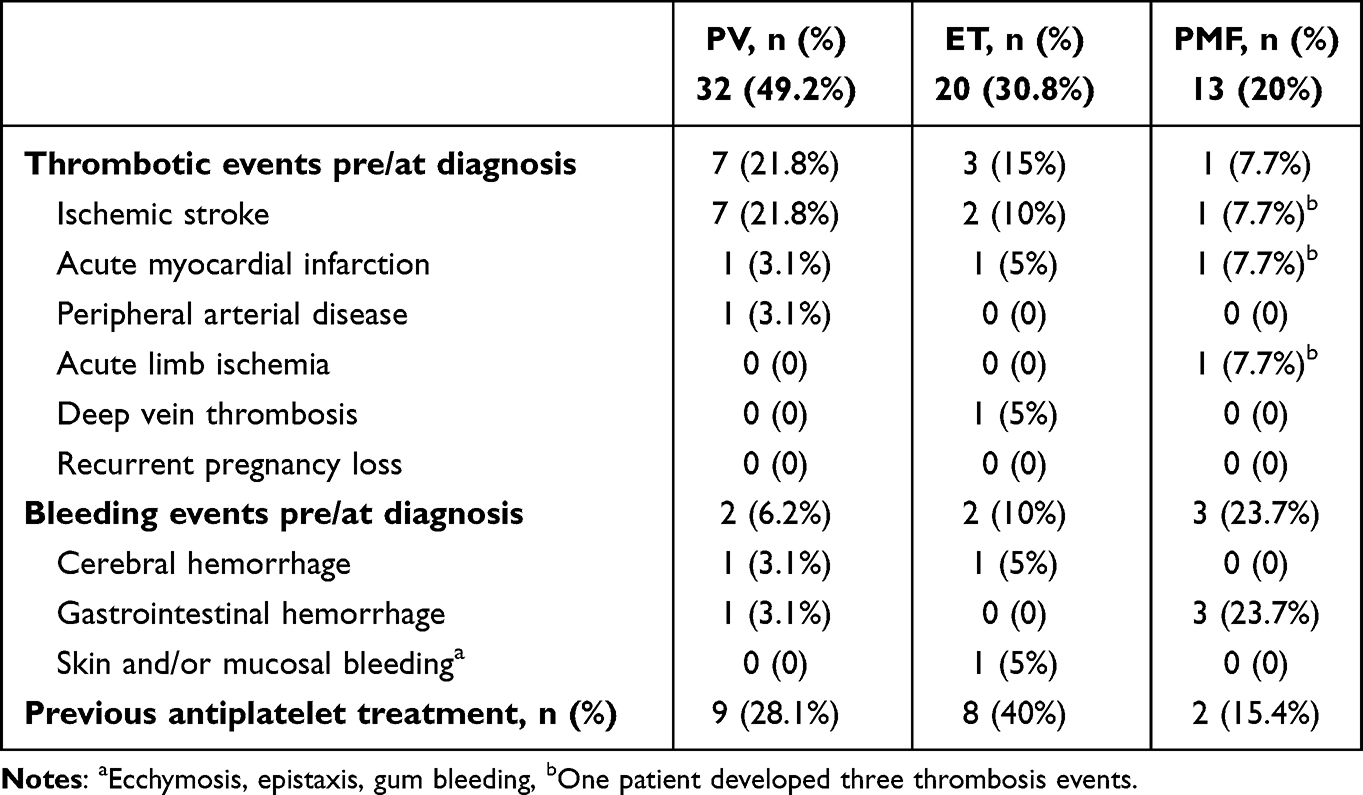 |
Table 3 Thrombotic and Bleeding Events of 65 JAK2V617F Mutation-Positive Subjects |
Tables 4–6 display the distribution of patients in the prognostic model risk category in PV, ET, and PMF subjects, respectively. By applying the IPSS stratification in PV subjects, most of the patients were distributed between intermediate- and high-risk groups (47.8% and 34.8% respectively). When patients with ET were classified using IPSET-thrombosis model, most of the patients were distributed between low- and high-risk groups (46.4% and 32.1% respectively). Using IPSS and DIPPS model in PMF, the majority of the patients were classified into the intermediate-2 risk group.
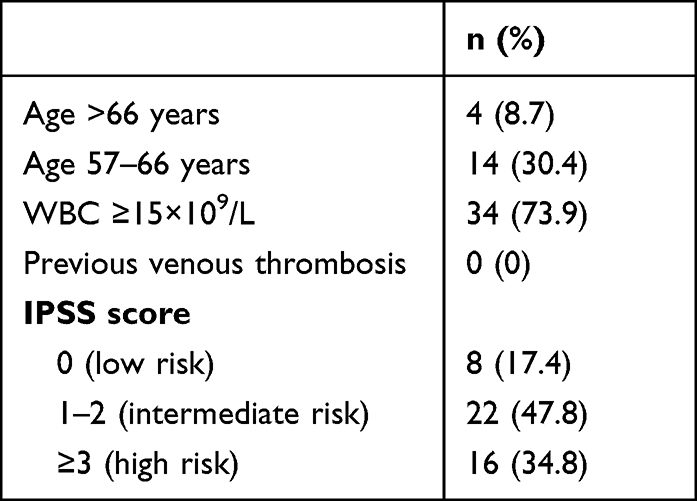 |
Table 4 IPSS in 46 PV Subjects |
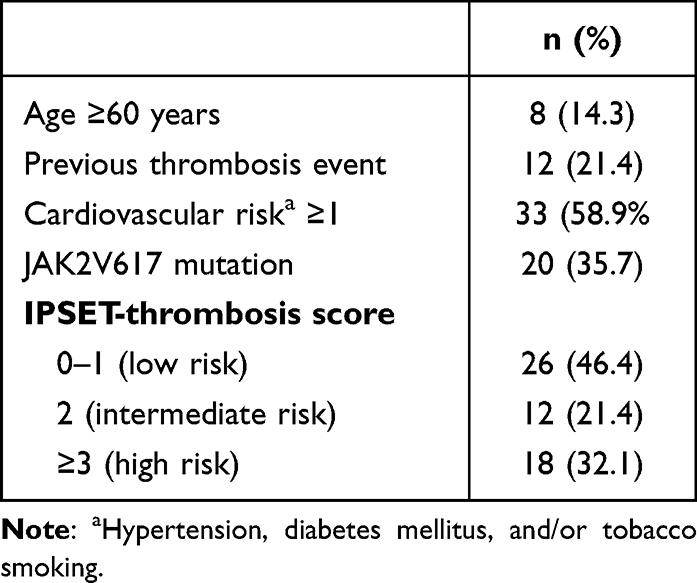 |
Table 5 IPSET-Thrombosis Model in 56 ET Subjects |
 |
Table 6 IPSS and DIPSS Model in 44 PMF Subjects |
Figure 1 shows the thrombotic and bleeding events of all PV subjects according to IPSS score. The thrombotic risk appears to be similar between low-risk, intermediate-risk and high-risk groups. Figure 2 shows that in all patients with ET, both the risk for thrombosis and bleeding rise gradually as the risk stratification increases.
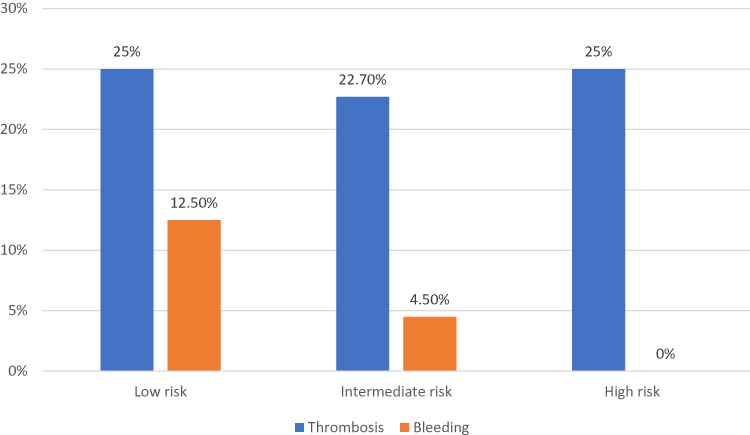 |
Figure 1 Thrombotic and bleeding events of 46 PV subjects according to IPSS score. |
 |
Figure 2 Thrombotic and bleeding events of 56 ET subjects according to IPSET-thrombosis score. |
Discussion
This study presents the clinical manifestation of Indonesian with Ph-negative Ph-MPNs. ET was the most frequent Ph-MPNs found in our study (38.4%). A study by Valladares et al also revealed similar results.19 There were 46 PV patients included in this study with the median age at diagnosis of 54 years, which was slightly lower than the median age from the International Study of PV (61 years).15 In addition, male-to-female ratio was close to 1:1, which was similar to the previous studies.15,20 For ET patients, the median age in our study was similar to the median age in IPSET-thrombosis study (53.5 vs 54.4 years).16
The median age of PMF in our study was 55 years, which was lower than the result of a study using the Mayo Clinic database for PMF and Porto-Soares et al (mean age of 65 and 64.7 years, respectively).21,22 The predominant symptoms of PMF were abdominal symptoms, fatigue, and splenomegaly. In the present study, patients with PMF, on average, had the greatest decrease in hemoglobin (anemia) and a wider range of WBC and platelet counts. Gangat et al and Duangnapasatit et al reported similar results.21,23 Cytopenias are essentially a difficult clinical problem in PMF. Anemia is typically presented in approximately 30% of the patients with PMF at diagnosis, and eventually all the patients develop anemia.24 The pathogenesis of anemia in PMF is incompletely understood, the contributing factors include ineffective erythropoiesis due to bone marrow scarring, aberrant inflammatory cytokine expression, and marked splenomegaly.25
The JAK2V617F mutation is an acquired somatic mutation that is found in the vast majority of PV patients and in half of ET and PMF patients.26 We found a lower percentage of JAK2 positive for PV and a higher percentage of JAK2 positive for PMF compared to multiple previous studies by Porto-Suares et al (93.5% of PV, 48% of ET, 60.8% of PMF), Valladares et al (86.6% of PV), and Gangat et al (66% of PMF).19,21,22 JAK2 is involved in cellular signaling processes, such as cell proliferation, differentiation, survival, and self-renewal. Mutated JAK2 cells confer hypersensitivity to several cytokines, including erythropoietin, thrombopoietin, granulocyte colony-stimulating factor. This may explain why patients with JAK2 mutation had a predominant rise in several blood parameters, including hemoglobin, hematocrit, and/or platelets, thus associated with thrombosis.27–29 In this study, we also evaluate thrombotic and bleeding events among 65 JAK2V617F mutation-positive subjects (Table 3) and found similar risk of thrombosis and bleeding compared to all patients regardless of their mutation status. Mutational analysis of JAK2V617F is not reimbursable by National Health Insurance, therefore not every patient was tested. Additionally, ischemic stroke is the most common thrombotic event in all population and JAK2V617F-positive population.
High hematocrit and WBC counts affect blood viscosity and blood flow, and further induce platelet activation and thrombosis.30,31 In addition, clinical factors, such as older age, obesity, history of thrombosis event, hypertension, dyslipidemia, contribute to the increased risk of thrombosis.11 In the present study, thrombotic events pre/at diagnosis were more pronounced in PV (26.1%) and followed by ET (21.4%) as PV patients had higher levels of hemoglobin, hematocrit, and WBC counts. Arterial thrombosis occurred at higher rates than venous thrombosis. This finding is in line with studies from Malaysia and the IPSET-thrombosis study, despite most studies in the Western countries revealed a higher rate of venous thrombosis.32,33 This is most probably because of a higher rate of arterial risk factors in older patients, such as hypertension, diabetes, or dyslipidemia. Hypertension dominated as the preexisting risk factor of thrombosis in our patients.
A prognostic model for PV with three-tiered categories was developed by the International Study of PV, and the median survivals were 26 years (in low risk), 15 years (in intermediate-risk), and 8.3 years (in high-risk).15 In our study, 47.8% of PV patients fell into intermediate-risk categories. The survival risk score was significantly associated with thrombosis.23 However, in our study, thrombotic risk did not differ significantly between IPSS risk groups (Figure 1). This is because the study population had very high prevalence of comorbidities (84.8%), including those with low risk. Therefore, thrombosis occurs even in low-risk individuals. Thrombotic complications are one of the most common causes of death in patients with PV. The International Study of PV reported that 32 out of 183 patients (20%) with a known cause of death deceased due to thrombotic complications.15 Therefore, cytoreductive therapy is recommended for high-risk PV.34
Advanced age (≥60 years), previous thrombotic event, cardiovascular risk factors (hypertension, diabetes mellitus, tobacco use), and JAK2V617F mutation are well-characterized predictors for thrombotic complications in patients with ET.34,35 The IPSET-thrombosis study incorporates those risk factors in the international prognostic scoring system for thrombosis. In the present study, when ET patients were stratified using IPSET-thrombosis model, 46.4%, 21.4%, and 32.1% of them were part of low-risk, intermediate-risk, and high-risk groups, respectively. Moreover, the risk for thrombosis and bleeding increased incrementally along the higher risk scores (Figure 2). Sevindik et al revealed that 33%, 26.8%, and 40.2% of the patients were categorized into low-, immediate-, and high-risk groups. Furthermore, one or more thrombotic episodes occurred in 5.4% of low-risk, 26.7% of intermediate-risk, and 66.2% of high-risk groups.36
The estimated median survival of PMF is 6 years, but it can be a few months to years. In clinical practice, IPSS is used at diagnosis to predict survival in patients with PMF. Based on the four risk categories, the median survivals were 135 months (in low risk), 95 months (in intermediate-1 risk), 48 months (in intermediate-2 risk), and 27 months (in high risk). During the disease course, patients may acquire additional risk factors that may interfere patient’s prognosis. In the present study, the proportion of patients in four prognostic groups using IPSS was similar to the previous study.17
In current practice, the therapy in PV and ET is intended to alleviate symptoms and prevent thrombosis. Aspirin, cytoreductive drug (in high-risk ET and high-risk PV), and phlebotomy (in PV with intended hematocrit target <45%) have been shown to have an antithrombotic effect.37 In our study, previous antiplatelet therapy was recorded in 21 (37.5%) patients with ET and 14 (30.4%) patients with PV. While there is a clear benefit of antiplatelet therapy in patients with PV, studies revealed inconsistent results regarding the benefit of antiplatelet therapy in patients with ET.38 Another study reported that antiplatelet therapy did not lower the thrombosis risk but increased the risk of bleeding in patients with ET.39 In the present study, bleeding events pre/at diagnosis occurred in 9 (20.5%) of PMF patients and 6 (10.7%) of ET patients, despite the low usage of antiplatelets in these groups compared with PV. Moreover, there was 5 (8.9%) patients with ET who were initially treated with phlebotomy due to increased hematocrit. After hematocrit reached its intended target, standard therapy for ET was administered.
This study provides a better understanding of the clinical characteristics and thrombotic events in Indonesian patients with Ph-negative MPN, who have been underreported in Indonesia. We used IPSS, DIPSS, IPSET-thrombosis in the analyses of prognosis of patients with MPN. However, some limitations of this study are needed to be acknowledged. First, this was a single-center study which only had a small number of subjects. Second, this was a retrospective study that resulted in incomplete several data. Third, JAK2V617F mutation status could not be evaluated for all the patients.
In conclusion, essential thrombocythemia was the most prevalent Ph-MPNs in Indonesia. Arterial thrombosis was the highest thrombosis event pre/at diagnosis in PV despite IPSET-thrombosis low risk being the most common risk group. In JAK2V617 mutation-positive MPN population, thrombosis event was also the highest in PV group.
Disclosure
The author reports no conflict of interest in this work.
References
1. Barbui T, Thiele J, Gisslinger H., et al. The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: document summary and in-depth discussion. Blood Cancer J. 2018;8(2):15. doi:10.1038/s41408-018-0054-y
2. Spivak JL, Longo DL. Myeloproliferative neoplasms. N Engl J Med. 2017;376(22):2168–2181. doi:10.1056/NEJMra1406186
3. Yassin MA, Taher A, Mathews V, et al. MERGE: a multinational, multicenter observational registry for myeloproliferative neoplasms in Asia, including Middle East, Turkey, and Algeria. Cancer Med. 2020;9(13):4512–4526. doi:10.1002/cam4.3004
4. Byun JM, Kim YJ, Youk T, Yang JJ, Yoo J, Park TS. Real world epidemiology of myeloproliferative neoplasms: a population based study in Korea 2004–2013. Ann Hematol. 2017;96(3):373–381. doi:10.1007/s00277-016-2902-9
5. McMullin MF, James G, Duncombe AS, et al. Patient perspectives of a diagnosis of myeloproliferative neoplasm in a case control study. Exp Hematol Oncol. 2015;5:14. doi:10.1186/s40164-016-0043-4
6. Barbui T, Thiele J, Vannucchi AM, Tefferi A. Problems and pitfalls regarding WHO-defined diagnosis of early/prefibrotic primary myelofibrosis versus essential thrombocythemia. Leukemia. 2013;27(10):1953–1958. doi:10.1038/leu.2013.74
7. Mesa R, Miller CB, Thyne M, et al. Myeloproliferative neoplasms (MPNs) have a significant impact on patients’ overall health and productivity: the MPN Landmark survey. BMC Cancer. 2016;16:167. doi:10.1186/s12885-016-2208-2
8. Anderson LA, James G, Duncombe AS, et al. Myeloproliferative neoplasm patient symptom burden and quality of life: evidence of significant impairment compared to controls. Am J Hematol. 2015;90(10):864–870. doi:10.1002/ajh.24098
9. Hultcrantz M, Kristinsson SY, Andersson TM-L, et al. Patterns of survival among patients with myeloproliferative neoplasms diagnosed in Sweden from 1973 to 2008: a population-based study. J Clin Oncol. 2012;30(24):2995–3001. doi:10.1200/JCO.2012.42.1925
10. Maynadié M, Girodon F, Manivet-Janoray I, et al. Twenty-five years of epidemiological recording on myeloid malignancies: data from the specialized registry of hematologic malignancies of Cote d’Or (Burgundy, France). Haematologica. 2011;96(1):55–61. doi:10.3324/haematol.2010.026252
11. Barbui T, Finazzi G, Falanga A. Myeloproliferative neoplasms and thrombosis. Blood. 2013;122(13):2176–2184. doi:10.1182/blood-2013-03-460154
12. Falanga A, Marchetti M. Thrombosis in myeloproliferative neoplasms. Semin Thromb Hemost. 2014;40(3):348–358. doi:10.1055/s-0034-1370794
13. Björkholm M, Hultcrantz M, Derolf ÅR. Leukemic transformation in myeloproliferative neoplasms: therapy-related or unrelated? Best Pract Res Clin Haematol. 2014;27(2):141–153. doi:10.1016/j.beha.2014.07.003
14. Yogarajah M, Tefferi A. Leukemic transformation in myeloproliferative neoplasms: a literature review on risk, characteristics, and outcome. Mayo Clin proc. 2017;92(7):1118–1128. doi:10.1016/j.mayocp.2017.05.010
15. Tefferi A, Rumi E, Finazzi G, et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia. 2013;27(9):1874–1881. doi:10.1038/leu.2013.163
16. Barbui T, Finazzi G, Carobbio A, et al. Development and validation of an International Prognostic Score of thrombosis in World Health Organization–essential thrombocythemia (IPSET-thrombosis). Blood. 2012;120(26):5128–5133. doi:10.1182/blood-2012-07-444067
17. Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009;113(13):2895–2901. doi:10.1182/blood-2008-07-170449
18. Passamonti F, Cervantes F, Vannucchi AM, et al. A dynamic prognostic model to predict survival in primary myelofibrosis: a study by the IWG-MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood. 2010;115(9):1703–1708. doi:10.1182/blood-2009-09-245837
19. Valladares X, Rojas C, Peña C, et al. Characterization of Philadelphia-negative myeloproliferative neoplasms in the Chilean public health system: multicentric study. Blood. 2018;132(Supplement 1):5479. doi:10.1182/blood-2018-99-117610
20. Barzilai M, Kirgner I, Avivi I, et al. Characteristics and outcomes of young adults with Philadelphia‐negative myeloproliferative neoplasms. Eur J Haematol;2019.
21. Gangat N, Caramazza D, Vaidya R, et al. DIPSS plus: a refined dynamic international prognostic scoring system for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. 2011;29(4):392–397. doi:10.1200/JCO.2010.32.2446
22. Porto-Soares MA, de Oliveira RD, Cortopassi GM, Machado-Neto JA, Palma LC, de Figueiredo-Pontes LL. Clinical and molecular profile of a Brazilian cohort of patients with classical BCR-ABL1-negative myeloproliferative neoplasms. Hematol Transfus Cell Ther. 2020;42(3):238–244. doi:10.1016/j.htct.2019.07.008
23. Duangnapasatit B, Rattarittamrong E, Rattanathammethee T, et al. Clinical manifestations and risk Factors for complications of Philadelphia chromosome-negative myeloproliferative neoplasms. Asian Pac J Cancer Prev. 2015;16(12):5013–5018. doi:10.7314/APJCP.2015.16.12.5013
24. Bose P. Myelofibrosis with cytopenias. Clin Lymphoma Myeloma Leuk. 2017;17:S30–S31. doi:10.1016/j.clml.2017.08.018
25. Tefferi A. Anemia in myelofibrosis—prevalence, the U2AF1 connection, new treatments. Blood Cancer J. 2017;7(12):648. doi:10.1038/s41408-017-0032-9
26. Nangalia J, Grinfeld J, Green AR. Pathogenesis of myeloproliferative disorders. Annu Rev Pathol Mech Dis. 2016;11(1):101–126. doi:10.1146/annurev-pathol-012615-044454
27. Levine RL, Wadleigh M, Cools J, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–397. doi:10.1016/j.ccr.2005.03.023
28. James C, Ugo V, Le Couédic J-P, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–1148. doi:10.1038/nature03546
29. Rampal R, Al-Shahrour F, Abdel-Wahab O, et al. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood. 2014;123(22):e123–33. doi:10.1182/blood-2014-02-554634
30. Skorczewski T, Erickson LC, Fogelson AL. Platelet motion near a vessel wall or thrombus surface in two-dimensional whole blood simulations. Biophys J. 2013;104(8):1764–1772. doi:10.1016/j.bpj.2013.01.061
31. Walton BL, Lehmann M, Skorczewski T, et al. Elevated hematocrit enhances platelet accumulation following vascular injury. Blood. 2017;129(18):2537–2546. doi:10.1182/blood-2016-10-746479
32. Yap YY, Law KB, Sathar J, et al. The epidemiology and clinical characteristics of myeloproliferative neoplasms in Malaysia. Exp Hematol Oncol. 2018;7(1):31. doi:10.1186/s40164-018-0124-7
33. Vannucchi A, Guglielmelli P. JAK2 mutation-related disease and thrombosis. Semin Thromb Hemost. 2013;39(05):496–506. doi:10.1055/s-0033-1343890
34. Tefferi A, Barbui T. Polycythemia vera and essential thrombocythemia: 2021 update on diagnosis, risk‐stratification and management. Am J Hematol. 2020;95(12):1599–1613. doi:10.1002/ajh.26008
35. Carobbio A, Thiele J, Passamonti F, et al. Risk factors for arterial and venous thrombosis in WHO-defined essential thrombocythemia: an international study of 891 patients. Blood. 2011;117(22):5857–5859. doi:10.1182/blood-2011-02-339002
36. Sevindik OG, Mersin S, Katgi A, et al. IPSET-thrombosis better identifies thrombosis-free survival: a Turkish cohort. Clin Lymphoma Myeloma Leuk. 2015;15(6):e101–e104. doi:10.1016/j.clml.2015.02.004
37. Tefferi A, Pardanani A. Myeloproliferative neoplasms: a contemporary review. JAMA Oncol. 2015;1(1):97. doi:10.1001/jamaoncol.2015.89
38. Chu DK, Hillis CM, Leong DP, Anand SS, Siegal DM. Benefits and risks of antithrombotic therapy in essential thrombocythemia: a systematic review. Ann Intern Med. 2017;167(3):170. doi:10.7326/M17-0284
39. Alvarez-Larran A, Pereira A, Guglielmelli P, et al. Antiplatelet therapy versus observation in low-risk essential thrombocythemia with a CALR mutation. Haematologica. 2016;101(8):926–931. doi:10.3324/haematol.2016.146654
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Clinical Characteristics and Prognosis of Acute Myeloid Leukemia Patients with Protein Tyrosine Phosphatase Non-Receptor Type 11 Gene Mutation

Huang R, Zhang YT, Lin Y, Pang RL, Yang Z, Zhao WH
Pharmacogenomics and Personalized Medicine 2023, 16:1011-1026
Published Date: 11 November 2023
Adolescent Non-Puerperal Mastitis: Risk Factors, Clinical Characteristics, and Prognosis Analysis
Tang H, Wu X, Feng J, Gao Q, Shao S, Qu W, Xie L, Sun J
Journal of Inflammation Research 2024, 17:487-495
Published Date: 24 January 2024
Clinical Implications and Novel Insights into Adolescent Primary Liver Cancer: A Nightmare for Adolescents?
Guo H, Chen X, Li R, Shen J, Gan D, Yin Y, Zhang H, Xie J, Xie L, Liu Y
Journal of Hepatocellular Carcinoma 2025, 12:2513-2540
Published Date: 7 November 2025