Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 19
Biofilm–Host Immune Crosstalk at the Diabetic Foot Ulcer Interface: Molecular Mechanisms, Immune Evasion, and Next-Generation Anti-Biofilm Strategies
Received 13 March 2026
Accepted for publication 24 April 2026
Published 22 May 2026 Volume 2026:19 608789
DOI https://doi.org/10.2147/DMSO.S608789
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Rebecca Baqiyyah Conway
Yu Han,1 Ye Yang2
1Department of Vascular and Thyroid Surgery, The First Hospital of China Medical University, Shenyang, Liaoning Province, 110001, People’s Republic of China; 2Department of Rehabilitation, The First Hospital of China Medical University, Shenyang, Liaoning Province, 110001, People’s Republic of China
Correspondence: Ye Yang, Department of Rehabilitation, The First Hospital of China Medical University, No. 155 Nanjing North Street, Heping District, Shenyang, Liaoning Province, People’s Republic of China, Email [email protected]
Abstract: Diabetic foot ulcers (DFUs) are chronic wounds in which microbial persistence and defective host defense interact to impair healing. This review examines DFU through the biofilm–host immune interface rather than viewing biofilm as a purely microbiological problem. We summarize how the diabetic wound milieu, including hyperglycemia, impaired perfusion, neuropathy, and polymicrobial community structure, favors persistent biofilm infection, and how DFU-relevant biofilms evade clearance through matrix shielding, altered innate recognition, virulence-associated host modulation, and intracellular Staphylococcus aureus persistence. We further highlight two major immune-dysregulation axes: excessive neutrophil extracellular trap formation with NLRP3-centered inflammatory amplification, and perforin-2 suppression linked to AIM2-mediated pyroptotic injury. We also appraise emerging immune-aware antibiofilm strategies, particularly quorum-sensing interference, enzymatic matrix disruption, phage therapy, and selected immune-directed interventions. Overall, current evidence supports a model in which non-healing DFU reflects failed host–pathogen resolution at the biofilm–immune interface, with important implications for mechanism-guided therapeutic development.
Keywords: diabetic foot ulcer, biofilm, innate immunity, immune evasion, neutrophil extracellular traps, pyroptosis, quorum sensing, phage therapy, inflammasome
Introduction
Diabetes mellitus remains a major global public health challenge, and diabetic foot ulcer (DFU) is one of its most serious complications. Approximately 19% to 34% of individuals with diabetes will develop a foot ulcer during their lifetime, and these ulcers precede most diabetes-related lower-extremity amputations and are a major contributor to non-traumatic limb loss.1,2 People with a diabetic foot ulcer have an approximately 30% 5-year mortality, rising to more than 70% after major amputation. The direct costs of DFU care in the United States alone are estimated at $9–13 billion annually, underscoring the substantial economic burden associated with hospitalization, limb-threatening infection, and amputation-related care.1
Diabetic foot infections are commonly polymicrobial, with Staphylococcus aureus remaining a frequent pathogen, but community composition varies substantially with wound depth, chronicity, and host context.3,4 Importantly, wound microorganisms are often organized, at least in part, within biofilm-structured communities, a mode of growth biologically linked to persistence, impaired clearance, and delayed healing in chronic wounds.5,6
Clinically, microbial colonization, overt infection, and biofilm presence in DFU should not be regarded as interchangeable states.4,7 Chronic wounds often harbor complex microbial communities without fulfilling criteria for clinical infection, and detection of biofilm reflects an organized mode of microbial persistence rather than, by itself, definitive evidence of clinically relevant pathogenic activity.6,7 The relevance of the biofilm–host immune interface therefore lies in clarifying when persistent host–microbe interactions shift a wound toward non-resolving inflammation, tissue injury, and impaired healing.8
The impact of biofilms on DFU chronicity therefore extends far beyond simple antimicrobial resistance. Biofilms actively engage the host immune system through a sophisticated repertoire of immune evasion strategies, subverting rather than merely resisting host defenses. The extracellular polymeric substance (EPS) matrix physically restricts leukocyte penetration, while biofilm-associated virulence factors directly modulate immune cell function.8 Recent work has shown that intracellular S. aureus can accumulate within DFU keratinocytes due to suppression of the antimicrobial effector molecule perforin-2, triggering absent in melanoma 2 (AIM2) inflammasome-mediated pyroptosis and contributing to prolonged inflammation and impaired healing.9 In parallel, excessive neutrophil extracellular traps (NET) formation has emerged as a pathologic feature of diabetic wounds and DFUs, with experimental and translational studies linking dysregulated NET responses to impaired healing and persistent inflammation. Mechanistic work further suggests that NET-derived extracellular DNA and associated mediators can reinforce NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome activation, establishing a feed-forward inflammatory loop.10–12 However, the extent to which NET material is structurally incorporated into mature DFU biofilms in vivo remains incompletely defined.
Although the importance of biofilm in DFU has been increasingly recognized, most DFU-focused reviews have primarily emphasized microbial composition, biofilm formation, diagnosis, clinical implications, or antibiofilm management. Immune-centered discussion does exist, but is often brief within DFU reviews or is developed in broader chronic-wound reviews rather than in a DFU-specific framework. Accordingly, a DFU-focused synthesis that integrates biofilm immune evasion, dysregulated host responses, and immune-informed therapeutic strategies still appears limited.4,8 This review addresses that gap by asking four related questions: how the diabetic wound microenvironment promotes persistent biofilm infection; how DFU-relevant biofilms evade host clearance; which immune-dysregulation pathways most strongly sustain non-resolving inflammation and impaired healing; and how these mechanisms may inform next-generation antibiofilm and immune-aware therapeutic strategies. Framed in this way, the biofilm–host immune interface is clinically relevant not only for explaining chronic wound persistence, but also for clarifying why some DFUs fail to resolve despite standard care and where mechanism-guided intervention may be most effective.
The Diabetic Wound Microenvironment: A Permissive Niche for Biofilm Establishment
Hyperglycemia as a Driver of Bacterial Persistence and Virulence
Among the metabolic abnormalities of the diabetic wound microenvironment, hyperglycemia is more firmly supported as an amplifier of staphylococcal persistence and virulence than as a definitively proven direct inducer of biofilm formation in human DFU tissue. In diabetic skin-infection models, excess glucose enhances agr-dependent virulence output and worsens infection severity, while hyperglycemia has also been linked to increased aureolysin-associated protease activity. Taken together, these findings support a mechanistic framework in which the diabetic milieu not only weakens host defense but also augments pathogen programs relevant to persistence; however, direct confirmation that glucose alone drives clinically meaningful biofilm expansion in human DFU remains limited.13,14
Microbial Heterogeneity, Functional Pathogroups, and Polymicrobial Interactions
Culture-independent profiling shows that DFU microbiomes are heterogeneous rather than compositionally fixed. Although S. aureus is commonly detected, deeper, longer-duration, or non-healing wounds often show greater diversity, including increased representation of anaerobes and Proteobacteria.3,15 Longitudinal work further suggests that non-healing wounds may differ not only in composition but also in strain-level variation and biofilm- or virulence-associated functional signatures.15,16 Importantly, these observations support a functional rather than purely taxonomic view of DFU pathogenicity: wound behavior is shaped not simply by the presence of individual species, but by the collective properties of microbial communities.
Within this framework, polymicrobial interactions can further reshape persistence and treatment response, although they should not be assumed to be uniformly synergistic across all DFU communities. The best-supported example is S. aureus–P. aeruginosa coexistence, which in experimental wound-related systems can drive persistence phenotypes such as small-colony-variant-like states and increased tolerance to eradication.17,18 For the present review, the main importance of polymicrobiality is therefore not that every interspecies interaction is mechanistically central, but that community-level interactions can complicate clearance and help sustain immune-dysregulated biofilm states.
Vascular and Neuropathic Contributions to Biofilm Persistence
These microbial-community features operate within a host environment that already favors persistence. Beyond metabolic dysregulation, vascular insufficiency and neuropathy create a poorly perfused, repeatedly traumatized, and immunologically disadvantaged wound bed. Reduced oxygen delivery, impaired clearance, and recurrent tissue injury together increase the likelihood that biofilm persistence becomes chronic rather than transient.2,19 In this sense, DFU chronicity reflects not only microbial complexity, but also a wound microenvironment that repeatedly fails to eliminate and resolve biofilm-associated infection.
Biofilm-Mediated Immune Evasion: Molecular Mechanisms at the Host–Pathogen Interface
The capacity of biofilms to persist in DFU wounds despite the presence of a functional (albeit impaired) immune system reflects a sophisticated array of immune evasion mechanisms that operate at multiple levels. These mechanisms can be conceptualized as a multi-layered defense system, with each layer targeting distinct components of the host immune response.
The EPS Matrix as a Physical and Chemical Barrier
The EPS matrix constitutes the first barrier to effective immune access. By organizing bacteria within a hydrated three-dimensional scaffold, the matrix spatially restricts efficient phagocyte engagement and favors persistent, low-efficiency inflammatory confrontation rather than sterilizing clearance. The biological consequence is sustained leukocyte activation with collateral oxidative and proteolytic tissue injury, helping to stabilize a non-resolving wound environment.6,20
The physical barrier function of EPS is complemented by chemical properties that further diminish antimicrobial efficacy. The EPS matrix can retard antibiotic penetration in a drug- and matrix-dependent manner rather than uniformly blocking diffusion.21,22 For positively charged antibiotics such as aminoglycosides, extracellular DNA and other anionic matrix components can sequester the drug and thereby further delay effective delivery to biofilm-embedded cells.23 Moreover, oxygen and nutrient gradients generate metabolically heterogeneous biofilm subpopulations, including slow-growing or dormant cells, thereby reducing susceptibility to antibiotics that preferentially target active bacterial processes.22,24
Pathogen-Associated Molecular Pattern Masking and Pattern Recognition Receptor Evasion
Beyond physical restriction of immune cell access, biofilms employ a subtler strategy of immune evasion by masking pathogen-associated molecular patterns (PAMP) that would otherwise activate pattern recognition receptors (PRR) on host immune cells. Biofilm growth can attenuate innate immune recognition of canonical PRR ligands by both shielding surface-exposed microbial structures within the extracellular matrix and altering the expression of selected immunostimulatory determinants; in P. aeruginosa biofilms, for example, classical PAMPs are less exposed to the immune system and flagellar expression may be reduced.25 In chronic wounds, the biological consequence is not immune silence but persistent, low-grade inflammatory activation coupled to inefficient clearance. Accordingly, the biofilm–host interface is better understood as a state of dysregulated innate sensing in which ongoing microbial and host-derived danger signaling sustains non-resolving inflammation without generating effective microbicidal eradication.8
In parallel with PAMP masking, S. aureus biofilms also deploy humoral immune-evasion strategies that further reduce opsonophagocytic clearance, even though these mechanisms are conceptually distinct from direct PRR escape. By binding the Fc region of IgG and interfering with IgG hexamer formation, SpA reduces C1q recruitment, complement activation, and opsonophagocytic killing.26 SpA can also be released from the bacterial surface into the extracellular milieu.27 However, direct evidence specifically establishing this as a DFU biofilm-matrix mechanism remains limited. Accordingly, this pathway is better interpreted as humoral immune evasion than as direct PRR escape.26
Virulence Factors as Immune Modulators
In parallel with physical shielding and PAMP masking, biofilm-associated bacteria continue to express and secrete virulence factors that actively modulate host immune function. In S. aureus, these include alpha-toxin, a pore-forming cytotoxin capable of activating NLRP3 inflammasome signaling in experimental infection models;28 phenol-soluble modulins (PSM), a family of amphipathic peptides with neutrophil-lytic and biofilm-remodeling activities;29 and surface adhesins such as ClfA, ClfB, and SdrC, which mediate attachment to host proteins and participate in biofilm organization.30
A critical finding with direct relevance to DFU is that the hyperglycemic wound environment can amplify bacterial virulence programs rather than merely weaken host defense. Elevated glucose has been shown to increase S. aureus aureolysin expression and activity; because aureolysin sits upstream of the extracellular protease cascade that includes SspA/V8 protease, these data are consistent with enhanced protease-mediated tissue injury under hyperglycemic conditions.13 More broadly, diabetic skin-infection models show that excess glucose enhances agr-dependent virulence output and worsens infection severity.14 Together, these findings support a feed-forward pathobiological loop in which metabolic dysregulation simultaneously weakens host defense and amplifies the virulence program of wound-colonizing S. aureus.
P. aeruginosa adds further immune-disruptive pressure to polymicrobial DFU biofilms through secreted effectors, quorum-linked host-active signals, and redox-active metabolites that impair host-cell function.31,32 In the polymicrobial setting, these mechanisms likely broaden tissue-damaging and immune-modulatory pressure, although their integrated effect in human DFU remains insufficiently quantified.6
Perforin-2 Suppression and Intracellular Staphylococcus aureus Accumulation
A DFU-specific study by Pastar et al showed intracellular accumulation of S. aureus within the epidermis of clinically uninfected DFUs, together with marked suppression of perforin-2 (P-2/MPEG1), a pore-forming innate effector involved in intracellular antibacterial defense. Using qPCR, single-cell analyses, immunohistochemistry, and confocal microscopy, the study further showed substantially reduced P-2 expression in DFU tissue compared with location-matched nonulcerated plantar foot-skin controls.9
Functionally, P-2 suppression is consistent with impaired intracellular clearance of internalized S. aureus, thereby favoring persistence of an epidermal intracellular reservoir. Rather than being categorically inaccessible to therapy, intracellular S. aureus is better regarded as a state associated with reduced susceptibility to antibiotic killing and enhanced persistence, which may contribute to recurrence and delayed resolution.33 This interpretation is supported by P-2-deficient mice, in which epicutaneous S. aureus challenge was associated with impaired bacterial control and enhanced dissemination, consistent with a critical role for P-2 in cutaneous intracellular antibacterial defense.9
The AIM2 Inflammasome–Caspase-1–Gasdermin D Pyroptotic Cascade
The intracellular accumulation of S. aureus associated with P-2 suppression is linked to activation of AIM2 inflammasome signaling rather than remaining immunologically silent. AIM2 is a cytosolic dsDNA sensor that recruits ASC and activates caspase-1, leading to IL-1β maturation; downstream, caspase-1 cleaves gasdermin D (GSDMD), and the liberated N-terminal fragment oligomerizes in the plasma membrane to form pores that execute pyroptosis.34,35
GSDMD pore formation drives pyroptotic membrane permeabilization and release of intracellular contents, thereby amplifying local inflammatory injury. In a four-week longitudinal clinical study, Pastar et al showed that AIM2 levels, ASC pyroptosome assembly, and cleaved GSDMD persisted or increased in nonhealing DFUs, whereas these pyroptosis-related readouts decreased during healing.9 These data support pyroptosis as a DFU-relevant mechanism linking intracellular S. aureus persistence to unresolved inflammation and impaired healing. Released intracellular contents from pyroptotic cells may further amplify local inflammatory signaling; however, a fully autonomous feed-forward loop independent of ongoing biofilm infection has not been directly demonstrated in human DFU tissue.
Collectively, these findings identify the P-2–AIM2–GSDMD axis as a DFU-relevant pathway linking intracellular S. aureus persistence with unresolved inflammation and impaired healing, while also suggesting—at a hypothesis-generating level—that restoration of P-2 activity or modulation of pyroptotic signaling may merit future therapeutic investigation.
Taken together, these immune-evasion strategies do not merely permit microbial persistence; they also reshape the host response into a chronically activated yet poorly resolving inflammatory state (Figure 1).
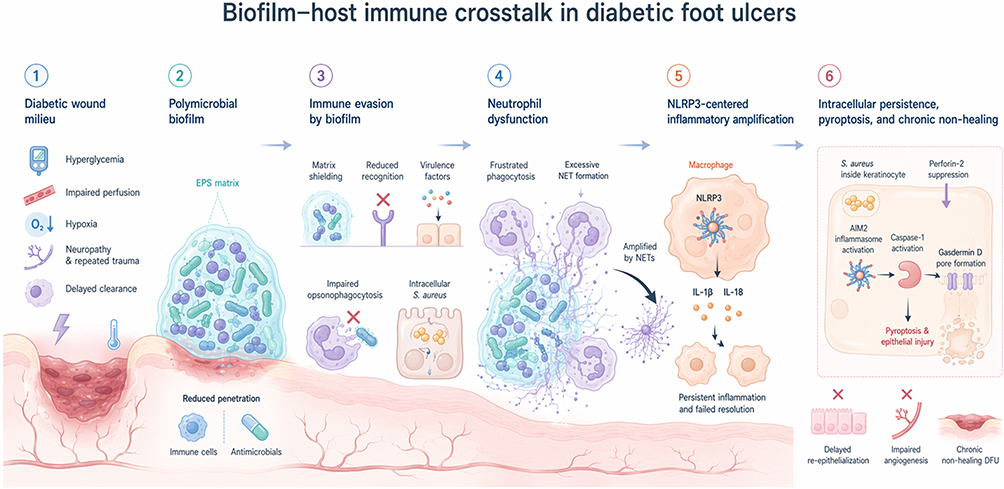 |
Figure 1 Biofilm–host immune crosstalk in diabetic foot ulcers. The diabetic wound milieu, characterized by hyperglycemia, impaired perfusion, hypoxia, neuropathy, repeated trauma, and delayed clearance, favors establishment of polymicrobial biofilms. Biofilm structure and virulence programs promote matrix shielding, reduced innate recognition, impaired opsonophagocytosis, neutrophil dysfunction, and excessive neutrophil extracellular trap (NET) formation. NET-associated signals amplify macrophage NLRP3 activation and IL-1β/IL-18-centered inflammatory responses. In parallel, intracellular Staphylococcus aureus persistence in keratinocytes, together with perforin-2 suppression, is linked to AIM2 inflammasome activation, caspase-1 signaling, gasdermin D pore formation, pyroptotic injury, and chronic non-healing. Overall, biofilm persistence and dysregulated host responses reinforce one another to sustain delayed re-epithelialization, impaired angiogenesis, and chronic diabetic foot ulceration. |
Host Innate Immune Responses to Biofilm and Their Dysregulation in DFU
Neutrophils: From First Responders to Inadvertent Accomplices
Neutrophils are among the earliest immune cells recruited to the wound bed and are central to early antimicrobial defense. In biofilm-associated DFU, however, neutrophil responses become functionally dysregulated and can contribute to persistent inflammation, tissue injury, and wound chronicity.8
The systemic effects of diabetes on neutrophil function are well documented. Overall, the balance of evidence supports reduced neutrophil chemotactic responsiveness and impaired phagocytic handling in diabetes, although these defects are not uniform across all assays and stimuli. In parallel, intracellular reactive oxygen species (ROS) generation required for efficient microbicidal killing is frequently diminished, whereas extracellular ROS release and other pro-inflammatory neutrophil outputs may be preserved or increased.36 Within diabetic wounds, these abnormalities are superimposed on a hypoxic and persistently inflammatory microenvironment that further hinders effective bacterial clearance.8
When neutrophils encounter biofilm aggregates, effective phagocytic clearance is limited, and sustained extracellular release of oxidants and proteases contributes more to tissue injury than to bacterial eradication.
Neutrophil Extracellular Traps: Defense Mechanism Turned Pathological Amplifier
Among the dysregulated neutrophil responses implicated at the DFU biofilm–host interface, one of the most consequential is the formation of NETs. NETs are extracellular chromatin structures decorated with antimicrobial proteins—including neutrophil elastase, myeloperoxidase, and cathelicidin (LL-37)—released by activated neutrophils. Although often discussed in the context of NETosis, NET release should not be framed as being restricted to a single cell-death program.37 NETs can immobilize microbes, but their effects are highly context-dependent; in experimental Staphylococcus aureus biofilms, biofilm growth can actively skew neutrophils toward NET formation while remaining poorly cleared by NET-mediated antimicrobial activity.38
Experimental evidence supports a pathological contribution of excessive NET formation to impaired diabetic wound repair. Wong et al showed that diabetes primes neutrophils for PAD4-dependent NET formation and that genetic or enzymatic disruption of NETs accelerates healing in diabetic wound models.10 Extending these observations to human DFU, Yang et al reported that circulating NET markers were increased in patients with DFU relative to diabetic patients without DFU and healthy controls, correlated positively with the Diabetic Ulcer Severity Score (DUSS) and Wound, Ischemia, and foot Infection (WIfI) severity indices, and identified citrullinated histone H3 (citH3) as a marker associated with impaired 12-month healing and higher amputation risk; patients in the highest citH3 quartile had significantly worse healing outcomes than those in the lower three quartiles.39 A complementary translational study by Sawaya et al further identified the FOXM1–TREM1 network as a regulator of ROS-dependent NET formation in DFU and showed that triggering receptor expressed on myeloid cells 1 (TREM1) expression correlated with clinical healing outcomes, reinforcing the clinical relevance of dysregulated neutrophil/NET biology in non-healing ulcers.11 Collectively, current human data support an association between increased NET burden and adverse DFU outcomes, but the available clinical evidence remains limited and should not be overinterpreted as establishing causality.
Mechanistic studies further suggest that NETs can impair repair programs beyond microbial trapping. In experimental diabetic wound systems, NET exposure has been linked to endothelial dysfunction, impaired angiogenic responses, and delayed closure, providing one plausible route by which excessive NETosis may worsen healing.40
The NLRP3 Inflammasome–NET Feed-Forward Loop
A mechanistically important aspect of NET biology in diabetic wounds is the amplification loop between excessive NET formation and macrophage NLRP3 inflammasome activation. This loop appears to be superimposed on a broader diabetic wound environment in which macrophage inflammasome activity is already persistently elevated.41 Liu et al showed that NETs generated in diabetic wounds can provide both a priming signal, through TLR-4/TLR-9/NF-κB-dependent upregulation of NLRP3 and pro-IL-1β, and an activation signal, through ROS-dependent promotion of TXNIP–NLRP3 association in macrophages.12 This mechanism was associated with enhanced IL-1β release and sustained inflammatory injury; however, its causal weight in biofilm-positive human DFUs has not yet been directly quantified.
Huang et al further used MFG-E8 as an experimental modulator of this axis. In Mfge8-deficient diabetic mice, wounds showed enhanced NET deposition, increased NLRP3 inflammasome activity with higher IL-1β/IL-18 output, impaired angiogenesis, and delayed closure.42 Recombinant MFG-E8 suppressed IL-18-primed NETosis and dampened NET/mCRAMP-induced inflammasome activation in macrophages; the authors further linked this effect to an integrin β3-restricted P2X7 signaling mechanism.42 These data support MFG-E8 as an endogenous negative regulator of the NLRP3–NET loop in experimental diabetic wound healing, rather than establishing it as a validated therapeutic target in human DFU.
In the same experimental line, DNase I-mediated NET digestion reduced NLRP3 inflammasome activation, modulated inflammatory-cell infiltration, and accelerated healing in a diabetic rat wound model. This finding supports proof-of-concept interruption of the NET–inflammasome axis, but remains preclinical.12 Taken together, current evidence supports the NLRP3–NET axis as a plausible inflammatory amplification loop in diabetic wounds and DFU-related experimental systems. Its quantitative contribution specifically in biofilm-positive human DFUs, however, remains to be defined by direct clinical studies.
At the biofilm interface, NETs also behave paradoxically: they may spatially constrain microbial spread yet still fail to eradicate established biofilm communities. In experimental systems, this interface is associated more with persistent inflammatory injury than with sterilizing clearance, whereas evidence that NET-derived DNA is routinely incorporated into mature DFU biofilms in vivo remains limited.38
Macrophage Polarization Failure and Biofilm Persistence
Macrophages play a pivotal role in coordinating the transition from inflammation to tissue repair during wound healing; operationally, this is often summarized as a shift from an early inflammatory M1-like program toward pro-resolving and pro-reparative M2-like states. In diabetic wounds, this transition is impaired, contributing to persistent inflammation and defective repair.43,44
In the setting of biofilm-associated infection, persistent microbial stimulation is more defensibly viewed as reinforcing a non-resolving inflammatory macrophage program and delaying timely transition toward reparative states; however, current high-quality evidence in DFU does not justify specifying NF-κB/STAT1 dominance or EPS-mediated loss of scavenger- or mannose-receptor engagement as established mechanisms of macrophage polarization failure.8 The resulting macrophage compartment is better described as persistently inflammatory and poorly pro-resolving, with increased expression of cytokines such as IL-1β, TNF-α, and IL-6 and reduced expression of healing-associated mediators including IL-10, IGF-1, and TGF-β. This non-resolving phenotype is consistent with sustained macrophage inflammasome activity and IL-1β-centered inflammatory circuitry documented in diabetic human and murine wounds.41,44
Furthermore, persistent NLRP3 inflammasome activity in wound macrophages represents an additional mechanism that may reinforce non-resolving inflammation in diabetic wounds. Available evidence supports associations with sustained IL-1β-centered inflammatory signaling, impaired healing, and experimental amplification by NET-derived stimuli; however, whether NLRP3 operates in biofilm-positive DFU as a discrete checkpoint governing the transition from inflammatory to reparative macrophage states remains unresolved.12,41
Quorum Sensing as a Regulatory Bridge at the Biofilm–Immune Interface
Quorum sensing (QS) is best understood here as a regulatory layer rather than a stand-alone endpoint. In DFU-relevant pathogens, QS coordinates biofilm maturation, dispersal, and virulence deployment; in P. aeruginosa this network is classically organized around las, rhl, and PQS signaling, whereas in S. aureus the agr system regulates the shift from surface-associated colonization toward secreted toxins, proteases, and phenol-soluble modulins.45,46
The key relevance of QS at the DFU biofilm–immune interface is that it links microbial coordination to host-active outputs. Experimental studies indicate that S. aureus agr signaling can suppress keratinocyte repair-associated programs and delay wound healing, while selected P. aeruginosa QS molecules can engage mammalian sensing pathways and reshape inflammatory or barrier responses. Direct DFU clinical validation, however, remains limited. Accordingly, QS should be viewed as a biologically plausible bridge between biofilm behavior and host dysfunction, whereas its therapeutic exploitation is considered separately below.
Next-Generation Anti-Biofilm Strategies: From Immune Evasion Disruption to Immune Restoration
The growing recognition that biofilm-mediated immune evasion, together with biofilm-associated antimicrobial tolerance and broader host–microbe dysregulation, is a major driver of DFU chronicity supports a reorientation of therapeutic strategy beyond conventional bactericidal approaches alone. Accordingly, next-generation approaches increasingly target the biofilm–immune interface, aiming either to weaken biofilm-associated immune evasion and structural protection or to restore effective host clearance and wound repair.6 The principal pathological axes discussed in this review and the major mechanism-guided therapeutic strategies are summarized in Figure 2.
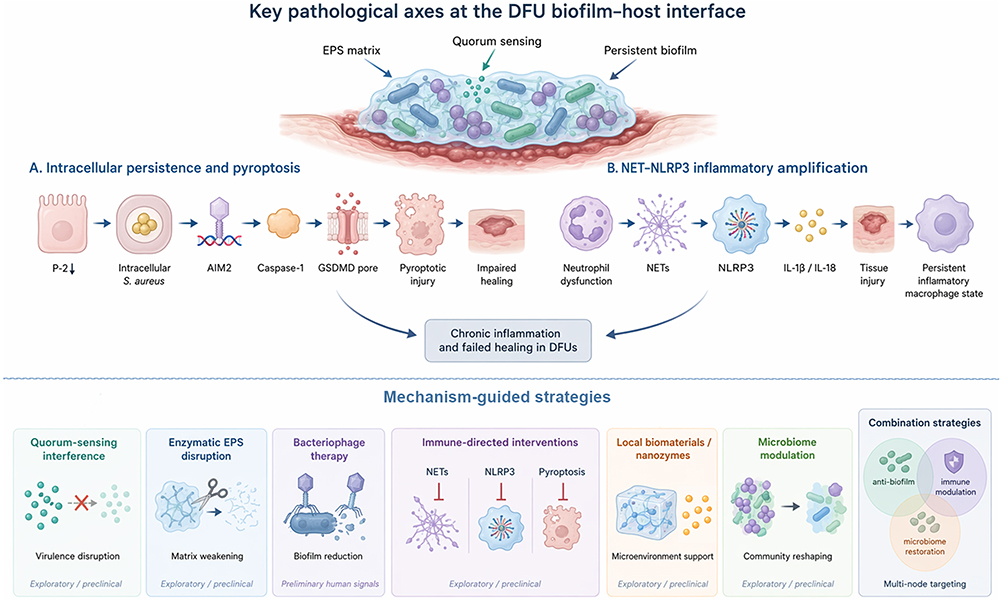 |
Figure 2 Key pathological axes at the DFU biofilm–host interface and mechanism-guided strategies. The upper panel summarizes two major innate immune pathobiological axes in diabetic foot ulcers: intracellular Staphylococcus aureus persistence with AIM2–caspase-1–gasdermin D-mediated pyroptosis, and NET-associated NLRP3 inflammatory amplification that sustains tissue injury and a persistent inflammatory macrophage state, converging on chronic inflammation and failed healing. The lower panel maps emerging intervention concepts to these mechanisms, including quorum-sensing interference, enzymatic EPS disruption, bacteriophage therapy, immune-directed interventions, local biomaterials/nanozymes, microbiome modulation, and multi-node combination strategies. Most approaches remain exploratory or preclinical, whereas bacteriophage therapy currently provides the most tangible preliminary human signal. |
Quorum Sensing Inhibitors
Quorum sensing inhibitors (QSI) represent a conceptually attractive anti-virulence strategy because they disrupt communication networks that regulate virulence expression and biofilm-associated phenotypes without necessarily relying on direct bactericidal activity. Mechanistically, QS interference can be achieved through inhibition of signal synthesis, enzymatic degradation of signal molecules, antagonism of signal receptors, or interruption of downstream quorum-responsive transcriptional programs.47 By attenuating QS-regulated phenotypes, these agents may enhance biofilm vulnerability to host clearance48 or to concomitant antibiotics in selected experimental systems,49 but such effects remain context-dependent and cannot yet be generalized across pathogens or infection settings.
Selected QS-disruption strategies have shown preclinical efficacy in wound-related infection models, but the evidence remains heterogeneous and largely non-DFU. At present, the strongest rationale for QS interference in DFU is mechanistic rather than clinical: quorum-sensing systems coordinate virulence deployment, biofilm behavior, and selected host-active outputs, making them plausible adjunctive targets rather than established therapies.31,45–47,50 Taken together, current data support QS disruption as a biologically plausible strategy for weakening persistence, but not yet as a clinically validated DFU therapy.
Despite these encouraging preclinical findings, translation of QSIs to DFU remains challenging. Because DFU biofilms are polymicrobial and may involve heterogeneous quorum-signaling architectures—single-pathway inhibition may provide incomplete coverage unless paired with species-matched or combination strategies.45–47
Enzymatic EPS Disruption Strategies
Direct enzymatic disruption of the EPS matrix remains one of the most mechanistically coherent strategies for converting an immune-excluded biofilm into an exposed, therapeutically accessible structure. DNase I targets extracellular DNA (eDNA), a widespread biofilm-matrix component whose degradation can destabilize biofilms and increase antimicrobial susceptibility.24,51 Dispersin B hydrolyzes PNAG/PIA and is therefore best framed as a composition-matched strategy for PNAG/PIA-rich staphylococcal biofilms rather than a universal enzyme for all staphylococcal matrices.52 Rather than functioning primarily as stand-alone bactericidal agents, matrix-degrading enzymes are better viewed as adjunctive biofilm-disruption tools. Current evidence most strongly supports their use in combination regimens, in which matrix weakening is paired with antibiotics or antisepsis to increase access to previously shielded bacteria, whereas direct restoration of host immune clearance remains less firmly established.24,53,54
Bacteriophage Therapy
Bacteriophage (phage) therapy is an investigational antibiofilm strategy with strong biological plausibility and a small but growing DFU-specific human evidence base; however, the available clinical data remain limited in scale and are insufficient to establish efficacy.55,56 Phages are bacterial viruses that selectively infect susceptible bacterial hosts. Their proposed relevance to biofilm infection lies in self-amplification where target bacteria are present, the capacity of some phages or phage-derived enzymes to disrupt matrix-associated barriers, and antibacterial activity that is mechanistically distinct from conventional antibiotics; however, activity remains isolate-specific and depends on phage susceptibility testing rather than being uniformly preserved across all antibiotic-resistant strains.56,57
Across preclinical studies and early translational reports, phage therapy in DFU has mainly focused on pathogen-matched treatment and phage–antibiotic combination strategies designed to broaden coverage and reduce resistance escape; however, superiority over optimized single-phage or standard-care regimens has not been established in DFU patients.55–57
Among peer-reviewed human datasets in DFU, the randomized double-blind TP-102 study is currently the most informative: 19 participants with infected or non-infected DFUs were randomized after susceptibility testing, 13 received TP-102, no treatment-related adverse events were reported, and microbiological reduction and wound-closure trends numerically favored TP-102; however, the study was explicitly underpowered to establish superiority.55 Accordingly, phage therapy in DFU is best regarded at present as a promising investigational adjunct rather than an established treatment modality, and further progress will require larger efficacy-defining trials together with standardized susceptibility testing, manufacturing, and delivery workflows.58
From an immunological perspective, phage-mediated reduction of bacterial burden and biofilm biomass could, in principle, lessen biofilm-driven inflammatory stimulation and thereby improve conditions for host clearance; however, this remains inferential rather than directly demonstrated in DFU patients.56 Current human DFU studies have assessed safety, microbiology, and healing outcomes rather than directly demonstrating reversal of NET- or inflammasome-centered inflammatory circuits; therefore, immune re-engagement should be framed as a mechanistically plausible but unproven translational hypothesis.55,56
Exploratory Local Adjunctive Platforms
Nanozyme-based platforms, peptide-enabled local biomaterials, and other natural-product-derived local approaches remain exploratory adjunctive strategies at the biofilm–immune interface.59 Across preclinical diabetic wound models, these systems have shown combinations of antibacterial, antibiofilm, ROS-modulating, and wound-repair-promoting effects; however, DFU-specific human evidence remains limited, and none can yet be regarded as an established therapy.60,61 Their current significance is therefore conceptual rather than practice-defining: they suggest that future local treatment platforms may need to combine bacterial control with microenvironmental or immune modulation. At present, these approaches are better viewed as future-oriented components of combination therapy than as standalone evidence-based DFU treatments.
Immune-Directed Interventions: Targeting NETosis and Pyroptosis
Given the increasingly recognized pathological roles of excessive NET formation and pyroptosis in diabetic wounds and DFU-related experimental systems, direct targeting of these processes represents a mechanistically rational therapeutic strategy. Several agents have shown preclinical proof-of-concept activity, although human efficacy data remain limited.
Disulfiram, a drug originally developed for alcohol use disorder, has been repurposed as a GSDMD-targeting agent; in DFU-related experimental systems, it suppressed NET formation and accelerated wound healing by inhibiting the NLRP3/caspase-1/GSDMD axis.62 In parallel, PAD4 inhibition—either pharmacologic or genetic—has reduced NET burden and improved diabetic wound healing in preclinical models.10,63
Recombinant MFG-E8, as discussed earlier, functioned as an experimental negative regulator of the NLRP3–NET inflammatory loop and promoted angiogenesis and wound closure in diabetic mice. In that study, it suppressed IL-18-primed NETosis and attenuated NET/mCRAMP-induced inflammasome activation in macrophages.42
DNase I represents a mechanistically straightforward NET-directed intervention. By digesting extracellular NET material, DNase I attenuated NET-driven NLRP3 inflammasome activation, modulated inflammatory-cell infiltration, and accelerated healing in diabetic wound models.12 Its therapeutic rationale lies in interrupting the NET–inflammasome amplification loop; notably, this rationale does not depend on assuming that NET-derived DNA is routinely incorporated into mature DFU biofilms in vivo, a point that remains unresolved.
Microbiome Modulation and Probiotic Approaches
Microbiome-directed intervention remains an exploratory but mechanistically appealing direction in DFU. Its rationale arises from growing recognition that wound behavior is shaped by community-level microbial functions rather than by single taxa alone, suggesting that future interventions may seek to reshape local ecology rather than merely suppress individual pathogens.6,15 At present, however, therapeutic evidence for probiotics or related microbiome-modulating strategies in DFU remains preclinical, heterogeneous, and insufficient to support efficacy claims in patients.
Methodological Advances in Studying the Biofilm–Immune Interface
Three-Dimensional Host–Biofilm Models
The study of biofilm–immune interactions has historically been limited by the reductionism of conventional in vitro models, which fail to recapitulate the three-dimensional architecture, cellular heterogeneity, and microenvironmental complexity of DFU wounds. Recent advances in three-dimensional tissue engineering have begun to address this limitation. Three-dimensional wound models are valuable not simply because they are more complex than conventional culture systems, but because they create an experimental bridge between microbial architecture and host cell state. Their greatest future value will likely lie in combining biofilm challenge with single-cell and spatial readouts, enabling direct mapping of how specific microbial structures correspond to epithelial stress states and, in future immune-competent versions, to macrophage polarization states and zones of failed repair.
Current three-dimensional epidermal biofilm platforms are best understood as tissue-engineered host–biofilm models: within a stratified human epidermal architecture, they can incorporate wounded-skin context and defined biofilm challenge, thereby enabling host–microbe interactions that are not accessible in conventional monolayer culture. More importantly, a human epidermis organoid model has been shown to support structured methicillin-resistant Staphylococcus aureus (MRSA) and P. aeruginosa biofilms while retaining measurable epithelial host readouts, including cytotoxicity and pro-inflammatory cytokine readouts in wounded-skin settings.64 In an MRSA-biofilm setting, transcriptomic analysis further showed impaired barrier function, extracellular-matrix remodelling, and activation of IL-17-, IL-12-family, and IL-6-family-associated inflammatory programs, making this platform experimentally tractable for linking biofilm architecture to epithelial injury and response to antibiofilm candidates.65 The next methodological step will be to extend current epidermal biofilm models toward immune-cell incorporation, spatially resolved host-response profiling, and patient-derived materials, rather than treating them simply as higher-complexity static culture systems.
Shotgun Metagenomics, Metatranscriptomics, and Spatially Resolved Profiling
While 16S rRNA amplicon sequencing has underpinned much of DFU microbiome characterization, it is primarily suited to inference of community composition and does not directly resolve strain-level gene content or transcriptional activity.7 By contrast, shotgun metagenomic sequencing extends DFU analysis from community composition to species- or strain-resolved functional potential, enabling direct profiling of antibiotic-resistance determinants together with biofilm- and virulence-associated gene repertoires from wound specimens.16 When further integrated with metatranscriptomic or dual-RNA-seq data, this framework moves beyond gene presence toward gene activity, helping prioritize transcriptionally active microbial populations and host–microbe response programs over taxa detected only at the DNA level. In DFU, however, such approaches are better viewed as emerging tools for biological stratification than as definitive clinical discriminators between colonization and pathogenicity.66
Spatially resolved platforms such as spatial transcriptomics and imaging mass cytometry are especially promising for dissecting the biofilm–immune interface. Spatial transcriptomics localizes wound-region-specific transcriptional states, whereas imaging mass cytometry resolves multiplex immune and stromal phenotypes together with their cellular neighborhoods at single-cell resolution. In DFU, spatial transcriptomics has already begun to identify region-specific fibroblast and macrophage states, but published work has thus far emphasized host tissue architecture rather than direct spatial co-mapping of microbial biofilm organization.67 Experience from other inflammatory skin disorders further suggests that these technologies can uncover disease-relevant cellular neighborhoods and signaling circuits that remain obscured in dissociative sequencing datasets.68
Challenges, Knowledge Gaps, and Future Directions
The Translational Gap: From in vitro Models to Clinical Complexity
Despite the considerable advances reviewed herein, a substantial translational gap persists between mechanistic insights obtained in vitro or in animal models and their application to clinical DFU management. In vitro biofilm models, even sophisticated three-dimensional systems, cannot fully recapitulate the polymicrobial complexity, oxygen gradients, immune cell dynamics, and chronic metabolic derangements of the human DFU wound.69 Commonly used diabetic wound animal models reproduce impaired healing and selected metabolic abnormalities, but no current preclinical model fully captures the combined neuropathic, vascular, infectious, immunological, and chronic features of human DFU.70 These limitations constrain the predictive validity of preclinical findings and underscore the need for clinical validation of mechanistic hypotheses. Recent DFU-focused literature further suggests that this translational challenge also includes the practical identification and management of wound biofilm in routine care.71 A recent review highlighted persistent limitations in clinically identifying biofilm in diabetic foot infections, whereas subsequent randomized clinical work has begun to evaluate biofilm-detection-based wound management in DFU.72 Together, these developments reinforce the need to connect mechanistic insight with clinically feasible diagnostic and therapeutic frameworks.
The Challenge of Polymicrobial Complexity
The polymicrobial nature of DFU biofilms represents a formidable challenge for targeted therapeutic strategies. Much of the mechanistic literature and preclinical therapeutic testing still relies on simplified systems centered on dominant pathogens such as S. aureus or P. aeruginosa, whereas clinical DFU wounds harbor more heterogeneous microbial communities and interspecies interactions.16 Cooperative and competitive interactions within polymicrobial biofilms can reshape collective phenotypes—including virulence, persistence, and treatment tolerance—such that findings from monospecies systems should not be assumed to translate directly to polymicrobial DFU biofilms.4 Future research must prioritize the development and validation of therapeutics in polymicrobial biofilm models that more faithfully represent clinical DFU microbiology.
Longitudinal, Stratified, and Clinically Actionable Human Studies
The current human evidence base remains largely cross-sectional, whereas the biofilm–immune interface is dynamic and patient-specific. Future progress depends on longitudinal serial-sampling studies that integrate microbial composition and function with host inflammatory phenotypes, NET- and inflammasome-related readouts, treatment exposure, and standardized healing outcomes within the same patient.7,16,66 This approach is needed not only to support biologically informed stratification, but also to identify clinically actionable windows for intervention and to determine which patients may plausibly benefit from precision antibiofilm strategies. Fungi, the wound virome, and adaptive immune pathways—including T-cell- and antibody-mediated responses—remain important but less mature lines of investigation, and their contribution to the broader biofilm–immune landscape in DFU requires more direct clarification.
Priority Research Directions
Based on the synthesis presented in this review, several research directions merit priority attention. First, the P-2 suppression–intracellular Staphylococcus aureus–AIM2–GSDMD axis identified by Pastar et al represents a high-priority target for mechanistic validation and therapeutic exploration. However, specific approaches to restore P-2 expression or activity in DFU keratinocytes remain unvalidated, and any therapeutic benefit should currently be regarded as hypothesis-generating rather than established.9 Second, because biofilm persistence is multifactorial, multi-targeted combination strategies that simultaneously weaken matrix protection, reduce bacterial burden, and temper maladaptive inflammation are biologically plausible and warrant systematic evaluation in polymicrobial and immune-relevant preclinical models. Third, the field needs validated biomarker panels that capture both microbial organization and host inflammatory state. Candidate platforms may include integrated host–microbe transcriptomic signatures and microbiologically informed molecular profiling, but no biomarker framework is yet established for DFU biofilm stratification. Fourth, future clinical trials of phage therapy and other biofilm-directed interventions in DFU should clearly distinguish early safety/tolerability studies from efficacy-defining trials and should incorporate standardized microbiologic or biofilm-centered endpoints; inclusion of mechanistically informative immune-related secondary outcomes would further strengthen trial interpretation. Recent systematic synthesis of the field likewise indicates that evidence on DFU biofilm prevalence, diagnostics, and treatment remains fragmented and that validated biofilm endpoints are still needed.73 Fifth, applying single-cell RNA sequencing and spatial transcriptomics to microbiologically annotated, ideally biofilm-positive, DFU tissue could move the field beyond descriptive microbiology by defining spatially resolved host response states. This would support a more precise framework for classifying DFUs according to dominant inflammatory circuitry, reparative failure, and microbial organization. However, direct host–microbe spatial co-mapping in DFU remains limited.
Conclusion
Non-healing DFU is best understood as a state of failed host–pathogen resolution in which biofilm persistence and innate immune dysregulation reinforce one another. Rather than acting solely as a structural barrier to antibiotics, biofilm helps sustain a chronically inflammatory, poorly resolving wound environment through matrix-mediated immune exclusion, intracellular Staphylococcus aureus persistence linked to inflammasome activation, excessive NET/NLRP3-driven inflammatory amplification, and delayed transition toward reparative macrophage states. These interlocking processes help explain why microbial persistence and tissue injury can coexist over prolonged periods instead of progressing toward effective clearance and healing. Therapeutically, this framework supports a shift beyond purely bactericidal logic toward combination strategies that weaken biofilm protection while restoring effective immune resolution. At present, phage therapy provides one of the most tangible preliminary human signals, whereas most other approaches—including quorum-sensing interference, enzymatic matrix disruption, and immune-directed interventions—remain preclinical or hypothesis-generating. Future progress will depend on longitudinal human studies, polymicrobial and immune-relevant models, and biomarker-guided translational strategies that can more precisely link microbial organization with host inflammatory state.
Highlights
● Non-healing DFU reflects failed biofilm-host immune resolution.
● Direct DFU evidence centers on perforin-2 suppression and intracellular Staphylococcus aureus persistence.
● Excessive NET formation and inflammasome signaling amplify tissue injury.
● Phage therapy provides one of the most tangible preliminary human signals, whereas most other approaches remain preclinical.
Declaration of Generative AI Use
Authors declare no AI use during the preparation of this work.
Abbreviations
AHL, acyl-homoserine lactone; AIM2, absent in melanoma 2; AIP, autoinducing peptide; DUSS, Diabetic Ulcer Severity Score; DFU, diabetic foot ulcer; eDNA, extracellular DNA; EPS, extracellular polymeric substances; GSDMD, gasdermin D; MFG-E8, milk fat globule-EGF factor 8; MRSA, methicillin-resistant Staphylococcus aureus; NETs, neutrophil extracellular traps; NLRP3, NOD-like receptor family pyrin domain containing 3; P-2, perforin-2; PAD4, peptidylarginine deiminase 4; PAMPs, pathogen-associated molecular patterns; PIA, polysaccharide intercellular adhesin; PNAG, poly-N-acetylglucosamine; PRRs, pattern recognition receptors; PQS, Pseudomonas quinolone signal; QS, quorum sensing; QSIs, quorum sensing inhibitors; ROS, reactive oxygen species; TREM1, triggering receptor expressed on myeloid cells 1; TXNIP, thioredoxin-interacting protein; WIfI, Wound, Ischemia, and foot Infection.
Data Sharing Statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.
Author Contributions
Yu Han: Conceptualization; Formal analysis; Investigation; Writing – original draft.
Ye Yang: Conceptualization; Supervision; Validation; Writing – review and editing.
All authors gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
No funding was received for this study.
Disclosure
The authors declare no competing interests.
References
1. Armstrong DG, Tan T-W, Boulton AJM, Bus SA. Diabetic Foot Ulcers: a Review. JAMA. 2023;330(1):62–15. doi:10.1001/jama.2023.10578
2. Armstrong DG, Boulton AJM, Bus SA. Diabetic Foot Ulcers and Their Recurrence. N Engl J Med. 2017;376:2367–2375. doi:10.1056/NEJMra1615439
3. Gardner SE, Hillis SL, Heilmann K, Segre JA, Grice EA. The neuropathic diabetic foot ulcer microbiome is associated with clinical factors. Diabetes. 2013;62:923–930. doi:10.2337/db12-0771
4. Buch PJ, Chai Y, Goluch ED. Treating Polymicrobial Infections in Chronic Diabetic Wounds. Clin Microbiol Rev. 2019;32:10–128. doi:10.1128/cmr.00091-18
5. Flemming HC, Wingender J. The biofilm matrix. Nat Rev Microbiol. 2010;8:623–633. doi:10.1038/nrmicro2415
6. Uberoi A, McCready-Vangi A, Grice EA. The wound microbiota: microbial mechanisms of impaired wound healing and infection. Nat Rev Microbiol. 2024;22:507–521. doi:10.1038/s41579-024-01035-z
7. Liu C, Ponsero AJ, Armstrong DG, Lipsky BA, Hurwitz BL. The dynamic wound microbiome. BMC Med. 2020;18:358. doi:10.1186/s12916-020-01820-6
8. Versey Z, da Cruz Nizer WS, Russell E, et al. Biofilm-Innate Immune Interface: contribution to Chronic Wound Formation. Front Immunol. 2021;12:648554. doi:10.3389/fimmu.2021.648554
9. Pastar I, Sawaya AP, Marjanovic J, et al. Intracellular Staphylococcus aureus triggers pyroptosis and contributes to inhibition of healing due to perforin-2 suppression. J Clin Invest. 2021;131:1. doi:10.1172/jci133727
10. Wong SL, Demers M, Martinod K, et al. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med. 2015;21(7):815–819. doi:10.1038/nm.3887
11. Sawaya AP, Stone RC, Mehdizadeh S, et al. FOXM1 network in association with TREM1 suppression regulates NET formation in diabetic foot ulcers. EMBO Rep. 2022;23(8):e54558. doi:10.15252/embr.202154558
12. Liu D, Yang P, Gao M, et al. NLRP3 activation induced by neutrophil extracellular traps sustains inflammatory response in the diabetic wound. Clin Sci. 2019;133:565–582. doi:10.1042/cs20180600
13. Wei R, Wang X, Wang Q, Qiang G, Zhang L, Hu HY. Hyperglycemia in Diabetic Skin Infections Promotes Staphylococcus aureus Virulence Factor Aureolysin: visualization by Molecular Imaging. ACS Sens. 2022;7:3416–3421. doi:10.1021/acssensors.2c01565
14. Thurlow LR, Stephens AC, Hurley KE, Richardson AR. Lack of nutritional immunity in diabetic skin infections promotes Staphylococcus aureus virulence. Sci Adv. 2020;6:5569. doi:10.1126/sciadv.abc5569.
15. Sachdeva C, Prasad SS, Shenoy KR, et al. A longitudinal profiling of microbiome of diabetic foot ulcers shows functional role of microbial communities in wound worsening and chronicity. Curr Res Microb Sci. 2026;10:100544. doi:10.1016/j.crmicr.2025.100544
16. Kalan LR, Meisel JS, Loesche MA, et al. Strain- and Species-Level Variation in the Microbiome of Diabetic Wounds Is Associated with Clinical Outcomes and Therapeutic Efficacy. Cell Host Microbe. 2019;25:641–655.e645. doi:10.1016/j.chom.2019.03.006
17. Hoffman LR, Déziel E, D’Argenio DA, et al. Selection for Staphylococcus aureus small-colony variants due to growth in the presence of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 2006;103:19890–19895. doi:10.1073/pnas.0606756104
18. DeLeon S, Clinton A, Fowler H, Everett J, Horswill AR, Rumbaugh KP. Synergistic interactions of Pseudomonas aeruginosa and Staphylococcus aureus in an in vitro wound model. Infect Immun. 2014;82:4718–4728. doi:10.1128/iai.02198-14
19. Prompers L, Schaper N, Apelqvist J, et al. Prediction of outcome in individuals with diabetic foot ulcers: focus on the differences between individuals with and without peripheral arterial disease. The EURODIALE Study. Diabetologia. 2008;51:747–755. doi:10.1007/s00125-008-0940-0
20. Flemming HC, Wingender J, Szewzyk U, Steinberg P, Rice SA, Kjelleberg S. Biofilms: an emergent form of bacterial life. Nat Rev Microbiol. 2016;14:563–575. doi:10.1038/nrmicro.2016.94
21. Walters III MC, Roe F, Bugnicourt A, Franklin MJ, Stewart PS. Contributions of antibiotic penetration, oxygen limitation, and low metabolic activity to tolerance of Pseudomonas aeruginosa biofilms to ciprofloxacin and tobramycin. Antimicrob Agents Chemother. 2003;47:317–323. doi:10.1128/aac.47.1.317-323.2003
22. Stewart PS, Costerton JW. Antibiotic resistance of bacteria in biofilms. Lancet. 2001;358:135–138. doi:10.1016/s0140-6736(01)05321-1
23. Chiang WC, Nilsson M, Jensen P, et al. Extracellular DNA shields against aminoglycosides in Pseudomonas aeruginosa biofilms. Antimicrob Agents Chemother. 2013;57:2352–2361. doi:10.1128/aac.00001-13
24. Koo H, Allan RN, Howlin RP, Stoodley P, Hall-Stoodley L. Targeting microbial biofilms: current and prospective therapeutic strategies. Nat Rev Microbiol. 2017;15:740–755. doi:10.1038/nrmicro.2017.99
25. Moser C, Jensen P, Thomsen K, et al. Immune Responses to Pseudomonas aeruginosa Biofilm Infections. Front Immunol. 2021;12:625597. doi:10.3389/fimmu.2021.625597
26. Cruz AR, Boer MAD, Strasser J, et al. Staphylococcal protein A inhibits complement activation by interfering with IgG hexamer formation. Proc Natl Acad Sci U S A. 2021;118. doi:10.1073/pnas.2016772118
27. Becker S, Frankel MB, Schneewind O, Missiakas D. Release of protein A from the cell wall of Staphylococcus aureus. Proc Natl Acad Sci U S A. 2014;111:1574–1579. doi:10.1073/pnas.1317181111
28. Kebaier C, Chamberland RR, Allen IC, et al. Staphylococcus aureus α-hemolysin mediates virulence in a murine model of severe pneumonia through activation of the NLRP3 inflammasome. J Infect Dis. 2012;205:807–817. doi:10.1093/infdis/jir846
29. Cheung GY, Joo HS, Chatterjee SS, Otto M. Phenol-soluble modulins--critical determinants of staphylococcal virulence. FEMS Microbiol Rev. 2014;38:698–719. doi:10.1111/1574-6976.12057
30. Speziale P, Pietrocola G, Foster TJ, Geoghegan JA. Protein-based biofilm matrices in Staphylococci. Front Cell Infect Microbiol. 2014;4:171. doi:10.3389/fcimb.2014.00171
31. Moura-Alves P, Puyskens A, Stinn A, et al. Host monitoring of quorum sensing during Pseudomonas aeruginosa infection. Science. 2019;366. doi:10.1126/science.aaw1629
32. Bianchi SM, Prince LR, McPhillips K, et al. Impairment of apoptotic cell engulfment by pyocyanin, a toxic metabolite of Pseudomonas aeruginosa. Am J Respir Crit Care Med. 2008;177:35–43. doi:10.1164/rccm.200612-1804OC
33. Al Kindi A, Alkahtani AM, Nalubega M, et al. Staphylococcus aureus Internalized by Skin Keratinocytes Evade Antibiotic Killing. Front Microbiol. 2019;10:2242. doi:10.3389/fmicb.2019.02242
34. Liu X, Zhang Z, Ruan J, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–158. doi:10.1038/nature18629
35. Rathinam VA, Jiang Z, Waggoner SN, et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol. 2010;11:395–402. doi:10.1038/ni.1864
36. Dowey R, Iqbal A, Heller SR, Sabroe I, Prince LR. A Bittersweet Response to Infection in Diabetes; Targeting Neutrophils to Modify Inflammation and Improve Host Immunity. Front Immunol. 2021;12:678771. doi:10.3389/fimmu.2021.678771
37. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. 2018;18:134–147. doi:10.1038/nri.2017.105
38. Bhattacharya M, Berends ETM, Chan R, et al. Staphylococcus aureus biofilms release leukocidins to elicit extracellular trap formation and evade neutrophil-mediated killing. Proc Natl Acad Sci U S A. 2018;115:7416–7421. doi:10.1073/pnas.1721949115
39. Yang S, Gu Z, Lu C, et al. Neutrophil Extracellular Traps Are Markers of Wound Healing Impairment in Patients with Diabetic Foot Ulcers Treated in a Multidisciplinary Setting. Adv Wound Care. 2020;9:16–27. doi:10.1089/wound.2019.0943
40. Yang S, Wang S, Chen L, et al. Neutrophil Extracellular Traps Delay Diabetic Wound Healing by Inducing Endothelial-to-Mesenchymal Transition via the Hippo pathway. Int J Biol Sci. 2023;19:347–361. doi:10.7150/ijbs.78046
41. Mirza RE, Fang MM, Weinheimer-Haus EM, Ennis WJ, Koh TJ. Sustained inflammasome activity in macrophages impairs wound healing in type 2 diabetic humans and mice. Diabetes. 2014;63:1103–1114. doi:10.2337/db13-0927
42. Huang W, Jiao J, Liu J, et al. MFG-E8accelerates wound healing in diabetes by regulating “NLRP3 inflammasome-neutrophil extracellular traps” axis. Cell Death Discov. 2020;6:84. doi:10.1038/s41420-020-00318-7
43. Louiselle AE, Niemiec SM, Zgheib C, Liechty KW. Macrophage polarization and diabetic wound healing. Transl Res. 2021;236:109–116. doi:10.1016/j.trsl.2021.05.006
44. Mirza RE, Fang MM, Ennis WJ, Koh TJ. Blocking interleukin-1β induces a healing-associated wound macrophage phenotype and improves healing in type 2 diabetes. Diabetes. 2013;62:2579–2587. doi:10.2337/db12-1450
45. Lee J, Zhang L. The hierarchy quorum sensing network in Pseudomonas aeruginosa. Protein Cell. 2015;6:26–41. doi:10.1007/s13238-014-0100-x
46. Schilcher K, Horswill AR. Staphylococcal Biofilm Development: structure, Regulation, and Treatment Strategies. Microbiol Mol Biol Rev. 2020. doi:10.1128/mmbr.00026-19
47. Defoirdt T. Quorum-Sensing Systems as Targets for Antivirulence Therapy. Trends Microbiol. 2018;26:313–328. doi:10.1016/j.tim.2017.10.005
48. Sully EK, Malachowa N, Elmore BO, et al. Selective chemical inhibition of agr quorum sensing in Staphylococcus aureus promotes host defense with minimal impact on resistance. PLoS Pathog. 2014;10:e1004174. doi:10.1371/journal.ppat.1004174
49. Rezzoagli C, Archetti M, Mignot I, Baumgartner M, Kümmerli R. Combining antibiotics with antivirulence compounds can have synergistic effects and reverse selection for antibiotic resistance in Pseudomonas aeruginosa. PLoS Biol. 2020;18:e3000805. doi:10.1371/journal.pbio.3000805
50. Bagood MD, Marjanovic J, Jiang N, et al. Staphylococcus aureus accessory gene regulator quorum-sensing system inhibits keratinocyte lipid enzymes and delays wound repair. J Clin Invest. 2025;135. doi:10.1172/jci190411
51. Okshevsky M, Regina VR, Meyer RL. Extracellular DNA as a target for biofilm control. Curr Opin Biotechnol. 2015;33:73–80. doi:10.1016/j.copbio.2014.12.002
52. Nguyen HTT, Nguyen TH, Otto M. The staphylococcal exopolysaccharide PIA - Biosynthesis and role in biofilm formation, colonization, and infection. Comput Struct Biotechnol J. 2020;18:3324–3334. doi:10.1016/j.csbj.2020.10.027
53. Fleming D, Chahin L, Rumbaugh K. Glycoside Hydrolases Degrade Polymicrobial Bacterial Biofilms in Wounds. Antimicrob Agents Chemother. 2017;61. doi:10.1128/aac.01998-16.
54. Redman WK, Welch GS, Williams AC, Damron AJ, Northcut WO, Rumbaugh KP. Efficacy and safety of biofilm dispersal by glycoside hydrolases in wounds. Biofilm. 2021;3:100061. doi:10.1016/j.bioflm.2021.100061
55. Nir-Paz R, Onallah H, Dekel M, et al. Randomized double-blind study on safety and tolerability of TP-102 phage cocktail in patients with infected and non-infected diabetic foot ulcers. Med. 2025;6:100565. doi:10.1016/j.medj.2024.11.018
56. Strathdee SA, Hatfull GF, Mutalik VK, Schooley RT. Phage therapy: from biological mechanisms to future directions. Cell. 2023;186:17–31. doi:10.1016/j.cell.2022.11.017
57. Azeredo J, García P, Drulis-Kawa Z. Targeting biofilms using phages and their enzymes. Curr Opin Biotechnol. 2021;68:251–261. doi:10.1016/j.copbio.2021.02.002
58. Suh GA, Lodise TP, Tamma PD, et al. Considerations for the Use of Phage Therapy in Clinical Practice. Antimicrob Agents Chemother. 2022;66:e0207121. doi:10.1128/aac.02071-21
59. Aburayyan WS, Seder N, Al-Fawares O, Fararjeh A, Majali IS, Al-Hajaya Y. Characterization of Antibiofilm and Antimicrobial Effects of Trigona Stingless Bee Honey Compared to Stinging Bee Centaurea hyalolepis and Citrus Honeys. J Evid Based Integr Med. 2024;29:2515690x241271978. doi:10.1177/2515690x241271978
60. Touqeer M, Siddiqui A, Haider MA, et al. Breaking the Vicious Cycle: nanozyme-Driven Multimodal Therapeutics for Diabetic Wound Regeneration. Adv Healthc Mater. 2025;e04482. doi:10.1002/adhm.202504482
61. Xing C, Zhu H, Dou X, et al. Infected Diabetic Wound Regeneration Using Peptide-Modified Chiral Dressing to Target Revascularization. ACS Nano. 2023;17:6275–6291. doi:10.1021/acsnano.2c10039
62. Yang S, Feng Y, Chen L, et al. Disulfiram accelerates diabetic foot ulcer healing by blocking NET formation via suppressing the NLRP3/Caspase-1/GSDMD pathway. Transl Res. 2023;254:115–127. doi:10.1016/j.trsl.2022.10.008
63. Fadini GP, Menegazzo L, Rigato M, et al. NETosis Delays Diabetic Wound Healing in Mice and Humans. Diabetes. 2016;65:1061–1071. doi:10.2337/db15-0863
64. Wu BC, Haney EF, Akhoundsadegh N, et al. Human organoid biofilm model for assessing antibiofilm activity of novel agents. NPJ Biofilms Microbiomes. 2021;7:8. doi:10.1038/s41522-020-00182-4
65. Wu BC, Blimkie TM, Haney EF, Falsafi R, Akhoundsadegh N, Hancock REW. Host Response of Human Epidermis to Methicillin-Resistant Staphylococcus aureus Biofilm Infection and Synthetic Antibiofilm Peptide Treatment. Cells. 2022;11. doi:10.3390/cells11213459.
66. Radzieta M, Sadeghpour-Heravi F, Peters TJ, et al. A multiomics approach to identify host-microbe alterations associated with infection severity in diabetic foot infections: a pilot study. NPJ Biofilms Microbiomes. 2021;7:29. doi:10.1038/s41522-021-00202-x
67. Theocharidis G, Thomas BE, Sarkar D, et al. Single cell transcriptomic landscape of diabetic foot ulcers. Nat Commun. 2022;13:181. doi:10.1038/s41467-021-27801-8
68. Eder L, Caucheteux SM, Afiuni-Zadeh S, et al. Imaging Mass Cytometry in Psoriatic Disease Reveals Immune Profile Heterogeneity in Skin and Synovial Tissue. J Invest Dermatol. 2025;145:1361–1370. doi:10.1016/j.jid.2024.08.039
69. Thaarup IC, Bjarnsholt T. Current In Vitro Biofilm-Infected Chronic Wound Models for Developing New Treatment Possibilities. Adv Wound Care. 2021;10:91–102. doi:10.1089/wound.2020.1176
70. Sanapalli BKR, Yele V, Singh MK, Thaggikuppe Krishnamurthy P, Karri V. Preclinical models of diabetic wound healing: a critical review. Biomed Pharmacother. 2021;142:111946. doi:10.1016/j.biopha.2021.111946
71. Astrada A, Nakagami G, Kashiwabara K, Sanada H. Biofilm detection-based wound management in diabetic foot ulcers: a randomised controlled trial. J Wound Care. 2025;34:514–524. doi:10.12968/jowc.2024.0051
72. Astrada A, Nakagami G, Sanada H. Challenges in Biofilm Identification in Diabetic Foot Infections: review of Literature. Int J Low Extrem Wounds. 2024;15347346241273112. doi:10.1177/15347346241273112
73. Theodorakopoulos G, Armstrong DG. Biofilm in Diabetic Foot Ulcers: a Systematic Narrative Review. Int Wound J. 2025;22:e70795. doi:10.1111/iwj.70795
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Exploring the Benefits of Metal Ions in Phage Cocktail for the Treatment of Methicillin-Resistant Staphylococcus aureus (MRSA) Infection
Li X, Chen Y, Wang S, Duan X, Zhang F, Guo A, Tao P, Chen H, Li X, Qian P
Infection and Drug Resistance 2022, 15:2689-2702
Published Date: 27 May 2022
Extracellular Histones Activate Endothelial NLRP3 Inflammasome and are Associated with a Severe Sepsis Phenotype

Beltrán-García J, Osca-Verdegal R, Pérez-Cremades D, Novella S, Hermenegildo C, Pallardó FV, García-Giménez JL
Journal of Inflammation Research 2022, 15:4217-4238
Published Date: 25 July 2022
Pyroptosis and Intervertebral Disc Degeneration: Mechanistic Insights and Therapeutic Implications
Ge Y, Chen Y, Guo C, Luo H, Fu F, Ji W, Wu C, Ruan H
Journal of Inflammation Research 2022, 15:5857-5871
Published Date: 17 October 2022
Pyroptosis and Inflammasome-Related Genes-NLRP3, NLRC4 and NLRP7 Polymorphisms Were Associated with Risk of Lung Cancer
Jing X, Yun Y, Ji X, Yang E, Li P
Pharmacogenomics and Personalized Medicine 2023, 16:795-804
Published Date: 25 August 2023
The Mechanism of Pyroptosis and Its Application Prospect in Diabetic Wound Healing
Al Mamun A, Shao C, Geng P, Wang S, Xiao J
Journal of Inflammation Research 2024, 17:1481-1501
Published Date: 6 March 2024