Back to Journals » Journal of Inflammation Research » Volume 15
Extracellular Histones Activate Endothelial NLRP3 Inflammasome and are Associated with a Severe Sepsis Phenotype
Authors Beltrán-García J ![]() , Osca-Verdegal R, Pérez-Cremades D, Novella S, Hermenegildo C, Pallardó FV, García-Giménez JL
, Osca-Verdegal R, Pérez-Cremades D, Novella S, Hermenegildo C, Pallardó FV, García-Giménez JL ![]()
Received 15 March 2022
Accepted for publication 29 June 2022
Published 25 July 2022 Volume 2022:15 Pages 4217—4238
DOI https://doi.org/10.2147/JIR.S363693
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Jesús Beltrán-García,1– 3 Rebeca Osca-Verdegal,1,3 Daniel Pérez-Cremades,2,3 Susana Novella,2,3 Carlos Hermenegildo,2,3 Federico V Pallardó,1– 3 José Luis García-Giménez1– 3
1Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), Instituto de Salud Carlos III, Madrid, Spain; 2Instituto de Investigación Sanitaria INCLIVA, Valencia, Spain; 3Departamento de Fisiología, Facultad de Medicina y Odontología, Universitat de València, València, Spain
Correspondence: José Luis García-Giménez, Departamento de Fisiología, Facultad de Medicina y Odontología, Universitat de València, València, 46010, Spain, Tel +34 963 864 646, Email [email protected]
Introduction: Circulating extracellular histones acquire relevance as cytotoxic mediators in sepsis. Extracellular histones act as damage-associated molecular patterns (DAMPs), which induce oxidative stress and NLRP3 inflammasome activation. Inflammasome mediates pyroptosis, a programmed cell death mechanism that produces inflammation. Despite evidence for inflammasome activation in immune cells during sepsis, it was unknown whether extracellular histones can produce endothelial inflammasomes activation.
Methods: We used human umbilical vein endothelial cells (HUVEC) to explore the activation of pyroptosis, endothelial function and inflammation by extracellular histones. We evaluated pyroptosis by flow cytometry, caspase-1 activity assay, and gene and protein expression analysis by RT-qPCR and Western blot, respectively. The upstream molecular responses involved in pyroptosis activation by extracellular histones were validated by means of using antioxidant glutathione ethyl ester and NLRP3 inflammasome inhibitors. Finally, using mass spectrometry, we measured circulating histones in blood from critically-ill patients and demonstrated that circulating histone levels correlated with the expression of pyroptosis-related cytokines, the release of endothelial adhesion factors and septic shock severity.
Results: We found that extracellular histones mediate the activation of NLRP3 inflammasome and pyroptosis in endothelial cells by contributing to endothelial dysfunction and the dysregulation of the immune response mediated by endothelium. Likewise, we demonstrated how the hyperacetylation of extracellular histones or the use of antioxidants decreased pyroptosis. In addition, we showed that pyroptosis is a feasible process occurring in septic shock patients.
Discussion: Circulating histone levels correlated with the expression of pro-inflammatory and pyroptosis-related cytokines, the release of endothelial adhesion factors and septic shock severity. We propose to block histone-mediated pyroptosis as a feasible therapeutic strategy in sepsis.
Keywords: sepsis, extracellular histones, histone acetylation, endothelium, inflammasome, NLRP3
Graphical Abstract:

Introduction
Loss of endothelial function is an important hallmark of sepsis because it alters vascular homeostasis and activates processes like thrombosis, inflammation, or vascular remodeling.1 Endothelial cells release mediators that control vascular relaxation and contraction, as well as enzymes that control blood clotting, immune function, leukocyte and platelet adhesion.3 In this regard, endothelial damage plays a central role in the progression of organ failure during sepsis and is a major contributor to sepsis mortality.4–6 Therefore, avoiding the endothelial dysfunction has been demonstrated to improve the sepsis outcome.7
Histones are the core structure for nucleosome assembly and contribute to the conformation of chromatin and gene expression regulation by post-translational modifications (PTMs). Histones have been proposed as antimicrobial agents,8 nevertheless, in recent years the role of extracellular histones as cytotoxic mediators has gained relevance in sepsis and other inflammation-related disorders.2,9–12
Particularly in sepsis, extracellular histones are released from neutrophils and macrophages during neutrophil extracellular traps formation in a process known as NETosis, and also from cells after cellular damage, thus contributing to endothelial dysfunction, organ failure and death.2,11,13,14 Abrams et al demonstrated that blood circulating histones act as mediators for distant organ damage (ie lungs, heart, etc.).5 Moreover, histones released into the bloodstream are found in patients with severe blunt trauma, pancreatitis, sepsis and septic shock (SS),2,9,15 and in pathological complications, such as disseminated intravascular coagulation (DIC).9 More recently, circulating histones have been found in COVID-19 as mediators of inflammation, coagulation activation and organ dysfunction.16,17
Previous studies performed by us and other authors have demonstrated that extracellular histones seriously damage human endothelial cells in culture by producing endothelial dysregulation and increasing apoptosis and autophagy,5,6,18 in a dose-dependent manner. We have also shown that extracellular histones are high in SS patients, mainly in those with fatal outcomes.9
Recent research has emphasized the role of histones in stimulating Toll-like receptors (TLRs) and NOD-Like Receptors (NLRs) in sepsis.2,7,19 Among NLRs, nucleotide-binding domain leucine-rich repeat-containing protein 3 (NLRP3) plays a critical role in innate immunity, and mediates the activation of the caspase-1 molecular complex termed “inflammasome”, which promotes pro-inflammatory IL-1β and IL-18 production.17,18 Importantly, inflammation plays a relevant role in sepsis-mediated cell death through intricate mechanisms, such as autophagy, apoptosis and a caspase-1 dependent form of programmed necrosis known as pyroptosis.2,20–24 Unlike apoptosis, pyroptosis is a form of necrotic and inflammatory programmed cell death induced by inflammatory caspases25 and other stimuli, such as oxidative stress,26,27 mitochondrial dysfunction, Ca+2 signaling and lysosomal rupture28 through the activation of the inflammasome.29
Several studies have also shown how histone deacetylase inhibitors (HDACis) play a protective role in sepsis by regulating the acute hyperinflammatory response and improving prognosis and survival in animal models.30,31 Importantly, treatment with HDACis can significantly ameliorate the phenotype of cells and tissues, and clinical features in animal models of sepsis.2,32,33 However, the molecular mechanisms underlying the improvement of sepsis-associated features remain unknown.
Here, we show a new pathway in endothelial cells mediated by extracellular histones, and how histone hyperacetylation and oxidative stress modulate NLRP3 inflammasome activation by triggering pyroptosis in human endothelial cells in a dose-dependent manner. Extracellular histones mediate a strong dysregulation of the endothelial function, antioxidant capacity and immune response mediated by the endothelium. We also show that pyroptosis occurs in SS patients, and pyroptosis-related cytokines are correlated with the levels of extracellular circulating histones in plasma. We propose histone-mediated pyroptosis as a new target to develop clinical interventions and therapies against sepsis.
Materials and Methods
Cell Culture and Experimental Design
Human umbilical veins were treated with 1% collagenase (Life Technologies, Carlsbad, CA, USA) to obtain HUVEC and were cultured in specific growth medium EGM-2 (Lonza, Cultek, Barcelona, Spain). The umbilical cords from neonates used to obtain HUVEC were obtained following the principles outlined in the Declaration of Helsinki, approved by the Ethical Committee of Clinical Research of the Hospital Clínico Universitario of Valencia (HCUV, Spain). Written informed consent was obtained from parents for the collection and use of umbilical cords from neonates from which HUVEC cells were obtained and utilized. Cells were identified as endothelial by their characteristic cobblestone morphology and the presence of von Willebrand factor by immunofluorescence using a specific antibody (ab6994; Abcam, Cambridge, UK). The cells used in this study were more than 95% vWF positive (Supplementary Figure 1). The HUVECs herein employed had 3–6 passages. Cells were cultured in culture medium 199 with Earle’s Balanced Salt Solution and L-Glutamine (Lonza, Verviers, Belgium), supplemented with 20% foetal bovine serum (FBS) (Invitrogen, San Diego, CA, USA), 1% penicillin/amphotericin (Invitrogen, San Diego, CA, USA) and growth factors (Sigma-Aldrich, Missouri, USA). HUVEC were incubated in a Heracell 150i CO2 incubator (Thermo Scientific, New York, NY, USA) in a humidified atmosphere (5% CO2) and 37℃.
In vitro Hyperacetylation and Purification of Histones
Histones were obtained from commercial HeLa cells (ATCC, Manassas, Virginia) with Iscove’s Dulbecco’s Modified Eagle Medium (DMEM) High Glucose (Invitrogen, CA, USA), supplemented with 10% FBS (Invitrogen, CA, USA) and 1% penicillin/streptomycin (Invitrogen, San Diego, USA).
Hyperacetylation of histones was achieved using suberanilohydroxamic acid (SAHA) (Cayman Chemical, Michigan, USA) (Supplementary Figure 2A). Different concentrations were tested (0–100μM) to identify the strongest hyperacetylation effect at the lowest cytotoxicity (Supplementary Figure 2B–D). Treatment was performed during 24 h.
Histones were purified using the protocol described by Shechter et al,34 with some modifications. Cells were washed with PBS, and the dry pellet was resuspended in Hypotonic Lysis Buffer (10 mM Tris-HCl pH = 8, 1 mM KCl, 1.5 mM MgCl2) with 10 μL/mL of orthovanadate, and 2 μL/mL of proteases inhibitor (Fisher Scientific, Hampton, USA). Afterwards, cells were kept in rotation 30 minutes at 4℃ and then were centrifuged 10 minutes at 4℃ at 10,000 rpm. Finally, the pellet was resuspended in 400 μL of 0.4N of H2SO4 and was kept in rotation overnight at 4 ℃. The next day, the sample was centrifuged 10 minutes at 4 ℃ at 13,000 rpm. The supernatant was kept. The following step was to add 132 μL of 100% tricarboxylic acid to the sample and incubate on ice 30 minutes. Then, the sample was centrifuged 10 minutes at 4℃ at 13,000 rpm and the pellet was washed with 0.5 mL ice-cold acetone. Finally, the sample was centrifuged 10 minutes at 13,000 rpm at 4℃ and the pellet was resuspended in H2O.
Cell Viability Determination and in vitro Inhibition of Apoptosis and Pyroptosis
Cell viability, apoptosis and necrosis were determined by flow cytometry using the Annexin-V kit (Immunostep, Salamanca, Spain) following the manufacturer’s specifications. HUVEC were exposed to different treatments of extracellular histones for 4 h (see Supplementary Methods). Z-LEHD-FMK (MBL Life Science, Japan), a specific inhibitor of caspase-9,35 was used to demonstrate the histone-mediated apoptosis mechanism. HUVEC were exposed to Z-LEHD-FMK 20 μM for 1 h. Cell viability and apoptosis were determined by flow cytometry using the Annexin-V kit (Immunostep, Salamanca, Spain) following the manufacturer’s specifications. MCC950 (Cayman Chemical, Michigan, USA), a specific inhibitor of the oligomerization of NLRP3 inflammasome,36 was used to demonstrate the histone-mediated pyroptosis mechanism. HUVEC were exposed to MCC950 10 μM during 2 h. Cells were incubated with both inhibitors, Z-LEHD-FMK and MCC950, before the 4-hour challenge with extracellular histones (50μg/mL).2 The cytotoxicity analysis of MCC950 was performed using the MTT-based test (Roche, Mannheim, Germany). Glutathione Ethyl Ester (GSH-EE) (Merck, Kenilworth, NJ, United States), a cell-permeable derivative of glutathione (GSH) that works protecting cells against damage from oxidative stress,37 was used to demonstrate that oxidative stress is the main mediator of histone-induced pyroptosis in endothelial cells, and an increase in the antioxidant levels avoid the pyroptosis cell-death mediated by histones. HUVEC were incubated with GSH-EE 1 hour before the 4-hour challenge with extracellular histones (50 μg/mL).2
Oxyblot and Western Blot
HUVEC were scraped on ice in a lysis buffer and centrifuged for 15 minutes at 13,000 rpm to recover the proteins. The lysis buffer used for protein extraction is made by mixing different compounds: 20mM Hepes, pH 7.4, 1% Triton X-100, 100mM NaCl, 50mM NaF, 10mM β-glycerophosphate, 1mM phenylmethylsulfonyl fluoride (PMSF), 1mM sodium orthovanadate, protease inhibitor cocktail (Roche Diagnostics, Basilea, Switzerland). Primary specific antibodies: PRX-SO3 (Abcam, ab16830), PRX6 (Abcam, ab59543), GPX1 (Abcam, ab22604), Catalase (Sigma-Aldrich, c0979), MnSOD (Abcam, ab13533), Cu/ZnSOD (Abcam, ab13498) and eNOS (Santa Cruz Biotechnology, sc-653), COX-1 (Santa Cruz Biotechnology, sc-19998), COX-2 (Santa Cruz Biotechnology, sc-19999), PGIS (Santa Cruz Biotechnology, sc-20933), TXAS (Santa Cruz Biotechnology, sc-79181), Caspase-3 (Cell Signaling, 9662), NLRP3 (Novus, NBP2-12446).2 Finally, β-actin (Sigma-Aldrich, a1978) (Sigma-Aldrich) as loading control.2 Detection was performed using peroxidase-linked secondary antibodies (anti-mouse (Sigma-Aldrich, NA931V), anti-goat (Santa Cruz Biotechnology sc2020), anti-rabbit (Sigma-Aldrich, NA934V).
Protein carbonyls were measured using the OxyBlot™ protein oxidation kit (Merck, Massachusetts, USA) following the manufacturer’s specifications.
Membrane Potential Determination
The analysis of the membrane potential was performed using the DIBAC probe (Bis-(1,3-Dibutylbarbituric Acid) Trimethine Oxonol), Thermo Fisher, Massachusetts, United States). Briefly, HUVEC were incubated during 4 hours with extracellular histones in native and hyperacetylated conditions.2 Therefore, HUVEC were incubated with DIBAC for 10 minutes and the membrane potential was analyzed mediating flow cytometry.
RNA Purification and Quantitative Real‐time PCR Assay (qRT‐PCR)
Extraction and purification of RNA from HUVEC were performed using the miRNeasy Mini Kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions. Reverse transcription was done using 200 ng of total RNA with the High‐Capacity cDNA Reverse Transcription Kit (Applied Biosystems, CA, USA). mRNA levels were determined by qRT‐PCR analysis with the ABI Prism 7900 HT Fast Real‐Time PCR System (Applied Biosystems) using the TaqMan Universal Mastermix (Thermo Fisher, Rockford, USA). The gene‐specific primers and probes used were as follows: PGIS-2 (Hs00919949_m1, Life Technologies), TBXAS (Hs01022706_m1, Life Technologies), NOS-3 (Hs01574665_m1, Life Technologies), V-CAM-1 (Hs01003372_m1, Life Technologies) I-CAM-1 (Hs00164932_m1, Life Technologies), SEL-E (Hs00174057_m1, Life Technologies), COX-1 (Hs00377726_m1, Life Technologies), COX-2 (Hs00153133_m1, Life Technologies), TRX-1 (Hs01555214_g1, Life Technologies), TRX-2 (Hs00429399_m1, Life Technologies), IL-1α (Hs00174092_m1, Life Technologies), IL-1β (Hs01555410_m1, Life Technologies), IL-6 (Hs00174131_m1, Life Technologies), IL-18 (Hs01038788_m1, Life Technologies) and GAPDH (Hs02758991_g1, Life Technologies).2 PCR conditions were 10 min. at 95℃ for enzyme activation, followed by 40 two‐step cycles (15 sec. 95℃; 1 min. 60℃). Data were analyzed with the SDS 2.2.2 software (Applied Biosystems) according to the 2−ΔΔCt methods.
Analysis of Caspase-1 Activity
Caspase-1 activity was measured using the Caspase-Glo®1 Reagent (Promega, Wisconsin, USA) following the manufacturer’s specifications. Briefly, 1:1 ratio of cell culture medium and Caspase-Glo® 1 Reagent previously prepared was mixed and incubated for 1 h at room temperature (RT). Next, luminosity was measured with Spectra max Gemini XPS (BioNova Científica, Madrid, Spain). To confirm that MG-132 inhibition and Ac-YVAD-CHO inhibition were complete, luminescence was measured at 60, 90 and 120 minutes. Data were acquired once having stabilized the signal when MG-132 inhibition and Ac-YVAD-CHO inhibition were complete using the SoftMax Pro 6.2.2 software.
Patients’ Selection
Plasma samples were analyzed from nine healthy volunteers, six critically ill patients admitted to the ICUs suffering from stroke, nine patients with sepsis and 15 SS patients. Acute physiology and chronic health evaluation II (APACHE II) and a sequential organ failure assessment (SOFA), different clinical, microbiological, hemodynamic, and biochemical determinations were evaluated in every patient. All the septic patients met the sepsis or SS definition according to the Sepsis-3 Consensus.38 Plasma samples from healthy subjects were considered the control group. The Intensive Care Unit (ICU) patients with non-septic-related process, sepsis and SS patients were obtained from INCLIVA’s Biobank. Informed consent was obtained from every participant. All the experimental protocols and methods were performed after obtaining approval from the HCUV’s Biomedical Research Ethics Committee. All participants signed the written informed consent.
All the methods were performed in line with relevant international guidelines and regulations.
Circulating Histone Levels Measured by Mass Spectrometry
Circulating histone levels were measured by MRM-MS using spike-in peptides (see Supplementary Table 1), as previously described,9,10 with some modifications to the method. The MRM experiments were performed with a 5500QTRAP hybrid triple quadrupole/linear ion trap mass spectrometer (ABSIEX), equipped with a Micro M3 MicroLC chromatographic system. 10 L of triptic digest (about 9 ug of protein and 25 fmol of each Spike-In peptide) was injected into a Trap column (10X0.3mm Trap Cartridge Chromxp C18CL 5um, ABSCIEX) and was then loaded and separated onto an analytical column (ChromXP C18, 120A, 3um, 150 × 0.3mm). Elution was carried out with a linear gradient of 0 a 35% B in A for 30min. (A: 0.1% FA; B: ACN, 0.1% FA) at a flow rate of 5μL/min.2
The 5500 QTRAP was operated in a MRM mode. MRM data were acquired in the positive mode with a spray voltage of 5500 V, curtain gas: 25 psi, ion source gas: 25 psi, entrance potential (EP): 10 and exit potential (EXP): 16. Collision energy (CE) and Declustering Potential (DP) were optimized previously for each transition. Relative quantification of each histone was done and the area ratios (light/heavy) for all transitions, which were calculated using the Skyline 4.2.1.19058 software (MacCoss lab) and light concentration estimated as fmol/μL of initial serum and converted to ng/mL of circulating histones.2
Analysis of Circulating Caspase-1 and Cytokine Levels in Plasma in Patients
Human caspase-1 was measured using the Human Caspase-1 ELISA (RayBiotech, Parkway, USA). The human IL-18 levels were measured by Human IL-18 ELISA (Thermo Fisher, Massachusetts, USA). Different levels of cytokines present in plasma from sepsis and SS patients were measured with the Human Inflammation 20-plex ProcartaPlex Panel (ThermoFisher, Massachusetts, USA), following manufacturer’s instructions.2
Statistical Analysis
Statistics were calculated with SPPS v23 and the Prism software (Software Inc. CA, USA) was used for graphics. Values are expressed as mean ± SEM. A one-way analysis of variance was used to determine the difference between groups in the in vitro experiments. When an interaction effect was found, multiple comparisons using the Scheffé method post hoc test were performed. Significance was considered at *p≤ 0.05, **p≤ 0.01 and ***p≤ 0.001, as indicated in each case.2
For the studies performed in human, normality of samples was determined with Kolmogorov–Smirnov normality test and samples did not follow a Gaussian distribution. The non-parametric Mann–Whitney test was used to analyze differences between two non-paired groups with a 0.05 significance level. The Kruskal–Wallis test was used to analyze differences among ICU controls, sepsis and SS patients, followed by a post hoc test using Bonferroni correction for α (0.05/3). Spearman’s rho test was used for the correlation analysis between variables.2
Results
Extracellular Histones Induced Pyroptosis Through NLRP3 in HUVEC
The cytotoxic effect of extracellular histones on HUVEC was evaluated using purified histone extracts obtained as described in methods section. Using a flow cytometry analysis, we studied cell death mechanisms activated by extracellular histones at 0, 2, 5, 10, 25 and 50 μg/mL concentrations, which fell within the range of concentrations described by us and others.6,18,39
Our results showed decreased cell viability that corresponded to an increase in Annexin V- (AV) and/or propidium iodide- (PI) positive cells from 10 μg/mL to 50 μg/mL after a HUVEC challenge with native or hyperacetylated histones compared to the control condition (0 μg/mL) (Figure 1A and B, respectively). Notably, HUVEC treated with 50 μg/mL of native histones showed increased AV/PI-positive cells compared to the HUVEC treated with hyperacetylated histones (Supplementary Figure 3).
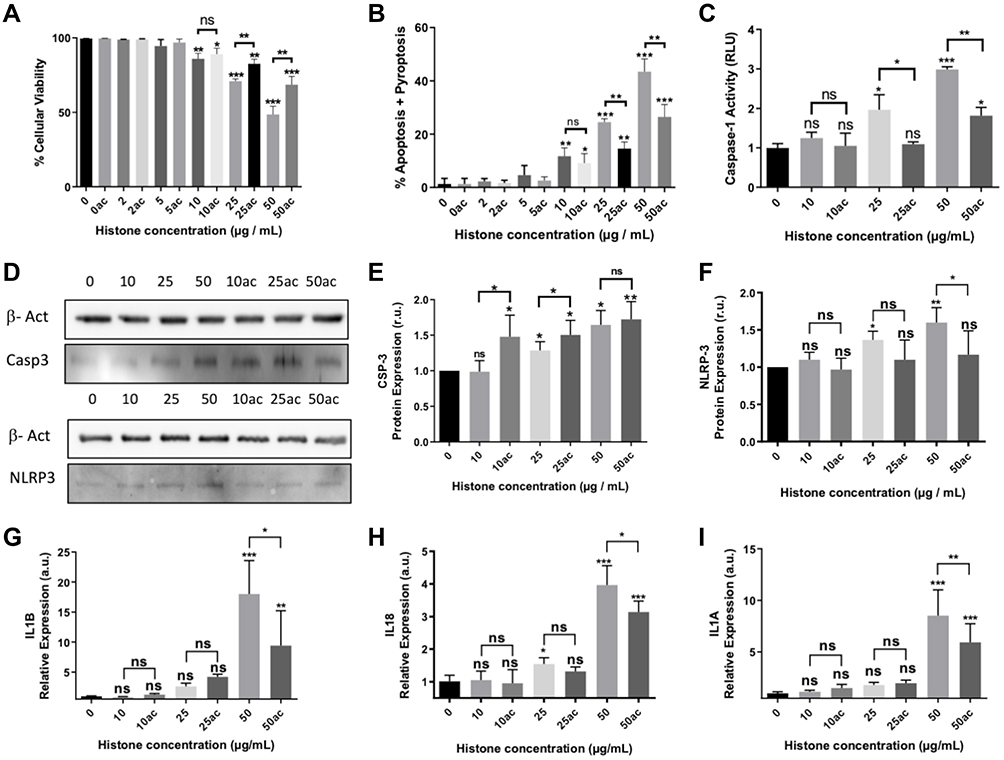 |
Figure 1 Effect of extracellular histones on apoptosis, pyroptosis and inflammasome after incubating with native or hyperacetylated extracellular histones: (A) The percentage of viable cells; (B) Annexin V/PI-positive cells after incubation with native histones and hyperacetylated histones determined by flow cytometry; (C) Caspase-1 activity measured using the Caspase-Glo® 1 Reagent kit (Promega); (D) Protein levels of caspase-3 and NLRP3 inflammasome determined by WB; (E) Caspase-3 protein levels in relation to β-actin determined by densitometry of specific bands in the WB membrane; (F) NLRP3 inflammasome levels in relation to β-actin determined by densitometry of specific bands in the WB membrane. Relative expression of inflammatory cytokines as markers of pyroptosis activation; (G) IL1B; (H) IL18; (I) IL1A gene expression determined by RT-qPCR. Data are expressed as mean±SEM from 3–5 independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001 compared to histones 0μg/mL The lines at the top of columns indicate differences between compared conditions. Abbreviation: ns, non-significant p value. |
To analyze the different cell death mechanisms mediated by extracellular histones, we analyzed the expression levels of Caspase-3, as the main effector of apoptosis, and the expression levels of Caspase 1, which triggers pyroptosis.40,41 Our results indicated that both mechanisms, pyroptosis and apoptosis, were induced by extracellular histones in HUVEC. When we used 50 μg/mL of extracellular histones (native or hyperacetylated histones), the levels of caspase-3 increased significantly (Figure 1D and E). Interestingly, caspase-1 activation was only observed when HUVEC were treated with 25μg/mL of native histones, but not with hyperacetylated histones. When cells were treated with 50 μg/mL of extracellular histones (native or hyperacetylated histones), caspase-1 was activated, although its activity was lower when cells were treated with hyperacetylated histones (Figure 1C). These results were accompanied by the overexpression of NLRP3 inflammasome (Figure 1D–F) and the increased expression of pro-inflammatory cytokines IL18 (Figure 1G), IL1B (Figure 1H) and IL1A (non-canonical pyroptosis) (Figure 1I), which are the products of the inflammasome and contribute to the inflammatory response.42
Extracellular Histones Induced Oxidative Stress, Membrane Depolarization in HUVEC
Several authors have demonstrated the interconnectivity among oxidative stress, NLRP3 activation and inflammation.43,44 Therefore, we measured oxidative stress markers, the membrane potential, as well as the antioxidant enzyme levels in HUVEC when these cells were challenged with native or hyperacetylated extracellular histones.
Our results showed that extracellular histones increased the levels of carbonylated proteins and the oxidized form of Peroxiredoxin-6 (Prdx6) in HUVEC (Figure 2A and B) and suggest that native histones induced more oxidative stress damage than hyperacetylated histones. This mechanism may involve the increase in the membrane potential, which induces a membrane depolarization in a dose-dependent manner, as showed in our results (Figure 2C). In this experiment, hyperacetylated histones did not modify the cell membrane potential. Moreover, HUVEC were sensitive to the levels of extracellular histones when we explored the response against the oxidative stress because the mRNA levels of the antioxidant enzymes (ie Superoxide dismutase 1 (SOD1), Superoxide dismutase 2 (SOD2), Catalase (CAT) and Glutathione peroxidase 1 (GPX1)) increased in a dose-dependent manner (Figure 2D). Antioxidant enzymes were higher in HUVEC challenged with native histones than in those treated with hyperacetylated histones (Figure 2E and F). These results suggest that hyperacetylation of histone cushions deleterious effects related to the oxidative stress induced by extracellular histones.
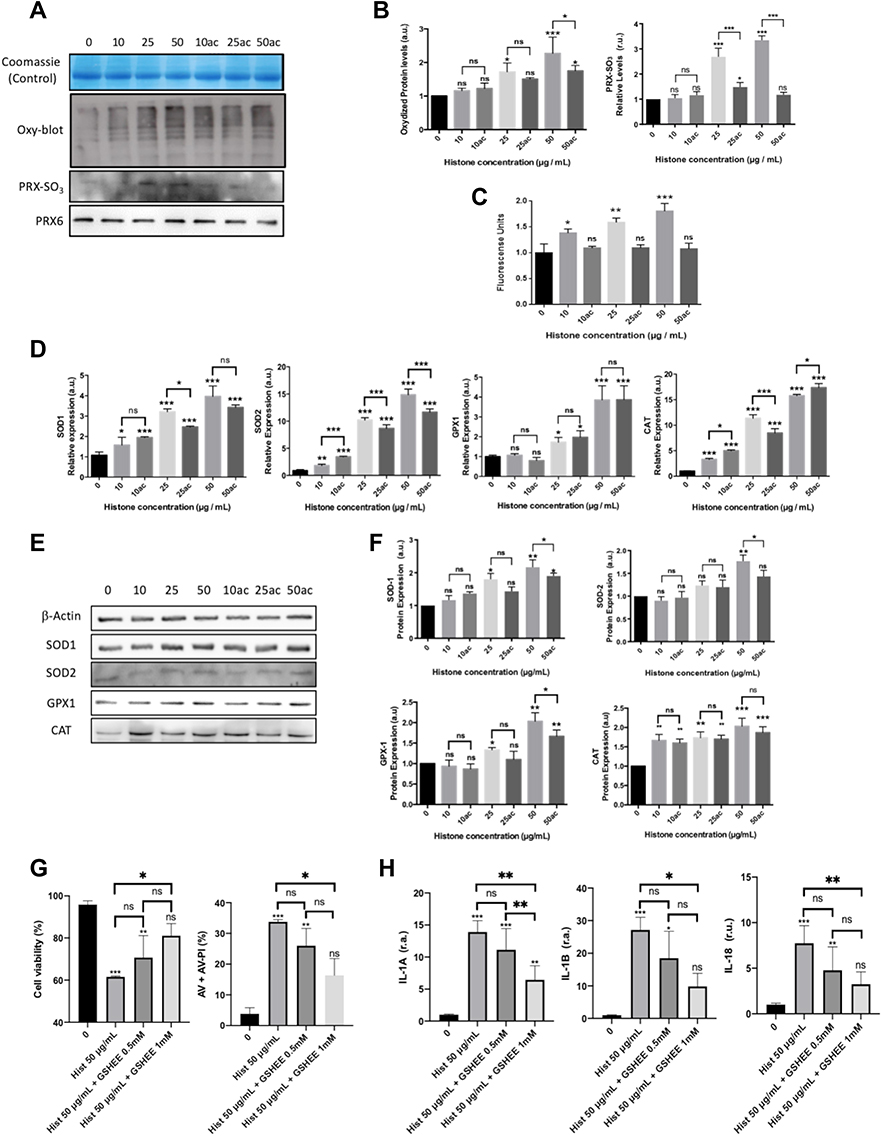 |
Figure 2 Analysis of oxidative stress markers, cell membrane potential and antioxidant defenses in HUVEC-treated with different concentrations of native or hyperacetylated extracellular histones. (A) Oxyblot of the carbonylated proteins and WB for oxidized Prx6-SO3, Coomassie blue gel was used as loading control for oxyblot and PRX6 total levels were used as reference of its peroxidized form; (B) Densitometry of the relative levels for carbonylated proteins and oxidized Prx6-SO3; (C) Cell membrane potential measured with the DIBAC probe by means the use of flow cytometry after counting 10,000 cells per each condition; (D) Relative expression of genes codifying for antioxidant enzymes (SOD1, SOD2, GPX1 and CAT) by RT-qPCR; (E) the WB of antioxidant enzymes (SOD1, SOD2, GPX1 and CAT); (F) Relative expression of the antioxidant enzymes quantified by densitometry of specific bands in the WB membrane and using β-actin as a loading control. (G) HUVEC cell viability and Annexin V/PI-positive cells after incubation with GSH-EE and native histones treatment analyzed by flow cytometry after counting 10,000 cells per each condition; (H) Relative expression levels of IL1A (left), IL1B (middle) and IL18 (right) determined by RT‐qPCR. Data are expressed as mean±SEM of 3–6 independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001 versus histones 0μg/mL. The lines at the top of columns indicate differences between compared conditions. Abbreviation: ns, non-significant p value. |
To demonstrate the control of redox switches involved in the inflammasome and pyroptosis activation, we pre-incubated HUVEC for 1 h with 0.5 mM and 1 mM of GSH-EE 1 hour prior to the 4-hour challenge with extracellular histones (50 μg/mL). Our findings demonstrated that GSH-EE 1 mM was able to reduce the histone-mediated pyroptosis and cell-death (Figure 2G) and decreased the production of the inflammasome-mediated IL-1α, IL-1β and IL-18 (Figure 2H).
Extracellular Histones Affected Key Prostanoid Biosynthesis Pathways in Human Endothelial Cells
Cyclooxygenase-1 (COX-1) participates in the prostanoids synthesis during vascular homeostasis. Cyclooxygenase-2 (COX-2) is induced by proinflammatory cytokines and stress and is a major source of prostanoids biosynthesis during inflammation.45 Our results showed that extracellular histones produced the overexpression of COX1 and COX2 in a dose‐dependent manner (Figure 3A–C). Interestingly, gene expression and protein levels of both cyclooxygenases were lower in HUVEC treated with hyperacetylated histones than native histones under the same conditions (Figure 3A–C).
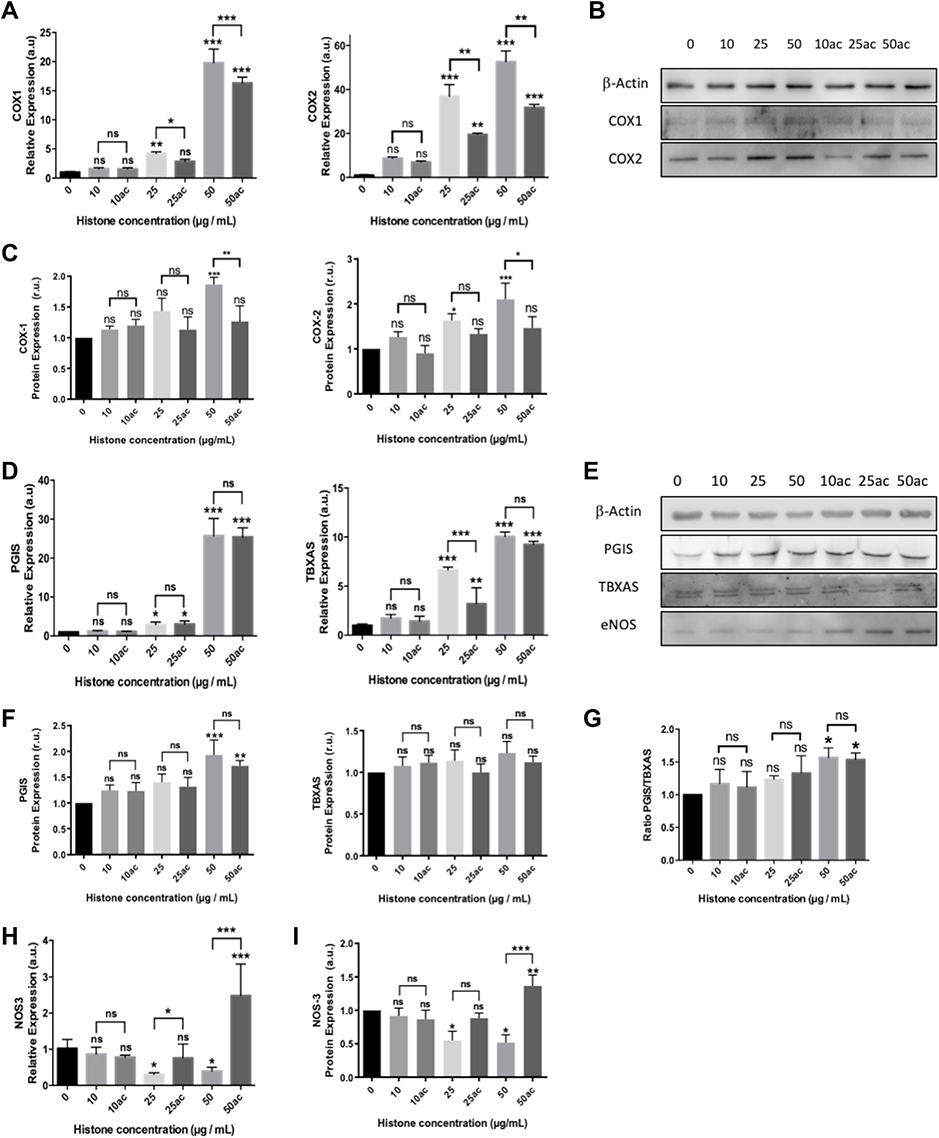 |
Figure 3 Extracellular histones (native and hyperacetylated) alter HUVEC prostanoid production enzymes through the up‐regulation of the COX‐Prostanoids pathway: (A) Relative COX1 and COX2 expressions determined by qRT‐PCR; (B) Representative WB images of COX-1 and COX-2 in HUVEC treated with extracellular histones for 4 h. β‐actin was used as the loading control; (C) Relative protein COX1 and COX2 levels quantified by densitometry of specific bands in the WB membrane; (D) Relative expression levels of PGIS (left) and TBXAS (right) determined by qRT‐PCR; (E) Protein levels analyzed by WB using anti‐PGIS, anti‐TBXAS and anti-eNOS. β‐actin was used as the loading control. One representative experiment of the three performed is shown; (F) Relative protein levels of PGIS and TBXAS quantified by densitometry of specific bands in the WB membrane; (G) The PGIS/TBXAS ratio of the relative levels of each protein assessed by densitometry of specific bands in the WB membrane; (H) NOS3 gene expression levels determined by RT-qPCR; (I) Relative protein eNOS levels quantified by densitometry of specific bands in the WB membrane. Densitometry was assessed by means of three independent WB experiments. Data are expressed as mean±SEM of three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001 compared to histones 0μg/mL. The lines at the top of columns indicate differences between compared conditions. Abbreviation: ns, non-significant p value. |
The vascular endothelium plays a key role in regulating cardiovascular homeostasis by controlling vascular tone. Therefore, it is likely that extracellular histones alter the appropriate function of HUVEC cells. Accordingly, PGE2 and TBXA2 are two of the main vascular prostanoids participating in the relaxation/contraction of blood vessels. PGE2 and TBXA2 are produced by prostacyclin synthase (PGIS) and thromboxane synthase (TBXAS), respectively. Vascular homeostasis also depends on the nitric oxide (NO), which is generated by endothelial nitric oxide synthase (eNOS).
Regarding prostanoids synthesis, the increased mRNA levels of PGIS (Figure 3D) were accompanied by a larger amount of protein expression at 50 μg/mL of extracellular histones (Figure 3E and F, respectively). Despite the increase in the gene expression of TBXAS, no changes in protein levels were found.
Although TBXAS levels were not affected, the elevated levels of PGIS (Figure 3E and F) produced changes in the PGIS/TBXAS ratio (Figure 3G) which in turn may induce changes in vasoactive compounds’ production. Moreover, native extracellular histones decreased NOS3 expression in HUVEC (Figure 3H). Conversely, the treatment of HUVEC with hyperacetylated histones did not decrease the expression of eNOS (Figure 3E–I) at concentrations of extracellular histones below 50 μg/mL, but at 50 μg/mL eNOS expression levels were increased.
Extracellular Histones Changed Endothelial Adhesion Molecules Expression and Induced Inflammatory Responses
NLRP3 inflammatory activation in endothelial cells after infection has been described as giving rise to inflammation and the dysregulation of ICAM-1 and VCAM-1.46
Our results show that concentrations above 25 μg/mL of extracellular histones produced IL6 overexpression (Figure 4A), which is related to the increase of ICAM1, VCAM1 and ESEL expression in a dose‐dependent manner (Figure 4B–D). However, treatment with hyperacetylated histones did not produce the same effect on the expression of IL6, ICAM1, VCAM1 and ESEL, indicating that hyperacetylation may reduce the cellular signaling involved in inflammation and the expression of these molecules.
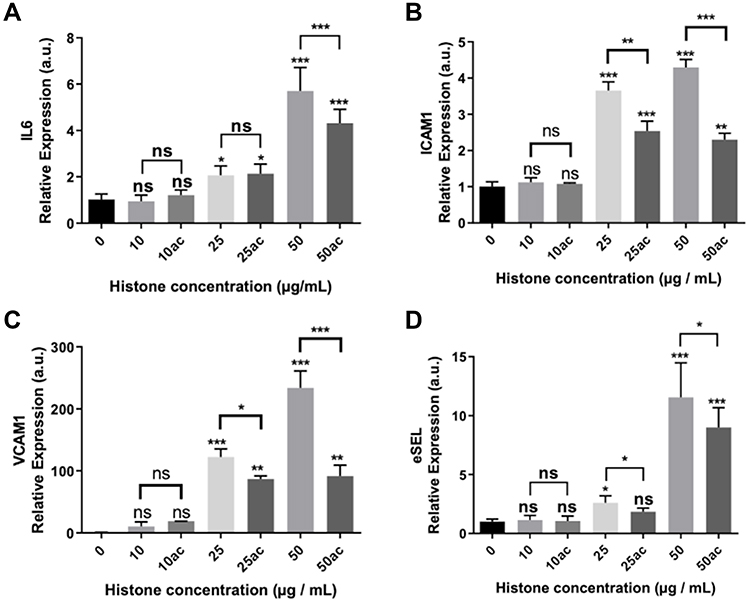 |
Figure 4 Expression of pro-inflammatory and endothelial adhesion factors in HUVEC exposed to 10–50μg/mL of extracellular histones (native and hyperacetylated) for 4 h. Relative expression of (A) IL6; (B) ICAM1; (C) VCAM1 and (D) eSEL gene expression levels determined by qRT‐PCR. Data are expressed as mean±SEM of three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001 versus histones 0μg/mL. The lines at the top of columns indicate differences between compared conditions. Abbreviation: ns, non-significant p value. |
Inflammasome Inhibition Prevents Subsequent Effects Mediated by Extracellular Histones in HUVEC
Both secondary apoptosis and pyroptosis showed positive Annexin V and PI signals. In this regard, to confirm pyroptosis in HUVEC exposed to extracellular histones, we used two specific inhibitors, Z-LEHD-FMK (LEHD) a specific inhibitor of apoptosis mediated by Caspase-9, and MCC950, which inhibits NLRP3-mediated pyroptosis.
HUVEC cells pre-incubated with LEHD recovered part of the cell viability when treated cells with native extracellular histones, indicating that other cell death mechanisms may be involved (Figure 5A). However, the highest cell viability was recovered when HUVEC cells were treated with MCC950, indicating the main mechanism involved in cell death was mostly pyroptosis (Figure 5B). Nevertheless, apoptosis plays a more important role than pyroptosis when HUVECs were exposed to hyperacetylated extracellular histones because MCC950 barely had an effect on cell viability and AV/PI-positive cells when MCC950 was used as inhibitor of pyroptosis (Figure 5B and C).
 |
Figure 5 Inflammasome inhibition prevents subsequent effects mediated by extracellular histones in HUVEC: (A) HUVEC cell viability treated with LEHD inhibitor; (B) HUVEC cell viability treated with MCC950 inhibitor (C) HUVEC apoptosis+pyroptosis (measured as Annexin V/PI positive cells) after the challenge with extracellular histones during 4 h and MCC950 for 6 h (2 h before the challenge with 50μg/mL of extracellular histones); (D) IL1A gene expression; (E) IL1B gene expression; (F) IL18; (G) PGIS gene expression; (H) TBXAS gene expression; (I) NOS3 gene expression; (J) COX1 gene expression; (K) COX2 gene expression; (L) IL6 gene expression; (M) ICAM1 gene expression; (N) VCAM1 gene expression; (O) ESEL gene expression measured by RT-qPCR. Data are expressed as mean±SEM of three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001 compared to histones 0μg/mL. The lines at the top of columns indicate differences between compared conditions. Abbreviation: ns, non-significant p value. |
To confirm the inhibition of pyroptosis when NLRP3 was inhibited, we evaluated the expression of inflammasome-released pro-inflammatory cytokines and demonstrated that HUVEC treated with MCC950 lowered the levels of pro-inflammatory interleukins IL1A, IL1B and IL18 (Figure 5D–F, respectively).
Regarding the antioxidant response, our results demonstrated only a statistically significant decrease in SOD2 expression in cells with inhibited NLRP3 inflammasome, suggesting the role of extracellular histones in mediating mitochondrial superoxide production (Supplementary Figure 4). Furthermore, we also studied downstream responses in the endothelial function and the immune response mediated by endothelium in HUVEC after blocking the NLRP3 inflammasome. Our results indicated decreased expression for PGIS and TBXAS and restored levels in NOS3 (Figure 5G–I, respectively), after blocking the inflammasome in HUVEC which were challenged with native histones. Accordingly, COX1 and COX2 were also downregulated when cells were treated with MCC950 (Figure 5J and K) and challenged with native histones, and to a lesser extent with hyperacetylated histones, which suggests a reduction of the pro-inflammatory profile in HUVEC. These results agree with the decreased pro-inflammatory cytokine IL6 expression observed after inhibiting the inflammasome (Figure 5L). Moreover, as IL6 overexpression positively correlated with the increase in endothelial adhesion molecules, we evaluated again the expression of endothelial adhesion factors after inhibiting the NLRP3 inflammasome and found that after inflammasome inhibition, native extracellular histones treatment of HUVEC did not produce any alteration in ICAM1 and VCAM1 levels (Figure 5M and N) and decreased the expression of NOS3 (Figure 5O).
Pyroptosis is Active in Septic Shock Patients
Healthy subjects’ mean age was 47 ± 14, with males accounting for 55.6% (see the Table 1); ICU patients (non-septic critically ill patients) mean age was 68 (68 ± 8) with males accounting for 50%; septic patients’ mean age was 68 (68 ± 11) with males accounting for 55.6%; SS patients’ mean age was 65 (65 ± 15) with males accounting for 73.3%. Table 1 provides the detailed clinical data. The APACHE II score showed no differences among patients. The median SOFA score value of 9 ± 3 was the highest for the SS patients versus the ICU controls (5 ± 3) and sepsis patients (6 ± 2). The highest values were found in SS for CRP, PCT and lactate in SS (see Table 1).
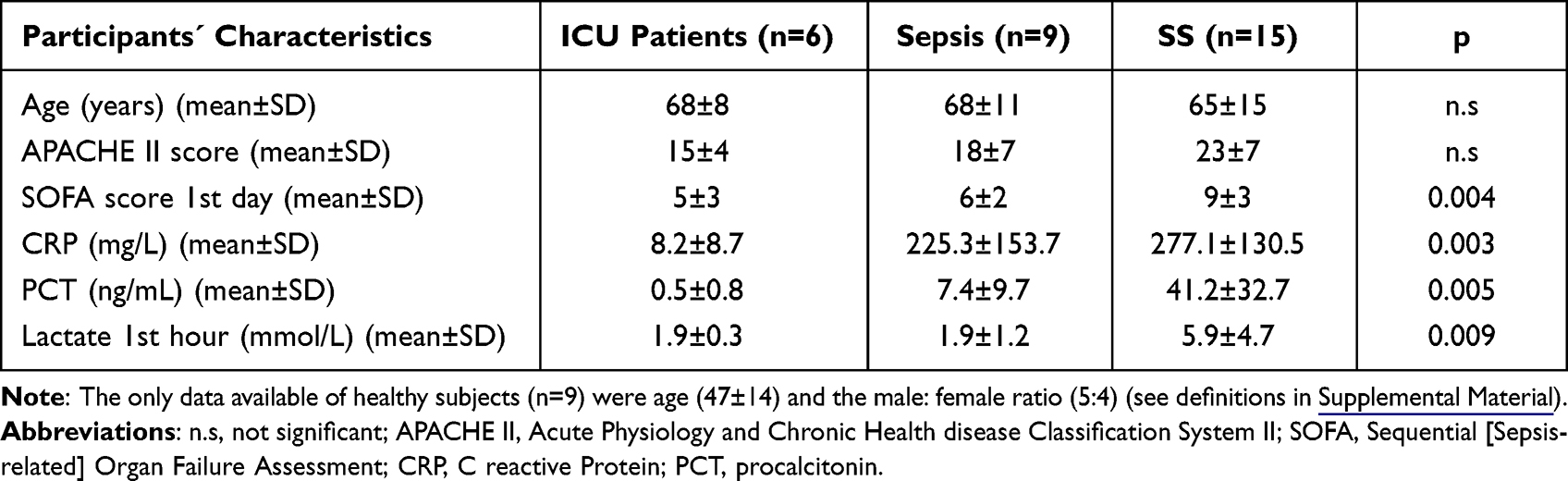 |
Table 1 Clinical Data of the ICU, Sepsis and SS Patients |
Since histones are inductors of pyroptosis, we measured circulating histones in plasma by mass spectrometry using QTRAP instrument, as we previously published.9 Our results demonstrated that the SS patients had the highest levels of blood circulating histones (Figure 6A). We found that the high levels of circulating histones were associated with sepsis severity and fatal outcome (mean values [H2B]: 49.44 (0.01–227.32) ng/mL in survivors vs 1392.90 (26.50–3414.16) ng/mL in no survivors; p-value 0.004 U Mann–Whitney test) (mean values [H3]: 4.33 (0.01–64.87) ng/mL in survivors vs 310.93 (0.01–1014.81) ng/mL in non-survivors; p-value 0.023 U Mann–Whitney test). Despite a tendency to rise, we did not observe statistically significant increased levels for IL-1β (Figure 6C). However, circulating caspase-1 (Figure 6B) and pyroptosis-released cytokine IL-18 (Figure 6D) and IL-1α (Figure 6E) were higher in SS patients when compared to sepsis and critically ill patients. The results demonstrated that both canonical (through IL-18) and non-canonical (through IL-1α) pyroptosis simultaneously occurred in sepsis.
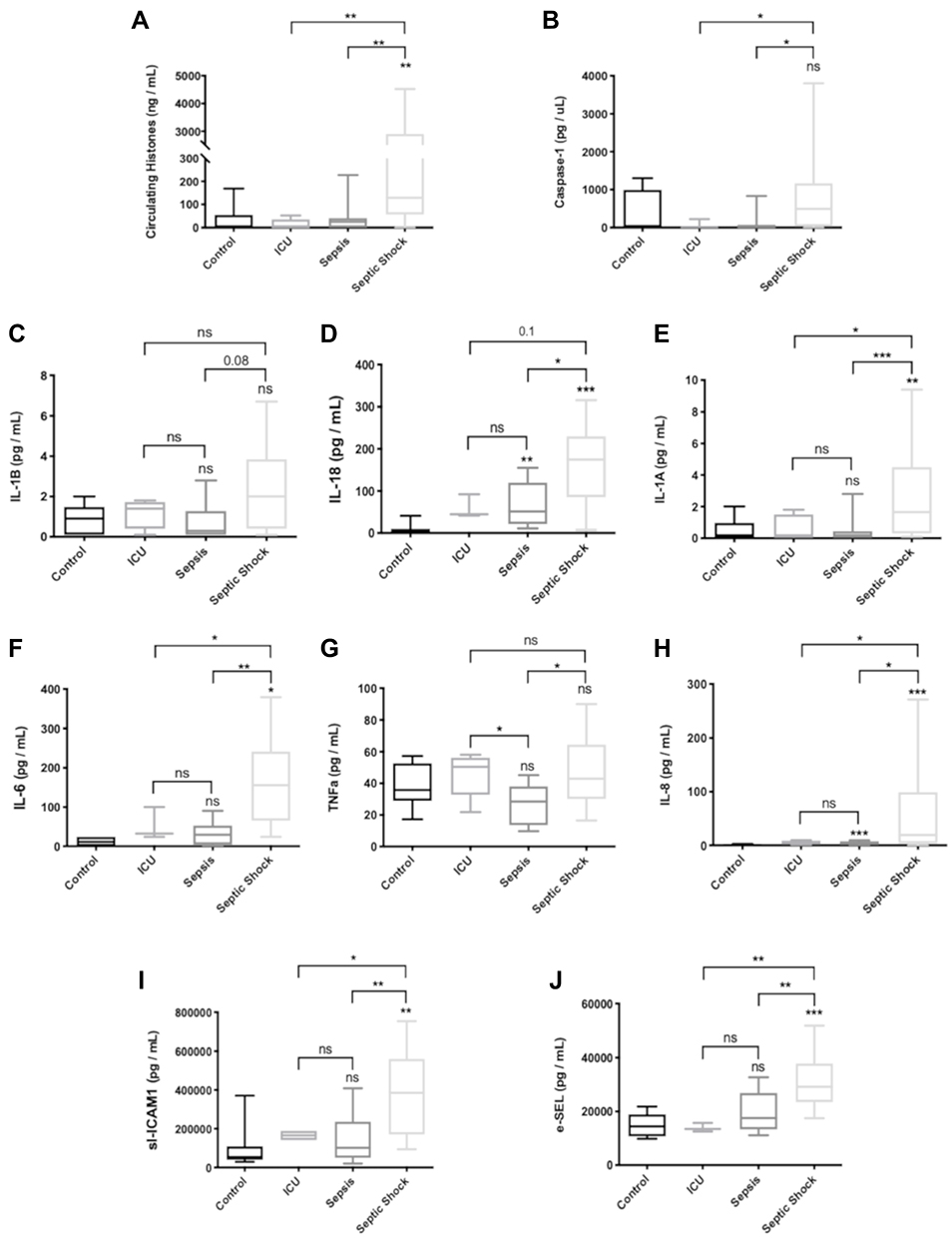 |
Figure 6 Levels of circulating markers in plasma of patients admitted in the ICU. (A) Circulating histones levels referred to as the sum of H2B, H3 and H4 concentrations in plasma determined by MRM-MS; (B) Caspase-1 levels; (C) IL-1β levels; (D) IL-18 levels; (E) IL-1α levels; (F) IL-6 levels; (G) TNF-α; (H) IL-8; (I) sl-ICAM-1 levels; (J) e-SEL levels. Data are expressed as mean±SEM from two independent analysis. *P < 0.05; **P < 0.01; ***P < 0.001. The lines at the top of columns indicate differences between compared conditions. Abbreviation: ns, non-significant p value. |
Moreover, we analyzed the correlations between the circulating histone levels, caspase-1, IL-18 and IL-1α and observed a statistical significance in the positive correlation among them and the circulating histone levels (Table 2). Blood circulating histone levels showed a statistically significant positive correlation with SOFA and lactate levels, which suggests the role of circulating histones with SS severity and poor prognosis. Blood circulating caspase-1 and IL-18, markers of pyroptosis, showed a significantly statistical positive correlation with the SOFA scores and lactate levels (Table 2).
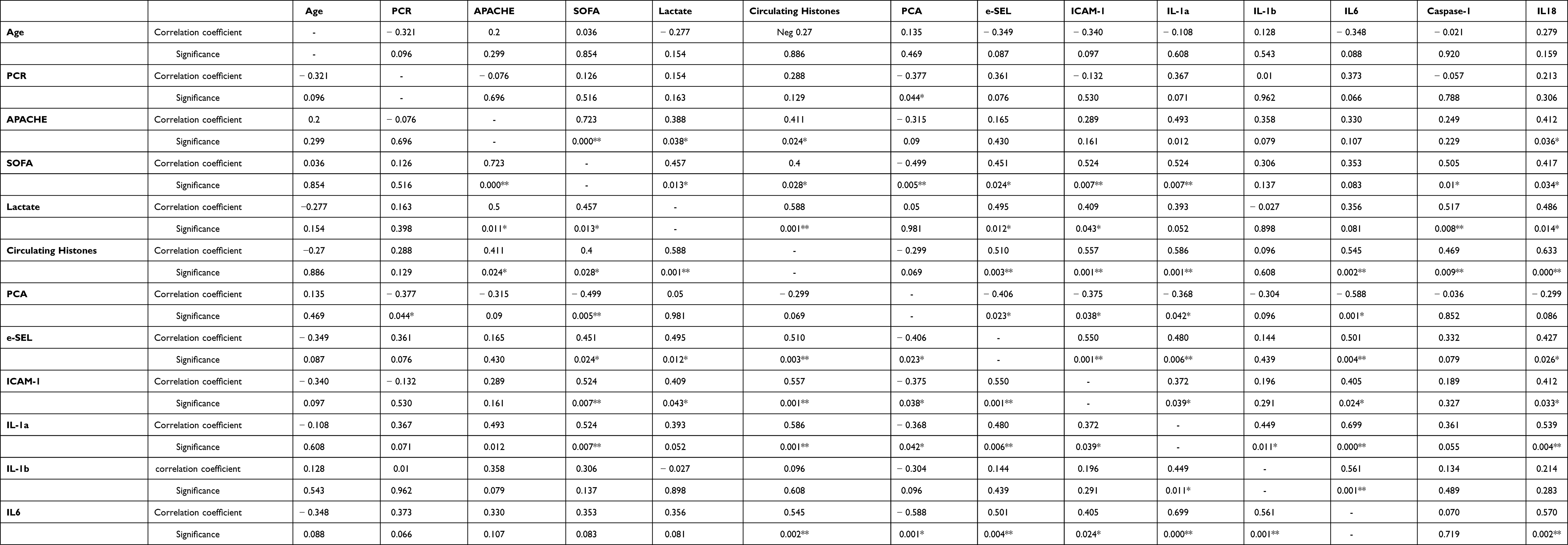 |
Table 2 Correlation Between Circulating Histones and Clinical Parameters Measured |
When analyzing the circulating levels of relevant pro-inflammatory cytokines in sepsis, such as IL-6 (Figure 6F), TNFα (Figure 6G) and IL-8 (Figure 6H), we observed that the SS patients had the highest levels. We also measured levels of endothelial adhesion factors in plasma, such as sl-ICAM-1 (Figure 6I) and e-SEL (Figure 6J) and found that the SS patients had the highest values for both mediators of endothelial function.
We also analyzed statistical correlations between circulating histones, pro-inflammatory cytokines and molecular adhesion factors. Our results revealed a correlation between circulating histones and IL-6, ICAM-1 and e-SEL (Table 2). We also observed a significant statistical positive correlation between pyroptosis-related cytokines IL-1α and IL-18 with pro-inflammatory IL-6 and endothelial adhesion factors (Table 2).
Discussion
Blood-circulating histones are DAMPS in sepsis and induce cellular toxicity through receptor-dependent and independent mechanisms.4,15 Whether the activation of endothelial inflammasomes by histones directly causes endothelial dysfunction or injury without recruiting immune cells remains unknown. The elucidation of the mechanisms mediated by extracellular histones activating the inflammasome, and subsequently pyroptosis, may unveil some complex events occurring in sepsis. Indeed, the activation of the NLRP3 inflammasome, inflammation, oxidative stress and alteration of the redox homeostasis, antioxidant enzymes expression, endothelial impairment and cell death are closely connected events. All these cellular responses contribute to vascular damage, endothelial dysfunction, and overexpression of adhesion molecules, which are all associated with sepsis.47,48
Previous research has demonstrated that ROS can activate endothelial NLRP3 inflammasomes under hyperglycemic conditions in vitro50 and in a mouse model, proposing that this mechanism may initiate endothelial dysregulation.49 Similarly, we suggest that oxidative stress induced by extracellular histones in a dose-dependent manner activates the NLRP3 inflammasome. ROS is a major driver of inflammation and vice versa, and the latest research suggests a key role of the NLRP3 inflammasome in the endothelium function and immunity.44,51–54 In fact, oxidative stress induces NLRP3 activation by triggering deglutathionylation of ASC (the adaptor molecule apoptosis-associated speck-like protein containing a CARD) to promote pyroptosis.55 Moreover, glutathionylation of NLRP3 has been described to occur in macrophages.56 This finding warrants further research, as GSH may by itself limit the inflammasome activation. In this regard, Guglielmo et al and Zhang et al demonstrated that GSH or GSSG strongly inhibited inflammasome activation in vitro and in vivo.56,57 In the same way, we demonstrated that GSH supply by pre-incubating HUVEC cells with GSH-EE prevented cell death through pyroptosis triggered by extracellular histones. Moreover, our results showed that histone-mediated oxidative stress is a key player in the activation of pyroptosis in HUVEC cells.
Histones may be embedded in the plasma membrane due to electrical charge affinity, so they can alter the membrane potential of endothelial cells. In this regard, it has been recently proposed that nucleosomes can enter the cytoplasm of mammalian cells via clathrin- or caveolin-dependent endocytosis.58 It is noteworthy that histones destabilize the mitochondrial membranes,59 so once histones are internalized into the cells through endocytosis, both linker histone H1 and core histones can permeabilize the outer mitochondrial membrane, therefore altering the mitochondrial membrane potential. We agree with this idea, and in our model, oxidative stress may be induced mainly by hydrogen peroxide (H2O2), which may produce the oxidation of proteins60 and generate the hyperoxidized form of Prdx6.61 Furthermore, our results suggest the antioxidant defense is not enough to attenuate the histone-mediated oxidative stress. Although our results indicate that oxidative stress takes place after extracellular histones challenge, to gain deeper insight into the mechanisms involved in the production of oxidative stress, NADPH oxidase and uncoupled mitochondria may be further explored as feasible sources of ROS. Furthermore, our results also suggest that antioxidant defense is not enough to attenuate the histone-mediated oxidative stress.
Interestingly, neutrophil pyroptosis is acquiring relevance in sepsis.62 However, as far as we know pyroptosis in endothelial cells has not yet been described in sepsis. Thus, here we describe a new pathway in which extracellular histones regulate NLRP3 inflammasome activation in human endothelial cells.
Our results showed that extracellular histones induce both pyroptosis and apoptosis. Activation of the NLRP3 inflammasome induces IL-1β and IL-18 expression, promoting many metabolic, physiological, inflammatory and immunological effects.51 In fact, it is known that IL-1β increases the generation of prostanoids, and mice lacking COX-2 are defective in producing IL-18.63 This suggests an interconnection between the inflammasome and the prostanoid biosynthesis pathway. Moreover, IL-1α, which is released in the non-canonical pyroptosis, induces the expression of COX genes.62 Our results showed extracellular histones above 50 μg/mL mediate the overexpression of IL1B, IL18 and IL1A genes and, in turn, COX1 and COX2, thus demonstrating the link between pyroptosis-released cytokines and the homeostasis of endothelium.
Sepsis alters vasoconstriction by regulating prostanoids levels.65 Importantly, it has been shown that the NLRP3 inflammasome activates PGE2 expression via IL-1β/IL-1R signaling.64 The COX-2-dependent generation of prostanoids in endothelial cells participates in vascular functions, which are altered during sepsis and SS.65 Our results reinforce the notion that extracellular histones alter vascular homeostasis by modifying the PGIS/TBXAS ratio and affecting eNOS expression and NO production, which dilates blood vessels by acting on smooth muscle cells and inhibiting platelet aggregation and leukocyte adhesion.66
In response to IL-1 (IL-1α and IL-1β), endothelial cells express eSEL, which serves to neutrophils and monocytes to be anchored to the endothelium. In addition to the interleukins overexpression, our results showed enhanced expression of endothelial cell adhesion molecules (eSEL, ICAM-1, and VCAM-1) after extracellular histone exposition. In response to IL-1 (IL-1α and IL-1β) and IL-6, endothelial cells express eSEL, which serves to neutrophils and monocytes to be anchored to the endothelium and stimulates the expression of ICAM-1 and VCAM-1, promoting the rolling of the leukocytes to the inflammation site.67,68 The activation of the NLRP3 inflammasome mediates the up-regulation of endothelial adhesion molecules, a process which is also contributed by ROS and IL-1β.69,70 Moreover, ICAM-1 and VCAM-1 are key players in neutrophil adhesion, transcellular migration and organ damage during sepsis and SS, and their overexpression is a common feature in sepsis.46,71,72 Our findings agree with these results and demonstrate that extracellular histones increase the expression of adhesion molecules in HUVEC, being their overexpression less pronounced when HUVEC were treated with hyperacetylated histones. It is noteworthy, IL-1β cleavage mediated by caspase-1 activation is related to platelet activation, aggregation and thrombus formation, contributing to hemostatic abnormalities in sepsis and SS.73 Our results agree with molecular processes with well-recognized endothelial dysfunction features, which contribute to severe sepsis.
To enlighten the role of extracellular histones-mediated inflammasome activation, we inhibited the NLRP3 inflammasome with MCC950. This process increased HUVEC viability and restored the levels of pro-inflammatory mediators and endothelial adhesion factors. Perera et al36 have described how MCC950 diminishes the activation of caspase-1, and reduced the release of IL-1β, IL-18, IL-1α and NO in colonic explants. Hence, we propose that the NLRP3 inflammasome is also a key mediator in endothelial activation by extracellular histones.
Finally, we confirmed that pyroptosis occurs in SS patients, in which high IL-18 and IL-1α levels in blood were found. We observed a positive correlation among caspase-1, IL-1α, IL-18 and circulating histones levels, which fell within the range of the previous levels detected by us and other authors.2,9,39 Our results indicate circulating histones mediate pyroptosis and endothelial dysfunction and the release of endothelial adhesion molecules, as we observed in the plasma of SS patients. In fact, circulating histones levels showed a positive correlation with the SOFA and lactate levels, and with the expression of other pro-inflammatory cytokines (eg, TNF-α and IL-8), and the highest levels of circulating histones were found in those patients who finally died.
Although circulating histones in blood may reflect those derived from immune and/or endothelial subsets, the final consequence of extracellular histones is to activate different mechanisms of cell death and tissue damage including pyroptosis. Based on our results, we suggest that blocking the upstream activation of the NLRP3 inflammasome mediated by circulating histones may set the basis for a feasible therapy. Indeed, our in vitro studies demonstrated that by blocking the NLRP3 inflammasome, it was possible to modulate the endothelium activation mediated by extracellular histones.
As far as we know, no one has previously analyzed the effect of post-translational modifications in histones-mediated HUVEC cell death and dysfunction. Our results demonstrated that hyperacetylated extracellular histones were less cytotoxic and were not able to induce pyroptosis in HUVEC. Accordingly, although hyperacetylated extracellular histones were also able to activate the NLRP3 inflammasome, their effect on the release of pyroptosis-related cytokines was less pronounced than that caused by native histones.
Some authors have speculated on the effect of negatively charged agents to neutralize histone-mediated effects.74 Aligning with this idea, we propose that the hyperacetylation of histone positive charged lysins may contribute to decrease the cytotoxic activity of extracellular histones in endothelial cells. From a future perspective, we propose that blocking the mechanisms stimulated by circulating histones such as pyroptosis, removing extracellular histones, or even the use of histone deacetylase inhibitors to reduce the cellular cytotoxicity mediated by circulating histones, are feasible therapeutic strategies in sepsis. In fact, the simultaneous blocking of NLRP3 and increased hyperacetylation of histones may set the basis for a promising strategy. This is consistent with recent results describing a new generation of HDACi, which have demonstrated its potential to inhibit NLRP3 inflammasome.75,76
In conclusion, our study provides further insights into the molecular mechanisms contributing to sepsis severity stimulated by circulating histones and their role in inducing pyroptosis in endothelial cells, which in turn contribute to inflammation, oxidative stress, altered the enzymatic antioxidant expression and endothelial dysfunction.
Conclusions
Extracellular histones induce a membrane depolarization, increasing oxidative stress, that in turn triggers endothelial pyroptosis via NLRP3 inflammasome in endothelial cells. Histone hyperacetylation plays a protective role, as well as, the use of the antioxidant glutathione ethyl ester and the NLRP3 inhibitor MCC950. Clinically, septic shock patients showed the highest levels of circulating histones, which correlated with pyroptosis-related cytokine and elevated endothelial mediators, which in turn was associated with sepsis severity and poor outcome.
Ethics Approval and Consent to Participate
The study was approved by the local Medical Ethical Committee (CEIm at Hospital Clínico Universitario de Valencia) on June 30th, 2016. Written informed consent was obtained from all participants. Informed consent was obtained from all individual participants included in the study.
Acknowledgments
We thank Beatriz Jávega (University of Valencia) who helped us in the flow cytometry experiments Luminex Panel measurements. Oreto Antúnez (University of Valencia (SCSIE) Proteomics lab) for measuring circulating histones in the plasma samples. We thank Eva M. Garcia-López (Department of Legal Medicine. University of Valencia) for her support in the statistical analysis. We especially thank MD. Nieves Carbonell, MD. María Rodríguez-Gimillo and MD José Ferreres for obtaining blood samples from patients. The abstract of this paper was presented at the SFRR-International 2021 Virtual Meeting as a poster presentation with interim findings. The poster’s abstract was published in Free Radical Biology and Medicine as part of a special issue.
Parts of this paper are included in a Thesis work of the main author, which was uploaded to the University of Valencia repository as a thesis in January 20th, 2022. https://roderic.uv.es/handle/10550/81440.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
J.B-G. is supported by a Contratos i-PFIS grant (IFI18/00015) and co-financed by the European Social Fund. R.O-V. is supported by Contratos PFIS grant (FI20/00202) from AES-ISCIII and co-financed by the European Regional Development Fund (ERDF). D.P-C. Thanks Spanish Ministerio de Ciencia e Innovación for the fellowship “Juan de la Cierva - Incorporación” (grant number IJC2019-040237-I). J.L.G-G. thanks INCLIVA and GVA for the starting grants GV/2014/132) and AES2016 (ISCIII) for grant numbers PI16/01036, DTS21/00193 and PI19/00994, co-financed by the European Regional Development Fund (ERDF). S.N. and C.H. thank AES2016 (ISCIII) for grant number PI16/00229 and PI19/01714, co-financed by the European Regional Development Fund (ERDF). J.L.G-G. and FVP thank Grand Challenges Canada and the Spanish Ministry of Science and Innovation, ISCIII through CIBERer (Biomedical Network Research Center for Rare Diseases). We would like to thank INCLIVA’s Biobank ISCIII, FEDER for grant PT13/0010/0004 and PT20/00029.
Disclosure
Dr José Luis García-Giménez reports grants from Spanish Institute of Health, grants from Ministerio de Ciencia e Innovación, during the conduct of the study; personal fees from EpiDisease S.L., outside the submitted work; In addition, Dr José Luis García-Giménez has a patent EP3535587 licensed to EpiDisease S.L. Dr Carlos Hermenegildo reports grants from University of Valencia, during the conduct of the study. Federico V Pallardó has a pending patent related to this work. The other authors report no conflicts of interest.
References
1. Goodwin JE, Feng Y, Velazquez H, Sessa WC. Endothelial glucocorticoid receptor is required for protection against sepsis. Proc Natl Acad Sci. 2013;110(1):306–311. doi:10.1073/pnas.1210200110
2. Beltrán-García J. Use of circulating histones and their post-translational modifications as biomarkers in sepsis and septic shock [dissertation]; 2022. Available from: https://roderic.uv.es/handle/10550/81440.
3. Vardon-Bounes F, Ruiz S, Gratacap M-P, Garcia C, Payrastre B, Minville V. Platelets are critical key players in sepsis. Int J Mol Sci. 2019;20(14):3494. doi:10.3390/ijms20143494
4. Nakahara M, Ito T, Kawahara K, et al. Recombinant thrombomodulin protects mice against histone-induced lethal thromboembolism. PLoS One. 2013;8(9):e75961. doi:10.1371/journal.pone.0075961
5. Abrams ST, Zhang N, Manson J, et al. Circulating histones are mediators of trauma-associated lung injury. Am J Respir Crit Care Med. 2013;187(1):160–169. doi:10.1164/rccm.201206-1037OC
6. Pérez-Cremades D, Bueno-Betí C, García-Giménez JL, et al. Extracellular histones disarrange vasoactive mediators release through a COX-NOS interaction in human endothelial cells. J Cell Mol Med. 2017;21(8):1584–1592. doi:10.1111/jcmm.13088
7. Opitz B, Eitel J, Meixenberger K, Suttorp N. Role of Toll-like receptors, NOD-like receptors and RIG-I-like receptors in endothelial cells and systemic infections. Thromb Haemost. 2009;102(12):1103–1109. doi:10.1160/TH09-05-0323
8. Hoeksema M, Van Eijk M, Haagsman HP, Hartshorn KL. Histones as mediators of host defense, inflammation and thrombosis. Future Microbiol. 2016;11:441–453. doi:10.2217/fmb.15.151
9. García-Giménez JL, Romá-Mateo C, Carbonell N, et al. A new mass spectrometry-based method for the quantification of histones in plasma from septic shock patients. Sci Rep. 2017;7(1):1–10. doi:10.1038/s41598-017-10830-z
10. Chen R, Kang R, Fan X-G, Tang D. Release and activity of histone in diseases. Cell Death Dis. 2014;5:1–9. doi:10.1038/cddis.2014.337
11. Silk E, Zhao H, Weng H, Ma D. The role of extracellular histone in organ injury. Cell Death Dis. 2017;8(5):2812. doi:10.1038/cddis.2017.52
12. Carson WF, Kunkel SL. Regulation of cellular immune responses in sepsis by histone modifications. Adv Protein Chem Struct Biol. 2017;106(1):191–225. doi:10.1016/bs.apcsb.2016.08.004
13. Yipp BG, Kubes P. NETosis: how vital is it? Blood. 2013;122(16):2784–2795. doi:10.1182/blood-2013-04-457671.In
14. Xu J, Zhang X, Pelayo R, et al. Extracellular histones are major mediators of death in sepsis. Nat Med. 2009;15:1318–1321. doi:10.1038/nm.2053
15. Allam R, Kumar SVR, Darispundi MN, Anders H-J. Extracellular histones in tissue injury and inflammation. J Mol Med. 2014;92:465–472. doi:10.1007/s00109-014-1148-z
16. Shaw RJ, Austin J, Taylor J, et al. Circulating histone levels correlate with the severity of COVID-19 and the extent of coagulation activation and inflammation. Blood. 2020;136(Supplement 1):19. doi:10.1182/blood-2020-142344
17. Ng H, Havervall S, Rosell A, et al. Circulating markers of neutrophil extracellular traps are of prognostic value in patients with COVID-19. Arterioscler Thromb Vasc Biol. 2021;41(2):988–994. doi:10.1161/ATVBAHA.120.315267
18. Ibañez-Cabellos JS, Aguado C, Pérez-Cremades D, et al. Extracellular histones activate autophagy and apoptosis via mTOR signaling in human endothelial cells. Biochim Biophys acta Mol basis Dis. 2018;1864(10):3234–3246. doi:10.1016/j.bbadis.2018.07.010
19. Huang H, Chen H-W, Evankovich J, et al. Histones activate the NLRP3 inflammasome in Kupffer cells during sterile inflammatory liver injury. J Immunol. 2013;191(5):2665–2679. doi:10.4049/jimmunol.1202733
20. Elliott EI, Sutterwala FS. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol Rev. 2015;265(1):35–52. doi:10.1111/imr.12286
21. Sutterwala FS, Haasken S, Cassel SL. Mechanism of NLRP3 inflammasome activation. Ann N Y Acad Sci. 2014;1319(1):82–95. doi:10.1111/nyas.12458
22. Jin L, Batra S, Jeyaseelan S. Deletion of Nlrp3 augments survival during polymicrobial sepsis by decreasing autophagy and enhancing phagocytosis. J Immunol. 2017;198(3):1253–1262. doi:10.4049/jimmunol.1601745
23. Fan Y, Du L, Fu Q, et al. Inhibiting the NLRP3 inflammasome with MCC950 ameliorates isoflurane-induced pyroptosis and cognitive impairment in aged mice. Front Cell Neurosci. 2018;12:426. doi:10.3389/fncel.2018.00426
24. Cheng KT, Xiong S, Ye Z, et al. Caspase-11–mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J Clin Invest. 2017;127(11):4124–4135. doi:10.1172/JCI94495
25. Vande Walle L, Lamkanfi M. Pyroptosis. Curr Biol. 2016;26(13):568–572. doi:10.1016/j.cub.2016.02.019
26. Liu S, Du J, Li D, et al. Oxidative stress induced pyroptosis leads to osteogenic dysfunction of MG63 cells. J Mol Histol. 2020;51(3):221–232. doi:10.1007/s10735-020-09874-9
27. Wang Y, Shi P, Chen Q, et al. Mitochondrial ROS promote macrophage pyroptosis by inducing GSDMD oxidation. J Mol Cell Biol. 2019;11(12):1069–1082. doi:10.1093/jmcb/mjz020
28. He Y, Hara H, Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. 2016;41(12):1012–1021. doi:10.1016/j.tibs.2016.09.002
29. Xue Y, Enosi Tuipulotu D, Tan WH, Kay C, Man SM. Emerging activators and regulators of inflammasomes and pyroptosis. Trends Immunol. 2019;40(11):1035–1052. doi:10.1016/j.it.2019.09.005
30. Li Y, Alam HB. Modulation of acetylation: creating a pro-survival and anti-inflammatory phenotype in lethal hemorrhagic and septic shock. J Biomed Biotechnol. 2011;2011:1–15. doi:10.1155/2011/523481
31. Zhao T, Li Y, Liu B, et al. Novel pharmacologic treatment attenuates septic shock and improves long-term survival. Surg. 2013;154(2):206–213. doi:10.1016/j.surg.2013.04.003
32. Bonizzio CR, Clara M, Rodrigues T, Soriano FG. The viability of using epigenetic drugs as a treatment of patients in sepsis - A translational perspective*. Rev Med. 2016;95(1):91–102. doi:10.11606/issn.1679-9836.v.95i2p91-102
33. Ciarlo E, Savva A, Roger T. Epigenetics in sepsis: targeting histone deacetylases. Int J Antimicrob Agents. 2013;42(1):8–12. doi:10.1016/j.ijantimicag.2013.04.004
34. Shechter D, Dormann HL, Allis CD, Hake SB. Extraction, purification and analysis of histones. Nat Protoc. 2007;2:1445–1457. doi:10.1038/nprot.2007.202
35. Wang L, Mehta S, Ahmed Y, Wallace S, Pape MC, Gill SE. Differential mechanisms of septic human pulmonary microvascular endothelial cell barrier dysfunction depending on the presence of neutrophils. Front Immunol. 2018;9:1743. doi:10.3389/fimmu.2018.01743
36. Perera AP, Fernando R, Shinde T, et al. MCC950, a specific small molecule inhibitor of NLRP3 inflammasome attenuates colonic inflammation in spontaneous colitis mice. Sci Rep. 2018;8(1):8618. doi:10.1038/s41598-018-26775-w
37. Franco R, Cidlowski JA. Glutathione efflux and cell death. Antioxid Redox Signal. 2012;17(12):1694–1713. doi:10.1089/ars.2012.4553
38. Singer M, Deutschman CS, Seymour C, et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). J Am Med Assoc. 2016;315:801–810. doi:10.1001/jama.2016.0287
39. Alhamdi Y, Abrams ST, Cheng Z, et al. Circulating histones are major mediators of cardiac injury in patients with sepsis. Crit Care Med. 2015;43(10):2094–2103. doi:10.1097/CCM.0000000000001162
40. Wang Y, Liu Q-X, Liu T, et al. Caspase-1-dependent pyroptosis of peripheral blood mononuclear cells predicts the development of sepsis in severe trauma patients: a prospective observational study. Medicine. 2018;97(8):e9859. doi:10.1097/MD.0000000000009859
41. Porter AG, Jänicke RU. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6(2):99–104. doi:10.1038/sj.cdd.4400476
42. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10(2):417–426. doi:10.1016/S1097-2765(02)00599-3
43. Cruz CM, Rinna A, Forman HJ, Ventura ALM, Persechini PM, Ojcius DM. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem. 2007;282(5):2871–2879. doi:10.1074/jbc.M608083200
44. Abais JM, Xia M, Zhang Y, Boini KM, Li P-L. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid Redox Signal. 2015;22(13):1111–1129. doi:10.1089/ars.2014.5994
45. Smyth EM, Grosser T, Wang M, Yu Y, FitzGerald GA. Prostanoids in health and disease. J Lipid Res. 2009;50(Supplement):S423–S428. doi:10.1194/jlr.R800094-JLR200
46. Chen Y, Li X, Boini KM, et al. Endothelial Nlrp3 inflammasome activation associated with lysosomal destabilization during coronary arteritis. Biochim Biophys Acta - Mol Cell Res. 2015;1853(2):396–408. doi:10.1016/j.bbamcr.2014.11.012
47. Xiong S, Hong Z, Huang LS, et al. IL-1β suppression of VE-cadherin transcription underlies sepsis-induced inflammatory lung injury. J Clin Invest. 2020;130:3684–3698. doi:10.1172/JCI136908
48. Wang J-G, Williams JC, Davis BK, et al. Monocytic microparticles activate endothelial cells in an IL-1 -dependent manner. Blood. 2011;118(8):2366–2374. doi:10.1182/blood-2011-01-330878
49. Allam R, Darisipudi MN, Tschopp J, Anders HJ. Histones trigger sterile inflammation by activating the NLRP3 inflammasome. Eur J Immunol. 2013;43:3336–3342. doi:10.1002/eji.201243224
50. Chen Y, Wang L, Pitzer AL, Li X, Li P-L, Zhang Y. Contribution of redox-dependent activation of endothelial Nlrp3 inflammasomes to hyperglycemia-induced endothelial dysfunction. J Mol Med. 2016;94(12):1335–1347. doi:10.1007/s00109-016-1481-5
51. Abderrazak A, Syrovets T, Couchie D, et al. NLRP3 inflammasome: from a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol. 2015;4:296–307. doi:10.1016/j.redox.2015.01.008
52. Sena CM, Leandro A, Azul L, Seiça R, Perry G. Vascular oxidative stress: impact and therapeutic approaches. Front Physiol. 2018;9:1668. doi:10.3389/fphys.2018.01668
53. Chen Y, Zhou Z, Min W. Mitochondria, oxidative stress and innate immunity. Front Physiol. 2018;9:1–25. doi:10.3389/fphys.2018.01487
54. Sharma A, Tate M, Mathew G, Vince JE, Ritchie RH, de Haan JB. Oxidative stress and NLRP3-inflammasome activity as significant drivers of diabetic cardiovascular complications: therapeutic implications. Front Physiol. 2018;9:114–149. doi:10.3389/fphys.2018.00114
55. Li S, Wang L, Xu Z, et al. ASC deglutathionylation is a checkpoint for NLRP3 inflammasome activation. J Exp Med. 2021;218(9). doi:10.1084/jem.20202637
56. Guglielmo A, Sabra A, Elbery M, et al. A mechanistic insight into curcumin modulation of the IL-1β secretion and NLRP3 S-glutathionylation induced by needle-like cationic cellulose nanocrystals in myeloid cells. Chem Biol Interact. 2017;274:1–12. doi:10.1016/j.cbi.2017.06.028
57. Zhang T, Tsutsuki H, Islam W, et al. ATP exposure stimulates glutathione efflux as a necessary switch for NLRP3 inflammasome activation. Redox Biol. 2021;41:101930. doi:10.1016/j.redox.2021.101930
58. Wang H, Shan X, Ren M, Shang M, Zhou C. Nucleosomes enter cells by clathrin- and caveolin-dependent endocytosis. Nucleic Acids Res. 2021;49(21):12306–12319. doi:10.1093/nar/gkab1121
59. Cascone A, Bruelle C, Lindholm D, Bernardi P, Eriksson O. Destabilization of the outer and inner mitochondrial membranes by core and linker histones. PLoS One. 2012;7(4):e35357. doi:10.1371/journal.pone.0035357
60. Costa VM, Amorim MA, Quintanilha A, Moradas-Ferreira P. Hydrogen peroxide-induced carbonylation of key metabolic enzymes in Saccharomyces cerevisiae: the involvement of the oxidative stress response regulators Yap1 and Skn7. Free Radic Biol Med. 2002;33(11):1507–1515. doi:10.1016/S0891-5849(02)01086-9
61. Kim SY, Jo H-Y, Kim MH, et al. H2O2-dependent hyperoxidation of peroxiredoxin 6 (Prdx6) plays a role in cellular toxicity via up-regulation of iPLA2 activity. J Biol Chem. 2008;283(48):33563–33568. doi:10.1074/jbc.M806578200
62. Liu L, Sun B. Neutrophil pyroptosis: new perspectives on sepsis. Cell Mol Life Sci. 2019;76(11):2031–2042. doi:10.1007/s00018-019-03060-1
63. Radeke HH, Martin M, Topley N, Kaever V, Resch K. Differential biological activities of human interleukin-1 alpha and interleukin-1 beta. Eur Cytokine Netw. 1991;2(1):51–59.
64. Maier JA, Hla T, Maciag T. Cyclooxygenase is an immediate-early gene induced by interleukin-1 in human endothelial cells. J Biol Chem. 1990;265(19):10805–10808. doi:10.1016/S0021-9258(19)38515-1
65. Kawabe T, Harris PD, Zakaria ELR, Garrison RN. Sepsis alters vessel contraction by adrenoceptor-induced nitric oxide and prostanoid. J Surg Res. 2003;110(2):352–359. doi:10.1016/S0022-4804(03)00029-5
66. Zoccal KF, Sorgi CA, Hori JI, et al. Opposing roles of LTB4 and PGE2 in regulating the inflammasome-dependent scorpion venom-induced mortality. Nat Commun. 2016;7(1):10760. doi:10.1038/ncomms10760
67. Li Z, Zhang Y, Liu B, Luo W, Li H, Zhou Y. Role of E-type prostaglandin receptor EP3 in the vasoconstrictor activity evoked by prostacyclin in thromboxane-prostanoid receptor deficient mice. Sci Rep. 2017;7(1):42167. doi:10.1038/srep42167
68. Pober JS, Min W, Bradley JR. Mechanisms of endothelial dysfunction, injury, and death. Annu Rev Pathol. 2009;4:71–95. doi:10.1146/annurev.pathol.4.110807.092155
69. Barnes TC, Anderson ME, Moots RJ. The many faces of interleukin-6: the role of IL-6 in inflammation, vasculopathy, and fibrosis in systemic sclerosis. Int J Rheumatol. 2011;2011:721608. doi:10.1155/2011/721608
70. Lin Y-M, Chang Z-L, Liao -Y-Y, Chou M-C, Tang C-H. IL-6 promotes ICAM-1 expression and cell motility in human osteosarcoma. Cancer Lett. 2013;328(1):135–143. doi:10.1016/j.canlet.2012.08.029
71. Habas K, Shang L. Alterations in intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) in human endothelial cells. Tissue Cell. 2018;54:139–143. doi:10.1016/j.tice.2018.09.002
72. Hashimoto M, Shingu M, Ezaki I, et al. Production of soluble ICAM-1 from human endothelial cells induced by IL-1 beta and TNF-alpha. Inflammation. 1994;18(2):163–173. doi:10.1007/BF01534557
73. Figueras-Aloy J, Gómez-López L, Rodríguez-Miguélez J-M, et al. Serum soluble ICAM-1, VCAM-1, L-selectin, and P-selectin levels as markers of infection and their relation to clinical severity in neonatal sepsis. Am J Perinatol. 2007;24(6):331–338. doi:10.1055/s-2007-981851
74. Yang L, Froio RM, Sciuto TE, Dvorak AM, Alon R, Luscinskas FW. ICAM-1 regulates neutrophil adhesion and transcellular migration of TNF- -activated vascular endothelium under flow. Blood. 2005;106(2):584–592. doi:10.1182/blood-2004-12-4942
75. Hotchkiss RS, Moldawer LL, Opal SM, Reinhart K, Turnbull IR, Vincent J-L. Sepsis and septic shock. Nat Rev Dis Prim. 2016;2:1–21. doi:10.1038/nrdp.2016.45
76. Kawai C, Kotani H, Miyao M, et al. Circulating extracellular histones are clinically relevant mediators of multiple organ injury. Am J Pathol. 2016;186:829–843. doi:10.1016/j.ajpath.2015.11.025
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Pyroptosis and Intervertebral Disc Degeneration: Mechanistic Insights and Therapeutic Implications
Ge Y, Chen Y, Guo C, Luo H, Fu F, Ji W, Wu C, Ruan H
Journal of Inflammation Research 2022, 15:5857-5871
Published Date: 17 October 2022
The Role of NLRP3 Inflammasome Signaling on Arrhythmias in Diabetes
Zhang L, Liu HH, Li F, Yang F, Qian LL, Wang RX
Journal of Inflammation Research 2022, 15:6883-6889
Published Date: 29 December 2022
The Mechanism of Pyroptosis and Its Application Prospect in Diabetic Wound Healing
Al Mamun A, Shao C, Geng P, Wang S, Xiao J
Journal of Inflammation Research 2024, 17:1481-1501
Published Date: 6 March 2024
NLRP3 Inflammasome Targeting Offers a Novel Therapeutic Paradigm for Sepsis-Induced Myocardial Injury
Jin Y, Fleishman JS, Ma Y, Jing X, Guo Q, Shang W, Wang H
Drug Design, Development and Therapy 2025, 19:1025-1041
Published Date: 14 February 2025
A Topical Chinese Herbal Alleviates Psoriasis by Regulating Keratinocytes Pyroptosis Through Inhibition of NLRP3 Inflammasome Activation
Wang X, Deng Y, Ren X, Li Y, Liu T, Hu B, Li Y
Clinical, Cosmetic and Investigational Dermatology 2026, 19:559712
Published Date: 15 January 2026