Back to Journals » Journal of Inflammation Research » Volume 19
Bidirectional Communication of the Gut–Brain Axis in Pain Regulation: From Microbial Metabolites to Neuroinflammation
Authors Song Z ![]() , Zhou K, Liang G, Liu Y, Li Q, Liu X
, Zhou K, Liang G, Liu Y, Li Q, Liu X
Received 17 April 2026
Accepted for publication 7 June 2026
Published 17 June 2026 Volume 2026:19 614847
DOI https://doi.org/10.2147/JIR.S614847
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Adam Bachstetter
Zhouyi Song,1– 3,* Kai Zhou,4,* Guangda Liang,1– 3 Yanfang Liu,1– 3 Qingmei Li,1– 3 Xingfeng Liu1– 3
1Guizhou Key Laboratory of Brain Science, Zunyi Medical University, Zunyi, Guizhou, People’s Republic of China; 2Guizhou Key Laboratory of Anesthesia and Organ Protection, Zunyi Medical University, Zunyi, Guizhou, People’s Republic of China; 3Key Laboratory of Anesthesia and Organ Protection, Ministry of Education (Cultivating Build), Zunyi Medical University, Zunyi, Guizhou, People’s Republic of China; 4Department of Oncology, The Second Affiliated Hospital of Zunyi Medical University, Zunyi, Guizhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xingfeng Liu, Email [email protected]
Abstract: Chronic pain is increasingly recognized not merely as a physiological symptom of tissue damage, but as a multidimensional pathological state involving sensory, emotional, and cognitive components. Central to its modulation is the gut–brain axis (GBA), a bidirectional communication network linking the enteric nervous system (ENS), the active intestinal epithelium, gut microbiota, and central nervous system (CNS) through neural, endocrine, immune, and metabolic pathways. Despite growing clinical evidence linking microbial dysbiosis to conditions such as irritable bowel syndrome (IBS), migraine, and fibromyalgia (FM), important gaps remain in understanding the molecular mechanisms that govern gut microenvironmental signaling in pain regulation. This review comprehensively summarizes the current literature on GBA-mediated pain regulation, with a focus on the molecular mechanisms by which microbial metabolites, such as short-chain fatty acids (SCFAs), and brain–gut peptides (BGPs) influence peripheral and central sensitization. Available evidence suggests that microbiota-derived inflammatory mediators, including lipopolysaccharide (LPS) and pro-inflammatory cytokines, contribute to neuroinflammation by activating glial cells and increasing blood–brain barrier (BBB) permeability. In addition, host intestinal epithelial and enteroendocrine cells (EECs), particularly enterochromaffin cells (ECs), are not merely passive barriers but active signaling interfaces, capable of releasing 5-hydroxytryptamine (5-HT), glutamate derived from neuropod cells, and multiple endocrine peptides involved in gut–brain communication and nociceptive regulation. The interplay between the hypothalamic–pituitary–adrenal (HPA) axis and the endogenous cannabinoid system (ECS) may act as an important regulatory “filter” in descending pain modulation. This review also discusses how reprogramming of the gut microbiota through probiotics and dietary interventions may influence the pain matrix and help alleviate comorbid affective symptoms. Overall, this review provides an integrated perspective on chronic pain as a disorder influenced by multi-level gut–brain interactions and potentially sustained by a bidirectional pathogenic feedback loop, thereby offering a theoretical basis for the development of gut microenvironment-targeted analgesic strategies.
Keywords: gut–brain axis, chronic pain, microbial metabolites, neuroinflammation, glial cell activation, gut microbiota
Introduction
Chronic pain is a severe global public health crisis, affecting approximately 20% to 45% of the population worldwide and representing a leading cause of disability.1 Beyond its profound impact on individual morbidity and quality of life, chronic pain imposes a staggering socioeconomic burden.2,3 In the United States alone, the annual direct healthcare costs and lost productivity are estimated to exceed $560 billion, surpassing the combined costs of heart disease, cancer, and diabetes.2 This enormous burden largely stems from the complex and multifactorial nature of the condition. As such, chronic pain is not merely a physiological signal of tissue damage, but a multidimensional pathological state integrating sensory, emotional, and cognitive components, and has increasingly been recognized as an independent disease entity.4–6 Due to the therapeutic bottlenecks and side effects of conventional analgesics, exploring broader mechanisms of pain modulation is urgent. As a bidirectional communication network connecting the central nervous system (CNS) and the gastrointestinal tract, the gut–brain axis (GBA) is composed of the brain, spinal cord, autonomic nervous system (ANS), enteric nervous system (ENS), the intestinal epithelium (which functions as a dynamic sensory, neuroimmune, and endocrine signaling interface rather than solely as a physical barrier), and the gut microbiota.7–9 Within this cross-talk, both the epithelial tissue and the microbiota play central modulatory roles in pain perception.7,10,11 Studies have confirmed that GBA dysfunction is closely associated with irritable bowel syndrome (IBS), migraine, fibromyalgia (FM), neuropathic pain (NP), and orofacial pain.7,12,13 At the peripheral level, signaling molecules produced by the gut ecosystem (such as microbial metabolites and enteroendocrine cell-derived neurotransmitters) amplify pain signals by modulating the excitability of primary nociceptive neurons.14 At the central level, gut microenvironmental inflammatory mediators can cross damaged intestinal and blood-brain barriers, activate glial cells in the spinal cord and brain, induce sustained neuroinflammation, and drive central sensitization.7,15
Scholarly consensus remains elusive concerning the definitive role of short-chain fatty acids (SCFAs) in pain modulation. While a body of evidence highlights their potent analgesic properties, countervailing research suggests a paradoxical capacity to exacerbate neural sensitization. These discrepancies underscore that GBA-mediated pain regulation is governed by profound concentration-dependency and pronounced inter-individual variability.14,16,17 Although microbiota remodeling has shown initial analgesic value in clinical practice, there is still a lack of consensus on whether there is a unified feature in different pain subtypes.12 This review is guided by the overarching hypothesis that chronic pain arises from a bidirectional pathogenic feedback loop within the GBA: peripheral microbial metabolites and immune mediators promote central neuroinflammation and sensitization. In contrast, central neural dysfunction exacerbates gut dysbiosis and barrier permeability. Building on this framework, we review the molecular mechanisms by which microbial metabolites influence peripheral and central sensitization through immune–nervous system interactions. We also discuss the potential of microecological agents and dietary interventions to modulate pain matrix networks, offering theoretical support for precision pain relief strategies.
The Neurobiology of Pain
Pain is not only a physiological signal of tissue damage but also a multi-dimensional perceptual experience that integrates sensations, emotions, and cognitive evaluations. The International Association for the Study of Pain (IASP) defines pain as “an unpleasant sensory and emotional experience associated with actual or potential tissue damage, or similar discomfort”.4–6 Chronic pain that persists or recurs for more than 3 months is often defined as an independent disease state in clinical practice, and its high prevalence and disability rate have posed a global public health challenge.1,5 From a neurobiological perspective, pain processing mainly follows four physiological processes: transduction, transmission, perception, and regulation. Pain perception begins with peripheral nociceptors (primary afferent nerve endings widely distributed in the skin, muscles, and internal organs), whose surface transient receptor potentials and acid-sensitive ion channels convert mechanical, thermal, or chemical stimuli into electrical signals.6
Meanwhile, inflammatory mediators released by damaged tissues trigger neurogenic inflammation through neural-immune interactions, such as the axonal reflex mediated by substance P (SP), thereby amplifying the signal. Subsequently, the pain signal travels along the Aδ fibers (fast pain) and C fibers (slow pain) to the dorsal horn of the spinal cord. Here, the primary afferent neurons release neurotransmitters such as glutamate, SP, and calcitonin gene-related peptide (CGRP) to complete synaptic transmission. The signal is projected via the spinothalamic tract and dorsal column pathways to the pain matrix, composed of the somatosensory cortex, anterior cingulate cortex (ACC), insula, and prefrontal cortex, mediating the perception and emotional-motivational processing of pain (Figure 1A). The body uses a descending modulation system centered around the periaqueductal gray (PAG) and the rostral ventromedial medulla (RVM) of the medulla oblongata to intervene in pain sensation in real-time through endogenous opioid peptides and monoamine neurotransmitters.6,9,18 Chronic pain originates from changes in neuroplasticity of the peripheral and CNS, driven by both peripheral and central sensitization, resulting in abnormal enhancement of reactivity.9 Among them, glutamate receptors (especially the NMDA type) located in the peripheral and central regions participate in the abnormal transmission of visceral sensitivity, which is a key mechanism for promoting the development of visceral hypersensitivity under pathological conditions (Figure 1A).6 They also work together with microglia and astrocytes to activate neuroinflammation, leading to an abnormal increase in excitatory synaptic transmission, which is a key pathological basis for promoting chronic pain.14,15 This process may represent a key biological basis through which the gut microenvironment regulates pain perception via the GBA.
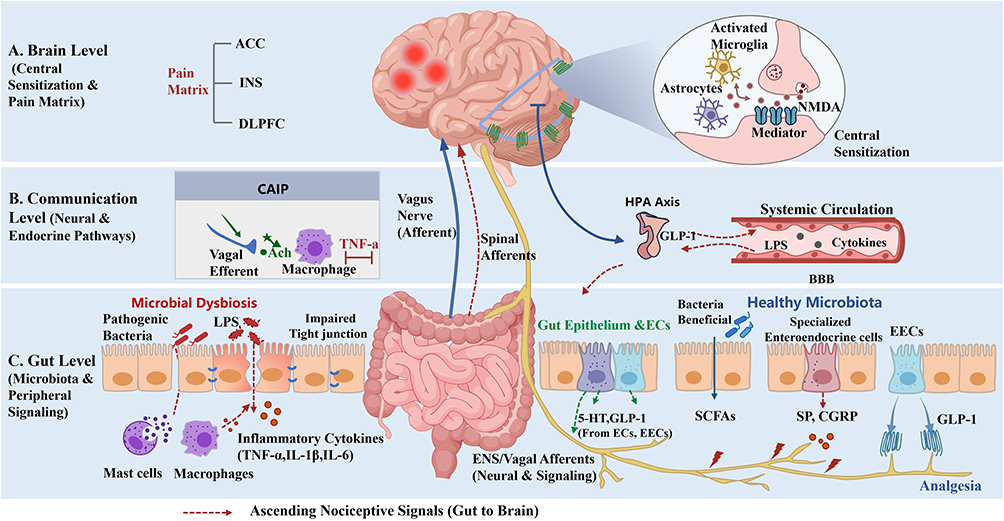 |
Figure 1 Integrated mechanisms of the gut-brain axis (GBA) in the regulation of chronic pain. The schematic illustrates the bidirectional communication between the gastrointestinal tract and the central nervous system across three functional levels: (A) Brain Level (Central Sensitization and Pain Matrix): Ascending signals lead to the hyperactivation of the Pain Matrix, consisting of the anterior cingulate cortex (ACC), insula (INS), and dorsolateral prefrontal cortex (DLPFC). The inset details the microscopic mechanism of central sensitization: activated microglia and astrocytes enhance synaptic transmission via NMDA receptors. (B) Communication Level (Neural and Endocrine Pathways): Ascending nociceptive signals are transmitted to the brain via the vagus nerve and spinal afferents. The Cholinergic Anti-inflammatory Pathway (CAIP) is highlighted, where vagal efferents release acetylcholine (ACh) to act on macrophages, inhibiting inflammatory responses and maintaining barrier integrity. Additionally, the HPA axis mediates stress feedback through the release of cortisol. (C) Gut Level (Microbiota and Peripheral Signaling): A comparative view of intestinal homeostasis and microbial dysbiosis. Microbial dysbiosis leads to the translocation of endotoxins (LPS) and pro-inflammatory cytokines (TNF-α, IL-1β, IL-6) through the “leaky gut” barrier (impaired tight junctions), which activates mast cells and macrophages to trigger peripheral sensitization. Specialized enteroendocrine cells release brain-gut peptides, such as Substance P (SP) and CGRP, to enhance nociception. In contrast, healthy microbiota produces SCFAs and GLP-1, which exert analgesic effects. (Red dashed arrows indicate ascending nociceptive signals). |
Gut–Brain Axis
The GBA is a complex bidirectional signaling network that connects the gastrointestinal tract to the CNS. Its core not only covers central structures such as the cerebral cortex, limbic system, hypothalamic-pituitary-adrenal axis (HPA), and brainstem, but also includes the autonomic nervous system (ANS) composed of sympathetic and parasympathetic branches, the enteric nervous system (ENS) known as the “second brain,” the intestinal epithelium and enteroendocrine system, as well as the vast gut microbiota. The functional implementation of this axis relies on multiple pathways, including neural, endocrine, and immune systems, which facilitate information exchange between the gut and brain through sensory and motor pathways.9,19–21 The vagus nerve serves as the main bidirectional connection between the intestine and the brain. Its afferent fibers can sense mechanical, chemical, and microbial signals in the intestine and transmit them to the solitary tract nucleus of the brainstem, which then projects them to the amygdala, locus coeruleus, and other higher levels that regulate emotions and pain. The efferent fibers mediate central regulation of gastrointestinal function. This mechanism is utilized by various probiotics, such as Lactobacillus rhamnosus, which relies on the vagus nerve pathway and oxytocin signaling to improve social deficits in mice with autism spectrum disorder.6,9,22,23 More importantly, the vagus nerve can also release acetylcholine (ACh) through the cholinergic anti-inflammatory pathway (CAIP) to regulate intestinal immunity and inhibit the release of pro-inflammatory factors such as TNF-α. This neuroimmune interaction not only maintains the integrity of the intestinal barrier but also directly blunts pain perception by regulating autonomic balance (Figure 1B).9
Firstly, endocrine and metabolic pathways are involved in the secretion of brain–gut peptides (BGPs), such as glucagon-like peptide-1 (GLP-1) and cholecystokinin (CCK), as well as the generation of gut microbiota metabolites, such as SCFAs, tryptophan metabolites, and secondary bile acids.6,9 Functioning as a highly active participant in the gut-brain axis rather than a mere barrier, the intestinal epithelium actively releases both neurotransmitters and endocrine signals to mediate this cross-talk.7,10,11 Specifically, microbial metabolites can directly stimulate host enterochromaffin cells (ECs) to synthesize the vast majority of peripheral 5-hydroxytryptamine (5-HT). This underscores that peripheral 5-HT signaling is predominantly microbe-regulated rather than solely microbe-derived.24 Subsequently, the released 5-HT activates vagal afferent fibers, transmitting peripheral signals to the solitary tract nucleus and higher central regions. Furthermore, these bioactive molecules can also regulate the levels of central neurotransmitters, such as gamma-aminobutyric acid and dopamine, via blood circulation or vagus nerve pathways, thereby participating in the regulation of pain and emotions.6,25 Secondly, there are immune and anti-inflammatory pathways, and the depth of cross-communication between the gut and brain depends on the maintenance of immune homeostasis. The immune pathway mainly mediates the interaction between the peripheral and CNS through the release of cytokines, chemokines, and activation of central microglia.17 In terms of physiological function, GBA not only maintains peristalsis and intestinal secretion, and the integrity of the intestinal barrier, but also plays a significant role in regulating emotional states (such as anxiety and depression), stress responses, cognitive functions, and visceral pain sensitivity. The disruption of its homeostasis is often the pathological basis for visceral hypersensitivity and the occurrence of various neurological and psychiatric diseases.26,27
The Mechanism of the Gut–Brain Axis and Pain Association
Gut Microbiota and Pain
The gut microbiota, as a core element of GBA, interacts extensively with the CNS through neural, endocrine, and immune pathways.28 The metabolites produced by it (such as SCFAs and tryptophan metabolites) and neurotransmitter analogs are key signaling molecules that regulate pain perception.23 Among them, SCFAs (such as butyric acid, acetic acid, and propionic acid) are the main products of bacterially fermented dietary fiber, and their anti-inflammatory and anti-immune effects as histone deacetylase inhibitors have been widely confirmed. In terms of immune regulation, SCFAs can induce the differentiation of regulatory T cells (Tregs) in the colon and maintain peripheral immune tolerance through the release of anti-inflammatory factors such as IL-10.17,23 In addition, SCFAs have both barrier protection and neural regulation functions and can upregulate the expression of tight junction proteins such as zonula occludens-1 (ZO-1) and occluding to repair the “leaky” intestinal barrier, reduce the production of pro-inflammatory cytokines such as TNF-α and IL-6, inhibit LPS-induced inflammation, and intervene in the inflammatory state of microglia by crossing the blood-brain barrier (BBB) (Figure 1B).17,27 Especially butyric acid, which has been proven to have significant anti-inflammatory and analgesic potential in various visceral pain models, suggests that SCFAs play an important role in GBA communication and neuroprotection.28 However, it must be pointed out that the current evidence regarding the “SCFA paradox” and its concentration dependence is almost entirely based on animal models. There is still a severe lack of direct clinical human data on the actual concentrations of SCFAs in the bodies of patients with chronic pain. Due to the complexity of in vivo metabolism, epithelial consumption, and cross-barrier transport, accurately quantifying the absolute concentrations of SCFAs in the peripheral circulation or central target tissues of human pain patients remains a bottleneck that urgently needs to be addressed in current clinical translational research.17,29,30
In addition, dysbiosis of the microbiota disrupts tryptophan metabolism and microbial regulation of ECs, thereby altering peripheral and central 5-HT signaling and reshaping pain transmission pathways.31 Increasing evidence suggests that intestinal flora imbalance, which refers to abnormal changes in the composition and function of the flora, is involved in the pathogenesis of various chronic pain conditions, such as IBS, NP, migraine, FM, and oral pain.6,7,12,13,32 For example, IBS patients often exhibit a decrease in microbial diversity and abundance of beneficial bacteria, such as lactobacilli, and the degree of imbalance is positively correlated with pain scores.33 In addition to bacterial communities, other microbial components of the gut ecosystem, including fungi, viruses, and archaea, may also participate in gut–brain communication and pain regulation.6,34 Emerging evidence suggests that the gut mycobiome and virome can influence host immune responses, intestinal barrier integrity, and microbial metabolic activity, thereby potentially contributing to neuroinflammation and chronic pain progression.35,36 However, the roles of these non-bacterial microorganisms in pain-related disorders remain largely underexplored.35
Further functional imaging studies have shown that dysbiosis of the gut microbiota significantly alters the activity of pain-related brain regions such as the ACC and amygdala.6 Correspondingly, reshaping the gut microbiota can effectively reduce the abnormal response of the brain to negative stimuli, objectively confirming the regulation of central pain encoding by gut intervention.33 The sterile mouse experiments, on the other hand, provided counter-evidence demonstrating the crucial role of the microbiota in pain perception. These models lacking microbial colonization typically exhibit abnormal changes in pain threshold and the absence of the development of visceral afferent nerves.14 Clinical practice has confirmed that FMT can significantly alleviate the clinical symptoms of some patients with chronic pain and improve their quality of life by reshaping the intestinal microecological structure.37 In summary, the gut microbiota significantly influences the perception and persistence of pain through multiple mechanisms, such as driving metabolic disorders, damaging the barrier, and inducing neuroinflammation. This multidimensional regulation from the distal gut to the central nervous system makes the gut microbiota not only an active participant in chronic pain but also a potential diagnostic marker and therapeutic target, especially in driving disease progression.
Intestinal Epithelial and Enteroendocrine Signaling in the Gut–Brain Axis
In exploring the regulation of the gut–brain axis by the gut microbiota, previous studies have often struggled to clarify how microbial metabolites exert their effects across the physical barrier of the intestine. In fact, the intestinal epithelium is not merely a passive barrier but a highly active signaling interface.10,11 Intestinal epithelial cells and enteroendocrine cells (EECs), particularly ECs, function as key “transducers” capable of directly sensing luminal microbial metabolites, such as short-chain fatty acids and secondary bile acids. For instance, ECs synthesize and release the majority of peripheral 5-HT under tight microbial regulation.24,38,39 Recent anatomical evidence demonstrates that EECs not only act through conventional endocrine and paracrine mechanisms but can also establish direct “synapse-like” contacts with vagal and other peripheral afferent fibers via basolateral axon-like projections (eg., L cell neuropods) or presynaptic marker expression, transmitting signals in a restricted, point-to-point manner.10,11,40 This provides a clearer mechanistic explanation for how peripheral luminal signals rapidly activate neural pathways.
Beyond rapid neural transmission, signaling molecules released by intestinal epithelial cells and EECs also serve as critical mediators linking humoral circulation with local immune responses. On the one hand, gut-derived hormones such as GLP-1, CCK, and peptide YY can locally modulate nociceptive sensitization of adjacent nerve terminals while also entering the circulation to exert central regulatory effects.9,10,40 On the other hand, molecules such as 5-HT directly regulate peripheral inflammatory states by binding to specific receptors on immune cells, including mast cells and macrophages.25,39 In turn, cytokines released by immune cells can reciprocally activate EECs to amplify peripheral signals; for example, IL-13 can regulate EC proliferation and 5-HT synthesis through its receptor, whereas IL-33 can trigger calcium influx and directly drive 5-HT release, forming a local feedback loop.39 These multidimensional humoral and immune interactions further reveal the coordinated regulation among the gut microbiota, neural networks, and the immune system, contributing to the development and maintenance of chronic pain.40
The Role of Brain-Gut Peptides (BGPs) and Their Related Signaling Systems in Pain Regulation
As the molecular basis for targeted action within the bidirectional communication network of the gut–brain axis (GBA), brain-gut peptides (BGPs) exert the dual functions of neurotransmitters and endocrine hormones. They are widely distributed throughout the CNS, ANS, and ENS, playing a core role in the transmission and sensitization of pain signals.39 At both the central and peripheral levels, BGPs not only enhance the transmission of pain signals but also serve as key molecular bridges linking pain amplification with emotional disorders. Within this framework, SP is particularly highlighted. As a typical excitatory BGP, SP not only regulates gastrointestinal motility but also serves as a key mediator driving both peripheral and central sensitization.38 At the peripheral level, under inflammatory or stress-related conditions, the activation of transient receptor potential channels, such as TRPA1, promotes the release of SP. Subsequently, the released SP exacerbates intestinal immune-inflammatory responses through the activation of neurokinin receptors, particularly the neurokinin-1 receptor (NK1R). This process is accompanied by the elevated expression of transient receptor potential vanilloid 1 (TRPV1), collectively contributing to the induction and maintenance of visceral hypersensitivity.41–44 At the central level, studies have shown that pathological increases in SP within the cerebrospinal fluid can induce the internalization and abnormal response of NK1 receptors within the prominent network of the dorsal anterior cingulate cortex, directly driving central sensitization.21
Next, CGRP acts as a key mediator in pain regulation. It mediates neurogenic inflammation in the periphery and promotes peripheral sensitization by lowering the threshold of nociceptors.18 It is worth noting that the pathological elevation of CGRP expression is deeply controlled by epigenetic remodeling, such as DNA methylation and miRNA regulation, which provides a molecular explanation for the persistence of chronic pain.45 Concurrently, peripheral nociceptive neurons enhance nociceptive transmission by releasing SP and CGRP, and mediate communication abnormalities in the striatum-thalamus-frontal lobe pathway by profoundly modulating neuronal activity in core hubs such as the prefrontal cortex and amygdala (Figure 1C). This high degree of overlap between GBA signals within pain perception and emotion regulation circuits explains the neurobiological basis for the coexistence of chronic pain and mental disorders such as anxiety.18,21
In addition to the classic peptides discussed above primarily as neuromodulators, a variety of endogenous endocrine peptides secreted by the gut also play a crucial regulatory role in pain responses. Although exogenous pharmacological interventions (such as glucagon-like peptide-1 receptor agonists, GLP-1 RAs) have demonstrated unique analgesic potential—alleviating pain by inhibiting microglial activation and regulating the central dopamine system through both neuroinflammatory and reward dimensions (Figure 1C)—the endogenous forms of these peptides are equally and extensively involved in the physiological regulation of nociception.9 For example, endogenously secreted GLP-1 not only possesses intrinsic analgesic properties but can also directly modulate pain signal transmission in the dorsal horn of the spinal cord. CCK is widely distributed throughout the central and peripheral nervous systems and participates in the regulation of endogenous pain signaling, while gastrin-releasing peptide mediates the transmission of pain and itch signals within the spinal cord.9,26,46 In addition, certain classical endocrine peptides and gut-derived signaling molecules can influence visceral sensitivity. For instance, glucagon has been shown to alleviate visceral spasms and pain by relaxing gastrointestinal smooth muscle and reducing abnormal motility.47 Furthermore, uroguanylin can inhibit nociceptor activity by stimulating epithelial cells to release cGMP, whereas 5-HT can modulate both local and systemic pain thresholds through the circulatory system or by activating vagal afferent fibers.24,25,38
Within the overall bidirectional regulatory mechanism of the GBA, these multidimensional peptide signals collectively serve as key dynamic feedback mediators. In the ascending pathway, intestinal chemical stimulation drives the release of SP and CGRP, which are transmitted via the vagus nerve to the central nervous system, triggering pain and negative emotions.6 In the descending pathway, the ANS and HPA axis regulate peripheral BGP levels. Furthermore, stress-induced hyperactivity of the HPA axis can, in turn, aggravate pain, leading to dysregulation of motility, inflammation spread, and barrier damage.48 Concurrently, the endogenous cannabinoid system (ECS), a crucial lipid signaling network within the GBA, plays a central “filtering” role. Its receptors (such as CB1 and CB2) are widely distributed throughout the GBA pathways. By fine-tuning the activity of the amygdala centrally and peripheral immune metabolism locally, they achieve negative feedback inhibition of abnormal peptide signals, thereby maintaining systemic homeostasis. This neuro-immune-endocrine network regulated by the ECS provides a promising therapeutic target for the management of chronic visceral pain conditions such as IBS.28,49
The Role of Inflammatory Factors and Neuro-Immune Regulation in Pain
In the complex network of GBA, inflammatory factors act as the core signaling molecules, mediating the cross-talk between the intestinal immune system and the CNS through neuro-immune communication. The intestinal microbiota and its metabolites, such as lipopolysaccharide (LPS) and SCFAs, can activate intestinal mucosal immune cells (such as mast cells and macrophages), inducing the release of pro-inflammatory cytokines (such as TNF-α, IL-1β, and IL-6) and chemokines.28,44,50,51 When the microbiota is imbalanced or the intestinal barrier is damaged, translocated LPS triggers a local immune cascade reaction, releasing a large number of inflammatory mediators and chemokines, thereby forming an “inflammatory soup” in the local microenvironment (Figure 1C).14,33,44,52 These mediators activate signaling pathways, such as the protein kinase A/mitogen-activated protein kinase (PKA/MAPK) pathways, in dorsal root ganglion (DRG) and ENS neurons, inducing the phosphorylation of transient receptor potential (TRP) channels and significantly reducing the threshold of nociceptors. This process drives visceral hypersensitivity and abdominal pain through peripheral sensitization.6,14 For instance, in a stress-induced IBS model, abnormal changes in the phosphorylation levels of the tight junction protein ZO-1 and IL-1RAP are key factors leading to increased intestinal epithelial permeability and an imbalance in GBA immune homeostasis.27,53,54 This barrier dysfunction and microbiota dysbiosis are mutually causal, jointly driving disease progressione.44 Furthermore, gut microenvironmental inflammatory factors can cross the damaged intestinal barrier into the circulatory system and infiltrate the brain via the BBB. Inflammatory factors and dysbiosis can compromise the integrity of the BBB, allowing peripheral inflammatory mediators to enter the brain more easily, thereby exacerbating neuroinflammation and pain by activating glial cells.28,50,51 This immune-driven imbalance of neurotransmitters and the corresponding changes in synaptic plasticity pathologically amplify pain signals and induce central sensitization, which is often accompanied by negative emotions such as depression or anxiety.15,27 Interventions such as ginger extract can alleviate pain by inhibiting this neuroinflammatory process, further confirming the crucial role of the GBA in the chronification of pain.15,55,56 In summary, through the complex neural input–immune regulation–central glial cell cascade of this neuro-immune axis, inflammatory factors constitute an important pathological basis for the development and maintenance of chronic pain.28
Central Sensitization and the Gut–Brain Axis
Central sensitization serves as the core pathological mechanism underlying the onset and persistence of chronic pain (such as IBS, FM, NP, and other nociceptive pain disorders). It is characterized by abnormally enhanced CNS responsiveness to nociceptive signals, clinically presenting as a reduced pain threshold and pathological expansion of the pain area.6,37 At the molecular level, this process involves neuronal hyperexcitability and synaptic plasticity remodeling in the spinal dorsal horn and pain-processing brain regions (such as the ACC cortex and periaqueductal gray): The activation of glutamate receptors (such as NMDA) triggers downstream signaling cascades, including protein kinase C (PKC) and calcium/calmodulin-dependent protein kinase II (CaMKII) pathways, which mediate ion channel phosphorylation and induce long-term potentiation.38 Meanwhile, the function of the descending inhibitory system (mediated by neurotransmitters such as GABA and glycine) is compromised. This impairment interacts with sustained injury stimulus-induced activation of microglia and astrocytes, collectively facilitating the release of pro-inflammatory factors and driving neurons into a hyper-excited state.15,55,56 In this pathological cascade, GBA exerts a decisive driving effect via immune, metabolic, and neuroendocrine pathways. Intestinal flora disruption triggers systemic inflammation by impairing intestinal barrier integrity. Its metabolites, including tryptophan metabolites, SCFAs, and LPS, can cross or increase BBB permeability, thereby directly inducing central neuroinflammation.9,37 Given the essential role of microbiota in regulating microglial maturation and homeostasis, abnormal microbiota can polarize microglia toward an inflammatory phenotype and promote reactive astrocyte proliferation. The subsequent release of pro-inflammatory factor cascades forms the core pathological foundation of central sensitization.15,23,37 Furthermore, GBA-associated neuroactive molecules, as key mediators of neuroendocrine signaling, contribute substantially to central sensitization progression. Specifically, CGRP enhances glutamatergic transmission in the spinal dorsal horn via presynaptic modulation, thereby reshaping synaptic plasticity and further facilitating central sensitization.18 Meanwhile, microbiota-regulated neurotransmitters (such as 5-HT) activate vagal afferent fibers, continuously projecting peripheral signals to the brainstem solitary nucleus and higher centers, disrupting the negative feedback loop of the HPA axis and leading to sustained cortisol hypersecretion.25 Such chronic neuroendocrine hyperactivity not only lowers peripheral pain thresholds but also further modulates limbic system function (such as the amygdala and hippocampus), promoting the emotional processing of pain.9,25 This GBA-driven neuro-immune-endocrine network crosstalk alters the conduction efficiency of dorsal horn neurons and remodels pain-related neural circuits, forming a vicious cycle from intestinal dysfunction to central pain amplification and subsequent emotional comorbidities such as anxiety and depression, which constitutes a critical biological basis for the central sensitization of chronic pain. Notably, placebo analgesia, which shares neural circuits with central sensitization, is primarily driven by cognitive expectations and the reward network, relying on dopaminergic and endogenous opioid systems to initiate the descending pain modulatory system (DPMS).57,58 Given that the microbiota regulates the central bioavailability of neurotransmitter precursors (tryptophan) and thus participates in brain serotonin and dopamine synthesis, gut dysbiosis may disrupt the neurochemical foundation required for an effective endogenous placebo response.59,60 This potential interaction could partially explain the significant inter-individual variability in placebo effects frequently observed in clinical trials for chronic pain conditions like IBS.61 It also suggests that the GBA modulates not only pain amplification but also the baseline therapeutic responsiveness to pain interventions.
Sexual Dimorphism of Gut Microbiota and the Regulatory Role of Sex Hormones in the Gut-Brain Axis
Accumulating evidence has demonstrated robust sexual dimorphism in GBA signaling and chronic pain susceptibility. Sex hormones, particularly estrogen and testosterone, markedly shape the composition, diversity, and metabolic activity of intestinal microbiota.38,50 Clinical and preclinical studies confirm that serum sex steroid hormone levels are tightly correlated with the diversity and compositional profiles of the human intestinal microbiota, and this bidirectional interplay profoundly modulates systemic immune and neural homeostasis.62 Consistent with this notion, visceral nociceptive responses to colorectal distension fluctuate across the estrous cycle in female mice, whereas such cyclic variability is completely ablated in germ-free animals, providing direct evidence for the causal linkage among sex hormones, intestinal microbiota, and visceral pain.50
Mechanistically, cyclic fluctuations in sex hormone levels directly modulate intestinal barrier integrity. Specifically, estrogen remodels tight junction proteins and the intestinal mucin layer, thereby altering intestinal permeability.50 In addition, sex hormone receptors are broadly distributed throughout the gut–brain network and participate in the bidirectional regulation of peripheral and central pain signaling. At the peripheral level, physiological estrogen concentrations downregulate the expression of TRPV1 channels in DRGs and promote their intracellular translocation, ultimately producing analgesic effects. At the central level, neuroendocrine regulation also exhibits distinct sexual dimorphism: corticotropin-releasing factor (CRF)-producing neurons in the bed nucleus of the stria terminalis (BNST) are predominately activated in females, and their neuronal activity is positively modulated by estrogen.32,38 Conversely, impaired central estrogen signaling contributes to the abnormal upregulation of spinal dorsal horn CGRP, which lowers the threshold for migraine attacks and exacerbates overall central sensitization.50
Such systematic disparities in the “hormone–microbiota–neural” interactive network elucidate the biological basis for the higher prevalence of GBA-associated chronic pain disorders in females. Epidemiological data verify that IBS and migraine disproportionately affect women, and FM also exhibits strong female predilection, with a male-to-female incidence ratio of approximately 1:3. The high comorbidity between FM and IBS is closely attributed to female-specific sympathetic dysfunction and exaggerated central sensitization.12,26,63 These significant sex differences suggest that in the future development of precise analgesic technologies targeting the GBA, sex should be regarded as a fundamental biological variable, and further exploration of sex-specific clinical intervention strategies is needed.
Research Progress on Pain Disorders Related to the Gut–Brain Axis
Irritable Bowel Syndrome (IBS) and the Gut–Brain Axis
In recent years, accumulating studies have demonstrated that IBS, as a typical disease of gut–brain interaction (DGBIs), develops chronic visceral pain driven by the combined effects of peripheral and central sensitization within the bidirectional GBA network.9,21 At the central level, functional magnetic resonance imaging findings have revealed prominent dysregulation of endogenous pain modulation in IBS patients. Upon exposure to actual or anticipated visceral stimuli, salience network brain regions (including the anterior insula and middle ACC) exhibit excessive activation, whereas the prefrontal cortex (PFC), a core region responsible for pain inhibition, shows insufficient activation. In addition, female patients display enhanced activation and connectivity within the emotional arousal network, along with more robust stimulus-induced responses.21 Meanwhile, persistent overactivation of the HPA axis under acute psychological stress further exacerbates abdominal pain symptoms.44
At the peripheral level, central and peripheral CRF-CRF1 receptor signaling systems are critical for mediating stress-induced visceral hypersensitivity.44 Intestinal barrier impairment triggers low-grade intestinal inflammation, which promotes mast cell infiltration and the subsequent release of immune mediators, including histamine and proteases. These mediators directly sensitize sensory neurons via the CRF-TLR4 pathway (Figure 2A). Continuous peripheral nociceptive input further activates central glial cells, ultimately sustaining and consolidating central sensitization.64 Furthermore, intestinal microecological imbalance, characterized by reduced Bifidobacteria and increased Firmicutes abundance, modulates 5-HT release from ECs through SCFAs, thereby lowering the visceral pain threshold (Figure 2A).33,39 Clinical studies have indicated that antibiotic intervention for IBS pain exhibits double-edged effects, with clinical outcomes dependent on the direction of specific bacterial remodeling and corresponding metabolic alterations.64 Antibiotics (eg., rifaximin) primarily reduce inflammatory mediator production by decreasing intestinal bacterial load and are commonly combined with probiotics to maintain intestinal microenvironmental stability.44,65,66 In comparison, probiotic supplementation combined with a low-FODMAP diet 68 shows greater potential in restoring intestinal metabolic homeostasis and suppressing excessive immune activation.67,68
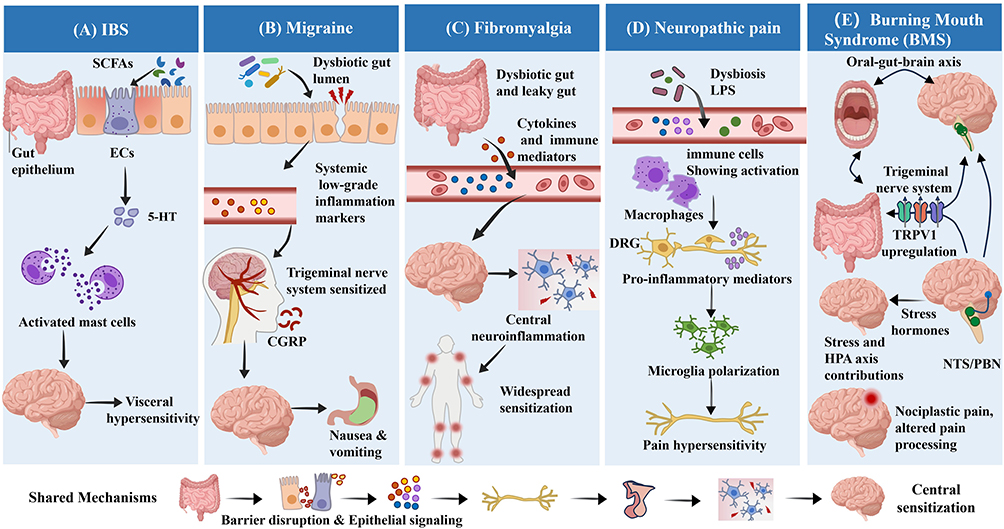 |
Figure 2 Overview of distinct and shared pathophysiological mechanisms of the gut–brain axis across specific chronic pain disorders. The figure summarizes GBA-mediated mechanisms in five representative chronic pain conditions. (A) Irritable Bowel Syndrome (IBS): Characterized by mast cell activation, dysbiosis, and abnormal 5-HT signaling mediated by enterochromaffin (EC) cells of the gut epithelium, leading to visceral hypersensitivity. (B) Migraine: Dysbiosis-induced systemic inflammation sensitizes the trigeminal nerve system, promoting CGRP release and associated nausea/vomiting. (C) Fibromyalgia: Leaky gut-derived immune activation drives central neuroinflammation and widespread sensitization. (D) Neuropathic Pain: Translocation of LPS induces inflammation in the dorsal root ganglia (DRG) and polarization of microglia. (E) Oral Pain / Burning Mouth Syndrome (BMS): Involves the oral-gut-brain axis, where stress and HPA axis dysfunction upregulate TRPV1 expression in the trigeminal system. Bottom Panel (Shared Mechanisms): Depicts the common pathological cascade underlying these disorders, progressing from dysbiosis, barrier disruption, and epithelium-mediated signaling to vagal dysfunction and culminating in central sensitization. |
To evaluate the animal models of GBA dysfunction (such as restraint stress or maternal-infant separation), induce rats to exhibit visceral hypersensitivity, abnormal defecation, and behavioral patterns resembling emotional disorders.27,69 Mechanistic studies further validate that the loss of tight junction protein ZO-1 and abnormal phosphorylation of IL-1RAP constitute the core molecular mechanisms underlying stress-induced intestinal barrier damage. Collectively, the severity of IBS abdominal pain is determined by the degree of GBA-mediated central sensitization and pain sensitivity, which can be quantitatively assessed via colorectal distension (CRD) and abdominal withdrawal reflex (AWR) assays.27 These findings lay a solid foundation for the development of novel microbiome and GBA-targeted analgesic strategies.
Migraine and the Gut–Brain Axis
Migraine, a chronic disorder characterized by recurrent sensitization of the trigeminovascular system, has been increasingly recognized to closely interact with the GBA. Intestinal flora dysbiosis serves as a core factor driving migraine chronification. Migraine patients frequently exhibit reduced microbial diversity, including decreased coliform abundance, and a large proportion of patients present comorbid IBS or Helicobacter pylori infection.13,50 The core pathological mechanisms are summarized as follows. First, microbiota imbalance disrupts intestinal barrier integrity, facilitating the entry of LPS and other metabolites into the circulation and triggering low-grade systemic inflammation. This cascade subsequently induces neurogenic inflammation within the trigeminovascular system and promotes the release of key pro-nociceptive peptides such as CGRP (Figure 2B).50 Second, gut microbiota directly modulates the biosynthesis of key neurotransmitters, including tryptophan-derived 5-HT and GABA, by providing precursors such as tryptophan and glutamine. Such remote microbial interference disturbs central pain perception and descending inhibitory pathways. This biochemical regulation not only induces migraine-associated central sensitization but also explains gastrointestinal dysfunctions, including nausea and vomiting, that accompany migraine attacks (Figure 2B), indicating that bidirectional gut–brain communication coordinately modulates pain processing and gastrointestinal homeostasis.13 Furthermore, microbial signals can also regulate the pain stress response by activating the vagus nerve or the HPA axis.31,50 This mechanism has been fully validated in animal models induced by nitroglycerin (NTG) and CSD. In particular, experiments on germ-free animals and microbiota transplantation further confirmed that the microbiota waves can directly regulate the expression of pro-inflammatory factors in the trigeminal ganglion and the degree of central sensitization, thus outlining the complete pathological chain of “microbiota imbalance-barrier damage-trigeminal nerve sensitization”.13,50,70
In terms of clinical intervention, numerous randomized controlled trials (RCTs) have confirmed that through intervention strategies such as probiotics/probiotics, eradication of Helicobacter pylori, fecal microbiota transplantation, or the Mediterranean diet, not only can the intestinal barrier be repaired and comorbidities of mood disorders be improved, but also the frequency of migraine attacks, pain intensity, and dependence on painkillers can be significantly reduced.13,15 In addition, pharmacological agents, such as 5-HT receptor modulators and CGRP receptor antagonists, combined with psychological interventions, achieve multi-target modulation of GBA function.13 Future research suggests that targeting intestinal microecological remodeling may serve as a promising non-pharmacological strategy to reverse the chronic progression of migraine.
Fibromyalgia (FM) and the Gut–Brain Axis
FM is a pain syndrome driven by central sensitization and injury-related neural plasticity and is characterized by chronic widespread pain. Accumulating clinical studies have confirmed that its pathogenesis is closely associated with GBA dysregulation and intestinal microecological disorders.12 Clinically, FM patients frequently present with gut–brain interaction disorders, most notably IBS. Such substantial symptom overlap implies shared GBA-centered pathological mechanisms between the two disorders. Existing studies have demonstrated that intestinal microecological imbalance in FM patients is mainly manifested as altered bacterial diversity and increased proportions of pathogenic bacteria, accompanied by intestinal mucosal barrier damage.12 This “intestinal leak” permits the entry of gut microenvironmental metabolites and immune mediators into the bloodstream, thereby triggering neuroimmune-mediated central neuroinflammation and subsequent widespread central sensitization (Figure 2C).31 Furthermore, clinical evidence has revealed a high degree of symptomatic similarity between FM and small intestinal bacterial overgrowth (SIBO), indicating that SIBO may serve as a potential driver of FM pathogenesis. Intestinal SIBO occurrence and abnormal intestinal hydrogen production are positively correlated with pain severity in FM patients. In addition, alterations in secondary bile acid profiles also participate in modulating pain intensity.12 Current mechanistic investigations primarily focus on the analgesic effects of intestinal interventions. For example, supplementation with SCFAs (eg., butyrate) or probiotics not only improves intestinal barrier integrity and reduces pro-inflammatory levels but also modulates central 5-HT synthesis by regulating tryptophan metabolism. Meanwhile, microbiota-regulated GABA and 5-HT modulate central pain perception via vagal afferent signaling to the central nervous system.24 Specifically, GABA suppresses nociceptive transmission more efficiently by binding to surface receptors on DRG neurons and inducing neuronal depolarization.9,71,72
The above pathological cascade has been validated in multiple animal models, including levodopa-induced and chronic unpredictable mild stress (CUMS) models. In particular, fecal microbiota transplantation from FM patients into germ-free mice successfully recapitulates hyperalgesic phenotypes, directly verifying the pathogenic role of gut microbiota in FM development.12,63 In terms of clinical translation, FMT significantly alleviates pain and fatigue symptoms by reshaping intestinal microbiota composition and reducing the levels of pro-inflammatory cytokines such as IL-6 and TNF-α.37 Collectively, FM pathogenesis is comprehensively modulated by the GBA. Targeted intestinal microecological remodeling has therefore emerged as a promising clinical strategy for intervening in FM progression and achieving precise pain relief.
Neuropathic Pain (NP) and the Gut–Brain Axis
The occurrence and progression of NP are tightly associated with homeostatic imbalance driven by intestinal immune activation. Under pathological conditions including nerve injury, chemotherapy exposure, and diabetes, intestinal microecological imbalance impairs the physical defense of the intestinal epithelial barrier. This process facilitates the translocation of endotoxins such as LPS into the circulation, thereby activating macrophages and increasing the excitability of primary DRG neurons (Figure 2D).7,15,64 Studies have demonstrated that gut microbiota modulates the development and activation of central microglia via microbial-derived SCFAs. Damaged nerve terminals and inflammatory stimuli induce the polarization of spinal microglia and the subsequent release of brain-derived neurotrophic factor, which drives the sensitization of spinal nociceptive neurons (Figure 2D).16 In the chronic NP state, persistent neuroinflammation and accumulated inflammatory mediators within brain regions including the medial prefrontal cortex and amygdala directly disrupt local neuronal function and circuit homeostasis. Such microenvironmental disturbances not only exacerbate central pain processing dysfunction but also underlie the high comorbidity of anxiety and depression in NP patients.56
His pathological cascade has been extensively validated in classic NP animal models, including chronic constriction injury (CCI), spinal nerve ligation (SNL), and chemotherapy-induced peripheral neuropathy (CIPN). Preclinical evidence confirms that preventive microbiota depletion or microbial remodeling via FMT effectively ameliorates hyperalgesia phenotypes.64 Clinical and mechanistic studies have further identified significant alterations in intestinal microbial composition in CIPN patients, which are closely correlated with pain sensitivity, indicating that gut microbiota drives NP progression by modulating neuroinflammatory responses.55 In terms of clinical intervention, supplementation with specific probiotic formulations and natural products such as ginger extracts has been proven to relieve mechanical pain by preserving intestinal barrier integrity, suppressing neuroinflammation, and restoring mitochondrial homeostasis.15,56 In summary, intestinal microecology profoundly participates in peripheral and central sensitization during NP pathogenesis through immune, metabolic, and neural pathways. Targeting GBA through microecological interventions is expected to become a new strategy for the clinical management of chronic neuropathic pain.
Oral Pain and the Gut–Brain Axis
Oral pain progression is uniquely mediated by the trigeminal nervous system, in which the oral–gut axis exerts pivotal immune and neural modulatory functions. Oral microbiota can alter intestinal homeostasis through saliva-mediated translocation and gastrointestinal colonization. Conversely, inflammatory mediators derived from intestinal dysbiosis act as crucial triggers for neurodegeneration in the orofacial region.32,73 Burning mouth syndrome (BMS) is a representative idiopathic chronic oral pain condition characterized by persistent burning or pricking sensations in the oral mucosa. Its underlying pathogenesis can be systematically interpreted from three dimensions: peripheral receptors, peripheral nerves, and central brain circuits. At the receptor level, gastrointestinal signals upregulate TRPV1 expression within the trigeminal system, thereby facilitating the generation of oral nociception (Figure 2E) 32,74. At the neural level, immune activation-induced intestinal inflammation markedly lowers the discharge threshold of orofacial nerves75. At the central circuit level, BMS is defined as injury-sensitized pain, which is primarily driven by enhanced central pain processing (Figure 2E).32,74 At the neural level, the inflammatory response driven by gut microenvironmental immune activation significantly reduces the threshold for neural discharge.75 At the brain circuitry level, BMS is classified as injury sensitization pain, involving the enhancement of central pain processing.32,74
Studies have validated that gut microbiota remotely modulates central pain transmission via two primary pathways: vagus nerve-mediated direct neural signaling and metabolite-mediated indirect humoral signaling across the BBB.75,76 For example, SCFAs modulate trigeminal sensitization by targeting key brain nuclei, including the nucleus tractus solitarius (NTS) and parabrachial nucleus (PBN). In addition, chronic psychological stress induces dual dysbiosis of oral and intestinal microbiota through HPA axis activation. Such oral–gut–brain axis dysfunction not only exacerbates pain in the temporomandibular joint and masticatory muscles but also provides a biological basis for prevalent comorbidities in BMS patients, including sleep disturbances, memory impairment, and anxiety (Figure 2E).73,76 Notably, BMS exhibits a high prevalence among perimenopausal women. Rapid estrogen decline during this period disrupts protective sex hormone-GBA crosstalk, relieving TRPV1 inhibition and amplifying central pain sensitization. This hormonal alteration substantially aggravates oral burning sensations and concomitant emotional disorders.32,74
Pain Intervention Strategies for the Brain-Gut Axis
Drug Intervention Targeting the Brain-Gut Axis
Current GBA-targeted pharmacological strategies primarily relieve pain by regulating intestinal microecological homeostasis and modulating neural signaling pathways (Figure 3). Microbial preparations, including probiotics and prebiotics, exert analgesic effects by reshaping intestinal flora composition, enhancing intestinal epithelial barrier integrity, and elevating the abundance of butyric acid and other SCFAs.14,33,44 By attenuating excessive HPA axis activation, these interventions prevent chronic stress-associated brain dysfunction and effectively improve visceral hypersensitivity in IBS and NP. Nevertheless, their clinical limitations remain non-negligible, as therapeutic efficacy is largely constrained by bacterial strain specificity and individual heterogeneity, accompanied by occasional adverse reactions.7,14 Antibiotics (such as rifaximin) mainly target the inhibition of pro-inflammatory mediators by regulating the intestinal flora, thereby alleviating mucosal inflammation. Clinically, a probiotic strategy is often combined to further restore the micro-ecological barrier and maintain intestinal homeostasis.65,66 However, clinical studies have shown that antibiotics have been controversial in pain intervention: although antibiotics are effective for some patients, the induced microbiota remodeling by them may increase pro-inflammatory metabolites such as LPS, thereby exacerbating pain sensitivity.40,64
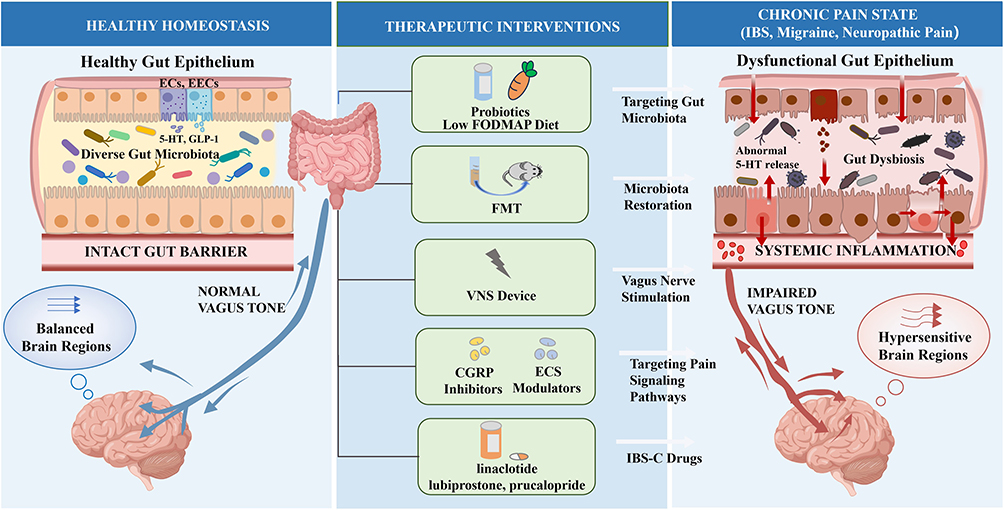 |
Figure 3 Therapeutic interventions targeting the gut-brain axis (GBA) for chronic pain management. The schematic illustrates the transition from healthy homeostasis to a chronic pain state and the corresponding multi-level therapeutic strategies. In Healthy Homeostasis (left), diverse gut microbiota and a healthy gut epithelium maintain normal vagus tone. Crucially, the epithelium acts as an active signaling interface, where enterochromaffin cells (ECs) and enteroendocrine cells (EECs) release neurotransmitters and endogenous peptides (e.g., 5-HT and GLP-1). In contrast, the Chronic Pain State (right) is characterized by gut dysbiosis, epithelial barrier disruption, and altered epithelial signaling (e.g., abnormal 5-HT release). These alterations drive systemic inflammation and an impaired vagus tone, leading to the development of hypersensitive brain regions. Therapeutic interventions (center) aim to restore GBA stability through diverse modalities: targeting gut microbiota via probiotics and a low FODMAP diet; microbiota restoration using FMT; vagus nerve stimulation employing VNS devices to modulate autonomic feedback; targeting pain signaling pathways using CGRP inhibitors and ECS modulators to suppress neuroinflammation and central sensitization; and targeting gut epithelial signaling with classic IBS-C drugs (e.g., linaclotide, lubiprostone, prucalopride). |
Apart from the above microbiota-based interventions, conventional treatments for constipation-predominant irritable bowel syndrome (IBS-C) have also been shown to exert analgesic and neuromodulatory effects through the GBA. For example, linaclotide, a guanylate cyclase-C receptor agonist, can promote the release of extracellular cGMP from intestinal epithelial cells, thereby inhibiting nociceptor activity and alleviating visceral hypersensitivity. Similarly, lubiprostone improves intestinal secretion and barrier function by activating chloride channels, blocking abnormal immune sensitization along the brain-gut axis.44,77 Prucalopride, a 5-HT4 receptor agonist, accelerates colonic motility and reflexes by inducing acetylcholine release from neurons, providing a solid neuropharmacological basis within the brain-gut axis for relieving constipation and related abdominal symptoms in patients.38,66
At the neural modulation level, central regulatory drugs (such as antidepressants) exert distal analgesic effects by strengthening the descending pain inhibition system.78,79 Meanwhile, GLP-1 RAs not only inhibit neuroinflammation but also alleviate chronic pain by regulating the central dopamine system.9 Furthermore, novel modulators targeting CGRP and the ECS show great potential for pain relief. The former have achieved breakthroughs in migraine treatment by blocking signaling cascades, while the latter regulate visceral perception by activating CB1 receptors, providing a new approach for precise intervention in oral facial pain and gastrointestinal diseases.13,28,49
Non-Pharmacological Intervention and Lifestyle Modification
Non-pharmacological approaches aim to achieve functional optimization of the GBA system through exogenous physical stimulation or lifestyle adjustments (Figure 3). Dietary intervention is the most direct way to regulate the intestinal microenvironment. A low FODMAP diet, by restricting colonic fermentation and lowering LPS levels, can restore intestinal barrier function and alleviate visceral pain. Additionally, functional foods such as ginger root extract have been proven to have the potential to inhibit neuroinflammation.7,14,56
Moreover, FMT demonstrates clinical potential in improving visceral pain, FM, and even delaying morphine tolerance by directly reshaping the gut microbiota structure of the recipients and regulating bile acid metabolism.14 Given the central role of the GBA in stress sensitivity, psychological interventions (such as cognitive behavioral therapy and meditation) weaken pain perception at the level of the brain’s higher centers by inhibiting the excessive activation of the HPA axis.78 Furthermore, emerging neuroregulation technologies, such as vagus nerve stimulation (VNS) and transcutaneous electrical stimulation (TENS), directly regulate the fibers that facilitate communication between the brain and the intestines through electrical signals, exerting a synergistic effect of anti-inflammation and pain relief.79 This multi-dimensional and non-invasive management model complements drug therapy and together forms a new framework for comprehensive treatment of chronic pain in the modern era.
Conclusions and Prospects
The GBA is far more than a background factor in the progression of chronic pain. Increasing evidence has demonstrated that gut microbial dysbiosis is not only a common clinical feature of IBS, migraine, and NP, but also an important systemic driver of central neuroinflammation. Within this complex communication network, the origins and mechanisms of multidimensional signaling molecules must be more precisely defined. On the one hand, microbiota-derived SCFAs and LPS can regulate the excitability of primary nociceptors and influence the polarization states of peripheral and central glial cells.6,16,29 On the other hand, host intestinal epithelial cells and EECs, particularly ECs, are capable of releasing 5-HT, glutamate derived from neuropod cells, and various classical endogenous endocrine peptides,39,80 including CCK, GLP-1, gastrin-releasing peptide, uroguanylin, and Apelin-13.9,26,38,46,81 These multi-level signals derived from both the microbiota and the host intestine not only cooperatively regulate peripheral nociceptors, but also establish a complex interactive network through neural, humoral, and immune pathways, thereby further strengthening the functional coupling between the gut microenvironment and the host neuroimmune system.
This persistent neuroinflammation and aberrant signal transmission further remodel pain transmission pathways and pathologically amplify physiological signals.17 By integrating multidisciplinary evidence, this review highlights the critical role of the intestinal microenvironment in chronic pain and suggests that chronic pain is not merely a localized abnormality within the nervous system, but is closely associated with systemic disruption of neurohomeostasis. Although the mechanisms by which the GBA regulates pain have gradually become clearer, many key questions remain unresolved. For example, do different types of chronic pain possess distinct microbial signatures? How does the microbiota influence individual susceptibility to pain and disease progression? More importantly, the key “molecular switches” regulating the bidirectional effects of metabolites—that is, the mechanisms driving the transition from maintaining physiological homeostasis to promoting pathological pro-inflammatory responses—still require further clarification. Current evidence suggests that SCFA-related G protein-coupled receptors (GPR41/43), the LPS-mediated microglial activation pathway TLR4, P2X7R involved in maintaining neuroinflammation, and lipid metabolite-associated signaling receptors such as FPR2 may all represent important candidate therapeutic targets.29,40,82 As the roles of intestinal epithelial cells, EECs, and neuropod structures in gut–brain communication continue to be uncovered, future studies should further elucidate how different gut-derived signals collectively participate in pain regulation through neural, immune, and endocrine pathways, as well as clarify their disease-specific mechanisms across different pain disorders.
Given the high comorbidity between chronic pain and emotional disorders such as anxiety and depression, interventions aimed at simultaneously improving emotional status and pain perception through microbial therapeutics, dietary modulation, or fecal microbiota transplantation may become highly promising future strategies.14,37,83 Meanwhile, recent advances in dietary interventions, GBA-targeted pharmacological therapies, and microbiota-targeted strategies have further demonstrated substantial potential in analgesia, anti-inflammatory regulation, and neuroprotection.84,85 Research on the GBA not only provides novel therapeutic targets for refractory chronic pain, but also suggests a future transition in pain management from “local symptom control” toward a precision medicine framework centered on “systemic ecological regulation”.86
Abbreviations
5-HT, 5-hydroxytryptamine; ACC, anterior cingulate cortex; ACh, acetylcholine; ANS, autonomic nervous system; AWR, abdominal withdrawal reflex; BBB, blood-brain barrier; BGPs, brain-gut peptides; BMS, burning mouth syndrome; BNST, bed nucleus of the stria terminalis; CAIP, cholinergic anti-inflammatory pathway; CaMKII, calcium/calmodulin-dependent protein kinase II; CB1/CB2, cannabinoid receptor type 1/2; CCI, chronic constriction injury; CCK, cholecystokinin; cGMP, cyclic guanosine monophosphate; CGRP, calcitonin gene-related peptide; CIPN, chemotherapy-induced peripheral neuropathy; CNS, central nervous system; CRD, colorectal distension; CRF, corticotropin-releasing factor; CSD, cortical spreading depression; CUMS, chronic unpredictable mild stress; DGBIs, disorders of gut–brain interaction; DLPFC, dorsolateral prefrontal cortex; DPMS, descending pain modulatory system; DRG, dorsal root ganglion; ECs, enterochromaffin cells; ECS, endogenous cannabinoid system; EECs, enteroendocrine cells; FM, fibromyalgia; FMT, fecal microbiota transplantation; FODMAP, fermentable oligosaccharides, disaccharides, monosaccharides, and polyols; FPR2, formyl peptide receptor 2; GABA, gamma-aminobutyric acid; GBA, gut–brain axis; GLP-1, glucagon-like peptide-1; GLP-1 RAs, glucagon-like peptide-1 receptor agonists; GPR41/43, G protein-coupled receptor 41/43; HPA, hypothalamic-pituitary-adrenal; IASP, International Association for the Study of Pain; IBS, irritable bowel syndrome; IBS-C, constipation-predominant irritable bowel syndrome; IL, interleukin; IL-1RAP, interleukin-1 receptor accessory protein; INS, insula; LPS, lipopolysaccharide; miRNA, microRNA; NK1R, neurokinin-1 receptor; NMDA, N-methyl-D-aspartate; NP, neuropathic pain; NTG, nitroglycerin; NTS, nucleus tractus solitarius; P2X7R, P2X7 receptor; PAG, periaqueductal gray; PBN, parabrachial nucleus; PFC, prefrontal cortex; PKA/MAPK, protein kinase A/mitogen-activated protein kinase; PKC, protein kinase C; RCTs, randomized controlled trials; RVM, rostral ventromedial medulla; SCFAs, short-chain fatty acids; SIBO, small intestinal bacterial overgrowth; SNL, spinal nerve ligation; SP, substance P; TENS, transcutaneous electrical stimulation; TLR4, Toll-like receptor 4; TNF-α, tumor necrosis factor-alpha; Tregs, regulatory T cells; TRP, transient receptor potential; TRPA1, transient receptor potential ankyrin 1; TRPV1, transient receptor potential vanilloid 1; VNS, vagus nerve stimulation; ZO-1, zonula occludens-1.
Data Sharing Statement
Data sharing is not applicable to this article as no data were created or analyzed in this study.
Acknowledgments
We thank all the participants for their contributions to this study. During the preparation of this manuscript, the authors used generative artificial intelligence tools to assist with language polishing and improvement of clarity and readability. The authors reviewed and edited the content as needed and take full responsibility for the final version of the manuscript.
Author Contributions
Zhouyi SONG: Conceptualization, Methodology, Data curation, Investigation, Visualization, Writing – original draft, Writing – review & editing;
Kai ZHOU: Investigation, Data curation, Writing – original draft, Writing – review & editing;
Guangda LIANG: Methodology, Investigation, Resources, Writing – review & editing;
Yanfang LIU: Methodology, Data curation, Visualization, Writing – review & editing;
Qingmei LI: Methodology, Investigation, Resources, Writing – review & editing;
Xingfeng LIU: Conceptualization, Supervision, Project administration, Funding acquisition, Writing – review & editing.
All authors took part in drafting, revising, or critically reviewing the article; gave final approval of the version to be published; agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the National Natural Science Foundation of China (82360232); Guizhou Provincial Science and Technology Planning Project (QianKeHe JiChu ZD[2025] 055).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Yongjun Z, Tingjie Z, Xiaoqiu Y, et al. A survey of chronic pain in China. Libyan J Med. 2020;15(1):1730550. doi:10.1080/19932820.2020.1730550
2. Gaskin DJ, Richard P. The economic costs of pain in the United States. J Pain. 2012;13(8):715–18. doi:10.1016/j.jpain.2012.03.009
3. Yong RJ, Mullins PM, Bhattacharyya N. Prevalence of chronic pain among adults in the United States. Pain. 2022;163(2):e328–e332. doi:10.1097/j.pain.0000000000002291
4. Raja SN, Carr DB, Cohen M, et al. The revised International Association for the Study of Pain definition of pain: concepts, challenges, and compromises. Pain. 2020;161(9):1976–1982. doi:10.1097/j.pain.0000000000001939
5. Raffaeli W, Tenti M, Corraro A, et al. Chronic pain: what does it mean? A review on the use of the term chronic pain in clinical practice. J Pain Res. 2021;14:827–835. doi:10.2147/JPR.S303186
6. Morreale C, Bresesti I, Bosi A, et al. Microbiota and pain: save your gut feeling. Cells. 2022;11(6). doi:10.3390/cells11060971
7. Lin B, Wang Y, Zhang P, et al. Gut microbiota regulates neuropathic pain: potential mechanisms and therapeutic strategy. J Headache Pain. 2020;21(1):103. doi:10.1186/s10194-020-01170-x
8. Ağagündüz D, Yılmaz B, Şahin T, et al. Dairy lactic acid bacteria and their potential function in dietetics: the food-gut-health axis. Foods. 2021;10(12):3099. doi:10.3390/foods10123099
9. Tim H, Ömer E, Lucy K, et al. The brain-gut axis and chronic pain: mechanisms and therapeutic opportunities. Front Neurosci. 2025;19:1545997. doi:10.3389/fnins.2025.1545997
10. Cryan JF, O’Riordan KJ, Cowan CSM, et al. The microbiota-gut-brain axis. Physiol Rev. 2019;99(4):1877–2013. doi:10.1152/physrev.00018.2018
11. Bellono NW, Bayrer JR, Leitch DB, et al. Enterochromaffin cells are gut chemosensors that couple to sensory neural pathways. Cell. 2017;170(1):185–198e116. doi:10.1016/j.cell.2017.05.034
12. Garofalo C, Cristiani CM, Ilari S, et al. Fibromyalgia and irritable bowel syndrome interaction: a possible role for gut microbiota and gut-brain axis. Biomedicines. 2023;11(6):1701. doi:10.3390/biomedicines11061701
13. Caroline WM, Ella C, Richard DH, et al. Unravelling the gut-brain connection: a systematic review of migraine and the gut microbiome. J Headache Pain. 2025;26(1):125. doi:10.1186/s10194-025-02039-7
14. Guo R, Chen L-H, Xing C, Liu T. Pain regulation by gut microbiota: molecular mechanisms and therapeutic potential. Br J Anaesth. 2019;123(5):637–654. doi:10.1016/j.bja.2019.07.026
15. Magni G, Riboldi B, Ceruti S. Modulation of glial cell functions by the gut–brain axis: a role in ne urodegenerative disorders and pain transmission. Cells. 2023;12(12):1612. doi:10.3390/cells12121612
16. Zhou F, Wang X, Han B, et al. Short-chain fatty acids contribute to neuropathic pain via regulating microglia activation and polarization. Mol Pain. 2021;17:1744806921996520. doi:10.1177/1744806921996520
17. Qingyu C, Mengmeng S, Ruoqiu L, et al. Elucidating the specific mechanisms of the gut-brain axis: the short-chain fatty acids-microglia pathway. J Neuroinflammation. 2025;22(1):133. doi:10.1186/s12974-025-03454-y
18. Russo AF, Hay DL. CGRP physiology, pharmacology, and therapeutic targets: migraine and beyond. Physiol Rev. 2023;103(2):1565–1644. doi:10.1152/physrev.00059.2021
19. Hattori N, Yamashiro Y. The gut-brain axis. Ann Nutr Metab. 2021;77(Suppl. 2):1–3. doi:10.1159/000512226
20. Awe T, Fasawe A, Sawe C, et al. The modulatory role of gut microbiota on host behavior: exploring the interaction between the brain-gut axis and the neuroendocrine system. AIMS Neuroscience. 2024;11(1):49–62. doi:10.3934/neuroscience.2024004
21. Mayer EA, Ryu HJ, Bhatt RR. The neurobiology of irritable bowel syndrome. Mol Psychiatry. 2023;28(4):1451–1465. doi:10.1038/s41380-023-01972-w
22. Shao P, Li H, Jiang J, et al. Role of vagus nerve stimulation in the treatment of chronic pain. Neuroimmunomodulation. 2023;30(1):167–183. doi:10.1159/000531626
23. Loh JS, Mak WQ, Tan LKS, et al. Microbiota–gut–brain axis and its therapeutic applications in neurodegenerative diseases. Signal Transduct Target Ther. 2024;9(1). doi:10.1038/s41392-024-01743-1
24. Yano JM, Yu K, Donaldson GP, et al. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell. 2015;161(2):264–276. doi:10.1016/j.cell.2015.02.047
25. Hwang YK, Oh JS. Interaction of the vagus nerve and serotonin in the gut-brain axis. Int J Mol Sci. 2025;26(3):1160. doi:10.3390/ijms26031160
26. Arzani M, Jahromi SR, Ghorbani Z, et al. Gut-brain Axis and migraine headache: a comprehensive review. J Headache Pain. 2020;21(1):15. doi:10.1186/s10194-020-1078-9
27. He Y-Q, Zhu J-R, Sun W-J, et al. ZO-1 and IL-1RAP phosphorylation: potential role in mediated brain-gut axis dysregulation in irritable bowel syndrome-like stressed mice. Int J Med Sci. 2024;21(9):1738–1755. doi:10.7150/ijms.95848
28. Roberto R, Claudia C, Carmen A, et al. Gut-brain axis: role of lipids in the regulation of inflammation, pain and CNS diseases. Curr Med Chem. 2017;25(32):3930–3952. doi:10.2174/0929867324666170216113756
29. Rodríguez-Cortés L, Pacheco R. A short-chain fatty acid triad in pain regulation. Exploration Neuroprotect Ther. 2026;6. doi:10.37349/ent.2026.1004151
30. Tang Y, Du J, Wu H, et al. Potential therapeutic effects of short-chain fatty acids on chronic pain. Curr Neuropharmacol. 2024;22(2):191–203. doi:10.2174/1570159X20666220927092016
31. Stasi C, Sadalla S, Milani S. The relationship between the serotonin metabolism, gut-microbiota and the gut-brain axis. Curr Drug Metab. 2019;20(8):646–655. doi:10.2174/1389200220666190725115503
32. Nagamine T. Burning mouth syndrome needs to consider the gut-brain axis from three types of pain: nociceptive, neuropathic, and nociplastic pain. J Gastrointest Liver Dis. 2023;32(4):558–559. doi:10.15403/jgld-5322
33. Aggeletopoulou I, Triantos C. Microbiome shifts and their impact on gut physiology in irritable bowel syndrome. Int J Mol Sci. 2024;25(22). doi:10.3390/ijms252212395
34. Matijašić M, Meštrović T, Čipčić Paljetak H, et al. Gut microbiota beyond bacteria—mycobiome, virome, archaeome, and eukaryotic parasites in IBD. Int J Mol Sci. 2020;21(8). doi:10.3390/ijms21082668
35. Botschuijver S, Roeselers G, Levin E, et al. Intestinal fungal dysbiosis is associated with visceral hypersensitivity in patients with irritable bowel syndrome and rats. Gastroenterology. 2017;153(4):1026–1039. doi:10.1053/j.gastro.2017.06.004
36. Tetz G, Brown SM, Hao Y, Tetz V. Parkinson’s disease and bacteriophages as its overlooked contributors. Sci Rep. 2018;8(1):10812. doi:10.1038/s41598-018-29173-4
37. Martín Pérez SE, Abdel Lah HAL, García NH, Reyes Carreño UA, Martín Pérez IM. Effectiveness of fecal microbiota transplantation in nociplastic pain management: a systematic review. Gastrointest Disorders. 2025;7(1):5.
38. Ford AC, Vanner S, Kashyap PC, Nasser Y. Chronic visceral pain: new peripheral mechanistic insights and resulting treatments. Gastroenterology. 2024;166(6):976–994. doi:10.1053/j.gastro.2024.01.045
39. Enfu T, Zhenya Z, Chenmin H, et al. Potential roles of enterochromaffin cells in early life stress-induced irritable bowel syndrome. Front Cell Neurosci. 2022;16:837166. doi:10.3389/fncel.2022.837166
40. Zhong S, Zhou Z, Liang Y, et al. Targeting strategies for chemotherapy-induced peripheral neuropathy: does gut microbiota play a role? Crit Rev Microbiol. 2019;45(4):369–393. doi:10.1080/1040841X.2019.1608905
41. Bradesi S, Kokkotou E, Simeonidis S, et al. The role of neurokinin 1 receptors in the maintenance of visceral hyperalgesia induced by repeated stress in rats. Gastroenterology. 2006;130(6):1729–1742. doi:10.1053/j.gastro.2006.01.037
42. Liang C, Luo H, Liu Y, Cao J, Xia H. Plasma hormones facilitated the hypermotility of the colon in a chronic stress rat model. PLoS One. 2012;7(2):e31774. doi:10.1371/journal.pone.0031774
43. Starinets A, Tyrtyshnaia A, Manzhulo I. Anti-inflammatory activity of synaptamide in the peripheral nervous system in a model of sciatic nerve injury. Int J Mol Sci. 2023;24(7):6273. doi:10.3390/ijms24076273
44. Chen Y, Feng S, Li Y, et al. Gut microbiota and intestinal immunity-A crosstalk in irritable bowel syndrome. Immunology. 2024;172(1):1–20. doi:10.1111/imm.13749
45. Fila M, Sobczuk A, Pawlowska E, Blasiak J. epigenetic connection of the calcitonin gene-related peptide and its potential in migraine. Int J Mol Sci. 2022;23(11):6151. doi:10.3390/ijms23116151
46. Chen Z-F. A neuropeptide code for itch. Nat Rev Neurosci. 2021;22(12):758–776. doi:10.1038/s41583-021-00526-9
47. Hammam H, Arvind A, Khalafallah S, et al. Effect of buscopan compared with glucagon during gastroscopy, colonoscopy, and ERCP- a narrative review. Int J Adv Res. 2024;12(08):310–314. doi:10.21474/ijar01/19256
48. Rusch JA, Layden BT, Dugas LR. Signalling cognition: the gut microbiota and hypothalamic-pituitary-adrenal axis. Front Endocrinol. 2023;14:1130689. doi:10.3389/fendo.2023.1130689
49. Brierley SM, Greenwood-Van Meerveld B, Sarnelli G, et al. Targeting the endocannabinoid system for the treatment of abdominal pain in irritable bowel syndrome. Nat Rev Gastroenterol Hepatol. 2023;20(1):5–25. doi:10.1038/s41575-022-00682-y
50. Sgro M, Ray J, Foster E, Mychasiuk R. Making migraine easier to stomach: the role of the gut-brain-immune axis in headache disorders. Eur J Neurol. 2023;30(11):3605–3621. doi:10.1111/ene.15934
51. Miaomiao Z, Xun Z, Jun C. Microbiota-gut-brain axis: interplay between microbiota, barrier function and lymphatic system. Gut Microbes. 2024;16(1):2387800. doi:10.1080/19490976.2024.2387800
52. Kraimi N, Ross T, Pujo J, De Palma G. The gut microbiome in disorders of gut-brain interaction. Gut Microbes. 2024;16(1):2360233. doi:10.1080/19490976.2024.2360233
53. Zhuang M, Zhang X, Cai J. Microbiota–gut–brain axis: interplay between microbiota, barrier funct ion and lymphatic system. Gut Microbes. 2024;16(1). doi:10.1080/19490976.2024.2387800
54. Chenghan M, Wanxin L, Bangcheng Z, et al. Short-chain fatty acids mediate gut microbiota–brain communication and protect the blood–brain barrier integrity. Ann NY Acad Sci. 2025;1545(1):116–131. doi:10.1111/nyas.15299
55. Ramakrishna C, Corleto J, Ruegger PM, et al. Dominant role of the gut microbiota in chemotherapy induced neuropathic pain. Sci Rep. 2019;9(1):20324. doi:10.1038/s41598-019-56832-x
56. Santos JM, Deshmukh H, Elmassry MM, et al. Beneficial effects of ginger root extract on pain behaviors, inflammat ion, and mitochondrial function in the colon and different brain regions of male and female neuropathic rats: a gut–brain axis study. Nutrients. 2024;16(20):3563. doi:10.3390/nu16203563
57. Wager TD, Atlas LY. The neuroscience of placebo effects: connecting context, learning and health. Nat Rev Neurosci. 2015;16(7):403–418. doi:10.1038/nrn3976
58. Livrizzi G, Liao J, Johnson DA, et al. Top-down control of the descending pain modulatory system drives placebo analgesia. bioRxiv. 2025. doi:10.1101/2025.02.13.638185
59. Xu M, Zhou EY, Shi H. Tryptophan and its metabolite serotonin impact metabolic and mental disorders via the brain-gut-microbiome axis: a focus on sex differences. cells. 2025;14(5). doi:10.3390/cells14050384
60. Xia HH, Huang JZ. Tryptophan metabolism at the crossroads of the neuro-immuno-microbial axis: implications for precision medicine in chronic diseases. Front Cell Infect Microbiol. 2025;15:1707850. doi:10.3389/fcimb.2025.1707850
61. Lackner JM, Quigley BM, Zilcha-Mano S, et al. Factors That Predict Magnitude, Timing, and Persistence of Placebo-Like Response in Patients With Irritable Bowel Syndrome. Gastro Hep Adv. 2024;3(2):221–229. doi:10.1016/j.gastha.2023.10.003
62. Shin JH, Park YH, Sim M, et al. Serum level of sex steroid hormone is associated with diversity and profiles of human gut microbiome. Res Microbiol. 2019;170(4–5):192–201. doi:10.1016/j.resmic.2019.03.003
63. Brum ES, Becker G, Fialho MFP, Oliveira SM. Animal models of fibromyalgia: what is the best choice? Pharmacol Ther. 2022;230:107959. doi:10.1016/j.pharmthera.2021.107959
64. Ma P, Mo R, Liao H, et al. Gut microbiota depletion by antibiotics ameliorates somatic neuropathic pain induced by nerve injury, chemotherapy, and diabetes in mice. J Neuroinflammation. 2022;19(1):169. doi:10.1186/s12974-022-02523-w
65. Chey WD, Shah ED, DuPont HL. Mechanism of action and therapeutic benefit of rifaximin in patients with irritable bowel syndrome: a narrative review. Therap Adv Gastroenterol. 2020;13:1756284819897531. doi:10.1177/1756284819897531
66. Mamieva Z, Poluektova E, Svistushkin V, et al. Antibiotics, gut microbiota, and irritable bowel syndrome: what are the relations? World J Gastroenterol. 2022;28(12):1204–1219. doi:10.3748/wjg.v28.i12.1204
67. Ford AC, Staudacher HM, Talley NJ. Postprandial symptoms in disorders of gut-brain interaction and their potential as a treatment target. Gut. 2024;73(7):1199–1211. doi:10.1136/gutjnl-2023-331833
68. Zhang T, Zhang C, Zhang J, Sun F, Duan L. Efficacy of probiotics for irritable bowel syndrome: a systematic review and network meta-analysis. Front Cell Infect Microbiol. 2022;12. doi:10.3389/fcimb.2022.859967
69. Chen Q, Zhang H, Sun CY, et al. Evaluation of two laboratory model methods for diarrheal irritable bowel syndrome. Mol Med. 2023;29(1):5. doi:10.1186/s10020-022-00599-x
70. Qianwen W, Yunfeng W, Qi P, et al. MicroRNA-155-5p promotes neuroinflammation and central sensitization via inhibiting SIRT1 in a nitroglycerin-induced chronic migraine mouse model. J Neuroinflammation. 2021;18(1):287. doi:10.1186/s12974-021-02342-5
71. Du X, Hao H, Yang Y, et al. Local GABAergic signaling within sensory ganglia controls peripheral nociceptive transmission. J Clin Invest. 2017;127(5):1741–1756. doi:10.1172/JCI86812
72. Bosi A, Banfi D, Bistoletti M, Giaroni C, Baj A. Tryptophan metabolites along the microbiota-gut-brain axis: an interkingdom communication system influencing the gut in health and disease. Int J Tryptophan Res. 2020;13:1178646920928984. doi:10.1177/1178646920928984
73. Paudel D, Uehara O, Giri S, et al. Effect of psychological stress on the oral-gut microbiota and the potential oral-gut-brain axis. Jpn Dent Sci Rev. 2022;58:859967. doi:10.1016/j.jdsr.2022.11.003
74. Sansores-Espana LD, Melgar-Rodriguez S, Olivares-Sagredo K, et al. Oral-Gut-brain axis in experimental models of periodontitis: associating gut dysbiosis with neurodegenerative diseases. Front Aging. 2021;2:781582. doi:10.3389/fragi.2021.781582
75. Tao R, Liu S, Crawford J, Tao F. Gut-brain crosstalk and the central mechanisms of orofacial pain. Brain Sci. 2023;13(10):1456. doi:10.3390/brainsci13101456
76. Krishnamoorthy G, Narayana A, Balakrishnan D. The gut-masticatory muscles-temporomandibular joint pain axis-a scoping review. J Oral Facial Pain Headache. 2025;39(1):24–33. doi:10.22514/jofph.2025.021
77. Mayer EA, Tillisch K. The brain-gut axis in abdominal pain syndromes. Annu Rev Med. 2011;62:381–396. doi:10.1146/annurev-med-012309-103958
78. Laurie K, Cynthia WK, Alexander CF. AGA clinical practice update on management of chronic gastrointestinal pain in disorders of gut-brain interaction: expert review. Clin Gastroenterol Hepatol. 2021;19(12):1518–1530. doi:10.1016/j.cgh.2021.07.006
79. Aljeradat B, Kumar D, Abdulmuizz S, et al. Neuromodulation and the gut-brain axis: therapeutic mechanisms and implications for gastrointestinal and neurological disorders. Pathophysiology. 2024;31(2):244–268. doi:10.3390/pathophysiology31020019
80. Erny D, Hrabe de Angelis AL, Jaitin D, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. 2015;18(7):965–977. doi:10.1038/nn.4030
81. Luo H, Xiang Y, Qu X, et al. Apelin-13 suppresses neuroinflammation against cognitive deficit in a streptozotocin-induced rat model of Alzheimer’s disease through activation of BDNF-TrkB signaling pathway. Front Pharmacol. 2019;10. doi:10.3389/fphar.2019.00395
82. Lin H, Ma C, Cai K, et al. Metabolic signaling of ceramides through the FPR2 receptor inhibits adipocyte thermogenesis. Science. 2025;388(6746). doi:10.1126/science.ado4188
83. Ustianowska K, Ustianowski Ł, Machaj F, et al. The role of the human microbiome in the pathogenesis of pain. Int J Mol Sci. 2022;23(21). doi:10.3390/ijms232113267
84. Zhang S, Wu Z, Zhang S, et al. The intricate microbial–gut–brain axis in Alzheimer’s disease: a review of microbiota-targeted strategies. Food Funct. 2025;16(21):8320–8344. doi:10.1039/d5fo03139g
85. Chen F, Wang Y, Wang K, et al. Effects of Litsea cubeba essential oil on growth performance, blood antioxidation, immune function, apparent digestibility of nutrients, and fecal microflora of pigs. Front Pharmacol. 2023;14. doi:10.3389/fphar.2023.1166022
86. Li Y-K, Li W-R, Ren H, et al. Gut microbiome-targeted therapeutics for chronic kidney disease: comparative efficacy of probiotic and microbial preparations. Inflammopharmacology. 2025;33(12):7569–7585. doi:10.1007/s10787-025-02044-x
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Normalization of Neuroinflammation: A New Strategy for Treatment of Persistent Pain and Memory/Emotional Deficits in Chronic Pain
Liu XG
Journal of Inflammation Research 2022, 15:5201-5233
Published Date: 9 September 2022
Transforming Chronic Pain Management: Integrating Neuromodulation with Advanced Technologies to Tackle Cognitive Dysfunction – A Narrative Review
Green M, Hayley A, Gunnersen JM, Nazemian V, Cabble A, Thompson S, Chakravarthy K
Journal of Pain Research 2025, 18:2497-2507
Published Date: 16 May 2025
Gut–Spinal Cord Axis in Spinal Cord Injury: Bidirectional Inflammatory Mechanisms and Microbiota-Targeted Therapeutic Strategies
Dong J, Xie T, Shi C, Feng G, Zhang H, Xu Z, Dong L
Journal of Inflammation Research 2025, 18:12549-12573
Published Date: 12 September 2025
Circulating MicroRNA Biomarkers for Chronic Pain and Acupuncture Response: An Exploratory High-Dimensional Small-Sample Study
Kang IH, Kim SN
Journal of Pain Research 2025, 18:6591-6605
Published Date: 6 December 2025