Back to Journals » Journal of Pain Research » Volume 18
Circulating MicroRNA Biomarkers for Chronic Pain and Acupuncture Response: An Exploratory High-Dimensional Small-Sample Study
Received 24 September 2025
Accepted for publication 2 December 2025
Published 6 December 2025 Volume 2025:18 Pages 6591—6605
DOI https://doi.org/10.2147/JPR.S567154
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Houman Danesh
In-Hye Kang, Seung-Nam Kim
College of Korean Medicine, Dongguk University, Goyang, 10326, South Korea
Correspondence: Seung-Nam Kim, Email [email protected]
Background: Chronic pain involves complex neuroplastic changes and neuroinflammatory processes that may be reflected in circulating microRNA profiles. Acupuncture analgesia operates through multiple neurobiological mechanisms including modulation of neurotransmitter release and synaptic plasticity. High-dimensional small-sample (HDSS) datasets pose significant challenges for biomarker discovery in clinical omics studies, where the number of features vastly exceeds sample size. Existing approaches often lack robust validation frameworks, leading to overfitted models with poor generalizability. We developed and validated a comprehensive framework for HDSS biomarker discovery, demonstrated on circulating microRNA data from a chronic neck pain acupuncture trial. Our objective was to establish a simulation-validated pipeline that addresses preprocessing instability, model selection uncertainty, and statistical significance assessment in extreme HDSS scenarios.
Methods: In this exploratory high-dimensional small-sample study, we analyzed plasma microRNA profiles from 6 participants in a chronic neck pain acupuncture trial, generating 9 paired measurements of microRNA fold-changes and pain reduction (ΔVAS) across baseline, 4-week, and 8-week time points. Our framework integrated: (1) robust preprocessing with Winsorization, arcsinh transformation, and median/MAD scaling; (2) nested leave-one-out cross-validation with LASSO/Elastic Net regularization; (3) permutation testing (10,000 iterations); and (4) comprehensive HDSS factorial simulation (19,440 runs across varying sample sizes, sparsity, signal-to-noise ratios, and preprocessing strategies).
Results: The observed model achieved MAE=10.58, RMSE=13.70, and Spearman ρ=− 0.217, with permutation p< 0.001. Simulation benchmarking revealed our correlation coefficient ranked at the 91st percentile compared to null-like HDSS scenarios. Three microRNAs (miR-3681-3p, miR-4743-5p, miR-6822-5p) emerged as a dual-function panel, consistently selected across cross-validation folds and associated with both pain prediction and acupuncture response. Pathway enrichment analysis revealed significant associations with PI3K-Akt/mTOR, TGF-β signaling, synaptic plasticity, and neuroinflammatory pathways.
Conclusion: We present a rigorously validated framework for HDSS biomarker discovery that demonstrates modest but statistically significant predictive signal in extremely challenging conditions. This methodology is broadly applicable to other HDSS omics problems where traditional validation approaches fail.
Keywords: microRNA, chronic pain, acupuncture, neuroinflammation, synaptic plasticity, biomarker discovery, high-dimensional data, permutation testing
Introduction
Pain biomarker discovery exemplifies analytical challenges at the intersection of clinical neuroscience and molecular biology. Chronic pain pathophysiology is characterized by peripheral and central sensitization processes.1 Peripheral sensitization involves increased nociceptor responsiveness following tissue injury, while central sensitization represents enhanced excitability of central pain pathways.2 These mechanisms are mediated by neurotransmitters including glutamate and neuropeptides such as substance P,3 driving neuroplastic changes in synaptic transmission and neuroinflammatory cascades that may be reflected in circulating molecular signatures. Despite growing interest in these molecular mechanisms, robust biomarker identification has been hindered by methodological difficulties in handling high-dimensional molecular profiling data from necessarily small clinical cohorts. Many studies employ standard statistical approaches that may not adequately account for multiple testing burdens and overfitting risks.4 Additionally, the lack of standardized validation frameworks makes it difficult to contextualize model performance and distinguish meaningful biological signals from statistical artifacts.
Circulating microRNAs (miRNAs) represent promising candidates for pain biomarkers due to their regulatory roles in neurobiological processes including synaptic plasticity, neuroinflammation, and nociceptive signaling, combined with their stability in biological fluids and potential for non-invasive measurement.5,6 Several studies have reported associations between miRNA expression patterns and various pain conditions,7 though findings have often been inconsistent across studies, potentially reflecting analytical challenges rather than biological variability.
Acupuncture analgesia operates through multiple neurobiological mechanisms including modulation of neurotransmitter release, activation of descending pain inhibitory pathways, and regulation of neuroinflammatory responses. These effects involve modulation of glial cell activity and suppression of neuroinflammatory cascades that contribute to pain sensitization and chronification. MicroRNAs, as key regulators of inflammatory pathways and synaptic plasticity, may mediate or reflect these therapeutic mechanisms. Our previous clinical trial demonstrated significant improvements in pain intensity and functional disability following acupuncture treatment in patients with chronic neck pain, accompanied by changes in serum substance P levels,8 indicating underlying molecular responses to treatment. This established clinical framework provides an opportunity to explore circulating miRNA profiles as potential molecular indicators of acupuncture’s anti-inflammatory and neuromodulatory effects. Accumulating preliminary evidence indicates that acupuncture may influence microRNA expression involved in pain-related pathways, suggesting a potential epigenetic mechanism underlying its therapeutic effects.9,10 However, systematic investigation of circulating miRNA biomarkers for acupuncture response prediction in chronic pain patients remains limited.
High-dimensional small-sample (HDSS) datasets pose particular challenges in clinical neuroscience studies, where comprehensive molecular profiling must be balanced against practical constraints of patient recruitment and intervention complexity.11 This scenario is especially common in pain research, where the number of molecular features (p) often vastly exceeds the sample size (n) due to cost, patient availability, or study feasibility constraints. While regularization methods such as LASSO and Elastic Net have been developed to address some aspects of the “large p, small n” problem,12,13 practical challenges remain in model validation, statistical significance assessment, and performance benchmarking when dealing with extreme HDSS scenarios.4 While recent advances have addressed specific aspects of HDSS analysis—including robust preprocessing,14 regularization with cross-validation,12,13,15 and permutation-based inference16—integrated frameworks combining these approaches with comprehensive simulation-based validation across realistic HDSS conditions remain limited.4,11
The development of practical analytical frameworks for HDSS biomarker discovery requires careful integration of preprocessing strategies, regularization methods, and validation approaches tailored to the specific challenges of extremely unbalanced datasets. Simulation-based validation offers a particularly useful approach for performance contextualization by providing empirical benchmarks against which observed results can be evaluated.17
In this study, we present a practical framework for HDSS biomarker discovery and demonstrate its application using circulating microRNA profiling data from our chronic neck pain acupuncture cohort. Our specific aims were: (1) to develop and validate a preprocessing and modeling pipeline suitable for extreme HDSS scenarios in clinical neuroscience; (2) to identify circulating microRNAs associated with both pain prediction and acupuncture treatment response; (3) to demonstrate a simulation-based validation approach for performance benchmarking in HDSS settings; and (4) to explore neurobiological pathways underlying the identified microRNA-pain associations. Using paired measurements of microRNA expression changes and pain outcomes (n=9 pairs, p=2330 features), we show how meaningful biological signals can be extracted from challenging datasets when appropriate analytical strategies are employed.
Methods
Study Design and Participants
This study utilized biospecimens and clinical data from a previously published chronic neck pain acupuncture trial.8 The original study enrolled female participants aged 40–60 years with chronic neck pain (>12 weeks duration, VAS ≥30 mm) and followed a waiting-list controlled design with a 4-week observation period followed by 4 weeks of acupuncture treatment. The acupuncture intervention utilized bilateral acupoints (LI4, TE5, GB20, GB21, SI3, and BL60) with disposable stainless steel needles (0.25×40 mm) inserted to depths of 15–25 mm depending on location. Manual stimulation was applied to achieve de-qi sensation, needles were retained for 20 minutes per session, and treatment was administered twice weekly for 4 weeks (8 sessions total). Detailed inclusion/exclusion criteria and complete clinical procedures have been described previously.8 For the current analysis, we selected a representative subset of 6 participants (3 control, 3 acupuncture group) to demonstrate our HDSS analytical framework under extreme sample size constraints.
The study generated 9 paired observations: control participants contributed baseline vs 4-week measurements (3 pairs), while acupuncture participants contributed both baseline vs 4-week and 4-week vs 8-week measurements (6 pairs). Each pair consisted of microRNA expression fold-changes and corresponding pain change scores (ΔVAS), providing independent data points for correlation analysis regardless of treatment group assignment.
MicroRNA Profiling
Plasma samples were collected at baseline, 4 weeks, and 8 weeks, and processed using standard protocols with storage at −80°C until analysis. Total RNA including small RNAs was extracted from plasma samples using the miRNeasy Serum/Plasma Kit (Qiagen) following the manufacturer’s protocol. RNA quality and quantity were assessed using a NanoDrop spectrophotometer. MicroRNA expression profiling was performed using Affymetrix microRNA GeneChip microarrays. CEL files were imported and processed using Affymetrix Power Tools (APT) Software with RMA+DABG-All analysis.18 Array data were filtered by probes annotated to human species, resulting in approximately 6000 initially detected features. Quality control procedures and probe filtering reduced the final dataset to 2330 microRNA features for downstream analysis.
For each paired measurement, microRNA fold-changes were calculated as log2 ratios between time points, while pain changes were calculated as ΔVAS (later time point VAS - earlier time point VAS). This approach generated 9 independent (fold-change, ΔVAS) observations for statistical modeling.
Preprocessing Pipeline
We developed a robust preprocessing pipeline specifically designed for HDSS scenarios with potential outliers and distributional challenges commonly encountered in microarray data. The primary pipeline consisted of the following sequential steps:
Step 1: Winsorization - Extreme values were capped at the 99.5th percentile (absolute values) to reduce the influence of outliers while preserving the overall data structure.19
Step 2: Variance-stabilizing transformation - The arcsinh transformation was applied to stabilize variance across the dynamic range of expression values and approximate normality.20
Step 3: Robust scaling - Features were scaled using median centering and median absolute deviation (MAD) normalization to provide robust alternatives to mean/standard deviation scaling.21
Step 4: Dispersion filtering - Features were ranked by MAD and the top 50% most variable features were retained, with a minimum threshold of max (50, 3×n) features to ensure sufficient feature diversity for modeling.
Step 5: Quality control - Defensive handling of missing values, infinite values, and zero-variance features was implemented with appropriate exclusion criteria.
To evaluate preprocessing sensitivity, we implemented two comparator pipelines: (1) standard z-score normalization, and (2) quantile normalization followed by z-score transformation with variance-based feature selection (top 50% by variance).22
Statistical Modeling
Regularized Regression
We employed LASSO and Elastic Net regularization using the glmnet package in R.12,23 Elastic Net was implemented with alpha parameters ranging from 0.1 to 1.0 using grid search. Both methods use L1 regularization to perform automatic feature selection while controlling overfitting in HDSS settings.
Cross-Validation Strategy
Given the extreme sample size (n=9), we implemented nested leave-one-out cross-validation (LOOCV) to maximize training data utilization while providing unbiased performance estimates.24 The nested structure consisted of:
- Outer loop: LOOCV for final performance estimation (9 iterations, each holding out 1 observation)
- Inner loop: LOOCV on the remaining 8 observations for hyperparameter tuning and model selection
- One-SE rule: Applied during inner cross-validation to select the most parsimonious model within one standard error of the minimum cross-validation error
This strategy ensures that each performance estimate is based on completely independent test data while maintaining rigorous separation between model selection and performance evaluation.15 All preprocessing steps (Winsorization, arcsinh transform, median/MAD scaling, and dispersion filtering) were fit within each inner training fold and applied to the held-out samples to avoid information leakage; the same fold-wise protocol was used for permutations and simulations.
Performance Metrics
Primary evaluation metrics included Mean Absolute Error (MAE), Root Mean Square Error (RMSE), and Spearman rank correlation coefficient (ρ). Scale-free versions (sMAE = MAE/SD(y), sRMSE = RMSE/SD(y)) were calculated to enable comparison across different simulation scenarios.
Validation Framework
Permutation Testing
Statistical significance was assessed using two-sided permutation testing with 10,000 random permutations of the outcome variable (ΔVAS) while maintaining the feature matrix structure.16 For each permutation, the complete modeling pipeline was re-run and the Spearman correlation coefficient was recalculated. P-values were calculated as (number of permutations with |correlation| ≥ |observed correlation| + 1) / (total permutations + 1) to provide conservative estimates. Random seed was set to 12345 for reproducibility, with Monte Carlo standard error approximately 0.001. This approach provides an empirical null distribution for the observed correlation coefficient under the assumption of no true association between microRNA patterns and pain outcomes.
HDSS Simulation Study
To contextualize our results within the broader landscape of HDSS performance, we conducted a comprehensive factorial simulation study spanning 19,440 runs across the following parameters:
- Sample sizes: n = 6, 9, 12
- Feature dimensionality: p = 2000 (approximately matching our dataset)
- Sparsity levels: 1–2% (proportion of features with true signal)
- Signal-to-noise ratios: 0.75, 1.0, 1.5
- Distributional assumptions: Normal vs heavy-tailed (t-distribution, df=3)
- Batch effects: Standard deviation = 0 (no batch effects) vs 0.5 (moderate batch effects)
- Preprocessing strategies: Robust (our primary pipeline), z-score, quantile normalization
- Validation strategies: Nested LOOCV, repeated k-fold, bootstrap 0.63225
- Replications: 30 independent runs per parameter combination
For each simulation run, synthetic datasets were generated with known ground truth, and the same modeling pipeline applied to our real data was used to generate performance metrics. This design allows direct comparison of our observed results against empirical distributions of performance under various HDSS scenarios.
Feature Selection and Biological Interpretation
MicroRNAs consistently selected across multiple cross-validation folds in both LASSO and Elastic Net were identified as candidate biomarkers. Selection frequency was tracked across all outer LOOCV iterations, and microRNAs appearing in both regularization methods were prioritized for biological interpretation.
Target prediction and pathway enrichment analysis were performed using the miRDB database for target prediction26 followed by pathway analysis using Enrichr.27 Significant pathways were identified using FDR < 0.05 as the significance threshold.
Statistical Software
All analyses were performed in R version 4.5.1. Key packages included glmnet for regularized regression, caret for cross-validation frameworks, and custom scripts for simulation studies. The complete analytical pipeline is available as supplementary code to ensure reproducibility.
Results
Our HDSS framework achieved modest but statistically significant predictive performance with MAE=10.58, RMSE=13.70, and Spearman ρ=−0.217. The observed MAE of 10.58 represents approximately 10.6% of the VAS scale range (0–100). In chronic pain research, VAS changes of 15–20 points are considered clinically significant; thus our prediction error falls within a potentially meaningful range for exploratory biomarker studies, though substantial validation is required. Detailed feature selection, biological validation, and methodological validation are presented below.
Regularized Feature Selection Identifies Consensus MicroRNA Panel
Nested leave-one-out cross-validation with LASSO and Elastic Net regularization consistently selected a core set of microRNAs across multiple cross-validation folds (Figure 1). Three microRNAs emerged as the most robust features: miR-3681-3p (selected in 69% of LASSO folds and 69% of Elastic Net folds), miR-933 (44% and 56% respectively), and miR-4743-5p (44% and 56% respectively). The high concordance between LASSO and Elastic Net selection patterns, particularly for the top-ranked features, demonstrates the robustness of these candidates despite the challenging HDSS conditions.
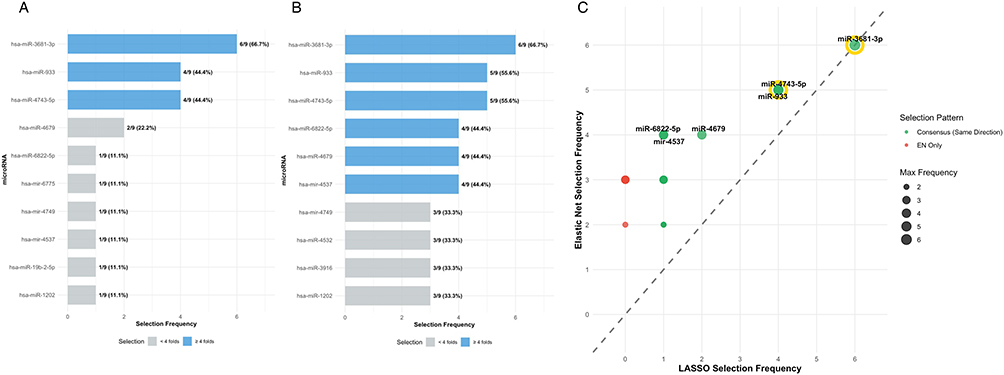 |
Figure 1 Regularized feature selection identifies consensus microRNA biomarkers. MicroRNA selection frequencies across nested leave-one-out cross-validation for LASSO (A) and Elastic Net (B) regression. Bars show percentage of folds in which each microRNA was selected. Scatter plot (C) compares selection frequencies between methods, with diagonal line indicating perfect agreement. Green circles represent consensus features with same directional selection; red circles indicate Elastic Net-specific selections. Point size reflects maximum selection frequency. Diagonal = perfect agreement; gold rings = core trio. |
Additional microRNAs showed moderate selection frequencies, including miR-4679 (22% LASSO, 44% Elastic Net), miR-6822-5p (11% LASSO, 44% Elastic Net), and several others with selection frequencies ranging from 11% to 33%. The selection frequency patterns revealed that while the extreme HDSS scenario resulted in variable feature selection across folds, a consensus panel of 3–5 microRNAs consistently emerged as the most predictive features.
Treatment Response Analysis Reveals Differential MicroRNA Patterns
Analysis of treatment response patterns identified substantial differences in microRNA expression changes between acupuncture and control groups (Figure 2). The top 50 microRNAs showing the greatest differential response to acupuncture versus control conditions demonstrated clear clustering patterns, with acupuncture subjects (ACU1-3) showing distinct expression profiles compared to control subjects (CON1-3).
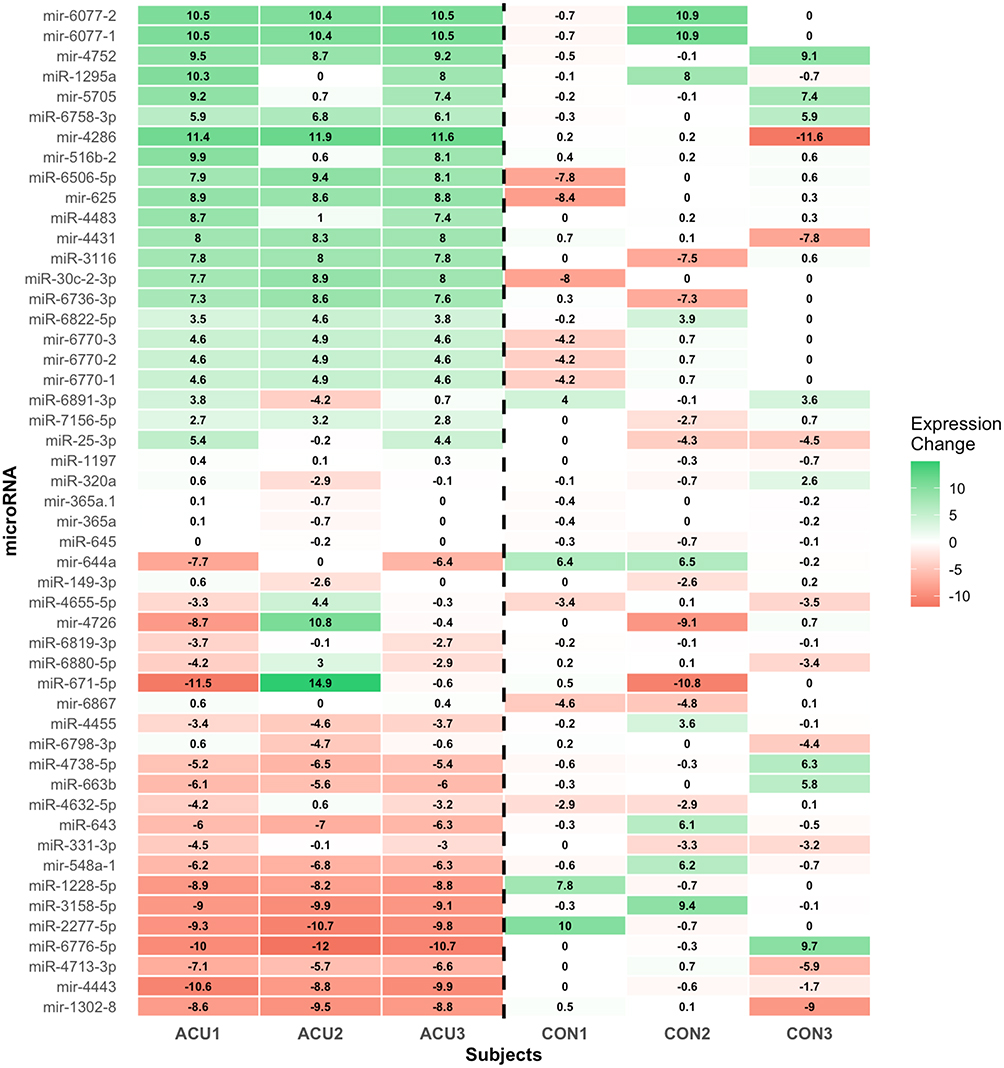 |
Figure 2 Treatment-specific microRNA expression changes distinguish acupuncture from control groups. Heatmap of the top 50 microRNAs ranked by correlation magnitude with treatment status (acupuncture vs control). Rows represent individual microRNAs; columns represent subjects (ACU1-3: acupuncture group baseline-to-4 weeks and 4 weeks-to-8 weeks measurements; CON1-3: control group baseline-to-4 weeks measurements). Color scale indicates expression fold-change values (green: upregulation, red: downregulation). |
Notable patterns included coordinate upregulation of multiple microRNAs in acupuncture responders, particularly miR-6077-2, miR-6077-1, and miR-4752, while control subjects showed more variable and generally smaller magnitude changes. Several microRNAs displayed opposing directional changes between groups, including miR-4286, miR-5186-2, and miR-6506-5p, suggesting treatment-specific regulatory responses.
The heatmap visualization revealed that acupuncture treatment induced coordinated changes across multiple microRNA families, while control subjects showed predominantly random fluctuations consistent with measurement noise or natural biological variation.
Dual-Function MicroRNA Panel Links Pain Prediction and Treatment Response
Integration of pain prediction capability with treatment response revealed a subset of microRNAs with dual functionality (Figure 3). The scatter plot analysis, plotting pain prediction strength (x-axis) against treatment response association (y-axis), identified microRNAs in three functional categories: pain-specific (high x, green), treatment-specific (high y, blue), and dual-function (high x, high y, red).
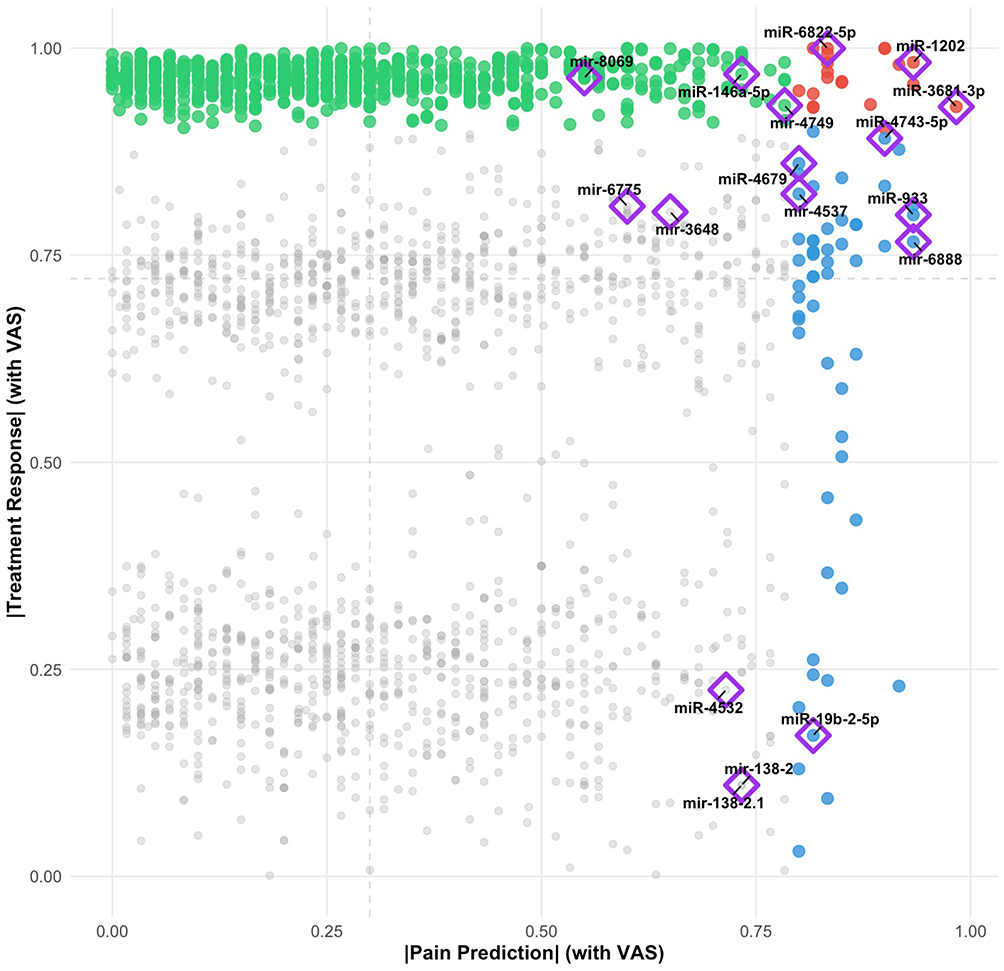 |
Figure 3 microRNA associations with pain prediction and treatment response. Scatter plot of microRNA associations with pain prediction (x-axis, correlation with VAS) versus treatment response (y-axis, correlation with treatment status). Each point represents one microRNA. Purple diamonds highlight microRNAs selected during regularized feature selection from Figure 1. Three representative dual-function candidates (miR-6822-5p, miR-4743-5p, miR-3681-3p) are labeled in the upper-right quadrant. Color coding: green = treatment-specific, blue = pain-specific, red = dual-function, gray = others. |
Three microRNAs emerged as representative dual-function biomarkers based on their position in the upper-right quadrant of the scatter plot and their appearance in feature selection results: miR-6822-5p, miR-4743-5p, and miR-3681-3p (highlighted with purple diamonds, indicating their selection during regularized feature selection). These candidates demonstrated both strong associations with pain prediction (|correlation with VAS| > 0.8) and clear differential response to acupuncture treatment (|treatment response| > 0.9). The purple diamond annotation indicates that these microRNAs were consistently selected across multiple cross-validation folds in the LASSO and/or Elastic Net procedures, providing additional validation of their predictive importance beyond their dual-function characteristics.
Additional microRNAs showed more specialized functions: miR-1202 and miR-3681-3p exhibited primarily pain prediction capabilities, while miR-4679, miR-4537, and miR-933 showed stronger associations with treatment response. The identification of microRNAs spanning both functional domains provides biological plausibility for their role as both predictive biomarkers and mechanistic indicators of therapeutic response.
Pathway Enrichment Analysis Reveals Biologically Plausible Mechanisms
Functional annotation of the three dual-function microRNAs (miR-3681, miR-6822, miR-4743) using miRDB target prediction followed by pathway enrichment analysis revealed significant associations with multiple pathways relevant to pain mechanisms and acupuncture response (Figure 4).
 |
Figure 4 Pathway enrichment analysis reveals biologically relevant mechanisms. Pathway enrichment results for three dual-function microRNAs (miR-3681, miR-6822, miR-4743) across four databases: (A) GO Biological Process, (B) KEGG Pathway Enrichment, (C) Reactome Enrichment, and (D) WikiPathways Enrichment. Bubble plots show -log10(p-value) on x-axis with different y-axis scales per database. Bubble size represents gene count; color intensity indicates combined enrichment score. Key enriched pathways include transcriptional regulation, PI3K-Akt signaling, and neuronal processes. |
Gene Ontology Biological Process enrichment identified significant associations with transcriptional regulation, particularly positive regulation of DNA-templated transcription (GO: 0045893) and regulation of transcription by RNA Polymerase II (GO: 0006357). Additional significant processes included peptidyl-serine modification and negative regulation of RNA biosynthetic processes, suggesting roles in post-translational modification and gene expression control.
KEGG pathway analysis revealed enrichment in several pain-relevant pathways, including AMPK signaling pathway, insulin resistance, and TGF-beta signaling pathway. The longevity regulating pathway and non-small cell lung cancer pathways were also significantly enriched, potentially reflecting broader cellular stress response mechanisms.28
Reactome pathway enrichment highlighted neuronal system pathways, signal transduction, and RNA Polymerase II transcription as key functional domains. Particularly relevant was the enrichment in protein-protein interactions at synapses and MET activates PI3K/AKT signaling, both directly relevant to pain processing and synaptic plasticity.29
WikiPathways analysis identified focal adhesion PI3K-Akt-mTOR signaling (WP3932) and VEGFA-VEGFR2 signaling (WP3888) as significantly enriched pathways. The PI3K-Akt-mTOR pathway is particularly relevant to pain chronification and neuroplasticity, while VEGF signaling has been implicated in both angiogenesis and neuronal survival responses.
Permutation Testing Confirms Statistical Significance
Permutation analysis using 10,000 random outcome permutations confirmed that the observed model performance was highly unlikely under the null hypothesis of no association (Figure 5). The empirical null distributions for MAE, RMSE, and Spearman correlation all demonstrated that our observed values were extreme outliers compared to the permuted results.
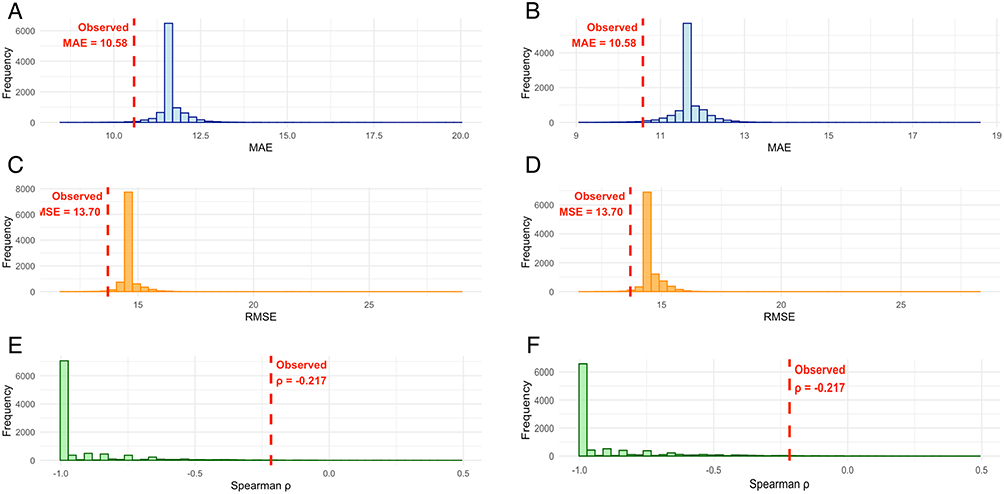 |
Figure 5 Permutation testing confirms statistical significance of model performance. Histograms show distribution of MAE (A and B), RMSE (C and D), and Spearman correlation (E and F) under null hypothesis. Empirical null distributions from 10,000 random permutations of outcome variables for LASSO (A, C, and E) and Elastic Net (B, D, and F) regression. Red dashed lines indicate observed values (MAE=10.58, RMSE=13.70, ρ=−0.217). All observed metrics fall in extreme tails of null distributions, confirming statistical significance (p<0.001). |
For MAE, the observed value of 10.58 fell well outside the permutation distribution (centered around 12.5), representing approximately the 5th percentile of the null distribution. Similarly, the RMSE of 13.70 was substantially lower than the permutation distribution (centered around 15.5), falling in the lower tail of the null distribution.
Most strikingly, the Spearman correlation of ρ=−0.217 was dramatically different from the permutation distribution, which was tightly centered around zero. The observed correlation represented an extreme outlier (p < 0.001), providing strong evidence against the null hypothesis of random associations between microRNA patterns and pain outcomes.
These results demonstrate that despite the challenging HDSS conditions (n=9, p=2330), the identified microRNA-pain associations represent genuine biological signals rather than statistical artifacts.
HDSS Simulation Study Provides Performance Context
Comprehensive simulation analysis across 19,440 HDSS scenarios provided empirical benchmarking for our observed results (Figure 6). The simulation study systematically varied sample sizes (n=6, 9, 12), sparsity levels (1–2%), signal-to-noise ratios (0.75–1.5), distributional assumptions (normal vs heavy-tailed), batch effects, preprocessing strategies, and validation approaches.
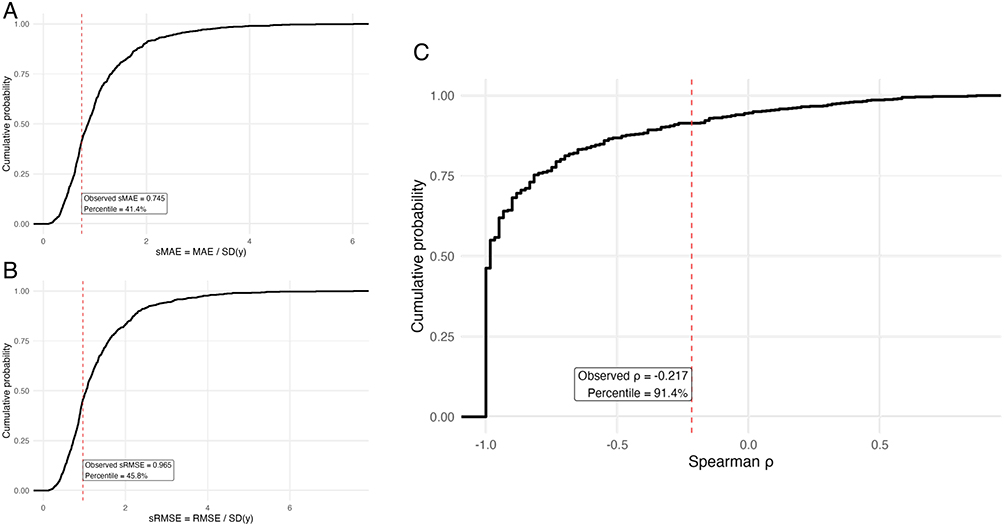 |
Figure 6 HDSS simulation study provides performance benchmarking context. Results from 19,440 HDSS simulation runs varying sample size, sparsity, signal-to-noise ratio, and other parameters. Left panels show empirical cumulative distribution functions (ECDF) for scale-free error metrics: sMAE (A) and sRMSE (B). Right panel shows Spearman correlation ECDF (C). Dashed vertical lines indicate observed values, with percentile rankings shown. The correlation coefficient (ρ=−0.217) ranks at 91st percentile, indicating stronger signal than most null-like HDSS scenarios. |
Scale-free error metrics positioned our results within expected ranges for HDSS scenarios: sMAE=0.745 (41st percentile) and sRMSE=0.965 (45th percentile). These percentiles indicate that our prediction errors were neither unusually large nor small compared to the broad range of HDSS simulation conditions, suggesting appropriate model complexity for the available data.
Spearman correlation benchmarking revealed more encouraging results: our observed ρ=−0.217 ranked at the 91st percentile of the simulation distribution for n=9 with robust preprocessing and nested LOOCV. This percentile ranking indicates that our observed correlation strength was substantially higher than expected under most null-like HDSS scenarios, providing additional evidence for meaningful biological signal.
The empirical cumulative distribution function (ECDF) curves demonstrate that while prediction errors remained within typical HDSS ranges, the correlation strength exceeded the vast majority of simulation scenarios with similar characteristics. This pattern suggests that although absolute prediction accuracy is limited by the extreme HDSS constraints, the rank-order relationships between microRNA patterns and pain outcomes represent robust biological associations.
The simulation results provide critical context for interpreting HDSS biomarker studies, demonstrating that meaningful biological signals can be detected even under challenging conditions when appropriate analytical frameworks are employed.
Discussion
Methodological Contributions to HDSS Biomarker Discovery
This study presents a comprehensive framework for addressing the analytical challenges inherent in high-dimensional small-sample biomarker discovery.30 Our approach integrates robust preprocessing, nested cross-validation, and simulation-based validation to provide a practical solution for extracting meaningful signals from extreme HDSS datasets. The key methodological innovation lies not in any single analytical component, but in the systematic integration of multiple validation strategies that collectively address the fundamental challenges of HDSS scenarios.
The nested leave-one-out cross-validation strategy proved essential for maximizing information utilization while maintaining rigorous separation between model selection and performance evaluation. Traditional approaches often fail in HDSS settings due to inadequate validation frameworks that cannot distinguish genuine biological signals from statistical artifacts.4 Our permutation testing and simulation benchmarking provide complementary perspectives on statistical significance: permutation testing confirms that observed associations are unlikely under the null hypothesis, while simulation benchmarking contextualizes performance within the broader landscape of HDSS scenarios.
The simulation study results reveal a critical insight for HDSS biomarker research: while absolute prediction accuracy remains limited by sample size constraints, rank-order relationships can capture meaningful biological associations. Our observed Spearman correlation of ρ=−0.217, ranking at the 91st percentile of simulation scenarios, demonstrates that correlation-based metrics may be more informative than absolute error metrics in extreme HDSS conditions. This finding has important implications for biomarker validation strategies, suggesting that correlation-based validation may be more appropriate than traditional prediction accuracy measures when sample sizes are severely constrained.
Biological Plausibility of Dual-Function MicroRNA Panel
Our findings should be interpreted within the broader context of emerging circulating microRNA research in chronic pain. Previous studies have identified circulating miRNAs associated with various chronic pain conditions. Bjersing et al reported altered cerebrospinal fluid miRNA profiles in fibromyalgia patients, with miR-145 correlating with pain and fatigue severity.31 McDonald et al demonstrated that macrophage-derived exosomes containing specific miRNAs could modulate inflammatory pain in preclinical models and that circulating miRNAs were altered in patients with complex regional pain syndrome.32 Recent comprehensive reviews have highlighted the regulatory roles of miRNAs in pain processing pathways and their potential as biomarkers across multiple chronic pain conditions.5,33 However, most prior studies focused on identifying diagnostic biomarkers for single pain conditions, with limited investigation of treatment-responsive miRNA profiles. To our knowledge, this represents the first study examining circulating miRNA patterns associated with acupuncture treatment response in chronic pain, extending the field toward predictive biomarkers for therapeutic outcomes.
The identification of microRNAs with dual functionality in both pain prediction and treatment response provides biological plausibility for their potential clinical utility. The three representative candidates—miR-3681-3p, miR-6822-5p, and miR-4743-5p—demonstrated convergent evidence from both machine learning-based feature selection and correlation-based functional analysis. This convergence strengthens confidence in their biological relevance despite the challenging analytical conditions.
Pathway enrichment analysis revealed significant associations with multiple mechanisms relevant to pain processing and acupuncture response. The enrichment of PI3K-Akt-mTOR signaling pathways is particularly noteworthy, as this pathway has been extensively implicated in chronic pain development and maintenance through its roles in synaptic plasticity, neuroinflammation, and central sensitization.29 Similarly, the enrichment of TGF-β signaling and transcriptional regulation pathways suggests involvement in the broader molecular responses to acupuncture intervention.
The neuronal system and synaptic plasticity pathways identified through Reactome analysis align with established mechanisms of acupuncture analgesia, including modulation of neurotransmitter release, synaptic transmission, and neuroplasticity.34 The identification of these pathways through unbiased computational analysis of microRNA targets provides independent support for the biological relevance of the selected biomarkers, despite the modest effect sizes observed in our small cohort.
Acupuncture may influence circulating miRNA profiles through multiple interconnected mechanisms. Central nervous system modulation, particularly in pain-processing regions such as the periaqueductal gray and rostral ventromedial medulla, could alter neuronal activity patterns that affect miRNA biogenesis and secretion. Peripherally, acupuncture’s documented anti-inflammatory effects may modify miRNA expression in immune cells and their release into circulation via extracellular vesicles. The identified miRNAs may thus reflect acupuncture’s neuromodulatory and anti-inflammatory actions, potentially serving as molecular indicators of treatment-induced changes in pain regulatory networks. These findings align with emerging evidence that epigenetic mechanisms, including microRNA-mediated post-transcriptional control, play important roles in the long-term maintenance and potentially the reversibility of chronic pain states.35,36 This epigenetic framework provides a biological rationale for investigating miRNA biomarkers not only for pain diagnosis but also for monitoring treatment responses, as therapeutic interventions may reverse maladaptive epigenetic changes underlying pain chronification.
However, it is important to acknowledge that pathway analysis based on predicted microRNA targets has inherent limitations. Target prediction algorithms are imperfect, and the biological significance of predicted interactions requires experimental validation. Moreover, this study establishes correlations between circulating miRNA profiles and pain outcomes but cannot establish causation. The identified miRNAs may represent direct regulators of pain pathophysiology, downstream consequences of pain-related processes, or general markers of systemic stress and inflammation that correlate with but do not directly drive pain states. Distinguishing these possibilities requires mechanistic studies in pain models and functional validation demonstrating that manipulating these miRNAs affects pain-related outcomes. The pathway enrichments should therefore be interpreted as supportive evidence for biological plausibility rather than definitive proof of mechanism.
Performance Interpretation in HDSS Context
The model performance metrics require careful interpretation within the context of extreme HDSS conditions. The observed MAE of 10.58 and RMSE of 13.70 represent moderate prediction errors relative to the VAS scale range (0–100) but must be evaluated against the fundamental limitations imposed by the p≫n scenario. The simulation benchmarking provides crucial context: our scale-free error metrics (sMAE=0.745, sRMSE=0.965) fell within the middle range of HDSS simulation scenarios, indicating that prediction accuracy was neither unusually poor nor optimistic given the analytical constraints.
The negative Spearman correlation (ρ=−0.217) reflects the expected relationship direction increases in certain microRNA expression patterns are associated with smaller pain reductions (higher ΔVAS values). This inverse relationship is biologically plausible, as microRNAs involved in pain chronification or treatment resistance would be expected to correlate negatively with therapeutic benefit.
The 91st percentile ranking of our correlation coefficient in the simulation distribution provides the most compelling evidence for meaningful signal detection. This ranking indicates that the observed associations are unlikely to arise from random chance alone, even under challenging HDSS conditions. The simulation approach offers a more nuanced view of statistical significance than traditional p-values, providing empirical context for performance evaluation in extreme analytical scenarios.
Limitations and Considerations
This study has several critical limitations requiring cautious interpretation. The extremely small sample size (n=9 paired observations) represents a proof-of-concept demonstration rather than definitive biomarker validation. Results should be viewed as exploratory and hypothesis-generating, requiring replication in larger, independent cohorts before any clinical consideration.
Second, the study employed convenience sampling from a subset of the original clinical trial participants, which may limit generalizability. The selection of participants with representative treatment response ranges was intended to maximize analytical power, but may introduce selection bias that could affect external validity. Future studies should employ systematic sampling strategies and larger cohorts to address this limitation.
Third, the pathway analysis relies on predicted microRNA targets rather than experimentally validated interactions. While the computational predictions provide valuable insights into potential mechanisms, functional validation studies are needed to confirm the biological roles of the identified microRNAs in pain processing and acupuncture response.
Fourth, the cross-sectional design of the biomarker analysis cannot establish causal relationships between microRNA expression changes and clinical outcomes. Longitudinal studies with repeated measurements would provide stronger evidence for causal inference and temporal relationships.
Clinical and Methodological Implications
Despite these limitations, the study demonstrates several important principles for HDSS biomarker research. The integration of multiple validation strategies provides a robust framework for signal detection under challenging analytical conditions. The simulation-based benchmarking approach offers a generalizable method for performance contextualization that could be applied to other HDSS omics studies.
From a clinical perspective, the identification of dual-function biomarkers suggests the possibility of developing personalized treatment approaches that combine prognostic and predictive capabilities. MicroRNAs that can both predict baseline pain severity and anticipate treatment response could inform clinical decision-making, though substantial additional validation would be required before clinical implementation.
The methodological framework presented here addresses a significant gap in the biomarker discovery pipeline, particularly for exploratory studies with necessarily small sample sizes. The emphasis on correlation-based validation rather than absolute prediction accuracy may be particularly relevant for early-stage biomarker development, where the goal is signal detection rather than clinical deployment.
Future Research Directions
Several research directions emerge from this work. First, independent replication in larger, systematically sampled cohorts is essential to confirm the generalizability of the identified biomarkers and validate the analytical framework. Multicenter studies with standardized protocols would strengthen external validity and provide opportunities for cross-validation.
Second, functional validation studies are needed to confirm the biological roles of the identified microRNAs in pain mechanisms and acupuncture response. In vitro and animal model studies could elucidate the specific molecular pathways involved and provide mechanistic insights beyond computational prediction.
Third, longitudinal studies with repeated biomarker measurements could provide insights into temporal relationships and causal inference. Understanding how microRNA expression patterns change over the course of treatment could inform optimal timing for biomarker assessment and reveal dynamic aspects of treatment response.
Fourth, the analytical framework could be extended to other HDSS omics applications beyond pain biomarkers. The simulation-based validation approach could be particularly valuable for rare disease research, personalized medicine studies, and other scenarios where sample sizes are inherently constrained.
Finally, integration with other omics data types (proteomics, metabolomics, transcriptomics) could provide a more comprehensive molecular profile of treatment response and improve prediction accuracy through multi-modal biomarker panels.
Conclusions
We have developed and validated a practical framework for HDSS biomarker discovery that addresses key analytical challenges through integrated preprocessing, validation, and performance benchmarking strategies. Applied to circulating microRNA profiling in chronic neck pain, this approach identified a dual-function biomarker panel with biological plausibility for both pain prediction and acupuncture response assessment. While effect sizes remain modest and replication in larger cohorts is needed, the study demonstrates that meaningful biological signals can be extracted from extreme HDSS datasets when appropriate analytical frameworks are employed. The methodological contributions provide a foundation for future HDSS biomarker studies and highlight the importance of simulation-based validation in challenging analytical scenarios. Future validation studies should target sample sizes of 50–100 participants per group to enable adequate statistical power for biomarker confirmation while maintaining feasibility for comprehensive molecular profiling. Integration of complementary omics data—including transcriptomics, proteomics, and metabolomics alongside circulating miRNA profiles—could yield multi-modal molecular signatures with improved predictive accuracy and mechanistic insights, facilitating translation toward clinical applications.
Data Sharing Statement
The datasets generated and analyzed during the current study are available from the corresponding author (Dr. Seung-Nam Kim, [email protected]) upon reasonable request.
Ethics Approval and Consent to Participate
Ethical approval for this study was obtained from the Institutional Review Board of Kyonggi University (KGU-20171222-HR-026). This study was also registered with the Korean Clinical Trial Registry and WHO Clinical Trial Registry (KCT0005363, registered April 3rd, 2018, https://cris.nih.go.kr/cris/en/). Although all authors are affiliated with Dongguk University, the study was conducted as a collaborative project with Kyonggi University. Recruitment, intervention, and biological sample collection were carried out in facilities under Kyonggi University’s oversight; therefore, IRB approval was obtained from Kyonggi University. All procedures followed the ethical standards of the responsible institutional committee and the Declaration of Helsinki. Written informed consent was obtained from all participants prior to their enrollment.
Funding
This study was supported by the National Research Foundation of Korea funded by the Korean government (MSIT) (RS-2025-25413539) and Ministry of Health & Welfare through the Korea Health Industry Development Institute (KHIDI) (Grant No. RS-2025-02263620).
Disclosure
The authors declare that they have no competing interests.
References
1. Woolf CJ. Central sensitization: implications for the diagnosis and treatment of pain. Pain. 2011;152(3 Suppl):S2–S15. doi:10.1016/j.pain.2010.09.030
2. Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain. 2009;10(9):895–926. doi:10.1016/j.jpain.2009.06.012
3. Todoroki K, Cipolla E, Kahler ZP, et al. The mechanisms and models of pain associated with substance use disorder. Biomedicines. 2023;11(7):1888. doi:10.3390/biomedicines11071888
4. Varma S, Simon R. Bias in error estimation when using cross-validation for model selection. BMC Bioinf. 2006;7:91. doi:10.1186/1471-2105-7-91
5. López-González MJ, Landry M, Favereaux A. MicroRNA and chronic pain: from mechanisms to therapeutic potential. Pharmacol Ther. 2017;180:1–15. doi:10.1016/j.pharmthera.2017.06.001
6. Bali KK, Kuner R, Thierry-Mieg J. MicroRNAs as modulators and biomarkers of inflammatory and neuropathic pain conditions. Mol Pain. 2014;10:44. doi:10.1186/1744-8069-10-44
7. McDonald MK, Ajit SK. MicroRNA biology and pain. Prog Mol Biol Transl Sci. 2015;131:215–249.
8. Ko JH, Kim SN. Effect of acupuncture on pain and substance P levels in middle-aged women with chronic neck pain. Front Neurol. 2023;14:1267952. doi:10.3389/fneur.2023.1267952
9. Tu W, Li S, Jiang X, et al. Electroacupuncture alleviates neuropathic pain through regulating miR-206-3p targeting BDNF after CCI. Neural Plast. 2022;2022:1489841. doi:10.1155/2022/1489841
10. Liu L, Qi W, Wang Y, et al. Circulating exosomal microRNA profiles in migraine patients receiving acupuncture treatment: a placebo-controlled clinical trial. Front Mol Neurosci. 2022;15:1098766. doi:10.3389/fnmol.2022.1098766
11. Johnstone IM, Titterington DM. Statistical challenges of high-dimensional data. Philos Trans a Math Phys Eng Sci. 2009;367(1906):4237–4253. doi:10.1098/rsta.2009.0159
12. Tibshirani R. Regression shrinkage and selection via the lasso. J R Stat Soc Series B. 1996;58(1):267–288. doi:10.1111/j.2517-6161.1996.tb02080.x
13. Zou H, Hastie T. Regularization and variable selection via the elastic net. J R Stat Soc Series B. 2005;67(2):301–320. doi:10.1111/j.1467-9868.2005.00503.x
14. Llamos-Paneque A, Sanabria-Díaz G, Perera-Linares N, et al. Omics data preprocessing for machine learning: a case study in childhood obesity. Genes. 2023;14(2):392. doi:10.3390/genes14020392
15. Hastie T, Tibshirani R, Friedman J. The Elements of Statistical Learning: Data Mining, Inference, and Prediction.
16. Good PI. Permutation Tests: A Practical Guide to Resampling Methods for Testing Hypotheses.
17. Budhraja S, Doborjeh M, Singh B, et al. Filter and Wrapper Stacking Ensemble (FWSE): a robust approach for reliable biomarker discovery in high-dimensional omics data. Brief Bioinform. 2023;24(6):bbad382. doi:10.1093/bib/bbad382
18. Liu WM, Mei R, Di X, et al. Analysis of high density expression microarrays with signed-rank call algorithms. Bioinformatics. 2002;18(12):1593–1599. doi:10.1093/bioinformatics/18.12.1593
19. Huber PJ. Robust estimation of a location parameter. Ann Math Stat. 1964;35(1):73–101. doi:10.1214/aoms/1177703732
20. Durbin BP, Hardin JS, Hawkins DM, Rocke DM. A variance-stabilizing transformation for gene-expression microarray data. Bioinformatics. 2002;18(Suppl 1):S105–110. doi:10.1093/bioinformatics/18.suppl_1.S105
21. Rousseeuw PJ, Croux C. Alternatives to the median absolute deviation. J Am Stat Assoc. 1993;88(424):1273–1283. doi:10.1080/01621459.1993.10476408
22. Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19(2):185–193. doi:10.1093/bioinformatics/19.2.185
23. Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J Stat Softw. 2010;33(1):1–22. doi:10.18637/jss.v033.i01
24. Stone M. Cross-validatory choice and assessment of statistical predictions. J R Stat Soc Series B. 1974;36(2):111–147. doi:10.1111/j.2517-6161.1974.tb00994.x
25. Efron B. Estimating the error rate of a prediction rule: improvement on cross-validation. J Am Stat Assoc. 1983;78(382):316–331. doi:10.1080/01621459.1983.10477973
26. Wang Y, Huang YS, Li JJ, Ostell J, Pruitt KD, Karsch-Mizrachi I. miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res. 2019;47(D1):D94–D99. doi:10.1093/nar/gky989
27. Kuleshov MV, Jones MR, Rouillard AD, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44(W1):W90–96. doi:10.1093/nar/gkw377
28. Kanehisa M, Furumichi M, Sato Y, Kawashima M, Ishiguro-Watanabe M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023;51(D1):D587–D592. doi:10.1093/nar/gkac963
29. Geranton SM, Jimenez-Diaz L, Torsney C, et al. A rapamycin-sensitive signaling pathway is essential for the full expression of persistent pain states. J Neurosci. 2009;29(47):15017–15027. doi:10.1523/JNEUROSCI.3451-09.2009
30. Christin C, Hoefsloot HC, Smilde AK, et al. A critical assessment of feature selection methods for biomarker discovery in clinical proteomics. Mol Cell Proteomics. 2013;12(1):263–276. doi:10.1074/mcp.M112.022566
31. Bjersing JL, Lundborg C, Bokarewa MI, Mannerkorpi K. Profile of cerebrospinal microRNAs in fibromyalgia. PLoS One. 2013;8(10):e78762. doi:10.1371/journal.pone.0078762
32. McDonald MK, Tian Y, Qureshi RA, et al. Functional significance of macrophage-derived exosomes in inflammation and pain. Pain. 2014;155(8):1527–1539. doi:10.1016/j.pain.2014.04.029
33. Sabina S, Panico A, Mincarone P, et al. Expression and biological functions of miRNAs in chronic pain: a review on human studies. Int J Mol Sci. 2022;23(11):6016. doi:10.3390/ijms23116016
34. Zhao ZQ. Neural mechanism underlying acupuncture analgesia. Prog Neurobiol. 2008;85(4):355–375. doi:10.1016/j.pneurobio.2008.05.004
35. Liang L, Lutz BM, Bekker A, Fan X. Epigenetic regulation of chronic pain. Epigenomics. 2015;7(2):235–242. doi:10.2217/epi.14.75
36. Mauceri D. Role of epigenetic mechanisms in chronic pain. Cells. 2022;11(16):2613. doi:10.3390/cells11162613
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Normalization of Neuroinflammation: A New Strategy for Treatment of Persistent Pain and Memory/Emotional Deficits in Chronic Pain
Liu XG
Journal of Inflammation Research 2022, 15:5201-5233
Published Date: 9 September 2022
Knowledge Mapping of International Research on Acupuncture for Chronic Pain: A Bibliometric Analysis

Jiao H, Ding R, Jin P, Wang J, Xu J, Tian W
Journal of Pain Research 2022, 15:3711-3728
Published Date: 2 December 2022
Research Trends of Acupuncture Therapy for Chronic Pain-Related Depression or Anxiety from 2003 to 2023: A Bibliometric Analysis
Yang P, Wang T, He YJ, Su SY
Journal of Pain Research 2023, 16:4301-4315
Published Date: 15 December 2023
Misinformation Persists in Complementary Health: Evaluating the Reliability and Quality of YouTube-Based Information on the Use of Acupuncture for Chronic Pain
Greer M, Kamma S, Tran H, Etumuse B, Shah JD, El-Abed Y, Onyemachi JO, Hussain N, Pittelkow TP, D'Souza RS
Journal of Pain Research 2024, 17:1509-1518
Published Date: 16 April 2024
Transforming Chronic Pain Management: Integrating Neuromodulation with Advanced Technologies to Tackle Cognitive Dysfunction – A Narrative Review
Green M, Hayley A, Gunnersen JM, Nazemian V, Cabble A, Thompson S, Chakravarthy K
Journal of Pain Research 2025, 18:2497-2507
Published Date: 16 May 2025