")
Back to Journals » Journal of Inflammation Research » Volume 15
Normalization of Neuroinflammation: A New Strategy for Treatment of Persistent Pain and Memory/Emotional Deficits in Chronic Pain
Authors Liu XG
Received 18 June 2022
Accepted for publication 18 August 2022
Published 9 September 2022 Volume 2022:15 Pages 5201—5233
DOI https://doi.org/10.2147/JIR.S379093
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Xian-Guo Liu
Pain Research Center and Department of Physiology, Zhongshan School of Medicine of Sun Yat-sen University, Guangzhou, People’s Republic of China
Correspondence: Xian-Guo Liu, Pain Research Center and Department of Physiology, Zhongshan School of Medicine of Sun Yat-sen University, 74 Zhongshan Road2, Hemulou 806, Guangzhou, 510080, People’s Republic of China, Tel/Fax +86 87331956, Email [email protected]
Abstract: Chronic pain, which affects around 1/3 of the world population and is often comorbid with memory deficit and mood depression, is a leading source of suffering and disability. Studies in past decades have shown that hyperexcitability of primary sensory neurons resulting from abnormal expression of ion channels and central sensitization mediated pathological synaptic plasticity, such as long-term potentiation in spinal dorsal horn, underlie the persistent pain. The memory/emotional deficits are associated with impaired synaptic connectivity in hippocampus. Dysregulation of numerous endogenous proteins including receptors and intracellular signaling molecules is involved in the pathological processes. However, increasing knowledge contributes little to clinical treatment. Emerging evidence has demonstrated that the neuroinflammation, characterized by overproduction of pro-inflammatory cytokines and glial activation, is reliably detected in humans and animals with chronic pain, and is sufficient to induce persistent pain and memory/emotional deficits. The abnormal expression of ion channels and pathological synaptic plasticity in spinal dorsal horn and in hippocampus are resulting from neuroinflammation. The neuroinflammation is initiated and maintained by the interactions of circulating monocytes, glial cells and neurons. Obviously, unlike infectious diseases and cancer, which are caused by pathogens or malignant cells, chronic pain is resulting from alterations of cells and molecules which have numerous physiological functions. Therefore, normalization (counterbalance) but not simple inhibition of the neuroinflammation is the right strategy for treating neuronal disorders. Currently, no such agent is available in clinic. While experimental studies have demonstrated that intracellular Mg2+ deficiency is a common feature of chronic pain in animal models and supplement Mg2+ are capable of normalizing the neuroinflammation, activation of upregulated proteins that promote recovery, such as translocator protein (18k Da) or liver X receptors, has a similar effect. In this article, relevant experimental and clinical evidence is reviewed and discussed.
Keywords: ion channels, synaptic plasticity, cytokine, glial cell, monocytes
Introduction
The prevalence of chronic pain is around 30% in the world population1–3 and around 2/3 chronic pain patients suffer from memory deficit and mood depression.4–7 In spite of intensive studies for decades worldwide, clinical treatment of chronic pain is still largely unmet. Understanding the mechanisms underlying the chronic pain and its comorbidities is important scientifically and clinically.
Definition, Classification and a Brief History of Chronic Pain
Pain, a most common chief complaint in clinic,8 is redefined as an unpleasant sensory and emotional experience associated with, or resembling that associated with, actual or potential tissue damage in 2020.9 According to physiological function, pain is classified into nociceptive (physiological) pain and pathological pain. Nociceptive pain is a pain triggered by activation of nociceptors. It serves as an alarm system and protects tissues from further damage, while pathological pain is a consequence of injury and disease, manifested as allodynia (decrease in pain threshold), hyperalgesia (increase in response to noxious stimuli) and spontaneous pain. Clinically, pain is divided into acute pain and chronic pain. The acute pain is identical to physiological pain. Whereas chronic pain is pain persisting beyond the expected healing period. In clinic practice, the pain that lasts or recurs for longer than 3 months is defined as chronic pain.10 The short-lasting pathological pain is also protective, such as the pain hypersensitivity induced by skin burning that favors recovery by avoiding touching the injured tissues. In contrast, chronic pain is a leading source of human suffering and disability without any beneficial effect.11
Mechanistically, chronic pain is mediated by nociceptive pain, neuropathic pain and nociplastic pain.12 Persistent nociceptive pain is most common form of chronic pain, such as osteoarthritis pain13 produced by mechanical stimuli due to bone destruction and chemical stimuli by substance P, K+ and inflammatory mediators released by damaged tissues. Neuropathic pain is redefined as pain arising as a direct consequence of a lesion or disease affecting the somatosensory system.14 In experimental studies, neuropathic pain is often produced by injury of peripheral nerves in rats and mice, such as chronic constriction of sciatic nerve (CCI),15 lumbar 5 and/or 6 spinal nerve ligations (L5/L6-SNL)16 and spared nerve injury (SNI), in which the two terminal branches of the sciatic nerve (tibial and common peroneal nerves) are lesioned, leaving the sural nerve intact.17 Neuropathic pain can be also produced by selective injury of motor fibers by L5 ventral root transection (L5-VRT),18 by application of anti-cancer agents, such as paclitaxel19 and vincristine,20 and by streptozotocin that induces type 1 diabetes.21 Nociplastic pain was first put forward in 2016 as the third mechanistic descriptor for chronic pain,22 based on the clinical observations that in some types of chronic pain, such as fibromyalgia, complex regional pain syndrome type 1 and nonspecific chronic lower back pain, neither obvious nociceptor activation nor neuropathy is evident and, therefore, cannot be described as nociceptive or neuropathic pain. The nociplastic pain is believed to be caused by alteration of nociceptive processing in central nervous system (CNS),23 such as changes in cerebral activation, synaptic connectivity and even neuronal structures. However, it is worth noting note that compelling experimental and clinical evidence has demonstrated that the functional and structural plastic changes in both peripheral nerves and CNS can be produced by many pathogenic causes of chronic pain, such as activation of nociceptors by intensive noxious stimuli,24 nerve injury,25,26 internal biochemical change, including overproduction of proinflammatory cytokines,27,28 and external chemical stimuli including anti-cancer agents29,30 and opioids.31 Thus, the neuroplastic change is probably a common mechanism of chronic pain (Figure 1).
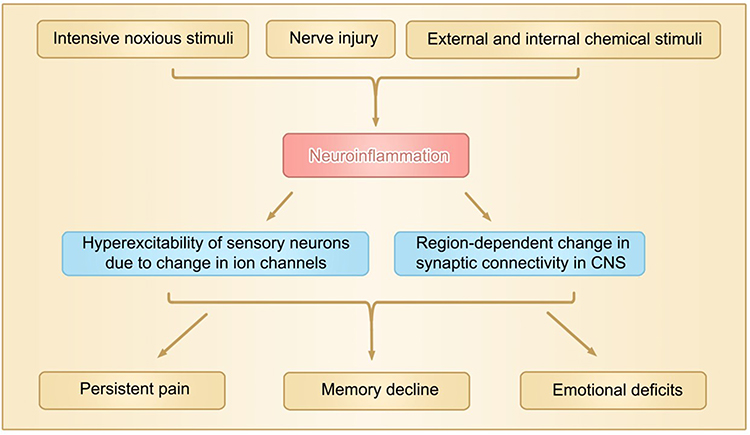 |
Figure 1 Neuroinflammation produced by various etiological factors plays a central role in functional and structural plastic changes in peripheral and central nervous system, leading to persistent pain, memory decline and emotional deficits. The region-dependent change in synaptic connectivity means differential changes in synaptic connection, such as synaptic connection is enhanced in spinal dorsal horn but reduced in hippocampus in neuropathic pain mice. Over-production of TNF-α is sufficient to induce the change. |
In medical history, pain was considered merely a symptom of other diseases for a long time. At that time, little is known about the difference in mechanisms of physiological pain and pathological pain. In 1974, Wall et al found that the injured sensory fibers generated action potentials spontaneously after sciatic nerve transection.32 The ongoing activity (ectopic discharge) in the injured afferents is considered to underlie persistent pain (see33,34 for reviews). In 1983, Woolf provided the first evidence for a central component of persistent pain by showing that the excitability of spinal cord neurons is persistently increased following peripheral injury, and the change is independent of ongoing activity from peripheral nerves.35 The phenomenon is termed central sensitization. In 1988, Bennett and Xie15 made the first rat neuropathic pain model, ie, CCI model. The animals exhibit the behavioral signs of neuropathic pain, including allodynia, hyperalgesia and spontaneous pain after surgery. In 1999, Basbaum first proposed that persistent pain should be considered a disease state of the nervous system, not merely a prolonged acute pain symptom of other disease conditions,36 according to the fact that the neurochemical features of persistent pain are distinct from acute pain revealed by many studies. Twenty years later, chronic pain was systematically classified in 11th revision of the International Classification of Diseases (ICD-11).37 In the ICD-11, chronic pain with unknown etiology is defined as chronic primary pain. The chronic primary pain is considered a disease of nervous system in its own right, including chronic widespread pain, complex regional pain syndrome, chronic primary headache or orofacial pain, chronic primary visceral pain and chronic musculoskeletal pain. Chronic pain produced by injury or other diseases is classified as chronic secondary pain syndromes, including chronic cancer-related pain, chronic neuropathic pain, chronic secondary visceral pain, chronic posttraumatic and postsurgical pain and chronic secondary musculoskeletal pain. The ICD-11 classification of chronic pain is continuously updated (see http://id.who.int/icd/entity/1581976053).
Increased Excitability of Primary Sensory Neurons and Pathological Synaptic Plasticity in Pain Pathways Underlie Persistent Pain
It has long been proposed that persistent pain is mediated by peripheral sensitization and central sensitization.35,38 Now, it is well established that the peripheral sensitization, an increased excitability of primary sensory neurons, is resulting from abnormal expression of a variety of ion channels, including voltage-gated sodium (Nav) channels,39,40 voltage-gated calcium (Cav) channels,41,42 potassium channels,43 transient receptor potential (TRP) channels,44,45 and acid-sensing ion channels (ASICs).46 The changes of some ion channels in sensory neurons in chronic pain are shown in Figure 2. The hyperexcitability of sensory neurons leads to spontaneous discharges and hypersensitivity to peripheral stimuli. The central sensitization is underlined primarily by long-lasting enhancement of synaptic transmission, called long-term potentiation (LTP), which amplifies signals in pain pathways (see47,48 for reviews).
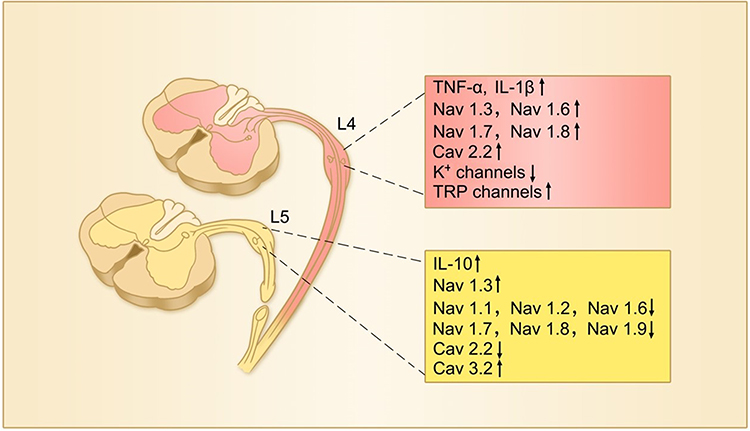 |
Figure 2 The changes in expression of TNF-α, IL-1β and IL-10 and various ion channels in injured and uninjured (intact) DRG neurons following peripheral nerve injury. In this L5-spinal nerve ligation model of neuropathic pain, L5 or both L5 and L6 spinal nerves are ligated and transected. Therefore, L5 DRG neurons are injured and L4 DRG neurons remain intact.16 |
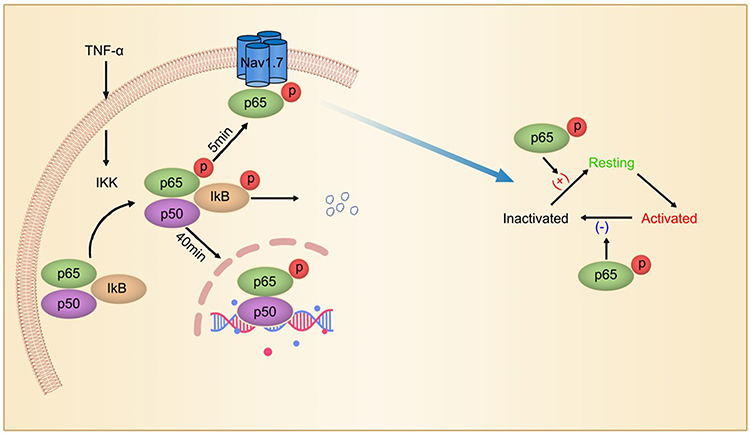 |
Figure 3 Non-transcriptional regulation of Nav1.7 by TNF-α/NF-ƘB signaling. NF-kB p50/p65/inhibitor of NF-kB (IkB) complex is located in the cytoplasm. On activation by TNF-α, both p65 and IkB are phosphorylated, and then p-IkB is degenerated after ubiquitination. p-p65 is translocated to membrane and increases the excitability of DRG neurons by interaction with Nav1.7 within 5 min, and then into the nucleus, where it regulates gene transcription. Adapted from iScience. Volume: 19. Xie MX, Zhang XL, Xu J, et al. Nuclear Factor-kappa B gates Nav1.7 channels in DRG neurons via protein-protein interaction. iScience.Page numbers: 623–633, Copyright (2019), with permission from Elsevier.150 |
Mechanisms Underlying the Comorbidity of Persistent Pain and Memory/Emotional Deficits are Under Debate
To explain the comorbidity of persistent pain and memory deficits, it was proposed that persistent pain might impair memory by attracting attention.49,50 However, human studies have shown that acute pain does not affect memory in healthy individuals51 and that relief of pain with opioids cannot improve the memory in chronic pain patients.52 Likewise, depression was also believed to be a consequence of persistent pain.53,54 It has also been proposed that persistent pain and depression may share common pathological mechanisms but are independent diseases without causal interaction.55,56
Together, chronic pain may be resulting from either chronification of acute pain,57 other diseases or internal molecular changes with unknown cause. In recent years, emerging evidence indicates that neuroinflammation, characterized by overproduction of proinflammatory cytokines and glial activation,58 is a common pathological mechanism of persistent pain, memory decline and mood depression59,60 (Figure 1). In this article, related evidence is reviewed and a new strategy for treating neuronal disorders, ie, normalization (or counterbalance) but not simply inhibition of neuroinflammation, is proposed.
Cytokine Microenvironment Hypothesis of Chronic Pain
The functions of the nervous system are exerted by the neuronal networks, in which the neurons are connected to each other via synapses. The efficacy of the network depends on the excitability of individual neurons and the strength and pattern of synaptic connections. Now, it is well established that both the excitability of neurons and the synaptic connectivity are not static; rather, they undergo activity- or experience-dependent plastic change in whole life. The numbers of excitatory and inhibitory synapses are also frequently changed, leading to remodulation of neuronal circuits (see61 for a review). The plastic change means that, once the change occurs, it persists for a period of time without automatic recovery. The plastic change is essential for many physiological and pathological processes in nervous system, such as memory storage62,63 and chronic pain.47 A key question is what is the root cause for the activity-dependent plastic change? Before discussing this, let us review a key physiological concept: internal environment homeostasis. The idea was first described by French physiologist Claude Bernard in 1865, and the word “homeostasis” was coined by American physiologist Walter Bradford Cannon in 1926.64 In this concept, extracellular fluid, in which the cells live, is defined as internal environment. To sustain the functions and survival of the cells, physical and chemical conditions, including temperature, pH, osmolality, the contents of ions, nutriment, oxygen and carbon dioxide etc., in the internal environment should be kept constant. In the last decades, studies from ourselves and others have demonstrated that the overproduction of proinflammatory cytokines plays a key role in persistent pain and accompanied memory/emotional deficits in chronic pain. In 2013, we put forward a cytokine microenvironment hypothesis of chronic pain.65 The hypothesis has been supported by more and more studies. The updated experimental and clinical evidence is listed below. (1) Overexpression of proinflammatory cytokines is reliably detected in humans and animals with chronic pain. (2) Administration of proinflammatory cytokines is sufficient to induce behavioral signs of persistent pain, memory deficit and depressive behaviors in naive animals. (3) The abnormal expression of numerous ion channels that causes hyperexcitability of sensory neurons is resulting from overproduction of inflammatory cytokines. (4) The inflammatory cytokines regulate the synaptic plasticity in a region-dependent manner, ie, enhance and reduce synaptic connectivity in spinal dorsal horn and in hippocampus, respectively, contributing to persistent pain and memory/emotional deficits. Therefore, the inflammatory microenvironment might be a common cause of persistent pain and memory/emotional deficits in chronic pain. In this subsection, the related evidence is reviewed and discussed.
Overexpression of Proinflammatory Cytokines is Reliably Detected in Humans and Animals with Chronic Pain and is Sufficient to Induce Persistent Pain and Memory/Emotional Deficits
Cytokines are proteins, peptides or glycoproteins with a molecular mass between 8 and 30 kDa, including tumor necrosis factor (TNF-α), interleukins, chemokines and many other signaling molecules. Cytokines play key roles in the interactions and communications between cells at nano- to pico-molar concentrations under normal or pathological conditions.66,67 The production of cytokines is primarily regulated by autocrine and paracrine, ie, the cytokines released by a cell act on itself or on nearby cells by activation of their specific receptors, leading to production of more cytokines, and positive feedback is important in many diseases.68–70 According to their immune functions, cytokines are divided into two classes: pro-inflammatory cytokines, including TNF-α, interleukin-1beta (IL-1β), IL-6, and anti-inflammatory cytokines such as IL-10 and transforming growth factor-beta (TGF-β).
The increase in proinflammatory cytokines is reliably detected in human patients with many forms of chronic pain. A recent systematic review indicates that plasma TNF-α is increased in patients with chronic non-specific lower back pain, which is one of the greatest contributors to suffering and disability in the world.71 Plasma TNF-α is also enhanced in patients with fibromyalgia, a syndrome characterized by widespread chronic pain,72 and with rheumatoid arthritis.73 TNF-α is increased while Il-10 is decreased in plasma of women with migraine.74
Cytokines are originally identified in peripheral immune cells, such as lymphocytes and monocytes,67 while emerging experimental evidence has shown that both TNF-α28,75 and IL-1β59,76 are expressed in nervous system and are persistently upregulated in dorsal root ganglion (DRG), spinal dorsal horn, hippocampus and many other brain regions in neuropathic pain condition, upregulation of TNF-α or IL-1β in sensory neurons is detected a few hours or even tens of minutes after nerve injury.59,77 It has been shown that knockout of either TNF-α or IL-1β only partially prevents the mechanical allodynia induced by peripheral nerve injury, while knockout of both of them completely abolishes the behavioral sign of neuropathic pain.78 Thus, both TNF-α and IL-1β may be needed for full expression of neuropathic pain. Upregulation of TNF-α in DRG neurons is also reported in vincristine- and bortezomib-induced chemical neuropathy,29,79 diabetic neuropathy80 and menopausal-induced chronic pain,27 while blockage of TNF-α synthesis or genetic deletion of TNF receptor 1 (TNFR1) substantially prevents neuropathic pain.75,81 Importantly, local application of TNF-α70,82 or IL-1β83 onto sciatic nerve at physiological concentrations in naive rats reliably induces the behavioral signs of neuropathic pain. Thus, overproduction of the inflammatory cytokines is not only necessary but also sufficient to induce neuropathic pain. Furthermore, pro-inflammatory cytokines are also critically involved in memory deficits (see84,85 for reviews) and major depression (see86,87 for reviews) in many diseases other than chronic pain. Likewise, the working memory and short-term memory impairments produced by SNI are prevented by genetic deletion of TNFR1 and mimicked by intracerebroventricular or intrahippocampal injection of TNF-α.28 SNI induced-depression-like behavior and upregulation of IL-1β mRNA in the frontal cortex are ameliorated by intracerebroventricular administration of IL-1 receptor antagonist.88 The SNI-induced persistent pain, memory decline and depression-like behaviors are mimicked in naive rats by repetitive intravenous injection of recombinant rat IL-1β at a pathological concentration, determined in SNI rats.59 Thus, upregulation of the proinflammatory cytokines is sufficient to induce memory/emotional deficits, too.
Role of Ion Channels in Sensory Neurons for Persistent Pain and Their Regulation by Cytokines
In peripheral nerves, nociceptive signals (action potentials) are initiated and conducted by myelinated Aδ- and unmyelinated C-fibers; their cell bodies are located in DRG and trigeminal ganglia. The peripheral terminals (nociceptors) of the sensory neurons are distributed in whole body and their central terminals make synapses with the second order neurons in spinal dorsal horn or in trigeminal subnucleus caudalis. The ion channels are essential for nociceptor transduction (conversion of peripheral noxious stimuli into electrical signals), generation and conduction of action potentials, and neurotransmitter release in central terminals. The sensory neurons that give rise to myelinated A-fibers are neurofilament-200 (NF-200)+, while the neurons that give rise to unmyelinated C-fibers are either calcitonin gene-related peptide (CGRP)+, called peptidergic C-fibers, or isolectin B4 (IB4)+, called non-peptidergic C-fibers. The ion channels are unevenly distributed in different types of sensory neurons. Emerging evidence shows that the ion channels in sensory neurons are regulated by cytokines. The roles of various ion channels in persistent pain and their regulation by cytokines are discussed in this subsection (Table 1).
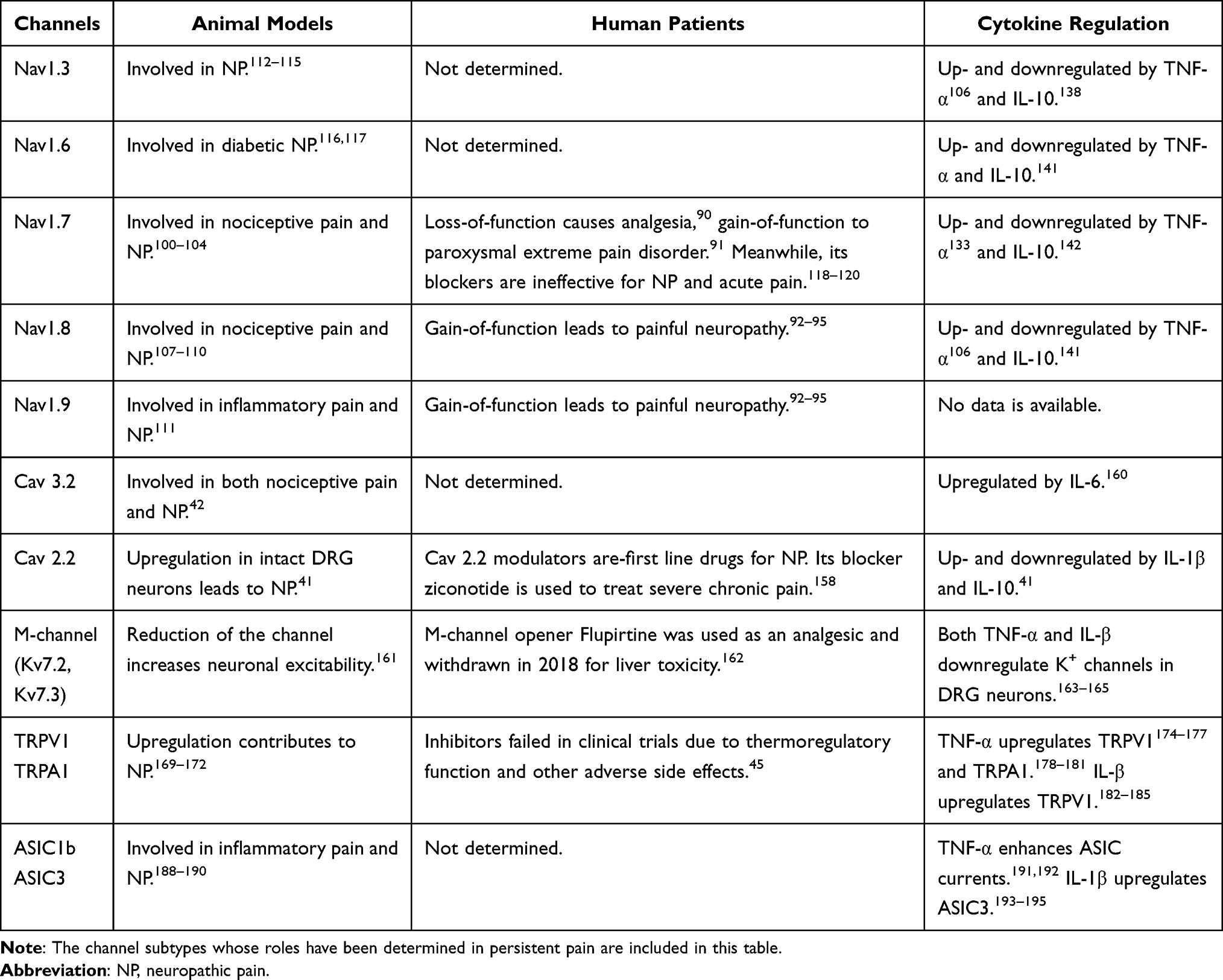 |
Table 1 The Roles of Ion Channels in Sensory Neurons for Persistent Pain and Their Regulation by Cytokines |
Voltage-Gated Sodium Channels
At least nine subtypes of sodium channels (Na1.1 to Nav1.9) have been identified, and most of them (except for Nav1.4 and Nav1.5) are expressed in DRG neurons. Nav1.8 and Nav1.9 are tetrodotoxin (TTX)-resistant and others are TTX-sensitive. Nav1.7, Nav1.8 and Nav1.9 are preferentially expressed in the afferent sensory neurons, including DRG and trigeminal ganglia (see89 for a review). To date, mutation of Nav1.7, Nav1.8 and Nav1.9 has been reported to be involved in human chronic pain. Loss-of-function mutation of Nav1.7 leads to complete inability to sense pain,90 while gain-of-function of Nav1.7 results in paroxysmal extreme pain disorder.91 Gain-of-function mutation of Nav1.8 and Nav1.9 leads to painful peripheral neuropathy92,93 and to painful neuropathy94 or familial episodic pain,95 respectively.
In rats, Nav1.7 is mainly expressed in nociceptive neurons that give rise to Aδ-fibers and C-fibers. Nav1.7 is detected in every part of DRG neurons, including cell body, peripheral axons in the sciatic nerve and peripheral terminals in skin and central terminals in superficial spinal dorsal horn.96 Nav1.7 is also expressed in sympathetic ganglion neurons97,98 and in spinal dorsal horn neurons.99 In rodents, deletion of Nav1.7 in DRG neurons attenuates nociceptive pain and the persistent pain induced by nerve injury, inflammation and skin burning.100–103 Blockage of Nav1.7 significantly alleviates neuropathic pain induced by the anti-cancer agent paclitaxel.104
Nav1.8 is expressed in all types of DRG neurons,105,106 and is involved in both nociceptive pain and neuropathic pain. In Nav1.8-null mutation mice, response to cold, mechanical stimuli, and thermal hyperalgesia are reduced.107 In rodents, selective knockdown of Nav1.8 with specific antisense oligodeoxynucleotides108 or blockage of Nav1.8 with different kinds of specific antagonists reverses neuropathic pain.109,110
Nav1.9 is also expressed in the soma, peripheral and central terminals of all types of DRG neurons and is involved in inflammatory pain and neuropathic pain in animal models (see111 for a review).
Nav1.3 is hardly detected in DRG neurons of adult rodents, but quickly re-expressed in injured and intact sensory neurons after SNI.112 Knockdown of Nav1.3 in L4 DRG by injection of virus-mediated shRNA directed against Nav1.3 attenuates SNI-induced neuropathic pain in rats.113 The same manipulation also alleviates tactile allodynia in streptozotocin-induced diabetic models of neuropathic pain.114 Nav1.3 is also re-expressed in dorsal horn neurons in rat CCI model. Intrathecal antisense oligodeoxynucleotides targeting Nav1.3 reduces both hypersensitivity of dorsal horn neurons and pain-related behavior.115 Thus, the re-expression of Nav1.3 DRG neurons may contribute to neuropathic pain. Nav.16 is upregulated in diabetic peripheral neuropathy rats.116 Nav1.6 activation in skin and gut leads to increased response to mechanical stimuli and mechanical allodynia but not thermal allodynia.117 The roles of Nav1.3 and Nav 1.6 in human chronic pain have not been determined.
Regardless of importance of Navs in nociceptive and neuropathic pain in humans and in rodents discussed above, to date no specific Nav subtype blocker is used in clinic for pain relief. In clinical trials, specific Nav1.7 blockers are ineffective for painful diabetic peripheral neuropathy,118 trigeminal neuralgia119 and acute pain induced by noxious stimuli in healthy subjects.120 As the nociceptive process, from nociceptor transduction to synaptic transmission in superficial spinal dorsal horn, is not dependent on a single Nav subtype, it was proposed that various Nav types, such as Nav1.7 and Nav1.8, should be blocked spontaneously to achieve effective analgesia (see40 for a review).
Because of the importance of Navs in the genesis and propagation of action potentials, it was hypothesized that upregulation of the sodium channels in injured sensory neurons might contribute to neuropathic pain. However, animal studies in rodents from different groups have revealed that, apart from Nav1.3, all other Nav subtypes, including Nav 1.1, Nav1.2, Na1.6, Nav1.7-Nav1.9, are downregulated in the injured DRG neurons in SNI and L5-SNL models of neuropathic pain (see121 for a review). Apparently, the experimental data seem contradictory to the clinical observation that sodium channel blockers (such as lidocaine) can effectively inhibit a variety of neuropathic pain syndromes in patients122 and to the long-standing general belief that the ectopic discharges in injured sensory neurons directly contribute to neuropathic pain.123–125
A previous work showed that the behavioral sign of neuropathic pain was not dependent on inputs from injured nerve fibers but on those from intact ones by showing that mechanical hyperalgesia induced by lesion of L5 spinal nerve was reversed by L4 dorsal rhizotomy but not by L5 dorsal rhizotomy.126 The data suggest that the intact DRG neurons may be responsible for abnormal pain behavior. Consistently, it has been shown that the intact DRG neurons also discharged spontaneously after nerve injury.127,128 Furthermore, selective injury of motor neurons, leaving the sensory neuron intact, by L5-VRT produces persistent mechanical allodynia and thermal hyperalgesia in bilateral hind-paws,18,75,106,129 while selective injury of sensory neurons by L5 dorsal root transection fails to induce the behavioral signs of neuropathic pain.129–134 The data suggest that damage of sensory neurons is neither necessary nor sufficient to induce neuropathic pain. And then, how could damage of motor fibers lead to neuropathic pain? We found that selective injury to motor neurons by L5-VRT upregulated TNF-α in the neurons of DRG and spinal dorsal horn. Blockage of TNF-α synthesis or genetic deletion of TNFR1 substantially prevents neuropathic pain.75,81 Furthermore, L5-VRT also persistently upregulates Nav1.3 and Nav1.8 at both mRNA and protein levels in the neurons of L4 and L5 DRGs. Importantly, the upregulation of sodium channels is substantially prevented by inhibition of TNF-α synthesis or genetic deletion of TNFR1, and is mimicked by peri-sciatic application of TNF-α.106 In cultured DRG neurons, TNF-α at 10–1000 pg/mL dose-dependently upregulates Nav1.3, Nav1.8106 and Nav1.7,135 which is in parallel with the increase in Na+ currents and the excitability of DRG neurons.81 IL-1β also increases the excitability of DRG and trigeminal sensory neurons by enhancing sodium current.136–139 Whereas, anti-inflammatory cytokine IL-10, which is effective to suppress neuropathic pain,140 downregulates Nav1.3, Nav1.6 Nav1.7 and Nav1.8, and reverses the upregulation of the sodium channels produced by TNF-α.141,142 The results indicate that TNF-α and IL-10 oppositely regulate Navs in sensory neurons. As IL-10 is upregulated in injured DRG neurons,41 the downregulation of Navs in injured DRG neurons may be resulting from IL-10 upregulation. The data demonstrate that the abnormal expression of Navs in sensory neurons is caused by imbalance of pro- and anti-inflammatory cytokines following peripheral nerve injury. The changes in expression of TNF-α, IL-1β, IL-10, Navs and other ion channels in injured and intact DRG neurons following peripheral nerve injury are summarized in Figure 2.
Nuclear factor-kappa B (NF-ƘB), a potent transcription factor, plays key roles in a wide variety of physiological and pathological processes by controlling inflammation response.143–145 NF-ƘB is also critically involved in chronic pain.146 Intrathecal injection of NF-ƘB inhibitor (pyrrolidine dithiocarbamate, PDTC) prevents the mechanical allodynia induced by peri-sciatic application of TNF-α70 and by L5-VRT. PDTC also prevents the upregulation of Nav1.3 in DRG neurons induced by L5-VRT and by TNF-α in cultured DRG neurons.147 Intrathecal injection of PDTC attenuates mechanical allodynia and thermal hyperalgesia, and Nav1.7 upregulation in DRG neurons in rats with diabetic neuropathy.148 The data suggest that TNF-α/NF-ƘB signaling contributes to persistent pain by transcriptional upregulation of sodium channels.
In addition, TNF-α is also capable of acutely enhancing excitability of sensory neurons. With a preparation of L4 and L5 DRGs attached with the sciatic nerve, Zhang et al have shown that application of TNF-α (1 ng/mL) for 15 min not only evokes discharges in silent fibers, but also enhances ongoing activity in spontaneously active fibers and sensitivity to electrical stimulation of the peripheral nerves.149 The mechanism underlying the acute effect of TNF-α is unclear. A recent work150 shows that phospho-p65 (p-p65), an active form of NF-ƘB subunit, reversibly interacts with Nav1.7 channels in the membrane of DRG neurons in rats with neuropathic pain induced by either antineoplastic agent vincristine or L5-SNL. The interaction enhances Nav1.7 currents via slowing inactivation of Nav1.7 channels and facilitating their recovery from inactivation. In cultured DRG neurons TNF-α increases the membrane p-p65 and enhances Nav1.7 currents within 5 min but does not affect nuclear p-p65 within 40 min, indicating that p-p65 is translocated to cell membrane at first, then to nucleus after NF-ƘB activation (Figure 3). This non-transcriptional effect on Nav1.7 may contribute to the acute effect of TNF-α on excitability of sensory neurons. Recently, we found that forkhead protein FOXO1, another transcription factor, also interacted with Nav1.7 in DRG neuron membrane and increased Nav1.7 currents, leading to mechanical allodynia.151 Although the effect of TNF-α on FOXO1 in DRG neurons has not been investigated, it has been shown that TNF-α increases FOXO1 activity in GnRH neuronal cell line GT1-7.152 The inflammatory cytokine-mediated non-transcriptional regulation of sodium channels by activation of traditional transcription factors may contribute to acute and persistent pain.
Voltage-Gated Calcium Channels
According to electrophysiological properties, Cavs are divided into low voltage activated (LVA) channels and high voltage activated (HVA) channels, which are activated at membrane potential near −60 mV and −30 mV, respectively. Based on the differences in pore-forming α1 subunits, the Cav channels are classified into Cav 1, Cav 2 and Cav 3. HVA channels, but not LVA channels, are associated with α2δ and β subunits. LVA channels are also called T-type channels including Cav 3.1-Cav 3.3. The HVA channels are further classified into L-type channels (Cav 1.1–Cav 1.4), P/Q-type (Cav 2.1), N-type (Cav 2.2), and R-type (Cav 2.3) (see153 for a review).
Emerging evidence indicates that Cav 3.2 and Cav 2.2, which are also expressed in all parts of nociceptive DRG neurons, are critically involved in nociceptive and neuropathic pain (see42 for a review). Cav 3.2 is upregulated in injured DRG neurons in many chronic pain models, such as chronic compression of DRG, SNL, CCI, partial sciatic nerve ligation and paclitaxel,154 and also in intact L4 DRG neurons in L5-SNL model.155 N-type channels, consisting of α1B (Cav 2.2), α2δ and β subunits, are exclusively expressed in neurons and neuroendocrine cells.156 Similar to sodium channels, Cav 2.2 is upregulated in intact L4 DRG neurons and downregulated in injured L5 DRG neurons in rat L5-SNL model.41 Activation of Cav 2.2 expressed in presynaptic terminals is essential for neurotransmitter release and, therefore, blockage of Cav 2.2 reduces the synaptic transmission, including nociceptive signals. Upregulation of Cav 2.241 and Cav 3.2155 in cell bodies of DRG neurons contributes to neuropathic pain by increasing neuronal excitability.
In clinic, anticonvulsants (pregabalin and gabapentin), which interrupt membrane trafficking of Cavs, especially N-type channels, by binding to α2δ1 subunit,157 are the first-line drug for treatment of neuropathic pain. Intrathecal injection of prialt (ziconotide), a potent and selective N-type calcium channel peptide blocker, was approved in United States and European Union to treat severe chronic pain patients who are intolerant or refractory to other treatment, such as systemic analgesics, adjunctive therapies, or intrathecal morphine.158 As the N-type channel is widely distributed in nervous system, its blockers have many severe side effects including cognitive impairment, hallucinations, and changes in mood and consciousness.159
In L5-SNL model, the upregulation of Cav 2.2 in intact L4 DRG neurons is accompanied by IL-1β upregulation, while its downregulation in injured L5 DRG neurons by IL-10 upregulation.41 The data suggest that the differential cytokine expression may lead to the opposite changes in Cav 2.2. The notion is supported by the data showing that intrathecal injection of IL-1β upregulates Cav 2.2 in both L4 and L5 DRG neurons in naive rats, while intrathecal injection of IL-10 reverses Cav 2.2 upregulation in intact L4 DRG neurons produced by L5-SNL. Furthermore, in cultured DRG neurons, IL-1β and IL-10 up- and downregulates Cav 2.2, dose-dependently. Therefore, the upregulation of Cav 2.2 in intact DRG neurons is resulting from increased IL-1β, and its downregulation in injured DRG neurons from increased IL-10. It has been shown that IL-6 is involved in the upregulation of Cav 3.2 in injured L5 DRG neurons in L5-SNL model.160
Potassium Channels
Potassium channel families are consisted of the voltage gated (Kv) channels, Ca2+ activated K+ channels, inwardly rectifying K+ channels and tandem pore domain channels. All of them are expressed in DRG neurons, and majority of them are downregulated in DRG neurons in neuropathic pain condition. K+ channel downregulation increases the excitability, axonal conduction and neurotransmitter release from primary afferent terminals in the spinal dorsal horn. Importantly, selective impairment of just one subtype of K+ channel in DRG neurons can produce signs of pain in vivo (see43 for a review).
Kv7.2 and Kv7.3 are the principal molecular components of the slow voltage-gated M-channel, which is named after its inhibition by muscarine acetylcholine receptor agonist. Reduction of M-channel function leads to neuronal hyperexcitability.161 The M-channel opener retigabine (ezogabine or Potiga) was approved by FDA in 2011 as an anticonvulsant used for an adjunctive treatment of partial epilepsies, and was discontinued in 2017 due to its side effects of blue-colored appearance of the skin and eyes after prolonged intake. Flupirtine, a structural derivative of retigabine, was used as a non-opioid analgesic and was also withdrawn in 2018 for liver toxicity (see162 for a review). As K+ channels are widely expressed in all excitable cells in the body, they are not good targets for treatment of any disease.
Unlike other channels, K+ channels are downregulated by TNF-alpha in DRG neurons.163 Prolonged (5–6 days) exposure of DRG neurons to IL-1β also reduces the function of K+ channels.164,165
Transient Receptor Potential Channels
The transient receptor potential (TRP) channels expressed in nociceptors sense various stimuli. In mammals, TRP superfamily has been divided into six subfamilies based on sequence homology: canonical (TRPC1 to TRPC7), vanilloid (TRPV1 to TRPV6), melastatin (TRPM1 to TRPM8), ankyrin (TRPA1), mucolipin (TRPML1 to TRPML3) and polycystin (TRPP1 to TRPP3) (see166 for a review). Among them, TRPV1, which responds to capsaicin and noxious heat (>43 °C),167 and TRPA1, which responds to cold (>18 °C) and endogenous and exogenous chemical stimuli are believed to be promising targets for analgesics.168 Animal studies show that upregulation of TRPV1 or TRPA1 in DRG neurons contributes to persistent pain in various neuropathic pain models, such as L5-SNL,169 CCI,170 and paclitaxel-induced peripheral neuropathy.171,172
Unfortunately, small-molecule inhibitors directly targeting TRPV1 and TRPA1 were unsuccessful due to their thermoregulatory function and other adverse side effects in various clinical trials.45 However, local application of capsaicin (a TRPV1 agonist), which induces desensitization of nociceptors on repetitive application, is in use for the treatment of arthritis, muscle pain, neuropathic pain and migraine (see173 for a review).
TNF-α is reported to upregulate TRPV1174–177 and TRPA1178–181 in sensory neurons in various pathological conditions. IL-β upregulates TRPV1 in cultured DRG neurons182 and injection of IL-β into rat hind-paw increases TRPV1+ DRG neurons.183 IL-β is also associated with upregulation of TRPV1 in rats with experimental autoimmune prostatitis184 and with the upregulation of TRPA1 in vincristine-induced peripheral neuropathy in rats.185
Acid-Sensing Ion Channels
Acid-sensing ion channel (ASIC) was cloned and identified in DRG neurons in 1997.186 ASICs are permeable to cations and are activated by extracellular acidosis. Now, six ASIC subunits (ASIC1a, ASIC1b, ASIC2a, ASIC2b, ASIC3 and ASIC4) have been identified. Apart from ASIC4, all of them are expressed in peripheral nerves (see46 for a review). A recent work187 using an in situ hybridization technique (RNAscope) showed that, in DRG neurons of naive mice, all five subunits are expressed in NF-200+ and CGRP+ neurons with different levels; while in IB4+ neurons, ASIC2a, ASIC2b and ASIC3 but not ASIC1a and ASIC1b were detected. At least half of sensory neurons express multiple types of ASIC subunits. In the nerve injury mice, overall expression levels of the different ASIC subunits are not altered, as assessed by real-time qPCR. Further analysis revealed that the expression of ASICs was changed in CGRP+ neurons but not in IB4+ neurons. Namely, the percentages of ASIC1b- and ASIC3-expressing CGRP+ neurons in L4 DRG and in L5 DRG are increased, while ASIC1a-expressing CGRP+ neurons are reduced in L4 DRG.187 Consistently, previous works show that inhibition of ASIC1b-containing channels underlies the opioid-independent inhibitory effect on inflammatory pain and neuropathic pain by mambalgins isolated from snake venom.188 ASIC3 is involved in the inflammatory pain189 and neuropathic pain.190 To date, no clinical trial using ASIC ligands for treating chronic pain is available.
A brief (5 min) application of TNF-α rapidly enhanced ASIC-mediated currents in rat DRG neurons in a dose-dependent manner.191,192 IL-1β upregulates ASIC3 mRNA in cultured fibroblast-like synoviocytes,193 and in DRG neurons of animals with musculoskeletal pain induced by ischemia and reperfusion injury.194,195
Taken together, the growing experimental and clinical studies have greatly increased our understanding of the role of ion channels in persistent pain, but knowledge contributes little to pain relief in clinic. Many, if not all, ion channels in primary sensory neurons are changed in chronic pain, especially in neuropathic pain. The functional and transcriptional changes of all ion channels may contribute to the persistent hyperexcitability of sensory neurons. This explains why some specific ion channel subtype blockers tested in clinic trials are ineffective for treatment of chronic pain. Fortunately, emerging evidence indicates that the plastic changes in ion channels are tightly regulated by pro- and anti-inflammatory cytokines, which are imbalanced in chronic pain condition. Therefore, normalization of cytokine production may prevent and/or reverse the dysregulation of the ion channels, and treat chronic pain. The roles of ion channels of sensory neurons in persistent pain and their regulation by cytokines are summarized in Table 1.
Inflammatory Cytokines and Glial Cells Region-Dependently Modulate Synaptic Plasticity, Leading to Persistent Pain and Memory/Emotional Deficits
As mentioned above, the plastic changes in synaptic transmission are critical for learning/memory and chronic pain. Emerging evidence has demonstrated that overproduction of proinflammatory cytokines and glial activation induce LTP and enhance excitatory synapses in spinal dorsal horn but impair LTP and reduce excitatory synapses in hippocampus. This intriguing region-dependent effect may contribute to persistent pain and memory/emotional deficits, respectively.
Long-Term Potentiation in Spinal Dorsal Horn Contributes to Persistent Pain
LTP, discovered in the hippocampus in 1973,196 has been intensively studied as a synaptic model of memory storage (see62,63 for reviews), while LTP at C-fiber synapses in spinal dorsal horn was first reported in 1995197 (Figure 4). The spinal LTP is considered as a synaptic model of persistent pain, based on following experimental and clinical data. (1) Afferent C-fiber conducts nociceptive signals and makes synapses with second order neurons in the superficial spinal dorsal horn.198,199 (2) Pathogenic factors that cause persistent pain can reliably induce the spinal LTP, such as activation of afferent C-fibers by electrical high frequency stimulation (HFS),200 low frequency stimulation201 or natural stimuli,202 peripheral nerve injury,203,204 tissue inflammation,201 opioid withdrawal,205 estrogen deficit produced by ovariotomy or aging27 and antineoplastic agent vincristine.29 (3) LTP-inducible HFS produces long-lasting behavioral signs of pathological pain in rodent24 and human subjects.206 (4) The drugs that are effective to attenuate persistent pain are capable of depressing the spinal LTP, such as N-type calcium channel blockers omega-conotoxin GVIA207 and gabapentin,208 clonidine,209 N-methyl-D-aspartic acid (NMDA) receptor antagonist210 and diazepam.211 The pathological significance of spinal LTP at C-fiber synapses is to amplify pain signals in the first order relay in pain pathway (see47 for a review).
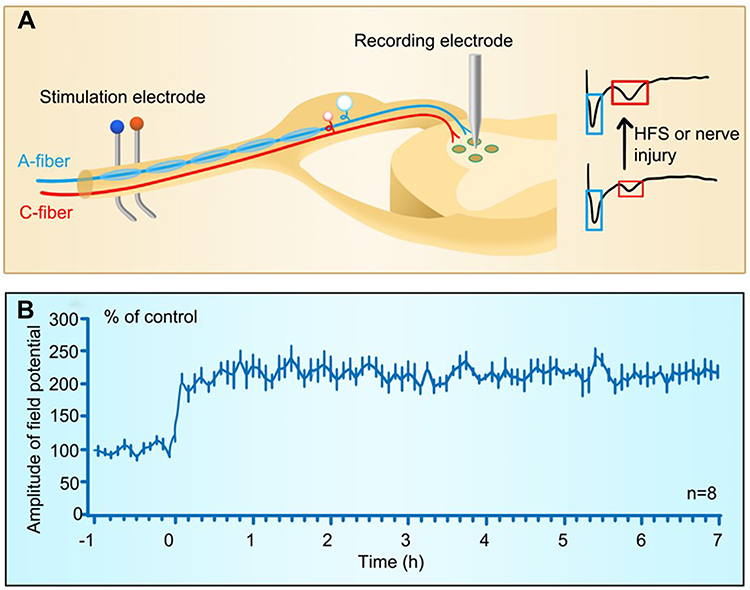 |
Figure 4 Long-term potentiation at C-fiber synapses in spinal dorsal horn. (A) Experimental setup for recording LTP of C-fiber evoked field potentials induced by electrical stimulation or injury of peripheral nerves. A-fiber and A-fiber-evoked field potentials are marked in blue and C-fiber and C-fiber-evoked field potentials in red. (B) The time course of the spinal LTP induced by high frequency stimulation (100 Hz, 100 pulses is given in 4 trains of 1-s duration at 10-s intervals, at the intensity sufficient to activate C-fibers) delivered to peripheral nerve. (B) Reprinted with permission from Liu XG, Sandkuhler J. Characterization of long-term potentiation of C-fiber-evoked potentials in spinal dorsal horn of adult rat: essential role of NK1 and NK2 receptors. J Neurophysiol. 1997;78(4):1973–1982.200 |
The synaptic transmission in spinal dorsal horn is strongly controlled by descending pathway from midbrain periaqueductal gray and the rostral ventromedial medulla. The descending system exerts both inhibitory and facilitatory effects on spinal nociception.212 Removing the descending control system by transection of spinal cord at cervical 3 level increases the amplitude of spinal C-fiber-evoked field potentials by up to 250% of control in anaesthetized adult rats.213 Prolonged high frequency burst stimulation of the sciatic nerve at Aδ-fiber strength produces long-term depression (LTD, a persistent decrease in efficacy of synaptic transmission) of C-fiber-evoked field potentials in intact rats, but induces LTP when the descending system is removed.213 Therefore, the descending system may not only tonically inhibit pain synaptic transmission but also determine the direction of synaptic plasticity in spinal dorsal horn. The descending system regulates the spinal nociception by releasing noradrenaline, serotonin (5-hydroxytryptamine) and dopamine.214 Noradrenaline inhibits pain transmission via activation of presynaptic α2-adrenergic receptors.215 Spinal application of α2-adrenergic receptor agonist clonidine not only blocks but also reverses spinal LTP at C-fiber synapses.209 Antidepressants (duloxetine, venlafaxine), which enhance the descending inhibition via inhibition of presynaptic serotonin-noradrenaline reuptake,216 are first-line drugs for neuropathic pain. The drugs have many side effects, including nausea, diarrhea, fatigue or somnolence, sexual dysfunction, increase in blood pressure, diaphoresis, tachycardia, tremors, and anxiety.217 Dopamine produces anti-hyperalgesia or hypoalgesia by acting on dopamine D2 receptors.214 Activation of D2 receptors by spinal application of quinpirole depresses spinal C-fiber synaptic transmission, while activation of D1/D5 receptors induces protein synthesis-dependent late-phase LTP of C-fiber-evoked field potentials in the absence of presynaptic activation.218 As the expression of D2 receptors is much high than D1/D5 receptors in spinal cord,219 the net effect of dopamine on spinal nociception is inhibition. Spinal D1/D5 receptors are critically involved in translation of acute pain to chronic pain.220,221
Overproduction of Proinflammatory Cytokines and Glial Activation Inhibit LTP in Hippocampus but is Essential for Induction of LTP at C-Fiber Synapses in Spinal Dorsal Horn
Compelling evidence has demonstrated that overproduction of proinflammatory cytokines, such as TNF-α28,222 and IL-1β,223 impairs memory and LTP in hippocampus (see224 for a review). TNF-α inhibits hippocampal LTP by activation of p38225 and c-Jun N-terminal kinase (JNK) signaling,226 while spinal application of either TNF-α or IL-1β at recording segments is able to induce LTP at C-fiber synapses in rats with neuropathic pain produced by L-5 VRT or SNI.227,228 Interestingly, the spinal LTP induced by both TNF-α and IL-1β are blocked by inhibition of p38 and JNK signaling. LTP at C-fiber synapses induced by HFS is completely blocked in TNFR1 knockout mice, and the inhibitory effect of microglia inhibitors on LTP can be rescued by spinal application of TNF-α.229 Thus, the proinflammatory cytokines inhibit LTP in hippocampus, but are essential for spinal LTP induction.
Likewise, in hippocampus activation of astrocytes230 and microglia231,232 impairs memory and LTP.233 While in spinal dorsal horn the glial activation is indispensable for induction of LTP at C-fiber synapses. Our primary work showed that inhibition of microglia by spinal application of either minocycline or inhibitors of Src-family kinases (SFKs), which are exclusively activated in spinal microglia after peripheral nerve injury,234 reversed the effect of HFS on synaptic plasticity. That is, HFS induces LTP in naive rats, but induces LTD in rats treated with microglia inhibitors.229 The data indicate that the direction of C-fiber synaptic plasticity is decided by spinal microglia. Furthermore, spinal application of either ATP,235 brain-derived neurotrophic factor (BDNF)236 or opioids205 is able to induce LTP at C-fiber synapses in the absence of conditioning activation of primary afferents. The chemical-induced spinal LTP is also dependent on activation of microglia. Sandkühler et al237 show that combined activation of microglia and astrocytes by P2X7 receptor agonist BzATP induces LTP at C-fiber synapses in spinal lamina I neurons in the absence of presynaptic activation, which is termed gliogenic LTP. Therefore, activation of spinal glial cells is sufficient to induce the spinal LTP. Interestingly, the gliogenic LTP can be transferred between individuals, ie, application of spinal superfusate collected from lumbar segments of animals, in which the spinal LTP has been induced by HFS, onto spinal dorsal dorsum of naive animals is capable of inducing LTP. The transferable LTP is prevented by blocking TNF-α, D-serine signaling and NMDA receptors but not by blocking glial activation in recipient animals. Accordingly, the spinal LTP is induced by the accumulated bioactive substances released by glial cells called gliotransmitters in activated site. The gliotransmitters may travel long distances via the cerebrospinal fluid and induce LTP in remote sites, and may, therefore, underlie some forms of widespread pain in intact sites. Together, glial activation is a common mechanism underlying spinal LTP induced by different approaches.238
Hippocampal LTP and spinal LTP at C-fiber synapses also share many common mechanisms. Receptors in cell membrane, including NMDA receptor,197,210 dopamine D1 receptor218 and TrkB receptor,204,239 and intracellular signaling molecules, such as calcium/calmodulin-dependent protein kinase II (CaMKII), protein kinase C (PKC), PKA,240 extracellular signal-regulated kinase (ERK)/cAMP response element binding protein (CREB) pathway,241 are critical for the induction and maintenance of LTP in both hippocampus and spinal dorsal horn (see47 for a review). The similarities and differences between hippocampal LTP and spinal LTP are summarized in Figure 5. The findings are of importance for the development of pain-relieving drugs. Targeting the molecules that are shared by hippocampal LTP and spinal LTP may impair the memory function of hippocampus. While targeting the neuroinflammation may not only treat persistent pain but also improve the memory function of hippocampus.
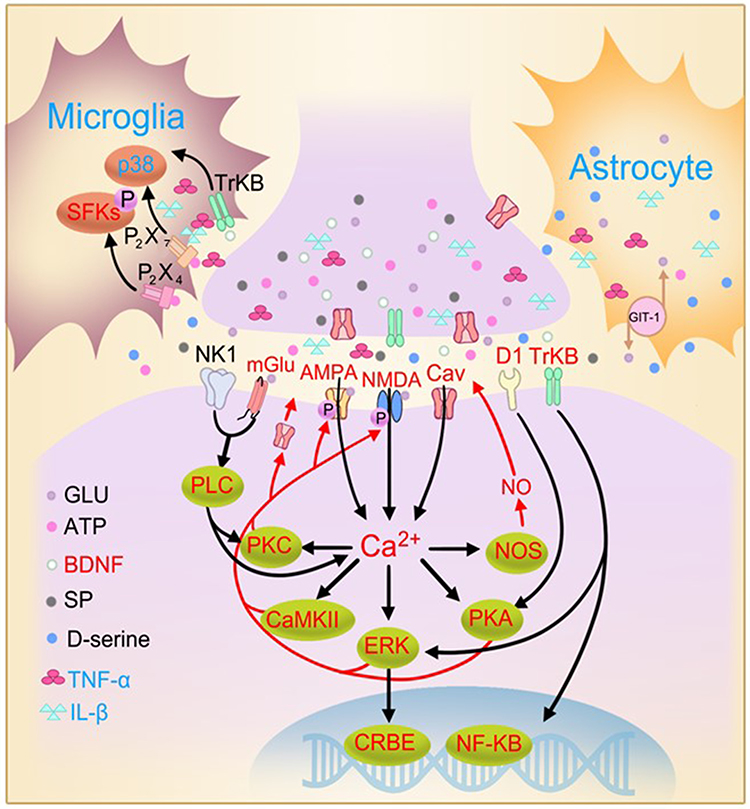 |
Figure 5 The similarities and differences between hippocampal LTP and spinal LTP. Similarities (indicated by red text): in both spinal dorsal and hippocampus, the induction of LTP depends on Ca2+ rise in postsynaptic neurons resulting from opening of NMDA receptors and Cavs; to a less extent, opening of α-amino-3hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor channels; and from Ca2+ release from intracellular store (not shown). The expression of LTP, referring to the enhanced efficacy of synaptic transmission manifested by persistent increase in magnitude of excitatory postsynaptic potentials, is primarily resulting from the enhancement of AMPA receptor currents produced by the receptor phosphorylation and the receptor insertion into postsynaptic membrane. The maintenance of spinal LTP is supported by activation of signal transduction pathways by Ca2+ rise in postsynaptic neurons. Early-phase LTP (<3 h) needs activation of PKA, PKC, CaMKII, ERK and phospholipase C (PLC) activated by neurokinin 1 (NK1) receptors and metabotropic glutamate (mGlu) receptors, and release of nitric oxide (NO). Late-phase LTP (>3 h) depends on gene transcription mediated by CREB and NF-κB and de novo protein synthesis (see47 for a review). Activation of both TrkB and dopamine D1 receptors induces late-phase LTP in both hippocampus and spinal dorsal horn. Disrupting glutamate transporter 1 (GLT-1) in astrocytes, which regulates synaptic transmission via reuptaking glutamate, blocks LTP in both hippocampus361 and spinal dorsal horn.362 Differences (blue text): activation of either microglia or astrocytes and overproduction of TNF-α or IL-1β inhibit hippocampal LTP but are essential for spinal LTP induction. Also, activation of ligand-gated ATP receptors (P2X4 and P2X7), p38 mitogen-activated protein kinase (MAPK) and Src-family kinases (SFKs) in microglia of spinal dorsal horn is essential for induction of spinal LTP (see47 for a review). Meanwhile, in hippocampus, blockage of P2X7 attenuates age-related LTP deficits.363 The roles of P2X4 and SFKs in glial cells for hippocampal LTP remain elusive. Adapted with permission from Liu XG, Zhou LJ. Long-term potentiation at spinal C-fiber synapses: a target for pathological pain. Curr Pharm Des. 2015;21(7):895–905.47 |
Peripheral Nerve Injury Impairs Working Memory and Hippocampal LTP by Upregulation of TNF-α
As discussed above, the spinal LTP induced by peripheral nerve injury may contribute to persistent pain. To investigate the mechanism underlying memory deficits in chronic pain, we tested effect of the nerve injury on LTP in hippocampus, a synaptic model of memory storage, in SNI model of neuropathic pain. The results show that SNI induces working memory and short-term memory deficits, and impairs the LTP at CA3-CA1 synapses in a time-dependent manner.28 Neither baseline synaptic transmission nor LTP induction by HFS are affected one hour after SNI, indicating peripheral nerve injury has no acute effect on synaptic transmission and LTP induction in hippocampus. However, the synaptic potentiation induced by HFS persists for less than one hour 18 to 20 h after SNI and for less than 30 min 6–10 d after SNI. The LTP impairment produced by SNI persists for at least 1.5 months, and is also evident in contralateral hippocampus after unilateral peripheral nerve injury. Namely, SNI leads to a delayed and long-lasting LTP impairment in bilateral hippocampus. We found that TNF-α was persistently increased in both plasma and cerebrospinal fluid, as well as in hippocampal tissue after SNI. The impairments of memory and LTP by SNI are prevented by genetic deletion of TNFR1, mimicked by intracerebroventricular or intrahippocampal injection of rat recombinant TNF-α.28 The results indicate that the memory deficits and dysfunction of hippocampus in neuropathic pain are not directly produced by peripheral nerve injury by the injury-induced overproduction of TNF-α.
Number of Excitatory Synapses is Enhanced in Spinal Dorsal Horn but Reduced in Hippocampus by TNF-α After Peripheral Nerve Injury
To elucidate the long-lasting persistent pain and memory/emotional deficits induced by peripheral nerve injury, we investigated the structural synaptic plasticity induced by SNI. The results show that SNI produces an opposite change in morphological synaptic connectivity in hippocampus and in spinal dorsal horn.25 The dendrite lengths and spine densities are reduced significantly in hippocampal CA1 pyramidal neurons, but increased in spinal neurokinin-1-positive projection neurons in SNI mice. As most excitatory synapses are located in spines,242 the data indicate that the excitatory synaptic connectivity is reduced in hippocampus but enhanced in spinal dorsal horn in neuropathic pain condition. Interestingly, the opposite morphological changes are again blocked by genetic deletion of TNFR1 or conditioning deletion of microglia, and are mimicked by TNF-α in cultured hippocampal and spinal cord slices.25 Furthermore, the length of dendrites of gamma-aminobutyric acid (GABA)-nergic inhibitory interneurons in lamina II of spinal dorsal horn is significantly reduced in CCI model of neuropathic pain.243 The opposite structural synaptic changes in spinal dorsal horn and in hippocampus may contribute to persistent pain and memory/emotional deficits, respectively (Figure 6).
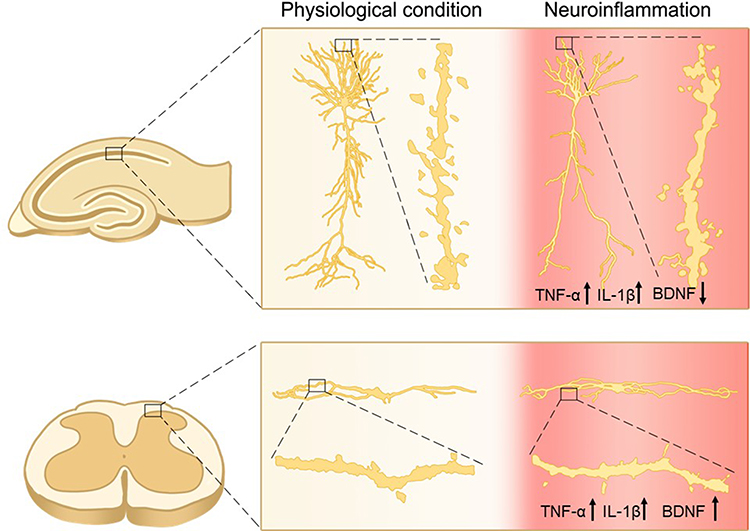 |
Figure 6 The opposite effect of spared nerve injury of the sciatic nerve on excitatory synaptic connectivity in hippocampus and spinal dorsal horn. The dendrite lengths and spine densities are reduced in hippocampal CA1 pyramidal neurons but increased in spinal projection neurons in SNI rats. The morphological changes are paralleled with upregulation of TNF-α and IL-1β in both hippocampus and spinal dorsal horn. Meanwhile, BDNF that is critical for synapse formation is downregulated in hippocampus and upregulated in spinal dorsal horn. These opposite changes in synaptic connectivity and BDNF expression are blocked by either deletion of TNFR1 or conditioned deletion of microglia, and are mimicked by TNF-α in cultured hippocampal and spinal cord slices. The data suggest that the differential regulation of BDNF by inflammatory cytokines may contribute to the opposite morphological changes in hippocampus and spinal dorsal horn. Adapted with permission from Liu Y, Zhou LJ, Wang J, et al.TNF-alpha differentially regulates synaptic plasticity in the hippocampus and spinal cord by microglia-dependent mechanisms after peripheral nerve injury. J Neurosci. 2017;37(4):871–881.25 |
Our finding that peripheral nerve injury induces cognitive deficits is repetitively confirmed by others.244–247 In consistence with reduction of dendrite length and spine density in hippocampus in SNI mice, Apkarian’ s group show that the volume of bilateral hippocampi is reduced in human patients with chronic back pain and complex regional pain syndrome, and the synaptic plasticity in hippocampus is impaired in SNI mice.248 SNI-induced working memory deficits are associated with reduced hippocampus–PFC connectivity.249 In consistence with the enhancement of excitatory synapses in spinal dorsal horn in neuropathic pain, we show that CGRP (a marker for peptidergic C-fiber) in spinal dorsal horn is increased in several animal models of chronic pain, including chemotherapy-induced neuropathy,29 high frequency noxious stimulation of peripheral nerve24 and estrogen decline produced ovariectomy and aging.27 Importantly, the CGRP increase is paralleled with LTP at C-fiber synapses and neuropathic pain behaviors. As CGRP is colocalized with p-p65, TNF-α and IL-1β in DRG neurons,27 the increased CGRP+ C-fibers may be also resulting from neuroinflammation.
Interestingly, our recent work shows that in rodents with either SNI or L5-SNL, activation of dorsal hippocampus with optogenetic and pharmacological methods relieves neuropathic pain behavior. The functional connectivity of the dorsal hippocampus with many brain regions in SNI rats and in chronic pain patients is reorganized. Therefore, the dysfunction of hippocampus may not only cause cognitive deficits but also aggravate persistent pain.26
Opposite Regulation of BDNF by Neuroinflammation Contributes to Region-Dependent Synaptic Connectivity in Hippocampus and Spinal Dorsal Horn
It has been shown that BDNF is essential for LTP in both hippocampus and spinal dorsal horn. Acute intrahippocampal infusion of BDNF induces LTP in hippocampus,250 and spinal application of BDNF at recording segments induces spinal LTP at C-fiber synapses.239 Following peripheral nerve injury, both TNF-α28,75 and IL-1β59 are upregulated in cerebral fluid, hippocampus and spinal dorsal horn, while BDNF is downregulated in hippocampus but upregulated in spinal dorsal horn.251–254 The opposite changes in BDNF are again blocked by genetic deletion of TNFR1 or by inhibition or deletion of microglia.24,25 Furthermore, TNF-α upregulates BDNF in spinal slices and downregulates BDNF in hippocampal slices in a dose-dependent manner.25 As BDNF is critical for synapse formation,255 TNF-α overproduction and glial activation may cause the opposite changes in synaptic connectivity in spinal dorsal horn and in hippocampus by the differential regulation of BDNF (Figure 6).
Overproduction of proinflammatory cytokines and glial activation are also evident in other brain regions closely related to memory and emotion in chronic pain condition. The upregulation of TNF-α is reported in the anterior cingulate cortex (ACC), a forebrain structure involving pain transmission and emotion, in persistent inflammation pain induced by hind-paw injection of complete Freund’s adjuvant256 and in SNI model of neuropathic pain.257 IL-1β258 and TNF-α259 are upregulated in the medial prefrontal cortex (mPFC) in neuropathic pain. The upregulation of TNF-α and p-p65 and microglial activation in the mPFC are also demonstrated in oxaliplatin-induced memory and emotional deficits.30 The roles of proinflammatory cytokines for synaptic plasticity in ACC and mPFC in neuropathic pain remain elusive.
Neuroinflammation May Underlie the Comorbidity of Persistent Pain and Memory/Emotional Deficits
It has long been proposed that persistent pain and depression are independent diseases but may share common pathological mechanisms.55,56 As discussed above, the overproduction of proinflammatory cytokines and glial activation are essential for both hypersensitivity of sensory neurons and the pathological synaptic plasticity in CNS. Previous works show that both proinflammatory cytokines and glial activation are also critically involved in memory deficits (see84,85 for reviews) and major depression (see86,87 for reviews) in many diseases other than chronic pain. Therefore, neuroinflammation might be the common cause for persistent pain and memory/emotional deficits in chronic pain condition. Indeed, in SNI rats the pain behaviors are co-related with neither memory scores assessed with novel object recognition test nor flatting times that measure depression-like behavior in force swimming test.59 The data suggest that the cognitive deficits may not be caused by persistent pain in neuropathic pain condition. This notion is supported by a later work.60 However, IL-1β is upregulated in the injured sciatic nerve and in plasma within hours, and then in the spinal dorsal horn and in the brain regions closely associated with memory and emotion, including ACC, mPFC, amygdala and hippocampus in SNI rats. The changes are accompanied by glial activation. Importantly, the SNI-induced persistent pain, memory decline and depressive behaviors, as well as the IL-1β upregulation and glial activation, are substantially prevented by local administration of IL-1β neutralizing antibody at injured nerve or deletion of IL-1R1. Furthermore, the behavioral changes and neuroinflammation induced by SNI are mimicked in naive rats by repetitive intravenous injection of recombinant rat IL-1β at a pathological concentration, determined in SNI rats.59 Therefore, peripheral nerve injury causes persistent pain, memory deficit and mood depression by triggering neuroinflammation. Our late studies showing that activation of TNF-α/NF-ƘB and glial cells in DRG and spinal dorsal horn in vincristine-treated rats29 and in hippocampus in oxaliplatin-treated rats30 contribute to persistant pain and memory/emotional deficits support this notion.
Interactions of Circulating Immune Cells with Glial Cells and Neurons are Critical for Initiation and Maintenance of Neuroinflammation
The data discussed above indicate that neuroinflammation is a common cause of persistent pain and memory/emotional deficits in chronic pain condition. The next key question is how the neuroinflammation is triggered and maintained in chronic pain.
Transmigration of Circulating Inflammatory Monocytes into Peripheral Nerve, Spinal Parenchyma and Brain Perivascular Space Leads to Neuroinflammation
In peripheral nerves, neuroinflammation produced by injury is resulting from increased resident and infiltrating macrophages. The upregulation of chemokine C-C motif ligand 2 (CCL2) in injured DRGs causes transmigration of circulating monocytes into DRGs through CCL2 receptor expressed on monocytes (see260 for a review).
In spinal dorsal horn, it has been shown that the colony-stimulating factor 1 (CSF1) in afferent neurons is upregulated after peripheral nerve injury (see261 for a review) or high frequency noxious stimulation of sciatic nerve.24 The CSF1 released from the central terminals of DRG neurons activates microglia in spinal dorsal horn by binding to its receptors, contributing to spinal neuroinflammation.261 In addition, the blood–spinal cord barrier (BSCB) breaks down following peripheral nerve injury. The disrupted BSCB leads to the influx of inflammatory mediators and the recruitment of circulating monocytes into spinal parenchyma. The BSCB impairment can be also produced by circulating IL-β and is shut down by anti-inflammatory cytokines IL-10 and TGF-β1.262 The CCL2/CCL2R system plays a role in the recruitment of circulating monocytes into spinal parenchyma.263 Accordingly, glial activation induced by both CSF1 released from sensory afferents and the recruitment of circulating monocytes due to BSCB disruption may lead to neuroinflammation in spinal cord.
How can peripheral nerve injury induce remote inflammation in the brain? Previous studies have shown that the infiltration of immune cells into brain parenchyma resulting from blood–brain barrier (BBB) interruption leads to neuroinflammation and cognitive deficits in a variety of pathological conditions.264–266 In SNI model of neuropathic pain, however, BBB is intact and no immune cell is detected in brain parenchyma, while circulating proinflammatory (classical) monocytes and C-X-C motif chemokine 12 (CXCL12) in the blood and in brain perivascular space are persistently increased.267 In parallel, the number of perivascular macrophages (PVMs) in the brain, particularly in hippocampus, is enhanced, and both microglia and astrocytes are activated. The experiments with the transgenic CCR2 (RFP/+) and CX3CR1 (GFP/+) mice reveal that at least some of the PVMs are derived from circulating monocytes. The SNI-induced PVM increase, glial activation and memory decline are substantially prevented by either depleting circulating monocytes via intravenous injection of clodronate liposomes or by blockade of CXCL12-CXCR4 signaling, whereas intravenous injection of CXCL12 at a pathological concentration in naïve mice mimics the changes induced by SNI. Because glial activation and PVM increase are in the same spatial and temporal fashion, and glial activation is prevented by obstruction of PVM recruitment by either deleting circulating monocytes or blocking CXCL12-CXCR4 signaling, glial activation may be resulting from an increase in PVMs. Accordingly, the brain neuroinflammation induced by peripheral nerve injury may be initiated at and distributed along the blood vessels. Importantly, in chronic pain patients circulating monocytes and plasma CXCL12 are elevated, and both of them are highly correlated with memory decline assessed by the Montreal Cognitive Assessment.267 The data indicate that CXCL12-mediated monocyte recruitment into the perivascular space is critical for brain neuroinflammation and the resultant cognitive impairment in neuropathic pain.
Overproduction of Inflammatory Cytokines in Both Neurons and Glial Cells Contributes to Persistent Neuroinflammation
It is generally believed that bioactive substances released by activated glial cells contribute to chronic pain.268 In DRG, activation of satellite glial cells enhances the excitability of sensory neurons by releasing ATP, glutamate and cytokines, such as TNF-α and IL-1β, and fractalkine.269 In spinal cord TNF-α and IL-1β released by the activated microglia and astrocytes enhance synaptic transmission.270 Glial activation is also believed to cause memory deficits in Alzheimer’s disease,271 major depression272 and postoperative cognitive dysfunction273 by sustained release of proinflammatory cytokines. In other words, the cytokines released by glial cells cause the neuronal disorders.
However, accumulating evidence has demonstrated that the neurons in peripheral and central nervous system are also capable of producing proinflammatory cytokines. TNF-α is shown to be upregulated in the neurons of DRG and spinal dorsal horn in both L5-VRT and SNI models,75,274 and in the neurons of bilateral hippocampi in SNI model.28,275 NF-ƘB that is critical for transcriptional upregulation of proinflammatory cytokines276 is upregulated in neurons of spinal dorsal horn277 and DRG in both inflammatory pain278 and neuropathic pain models.279 Tnf-α mRNA is upregulated in the neurons of DRG and spinal dorsal horn after coronary artery occlusion in rats.280 TNF-α and p-p65 are upregulated in the neurons of DRG and spinal dorsal horn in vincristine-induced peripheral neuropathy.29 Furthermore, p-p65 and TNF-α are upregulated in the neurons but not in microglia and astrocytes of hippocampus and mPFC in rats with neuropathic pain induced by oxaliplatin.30 A recent work shows that p-p65, TNF-α and IL-β are upregulated predominantly in the neurons and sparsely in glial cells of spinal dorsal horn and hippocampus in ovariectomized and aged female mice, and the change is essential for both glial activation and the genesis of persistent pain, memory/emotional deficits induced by estrogen decline. The results of in situ hybridization show that Tnf and Il-1β mRNAs are expressed in the neurons of DRGs and spinal dorsal horn.27 As the proinflammatory cytokines can potently activate glial cells,59,281–284 the cytokines produced by neurons may initiate or maintain glial activation in pathological conditions. The neuron–glia interaction mediated by proinflammatory cytokines may contribute to neuronal disorders by producing persistent neuroinflammation.
A New Strategy for Treatment of Chronic Pain
In spite of accumulating knowledge on mechanisms of chronic pain, as discussed above, the pharmacological treatment of chronic pain is still a big challenge in clinic. According to a systematic review and meta-analysis285 and a recent guideline,286 antidepressants (duloxetine, venlafaxine) and anticonvulsants (pregabalin and gabapentin) are still the first-line drugs for treatment of neuropathic pain. Pain relief is only achieved in <50% of chronic pain patients with the drugs, and is particularly low for patients with neuropathic pain. In recent decades, exponential growth of experimental studies on neuropathic pain has not yet led to any major clinical applications.287 The reasons for the insufficient translation of basic research into clinical application are complicated. Unlike infectious diseases and cancer, which are caused by invading pathogens or malignant cells, chronic pain results from the alteration of endogenous molecules, such as ion channels, cytokines and intracellular signals, which are critically involved in many physiological functions. Elimination or inhibition of pathogens and cancer cells are successful for treatment of infectious diseases and cancer, while the same strategy is inappropriate for treating chronic pain. The drugs targeting pain-related molecules, which are widely expressed in the nervous system or even the whole body, will certainly cause severe side effects, such as N-type calcium channels159 and K+ channels162 discussed above. Meanwhile, the drugs targeting the molecules that are preferably expressed in sensory neurons are ineffective for pain relief, such as Nav1.7 blockers.118–120 In recent years, the single-cell and/or spatial transcriptomics of DRG neurons in mice,288 nonhuman primates289 and humans290,291 have identified many subtypes of nociceptors, the species differences in molecular phenotypes and new potential drug targets. Although the works have greatly increased our knowledges of nociception, we have a long way to go for determining efficacy and side effects of the new drug targets.
Compelling evidence has demonstrated that inflammation plays a key role in many, if not all, chronic diseases (see292 for a review). As discussed above, overproduction of inflammatory cytokines causes persistent pain and memory/emotional deficits by enhancing the excitability of sensory neurons via regulation of ion channels and region-dependent regulation of synaptic connectivity in CNS (Figure 1). Persistent inflammatory cytokine overproduction is initiated and maintained by the interactions of immune cells, glial cells and neurons. Therefore, neuroinflammation is believed to be a promising target for treatment of chronic pain.293 However, proinflammatory cytokines and glial cells also have many important physiological functions, such as protecting the host from infection and promoting tissue repair and recovery.294,295 In the nervous system, TNF-α/NF-κB signaling is important for memory storage.143 Glial cells are crucial for regulating neuronal structures and functions296 as well as for neurogenesis.297 Thus, normal inflammation response is essential for the structures and functions of nervous system. However, persistent overproduction of inflammatory cytokines and glial activation play key roles in neuropathic pain,298 Alzheimer’s disease299 and mood depression.300,301 Clearly, the function of nervous system and inflammation exhibits a reverted-U relationship. Therefore, the right strategy for treating the neuronal disorders is normalization (counterbalance) of the neuroinflammation, ie, to bring it back to normal level. This cannot be achieved with the agents that simply inhibit inflammatory cytokinse and glial cells, which may lead to either insufficient or excessive inhibition of inflammation response. Consistently, clinical data show that anti-TNF agents are effective for treating osteoarthritis pain,302 lower back pain,303 and rheumatoid arthritis.304 Systematic review and meta-analysis indicate the agents have many side effects, including infection305,306 and psoriasis.307 To date, no agent for normalization of neuroinflammation is available in clinic. However, in recent years experimental studies have brought light to this issue.
Supplement Mg2+ by Oral Magnesium-L-Threonate Normalizes Neuroinflammation in Rodents
Clinical data have shown that Mg2+ deficiency is involved in many chronic diseases, such as hypertension, ischemic heart disease, stroke, metabolic syndrome, diabetes, colorectal cancer (see308 for a review) and postmenopausal symptoms.309,310 In nervous system, Mg2+ deficiency results in anxiety and depression in animals and humans (see311 for a review). A clinical trial shows that the global cognitive ability of older adults is improved by oral application of magnesium-L-threonate (L-TAMS, also called MgT).312 A recent review article by Morel et al313 indicates that supplementing Mg2+ by intravenous administration of MgSO4 is effective for pain relief and reduction of analgesic consumption, based on 80 randomized controlled trials and 8 systematic reviews.
In experimental studies, supplementing Mg2+ with chronic oral administration of L-TAMS improves spatial learning in naive rats314 and in mice with Alzheimer’s disease,315 and prevents and restores the memory deficit induced by SNI.275 Mg2+ deficiency contributes to the chronic pain and memory/emotional deficits induced by antineoplastic agents, including vincristine, cyclophosphamide and oxaliplatin, and the effects are prevented by oral L-TAMS.29,30,316 Oral L-TAMS also prevents or reverses the chronic pain and memory/emotional deficits induced by estrogen decline in ovariectomized or aging female mice.27
The mechanisms underlying therapeutic effects of Mg2+ are still in debate. It has long been shown that Mg2+ voltage-dependently blocks NMDA receptor in spinal dorsal horn.317 As NMDA receptor activation is essential for neuronal hypersensitivity and LTP at C-fiber synapses in spinal dorsal horn,197,318 blockade of central sensitization is believed to underlie the pain relief of Mg2+.319 If this is true, supplementing Mg2+ would impair memory function, as NMDA receptor is also indispensable for the memory function and LTP in hippocampus.320 In contrast, it has been shown that elevating brain Mg2+ by oral application of L-TAMS improves memory function and enhances NMDA receptor activity by upregulation of the NR2B subunit of NMDA receptors in hippocampus of naive rats.314 It has been repetitively shown that overexpression of NR2B in forebrain facilitates synaptic potentiation and enhances memory function in mice,321,322 while upregulation of NR2B in spinal dorsal horn is critical for development of neuropathic pain.323,324 Obviously, blocking NMDA receptor cannot explain the fact that Mg2+supplement can relieve pain and improve memory, simultaneously.
We addressed this contradictory issue with animal model of chemotherapy-induced neuropathy, which exhibits both persistent pain325 and memory/emotional deficits.326,327 We found that NR2B expression was downregulated in hippocampus but upregulated in spinal dorsal horn in vincristine-treated animals, and the opposite changes were prevented by oral L-TAMS.29 How could supplementing Mg2+ regulate NR2B in hippocampus and in spinal dorsal horn in the opposite direction? Experimental studies have demonstrated that Mg2+ deficiency in the rat induces an inflammatory syndrome characterized by leukocyte and macrophage activation, and release of inflammatory cytokines.328 A recent meta-analysis of randomized controlled trials indicates that Mg2+ supplementation significantly reduces different human inflammatory markers.329 Therefore, neuroinflammation induced by Mg2+ deficiency may region-dependently regulate NR2B. Indeed, in the rats treated with vincristine, intracellular Mg2+ in DRG neurons is significantly reduced, and free Mg2+ in blood and CSF is decreased from 8 mM to 6 mM. In cultured DRG neurons, reducing Mg2+ from 8 mM to 6 mM in medium upregulates TNF-α and p-p65.29 In cultured hippocampal slices, reducing Mg2+ to 6 mM also upregulates TNF-α and p-p65, but downregulates NR2B subunit of NMDA receptor.30 Furthermore, single intrathecal injection of rat recombination TNF-α in naive rats upregulates TNF-α in both hippocampus and spinal dorsal horn, but down- and upregulates NR2B in hippocampus and in spinal dorsal horn, respectively. Therefore, intracellular Mg2+ deficiency leads to the differential expression of NR2B in hippocampus and in spinal dorsal horn by upregulation of TNF-α, contributing to persistent pain and memory/emotional neuronal deficits. It has been shown that 31% of Mg2+ in the body is distributed inside cells, where it functions as a co-activator for around 600 enzymes.330 Dysfunction of the enzymes may be responsible for Mg2+ deficiency-induced neuroinflammation, while the underlying mechanisms remain elusive.
We used L-TAMS in our experiments because only L-TAMS, but no other magnesium compound, including MgCl2, Mg citrate and Mg gluconate, is able to elevate Mg2+ in the CSF of rats.314 In human patients, increasing the plasma Mg2+ three-fold via intravenous infusion of MgSO4 does not elevate Mg2+ in the CSF.331 This may explain why supplementing Mg2+ with MgSO4 has only modest effect on pain relief,313 as the central mechanisms of chronic pain may not be affected by intravenous MgSO4. To date, effect of oral L-TAMS on chronic pain has not been tested in clinical trial. As Mg2+ deficiency causes neuroinflammation, supplementing Mg2+ may bring the neuroinflammation back to but never below normal level.
Upregulated Proteins in Neuropathic Condition Promote Recovery of Persistent Pain and Memory/Emotional Deficits by Normalization of Neuroinflammation
In disease conditions, such as neuropathic pain, many proteins are upregulated (see332 for a review). Some of them contribute directly to neuropathic pain. Till now, all drugs are designed to target the upregulated proteins that directly cause disease, such as blockers of ion channels. As many of the upregulated proteins are widely distributed in the body and exert many physiological functions, side effects are inevitable. In recent years, studies have shown that some upregulated proteins, such as translocator protein (TSPO, 18 kDa) and liver X receptors (LXRs), promote recovery of neuropathic pain by inhibition of neuroinflammation. When persistent pain is healed, their expression returns to normal level (see below). More and more such kinds of proteins may be discovered in the future. Accordingly, a natural cure may occur when endogenous agonists for such proteins are sufficient in the body; however, in majority of cases they are probably deficient and exogenous ones are needed for recovery. A novel approach is developing agents that activate the upregulated proteins that promote recovery from diseases.
Translocator Protein (18 kDa)
Early experimental studies show that diazepam inhibits morphine tolerance333 and nociceptive pain.334 Clinical data have demonstrated that diazepam is effective for treatment of chronic pain associated with spinal cord injury (see335 for a review). Our electrophysiological data211 reveal that spinal application of diazepam not only prevents LTP at C-fiber synapses but also potently depresses the late-phase LTP (>3 h), which is protein synthesis-dependent.336 The pharmacological effects of diazepam are mediated by the GABAA receptors in CNS and so-called peripheral benzodiazepine receptors.337 The peripheral receptor, now called TSPO (18 kDa), localized primarily in the outer mitochondrial membrane, is critically involved in steroid synthesis by transporting the substrate cholesterol into mitochondria. In the brain, TSPO is mainly expressed in microglia and astrocytes, and is upregulated in response to injury, inflammation and disease. Therefore, TSPO is used as a biomarker for brain inflammation and reactive gliosis.338 Meanwhile, activation of TSPO also improves neurological and psychiatric disorders by controlling neuroinflammation via enhancing neurosteroidogenesis (see339 for a review). In spinal dorsal horn, TSPO is upregulated predominately in astrocytes, sparsely in microglia but not in neurons after L5-SNL.340 The TSPO expression returns to normal level when behavioral signs of neuropathic pain are naturally extinct (approximately 50 d after L5-SNL). Interestingly, a single intrathecal injection of specific TSPO agonists Ro5-4864 or FGIN-1-27 at 7 and 21 d after L5-SNL reverses the established mechanical allodynia and thermal hyperalgesia. Importantly, TSPO upregulation also returns to normal level when neuropathic pain behavior is reversed by Ro5-4864. These findings are confirmed by another work.341 The data suggest that the role of TSPO upregulation is to promote recovery from the neuronal disorder. Mechanically, Ro5-4864 substantially inhibits spinal astrocytes and reduces the production of TNF-α in vivo and in vitro. The effects are prevented by a neurosteroid synthesis inhibitor (AMG), suggesting that enhancing neurosteroid production contributes to the anti-neuroinflammatory effect of TSPO agonists. In addition, TSPO upregulation has been shown in many other animal models of chronic pain, such as arthritis342 and complex regional pain syndrome.343
Importantly, TSPO is also upregulated in patients with major depression,344,345 Alzheimer’s disease and lumbar radiculopathy.346 The increased TSPO is believed to improve memory by repairing cellular damage and preventing further neuronal degeneration (see347 for a review). Recently a randomized, double-blind clinical trial showed that etifoxine (a TSPO ligand) is effective for reduction of anxiety symptoms in patients with anxiety disorder.348 To date, no clinal study that treats chronic pain with TSPO ligand is available.
Liver X Receptors
Liver X receptors (LXR-α and LXR-β) are ligand-activated transcription factors. LXR-α is upregulated in the neurons and oligodendrocytes of spinal dorsal horn in SNI model, and a single intrathecal injection of a specific liver X receptor agonist (T0901317 or GW3965) reverses mechanical allodynia in both rats and mice.349 GW3965 inhibits glial activation and downregulates TNF-α and IL-β but upregulates IL-10 in the spinal dorsal horn. Genetic deletion of LXR-α abolishes all the effects of GW3965, and exacerbates the neuroinflammation induced by SNI. The effects of liver X receptor agonist also depend on neurosteroid synthesis. Thus, activation of LXRs depresses neuropathic pain by normalization of neuroinflammation via neurosteroids. The similar effect of LXRs is also observed in skin/muscle incision and retraction (SMIR) model of postoperative pain. SMIR that produces a lasting mechanical allodynia in the hind-paw350 induces LTP at C-fiber synapses.351 This spinal LTP is accompanied by the upregulations of TNF-α, acetylated NF-κB p65, LXRα and LXRβ and the downregulation of silent information regulator 1 (SIRT1), which activates NF-κB by the direct deacetylation of NF-κB p65, in dorsal horn neurons. The spinal LTP induced by SMIR is blocked by spinal application of either TNF-α neutralization antibody, NF-κB inhibitor (PDTC) or LXRs agonist (T0901317). The effects of T0901317 on spinal LTP and acetylated NF-κB p65 and TNF-α are blocked by SIRT1 antagonist. Therefore, activation of LXRs prevents spinal LTP by inhibiting NF-κB/TNFα pathway via activating SIRT1. In LXR-α knockout mice, the mechanical allodynia induced by sciatic nerve crush lasts much longer than in wild-type mice.352
Recent works show that conditional deletion of LXR-β in astrocytes induces anxiety-like behaviors and increases the frequency of spontaneous excitatory postsynaptic currents (sEPSCs) and dendritic complexity of layer V pyramidal neurons in mPFC.353 Meanwhile, activation of LXRs by GW3965 prevents emotional and cognitive deficits induced by either chronic unpredictable mild stress or lipopolysaccharide. The activation of LXRs significantly alleviates the impairment of synaptic plasticity, prevents the upregulation of inflammatory factors and inhibits activation of NF-κB and microglial M1-polarization in both models.354
These data suggest that both TSPO and LXRs may be promising therapeutic targets for the treatment of persistent pain, memory decline and mood depression by anti-neuroinflammation. As their upregulations return to normal levels after cure of chronic pain, activation of such proteins may have less side effects.
In clinical practice, it is practical to monitor neuroinflammation with currently available technologies. As mentioned above, neuroinflammation is characterized by upregulation of proinflammatory cytokines and glial activation,58 and is initiated by immune cell infiltration into nervous system. In human patients, the level of cytokines in plasma and in cerebrospinal fluid can be measured with different methods, such as ELLESA. Microglial and astrocytic activation can be detected with positron emission tomography and magnetic resonance imaging, which track TSPO ligands and mono-amine oxidase type B ligands, respectively.338 Immune cell infiltration into nervous system is accompanied by increased circulating immune cells and chemokines. As discussed above, the increased monocytes, CCL2 and CXCL12, in blood are paralleled with monocyte infiltration into nervous system in animals with neuropathic pain. In chronic pain patients, monocytes and CXCL12 in blood are also elevated, and both of them are highly correlated with memory decline.267 The increase in plasma CCL2 is also detected in human patients with chronic and recurrent mechanical neck pain syndrome355 and with nonspecific lower back pain.356 The increased circulating monocytes, CCL2 and CXCL12, may predict infiltration of monocyte into nervous system.
Conclusions and Perspectives
It is increasingly clear that conventional strategies for treatment of chronic pain are less than successful. Targeting the changed ion channels that result in hyperexcitability of sensory neurons or the receptors and intracellular signaling molecules that lead to pathological synaptic plasticity is either ineffective or causes many serious side effects. Emerging evidence indicates that the dysfunction of ion channels in sensory neurons and the pathological plasticity in CNS, which lead to persistent pain and memory/emotional deficits, result from overproduction of inflammatory cytokines. The persistent upregulation of inflammatory cytokines is initiated and maintained by interactions of circulating immune cells, glial cells and neurons. Because all the molecules and cells that contribute to the neuroinflammation have many physiological functions, normalization (counterbalance) but not simple inhibition of the neuroinflammation is the right strategy for treating chronic pain. Experimental studies suggest that this can be achieved by supplementing Mg2+ with L-TAMS or activation of some upregulated proteins, such as TSPO and LXRs.
Answers to the following questions remain elusive. Mechanisms underlying opposite regulation of ion channels by pro- and anti-inflammatory cytokines in sensory neurons remain unknown. The excitability of injured DRG neurons is increased, while all Nav subtypes except for Nav1.3 are downregulated in the neurons. Although both Cav 3.2 and the auxiliary β2 subunit of Navs,357 which plays a role in the regulation of sodium channel density and for action potential generation,358 are reported to be upregulated, the data may be insufficient to explain the hyperexcitability of the injured DRG neurons. The mechanisms underlying the opposite regulation of BDNF, NR2B subunit of NMDA receptor and synaptic connectivity in spinal dorsal horn and in hippocampus by proinflammatory cytokines and glial activation are largely unknown. Clarifying the region-dependent effects is important for understanding and treating the neuronal disorders related to neuroinflammation. The upregulated proteins that promote recovery in disease conditions are optimal targets for treating neuroinflammation-related disorders. Further study is needed to discover more such kinds of proteins and to clarify the mechanisms of their effects with advanced techniques. It has been well established that intracellular Mg2+ deficiency induces inflammation, while the underlying mechanism remains unknown. As only <2% Mg2+ is distributed in extracellular fluid, the routinely measured serum Mg2+ levels do not always reflect total body magnesium status and are insensitive for detecting Mg2+ deficiency,359 a simple method that can quickly measure intracellular Mg2+ is required. The cytokines are short lived, for instance the half-time of TNF-α in plasma is only 12 min.360 The concentration of the cytokines should be much higher in interstitial fluid, where infiltrated immune cells, glial cells and neurons live. To investigate the function of cytokines and monitor local inflammation, a method for assessing the cytokine levels in interstitial fluid is highly desired.
Abbreviations
ACC, anterior cingulate cortex; ASIC, acid-sensing ion channels; BBB, blood–brain barrier; BDNF, brain-derived neurotrophic factor; BSCB, blood–spinal cord barrier; Cav, voltage-gated calcium channel; CCI, chronic constriction of sciatic nerve; CCL2, C-C motif ligand 2; CGRP, calcitonin gene-related peptide; CSF1, colony-stimulating factor 1; CNS, central nervous system; CXCL12, C-X-C motif chemokine 12; DRG, dorsal root ganglion; HFS, high frequency stimulation; HVA, high voltage activated; IB4, isolectin B4; ICD-11,11th revision of the International Classification of Diseases; JNK, c-Jun N-terminal kinase; L5/L6-SNL, lumbar 5 and /or 6 spinal nerve ligation; L5-VRT, L5 ventral root transection; IL-1β, interleukin-1beta; IL-10, interleukin-10; LTP, long-term potentiation; LTD, long-term depression; L-TAMS, magnesium-L-threonate; LVA, low voltage activated; LXR, liver X receptor; mPFC, medial prefrontal cortex; Nav, voltage-gated sodium channel; NF-200, neurofilament-200; NF-ƘB, nuclear factor-kappa B; NMDA, N-methyl-D-aspartic acid; PDTC, pyrrolidine dithiocarbamate; NP, neuropathic pain; PVM, perivascular macrophage; SIRT1, silent information regulator 1; SMIR, skin/muscle incision and retraction; SNI, spared nerve injury; TGF-β, transforming growth factor-beta; TNF-α, tumor necrosis factor-alpha; TNFR1,TNF receptor 1; TRP, transient receptor potential channels; TSPO, translocator protein; TTX, tetrodotoxin.
Acknowledgments
We are grateful for the support of the Natural Science Foundation of China (31771166, U1201223).
Disclosure
Prof. Dr. Xian-Guo Liu reports grants from National Natural Science Foundation of China, during the conduct of the study. The author declares no other competing interests in this work.
References
1. Toth C, Lander J, Wiebe S. The prevalence and impact of chronic pain with neuropathic pain symptoms in the general population. Pain Med. 2009;10(5):918–929. doi:10.1111/j.1526-4637.2009.00655.x
2. Mohamed Zaki LR, Hairi NN. A systematic review of the prevalence and measurement of chronic pain in Asian adults. Pain Manage Nurs. 2015;16(3):440–452. doi:10.1016/j.pmn.2014.08.012
3. Jank R, Gallee A, Boeckle M, Fiegl S, Pieh C. Chronic pain and sleep disorders in primary care. Pain Res Treat. 2017;2017:9081802. doi:10.1155/2017/9081802
4. Means-Christensen AJ, Roy-Byrne PP, Sherbourne CD, Craske MG, Stein MB. Relationships among pain, anxiety, and depression in primary care. Depress Anxiety. 2008;25(7):593–600. doi:10.1002/da.20342
5. Hart RP, Wade JB, Martelli MF. Cognitive impairment in patients with chronic pain: the significance of stress. CurrPain Headache Rep. 2003;7(2):116–126. doi:10.1007/s11916-003-0021-5
6. Whitlock EL, Diaz-Ramirez LG, Glymour MM, Boscardin WJ, Covinsky KE, Smith AK. Association between persistent pain and memory decline and dementia in a longitudinal cohort of elders. JAMA Intern Med. 2017;177(8):1146–1153. doi:10.1001/jamainternmed.2017.1622
7. Nahin RL, DeKosky ST. Comorbid pain and cognitive impairment in a nationally representative adult population: prevalence and associations with health status, health care utilization, and satisfaction with care. Clin J Pain. 2020;36(10):725–739. doi:10.1097/AJP.0000000000000863
8. Mäntyselkä P, Kumpusalo E, Ahonen R, et al. Pain as a reason to visit the doctor: a study in Finnish primary health care. Pain. 2001;89(2–3):175–180. doi:10.1016/S0304-3959(00)00361-4
9. Raja SN, Carr DB, Cohen M, et al. The revised International Association for the Study of Pain definition of pain: concepts, challenges, and compromises. Pain. 2020;161:1976–1982. doi:10.1097/j.pain.0000000000001939
10. Treede RD, Rief W, Barke A, et al. A classification of chronic pain for ICD-11. Pain. 2015;156(6):1003–1007. doi:10.1097/j.pain.0000000000000160
11. Goldberg DS, McGee SJ. Pain as a global public health priority. BMC Public Health. 2011;11:770. doi:10.1186/1471-2458-11-770
12. Cohen SP, Vase L, Hooten WM. Chronic pain: an update on burden, best practices, and new advances. Lancet. 2021;397(10289):2082–2097. doi:10.1016/S0140-6736(21)00393-7
13. Dieppe PA, Lohmander LS. Pathogenesis and management of pain in osteoarthritis. Lancet. 2005;365(9463):965–973. doi:10.1016/S0140-6736(05)71086-2
14. Treede RD, Jensen TS, Campbell JN, et al. Neuropathic pain: redefinition and a grading system for clinical and research purposes. Neurology. 2008;70(18):1630–1635. doi:10.1212/01.wnl.0000282763.29778.59
15. Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33(1):87–107. doi:10.1016/0304-3959(88)90209-6
16. Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50(3):355–363. doi:10.1016/0304-3959(92)90041-9
17. Decosterd I, Woolf CJ. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain. 2000;87(2):149–158. doi:10.1016/S0304-3959(00)00276-1
18. Li L, Xian CJ, Zhong JH, Zhou XF. Effect of lumbar 5 ventral root transection on pain behaviors: a novel rat model for neuropathic pain without axotomy of primary sensory neurons. ExpNeurol. 2002;175(1):23–34.
19. Polomano RC, Mannes AJ, Clark US, Bennett GJ. A painful peripheral neuropathy in the rat produced by the chemotherapeutic drug, paclitaxel. Pain. 2001;94(3):293–304. doi:10.1016/S0304-3959(01)00363-3
20. Siau C, Bennett GJ. Dysregulation of cellular calcium homeostasis in chemotherapy-evoked painful peripheral neuropathy. Anesth Analg. 2006;102(5):1485–1490. doi:10.1213/01.ane.0000204318.35194.ed
21. Zychowska M, Rojewska E, Przewlocka B, Mika J. Mechanisms and pharmacology of diabetic neuropathy - experimental and clinical studies. Pharmacol Rep. 2013;65(6):1601–1610. doi:10.1016/S1734-1140(13)71521-4
22. Kosek E, Cohen M, Baron R, et al. Do we need a third mechanistic descriptor for chronic pain states? Pain. 2016;157(7):1382–1386. doi:10.1097/j.pain.0000000000000507
23. Fitzcharles MA, Cohen SP, Clauw DJ, Littlejohn G, Usui C, Häuser W. Nociplastic pain: towards an understanding of prevalent pain conditions. Lancet. 2021;397(10289):2098–2110. doi:10.1016/S0140-6736(21)00392-5
24. Zhou LJ, Peng J, Xu YN, et al. Microglia are indispensable for synaptic plasticity in the spinal dorsal horn and chronic pain. Cell Rep. 2019;27(13):3844–3859 e3846. doi:10.1016/j.celrep.2019.05.087
25. Liu Y, Zhou LJ, Wang J, et al. TNF-alpha differentially regulates synaptic plasticity in the hippocampus and spinal cord by microglia-dependent mechanisms after peripheral nerve injury. J Neurosci. 2017;37(4):871–881. doi:10.1523/JNEUROSCI.2235-16.2016
26. Wei XH, Centeno MV, Ren WJ, et al. Activation of the dorsal, but not the ventral, hippocampus relieves neuropathic pain in rodents. Pain. 2021;162(12):2865–2880. doi:10.1097/j.pain.0000000000002279
27. Zhang J, Mai CL, Xiong Y, et al. The causal role of magnesium deficiency in the neuroinflammation, pain hypersensitivity and memory/emotional deficits in ovariectomized and aged female mice. J Inflamm Res. 2021;14:6633–6656. doi:10.2147/JIR.S330894
28. Ren WJ, Liu Y, Zhou LJ, et al. Peripheral nerve injury leads to working memory deficits and dysfunction of the hippocampus by upregulation of TNF-alpha in rodents. Neuropsychopharmacology. 2011;36(5):979–992. doi:10.1038/npp.2010.236
29. Xu T, Li D, Zhou X, et al. Oral application of magnesium-l-threonate attenuates vincristine-induced allodynia and hyperalgesia by normalization of tumor necrosis factor-alpha/nuclear factor-kappaB signaling. Anesthesiology. 2017;126(6):1151–1168. doi:10.1097/ALN.0000000000001601
30. Zhou X, Huang Z, Zhang J, et al. Chronic oral administration of magnesium-L-threonate prevents oxaliplatin-induced memory and emotional deficits by normalization of TNF-α/NF-κB signaling in rats. Neurosci Bull. 2020;37(1):55–69. doi:10.1007/s12264-020-00563-x
31. Drdla-Schutting R, Heinl C, Hadschieff V, Sandkühler J. Withdrawal from an opioid induces a transferable memory trace in the cerebrospinal fluid. Pain. 2019;160(12):2819–2828. doi:10.1097/j.pain.0000000000001688
32. Wall PD, Gutnick M. Ongoing activity in peripheral nerves: the Physiology and pharmacology of impulses originating from a neuroma. Exp Neurol. 1974;43(3):580–593. doi:10.1016/0014-4886(74)90197-6
33. Devor M. Ectopic discharge in Abeta afferents as a source of neuropathic pain. ExpBrain Res. 2009;196(1):115–128.
34. Roza C, Bernal L. Electrophysiological characterization of ectopic spontaneous discharge in axotomized and intact fibers upon nerve transection: a role in spontaneous pain? Pflugers Archiv. 2022;474(4):387–396. doi:10.1007/s00424-021-02655-7
35. Woolf CJ. Evidence for a central component of post-injury pain hypersensitivity. Nature. 1983;306(5944):686–688. doi:10.1038/306686a0
36. Basbaum AI. Distinct neurochemical features of acute and persistent pain. ProcNatlAcadSciUSA. 1999;96(14):7739–7743. doi:10.1073/pnas.96.14.7739
37. Treede RD, Rief W, Barke A, et al. Chronic pain as a symptom or a disease: the IASP classification of chronic pain for the International Classification of Diseases (ICD-11). Pain. 2019;160(1):19–27. doi:10.1097/j.pain.0000000000001384
38. Scholz J, Woolf CJ. Can we conquer pain? NatNeurosci. 2002;5:1062–1067.
39. Dib-Hajj SD, Waxman SG. Sodium channels in human pain disorders: genetics and pharmacogenomics. Annu Rev Neurosci. 2019;42:87–106. doi:10.1146/annurev-neuro-070918-050144
40. Goodwin G, McMahon SB. The physiological function of different voltage-gated sodium channels in pain. Nat Rev Neurosci. 2021;22(5):263–274. doi:10.1038/s41583-021-00444-w
41. Yang J, Xie MX, Hu L, et al. Upregulation of N-type calcium channels in the soma of uninjured dorsal root ganglion neurons contributes to neuropathic pain by increasing neuronal excitability following peripheral nerve injury. Brain Behav Immun. 2018;71:52–65. doi:10.1016/j.bbi.2018.04.016
42. Hoppanova L, Lacinova L. Voltage-dependent Ca(V)3.2 and Ca(V)2.2 channels in nociceptive pathways. Pflugers Archiv. 2022;474:421–434. doi:10.1007/s00424-022-02666-y
43. Smith PA. K(+) channels in primary afferents and their role in nerve injury-induced pain. Front Cell Neurosci. 2020;14:566418. doi:10.3389/fncel.2020.566418
44. Iannone LF, De Logu F, Geppetti P, De Cesaris F. The role of TRP ion channels in migraine and headache. Neurosci Lett. 2022;768:136380. doi:10.1016/j.neulet.2021.136380
45. Go EJ, Ji J, Kim YH, Berta T, Park CK. Transient receptor potential channels and botulinum neurotoxins in chronic pain. Front Mol Neurosci. 2021;14:772719. doi:10.3389/fnmol.2021.772719
46. Wemmie JA, Taugher RJ, Kreple CJ. Acid-sensing ion channels in pain and disease. Nat Rev Neurosci. 2013;14(7):461–471. doi:10.1038/nrn3529
47. Liu XG, Zhou LJ. Long-term potentiation at spinal C-fiber synapses: a target for pathological pain. Curr Pharm Des. 2015;21(7):895–905. doi:10.2174/1381612820666141027115949
48. Ji RR, Kohno T, Moore KA, Woolf CJ. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci. 2003;26(12):696–705. doi:10.1016/j.tins.2003.09.017
49. Eccleston C. Chronic pain and distraction: an experimental investigation into the role of sustained and shifting attention in the processing of chronic persistent pain. Behav Res Ther. 1995;33(4):391–405. doi:10.1016/0005-7967(94)00057-Q
50. Legrain V, Damme SV, Eccleston C, Davis KD, Seminowicz DA, Crombez G. A neurocognitive model of attention to pain: behavioral and neuroimaging evidence. Pain. 2009;144(3):230–232. doi:10.1016/j.pain.2009.03.020
51. Etherton JL, Bianchini KJ, Ciota MA, Heinly MT, Greve KW. Pain, malingering and the WAIS-III working memory index. Spine J. 2006;6(1):61–71. doi:10.1016/j.spinee.2005.05.382
52. Dick BD, Rashiq S. Disruption of attention and working memory traces in individuals with chronic pain. Anesthesia Analgesia. 2007;104(5):1223–1229. doi:10.1213/01.ane.0000263280.49786.f5
53. Blackburn-Munro G, Blackburn-Munro RE. Chronic pain, chronic stress and depression: coincidence or consequence? J Neuroendocrinol. 2001;13(12):1009–1023. doi:10.1046/j.0007-1331.2001.00727.x
54. Yalcin I, Barrot M. The anxiodepressive comorbidity in chronic pain. Curr Opin Anaesthesiol. 2014;27(5):520–527. doi:10.1097/ACO.0000000000000116
55. Fishbain DA, Cutler R, Rosomoff HL, Rosomoff RS. Chronic pain-associated depression: antecedent or consequence of chronic pain? A review. Clin J Pain. 1997;13(2):116–137. doi:10.1097/00002508-199706000-00006
56. Magni G, Moreschi C, Rigatti-Luchini S, Merskey H. Prospective study on the relationship between depressive symptoms and chronic musculoskeletal pain. Pain. 1994;56(3):289–297. doi:10.1016/0304-3959(94)90167-8
57. Kuner R. Central mechanisms of pathological pain. Nat Med. 2010;16(11):1258–1266. doi:10.1038/nm.2231
58. Schwartz M, Deczkowska A. Neurological disease as a failure of brain-immune crosstalk: the multiple faces of neuroinflammation. Trends Immunol. 2016;37(10):668–679. doi:10.1016/j.it.2016.08.001
59. Gui WS, Wei X, Mai CL, et al. Interleukin-1beta overproduction is a common cause for neuropathic pain, memory deficit, and depression following peripheral nerve injury in rodents. Mol Pain. 2016;12:174480691664678. doi:10.1177/1744806916646784
60. Guimarães MR, Soares AR, Cunha AM, et al. Evidence for lack of direct causality between pain and affective disturbances in a rat peripheral neuropathy model. Genes Brain Behav. 2019;18(6):e12542. doi:10.1111/gbb.12542
61. Holtmaat A, Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci. 2009;10(9):647–658. doi:10.1038/nrn2699
62. Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361(6407):31–39. doi:10.1038/361031a0
63. Neves G, Cooke SF, Bliss TVP. Synaptic plasticity, memory and the hippocampus: a neural network approach to causality. Nat Rev Neurosci. 2008;9(1):65–75. doi:10.1038/nrn2303
64. Cannon BW. The Wisdom of the Body. New York: Norton \& Co.; 1932.
65. Zhou LJ, Xin WJ, Pang RP, Liu XG. Cytokine microenvironment hypothesis of chronic pain. Chin J Pain Med. 2013;19(11):679–684.
66. Zhang JM, An J. Cytokines, inflammation, and pain. Int Anesthesiol Clin. 2007;45(2):27–37. doi:10.1097/AIA.0b013e318034194e
67. Dinarello CA. Historical insights into cytokines. Eur J Immunol. 2007;37(Suppl1):S34–45. doi:10.1002/eji.200737772
68. Baeuerle PA, Henkel T. Function and activation of NF-kappa B in the immune system. Annu Rev Immunol. 1994;12:141–179. doi:10.1146/annurev.iy.12.040194.001041
69. Kabata H, Artis D. Neuro-immune crosstalk and allergic inflammation. J Clin Invest. 2019;129(4):1475–1482. doi:10.1172/JCI124609
70. Wei XH, Zang Y, Wu CY, Xu JT, Xin WJ, Liu XG. Peri-sciatic administration of recombinant rat TNF-alpha induces mechanical allodynia via upregulation of TNF-alpha in dorsal root ganglia and in spinal dorsal horn: the role of NF-kappa B pathway. ExpNeurol. 2007;205(2):471–484.
71. Morris P, Ali K, Merritt M, Pelletier J, Macedo LG. A systematic review of the role of inflammatory biomarkers in acute, subacute and chronic non-specific low back pain. BMC Musculoskelet Disord. 2020;21(1):142. doi:10.1186/s12891-020-3154-3
72. Ernberg M, Christidis N, Ghafouri B, et al. Plasma cytokine levels in fibromyalgia and their response to 15 weeks of progressive resistance exercise or relaxation therapy. Mediators Inflamm. 2018;2018:3985154. doi:10.1155/2018/3985154
73. Ingegnoli F, Fantini F, Favalli EG, et al. Inflammatory and prothrombotic biomarkers in patients with rheumatoid arthritis: effects of tumor necrosis factor-alpha blockade. J Autoimmun. 2008;31(2):175–179. doi:10.1016/j.jaut.2008.07.002
74. Oliveira AB, Bachi ALL, Ribeiro RT, Mello MT, Tufik S, Peres MFP. Unbalanced plasma TNF-α and IL-12/IL-10 profile in women with migraine is associated with psychological and physiological outcomes. J Neuroimmunol. 2017;313:138–144. doi:10.1016/j.jneuroim.2017.09.008
75. Xu JT, Xin WJ, Zang Y, Wu CY, Liu XG. The role of tumor necrosis factor-alpha in the neuropathic pain induced by Lumbar 5 ventral root transection in rat. Pain. 2006;123(3):306–321. doi:10.1016/j.pain.2006.03.011
76. Watkins LR, Maier SF, Goehler LE. Immune activation: the role of pro-inflammatory cytokines in inflammation, illness responses and pathological pain states. Pain. 1995;63(3):289–302. doi:10.1016/0304-3959(95)00186-7
77. Chen SX, Liao GJ, Yao PW, et al. Calpain-2 regulates TNF-alpha expression associated with neuropathic pain following motor nerve injury. Neuroscience. 2018;376:142–151. doi:10.1016/j.neuroscience.2018.02.023
78. Nadeau S, Filali M, Zhang J, et al. Functional recovery after peripheral nerve injury is dependent on the pro-inflammatory cytokines IL-1β and TNF: implications for neuropathic pain. J Neurosci. 2011;31(35):12533–12542. doi:10.1523/JNEUROSCI.2840-11.2011
79. Zhang J, Su YM, Li D, et al. TNF-alpha-mediated JNK activation in the dorsal root ganglion neurons contributes to Bortezomib-induced peripheral neuropathy. Brain Behav Immun. 2014;38:185–191. doi:10.1016/j.bbi.2014.01.020
80. Yamakawa I, Kojima H, Terashima T, et al. Inactivation of TNF-α ameliorates diabetic neuropathy in mice. Am J Physiol Endocrinol Metab. 2011;301(5):E844–852. doi:10.1152/ajpendo.00029.2011
81. Chen X, Pang RP, Shen KF, et al. TNF-alpha enhances the currents of voltage gated sodium channels in uninjured dorsal root ganglion neurons following motor nerve injury. Exp Neurol. 2011;227:279–286. doi:10.1016/j.expneurol.2010.11.017
82. Zelenka M, Schafers M, Sommer C. Intraneural injection of interleukin-1beta and tumor necrosis factor-alpha into rat sciatic nerve at physiological doses induces signs of neuropathic pain. Pain. 2005;116(3):257–263. doi:10.1016/j.pain.2005.04.018
83. Wei XH, Yang T, Wu Q, et al. Peri-sciatic administration of recombinant rat IL-1beta induces mechanical allodynia by activation of src-family kinases in spinal microglia in rats. Exp Neurol. 2012;234(2):389–397. doi:10.1016/j.expneurol.2012.01.001
84. Muscat SM, Barrientos RM. The perfect cytokine storm: how peripheral immune challenges impact brain plasticity & memory function in aging. Brain plast. 2021;7(1):47–60. doi:10.3233/BPL-210127
85. Decourt B, Lahiri DK, Sabbagh MN. Targeting tumor necrosis factor alpha for Alzheimer’s disease. Curr Alzheimer Res. 2017;14(4):412–425. doi:10.2174/1567205013666160930110551
86. Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 2009;65(9):732–741. doi:10.1016/j.biopsych.2008.11.029
87. Jia X, Gao Z, Hu H. Microglia in depression: current perspectives. Sci China Life Sci. 2021;64(6):911–925. doi:10.1007/s11427-020-1815-6
88. Norman GJ, Karelina K, Zhang N, Walton JC, Morris JS, Devries AC. Stress and IL-1beta contribute to the development of depressive-like behavior following peripheral nerve injury. Mol Psychiatry. 2010;15(4):404–414. doi:10.1038/mp.2009.91
89. Bennett DL, Clark AJ, Huang J, Waxman SG, Dib-Hajj SD. The role of voltage-gated sodium channels in pain signaling. Physiol Rev. 2019;99(2):1079–1151. doi:10.1152/physrev.00052.2017
90. Cox JJ, Reimann F, Nicholas AK, et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444(7121):894–898. doi:10.1038/nature05413
91. Fertleman CR, Baker MD, Parker KA, et al. SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron. 2006;52(5):767–774. doi:10.1016/j.neuron.2006.10.006
92. Faber CG, Lauria G, Merkies IS, et al. Gain-of-function Nav1.8 mutations in painful neuropathy. Proc Natl Acad Sci U S A. 2012;109(47):19444–19449. doi:10.1073/pnas.1216080109
93. Han C, Vasylyev D, Macala LJ, et al. The G1662S NaV1.8 mutation in small fibre neuropathy: impaired inactivation underlying DRG neuron hyperexcitability. J Neurol Neurosurg Psychiatry. 2014;85(5):499–505. doi:10.1136/jnnp-2013-306095
94. Huang J, Han C, Estacion M, et al. Gain-of-function mutations in sodium channel Na(v)1.9 in painful neuropathy. Brain. 2014;137(Pt 6):1627–1642. doi:10.1093/brain/awu079
95. Zhang XY, Wen J, Yang W, et al. Gain-of-function mutations in SCN11A cause familial episodic pain. Am J Hum Genet. 2013;93(5):957–966. doi:10.1016/j.ajhg.2013.09.016
96. Black JA, Frezel N, Dib-Hajj SD, Waxman SG. Expression of Nav1.7 in DRG neurons extends from peripheral terminals in the skin to central preterminal branches and terminals in the dorsal horn. Mol Pain. 2012;8:82. doi:10.1186/1744-8069-8-82
97. Toledo-Aral JJ, Moss BL, He ZJ, et al. Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons. Proc Natl Acad Sci U S A. 1997;94(4):1527–1532. doi:10.1073/pnas.94.4.1527
98. Rush AM, Dib-Hajj SD, Liu S, Cummins TR, Black JA, Waxman SG. A single sodium channel mutation produces hyper- or hypoexcitability in different types of neurons. Proc Natl Acad Sci U S A. 2006;103(21):8245–8250. doi:10.1073/pnas.0602813103
99. Alles SRA, Nascimento F, Luján R, et al. Sensory neuron-derived Na(V)1.7 contributes to dorsal horn neuron excitability. Sci Adv. 2020;6(8):eaax4568. doi:10.1126/sciadv.aax4568
100. Minett MS, Nassar MA, Clark AK, et al. Distinct Nav1.7-dependent pain sensations require different sets of sensory and sympathetic neurons. Nat Commun. 2012;3:791. doi:10.1038/ncomms1795
101. Nassar MA, Stirling LC, Forlani G, et al. Nociceptor-specific gene deletion reveals a major role for Nav1.7 (PN1) in acute and inflammatory pain. Proc Natl Acad Sci U S A. 2004;101(34):12706–12711. doi:10.1073/pnas.0404915101
102. Shields SD, Cheng X, Uceyler N, Sommer C, Dib-Hajj SD, Waxman SG. Sodium channel Na(v)1.7 is essential for lowering heat pain threshold after burn injury. J Neurosci. 2012;32(32):10819–10832. doi:10.1523/JNEUROSCI.0304-12.2012
103. Minett MS, Falk S, Santana-Varela S, et al. Pain without nociceptors? Nav1.7-independent pain mechanisms. Cell Rep. 2014;6(2):301–312. doi:10.1016/j.celrep.2013.12.033
104. Li Y, North RY, Rhines LD, et al. DRG voltage-gated sodium Channel 1.7 is upregulated in paclitaxel-induced neuropathy in rats and in humans with neuropathic pain. J Neurosci. 2018;38(5):1124–1136. doi:10.1523/JNEUROSCI.0899-17.2017
105. Shields SD, Ahn HS, Yang Y, et al. Nav1.8 expression is not restricted to nociceptors in mouse peripheral nervous system. Pain. 2012;153(10):2017–2030. doi:10.1016/j.pain.2012.04.022
106. He XH, Zang Y, Chen X, et al. TNF-alpha contributes to up-regulation of Nav1.3 and Nav1.8 in DRG neurons following motor fiber injury. Pain. 2010;151:266–279. doi:10.1016/j.pain.2010.06.005
107. Akopian AN, Souslova V, England S, et al. The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat Neurosci. 1999;2(6):541–548. doi:10.1038/9195
108. Lai J, Gold MS, Kim CS, et al. Inhibition of neuropathic pain by decreased expression of the tetrodotoxin-resistant sodium channel, NaV1.8. Pain. 2002;95(1–2):143–152. doi:10.1016/S0304-3959(01)00391-8
109. Ekberg J, Jayamanne A, Vaughan CW, et al. muO-conotoxin MrVIB selectively blocks Nav1.8 sensory neuron specific sodium channels and chronic pain behavior without motor deficits. ProcNatlAcadSciUSA. 2006;103(45):17030–17035. doi:10.1073/pnas.0601819103
110. Jarvis MF, Honore P, Shieh CC, et al. A-803467, a potent and selective Nav1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc Nat Acad Sci. 2007;104(20):8520–8525. doi:10.1073/pnas.0611364104
111. Dib-Hajj SD, Black JA, Waxman SG. NaV1.9: a sodium channel linked to human pain. Nat Rev Neurosci. 2015;16(9):511–519. doi:10.1038/nrn3977
112. Lindia JA, Kohler MG, Martin WJ, Abbadie C. Relationship between sodium channel NaV1.3 expression and neuropathic pain behavior in rats. Pain. 2005;117(1–2):145–153. doi:10.1016/j.pain.2005.05.027
113. Samad OA, Tan AM, Cheng X, Foster E, Dib-Hajj SD, Waxman SG. Virus-mediated shRNA knockdown of Na(v)1.3 in rat dorsal root ganglion attenuates nerve injury-induced neuropathic pain. Mol ther. 2013;21(1):49–56. doi:10.1038/mt.2012.169
114. Tan AM, Samad OA, Dib-Hajj SD, Waxman SG. Virus-mediated knockdown of Nav1.3 in dorsal root ganglia of STZ-induced diabetic rats alleviates tactile allodynia. Mol Med. 2015;21(1):544–552. doi:10.2119/molmed.2015.00063
115. Hains BC, Saab CY, Klein JP, Craner MJ, Waxman SG. Altered sodium channel expression in second-order spinal sensory neurons contributes to pain after peripheral nerve injury. J Neurosci. 2004;24(20):4832–4839. doi:10.1523/JNEUROSCI.0300-04.2004
116. Cheng KI, Wang HC, Tseng KY, et al. Cilostazol ameliorates peripheral neuropathic pain in streptozotocin-induced type I diabetic rats. Front Pharmacol. 2021;12:771271. doi:10.3389/fphar.2021.771271
117. Israel MR, Tanaka BS, Castro J, et al. Na(V) 1.6 regulates excitability of mechanosensitive sensory neurons. J Physiol. 2019;597(14):3751–3768. doi:10.1113/JP278148
118. McDonnell A, Collins S, Ali Z, et al. Efficacy of the Nav1.7 blocker PF-05089771 in a randomised, placebo-controlled, double-blind clinical study in subjects with painful diabetic peripheral neuropathy. Pain. 2018;159(8):1465–1476. doi:10.1097/j.pain.0000000000001227
119. Zakrzewska JM, Palmer J, Morisset V, et al. Safety and efficacy of a Nav1.7 selective sodium channel blocker in patients with trigeminal neuralgia: a double-blind, placebo-controlled, randomised withdrawal phase 2a trial. Lancet Neurol. 2017;16(4):291–300. doi:10.1016/S1474-4422(17)30005-4
120. Siebenga P, van Amerongen G, Hay JL, et al. Lack of detection of the analgesic properties of PF-05089771, a Selective Na(v) 1.7 inhibitor, using a battery of pain models in healthy subjects. Clin Transl Sci. 2020;13(2):318–324. doi:10.1111/cts.12712
121. Wang W, Gu J, Li YQ, Tao YX. Are voltage-gated sodium channels on the dorsal root ganglion involved in the development of neuropathic pain? Mol Pain. 2011;7:16. doi:10.1186/1744-8069-7-16
122. Priest BT, Kaczorowski GJ. Blocking sodium channels to treat neuropathic pain. Expert Opin Ther Targets. 2007;11(3):291–306. doi:10.1517/14728222.11.3.291
123. Wall PD, Waxman S, Basbaum AI. Ongoing activity in peripheral nerve: injury discharge. Exp Neurol. 1974;45(3):576–589. doi:10.1016/0014-4886(74)90163-0
124. Blumberg H, Janig W. Discharge pattern of afferent fibers from a neuroma. Pain. 1984;20(4):335–353. doi:10.1016/0304-3959(84)90111-8
125. Govrin-Lippmann R, Devor M. Ongoing activity in severed nerves: source and variation with time. Brain Res. 1978;159(2):406–410. doi:10.1016/0006-8993(78)90548-6
126. Li Y, Dorsi MJ, Meyer RA, Belzberg AJ. Mechanical hyperalgesia after an L5 spinal nerve lesion in the rat is not dependent on input from injured nerve fibers. Pain. 2000;85(3):493–502. doi:10.1016/S0304-3959(00)00250-5
127. Michaelis M, Liu XG, Janig W. Axotomized and intact muscle afferents but no skin afferents develop ongoing discharges of dorsal root ganglion origin after peripheral nerve lesion. J Neurosci. 2000;20(7):2742–2748. doi:10.1523/JNEUROSCI.20-07-02742.2000
128. Wu G, Ringkamp M, Hartke TV, et al. Early onset of spontaneous activity in uninjured C-fiber nociceptors after injury to neighboring nerve fibers. J Neurosci. 2001;21(8):RC140. doi:10.1523/JNEUROSCI.21-08-j0002.2001
129. Xu JT, Xin WJ, Wei XH, et al. p38 activation in uninjured primary afferent neurons and in spinal microglia contributes to the development of neuropathic pain induced by selective motor fiber injury. Exp Neurol. 2007;204(1):355–365. doi:10.1016/j.expneurol.2006.11.016
130. Obata K, Yamanaka H, Kobayashi K, et al. The effect of site and type of nerve injury on the expression of brain-derived neurotrophic factor in the dorsal root ganglion and on neuropathic pain behavior. Neuroscience. 2006;137(3):961–970. doi:10.1016/j.neuroscience.2005.10.015
131. Black JA, Cummins TR, Plumpton C, et al. Upregulation of a silent sodium channel after peripheral, but not central, nerve injury in DRG neurons. JNeurophysiol. 1999;82(5):2776–2785. doi:10.1152/jn.1999.82.5.2776
132. Sheen K, Chung JM. Signs of neuropathic pain depend on signals from injured nerve fibers in a rat model. Brain Res. 1993;610(1):62–68. doi:10.1016/0006-8993(93)91217-G
133. Luo ZD, Chaplan SR, Higuera ES, et al. Upregulation of dorsal root ganglion (alpha)2(delta) calcium channel subunit and its correlation with allodynia in spinal nerve-injured rats. J Neurosci. 2001;21(6):1868–1875. doi:10.1523/JNEUROSCI.21-06-01868.2001
134. Liu XG, Pang RP, Zhou LJ, Wei XH, Zang Y. Neuropathic pain: sensory nerve injury or motor nerve injury? Adv Exp Med Biol. 2016;904:59–75.
135. Tamura R, Nemoto T, Maruta T, et al. Up-regulation of NaV1.7 sodium channels expression by tumor necrosis factor-alpha in cultured bovine adrenal chromaffin cells and rat dorsal root ganglion neurons. Anesth Analg. 2014;118(2):318–324. doi:10.1213/ANE.0000000000000085
136. Noh MC, Stemkowski PL, Smith PA. Long-term actions of interleukin-1β on K(+), Na(+) and Ca(2+) channel currents in small, IB(4)-positive dorsal root ganglion neurons; possible relevance to the etiology of neuropathic pain. J Neuroimmunol. 2019;332:198–211. doi:10.1016/j.jneuroim.2019.05.002
137. Binshtok AM, Wang H, Zimmermann K, et al. Nociceptors are interleukin-1beta sensors. J Neurosci. 2008;28(52):14062–14073. doi:10.1523/JNEUROSCI.3795-08.2008
138. Goldstein RH, Barkai O, Íñigo-portugués A, Katz B, Lev S, Binshtok AM. Location and plasticity of the sodium spike initiation zone in nociceptive terminals in vivo. Neuron. 2019;102(4):801–812.e805. doi:10.1016/j.neuron.2019.03.005
139. Liu L, Yang TM, Liedtke W, Simon SA. Chronic IL-1beta signaling potentiates voltage-dependent sodium currents in trigeminal nociceptive neurons. J Neurophysiol. 2006;95(3):1478–1490. doi:10.1152/jn.00509.2005
140. Milligan ED, Penzkover KR, Soderquist RG, Mahoney MJ. Spinal interleukin-10 therapy to treat peripheral neuropathic pain. Neuromodulation. 2012;15(6):
141. Shen KF, Zhu HQ, Wei XH, et al. Interleukin-10 down-regulates voltage gated sodium channels in rat dorsal root ganglion neurons. Exp Neurol. 2013;247:466–475. doi:10.1016/j.expneurol.2013.01.018
142. Huang Y, Zhu L, Zhang W, Tang Q, Zhong Y. IL-10 alleviates radicular pain by inhibiting TNF-α/p65 dependent Nav1.7 up-regulation in DRG neurons of rats. Brain Res. 2022;1791:147997. doi:10.1016/j.brainres.2022.147997
143. Meffert MK, Baltimore D. Physiological functions for brain NF-kappaB. Trends Neurosci. 2005;28(1):37–43. doi:10.1016/j.tins.2004.11.002
144. Taniguchi K, Karin M. NF-kappaB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol. 2018;18(5):309–324. doi:10.1038/nri.2017.142
145. Srinivasan M, Lahiri DK. Significance of NF-kappaB as a pivotal therapeutic target in the neurodegenerative pathologies of Alzheimer’s disease and multiple sclerosis. Expert Opin Ther Targets. 2015;19(4):471–487. doi:10.1517/14728222.2014.989834
146. Niederberger E, Geisslinger G. The IKK-NF-kappaB pathway: a source for novel molecular drug targets in pain therapy? FASEB J. 2008;22(10):3432–3442. doi:10.1096/fj.08-109355
147. Zang Y, He XH, Xin WJ, et al. Inhibition of NF-kappaB prevents mechanical allodynia induced by spinal ventral root transection and suppresses the re-expression of Nav1.3 in DRG neurons in vivo and in vitro. Brain Res. 2010;1363:151–158. doi:10.1016/j.brainres.2010.09.048
148. Huang Y, Zang Y, Zhou L, Gui W, Liu X, Zhong Y. The role of TNF-alpha/NF-kappa B pathway on the up-regulation of voltage-gated sodium channel Nav1.7 in DRG neurons of rats with diabetic neuropathy. Neurochem Int. 2014;75:112–119. doi:10.1016/j.neuint.2014.05.012
149. Zhang JM, Li H, Liu B, Brull SJ. Acute topical application of tumor necrosis factor alpha evokes protein kinase A-dependent responses in rat sensory neurons. JNeurophysiol. 2002;88(3):1387–1392. doi:10.1152/jn.2002.88.3.1387
150. Xie MX, Zhang XL, Xu J, et al. Nuclear Factor-kappaB gates Nav1.7 channels in DRG neurons via protein-protein interaction. iScience. 2019;19:623–633. doi:10.1016/j.isci.2019.08.017
151. Zhang XL, Lai RC, Chen ZH, et al. Foxo1 selectively regulates static mechanical pain by interacting with Nav1.7. Pain. 2021;162(2):490–502. doi:10.1097/j.pain.0000000000002055
152. Shi C, Shi R, Guo H. Tumor necrosis factor α reduces gonadotropin-releasing hormone release through increase of forkhead box protein O1 activity. Neuroreport. 2020;31(6):473–477. doi:10.1097/WNR.0000000000001424
153. Dolphin AC. Calcium channel auxiliary alpha2delta and beta subunits: trafficking and one step beyond. Nat Rev Neurosci. 2012;13(8):542–555. doi:10.1038/nrn3311
154. Cai S, Gomez K, Moutal A, Khanna R. Targeting T-type/CaV3.2 channels for chronic pain. Transl Res. 2021;234:20–30. doi:10.1016/j.trsl.2021.01.002
155. Liu QY, Chen W, Cui S, et al. Upregulation of Ca(v)3.2 T-type calcium channels in adjacent intact L4 dorsal root ganglion neurons in neuropathic pain rats with L5 spinal nerve ligation. Neurosci Res. 2019;142:30–37. doi:10.1016/j.neures.2018.04.002
156. Dubel SJ, Starr TV, Hell J, et al. Molecular cloning of the alpha-1 subunit of an omega-conotoxin-sensitive calcium channel. Proc Natl Acad Sci U S A. 1992;89(11):5058–5062. doi:10.1073/pnas.89.11.5058
157. Field MJ, Cox PJ, Stott E, et al. Identification of the alpha2-delta-1 subunit of voltage-dependent calcium channels as a molecular target for pain mediating the analgesic actions of pregabalin. Proc Natl Acad Sci U S A. 2006;103(46):17537–17542. doi:10.1073/pnas.0409066103
158. Skov MJ, Beck JC, de Kater AW, Shopp GM. Nonclinical safety of ziconotide: an intrathecal analgesic of a new pharmaceutical class. Int J Toxicol. 2007;26(5):411–421. doi:10.1080/10915810701582970
159. Snutch TP. Targeting chronic and neuropathic pain: the N-type calcium channel comes of age. NeuroRx. 2005;2(4):662–670. doi:10.1602/neurorx.2.4.662
160. Liu Q, Chen W, Fan X, et al. Upregulation of interleukin-6 on Ca(v)3.2 T-type calcium channels in dorsal root ganglion neurons contributes to neuropathic pain in rats with spinal nerve ligation. Exp Neurol. 2019;317:226–243. doi:10.1016/j.expneurol.2019.03.005
161. Brown DA, Passmore GM. Neural KCNQ (Kv7) channels. Br J Pharmacol. 2009;156(8):1185–1195. doi:10.1111/j.1476-5381.2009.00111.x
162. Liu Y, Bian X, Wang K. Pharmacological activation of neuronal voltage-gated Kv7/KCNQ/M-channels for potential therapy of epilepsy and pain. Handb Exp Pharmacol. 2021;267:231–251.
163. Ibeakanma C, Vanner S. TNFalpha is a key mediator of the pronociceptive effects of mucosal supernatant from human ulcerative colitis on colonic DRG neurons. Gut. 2010;59(5):612–621. doi:10.1136/gut.2009.190439
164. Stemkowski PL, Noh MC, Chen Y, Smith PA. Increased excitability of medium-sized dorsal root ganglion neurons by prolonged interleukin-1β exposure is K(+) channel dependent and reversible. J Physiol. 2015;593(16):3739–3755. doi:10.1113/JP270905
165. Stemkowski PL, Bukhanova-Schulz N, Baldwin T, de Chaves EP, Smith PA. Are sensory neurons exquisitely sensitive to interleukin 1β? J Neuroimmunol. 2021;354:577529. doi:10.1016/j.jneuroim.2021.577529
166. Moran MM, Szallasi A. Targeting nociceptive transient receptor potential channels to treat chronic pain: current state of the field. Br J Pharmacol. 2018;175(12):2185–2203. doi:10.1111/bph.14044
167. Rosenbaum T, Simon SA. Frontiers in neuroscience TRPV1 receptors and signal transduction. In: Liedtke WB, Heller S, editors. TRP Ion Channel Function in Sensory Transduction and Cellular Signaling Cascades. Boca Raton (FL): CRC Press/Taylor & Francis Copyright © 2007, Taylor & Francis Group, LLC.; 2007.
168. Hu F, Song X, Long D. Transient receptor potential ankyrin 1 and calcium: interactions and association with disease (Review). Exp Ther Med. 2021;22(6):1462. doi:10.3892/etm.2021.10897
169. Ji G, Zhou S, Carlton SM. Intact Adelta-fibers up-regulate transient receptor potential A1 and contribute to cold hypersensitivity in neuropathic rats. Neuroscience. 2008;154(3):1054–1066. doi:10.1016/j.neuroscience.2008.04.039
170. Wang Z, Ling D, Wu C, Han J, Zhao Y. Baicalin prevents the up-regulation of TRPV1 in dorsal root ganglion and attenuates chronic neuropathic pain. Veter med sci. 2020;6(4):1034–1040. doi:10.1002/vms3.318
171. Hara T, Chiba T, Abe K, et al. Effect of paclitaxel on transient receptor potential vanilloid 1 in rat dorsal root ganglion. Pain. 2013;154(6):882–889. doi:10.1016/j.pain.2013.02.023
172. Ba X, Wang J, Zhou S, et al. Cinobufacini protects against paclitaxel-induced peripheral neuropathic pain and suppresses TRPV1 up-regulation and spinal astrocyte activation in rats. Biomed Pharmacother. 2018;108:76–84. doi:10.1016/j.biopha.2018.09.018
173. Iftinca M, Defaye M, Altier C. TRPV1-targeted drugs in development for human pain conditions. Drugs. 2021;81(1):7–27. doi:10.1007/s40265-020-01429-2
174. Wang Y, Feng C, He H, et al. Sensitization of TRPV1 receptors by TNF-α orchestrates the development of vincristine-induced pain. Oncol Lett. 2018;15(4):5013–5019. doi:10.3892/ol.2018.7986
175. Leo M, Schulte M, Schmitt LI, Schäfers M, Kleinschnitz C, Hagenacker T. Intrathecal resiniferatoxin modulates TRPV1 in DRG neurons and reduces TNF-induced pain-related behavior. Mediators Inflamm. 2017;2017:2786427. doi:10.1155/2017/2786427
176. Malek N, Pajak A, Kolosowska N, Kucharczyk M, Starowicz K. The importance of TRPV1-sensitisation factors for the development of neuropathic pain. Mol Cell Neurosci. 2015;65:1–10. doi:10.1016/j.mcn.2015.02.001
177. Hensellek S, Brell P, Schaible HG, Bräuer R, Segond von Banchet G. Segond von Banchet G: the cytokine TNFalpha increases the proportion of DRG neurones expressing the TRPV1 receptor via the TNFR1 receptor and ERK activation. Mol Cell Neurosci. 2007;36(3):381–391. doi:10.1016/j.mcn.2007.07.010
178. Nugent M, Yusef YR, Meng J, Wang J, Dolly JO. A SNAP-25 cleaving chimera of botulinum neurotoxin /A and /E prevents TNFα-induced elevation of the activities of native TRP channels on early postnatal rat dorsal root ganglion neurons. Neuropharmacology. 2018;138:257–266. doi:10.1016/j.neuropharm.2018.06.016
179. Zhao D, Han DF, Wang SS, Lv B, Wang X, Ma C. Roles of tumor necrosis factor-α and interleukin-6 in regulating bone cancer pain via TRPA1 signal pathway and beneficial effects of inhibition of neuro-inflammation and TRPA1. Mol Pain. 2019;15:1744806919857981. doi:10.1177/1744806919857981
180. El Karim I, McCrudden MT, Linden GJ, et al. TNF-α-induced p38MAPK activation regulates TRPA1 and TRPV4 activity in odontoblast-like cells. Am J Pathol. 2015;185(11):2994–3002. doi:10.1016/j.ajpath.2015.07.020
181. Wu Z, Wang S, Wu I, Mata M, Fink DJ. Activation of TLR-4 to produce tumour necrosis factor-α in neuropathic pain caused by paclitaxel. Eur J Pain. 2015;19(7):889–898. doi:10.1002/ejp.613
182. Ebbinghaus M, Uhlig B, Richter F, et al. The role of interleukin-1β in arthritic pain: main involvement in thermal, but not mechanical, hyperalgesia in rat antigen-induced arthritis. Arthritis Rheum. 2012;64(12):3897–3907. doi:10.1002/art.34675
183. Araldi D, Ferrari LF, Lotufo CM, et al. Peripheral inflammatory hyperalgesia depends on the COX increase in the dorsal root ganglion. Proc Natl Acad Sci U S A. 2013;110(9):3603–3608. doi:10.1073/pnas.1220668110
184. Zhang J, Yi QT, Gong M, Zhang YQ, Liu D, Zhu RJ. Upregulation of TRPV1 in spinal dorsal root ganglion by activating NGF-TrkA pathway contributes to pelvic organ cross-sensitisation in rats with experimental autoimmune prostatitis. Andrologia. 2019;51(8):e13302. doi:10.1111/and.13302
185. Amirkhanloo F, Karimi G, Yousefi-Manesh H, Abdollahi A, Roohbakhsh A, Dehpour AR. The protective effect of modafinil on vincristine-induced peripheral neuropathy in rats: a possible role for TRPA1 receptors. Basic Clin Pharmacol Toxicol. 2020;127(5):405–418. doi:10.1111/bcpt.13454
186. Waldmann R, Champigny G, Bassilana F, Heurteaux C, Lazdunski M. A proton-gated cation channel involved in acid-sensing. Nature. 1997;386(6621):173–177. doi:10.1038/386173a0
187. Papalampropoulou-Tsiridou M, Labrecque S, Godin AG, De Koninck Y, Wang F. Differential expression of acid - sensing ion channels in mouse primary afferents in naïve and injured conditions. Front Cell Neurosci. 2020;14:103. doi:10.3389/fncel.2020.00103
188. Diochot S, Alloui A, Rodrigues P, et al. Analgesic effects of mambalgin peptide inhibitors of acid-sensing ion channels in inflammatory and neuropathic pain. Pain. 2016;157(3):552–559. doi:10.1097/j.pain.0000000000000397
189. Deval E, Gasull X, Noël J, et al. Acid-sensing ion channels (ASICs): pharmacology and implication in pain. Pharmacol Ther. 2010;128(3):549–558. doi:10.1016/j.pharmthera.2010.08.006
190. Kung CC, Huang YC, Hung TY, Teng CY, Lee TY, Sun WH. Deletion of acid-sensing ion channel 3 relieves the late phase of neuropathic pain by preventing neuron degeneration and promoting neuron repair. Cells. 2020;9(11):2355. doi:10.3390/cells9112355
191. Wei S, Qiu CY, Jin Y, Liu TT, Hu WP. TNF-α acutely enhances acid-sensing ion channel currents in rat dorsal root ganglion neurons via a p38 MAPK pathway. J Neuroinflammation. 2021;18(1):92. doi:10.1186/s12974-021-02151-w
192. Verkest C, Diochot S, Lingueglia E, Baron A. C-Jun N-terminal kinase post-translational regulation of pain-related acid-sensing ion channels 1b and 3. J Neurosci. 2021;41(42):8673–8685. doi:10.1523/JNEUROSCI.0570-21.2021
193. Gong W, Kolker SJ, Usachev Y, et al. Acid-sensing ion channel 3 decreases phosphorylation of extracellular signal-regulated kinases and induces synoviocyte cell death by increasing intracellular calcium. Arthritis Res Ther. 2014;16(3):R121. doi:10.1186/ar4577
194. Ross JL, Queme LF, Cohen ER, et al. Muscle IL1β drives ischemic myalgia via ASIC3-mediated sensory neuron sensitization. J Neurosci. 2016;36(26):6857–6871. doi:10.1523/JNEUROSCI.4582-15.2016
195. Ross JL, Queme LF, Lamb JE, Green KJ, Ford ZK, Jankowski MP. Interleukin 1β inhibition contributes to the antinociceptive effects of voluntary exercise on ischemia/reperfusion-induced hypersensitivity. Pain. 2018;159(2):380–392. doi:10.1097/j.pain.0000000000001094
196. Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol. 1973;232(2):331–356. doi:10.1113/jphysiol.1973.sp010273
197. Liu XG, Sandkuhler J. Long-term potentiation of C-fiber-evoked potentials in the rat spinal dorsal horn is prevented by spinal N-methyl-D-aspartic acid receptor blockage. Neurosci Lett. 1995;191(1–2):43–46. doi:10.1016/0304-3940(95)11553-0
198. Gobel S, Falls WM. Anatomical observations of horseradish peroxidase-filled terminal primary axonal arborizations in layer II of the substantia gelatinosa of Rolando. Brain Res. 1979;175(2):335–340. doi:10.1016/0006-8993(79)91012-6
199. Light AR, Trevino DL, Perl ER. Morphological features of functionally defined neurons in the marginal zone and substantia gelatinosa of the spinal dorsal horn. J Comp Neurol. 1979;186(2):151–171. doi:10.1002/cne.901860204
200. Liu XG, Sandkuhler J. Characterization of long-term potentiation of C-fiber-evoked potentials in spinal dorsal horn of adult rat: essential role of NK1 and NK2 receptors. J Neurophysiol. 1997;78(4):1973–1982. doi:10.1152/jn.1997.78.4.1973
201. Ikeda H, Stark J, Fischer H, et al. Synaptic amplifier of inflammatory pain in the spinal dorsal horn. Science. 2006;312(5780):1659–1662. doi:10.1126/science.1127233
202. Sandkuhler J, Liu XG. Induction of long-term potentiation at spinal synapses by noxious stimulation or nerve injury. Eur J Neurosci. 1998;10(7):2476–2480. doi:10.1046/j.1460-9568.1998.00278.x
203. Zhang HM, Zhou LJ, Hu XD, Hu NW, Zhang T, Liu XG. Acute nerve injury induces long-term potentiation of C-fiber evoked field potentials in spinal dorsal horn of intact rat. Sheng Li XueBao. 2004;56(5):591–596.
204. Zhou LJ, Ren WJ, Zhong Y, et al. Limited BDNF contributes to the failure of injury to skin afferents to produce a neuropathic pain condition. Pain. 2010;148(1):148–157. doi:10.1016/j.pain.2009.10.032
205. Drdla R, Gassner M, Gingl E, Sandkuhler J. Induction of synaptic long-term potentiation after opioid withdrawal. Science. 2009;325(5937):207–210. doi:10.1126/science.1171759
206. Klein T, Magerl W, Hopf HC, Sandkuhler J, Treede RD. Perceptual correlates of nociceptive long-term potentiation and long-term depression in humans. J Neurosci. 2004;24(4):964–971. doi:10.1523/JNEUROSCI.1222-03.2004
207. Ohnami S, Tanabe M, Shinohara S, Takasu K, Kato A, Ono H. Role of voltage-dependent calcium channel subtypes in spinal long-term potentiation of C-fiber-evoked field potentials. Pain. 2011;152(3):623–631. doi:10.1016/j.pain.2010.12.004
208. Tanabe M, Murakami H, Honda M, Ono H. Gabapentin depresses C-fiber-evoked field potentials in rat spinal dorsal horn only after induction of long-term potentiation. ExpNeurol. 2006;202(2):280–286.
209. Ge YX, Xin WJ, Hu NW, Zhang T, Xu JT, Liu XG. Clonidine depresses LTP of C-fiber evoked field potentials in spinal dorsal horn via NO-cGMP pathway. Brain Res. 2006;1118:58–65. doi:10.1016/j.brainres.2006.08.009
210. Zhang HM, Zhou LJ, Hu NW, Zhang T, Liu XG. NMDA receptor channels are involved in the expression of long-term potentiation of C-fiber evoked field potentials in rat spinal dorsal horn. Prog Biochem Biophy. 2006;33(12):1183–1189.
211. Hu XD, Ge YX, Hu NW, et al. Diazepam inhibits the induction and maintenance of LTP of C-fiber evoked field potentials in spinal dorsal horn of rats. Neuropharmacology. 2006;50(2):238–244. doi:10.1016/j.neuropharm.2005.09.010
212. Heinricher MM, Tavares I, Leith JL, Lumb BM. Descending control of nociception: specificity, recruitment and plasticity. Brain ResRev. 2009;60(1):214–225. doi:10.1016/j.brainresrev.2008.12.009
213. Liu XG, Morton CR, Azkue JJ, Zimmermann M, Sandkuhler J. Long-term depression of C-fibre-evoked spinal field potentials by stimulation of primary afferent A delta-fibres in the adult rat. EurJNeurosci. 1998;10(10):3069–3075.
214. Wood PB. Role of central dopamine in pain and analgesia. ExpertRevNeurother. 2008;8(5):781–797.
215. Pertovaara A. Noradrenergic pain modulation. ProgNeurobiol. 2006;80(2):53–83.
216. Sindrup SH, Otto M, Finnerup NB, Jensen TS. Antidepressants in the treatment of neuropathic pain. Basic ClinPharmacolToxicol. 2005;96(6):399–409.
217. Shelton RC. Serotonin and norepinephrine reuptake inhibitors. Handb Exp Pharmacol. 2019;250:145–180.
218. Yang HW, Zhou LJ, Hu NW, Xin WJ, Liu XG. Activation of spinal D1/D5 receptors induces late-phase LTP of C-fiber-evoked field potentials in rat spinal dorsal horn. J Neurophysiol. 2005;94(2):961–967. doi:10.1152/jn.01324.2004
219. Zhu H, Clemens S, Sawchuk M, Hochman S. Expression and distribution of all dopamine receptor subtypes (D(1)-D(5)) in the mouse lumbar spinal cord: a real-time polymerase chain reaction and non-autoradiographic in situ hybridization study. Neuroscience. 2007;149(4):885–897. doi:10.1016/j.neuroscience.2007.07.052
220. Kim JY, Tillu DV, Quinn TL, et al. Spinal dopaminergic projections control the transition to pathological pain plasticity via a D1/D5-mediated mechanism. J Neurosci. 2015;35(16):6307–6317. doi:10.1523/JNEUROSCI.3481-14.2015
221. Megat S, Shiers S, Moy JK, et al. A critical role for dopamine d5 receptors in pain chronicity in male mice. J Neurosci. 2018;38(2):379–397. doi:10.1523/JNEUROSCI.2110-17.2017
222. Pickering M, Cumiskey D, O’Connor JJ. Actions of TNF-alpha on glutamatergic synaptic transmission in the central nervous system. ExpPhysiol. 2005;90(5):663–670.
223. Katsuki H, Nakai S, Hirai Y, Akaji K, Kiso Y, Satoh M. Interleukin-1 beta inhibits long-term potentiation in the CA3 region of mouse hippocampal slices. Eur J Pharmacol. 1990;181(3):323–326. doi:10.1016/0014-2999(90)90099-R
224. Pickering M, O’Connor JJ. Pro-inflammatory cytokines and their effects in the dentate gyrus. In: Helen ES, editor. Progress in Brain Research the Dentate Gyrus: A Comprehensive Guide to Structure, Function, and Clinical Implications. Vol. 163. Elsevier; 2007:339–354.
225. Butler MP, O’Connor JJ, Moynagh PN. Dissection of tumor-necrosis factor-alpha inhibition of long-term potentiation (LTP) reveals a p38 mitogen-activated protein kinase-dependent mechanism which maps to early-but not late-phase LTP. Neuroscience. 2004;124(2):319–326. doi:10.1016/j.neuroscience.2003.11.040
226. Wang Q, Walsh DM, Rowan MJ, Selkoe DJ, Anwyl R. Block of long-term potentiation by naturally secreted and synthetic amyloid beta-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J Neurosci. 2004;24(13):3370–3378. doi:10.1523/JNEUROSCI.1633-03.2004
227. Zhong Y, Zhou LJ, Ren WJ, et al. Interleukin-1beta induces long-term potentiation of C-fiber evoked field potentials in spinal dorsal horn in rats with neuropathic pain. Open Pain J. 2009;2:18–23. doi:10.2174/1876386300902010018
228. Liu YL, Zhou LJ, Hu NW, et al. Tumor necrosis factor-alpha induces long-term potentiation of C-fiber evoked field potentials in spinal dorsal horn in rats with nerve injury: the role of NF-kappa B, JNK and p38 MAPK. Neuropharmacology. 2007;52(3):708–715. doi:10.1016/j.neuropharm.2006.09.011
229. Zhong Y, Zhou LJ, Ren WJ, et al. The direction of synaptic plasticity mediated by C-fibers in spinal dorsal horn is decided by Src-family kinases in microglia: the role of tumor necrosis factor-alpha. Brain BehavImmun. 2010;24:874–880.
230. Cowley TR, O’Sullivan J, Blau C, et al. Rosiglitazone attenuates the age-related changes in astrocytosis and the deficit in LTP. Neurobiol Aging. 2012;33(1):162–175. doi:10.1016/j.neurobiolaging.2010.02.002
231. Griffin R, Nally R, Nolan Y, McCartney Y, Linden J, Lynch MA. The age-related attenuation in long-term potentiation is associated with microglial activation. J Neurochem. 2006;99(4):1263–1272. doi:10.1111/j.1471-4159.2006.04165.x
232. Hayashi Y, Yoshida M, Yamato M, et al. Reverse of age-dependent memory impairment and mitochondrial DNA damage in microglia by an overexpression of human mitochondrial transcription factor a in mice. J Neurosci. 2008;28(34):8624–8634. doi:10.1523/JNEUROSCI.1957-08.2008
233. VanItallie TB. Alzheimer’s disease: innate immunity gone awry? Metabolism. 2017;69s:S41–s49. doi:10.1016/j.metabol.2017.01.014
234. Katsura H, Obata K, Mizushima T, et al. Activation of Src-family kinases in spinal microglia contributes to mechanical hypersensitivity after nerve injury. J Neurosci. 2006;26(34):8680–8690. doi:10.1523/JNEUROSCI.1771-06.2006
235. Gong QJ, Li YY, Xin WJ, et al. ATP induces long-term potentiation of C-fiber-evoked field potentials in spinal dorsal horn: the roles of P2X(4) receptors and p38 MAPK in microglia. Glia. 2009;57:583–591. doi:10.1002/glia.20786
236. Zhou LJ, Yang T, Wei XA, et al. Brain-derived neurotrophic factor contributes to spinal long-term potentiation and mechanical hypersensitivity by activation of spinal microglia in rat. Brain Behav Immun. 2011;25(2):322–334. doi:10.1016/j.bbi.2010.09.025
237. Kronschlager MT, Drdla-Schutting R, Gassner M, Honsek SD, Teuchmann HL, Sandkuhler J. Gliogenic LTP spreads widely in nociceptive pathways. Science. 2016;354(6316):1144–1148. doi:10.1126/science.aah5715
238. Zhou LJ, Liu XG. Glial activation, A common mechanism underlying spinal synaptic plasticity? Neurosci Bull. 2017;33(1):121–123. doi:10.1007/s12264-016-0091-0
239. Zhou LJ, Zhong Y, Ren WJ, Li YY, Zhang T, Liu XG. BDNF induces late-phase LTP of C-fiber evoked field potentials in rat spinal dorsal horn. ExpNeurol. 2008;212(2):507–514.
240. Yang HW, Hu XD, Zhang HM, et al. The roles of CaMKII, PKA and PKC in the induction and maintenance of LTP of C-fiber evoked field potentials in rat spinal dorsal horn. JNeurophysiol. 2004;91(3):1122–1133. doi:10.1152/jn.00735.2003
241. Xin WJ, Gong QJ, Xu JT, et al. Role of phosphorylation of ERK in induction and maintenance of LTP of the C-fiber evoked field potentials in spinal dorsal horn. J NeurosciRes. 2006;84(5):934–943.
242. Sorra KE, Harris KM. Overview on the structure, composition, function, development, and plasticity of hippocampal dendritic spines. Hippocampus. 2000;10(5):501–511. doi:10.1002/1098-1063(2000)10:5<501::AID-HIPO1>3.0.CO;2-T
243. Zhang HM, Li Y, Yang Q, Liu XG, Dougherty PM. Morphological and physiological plasticity of spinal lamina II GABA neurons is induced by sciatic nerve chronic constriction injury in mice. Front Cell Neurosci. 2018;12:143. doi:10.3389/fncel.2018.00143
244. Tyrtyshnaia A, Bondar A, Konovalova S, Manzhulo I. Synaptamide improves cognitive functions and neuronal plasticity in neuropathic pain. Int J Mol Sci. 2021;22(23):12779. doi:10.3390/ijms222312779
245. Cardoso-Cruz H, Dourado M, Monteiro C, Matos MR, Galhardo V. Activation of dopaminergic D2/D3 receptors modulates dorsoventral connectivity in the hippocampus and reverses the impairment of working memory after nerve injury. J Neurosci. 2014;34(17):5861–5873. doi:10.1523/JNEUROSCI.0021-14.2014
246. Fonseca-Rodrigues D, Amorim D, Almeida A, Pinto-Ribeiro F. Emotional and cognitive impairments in the peripheral nerve chronic constriction injury model (CCI) of neuropathic pain: a systematic review. Behav Brain Res. 2021;399:113008. doi:10.1016/j.bbr.2020.113008
247. Xiong B, Zhang W, Zhang L, et al. Hippocampal glutamatergic synapses impairment mediated novel-object recognition dysfunction in rats with neuropathic pain. Pain. 2020;161(8):1824–1836. doi:10.1097/j.pain.0000000000001878
248. Mutso AA, Radzicki D, Baliki MN, et al. Abnormalities in hippocampal functioning with persistent pain. J Neurosci. 2012;32(17):5747–5756. doi:10.1523/JNEUROSCI.0587-12.2012
249. Cardoso-Cruz H, Lima D, Galhardo V. Impaired spatial memory performance in a rat model of neuropathic pain is associated with reduced hippocampus-prefrontal cortex connectivity. J Neurosci. 2013;33(6):2465–2480. doi:10.1523/JNEUROSCI.5197-12.2013
250. Messaoudi E, Ying SW, Kanhema T, Croll SD, Bramham CR. Brain-derived neurotrophic factor triggers transcription-dependent, late phase long-term potentiation in vivo. J Neurosci. 2002;22(17):7453–7461. doi:10.1523/JNEUROSCI.22-17-07453.2002
251. Geng SJ, Liao FF, Dang WH, et al. Contribution of the spinal cord BDNF to the development of neuropathic pain by activation of the NR2B-containing NMDA receptors in rats with spinal nerve ligation. ExpNeurol. 2010;222(2):256–266.
252. Zhou W, Xie Z, Li C, et al. Driving effect of BDNF in the spinal dorsal horn on neuropathic pain. Neurosci Lett. 2021;756:135965. doi:10.1016/j.neulet.2021.135965
253. Mai CL, Wei X, Gui WS, et al. Differential regulation of GSK-3beta in spinal dorsal horn and in hippocampus mediated by interleukin-1beta contributes to pain hypersensitivity and memory deficits following peripheral nerve injury. Mol Pain. 2019;15:1744806919826789. doi:10.1177/1744806919826789
254. Xue M, Sun YL, Xia YY, Huang ZH, Huang C, Xing GG. Electroacupuncture modulates spinal BDNF/TrκB signaling pathway and ameliorates the sensitization of dorsal horn WDR neurons in spared nerve injury rats. Int J Mol Sci. 2020;21(18):6524. doi:10.3390/ijms21186524
255. Park H, Poo MM. Neurotrophin regulation of neural circuit development and function. Nat Rev Neurosci. 2013;14(1):7–23. doi:10.1038/nrn3379
256. Jia D, Gao GD, Liu Y, et al. TNF-alpha involves in altered prefrontal synaptic transmission in mice with persistent inflammatory pain. Neurosci Lett. 2007;415(1):1–5. doi:10.1016/j.neulet.2006.12.032
257. Yao PW, Wang SK, Chen SX, Xin WJ, Liu XG, Zang Y. Upregulation of tumor necrosis factor-alpha in the anterior cingulate cortex contributes to neuropathic pain and pain-associated aversion. Neurobiol Dis. 2019;130:104456. doi:10.1016/j.nbd.2019.04.012
258. Fiore NT, Austin PJ. Peripheral nerve injury triggers neuroinflammation in the medial prefrontal cortex and ventral hippocampus in a subgroup of rats with coincident affective behavioural changes. Neuroscience. 2019;416:147–167. doi:10.1016/j.neuroscience.2019.08.005
259. Nascimento FP, Macedo-Júnior SJ, Borges FR, et al. Thalidomide reduces mechanical hyperalgesia and depressive-like behavior induced by peripheral nerve crush in mice. Neuroscience. 2015;303:51–58. doi:10.1016/j.neuroscience.2015.06.044
260. Zigmond RE, Echevarria FD. Macrophage biology in the peripheral nervous system after injury. Prog Neurobiol. 2019;173:102–121. doi:10.1016/j.pneurobio.2018.12.001
261. Yu X, Basbaum A, Guan Z. Contribution of colony-stimulating factor 1 to neuropathic pain. Pain Rep. 2021;6(1):e883. doi:10.1097/PR9.0000000000000883
262. Echeverry S, Shi XQ, Rivest S, Zhang J. Peripheral nerve injury alters blood-spinal cord barrier functional and molecular integrity through a selective inflammatory pathway. J Neurosci. 2011;31(30):10819–10828. doi:10.1523/JNEUROSCI.1642-11.2011
263. Zhang J, Shi XQ, Echeverry S, Mogil JS, De Koninck Y, Rivest S. Expression of CCR2 in both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. J Neurosci. 2007;27(45):12396–12406. doi:10.1523/JNEUROSCI.3016-07.2007
264. Prinz M, Priller J, Sisodia SS, Ransohoff RM. Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat Neurosci. 2011;14(10):1227–1235. doi:10.1038/nn.2923
265. Fife BT, Huffnagle GB, Kuziel WA, Karpus WJ. CC chemokine receptor 2 is critical for induction of experimental autoimmune encephalomyelitis. J Exp Med. 2000;192(6):899–905. doi:10.1084/jem.192.6.899
266. Fang W, Zhai X, Han D, et al. CCR2-dependent monocytes/macrophages exacerbate acute brain injury but promote functional recovery after ischemic stroke in mice. Theranostics. 2018;8(13):3530–3543. doi:10.7150/thno.24475
267. Mai CL, Tan Z, Xu YN, et al. CXCL12-mediated monocyte transmigration into brain perivascular space leads to neuroinflammation and memory deficit in neuropathic pain. Theranostics. 2021;11(3):1059–1078. doi:10.7150/thno.44364
268. Donnelly CR, Andriessen AS, Chen G, et al. Central nervous system targets: glial cell mechanisms in chronic pain. Neurotherapeutics. 2020;17(3):846–860. doi:10.1007/s13311-020-00905-7
269. Hanani M, Spray DC. Emerging importance of satellite glia in nervous system function and dysfunction. Nat Rev Neurosci. 2020;21(9):485–498. doi:10.1038/s41583-020-0333-z
270. Grace PM, Hutchinson MR, Maier SF, Watkins LR. Pathological pain and the neuroimmune interface. Nat Rev Immunol. 2014;14(4):217–231. doi:10.1038/nri3621
271. Regen F, Hellmann-Regen J, Costantini E, Reale M. Neuroinflammation and Alzheimer’s disease: implications for microglial activation. Curr Alzheimer Res. 2017;14(11):1140–1148. doi:10.2174/1567205014666170203141717
272. Santos LE, Beckman D, Ferreira ST. Microglial dysfunction connects depression and Alzheimer’s disease. Brain Behav Immun. 2016;55:151–165. doi:10.1016/j.bbi.2015.11.011
273. Liu Y, Yin Y. Emerging roles of immune cells in postoperative cognitive dysfunction. Mediators Inflamm. 2018;2018:6215350. doi:10.1155/2018/6215350
274. Yang CP, Cherng CH, Wu CT, et al. Intrathecal ultra-low dose naloxone enhances the antihyperalgesic effects of morphine and attenuates tumor necrosis factor-α and tumor necrosis factor-α receptor 1 expression in the dorsal horn of rats with partial sciatic nerve transection. Anesth Analg. 2013;117(6):1493–1502. doi:10.1213/ANE.0000000000000020
275. Wang J, Liu Y, Zhou LJ, et al. Magnesium L-threonate prevents and restores memory deficits associated with neuropathic pain by inhibition of TNF-alpha. Pain Phys. 2013;16(5):E563–575.
276. Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1(6):a001651. doi:10.1101/cshperspect.a001651
277. Pollock G, Pennypacker KR, Mémet S, Israël A, Saporta S. Activation of NF-kappaB in the mouse spinal cord following sciatic nerve transection. Exp Brain Res. 2005;165(4):470–477. doi:10.1007/s00221-005-2318-6
278. Möser CV, Kynast K, Baatz K, et al. The protein kinase IKKε is a potential target for the treatment of inflammatory hyperalgesia. J Immunol. 2011;187(5):2617–2625. doi:10.4049/jimmunol.1004088
279. Kanngiesser M, Häussler A, Myrczek T, et al. Inhibitor kappa B kinase beta dependent cytokine upregulation in nociceptive neurons contributes to nociceptive hypersensitivity after sciatic nerve injury. J Pain. 2012;13(5):485–497. doi:10.1016/j.jpain.2012.02.010
280. Niu YL, Guo Z, Zhou RH. Up-regulation of TNF-alpha in neurons of dorsal root ganglia and spinal cord during coronary artery occlusion in rats. Cytokine. 2009;47(1):23–29. doi:10.1016/j.cyto.2009.04.003
281. Gruber-Schoffnegger D, Drdla-Schutting R, Hönigsperger C, Wunderbaldinger G, Gassner M, Sandkühler J. Induction of thermal hyperalgesia and synaptic long-term potentiation in the spinal cord lamina I by TNF-α and IL-1β is mediated by glial cells. J Neurosci. 2013;33(15):6540–6551. doi:10.1523/JNEUROSCI.5087-12.2013
282. Brás JP, Bravo J, Freitas J, et al. TNF-alpha-induced microglia activation requires miR-342: impact on NF-kB signaling and neurotoxicity. Cell Death Dis. 2020;11(6):415. doi:10.1038/s41419-020-2626-6
283. Basu A, Krady JK, Levison SW. Interleukin-1: a master regulator of neuroinflammation. J Neurosci Res. 2004;78(2):151–156. doi:10.1002/jnr.20266
284. Kaushik DK, Thounaojam MC, Kumawat KL, Gupta M, Basu A. Interleukin-1β orchestrates underlying inflammatory responses in microglia via Krüppel-like factor 4. J Neurochem. 2013;127(2):233–244. doi:10.1111/jnc.12382
285. Finnerup NB, Attal N, Haroutounian S, et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol. 2015;14(2):162–173. doi:10.1016/S1474-4422(14)70251-0
286. Moisset X, Bouhassira D, Attal N. French guidelines for neuropathic pain: an update and commentary. Rev Neurol (Paris). 2021;177(7):834–837. doi:10.1016/j.neurol.2021.07.004
287. Bouhassira D, Attal N. Translational neuropathic pain research: a clinical perspective. Neuroscience. 2016;338:27–35. doi:10.1016/j.neuroscience.2016.03.029
288. Li CL, Li KC, Wu D, et al. Somatosensory neuron types identified by high-coverage single-cell RNA-sequencing and functional heterogeneity. Cell Res. 2016;26(1):83–102. doi:10.1038/cr.2015.149
289. Kupari J, Usoskin D, Parisien M, et al. Single cell transcriptomics of primate sensory neurons identifies cell types associated with chronic pain. Nat Commun. 2021;12(1):1510. doi:10.1038/s41467-021-21725-z
290. Nguyen MQ, von Buchholtz LJ, Reker AN, Ryba NJ, Davidson S. Single-nucleus transcriptomic analysis of human dorsal root ganglion neurons. eLife. 2021;10. doi:10.7554/eLife.71752
291. Tavares-Ferreira D, Shiers S, Ray PR, et al. Spatial transcriptomics of dorsal root ganglia identifies molecular signatures of human nociceptors. Sci Transl Med. 2022;14(632):eabj8186. doi:10.1126/scitranslmed.abj8186
292. Furman D, Campisi J, Verdin E, et al. Chronic inflammation in the etiology of disease across the life span. Nat Med. 2019;25(12):1822–1832. doi:10.1038/s41591-019-0675-0
293. Ji RR, Xu ZZ, Gao YJ. Emerging targets in neuroinflammation-driven chronic pain. Nat Rev Drug Discov. 2014;13(7):533–548. doi:10.1038/nrd4334
294. Netea MG, Balkwill F, Chonchol M, et al. A guiding map for inflammation. Nat Immunol. 2017;18(8):826–831. doi:10.1038/ni.3790
295. Kotas ME, Medzhitov R. Homeostasis, inflammation, and disease susceptibility. Cell. 2015;160(5):816–827. doi:10.1016/j.cell.2015.02.010
296. Kato D, Eto K, Nabekura J, Wake H. Activity-dependent functions of non-electrical glial cells. J Biochem. 2018;163(6):457–464. doi:10.1093/jb/mvy023
297. Falk S, Götz M. Glial control of neurogenesis. Curr Opin Neurobiol. 2017;47:188–195. doi:10.1016/j.conb.2017.10.025
298. Ellis A, Bennett DL. Neuroinflammation and the generation of neuropathic pain. Br J Anaesth. 2013;111(1):26–37. doi:10.1093/bja/aet128
299. Calsolaro V, Edison P. Neuroinflammation in Alzheimer’s disease: current evidence and future directions. Alzheimer’s Dementia. 2016;12(6):719–732. doi:10.1016/j.jalz.2016.02.010
300. Walker AK, Kavelaars A, Heijnen CJ, Dantzer R. Neuroinflammation and comorbidity of pain and depression. Pharmacol Rev. 2014;66(1):80–101. doi:10.1124/pr.113.008144
301. Benedetti F, Aggio V, Pratesi ML, Greco G, Furlan R. Neuroinflammation in bipolar depression. Front Psych. 2020;11:71. doi:10.3389/fpsyt.2020.00071
302. Conaghan PG, Cook AD, Hamilton JA, Tak PP. Therapeutic options for targeting inflammatory osteoarthritis pain. Nat Rev Rheumatol. 2019;15(6):355–363. doi:10.1038/s41584-019-0221-y
303. Sánchez-Robles EM, Girón R, Paniagua N, Rodríguez-Rivera C, Pascual D, Goicoechea C. Monoclonal antibodies for chronic pain treatment: present and future. Int J Mol Sci. 2021;22(19):10325. doi:10.3390/ijms221910325
304. Zamri F, de Vries TJ. Use of TNF inhibitors in rheumatoid arthritis and implications for the periodontal status: for the benefit of both? Front Immunol. 2020;11:591365. doi:10.3389/fimmu.2020.591365
305. Piski Z, Gerlinger I, Nepp N, Farkas K, Weber R. TNF-alpha inhibitors and rhinosinusitis-a systematic review and meta-analysis. Am J Rhinol Allergy. 2020;34(3):436–442. doi:10.1177/1945892419898988
306. Minozzi S, Bonovas S, Lytras T, et al. Risk of infections using anti-TNF agents in rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis: a systematic review and meta-analysis. Expert Opin Drug Saf. 2016;15(sup1):11–34. doi:10.1080/14740338.2016.1240783
307. Nigam GB, Bhandare AP, Antoniou GA, Limdi JK. Systematic review and meta-analysis of dermatological reactions in patients with inflammatory bowel disease treated with anti-tumour necrosis factor therapy. Eur J Gastroenterol Hepatol. 2021;33(3):346–357. doi:10.1097/MEG.0000000000001917
308. Nielsen FH. Dietary magnesium and chronic disease. Adv Chronic Kidney Dis. 2018;25(3):230–235. doi:10.1053/j.ackd.2017.11.005
309. Reginster JY, Strause L, Deroisy R, Lecart MP, Saltman P, Franchimont P. Preliminary report of decreased serum magnesium in postmenopausal osteoporosis. Magnesium. 1989;8(2):106–109.
310. Steidl L, Ditmar R. Blood magnesium, calcium and zinc in osteoporosis. Acta Univ Palacki Olomuc Fac Med. 1991;129:91–98.
311. Serefko A, Szopa A, Poleszak E. Magnesium and depression. Magn res. 2016;29(3):112–119. doi:10.1684/mrh.2016.0407
312. Liu G, Weinger JG, Lu ZL, Xue F, Sadeghpour S. Efficacy and safety of MMFS-01, a synapse density enhancer, for treating cognitive impairment in older adults: a randomized, double-blind, placebo-controlled trial. J Alzheimer’s Dis. 2016;49(4):971–990. doi:10.3233/JAD-150538
313. Morel V, Pickering ME, Goubayon J, Djobo M, Macian N, Pickering G. Magnesium for pain treatment in 2021? state of the art. Nutrients. 2021;13(5):1397. doi:10.3390/nu13051397
314. Slutsky I, Abumaria N, Wu LJ, et al. Enhancement of learning and memory by elevating brain magnesium. Neuron. 2010;65(2):165–177. doi:10.1016/j.neuron.2009.12.026
315. Ying YL, Wei XH, Xu XB, et al. Over-expression of P2X7 receptors in spinal glial cells contributes to the development of chronic postsurgical pain induced by skin/muscle incision and retraction (SMIR) in rats. Exp Neurol. 2014;261:836–843. doi:10.1016/j.expneurol.2014.09.007
316. Chen JL, Zhou X, Liu BL, et al. Normalization of magnesium deficiency attenuated mechanical allodynia, depressive-like behaviors, and memory deficits associated with cyclophosphamide-induced cystitis by inhibiting TNF-alpha/NF-kappaB signaling in female rats. J Neuroinflammation. 2020;17(1):99. doi:10.1186/s12974-020-01786-5
317. Mayer ML, Westbrook GL, Guthrie PB. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature. 1984;309(5965):261–263. doi:10.1038/309261a0
318. Woolf CJ, Thompson SW. The induction and maintenance of central sensitization is dependent on N-methyl-D-aspartic acid receptor activation; implications for the treatment of post-injury pain hypersensitivity states. Pain. 1991;44(3):293–299. doi:10.1016/0304-3959(91)90100-C
319. Shin HJ, Na HS, Do SH. Magnesium and Pain. Nutrients. 2020;12(8):2184. doi:10.3390/nu12082184
320. Tsien JZ, Huerta PT, Tonegawa S. The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell. 1996;87(7):1327–1338. doi:10.1016/S0092-8674(00)81827-9
321. Tang YP, Shimizu E, Dube GR, et al. Genetic enhancement of learning and memory in mice. Nature. 1999;401(6748):63–69. doi:10.1038/43432
322. Wang D, Jacobs SA, Tsien JZ. Targeting the NMDA receptor subunit NR2B for treating or preventing age-related memory decline. Expert Opin Ther Targets. 2014;18(10):1121–1130. doi:10.1517/14728222.2014.941286
323. Wilson JA, Garry EM, Anderson HA, et al. NMDA receptor antagonist treatment at the time of nerve injury prevents injury-induced changes in spinal NR1 and NR2B subunit expression and increases the sensitivity of residual pain behaviours to subsequently administered NMDA receptor antagonists. Pain. 2005;117(3):421–432. doi:10.1016/j.pain.2005.07.005
324. Qu XX, Cai J, Li MJ, et al. Role of the spinal cord NR2B-containing NMDA receptors in the development of neuropathic pain. Exp Neurol. 2009;215(2):298–307. doi:10.1016/j.expneurol.2008.10.018
325. Wolf S, Barton D, Kottschade L, Grothey A, Loprinzi C. Chemotherapy-induced peripheral neuropathy: prevention and treatment strategies. Eur J Cancer. 2008;44(11):1507–1515. doi:10.1016/j.ejca.2008.04.018
326. Tannock IF, Ahles TA, Ganz PA, Van Dam FS. Cognitive impairment associated with chemotherapy for cancer: report of a workshop. J clin Oncol. 2004;22(11):2233–2239. doi:10.1200/JCO.2004.08.094
327. Ballenger JC, Davidson JR, Lecrubier Y, Nutt DJ, Jones RD, Berard RM. Consensus statement on depression, anxiety, and oncology. J Clin Psychiatry. 2001;62(Suppl 8):64–67.
328. Mazur A, Maier JA, Rock E, Gueux E, Nowacki W, Rayssiguier Y. Magnesium and the inflammatory response: potential physiopathological implications. Arch Biochem Biophys. 2007;458(1):48–56. doi:10.1016/j.abb.2006.03.031
329. Veronese N, Pizzol D, Smith L, Dominguez LJ, Barbagallo M. Effect of magnesium supplementation on inflammatory parameters: a meta-analysis of randomized controlled trials. Nutrients. 2022;14(3):679. doi:10.3390/nu14030679
330. Gile J, Ruan G, Abeykoon J, McMahon MM, Witzig T. Magnesium: the overlooked electrolyte in blood cancers? Blood Rev. 2020;3:100676.
331. McKee JA, Brewer RP, Macy GE, Borel CO, Reynolds JD, Warner DS. Magnesium neuroprotection is limited in humans with acute brain injury. Neurocrit Care. 2005;2(3):342–351. doi:10.1385/NCC:2:3:342
332. Niederberger E, Geisslinger G, Warner D, Warner M. Proteomics in neuropathic pain research. Anesthesiology. 2008;108(2):314–323. doi:10.1097/01.anes.0000299838.13368.6e
333. Sribanditmongkol P, Sheu MJ, Tejwani GA. Inhibition of morphine tolerance and dependence by diazepam and its relation to the CNS Met-enkephalin levels. Brain Res. 1994;645(1–2):1–12. doi:10.1016/0006-8993(94)91631-4
334. Zambotti F, Zonta N, Tammiso R, et al. Effects of diazepam on nociception in rats. Naunyn Schmiedebergs Arch Pharmacol. 1991;344(1):84–89. doi:10.1007/BF00167386
335. Warms CA, Turner JA, Marshall HM, Cardenas DD. Treatments for chronic pain associated with spinal cord injuries: many are tried, few are helpful. ClinJPain. 2002;18(3):154–163.
336. Hu NW, Zhang HM, Hu XD, et al. Protein synthesis inhibition blocks the late-phase LTP of C-fiber evoked field potentials in rat spinal dorsal horn. JNeurophysiol. 2003;89(5):2354–2359. doi:10.1152/jn.01027.2002
337. Woods MJ, Williams DC. Multiple forms and locations for the peripheral-type benzodiazepine receptor. Biochem Pharmacol. 1996;52(12):1805–1814. doi:10.1016/S0006-2952(96)00558-8
338. Cavaliere C, Tramontano L, Fiorenza D, Alfano V, Aiello M, Salvatore M. Gliosis and neurodegenerative diseases: the role of PET and MR Imaging. Front Cell Neurosci. 2020;14:75. doi:10.3389/fncel.2020.00075
339. Rupprecht R, Papadopoulos V, Rammes G, et al. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat Rev Drug Discov. 2010;9(12):971–988. doi:10.1038/nrd3295
340. Wei XH, Wei X, Chen FY, et al. The upregulation of translocator protein (18 kDa) promotes recovery from neuropathic pain in rats. J Neurosci. 2013;33(4):1540–1551. doi:10.1523/JNEUROSCI.0324-12.2013
341. Liu X, Liu H, Xu S, et al. Spinal translocator protein alleviates chronic neuropathic pain behavior and modulates spinal astrocyte-neuronal function in rats with L5 spinal nerve ligation model. Pain. 2016;157(1):103–116. doi:10.1097/j.pain.0000000000000339
342. Hernstadt H, Wang S, Lim G, Mao J. Spinal translocator protein (TSPO) modulates pain behavior in rats with CFA-induced monoarthritis. Brain Res. 2009;1286:42–52. doi:10.1016/j.brainres.2009.06.043
343. Cropper HC, Johnson EM, Haight ES, et al. Longitudinal translocator protein-18 kDa-positron emission tomography imaging of peripheral and central myeloid cells in a mouse model of complex regional pain syndrome. Pain. 2019;160(9):2136–2148. doi:10.1097/j.pain.0000000000001607
344. Setiawan E, Wilson AA, Mizrahi R, et al. Role of translocator protein density, a marker of neuroinflammation, in the brain during major depressive episodes. JAMA psych. 2015;72(3):268–275. doi:10.1001/jamapsychiatry.2014.2427
345. Attwells S, Setiawan E, Rusjan PM, et al. A double-blind placebo-controlled trial of minocycline on translocator protein distribution volume in treatment-resistant major depressive disorder. Transl Psychiatry. 2021;11(1):334. doi:10.1038/s41398-021-01450-3
346. Albrecht DS, Ahmed SU, Kettner NW, et al. Neuroinflammation of the spinal cord and nerve roots in chronic radicular pain patients. Pain. 2018;159(5):968–977. doi:10.1097/j.pain.0000000000001171
347. Jung ME, Protective A. Role of translocator protein in Alzheimer’s disease brain. Curr Alzheimer Res. 2020;17(1):3–15. doi:10.2174/1567205017666200217105950
348. Vicente B, Saldivia S, Hormazabal N, Bustos C, Rubí P. Etifoxine is non-inferior than clonazepam for reduction of anxiety symptoms in the treatment of anxiety disorders: a randomized, double blind, non-inferiority trial. Psychopharmacology. 2020;237(11):3357–3367. doi:10.1007/s00213-020-05617-6
349. Xu J, Feng YW, Liu L, et al. Liver X receptor alpha is involved in counteracting mechanical allodynia by inhibiting neuroinflammation in the spinal dorsal horn. Anesthesiology. 2017;127(3):534–547. doi:10.1097/ALN.0000000000001718
350. Flatters SJ. Characterization of a model of persistent postoperative pain evoked by skin/muscle incision and retraction (SMIR). Pain. 2008;135(1–2):119–130. doi:10.1016/j.pain.2007.05.013
351. Zhong X, Wang W, Mao Z, et al. Activation of liver x receptors prevents the spinal LTP induced by skin/muscle retraction in the thigh via SIRT1/NF-Κb pathway. Neurochem Int. 2019;128:106–114. doi:10.1016/j.neuint.2019.04.002
352. Mao Z, Huang R, Xu J, Guo R, Wei X. Liver X receptor α in sciatic nerve exerts an alleviating effect on neuropathic pain behaviors induced by crush injury. Neurochem Res. 2021;46(2):358–366. doi:10.1007/s11064-020-03171-3
353. Li X, Zhong H, Wang Z, et al. Loss of liver X receptor β in astrocytes leads to anxiety-like behaviors via regulating synaptic transmission in the medial prefrontal cortex in mice. Mol Psychiatry. 2021;26:6380–6393. doi:10.1038/s41380-021-01139-5
354. Xu X, Xiao X, Yan Y, Zhang T. Activation of liver X receptors prevents emotional and cognitive dysfunction by suppressing microglial M1-polarization and restoring synaptic plasticity in the hippocampus of mice. Brain Behav Immun. 2021;94:111–124. doi:10.1016/j.bbi.2021.02.026
355. Teodorczyk-Injeyan JA, Triano JJ, McGregor M, Woodhouse L, Injeyan HS. Effect of interactive neurostimulation therapy on inflammatory response in patients with chronic and recurrent mechanical neck pain. J Manipulative Physiol Ther. 2015;38(8):545–554. doi:10.1016/j.jmpt.2015.08.006
356. Teodorczyk-Injeyan JA, McGregor M, Triano JJ, Injeyan SH. Elevated production of nociceptive CC chemokines and sE-selectin in patients with low back pain and the effects of spinal manipulation: a nonrandomized clinical trial. Clin J Pain. 2018;34(1):68–75. doi:10.1097/AJP.0000000000000507
357. Pertin M, Ji RR, Berta T, et al. Upregulation of the voltage-gated sodium channel beta2 subunit in neuropathic pain models: characterization of expression in injured and non-injured primary sensory neurons. J Neurosci. 2005;25(47):10970–10980. doi:10.1523/JNEUROSCI.3066-05.2005
358. Chen C, Bharucha V, Chen Y, et al. Reduced sodium channel density, altered voltage dependence of inactivation, and increased susceptibility to seizures in mice lacking sodium channel beta 2-subunits. Proc Natl Acad Sci U S A. 2002;99(26):17072–17077. doi:10.1073/pnas.212638099
359. Razzaque MS. Magnesium: are we consuming enough? Nutrients. 2018;10(12):1863. doi:10.3390/nu10121863
360. Shibata H, Yoshioka Y, Ikemizu S, et al. Functionalization of tumor necrosis factor-alpha using phage display technique and PEGylation improves its antitumor therapeutic window. Clin Cancer Res. 2004;10(24):8293–8300.
361. Katagiri H, Tanaka K, Manabe T. Requirement of appropriate glutamate concentrations in the synaptic cleft for hippocampal LTP induction. Eur J Neurosci. 2001;14(3):547–553. doi:10.1046/j.0953-816x.2001.01664.x
362. Wang ZY, Zhang YQ, Zhao ZQ. Inhibition of tetanically sciatic stimulation-induced LTP of spinal neurons and Fos expression by disrupting glutamate transporter GLT-1. Neuropharmacology. 2006;51(4):764–772. doi:10.1016/j.neuropharm.2006.05.024
363. Murphy N, Cowley TR, Richardson JC, et al. The neuroprotective effect of a specific P2X7 receptor antagonist derives from its ability to inhibit assembly of the NLRP3 inflammasome in glial cells. Brain Pathol. 2012;22(3):295–306. doi:10.1111/j.1750-3639.2011.00531.x
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.