Back to Journals » Journal of Inflammation Research » Volume 14
An Indole-2-Carboxamide Derivative, LG4, Alleviates Diabetic Kidney Disease Through Inhibiting MAPK-Mediated Inflammatory Responses
Authors Qian J, Yin S, Ye L, Wang Z ![]() , Shu S, Mou Z, Xu M, Chattipakorn N, Liu Z, Liang G
, Shu S, Mou Z, Xu M, Chattipakorn N, Liu Z, Liang G
Received 25 February 2021
Accepted for publication 2 April 2021
Published 27 April 2021 Volume 2021:14 Pages 1633—1645
DOI https://doi.org/10.2147/JIR.S308353
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Jianchang Qian,1,* Sihui Yin,1,* Lin Ye,1 Zhe Wang,2 Sheng Shu,1,3 Zhenxin Mou,1,3 Mingjiang Xu,1 Nipon Chattipakorn,4 Zhiguo Liu,1 Guang Liang1,3,5
1Chemical Biology Research Center, School of Pharmaceutical Sciences, Wenzhou Medical University, Wenzhou, Zhejiang, People’s Republic of China; 2Department of Pharmacy, The Second Affiliated Hospital of Wenzhou Medical University, Wenzhou, Zhejiang, People’s Republic of China; 3School of Pharmacy, Hangzhou Medical College, Hangzhou, Zhejiang, People’s Republic of China; 4Cardiac Electrophysiology Research and Training Center, Faculty of Medicine, Chiang Mai University, Chiang Mai, Thailand; 5Wenzhou Institute, University of Chinese Academy of Sciences, Wenzhou, Zhejiang, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Guang Liang; Zhiguo Liu
Chemical Biology Research Center, School of Pharmaceutical Sciences, Wenzhou Medical University, Wenzhou, 325035, People’s Republic of China
Tel/Fax +86-577-86699396
Email [email protected]; [email protected]
Aim: Elevated inflammatory signaling has been shown to play an important role in diabetic kidney disease (DKD). We previously developed a new anti-inflammatory compound LG4. In the present study, we have tested the hypothesis that LG4 could prevent DKD by suppressing inflammation and identified the underlying mechanism.
Methods: Streptozotocin-induced type 1 diabetic mice were used to develop DKD and evaluate the effects of LG4 against DKD. To identify the potential targets of LG4, biotin-linked LG4 was synthesized and subjected to proteome microarray screening. The cellular mechanism of LG4 was investigated in HG-challenged SV40MES13 cells.
Results: Although LG4 treatment had no effect on the body weight and blood glucose levels, it remarkably reversed the hyperglycemia-induced pathological changes and fibrosis in the kidneys of T1DM mice. Importantly, hyperglycemia-induced renal inflammation evidenced by NF-κB activation and TNFα and IL-6 overexpression was greatly ameliorated with LG4 treatment. Proteosome microarray screening revealed that JNK and ERK were the direct binding proteins of LG4. LG4 significantly reduced HG-induced JNK and ERK phosphorylation and subsequent NF-κB activation in vivo and in vitro. In addition, LG4 did not show further anti-inflammatory effect in HG-challenged mesangial cells with the presence of JNK or ERK inhibitor.
Conclusion: LG4 showed renoprotective activity through inhibiting ERK/JNK-mediated inflammation in diabetic mice, indicating that LG4 may be a therapeutic agent for DKD.
Keywords: indole-2-carboxamide derivative, diabetic kidney disease, inflammation, MAPK, NF-κB
Introduction
Although a great improvement has been made in blood glucose management, the number of patients with diabetic kidney disease (DKD) is still growing due to the increasing of diabetic population.1 Microalbuminuria is a key feature in the initial stage of DKD (30–300 mg/day), and macroalbuminuria (>300 mg/day) and overt nephropathy occur as the disease progressed.2–4 Meanwhile, glomerular dysfunction and morphological changes that lead to subsequent development of glomerulosclerosis and nephron dropout will be seen with or without tubular atrophy, tubulointerstitial fibrosis and renal arteriolar hyalinosis.5–8 Although the progression of DKD could be slowed with tight glycemic control and antihypertensive/lipid-lowering therapies, these interventions do not prevent the progress of DKD to end stage of renal disease (ESRD) or renal failure.9
Chronic inflammation in the kidney plays a key role in the development and progression of DKD.10,11 Hyperglycemia induces glomerular hyperfiltration, mechanical stress, and glycocalyx dysfunction that result in the overexpression of proinflammatory cytokines such as tumor necrosis factor α (TNF-α), interleukin-6 (IL-6) and interleukin-1β (IL-1β) through activating mitogen-activated protein kinases (MAPKs) and NF-κB signaling pathways in the kidney.12,13 Stress-activated protein kinases, especially p38 mitogen activated protein kinase and the c-Jun amino terminal kinase signaling, are the validated upstream transductor that modulate activation of NF-κB in hyperglycemia.14 These inflammatory responses are also causal factors of glomerulosclerosis and tubulointerstitial fibrosis by inducing extracellular matrix deposition and differentiation/proliferation of myofibroblast. The underlying signaling pathways include NF-κB, JAK/STAT, TGF-β/Smad, etc.15,16 Therefore, targeting MAPK/NF-κB transduction could be a promising strategy to block hyperglycemia-induced inflammation for DKD treatment.
Indole is an important scaffold in the field of medicinal chemistry. Many indole derivatives possess significant pharmacological activities such as anti-inflammatory, analgesic and antipyretic effects.17–20 For instance, indomethacin is one of the indole acetic acid derivative and a non-steroidal anti-inflammatory drug (NSAID), it however is known to cause ulcers for the users, and its safety profile has been improved by chemical modifications.21 We had designed and synthesized a series of indole derivatives and evaluated their anti-inflammatory property by measuring the production of TNF-α and IL-6 in activated macrophages and LPS-induced sepsis in mice. Among them, an indole-2-carboxamide derivative, LG4, exhibited significant anti-inflammatory effects in vitro and in vivo.22 In this study, we explored the potential effect of LG4 on chronic inflammation and explored the underlying molecular mechanisms in a mouse DKD model and mesangial cells.
Materials and Methods
Chemicals and Reagents
Compound LG4 was synthesized as described previously.22 The chemical synthesis of biotin-labeled LG4 was described in the Supplementary File. UO126 and SP600125 were purchased from MedChemExpress (Monmouth Junction, NJ). The compounds used in the study were dissolved in dimethyl sulfoxide (DMSO) at 20 mM as stock solutions. Other reagents used in this study were purchased from Sigma-Aldrich (Burlington, MA, USA).
Cell Culture
A mouse glomerular mesangial cell line, SV40MES13, was obtained from Shanghai Institute of Biochemistry and Cell Biology (Shanghai, China) and cultured in low-glucose (5.5 mM) DMEM (Gibco, Eggenstein, Germany) containing 5% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 mg/mL streptomycin at 37°C with 5% CO2. When cells are treated with high-concentration glucose (HG), DMEM with 33 mM glucose was used.
Animal Model
Male C57BL/6 mice were obtained from Animal Center of Wenzhou Medical University and housed at a constant room temperature with a 12:12 h light-dark cycle, chow diet and water ad libitum. Animal study protocols were approved by Wenzhou Medical University Animal Policy and Welfare Committee in accordance with the NIH guidelines (Guide for the care and use of laboratory animals). Type 1 diabetic (T1DM) mouse model was prepared as previously described.23 Briefly, diabetes was induced in the mice at 8 weeks of age by intraperitoneal injection of 50 mg/kg streptozotocin (STZ, 10 mL/kg in citrate buffer, pH 4.5) for five consecutive days, while the control animals were received citrate buffer in the same schedule. One week later after first STZ dosing, blood glucose was measured through tail vein using a glucometer. Mice with fasting blood glucose >300 mg/dL were considered as T1DM.
Eight weeks after setting up the T1DM, randomly selected diabetic animals were divided into three groups, ie, T1DM+vehicle, T1DM+LG4 (5 mg/kg), and T1DM+LG4 (10 mg/kg); n = 5 mice per group. LG4 (5 and 10 mg/kg) was administered by oral gavage once every two days for 10 weeks. The T1DM+vehicle and age-matched control group (n = 5) was given with vehicle (0.5% CMC-Na solution) in the same schedule. Blood glucose and body weight were recorded every two weeks. After 10 weeks of treatment, the mice were sacrificed under sodium pentobarbital anesthesia (i.p. injection of 0.2 mL sodium pentobarbital at 100 mg/mL), and blood samples and kidney tissues were collected and processed for further analyses.
Real-Time Quantitative PCR
Cells or kidney tissues were homogenized in TRIZOL (Invitrogen, Carlsbad, CA) for total RNA extraction. Reverse transcription and quantitative PCR (RT-qPCR) were performed using M-MLV Platinum RT-qPCR Kit (Invitrogen, Carlsbad, CA). Primers for genes including Tnf-a, Il-6 and β-actin were synthesized by Invitrogen (Invitrogen, Shanghai, China), supplementary table s1. The relative amount of target genes was normalized to the amount of β-actin.
Immunoblotting Assay
Cell and tissue lysates were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene fluoride membrane (Bio-Rad Laboratory, Hercules, CA). The membrane was blocked for 2 hours at room temperature, and then incubated with primary antibodies overnight, followed with 2-h incubation with secondary antibody that conjugated with horseradish peroxidase. Finally, the specific bands were visualized using enhanced chemiluminescence reagents (Bio-Rad, Hercules, CA, USA). Densitometric quantification for blots was performed using ImageJ software.
The primary antibodies against BCL-2 (Cat. sc-7382, 1:200) and GAPDH (Cat. sc-47724, 1:200) were purchased from Santa Cruz Technology (Santa Cruz, CA, USA). Anti-TGF-β (Cat. ab92486, 1:1000), anti-Collagen IV antibodies (Cat. ab6586, 1:1000) and BAX (Cat. ab32503, 1:1000) were obtained from Abcam (Cambridge, MA, USA). Rabbit mAbs against p-p44/42 (Erk1/2)_Thr202/Tyr204 (Cat. 4370S, 1:1000), p-SAPK/JNK_Thr183/Tyr185 (Cat. 4668, 1:1000), P-IKK (Cat. 2697T, 1:1000) and IκBα (Cat. 4812S, 1:1000) were purchased from Cell Signaling (Beverly, MA, USA). These antibodies were all diluted with primary antibody dilution buffer (Beyotime, Cat. P0023).
Histological Analyses
Kidney was fixed in 4% paraformaldehyde solution and paraffin embedded. Tissues were cut into 5 μm sections and stained with Hematoxylin and Eosin (H&E), Masson’s trichrome or anti-TNF-α antibody (Cat. ab6671, Abcam), respectively. Subsequently, the slides were imaged and captured under light microscope (200× amplification; Nikon, Tokyo, Japan). The ImageJ software (NIH) was applied to quantify the level of positive staining area. At least 6 non-overlapping fields in the view were scored and the value determined was related to total tissue area in the field.
Glomerulosclerosis was evaluated using Masson’s trichrome staining images. Glomerular sclerosis was assigned score from 0 to 4 based on sclerotic area. Grade 0 means normal; grade 1, area up to 25%; grade 2, area from 25% to 50%; grade 3, area from 50% to 75%; grade 4, area from 75% to 100%. The Glomerular sclerosis score was determined using formula: (1×A1+2×A2+3×A3+4×A4)/(A0+A1+A2+A3+A4), where Ax is the number of glomeruli in each grade.
Assessment of Kidney Function
The levels of urine albumin creatinine ratio (UACR), serum creatinine (SCR), and blood urea nitrogen (BUN) were detected using commercial kits (Nanjing, Jiancheng, Jiangsu, China).
Immunofluorescence Staining
The paraffin sections (5 µm thickness) from kidney were dewaxed and dehydrated. After antigen retrieval and blocking with 5% BSA, the sections were incubated with anti-collagen IV antibody overnight at 4°C, followed by incubation with a FITC-conjugated secondary antibody. After counterstaining with DAPI for 5 min, the slides were viewed by a fluorescent microscope (200× amplification; Nikon, Tokyo, Japan). The semiquantitation was performed on ImageJ to determine relative fluorescence intensity.
HuProtTM Human Proteome Chip
HuProtTM chip was blocked (5% BSA in 0.1% Tween 20, 1×TBST, pH=7.4) for 2 hours at room temperature with gentle agitation, and then washed with TBST (pH 7.5) for 5 minutes and repeated for three times. The chips were then incubated with D-biotin or biotin-labeled LG4 (10 μM) in blocking buffer for 1 hour at room temperature. Rinse the chip for 3 times with TBST, and then incubate with 1‰ Cy5-Streptavidin solution for 20 minutes at room temperature protected from light. The microarrays were spun dried for 2 minutes and scanned with a GenePix 4000B (Axon Instruments, USA). The GenePix Pro 6.0 was used for data analysis.
Statistical Analysis
All experiments were randomized and blinded. In all in vitro experiments, data represented 3 independent experiments and expressed as means ± SEM. Statistical analysis was performed with GraphPad Prism 6.0 software (San Diego, CA, USA). We used one-way ANOVA followed by Dunnett’s post hoc test when comparing more than two groups of data and one-way ANOVA, non-parametric Kruskal–Wallis test, followed by Dunn’s post hoc test when comparing multiple independent groups. P values of ˂ 0.05 were considered to be statistically significant. Post-tests were run only if F achieved P< 0.05 and there was no significant variance inhomogeneity.
Results
LG4 Treatment Improved Renal Function in T1DM Mice
The chemical structure of LG4 is shown in Figure 1A. To evaluate the effect of LG4 on DKD, we treated STZ-induced T1DM mice with LG4 (at 5 and 10 mg/kg/2 days) for 10 weeks. As shown in Figure 1B, the blood glucose levels of T1DM mice were higher than control group, and there was no difference in blood glucose between vehicle and LG4 treatment groups. During the administration period, mouse body weight was stable, and LG4 treatment showed no effect on the body weight of the T1DM mice (Figure 1C). Interestingly, BUN, UACR, and SCR, which are important indicators in assessing DKD, were significantly increased in the vehicle-treated group compared to the control group, and they were significantly decreased with LG4 treatment for 10 weeks in the T1DM mice (Figure 1D–F).
 |
Figure 1 LG4 treatment improved renal function in T1DM mice. (A) Chemical structure of LG4. (B and C) Type I diabetes mellitus mice (T1DM) were orally administrated with vehicle (Veh) or LG4 (5, 10 mg/kg, every other day) from week 8 to week 18. Blood glucose and body weight were measured every other week. (D–F) Blood Urea Nitrogen (BUN), Urine Albumin Creatinine Ratio (UACR), and serum creatinine (SCR) of each group were determined at 18th week. **p < 0.01; ***p < 0.001 vs Ctrl; #p < 0.05, ##p < 0.01 vs T1DM, n = 5. |
LG4 Attenuates Renal Fibrosis in T1DM Mice
To further evaluate the effect of LG4 on kidney morphological changes of the T1DM mice, H&E and Masson trichrome staining were conducted using the kidney tissues. Firstly, H&E staining showed classic DKD morphological changes in the kidney of the T1DM mice, including mesangial expansion, and LG4 treatment significantly improved these morphological changes (Figure 2A). Secondary, Masson trichrome staining and anti-collagen IV (COL-4) staining showed obvious accumulation of collagen in the glomerulus and tubulointerstitium of the T1DM mice, and LG4 treatment greatly mitigated these collagen accumulation in the kidney (Figure 2A–C). Meanwhile, glomerulosclerosis was assessed and scored according to Masson trichrome staining images. As shown in Figure 2D, the glomerular sclerosis index of T1DM group was up to 2.6 which means moderate sclerosis compared with control group. However, treatment with LG4 significantly downgraded the glomerulosclerosis. Furthermore, Western blot showed significant increases in COL-4 and TGF-β expression in the kidneys of the vehicle-treated T1DM mice, and they were ameliorated with LG4 administration (Figure 2E and F).
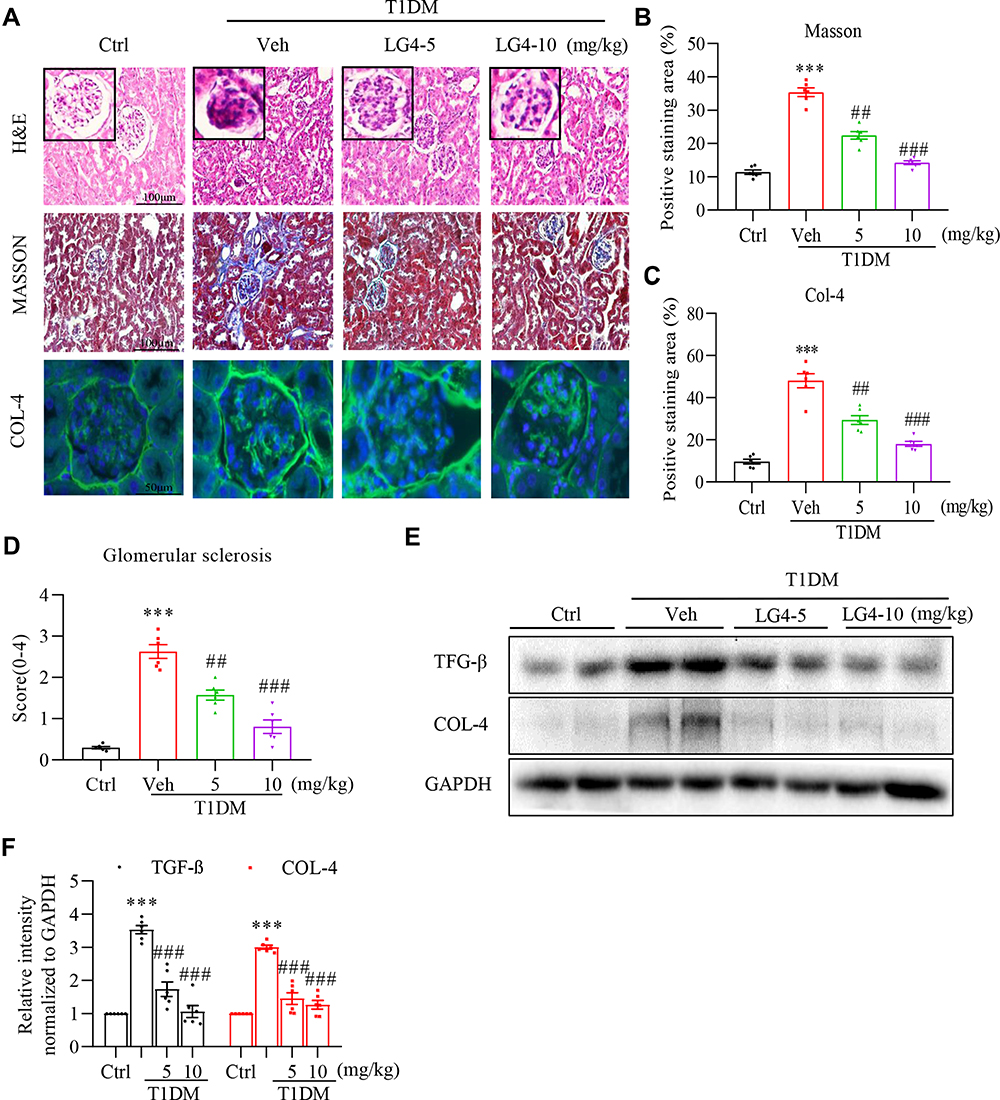 |
Figure 2 LG4 attenuates renal fibrosis in T1DM mice. (A) Kidney tissue sections were from the control, T1DM, and T1DM+LG4 mice. Representative images of H&E staining (200× magnification), Masson’s staining (200× magnification), and anti-COL-4 immunofluorescence staining (green, 400× magnification) were shown. (B and C) Quantitative date of the Masson’s (expressed as percentage of positive area) and COL–4 staining (expressed as relative fluorescence intensity). (D) Glomerular sclerosis score (0–4) was determined and plotted. (E and F) Western blot showed COL-4 and TGF-β levels in the kidney tissues, and relative protein levels to GAPDH were shown. ***p < 0.001 vs Ctrl; ##p < 0.01, ###p < 0.001 vs T1DM, n = 5. |
LG4 Treatment Inhibits Renal Inflammation in T1DM Mice
Chronic inflammation is one of the causing reasons for renal fibrosis in DKD. We then tested the effect of LG4 on pro-inflammatory cytokine expression in the kidney. Firstly, immunohistochemical staining showed increased TNF-α expression in the kidney of T1DM mice compared to control group (Figure 3A and B), and it was significantly blunted with LG4 treatment. Similarly, LG4 treatment significantly prevented the increases of transcription levels of Tnf-α and Il-6 in the kidney of T1DM mice (Figure 3C and D). Furthermore, the protein level of p-IκBα was enhanced in vehicle-treated T1DM mice, which was obviously suppressed with LG4 administration. The changes of IκBα and p-IκBα indicated LG4 inhibited diabetes-induced activation of NF-κB in diabetic mouse kidneys (Figure 3E and F).
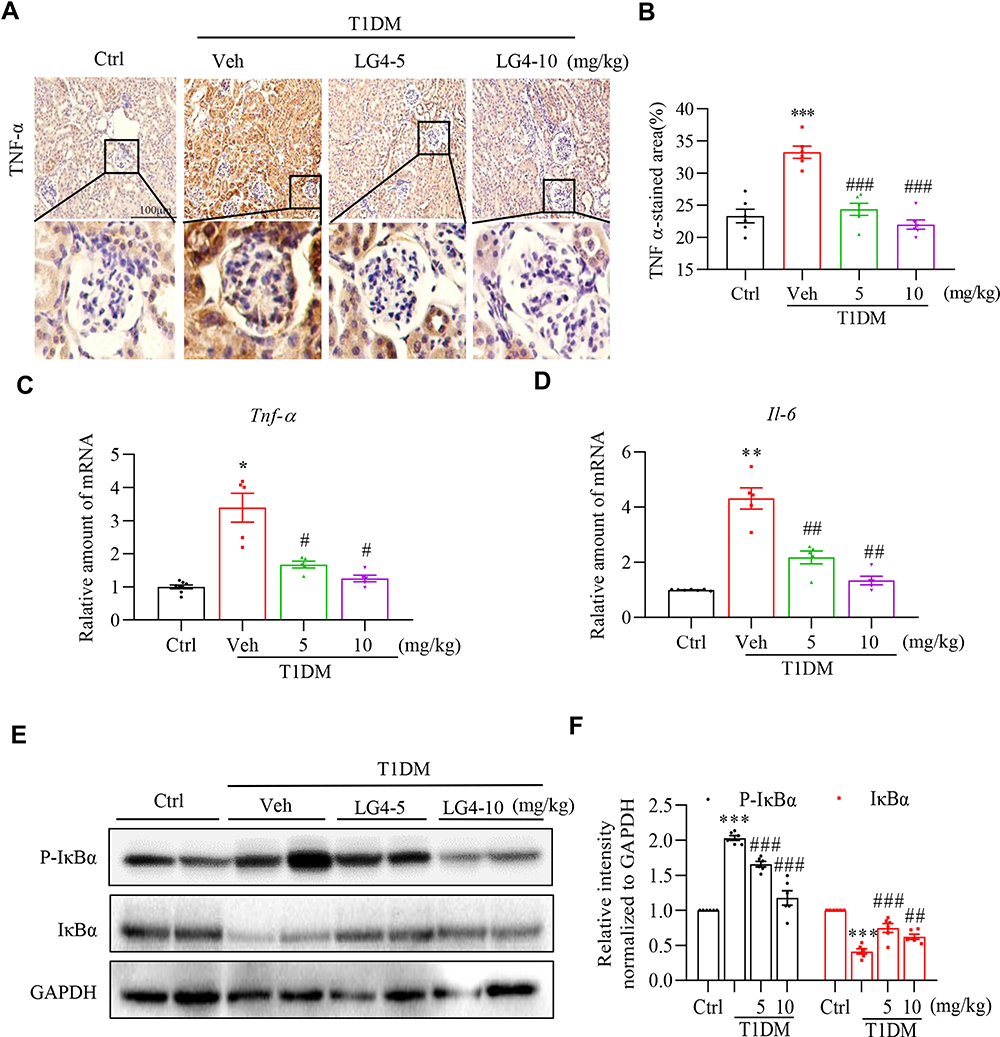 |
Figure 3 LG4 treatment inhibits renal inflammation in T1DM mice. (A and B) Representative images and quantitation of immunohistochemistry staining against TNF-α in the kidneys (200×). A typical view of glomerulus was selected and showed. (C and D) Real-time qPCR analyzed gene expression levels of Tnf-α and Il-6 in the kidneys. (E and F) Western blot assay detected p-IκBα and IκBα level in the kidneys. Relative densitometric quantification to GAPDH was shown. *p < 0.05, **p < 0.01, ***p < 0.001, T1DM vs control (Ctrl); #p < 0.05, ##p < 0.01, ###p < 0.001, T1DM + LG4 vs Veh, n =5. |
LG4 Directly Binds to Proteins of MAPKs Pathways
To explore the underlying mechanism of the renoprotection of LG4 in DKD, HuProtTM human proteasome microarray was used to screen potential targets of LG4. This methodology has been used to finding the molecular target of natural product in our previous study.24 Firstly, the biotin-labeled LG4 (LG4-biotin) (Figure 4A) was synthesized and structurally characterized as described in the supplementary file (Supplementary Figure S1–7). Next, the anti-inflammatory activity of LG4-biotin was confirmed in LPS-challenged mouse primary peritoneal macrophage. LG4-biotin and LG4 showed equal effect on suppressing LPS-induced IL-6 and TNF-α expression, indicating the biotin conjugation did not affect the bioactive structure of LG4 (Supplementary Figure S8). Then, the LG4-biotin and D-biotin were binding to the proteasome microarray, Figure 4B. Compared to the D-biotin control, a total of 406 proteins were identified with SNR >2 (p < 0.05). Pathway enrichment of the differential binding proteins are shown in Figure 4C. Among them, MAPKs were the top 2 signaling pathway that had direct binding activity to LG4. As shown in Figure 4D, LG4 could bind to MAPK10 (JNK3), MAPK9 (JNK2), MAPK8 (JNK1) and MAPK1 (ERK) that are key signal cascades of NF-κB pathway in response to external stimuli and production of pro-inflammatory cytokines.25,26
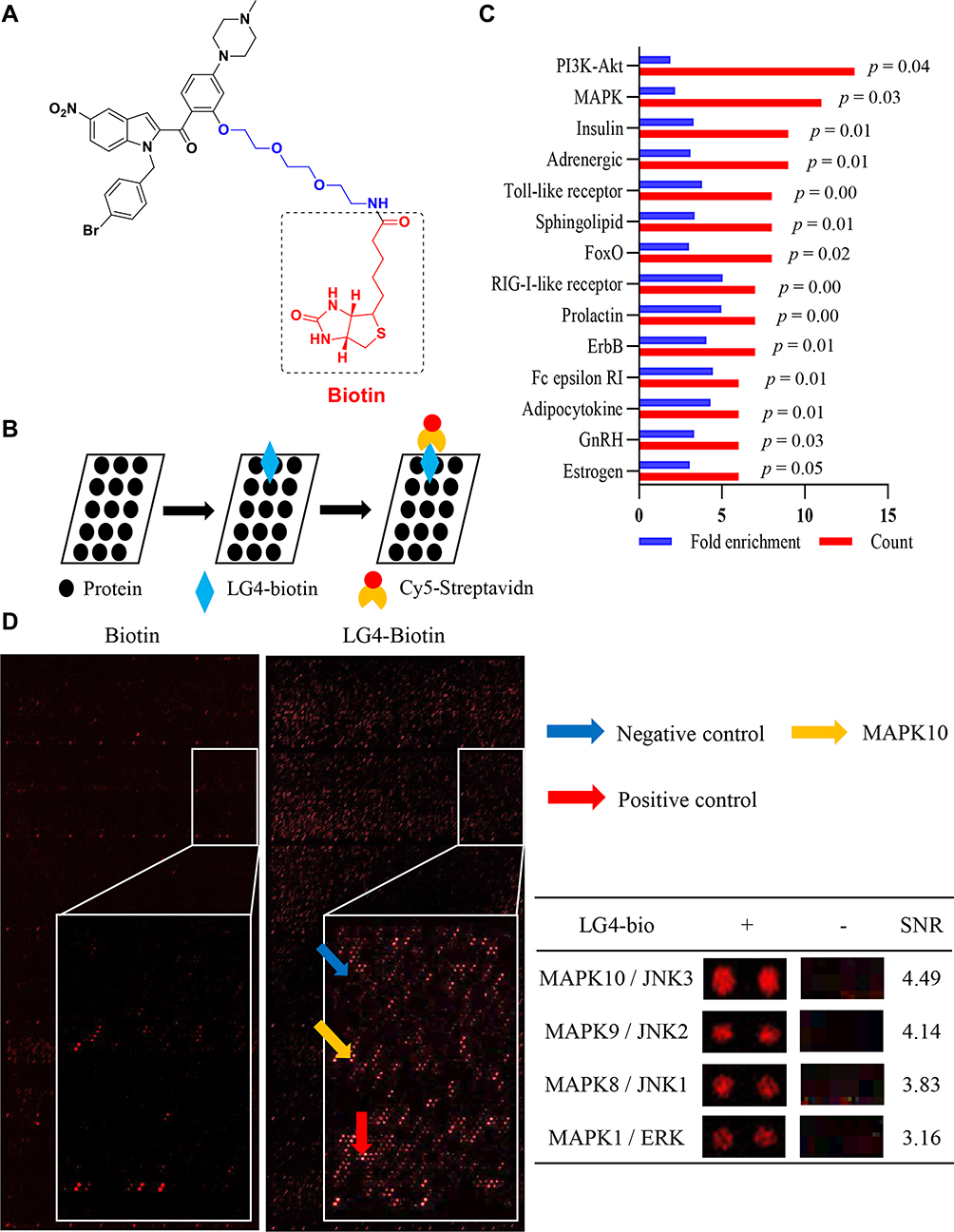 |
Figure 4 LG4 directly binding to proteins of MAPK pathways. (A) Chemical structure of biotin-labelled LG4. (B) Schematic showing the main steps for biotin or LG4-biotin using HuProtTM microarray. (C) Pathway enrichment of the differential binding proteins (fold enrichment ≥2 and p value < 0.05) were listed. (D) Representative image of the microarray showing positive (red arrow), negative (blue arrow) and MAPK10 (yellow arrow) spots. The table showed the magnified images of LG4-biotin binding to MAPKs spots and signal-to-noise ratios (SNR). |
LG4 Inhibits JNK and ERK Signaling in vitro and in vivo
To determine the effect of LG4 on modulating MAPKs activation, high glucose (HG)-treated SV40MES13 cells were pretreated with LG4 at 2.5 and 5 μM. HG significantly increased JNK and ERK phosphorylation, and these were blunted with LG4 pretreatment at 2.5 and 5 μM (Figure 5A and B). Similarly, phosphorylated JNK and ERK levels were significantly higher in the kidney of the vehicle-treated group than the control group, and LG4 treatment significantly suppressed this phosphorylation in the T1DM mice (Figure 5C and D).
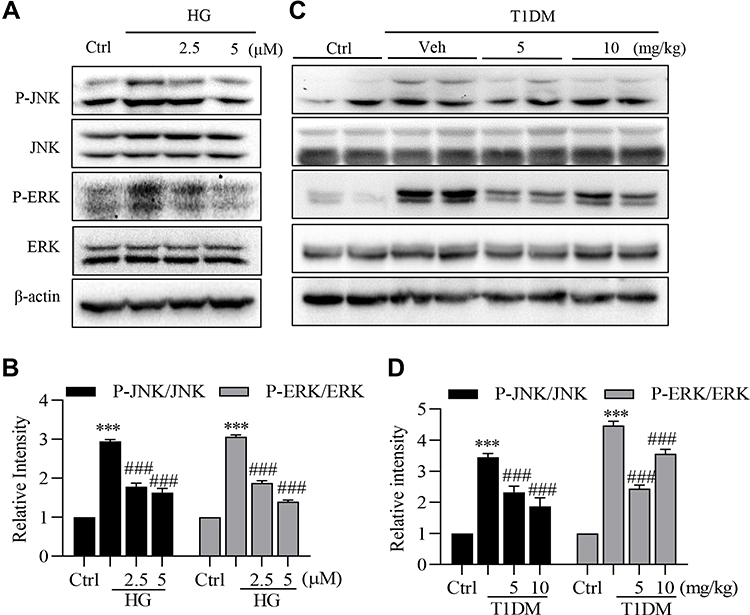 |
Figure 5 LG4 inhibits the activation of JNK and ERK in vivo and in vitro. (A and B) SV40MES13 cells were pretreated with LG4 (2.5 and 5 μM) for 1 h and incubated with HG (33 mM) for 2 h. Cells were harvested and processed for phosphorylated JNK and ERK detection by Western blot. GAPDH, total JNK and ERK were used as loading controls. Corresponding densitometric quantifications were shown. (C and D) Western blots detected p-JNK and p-ERK levels in the kidneys. Corresponding densitometric quantifications were shown. ***p < 0.001 vs Ctrl; ###p < 0.001 vs HG or Veh. |
LG4 Inhibits HG-Induced Inflammation via Targeting JNK and ERK Pathways
Next, we tested the role of LG4 in HG-induced inflammation in vitro. Firstly, HG induced IKK and IκBα phosphorylation and IκBα degradation in SV40MES13 cells, while these changes were prevented by LG4 pretreatment at 2.5 and 5 μM (Figure 6A), indicating that LG4 inhibited HG-induced NF-κB activation. Accordingly, LG4 pretreatment significantly inhibited HG-upregulated Il-6 and Tnf-α gene transcription in SV40MES13 cells (Figure 6B). As expected, JNK and ERK inhibition with SP600125 (10 μM) and U0126 (10 μM), respectively, significantly reduced HG-induced Il-6 and Tnf-α expression in SV40MES13 cells (Figure 6C and D), and LG4 had no further inhibitory effect on HG-induced Il-6 and Tnf-a expression at the presence of SP600125 or U0126. These data validated that the anti-inflammatory effect of LG4 might be through inhibiting MAPKs signaling.
 |
Figure 6 LG4 inhibits high glucose-induced inflammation via targeting JNK and ERK pathways. (A) SV40MES13 cells were pretreated with LG4 (2.5 and 5μM) for 1 h and incubated with HG (33 mM) for 2 h. Cells were harvested for p-IKK and Iκ-Bα analysis with Western blot. Corresponding densitometric quantifications were shown. (B) SV40MES13 cells were pretreated with LG4 (2.5 and 5 μM) for 1 h and incubated with HG (33 mM) for 8 h. Gene expression levels of Tnfa and Il6 were analyzed with real-time qPCR. (C and D) SV40MES13 cells were pretreated with LG4 (5 μM) with/without SP600125 (10 μM) or U0126 (10 μM) for 1 h, followed by incubation with 33 mmol/L HG for 8 h. Gene expression levels of Tnfa and Il6 were examined by RT-qPCR. Data were normalized by β-Actin level. **p < 0.01; ***p < 0.001 vs Ctrl. #p<0.05, ##p<0.01, ###p<0.001 vs HG, ns: no significant difference. |
Discussion
Inflammation plays a key role in the development and progression of DKD.27,28 Inflammatory responses are causal factors of glomerulosclerosis and tubulointerstitial fibrosis by inducing extracellular matrix deposition and differentiation/proliferation of myofibroblast. Therefore, blocking inflammation in kidney could be a strategy for the treatment of DKD. Hyperglycemia induces glomerular hyperfiltration, mechanical stress, and glycocalyx dysfunction that result in the overexpression of proinflammatory cytokines such as TNF-α, IL-6, and IL-1β through activating MAPKs and NF-κB signaling pathways in the kidney.12,13 From the prospective of molecular mechanism, MAPKs have been validated as upstream transductor of NF-κB activation in hyperglycemia.14 As a key transcription factor mediating inflammatory responses, NF-κB has been demonstrated to regulate the expression of a variety of inflammatory cytokines, chemokines, and profibrotic factors.29,30 Of note, NF-κB, JAK/STAT, and TGFβ/Smad signaling pathways have been demonstrated involving in the fibrotic process.15,16 Therefore, MAPKs/ NF-κB axis could be the potential therapeutic target for DKD.
Indole is one of the important structural motifs in medicinal chemistry and has shown considerable pharmacological activities against inflammatory conditions, hyperlipidemia, and tumor growth.31–33 LG4, a novel indole compound developed in our laboratory, has been shown to exert anti-inflammatory effects both in vitro and in vivo.34 In this study, we found that LG4 treatment remarkably reversed the function, histological abnormalities, fibrosis, and inflammation in the kidneys of T1DM mice without affecting their blood glucose and body weight. The effect of LG4 is highly related to its anti-inflammation property. However, a limitation of this study is the absence of in vivo pharmacokinetic data of LG4. The combination of pharmacokinetic (PK) and pharmacodynamic (PD) study will help to determine the potency of LG4 as a candidate for drug development. Our preliminary in vitro PK study demonstrated that LG4 was probably not metabolized by cytochrome P450, indicating that the first pass elimination effect is weak (Supplementary Figure S9). Our data show that LG4 is a candidate compound with potential clinical utility in prevention of DKD. Further in vivo study is necessary for the pre-clinical study of LG4.
Based on the proteasome microarray assay, LG4 showed significant interactions with proteins of multiple signaling pathways including PI3K-AKT, MAPK, toll–like receptor, etc. There may be multiple signaling pathways mediating the therapeutic effect of LG4. We focused on MAPK pathways in this study, because MAPKs are functionally connected kinases that regulate key cellular process involved in kidney disease such as all survival, death, differentiation and proliferation, and MAPKs also have been proved to mediate inflammation in the kidney injury.35–37 In this study, phosphorylation of MAPKs (including JNK and ERK1/2) was increased in HG-challenged SV40MES13 cells and diabetic mouse kidneys. Importantly, LG4 could directly bind to MAPKs (JNK and ERK) and inhibit HG/diabetes-induced MAPKs activation. Because LG4 did not show further anti-inflammatory effect in HG-challenged mesangial cells with the presence of JNK and ERK inhibitors, LG4 might exhibit anti-inflammatory effect through inhibiting MAPKs and NF-κB signaling pathways in kidney of T1DM mice.
However, a limitation of this study is that only a mouse model of STZ-induced type 1 DKD was used. DKD occurs in many patients with type 2 diabetes and a progression of DKD may be better reflected in type 2 diabetic mice.38,39 The renal protective effect of LG4 on DKD of type 2 diabetic mice needs to be further explored.
Conclusion
In conclusion, this study demonstrated a therapeutic role of LG4 in DKD through inhibiting renal inflammation and fibrosis in T1DM mice. Mechanistically, LG4 may exhibit anti-inflammatory and anti-fibrotic effects through directly binding to MAPKs and subsequent MAPK/NF-κB signaling inhibition.
Abbreviations
BAX, BCL2-Associated X; BCL2, B-cell lymphoma-2; BSA, Bovine serum albumin; BUN, blood urea nitrogen; CMC-Na, carboxymethyl cellulose sodium; COL-4, collagen IV; DAPI, 4’,6-Diamidino-2-Phenylindole, Dihydrochloride; DKD, diabetic kidney disease; DMEM, Dulbecco’s Modified Eagle Medium; DMSO, dimethyl sulfoxide; ERK, extracellular regulated protein kinases; ESRD, end stage of renal disease; FBS, fetal bovine serum; FITC, Fluorescein Isothiocyanate; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; H&E, Hematoxylin and Eosin; HG, high glucose; IKK, inhibitor of nuclear factor kappa-B kinase; IL-1β, interleukin-1β; IL-6, interleukin-6; IκBα, inhibitor of NF-κB; JAK, Janus Kinase; JNK, c-Jun N-terminal kinase; MAPK, activating mitogen-activated protein kinases; NF-κB, nuclear factor-kappa; NSAID, non-steroidal anti-inflammatory drug; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis; STAT, signal transducer and activator of transcription; STZ, streptozotocin; T1DM, type 1 diabetic mellitus; TGF-β, transforming growth factor-β; TNF-α, tumor necrosis factor α; UACR, urine albumin creatinine ratio.
Acknowledgments
This study was supported by the National Key Research Project (2017YFA0506000 to G.L.), National Natural Science Foundation of China (81803600 to J.Q., 21961142009 to G.L., 81970323 to M.X., 81973168 to Z.L. and 81900331 to Z.W.), Thailand Research Fund Grant (DBG6280006 to N.C.), and Zhejiang Provincial Key Scientific Project (2018C03068 to G.L. and 2021C03041 to G.L.).
Disclosure
The authors declare that they have no conflicts of interest.
References
1. Anders HJ, Huber TB, Isermann B, Schiffer M. CKD in diabetes: diabetic kidney disease versus nondiabetic kidney disease. Nat Rev Nephrol. 2018;14(6):361–377. doi:10.1038/s41581-018-0001-y
2. Tuttle KR, Bakris GL, Bilous RW, et al. Diabetic kidney disease: a report from an ADA Consensus Conference. Am J Kidney Dis. 2014;64(4):510–533. doi:10.1053/j.ajkd.2014.08.001
3. Wada J, Makino H. Inflammation and the pathogenesis of diabetic nephropathy. Clin Sci. 2013;124(3):139–152. doi:10.1042/CS20120198
4. Hasslacher C. Diabetic nephropathy: structural-functional relationships. Contrib Nephrol. 1989;73:24–28.
5. Lehman R, Gerber PA. End-stage nephropathy in type 1-diabetes mellitus - kidney transplantation alone or combined with islet or pancreas transplantation? Ther Umsch. 2011;68(12):699–706. doi:10.1024/0040-5930/a000233
6. Grande MT, Pérez-Barriocanal F, López-Novoa JM. Role of inflammation in túbulo-interstitial damage associated to obstructive nephropathy. J Inflamm. 2010;7(1):19. doi:10.1186/1476-9255-7-19
7. Schelling JR. Tubular atrophy in the pathogenesis of chronic kidney disease progression. Pediatr Nephrol. 2016;31(5):693–706. doi:10.1007/s00467-015-3169-4
8. Wee YM, Lee HW, Choi MY, et al. A composite of urinary biomarkers for differentiating between tubulointerstitial inflammation and interstitial fibrosis/tubular atrophy in kidney allografts. Ann Hepatobiliary Pancreat Surg. 2018;22(4):310–320. doi:10.14701/ahbps.2018.22.4.310
9. Abdel-Rahman EM, Saadulla L, Reeves WB, Awad AS. Therapeutic modalities in diabetic nephropathy: standard and emerging approaches. J Gen Intern Med. 2012;27(4):458–468. doi:10.1007/s11606-011-1912-5
10. Ogawa S, Mori T, Nako K, Ito S. Combination therapy with renin-angiotensin system inhibitors and the calcium channel blocker azelnidipine decreases plasma inflammatory markers and urinary oxidative stress markers in patients with diabetic nephropathy. Hypertens Res. 2008;31(6):1147–1155. doi:10.1291/hypres.31.1147
11. Perlman AS, Chevalier JM, Wilkinson P, et al. Serum inflammatory and immune mediators are elevated in early stage diabetic nephropathy. Ann Clin Lab Sci Spring. 2015;45(3):256–263.
12. Hamid T, Gu Y, Ortines RV, et al. Divergent tumor necrosis factor receptor-related remodeling responses in heart failure: role of nuclear factor-kappaB and inflammatory activation. Circulation. 2009;119(10):1386–1397. doi:10.1161/CIRCULATIONAHA.108.802918
13. Tamada S, Asai T, Kuwabara N, et al. Molecular mechanisms and therapeutic strategies of chronic renal injury: the role of nuclear factor kappaB activation in the development of renal fibrosis. J Pharmacol Sci. 2006;100(1):17–21. doi:10.1254/jphs.FMJ05003X4
14. Bantel H, Schmitz ML, Raible A, Gregor M, Schulze-Osthoff K. Critical role of NF-kappaB and stress-activated protein kinases in steroid unresponsiveness. FASEB j. 2002;16(13):1832–1834. doi:10.1096/fj.02-0223fje
15. Rayego-Mateos S, Morgado-Pascual JL, Opazo-Ríos L, et al. Pathogenic pathways and therapeutic approaches targeting inflammation in diabetic nephropathy. Int J Mol Sci. 2020;21(11):3798. doi:10.3390/ijms21113798
16. Yaribeygi H, Butler AE, Barreto GE, Sahebkar A. Antioxidative potential of antidiabetic agents: a possible protective mechanism against vascular complications in diabetic patients. J Cell Physiol. 2019;234(3):2436–2446. doi:10.1002/jcp.27278
17. Lamie PF, Ali WAM, Bazgier V, Rárová L. Novel N-substituted indole Schiff bases as dual inhibitors of cyclooxygenase-2 and 5-lipoxygenase enzymes: synthesis, biological activities in vitro and docking study. Eur J Med Chem. 2016;123:803–813. doi:10.1016/j.ejmech.2016.08.013
18. Abdellatif KR, Lamie PF, Omar HA. 3-methyl-2-phenyl-1-substituted-indole derivatives as indomethacin analogs: design, synthesis and biological evaluation as potential anti-inflammatory and analgesic agents. J Enzyme Inhib Med Chem. 2016;31(2):318–324. doi:10.3109/14756366.2015.1022174
19. Rani P, Srivastava VK, Kumar A. Synthesis and antiinflammatory activity of heterocyclic indole derivatives. Eur J Med Chem. 2004;39(5):449–452. doi:10.1016/j.ejmech.2003.11.002
20. Guerra AS, Malta DJ, Laranjeira LP, et al. Anti-inflammatory and antinociceptive activities of indole-imidazolidine derivatives. Int Immunopharmacol. 2011;11(11):1816–1822. doi:10.1016/j.intimp.2011.07.010
21. Amir M, Kumar S. Anti-inflammatory and gastro sparing activity of some new indomethacin derivatives. Arch Pharm. 2005;338(1):24–31. doi:10.1002/ardp.200400891
22. Liu Z, Tang L, Zhu H, et al. Design, synthesis, and structure-activity relationship study of novel indole-2-carboxamide derivatives as anti-inflammatory agents for the treatment of sepsis. J Med Chem. 2016;59(10):4637–4650. doi:10.1021/acs.jmedchem.5b02006
23. Al-Awar A, Kupai K, Veszelka M, et al. Experimental diabetes mellitus in different animal models. J Diabetes Res. 2016;2016:9051426. doi:10.1155/2016/9051426
24. Ye S, Luo W, Khan ZA, et al. Celastrol attenuates angiotensin ii-induced cardiac remodeling by targeting STAT3. Circ Res. 2020;126(8):1007–1023. doi:10.1161/CIRCRESAHA.119.315861
25. Lee MY, Yuk JE, Kwon OK, Oh SR, Lee HK, Ahn KS. Zuonin B inhibits lipopolysaccharide-induced inflammation via downregulation of the ERK1/2 and JNK pathways in RAW264.7 macrophages. Evid Based Complement Alternat Med. 2012;2012:728196.
26. Thalhamer T, McGrath MA, Harnett MM. MAPKs and their relevance to arthritis and inflammation. Rheumatology. 2008;47(4):409–414. doi:10.1093/rheumatology/kem297
27. Reidy K, Kang HM, Hostetter T, Susztak K. Molecular mechanisms of diabetic kidney disease. J Clin Invest. 2014;124(6):2333–2340. doi:10.1172/JCI72271
28. Wada J, Makino H. Innate immunity in diabetes and diabetic nephropathy. Nat Rev Nephrol. 2016;12(1):13–26.
29. Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441(7092):431–436. doi:10.1038/nature04870
30. Romeo G, Liu WH, Asnaghi V, Kern TS, Lorenzi M. Activation of nuclear factor-kappaB induced by diabetes and high glucose regulates a proapoptotic program in retinal pericytes. Diabetes. 2002;51(7):2241–2248. doi:10.2337/diabetes.51.7.2241
31. Chadha N, Silakari O. Indoles as therapeutics of interest in medicinal chemistry: bird’s eye view. Eur J Med Chem. 2017;134:159–184. doi:10.1016/j.ejmech.2017.04.003
32. Kaushik NK, Kaushik N, Attri P, et al. Biomedical importance of indoles. Molecules. 2013;18(6):6620–6662. doi:10.3390/molecules18066620
33. Shattat G, Al-Qirim T, Sheikha GA, et al. The pharmacological effects of novel 5-fluoro-N-(9,10-dihydro-9,10-dioxoanthracen-8-yl)-1H-indole-2-carboxamide derivatives on plasma lipid profile of Triton-WR-1339-induced Wistar rats. J Enzyme Inhib Med Chem. 2013;28(4):863–869. doi:10.3109/14756366.2012.692085
34. Liu Z, Chen L, Yu P, et al. Discovery of 3-(Indol-5-yl)-indazole derivatives as novel myeloid differentiation protein 2/toll-like receptor 4 antagonists for treatment of acute lung injury. J Med Chem. 2019;62(11):5453–5469. doi:10.1021/acs.jmedchem.9b00316
35. Cuarental L, Sucunza-Sáenz D, Valiño-Rivas L, et al. MAP3K kinases and kidney injury. Nefrologia. 2019;39(6):568–580. doi:10.1016/j.nefro.2019.03.004
36. Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev. 2012;92(2):689–737. doi:10.1152/physrev.00028.2011
37. Yeung YT, Aziz F, Guerrero-Castilla A, Arguelles S. Signaling pathways in inflammation and anti-inflammatory therapies. Curr Pharm Des. 2018;24(14):1449–1484. doi:10.2174/1381612824666180327165604
38. Flyvbjerg A. The role of the complement system in diabetic nephropathy. Nat Rev Nephrol. 2017;13(5):311–318. doi:10.1038/nrneph.2017.31
39. Greco EV, Russo G, Giandalia A, Viazzi F, Pontremoli R, De Cosmo S. GLP-1 receptor agonists and kidney protection. Medicina. 2019;55(6):233. doi:10.3390/medicina55060233
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.